

Abstract
The GACKIX activator binding domain has been a compelling target for small-molecule probe discovery because of the central role of activator–GACKIX complexes in diseases ranging from leukemia to memory disorders. Additionally, GACKIX is an ideal model to dissect the context-dependent function of activator–coactivator complexes. However, the dynamic and transient protein–protein interactions (PPIs) formed by GACKIX are difficult targets for small molecules. An additional complication is that activator-binding motifs, such as GACKIX, are found in multiple coactivators, making specificity difficult to attain. In this study, we demonstrate that the strategy of tethering can be used to rapidly discover highly specific covalent modulators of the dynamic PPIs between activators and coactivators. These serve as both ortho- and allosteric modulators, enabling the tunable assembly or disassembly of the activator–coactivator complexes formed between the KIX domain and its cognate activator binding partners MLL and CREB. The molecules maintain their function and selectivity, even in human cell lysates and in bacterial cells, and thus, will ultimately be highly useful probes for cellular studies.
Keywords: chaperone proteins, inhibitors, protein–protein interactions, sulfur, transcription factors
Introduction
GACKIX is one of several conformationally plastic domains found in the master coactivators CBP and p300.[1,2] It interacts with more than 15 transcriptional activators at two distinct interfaces.[2] NMR spectroscopy studies of GACKIX bound with native transcriptional activation domains (TADs) have shown that the endogenous partners, such as MLL and c-Jun, interact at a deeper and smaller site (an area of approximately 900 a2)[3,4] whereas the TADs of CREB (pKID) and c-Myb interact with a shallower and larger surface on the opposite side.[5] The two interfaces are allosterically connected, such that binding of the TAD of MLL enhances the interaction of c-Myb or pKID at the other binding site by approximately twofold.[4,6] This allosteric enhancement is thought to play a key role in the recruitment of p300 or CBP to gene promoters.[4,6,7]
The activator–KIX complexes participate in fundamental processes, such as hematopoiesis and memory formation.[8–10] The discovery of small-molecule modulators for these processes has been of high priority in the selection, screening, and top-down approaches to yield KIX inhibitors.[11–15] One early success identified Naphthol AS-E; a molecule that disrupts oncogenic responses in cancer cell models, with the CREB-GACKIX and Myb-GACKIX complexes as its intended targets.[11,16,17] As this example illustrates, small molecules that target GACKIX have the potential to delineate activator–coactivator functions on a phenotypic level. However, even the most specific modulators for GACKIX have limited selectivity due to the level of redundancy in the protein interaction network.[14,18–20] These binding surfaces that coactivators use to interact with activators are often similar. For instance, activator p53 interacts with GACKIX, as well as three other domains within CBP and p300, which illustrates similarities in these binding surfaces.[21,22] Finally, the GACKIX motif within CBP has been found in at least four other eukaryotic coactivators, including closely related p300, but also in unrelated coactivators, such as MED15 and ARC105.[23–25] Therefore, even the unusually specific inhibitor sekikaic acid, a natural product that exhibits high specificity for the GACKIX domain, has the undesirable potential to target other coactivators with this motif in a cellular setting.[14] This severely limits the utility of these probe molecules to dissect individual activator–GACKIX interactions and to understand their role in normal and pathological processes.[24]
Recently, we reported that the covalent fragment discovery strategy of tethering yielded small-molecule modulators for GACKIX, termed chemical cochaperones, that stabilized distinct conformations of this plastic motif and, in doing so, modulated its ability to form binary and ternary complexes.[26] Although useful for structural and in vitro biophysical studies, these molecules react with GACKIX through disulfide exchange under reversible conditions and are thus not usable in more complex environments, such as those of cells and cell lysates. Here, we show that, despite the shallow and poorly defined binding surfaces in GACKIX, irreversible covalent cochaperones highly selective for their cognate binding site can be readily accessed by replacement of disulfides in the tethering hits with more reactive moieties (Scheme 1). Furthermore, the cochaperones show high selectivity for their cognate binding site, even in the presence of many potential reactive partners. Finally, changes in the thiophile enable tuning of the assembly behavior of GACKIX, leading to allosteric enhancement or inhibition of binding with a subset of partners.
Scheme 1.

Schematic representation for the development of irreversible chemical cochaperones to target the GACKIX domain of CBP. The structure of GACKIX is shown as a cartoon in gray derived from PDB ID: 4I9O.[29] The black sphere represents the position of the N627C mutant used to target GACKIX with chemical cochaperones. The standard tethering scheme involves in the reversible formation of a mixed disulfide with the target if the fragment favorably interacts with the regions surrounding the cysteine residue. By replacing the disulfide of the fragment with an alkylating moiety, chemical cochaperones of the 1–10 fragment irreversibly bind to GACKIX N627C, which is represented in the structure at the far right.
Results and Discussion
In a study of the oncogenic K-RAS variant K-RAS(G12C), Shokat and co-workers found that inhibitors discovered through tethering could be converted into irreversible modulators through replacement of the disulfide moiety with thiophilic moieties.[27] Encouraged by these results, we hypothesized that disulphide fragments identified in a screening of the CBP/p300 GACKIX motif could serve as starting points for irreversible covalent cochaperones.[28–30] However, specificity of the resulting structures was a major concern. The GACKIX motif does not contain attractive small-molecule binding pockets, but rather binding surfaces that are relatively featureless. These GACKIX binding surfaces adapt upon interacting with their binding partners, so the conversion of the disulfide fragments into irreversible cochaperones can be challenging. The irreversible cochaperones also need to orient the fragment at the selected point of tethering and effectively modulate the cysteine-containing GACKIX motif interactions. We chose to start with the GACKIX N627C mutant located in proximity to the MLL-binding site because the GACKIX N627C mutant had a negligible effect on the endogenous GACKIX-binding partners.[30,31] Therefore, any alterations in the binding of GACKIX to its partners was attributed to chemical cochaperones that tethered to this mutant.
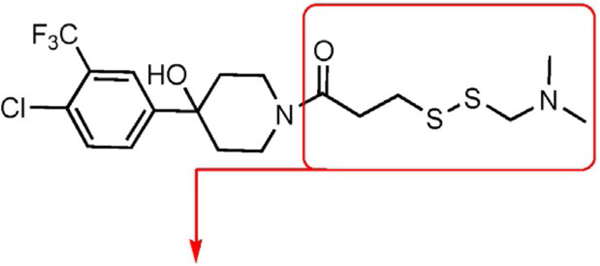
We sought a fragment that was known to impact on the conformations and interactions of the GACKIX motif. Fragment 1–10 tethered to GACKIX N627C stands out as an effective modulator of GACKIX interactions by both inhibiting MLL binding (IC50 68 μm)[31] and enhancing pKID binding.[30] Without the tether, fragment 1–10 binds to the GACKIX motif at the MLL-binding site with modest affinity.[28]
To initiate the investigation, the disulfide moiety of 1–10 was replaced with three distinct thiophiles with or without a glycine linker to produce 1–10a–f.[32–37] These molecules were then assessed for dose-dependent alkylation of the GACKIX N627C mutant and the dose–response () values varied significantly with the nature and linkage of thiophilic group (Table 1). The least effective reactive group in this series is the α,β-unsaturated amide, including 1–10e and 1–10 f, with la-beling of the protein occurring only under forcing conditions. Both 1–10a and 1–10b, with an α-chloroamide, and 1–10c and 1–10d, from the vinyl sulfonamide series, were more effective, with values ranging from 4.6 to 150 μm.
Table 1.
Comparing irreversible analogues of 1–10 against GACKIX N627C.[a]
| ||||
|---|---|---|---|---|
| Compound | Dose-response | Fold inhibition | ||
| [mM] | MLL | pKID | ||
|
| ||||
| 1–10a |
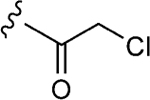
|
25 | 12±1 | 0.9±0.1 |
| 1–10b |
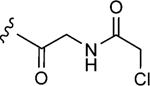
|
150 | 13±1 | 1.4±0.2 |
| 1–10c |
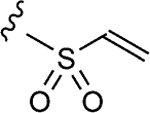
|
6.8 | 17±2 | 1.5±0.2 |
| 1–d |
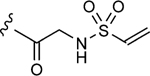
|
4.6 | 17±2 | 0.65±0.08 |
| 1–e |
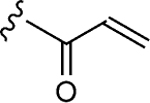
|
>500 | 14±2 | 0.88±0.09 |
| 1–f |
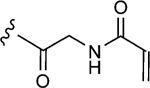
|
>500 | 5.9±0.6 | 0.66±0.07 |
The values were assessed by measuring the extent of GACKIX N627C labeled by means of Q-TOF LC-MS at various concentrations of compound. The fold inhibition values were obtained by comparing the dissociation constants () for the unlabeled (DMSO) KIX N627C construct with the values for the labeled KIX N627C–1–10 alkylator complex for both the MLL and pKID tracers. The values were measured from fluorescence polarization (FP) experiments that were performed in triplicate and errors reflect the standard deviation (SD) error.
The impact of these fragments on GACKIX N627C interactions was considered to be unique because labeling this mutant with the alkylator, iodoacetamide, had minimal impact on their affinity (Figure S1 in the Supporting Information). Upon covalent attachment of each of these structures to GACKIX, compounds 1–10a–d competitively blocked MLL from interacting with its cognate binding site analogous to the parent disulfide 1–10. More remarkable are the effects at the distal binding site, with fragments 1–10b and 1–10c allosterically inhibiting the binding of pKID and 1–10d enhancing the binding nearly twofold, comparable to the effects observed with the native ligands MLL and pKID. The affinity differences can be reasonably associated with how the 1–10 cochaperones interact with GACKIX. Previously, the 1–10 disulfide was shown to impact the distal site by altering the conformational dynamics of GACKIX.[29] Therefore, we hypothesize that the spacing between the 1–10 fragment and the reactive group influences the dynamics within GACKIX and these structural changes are reflected by how the 1–10 cochaperones effect MLL and pKID binding.
Assessing target engagement of two 1–10 irreversible probes
A further examination of the reactivity of the covalent modifiers revealed a remarkable degree of selectivity for the MLL-binding site within GACKIX. None of the molecules (1–10a–f) react with the KIX domain in the absence of a cysteine residue (native GACKIX). In addition, covalent modification selectively occurs only in the MLL-binding site, even if cysteine residues are introduced around other activator binding sites. As shown in Figure 1A, covalent modifiers 1–10a and 1–10d, both of which are reactive electrophiles, only form covalent bonds with GACKIX if a cysteine is present at the cognate (MLL) binding site. In a more complex environment, Escherichia coli expressing GACKIX N627C protein was dosed with the irreversible probes, and the resulting purified protein was quantitatively labeled by the irreversible probes, as determined by Q-TOF LC-MS (Figure 1B). The affinity of the core scaffold of 1–10 (no electrophilic moiety) has been measured to be approximately 250 μm; thus, it is unlikely 1–10d, at concentrations under 10 αm, will produce effects due to noncovalent binding interactions.
Figure 1.

A) 1–10a (■) and 1–10d (■) show preference for the GACKIX N627C mutant located near the MLL-binding site. N644C is located in the loop and the remaining mutants, K662C, H651C, Q609C, and K606C, are located within the pKID-binding site. The error bars represent SD of the average of two separate experiments. B) The 1–10 chemical cochaperones were dosed into E. coli during the expression of GACKIX N627C and after nickel affinity purification; the extent of labeling was assessed by means of LC-MS. The error bars represent SD of the average of three values from separate experiments. *0.01<p<0.05, **0.05<p<0.001,***0.001<p<0.001, ****p<0.001; p values are calculated by using the GraphPad Prism 7.0 program.
The concern with the use of irreversible probes is that they may target critical cellular components in mammalian cells and cause adverse effects due to their reactivity.[38–40] Preliminary results show that 1–10d readily labels GACKIX N627C in the presence of excess glutathione, with no observed alkylator–glutathione adducts after 1 h; this suggests it could function selectively in cells (Figure S2). Thus, we decided to visualize the potential targets of these probes in a cellular environment. In cells, the purified GACKIX N627C mutant was added to HEK 293T lysate and then dosed with a biotinylated variant of the 1–10d probe. Biotin-containing components were pulled out of the lysate by using NeutrAvidin resin and analyzed by western blot with streptavidin–HRP (HRP: horseradish peroxidase; Figure 2). Once again, the 1–10d alkylator displayed significant selectivity for the target.
Figure 2.

The structure of the biotinylated 1–10d probe is shown above. This probe was added to HEK293T lysate (100 μg) containing purified GACKIX N627C (25 nm). Any biotin-containing proteins isolated on NeutrAvidin agarose beads were visualized by means of western blotting through chemiluminescence with streptavidin–HRP. The bands within the red box represent the expected mass for GACKIX N627C (12 kDa).
Conclusion
The purpose of targeting the GACKIX motif is to understand what roles its interactions with activator complexes play in transcriptional events. However, it is difficult to distinguish the role of the GACKIX motif in CBP versus p300 function due to their significant homology (90%).[38] From our results, the irreversible 1–10 derivatives displayed a preference for the cysteine-containing GACKIX N627C mutant. They selectively enhanced or disrupted interactions between GACKIX N627C and its activators only if the cognate cysteine residue were present. Because these probes are irreversible, nonspecific targets can drastically weaken their fitness as cellular probes.[36,43–46] Fortunately, our preliminary evidence shows that 1–10d engages the KIX N627C target with no prominent off-targets in different complex environments.
With our strategy, one fragment, known as 1–10, was taken from the reversible disulfide tethering screen to provide a framework for a suite of irreversible modulators that targeted the cysteine-containing GACKIX N627C domain. Because the 1–10 irreversible cochaperones target GACKIX in cellular environments, they have the potential to examine how GACKIX recognizes different activator binding partners to regulate gene expression. Our chemical cochaperones were able to change the assembly or disassembly of GACKIX complexes at the distal site. For instance, cochaperones 1–10c and 1–10d both inhibit MLL, but they either disrupt or enhance the pKID–GACKIX interaction. Previously, we showed that 1–10 tethered to GACKIX N627C prolonged the residence time of pKID to cause allosteric enhancement. The 1–10 derivatives exhibit either positive or negative cooperativity by how they alter the conformational dynamics with GACKIX N627C. Finally, these cochaperones, 1–10c and 1–10d, could also provide novel, contrasting effects on specific pKID (CREB)-dependent genes.
Studies in cells bearing cysteine mutants of p300 and CBP are a current focus to connect the biophysically derived model of GACKIX binding and function with cellular function.
Experimental Section
Protein expression and purification:
As previously described,[47] the DNA sequence encoding the GACKIX domain from mouse CBP residues 586–672 was cloned into the bacterial expression pRSETB vector with an additional hexahistidine tag and a short polar linker fused to the N terminus of GACKIX resulting in a protein with the following sequence (tag and linker residues are shown in lower case): mrgshhhhhhgmasGVRKGWHEHVTQDLRSHLVHKLVQAIFPTPD-PAALKDRRMENLVAYAKKVEGDMYESANSRDEYYHLLAEKIYKIQKELEEKRRSRL.
The human MED15 KIX circular DNA (cDNA) encoding amino acids 1–78 was synthesized by GenScript USA, Inc. and cloned into the pET-15b plasmid (Novagen, EMD Millipore) by using the Nde1 and Xho1 cloning sites. The resulting recombinant wild-type MED15 KIX protein sequence contained the N-terminal hexahistidine tag and a thrombin cleavage site, as shown in lower case letters: mgsshhhhhhssglvprgsHMDVSGQETDWRSTAFRQKLVSQIEDAMRKA-GVAHSKSSKDMESHVFLKAKTRDEYLSLVARLIIHFRDIHNKKSQASV.
The cysteine mutants at N627, R644, K662, H651, Q609, and K606 of CBP GACKIX and at R67 and H70 of MED15 GACKIX were generated through site-directed mutagenesis, as previously described.[28]
The GACKIX protein was expressed in Rosetta2(DE3) pLysS E. coli (Novagen). Cells were grown to an optical density at λ = 600 nm (OD600nm) of 0.8–1.0 (37°C, 250 rpm), induced with 0.25 mm isopropyl β-d−1-thiogalactopyranoside (IPTG) for 4 h at 25°C, harvested by centrifugation, and stored at −80°C. The His6-tagged GACKIX protein was affinity purified by using a batch method with Ni-NTA beads (QIAGEN) by following the manufacturer’s instructions and eluted with 400 mm imidazole. GACKIX was purified by means of ion-exchange chromatography on a Source S column (GE Health-care) in phosphate buffer (50 mm, pH 7.2) by eluting with a NaCl gradient from 0 to 1m. Purified protein was buffer-exchanged into 10 mm sodium phosphate, 100 mm NaCl, pH 6.8, by using a PD-10 column (GE Healthcare) and stored at −80°C.
The MED15 KIX (1–78) protein was also expressed in Rosetta2(DE3) pLysS E. coli (Novagen). After the cultures reached an OD600nm of between 0.8 and 1.0, the cultures were cooled to 20°C and MED15 KIX expression was induced with the addition of 125 μm IPTG. After 12–18 h, the cells were harvested by centrifugation for 15 min at 7903 g in a Sorvall LYNX superspeed centrifuge with a Fiberlight F6−6×1000 LEX carbon fiber rotor (ThermoFisher Scientific), collected in a 50 mL falcon tube, and stored at −80°C.
The MED15 KIX protein was purified by first suspending bacterial cells in lysis buffer (≈25 mL; 50 mm sodium phosphate, 300 mm NaCl, 5 mm imidazole, 1 mm 2-mercaptoethanol (β-ME) pH 7.2, complete ethylenediaminetetraacetic acid (EDTA)-free protease inhibitor cocktail tablet (Roche)). The cells were lysed by sonication at 50% amplitude on ice by using a 6 mm tip with pulsing cycles of 3 s on and 6 s off for at least 3 min of total pulsing time. The soluble lysate was collected by centrifugation for 30 min at 9500 rpm (9299g) in an Allegra X-22R centrifuge (Beckman Coulter) with a C0650 fixed-angle rotor, and then incubated with suspended Ni-NTA agarose resin (2 mL; Qiagen) for 1 to 2 h rotating at 4°C. The resin was washed with washing buffer (5×5 mL; 50 mm sodium phosphate, 300 mm NaCl, 30 mm imidazole, 1 mm β-ME, pH 7.2). The nickel-bound protein was eluted with 300 mm imidazole and diluted into 10 mm sodium phosphate, 100 mm NaCl, 10% glycerol, 0.01% NP-40, pH 6.8. The hexahistidine affinity tag was cleaved overnight with thrombin (restriction grade; Novagen 69671) according to the manufacturer’s instructions. The cleaved protein was further purified on an AKTA FPLC purifier (GE Healthcare) with strong cation exchanger Source 15S media (GE Healthcare) packed to a 17 mL column volume. After loading the sample, the column was washed for 1.5 column volumes with buffer A (50 mm sodium phosphate, 1 mm dithiothreitol (DTT), pH 6.8). The protein was eluted from the column with a gradient of 0 to 60% of buffer B (50 mm sodium phosphate,1 mm DTT, 1m NaCl, pH 6.8) over four column volumes. With a 3 kDa molecular-weight cutoff Amicon Ultra-15 centrifuge filter units (EMD Millipore), MED15 KIX was concentrated and buffer-exchanged into 10 mm sodium phosphate, sodium chloride (100 mL), 10% glycerol, 0.01% NP-40, pH 6.8. Aliquots of protein were flash frozen in liquid nitrogen and stored at −80°C. The concentration of the protein was measured by using the absorbance at λ =280 nm on a NanoDrop 1000 Spectrophotometer (Thermo Scientific).
Peptide synthesis and purification:
All peptides were synthesized by standard N-9-fluorenylmethoxycarbonyl (Fmoc) solid-phase synthesis methods,[48] as previously described.[47] The peptide sequences, written as single-letter amino acid abbreviations, were as follows:
MLL: βA-DAGNILPSDIMDFVLKNTP-CONH2
pKID: βA-DSQKRREILSRRPS(Phos)YRKILNDLSSDAPG-CONH2
βA represents β-alanine and S(Phos) is phosphoserine. The fluorescent fluorescein isothiocyanate (FITC) tag was added at the amino terminus of the peptide before the β-alanine residue.
assessment:
For the values, 5 μm GACKIX N627C was incubated with varying concentrations (500–0.2 μm) of the compounds (at 1 mm β-ME) for 45 min at RT. Then the samples were incubated for 15 min at 4°C. The percent of protein tethered to fragment molecules was determined by means of Q-TOF LC-MS (Agilent).[49] The concentration of fragment molecule required for 50% maximum tethering () was determined by using GraphPad Prism software 4.00, fitting to Equation (1), in which is the log of fragment molecule concentration and is the normalized response from 1 to 100 (percent of protein tethered to fragment molecule).
| (1) |
Alkylation of GACKIX:
The GACKIX N627C mutant was incubated with small molecule (5–10 equiv) in 10 mm phosphate buffer, 100 mm NaCl, pH 6.8, overnight at RT. Excess small molecule was removed and small-molecule–protein complexes were concentrated by using 10 kDa molecular-weight cutoff centrifugal concentrators (Vivascience). The extent of labeling was measured by means of Q-TOF LC-MS (Agilent). Protein complexes that were at least 95% alkylated were flash frozen in liquid nitrogen and stored at −80°C.
Fluorescent anisotropy assays:
The fluorescent anisotropy was measured in triplicate with a final sample volume of 10 μL in a low volume, non-binding, black, 384-well plate (Corning). For each experiment, 25 nm fluorescently labeled peptide tracers, FITC-MLL, and FITC-pKID were incubated with varying concentrations of the small-molecule–GACKIX mutant complexes in binding buffer (10 mm phosphate, 100 mm NaCl, pH 6.8) for 30 min at RT. The plates were read by using a Tecan Genios Pro plate reader with polarized excitation at nm and emission intensity measured through a parallel and perpendicularly polarized nm filter. A binding isotherm that accounted for ligand depletion (assuming a 1:1 binding model of peptide to GACKIX) was fitted to the observed anisotropy values as a function of GACKIX to obtain the apparent equilibrium dissociation constant, [Eq. (2)]:
| (2) |
in which and are the total concentrations of fluorescent peptide and GACKIX, respectively; is the observed anisotropy at any GACKIX concentration; is the maximum observed anisotropy value; and is the minimum observed anisotropy value. Data analysis was performed by using GraphPad Prism 7.0 software.
Labeling GACKIX cysteine mutants:
Cysteine mutants (5 μm) at CBP KIX N627C, N644C, K662C, H651C, Q609C, and K606C, and MED15 KIX R67C and H70C, were incubated with small molecule (100 μm) in DMSO in the presence of β-ME (1 mm) for 1 h at RT. The extent of labeling was measured by means of Q-TOF LC-MS (Agilent) and each labeling reaction was duplicated.
Alkylation in growing E. coli:
The protein expression system for the GACKIX N627C protein was initiated as described above. Three hours after induction with 0.25 mm IPTG, the culture was concentrated by centrifugation, in which the cell pellet from 50 mL of culture was suspended in 1 mL of media. For a 1 mL alkylation reaction, 10 μL of compound in DMSO was added to concentrated cells to obtain concentrations of 1000, 500, 250, 62.5, and 15.6 αm. The mixtures were incubated for 1 h at 25°C (250 rpm). The cell pellets were washed three times with 10 mm phosphate, 100 mm NaCl, pH 6.8, and stored at −80°C. Purification was performed as previously described by using Ni-NTA resin.[29] The elutions were buffer-exchanged into 10 mm phosphate, 100 mm NaCl, pH 6.8, and concentrated by using 5 kDa molecular-weight cutoff concentrators (Vivascience). The samples were analyzed by means of Q-TOF LC-MS (Agilent). The extent of labeling was determined by comparing the peak intensity of small-molecule–GACKIX N627C complex versus unlabeled GACKIX N627C.
NeutrAvidin pull-down assay with the biotinylated 1–10 probe:
HEK 293T cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS). The cells were lysed in lysis buffer (750 μL; 150 mm NaCl, 50 mm Tris (pH 8.0), 0.1% NP-40) containing halt protease inhibitor cocktail (Thermo Scientific) and soluble lysate was isolated by centrifugation. HEK 293T lysate (100 μg total protein) and 25 nm of purified GACKIX N627C were incubated with DMSO or various concentrations of the biotinylated 1–10 d probe (5 μm, 1 μm, 200 nm, 40 nm, and 8 nm) at RT for 1 h. Following incubation with NeutrAvidin agarose resin (50 μL, Thermo Scientific) for 1 h at 4°C, the beads were washed with washing buffer (2×1 mL; 10 mm phosphate, 100 mm sodium chloride, 10% glycerol, 0.1% NP-40, pH 7.2) and the resin-bound complexes were eluted by boiling in NuPAGE LDS sample buffer (Invitrogen) containing DTT. The samples (15 μL) were resolved on a 12% SDS polyacrylamide gel by electrophoresis. The proteins were transferred onto a polyvinylidene fluoride membrane and incubated with streptavidin conjugated to HRP enzyme (ab7403, Abcam) at 1:10000 dilution in 10 mm phosphate-buffered saline containing 0.2% Tween-20. The membrane was developed by using Amersham ECL prime western blotting detection reagent (GE Healthcare) and the image was captured on X-ray film.
Supplementary Material
Footnotes
Supporting information and the ORCID identification numbers for the authors of this article can be found under https://doi.org/10.1002/cbic.201800173.
Conflict of Interest
The authors declare no conflict of interest.
References
- [1].Dyson HJ, Wright PE, J. Biol. Chem 2016, 291, 6714–6722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Thakur JK, Yadav A, Yadav G, Nucleic Acids Res. 2014, 42, 2112–2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Campbell KM, Lumb KJ, Biochemistry 2002, 41, 13956–13964. [DOI] [PubMed] [Google Scholar]
- [4].De Guzman RN, Goto NK, Dyson HJ, Wright PE, J. Mol. Biol 2006, 355, 1005–1013. [DOI] [PubMed] [Google Scholar]
- [5].Zor T, De Guzman RN, Dyson HJ, Wright PE, J. Mol. Biol 2004, 337, 521–534. [DOI] [PubMed] [Google Scholar]
- [6].Goto NK, Zor T, Martinez-Yamout M, Dyson HJ, Wright PE, J. Biol. Chem 2002, 277, 43168–43174. [DOI] [PubMed] [Google Scholar]
- [7].Brüschweiler S, Schanda P, Kloiber K, Brutscher B, Kontaxis G, Konrat R, Tollinger M, J. Am. Chem. Soc 2009, 131, 3063–3068. [DOI] [PubMed] [Google Scholar]
- [8].Wood MA, Attner MA, Oliveira AMM, Brindle PK, Abel T, Learn Memory 2006, 13, 609–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Sandberg ML, Sutton SE, Pletcher MT, Wiltshire T, Tarantino LM, Hogenesch JB, Cooke MP, Dev. Cell 2005, 8, 153–166. [DOI] [PubMed] [Google Scholar]
- [10].Pattabiraman DR, Sun J, Dowhan DH, Ishii S, Gonda TJ, Mol. Cancer Res. 2009, 7, 1477–1486. [DOI] [PubMed] [Google Scholar]
- [11].Best JL, Amezcua CA, Mayr B, Flechner L, Murawsky CM, Emerson B, Zor T, Gardner KH, Montminy M, Proc. Natl. Acad. Sci. USA 2004, 101, 17622–17627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Li BX, Xiao X, ChemBioChem 2009, 10, 2721–2724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Gee CT, Koleski EJ, Pomerantz WCK, Angew. Chem. Int. Ed 2015, 54, 3735–3739; Angew. Chem. 2015, 127, 3806–3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Majmudar CY, Højfeldt JW, Arevang CJ, Pomerantz WC, Gagnon JK, Schultz PJ, Cesa LC, Doss CH, Rowe SP, Vásquez V, Tamayo-Castillo G, Cierpicki T, Brooks III CL, Sherman DH, Mapp AK, Angew. Chem. Int. Ed 2012, 51, 11258–11262; Angew. Chem. 2012, 124, 11420–11424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Frangioni JV, LaRiccia LM, Cantley LC, Montminy MR, Nat. Biotechnol 2000, 18, 1080–1085. [DOI] [PubMed] [Google Scholar]
- [16].Mitton B, Chae HD, Hsu K, Dutta R, Aldana-Masangkay G, Ferrari R, Davis K, Tiu BC, Kaul A, Lacayo N, Dahl G, Xie F, Li BX, Breese MR, Landaw EM, Nolan G, Pellegrini M, Romanov S, Xiao X, Sakamoto KM, Leukemia 2016, 30, 2302–2311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Uttarkar S, Dukare S, Bopp B, Goblirsch M, Jose J, Klempnauer K-H, Mol. Cancer Ther. 2015, 14, 1276–1285. [DOI] [PubMed] [Google Scholar]
- [18].Majmudar CY, Mapp AK, Curr. Opin. Chem. Biol 2005, 9, 467–474. [DOI] [PubMed] [Google Scholar]
- [19].Lee LW, Mapp AK, J. Biol. Chem 2010, 285, 11033–11038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Schreiber G, Keating AE, Curr. Opin. Struct. Biol 2011, 21, 50–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Teufel DP, Freund SM, Bycroft M, Fersht AR, Proc. Natl. Acad. Sci. USA 2007, 104, 7009–7014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Lee CW, Arai M, Martinez-Yamout MA, Dyson HJ, Wright PE, Biochemistry 2009, 48, 2115–2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Novatchkova M, Eisenhaber F, Curr. Biol 2004, 14, R54–R55. [DOI] [PubMed] [Google Scholar]
- [24].Yang F, Vought BW, Satterlee JS, Walker AK, Jim Sun ZY, Watts JL, DeBeaumont R, Saito RM, Hyberts SG, Yang S, Macol C, Iyer L, Tjian R, van den Heuvel S, Hart AC, Wagner G, Näär AM, Nature 2006, 442, 700–704. [DOI] [PubMed] [Google Scholar]
- [25].Kassube SA, Jinek M, Fang J, Tsutakawa S, Nogales E, Nat. Struct. Mol. Biol 2013, 20, 892–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Erlanson DA, Braisted AC, Raphael DR, Randal M, Stroud RM, Gordon EM, Wells JA, Proc. Natl. Acad. Sci. USA 2000, 97, 9367–9372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM, Nature 2013, 503, 548–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Pomerantz WC, Wang N, Lipinski AK, Wang R, Cierpicki T, Mapp AK, ACS Chem. Biol 2012, 7, 1345–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Wang N, Majmudar CY, Pomerantz WC, Gagnon JK, Sadowsky JD, Meagher JL, Johnson TK, Stuckey JA, Brooks III CL, Wells JA, Mapp AK, J. Am. Chem. Soc 2013, 135, 3363–3366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Wang N, Lodge J, Fierke CA, Mapp AK, Proc. Natl. Acad. Sci. USA 2014, 111, 12061–12066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Lodge JM, Rettenmaier TJ, Wells JA, Pomerantz WC, Mapp AK, MedChemComm 2014, 5, 370–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Kluter S, Simard JR, Rode HB, Gretter C, Pawar V, Raaijmakers HC, Barf TA, Rabiller M, van Otterlo WA, Rauh D, ChemBioChem 2010, 11, 2557–2566. [DOI] [PubMed] [Google Scholar]
- [33].Liu Q, Sabnis Y, Zhao Z, Zhang T, Buhrlage SJ, Jones LH, Gray NS, Chem. Biol 2013, 20, 146–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Hu X, Manetsch R, Chem. Soc. Rev 2010, 39, 1316–1324. [DOI] [PubMed] [Google Scholar]
- [35].Reddick JJ, Cheng J, Roush WR, Org. Lett 2003, 5, 1967–1970. [DOI] [PubMed] [Google Scholar]
- [36].Jöst C, Nitsche C, Scholz T, Roux L, Klein CD, J. Med. Chem 2014, 57, 7590–7599. [DOI] [PubMed] [Google Scholar]
- [37].Kathman SG, Xu Z, Statsyuk AV, J. Med. Chem 2014, 57, 4969–4974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Novatchkova M, Eisenhaber F, Curr. Biol 2004, 14, R54–R55. [DOI] [PubMed] [Google Scholar]
- [39].Reddick JJ, Cheng J, Roush WR, Org. Lett 2003, 5, 1967–1970. [DOI] [PubMed] [Google Scholar]
- [40].Kathman SG, Statsyuk AV, MedChemComm 2016, 7, 576–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Mah R, Thomas JR, Shafer CM, Bioorg. Med. Chem. Lett 2014, 24, 33–39. [DOI] [PubMed] [Google Scholar]
- [42].Nonoo RH, Armstrong A, Mann DJ, ChemMedChem 2012, 7, 2082–2086. [DOI] [PubMed] [Google Scholar]
- [43].González-Bello C, ChemMedChem 2015, 10, 22–30. [DOI] [PubMed] [Google Scholar]
- [44].Schwartz PA, Kuzmic P, Solowiej J, Bergqvist S, Bolanos B, Almaden C, Nagata A, Ryan K, Feng J, Dalvie D, Kath JC, Xu M, Wani R, Murray BW, Proc. Natl. Acad. Sci. USA 2014, 111, 173–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Singh J, Petter RC, Baillie TA, Whitty A, Nat. Rev. Drug Discovery 2011, 10, 307–317. [DOI] [PubMed] [Google Scholar]
- [46].Johnson DS, Weerapana E, Cravatt BF, Future Med. Chem 2010, 2, 949–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Buhrlage SJ, Bates CA, Rowe SP, Minter AR, Brennan BB, Majmudar CY, Wemmer DE, Al-Hashimi H, Mapp AK, ACS Chem. Biol 2009, 4, 335–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Fmoc Solid Phase Peptide Synthesis: A Practical Approach (Eds.: Chan WC, White PD), Oxford University Press, Oxford, 2000. [Google Scholar]
- [49].Sadowsky JD, Burlingame MA, Wolan DW, McClendon CL, Jacobson MP, Wells JA, Proc. Natl. Acad. Sci. USA 2011, 108, 6056–6061. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.