

Abstract
Integrative analyses of genome-wide association studies and gene expression data have implicated many disease-critical tissues. However, co-regulation of genetic effects on gene expression across tissues impedes distinguishing biologically causal tissues from tagging tissues. Here, we introduce tissue co-regulation score regression (TCSC), which disentangles causal tissues from tagging tissues by regressing gene-disease association statistics (from transcriptome-wide association studies) on tissue co-regulation scores reflecting correlations of predicted gene expression across genes and tissues. We applied TCSC to 78 diseases/traits (average N = 302K) and gene expression prediction models for 48 GTEx tissues. TCSC identified 21 causal tissue-trait pairs at 5% FDR, including well-established findings, biologically plausible novel findings (e.g. aorta artery and glaucoma), and increased specificity of known tissue-trait associations (e.g. subcutaneous adipose, but not visceral adipose, and HDL). TCSC also identified 17 causal tissue-trait covariance pairs at 5% FDR. In conclusion, TCSC is a precise method for distinguishing causal tissues from tagging tissues.
Introduction
Most diseases are driven by tissue-specific or cell-type-specific mechanisms, thus the inference of causal disease tissues is an important goal1. For many polygenic diseases and complex traits, disease-associated tissues have previously been identified via the integration of genome-wide association studies (GWAS) with tissue-level functional data characterizing expression quantitative trait loci (eQTLs)2–5, gene expression6–9, or epigenetic features10–17. However, it is likely that most disease-associated tissues are not actually causal, due to the high correlation of eQTL effects (resp. gene expression or epigenetic features) across tissues; the correlation of eQTL effects across tissues, i.e. tissue co-regulation, can arise due to shared eQTLs or distinct eQTLs in linkage disequilibrium (LD)2,18,19,5. One approach to address this involves comparing eQTL-disease colocalizations across different tissues2 while another approach leverages multi-trait fine-mapping to simultaneously evaluate all tissues for disease colocalization5; both methods risk implicating co-regulated tagging tissues that colocalize with disease. To our knowledge, no previous study has formally modeled genetic co-regulation across tissues to statistically disentangle causal from tagging tissues.
Here, we introduce a new method, tissue co-regulation score regression (TCSC), that disentangles causal tissues from tagging tissues and partitions disease heritability (or genetic covariance of two diseases/traits) into tissue-specific components. TCSC leverages gene-disease association statistics across tissues from transcriptome-wide association studies (TWAS)20,21,18. A challenge is that TWAS association statistics include the effects of both co-regulated tissues (see above) and co-regulated genes18,22. To address this, TCSC regresses TWAS chi-square statistics (or products of z-scores for two diseases/traits) on tissue co-regulation scores reflecting correlations of predicted gene expression across genes and tissues. Distinct from previous methods that analyze each tissue marginally, TCSC jointly models contributions from each tissue to identify causal tissues (analogous to the distinction in GWAS between marginal association and fine-mapping23). We validate TCSC using extensive simulations using real genotypes with LD, including comparisons to RTC Coloc2, RolyPoly6, LDSC-SEG7, and CoCoNet9 (reviewed in 1,24). We apply TCSC to 78 diseases and complex traits (average N = 302K) and 48 GTEx tissues19, showing that TCSC recapitulates known biology and identifies biologically plausible novel tissue-trait pairs (or tissue-trait covariance pairs) while attaining increased specificity relative to previous methods.
Results
Overview of TCSC regression
TCSC estimates the disease heritability explained by cis-genetic components of gene expression in each tissue when jointly modeling contributions from each tissue. We refer to tissues with nonzero contributions as “causal” tissues (with the caveat that joint-fit effects of gene expression on disease may not reflect biological causality; see Discussion). TCSC assumes that gene expression-disease effect sizes are independent and identically distributed (i.i.d.) across genes and tissues (while accounting for the fact that cis-genetic components of gene expression are correlated across genes and tissues); violations of this model assumption are explored via simulations below. TCSC leverages the fact that TWAS statistics for each gene and tissue include both causal effects of that gene and tissue on disease and tagging effects of co-regulated genes and tissues. We define co-regulation based on squared correlations in cis-genetic expression, which can arise due to shared causal eQTLs and/or LD between causal eQTLs18. TCSC determines that a tissue is causal for disease if genes and tissues with high co-regulation to that tissue have higher TWAS statistics than genes and tissues with low co-regulation to that tissue.
In detail, let denote the disease heritability explained by the cis-genetic component of gene expression in tissue . The expected TWAS statistic for gene and tagging tissue is
| (1) |
where is GWAS sample size, indexes causal tissues, are tissue co-regulation scores (defined as , where denotes the cis-genetic component of gene expression for a gene-tissue pair across individuals, denotes the cis-predicted expression for a gene-tissue pair, the sum is over genes within +/− 1 Mb to gene ), and is the number of significantly cis-heritable genes in tissue . A derivation of Equation (1) is provided in the Methods section. Equation (1) allows us to estimate via a multiple linear regression of TWAS statistics (for each gene and tagging tissue) on tissue co-regulation scores (Fig. 1).
Figure 1. Overview of TCSC regression.
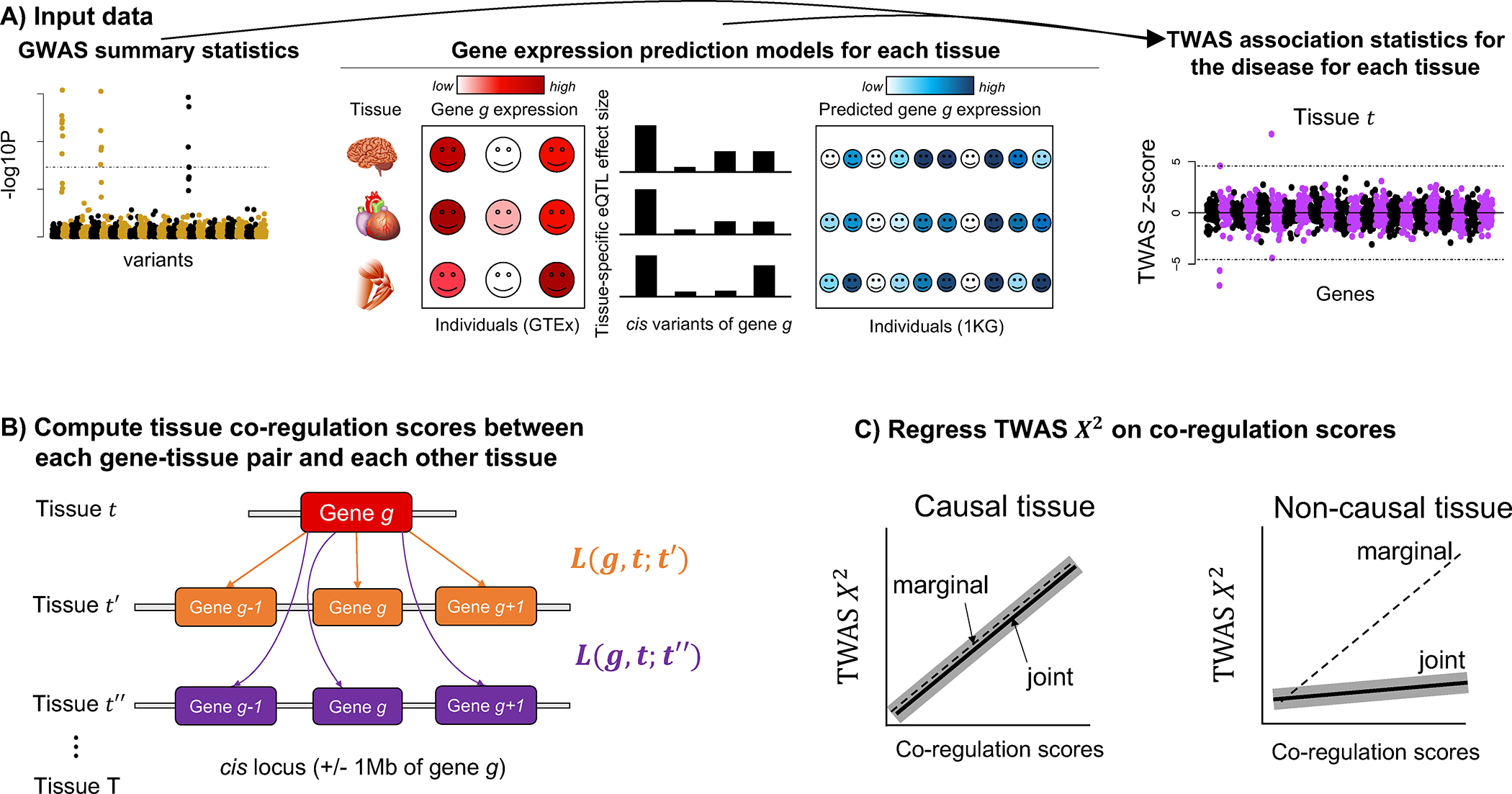
(A) Input data to TCSC includes (1) GWAS summary statistics for a disease and (2) gene expression prediction models for each tissue, which are used to produce (3) TWAS summary statistics for the disease for each tissue. (B) TCSC computes tissue co-regulation scores for each gene-tissue pair with potentially causal tissues . (C) TCSC regresses TWAS chi-squares on tissue co-regulation scores to estimate tissue-specific contributions to disease. Solid lines represent hypothetical TCSC estimates; dashed lines represent hypothetical TWAS associations; shadows represent hypothetical TCSC estimates +/− jackknife standard errors (joint models only).
TCSC can also estimate the genetic covariance between two diseases explained by cis-genetic components of gene expression in each tissue, using products of TWAS z-scores. In detail, let denote the genetic covariance explained by the cis-genetic component of gene expression in tissue (Methods). The expected product of TWAS z-scores in disease 1 and disease 2 for gene and tagging tissue is
| (2) |
where is GWAS sample size for disease 1, is GWAS sample size for disease 2, indexes causal tissues, are tissue co-regulation scores, is the number of significantly cis-heritable genes in tissue (Methods), is the phenotypic correlation between disease 1 and disease 2, and is the number of overlapping GWAS samples between disease 1 and disease 2. Equation (2) allows us to estimate via a multiple linear regression of products of TWAS z-scores in disease 1 and disease 2 (for each gene and tagging tissue) on tissue co-regulation scores. We note that the last term in Equation (2) is not known a priori but is accounted for via the regression intercept, analogous to previous work25.
Further details, including a formal definition of in terms of SNP-level effects, quality control of gene expression models and TWAS statistics, estimation of standard errors, correcting for bias in tissue co-regulation scores (analogous to GCSC22), are provided in the Methods section. We have publicly released open-source software implementing TCSC regression (see Code Availability), as well as all GWAS summary statistics, TWAS association statistics, tissue co-regulation scores, and TCSC output from this study (see Data Availability).
Simulations
We performed extensive simulations to evaluate the robustness and power of TCSC, using the TWAS simulator of Mancuso et al.26 (see Code Availability). We used real genotypes from the 1000 Genomes Project27 to simulate gene expression values (for each gene and tissue) and complex trait phenotypes, and then computed TWAS association statistics for each gene and tissue and estimated co-regulation scores. Primary simulations partitioned trait heritability across 10 highly correlated tissues (one of which contained causal gene-trait effects explaining 100% of trait heritability). We evaluated TCSC while varying many parameters including trait heritability explained by the causal tissue, trait heritability not explained by gene expression, and the number of causal tissues and the number of tagging tissues; more details are provided in the Supplementary Note: Simulation Framework. We also compared the type I error and power of TCSC to four previously published methods: RTC Coloc2, RolyPoly6, LDSC-SEG7, and CoCoNet9.
We first evaluated the bias in TCSC estimates of the disease heritability explained by the cis-genetic component of gene expression in tissue , for both causal and non-causal tissues. For causal tissues, TCSC produced unbiased estimates of (Fig. 2A, Supplementary Table 1); this implies that error in eQTL effect size estimates, which impacts TWAS statistics and co-regulation scores, does not bias TCSC estimates for causal tissues. A subtlety is that, as noted above, estimates of are impacted by the number of significantly cis-heritable genes in tissue ), which may be sensitive to eQTL sample size. For non-causal tissues, TCSC produced estimates of , that were significantly positive when averaged across all simulations, but not large enough to substantially impact type I error (see below).
Figure 2. Robustness and power of TCSC regression in simulations.
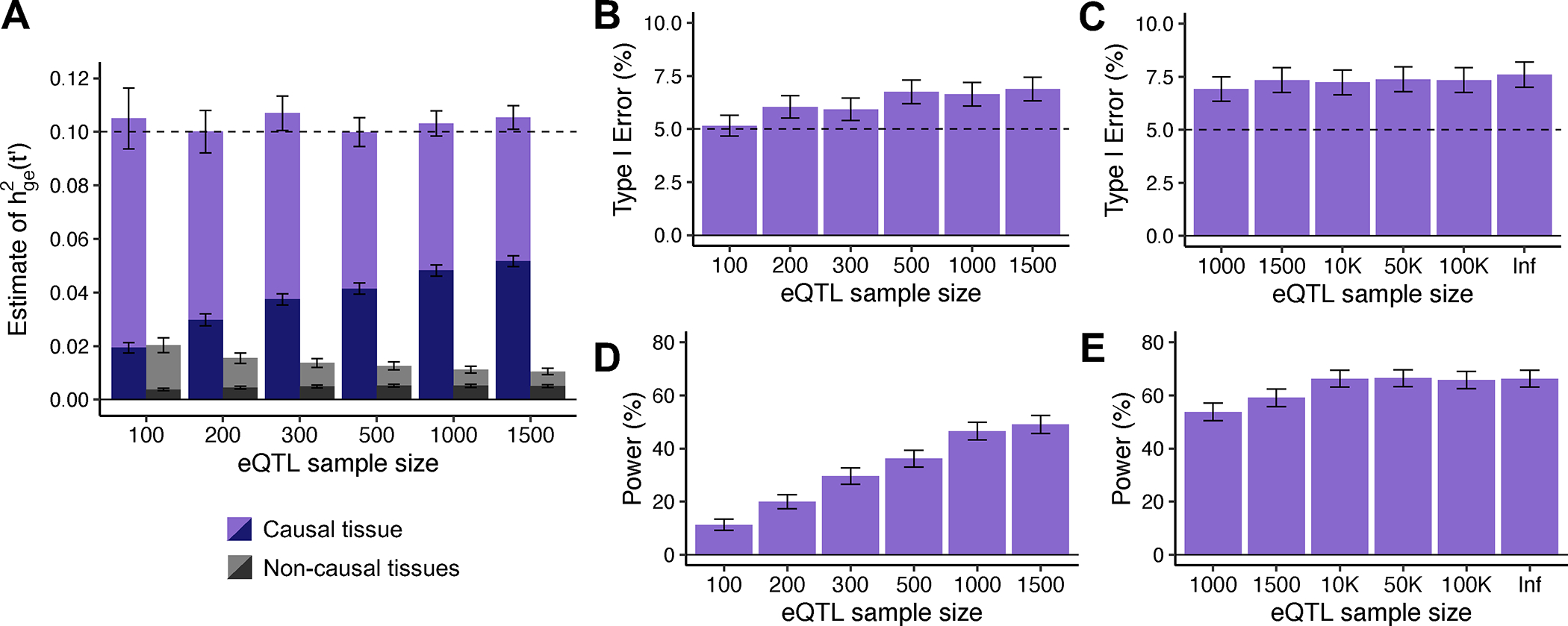
(A) Bias in estimates of disease heritability explained by the cis-genetic component of gene expression in tissue for causal (purples) and non-causal (grays) tissues. was set to the total number of true cis-heritable genes across tissues for light purple (resp. gray) or to the number of significantly cis-heritable genes in each tissue for dark purple (resp. gray). The dashed line indicates the true value of for causal tissues. (B) Percentage of estimates of for non-causal tissues that were significantly positive at p < 0.05 per eQTL sample size (n = 100, 200, 300, 500, 1000, 1500). (C) Percentage of estimates of for non-causal tissues that were significantly positive at p < 0.05 per larger eQTL sample size (n = 1000, 1500, 10K, 50K, 100K, Infinite). (D) Percentage of estimates of for causal tissues that were significantly positive at p < 0.05 per eQTL sample size (n = 100, 200, 300, 500, 1000, 1500). (E) Percentage of estimates of for causal tissues that were significantly positive at p < 0.05 per larger eQTL sample size (n = 1000, 1500, 10K, 50K, 100K, Infinite). For panels B-E, we used a one-sided z-test to obtain p-values. For panels A, B and D, we performed n = 1,000 independent simulations per eQTL sample size. For panels C and E, we performed n = 2,000 independent simulations per eQTL sample size and used cross-validation adjusted-R2 > 0 instead of GCTA to define significantly cis-heritable genes in these analyses. In all panels, data are presented as mean values +/− 1.96 × SEM. Numerical results are reported in Supplementary Table 1.
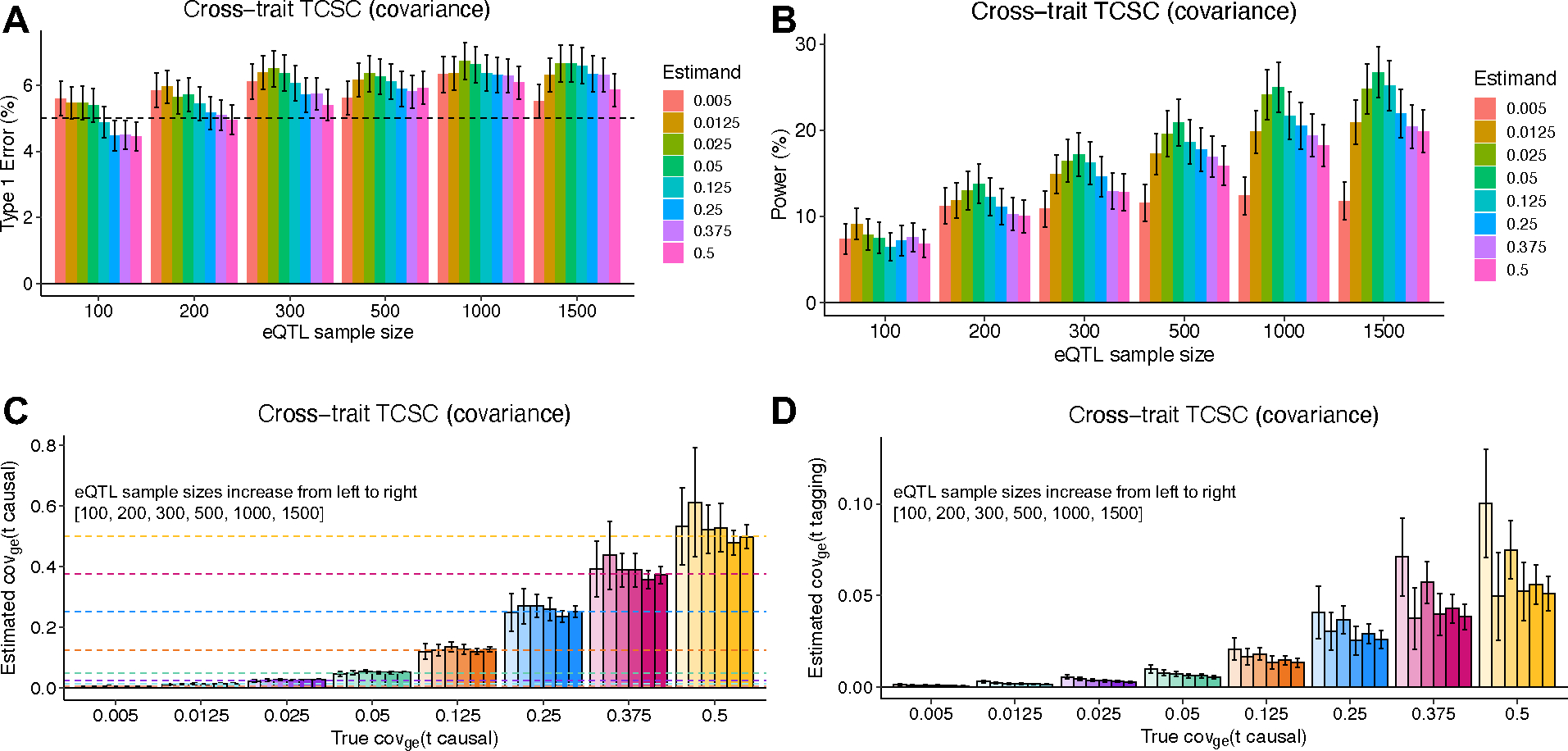
We next evaluated the type I error of TCSC for non-causal tissues. The type I error of TCSC was approximately well-calibrated, ranging from 5.2% to 6.9% across eQTL sample sizes of 100–1,500 at a significance threshold of p = 0.05 (Fig. 2B, Supplementary Table 1). In comparison, we observed type I errors from 53%–86% for RTC Coloc, 32%–33% for LDSC-SEG, 11%–12% for RolyPoly, and 32%–38% for CoCoNet, substantially greater than the type I error of TCSC (Extended Data Fig. 1, Supplementary Table 2). We confirmed that the type I error of TCSC does not increase further at larger gene expression sample sizes (Fig. 2C, Supplementary Note: Single-trait simulation analysis at large sample size).
We next evaluated the power of TCSC for causal tissues. We determined that TCSC was moderately well-powered to detect causal tissues, with power ranging from 11%–49% across eQTL sample sizes at a nominal significance threshold of p < 0.05 (Fig. 2D, Supplementary Table 1). As noted above, the power of TCSC varies greatly with the choice of parameter settings (see below), thus the power of TCSC in real-world settings is best evaluated using real trait analysis. As expected, power increased at larger eQTL sample sizes, due to lower standard errors on point estimates of (Fig. 2A). We also evaluated the power of RTC Coloc, RolyPoly, LDSC-SEG, and CoCoNet. For the only other method with type I error less than 15% (RolyPoly), power ranged from 14%–17% across eQTL sample sizes, substantially lower than TCSC (Extended Data Fig. 1, Supplementary Table 2). We also used ROC curves to assess the relationship between the sensitivity (power) and specificity (one minus the false positive rate) of all 5 methods across 1,000 uniformly spaced p-value thresholds. TCSC attained the largest AUC (0.78, vs. 0.54–0.59 for other methods) (Extended Data Fig. 1). We also evaluated the power of TCSC at four larger gene expression sample sizes (see above) and determined that power plateaus at 10,000 individuals (Fig. 2E).
We similarly evaluated cross-trait TCSC for simulated traits with a genetic correlation of 0.5. Cross-trait simulations produced similar conclusions to the single trait analysis above (Extended Data Figs. 1–2, Supplementary Table 3, Supplementary Note). We also performed 12 secondary simulation analyses showing the performance of TCSC across a diverse parameter space (Extended Data Figs. 3–6, Supplementary Figs. 2–21, Supplementary Note).
Tissue-specific contributions to 78 diseases/traits
We applied TCSC to publicly available GWAS summary statistics for 78 diseases and complex traits (average N = 302K; Methods, Supplementary Table 4) and gene expression data for 48 GTEx tissues19 (Table 1). The 48 GTEx tissues were aggregated into 39 meta-tissues (average N = 266, range: N = 101–320 individuals, 23 meta-tissues with N = 320) in order to reduce variation in eQTL sample size across tissues (Table 1 and Supplementary Note: Analyzing GTEx tissues) and referred to as “tissues” below. We constructed gene expression prediction models for an average of 3,993 significantly cis-heritable protein-coding genes (as defined above) in each tissue. We primarily report the proportion of disease heritability explained by the cis-genetic component of gene expression in tissue , where is the common variant SNP-heritability estimated by S-LDSC13,28,29.
Table 1. GTEx meta-tissues and constituent tissues analyzed.
For each meta-tissue we list the constituent tissue(s) and total sample size. Daggers denote meta-tissues with more than one constituent tissue; for these meta-tissues, each constituent tissue has equal sample size up to rounding error (an exception is the transverse intestine meta-tissue, which includes 176 transverse colon samples and all 144 small intestine samples).
| Meta-tissue | Constituent Tissue(s) | Sample Size |
|---|---|---|
| Adipose Subcutaneous | Adipose Subcutaneous | 320 |
| Adipose Visceral Omentum | Adipose Visceral Omentum | 320 |
| Adrenal Gland | Adrenal Gland | 200 |
| Aorta Artery | Aorta Artery | 320 |
| † Brain Basal Ganglia | Putamen, Caudate, Nucleus Accumbens | 320 |
| † Brain Cereb. | Cerebellum, Cerebellar Hemisphere | 320 |
| † Brain Cortex | Frontal, Anterior, Cingulate | 320 |
| † Brain Limbic | Amygdala, Hippocampus, Hypothalamus | 320 |
| Brain Spinal Cord | Brain Spinal Cord | 115 |
| Brain Substantia Nigra | Brain Substantia Nigra | 101 |
| Breast Mammary Gland | Breast Mammary Gland | 320 |
| Coronary Artery | Coronary Artery | 180 |
| Cultured Fibroblasts | Cultured Fibroblasts | 320 |
| Esophagus Mucosa | Esophagus Mucosa | 320 |
| Esophagus Muscularis | Esophagus Muscularis | 320 |
| Heart Atrial Appendage | Heart Atrial Appendage | 320 |
| Heart Left Ventricle | Heart Left Ventricle | 320 |
| LCLs | LCLs | 116 |
| Liver | Liver | 183 |
| Lung | Lung | 320 |
| Minor Salivary Gland | Minor Salivary Gland | 118 |
| Muscle Skeletal | Muscle Skeletal | 320 |
| Ovary | Ovary | 140 |
| Pancreas | Pancreas | 252 |
| Pituitary | Pituitary | 220 |
| Prostate | Prostate | 186 |
| Skin (sun exposed) | Skin (sun exposed) | 320 |
| Skin (sun unexposed) | Skin (sun unexposed) | 320 |
| † Sigmoid Intestine | Sigmoid Colon, Gastroesophageal Junction | 320 |
| Spleen | Spleen | 185 |
| Stomach | Stomach | 269 |
| Tibial Artery | Tibial Artery | 320 |
| Tibial Nerve | Tibial Nerve | 320 |
| Testis | Testis | 277 |
| Thyroid | Thyroid | 320 |
| † Transverse Intestine | Transverse Colon, Small Intestine | 320 |
| Uterus | Uterus | 108 |
| Vagina | Vagina | 122 |
| Whole Blood | Whole Blood | 320 |
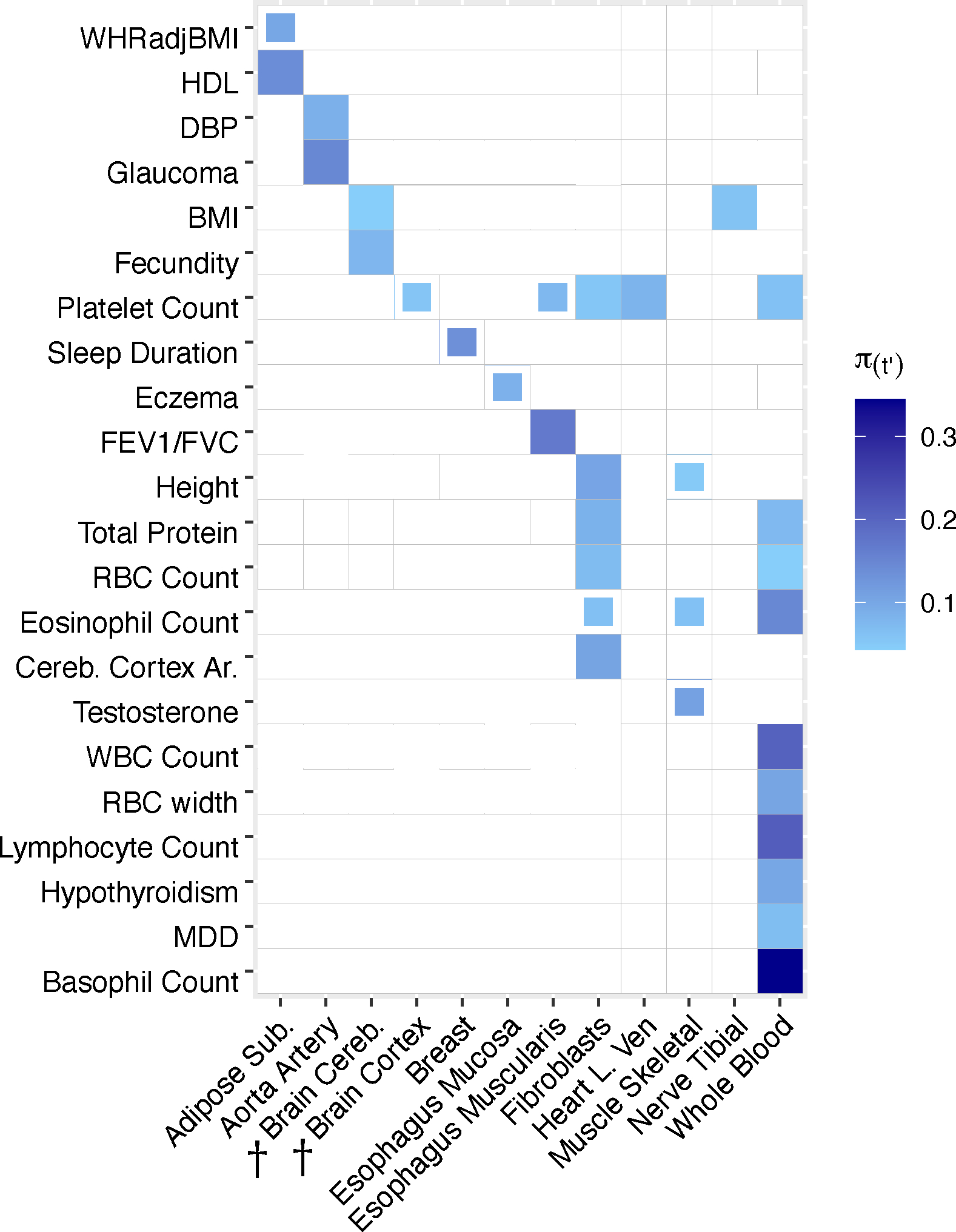
TCSC identified 21 causal tissue-trait pairs with significantly positive contributions to disease/trait heritability at 5% per-trait FDR, spanning 7 distinct tissues and 17 distinct diseases/traits (Fig. 3, Methods, Supplementary Table 5, Supplementary Fig. 22). Many of the significant findings recapitulated known biology, including associations of whole blood with blood cell traits such as white blood cell count (, s.e. = 0.064, ) and liver with lipid traits such as LDL (, s.e. = 0.050, ). TCSC also identified several biologically plausible findings not previously reported in the genetics literature. First, aorta artery was associated with glaucoma (, s.e. = 0.051, ) and also with diastolic blood pressure (DBP) (, s.e. = 0.024, ), which is consistent with DBP measuring the pressure exerted on the aorta30 and high blood pressure being a known risk factor for glaucoma31–35. Second, TCSC identified heart left ventricle as a causal tissue for platelet count (, s.e. = 0.031, ), consistent with the role of platelets in the formation of blood clots in cardiovascular disease36–39; in fact, antiplatelet drugs have been successful at reducing adverse cardiovascular outcomes40. Moreover, the left ventricle serves as a muscle to pump blood throughout the body41, likely modulating platelet and other blood cell counts. Other significant findings are reported in Supplementary Table 6 and the Supplementary Note.
Figure 3. TCSC estimates tissue-specific contributions to disease and complex trait heritability.
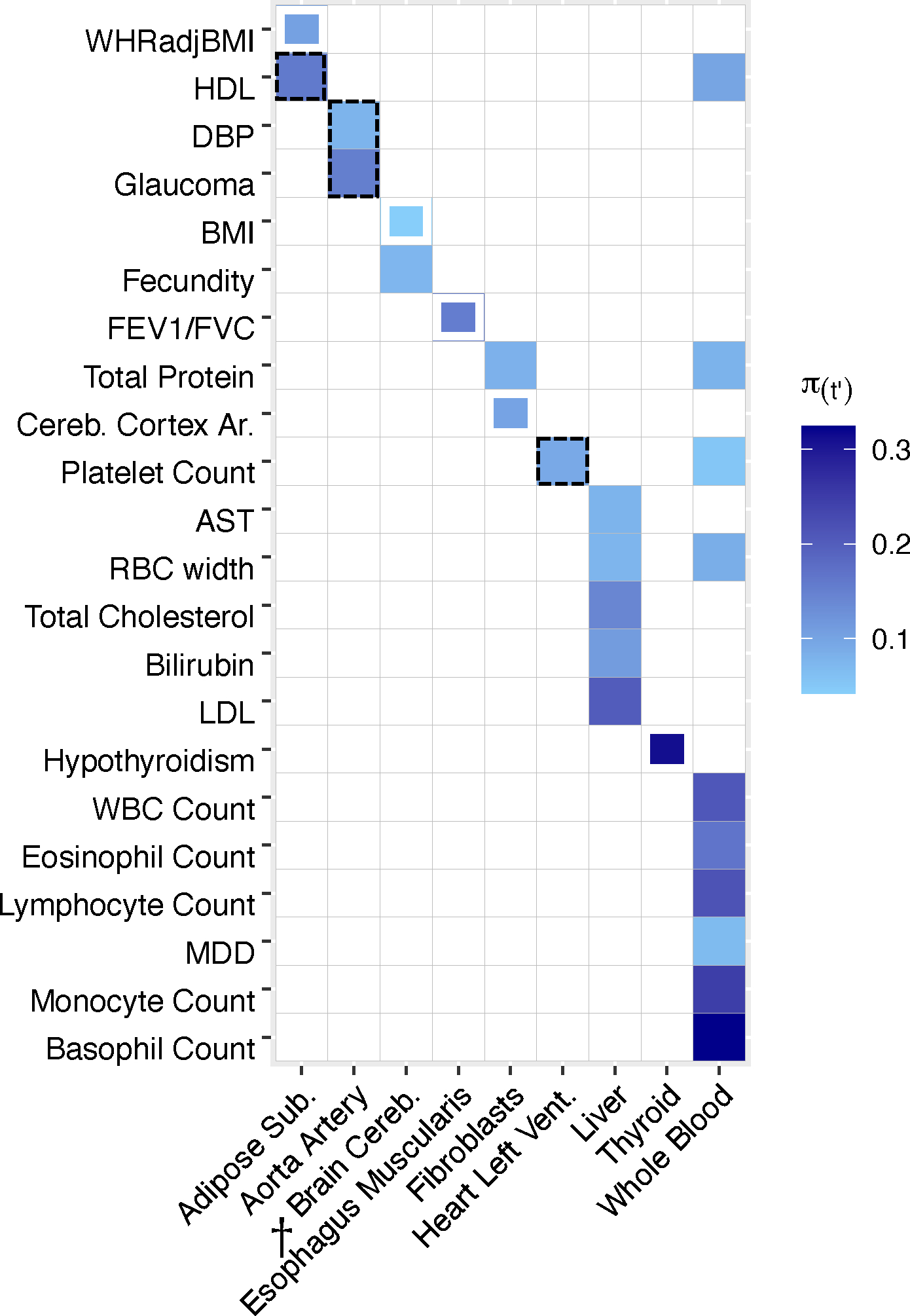
We report estimates of the proportion of disease heritability explained by the cis-genetic component of gene expression in tissue ). We report tissue-trait pairs with FDR of 10% or lower, where full boxes denote FDR of 5% or lower and partial boxes denote FDR between 5% and 10%. Dashed boxes denote results that are highlighted in the main text. Tissues are ordered alphabetically. Daggers denote meta-tissues with more than one constituent tissue. Diseases/traits are ordered with respect to causal tissues. Numerical results are reported in Supplementary Tables 5–6. WHRadjBMI: waist-hip-ratio adjusted for body mass index. HDL: high-density lipoprotein. DBP: diastolic blood pressure. BMI: body mass index. FEV1/FVC: forced expiratory volume in one second divided by forced vital capacity. Cereb. Cortex Ar.: cerebral cortex surface area. AST: aspartate aminotransferase. LDL: low-density lipoprotein. WBC Count: white blood cell count. MDD: major depressive disorder.
TCSC also increased the specificity of known tissue-trait associations. For high density lipoprotein (HDL), previous studies reported that deletion of a cholesterol transporter gene in adipose tissue reduces HDL levels42,43. TCSC specifically identified subcutaneous adipose (, s.e. = 0.054, ; Fig. 3), but not visceral adipose or breast adipose tissue (P > 0.05; Supplementary Table 6), as a causal tissue for HDL. Previous studies have established that levels of adiponectin, a hormone released by adipose tissue to regulate insulin, are positively correlated with HDL44–46 and more recently, a study has reported that adiponectin levels are associated specifically with subcutaneous adipose tissue and not visceral adipose tissue47. We note that TCSC did not identify liver as a causal tissue for HDL (FDR > 5%), which may be due to limited power in liver due to smaller eQTL sample size. For waist-hip ratio adjusted for BMI (WHRadjBMI), previous studies reported colocalization of WHRadjBMI GWAS variants with cis-eQTLs in subcutaneous adipose, visceral adipose, liver, and whole blood48, consistent with WHRadjBMI measuring adiposity in the intraabdominal space, which is likely regulated by metabolically active tissues49. TCSC specifically identified subcutaneous adipose as a suggestive finding (, s.e. = 0.037, , 5% < FDR < 10%; Fig. 3), but not visceral adipose, breast, liver, or whole blood (P > 0.05; Supplementary Table 6). The causal mechanism may again involve adiponectin, which is negatively correlated with WHRadjBMI50. We note that the P value distributions across traits are similar for adipose tissues (Supplementary Table 7). For BMI, previous studies have broadly implicated the central nervous system, but did not reveal more precise contributions51,13,52,53,7,54. TCSC specifically identified brain cereb. as a suggestive finding (, s.e. = 0.015, , 5% < FDR < 10%), but not brain cortex or brain limbic (P > 0.05; Supplementary Table 6), as a causal tissue for BMI. This finding is consistent with a known role for brain cerebellum in biological processes related to obesity including endocrine homeostasis55 and feeding control56; recently, a multi-omics approach has revealed cerebellar activation in mice upon feeding57.
We performed several secondary analyses. First, we removed tissues with eQTL sample size less than 320 individuals, as these tissues may often be underpowered (Fig. 2C). We identified 23 significant tissue-trait pairs, reflecting a gain of 8 newly significant tissue-trait pairs (Extended Data Fig. 7, Supplementary Table 8, Supplementary Note). We also performed a brain-specific analysis in which we applied TCSC to 41 brain traits (average N = 226K) while restricting to 13 individual GTEx brain tissues (Supplementary Tables 10–11, Supplementary Note). TCSC identified 8 brain tissue-brain trait pairs at 5% FDR, including the brain hippocampus as a causal tissue for ADHD consistent with dopamine regulation58–60 (Extended Data Fig. 8, Supplementary Table 12).
Comparisons of TCSC to other methods
We compared TCSC to two methods, RTC Coloc2 and LDSC-SEG7, that use gene expression data to identify disease-critical tissues based on tissue specificity of eQTL-GWAS colocalizations or heritability enrichment of specifically expressed genes, respectively. Both methods analyze each tissue marginally; thus, RTC Coloc and LDSC-SEG may output more significantly associated tissues per trait than TCSC (analogous to the distinction in GWAS between marginal association and fine-mapping23). To assess whether TCSC attains higher specificity, we applied each method to 7 traits with at least one tissue-trait association for each of the three methods (Methods) and compared results across causal tissues identified by TCSC and across the most strongly co-regulated tagging tissue (based on Spearman for estimated eQTL effect sizes, averaged across genes, from ref.19).
Results for the 7 traits are reported in Fig. 4 and Supplementary Table 13; results for all 17 diseases/traits with causal tissue-trait associations identified by TCSC are reported in Extended Data Fig. 9 and Supplementary Table 14, and results for all diseases/traits and tissues are in Supplementary Table 15. We reached three main conclusions. First, RTC Coloc implicates a broad set of tissues (not just strongly co-regulated tissues) for each trait (Fig. 4A); for example, for WBC count, RTC Coloc implicated 8 of 10 tissues. This is consistent with our simulations, in which RTC Coloc suffered a high type I error rate and had a substantially lower AUC than TCSC (Extended Data Fig. 1). Second, LDSC-SEG implicates a small set of strongly co-regulated tissues for each trait (Fig. 4B); for WBC count, LDSC-SEG typically implicated 3 of 10 tissues, consisting of whole blood, spleen, and breast tissue. This is consistent with our simulations, in which LDSC-SEG suffered a substantial type I error rate and had a substantially lower AUC than TCSC (Extended Data Fig. 1). Third, TCSC typically implicates one causal tissue per trait (Fig. 4C); for WBC count, TCSC implicated only whole blood as a causal tissue. This is consistent with our simulations, in which TCSC attained moderate power to identify causal tissues with approximately well-calibrated type I error. However, the higher specificity of TCSC may be accompanied by incomplete power to identify secondary causal tissues; we observed less significant (lower −log10P-value and lower −log10FDR) results for causal tissues from TCSC than by RTC Coloc or LDSC-SEG (Supplementary Table 14). We also observed similar patterns when comparing TCSC to RTC Coloc and LDSC-SEG in the brain-specific analysis (Extended Data Fig. 10, Supplementary Table 16, Supplementary Note).
Figure 4. Comparison of disease-critical tissues identified by RTC Coloc, LDSC-SEG and TCSC.
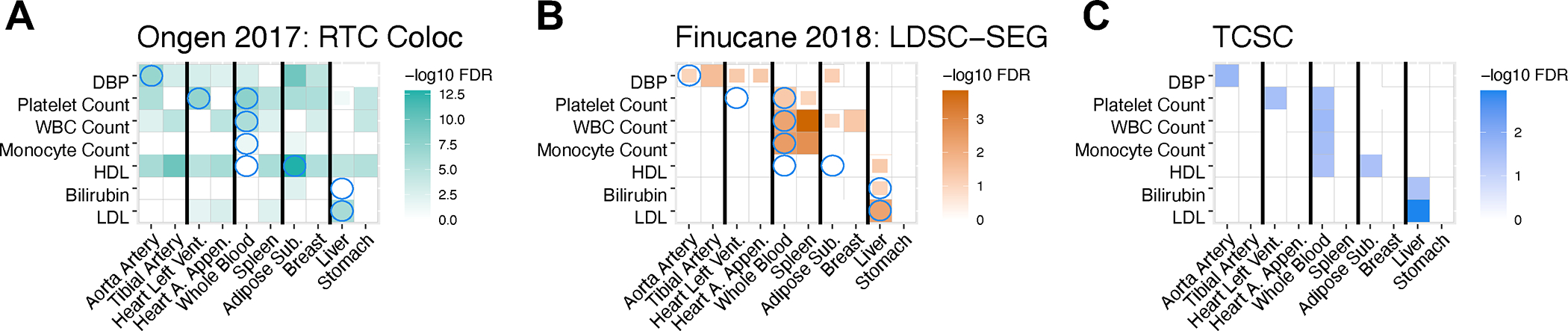
We report -log10FDR values for (A) RTC Coloc, (B) LDSC-SEG, (C) TCSC, across 7 traits with at least one significantly associated tissues (at FDR 5%) for each of the three methods and 10 tissues consisting of the causal tissues identified by TCSC and the most strongly co-regulated tagging tissues, ordered consecutively. We report tissue-trait pairs with FDR of 10% or lower, where full boxes denote FDR of 5% or lower and partial boxes denote FDR between 5% and 10%. Blue circles in panels (A) and (B) denote the causal tissue-trait pairs identified by TCSC. Numerical results are reported in Supplementary Table 13.
Tissue-specific contributions to cross-trait covariance
We applied cross-trait TCSC to 262 pairs of disease/traits (e.g. pairs with significantly nonzero genetic correlation, Methods, Supplementary Table 17) and gene expression data for 39 meta-tissues (Table 1). We primarily report the signed proportion of genetic covariance explained by the cis-genetic component of gene expression in tissue , where is the common variant genetic covariance estimated by cross-trait LDSC61. TCSC identified 17 causal tissue-trait covariance pairs with significant contributions to trait covariance at 5% per-trait FDR, spanning 12 distinct tissues and 13 distinct trait pairs (Fig. 5A, Supplementary Table 18).
Figure 5. Cross-trait TCSC estimates tissue-specific contributions to the genetic covariance of two diseases/traits.
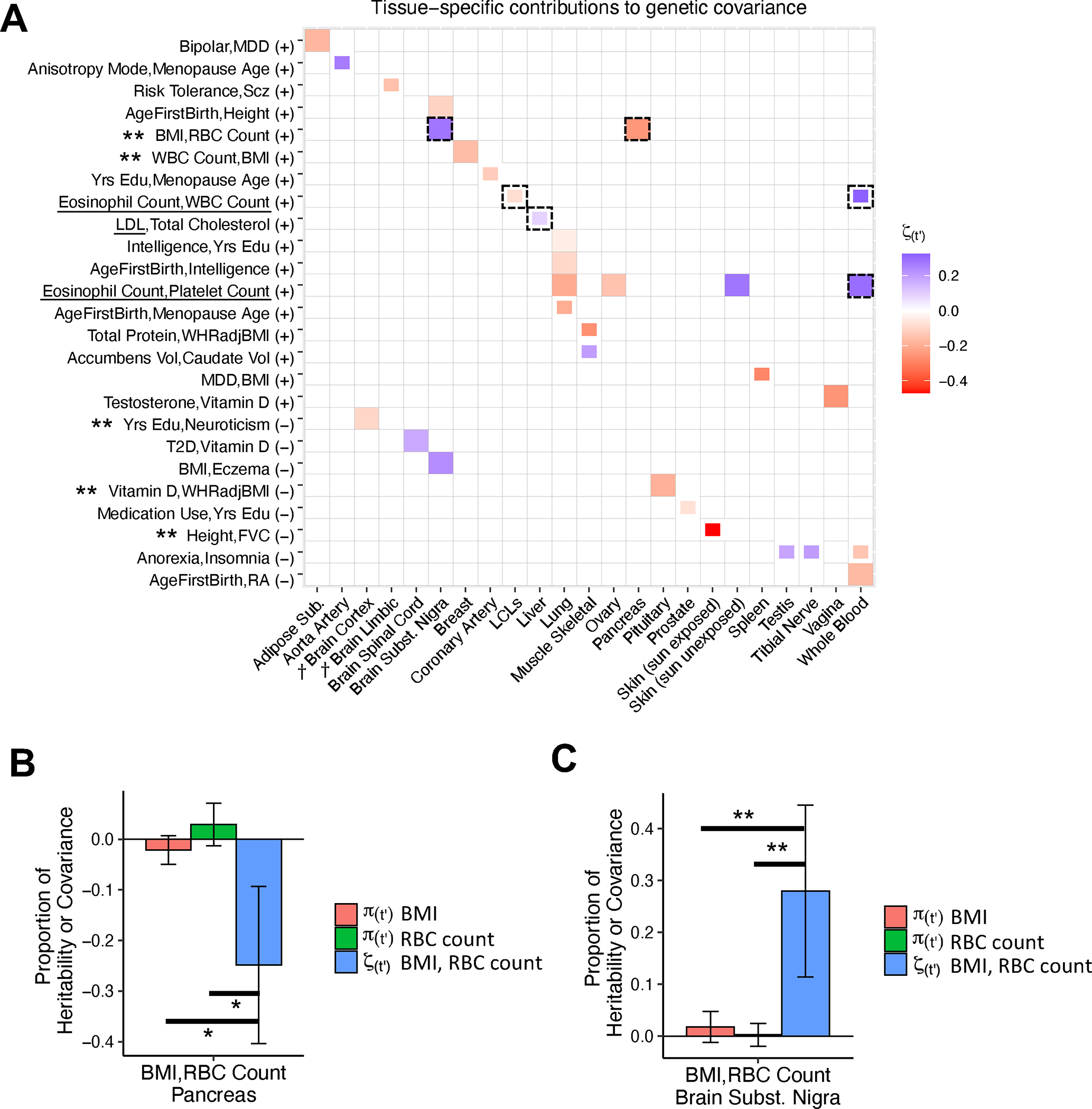
(A) Estimates of the signed proportion of genetic covariance explained by the cis-genetic component of gene expression in tissue ). Full boxes denote FDR < 5% and partial boxes denote 5% < FDR < 10%. Dashed boxes are highlighted in the main text. Tissues are ordered alphabetically. Daggers denote meta-tissues with >1 constituent tissue. Trait pairs are ordered by positive (+) or negative (−) genetic covariance, and then with respect to causal tissues. Underlined traits were shown in Fig. 3. Double asterisks denote trait pairs with significant differences between tissue-specific contributions to covariance and heritability, the latter of which was non-significant for both traits. Numerical results are reported in Supplementary Tables 18–19. BMI: body mass index. RBC Count: red blood cell count. WBC Count: white blood cell count. LDL: low-density lipoprotein. Yrs Edu: years of education. WHRadjBMI: waist-hip-ratio adjusted for body mass index. Accumbens Vol: brain accumbens volume. Caudate Vol: brain caudate volume. MDD: major depressive disorder. Scz: Schizophrenia. T2D: type 2 diabetes. FVC: forced vital capacity. RA: rheumatoid arthritis. (B) For BMI and RBC Count, estimates of the proportion of trait heritability and covariance explained by the cis-genetic component of gene expression in pancreas. Asterisks denote the difference between the covariance and heritability estimates was significantly < 0 at 10% FDR using a genomic block jackknife to estimate the standard error. (C) Same as (B), but for the brain substantia nigra. Double asterisks denote the difference between the covariance and heritability estimates was significantly > 0 at 5% FDR using a genomic block jackknife to estimate the standard error. Data in (B) and (C) are presented as TCSC estimates +/− 1.96 × the jackknife standard error. Numerical results are reported in Supplementary Table 21.
Several results recapitulated known biology. Consistent with the significant contribution of liver to LDL heritability in Fig. 3, TCSC identified a suggestive positive contribution of liver to the covariance of LDL and total cholesterol (, s.e. = 0.029, , 5% < FDR < 10%), and consistent with the positive contributions of whole blood to blood cell count heritabilities in Fig. 3, TCSC identified a significant positive contribution of whole blood to the covariance of eosinophil and platelet counts (, s.e. = 0.10, ).
TCSC identified several biologically plausible findings not previously reported in the genetics literature. First, brain substantia nigra had a significantly positive contribution to the genetic covariance of BMI and red blood cell count (RBC count) (, s.e. = 0.084, ), while pancreas had a significantly negative contribution (, s.e. = 0.079, ). In the brain, energy metabolism is regulated by oxidation and previous work has shown that red blood cells play a large role in these metabolic processes as oxygen sensors62; in addition, previous studies have reported differences in the level of oxidative enzymes in red blood cells between individuals with high BMI and low BMI63,64, suggesting that genes regulating oxidative processes might have pleiotropic effects on RBC count and BMI. In the pancreas, pancreatic inflammation (specifically acute pancreatitis) is associated with reduced levels of red blood cells, or anemia65, while pancreatic fat is associated with metabolic disease and increased BMI66. Second, LCLs had a suggestive negative contribution to the genetic covariance of eosinophil count and white blood cell count (, s.e. = 0.028, 5% < FDR < 10%, in contrast to the suggestive positive contribution of whole blood: , s.e. = 0.12, , 5% < FDR < 10%). This is plausible as previous studies have reported the suppression of proliferation of lymphocytes (the white blood cell hematopoietic lineage from which LCLs are derived) by molecules secreted from eosinophils67–69. Other significant findings are reported in Supplementary Table 19 and the Supplementary Note.
For 16 of the 17 causal tissue-trait covariance pairs, the causal tissue was non-significant for both constituent traits. We sought to formally assess whether tissue-specific contributions to covariance could exceed contributions to heritability. Specifically, for each causal tissue-trait covariance pair, we estimated the differences between the tissue-specific contribution to covariance () and the tissue-specific contributions to heritability for each constituent trait () (and estimated standard errors by jackknifing differences across the genome). We identified five tissue-trait covariance pairs for which these differences were statistically significant at 5% FDR for both traits and was non-significant for both traits (marked by double asterisks in Fig. 5A, results reported in Supplementary Table 20 and the Supplementary Note). The negative contribution of pancreas (Fig. 5B) and the positive contribution of brain substantia nigra (Fig. 5C) to the covariance of BMI and RBC count were larger than tissue-specific contributions to heritability, which were each non-significant. Even in simulations, TCSC often detected tissue-specific contributions to covariance without detecting tissue-specific contributions to heritability for both traits, especially when contributions to heritability substantially differed (Supplementary Table 21).
Discussion
We developed a new method, tissue co-regulation score regression (TCSC), that disentangles causal tissues from tagging tissues and partitions disease heritability (or cross-trait covariance) into tissue-specific components. We applied TCSC to 78 diseases and complex traits and 48 GTEx tissues, identifying 21 tissue-trait pairs (and 17 tissue-trait covariance pairs) with significant tissue-specific contributions. TCSC identified biologically plausible novel tissue-trait pairs, including associations of aorta artery with glaucoma, heart left ventricle with platelet count, and brain hippocampus with ADHD. TCSC also identified biologically plausible novel tissue-trait covariance pairs, including a positive contribution of brain substantia nigra and a negative contribution of pancreas to the covariance of BMI and red blood cell count; in particular, our findings suggest that genetic covariance may reflect distinct tissue-specific contributions.
TCSC differs from previous methods in jointly modeling contributions from each tissue to disentangle causal tissues from tagging tissues (analogous to the distinction in GWAS between marginal association and fine-mapping23). We briefly discuss several other methods that use eQTL or gene expression data to identify disease-associated tissues. RTC Coloc identifies disease-associated tissues based on tissue specificity of eQTL-GWAS colocalizations2; this study made a valuable contribution in emphasizing the importance of tissue co-regulation, but did not model tissue-specific effects, such that RTC Coloc may implicate many tissues (Fig. 4A). LDSC-SEG identifies disease-critical tissues based on heritability enrichment of specifically expressed genes7; this distinguishes a focal tissue from the set of all tissues analyzed but does not distinguish closely co-regulated tissues (Fig. 4B). We discuss four other methods in the Supplementary Note: Other tissue-trait association methods.
We note several limitations of our work. First, TCSC requires tissue-specific eQTL data (thus requiring genotype/gene expression data in substantial sample size), whereas some methods (LDSC-SEG7, RolyPoly6, and CoCoNet9) only require gene expression data in limited sample size. Second, joint-fit effects of gene expression on disease may not reflect biological causality; if a causal tissue or cell type is not assayed, TCSC may identify a co-regulated tissue (e.g. a tissue whose cell type composition favors a causal cell type) as causal. We anticipate that this limitation will become less severe as potentially causal tissues, cell types and contexts are more comprehensively assayed. Third, TCSC does not achieve a strict definition or estimation of mediated effects; this is conceptually appealing and can, in principle, be achieved by modeling non-mediated effects, but may result in limited power to distinguish disease-critical tissues70. Fourth, TCSC performs less well in the presence of disease heritability that is not mediated through gene expression (Supplementary Figs. 16–19). We discuss other limitations in the Supplementary Note: Other limitations. Despite these limitations, TCSC is a powerful and generalizable approach for modeling tissue co-regulation to estimate tissue-specific contributions to disease.
Methods
Our research complies with all relevant ethnical regulations; no specific approval was needed.
TCSC regression
TCSC leverages the fact that the TWAS statistic for a gene-tissue pair includes the direct effects of the gene on the disease as well as the tagging effects of co-regulated tissues and genes with shared eQTLs or eQTLs in LD. Thus, genes that are co-regulated across many tissues will tend to have higher statistics than genes regulated in a single tissue. TCSC determines that a tissue causally contributes to disease if genes with high co-regulation to the tissue have higher TWAS statistics than genes with low co-regulation to the tissue.
We model the genetic component of gene expression as a linear combination of SNP-level effects:
| (3) |
where is the cis-genetic component of gene expression in individual for gene and tissue is the standardized genotype of individual for SNP , and is the standardized effect of the mth SNP on the cis-genetic component of gene expression of gene in tissue . We define the cis-genetic component of gene expression to have mean 0 and variance 1 and to have mean 0 and variance , where is the number of cis variants for gene g.
TCSC assumes that true gene-disease effects are identically distributed (i.i.d.) across genes and tissues while accounting for the fact that cis-genetic components of gene expression (and cis-genetic predictions of gene expression) are correlated1 (see Supplementary Figs. 12–15 for simulations where gene expression-trait effect sizes are not i.i.d. across genes and tissues; TCSC performs well despite violations of model assumptions). The high correlation of cis-eQTLs across tissues leads to tagging from co-regulated tissues2. We model phenotype as a linear combination of genetic components of gene expression across genes in different tissues:
| (4) |
where is the (binary or continuous-valued) phenotype of individual is the standardized effect size of the cis-genetic component of gene expression on disease and is the component of phenotype not explained by cis-genetic components of gene expression. We emphasize that we model disease as a function of the unobserved true cis-genetic component of gene expression , not the genetically predicted value obtained from gene expression prediction models. Equation (4) can be rewritten in terms of SNP-level effects:
| (5) |
where are direct SNP-disease effects not mediated through gene expression.
We define the disease heritability explained by cis-genetic expression across all tissues as follows:
| (6) |
Because has mean 0 and variance 1 and are assumed to be i.i.d. across genes and tissues (see above), Equation (5) implies that:
| (7) |
analogous to the relationship between SNP effect sizes and SNP-heritability25: . We emphasize that the respective terms in Equation (5) for each tissue t’ are independent as are assumed to be i.i.d. across genes and tissues. It follows that the disease heritability explained by a particular tissue is
| (8) |
which given that has mean 0 and variance 1 and is i.i.d. across genes, reduces to:
| (9) |
Equation (7) and Equation (9) imply that Now, let be a random variable drawn from a normal distribution with mean zero and tissue-specific variance Then
| (10) |
where is the number of significantly cis-heritable genes in the model. In simulations, we demonstrate that when there are similar numbers of cis-heritable genes across tissues, setting to the total number of unique cis-heritable genes produces unbiased estimates in TCSC for the causal tissue; however, when there are varying numbers of cis-heritable genes across tissues (fewer in the causal tissue), this produces upward biased estimates (Supplementary Fig. 3–4) and thus setting to the number of significantly cis-heritable genes in tissue t’ is recommended. With this variance term, we can define a polygenic model that relates TWAS statistics to co-regulation scores, which explicitly model the covariance structure of the statistics. This strategy is analogous to modeling the dependence of GWAS statistics on LD scores25.
In a TWAS, the estimated value of the gene-disease effect size is proportional to the correlation of the cis-genetic components of gene expression and their true gene-disease effect sizes for nearby genes across tissues, analogous to GCSC22:
| (11) |
where is the estimated correlation in cis-genetic predicted expression between gene in tissue and genes in tissue is the component of phenotype not explained by cis-genetic components of gene expression, with mean 0 and variance .
The value of the TWAS is proportional to the squared estimated disease-gene effect size and the GWAS sample size N as follows:
| (12) |
Using the equations (9) and (10), we can write the expectation of TWAS as follows:
| (13) |
| (14) |
| (15) |
| (16) |
| (17) |
| (18) |
| (19) |
| (20) |
| (1) |
To go from Equation (15) to Equation (16) we use the following relationship from the derivation of LDSC13:
| (21) |
We go from Equation (19) to Equation (20) because the variance of the phenotype is and is equal to one. We also introduce the notation that are the tissue and gene co-regulation scores see below. We are interested in estimating , the per-gene disease heritability explained by the cis-genetic component of gene expression in tissue . From the derivation, the genome-wide tissue-specific contribution to disease heritability is estimated as
| (22) |
For the analysis of tissue-specific contributions to the covariance between two diseases, we can extend TCSC by using products of TWAS z-scores. Following the polygenic model described above, the expected product of TWAS z-scores in disease 1 and disease 2 for gene and tagging tissue is
| (23) |
where is GWAS sample size for disease 1, is GWAS sample size for disease 2, indexes causal tissues, are tissue co-regulation scores (see below), is the genetic covariance explained by the cis-genetic component of gene expression in tissue is the number of significantly cis-heritable genes in tissue (see below), is the phenotypic correlation between disease 1 and disease 2, and is the number of overlapping GWAS samples between disease 1 and disease 2. The last term represents the intercept61, and while we use a free intercept in the multivariate regression on co-regulation scores, the estimation of this term only plays a role in the estimation of regression weights (see below).
For estimates of and we use a free intercept; the estimation of serves only to inform the heteroscedasticity weights (see below) and is not used in the multivariate TCSC regression to estimate To estimate standard errors, we use a genomic block jackknife over 200 genomic blocks with an equal number of genes in each. The standard deviation is computed as the square root of the weighted variance across the jackknife estimates (where the weight of each block is equal to the sum of the regression weights for the genes in that block) multiplied by 200 blocks. We expect that the jackknife standard error will be conservative relative to the empirical standard error across estimates due to variation in causal signal across loci71.
Estimating tissue co-regulation scores and correcting for bias
We define the co-regulation score of gene with tissues and as
| (24) |
where denotes the cis-genetic component of gene expression for a gene-tissue pair across individuals, denotes the cis-predicted expression for a gene-tissue pair, and genes are within +/− 1 Mb of the focal gene . TCSC corrects for bias in tissue co-regulation scores arising from differences between cis-genetic vs. cis-predicted expression (analogous to GCSC22). We apply bias correction to co-regulation scores in the special case when and . While co-regulation scores aim to estimate , the squared correlation of the predicted cis-genetic component of expression of gene and tissue (corresponding to the TWAS statistic) with the actual cis-genetic component of gene expression of gene in tissue , when and , the estimated value of will always equals one because the estimate is based on . However, this implies that predictions of the cis-genetic component of expression are perfectly accurate, which is unlikely to be the case. Therefore, the estimated value of if left to equal one will cause co-regulation scores to be systematically inflated.
Therefore, when and , we set
| (25) |
where is the cross-validation prediction statistic of the gene expression model for gene in tissue and is the GCTA-estimated cis-heritability of gene expression for gene in tissue . The quotient is the accuracy of the gene expression prediction model, which reflects the upper bound on how much the cis-predicted expression can be correlated with the true cis-genetic component of gene expression. While we only consider genes with p < 0.01, the uncertainty in estimates should be modest and therefore not greatly impact our bias correction. We note that TWAS tests the null hypothesis that a specific weighted linear combination of SNPs is not associated with disease (and does not test the null hypothesis that the cis-genetic component of gene expression is not associated with disease).
TCSC regression weights
TCSC uses three sets of regression weights to increase power (analogous to GCSC22). The first regression weight is inversely proportional to the total co-regulation score of each gene-tissue pair summed across tissues :
| (26) |
(without applying bias correction; see above), which allows TCSC to properly account for redundant contributions of co-regulated genes to TWAS statistics.
The second regression weight is inversely proportional to the number of tissues in which a gene is significantly cis-heritable:
| (27) |
thereby up-weighting signal from genes that are regulated in a limited number of tissues and preventing TCSC from attributing more weight to genes that are co-regulated across many tissues.
The third regression weight is inversely proportional to , the heteroscedasticity of statistics, and is computed differently for estimates of than for estimates of (analogous to GCSC22 and cross-trait LDSC61, respectively).
For estimates of we estimate in two steps. First, we make a crude estimate of heritability explained by predicted expression () as follows:
| (28) |
where is the mean statistic:
| (29) |
where is the GWAS sample size, iterates over significantly cis-heritable genes and iterates over tissues, and is the mean value of total co-regulation across tissues ,
| (30) |
Then, we compute the heteroscedasticity for each significantly cis-heritable gene-tissue pair as
| (31) |
Finally, we combine the three regression weights as follows:
| (32) |
For estimates of we estimate in two steps. First, we regress the products of TWAS z-scores on total tissue co-regulation scores, , using regression weights, computed as follows:
| (33) |
where is first estimated as follows:
| (34) |
where is the crude heritability estimate for trait 1 and is the crude heritability estimate for trait 2, is estimated as is the sample size of the first GWAS, is the sample size of the second GWAS, and is the total number of tissues in the regression.
Second, we use the regression intercept to estimate the product
| (35) |
where represents the phenotypic correlation between trait 1 and 2 and represents the number of shared samples between GWAS 1 and 2. We also use the coefficient of the regression to update our estimate of , such that we may update the heteroscedasticity weight as follows:
| (36) |
Finally, we combine the three regression weights as follows:
| (37) |
Gene expression prediction models and tissue co-regulation scores in GTEx data
To construct gene expression prediction models, we applied FUSION21 (Code Availability) to individual-level GTEx data by regressing measured gene expression on genotypes of common variants (MAF > 0.05) and covariates provided by GTEx19. FUSION uses several different regression models: single eQTL, elastic net, lasso, and BLUP and the following covariates: sex, 5 genotyping principal components, PEER factors72, and assay type. We defined significantly cis-heritable genes as protein-coding genes with GCTA heritability p < 0.0121, heritability estimate > 0, and adjusted-R2 > 0 in cross-validation prediction.
We used gene expression prediction models of significantly cis-heritable genes to predict expression into 489 European individuals from 1000 Genomes27. We then estimated tissue co-regulation scores using Equation (24) and Equation (25)73, where cis-predicted gene expression is used to estimate the cis-genetic component of gene expression.
GWAS summary statistics and TWAS association statistics
We collected GWAS summary statistics from 78 relatively independent heritable complex diseases and traits (average N = 302K) with heritability z-score > 6, including 33 diseases/traits from UK Biobank74. We estimated the heritability of all summary statistics and genetic correlation of all pairs of summary statistics. We excluded traits with heritability z-score < 6, using S-LDSC with the baseline-LD v2.2 model13,28,29 and as done previously28. We excluded one of each pair of traits that are both genetically correlated and have significantly overlapping samples. Specifically, for any pair of non-UK Biobank traits with an estimated sample overlap greater than the following threshold -- squared cross-trait LDSC intercept / (trait 1 S-LDSC intercept * trait 2 S-LDSC intercept) > 0.161 -- the trait with the larger SNP heritability z-score was retained. For any pair of UK Biobank traits with a squared genetic correlation > 0.1, the trait with the larger SNP heritability z-score was retained75. In total, this procedure resulted in 78 sets of relatively independent GWAS summary statistics. We limited all analyses (including cross-trait analyses) to the 78 relatively independent traits in order to avoid redundant findings across single-trait (and cross-trait) analyses. For the brain-specific analysis, we first selected brain-related diseases and complex traits, e.g. psychiatric disorders and behavioral phenotypes, excluding multi case-control studies and case vs case studies. Then, we applied our standard filters as described above, but relaxing the threshold of squared genetic correlation to 0.25.
We used FUSION21 (Code Availability) to compute TWAS association statistics for each pair of signed GWAS summary statistics and each significantly cis-heritable gene-tissue pair, across the two scenarios described above. We restrict gene expression prediction models and TWAS association statistics for each tissue to significantly cis-heritable genes in that tissue, defined as genes with significantly positive cis-heritability (2-sided p < 0.01; estimated using GCTA76) and positive adjusted- in cross-validation prediction. We further removed genes within the MHC (chromosome 6, 29 Mb - 33 Mb) and TWAS or , where is the GWAS sample size, as previously used for quality control in the heritability analysis of GWAS summary statistics13. TCSC scales linearly with the number of genes and quadratically with the number of tissues. After all input datasets are created and processed, running TCSC on a single real GWAS trait with 39 tissues takes about two minutes73. We employ a per-trait FDR (as in ref.77,78) rather than a global FDR (as in ref.7), because power is likely to vary across traits and there are a sufficiently large number of independent quantities estimated per trait ( jointly estimated across 39 tissues); a global FDR is more appropriate when there are far fewer independent quantities estimated per trait, e.g. due to non-independent, marginal tissue associations in ref.7 For cross-trait analysis, we estimated the genetic correlation of each of 3,003 pairs of the 78 disease/traits analyzed in this study; the 262 pairs analyzed by cross-trait analysis had significantly nonzero genetic correlation (p < 0.05 / 3,003).
RTC Coloc and LDSC-SEG analysis of GWAS summary statistics and GTEx tissues
We downloaded supplementary tables for the RTC coloc method2 and for LDSC-SEG7. For traits in our set of 78 GWAS summary statistics that were not analyzed by the LDSC-SEG study and for traits that are inherently brain-related (as these traits require a different procedure for generating tissue-specific gene sets), we ran LDSC-SEG ourselves. To this end, we downloaded LD scores for GTEx tissues and specifically expressed gene set SNP-level annotations (https://alkesgroup.broadinstitute.org/LDSCORE/LDSC_SEG_ldscores/) and ran LDSC-SEG7 using S-LDSC v1.0.0. For brain-related traits, we additionally ran a brain-specific analysis using LDSC-SEG7. Briefly, specifically expressed genes were determined via a t-test of the sentinel brain tissue against all other brain tissues, rather than against all other non-brain GTEx tissues, as done in the primary analysis of the LDSC-SEG study. For traits in our set that were not analyzed by the RTC Coloc study, of which there were few, we did not apply their method, as it was too computationally intensive to apply to real trait data.
Statistics and Reproducibility
First, as described above, we downsampled or meta-analyzed GTEx tissues to reduce the potential for type I error in our analysis; downsampling was performed randomly. Second, as described above, GWAS summary statistics with a SNP heritability z-score < 6 were excluded, due to the potential to have low power. Third, in analyses of real diseases/traits, we verified that we obtained consistent results when analyzing different GWAS cohorts. We obtained independent GWAS summary statistics for 10 traits implicated in 13 significant tissue-trait pairs (Supplementary Table 4) and confirmed the same direction of effect for 13 of 13 tissue-trait pairs (including FDR < 5% for 7 of 13 tissue-trait pairs, FDR < 10% for 9 of 13 tissue-trait pairs) (Supplementary Table 5). Fourth, 1,000 independent genetic architectures were simulated to robustly evaluate TCSC performance. Randomization and blinding were not pertinent to our study.
Extended Data
Extended Data Figure 1. Comparison of tissue-trait association methods with TCSC in simulations.
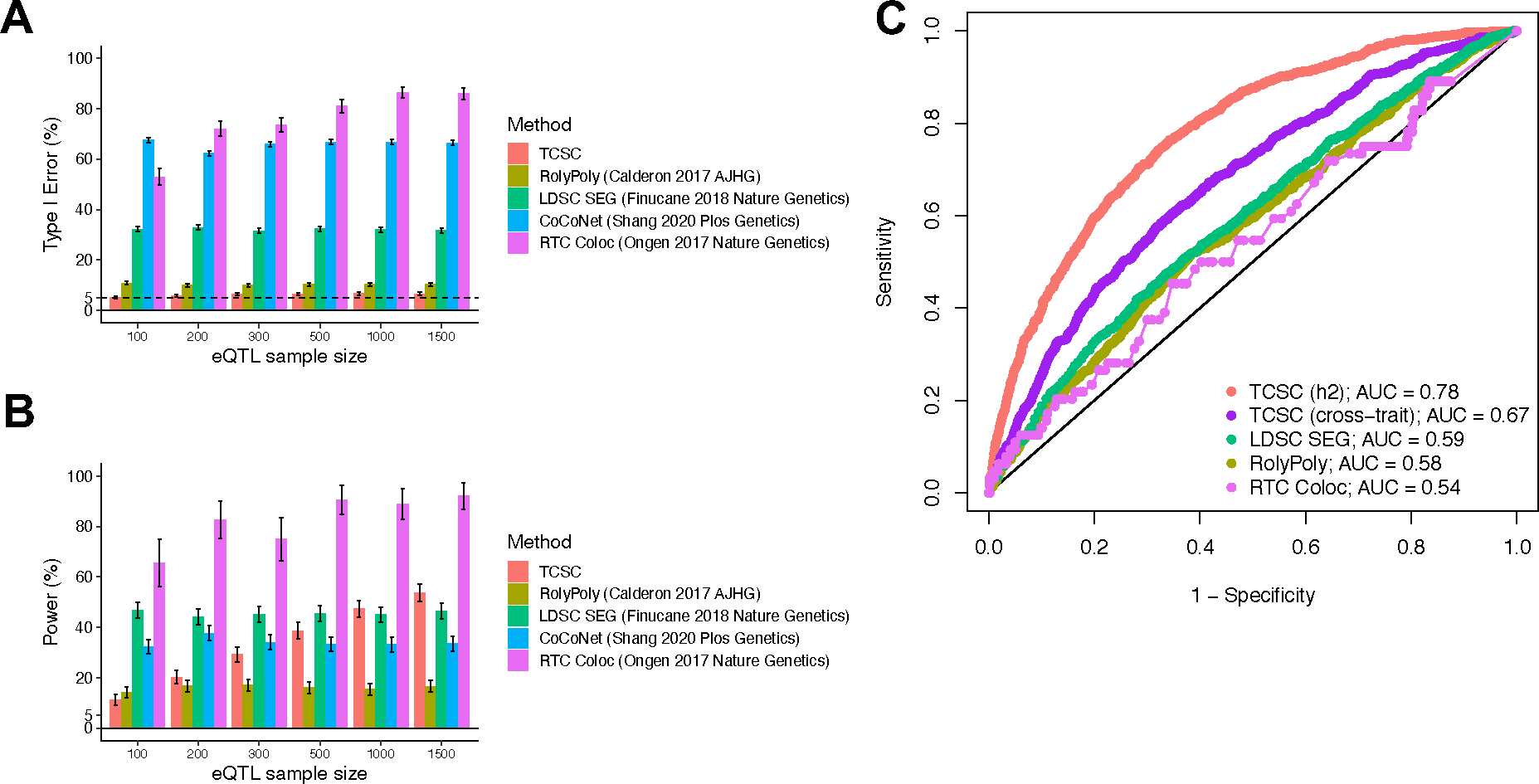
(A) Percentage of estimates of for non-causal tissues that were significantly positive at p < 0.05. is set to 10% and GWAS sample size is set to 10,000. (B) Percentage of estimates of for causal tissues that were significantly positive at p < 0.05. In panels A and B, we performed n = 1,000 independent simulated genetic architectures for TCSC, LDSC-SEG, CoCoNet, and RolyPoly and n = 100 independent simulated genetic architectures for RTC Coloc due to the computationally intensive nature of the method across each eQTL sample size (n = 100, 200, 300, 500, 1000, 1500); we used a one-sided z-test and the genomic block jackknife standard error to obtain p-values and data are presented as mean values +/− 1.96 × SEM. (C) Receiver operating characteristic (ROC) curves for each method, including cross-trait TCSC, across 1,000 uniformly spaced p-values between 0 and 1 used as the threshold to identify a causal tissue at a simulation eQTL sample size of 300, most closely matching real data analysis. We note that CoCoNet cannot be compared here because the method identifies the single most likely causal tissue using maximum likelihood estimation rather than via p-value. Numerical results are provided in Supplementary Tables 1 and 2.
Extended Data Figure 2. Robustness and power of cross-trait TCSC in simulations.
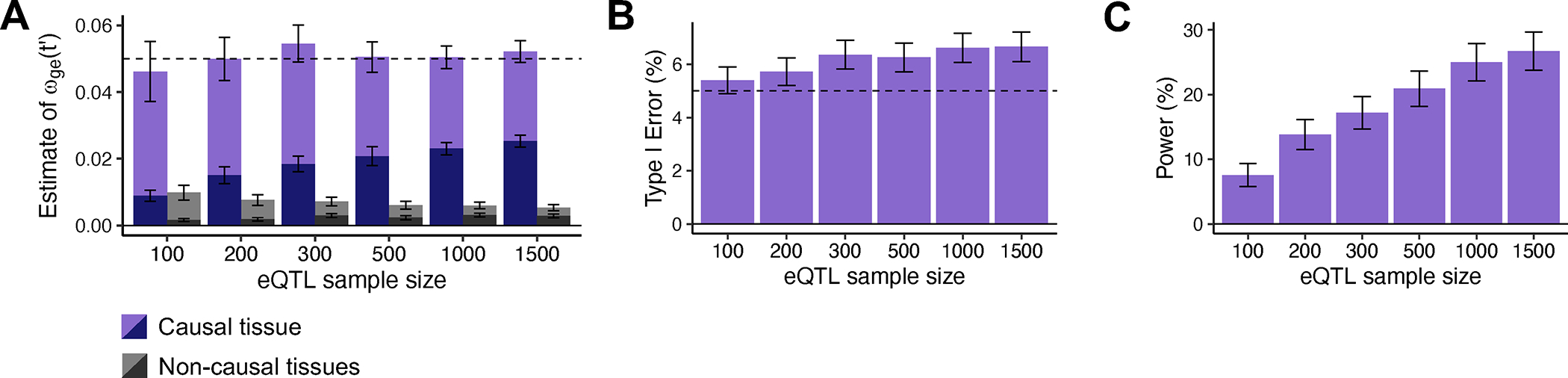
(A) Bias in estimates of genetic covariance explained by the cis-genetic component of gene expression in tissue ) for causal (purples) and non-causal (grays) tissues, across n = 1,000 independent simulations per eQTL sample size (n = 100, 200, 300, 500, 1000, 1500). Light purple (resp. gray) indicates that was set to the total number of true cis-heritable genes across tissues, dark purple (resp. gray) indicates that was set to the number of significantly cis-heritable genes detected in each tissue. The dashed line indicates the true value of for causal tissues. (B) Percentage of estimates of for non-causal tissues that were significantly positive at p < 0.05. (C) Percentage of estimates of for causal tissues that were significantly positive at p < 0.05. For panels B and C, we performed n = 1,000 independent simulations per eQTL sample size (n = 100, 200, 300, 500, 1000, 1500) and used a one-sided z-test to obtain p-values. In all panels, data are presented as mean values +/− 1.96 × SEM. Numerical results are reported in Supplementary Table 3.
Extended Data Figure 3. Robustness and power of TCSC regression in simulations with different values of
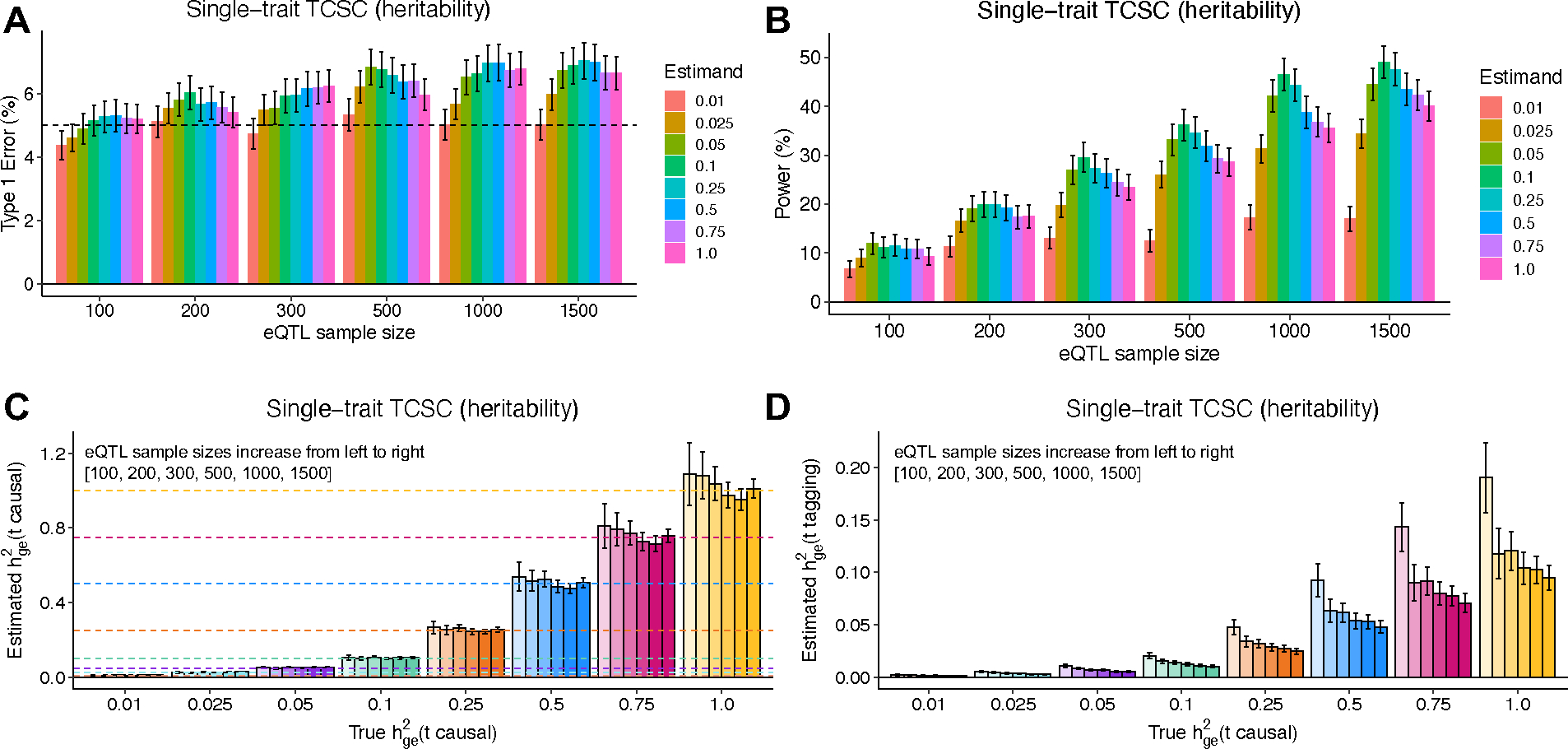
(A) Type I error per true value of in the causal tissue. False positive event is defined as for non-causal tissues at p < 0.05. (B) Power to detect the causal tissue per true value of in the causal tissue. A true positive event is defined as > 0 for causal tissues at p < 0.05. (C) Bias on causal estimates of for different true values of the causal tissue . Dashed lines indicate true values of . (D) Bias on non-causal estimates of for different true values of the causal tissue . In all panels, we performed n = 1,000 independent simulated genetic architectures across different eQTL sample sizes (n = 100, 200, 300, 500, 1000, 1500); we used a one-sided z-test and the genomic block jackknife standard error to obtain p-values and data are presented as mean values +/− 1.96 × SEM. The value of is set to the total number of unique cis-heritable genes across all tissues.
Extended Data Figure 4. Robustness and power of cross-trait TCSC regression in simulations with different values of .
(A) Type I error per true value of in the causal tissue. False positive event is defined as for non-causal tissues at p < 0.05. (B) Power to detect the causal tissue per true value of in the causal tissue. A true positive event is defined as for causal tissues at p < 0.05. (C) Bias on causal estimates of for different true values of the causal tissue . Dashed lines indicate true values of . (D) Bias on non-causal estimates of for different true values of the causal tissue . In all panels, we performed n = 1,000 independent simulated genetic architectures across different eQTL sample sizes (n = 100, 200, 300, 500, 1000, 1500); we used a one-sided z-test and the genomic block jackknife standard error to obtain p-values and data are presented as mean values +/− 1.96 × SEM. The value of is set to the total number of unique cis-heritable genes across all tissues.
Extended Data Figure 5. Robustness and power of TCSC regression in simulations with different numbers of non-causal tissues.
(A) Type I error involving a variable number of non-causal tissues in the presence of a single causal tissue. False positive event defined as > 0 for non-causal tissues at p < 0.05. Note, when there are 0 tagging tissues, there is no measurement of type I error. (B) Power to detect the causal tissue in which for causal tissues at p < 0.05. (C) Bias on estimates of for the causal tissue, while changing the number of non-causal tissues in the model. The dashed line indicates that the true value of . (D) Bias on estimates of for non-causal tissues, while changing the number of non-causal tissues in the model. In all panels, we performed n = 1,000 independent simulated genetic architectures across different eQTL sample sizes (n = 100, 200, 300, 500, 1000, 1500); we used a one-sided z-test and the genomic block jackknife standard error to obtain p-values and data are presented as mean values +/− 1.96 × SEM. The value of is set to the total number of unique cis-heritable genes across all tissues.
Extended Data Figure 6. Robustness and power of cross-trait TCSC regression in simulations with different numbers of non-causal tissues.
(A) Type I error involving a variable number of non-causal tissues in the presence of a single causal tissue. False positive event defined as > 0 for non-causal tissues at p < 0.05. Note, when there are 0 tagging tissues, there is no measurement of type I error. (B) Power to detect the causal tissue in which for causal tissues at p < 0.05. (C) Bias on estimates of for the causal tissue, while changing the number of non-causal tissues in the model. The dashed line indicates that the true value of . (D) Bias on estimates of for non-causal tissues, while changing the number of non-causal tissues in the model. In all panels, we performed n = 1,000 independent simulated genetic architectures across different eQTL sample sizes (n = 100, 200, 300, 500, 1000, 1500); we used a one-sided z-test and the genomic block jackknife standard error to obtain p-values and data are presented as mean values +/− 1.96 × SEM. The value of is set to the total number of unique cis-heritable genes across all tissues.
Extended Data Figure 7. Tissue-specific contributions to disease and complex trait heritability in secondary analysis of 23 tissues, removing tissues with small eQTL sample size.
We report estimates of the proportion of disease heritability explained by the cis-genetic component of gene expression in tissue ). Results are shown for tissue-trait pairs with FDR <= 10%; full boxes denote FDR of 5% or lower and partial boxes denote FDR between 5% and 10%. Tissues are ordered alphabetically. Color corresponds to , the proportion of common variant heritability causally explained by predicted gene expression in tissue . These results are largely consistent with the analysis of 39 GTEx tissues (Figure 4). WHRadjBMI: waist-hip-ratio adjusted for body mass index. HDL: high-density lipoprotein. DBP: diastolic blood pressure. BMI: body mass index. FEV1/FVC: forced expiratory volume in one second divided by forced vital capacity. Cereb. Cortex Ar.: cerebral cortex surface area. WBC Count: white blood cell count. RBC Count: red blood cell count. MDD: major depressive disorder. Daggers next to a tissue indicate a meta-tissue.
Extended Data Figure 8. Tissue-specific contributions to disease and complex trait heritability in brain-specific analysis.
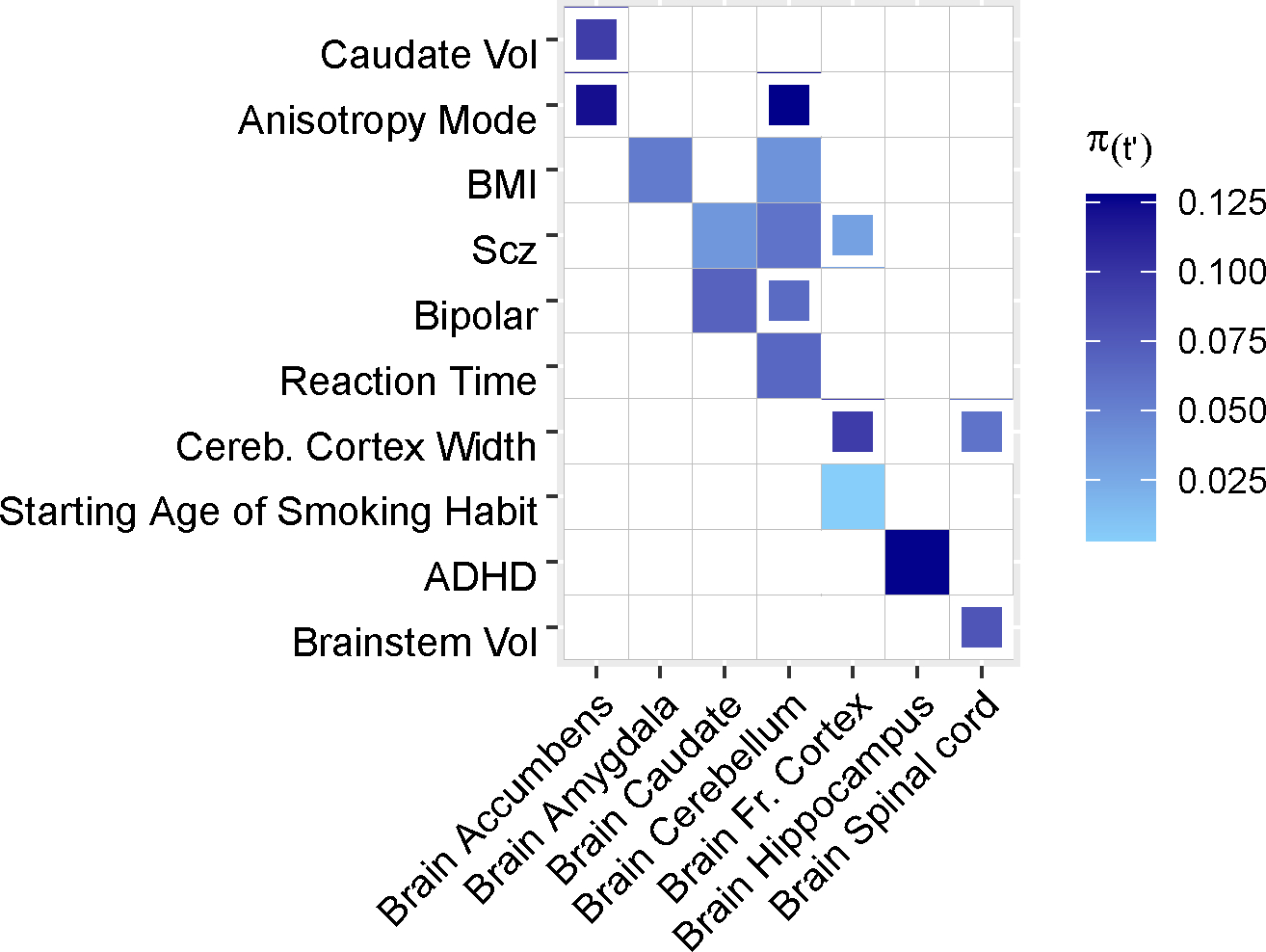
We separately analyzed results for 41 brain-related diseases and complex traits and 13 brain tissues. Results are shown for tissue-trait pairs with FDR <= 10%; full boxes denote FDR of 5% or lower and partial boxes denote FDR between 5% and 10%. Tissues are ordered alphabetically. Each tissue has an eQTL sample size ranging from 101 to 189 individuals. Color corresponds to , the proportion of common variant heritability causally explained by predicted gene expression in tissue . Caudate Vol: caudate volume. BMI: body mass index. Scz: schizophrenia. Bipolar: bipolar disorder. Brainstem Vol: brainstem volume. Cereb. Cortex Width: cerebral cortex width. ADHD: attention-deficit/hyperactivity disorder. Brainstem Vol: brainstem volume.
Extended Data Figure 9. Comparison of disease-critical tissues identified by RTC Coloc, LDSC-SEG and TCSC for all 17 disease/traits with causal tissue-trait associations identified by TCSC.
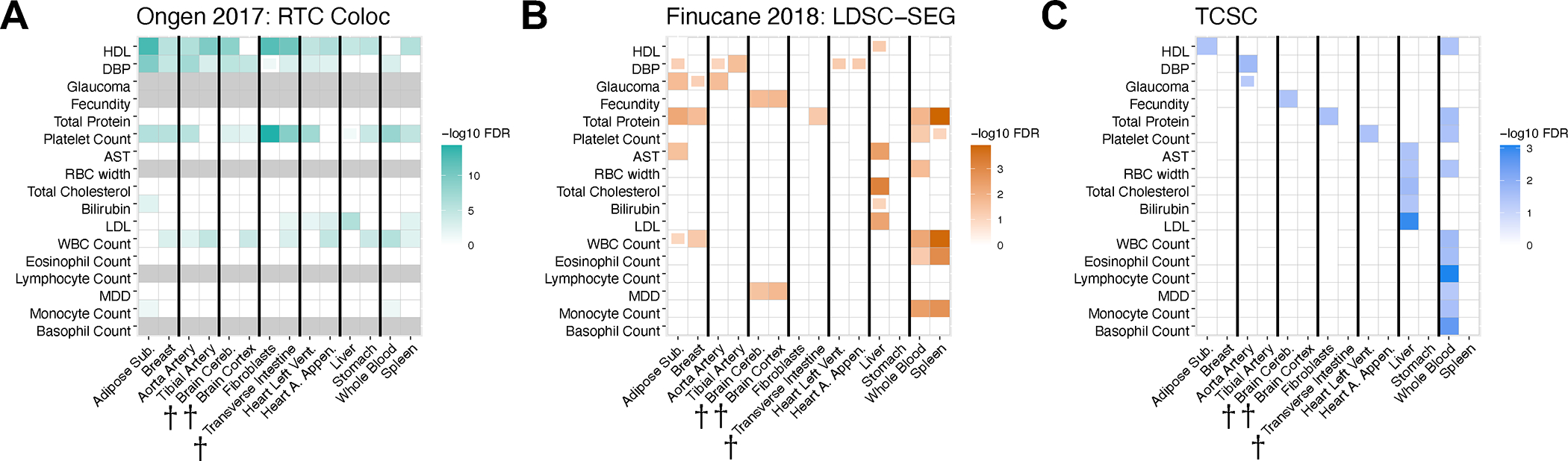
FDR significance of trait-tissue associations across three different methods for each of 17 traits with a significant tissue found by TCSC from Fig. 3 and 7 causal tissues, plus each tissue’s most highly genetically correlated GTEx tissue. Full boxes denote FDR of 5% or lower and partial boxes denote FDR between 5% and 10%. Thicker black lines separate the causal tissue found in the primary TCSC analysis (left) from its most highly genetically correlated GTEx tissue (right), with two exceptions. First, breast tissue was the most highly genetically correlated tissue for two causal tissues, adipose subcutaneous and thyroid; therefore, these three tissues appear as a trio. Second, the aorta artery and tibial artery are each other’s most highly genetically correlated tissue and both are a causal tissue different traits by TCSC. (A) RTC Coloc (Ongen 2017 Nat Genet), (B) LDSC-SEG (Finucane 2018 Nat Genet), (C) TCSC. Per-trait FDR in panels A and C, FDR across traits and tissues in panel B. WHRadjBMI: waist-hip-ratio conditional on body mass index. HDL: high-density lipoprotein. DBP: diastolic blood pressure. BMI: body mass index. FEV1/FVC: forced expiratory volume in one second divided by forced vital capacity. Cereb. Cortex Ar.: cerebral cortex surface area. AST: aspartate aminotransferase. LDL: low-density lipoprotein. WBC: white blood cell count. MDD: major depressive disorder. Daggers next to a tissue indicate a meta-tissue. For BMI, fecundity, and cereb. cortex ar., LDSC-SEG brain-specific analysis results are used.
Extended Data Figure 10. Comparison of disease-critical tissues identified by RTC Coloc, LDSC-SEG and TCSC for brain-specific analysis.
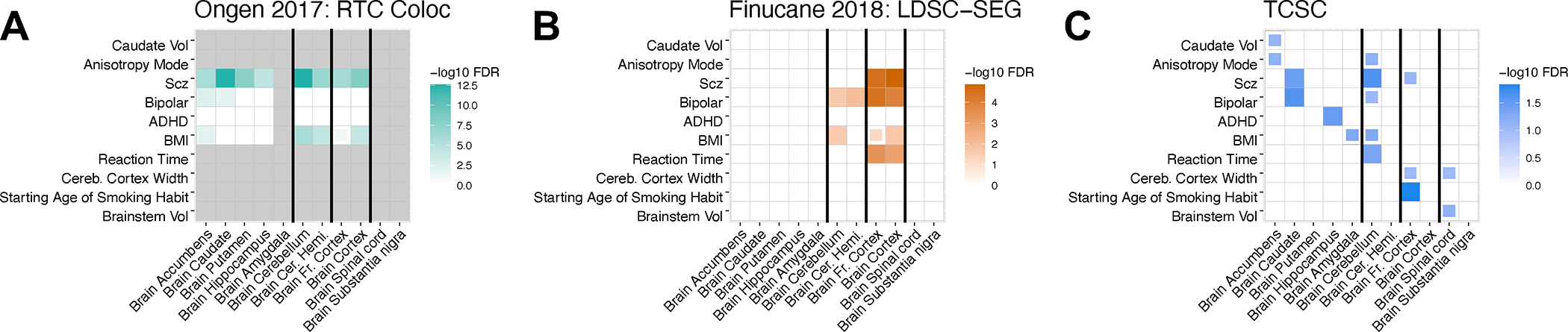
Brain-specific tissue-trait pairs identified by three methods: RTC Coloc, LDSC-SEG, and TCSC. Full boxes denote FDR of 5% or lower and partial boxes denote FDR between 5% and 10%. Each tissue has an eQTL sample size ranging from 100 to 189 individuals. Additionally, we include each tissue’s most highly genetically correlated GTEx tissue. Thicker black lines separate the causal tissue found in the primary TCSC analysis (left) from its most highly genetically correlated GTEx tissue (right), with one exception: brain accumbens, caudate, putamen, hippocampus, and amygdala are all highly genetically correlated, with no pairs of exclusively high genetic correlation. Caudate Vol: caudate volume. BMI: body mass index. Scz: schizophrenia. Bipolar: bipolar disorder. Brainstem Vol: brainstem volume. Cereb. Cortex Width: cerebral cortex width. ADHD: attention-deficit/hyperactivity disorder. Brainstem Vol: brainstem volume.
Supplementary Material
Acknowledgements
We thank Huwenbo Shi, Martin Zhang, and Benjamin Strober for helpful discussions. This work was funded by NIH grants U01 HG009379, R01 MH101244, R37 MH107649, R01 HG006399, R01 MH115676 and U01 HG012009 awarded to A.L.P. The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Footnotes
Competing interests
The authors declare no competing interests.
Code Availability
TCSC software including a quick start tutorial: https://github.com/TiffanyAmariuta/TCSC/ (DOI: 10.5281/zenodo.8030594)73
Mancuso Lab TWAS Simulator: https://github.com/mancusolab/twas_sim.
FUSION software: http://gusevlab.org/projects/fusion/.
Simulation code for RTC Coloc: https://github.com/TiffanyAmariuta/TCSC/tree/main/simulation_analysis.
Data Availability
We have made 78 GWAS summary statistics and 41 brain-specific summary statistics publicly available at https://github.com/TiffanyAmariuta/TCSC/tree/main/sumstats (DOI: 10.5281/zenodo.8030594)73, gene expression prediction models publicly available at https://alkesgroup.broadinstitute.org/TCSC/GeneExpressionModels/, TWAS association statistics publicly available at https://alkesgroup.broadinstitute.org/TCSC/TWAS_sumstats/, tissue co-regulation scores publicly available at https://github.com/TiffanyAmariuta/TCSC/tree/main/coregulation_scores, and TCSC output publicly available at https://github.com/TiffanyAmariuta/TCSC/tree/main/results. Gene expression and genotype data were acquired from the GTEx v8 eQTL dataset (dbGaP Accession phs000424.v8.p2) and 1000 Genomes phase 3 data were downloaded from https://data.broadinstitute.org/alkesgroup/FUSION/LDREF.tar.bz2.
References
- 1.Hekselman I & Yeger-Lotem E Mechanisms of tissue and cell-type specificity in heritable traits and diseases. Nat Rev Genet 21, 137–150 (2020). [DOI] [PubMed] [Google Scholar]
- 2.Ongen H et al. Estimating the causal tissues for complex traits and diseases. Nat Genet 49, 1676–1683 (2017). [DOI] [PubMed] [Google Scholar]
- 3.Gamazon ER et al. Using an atlas of gene regulation across 44 human tissues to inform complex disease- and trait-associated variation. Nat Genet 50, 956–967 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hormozdiari F et al. Leveraging molecular quantitative trait loci to understand the genetic architecture of diseases and complex traits. Nat Genet 50, 1041–1047 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arvanitis M, Tayeb K, Strober BJ & Battle A Redefining tissue specificity of genetic regulation of gene expression in the presence of allelic heterogeneity. Am J Hum Genet 109, 223–239 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Calderon D et al. Inferring Relevant Cell Types for Complex Traits by Using Single-Cell Gene Expression. Am J Hum Genet 101, 686–699 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Finucane HK et al. Heritability enrichment of specifically expressed genes identifies disease-relevant tissues and cell types. Nat Genet 50, 621–629 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bryois J et al. Genetic identification of cell types underlying brain complex traits yields insights into the etiology of Parkinson’s disease. Nat Genet 52, 482–493 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shang L, Smith JA & Zhou X Leveraging gene co-expression patterns to infer trait-relevant tissues in genome-wide association studies. PLoS Genet 16, e1008734 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maurano MT et al. Systematic localization of common disease-associated variation in regulatory DNA. Science 337, 1190–5 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Trynka G et al. Chromatin marks identify critical cell types for fine mapping complex trait variants. Nat Genet 45, 124–30 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pickrell JK Joint analysis of functional genomic data and genome-wide association studies of 18 human traits. Am J Hum Genet 94, 559–73 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Finucane HK et al. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat Genet 47, 1228–35 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roadmap Epigenomics C et al. Integrative analysis of 111 reference human epigenomes. Nature 518, 317–30 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Backenroth D et al. FUN-LDA: A Latent Dirichlet Allocation Model for Predicting Tissue-Specific Functional Effects of Noncoding Variation: Methods and Applications. Am J Hum Genet 102, 920–942 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Amariuta T et al. IMPACT: Genomic Annotation of Cell-State-Specific Regulatory Elements Inferred from the Epigenome of Bound Transcription Factors. Am J Hum Genet 104, 879–895 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boix CA, James BT, Park YP, Meuleman W & Kellis M Regulatory genomic circuitry of human disease loci by integrative epigenomics. Nature 590, 300–307 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wainberg M et al. Opportunities and challenges for transcriptome-wide association studies. Nat Genet 51, 592–599 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Consortium GT The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 369, 1318–1330 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gamazon ER et al. A gene-based association method for mapping traits using reference transcriptome data. Nat Genet 47, 1091–8 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gusev A et al. Integrative approaches for large-scale transcriptome-wide association studies. Nat Genet 48, 245–52 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Siewert-Rocks KM, Kim SS, Yao DW, Shi H & Price AL Leveraging gene co-regulation to identify gene sets enriched for disease heritability. Am J Hum Genet 109, 393–404 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schaid DJ, Chen W & Larson NB From genome-wide associations to candidate causal variants by statistical fine-mapping. Nat Rev Genet 19, 491–504 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhu H, Shang L & Zhou X A Review of Statistical Methods for Identifying Trait-Relevant Tissues and Cell Types. Front Genet 11, 587887 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bulik-Sullivan BK et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat Genet 47, 291–5 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mancuso N et al. Probabilistic fine-mapping of transcriptome-wide association studies. Nat Genet 51, 675–682 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Genomes Project C et al. An integrated map of genetic variation from 1,092 human genomes. Nature 491, 56–65 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gazal S et al. Linkage disequilibrium-dependent architecture of human complex traits shows action of negative selection. Nat Genet 49, 1421–1427 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gazal S, Marquez-Luna C, Finucane HK & Price AL Reconciling S-LDSC and LDAK functional enrichment estimates. Nat Genet 51, 1202–1204 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Homan TD, Bordes S & Cichowski E Physiology, Pulse Pressure. in StatPearls; (Treasure Island (FL), 2022). [PubMed] [Google Scholar]
- 31.Kass MA et al. The Ocular Hypertension Treatment Study: a randomized trial determines that topical ocular hypotensive medication delays or prevents the onset of primary open-angle glaucoma. Arch Ophthalmol 120, 701–13; discussion 829–30 (2002). [DOI] [PubMed] [Google Scholar]
- 32.Zhao D, Cho J, Kim MH & Guallar E The association of blood pressure and primary open-angle glaucoma: a meta-analysis. Am J Ophthalmol 158, 615–27 e9 (2014). [DOI] [PubMed] [Google Scholar]
- 33.Levine RM, Yang A, Brahma V & Martone JF Management of Blood Pressure in Patients with Glaucoma. Curr Cardiol Rep 19, 109 (2017). [DOI] [PubMed] [Google Scholar]
- 34.De Moraes CG, Cioffi GA, Weinreb RN & Liebmann JM New Recommendations for the Treatment of Systemic Hypertension and their Potential Implications for Glaucoma Management. J Glaucoma 27, 567–571 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Leeman M & Kestelyn P Glaucoma and Blood Pressure. Hypertension 73, 944–950 (2019). [DOI] [PubMed] [Google Scholar]
- 36.Gregg D & Goldschmidt-Clermont PJ Cardiology patient page. Platelets and cardiovascular disease. Circulation 108, e88–90 (2003). [DOI] [PubMed] [Google Scholar]
- 37.Coppinger JA et al. Characterization of the proteins released from activated platelets leads to localization of novel platelet proteins in human atherosclerotic lesions. Blood 103, 2096–104 (2004). [DOI] [PubMed] [Google Scholar]
- 38.Gawaz M, Langer H & May AE Platelets in inflammation and atherogenesis. J Clin Invest 115, 3378–84 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Davi G & Patrono C Platelet activation and atherothrombosis. N Engl J Med 357, 2482–94 (2007). [DOI] [PubMed] [Google Scholar]
- 40.Meadows TA & Bhatt DL Clinical aspects of platelet inhibitors and thrombus formation. Circ Res 100, 1261–75 (2007). [DOI] [PubMed] [Google Scholar]
- 41.Berman MN, Tupper C & Bhardwaj A Physiology, Left Ventricular Function. in StatPearls; (Treasure Island (FL), 2022). [PubMed] [Google Scholar]
- 42.Chung S, Sawyer JK, Gebre AK, Maeda N & Parks JS Adipose tissue ATP binding cassette transporter A1 contributes to high-density lipoprotein biogenesis in vivo. Circulation 124, 1663–72 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McGillicuddy FC, Reilly MP & Rader DJ Adipose modulation of high-density lipoprotein cholesterol: implications for obesity, high-density lipoprotein metabolism, and cardiovascular disease. Circulation 124, 1602–5 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zoccali C et al. Adiponectin, metabolic risk factors, and cardiovascular events among patients with end-stage renal disease. J Am Soc Nephrol 13, 134–141 (2002). [DOI] [PubMed] [Google Scholar]
- 45.Ryo M et al. Adiponectin as a biomarker of the metabolic syndrome. Circ J 68, 975–81 (2004). [DOI] [PubMed] [Google Scholar]
- 46.Toth PP Adiponectin and high-density lipoprotein: a metabolic association through thick and thin. Eur Heart J 26, 1579–81 (2005). [DOI] [PubMed] [Google Scholar]
- 47.Van Linthout S et al. Impact of HDL on adipose tissue metabolism and adiponectin expression. Atherosclerosis 210, 438–44 (2010). [DOI] [PubMed] [Google Scholar]
- 48.Shungin D et al. New genetic loci link adipose and insulin biology to body fat distribution. Nature 518, 187–196 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Emdin CA et al. Genetic Association of Waist-to-Hip Ratio With Cardiometabolic Traits, Type 2 Diabetes, and Coronary Heart Disease. JAMA 317, 626–634 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Smith J, Al-Amri M, Sniderman A & Cianflone K Leptin and adiponectin in relation to body fat percentage, waist to hip ratio and the apoB/apoA1 ratio in Asian Indian and Caucasian men and women. Nutr Metab (Lond) 3, 18 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Farooqi IS Defining the neural basis of appetite and obesity: from genes to behaviour. Clin Med (Lond) 14, 286–9 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Locke AE et al. Genetic studies of body mass index yield new insights for obesity biology. Nature 518, 197–206 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Medic N et al. Increased body mass index is associated with specific regional alterations in brain structure. Int J Obes (Lond) 40, 1177–82 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yengo L et al. Meta-analysis of genome-wide association studies for height and body mass index in approximately 700000 individuals of European ancestry. Hum Mol Genet 27, 3641–3649 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Onat F & Cavdar S Cerebellar connections: hypothalamus. Cerebellum 2, 263–9 (2003). [DOI] [PubMed] [Google Scholar]
- 56.Zhu JN & Wang JJ The cerebellum in feeding control: possible function and mechanism. Cell Mol Neurobiol 28, 469–78 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Low AYT et al. Reverse-translational identification of a cerebellar satiation network. Nature 600, 269–273 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Floresco SB, Todd CL & Grace AA Glutamatergic afferents from the hippocampus to the nucleus accumbens regulate activity of ventral tegmental area dopamine neurons. J Neurosci 21, 4915–22 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Enard W et al. A humanized version of Foxp2 affects cortico-basal ganglia circuits in mice. Cell 137, 961–71 (2009). [DOI] [PubMed] [Google Scholar]
- 60.Demontis D et al. Discovery of the first genome-wide significant risk loci for attention deficit/hyperactivity disorder. Nat Genet 51, 63–75 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bulik-Sullivan B et al. An atlas of genetic correlations across human diseases and traits. Nat Genet 47, 1236–41 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wei HS et al. Erythrocytes Are Oxygen-Sensing Regulators of the Cerebral Microcirculation. Neuron 91, 851–862 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Olusi SO Obesity is an independent risk factor for plasma lipid peroxidation and depletion of erythrocyte cytoprotectic enzymes in humans. Int J Obes Relat Metab Disord 26, 1159–64 (2002). [DOI] [PubMed] [Google Scholar]
- 64.Ozata M et al. Increased oxidative stress and hypozincemia in male obesity. Clin Biochem 35, 627–31 (2002). [DOI] [PubMed] [Google Scholar]
- 65.Druml W, Laggner AN, Lenz K, Grimm G & Schneeweiss B Pancreatitis in acute hemolysis. Ann Hematol 63, 39–41 (1991). [DOI] [PubMed] [Google Scholar]
- 66.Sakai NS, Taylor SA & Chouhan MD Obesity, metabolic disease and the pancreas-Quantitative imaging of pancreatic fat. Br J Radiol 91, 20180267 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Peterson CG, Skoog V & Venge P Human eosinophil cationic proteins (ECP and EPX) and their suppressive effects on lymphocyte proliferation. Immunobiology 171, 1–13 (1986). [DOI] [PubMed] [Google Scholar]
- 68.Nakagome K et al. IL-5-induced hypereosinophilia suppresses the antigen-induced immune response via a TGF-beta-dependent mechanism. J Immunol 179, 284–94 (2007). [DOI] [PubMed] [Google Scholar]
- 69.Onyema OO et al. Eosinophils downregulate lung alloimmunity by decreasing TCR signal transduction. JCI Insight 4(2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yao DW, O’Connor LJ, Price AL & Gusev A Quantifying genetic effects on disease mediated by assayed gene expression levels. Nat Genet 52, 626–633 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Weissbrod O et al. Leveraging fine-mapping and multipopulation training data to improve cross-population polygenic risk scores. Nat Genet 54, 450–458 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Stegle O, Parts L, Durbin R & Winn J A Bayesian framework to account for complex non-genetic factors in gene expression levels greatly increases power in eQTL studies. PLoS Comput Biol 6, e1000770 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Amariuta T TCSC GitHub Repository. (2023). [Google Scholar]
- 74.Bycroft C et al. The UK Biobank resource with deep phenotyping and genomic data. Nature 562, 203–209 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gazal S et al. Functional architecture of low-frequency variants highlights strength of negative selection across coding and non-coding annotations. Nat Genet 50, 1600–1607 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yang J, Lee SH, Goddard ME & Visscher PM GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet 88, 76–82 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Reshef YA et al. Detecting genome-wide directional effects of transcription factor binding on polygenic disease risk. Nat Genet 50, 1483–1493 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhang MJ et al. Polygenic enrichment distinguishes disease associations of individual cells in single-cell RNA-seq data. Nat Genet 54, 1572–1580 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
We have made 78 GWAS summary statistics and 41 brain-specific summary statistics publicly available at https://github.com/TiffanyAmariuta/TCSC/tree/main/sumstats (DOI: 10.5281/zenodo.8030594)73, gene expression prediction models publicly available at https://alkesgroup.broadinstitute.org/TCSC/GeneExpressionModels/, TWAS association statistics publicly available at https://alkesgroup.broadinstitute.org/TCSC/TWAS_sumstats/, tissue co-regulation scores publicly available at https://github.com/TiffanyAmariuta/TCSC/tree/main/coregulation_scores, and TCSC output publicly available at https://github.com/TiffanyAmariuta/TCSC/tree/main/results. Gene expression and genotype data were acquired from the GTEx v8 eQTL dataset (dbGaP Accession phs000424.v8.p2) and 1000 Genomes phase 3 data were downloaded from https://data.broadinstitute.org/alkesgroup/FUSION/LDREF.tar.bz2.