

Summary
In the embryonic heart, the activation of the mitochondrial electron transport chain (ETC) coincides with the closure of the cyclophilin D (CypD) regulated mitochondrial permeability transition pore (mPTP). However, it remains to be established whether the absence of CypD has a regulatory effect on mitochondria during cardiac development. Using a variety of assays to analyze cardiac tissue from wildtype and CypD knockout mice from embryonic day (E)9.5 to adult, we found that mitochondrial structure, function, and metabolism show distinct transitions. Deletion of CypD altered the timing of these transitions as the mPTP was closed at all ages, leading to coupled ETC activity in the early embryo, decreased citrate synthase activity, and an altered metabolome particularly after birth. Our results suggest that manipulating CypD activity may control myocyte proliferation and differentiation and could be a tool to increase ATP production and cardiac function in immature hearts.
Subject areas: Biological sciences, Physiology, Cell biology, Metabolomics
Graphical abstract
Highlights
-
•
Bioenergetic capacity increases during cardiac development
-
•
These changes correlate to the physiologic state and energetic demands
-
•
CypD deletion alters this metabolic trajectory, notably in the embryo and newborn
-
•
Manipulation of CypD activity might increase cardiac function in immature hearts
Biological sciences; Physiology; Cell biology; Metabolomics
Introduction
Mouse cardiac progenitor cells are present by about embryonic (E) day 8.0, cardiac contractions are first seen at E8.25, and cardiac morphogenesis is relatively complete by E14.1,2 During this time, cardiomyocytes undergo dramatic changes including the maturation of the cytoskeleton,3 closure of the mitochondrial permeability transition pore (mPTP;4), initiation of electron transport chain (ETC) activity, and the formation of mitochondrial supercomplexes.5 Further myocyte differentiation and bioenergetic maturation occurs during the fetal stages of development.1,2 Birth profoundly alters the physiologic environment, acutely exposing the heart and other tissues to higher oxygen levels, while dramatically increasing demands on the heart to perfuse the body. These changes induce a final burst of myocyte proliferation followed by terminal differentiation (now called cardiomyocyte maturation), which coincides with a transition from glycolytic and lactate metabolism to fatty acid oxidation as a source of ATP production.1,2,6,7,8
Published data support the idea that bioenergetic changes actually regulate cardiac development. Gestational/neonatal hypoxia carries an increased risk for congenital heart disease.9 The transition from the relatively hypoxic uterine environment to atmospheric O2 at birth is a switch that regulates changes in metabolism and differentiation.8,10,11 Hypoxic signaling and regulation of mitochondrial dynamics control the switch to fatty acid metabolism, mitochondrial biogenesis, morphology, function, and ROS production,10,11,12 which in turn regulates the balance of myocyte proliferation and differentiation.7,8 Furthermore, models of hypoplastic left heart syndrome, a severe form of congenital heart disease, have defects in mitochondrial structure and function.13,14 Additional data suggests that mitochondria play a major role in cardiac development. Mitochondria of early embryonic cardiomyocytes (E9.5) are fragmented and have little structural complexity, a low membrane potential, and high levels of reactive oxygen species (ROS), but mitochondria in late-embryonic (E13.5) cardiomyocytes contain a filamentous network, high membrane potential, and low ROS levels.4 These changes correlate with an increased in myocyte differentiation,4 and activation of ETC activity at about E11.5.5 The four ETC complexes (Cx I, II, III, and IV) transport electrons generated by the oxidation of NADH and FADH2 to create a membrane potential and proton gradient across the inner mitochondrial membrane that is used by ATP synthase (ETC Cx V) to make ATP. In WT hearts, ETC activity was not observed in E9.5, but around E11.5, Cx II and then Cx I activity became apparent, and by E13.5 the ETC was as active as in adult mitochondria.5
One mechanism that could explain low mitochondrial membrane potential in embryonic myocytes is sustained opening of the mPTP, which is regulated by the mitochondrial peptidyl-prolyl, cis-trans isomerase cyclophilin D (CypD, Ppif). The mPTP is a large conductance pore in the inner mitochondrial membrane whose identity remains unclear, although data from the last 10 years suggests that it is derived from ATP synthase and perhaps adenine nucleotide translocase (ANT, Slc25a4-6).15,16,17 Over the years, a number of proteins have been proposed to be components of the mPTP, including CypD, mitochondrial phosphate carrier (PiC, Slc25a3), voltage-dependent anion channel (VDAC, Vdac1-3), and paraplegin (Spg7), but these were found to be regulators and not actual pore forming proteins due to genetic deletion studies (reviewed in16). Our work has concentrated on the role of CypD in the regulation of the mPTP, although we have also used inhibitors and activators of ANT to study mechanisms of mPTP-mediated regulation of myocyte differentiation.4,18,19,20 For example, when mPTP closure was induced by deleting or inhibiting CypD, E9.5 myocytes quickly developed a more mature mitochondrial phenotype and increased differentiation compared to wildtype (WT) controls.4 Furthermore, genetic or pharmacologic inhibition of CypD in neonatal hearts and cultured myocytes increased cardiac systolic function in vivo and myocyte differentiation in vitro.19 From these studies, we concluded that the regulation of mPTP closure plays an important role during cardiac myocyte differentiation at specific stages of development, but details and continuity of this process are only superficially understood.
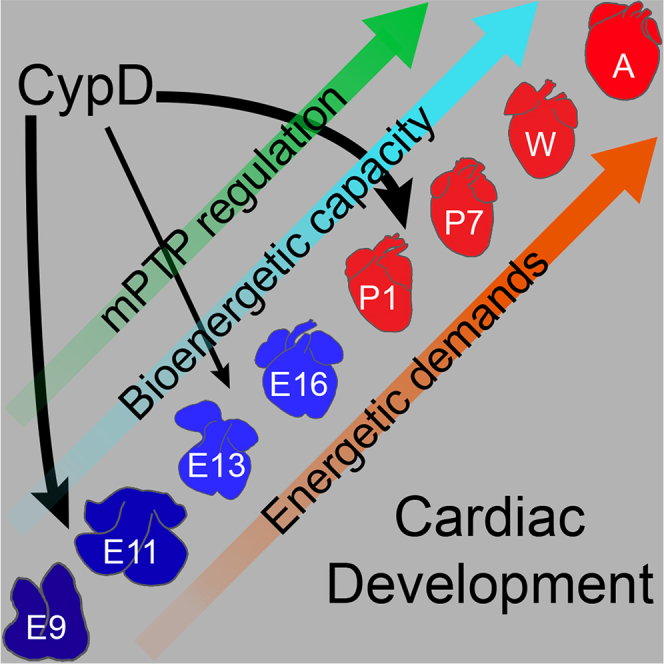
In summary, recent data suggests that myocyte differentiation and cardiac function during development are controlled by metabolism and mitochondria. We and others have studied this subject during limited windows of development.4,5,7,8,11,12,19,21,22,23,24,25 Herein, a detailed and comprehensive study of cardiac mitochondrial bioenergetics during the entire period of cardiac development from the early embryo to the adult demonstrates that structural, bioenergetic, and metabolic maturation occurs gradually with more abrupt transitions at certain stages of development. Furthermore, this maturation is altered by the deletion of CypD.
Results
Mitochondrial morphology: More complex mitochondria in the early embryo of cyclophilin D KO hearts
To comprehensively examine cell morphometry throughout cardiac development, electron micrographs of left ventricular myocytes from WT and CypD KO mice were analyzed for cell size and the cellular area composed of mitochondria, myofibrils, and nuclei (Figures 1 and S1–S6). Myocyte size was stable from E9.5 to P1 and then progressively increased, and differences were observed between WT and CypD null myocytes only in the adult (Figures 1C and S4, adult WT: 117 versus 760 μm2, CypD KO: 142 versus 1101 μm2). The area of the myocyte containing contractile apparatus/myofibrils was about 14% of the cell at E9.5 and rose to ∼50% at the weaning and adult stages (Figures 1D and S4). In contrast, nuclei composed 20–30% of the prenatal cell and decreased from P1 to adult, likely due to the increase in cell size (Figures S1, S2A, S3, S4, and S6A). Throughout development, mitochondrial area and number increased, but not in a coordinated manner; mitochondrial area (as % cell area) rose at E16.5 and P1 and further thereafter, while mitochondrial number rose slowly before birth and then more rapidly thereafter (Figures 1E, 1F, and S4). CypD deletion did not change mitochondrial number, but CypD KO myocytes had a larger mitochondrial area in adult hearts.
Figure 1.

Myocyte and mitochondrial morphology and biogenesis during cardiac development
(A) Graphical representation of work flow with silhouettes of hearts at various ages. Birth is marked in red on the timeline.
(B) Representative electron micrographs of E9.5, P1, and Adult (A) WT cardiac myocytes.
(C–J) Morphometry was performed using low- and high-powered electron micrographs, a tablet to outline cellular structures, and Fiji/ImageJ. Shape descriptors were copied into Excel spreadsheets and data was compiled for statistical analysis. Myocyte morphometry from electron micrographs shows changes in cell size (C), contractile apparatus area (D) and mitochondrial area (E) as a percentage of cell area, and mitochondrial number per cell (F) at each age and genotype (WT, CypD KO). Morphometry of individual mitochondria shows changes in mitochondrial area (G) and form factor (H) during development. Plot of mitochondria subgroups (% of total mitochondrial number/cell (D)) in WT (I) and CypD KO (J) mitochondria (C-cytoplasmic, IM-intermyofibrillar, PN-perinuclear, SS-subsarcolemmal).
(K) Mitochondrial to nuclear (mt/nuc) DNA ratio of WT and CypD KO hearts at different ages using primers to mt-CO1 and 18s rRNA for qPCR.
(L) Expression of the voltage-dependent anion channel (VDAC) in homogenates from hearts at each age and genotype analyzed by densitometry of denaturing immunoblots normalized to protein loading (A.U., arbitrary units). Data presented as Mean ± SEM and analyzed by one-way ANOVA with Sidak’s multiple comparison test (C-F, I-L) or Median +/− interquartile (IQ) range and Kruskal-Wallis with Dunn’s multiple comparison test (G, H) to compare data between WT and CypD KO cells at each age (red stars, C-H, K, L) and for differences between successive ages of the same genotype (black stars/bar, K). Only significant differences noted, ∗p <0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001; N: C-F, I, J = 9 cells or 18 cells for E11.5 and E13.5, G-H presented in G above the X axis, K = 3–6, L = 3. See also Figures S1–S6.
Mitochondrial function is related to mitochondrial shape in a complex relationship, so we analyzed mitochondrial shapes using morphometry (Figures 1 and S1–S6). The size of individual WT mitochondria, represented by area, perimeter, and Feret diameter, generally increased from E13.5 to E16.5, decreased at P1, increased at P7 and weaning, and decreased in the adult (Figures 1E, 1H, S2B, S2C, S5, and S6B). Deletion of CypD yielded larger mitochondria at E13.5 and in the adult. Mitochondrial shape descriptors (aspect ratio (AR), form factor (FF), and Feret diameter) were analyzed; higher values correlate to increased mitochondrial elongation and complexity (Figures 1H, S2C, S2D, S5, and S6B). In WT myocytes, AR and FF rise between E13.5 and E16.5, fall at P1, rise again at P7 and then are stable (FF) or fall (AR) in the adult. CypD deletion made embryonic mitochondria more complex, particularly at E11.5, and lessened the decrease in AR and FF at birth. However, although CypD mitochondria were larger in mature cells, their morphometry was less complex (Figures 1E–1H, S2B, S2C, S5, and S6).
We then compared the cell and mitochondrial parameters of myocytes from the myocardial wall and trabeculae in E11.5 and E13.5 hearts. These two populations are thought to be more proliferative (wall) or more differentiated (trabeculae). We found limited differences in these groups (Figure S6). We then compared mitochondrial parameters in different cellular locations, as location may dictate structure and function (Figures 1I and 1J). Mitochondrial locations in mature myocytes are usually described as intermyofibrillar, subsarcolemmal, and perinuclear, but we added a “cytoplasmic” category, as these mitochondria were not associated with any particular structure. In embryonic myocytes, cytoplasmic mitochondria predominate but disappear by P1 (Figure 1I). Intermyofibrillar mitochondria peak first in the later embryo and then again in weanling and adult cells, while perinuclear mitochondria peak in the fetus (E16.5). Subsarcolemmal mitochondria are more abundant in postnatal myocytes. Deletion of CypD did not seem to affect overall trends in mitochondrial localization (Figure 1J). Analysis of mitochondrial size and shape by subgroups revealed no other major or consistent differences between groups or genotypes.
Electron transport chain expression during cardiac development
In addition to the structural maturation of the cardiac mitochondria, mitochondrial DNA copy number and ETC gene expression should increase to accommodate mitochondrial respiration as the heart develops. The ratio of the mitochondrial/nuclear (mt/nuc) DNA is stable throughout the entire developmental period except for a marked increase in the WT adult that is not seen in the CypD KO adult; this does not follow the same trajectory as mitochondrial mass from electron micrographs (Figures 1E, 1F, and 1K). Note that our data on this ratio are similar to previous reports during this period of mouse, bovine, and human development.11,26,27,28 Quantitative analysis of VDAC expression by immunoblotting, to measure mitochondrial mass, revealed stable expression throughout development in WT hearts but continuously increased in CypD KO hearts (Figure 1L) and is therefore not suitable for normalization.
Analyzing ETC expression by qRT-PCR and immunoblotting showed that the expression of the genes Ndufa1 (Cx I), Sdha (Cx II), Uqcrq (Cx III), and Atp5e (Cx V) was relatively stable from E16.5 to P7 in both WT and CypD KO hearts and increased significantly in the weaning and adult hearts of WT but not CypD KO mice (Figure 2A). The Cx IV protein COX8 undergoes isoform switching during cardiac maturation,29 and this is reflected in a similar pattern of the Cox8b/Cox8a ratio, although this switch was not present in postnatal CypD KO hearts (Figure 2A). Expression of the CypD gene (Ppif) increased in the postnatal period (Figure 2A). Finally, we found that the neonatal switch in myosin heavy chain gene expression (Myh6/7) was exaggerated in postnatal CypD KO hearts, but there was no change in the troponin I gene (Tnni3/1) switch between genotypes (Figure 2A).
Figure 2.

CypD deletion alters mitochondrial gene and protein expression in the developing heart
(A) qRT-PCR analyses of mitochondrial (Ndufa1, Sdha, Uqcrq, Cox8a, Cox8b, Atp5e, and Ppif) and contractile apparatus (Myh6, Myh7, Tnni1, Tnni3) gene expression. Data for Cox8b/a, Myh6/7, and Tnni3/1 are expressed as ratios and all data was normalized to the expression of Polr2A.
(B–G) Representative immunoblot images (B) and graphs of the expression of NDUFB6 (C), SDHB (D), UQCRC2 (E), MtCO1 (F), and ATP5A (G) and CypD (H) demonstrate changes in proteins levels during development in tissue homogenates. Data from immunoblots for ETC proteins (C-G) was normalized to protein loading (A.U., arbitrary units) and not VDAC labeling due to data in Figure 1L. Note: an additional lane of adult homogenate in the top WT panel was removed from between E13.5 and E16.5 for clarity.
(H) CypD protein expression normalized to VDAC expression is stable during cardiac development in WT hearts. Data is presented as Mean ± SEM and analyzed by one-way ANOVA with Tukey’s or Sidak’s multiple comparison test to compare data between WT and CypD KO cells at each age (red stars in A) and also between successive ages of the same genotype (WT-underlined black stars, CypD KO-underlined gray stars). Only significant differences noted, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001; N: A = 3–6, C-H = 3 hearts/age and genotype.
In contrast to gene expression, proteins from each ETC complex, as analyzed by the immunoblotting of homogenates (Figures 2B–2G), showed a continuous increase in expression in WT hearts for NDUFB8 (Cx I) with a plateau at later ages for SDHB (Cx II), UQCRC2 (Cx III), MtCO1 (Cx IV), and ATP5A (Cx V). The pattern was similar with CypD deletion, except for a generally lower expression of the nuclear encoded Cx I subunit NDUFB8 in CypD KO hearts after E11.5, which matches the gene expression of Ndufa1 (Figures 2A and 2C). Conversely, SDHB detection was always higher in CypD KO hearts (Figures 2B and 2D). CypD protein expression was stable throughout the span of cardiac development in WT hearts (Figures 2B and 2H).
Electron transport chain activity: coupled oxidative phosphorylation at E9.5 in cyclophilin D KO hearts
The enzymatic activities of Cx I and II increase from E9.5 to E13.5 in tissue homogenates from WT hearts.5 New data confirm these experiments, but also show that the deletion of CypD increases the activity of NADH-ubiquinone dehydrogenase and succinate-ubiquinone dehydrogenase at E9.5 compared to WT (Figures 3A–3E), while NADH-cytochrome c DH activity was measureable only in CypD KO homogenates (Figure 3D). Isolated mitochondria show relatively stable Cx I, II, and V activities from E16.5 to A in both WT and CypD KO hearts, with no age-specific differences between the genotypes (Figures 3B–3G). Citrate synthase activity, the entry point for acetyl-coenzyme A (Ac-CoA) into the tricarboxylic acid (TCA) cycle, increased significantly in WT hearts from E13.5 to E16.5 in homogenates and was stable from E16.5 to adults in isolated mitochondria (Figure 3H). Interestingly, citrate synthase activity was consistently about 50% lower in CypD KO hearts from E16.5 to adult (Figure 3H), while citrate synthase protein levels did not change (Figure 3I); therefore, we did not use this activity to normalize ETC activity between WT and CypD KO samples.30,31
Figure 3.

Deletion of CypD alters embryonic ETC, OXPHOS, and citrate synthase activity throughout development
(A) Schematic of ETC activity and electron flux (e−); c/Cytc: cytochrome c, ETC complexes: I, II, III, IV and V, ubiquinone: q.
(B–G) Assays of ETC Cx I, II, and V activity in embryonic heart homogenates or fetal and postnatal isolated heart mitochondria (left and right side of panels, respectively) were performed in a spectrophotometer and represent: NADH oxidase (B, NADH->NAD+ in Cx I), NADH:Ubiquinone dehydrogenase (DH) (C, electron flux from NADH through Cx I to q), NADH:Cytochrome c DH (D, electron flux from NADH through Cx I to Cytc), Succinate:Ubiquinone DH (E, electron flux from succinate to q via Cx II), Succinate:Cytc DH (F, electron flux from succinate to Cytc), and ATP hydrolase (G, reverse reaction of ATP synthase/Cx V).
(H) Citrate synthase activity in embryonic homogenates (left) and isolated mitochondria from fetal to adult hearts (right).
(I) Citrate synthase protein expression in homogenates from embryonic to adult hearts by immunoblotting and normalized to protein loading (au, arbitrary units).
(J–N) OXPHOS activity was measured in a Clark oxygen electrode in homogenates from hearts throughout development.
(J–L) Substrate-mediated oxygen consumption (V0, succinate for E9.5-E11.5, malate/glutamate E13.5-A), maximal oxygen consumption (Vmax, substrate +ADP), and respiratory control ratio (RCR, = Vmax/V0, a measure of coupling electron transport to ATP generation) values at each age and genotype.
(M and N) Composite RCR data (M) and representative traces (N) of oxygen consumption in WT (+/−1 μm CsA) and CypD KO E9.5 heart homogenates; arrows in N denote the addition of 10 mM succinate (Suc), 1 mM ADP, and 0.1 mM atractyloside (ATR). V0 was measured after adding succinate, Vmax was measured after adding ADP, and rates of oxygen consumption are denoted by yellow bars. Data is presented as Mean ± SEM and analyzed by one-way ANOVA with Tukey’s or Sidak’s multiple comparison test to compare data between WT and CypD KO hearts (red stars) at each age and also between successive ages of the same genotype (WT-underlined black stars, CypD KO-underlined gray stars). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001. N: (B–H) 3–12 (I) 3, and (J-M) 6–15 samples per age/genotype.
We next examined oxidative phosphorylation (OXPHOS) activity in tissue homogenates. Even though the proteins of the ETC are expressed at E9.5 (Figures 2B–2G), OXPHOS was uncoupled with indistinguishable V0 (substrate mediated) and Vmax (maximal respiration) and a low respiratory control ratio (RCR = Vmax/V0) in WT but not in CypD KO hearts (Figures 3J–3L). Throughout the rest of development, the RCR was relatively stable due to a gradual increase in maximal respiration (Vmax) after the fetal stage and an equivalent increase in V0 (Figures 3J–3L). Inhibition of CypD at E9.5 with 1 μM cyclosporin A (CsA) in WT homogenates caused a spontaneous decrease of V0, while Vmax remained unchanged, and the RCR increased to that of E9.5 CypD KO homogenates or later ages (Figures 3J–3N). Note that succinate (Cx II substrate) was used in experiments using E9.5 and E11.5 hearts as there was inconsistent V0 respiration at these ages using malate and glutamate (Cx I substrate).
The probability of mitochondrial permeability transition pore activity decreases as the heart matures
The effect of CsA on RCR in WT E9.5 hearts suggested an acute regulation of CypD and possibly the mPTP (Figures 3M and 3N;5). Therefore, we determined the CRC in isolated mitochondria from E16.5 to adult hearts. The Ca2+ concentration required to open the mPTP was low in E16.5 WT mitochondria and steadily increased (Figures 4A–4D). Interestingly, Ca2+-induced mPTP opening was not as sensitive to CsA in the pre- and neonatal periods compared to older hearts (W and A, Figure 4D). The CRC was significantly higher in CypD KO hearts at E16.5, while the inhibition of CypD by CsA in E16.5 WT hearts did not result in a significant increase (Figures 4B–4E). Inhibition of CypD by CsA increased the CRC in adult WT hearts to that of adult CypD KO hearts (Figures 4E and 4F). These results suggest that the probability of mPTP activity decreases during differentiation as its acute regulation by CypD increases.
Figure 4.

mPTP open probability decreases during cardiac development
The calcium retention capacity (CRC) is a measure of mPTP. Mitochondria are exposed to pulses of calcium in the presence of arsenazo III in the solution, which fluoresces upon the binding of calcium. With each pulse, fluorescence increases and then decreases as mitochondria sequester the calcium until the calcium concentration in the mitochondrial matrix rises to a level that opens the mPTP (the CRC [nmol Ca2+/mg mitochondrial protein]), releasing all the calcium and causing a plateau in the arsenazo fluorescence.
(A–C) Representative CRC traces from E16.5 WT +/−1 μM CsA (A), E16.5 CypD KO +/−1 μM CsA (B), and Adult WT and CypD KO (C) heart mitochondria. Calcium pulses were 10 μM in E16.5 and 20 μM in Adult experiments.
(D) CRC of WT mitochondria from E16.5, P1, P7, weanling (W) and adult (A) hearts, +/− 1 μM CsA.
(E and F) CRC levels in mitochondria from WT and CypD KO E16.5 (E) and adult (F) hearts +/− treatment with 1 μM CsA. Data in D-F is presented as Mean ± SEM and analyzed by one-way ANOVA with Tukey’s or Sidak’s multiple comparison test. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗∗p < 0.0001. N: 3–5 samples per condition.
Metabolomic pathway analysis: Earlier metabolic maturation after birth in cyclophilin D KO hearts
Steady-state metabolomic analysis to examine changes in metabolic pathways during cardiac development identified 64 metabolites that were consistently recovered at all ages (Figures 5 and S7–S14). Sparse partial least squares-discriminant analysis (sPLS-DA) demonstrated a developmental stage-dependent sorting of WT samples (Figures 5B and S7C). Component 1 accounted for changes between the prenatal and postnatal samples, while Component 2 accounted for changes between E9.5 and E13.5/E16.5; no specific metabolic pathways were highly represented in these two components (Figure S7B). Testing for significant changes between ages of WT hearts demonstrated that the greatest changes in specific metabolite levels occur between E11.5 and E13.5, and from E16.5-weaning (Figures S8 and S10–S14). Notable changes in specific pathways include: The upper half of glycolysis increased throughout development, while the lower half was generally stable with significant changes in some metabolites at the embryonic/fetal transition (E13.5/E16.5) and after P1. UDP-glucose and UDP-Glc-Nac were lower after P7, suggesting decreased glycogen production and Glc-N-acylation. TCA cycle metabolite levels were generally lowest from E13.5 to P1. 2-hydroxyglutarate was elevated prenatally, likely due to its role in hypoxic signaling.32 For redox metabolites, NAD+ and NADP+ rose during development, and the ratios of reduced to oxidized NAD(P) were generally very low except at P1. In contrast, GSH levels were relatively stable while GSSG levels rose leading to high oxidized/reduced glutathione (GSSG/GSH) levels after the embryonic period. However, ascorbate levels varied during development but were almost non-existent at birth, while carnosine was not found until P7. In general, amino acids tended to be higher prenatally. Ketone levels were high in the embryo with a minor peak around birth, while fatty acid metabolites were also higher in the embryo.
Figure 5.

Metabolomic profile of WT and CypD KO hearts during development
Sixty four metabolites were recovered by steady state, LC MS/MS metabolomics from 7 hearts at each indicated age (E9.5, E11.5, E13.5, E16.5, P1, P7, W, A) and each genotype (WT, CypD KO).
(A) Heatmap of mean data for each age and genotype. Metabolites are grouped along the Y axis as metabolic pathways.
(B) Sparse partial least squares-discriminant analysis plots of all WT groups and P1 and P7 CypD KO groups shows individual specimens represented as small circles and groups as larger, similarly colored ovals, while each group is marked by age. Percentage of variability explained for Components 2 and 1 are as labeled. All metabolomics data were analyzed together in MetaboAnalyst to create this plot, but some groups (CypD KO E9.5, E11.5, E13.5, E16.5, W, A) were selectively removed to make the plot legible. WT and CypD KO data are shown separately in additional plots in Figure S7C.
(C) Volcano plots of metabolite changes in WT versus CypD KO hearts at each age during development, reported as the -log p value versus the log FC (fold change, CypD KO values/WT values) with green dashed lines demarcating a q value of 0.05 (horizontal) and FC of 0.75 and 1.5 (vertical). Significantly different metabolites are marked by red circles with their identity indicated. “Other” metabolites indicate that one group had essentially a 0 value while the other group had a measurable value.
(D) Depiction of changes in cardiac myocyte physiology, maturation, fuel supply and mitochondrial function observed during the development of the WT heart from E9.5 to adult. ∗ denotes the timing of significant changes in CypD KO compared to WT hearts (note that probability of mPTP opening is different throughout development between the two genotypes). See also Figures S7–S14.
Next, we determined the effects of CypD deletion on metabolite levels. The sPLS-DA analysis demonstrated no outstanding differences from WT in metabolic patterns prenatally and in mature hearts, but the P1 and P7 CypD KO hearts were shifted leftward along the Component 1 axis, suggesting that the deletion of CypD induces earlier metabolic maturation after birth (Figures 5B and S7C). Comparing WT and CypD KO metabolites revealed no or only scattered differences in glycolysis, the glycogen and hexose biosynthesis pathways, nucleotides, Ac-CoA biosynthesis, ketones, and fatty acid metabolism (Figures 5C and S9–S14). However, levels of pentose-5-phosphate were higher after P1 in the CypD KO hearts. TCA metabolites were generally similar in the two mouse lines, but with decreased succinyl-CoA and increased fumarate at E13.5 and decreased aconitate and succinate and increased oxaloacetate in mature (W, A) CypD KO hearts. There were no significant differences in most amino acids in CypD KO hearts, but changes were seen for aspartate, serine, and threonine during development. Most redox metabolites were unchanged in KO hearts, except for ascorbate (increased at E9.5 and E11.5, decreased at P1) and carnosine (earlier and higher peak at P1-P7).
Discussion
During cardiac morphogenesis, the heart undergoes many physiologic transitions that are dictated by its changing environment.1,2,6,7,8,11,24,25 The comprehensive survey of bioenergetics presented here demonstrates a gradual increase of the bioenergetic potential during cardiac development with distinct points of bioenergetic transitions in the early embryo, the late embryo, and the neonatal heart (Figure 5D). Thus, the changes in cardiac physiology during development are reflected in concordant changes in cardiac bioenergetics. Furthermore, we demonstrate that the deletion of CypD alters this developmental bioenergetic trajectory to enhance mitochondrial function and myocyte differentiation (Figure 5D).
A gradual increase in bioenergetic potential during cardiac development
The demands placed on the heart to perfuse the body gradually increase as the body grows and becomes more active, and this is reflected in most parameters of mitochondrial biology we report herein. Our data suggest that mitochondrial activity and coupling reaches a maximum well before birth, likely due to the fact that OXPHOS is the predominate source of ATP in the fetus, regardless of the substrates used. Changes are likely regulated by oxygen availability, oxidative stress, hypoxia inducible factor (HIF) and other related mechanisms that are being actively investigated by ours and other labs.7,8,11
During the entire period of cardiac development, there is an increase in cell size and myofibrillar area/volume, mitochondrial mass (both as percent area of and number of mitochondria in the cell) as well as size and complexity of individual mitochondria (Figures 1 and S1–S6) that we and other have observed.22,33 Changes in mitochondrial subcellular localization may reflect changes in cellular structure and mitochondrial demands as myocytes differentiate. “Cytoplasmic” mitochondria are more prevalent in the prenatal heart, when the cell has fewer mitochondria, a less-constraining cellular structure, and a reliance on mitochondria for cataplerosis. Subsarcolemmal mitochondria are always present, likely to provide energy for sarcolemmal transporters. Perinuclear mitochondria are more prominent during times of rapid proliferation and may provide metabolites for nucleotide synthesis and epigenetic regulation. Intermyofibrillar mitochondria peak in the late embryo and then again in mature hearts as might be expected to provide ATP for contraction, but the reason for lower levels at E16.5 are unclear.
Consistent with an increase in cell size and mitochondrial mass is a continuous increase in the expression of ETC proteins throughout development, while VDAC and CypD remained relatively unchanged (Figure 2). However, gene expression for these ETC proteins is relatively low until after birth and increases after P7 (Figure 2), suggesting a low turnover rate of ETC proteins during development. After the embryonic period, the enzymatic activity of Cx I, II, and V are relatively stable, which is consistent with the increase of V0 and Vmax after E16.5 (Figure 3) and with previous work for Cx V but not for NADH oxidase activity.31,32 We also find that the probability of mPTP opening slowly decreases during cardiac development, consistent with previous work.4,5,19 However, this does not necessarily correlate with a change in OXPHOS coupling other than at E9.5 (Figure 4;4,5), suggesting a complex regulation of the mPTP (discussed later in discussion).
From a broader metabolomic standpoint, sPLS-DA analysis reveals a clear developmental trajectory in WT and CypD KO mice with significant changes between hearts of different ages, but significant changes in individual pathways were not observed (Figures 5 and S7–S14). However, changes in some pathways and metabolites may represent competition between energy production and anaplerosis/cataplerosis,34 as discussed later in discussion.
The early embryonic transition
The most significant transition in mitochondrial function occurred between E9.5 and E11.5 in the WT heart, due to a drop in the probability of mPTP opening associated with the activation of the ETC. NADH oxidase activity (Figure 3B) and the expression of Cx I and II mRNA and proteins (Figures 2A–2D) were stable from E9.5 to 11.5, but Cx I and II activity (Figures 3C–3F) increased significantly over this time. Furthermore, E9.5 and E11.5 mitochondria did not respond to the Cx I substrates, malate and glutamate, in oxygen consumption experiments, while E11.5, but not E9.5, mitochondria respired using the Cx II substrate, succinate (Figures 3J–3N). Combined with our previous work,4,5 this suggests that Cx II, and then Cx I, activates and begins to transport electrons to Cx III and IV at around E11.5, when oxygen levels rise in the mouse embryo due to the development of the placenta.
This activation of the ETC is reflected in the differences in mitochondrial cristae morphology4 but not in overall mitochondrial morphology (Figures 1 and S1–S6). However, we note that this bioenergetic transition is associated with an increase in contractile apparatus area between E9.5 and 11.5 (Figure 1D), which likely reflects increased myocyte differentiation, as seen in cultured myocytes from these two ages.4 However, in E11.5 and E13.5 hearts, we did not see major differences in the morphometry of trabecular and wall myocytes, which are reported to be more and less differentiated, respectively (Figure S6).
Metabolomic analysis revealed a rapid decline in ascorbate (vitamin C) and GSH/GSSH levels from E9.5 to E11.5 that may reflect the fall in mitochondrial ROS levels we have observed in early cardiac myocytes.4 In addition, the fall in succinate levels could reflect the initiation of ETC activity through Cx II, while alterations in other metabolites may reflect changes in proliferation, as discussed later in discussion (Figures 5 and S7–S14).
Thus, increased embryonic oxygen concentrations at E11.5 due to the establishment of placental circulation are associated with an increase in aerobic glycolysis reflected by the activation of the ETC, alterations in mitochondrial structure, and changes in metabolic intermediates. Combined with our previous data and recent reports using advanced metabolic flux analysis, our results support the importance of this early embryonic transition.4,5,35
The embryonic-to-fetal transition
A second transition in cellular and mitochondrial structure, but not of mitochondrial function, occurs between E13.5-E16.5 when the mouse embryo becomes a fetus. Total mitochondrial area increases and individual mitochondria become larger and more complex (Figures 1 and S1–S6). These structural changes do not correspond with major changes in ETC gene or protein expression or mitochondrial oxygen consumption. Whether the activity of ETC Cx I and II or mPTP opening changes at this time is not clear due to limitations in the ability to isolate enough purified mitochondria at E13.5 and earlier (Figure 3). Furthermore, metabolomic results may indicate a shift in metabolism at this transition; after E13.5 changes in component 1 of the sPLS-DA analysis predominate (Figures 5 and S7C). However, we acknowledge that neither the individual metabolites in this component nor volcano plots of metabolomics data reveal specific metabolic pathways that are altered at this transition (Figures S7B, S7C, S8, and S10–S14).
The postnatal transition/neonatal myocyte maturation
Birth acutely exposes the heart to increased oxygen supply and subsequent oxidative stress as well as increased functional and bioenergetic demands that necessitate changes in myocyte and mitochondrial structure and function.7,10,11,12 Many of the parameters we studied (increased mitochondrial area, ETC protein expression, V0, and Vmax, most metabolomics data) demonstrated a gradual increase in bioenergetic capacity that likely reflect the gradual increase in mitochondrial mass and activity required for increased cardiac function after birth.
However, a few notable transitions occurred in the immediate postnatal period. First, the area and structural complexity of individual mitochondria transient decreased at P1 (Figures 1G, 1H, S2B–S2D, S5, and S6B). Second, ETC gene expression dramatically increased after P7 (Figure 2A).
Third, a few notable changes in individual metabolite levels may reflect metabolic control of important pathways that regulate myocyte maturation. 1) Ketone levels, which were very high in the embryo, peak again at birth, perhaps reflecting their role in postnatal myocyte maturation.11,21,24 2) Changes in a number of metabolites may reflect their role in cardiac myocyte proliferation, differentiation, and epigenetics. Carnosine levels, which are inversely proportional to proliferation and proportional to differentiation,36 peaked at P7 in WT hearts and suggest a regulatory role for carnosine during myocyte maturation.7 Urate has also been associated with myocyte differentiation,37 and high levels in the more mature hearts may reflect the terminal differentiation state. Elevated aspartate levels from E11.5 to P7 may reflect its role in proliferation.38,39 Total Ac-CoA levels were low until P7, likely reflecting the metabolic switch to fatty acid oxidation around this age. 3) Metabolites involved in one-carbon metabolism (methionine, serine, and threonine) show no clear patterns that suggest a metabolic regulation of histone and DNA methylation during cardiac development, although changes in ascorbate levels may regulate DNA demethylation to modulate myocyte differentiation and were lower after P1.40,41 4) Low ATP levels and energy charge after P1 could indicate that the creatine phosphate shuttle system may be active.6
Mechanisms of bioenergetic maturation in the developing heart
Our data demonstrate that the bioenergetic maturation of the heart, from the early embryo to the mature animal, occurs gradually with a few important periods of transition (Figure 5D). These acute and chronic changes in mitochondrial function and metabolomics are undoubtedly linked to the changing physiology that occurs during development. The gradual increase in mitochondrial function and capacity provides the energy for increased cardiac function that supports the growing organism. However, at a few notable stages of development, changes in the physiologic environment appear to drive metabolic transitions that acutely increase bioenergetic capacity and support this gradual increase in bioenergetic capacity. At the early embryonic transition (E9.5-E11.5), when the embryo requires a functioning placenta and heart for survival, and cardiac metabolism becomes more aerobic as oxygen levels rise, there is an acute activation of the ETC and changes in mitochondrial cristae structure (demonstrated herein and in previous articles4,5). At the embryonic-fetal transition (E13.5–16.5), when the development of the coronary arteries allows the growth of the compact myocardium to increase cardiac output, there is an increase in mitochondrial mass (area and number) per cell and perhaps a change in metabolic programming likely reflect increased the differentiation of the fetal myocytes. At the final transition at birth, acute increases in oxygen levels, oxidative stress, metabolic and cardiac demands, and changes driving myocyte maturation likely drive changes in mitochondrial structure and gene expression and levels of particular metabolites.
There are many cellular mechanisms that may drive and control these acute and chronic changes in mitochondrial biology and metabolism. Changes in oxygen levels discussed above likely regulate cellular redox balance that can regulate many intracellular signaling pathways, cardiac transcription factors, and epigenetics to regulate mitochondrial gene expression and function as well as myocyte differentiation. A number of important metabolic enzymes are regulated by ROS (e.g., aconitase), while ROS regulates the activity of important transcription factors (e.g., HIF and nuclear factor erythroid 2-related factor 2 (Nrf2)) to regulate metabolic gene transcription. Since mitochondria are a major source of ROS, there must be some level of feedback control of metabolism, but other cellular factors may modify this control. Indeed, we have shown that mitochondrial-regulated ROS is an important regulator of myocyte differentiation,4,19 but the specific mechanisms by which this occurs during cardiac development remain unclear and are the subject of current investigations.
Metabolism is rife with feedback regulation, and the changes in different metabolites likely regulate the changes in bioenergetics we have observed. First, changing substrate availability during development may play a role in this regulation. For example, it is thought that the early embryo relies on glucose as a fuel in part because of its easy diffability through the maternal tissue and the lack of oxygen to enable OXPHOS. In addition, it was recently found that γ-linolenic acid, via the regulation of retinoid X receptors, controls the balance of glucose and lipid oxidation during cardiac development.42 Second, changes in metabolite levels at each stage of development can regulate metabolism in the next stage of development. For example, changes in placental arginine may regulate urea cycle metabolites in the embryonic placenta, and different organs may respond to these stimuli in different manners.35
Finally, although the regulation of gene expression likely plays a role in the developmental trajectory of metabolism, there is an interesting lack of correlation between the gradual changes in mitochondrial mass, function, and protein expression and the changes in ETC gene expression and the mtDNA-to-nuclear DNA ratio we observed. It was recently demonstrated the mRNA expression does not track with protein expression in the developing heart,25 and changes in mitophagy in the postnatal period could explain changes in mitochondrial biogenesis.12 Perhaps future experiments will explore whether different mechanisms control different aspects of mitochondrial function during cardiac development.
Cyclophilin D deletion alters the developmental trajectory of bioenergetics during cardiac development
We specifically explored another mechanism that controls mitochondrial biology in the developing heart: Regulation of the mPTP by the mitochondrial chaperone, CypD. We previously demonstrated that the inhibition of CypD closed the embryonic and neonatal mPTP to decrease mitochondrial ROS production and increase mitochondrial membrane potential, myocyte differentiation, and postnatal cardiac function.4,19
The results reported here suggest that CypD deletion generally has subtle effects on the gradual changes in the metabolic trajectory of the developing heart, but a number of more dramatic changes are worthy of discussion. At the early embryonic metabolic transition, CypD deletion increased electron flux from both Cx I and II, decreased V0, and increased RCR (Figures 3 and 4), supporting our hypothesis that CypD controls not only the closure of the mPTP, but also the assembly of ETC complexes and possibly supercomplexes in the embryonic heart.4,5 The embryonic-fetal transition was not greatly affected by CypD deletion, but there was less of a gap in the sPLS-DA analysis between E11.5 and E13.5 CypD KO hearts compared to controls and aspartate levels were lower (Figures 5B and S7C).
Other significant changes occurred in the postnatal period, where the deletion of CypD caused a leftward shift along the Component 1 axis of P1 and P7 hearts, suggests an earlier metabolic maturation after birth (Figures 5B and S7C). This was not reflected in mitochondrial ETC activity or in cellular and mitochondrial morphometry data, where differences were seen only between adult WT and CypD KO hearts. However, compared to WT hearts, CypD deletion caused a significant decrease in mitochondrial gene expression at weaning and adult ages; similar changes were apparent for mtDNA levels and Cx I, but not other ETC, protein levels (Figures 1 and 2). Furthermore, consistent with our previous data showing increased function and myocyte differentiation of neonatal hearts when CypD was inhibited,19 we found an increase in Myh6/Myh7 (but not Tnni3/Tnni1) gene expression and cell size in postnatal CypD null hearts (Figures 1C and 2A). In addition, earlier changes in carnosine levels, which peaked at P7 in WT hearts and slightly earlier in CypD KO hearts, may indicate that this switch occurs early with CypD deletion/inhibition (Figures 5C and S11).
To determine whether the deletion or inhibition of CypD, we isolated mitochondria and tested for sensitivity to calcium-induced mPTP opening using CRC assays. First, we found a decrease in the probability of mPTP opening from E16.5 to adult in WT mitochondria. The same pattern was seen in CypD KO mitochondria or when mitochondria were exposed to the CypD inhibitor, CsA. However, at each age, the CRC was higher with CypD inhibition, but the sensitivity to CsA increased with increasing age (Figure 4), consistent with a complex regulation of CypD itself.43
Potential mechanisms of cyclophilin D’s regulation of mitochondrial function during cardiac development
Based on these data, CypD appears to regulate OXPHOS during cardiac development, perhaps via a number of mechanisms. As far as has been determined, the major function of CypD is to control the mPTP, an important regulator of mitochondrial function and cell death. Therefore, one might expect a significant phenotype with CypD deletion, but this does not cause embryonic lethality or shorten life-span. However, CypD deletion is not benign: 1. Global deletion of CypD decreases cardiac metabolic flexibility and increases heart failure and mortality under stressful conditions,44 and 2. Embryonic litters of CypD KO mice have a wider range of apparent development than wild type (WT) litters.4
Previous work suggested that elevated mitochondrial Ca2+ levels in CypD KO mice enhanced TCA cycle activity to increase basal OXPHOS activity, but this led to a detrimental metabolic inflexibility, while gene expression and subsequent proteomics data demonstrates the disruption of metabolic pathways.44,45 We proposed an alternative mechanism: CypD increases the assembly of ETC supercomplexes, increasing ETC efficiency but causing detrimental counter-regulation of OXPHOS and the mPTP.46 Since mPTP opening appears to be less sensitive to the pharmacologic inhibition of CypD by CsA in the fetal heart, CypD may exhibit less control of mPTP opening early compared to later in development.43 Despite this, CypD inhibition can effectively regulate myocyte differentiation and cardiac function in the newborn,19 making CypD a potential therapy target for neonatal cardiomyopathies.
It is also possible that CypD performs other, more cryptic regulatory functions in mitochondria. We have demonstrated that it regulates supercomplex assembly in the mature heart via unclear mechanisms.5 Furthermore, CypD is known to bind to other important mitochondrial proteins, such as ANT, PiC, and p53,16,20 the latter of which could mediate CypD’s control of mitochondrial gene transcription in the postnatal period. CypD deletion has also been shown to increase protein acetylation in the mature heart, which could alter mitochondrial function.47 The low citrate synthase activity in CypD KO hearts after E13.5 (Figure 3H) may indicate effects on TCA cycle activity due to increased efficiency of the ETC or direct effects on TCA cycle enzymes. However, this result is not congruent with increased pyruvate and α-ketoglutarate dehydrogenase activities observed previously in this mouse line.44 For now, these data suggest that citrate synthase activity, which is generally regulated by product inhibition and substrate availability,31 may not be a necessarily suitable marker for mitochondrial content and ETC activity between samples.30,31 Further work is ongoing to better understand these and other mechanisms of CypD’s regulation of cardiac myocyte physiology as well as its regulation by post-translational modifications such as oxidation and acetylation.
It is possible that other mPTP regulators may play a role in mPTP regulation during cardiac development. ANT and PiC were shown to regulate but not comprise the mPTP, but more recent data suggests that ANT may compose a low-conductance form of the mPTP.16 Paraplegin was found to link VDAC to CypD and regulate the mPTP,48 but no study demonstrates cardiac developmental issues related to its expression or function, although iPSC-derived neurons containing patient-specific mutations in paraplegin have abnormal cellular and mitochondrial morphology that is rescued by the mPTP activator, Bz-423.49 VDAC is an outer membrane protein commonly used to normalize mitochondrial protein expression in immunoblotting experiments. Genetic deletion experiments demonstrated that it is not essential for mPTP formation (reviewed in16), but questions linger as to its role in mPTP regulation.48 Postnatal cardiac ventricular myocyte deletion of Vdac2 caused a lethal, dilated cardiomyopathy in adult mice via changes in calcium homeostasis,50 but there are no other reports of cardiac developmental defects with the deletion of any other VDAC isoforms. Finally, global CypD deletion in the heart decreased the expression of VDAC345 and VDAC1/2 (Figure 1L), but the developmental significance of this remains unclear. Thus, the role of these other mPTP-associated proteins in the changes we have seen in bioenergetic during cardiac is unclear and may warrant further study.
Conclusions
The data presented here provide a broad profile of bioenergetics and describe the changes that occur in metabolism and mitochondrial structure and function during cardiac morphogenesis, a complex process that begins in the early embryo and ends well after birth. We also show the coordinated metabolic effects of CypD deletion, which highlight its importance in regulating OXPHOS, the mPTP, and myocyte differentiation (Figure 5D). Finally, these results suggest that the manipulation of CypD activity could be useful to increase ATP production and cardiac function in immature hearts.
Limitations of the study
There are several limitations of these experiments. The range of material obtained from hearts at different ages varied greatly, so some experiments in embryonic hearts were not performed or required pooling of hearts. Three-dimensional electron microscopic imaging would give additional information on cellular and mitochondrial morphometry, but this was not feasible given the scope of this work. Electron microscopy allowed us to specifically evaluate myocytes, but such specificity was not possible in other experiments. Metabolomic experiments were performed at steady state, but technical difficulties (e.g., limited biomass, inability to perfuse immature hearts) precluded utilizing metabolomics flux experiments.35,51 Finally, we did not systematically relate the changes in metabolism to changes in cardiac function, although we have reported this for P1 and P7 WT and CypD KO heart,19 and future work will explore changes in cardiac function in other models of CypD manipulation.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Citrate synthase (rabbit anti-) | Thermo Fisher Scientific | Cat #PA5-22126; RRID: AB_11154268 |
| Cyclophilin D (CypD, mouse anti-) | Thermo Fisher Scientific | Cat #45-5900; RRID: AB_2533820 |
| Oxphos Rodent Cocktail (mouse anti-) | Abcam | Cat #110413; RRID:AB_2629281 |
| Voltage dependent anion channel (VDAC) (rabbit anti-) | Abcam | Cat #154856; RRID: AB_2687466 |
| Goat ANTI-MOUSE IgG StarBright™ Blue 700 | Biorad | Cat #12004158; RRID: AB_2884948 |
| Goat ANTI-MOUSE IgG StarBright™ Blue 520 | Biorad | Cat #12005866; RRID: AB_2934034 |
| Goat ANTI-RABBIT IgG StarBright™ Blue 700 | Biorad | Cat #12004161; RRID: AB_2721073 |
| Goat ANTI-RABBIT IgG StarBright™ Blue 520 | Biorad | Cat #12005869; RRID: AB_2884949 |
| Chemicals, peptides, and recombinant proteins | ||
| Power Sybr™ Green PCR Master Mix | Thermo Fisher Scientific | Cat #4367659 |
| Arsenazo III | Sigma-Aldrich | Cat #A92775 |
| NADH | Sigma-Aldrich | Cat #N8129 |
| DCPIP | Sigma-Aldrich | Cat #1878 |
| Cytochrome c | Sigma-Aldrich | Cat #C2037 |
| Acetyl CoA | Sigma-Aldrich | Cat #A2056 |
| Ubiquinone/Coenzyme Q | Sigma-Aldrich | Cat #C7956 |
| Blocking reagent | Biorad | Cat #1706404 |
| Bovine Serum Albumin | Millipore | Cat #126575 |
| Potassium Cyanide | Sigma-Aldrich | Cat #60178 |
| Rotenone | Sigma-Aldrich | Cat #R8875 |
| Triton X-100 | Sigma-Aldrich | Cat #28314 |
| Cyclosporin A | Cayman | Cat #12088 |
| Critical commercial assays | ||
| BCA kit | Thermo Fisher Scientific | Cat #23225 |
| Direct-Zol™ RNA/DNA Miniprep kit | Zymo Research | Cat #R2089 |
| Maxima First Strand cDNA Synthesis Kit | Thermo Scientific | Cat #K1641 |
| Experimental models: Organisms/strains | ||
| Mouse (Wild Type): C57Bl6/N | Charles River | RRID:MGI:2159965 |
| Mouse (CypD KO): B6;129-Ppiftm1Jmol/J | JD Molkentin4 | RRID:IMSR_JAX:009071 |
| Deposited data | ||
| Electron micrographs: Adult Hearts (.jpg) | This paper; Figshare | https://doi.org/10.6084/m9.figshare.25050323 |
| Electron micrographs: Weaning Hearts (.jpg) | This paper; Figshare | https://doi.org/10.6084/m9.figshare.25050407 |
| Electron micrographs: P7 Hearts (.jpg) | This paper; Figshare | https://doi.org/10.6084/m9.figshare.25050437 |
| Electron micrographs: P1 Hearts (.jpg) | This paper; Figshare | https://doi.org/10.6084/m9.figshare.25050449 |
| Electron micrographs: E16.5 Hearts (.jpg) | This paper; Figshare | https://doi.org/10.6084/m9.figshare.25050452 |
| Electron micrographs: E13.5 Hearts (.jpg) | This paper; Figshare | https://doi.org/10.6084/m9.figshare.25050461 |
| Electron micrographs: E11.5 Hearts (.jpg) | This paper; Figshare | https://doi.org/10.6084/m9.figshare.25050467 |
| Electron micrographs: E9.5 Hearts (.jpg) | This paper; Figshare | https://doi.org/10.6084/m9.figshare.25050473 |
| Spreadsheets of morphometry data (.xlsx) | This paper; Figshare | https://doi.org/10.6084/m9.figshare.25050044 |
| Immunoblots (.pdf) | This paper; Figshare | https://doi.org/10.6084/m9.figshare.25062779 |
| Metabolomics analyses (.xlsx) | This paper; Figshare | https://doi.org/10.6084/m9.figshare.25050284 |
| Oligonucleotides | ||
| See Table S1 | ||
| Software and algorithms | ||
| GraphPad Prizm 9.5.1 | Graphpad | https://www.graphpad.com/ |
| Image Lab 6.1 | Biorad | https://www.bio-rad.com/en-us/product/image-lab-software?ID=KRE6P5E8Z#fragment-6 |
| Fiji/Image J | NIH | https://imagej.nih.gov/ij/index.html |
| Photoshop | Adobe | https://www.adobe.com/creativecloud.html |
| Illustrator | Adobe | https://www.adobe.com/creativecloud.html |
| XCaliber 4.2 | Thermo | Xcalibur™ Software (thermofisher.com) |
| MetaboAnalyst v5.0 | MetaboAnalyst | |
| Softmax Pro 7 | Molecular Devices | https://www.moleculardevices.com/products/microplate-readers/acquisition-and-analysis-software/softmax-pro-software |
| Other | ||
| Potter-Elvehjem Type Tissue Grinder, 10 mL | VWR | VWR®, Tissue Grinders, Potter-Elvehjem Type | VWR |
| ChemiDoc MP | Biorad | ChemiDoc™ MP Imaging System #12003154 | Bio-Rad |
| Trans-Blot Turbo Transfer System | Biorad | Trans-Blot Turbo Transfer System – Bio-Rad |
| Spectramax 384 Plus | Molecular Devices/VWR | SpectraMax® Plus 384 Absorbance Plate Reader, Molecular Devices | VWR |
| CFX384 Touch Real-Time PCR detection system | Biorad | CFX384 Touch Real-Time PCR Detection System | Bio-Rad |
| Hansatech Oxygraph | PP Systems | https://ppsystems.com/oxygen-measurement/ |
| Hitachi 7650 TEM | Hitachi | 817-9624 TG-S |
| Erlangshen 1000W Camera | Gatan | Model 785 |
| Shimadzu Prominence 20 LC system | Shimadzu | https://www.shimadzu.com/an/products/liquid-chromatograph-mass-spectrometry |
| Phenomenex Synergi RP-Fusion column | Phenomenex | https://www.phenomenex.com/Products/Synergi-hplc-column |
| Cintiq tablet | Wacom, Saitama, Japan | https://www.wacom.com/en-us/products/pen-tablets |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, George A. Porter, Jr. (george_porter@urmc.rochester.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
-
•
Original data (immunoblots, spreadsheets, and electron micrographs) have been deposited at https://figshare.com/ and are publicly available as of the date of publication. DOIs are listed in the key resources table.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental model and study participant details
Animals
Ethics Procedures were in strict accordance with the Division of Laboratory Animal Medicine, University of Rochester, in compliance with state law, federal statute, and NIH policy and were approved by the Institutional Animal Care and Use Committee of the University of Rochester (University Committee on Animal Resources (UCAR) protocol 2011-003). C57Bl/6N wild type (Charles River, RRID:MGI:2159965) and mixed background CypD global KO (local colony originally received from Dr. Jeffrey Molkentin, RRID:IMSR_JAX:009071) female mice were paired to male mice of the same strain. Pregnant female mice were sacrificed by cervical dislocation to prevent bioenergetic changes induced by CO2 or anesthetics, and the embryos were harvested from the mother immediately. Embryos were harvested on embryonic day (E)9.5, E11.5 and E13.5 and E16.5 (range E15.5-17.5) based on timed mating and the embryo morphology. Mouse pups at postnatal day (P)1 and P7 were sacrificed by decapitation and the hearts were immediately removed. Adult (A, 2-3 months old) and weanling (W, P21-28) mice were sacrificed by cervical dislocation and the hearts were harvested immediately. Weights of animals/hearts harvested were not obtained, and all animals appeared healthy at the time of harvest. Animals were stored in standard housing with male mice were kept in single cages between matings, which were timed to create litters of embryonic and fetal mice. Samples from each litter were randomized for further analyses, but there were no treatments that required randomization. Animals were not used in previous experiments. Unless indicated, experiments used tissue from the ventricles and the outflow tract. Sex as a biological variable was not examined in this manuscript, as gender could not be rapidly determined in time to harvest and study samples in many experiments in mice from most ages examined. This may limit interpretation of these data in terms of gender differences at older ages, but it is unclear if this limits gender differences in embryonic and fetal specimens.
Method details
Isolation of mitochondria
Mitochondria were isolated from hearts of WT and CypD KO mice by differential centrifugation, as described.46 Briefly, tissue was homogenized with a Potter-Elvehjem Type Tissue Grinder in a buffer containing mannitol/sucrose/Tris buffer (210 mM mannitol, 65 mM sucrose, 20 mM Tris-base, 0.5 mM EGTA, and 0.5 mM EDTA, pH 7.2). After centrifugation at 500 x g, the supernatant was centrifuged at 10,000 x g and the final mitochondrial pellet was resuspended in the same buffer, but without EGTA and EDTA. For functional experiments, mitochondria were used within 2-3 after isolation. For other experiments, mitochondria were frozen and stored at -80°C. Due to the limited size of the hearts at the early embryonic ages, mitochondria were not isolated from E9.5-E13.5 hearts and tissues homogenates of pooled samples were used.
Preparation of tissue homogenates
Tissue homogenates from E9.5 to E16.5 embryos were obtained by homogenizing 1-10 hearts manually in 0.1 ml ice-cold mannitol/sucrose/Tris buffer in microtubes with a fitted Teflon pestle. The hearts from older mice were first minced and then homogenized. Tissue homogenates were kept on ice and used within 30 minutes of preparation. For other experiments, homogenates were frozen and stored at -80°C.
Protein determination
Samples were assayed for protein content using the BCA assay kit from Thermo Fisher Scientific.
PCR
Gene expression and mitochondrial to nuclear DNA ratios were examined in individual hearts of wild type and CypD-/- mice at all timepoints except E9.5, when 3-4 hearts were pooled per sample. Total RNA and DNA was isolated using the Direct-Zol™ RNA/DNA miniprep kit (Zymo Research, R2080). RNA was treated with DNase 1 (K1641) and reverse transcribed with the Maxima First Strand cDNA Synthesis Kit (Thermo Scientific, K1641). Quantitative RT-PCR and PCR were performed using Power Sybr™ Green PCR Master Mix (4367659), primers, and a Bio-Rad CFX384 thermocycler. Ct values were adjusted for primer efficiency, and the levels of each mRNA and DNA determined using the ΔΔCt method with primers for Pol2ra (mRNA) and 18s (DNA) used as endogenous controls. Levels of mRNAs were normalized to the levels in the hearts of E9.5 wild type embryos. Oligonucleotides are listed in Table S1.
Oxygen consumption experiments
Mitochondrial oxygen consumption was performed using a Clark-type oxygraph (Hansatech, PPI Systems), as described.5 V0 respiration was measured in the presence of substrates (10 mM succinate at E9.5 and E11.5 and 3 mM malate/5 mM glutamate at E13.5 and older), while Vmax respiration was measured in the presence of substrate and 1 mM ADP. The respiratory control ratio (RCR) was calculated as the ratio of Vmax/V0. To access oxygen consumption during the early embryonic development 7-10 hearts from E9.5 embryos (5-10 μg protein) or 3-4 hearts from E11.5 embryos (≈ 20 μg protein) were directly transferred into the measuring chamber. For all other developmental stages tissue homogenate were used (50-150 μg protein/experiment).
Calcium retention capacity (CRC)
The CRC was measured with arsenazo III at room temperature as the difference in absorbance at 662 nm and the background at 692 nm using a Spectramax Plus 384 UV/Vis spectrophotometer (Molecular Devices), as described.52 Briefly, isolated mitochondria (≈ 100 μg of mitochondrial protein) were added to 0.5 ml CRC buffer (120 mM KCl, 70 mM mannitol, 25 mM sucrose, 20 mM HEPES pH 7.4 at room temperature, 5 mM KH2PO4, 5 mM glutamate, 3 mM malate). The change of absorbance upon addition of pulses of free Ca2+ (5-40 μM, every 2 minutes) was recorded every 15 sec until the CRC was reached.
Enzyme assays
The enzymatic activity of the ETC complexes was accessed, as described.53,54 Complex I activity was measured as NADH-ubiquinone dehydrogenase (DH, 340 nm; ε for NADH = 6.81 mM-1cm-1) or by its ability to transfer electrons to Complex III (NADH-cytochrome c DH (550 nm, ε for cytochrome c 18.7 mM-1cm-1) using potassium cyanide to prevent oxidation of cytochrome c by Complex IV. Where necessary, the assay was repeated with 2 mg/ml rotenone to assess mitochondria specific, rotenone-sensitive activity. Complex II was measured by its ability to oxidize DCPIP (412 nm, ε = 21 mM-1cm-1 53) or by its ability to transfer electrons to Complex III (succinate-cytochrome c DH.54 Complex V was measured by its reverse function to hydrolyze ATP in the presence or absence of oligomycin. This test becomes unreliable in tissue homogenates because of non-mitochondrial ATPases, so this was only performed with purified mitochondrial preparations.53 Citrate synthase was measured by the generation of citrate from oxaloacetate and acetyl-CoA (Ac-CoA) in a phosphate buffered saline containing 0.01% Triton X-100.53 The reduced CoA-SH is oxidized by 5’,5’Dithiobis 2-nitrobenzoic acid and the linear increase of absorbance was measured at 412 nm, ε = 13.6 mM-1cm-1.
Electrophoresis and immunoblotting
Denaturing sodium dodecyl sulfate (SDS) was performed, as published.18,55 5-20 μg protein per lane were separated on a 15% SDS polyacrylamide gels using Tris/glycine buffer using 130 V for 1.5-2 hours. Proteins were transferred onto nitrocellulose membranes using a Trans-Blot Turbo Transfer System (Biorad). Membranes were blocked with Biorad Blocking reagent, incubated with primary antibodies dissolved in 3% bovine serum albumin in Tris-buffered saline. followed by fluorescent labeled secondary antibodies (Starbright 520 and Starbright 700 from Biorad; dilution 1:10,000). Signals were detected using a ChemiDoc station from Biorad. Gel and immunoblot images were processed using Image Lab (Biorad) and Adobe Photoshop and Illustrator. Densitometry was determined using Fiji/Image J.
Electron microscopy
Pregnant dams were sacrificed at E9.5, 11.5, 13.5 and E16.5 by cervical dislocation and uterine segments were rapidly removed and dissected in EM fixative (4% paraformaldehyde, 2% glutaraldehyde, 0.1 M Na cacodylate) such that the embryo was exposed to fixative within 40 seconds of maternal cervical dislocation.4 Postnatal animals were sacrificed by the same method and E16.5 to adult hearts were rapidly perfused with EM fixative via 26-gauge syringe inserted into the left ventricle while clamping the ascending aorta with forceps. Specimens were stored in fixative at 4°C, rinsed in 0.1 M Na cacodylate, post-fixed in 1 % osmium tetroxide, dehydrated in a graded series of ethanol, embedded into EPON/Araldite resin and polymerized at 70°C for two days. Thin sections were cut with a diamond knife at 70nm, placed onto nickel slot grids and stained with aqueous uranyl acetate and lead citrate. The grids were examined using a Hitachi 7650 TEM with an attached Gatan 11 megapixel Erlangshen digital camera. Low power images were obtained to identify random left ventricular cardiac myocytes with the cell and contractile apparatus orientation a longitudinal orientation in more mature cells. All cells had at least one nucleus to ensure that a more central plane of the cell was examined. Higher power images were then taken to make a virtual montage of the entire cell for morphometry. Nine left ventricular myocytes were analyzed for each age and genotype (3 hearts x 3 myocytes per heart). However, for E11.5 and E13.5 hearts, analysis was performed on 9 cells from the myocardial wall and 9 from the trabeculae; a subgroup analysis found little difference between the two regions, so these data were combined for final analyses.
Both low- and high-power images were analyzed in Fiji (ImageJ 1.53f51) using a Cintiq tablet (Wacom, Saitama, Japan) to outline perimeters of the cell, nucleus, myofibrils, and individual mitochondria to measure shape descriptors, and these data were transferred into Excel spreadsheets for calculations and combining data from all cells at each age and genotype, as described.13 These measurements include mitochondrial area (μm2), perimeter (μm), aspect ratio (major axis/minor axis), form factor (perimeter2/4π x surface area), and Feret diameter (longest distance (μm) between any two points within a mitochondrion).
Mitochondria also underwent subgroup analysis to determine differences in cytoplasmic, intermyofibrillar, perinuclear, and subsarcolemmal mitochondria within and between ages/genotypes. Cytoplasmic mitochondria were defined as mitochondria not associated with nuclei or the sarcolemma and not between myofibrils and were only observed at earlier stages of development. Intermyofibrillar mitochondria were those that were surrounded by myofibrils or closely associated with myofibrils. Perinuclear mitochondria were defined as those touching nuclei or within the defined perinuclear region; if myofibrils enclosed this region, then mitochondria were considered intermyofibrillar when the “perinuclear” extension narrowed to <2 mitochondria in width. Similarly, subsarcolemmal mitochondria were defined as lying close to the sarcolemma, bounded intracellularly by myofibrils and/or nuclei, but mitochondria near the sarcolemma were considered intermyofibrillar using the same definition as above for the boundary of perinuclear and intermyofibrillar mitochondria.
Metabolomics
Animals were sacrificed by cervical dislocation and hearts or embryos/fetus were rapidly removed from the thorax, dissected to remove atria and great vessels, blotted on paper to remove blood, and frozen within 30 seconds of cervical dislocation by plunging in liquid nitrogen or placement on dry ice covered with aluminum foil. Samples were stored at -80 degrees. Hearts at E9.5 (7-11 hearts/sample), E11.5 (2), and E13.5 (2) were pooled to provide sufficient material for processing and analysis. The essential metabolomics workflow was as previously described.23,56,57,58 Pulverized heart powder was extracted 3x in 80% MeOH, dried under N2 and reconstituted in 50% MeOH for analysis. Samples were analyzed by high performance liquid chromatography-tandem mass spectrometry (LC-MS/MS) comprising reverse phase HPLC on a Phenomenex Synergi RP-Fusion column in a Shimadzu Prominence 20 LC system. Column effluent (150μL/min) was fed to a Thermo Quantum Access Max triple quadrupole mass spectrometer, with heated electrospray ionization (HESI-II) probe operating in negative mode. Metabolites were identified by retention time, precursor m/z, and at least 2 product ion m/z, against a custom SRM library with ∼110 metabolites prepared from authentic standards. Peak ID and integration were performed using Thermo XCaliber 4.2 software.
Metabolite abundances were normalized to the sum of all peak values for each run. Outliers were identified as those falling outside the 99.99th percent confidence intervals. Missing values were then imputed from the median of the remaining true values for each group.59 The total number of missing values and discarded outliers was 741 (10.3 % of 7168), and for each group of 8, a minimum of 3 true values were required, unless all values at that age were at or close to 0 (usually at E9.5 and 11.5, but also carnosine before birth). All data were then analyzed together using MetaboAnalyst (v5.0, https://www.metaboanalyst.ca/home.xhtml).
Quantification and statistical analysis
Data was obtained, processed, and prepared for presentation using Microsoft Excel, Adobe Illustrator and Photoshop, Fiji/Image J (NIH), Image Lab (v6.0.1, Biorad), Softmax Pro 7 (Molecular Devices), Xcalibur (v4.2, Thermo), Metaboanalyst (v5.0), and Prism (9.3.1, GraphPad). In general, data was analyzed for normality. Parametric data was analyzed using one-way ANOVA with Tukey’s or Sidak’s multiple comparison test and presented as mean +/- standard error of the mean (SEM). Non-parametric data was analyzed using Kruskal-Wallis with Dunn’s multiple comparison test and reported as median +/- interquartile range. In some cases, outliers were identified by GraphPad using ROUT (robust regression and outlier removal) and Q was set to 1% (medium aggressiveness). Significance was generally defined as p<0.05; Only significant differences are noted in figures: ∗P<0.05, ∗∗P<0.01, ∗∗∗P<0.001, ∗∗∗∗P<0.0001. For metabolomics data, statistical differences between WT and CypD KO samples at each age were determined as false discovery rate (FDR) corrected p values, using the 2-stage step-up method of Benjamini, Krieger, and Yekutieli and assuming an FDR of 5 or 10%, as indicated. Description of the test used and N for each data set/figure panel is present in Figure legends. Specimens from each litter were randomized at the time of harvest to the experiments to be done that day, but, as there were no treatments, randomization based on treatments was not performed. By the nature of the study, biochemical and metabolomics experiments were not done in a blinded manner. Electron micrographs were obtained by one investigator (KdMB), while morphometry was performed by another investigator (GAPJr) in an unblinded manner.
Acknowledgments
The authors thank the University of Rochester Mitochondrial Research and Innovation Group for constructive discussions, Denise Hanvi Fangnibo for technical assistance, and the URMC Electron Microscopy Resource (RRID:SCR_012366) in the Center for Advanced Research Technologies (CART) for technical support. This work was supported by the NIH (R01-HL144776 to GAPJr). CAK was supported by American Heart Association Postdoctoral Fellowship (#19POST34380212).
Author contributions
GB and GAPJr designed the project and prepared the article. GB, JRB, and MPC performed enzyme assays. GB performed OXPHOS and CRC experiments, SDS electrophoresis, and immunoblot analysis. KdMB and GAP Jr performed and analyzed electron microscopy and morphometry. EDC performed PCR experiments. CAK, SMN, PSB, and GAPJr performed metabolomics experiments. All authors performed data analysis and proof reading of the article.
Declaration of interests
GAPJr is the Inventor and the University of Rochester is the Assignee of United States Patent No. “US 10,179,161 B2” dated 1/15/19 and entitled “Compositions and methods for enhancing cardiac function in the neonate” that is related to the work reported herein. All other authors declare no competing interests.
Published: February 9, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2024.109157.
Supplemental information
References
- 1.Baker C.N., Ebert S.N. Development of aerobic metabolism in utero: Requirement for mitochondrial function during embryonic and fetal periods. OA Biotechnol. 2013;2:16. [Google Scholar]
- 2.Porter G.A., Jr., Hom J., Hoffman D., Quintanilla R., de Mesy Bentley K., Sheu S.S. Bioenergetics, mitochondria, and cardiac myocyte differentiation. Prog. Pediatr. Cardiol. 2011;31:75–81. doi: 10.1016/j.ppedcard.2011.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ali H., Braga L., Giacca M. Cardiac regeneration and remodelling of the cardiomyocyte cytoarchitecture. FEBS J. 2020;287:417–438. doi: 10.1111/febs.15146. [DOI] [PubMed] [Google Scholar]
- 4.Hom J.R., Quintanilla R.A., Hoffman D.L., de Mesy Bentley K.L., Molkentin J.D., Sheu S.S., Porter G.A., Jr. The permeability transition pore controls cardiac mitochondrial maturation and myocyte differentiation. Dev. Cell. 2011;21:469–478. doi: 10.1016/j.devcel.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beutner G., Eliseev R.A., Porter G.A., Jr. Initiation of electron transport chain activity in the embryonic heart coincides with the activation of mitochondrial complex 1 and the formation of supercomplexes. PLoS One. 2014;9 doi: 10.1371/journal.pone.0113330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Piquereau J., Ventura-Clapier R. Maturation of Cardiac Energy Metabolism During Perinatal Development. Front. Physiol. 2018;9:959. doi: 10.3389/fphys.2018.00959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Puente B.N., Kimura W., Muralidhar S.A., Moon J., Amatruda J.F., Phelps K.L., Grinsfelder D., Rothermel B.A., Chen R., Garcia J.A., et al. The oxygen-rich postnatal environment induces cardiomyocyte cell-cycle arrest through DNA damage response. Cell. 2014;157:565–579. doi: 10.1016/j.cell.2014.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang D., Li Y., Heims-Waldron D., Bezzerides V., Guatimosim S., Guo Y., Gu F., Zhou P., Lin Z., Ma Q., et al. Mitochondrial Cardiomyopathy Caused by Elevated Reactive Oxygen Species and Impaired Cardiomyocyte Proliferation. Circ. Res. 2018;122:74–87. doi: 10.1161/CIRCRESAHA.117.311349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.García A., Moreno K., Ronderos M., Sandoval N., Caicedo M., Dennis R.J. Differences by Altitude in the Frequency of Congenital Heart Defects in Colombia. Pediatr. Cardiol. 2016;37:1507–1515. doi: 10.1007/s00246-016-1464-x. [DOI] [PubMed] [Google Scholar]
- 10.Breckenridge R.A., Piotrowska I., Ng K.E., Ragan T.J., West J.A., Kotecha S., Towers N., Bennett M., Kienesberger P.C., Smolenski R.T., et al. Hypoxic regulation of hand1 controls the fetal-neonatal switch in cardiac metabolism. PLoS Biol. 2013;11 doi: 10.1371/journal.pbio.1001666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Neary M.T., Ng K.E., Ludtmann M.H.R., Hall A.R., Piotrowska I., Ong S.B., Hausenloy D.J., Mohun T.J., Abramov A.Y., Breckenridge R.A. Hypoxia signaling controls postnatal changes in cardiac mitochondrial morphology and function. J. Mol. Cell. Cardiol. 2014;74:340–352. doi: 10.1016/j.yjmcc.2014.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gong G., Song M., Csordas G., Kelly D.P., Matkovich S.J., Dorn G.W., 2nd Parkin-mediated mitophagy directs perinatal cardiac metabolic maturation in mice. Science. 2015;350:aad2459. doi: 10.1126/science.aad2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu X., Yagi H., Saeed S., Bais A.S., Gabriel G.C., Chen Z., Peterson K.A., Li Y., Schwartz M.C., Reynolds W.T., et al. The complex genetics of hypoplastic left heart syndrome. Nat. Genet. 2017;49:1152–1159. doi: 10.1038/ng.3870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu X., Jin K., Bais A.S., Zhu W., Yagi H., Feinstein T.N., Nguyen P.K., Criscione J.D., Liu X., Beutner G., et al. Uncompensated mitochondrial oxidative stress underlies heart failure in an iPSC-derived model of congenital heart disease. Cell Stem Cell. 2022;29:840–855.e7. doi: 10.1016/j.stem.2022.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alavian K.N., Beutner G., Lazrove E., Sacchetti S., Park H.A., Licznerski P., Li H., Nabili P., Hockensmith K., Graham M., et al. An uncoupling channel within the c-subunit ring of the F1FO ATP synthase is the mitochondrial permeability transition pore. Proc. Natl. Acad. Sci. USA. 2014;111:10580–10585. doi: 10.1073/pnas.1401591111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bernardi P., Gerle C., Halestrap A.P., Jonas E.A., Karch J., Mnatsakanyan N., Pavlov E., Sheu S.S., Soukas A.A. Identity, structure, and function of the mitochondrial permeability transition pore: controversies, consensus, recent advances, and future directions. Cell Death Differ. 2023;30:1869–1885. doi: 10.1038/s41418-023-01187-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bonora M., Giorgi C., Pinton P. Molecular mechanisms and consequences of mitochondrial permeability transition. Nat. Rev. Mol. Cell Biol. 2022;23:266–285. doi: 10.1038/s41580-021-00433-y. [DOI] [PubMed] [Google Scholar]
- 18.Beutner G., Porter G.A., Jr. Analyzing Supercomplexes of the Mitochondrial Electron Transport Chain with Native Electrophoresis, In-gel Assays, and Electroelution. J. Vis. Exp. 2017:55738. doi: 10.3791/55738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lingan J.V., Alanzalon R.E., Porter G.A., Jr. Preventing permeability transition pore opening increases mitochondrial maturation, myocyte differentiation and cardiac function in the neonatal mouse heart. Pediatr. Res. 2017;81:932–941. doi: 10.1038/pr.2017.19. [DOI] [PubMed] [Google Scholar]
- 20.Porter G.A., Jr., Beutner G. Cyclophilin D, Somehow a Master Regulator of Mitochondrial Function. Biomolecules. 2018;8 doi: 10.3390/biom8040176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chong D., Gu Y., Zhang T., Xu Y., Bu D., Chen Z., Xu N., Li L., Zhu X., Wang H., et al. Neonatal ketone body elevation regulates postnatal heart development by promoting cardiomyocyte mitochondrial maturation and metabolic reprogramming. Cell Discov. 2022;8:106. doi: 10.1038/s41421-022-00447-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hirakow R., Gotoh T., Watanabe T. Quantitative studies on the ultrastructural differentiation and growth of mammalian cardiac muscle cells. I. The atria and ventricles of the rat. Acta Anat. 1980;108:144–152. doi: 10.1159/000145293. [DOI] [PubMed] [Google Scholar]
- 23.Nadtochiy S.M., Wang Y.T., Nehrke K., Munger J., Brookes P.S. Cardioprotection by nicotinamide mononucleotide (NMN): Involvement of glycolysis and acidic pH. J. Mol. Cell. Cardiol. 2018;121:155–162. doi: 10.1016/j.yjmcc.2018.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Talman V., Teppo J., Pöhö P., Movahedi P., Vaikkinen A., Karhu S.T., Trošt K., Suvitaival T., Heikkonen J., Pahikkala T., et al. Molecular Atlas of Postnatal Mouse Heart Development. J. Am. Heart Assoc. 2018;7 doi: 10.1161/JAHA.118.010378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Edwards W., Greco T.M., Miner G.E., Barker N.K., Herring L., Cohen S., Cristea I.M., Conlon F.L. Quantitative proteomic profiling identifies global protein network dynamics in murine embryonic heart development. Dev. Cell. 2023;58:1087–1105.e4. doi: 10.1016/j.devcel.2023.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hays M.D., Gillette P.C. Atrial automatic ectopic tachycardia successfully treated with encainide in a 4-week-old infant after Senning procedure. Am. Heart J. 1989;117:489–492. doi: 10.1016/0002-8703(89)90801-6. [DOI] [PubMed] [Google Scholar]
- 27.Marin-Garcia J., Ananthakrishnan R., Agrawal N., Goldenthal M.J. Mitochondrial gene expression during bovine cardiac growth and development. J. Mol. Cell. Cardiol. 1994;26:1029–1036. doi: 10.1006/jmcc.1994.1123. [DOI] [PubMed] [Google Scholar]
- 28.Marin-Garcia J., Ananthakrishnan R., Goldenthal M.J. Heart mitochondrial DNA and enzyme changes during early human development. Mol. Cell. Biochem. 2000;210:47–52. doi: 10.1023/a:1007031919298. [DOI] [PubMed] [Google Scholar]
- 29.DeLaughter D.M., Bick A.G., Wakimoto H., McKean D., Gorham J.M., Kathiriya I.S., Hinson J.T., Homsy J., Gray J., Pu W., et al. Single-Cell Resolution of Temporal Gene Expression during Heart Development. Dev. Cell. 2016;39:480–490. doi: 10.1016/j.devcel.2016.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Larsen S., Nielsen J., Hansen C.N., Nielsen L.B., Wibrand F., Stride N., Schroder H.D., Boushel R., Helge J.W., Dela F., Hey-Mogensen M. Biomarkers of mitochondrial content in skeletal muscle of healthy young human subjects. J. Physiol. 2012;590:3349–3360. doi: 10.1113/jphysiol.2012.230185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McLaughlin K.L., Hagen J.T., Coalson H.S., Nelson M.A.M., Kew K.A., Wooten A.R., Fisher-Wellman K.H. Novel approach to quantify mitochondrial content and intrinsic bioenergetic efficiency across organs. Sci. Rep. 2020;10 doi: 10.1038/s41598-020-74718-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Intlekofer A.M., Dematteo R.G., Venneti S., Finley L.W.S., Lu C., Judkins A.R., Rustenburg A.S., Grinaway P.B., Chodera J.D., Cross J.R., Thompson C.B. Hypoxia Induces Production of L-2-Hydroxyglutarate. Cell Metabol. 2015;22:304–311. doi: 10.1016/j.cmet.2015.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Smith H.E., Page E. Ultrastructural changes in rabbit heart mitochondria during the perinatal period. Neonatal transition to aerobic metabolism. Dev. Biol. 1977;57:109–117. doi: 10.1016/0012-1606(77)90358-x. [DOI] [PubMed] [Google Scholar]
- 34.Martínez-Reyes I., Chandel N.S. Mitochondrial TCA cycle metabolites control physiology and disease. Nat. Commun. 2020;11:102. doi: 10.1038/s41467-019-13668-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Solmonson A., Faubert B., Gu W., Rao A., Cowdin M.A., Menendez-Montes I., Kelekar S., Rogers T.J., Pan C., Guevara G., et al. Compartmentalized metabolism supports midgestation mammalian development. Nature. 2022;604:349–353. doi: 10.1038/s41586-022-04557-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Severin E.S., Kondratyev A.D. Regulation of differentiation of PC12 cells by nerve growth factor. Adv. Enzym. Regul. 1988;27:357–370. doi: 10.1016/0065-2571(88)90026-x. [DOI] [PubMed] [Google Scholar]
- 37.Ke B., Zeng Y., Zhao Z., Han F., Liu T., Wang J., Khalique A., Lu W.J., Chong J., Lan F., He H. Uric acid: a potent molecular contributor to pluripotent stem cell cardiac differentiation via mesoderm specification. Cell Death Differ. 2019;26:826–842. doi: 10.1038/s41418-018-0157-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Birsoy K., Wang T., Chen W.W., Freinkman E., Abu-Remaileh M., Sabatini D.M. An Essential Role of the Mitochondrial Electron Transport Chain in Cell Proliferation Is to Enable Aspartate Synthesis. Cell. 2015;162:540–551. doi: 10.1016/j.cell.2015.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sullivan L.B., Gui D.Y., Hosios A.M., Bush L.N., Freinkman E., Vander Heiden M.G. Supporting Aspartate Biosynthesis Is an Essential Function of Respiration in Proliferating Cells. Cell. 2015;162:552–563. doi: 10.1016/j.cell.2015.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Abbey D., Seshagiri P.B. Ascorbic acid-mediated enhanced cardiomyocyte differentiation of mouse ES-cells involves interplay of DNA methylation and multiple-signals. Differentiation. 2017;96:1–14. doi: 10.1016/j.diff.2017.04.001. [DOI] [PubMed] [Google Scholar]
- 41.Cao N., Liu Z., Chen Z., Wang J., Chen T., Zhao X., Ma Y., Qin L., Kang J., Wei B., et al. Ascorbic acid enhances the cardiac differentiation of induced pluripotent stem cells through promoting the proliferation of cardiac progenitor cells. Cell Res. 2012;22:219–236. doi: 10.1038/cr.2011.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Paredes A., Justo-Méndez R., Jiménez-Blasco D., Núñez V., Calero I., Villalba-Orero M., Alegre-Martí A., Fischer T., Gradillas A., Sant'Anna V.A.R., et al. γ-Linolenic acid in maternal milk drives cardiac metabolic maturation. Nature. 2023;618:365–373. doi: 10.1038/s41586-023-06068-7. [DOI] [PubMed] [Google Scholar]
- 43.Gutiérrez-Aguilar M., Baines C.P. Structural mechanisms of cyclophilin D-dependent control of the mitochondrial permeability transition pore. Biochim. Biophys. Acta. 2015;1850:2041–2047. doi: 10.1016/j.bbagen.2014.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Elrod J.W., Wong R., Mishra S., Vagnozzi R.J., Sakthievel B., Goonasekera S.A., Karch J., Gabel S., Farber J., Force T., et al. Cyclophilin D controls mitochondrial pore-dependent Ca(2+) exchange, metabolic flexibility, and propensity for heart failure in mice. J. Clin. Invest. 2010;120:3680–3687. doi: 10.1172/JCI43171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Menazza S., Wong R., Nguyen T., Wang G., Gucek M., Murphy E. CypD(-/-) hearts have altered levels of proteins involved in Krebs cycle, branch chain amino acid degradation and pyruvate metabolism. J. Mol. Cell. Cardiol. 2013;56:81–90. doi: 10.1016/j.yjmcc.2012.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Beutner G., Alanzalon R.E., Porter G.A., Jr. Cyclophilin D regulates the dynamic assembly of mitochondrial ATP synthase into synthasomes. Sci. Rep. 2017;7 doi: 10.1038/s41598-017-14795-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nguyen T.T.M., Wong R., Menazza S., Sun J., Chen Y., Wang G., Gucek M., Steenbergen C., Sack M.N., Murphy E. Cyclophilin D modulates mitochondrial acetylome. Circ. Res. 2013;113:1308–1319. doi: 10.1161/CIRCRESAHA.113.301867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shanmughapriya S., Rajan S., Hoffman N.E., Higgins A.M., Tomar D., Nemani N., Hines K.J., Smith D.J., Eguchi A., Vallem S., et al. SPG7 Is an Essential and Conserved Component of the Mitochondrial Permeability Transition Pore. Mol. Cell. 2015;60:47–62. doi: 10.1016/j.molcel.2015.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wali G., Li Y., Liyanage E., Kumar K.R., Day M.L., Sue C.M. Pharmacological rescue of mitochondrial and neuronal defects in SPG7 hereditary spastic paraplegia patient neurons using high throughput assays. Front. Neurosci. 2023;17 doi: 10.3389/fnins.2023.1231584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shankar T.S., Ramadurai D.K.A., Steinhorst K., Sommakia S., Badolia R., Thodou Krokidi A., Calder D., Navankasattusas S., Sander P., Kwon O.S., et al. Cardiac-specific deletion of voltage dependent anion channel 2 leads to dilated cardiomyopathy by altering calcium homeostasis. Nat. Commun. 2021;12:4583. doi: 10.1038/s41467-021-24869-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang J., Wang Y.T., Miller J.H., Day M.M., Munger J.C., Brookes P.S. Accumulation of Succinate in Cardiac Ischemia Primarily Occurs via Canonical Krebs Cycle Activity. Cell Rep. 2018;23:2617–2628. doi: 10.1016/j.celrep.2018.04.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sokolova N., Pan S., Provazza S., Beutner G., Vendelin M., Birkedal R., Sheu S.S. ADP Protects Cardiac Mitochondria under Severe Oxidative Stress. PLoS One. 2013;8 doi: 10.1371/journal.pone.0083214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kirby D.M., Thorburn D.R., Turnbull D.M., Taylor R.W. Biochemical assays of respiratory chain complex activity. Methods Cell Biol. 2007;80:93–119. doi: 10.1016/S0091-679X(06)80004-X. [DOI] [PubMed] [Google Scholar]
- 54.Spinazzi M., Casarin A., Pertegato V., Salviati L., Angelini C. Assessment of mitochondrial respiratory chain enzymatic activities on tissues and cultured cells. Nat. Protoc. 2012;7:1235–1246. doi: 10.1038/nprot.2012.058. [DOI] [PubMed] [Google Scholar]
- 55.Beutner G., Porter G.A., Jr. Native Gel Electrophoresis and Immunoblotting to Analyze Electron Transport Chain Complexes. Methods Mol. Biol. 2021;2276:103–112. doi: 10.1007/978-1-0716-1266-8_7. [DOI] [PubMed] [Google Scholar]
- 56.Brookes P.S., Jimenez A.G. Metabolomics of aging in primary fibroblasts from small and large breed dogs. Geroscience. 2021;43:1683–1696. doi: 10.1007/s11357-021-00388-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kulkarni C.A., Nadtochiy S.M., Kennedy L., Zhang J., Chhim S., Alwaseem H., Murphy E., Fu D., Brookes P.S. ALKBH7 mediates necrosis via rewiring of glyoxal metabolism. Elife. 2020;9 doi: 10.7554/eLife.58573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nadtochiy S.M., Urciuoli W., Zhang J., Schafer X., Munger J., Brookes P.S. Metabolomic profiling of the heart during acute ischemic preconditioning reveals a role for SIRT1 in rapid cardioprotective metabolic adaptation. J. Mol. Cell. Cardiol. 2015;88:64–72. doi: 10.1016/j.yjmcc.2015.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Aittokallio T. Dealing with missing values in large-scale studies: microarray data imputation and beyond. Briefings Bioinf. 2010;11:253–264. doi: 10.1093/bib/bbp059. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Original data (immunoblots, spreadsheets, and electron micrographs) have been deposited at https://figshare.com/ and are publicly available as of the date of publication. DOIs are listed in the key resources table.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.