

Abstract
Plasmodium species causing malaria in humans are not monophyletic, sharing common ancestors with nonhuman primate parasites. Plasmodium gonderi is one of the few known Plasmodium species infecting African old-world monkeys that are not found in apes. This study reports a de novo assembled P. gonderi genome with complete chromosomes. The P. gonderi genome shares codon usage, syntenic blocks, and other characteristics with the human parasites Plasmodium ovale s.l. and Plasmodium malariae, also of African origin, and the human parasite Plasmodium vivax and species found in nonhuman primates from Southeast Asia. Using phylogenetically aware methods, newly identified syntenic blocks were found enriched with conserved metabolic genes. Regions outside those blocks harbored genes encoding proteins involved in the vertebrate host-Plasmodium relationship undergoing faster evolution. Such genome architecture may have facilitated colonizing vertebrate hosts. Phylogenomic analyses estimated the common ancestor between P. vivax and an African ape parasite P. vivax-like, within the Asian nonhuman primates parasites clade. Time estimates incorporating P. gonderi placed the P. vivax and P. vivax-like common ancestor in the late Pleistocene, a time of active migration of hominids between Africa and Asia. Thus, phylogenomic and time-tree analyses are consistent with an Asian origin for P. vivax and an introduction of P. vivax-like into Africa. Unlike other studies, time estimates for the clade with Plasmodium falciparum, the most lethal human malaria parasite, coincide with their host species radiation, African hominids. Overall, the newly assembled genome presented here has the quality to support comparative genomic investigations in Plasmodium.
Keywords: phylogenomics, GC content, Plasmodium vivax, Plasmodium falciparum, molecular clock, synteny
Significance.
The Plasmodium causing human malaria originated from species found in nonhuman primates. Here, a new Plasmodium gonderi genome assembly is presented; this parasite is related to Plasmodium vivax, the most prevalent human parasite outside Africa. In addition to informing about the evolution of Plasmodium genome fissures, such as synteny and codon usage, phylogenomic analyses and time estimates provide scenarios for the origins of Plasmodium species infecting humans. Plasmodium vivax's most recent common ancestor was found among nonhuman primate parasites from Southeast Asia. The species related to Plasmodium falciparum, the agent causing the most severe human malaria, have a complex history with host switches but not necessarily among extant ape species, as these parasites could be as old as African apes.
Introduction
The genus Plasmodium is a diverse taxon of vector-borne parasitic protozoa, some of which cause malaria in their vertebrate hosts, including humans (Garnham 1966; Coatney et al. 1971; Telford 2009; Valkiūnas and Iezhova 2018; Pacheco and Escalante 2023). The 5 species that typically infect humans do not form a monophyletic group (Escalante et al. 2022). Each shares its most recent common ancestors with different Plasmodium species found in nonhuman primates. Such distinct human parasites’ evolutionary histories translate into differences in phenotypic traits and clinical manifestations of the disease, requiring different therapeutics and control interventions (Garnham 1966; Coatney et al. 1971; Escalante et al. 2022). As a result, understanding the origins and biology of human malaria parasites has propelled genomic studies on their related species infecting nonhuman primates (Tachibana et al. 2012; Chien et al. 2016; Pasini et al. 2017; Rutledge et al. 2017; Lapp et al. 2018; Otto et al. 2018).
Taxonomists early recognized differences among Plasmodium species found in primates (Garnham 1966; Coatney et al. 1971), creating subgenera. E.g., Plasmodium falciparum, the agent of the most severe form of human malaria, was separated from the other human parasites in the subgenus Laverania (Garnham 1966; Coatney et al. 1971; Sinden et al. 1978). Nowadays, it is known that P. falciparum is part of a diverse clade of parasites infecting African apes, sometimes referred to as Laverania following the classical taxonomy (Ollomo et al. 2009; Krief et al. 2010; Liu et al. 2010; Pacheco et al. 2013; Liu et al. 2017). Given the interest in P. falciparum, most species in this clade have reference genomes (Otto et al. 2018).
The other parasites that primarily infect humans, Plasmodium ovale s.l., Plasmodium malariae, and P. vivax, were placed in a subgenus called Plasmodium, together with species found in catarrhine monkeys (Garnham 1966; Coatney et al. 1971). Although such a subgenus is likely paraphyletic (Pacheco et al. 2011, 2022), evidence suggests a clade of primate parasites with all nonfalciparum-related Plasmodium species, including species found in lemurs (Pacheco et al. 2018, 2022). Still, species infecting humans within this putative primate parasite clade do not form a monophyletic group.
Evidence shows that species infecting African apes share common ancestors with P. malariae and P. ovale s.l. (Duval et al. 2009; Rutledge et al. 2017; Mapua et al. 2018; Fuehrer et al. 2022; Plenderleith et al. 2022). Although P. malariae undergoes anthropozoonotic cycles between humans and platyrrhine primates in South America (Bajic et al. 2022; Fuehrer et al. 2022), its origin has been established in Africa (Rutledge et al. 2017; Plenderleith et al. 2022). Unlike other human parasites, the origin of P. vivax, the most common human malaria agent outside Africa, has been debated. Plasmodium vivax shares a common ancestor with parasite species from African and Southeast Asian nonhuman primates. These parasites exhibit extraordinary phenotypic diversity (Garnham 1966; Coatney et al. 1971) and will be further referred to as the “vivax-clade.” Also, within this vivax-clade are lineages related to P. vivax but infecting African apes. Such species are loosely called P. vivax-like, and their complex evolutionary history has driven the discussion about the origin of P. vivax as a human parasite (Prugnolle et al. 2013; Liu et al. 2014; Daron et al. 2021).
In addition to P. vivax and P. vivax-like, there are genomic data on some vivax-clade species found in Southeast Asia nonhuman primates as they have been used as models to understand malaria in humans, and some cause zoonotic malaria, such as Plasmodium knowlesi (Coatney et al. 1971; Cox-Sing et al. 2008; Tachibana et al. 2012; Pasini et al. 2017; Lapp et al. 2018; Galinski 2022). However, a complete picture regarding the origin of P. vivax requires understanding its phylogenetic relationships with the species infecting nonhuman primates in Southeast Asia and those found in African monkeys (Coatney et al. 1971; Poirriez et al. 1995), such as Plasmodium gonderi.
Plasmodium gonderi was first found by Gonder and von Berenberg-Cossler in 1908, infecting mangabeys (Coatney et al. 1971; Poirriez et al. 1995), and then described by Sinton and Mulligan in 1933. This parasite has also been found infecting drills (Mandrillus leucophaeus; Garnham et al. 1958) and mandrills (Mandrillus sphinx, Charpentier et al. 2019). Based on its biological traits, P. gonderi was considered “related” to nonhuman parasites in Southeast Asia and P. vivax (Coatney et al. 1971), an assumption confirmed by molecular phylogenetic studies using a single locus or multiple loci (Escalante et al. 1998; Leclerc et al. 2004; Escalante et al. 2005; Hayakawa et al. 2008; Nishimoto et al. 2008; Pacheco et al. 2018). However, the relative position of P. gonderi in the Plasmodium phylogeny has been controversial (Leclerc et al. 2004; Escalante et al. 2005; Nishimoto et al. 2008; Pacheco et al. 2018, Pacheco et al. 2020; Sharp et al. 2020; Cepeda et al. 2021; Escalante et al. 2022; Arisue et al. 2022). There is a debate about whether P. gonderi places the most recent common ancestor of P. vivax and P. vivax-like outside the clade of nonhuman primate parasites from Southeast Asia (Escalante et al. 2005; Pacheco et al. 2018; Arisue et al. 2019; Pacheco et al. 2020; Sharp et al. 2020; Cepeda et al. 2021; Arisue et al. 2022; Escalante et al. 2022; Pacheco et al. 2022; Pacheco and Escalante 2023). Such evidence has been interpreted as indicative of an African origin for the lineage leading to P. vivax. Beyond such controversies surrounding the origin of P. vivax, P. gonderi is one of the few known Plasmodium found in African catarrhine primates that are not apes. Thus, a good-quality genome for this species is essential for comparative studies in Plasmodium.
A P. gonderi isolate (Garnham strain, ATCC 30045) has been maintained (Coatney et al. 1971; Collins and Contacos 1980), facilitating a first draft genome (Honma et al. 2017). Although valuable (Sharp et al. 2020; Arisue et al. 2022; Escalante et al. 2022), it lacked the quality to assemble complete chromosomes without a reference, and annotating gene families was also challenging. A chromosome-level de novo assembled genome of P. gonderi ATCC 30045 is presented after incorporating new sequence data. This investigation offers new insights into the evolution of malaria parasites in primates, including the origins of those species infecting humans.
Results and Discussion
Assembly of Plasmodium gonderi Genome
The reference genome presented here will be referred to as P. gonderi v2 (supplementary fig. S1, Supplementary Material online). Its assembly statistics were compared with the previous draft genome, P. gonderi v1 (Honma et al. 2017), and genomes from other Plasmodium species (supplementary table S1, Supplementary Material online). The average coverage of P. gonderi v2 was 1270 × (min = 729 × and max = 4022 × ; supplementary fig. S1, Supplementary Material online), an improvement over P. gonderi v1, where the average coverage was 250 × (Honma et al. 2017). As a result, P. gonderi v2 reduced the number of scaffolds to 72 (compared to 743 for P. gonderi v1) and an ambiguous nucleotide ratio of 0.86 (compared to 283.03 for P. gonderi v1; supplementary table S1, Supplementary Material online). The N50 of P. gonderi v2 is 2,125,246 versus 1,639,955 in the previous version (supplementary table S1, Supplementary Material online). Each of the P. gonderi v2 largest 14 scaffolds represented a chromosome, while 56 smaller scaffolds could not be assigned to specific chromosomes. Two scaffolds contain the mitochondrial and apicoplast genomes. In particular, a 173 kb scaffold contained at least 53 copies of the linear mitochondrial genome (mtDNA) (sequence ID: Pgonderi_v2_iGEM_MIT), common in Plasmodium spp. (Vaidya et al. 1989) and a 34.5 kb scaffold contained the genome of the apicoplast, a nonphotosynthetic plastid-like organelle homologous to the photosynthetic plastids found in related protists (Alveolate, a superphylum that includes dinoflagellates) (Wilson et al. 1996) (Pgonderi_v2_iGEM_API). The organelle sequences were identical in both P. gonderi assembly versions.
Plasmodium gonderi v2 reached metrics of other high-quality genome assemblies, such as Plasmodium cynomolgi (Pasini et al. 2017) and P. vivax (Auburn et al. 2016; supplementary table S1, Supplementary Material online). As a result, there is an impact on the structural annotation, resulting in 6,412 CoDing sequences (CDSs) (399 more than the previous version; supplementary table S1, Supplementary Material online), excluding those in mitochondrial and apicoplast genomes. The genes were functionally annotated, and Gene Ontology (GO) terms are shown in supplementary table S2, Supplementary Material online.
Plasmodium Gonderi pir Gene Family
An example of this genome-improved annotation is the Plasmodium interspersed repeat (pir) family, which has a suspected role in virulence and chronic infection, particularly in nonfalciparum Plasmodium species infecting mammals (Harrison et al. 2020; Little et al. 2021). Many pir genes are species-specific, requiring high-quality genome assemblies to characterize them. Given the limited high-quality genome data for many Plasmodium species, the number of pir genes shared between closely related species is usually underestimated. Nevertheless, it is worth noting that it is not possible to find Pir orthologous genes across the genus (Harrison et al. 2020).
The P. gonderi v1 assembly shows 298 (of 914) annotated pir genes on chromosomes with a scattered distribution (supplementary fig. S2, Supplementary Material online). In contrast, the P. gonderi v2 assembly has 661 (out of 1160) pir genes on chromosomes distributed mainly in subtelomeric regions (supplementary fig. S2, Supplementary Material online), as observed in other Plasmodium assemblies with high coverage. The remaining pir genes from P. gonderi v2 are on 9 contigs not assigned to specific chromosomes. A similar pattern was previously reported in the P. cynomolgi genome (Pasini et al. 2017; supplementary fig. S3, Supplementary Material online).
Figure 1 shows clusters obtained using reciprocal Basic Local Alignment Search Tool (BLAST) of pir genes from P. gonderi v2 and those found in other related Plasmodium species expected to be in the same clade; several pir paralogs form clusters within rather than between species. This pattern indicates a fast rate of evolution in terms of contraction/expansion of the gene family, as reported elsewhere (Pasini et al. 2017). Clusters with 1 or more pir genes from more than one species suggest they may be conserved (Neafsey et al. 2012; Little et al. 2021), but those are usually considered very few. However, approximately 10% of P. gonderi pir genes reported in this new genome (114 out of 1160) form small clusters of 5 to 8 pirs, with genes from 5 or all 6 species included in this comparison (Fig. 1). Pirs from P. vivax-like are usually missing from those clusters where P. gonderi and the other species are represented, perhaps due to genome quality issues as will be shown later. Notably, most putatively conserved pir genes (105 out of 114 in P. gonderi) are mapped in chromosome 1. A similar pattern is observed in P. vivax and related species with assemblies carried out independently from this study (e.g. 115 out of 120 conserved pir genes in P. vivax chromosome 1) (Auburn et al. 2016; Chien et al. 2016; Pasini et al. 2017; Lapp et al. 2018). Whether the conservation regarding similarity and chromosome allocation of these pir genes within this clade reflects their functional importance needs to be determined (Little et al. 2021). Overall, improving the genome assembly (supplementary table S1, Supplementary Material online) allowed us to describe this gene family better as an example of how this resource will facilitate comparative genomic studies.
Fig. 1.
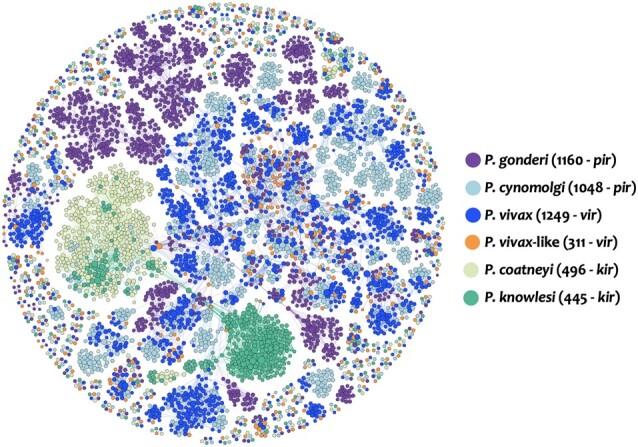
Network plotting the pir gene proteins similarity between P. gonderi (1160 pir), P. cynomolgi M (1048 pir), P. coatneyi Hackeri (496 kir), P. knowlesi H (445 kir), P. vivax-like Pvl01 (311 vir), and P. vivax P01 (1249 vir). The size of each node is determined by the number of edges per node (or gene). The more edges a node has, the larger its size.
GC Content among Plasmodium Genomes
There are 26 Plasmodium species with genomic assemblies of various qualities (supplementary table S1, Supplementary Material online). Two sets of loci were considered to investigate the GC content and the Relative Synonymous Codon Usage (RSCU). One is limited to 1,961 single-copy orthologous genes (SC-OGs) found in all species, and the other includes all CDSs available for each species regardless of the quality of the genome data (supplementary table S3, Supplementary Material online). The GC content and cluster analysis with their RSCU values on the SC-OGs are depicted in Fig. 2. All Plasmodium sequences have low GC content. However, there were marked differences among the species. For example, GC was twice as high in P. vivax (45.76% for CDS and 34.45% in SC-OGs) as in P. gallinaceum (22.54% for CDS and 15.20% in SC-OGs). The same differences were observed in total genomic GC content and noncoding regions (Fig. 2, supplementary fig. S4 and table S1, Supplementary Material online): a lower GC content in the subgenus Laverania and a distinct relatively high GC pattern in P. vivax and species found in Southeast Asia (subgenus Plasmodium), setting them apart from other nonfalciparum primate parasites from Africa, including P. gonderi. Such a pattern suggests some form of mutational bias. However, more data is needed to test specific hypotheses, such as GC content association with recombination rates per time unit across the genus Plasmodium (Romiguier and Roux 2017).
Fig. 2.

Hierarchical cluster by average and heat map of RSCU values of each codon in the SC-OGs of haemosporidian genomes. Each square in the heat map represents each codon RSCU value (in rows) within the SC-OG of each genome (in columns). Colors indicate the magnitude of RSCU values. A color bar indicates the species genome GC content.
Although not a phylogenetic analysis, the clustering of RSCU per codon using SC-OGs (Fig. 2) or all CDS (supplementary fig. S4, Supplementary Material online) showed patterns consistent with a phylogenetic signal. The previously reported monophyletic group that includes P. falciparum, which will henceforth be called Laverania, exhibited a similar low GC content and codon bias. Laverania has similar GC content and codon bias compared to the 2 avian parasites (Plasmodium relictum and Plasmodium gallinaceum) (Böhme et al. 2018) and the rodent parasites (supplementary table S4, Supplementary Material online). Hepatocystis, a genus of haemosporidian parasites with distinct life cycles (Garnham 1966), was placed within this group in the RSCU cluster analysis, presenting similarly low GC content.
The clustering of RSCU per codon also recovered 2 groups of primate parasites that are not related to P. falciparum. One cluster shows P. gonderi with P. malariae and P. ovale s.l. However, clearly outside the cluster that includes species from Southeast Asia, P. vivax, and P. vivax-like (Fig. 2). Plasmodium vivax (45.76%) and P. vivax-like were more GC-rich than other species from Southeast Asia (38.89% to 42.95%, supplementary table S1, Supplementary Material online). They were also biased toward codons CUG, CCC, GUG, and UCC, which correspond to the amino acids Leu, Pro, Val, and Ser (supplementary table S4, Supplementary Material online). Such patterns may translate into the rate heterogeneity reported in other studies affecting phylogenetic inferences with a limited number of genes and sites per gene (Galen et al. 2018). The same clustering emerged when analyzing the second set of loci that included all the CDSs available for each species (supplementary fig. S4, Supplementary Material online).
It is worth noting that the SC-OGs (found in all species) have lower GC content than all CDSs (supplementary table S1, Supplementary Material online). Such a difference might indicate that genes conserved across Plasmodium may have higher codon bias due to selected efficiency in their expression because they are essential (Zhou et al. 2016; Oberstaller et al. 2021). However, the low quality of some genomes and the lack of functional genomic data in many parasites do not allow comprehensive analyses.
Plasmodium gonderi clustered with P. malariae when analyzing the frequency of amino acids in the proteome and SC-OGs (supplementary fig. S5 and table S5, Supplementary Material online). In the same analysis, P. ovale s.l. was placed in the same cluster as the rodent malaria parasites and Hepatocystis sp., parasites found in nonhuman primates and bats. Although cluster analyses are not phylogenies, these patterns on amino acids may explain the findings of previous studies, where P. ovale seems to share a common ancestor with rodent parasites (Sharp et al. 2022), mainly when working with proteins instead of DNA.
In summary, cluster analyses of codon usage and proteome place P. gonderi with other primate-infecting, nonfalciparum-related Plasmodium species that originated in Africa but set it apart from Southeast Asian parasites. Discrepancies between the clustering in the proteome and the codon usage analyses may indicate underlying processes that affect phylogenetic inferences if those differences are not considered a priori.
Synteny among Plasmodium Genomes
Synteny among the Plasmodium genomes was studied on 18 species out of 26 genomes since they have chromosome-length or less fragmented assemblies (supplementary table S1, Supplementary Material online). Visual inspection indicated major syntenic blocks shared between Plasmodium species (Fig. 3). However, rearrangements that separated species clades were evident as the rodent malaria, the Laverania clade containing P. falciparum, and a primate parasite clade with all the other human malaria showing apparent differences in synteny. There were a few translocations, insertions, and deletions of large syntenic blocks between P. gonderi and other Plasmodium species found in primates from Southeast Asia and P. vivax. Further exploration requires determining whether there are still quality issues in some genome assemblies that could lead to the generation of putative chromosome rearrangements. E.g., the P. coatneyi genome, as available in databases, showed genome rearrangements in chromosomes 2, 13, and 14 compared with the other species found in macaques (supplementary fig. S6, Supplementary Material online). Such rearrangements are likely artifacts. Indeed, they disappeared when the P. coatneyi genome was assembled de novo using the same methodologies reported here for P. gonderi (Fig. 3 and supplementary fig. S6, Supplementary Material online). Putative assembly artifacts also could be observed among the rodent malaria parasites and the macaque parasite P. knowlesi (inversions in chromosome 12).
Fig. 3.

Phylogenomic reconstruction based on synteny and graphical representation of the syntenic blocks across the plasmodium genus. Phylogenetic reconstruction based on synteny was inferred by maximum likelihood methods implemented in IQ-tree software (100 bootstrap replicates) from the synteny matrix obtained following the protocol of Zhao et al. (2021). The synthetic blocks were graphed with the R package GeneSpace (Lovell et al. 2022). The corrected assembly of P. coatneyi Hackeri is used here.
Regardless of assembly concerns, a synthetic-based phylogenomic reconstruction (Fig. 3 and supplementary fig. S6, Supplementary Material online) supported previous phylogenetic inferences (Escalante and Ayala 1994; Escalante et al. 1995; Nishimoto et al. 2008; Pacheco et al. 2018; Escalante et al. 2022; Pacheco et al. 2022). The avian parasite Plasmodium relictum was used as the outgroup based on findings from single locus and multilocus phylogenies estimated on several Haemosporida species (Martinsen et al. 2008; Galen et al. 2018; Pacheco et al. 2018, 2022). Plasmodium gonderi shares a common ancestor with Southeast Asian parasites, and P. ovale and P. malariae form a clade with other primate parasites. However, the synteny phylogeny presented here lacks critical species such as P. vivax-like. Although a genome is available, P. vivax-like produced peculiar patterns likely due to assembly problems (see supplementary fig. S7, Supplementary Material online, which includes both assemblies of the 2 available P. vivax-like lineages).
Plasmodium relictum, the only avian parasite with a high-quality genome (Böhme et al. 2018), showed rearrangements not observed in parasites found in primates and rodents. However, many other avian genomes are needed to identify patterns since the available evidence indicates that these parasites are more diverse than those found in mammals (Valkiūnas, 2005; Valkiūnas and Iezhova 2018).
A previous study (Zhang et al. 2018b) hypothesized that syntenic regions have more critical genes based on the growth of P. falciparum asexual stage in vitro. In contrast, nonsyntenic blocks were enriched with “dispensable” genes, including genes encoding proteins considered virulence factors. The syntenic regions were relatively few inferred in a comparative genomic study with limited well-annotated Plasmodium genomes (DeBarry and Kissinger 2011). This hypothesis was further tested using a parsimony-based algorithm implemented in the software AGORA (Muffato et al. 2023) that estimated the content and gene order in the ancestor from the 18 complete genomes of Plasmodium species, defining syntenic block across all species, including P. gonderi. A GO enrichment analysis (using a Fisher's exact test and Bonferroni correction) identified the functional categories of each gene present in both the syntenic blocks and the breakpoints based on the classification from Zhang et al. (2018b) as essential genes (supplementary table S6, Supplementary Material online). The pattern was explored on the common ancestors of Laverania, as was P. falciparum where the functional analyses were performed, then all mammalian Plasmodium, and finally all Plasmodium species were included in the analysis. In all these 3 comparisons, syntenic blocks harbor more putatively essential genes, as defined by Zhang et al. (2018b), than expected by chance (supplementary fig. S8, Supplementary Material online).
However, a bias exists in focusing on P. falciparum data from an in vitro culture. Alternatively, it can be posited that orthologous genes among all Plasmodium species are putatively essential (Kooij et al. 2005; Oberstaller et al. 2021). The putative Plasmodium “core genome” comprised 3,408 SC-Ogs in these 18 Plasmodium reference genomes. Of those, >98.9% of the genes are located in the syntenic blocks depending on the clade (supplementary table S7 and fig. S9, Supplementary Material online). In contrast, several genes involved in the vertebrate host-parasite relationship were in the more plastic part of the genome or breakpoints outside the syntenic blocks (supplementary table S8 and fig. S9, Supplementary Material online). This pattern may reflect the observed biological diversity of Plasmodium in mechanisms to invade the vertebrate host cell and evade the host immune response, as previously suggested with a limited dataset (Kooij et al. 2005) (supplementary table S8, Supplementary Material online).
Although identifying conserved genes across Plasmodium seems a more robust approximation to define an essential gene than just data from one species in culture, it still overlooks Plasmodium species’ divergent life histories resulting in clade-specific genes. For example, Fig. 4 shows a small sample of functionally important genes expressed in different life cycle stages. Those genes were chosen because they are involved in processes considered fitness components, like the invasion of the red blood cell or passage of the oocyst (vector stage) through the Anopheles midgut in the different Plasmodium species (see Graumans et al. (2020), Kaur et al. (2022), and Wright and Rayner (2014) for genes expressed in the ookinete while in the mosquito vector, the sporozoite or infectious stage, and the blood stage or merozoite, respectively). As can be observed, some genes, particularly those involved in the invasion of red blood cells, do not have orthologs across the genus, such as the gene encoding RH5, essential in the infection of the red blood cell by P. falciparum (Wright and Rayner 2014; Plenderleith et al. 2018). Thus, the observed pattern of conserved genes in syntenic blocks must be interpreted considering that it does not account for clade-specific essential genes.
Fig. 4.

Mapping of genes involved in the parasitic stages of cell invasion. The phylogeny used corresponds to the species tree previously reported (Escalante et al. 2022). Each cell in the parasitic stages heatmaps corresponds to the number of gene copies for each gene per species. (*) Genes that are present but incomplete and (?) missing genes in the genome. The genes functions are described in Kaur et al. (2022), Graumans et al. (2020), and Wright and Rayner (2014) for the ookinete, sporozoite, and merozoite, respectively.
Overall, syntenic blocks provide phylogenetic information that places P. gonderi in a clade with P. vivax and other nonhuman primate parasites from Southeast Asia. Syntenic blocks are enriched with conserved genes detected across Plasmodium species. In contrast, genes associated with the host-parasite interaction, such as gene families involved in virulence, are outside such blocks (Kooij et al. 2005). This observation suggests that rearrangements in nonsyntenic regions are critical in the vertebrate host-parasite relationships.
Phylogenomic Reconstructions
A total of 1,961 single-copy gene ortho groups (SC-Ogs) found across all 26 species were included in this study (supplementary table S3, Supplementary Material online). Given that 3,408 SC-Ogs were recovered from 18 complete genomes, it is clear that some draft genomes were not only fragmented but incomplete (e.g. Hepatocistys sp., P. vivax-like, and P. fragile). As IQ-tree (Nguyen et al. 2015; Minh et al. 2020) estimated, 3 substitution models accounted for 91.82% of the total gene alignments (56.85% general time reversible (GTR), 29.57% TIM3, and 5.4% TVM; supplementary table S3, Supplementary Material online).
Plasmodium species trees were first inferred from a concatenated alignment (2.4 Mb without gaps) using Bayesian and Maximum Likelihood (ML) methods (Fig. 5a) with the GTR + I + G4 substitution model, as determined using IQ-tree. The species tree was also inferred using the multispecies coalescent model (Fig. 5b) implemented in Astral III using SC-OG trees estimated with IQ-tree under the best-fitted model (supplementary table S3, Supplementary Material online). The phylogenies obtained were almost identical and well-supported (Fig. 5); the exception was the fragile position of Plasmodium. The results were reproduced using codon (not included) instead of nucleotide models yielding the phylogeny of the concatenated alignment. It is worth noting that using normalized Robinson–Foulds distances (nRF), a paired comparison was made between each gene tree topology calculated by IQ-tree and the species tree (supplementary table S3, Supplementary Material online). The mean of these distances was 13.25 (s2 = 17.3), indicating that, on average, there are 8 changes between a gene tree and the estimated species tree.
Fig. 5.

Species trees: a) phylogeny based on the concatenated alignment of the 1,961 SC-OGs using the Bayesian methods implemented in MrBayes v3.2.6 with the default priors (Ronquist and Huelsenbeck 2003) and the ML method in IQ-Tree (Minh et al. 2020). All nodes show posterior probabilities and bootstrap values as a percentage obtained for 1,000 pseudoreplicates. b) Phylogeny based on the multispecies coalescent model implemented in ASTRAL-III (Zhang et al. 2018a, 2018b) from the 1961 SC-GO trees. All nodes show values of posterior probabilities.
Non-Laverania Plasmodium species found in primates, including P. gonderi, belonged to a monophyletic group in this analysis. This clade shared a common ancestor with rodent malarias and Hepatocystis sp. (Fig. 5). The relative position of Hepatocystis sp. is consistent with previous studies (Aunin et al. 2020; Escalante et al. 2022). However, it is worth reiterating that several species of Plasmodium in nonhuman primates have no genomic data available including all parasites from gibbons, orangutans, and lemurs (Coatney et al. 1971; Peters 1976; Pacheco et al. 2022). Other Plasmodium species found in bats and adding more species of Hepatocystis sp. may also change this result, particularly the rodent parasites clade (Schaer et al. 2013).
The phylogenomic analyses (Fig. 5) placed the common ancestor of P. vivax and P. vivax-like as part of the parasite radiation of nonhuman primates from Asia, supporting earlier studies that only included P. vivax (Escalante et al. 2005; Mu et al. 2005; Pacheco et al. 2012; Prugnolle et al. 2013; Muehlenbein et al. 2015; Pacheco et al. 2018). Although information from other parasite species found in macaques or gibbons could change this phylogeny, the tree topology is consistent with other studies with more Plasmodium species but that used single gene or multilocus approaches (Escalante et al. 2005; Nishimoto et al. 2008; Mitsui et al. 2010; Pacheco et al. 2018; Pacheco et al. 2022; Pacheco and Escalante 2023). It is consistent with the phylogenetic hypothesis emerging from studying syntenic blocks. Still, it differs from other studies where the vivax lineage appears as a sister clade to the other macaque parasites (Sharp et al. 2020; Arisue et al. 2022). The discrepancy may be in the methods used to reconstruct the phylogeny and the data considered. Here, DNA sequences were chosen a priori as they are more informative (Hall 2005; Quek and Huang 2019; Young and Gillung 2020).
Plasmodium cynomolgi has been considered the species from Southeast Asia that shared the most recent common ancestor with the lineage leading to P. vivax, an inference based on biological traits (Collins and Contacos 1980; Tachibana et al. 2012; Pasini et al. 2017; Galinski 2022). Finding Plasmodium inui sharing a more recent common ancestor with the P. vivax lineage contradicts that hypothesis. A better species sampling may be required (Muehlenbein et al. 2015). The lack of genomic data on parasites from gibbons or orangutans may have affected this phylogeny (Pacheco et al. 2012; Muehlenbein et al. 2015; Pacheco et al. 2018).
The inclusion of P. gonderi in the phylogenies inferred here showed that the common ancestor between P. vivax and P. vivax-like may have arisen during the radiation of the species currently found in nonhuman primates from Southeast Asia. These results coincide with population genomic and single gene studies indicating that P. vivax originated in Asia (Cornejo and Escalante 2006; Prugnolle et al. 2013; Daron et al. 2021). Thus, based on the phylogenies presented here, the species called P. vivax-like is likely an early introduction of the P. vivax lineage from Asia into Africa.
Time-Tree Analyses
Estimating time trees with several hundreds of genes is impractical (Battistuzzi et al. 2018; Tamura et al. 2018). Identifying genes with similar GC content (Romiguier and Roux 2017; Quek and Huang 2019; Kapli et al. 2020) among Plasmodium species was impossible (see supplementary tables S3 and S9, Supplementary Material online). Thus, 110 genes were chosen using the smaller nRF distances between gene and species trees. This subset yields a similar phylogeny obtained with the 1,961 SCGOs (Fig. 5).
Informative calibrations are critical to obtaining reliable absolute timescales in relaxed molecular clock methods (dos Reis et al. 2016). A problem when timing Plasmodium in primates, in addition to differences in GC content among clades, is that no parasite fossils can provide primary calibrations (Pacheco and Escalante 2023). Thus, assumptions based on the association between parasite clades and hosts’ events must be made, making such constraints a form of secondary calibrations. The use of secondary calibrations is controversial but, in this case, unavoidable (Powell et al. 2020). Potential problems can be mitigated by comparing different calibration strategies (Pacheco et al. 2018, 2022). Overall, calibrations using the host should be as inclusive as possible to avoid unrealistic precision (Graur and Martin 2004; Pacheco et al. 2018).
Relaxed clock methods differ in how evolutionary rate variation among lineages is modeled as autocorrelated or independent (uncorrelated) (dos Reis et al. 2016; Battistuzzi et al. 2018). Such differences have proven to affect time estimates in Plasmodium (Pacheco et al. 2018). Here, the effect of rate variation models was explored using MCMCTree and 3 different calibration scenarios (with one, two, and 3 calibration constraints). Then, MCMCTree estimates were compared against the results from a non-Bayesian dating method that estimates relative divergence times, RelTime (Tamura et al. 2012). Supplementary table S10, Supplementary Material online shows all the estimated node ages in MCMCTree and their credible intervals (CrI) under the different calibration scenarios. All calibration scenarios were compared using autocorrelated and uncorrelated models (supplementary fig. S10, Supplementary Material online indicates the node numbering in supplementary table S10, Supplementary Material online).
The first scenario used a single calibration prior that considers the divergence of African parasites found in Mandrillus spp. and Cercocebus spp. (P. gonderi) from those Plasmodium species found in Southeast Asian macaques (Mu et al. 2005; Pacheco et al. 2011). The second scenario (supplementary table S10, Supplementary Material online) also added a calibration at the origin of the monophyletic group that includes P. malariae (a parasite found in humans). In particular, a minimum of 23.5 MYA, which corresponds to the human/Macaca split (Benton and Donoghue 2007), with a maximum of 65 MYA, this calibration allows this clade to be as old as the origin of primates (Perelman et al. 2011; Ksepka et al. 2015; Álvarez-Carretero et al. 2022). Both scenarios yield consistent time estimates whether the independent (R2 = 1.0) or autocorrelated models were used (R2 = 1.0) (supplementary figs. S11 and S12, Supplementary Material online). Furthermore, RelTime and MCMCtree yield consistent time estimates (R2 = 0.99 when using both scenarios with the 2 Bayesian models). Thus, the scenario using 2 calibrations on Bayesian models is discussed in detail (Table 1, supplementary fig. S12, Supplementary Material online).
Table 1.
Molecular time estimates for major events during the evolution of Plasmodium in primates contrasting autocorrelated and independent (uncorrelated) rate models
| Scenarios | ||||||||
|---|---|---|---|---|---|---|---|---|
| Correlated | Independent | Correlated | Independent | |||||
| Calibrations (Ma) | Scenario I: 1: 6–14.2 2: 24.44–34.0 |
Scenario II 1: 6–14.2 2: 24.44–34.0 3: 6.5-15.2 |
||||||
| Divergence | Node age | 95% CrI | Node age | 95% CrI | Node age | 95% CrI | Node age | 95% CrI |
| Origin of Plasmodium spp./Plasmodium vivax (human and Asian NHP) | 3.44 | 3.30 to 1.63 | 5.77 | 7.92 to 3.83 | 4.11 | 5.77 to 2.55 | 6.0 | 7.38 to 4.56 |
| Divergence between P. vivax and P. vivax-like | 0.29 | 0.50 to 0.11 | 0.76 | 1.33 to 0.29 | 0.34 | 0.56 to 0.17 | 0.80 | 1.35 to 0.33 |
| Origin of Plasmodium gonderi (African NHP) | 11.14 | 14.42 to 7.37 | 10.88 | 14.28 to 7.38 | 13.31 | 14.82 to 11.09 | 13.29 | 14.81 to 11.13 |
| Origin of Plasmodium ovale (human) | 23.95 | 29.39 to 19.06 | 23.75 | 29.80 to 18.29 | 24.63 | 29.29 to 20.60 | 25.19 | 30.28 to 20.20 |
| Plasmodium ovale subspecies divergence | 2.88 | 5.83 to 0.62 | 2.37 | 4.26 to 0.87 | 2.82 | 5.42 to 0.76 | 2.52 | 4.33 to 1.04 |
| Origin of Plasmodium malariae (human and NHP) | 28.71 | 33.69 to 24.30 | 29.07 | 33.79 to 24.38 | 28.95 | 33.79 to 24.42 | 30.39 | 34.28 to 25.51 |
| Plasmodium malariae—malariae -like divergence | 1.00 | 2.42 to 0.11 | 0.38 | 0.69 to 0.13 | 0.92 | 2.17 to 0.12 | 0.41 | 0.71 to 0.16 |
| Origin of Hepatocystis spp. (NHP) | 28.08 | 34.34 to 22.61 | 28.44 | 35.89 to 20.95 | 28.02 | 34.21 to 22.57 | 29.75 | 37.18 to 21.95 |
| Origin of Plasmodium spp. (rodents) | 6.46 | 11.06 to 2.24 | 6.07 | 9.21 to 3.44 | 5.23 | 8.30 to 2.13 | 5.81 | 7.87 to 3.70 |
| Origin of Laverania genus | 9.75 | 15.02 to 4.63 | 5.27 | 7.93 to 3.0 | 6.98 | 8.84 to 5.56 | 6.68 | 7.96 to 5.55 |
| Origin of Plasmodium genus + Hepatocystis spp. (primates and rodents) | 40.75 | 50.19 to 31.83 | 40.4 | 49.72 to 31.79 | 42.24 | 55.75 to 30.52 | 43.03 | 52.57 to 33.28 |
The node ages and their credible intervals are provided for the 2 and 3 calibration scenarios.
Time estimates derived from nuclear genes did not deviate from those based on organelle genomes for several nodes, even when there are differences in GC content among clades in the nuclear genes. The estimates for the origin of Plasmodium found in primates and rodents were similar between the autocorrelated and independent models, 40.75 MYA (CrI = 50.20 to 31.83) and 40.40 MYA (CrI = 49.72 to 31.79), respectively. These time estimates were consistent with studies using the parasite mtDNA (Pacheco et al. 2018; Pacheco et al. 2020) and combining the mtDNA and loci from the apicoplast (≅6 kb) (Pacheco et al. 2022), a plastid-like organelle found in Haemosporida, which has a circular genome with similar AT content to the mtDNA genome (Wilson et al. 1996).
The split of the lineage leading to P. ovale s.l. species is dated at 23.95 MYA (CrI = 29.39 to 19.06) for autocorrelated and 23.75 MYA (CrI = 29.80 to 18.29) for uncorrelated (Table 1, supplementary fig. S12, Supplementary Material online) rate models. These estimates are consistent with target gene approaches on organelles using different assumptions (Hayakawa et al. 2008; Sutherland et al. 2010; Pacheco et al. 2018, Pacheco et al. 2022) and suggest that the lineage leading to P. ovale s.l. diverged from the other Plasmodium included in this analysis early during the origin of Catharrini primates (Benton and Donoghue 2007). The mean posterior divergence time between the 2 P. ovale cryptic species was estimated to be between 2.52 and 2.88 MYA, depending on the substitution rate model and calibration scenario (Table 1, supplementary fig. S12a, Supplementary Material online). These coincide with estimates based on mitochondrial loci using Laverania species as calibration (Sutherland et al. 2010) and are consistent with the proposed timeframes for the origin of the genus Homo (Püschel et al. 2021). These results contrast with a genomic study that estimated the split of the P. ovale s.l. species at 20.3 MYA (Rutledge et al. 2017). Although the methods and assumptions could partially explain the differences, the estimate seems unrealistic, considering the 2 cryptic species’ genomic divergence and biological similarities.
The mean posterior divergence time for the split of P. vivax and P. vivax-like is between 0.29 MYA (CrI = 0.50 to 0.11) in the autocorrelated model and 0.76 MYA (CrI = 1.33 to 0.29) in the independent model (Table 1). These estimates place this divergence at a time when environmental changes connected Asia and Africa, allowing hominids to migrate across (Roberts and Stewart 2018; Louys and Roberts 2020; Groucutt et al. 2021; Lee and Hudock 2021). Thus, the inferred divergence time between P. vivax and P. vivax-like, together with the placement of their common ancestor within the radiation of the Plasmodium species found in Southeast Asia, is consistent with a scenario where P. vivax originated in Asia as a human parasite as suggested by population genetics and genomic analyses on the extant populations of P. vivax (Cornejo and Escalante 2006; Benavente et al. 2021; Daron et al. 2021). Although an African origin (Loy et al. 2018) seems less parsimonious in light of the population genetics and phylogenetic data, it does not make it impossible. The P. vivax-like genomic data are still scarce, and data from P. vivax infections in Africa are still being studied by the scientific community. However, data indicating a more ancient P. vivax population in Africa is still not available so the African scenario seems less parsimonious just when considering the available data.
An Asian origin hyphotesis for P. vivax is usually criticized because it cannot explain the high prevalence of Duffy-negative in African human populations. Duffy-negative individuals are protected against the blood stage of P. vivax, and it is considered to have been selected by the parasite (revised in McManus et al. 2017) and explained the apparent absence of P. vivax in sub-Saharan Africa. However, unlike polymorphism protecting against P. falciparum malaria (Malaria Genomic Epidemiology Network 2019), the expected patterns consistent with the strong selection of these Duffy-negative alleles remain elusive, and the origins of all Duffy-negative haplotypes seem consistent with the timeframes estimated in this study (McManus et al. 2017). The situation is further complicated because P. vivax can be maintained in Duffy-negative African populations (Ménard et al. 2010; Gunalan et al. 2018). Although Duffy-negative polymorphisms in Africa are driven by evolutionary processes that likely correlate with what we observe in P. vivax today, establishing causation is not obvious (Gould and Lewontin 1979; Hughes and Verra 2010) so it cannot be used as evidence supporting a particular scenario for the origin of P. vivax as a human parasite.
The estimates for the origin of Laverania, the clade of P. falciparum and related species, differed between the autocorrelated (9.75 MYA, CrI = 15.02 to 4.63) and independent (5.27 MYA, CrI = 7.93 to 3.00) models. However, their CrI overlapped (Table 1, supplementary fig. S12, Supplementary Material online). These estimates coincided with the origins of their hosts (e.g. median 8.6 MYA, CrI = 8.3 to 9.3 MYA for the common ancestor of Gorilla and Homo, based on the TimeTree database accessed on August 2023, Kumar et al. 2022). Similar time estimates were also obtained when combining organelle loci from mtDNA and apicoplast, or studies on nuclear loci (Hughes and Verra 2010; Pacheco et al. 2022). These results differ from studies proposing that the Laverania clade originated less than 1 MYA ago (Otto et al. 2018) when Homo, Gorilla, and Pan coexisted (Bobe and Reynolds 2022; Urciuoli and Alba 2023). It implies that the local ecology drove the clade speciation (Makanga et al. 2016; Liu et al. 2017). However, Otto et al. (2018) used different assumptions in a coalescent-based approach suitable for intra-species variation rather than species divergence. Under such a model, all Laverania seems to be treated as a single species. In contrast, this investigation using models to study species divergence supports a scenario where the African apes’ evolutionary histories may have played a role in the radiation of the P. falciparum clade. This study is consistent with the time proposed for the origin of the extant Laverania species but without formal molecular clock analyses being presented (Sharp et al. 2020). It is worth emphasizing that no calibration within Laverania was used to obtain the estimates. Such a calibration (e.g. P. falciparum and P. reichenowi divergence) is controversial (Sharp et al. 2020; Pacheco et al. 2022).
Given the differences in time estimates between the Bayesian models, the method implemented in CorrTest (Tao et al. 2019), based on machine learning, was used to determine whether there was autocorrelation in substitution rates. CorrTest indicated that evolutionary rates are likely autocorrelated (CorrScore = 0.99845, P < 0.001). Considering that all the calibrations are in the “GC-rich” genomes, the effect of lacking calibrations within the Laverania subgenus, or P. falciparum clade, may have explained, at least in part, the difference between the 2 models. Thus, a third calibration was included at the origin of Laverania (Table 1 and Fig. 6) to explore its effect on the models. In particular, a minimum based on the fossil calibration between Chimps-Homo (6.5 MYA) and a maximum (15.2 MYA) using estimates for Homo-Pongo divergence (TimeTree database, Kumar et al. 2022). This calibration does not favor any particular event (uniform distribution). Such an interval includes recent estimates for the split between Homo-Gorilla and their crown groups (11.49 to 10 MYA; Püschel et al. 2021). It also allows the Laverania clade species to be as old as the origin of Hominidae, an unlikely scenario considering the extant Plasmodium species found in orangutans (Peters 1976; Pacheco et al. 2012). Adding this calibration yields an estimate of 6.98 MYA (CrI = 8.84 to 5.56) with the autocorrelated and 6.68 (CrI = 7.96 to 5.55) with the independent rate model. Thus, the uncertainty across the time tree decreases when a calibration constraint is added (even a broad one) in a clade that may be diverging from the others, Laverania, in this case, the one with lower GC content.
Fig. 6.

Time tree of the divergence of primate malarias using 110 genes. Divergence times were estimated using MCMCTree under the a) autocorrelated and b) independent rate models, using 3 calibration constraints that included a calibration for the origin of the Laverania clade. Calibrations priors were uniform, as explained in the text. Times are shown in MYA. 95% CrIs for the major clades are shown in parentheses next to the nodes.
The medians of time estimates seem to favor the origin of Laverania during the Homo-Pan divergence followed by host switches (fossils: 6.5 to 10 MYA sensu Benton and Donoghue 2007, http://fossilcalibrations.org last accessed on June 2022 and molecular estimates, median time = 6.4 MYA, CrI = 6.1 to 6.7 MYA; TimeTree accessed September 2022, Kumar et al. 2022). Such a scenario was proposed by others (Krief et al. 2010). The credible intervals, however, include the Homo-Gorilla divergence, particularly in the autocorrelated model that better fits the data. Also, estimates consistent with the Homo-Gorilla divergence emerged without calibration on Laverania under the autocorrelated model, which is a more conservative approach. Thus, the time frame presented here is consistent with a scenario where the extant host speciation played a role in the divergence of the Laverania parasites, masked partly by host switches during the evolutionary history of African apes.
Overall Conclusion
The P. gonderi v.2 genome assembly presented here is a valuable resource for Plasmodium comparative genomics. Plasmodium species in primates and rodents have conserved syntenic blocks enriched by putatively essential genes. Notably, the genes outside such syntenic blocks seem critical to exploit their vertebrate hosts, as those with functional information are associated with virulence or host cell invasion. The observation is consistent in the diversity of strategies observed to circumvent the barriers involved in the infection of the vertebrate host.
Although contingent on the assumptions of this analysis, the emerging picture from this investigation is that the primate parasite host ranges and speciation, including the lineages leading to extant human parasites, may have been driven by their vertebrate hosts’ biogeographical and speciation processes. In particular, whereas the distribution and speciation of African apes may have driven the evolution of P. falciparum and related species, the origin of P. vivax is associated with the dynamics of primates in Southeast Asia and the connectivity of Homo populations between Asia and Africa. Several questions remain unanswered due to the absence of genome data from critical species found in macaques, gibbons, and lemurs. Nevertheless, the genome of P. gonderi has enriched our understanding of the evolution of Plasmodium in primates, particularly those infecting humans as their primary vertebrate host.
Materials and Methods
Sample and Ethical Considerations
Plasmodium gonderi was maintained in the Centers for Disease Control and Prevention by W.E. Collins. Strain Garnham is the only strain available (Collins and Contacos 1980). It can be found in the BEI Resources, NIAID, NIH, under the label: P. gonderi, Strain Garnham, MRA-447, and W. E. Collins contributed it. The DNA was extracted from a Centers for Disease Control and Prevention sample using QIAamp DNA Blood Mini Kit (QIAGEN, Germany), following protocols used in previous studies (Tachibana et al. 2012).
Sequencing, Raw Data, Preprocessing, Assembly, and Annotation of Plasmodium gonderi Genome
The raw sequence data from the P. gonderi Sequence Read Archive (SRA) accession number SAMD00076127 (Honma et al. 2017) was combined with 3 paired-end next-generation sequencing libraries from the same strain; 2 with 100 bp (Illumina 2000) and 1 with 150 bp (Illumina 2500) read length (BioProject GenBank accession no. PRJNA928704). Low-quality read ends, and adapter sequences were removed using Trimmomatic software (Bolger et al. 2014). Trimmed reads were mapped to the Macaca fuscata genome (accession number GCA_003118495.1) using Bowtie2 with the default parameters (Langmead and Salzberg 2012) to eliminate reads that were likely from the host genome. Only unmapped reads against the host genome were used to run the assembly; ∼183 million reads were added from the 3 Illumina libraries produced in this study.
The genomic assembly (Genome Whole Sequence GenBank accession no. JARBEB000000000.1) was carried out by a de novo hybrid assembly strategy implemented in MaSuRCA software (Zimin et al. 2013). MaSuRCA was used with the following parameters: graph_kmer_size = 77, jf_size = 50000000, soap_assembly = 0, cgwerrorrate = 0.15, close_gaps = 1. This approach leads to fully assembled chromosomes (supplementary fig. S1, Supplementary Material online; Graphical representation was made using Circos software (Krzywinski et al. 2009)) and fully reproduced the synteny with P. cynomolgi without using any reference. The quality of the P. gonderi version 2 (P. gonderi v.2) genome assembly was evaluated and compared with 26 other haemosporidian genomes (including the draft assembly of P. gonderi, Honma et al. 2017) with Quast software (Gurevich et al. 2013; supplementary table S1, Supplementary Material online).
Structural annotation of genes was made using the Augustus gene prediction software (Stanke et al. 2006). Four genome annotations were used and compared to train Augustus: P. cynomolgi M (Pasini et al. 2017), P. malariae UG01 (Rutledge et al. 2017), P. vivax P01 (Auburn et al. 2016), and P. knowlesi H (Pain et al. 2008). However, training with P. cynomolgi showed the best results due to the number and length of genes. Manual corrections were made using Artemis software (Carver et al. 2005) comparing with closely related Plasmodium orthologous gene sequences. Functional and GO annotations were performed using the stand-alone software PANNZER2 (Protein ANNotation with Z-scoRE; Törönen et al. 2018; supplementary table S2, Supplementary Material online).
Annotation of Pir Genes in Subtelomeric Regions
Plasmodium-interspersed repeat (pir) genes mapped on the chromosomes of the 2 P. gonderi genome assemblies were made employing the R package karyoploteR (supplementary fig. S2, Supplementary Material online; Gel and Serra 2017). Whereas there are multiple gene families, only pir gene family was included since it is informative about the genome quality. The following 6 genomes were used as they are part of the clase that includes P. gonderi: P. coatneyi Hackeri (Chien et al. 2016), P. cynomolgi M (Pasini et al. 2017), P. gonderi v2 (this study), P. knowlesi H (Pain et al. 2008), P. vivax P01 (Auburn et al. 2016), and P. vivax-like Pvl01 (Gilabert et al. 2018) (supplementary table S11, Supplementary Material online). The Plasmodium interspaced repeat (pir) protein sequences were extracted from the proteomes. Then, a reciprocal BLASTp comparison was run with an e-value equal to 1^e-6, considered a good hit for homology matches (Madden 2008). Results were visualized in Gephi, clustered with the force field and the Reingold–Watermann algorithm (Fig. 1). Graphical representation of the distribution of pir gene family and blast reciprocal genes between P. gonderi v2 and P. cynomolgi was performed with Circos software (Krzywinski et al. 2009).
Orthologous Groups, GC Content, and Codon Usage
The following 26 reference genomes were used for these analyses: P. adleri G01 (Otto et al. 2018), P. berghei ANKA (Fougère et al. 2016), P. billcolinsi G01 (Otto et al. 2018), P. blacklocki G01 (Otto et al. 2018), P. chabaudi chabaudi (Otto et al. 2014), P. coatneyi Hackeri (Chien et al. 2016), P. cynomolgi M (Pasini et al. 2017), P. falciparum 3D7 (Böhme et al. 2018), P. gaboni SY75 (Otto et al. 2018), P. gonderi v2 (this study), P. knowlesi H (Pain et al. 2008), P. malariae UG01 (Rutledge et al. 2017), P. ovale curtisi G01 (Rutledge et al. 2017), P. praefalciparum G01 (Otto et al. 2018), P. reichenowi Centers for Disease Control and Prevention (CDC) (Otto et al. 2014), P. relictum SGS1-like (Böhme et al. 2018), P. vivax P01 (Auburn et al. 2016), P. yoelii yoelii 17 × (Otto et al. 2014), Hepatocistys sp. (Aunin et al. 2020), P. fragile nilgiri (GCA_000956335.1), P. gallinaceum 8A (Böhme et al. 2018), P. inui San Antonio 1 (GCA_000524495.1), P. malariae-like GA01 (Rutledge et al. 2017), P. ovale walkeri CR01 (Rutledge et al. 2017), and Plasmodium vinckei vinckei (Ramaprasad et al. 2021) (supplementary table S11, Supplementary Material online).
Orthofinder2 software (Emms and Kelly 2019) was used with default parameters to identify orthogroups from 26 haemosporidian genomes, leading to 8,465 orthogroups. Of those, 1,961 orthogroups were SC-GO found in all the species, including those with limited genomic data (supplementary table S3, Supplementary Material online).
Each SC-OG was aligned at the protein level using Muscle v.3.8 (default parameters). Once aligned, they were reverse-translated with their respective nucleotide sequence as a template to obtain nucleotide alignments. The length of each SC-GO nucleotide alignment, excluding gaps, is reported in supplementary table S3, Supplementary Material online. In addition, all SC-GO alignments were concatenated to obtain an alignment of 2,498,205 bp (excluding gaps). Using SC-OGs allows comparing the species independently of their assembly quality (some are highly fragmented and have multiple scaffolds, such as the nonhuman primate parasites Plasmodium fragile and Plasmodium inui). However, all analyses were also carried out with all CDSs.
All CDSs from the 26 haemosporidian reference genomes were analyzed using the RSCU (Fig. 2, ssupplementary fig. S4 and table S4, Supplementary Material online), and the frequency of each amino acid (supplementary fig. S5 and table S5, Supplementary Material online). Both analyses were carried out using the Dambe6 software (Xia 2017). RSCU displays the difference between synonymous codon use and their actual usage. The ratio between the observed frequency of synonymous codons and the predicted frequency of that codon, when all codons are utilized without bias at that specific amino acid, was measured by RSCU. RSCU > 1 shows a preference for the codon, RSCU < 1 indicates that the codon is used less frequently than predicted, and RSCU = 1 indicates no bias in the codon usage (Goldman and Yang 1994).
Analyzing Synteny Among Plasmodium Species
Due to their assembly quality, only 18 of the 26 genomes mentioned in the previous section were used for synteny-based analyses. However, to obtain more reliable results, problems associated with the assembly of P. coatneyi Hackeri were corrected (Chien et al. 2016). For the above, more than 300 million reads from 3 new Illumina libraries of the same strain were included in the data of Chien et al. (2016) (SRA accession number SAMN02595587 and SAMN04571750). The assembly followed the same methodology as P. gonderi v.2 (see above). The de novo structural annotation for this assembled genome did not differ from the available version, so RATT software (Otto et al. 2011) was used to transfer and update the annotation and gene order in the P. coatneyi genome. Orthofinder (see above) also detected the SC-OGs considered the “syntenic core genome” across the 18 genomes. The graphical representation of synteny was made using the R package GeneSpace (Lovell et al. 2022).
For synteny-based phylogenomic reconstruction, the pipeline of Zhao et al. (2021) (https://github.com/zhaotao1987/SynNet-Pipeline; Haug-Baltzell et al. 2017) was used, which includes 4 main steps: (i) Phylogenomic synteny network construction by reciprocal sequence similarity search of all 20 annotated parasitic proteomes using DIAMOND (version 0.9.14.115; default parameters; Buchfink et al. 2015), and pairwise synteny block detection by MCScanX36 (Wang et al. 2012) under these setting parameters b5s7m25 (b: number of top homologous pairs, s: number of minimum matched syntenic anchors, m: number of max gene gaps); this combination out of 27 (b: 5, 10, 15; s: 3, 5, 7; and m: 15, 25, 35) obtained the best result in terms of nodes and edges numbers (data not shown). (ii) Network clustering by R script available in Zhao's repository. (iii) Matrix representation, and (iv) maximum likelihood-based binary-tree inference in IQ-Tree (100 bootstrap replicates; Minh et al. 2020). Thus, the complete synteny network summarizes 729,572 pairwise synteny blocks and contains 92,498 nodes and 727,219 edges. Nodes are genes in synthetic blocks in the resulting synteny network, while edges connect synthetic anchor pairs (Fig. 3 and supplementary fig. S6, Supplementary Material online). As presented in the results (supplementary fig. S7, Supplementary Material online), the P. vivax-like Pv101 genome (Gilabert et al. 2018) was not used in the primary analyses due to the lack of several syntenic regions, but it was presented in Supplementary Materials.
Reconstruction of ancestral genomes across the Plasmodium genus was made using AGORA (Algorithm for Ancestral Gene Order Reconstruction; Muffato et al. 2023)). AGORA estimates all relevant pairwise gene order comparisons (avoiding gene gains, losses, and duplications). It assigns a weight to each possible gene-gene adjacency, corresponding to the number of pairwise comparisons that support this adjacency as ancestral at the node of interest. These weights are then used to solve a gene graph and extract the most likely gene order at the ancestor of interest. The ortholog groups were inferred for each internal node of the species tree using Orthofinder2 (see above).
Genes defined as essential genes characterized by Zhou et al. (2016) and “syntenic core genome” mapped to AGORA-defined syntenic blocks were used for GO enrichment analysis (Supek et al. 2011). These GO terms analyses were performed in PlasmoDB (The Plasmodium Genome Database Collaborative 2001) using P. falciparum 3D7 as a reference, with a P-value > 0.05 with Bonferroni correction (supplementary table S6 and table S7; supplementary supplementary table S7, figs. S8 and S9, Supplementary Material online).
Phylogenomic Analyses
Phylogenetic relationships were inferred on each SC-GO nucleotide alignment using the ML method implemented in IQ-Tree (Minh et al. 2020) under the best-fit substitution model (supplementary table S3, Supplementary Material online), and 200 bootstrap replicates. Other parameters were set by default.
ASTRAL-III (Zhang et al. 2018a, 2018b) was employed to estimate a species tree under the multispecies coalescent model. It allows estimating an unrooted species tree given a set of unrooted gene trees; in this case, those gene trees were estimated with IQ-Tree (supplementary table S3, Supplementary Material online) from the SC-GOs. Normalized Robinson–Foulds distances between pairs of trees were calculated using the RF.dist function of the phangorn package in R with the parameter normalize = TRUE to characterize the degree of (mis)agreement between tree topologies (Schliep 2011).
Phylogenomic reconstructions were also inferred on a concatenated alignment with all the SC-GOs. This analysis was carried out using the Bayesian methods implemented in MrBayes v3.2.6 with the default priors (Ronquist and Huelsenbeck 2003) and the ML method in IQ-Tree (Minh et al. 2020). A general time-reversible model with gamma-distributed substitution rates and a proportion of invariant sites (GTR + Γ+I + G4) was used for both analyses, as it was determined using IQ-TREE, which searches for all nucleotide substitution models implemented in ModelFinder. The optimal model is selected based on its Bayesian Information Criterion (BIC) value.
Bayesian support for all nodes was inferred by sampling every 1,000 generations from 2 independent chains lasting 107 Markov Chain Monte Carlo (MCMC) steps. The chains were assumed to have converged once the average standard deviation (SD) of the posterior probability was <0.01 and the value of the potential scale reduction factor (PSRF) was between 1.00 and 1.02 (Ronquist and Huelsenbeck 2003). As a “burn-in,” 50% of the sample was discarded once convergence was reached. In the case of ML analyses, 1,000 bootstrap replicates were evaluated for statistical confidence.
Divergence Dating Analyses
Molecular clock analyses were performed using an alignment with 110 genes. These 110 genes were chosen based on the nRF distances between their topologies and the species tree. The threshold was 8 (or 4 changes between the gene tree and the species tree topologies; supplementary table S9, Supplementary Material online). Bayesian methods implemented in MCMCTree (Yang and Rannala 2006; Rannala and Yang 2007; Yang 2007) were used to estimate time trees and nucleotide substitution rates/My per partition. Two independent chains lasting 108 MCMC steps were run for each analysis until convergence. Two scenarios were compared using a combination of calibration constraints. The calibration constraints include information on vertebrate hosts’ fossils (see Paleobiology Database at http://www.paleodb.org/). Although those are secondary calibrations, they are the only ones available, given the lack of Plasmodium fossils (Pacheco and Escalante 2023). In all cases, uniform priors were used to calibrate divergences. No particular time point was favored within a given time interval. No punctual calibrations were used.
The first calibration constraint considered that P. gonderi found in Mandrillus spp. and Cercocebus spp. diverged from those Plasmodium species found in Southeast Asia macaques when Macaca branched from Papio (Mu et al. 2005; Pacheco et al. 2011). Macaca spp. fossils indicate that such events took place 6 to 8 MYA as minimum and maximum boundaries, respectively (Delson 1980). However, these fossils are used as minimum time boundaries when studying primates (Perelman et al. 2011). Thus, a more inclusive alternative calibration constraint was proposed around this event, assuming that the divergence at the same node occurred between 6 and 14.2 MYA. Thus, this interval was used as a uniform prior (no particular time point was favored). This inclusive time interval incorporates molecular estimates for the same Papio-Macaca divergence event (Pacheco et al. 2011), and is also consistent with older fossils reported for Macaca spp. (see Paleobiology Database at http://www.paleodb.org/), and considers that P. gonderi has also been reported in Chlorocebus thuscopithecini.
A second fossil-based calibration constraint is the minimum of 23.5 MYA for the human/Macaca split (Benton and Donoghue 2007), which is assumed to be the minimum time when the monophyletic group, including P. malariae (a parasite found in humans) and all the Asian parasites, originated. This event is used to inform a calibration prior with a maximum of 65 MYA that allows this parasite clade to be as old as the origin of primates if the data support that (Perelman et al. 2011, Ksepka et al. 2015).
These calibrations were organized into 2 scenarios. Scenario 1 considers only the relaxed 6 to 14.2 MYA calibration of the Papio/Macaca parasite divergence (95% quantile = 13.8) (Delson 1980; Mu et al. 2005). The second scenario included the previous calibration and the one with a minimum of 23.5 and a maximum of 34.0 MYA for the human/Macaca split (95% quantile = 33.5). A third scenario was added to explore the effect of calibration in the clade with higher AT content, Laverania. In particular, the origin of the Laverania subgenus, a monophyletic group of parasites found in African Apes, was assumed to be the divergence of all the extant hosts of these parasites (Homininae that includes Homo-Pan-Gorilla) with a maximum in the Hominidae (Great Apes including Pongo). The calibration was uniform, with a minimum of 6.5 MYA and a maximum of 15.2 MYA.
The MCMC algorithm was used to estimate divergence times on the constrained tree topology in 2 separate runs using the approximate likelihood calculation (dos Reis and Yang 2011). We used baseml under the strict clock model to drive a prior for the overall substitution rate G(1.2,1.0). Given the lack of prior information, both independent and autocorrelated rate models (Rannala and Yang 2007) were employed with the following default parameters: a rate drift parameter σ2 G(1,1); and the parameters of the birth-death process with species sampling fixed at λ = μ = 1 and ρ = 0. The chains were run until effective sample size (ESS) values were higher than 200 after discarding the burn-in, sampling every 1000th cycle. As MCMCTree requires some prior information at the root age, a loose maximum of 86.8 MYA was provided to limit the age of the most recent common ancestor of Plasmodium. All the calibrations were uniform priors with soft bounds.
Besides the Bayesian inferences, divergence times were also estimated in a non-Bayesian framework with the RelTime method (Tamura et al. 2012). RelTime algorithm does not assume any statistical model for the variation in rates of evolution across lineages as the Bayesian methods do (Tamura et al. 2012). Although it estimates relative times, it is not free from assumptions. RelTime computes the rate of the ancestral branch as the average of those in the 2 immediate descendant branches (Tamura et al. 2018). In this case, calculations were carried out on the command-line version of MEGA11 (Tamura et al. 2021). The substitution model was the same as for Bayesian analyses (GTR+Γ+I). Calibration information was provided as uniform distributions in RelTime, using the same minimum and maximum boundaries used in MCMCTree. As RelTime does not allow calibrations to be placed at the root of the phylogenetic tree (Tamura et al. 2021), we used a maximum of 86.8 MYA at the root of the ingroup clade. The same calibration scenarios employed in MCMCTree were used to infer divergence times in RelTime.
Supplementary Material
Acknowledgments
We thank John Barnwell for his support while studying nonhuman primate malarias. The authors express their gratitude to Dr. William Collins (“Bill”), with whom some of us were fortunate to chat about “monkey malarias” while in Chamblee, GA.
Contributor Information
Axl S Cepeda, Biology Department/Institute of Genomics and Evolutionary Medicine (iGEM), Temple University, Philadelphia, PA 19122-1801, USA.
Beatriz Mello, Departamento de Genética, Universidade Federal do Rio de Janeiro, Rio de Janeiro, Brazil.
M Andreína Pacheco, Biology Department/Institute of Genomics and Evolutionary Medicine (iGEM), Temple University, Philadelphia, PA 19122-1801, USA.
Zunping Luo, Center for Genomics & Systems Biology, Department of Biology, New York University, New York, NY 10003, USA.
Steven A Sullivan, Center for Genomics & Systems Biology, Department of Biology, New York University, New York, NY 10003, USA.
Jane M Carlton, Center for Genomics & Systems Biology, Department of Biology, New York University, New York, NY 10003, USA.
Ananias A Escalante, Biology Department/Institute of Genomics and Evolutionary Medicine (iGEM), Temple University, Philadelphia, PA 19122-1801, USA.
Supplementary Material
Supplementary material is available at Genome Biology and Evolution online.
Authors Contributions
A.A.E., J.M.C., and M.A.P. conceived the study. J.M.C., S.A.S., and Z.L. generated the genomic data. A.S.C. curated and generated the assembly. A.S.C., M.A.P., and B.M. performed the downstream analyses of the data. A.A.E., A.S.C., B.M., and M.A.P. wrote the first draft of the manuscript. All authors edited and participated in the final version.
Funding
A.A.E., A.S.C., and M.A.P. were supported by grants from the National Institute of Allergy and Infectious Diseases (U19AI089681) and the National Science Foundation (NSF) (DEB 2146653). J.M.C., Z.L., and S.A.S. were supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under Award Number U19AI089676. B.M. is supported by the Fundação Carlos Chagas Filho de Amparo à Pesquisa do Estado do Rio de Janeiro, grants E-26/211.248/2019 and E-26/201.446/2022. The content is solely the authors’ responsibility and does not necessarily represent the official views of the agencies providing funding.
Data Availability
Scripts used to perform the analyses reported in this article are available at https://github.com/EscalanteLab/Plasmodium-gonderi-genome-analysis. The data used in this work is available under the BioProject GenBank accession no. PRJNA928704 and the Genome Whole Sequence GenBank accession no. JARBEB000000000.1.
Literature Cited
- Álvarez-Carretero S, Tamuri AU, Battini M, Nascimento FF, Carlisle E, Asher RJ, Yang Z, Donoghue PCJ, Dos Reis M. A species-level timeline of mammal evolution integrating phylogenomic data. Nature. 2022:602(7896):263–267. 10.1038/s41586-021-04341-1. [DOI] [PubMed] [Google Scholar]
- Arisue N, Hashimoto T, Kawai S, Honma H, Kume K, Horii T. Apicoplast phylogeny reveals the position of Plasmodium vivax basal to the Asian primate malaria parasite clade. Sci Rep. 2019:9(1):7274. 10.1038/s41598-019-43831-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arisue N, Honma H, Kume K, Hashimoto T. Progress in understanding the phylogeny of the Plasmodium vivax lineage. Parasitol Int. 2022:87:102507. 10.1016/j.parint.2021.102507. [DOI] [PubMed] [Google Scholar]
- Auburn S, Böhme U, Steinbiss S, Trimarsanto H, Hostetler J, Sanders M, Gao Q, Nosten F, Newbold CI, Berriman M, et al. A new Plasmodium vivax reference sequence with improved assembly of the subtelomeres reveals an abundance of pir genes. Wellcome Open Res. 2016:1:4. 10.12688/wellcomeopenres.9876.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aunin E, Böhme U, Sanderson T, Simons ND, Goldberg TL, Ting N, Chapman CA, Newbold CI, Berriman M, Reid AJ. Genomic and transcriptomic evidence for descent from Plasmodium and loss of blood schizogony in Hepatocystis parasites from naturally infected red colobus monkeys. PLoS Pathog. 2020:16(8):e1008717. 10.1371/journal.ppat.1008717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajic M, Ravishankar S, Sheth M, Rowe LA, Pacheco MA, Patel DS, Batra D, Loparev V, Olsen C, Escalante AA, et al. The first complete genome of the simian malaria parasite Plasmodium brasilianum. Sci Rep. 2022:12:19802. 10.1038/s41598-022-20706-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battistuzzi FU, Tao Q, Jones L, Tamura K, Kumar S. RelTime relaxes the strict molecular clock throughout the phylogeny. Genome Biol Evol. 2018:10(6):1631–1636. 10.1093/gbe/evy118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benavente ED, Manko E, Phelan J, Campos M, Nolder D, Fernandez D, Velez-Tobon G, Castaño AT, Dombrowski JG, Marinho CRF, et al. Distinctive genetic structure and selection patterns in Plasmodium vivax from South Asia and East Africa. Nat Commun. 2021:12(1):3160. 10.1038/s41467-021-23422-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benton MJ, Donoghue PC. Paleontological evidence to date the tree of life. Mol Biol Evol. 2007:24(1):26–53. 10.1093/molbev/msl150. [DOI] [PubMed] [Google Scholar]
- Bobe R, Reynolds S. Hominid paleoenvironments in tropical Africa from the late miocene to the early pleistocene. In: Reynolds S, Bobe R, editors. African Paleoecology and human evolution. Cambridge: Cambridge University Press; 2022. p. 161–186. [Google Scholar]
- Böhme U, Otto TD, Cotton JA, Steinbiss S, Sanders M, Oyola SO, Nicot A, Gandon S, Patra KP, Herd C, et al. Complete avian malaria parasite genomes reveal features associated with lineage-specific evolution in birds and mammals. Genome Res. 2018:28(4):547–560. 10.1101/gr.218123.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics. 2014:30(15):2114–2120. 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchfink B, Xie C, Huson DH. Fast and sensitive protein alignment using DIAMOND. Nat Methods. 2015:12(1):59–60. 10.1038/nmeth.3176. [DOI] [PubMed] [Google Scholar]
- Carver TJ, Rutherford KM, Berriman M, Rajandream MA, Barrell BG, Parkhill J. ACT: the artemis comparison tool. Bioinformatics. 2005:21(16):3422–3423. 10.1093/bioinformatics/bti553. [DOI] [PubMed] [Google Scholar]
- Cepeda AS, Andreína Pacheco M, Escalante AA, Alzate JF, Matta NE. The apicoplast of Haemoproteus columbae: a comparative study of this organelle genome in haemosporida. Mol Phylogenet Evol. 2021:161:107185. 10.1016/j.ympev.2021.107185. [DOI] [PubMed] [Google Scholar]
- Charpentier MJE, Boundenga L, Beaulieu M, Dibakou SE, Arnathau C, Sidobre C, Willaume E, Mercier-Delarue S, Simon F, Rougeron V, et al. A longitudinal molecular study of the ecology of malaria infections in free-ranging mandrills. Int J Parasitol Parasites Wildl. 2019:10:241–251. 10.1016/j.ijppaw.2019.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien JT, Pakala SB, Geraldo JA, Lapp SA, Humphrey JC, Barnwell JW, Kissinger JC, Galinski MR. High-Quality genome assembly and annotation for plasmodium coatneyi, generated using single-molecule real-time PacBio technology. Genome Announc. 2016:4(5):e00883-16. 10.1128/genomeA.00883-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coatney GR, Collins WE, Warren M, Contacos PG. The primate malarias. Bethesda: US National Institute of Allergy and Infectious Diseases; 1971. [Google Scholar]
- Collins WE, Contacos PG. Infection and transmission studies with Plasmodium gonderi in the Macaca mulatta monkey. J Parasitol. 1980:66(6):998–1002. 10.2307/3280404. [DOI] [PubMed] [Google Scholar]
- Cornejo OE, Escalante AA. The origin and age of Plasmodium vivax. Trends Parasitol. 2006:22(12):558–563. 10.1016/j.pt.2006.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox-Singh J, Davis TME, Lee KS, Shamsul SS, Matusop A, Ratnam S, Rahman HA, Conway DJ, Singh B. Plasmodium knowlesi malaria in humans is widely distributed and potentially life threatening. Clin Infect Dis. 2008:46(2):165–171. 10.1086/524888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daron J, Boissière A, Boundenga L, Ngoubangoye B, Houze S, Arnathau C, Sidobre C, Trape JF, Durand P, Renaud F, et al. Population genomic evidence of Plasmodium vivax Southeast Asian origin. Sci Adv. 2021:7(18):eabc3713. 10.1126/sciadv.abc3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBarry JD, Kissinger JC. Jumbled genomes: missing apicomplexan synteny. Mol Biol Evol. 2011:28(10):2855–2871. 10.1093/molbev/msr103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delson E. Fossil macaques, phyletic relationships and a scenario of deployment. In: Lindburg DG, editor. The macaques—studies in ecology, behavior, and evolution. New York: Van Nostrand Reinhold Co; 1980. p. 10–29. [Google Scholar]
- dos Reis M, Donoghue PC, Yang Z. Bayesian molecular clock dating of species divergences in the genomics era. Nat Rev Genet. 2016:17(2):71–80. 10.1038/nrg.2015.8. [DOI] [PubMed] [Google Scholar]
- dos Reis M, Yang Z. Approximate likelihood calculation for Bayesian estimation of divergence times. Mol Biol Evol. 2011:28(7):2161–2172. 10.1093/molbev/msr045. [DOI] [PubMed] [Google Scholar]
- Duval L, Nerrienet E, Rousset D, Sadeuh Mba SA, Houze S, Fourment M, Le Bras J, Robert V, Ariey F. Chimpanzee malaria parasites related to Plasmodium ovale in Africa. PLoS One. 2009:4(5):e5520. 10.1371/journal.pone.0005520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emms DM, Kelly S. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 2019:20(1):238. 10.1186/s13059-019-1832-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escalante AA, Ayala FJ. Phylogeny of the malarial genus Plasmodium, derived from rRNA gene sequences. Proc Natl Acad Sci USA. 1994:91(24):11373–11377. 10.1073/pnas.91.24.11373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escalante AA, Barrio E, Ayala FJ. Evolutionary origin of human and primate malarias: evidence from the circumsporozoite protein gene. Mol Biol Evol. 1995:12(4):616–626. 10.1093/oxfordjournals.molbev.a040241. [DOI] [PubMed] [Google Scholar]
- Escalante AA, Cepeda AS, Pacheco MA. Why Plasmodium vivax and Plasmodium falciparum are so different? A tale of two clades and their species diversities. Malar J. 2022:21(1):139. 10.1186/s12936-022-04130-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escalante AA, Cornejo OE, Freeland DE, Poe AC, Durrego E, Collins WE, Lal AA. A monkey's tale: the origin of Plasmodium vivax as a human malaria parasite. Proc Natl Acad Sci USA. 2005:102(6):1980–1985. 10.1073/pnas.0409652102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escalante AA, Freeland DE, Collins WE, Lal AA. The evolution of primate malaria parasites based on the gene encoding cytochrome b from the linear mitochondrial genome. Proc Natl Acad Sci USA. 1998:95(14):8124–8129. 10.1073/pnas.95.14.8124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fougère A, Jackson AP, Paraskevi Bechtsi D, Braks JAM, Annoura T, Fonager J, Spaccapelo R, Ramesar J, Chevalley-Maurel S, Klop O, et al. Variant exported blood-stage proteins encoded by Plasmodium multigene families are expressed in liver stages where they are exported into the parasitophorous vacuole. PLoS Pathog. 2016:12(11):e1005917. 10.1371/journal.ppat.1005917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuehrer HP, Campino S, Sutherland CJ. The primate malaria parasites Plasmodium malariae, Plasmodium brasilianum and Plasmodium ovale spp.: genomic insights into distribution, dispersal and host transitions. Malar J. 2022:21(1):138. 10.1186/s12936-022-04151-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galen SC, Borner J, Martinsen ES, Schaer J, Austin CC, West CJ, Perkins SL. The polyphyly of Plasmodium: comprehensive phylogenetic analyses of the malaria parasites (order haemosporida) reveal widespread taxonomic conflict. R Soc Open Sci. 2018:5(5):171780. 10.1098/rsos.171780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galinski MR. Systems biology of malaria explored with nonhuman primates. Malar J. 2022:21(1):177. 10.1186/s12936-022-04199-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garnham PCC. Malaria parasites and other haemosporidia. Oxford: Blackwell Science Ltd; 1966. [Google Scholar]
- Garnham PCC, Lainson R, Cooper W. The complete life cycle of a new strain of Plasmodium gonderi from the drill (Mandrillus leucophaeus), including its sporogony in Anopheles aztecus and its pre-erythrocytic schizogony in the rhesus monkey. Trans R Soc Trop Med Hyg. 1958:52(6):509–517. 10.1016/0035-9203(58)90100-7. [DOI] [PubMed] [Google Scholar]
- Gel B, Serra E. karyoploteR: an R/Bioconductor package to plot customizable genomes displaying arbitrary data. Bioinformatics. 2017:33(19):3088–3090. 10.1093/bioinformatics/btx346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilabert A, Otto TD, Rutledge GG, Franzon B, Ollomo B, Arnathau C, Durand P, Moukodoum ND, Okouga AP, Ngoubangoye B, et al. Plasmodium vivax-like genome sequences shed new insights into Plasmodium vivax biology and evolution. PLoS Biol. 2018:16(8):e2006035. 10.1371/journal.pbio.2006035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman N, Yang Z. A codon-based model of nucleotide substitution for protein-coding DNA sequences. Mol Biol Evol. 1994:11(5):725–736. 10.1093/oxfordjournals.molbev.a040153. [DOI] [PubMed] [Google Scholar]
- Gould SJ, Lewontin RC. The spandrels of san marco and the panglossian paradigm: a critique of the adaptationist programme. Proc R Soc Lond B Biol Sci. 1979:205(1161):581–598. 10.1098/rspb.1979.0086. [DOI] [PubMed] [Google Scholar]
- Graumans W, Jacobs E, Bousema T, Sinnis P. When is a Plasmodium-infected mosquito an infectious mosquito? Trends Parasitol. 2020:36(8):705–716. 10.1016/j.pt.2020.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graur D, Martin W. Reading the entrails of chickens: molecular timescales of evolution and the illusion of precision. Trends Genet. 2004:20(2):80–86. 10.1016/j.tig.2003.12.003. [DOI] [PubMed] [Google Scholar]
- Groucutt HS, White TS, Scerri EML, Andrieux E, Clark-Wilson R, Breeze PS, Armitage SJ, Stewart M, Drake N, Louys J, et al. Multiple hominin dispersals into southwest Asia over the past 400,000 years. Nature. 2021:597(7876):376–380. 10.1038/s41586-021-03863-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunalan K, Niangaly A, Thera MA, Doumbo OK, Miller LH. Plasmodium vivax infections of duffy-negative erythrocytes: historically undetected or a recent adaptation? Trends Parasitol. 2018:34(5):420–429. 10.1016/j.pt.2018.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurevich A, Saveliev V, Vyahhi N, Tesler G. QUAST: quality assessment tool for genome assemblies. Bioinformatics. 2013:29(8):1072–1075. 10.1093/bioinformatics/btt086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall BG. Comparison of the accuracies of several phylogenetic methods using protein and DNA sequences. Mol Biol Evol. 2005:22(3):792–802. 10.1093/molbev/msi066. [DOI] [PubMed] [Google Scholar]
- Harrison TE, Reid AJ, Cunningham D, Langhorne J, Higgins MK. Structure of the Plasmodium-interspersed repeat proteins of the malaria parasite. Proc Natl Acad Sci U S A. 2020:117(50):32098–32104. 10.1073/pnas.2016775117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haug-Baltzell A, Stephens SA, Davey S, Scheidegger CE, Lyons E. SynMap2 and SynMap3D: web-based whole-genome synteny browsers. Bioinformatics. 2017:33(14):2197–2198. 10.1093/bioinformatics/btx144. [DOI] [PubMed] [Google Scholar]
- Hayakawa T, Culleton R, Otani H, Horii T, Tanabe K. Big bang in the evolution of extant malaria parasites. Mol Biol Evol. 2008:25(10):2233–2239. 10.1093/molbev/msn171. [DOI] [PubMed] [Google Scholar]
- Honma H, Kawai S, Motooka D, Nakamura S, Tougan T, Horii T, Arisue N. Draft genome sequence of Plasmodium gonderi, a malaria parasite of African old world monkeys. Genome Announc. 2017:5(28):e00612–e00617. 10.1128/genomeA.00612-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes AL, Verra F. Malaria parasite sequences from chimpanzee support the co-speciation hypothesis for the origin of virulent human malaria (Plasmodium falciparum). Mol Phylogenet Evol. 2010:57(1):135–143. 10.1016/j.ympev.2010.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapli P, Yang Z, Telford MJ. Phylogenetic tree building in the genomic age. Nat Rev Genet. 2020:21(7):428–444. 10.1038/s41576-020-0233-0. [DOI] [PubMed] [Google Scholar]
- Kaur H, Pacheco MA, Garber L, Escalante AA, Vinetz JM. Evolutionary insights into the microneme-secreted, chitinase-containing high-molecular-weight protein complexes involved in plasmodium invasion of the mosquito midgut. Infect Immun. 2022:90(1):e0031421. 10.1128/IAI.00314-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kooij TWA, Carlton JM, Bidwell SL, Hall N, Ramesar J, Janse CJ, Waters AP. A Plasmodium whole-genome synteny map: indels and synteny breakpoints as foci for species-specific genes. PLoS Pathog. 2005:1(4):e44. 10.1371/journal.ppat.0010044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krief S, Escalante AA, Pacheco MA, Mugisha L, André C, Halbwax M, Fischer A, Krief JM, Kasenene JM, Crandfield M, et al. On the diversity of malaria parasites in African apes and the origin of Plasmodium falciparum from bonobos. PLoS Pathog. 2010:6(2):e1000765. 10.1371/journal.ppat.1000765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA. Circos: an information aesthetic for comparative genomics. Genome Res. 2009:19(9):1639–1645. 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ksepka DT, Parham JF, Allman JF, Benton MJ, Carrano MT, Cranston KA, Donoghue PC, Head JJ, Hermsen EJ, Irmis RB, et al. The fossil calibration database-A new resource for divergence dating. Syst Biol. 2015:64(5):853–859. 10.1093/sysbio/syv025. [DOI] [PubMed] [Google Scholar]
- Kumar S, Suleski M, Craig JE, Kasprowicz AE, Sanderford M, Li M, Stecher G, Hedges SB. TimeTree 5: an expanded resource for Species divergence times. Mol Biol Evol. 2022:39(8):msac174. 10.1093/molbev/msac174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. Fast gapped-read alignment with bowtie 2. Nat Methods. 2012:9(4):357–359. 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapp SA, Geraldo JA, Chien JT, Ay F, Pakala SB, Batugedara G, Humphrey J; MaHPIC consortium, DeBarry YJD, Le Roch KG, et al. PacBio assembly of a Plasmodium knowlesi genome sequence with Hi-C correction and manual annotation of the SICAvar gene family. Parasitology. 2018:145(1):71–84. 10.1017/S0031182017001329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leclerc MC, Hugot JP, Durand P, Renaud F. Evolutionary relationships between 15 Plasmodium species from new and old world primates (including humans): an 18S rDNA cladistic analysis. Parasitology. 2004:129(6):677–684. 10.1017/S0031182004006146. [DOI] [PubMed] [Google Scholar]
- Lee SH, Hudock A. Human evolution in Asia: taking stock and looking forward. Annu Rev Anthropol. 2021:50(1):145–166. 10.1146/annurev-anthro-101819-110230. [DOI] [Google Scholar]
- Little TS, Cunningham DA, Vandomme A, Lopez CT, Amis S, Alder C, Addy JWG, McLaughlin S, Hosking C, Christophides G, et al. Analysis of pir gene expression across the Plasmodium life cycle. Malar J. 2021:20(1):445. 10.1186/s12936-021-03979-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Li Y, Learn GH, Rudicell RS, Robertson JD, Keele BF, Ndjango JB, Sanz CM, Morgan DB, Locatelli S, et al. Origin of the human malaria parasite plasmodium falciparum in gorillas. Nature. 2010:467(7314):420–425. 10.1038/nature09442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Li Y, Shaw KS, Learn GH, Plenderleith LJ, Malenke JA, Sundararaman SA, Ramirez MA, Crystal PA, Smith AG, et al. African origin of the malaria parasite Plasmodium vivax. Nat Commun. 2014:5(1):3346. 10.1038/ncomms4346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Sherrill-Mix S, Learn GH, Scully EJ, Li Y, Avitto AN, Loy DE, Lauder AP, Sundararaman SA, Plenderleith LJ, et al. Wild bonobos host geographically restricted malaria parasites including a putative new laverania species. Nat Commun. 2017:8(1):1635. 10.1038/s41467-017-01798-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louys J, Roberts P. Environmental drivers of megafauna and hominin extinction in Southeast Asia. Nature. 2020:586(7829):402–406. 10.1038/s41586-020-2810-y. [DOI] [PubMed] [Google Scholar]
- Lovell JT, Sreedasyam A, Schranz ME, Wilson M, Carlson JW, Harkess A, Emms D, Goodstein DM, Schmutz J. GENESPACE tracks regions of interest and gene copy number variation across multiple genomes. Elife. 2022:11:e78526. 10.7554/eLife.78526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loy DE, Plenderleith LJ, Sundararaman SA, Liu W, Gruszczyk J, Chen YJ, Trimboli S, Learn GH, MacLean OA, Morgan ALK, et al. Evolutionary history of human Plasmodium vivax revealed by genome-wide analyses of related ape parasites. Proc Natl Acad Sci U S A. 2018:115(36):E8450–E8459. 10.1073/pnas.1810053115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madden T. User Manual, Updated 2020. In: BLAST Command Line Applications User Manual. Bethesda: National Center for Biotechnology Information. 2008. Available from: https://www.ncbi.nlm.nih.gov/books/NBK279691/
- Makanga B, Yangari P, Rahola N, Rougeron V, Elguero E, Boundenga L, Moukodoum ND, Okouga AP, Arnathau C, Durand P, et al. Ape malaria transmission and potential for ape-to-human transfers in Africa. Proc Natl Acad Sci U S A. 2016:113(19):5329–5334. 10.1073/pnas.1603008113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malaria Genomic Epidemiology Network . Insights into malaria susceptibility using genome-wide data on 17,000 individuals from Africa, Asia and oceania. Nat Commun. 2019:10(1):5732. 10.1038/s41467-019-13480-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mapua MI, Fuehrer HP, Petrželková KJ, Todd A, Noedl H, Qablan MA, Modrý D. Plasmodium ovale wallikeri in western lowland gorillas and humans, Central African Republic. Emerg Infect Dis. 2018:24(8):1581–1583. 10.3201/eid2408.180010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinsen ES, Perkins SL, Schall JJ. A three-genome phylogeny of malaria parasites (Plasmodium and closely related genera): evolution of life-history traits and host switches. Mol Phylogenet Evol. 2008:47(1):261–273. 10.1016/j.ympev.2007.11.012. [DOI] [PubMed] [Google Scholar]
- McManus KF, Taravella AM, Henn BM, Bustamante CD, Sikora M, Cornejo OE. Population genetic analysis of the DARC locus (duffy) reveals adaptation from standing variation associated with malaria resistance in humans. PLoS Genet. 2017:13(3):e1006560. 10.1371/journal.pgen.1006560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ménard D, Barnadas C, Bouchier C, Henry-Halldin C, Gray LR, Ratsimbasoa A, Thonier V, Carod JF, Domarle O, Colin Y, et al. Plasmodium vivax clinical malaria is commonly observed in duffy-negative Malagasy people. Proc Natl Acad Sci U S A. 2010:107(13):5967–5971. 10.1073/pnas.0912496107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minh BQ, Schmidt HA, Chernomor O, Schrempf D, Woodhams MD, von Haeseler A, Lanfear R. IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol Biol Evol. 2020:37(5):1530–1534. 10.1093/molbev/msaa015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsui H, Arisue N, Sakihama N, Inagaki Y, Horii T, Hasegawa M, Tanabe K, Hashimoto T. Phylogeny of Asian primate malaria parasites inferred from apicoplast genome-encoded genes with special emphasis on the positions of Plasmodium vivax and P. fragile. Gene. 2010:450(1-2):32–38. 10.1016/j.gene.2009.10.001. [DOI] [PubMed] [Google Scholar]
- Mu J, Joy DA, Duan J, Huang Y, Carlton J, Walker J, Barnwell J, Beerli P, Charleston MA, Pybus OG, et al. Host switch leads to emergence of Plasmodium vivax malaria in humans. Mol Biol Evol. 2005:22(8):1686–1693. 10.1093/molbev/msi160. [DOI] [PubMed] [Google Scholar]
- Muehlenbein MP, Pacheco MA, Taylor JE, Prall SP, Ambu L, Nathan S, Alsisto S, Ramirez D, Escalante AA. Accelerated diversification of nonhuman primate malarias in Southeast Asia: adaptive radiation or geographic speciation? Mol Biol Evol. 2015:32(2):422–439. 10.1093/molbev/msu310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muffato M, Louis A, Nguyen NTT, Lucas J, Berthelot C, Roest Crollius H. Reconstruction of hundreds of reference ancestral genomes across the eukaryotic kingdom. Nat Ecol Evol. 2023:7(3):355–366. 10.1038/s41559-022-01956-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neafsey DE, Galinsky K, Jiang RHY, Young L, Sykes SM, Saif S, Gujja S, Goldberg JM, Young S, Zeng Q, et al. The malaria parasite Plasmodium vivax exhibits greater genetic diversity than plasmodium falciparum. Nat Genet. 2012:44(9):1046–1050. 10.1038/ng.2373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen LT, Schmidt HA, von Haeseler A, Minh BQ. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 2015:32(1):268–274. 10.1093/molbev/msu300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimoto Y, Arisue N, Kawai S, Escalante AA, Horii T, Tanabe K, Hashimoto T. Evolution and phylogeny of the heterogeneous cytosolic SSU rRNA genes in the genus Plasmodium. Mol Phylogenet Evol. 2008:47(1):45–53. 10.1016/j.ympev.2008.01.031. [DOI] [PubMed] [Google Scholar]
- Oberstaller J, Otto TD, Rayner JC, Adams JH. Essential genes of the parasitic apicomplexa. Trends Parasitol. 2021:37(4):304–316. 10.1016/j.pt.2020.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ollomo B, Durand P, Prugnolle F, Douzery E, Arnathau C, Nkoghe D, Leroy E, Renaud F. A new malaria agent in African hominids. PLoS Pathog. 2009:5(5):e1000446. 10.1371/journal.ppat.1000446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto TD, Böhme U, Jackson AP, Hunt M, Franke-Fayard B, Hoeijmakers WAM, Religa AA, Robertson L, Sanders M, Ogun SA, et al. A comprehensive evaluation of rodent malaria parasite genomes and gene expression. BMC Biol. 2014:12(1):86. 10.1186/s12915-014-0086-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto TD, Dillon GP, Degrave WS, Berriman M. RATT: rapid annotation transfer tool. Nucleic Acids Res. 2011:39(9):e57. 10.1093/nar/gkq1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto TD, Gilabert A, Crellen T, Böhme U, Arnathau C, Sanders M, Oyola SO, Okouga AP, Boundenga L, Willaume E, et al. Genomes of all known members of a Plasmodium subgenus reveal paths to virulent human malaria. Nat Microbiol. 2018:3(6):687–697. 10.1038/s41564-018-0162-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacheco MA, Battistuzzi FU, Junge RE, Cornejo OE, Williams CV, Landau I, Rabetafika L, Snounou G, Jones-Engel L, Escalante AA. Timing the origin of human malarias: the lemur puzzle. BMC Evol Biol. 2011:11(1):299. 10.1186/1471-2148-11-299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacheco MA, Ceríaco LMP, Matta NE, Vargas-Ramírez M, Bauer AM, Escalante AA. A phylogenetic study of Haemocystidium parasites and other Haemosporida using complete mitochondrial genome sequences. Infect Genet Evol. 2020:85:104576. [DOI] [PubMed] [Google Scholar]
- Pacheco MA, Cranfield M, Cameron K, Escalante AA. Malarial parasite diversity in chimpanzees: the value of comparative approaches to ascertain the evolution of Plasmodium falciparum antigens. Malar J. 2013:12(1):328. 10.1186/1475-2875-12-328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacheco MA, Escalante AA. Origin and diversity of malaria parasites and other haemosporida. Trends Parasitol. 2023:39(7):501–516. 10.1016/j.pt.2023.04.004. [DOI] [PubMed] [Google Scholar]
- Pacheco MA, Junge RE, Menon A, McRoberts J, Valkiunas G, Escalante AA. The evolution of primate malaria parasites: a study on the origin and diversification of Plasmodium in lemurs. Mol Phylogenet Evol. 2022:174:107551. 10.1016/j.ympev.2022.107551. [DOI] [PubMed] [Google Scholar]
- Pacheco MA, Matta NE, Valkiūnas G, Parker PG, Mello B, Stanley CE Jr, Lentino M, Garcia-Amado MA, Cranfield M, Kosakovsky Pond SL, et al. Mode and rate of evolution of haemosporidian mitochondrial genomes: timing the radiation of avian parasites. Mol Biol Evol. 2018:35(2):383–403. 10.1093/molbev/msx285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacheco MA, Reid MJC, Schillaci MA, Lowenberger CA, Galdikas BMF, Jones-Engel L, Escalante AA. The origin of malarial parasites in orangutans. PLoS One. 2012:7(4):e34990. 10.1371/journal.pone.0034990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pain A, Böhme U, Berry AE, Mungall K, Finn RD, Jackson AP, Mourier T, Mistry J, Pasini EM, Aslett MA, et al. The genome of the simian and human malaria parasite Plasmodium knowlesi. Nature. 2008:455(7214):799–803. 10.1038/nature07306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasini EM, Böhme U, Rutledge GG, Voorberg-Van der Wel A, Sanders M, Berriman M, Kocken CH, Otto TD. An improved Plasmodium cynomolgi genome assembly reveals an unexpected methyltransferase gene expansion. Wellcome Open Res. 2017:2:42. 10.12688/wellcomeopenres.11864.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perelman P, Johnson WE, Roos C, Seuánez HN, Horvath JE, Moreira MAM, Kessing B, Pontius J, Roelke M, Rumpler Y, et al. A molecular phylogeny of living primates. PLoS Genet. 2011:7(3):e1001342. 10.1371/journal.pgen.1001342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters W. Malaria of the orangutan (Pongo pygmaeus) in borneo. Philos Trans R Soc Lond B Biol Sci. 1976:275(941):439–482. 10.1098/rstb.1976.0089. [DOI] [PubMed] [Google Scholar]
- Plasmodium Genome Database Collaborative . PlasmoDB: an integrative database of the Plasmodium falciparum genome. Tools for accessing and analyzing finished and unfinished sequence data. The plasmodium genome database collaborative. Nucleic Acids Res. 2001:29(1):66–69. 10.1093/nar/29.1.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plenderleith LJ, Liu W, Li Y, Loy DE, Mollison E, Connell J, Ayouba A, Esteban A, Peeters M, Sanz CM. Zoonotic origin of the human malaria parasite Plasmodium malariae from African apes. Nat Commun. 2022:13(1):1868. 10.1038/s41467-022-29306-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plenderleith LJ, Liu W, MacLean OA, Li Y, Loy DE, Sundararaman SA, Bibollet-Ruche F, Learn GH, Hahn BH, Sharp PM. Adaptive evolution of RH5 in ape Plasmodium species of the Laverania subgenus. mBio. 2018:9(1):e02237-17. 10.1128/mBio.02237-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poirriez J, Dei-Cas E, Dujardin L, Landau I. The blood-stages of Plasmodium georgesi, P. gonderi and P. petersi: course of untreated infection in their natural hosts and additional morphological distinctive features. Parasitology. 1995:111(5):547–554. 10.1017/S0031182000077015. [DOI] [PubMed] [Google Scholar]
- Powell CLE, Waskin S, Battistuzzi FU. Quantifying the error of secondary vs. Distant primary calibrations in a simulated environment. Front Genet. 2020:11:252. 10.3389/fgene.2020.00252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prugnolle F, Rougeron V, Becquart P, Berry A, Makanga B, Rahola N, Arnathau C, Ngoubangoye B, Menard S, Willaume E, et al. Diversity, host switching and evolution of Plasmodium vivax infecting African great apes. Proc Natl Acad Sci USA. 2013:110(20):8123–8128. 10.1073/pnas.1306004110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Püschel HP, Bertrand OC, O'Reilly JE, Bobe R, Püschel TA. Divergence-time estimates for hominins provide insight into encephalization and body mass trends in human evolution. Nat Ecol Evol. 2021:5(6):808–819. 10.1038/s41559-021-01431-1. [DOI] [PubMed] [Google Scholar]
- Quek ZBR, Huang D. Effects of missing data and data type on phylotranscriptomic analysis of stony corals (cnidaria: anthozoa: scleractinia). Mol Phylogenet Evol. 2019:134:12–23. 10.1016/j.ympev.2019.01.012. [DOI] [PubMed] [Google Scholar]
- Ramaprasad A, Klaus S, Douvropoulou O, Culleton R, Pain A. Plasmodium vinckei genomes provide insights into the pan-genome and evolution of rodent malaria parasites. BMC Biol. 2021:19(1):69. 10.1186/s12915-021-00995-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rannala B, Yang Z. Inferring speciation times under an episodic molecular clock. Syst Biol. 2007:56(3):453–466. 10.1080/10635150701420643. [DOI] [PubMed] [Google Scholar]
- Roberts P, Stewart BA. Defining the ‘generalist specialist’ niche for pleistocene Homo sapiens. Nat Hum Behav. 2018:2(8):542–550. 10.1038/s41562-018-0394-4. [DOI] [PubMed] [Google Scholar]
- Romiguier J, Roux C. Analytical biases associated with GC-content in molecular evolution. Front Genet. 2017:8:16. 10.3389/fgene.2017.00016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronquist F, Huelsenbeck JP. Mrbayes 3: bayesian phylogenetic inference under mixed models. Bioinformatics. 2003:19(12):1572–1574. 10.1093/bioinformatics/btg180. [DOI] [PubMed] [Google Scholar]
- Rutledge GG, Böhme U, Sanders M, Reid AJ, Cotton JA, Maiga-Ascofare O, Djimdé AA, Apinjoh TO, Amenga-Etego L, Manske M, et al. Plasmodium malariae and P. ovale genomes provide insights into malaria parasite evolution. Nature. 2017:542(7639):101–104. 10.1038/nature21038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaer J, Perkins SL, Decher J, Leendertz FH, Fahr J, Weber N, Matuschewski K. High diversity of west African bat malaria parasites and a tight link with rodent Plasmodium taxa. Proc Natl Acad Sci U S A. 2013:110(43):17415–17419. 10.1073/pnas.1311016110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schliep KP. Phangorn: phylogenetic analysis in R. Bioinformatics. 2011:27(4):592–593. 10.1093/bioinformatics/btq706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp PM, Plenderleith LJ, Culleton R, Hahn BH. The African origin of Plasmodium vivax. FEMS Microbiol Rev. 2022:46(4):fuac015. 10.1093/femsre/fuac015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp PM, Plenderleith LJ, Hahn BH. Ape origins of human malaria. Annu Rev Microbiol. 2020:74(1):39–63. 10.1146/annurev-micro-020518-115628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinden RE, Canning EU, Bray RS, Smalley ME. Gametocyte and gamete development in Plasmodium falciparum. Proc R Soc Lond B Biol Sci. 1978:201(1145):375–399. 10.1098/rspb.1978.0051. [DOI] [PubMed] [Google Scholar]
- Stanke M, Keller O, Gunduz I, Hayes A, Waack S, Morgenstern B. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res. 2006:2006(Web Server):W435–W439. 10.1093/nar/gkl200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supek F, Bošnjak M, Škunca N, Šmuc T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS One. 2011:6(7):e21800. 10.1371/journal.pone.0021800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland CJ, Tanomsing N, Nolder D, Oguike M, Jennison C, Pukrittayakamee S, Dolecek C, Hien TT, do Rosário VE, Arez AP, et al. Two nonrecombining sympatric forms of the human malaria parasite Plasmodium ovale occur globally. J Infect Dis. 2010:201(10):1544–1550. 10.1086/652240. [DOI] [PubMed] [Google Scholar]
- Tachibana S, Sullivan SA, Kawai S, Nakamura S, Kim HR, Goto N, Arisue N, Palacpac NMQ, Honma H, Yagi M, et al. Plasmodium cynomolgi genome sequences provide insight into Plasmodium vivax and the monkey malaria clade. Nat Genet. 2012:44(9):1051–1055. 10.1038/ng.2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Battistuzzi FU, Billing-Ross P, Murillo O, Filipski A, Kumar S. Estimating divergence times in large molecular phylogenies. Proc Natl Acad Sci USA. 2012:109(47):19333–19338. 10.1073/pnas.1213199109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Stecher G, Kumar S. MEGA11: molecular evolutionary genetics analysis version 11. Mol Biol Evol. 2021:38(7):3022–3027. 10.1093/molbev/msab120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Tao Q, Kumar S. Theoretical foundation of the RelTime method for estimating divergence times from Variable evolutionary rates. Mol Biol Evol. 2018:35(7):1770–1782. 10.1093/molbev/msy044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao Q, Tamura K, Battistuzzi UF, Kumar S. A machine learning method for detecting autocorrelation of evolutionary rates in large phylogenies. Mol Biol Evol. 2019:36(4):811–824. 10.1093/molbev/msz014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telford SR Jr. 2009. Hemoparasites of the reptilia. Boca Raton: CRC Press. [Google Scholar]
- Törönen P, Medlar A, Holm L. PANNZER2: a rapid functional annotation web server. Nucleic Acids Res. 2018:46(W1):W84–W88. 10.1093/nar/gky350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urciuoli A, Alba DM. Systematics of miocene apes: state of the art of a neverending controversy. J Hum Evol. 2023:175:103309. 10.1016/j.jhevol.2022.103309. [DOI] [PubMed] [Google Scholar]
- Vaidya AB, Akella R, Suplick K. Sequences similar to genes for two mitochondrial proteins and portions of ribosomal RNA in tandemly arrayed 6-kb-pair DNA of a malarial parasite. Mol Biochem Parasitol. 1989:35(2):97–107. 10.1016/0166-6851(89)90112-6. [DOI] [PubMed] [Google Scholar]
- Valkiūnas G. Avian Malaria parasites and other Haemosporidian. CRC Press; 2005. [Google Scholar]
- Valkiūnas G, Iezhova TA. Keys to the avian malaria parasites. Malar J. 2018:17(1):212. 10.1186/s12936-018-2359-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Tang H, Debarry JD, Tan X, Li J, Wang X, Lee TH, Jin H, Marler B, Guo H, et al. MCScanx: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012:40(7):e49. 10.1093/nar/gkr1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson IRJM, Denny PW, Preiser PR, Rangachari K, Roberts K, Roy A, Whyte A, Strath M, Moore DJ, Moore PW, et al. Complete gene map of the plastid-like DNA of the malaria parasite Plasmodium falciparum. J Mol Biol. 1996:261(2):155–172. 10.1006/jmbi.1996.0449. [DOI] [PubMed] [Google Scholar]
- Wright GJ, Rayner JC. Plasmodium falciparum erythrocyte invasion: combining function with immune evasion. PLoS Pathog. 2014:10(3):e1003943. 10.1371/journal.ppat.1003943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia X. DAMBE6: new tools for microbial genomics, phylogenetics, and molecular evolution. J Hered. 2017:108(4):431–437. 10.1093/jhered/esx033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007:24(8):1586–1591. 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- Yang Z, Rannala B. Bayesian estimation of species divergence times under a molecular clock using multiple fossil calibrations with soft bounds. Mol Biol Evol. 2006:23(1):212–226. 10.1093/molbev/msj024. [DOI] [PubMed] [Google Scholar]
- Young AD, Gillung JP. Phylogenomics—principles, opportunities and pitfalls of big-data phylogenetics. Syst Entomol. 2020:45(2):225–247. 10.1111/syen.12406. [DOI] [Google Scholar]
- Zhang C, Rabiee M, Sayyari E, Mirarab S. ASTRAL-III: polynomial time species tree reconstruction from partially resolved gene trees. BMC Bioinformatics. 2018a:19(S6):153. 10.1186/s12859-018-2129-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Wang C, Otto TD, Oberstaller J, Liao X, Adapa SR, Udenze K, Bronner IF, Casandra D, Mayho M, et al. Uncovering the essential genes of the human malaria parasite Plasmodium falciparum by saturation mutagenesis. Science. 2018b:360(6388):eaap7847. 10.1126/science.aap7847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao T, Zwaenepoel A, Xue JY, Kao SM, Li Z, Schranz ME, Van de Peer Y. Whole-genome microsynteny-based phylogeny of angiosperms. Nat Commun. 2021:12(1):3498. 10.1038/s41467-021-23665-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Dang Y, Zhou M, Li L, Yu CH, Fu J, Chen S, Liu Y. Codon usage is an important determinant of gene expression levels largely through its effects on transcription. Proc Natl Acad Sci USA. 2016:113(41):E6117–E6125. 10.1073/pnas.1606724113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimin AV, Marçais G, Puiu D, Roberts M, Salzberg SL, Yorke JA. The MaSuRCA genome assembler. Bioinformatics. 2013:29(21):2669–2677. 10.1093/bioinformatics/btt476. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Scripts used to perform the analyses reported in this article are available at https://github.com/EscalanteLab/Plasmodium-gonderi-genome-analysis. The data used in this work is available under the BioProject GenBank accession no. PRJNA928704 and the Genome Whole Sequence GenBank accession no. JARBEB000000000.1.