

Abstract
Diastolic heart failure (DHF), in which impaired ventricular filling leads to typical heart failure symptoms, represents over 50% of all heart failure cases and is linked with risk factors, including metabolic syndrome, hypertension, diabetes, and aging. A substantial proportion of patients with this disorder maintain normal left ventricular systolic function, as assessed by ejection fraction. Despite the high prevalence of DHF, no effective therapeutic agents are available to treat this condition, partially because the molecular mechanisms of diastolic dysfunction remain poorly understood. As such, by focusing on the underlying molecular and cellular processes contributing to DHF can yield new insights that can represent an exciting new avenue and propose a novel therapeutic approach for DHF treatment. This review discusses new developments from basic and clinical/translational research to highlight current knowledge gaps, help define molecular determinants of diastolic dysfunction, and clarify new targets for treatment.
Keywords: Cardiomyocyte, Diastolic dysfunction, Myofilament, Relaxation
Introduction
Heart failure (HF), a primary cause of mortality in the USA, is a chronic progressive disorder in which the heart is incapable of pumping (systolic) or filling (diastolic) sufficiently. HF occurs due to either systolic or diastolic dysfunction, and many patients have a considerable overlap of both systolic and diastolic dysfunction. Greater than 50% of heart failure patients have impaired relaxation or predominant abnormalities in diastolic function, with relatively normal or minor depression in systolic ventricular performance; this condition is identified as diastolic heart failure (DHF) [1, 2].
DHF is a clinically distinct syndrome from systolic heart failure (SHF) with specific morphologic and functional changes [3]. Patients with SHF exhibit a distinctive metabolic profile, which becomes further pronounced when coupled with comorbidities like diabetes and kidney dysfunction. DHF is linked to elevated inflammation and oxidative stress markers, compromised lipid metabolism, heightened collagen production, and a reduction in nitric oxide signaling [4]. DHF occurs when the ventricle is unable to accommodate sufficient blood volume to maintain appropriate stroke volume during diastole despite normal diastolic pressure [5]. These abnormalities develop because of elevated ventricular stiffness and/or impaired ventricular relaxation, distinguished by decreased contraction velocity, lengthy relaxation, elevated filling pressure, and reduced cardiac output [6].
Although considerable progress has been made in managing and controlling systolic heart failure, the mechanisms contributing to DHF remain insufficiently investigated. Thus, DHF-specific treatments have lagged because of a gap in knowledge of the molecular and biochemical mechanisms leading to myocardial functional and structural modifications in this disorder. To address this knowledge gap, the current review discusses the pathophysiologic mechanisms causing diastolic dysfunction (DD) and the molecular mechanisms that control relaxation at the cardiomyocyte level. We specifically focus on mechanisms regulating cardiac relaxation which can promote DHF.
Pathophysiology of diastolic dysfunction
The cardiac cycle involves two phases: (1) diastole, in which the heart relaxes and refills with blood; and (2) systole, characterized by robust cardiac contraction [7]. One cardiac cycle at a regular heart rate of 75 beats per minute and under resting conditions lasts 0.8 s; systole occupies one third and diastole two thirds of the cardiac cycle duration. However, during intensive muscle work, the duration of diastole decreases much more and usually lasts for approximately half a cardiac cycle time at the regular resting heart rate [8]. In DHF, there is a change in the balance of left ventricular filling pressures, causing them to increase unevenly in proportion to the magnitude of LV dilation. This change can result in an inappropriately rapid heart rate (tachycardia), reduced ability of the ventricle to relax and fill during the diastolic phase of the cardiac cycle (ventricular diastolic compliance), and/or compromised relaxation of the ventricle itself [9].
Both structural and biochemical modifications in cardiomyocytes are responsible for impaired ventricular relaxation. In addition, compliance and impaired relaxation can be caused by extra-cardiomyocyte factors in the extracellular matrix surrounding the cardiomyocytes and hormones regulating both structure and function [9]. Most often, DD is linked to cardiac hypertrophy and stiffening of ventricular tissue, which can cause the inability of the heart muscle fibers to return to regular size and inadequate ventricular filling volume under low pressure [10].
Changes in any of the intrinsic characteristics of cardiomyocytes, such as calcium homeostasis, myofilament function, cardiac metabolism, and cytoskeletal structure, can cause irregularities in both active relaxation and passive stiffness. Although impaired filling can be caused by anatomical abnormalities that delay filling, the current review will focus on decreased ventricular compliance resulting from cytoskeletal dysfunction. The comprehensive studies have adequately addressed other crucial elements of DHF, including cardiac metabolism [11], the renin–angiotensin axis [12], mitochondrial function [13], and animal research [14]. Within the subsequent sections, we will delve into several pivotal processes associated with cardiac relaxation.
Reduced intracellular calcium
Any process that interferes with calcium removal from the cytosol or cross-bridge detachment can potentially delay cardiac relaxation [7]. Depolarization of the plasma membrane is the initial phase of the cardiac cycle that opens L-type voltage-gated, dihydropyridine-sensitive sarcolemmal calcium channels known as dihydropyridine receptors to the influx of calcium into the myocyte [15]. The process involves the release of calcium from the sarcoplasmic reticulum (SR) through a channel called the cardiac ryanodine receptor (RyR2), facilitated in part by the close proximity between sarcolemmal calcium channels and RyR2 (as shown in Fig. 1) [15]. The sarcoplasmic reticulum handles calcium transportation within the cell and maintains the calcium concentration during the contraction–relaxation cycle. This process is crucial in regulating the contraction and relaxation of cardiac muscle [16–18]. Also, this relationship is supported by a mouse model study, which demonstrated that amplifying the presence of the α1-subunit of the L-type calcium channel (α1CTG) leads to dilated cardiomyopathy. This condition exhibits systolic and diastolic heart failure symptoms by the time the mice reach 4 months of age [19].
Fig. 1.

a Hierarchical scheme of cardiac structures. Bundles of myofibrils group together to form heart tissue. A bundle of myofibrils wrapped by sarcolemma and composed of the repeats of sarcomeres. SR establishes a mesh-like structure to cover the myofibers. Invagination of the sarcolemma, known as the T-tubule, is located transversely to the sarcomere and forms close contact with the SR. Mitochondria located next to the myofibers along the t-tubules and SR. b Intracellular Ca2+ cycling in cardiomyocytes. After depolarizing the sarcolemma, a small quantity of Ca2+ enters the sarcoplasm via the DHPR of the T-tubule, activating a rapid and large influx of Ca2+ release from inside SR via the RyR2. NCX is the main mechanism by which Ca2+ is released from cells. Activation of the SERCA2 pump regulated by its binding partner PLN promotes the reuptake of cytosolic Ca2+ into the SR. Ca+2, calcium; DHPR, dihydropyridine receptor; K, potassium; Na/K, Na+/K+-pump membrane receptor; NCX, sodium–calcium exchanger; PLN, phospholamban; SERCA-2, sarco/endoplasmic reticulum Ca2+-ATPase; SR, sarcoplasmic reticulum
Increased concentration of intracellular calcium serves as the internal signal initiating the activation of the contractile response within sarcomeres [20]. Consequently, the elevation in calcium levels promptly instigates the process of calcium removal, leading to the subsequent deactivation of the contractile machinery [21]. Calcium removal from the cardiomyocyte cytosol is essential to initiate relaxation [10]. This process is regulated by the action of a calcium pump (SERCA2) and the sarcolemmal sodium–calcium exchanger [9]. SERCA2 is a significant regulator of cardiac relaxation since it largely determines the removal rate of more than 70% of cytosolic Ca2+ in human cardiomyocytes [22].
Consequently, SERCA2 activity reduction is linked to impaired relaxation and LV hypertrophy in HF due to decreased gene expression and phosphorylation of its repressive modulatory protein phospholamban [23, 24]. In addition, a significant decline in SERCA2 expression in hearts causes immediate severe myocardial systolic and DD and death from HF. For example, Serca2null mutant mice die in utero, which shows the importance of a significant reduction in SERCA2 for cardiac function in the adult mouse (Table 1) [25, 26].
Table 1.
Animal models of myofilament mutations causing diastolic heart failure
| Protein | Gene | Protein Name | Sarcomere component | Animal model | Reference |
|---|---|---|---|---|---|
| ATPase Sarco/endoplasmic reticulum Ca2+ transporting 2 | ATP2A2 | SERCA2 | sarco(endo)plasmic reticulum | SERCA2 whole body (WB) knock out (KO) & cardiomyocyte-specific excision (CMs) | [25, 26] |
| Myosin heavy chain | MYH6 | α-MyHC | Thick filament (Atria) | WB Heterozygous aMHC403/+ mice (strain 129/BS) | [37] |
| MYH7 | β-MyHC (myosin mutation R723G) | Thick filament (Cardiac ventricles; slow-twitch skeletal muscle) | mutant β-myosin heavy chain-glutamic acid403 transgenic rabbit model of human HCM | [40] | |
| Myosin-binding protein C | MYBPC33 | cMyBP-C | Thick filament | CMs MyBP-C(-/-, Ex3-10) | [84] |
| Myosin light essential chain | MYL3 | MLC1SB | Thick filament | Cardio-specific Tg-Δ43 mice | [42] |
| Myosin light chain 2, regulatory, cardiac, slow | MYL2 | MLC-2A | Thick filament | Transgenic Lys104Glu-RLC-cardiomycocyte-specific | [41] |
| Actin | ACTC | α-cardiac actin | Thin filament | Transgenic mice model mutation ACTC E99K | [43] |
| Troponin complex and tropomuosin | TNNT2 | Troponin T | Thin filament | CMs Transgenic rats with mutated human cTnT | [54] |
| TNNI3 | Troponin I | Thin filament | CMs Tnl KO mice | [51, 52] | |
| TPM1 | α-Tropomyosin | Thin filament | CMs FHC 180 α-TM trangenic mice46 | [56] | |
| Titin | TTN | Titin | Thick filament/Z-Disc | CMs Murince Mode (TtnΔlAjxn) | [87] |
| Myosin light chain kinase 3 | MYLK3 | cMLCK/MLCK | Phosphoryl of cardiac myosin heavy chains | WB knockout (Mylk3 wild/-) | [65] |
| Myosin light chain phosphatase | PP1CB | PP1 catalytic subunit beta | Thin filament | Not available | - |
| PPP1R12C | PP 1 regulatory subunit | Regulates the catalytic activity | Not available | - | |
| Muscle LIM protein | CSRP3 | cysteine and glycine rich protein 3 | Z-Disc | csrp3 knockout zebrafish | [91, 92] |
| Telethonin | TCAP | titin-cap | Z-Disc | Telethonin-deficient mice | [93] |
| Myozenin 2 | MYOZ2 | myozenin 2 | Z-Disc | WB Mutant MYOZ2-P48 mouse | [95] |
| Vinculin | VCL | vinculin | Intercalated disc | CMs Vinculin-Δln20/21 Mouse: | [98] |
As mentioned above, Ca2+ cycling alterations have been shown in the last two decades that are closely linked to heart failure. In that case, Ca2+ overload during diastole via RyR2 can compromise sarcoplasmic reticulum Ca2+ storage capacity, impairing systolic contractility and possibly diastolic cardiac function. There is much to discuss regarding the clinical consequences of “leaky RyR2” and possible therapeutic strategies to correct RyR2 dysfunction [27]; however, the current review focuses on the role of myofilaments.
Role of myofilament proteins in diastolic dysfunction
Regulation of myofilament function plays a pivotal role in overseeing the extent and rate of cardiac relaxation. While the leading phase in relaxation begins with a reduction in intracellular Ca2+ concentration to initiate relaxation, ventricular relaxation is predominantly regulated by the biophysical properties of the myofilament proteins. These proteins include actin, myosin heavy chain, troponins, tropomyosin, and regulatory enzymes such as myosin light chain kinase (MLCK) and myosin light chain phosphatase (MLCP). A discussion of the contribution of each protein is included below.
Actin and myosin
To accomplish rapid and efficient contraction, cardiomyocytes are arrayed as a tubular assembly composed of chains of myofibrils (Fig. 1). The myofibrils consist of repeating segments of sarcomeres, an important contractile unit. Sarcomeres are made up of thick (myosin) and thin filaments (actin) [28, 29]. Myosin is composed of two globular myosin heads (S1), a myosin neck region (S2), and a tail or rod section [30] (Fig. 2). A tail domain consists of two heavy chains twisted together to shape a long coiled-coil α-helical tail domain. However, two round myosin heads develop from the N terminus, which is attached to the neck with two essential light chains (ELC) and the regulatory light chain (RLC) (Fig. 2) [28]. Thin filaments contain three main proteins: actin monomer, two strings of tropomyosin molecules, and troponin complex (Fig. 3).
Fig. 2.

Schematic illustration of the cardiac β-myosin heavy chain (β-MHC). β-MHC contains two heavy chains (tail), an essential light chain (ELC), a regulatory light chain (RLC), a myosin neck chain (S2), and a head domain (S1). The two light chains of myosin are located near the myosin head and facilitate calcium-dependent force transduction by the myosin head domain. In addition, myosin ATPase can cause hydrolysis ATP to provide energy for actomyosin contraction
Fig. 3.

A schematic representation of contraction and relaxation with the interaction between troponin and other thin filament components. CaM, Calmodulin; cMyBP-C, cardiac myosin binding protein-C; CPI-17, C-kinase-potentiated protein phosphatase 1 inhibitor of 17 kDa; MLCK, myosin light-chain kinase; MLCP, myosin light-chain phosphatase; PKA, protein kinase A; TnI, troponin I; TnC, troponin C; TnT, troponin T; Tm, tropomyosin
Muscle contraction involves the interaction between the thick filament of myosin heads and the thin filament of actin subunits. However, relaxation occurs when molecular switches block this interaction on these filaments [31]. The sliding of myosin head on actin monomers produces the formation of “cross-bridges,” which causes heart contraction and generation of force [32, 33].
Actin–myosin interaction and force generation are fundamental to the pathophysiology of HF. Usually, the β-myosin isoform is expressed mainly in the ventricles, while the atria mostly express the α-isoform in the human heart [34]. Alterations in myofilament isoforms themselves can also delay relaxation. An illustration of this concept is comparing the α- and β-myosin heavy chain (MHC) isoforms, where the α-isoform is associated with a prolonged increase in force during contraction and relaxation phases [35]. However, this modification is observed mainly in larger mammals and fewer humans because the β-MHC isoform dominates under normal conditions.
Mutations in genes encoding myosin and actin are linked to various types of cardiomyopathies. Specific mutations in myosin can directly affect its interaction with actin. Among the most common causes of cardiomyopathy are mutations in myosin. A summary of known mutations in both actin and myosin is provided in Table 1 [36]. For instance, a mutation in the α-myosin heavy chain (α-MHC) at position 403 (arginine to glutamine) is associated with familial hypertrophic cardiomyopathy (FHC). In mice with the same missense mutation (αMHC403/+) as in humans, a model of this genetic disease shows diastolic dysfunction similar to human FHC [37].
Mutations in the β-cardiac myosin heavy chain (β-MyHC) gene (MYH7) can lead to ventricular wall hypertrophy and changes in diastolic filling volumes. For example, a mutation in the myosin heavy chain, R723G (MyHC723), reduces calcium sensitivity in cardiomyocytes. Calcium sensitivity, crucial for contractile function, is regulated independently from the actin regulatory protein complex [38, 39]. In addition, a transgenic rabbit model of mutant β-myosin heavy chain-glutamic acid at amino acid 403 also reduced myocardial contraction and relaxation [40].
Mutations in the Regulatory Light Chain (RLC) can impair ventricular relaxation, affecting indicators of left ventricular (LV) function like the E/A ratio (a measure of diastolic filling). This mutation can lead to energy inefficiency due to decreased contractile force and quicker ATP consumption [41]. The Myosin Essential Light Chain (ELC), encoded by the MYL3 gene, is essential for cardiac contraction. Transgenic mice expressing a mutation in this gene (Tg-D43) exhibit pathological cardiac hypertrophy [42].
Song et al. discovered that a mutation in the cardiac actin gene (Actin Alpha Cardiac Muscle 1, ACTC) in a transgenic mouse led to dilated cardiomyopathy. This condition was characterized by increased end-systolic capacity, end-diastolic pressure, and extended relaxation and contraction rates [43]. Over time, these symptoms could progress to apical hypertrophic cardiomyopathy, marked by heightened myofibrillar calcium sensitivity responsible for apical hypertrophy. This progression further leads to heart failure’s eventual development in mice and humans [43].
Troponin complex and tropomyosin
The troponin complex regulates myofilament response to Ca2+ and cardiac muscle contraction. The hetero-trimeric troponin complex comprises three regulatory proteins: cardiac troponin T (cTnT), troponin I (cTnI), and troponin C (cTnC). Together with tropomyosin 1 (TPM1), a contractile protein, they are positioned on the actin filament [44]. cTnI, commonly used as a marker for myocardial damage, plays a crucial role in regulating cardiac function. cTnI is a protein that helps regulate the interaction between actin and myosin in the heart. It acts as an inhibitor to prevent the binding of myosin to actin, thereby stopping the formation of cross-bridges and allowing for the relaxation of the heart muscle [45].
In systole and at higher Ca2+ concentrations, cTnI goes through major changes both in conformation and position to be disconnected from actin–tropomyosin activation of the thin filament, allowing for a strong cross-bridge binding with myosin [46], linked to the force generation and elevated rate of ATP hydrolysis (Fig. 3). However, in diastole and lower Ca2+ concentrations, cross-bridge bindings are weakened or blocked from interacting with actin without generating force. cTnI plays a significant role in regulating the actin and tropomyosin complex on the actin filament. It attaches to tropomyosin and actin, forming a complex that helps regulate muscle contraction. The binding of cTnI to tropomyosin stabilizes the position of tropomyosin on the actin filament, preventing myosin from binding to actin and inhibiting muscle contraction. This interaction ensures proper muscle contraction and relaxation control, allowing the heart to function effectively [47, 48].
Troponin mutations have been shown to cause dilated cardiomyopathy and diastolic dysfunction in transgenic mice. The deficiency of cTnI, or modifications in cTnI, in pathological conditions, especially in the C terminus of cTnI, is connected to DD triggered by myofibril hypersensitivity to Ca2+ [49].
Mutations of cTnI in the region of PKA-targeted phosphorylation sites are associated with HF in which force-frequency modulation is lessened and afterload relaxation sensitivity rises in connection with reduced PKA TnI phosphorylation (Table 1) [50]. However, when cTnI is overexpressed or constitutively phosphorylated, it can impair heart relaxation [51]. Much of this mechanism is still unknown. Huang et al. conducted a study using a cardiac troponin I (cTnI) gene knockout mouse model created by targeting murine embryonic stem cells [52]. This model aimed to investigate the effects of troponin I deficiency on cardiac function and survival. The study results showed that mice lacking cardiac troponin I were born healthy with normal heart and body weight, as a fetal troponin I isoform compensated for the absence of cardiac troponin I. However, this compensation was temporary, and 15 days after birth, the expression of the compensatory isoform began to decline. This decline led to a troponin I deficiency, which resulted in a lethal form of acute heart failure. The mice died on day 18, demonstrating that the troponin I is necessary for normal cardiac function and survival [52]. A similar effect was observed in which mutations and cTnI deficiency caused DD and restrictive cardiomyopathies [53]. In addition, a human cTnT obliteration in transgenic rats exerts and mimics the phenotype of FHC with DD and arrhythmias [54]. These genetic modifications are distinguished by relatively mild and sometimes clinical hypertrophy but a high incidence of sudden death. Therefore, genetic testing may be necessary for this group [55].
In addition, mutations in ɑ-tropomyosin, an essential sarcomere component, regulate muscle contraction through calcium-mediated activation and can severely disrupt sarcomeric function (Table 1). Physiological analyses of mice with ɑ-tropomyosin mutation reveal functional differences in diastolic performance and increased calcium sensitivity, which can severely disrupt sarcomeric function. This mutation can cause ventricular hypertrophy, atrial enlargement, fibrosis, and culminating death in 5 months [56]. Despite the lethality of this mutation, there are no suggested treatments to modulate this activity.
Cardiac myosin light-chain kinase
Myosin light-chain kinase (MYLK or MLCK) is a segment of a Ca2+/calmodulin (CaM)-dependent protein kinases group. It is a serine/threonine-specific protein kinase that regulates the light chain of myosin II phosphorylation [57]. Four distinct versions of the MYLK gene exist, each producing a specific MYLK isoform: MYLK1, MYLK2, MYLK3, and MYLK4 [58]. MLCK3 and MLCK4 are expressed in cardiac muscle based on their muscle type, known explicitly as cMLCK [59]. cMLCK is required for normal RLC phosphorylation and physiological cardiac operation [59]. cMLCK is essential in cardiomyocyte contraction [60] and is involved in efficiently coupling energy sources and force development [61]. Contraction is initiated with an influx of Ca2+ into the cardiac muscles from the sarcoplasmic reticulum (SR) and the extracellular space and binding to calmodulin [62]. Once Ca2+ levels are elevated, Ca2+ binds to calmodulin (CaM), which then Ca2+/CaM causes a conformation change in MLCK and activation (Fig. 3). Activated MLCK can increase the regulatory RLC phosphorylation of serine residue 19. Phosphorylated myosin and ADP and inorganic phosphate (Pi) can create connections called cross-bridges with actin. Releasing ADP and Pi triggers a power stroke that triggers muscle contraction [63]. This force leads the thin filament to glide past the thick filament and compresses the sarcomeres. Consequently, MLC phosphorylation lets myosin cross-bridges bind to the actin and enable contraction to initiate. Finally, the ATP binds to myosin and then hydrolyzes, which can release myosin from actin and repeat the process (Fig. 3).
Recent studies identified that cMLCK is associated with familial dilated cardiomyopathy. Heterozygous Mylk3 knockout mice indicate a mild decrease in cardiac contractility by 4 months of age. These mice partially resemble humans with the heterozygous MYLK3 mutation, but the diminution in cardiac contractility was slighter [64]. Also, the outcomes on Tg-D166V mice recommended that a mutation-induced reserve of RLC phosphorylation might cause the development of cardiomyopathy phenotype [65].
Reduction of intracellular calcium concentration inactivates MLCK but does not stop muscle contraction since the MLC has been physically adjusted through phosphorylation and not through ATPase activity. Therefore, to halt muscle contraction, these changes need to be reversed. Dephosphorylation of the MLC and following termination of muscle contraction happens through the action of a second enzyme identified as myosin light-chain phosphatase (MLCP).
Cardiac myosin light-chain phosphatase
Cardiac myosin light-chain phosphatase (cMLCP) is a serine (Ser)-threonine (Thr) phosphatase that is accountable for the dephosphorylation of the RLC and so regulates relaxation in myofilament [66]. The typical structure of MLCP is a holoenzyme with three following subunits: (1) a catalytic phosphoprotein phosphatase (PP1c) type-I Ser/Thr-phosphatase PP1c (37 kDa), (2) a sizeable regulatory subunit called myosin phosphatase target subunit (MYPT) or myosin-binding subunit (MBS), in the range of from 115 kDa in MYPT1 to 58 kDa to in MYPT3, and (3) a small accessory subunit of 20 kDa called M20 that is tightly bound MYPT with unknown function [67]. MYPT plays a strategic role in regulating the functional and physical integrity of the trimeric MLCP holoenzyme (Fig. 4).
Fig. 4.

Subunit structure of cardiac heart myosin target phosphatase. KVKF motif, M20, 20 kDa small regulatory subunit; PP1cβ binding site; P, phosphorylation site; PPP1R12C, protein phosphatase 1 regulatory subunit 12C; PPPP1Cβ; protein phosphatase 1 catalytic subunit beta
The catalytic subunits of type 1 phosphatase (PP1C) are outcomes of three genes α (PPP1CA), β/δ (PPP1CB), and γ (PPP1CC). Of these isoforms, MYPT1 and MBS85 attach specifically to PP1cβ/ [68]. A sizeable regulatory subunit that was recently re-classified as an RRR1R1 contains the outcomes of five different genes, including MYPT1 (PPP1R12A), MYPT2 (PPP1R12B), MBS85 (PPP1R12C), MYPT3 (PPP1R16A), and TIMAP (PPP1R16B). These regulatory subunits share several preserved domains, including multiple ankyrin repeat domains and an RVxF motif for PP1c binding to mediate protein–protein interactions [69]. MYPT1, the regulatory subunit of MLCP, has a role in the phosphatase activity in smooth muscles. MYPT2 and MBS85 (PPP1R12C), other myosin-targeting subunits of MLCP, are the critical phosphatase in skeletal muscles and cardiac tissues [69, 70] and are likely to subcellular structures such as myofilaments. Despite the critical role of cMLCP in relaxation, few genetic models are available to study the effect of this enzyme’s consequence mutation and depletion in DHF. MLCP activity may alter the phosphorylation rate independent of MLCK activity. Thus, the activation of MLCP may upgrade relaxation. On the other hand, MLCP inhibition may also contribute to a worsening of cardiomyocyte contraction by increasing MLC phosphorylation to induce hypercontractility. Since the MLCP regulation in cardiomyocytes is mediated mainly by PP1C and PPP1R12C subunits without any significant overlap with other MYPT families [71], these two proteins may be involved in cardiomyocyte relaxation and, in general, contribute to DHF. In addition, the DHF model of a high-fat diet (HFD) with nitroarginine, as an inhibitor of nitric oxide synthase, is associated with endothelial dysfunction and reduction in MYPT1, thus altering MLC2v phosphorylation [72].
Sarcomere lengthening
Another key modulator of cardiac relaxation is the direction and amplitude in which sarcomeric proteins move. Rapid post-systole relaxation is necessary to efficiently fill the left and right ventricles [66]. Conversely, the ventricular filling is impeded when relaxation occurs too slowly or incompletely, leading to clinical DHF [73].
Relaxation has been shown to accelerate significantly when a sarcomere length is lengthened at the end of the systole [70]. This is a Frank–Starling effect as it is a fundamental biophysical property of the myofilaments. Generally, an enlargement of the sarcomere may provide a slighter chance for the myosin head to bind to the actin-binding site compared to an isometric or shortening sarcomere based on an estimate of filament compliance on the requirement of fiber stiffness on sarcomere length [74, 75]. This phenomenon also explains why isovolumic relaxation is prolonged during early filling in patients with DD [76]. Once the myocardium relaxes and sarcomeres stretch, it may suggest a mechanical way to terminate other cross-bridge formations, regardless of a potentially activated thin filament [77]. Cardiac Myosin Binding Protein-C (cMyBP-C) (Fig. 3), a possible activator of the thin filament, is a regulatory protein positioned on a thick filament [78]. Mutations and modifications in the cardiac MyBP-C gene (MyBP-C-/- knockout mice) are associated with DHF [79]. Since MyBP-C has been indicated to adjust the cross-bridge cycling kinetics and could change contraction–relaxation coupling by the amount and direction of expansion, the exact overall sarcomere size can influence myocardial relaxation [80]. Consequently, mutations in cMyBP-C have the potential to lead to DD [78, 81, 82]. Individuals carrying cMyBP-C mutations might display diastolic dysfunction, characterized by decreased relaxation velocity in the heart muscle. Importantly, this effect is observed regardless of hypertrophy [83]. A cardiac-specific mouse model that targets exons 3–10 (cMyBP-C(-/-, Ex3-10)) exhibits DD with a more significant E/E′ ratio similar to human patients [84]. In addition, cMyBP-C phosphorylation inhibits cardiac dysfunction linked to aging. This study presented that aging is associated with reducing cMyBP-C phosphorylation and deteriorating cardiac dysfunction, and cMyBP-C phosphorylation can adjust diastolic function [85].
Moreover, genetic obliteration of the I-band–A-band junction (IAjxn) in titin protein encoded by TTN in cardiac-specific TtnΔIAjxn mice can cause HF with normal ejection fraction (HFNEF) showing higher filling pressures and lowered ventricular compliance [86]. Bull et al. showed that genetic removal of the I-band–A-band junction (IAjxn) in titin raises force on the spring region, leading to an HFNEF-like syndrome in the mouse [87]. In addition, the TtnΔIAjxn mouse model showed higher diastolic stiffness and lower exercise tolerance, similar to HFNEF symptoms observed in patients [88]. Titin is a massive protein in the thick filament that connects the Z and M lines in the sarcomere segment, causing force transmission to the Z line and friction in the I band section. Soetkamp et al. showed that increased titin phosphorylation at the junction of the Z-disk affects the myofilament structure and contractility performance and is associated with DD [89]. Also, higher PKCα activity is a crucial modulator of cardiomyocyte stiffening in diabetic hearts due to titin-based modifications [90].
In addition to titin, mutations in other sarcomere genes are also causative of human cardiomyopathies. Chang et al. showed that cysteine and glycine-rich protein 3 (CSRP3) play a role in cardiac stretch sensing. Genetic mutations in the CSRP3 gene can result in cardiomyopathies, paralleled to human patients with downregulation of CSRP3 showing HF [91, 92]. Telethonin whole-body knockout mice develop HF following biomechanical stress, owing at least in part to apoptosis of cardiomyocytes [93]. This effect may also affect human heart failure [94].
Ruggiero et al. demonstrated in a human study that calcineurin protein phosphatase 2B (PP2B) plays a pivotal role in the development of hypertrophic cardiomyopathy. Moreover, they highlighted the significance of mutations in myozenin 2 (MYOZ2) as calcineurin inhibitors, underscoring their contribution to the onset of cardiomyopathy. These findings shed crucial light on the disorder’s molecular mechanisms, enhancing our understanding of its pathogenesis and potentially guiding future therapeutic interventions [95].
Finally, Vinculin (Vcl), a membrane-associated protein expressed in intercalated disks, is a crucial structural part of forming costamere protein complexes. Vcl connects the actin to integrins on the cell surface of cardiomyocytes [96]. Vcl mutations have been linked with dilated and hypertrophic cardiomyopathies in human knockout, resulting in heart and brain defects during embryonic [97]. In addition, the knockout mice model of cardiomyocyte-specific Vcl (cVclKO) exhibited a significant reduction in membrane cortical stiffness because of the expanded lattice spacing, which might explain the systolic wall strains before the beginning of ventricular dysfunction [98].
In addition to the genetic model, other available surgical and metabolic mouse models of diastolic heart failure have already been addressed [99, 100]. Beyond the crucial role myofilaments play, other factors can also significantly contribute to the development of DHF, which will be discussed in the upcoming sections, shedding light on the intricate nature of DHF and its underlying mechanisms.
Extracellular matrix
Abnormal extracellular matrix (ECM) remodeling contributes to diastolic dysfunction and impaired ventricular relaxation [7]. Changes in the architecture, composition, and distribution of the ECM play a central role in the pathophysiology of DHF [101, 102]. The ECM forms a dynamic network of molecules and proteins that offer structural support to cardiomyocytes [103]. ECM composition and organization alterations contribute to the impaired relaxation and increased stiffness characteristic of the disorder. Excessive deposition of collagen and other matrix proteins can lead to fibrosis, compromising the compliance of the myocardium during diastole [104, 105]. Changes in ECM properties can also influence the behavior of resident cardiac cells, including fibroblasts, cardiomyocytes, and endothelial cells, further impacting cardiac structure and function [106, 107]. The complex relationship between ECM and DHF has valuable therapeutic implications. By targeting ECM pathways, novel strategies can be developed to restore cardiac compliance, reduce fibrosis, and enhance prognosis for those with DHF.
Collagen, a fundamental structural protein in the ECM of the heart, plays a critical role in maintaining the integrity and function of cardiac tissue. Its dynamic balance, referred to as collagen homeostasis, is pivotal for maintaining appropriate cardiac structure and function [108]. Recent research has revealed a compelling connection between collagen homeostasis and the development of DHF [108]. In cases of DHF, the relaxation phase of the heart’s rhythm is compromised, leading to heightened stiffness in the cardiac muscle. This stiffness is frequently attributed to modifications in the composition and distribution of collagen throughout the myocardium. Disruptions in collagen synthesis, degradation, and cross-linking processes can induce fibrotic alterations that impede cardiac flexibility and relaxation [109, 110]. Understanding the intricate interplay between collagen homeostasis and DHF presents promising opportunities for therapeutic interventions aimed at reinstating normal cardiac function and enhancing patient outcomes.
cGMP/PKG signaling
The cGMP/PKG signaling pathway has emerged as a significant player in DHF.
Numerous investigations have indicated that enhancing the pathways involving cyclic guanosine monophosphate (cGMP)-dependent protein kinase or protein kinase G (cGMP–PKG) holds significant potential as a target for enhancing diastolic function in patients with DHF [111]. The presence of inflammation disrupts the intricate local communication between endothelial cells and adjacent cardiomyocytes, significantly impacting the nitric oxide (NO)–cGMP–PKG pathway and NO production. This diminishing signaling pathway lays the groundwork for cardiomyocytes to undergo hypertrophy and elevate diastolic resting tension [112]. Delayed relaxation and increased diastolic stiffness as a predominant clinical feature of DHF have demonstrated positive responses to elevated PKG activity [113]. A recent study showed that the cardiac-specific deletion of STAT3 leads to the creation of a mouse model for DHF, mirroring clinical traits, partially achieved by impacting the cardiac levels of PKG [114]. Empagliflozin, by inhibiting the sodium-dependent glucose co-transporter 2, reduces inflammatory and oxidative stress, leading to an enhancement of the NO–sGC–cGMP pathway. This improvement results in heightened PKGIα activity, achieved by mitigating PKGIα oxidation and polymerization, ultimately leading to decreased pathological stiffness in cardiomyocytes [115].
On the other hand, nitrosative stress emerges as an overabundance of reactive nitrogen species (RNS), prominently including NO, a crucial signaling molecule renowned for its contributions to vasodilation and cardiac relaxation [116, 117]. Post-translational modifications of the sarcomeric protein, such as titin, are pivotal in enhancing cardiomyocyte stiffness, contributing significantly to diastolic dysfunction. Elevated nitrosative and oxidative stress, impaired NO availability, and suppressed cGMP and PKG signaling pathways initiate alterations in titin's structure and function. These changes, affecting titin isoform expression and phosphorylation patterns, lead to amplified cardiomyocyte stiffness [117]. Shifts in titin isoforms toward stiffer forms and disrupted phosphorylation patterns due to oxidative stress compromise titin’s compliance, collectively intensifying cardiomyocyte stiffness. This interplay between stress factors, signaling attenuation, and titin modifications highlights their complex role in shaping left ventricular diastolic stiffness, offering insights into diastolic dysfunction within the context of heart failure [103].
Nitrosative stress can also activate the unfolded protein response (UPR) within cells [118]. The UPR is a cellular signaling pathway activated in response to the accumulation of misfolded or unfolded proteins within the endoplasmic reticulum (ER), a cellular compartment responsible for protein folding and quality control [118]. Nitrosative stress can impact ER homeostasis and protein folding, leading to the induction of the UPR. Nitrosative stress can trigger the UPR through protein nitrosylation [119], ER disruption [120], calcium dysregulation [121], and induction of ER stress sensors [122]. Unraveling the intricate relationship between nitrosative stress, the unfolded protein response, and DHF holds promise for advancing the understanding of disease progression and identifying new therapeutic avenues to restore proper cardiac function and improve patient outcomes.
Diagnostic
As delineated above, DHF is the hemodynamic manifestation of impaired LV filling volume due to myocardium abnormalities identified in relaxation and stiffness of the impaired active ventricular relaxation due to cellular anomalies during diastole, producing a stiff ventricle, thereby increasing LV end-diastolic pressure. Based on a European Study Group guideline of diastolic HF, the concurrent presence of three following criteria can be considered for establishing a diagnosis of DHF: (1) indication of heart failure symptoms, (2) LV systolic function may be normal or slightly abnormal, and (3) LV relaxation function is abnormal due to diastolic stiffness, or filling diastolic capacity [123]. Based on this guideline, a patient who meets the following criteria is defined as a DHF (Fig. 5).
Fig. 5.
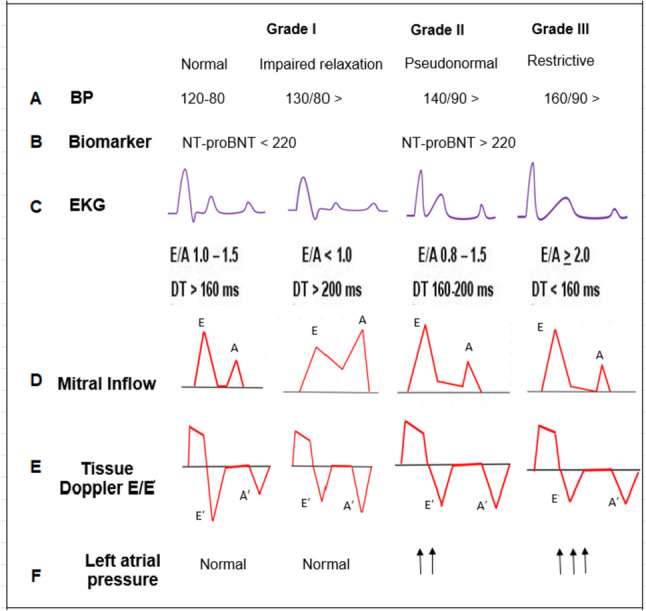
Characterization of diastolic dysfunction by echocardiography. BP, Blood pressure; EKG, electrocardiogram; TD, tissue Doppler; E, the velocity of blood flow across the mitral valve during early diastolic; A, velocity of blood flow across the mitral valve during atrial contraction; Aʹ, velocity of myocardial tissue relaxation during atrial contraction; Eʹ, the velocity of myocardial tissue relaxation during early diastole
DHF is also related to increased age and other cardiovascular risk factors, such as diabetes and high blood pressure (BP). BP is the most common risk factor and the fundamental precursor of HF [124]. The risk for progressing HF in hypertensive in comparison to normal BP individuals is about twofold in men and threefold in women [125]. In patients with HF with normal ejection fraction, guidelines recommend hypertension with a target BP of 130/80 mmHg [126].
Diagnostic evidence of DHF can be acquired by invasive measurement of LV pressure during cardiac catheterization by measuring the timing and rate of the cardiac cycle [127, 128]. Cardiac catheterization, or catheter insertion into a heart chamber, is the latest method for indicating the characteristics of DHF [129]. However, because of the risks and costs of invasive hemodynamic assessment, it is not practical to diagnose DHF [130]. On the other hand, non-invasive imaging methods provided by echocardiography, with less risk than heart catheterization, are beneficial for showing diastolic abnormalities by evaluating myocardial tissue motion, LV dimensions, and filling dynamics [131–133]. The mitral inflow velocities are the most familiar by measuring the E and A waves corresponding to the blood flow velocity during LV relaxation and atrial contraction, respectively [134]. In normal diastolic function, E wave surpasses A wave velocity. However, with Grade I (impaired relaxation), A will exceed E and E/A ratio < 1.0, as the atrial contraction is more responsible for ventricular filling, and the deceleration time of E > 200 ms (Fig. 5). Therefore, if the E/E′ ratio indicates diastolic dysfunction, extra non-invasive examinations are essential to diagnose the evidence of diastolic LV dysfunction [135]. Also, electrocardiographic evidence of atrial fibrillation or plasma levels of atrial natriuretic peptide (ANP) and brain natriuretic peptide (BNP) could be other options [136, 137]. However, echocardiography has limitations associated with poor quality images, fewer details in pixels of images, geometric assumptions, and highly depends on the operator’s skills to accurately assess and calculate diastolic function [102].
Cardiac MRI evaluations are better than echocardiography in accuracy for evaluating DHF, especially in patients with cardiovascular symptoms, including hypertrophic cardiomyopathy, hypertension, and congestive heart failure [138]. In addition, MRI gives several parameters that evaluate heart function and morphology, hemodynamic parameters, myocardial contractility, and tissue characterization [139]. Therefore, the recent developments in MRI technology are supposed to develop in wider clinical adoption combined with other clinical practice techniques.
Treatment
Although there have been notable improvements in managing DHF, the unfavorable outlook for patients underscores the need to investigate novel pathways for drug testing. These approaches should extend beyond conventional methods and instead concentrate on inventive techniques that address cardiomyocytes and myofilament function. Current treatment for DHF is based on reducing symptomatic using drugs such as β blockers, disopyramide, and nondihydropyridine Ca+2 channel blockers. Nevertheless, these nonspecific drugs are frequently deficient or not tolerated and do not target the underlying molecular mechanisms [140]. On the other hand, invasive surgical therapy can successfully benefit patients with drug resistance [140]. However, it can involve risks due to the nature of procedures and requires expertise that is not commonly available [141].
Finding a new therapeutic approach that targets the underlying mechanisms of DHF, such as gene therapy [142] or drugs that specifically target/inhibit specific pathways leading to DHF, would be invaluable in treating DHF. Mavacamten is an example of a recently available drug that suppresses cardiac myosin ATPase by diminishing actin-myosin crossbridge formation. As a result, it addresses the underlying pathophysiology of hypertrophic cardiomyopathy [119], decreasing contractility and enhancing myocardial energetics [143]. In addition, because Mavacamten is a cardiac myosin inhibitor, it improves ventricular compliance and favorably impacts diastolic function [140, 144].
Other recently suggested DHF treatments include sodium-glucose co-transporter 2 inhibitors (SGLT2is) and mineralocorticoid receptor antagonists. These medications have shown promise in managing DHF, although the precise mechanisms of action are not fully understood. SGLT2is, such as empagliflozin, can benefit DHF management by promoting euvolemic diuresis, improving hemodynamics, and stabilizing heart function. A randomized, double-blind clinical trial was orchestrated to investigate the hypothesis surrounding SGLT2 inhibitors and their potential to mitigate the risk of hospitalization involving a cohort of 5988 patients divided between the treatment and placebo groups. The empirical data revealed a noteworthy discrepancy. The total occurrences of heart failure-related hospitalizations were notably fewer in the empagliflozin-administered group (13.8%) compared to the placebo group (17.1%). These findings significantly enhanced the condition of patients treated with empagliflozin compared to those in the control (placebo) group [145, 146]. Also, another clinical trial showed a decrease in the combined risk of cardiovascular death or hospitalization for DHF patients [147].
Mineralocorticoid receptor antagonists, which counteract aldosterone’s effects on water and electrolyte balance, have remarkably reduced morbidity and mortality among patients with congestive heart failure and left ventricular dysfunction [148]. Recent research highlighted spironolactone’s positive impact on LV diastolic function and myocardial fibrosis regression [149]. In addition, in DHF patients, using mineralocorticoid receptor antagonists was associated with reduced heart failure hospitalizations [150, 151]. In addition, S-glutathionylation of cMyBP-C is an essential alteration in cross-bridge kinetics and, accordingly, the development of DD. Therefore, sphingosine-1-receptor modulator FTY720 can partially reverse established DD and reduce left atrial enlargement connected with decreased oxidative alteration of cMyBP-C; it may help DHF [152, 153]. Thus, understanding and targeting the underlying mechanism of DHF can help develop effective pharmacological therapy for DHF, which is a significant unmet need.
Conclusions
The management of DHF demands potential novel drugs beyond the existing treatments. Based on pre-clinical studies of DHF in transgenic models of myofilament disease, new approaches to intervention should focus on myofilament signaling, mitochondrial dysfunction, and myofilament structure/function. Deeper investigations will be essential to unveil the core mechanisms driving DHF, coupled with the development of animal models mirroring DHF symptoms. Such studies are key to identifying fresh therapeutic avenues targeting novel DHF treatment targets.
Abbreviations
- β-MHC
β-Myosin heavy chain
- BP
Blood pressure
- Ca2+
Intracellular calcium
- CaM
Calmodulin
- CPI-17
C-Kinase-potentiated protein phosphatase 1 inhibitor of 17 kDa
- DHF
Diastolic heart failure
- DD
Diastolic dysfunction
- DHPR
Dihydropyridine receptor
- ECG
Electrocardiogram
- ELC
Essential light chain
- FHC
Familial hypertrophic cardiomyopathy
- HF
Heart failure
- K+
Potassium
- MLCK
Myosin light-chain kinase
- MLCP
Myosin light-chain phosphatase
- Na/K
Na+/K+ ATPase pump
- NCX
Sodium–calcium exchanger
- PLN
Phospholamban
- RLC
Regulatory light chain
- RyR2
Ryanodine receptor 2
- SERCA2
Sarco/endoplasmic reticulum Ca2+-ATPase
- SR
Sarcoplasmic reticulum
- TD
Tissue Doppler
- TnI
Troponin I
- TnT
Troponin T
- TnC
Troponin C
- Tm
Tropomyosin
- T-tubule
Transverse-tubule
- SERCA-2
Sarco/endoplasmic reticulum Ca2+-ATPase
Author contribution
AA conceptualized the topic and idea and prepared the first draft and all figures. MM read and edited the first draft and finalized the manuscript with AA. Both authors approved the final version.
Funding
M.DM. is funded by NIH R01 HL151508 and VA Merit I01 BX004918.
Declarations
Conflict of interest
The authors declare no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Anahita Aboonabi, Email: anahita@uic.edu.
Mark D. McCauley, Email: mcaule1@uic.edu
References
- 1.Xu Y, Tian J, Huang X. Troponin mutation caused diastolic dysfunction and experimental treatment in transgenic mice with cardiomyopathy. GSTF J Adv Med Res. 2014;1(2):17. doi: 10.7603/s40782-014-0017-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Heidenreich PA, et al. 2022 AHA/ACC/HFSA Guideline for the Management of Heart Failure: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation. 2022;145(18):e895–e1032. doi: 10.1161/CIR.0000000000001063. [DOI] [PubMed] [Google Scholar]
- 3.Chatterjee K, Massie B. Systolic and diastolic heart failure: differences and similarities. J Card Fail. 2007;13(7):569–576. doi: 10.1016/j.cardfail.2007.04.006. [DOI] [PubMed] [Google Scholar]
- 4.Hage C, et al. Metabolomic profile in HFpEF vs HFrEF patients. J Cardiac Fail. 2020;26(12):1050–1059. doi: 10.1016/j.cardfail.2020.07.010. [DOI] [PubMed] [Google Scholar]
- 5.Wake R, Yoshikawa J, Yoshiyama M (2012) Diastolic heart failure. In: Echocardiography-in specific diseases. IntechOpen
- 6.Periasamy M, Janssen PM. Molecular basis of diastolic dysfunction. Heart Fail Clin. 2008;4(1):13–21. doi: 10.1016/j.hfc.2007.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kass DA, Bronzwaer JGF, Paulus WJ. What mechanisms underlie diastolic dysfunction in heart failure? Circ Res. 2004;94(12):1533–1542. doi: 10.1161/01.RES.0000129254.25507.d6. [DOI] [PubMed] [Google Scholar]
- 8.Mostafa SS et al (2011) Correlation of heart-rate and cardiac cycle duration under different body positions and breathing. In: International Conference on Advances in Electrical Engineering (ICAEE)
- 9.(2012) Diastolic dysfunction. In: Vincent J-L, Hall JB (eds) Encyclopedia of intensive care medicine. Berlin, Heidelberg, Springer Berlin Heidelberg, pp 718–718
- 10.Zile MR, Baicu CF, Bonnema DD. Diastolic heart failure: definitions and terminology. Prog Cardiovasc Dis. 2005;47(5):307–313. doi: 10.1016/j.pcad.2005.02.006. [DOI] [PubMed] [Google Scholar]
- 11.Abdellatif M, Sedej S, Kroemer G. NAD+ metabolism in cardiac health, aging, and disease. Circulation. 2021;144(22):1795–1817. doi: 10.1161/CIRCULATIONAHA.121.056589. [DOI] [PubMed] [Google Scholar]
- 12.Mori J, et al. Impact of the renin–angiotensin system on cardiac energy metabolism in heart failure. J Mol Cell Cardiol. 2013;63:98–106. doi: 10.1016/j.yjmcc.2013.07.010. [DOI] [PubMed] [Google Scholar]
- 13.Kumar AA, Kelly DP, Chirinos JA. Mitochondrial dysfunction in heart failure with preserved ejection fraction. Circulation. 2019;139(11):1435–1450. doi: 10.1161/CIRCULATIONAHA.118.036259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shah SJ, et al. Research priorities for heart failure with preserved ejection fraction: National Heart, Lung, and Blood Institute working group summary. Circulation. 2020;141(12):1001–1026. doi: 10.1161/CIRCULATIONAHA.119.041886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barry WH, Bridge JH. Intracellular calcium homeostasis in cardiac myocytes. Circulation. 1993;87(6):1806–1815. doi: 10.1161/01.CIR.87.6.1806. [DOI] [PubMed] [Google Scholar]
- 16.Fabiato A, Fabiato F. Calcium and cardiac excitation-contraction coupling. Annu Rev Physiol. 1979;41(1):473–484. doi: 10.1146/annurev.ph.41.030179.002353. [DOI] [PubMed] [Google Scholar]
- 17.Bers DM. Cardiac excitation–contraction coupling. Nature. 2002;415(6868):198–205. doi: 10.1038/415198a. [DOI] [PubMed] [Google Scholar]
- 18.Fabiato A. Calcium-induced release of calcium from the cardiac sarcoplasmic reticulum. Am J Physiol Cell Physiol. 1983;245(1):C1–C14. doi: 10.1152/ajpcell.1983.245.1.C1. [DOI] [PubMed] [Google Scholar]
- 19.Wang S, et al. Dilated cardiomyopathy with increased SR Ca2+ loading preceded by a hypercontractile state and diastolic failure in the α1CTG mouse. PLoS ONE. 2009;4(1):e4133. doi: 10.1371/journal.pone.0004133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cheng H, Lederer WJ, Cannell MB. Calcium sparks: elementary events underlying excitation-contraction coupling in heart muscle. Science. 1993;262(5134):740–744. doi: 10.1126/science.8235594. [DOI] [PubMed] [Google Scholar]
- 21.Hasenfuss G, Pieske B. Calcium cycling in congestive heart failure. J Mol Cell Cardiol. 2002;34(8):951–969. doi: 10.1006/jmcc.2002.2037. [DOI] [PubMed] [Google Scholar]
- 22.MacLennan DH, Kranias EG. Phospholamban: a crucial regulator of cardiac contractility. Nat Rev Mol Cell Biol. 2003;4(7):566–577. doi: 10.1038/nrm1151. [DOI] [PubMed] [Google Scholar]
- 23.Frank KF, et al. Modulation of SERCA: implications for the failing human heart. Basic Res Cardiol. 2002;97(1):I72–I78. doi: 10.1007/s003950200033. [DOI] [PubMed] [Google Scholar]
- 24.del Monte F, et al. Targeting phospholamban by gene transfer in human heart failure. Circulation. 2002;105(8):904–907. doi: 10.1161/hc0802.105564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Periasamy M, et al. Impaired cardiac performance in heterozygous mice with a null mutation in the sarco (endo) plasmic reticulum Ca2+-ATPase isoform 2 (SERCA2) gene. J Biol Chem. 1999;274(4):2556–2562. doi: 10.1074/jbc.274.4.2556. [DOI] [PubMed] [Google Scholar]
- 26.Andersson KB, et al. Moderate heart dysfunction in mice with inducible cardiomyocyte-specific excision of the Serca2 gene. J Mol Cell Cardiol. 2009;47(2):180–187. doi: 10.1016/j.yjmcc.2009.03.013. [DOI] [PubMed] [Google Scholar]
- 27.van Oort RJ, et al. Ryanodine receptor phosphorylation by CaMKII promotes life-threatening ventricular arrhythmias in mice with heart failure. Circulation. 2010;122(25):2669. doi: 10.1161/CIRCULATIONAHA.110.982298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Al-Khayat HA. Three-dimensional structure of the human myosin thick filament: clinical implications. Glob Cardiol Sci Pract. 2013;2013(3):280–302. doi: 10.5339/gcsp.2013.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kensler RW. The mammalian cardiac muscle thick filament: crossbridge arrangement. J Struct Biol. 2005;149(3):303–312. doi: 10.1016/j.jsb.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 30.Rayment I, et al. Three-dimensional structure of myosin subfragment-1: a molecular motor. Science. 1993;261(5117):50–58. doi: 10.1126/science.8316857. [DOI] [PubMed] [Google Scholar]
- 31.Alamo L, et al. Three-dimensional reconstruction of tarantula myosin filaments suggests how phosphorylation may regulate myosin activity. J Mol Biol. 2008;384(4):780–797. doi: 10.1016/j.jmb.2008.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Saxton A, Tariq MA, Bordoni B (2021) Anatomy, thorax, cardiac muscle. In: StatPearls [Internet]. StatPearls Publishing [PubMed]
- 33.Ripa R, George T, Sattar Y (2022) Physiology, cardiac muscle. In: StatPearls [Internet]. StatPearls Publishing [PubMed]
- 34.Weiss A, Schiaffino S, Leinwand LA. Comparative sequence analysis of the complete human sarcomeric myosin heavy chain family: implications for functional diversity. J Mol Biol. 1999;290(1):61–75. doi: 10.1006/jmbi.1999.2865. [DOI] [PubMed] [Google Scholar]
- 35.Fitzsimons DP, Patel JR, Moss RL. Role of myosin heavy chain composition in kinetics of force development and relaxation in rat myocardium. J Physiol. 1998;513(1):171–183. doi: 10.1111/j.1469-7793.1998.171by.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Volkmann N, et al. The R403Q myosin mutation implicated in familial hypertrophic cardiomyopathy causes disorder at the actomyosin interface. PLoS ONE. 2007;2(11):e1123. doi: 10.1371/journal.pone.0001123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Spindler M, et al. Diastolic dysfunction and altered energetics in the alphaMHC403/+ mouse model of familial hypertrophic cardiomyopathy. J Clin Investig. 1998;101(8):1775–1783. doi: 10.1172/JCI1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brenner B. Effect of Ca2+ on cross-bridge turnover kinetics in skinned single rabbit psoas fibers: implications for regulation of muscle contraction. Proc Natl Acad Sci. 1988;85(9):3265–3269. doi: 10.1073/pnas.85.9.3265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kraft T, et al. Familial hypertrophic cardiomyopathy: functional effects of myosin mutation R723G in cardiomyocytes. J Mol Cell Cardiol. 2013;57:13–22. doi: 10.1016/j.yjmcc.2013.01.001. [DOI] [PubMed] [Google Scholar]
- 40.Nagueh SF, et al. Tissue Doppler imaging consistently detects myocardial contraction and relaxation abnormalities, irrespective of cardiac hypertrophy, in a transgenic rabbit model of human hypertrophic cardiomyopathy. Circulation. 2000;102(12):1346–1350. doi: 10.1161/01.CIR.102.12.1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huang W, et al. Hypertrophic cardiomyopathy associated Lys104Glu mutation in the myosin regulatory light chain causes diastolic disturbance in mice. J Mol Cell Cardiol. 2014;74:318–329. doi: 10.1016/j.yjmcc.2014.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sitbon YH, et al. Ablation of the N terminus of cardiac essential light chain promotes the super-relaxed state of myosin and counteracts hypercontractility in hypertrophic cardiomyopathy mutant mice. FEBS J. 2020;287(18):3989–4004. doi: 10.1111/febs.15243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Song W, et al. Molecular mechanism of the E99K mutation in cardiac actin (ACTC gene) that causes apical hypertrophy in man and mouse. J Biol Chem. 2011;286(31):27582–27593. doi: 10.1074/jbc.M111.252320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Takeda S, et al. Structure of the core domain of human cardiac troponin in the Ca2+-saturated form. Nature. 2003;424(6944):35–41. doi: 10.1038/nature01780. [DOI] [PubMed] [Google Scholar]
- 45.Sharma S, Jackson PG, Makan J. Cardiac troponins. J Clin Pathol. 2004;57(10):1025–1026. doi: 10.1136/jcp.2003.015420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Solaro RJ, Rarick HM. Troponin and tropomyosin: proteins that switch on and tune in the activity of cardiac myofilaments. Circ Res. 1998;83(5):471–480. doi: 10.1161/01.RES.83.5.471. [DOI] [PubMed] [Google Scholar]
- 47.Farah CS, et al. Structural and regulatory functions of the NH2- and COOH-terminal regions of skeletal muscle troponin I. J Biol Chem. 1994;269(7):5230–5240. doi: 10.1016/S0021-9258(17)37679-2. [DOI] [PubMed] [Google Scholar]
- 48.Takeda S, et al. Structural and functional domains of the troponin complex revealed by limited digestion. Eur J Biochem. 1997;246(3):611–617. doi: 10.1111/j.1432-1033.1997.00611.x. [DOI] [PubMed] [Google Scholar]
- 49.Liu X, et al. Restrictive cardiomyopathy caused by troponin mutations: application of disease animal models in translational studies. Front Physiol. 2016;7:629. doi: 10.3389/fphys.2016.00629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wolska BM, et al. Expression of slow skeletal troponin I in adult transgenic mouse heart muscle reduces the force decline observed during acidic conditions. J Physiol. 2001;536(3):863–870. doi: 10.1111/j.1469-7793.2001.00863.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Takimoto E, et al. Frequency- and afterload-dependent cardiac modulation in vivo by troponin I with constitutively active protein kinase A phosphorylation sites. Circ Res. 2004;94(4):496–504. doi: 10.1161/01.RES.0000117307.57798.F5. [DOI] [PubMed] [Google Scholar]
- 52.Huang X, et al. Cardiac troponin I gene knockout: a mouse model of myocardial troponin I deficiency. Circ Res. 1999;84(1):1–8. doi: 10.1161/01.RES.84.1.1. [DOI] [PubMed] [Google Scholar]
- 53.Huang X-P, Du J-F. Troponin I, cardiac diastolic dysfunction and restrictive cardiomyopathy. Acta Pharmacol Sin. 2004;25:1569–1575. [PubMed] [Google Scholar]
- 54.Frey N et al (1998) Transgenic rats overexpressing a human cardiac troponin T deletion exhibit diastolic dysfunction in the "working heart model". In: Circulation. Lippincott Williams & Wilkins 227 East Washington Sq, Philadelphia, PA 19106 USA
- 55.Watkins H, et al. Mutations in the genes for cardiac troponin T and α-tropomyosin in hypertrophic cardiomyopathy. N Engl J Med. 1995;332(16):1058–1065. doi: 10.1056/NEJM199504203321603. [DOI] [PubMed] [Google Scholar]
- 56.Prabhakar R, et al. A familial hypertrophic cardiomyopathy α-tropomyosin mutation causes severe cardiac hypertrophy and death in mice. J Mol Cell Cardiol. 2001;33(10):1815–1828. doi: 10.1006/jmcc.2001.1445. [DOI] [PubMed] [Google Scholar]
- 57.Gao Y, et al. Myosin light chain kinase as a multifunctional regulatory protein of smooth muscle contraction. IUBMB Life. 2001;51(6):337–344. doi: 10.1080/152165401753366087. [DOI] [PubMed] [Google Scholar]
- 58.Manning G, et al. The protein kinase complement of the human genome. Science. 2002;298(5600):1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- 59.Chang AN, et al. Cardiac myosin light chain is phosphorylated by Ca2+/calmodulin-dependent and -independent kinase activities. Proc Natl Acad Sci. 2016;113(27):E3824–E3833. doi: 10.1073/pnas.1600633113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sheikh F, Lyon RC, Chen J. Getting the skinny on thick filament regulation in cardiac muscle biology and disease. Trends Cardiovasc Med. 2014;24(4):133–141. doi: 10.1016/j.tcm.2013.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shen Q, et al. Myosin light chain kinase in microvascular endothelial barrier function. Cardiovasc Res. 2010;87(2):272–280. doi: 10.1093/cvr/cvq144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Colbran R, et al. Calcium/calmodulin-dependent protein kinase II. Biochemical Journal. 1989;258(2):313. doi: 10.1042/bj2580313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kuo IY, Ehrlich BE. Signaling in muscle contraction. Cold Spring Harb Perspect Biol. 2015;7(2):a006023. doi: 10.1101/cshperspect.a006023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tougas CL, et al. Heterozygous Mylk3 knockout mice partially recapitulate human DCM with heterozygous MYLK3 mutations. Front Physiol. 2019;10:696. doi: 10.3389/fphys.2019.00696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yuan C-C, et al. Constitutive phosphorylation of cardiac myosin regulatory light chain prevents development of hypertrophic cardiomyopathy in mice. Proc Natl Acad Sci. 2015;112(30):E4138–E4146. doi: 10.1073/pnas.1505819112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schaub MC. Myosin light chain phosphatase. In: Enna SJ, Bylund DB, editors. xPharm: The Comprehensive Pharmacology Reference. New York: Elsevier; 2007. pp. 1–3. [Google Scholar]
- 67.Barford D, Das AK, Egloff MP (1998) The structure and mechanism of protein phosphatases: insights into catalysis and regulation. Annu Rev Biophys Biomol Struct 27 [DOI] [PubMed]
- 68.Peti W, Nairn AC, Page R. Structural basis for protein phosphatase 1 regulation and specificity. FEBS J. 2013;280(2):596–611. doi: 10.1111/j.1742-4658.2012.08509.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fujioka M, et al. A new isoform of human myosin phosphatase targeting/regulatory subunit (MYPT2): cDNA cloning, tissue expression, and chromosomal mapping. Genomics. 1998;49(1):59–68. doi: 10.1006/geno.1998.5222. [DOI] [PubMed] [Google Scholar]
- 70.Tan I, et al. Phosphorylation of a novel myosin binding subunit of protein phosphatase 1 reveals a conserved mechanism in the regulation of actin cytoskeleton. J Biol Chem. 2001;276(24):21209–21216. doi: 10.1074/jbc.M102615200. [DOI] [PubMed] [Google Scholar]
- 71.Csortos C, et al. TIMAP is a positive regulator of pulmonary endothelial barrier function. American Journal of Physiology-Lung Cellular and Molecular Physiology. 2008;295(3):L440–L450. doi: 10.1152/ajplung.00325.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Han YS, et al. Rat model of heart failure with preserved ejection fraction: changes in contractile proteins regulating Ca2+ cycling and vascular reactivity. Circulation. 2021;144(16):1355–1358. doi: 10.1161/CIRCULATIONAHA.121.054465. [DOI] [PubMed] [Google Scholar]
- 73.Paulus WJ, et al. How to diagnose diastolic heart failure: a consensus statement on the diagnosis of heart failure with normal left ventricular ejection fraction by the Heart Failure and Echocardiography Associations of the European Society of Cardiology. Eur Heart J. 2007;28(20):2539–2550. doi: 10.1093/eurheartj/ehm037. [DOI] [PubMed] [Google Scholar]
- 74.Ford L, Huxley A, Simmons R. The relation between stiffness and filament overlap in stimulated frog muscle fibres. J Physiol. 1981;311(1):219–249. doi: 10.1113/jphysiol.1981.sp013582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bagni M, et al. Tension and stiffness of frog muscle fibres at full filament overlap. J Muscle Res Cell Motil. 1990;11(5):371–377. doi: 10.1007/BF01739758. [DOI] [PubMed] [Google Scholar]
- 76.Stoddard MF, et al. Left ventricular diastolic function: comparison of pulsed Doppler echocardiographic and hemodynamic indexes in subjects with and without coronary artery disease. J Am Coll Cardiol. 1989;13(2):327–336. doi: 10.1016/0735-1097(89)90507-X. [DOI] [PubMed] [Google Scholar]
- 77.Janssen PM. Myocardial relaxation in human heart failure: why sarcomere kinetics should be center-stage. Arch Biochem Biophys. 2019;661:145–148. doi: 10.1016/j.abb.2018.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Barefield D, Sadayappan S. Phosphorylation and function of cardiac myosin binding protein-C in health and disease. J Mol Cell Cardiol. 2010;48(5):866–875. doi: 10.1016/j.yjmcc.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Korte FS, et al. Loaded shortening, power output, and rate of force redevelopment are increased with knockout of cardiac myosin binding protein-C. Circ Res. 2003;93(8):752–758. doi: 10.1161/01.RES.0000096363.85588.9A. [DOI] [PubMed] [Google Scholar]
- 80.Previs MJ, et al. Myosin-binding protein C corrects an intrinsic inhomogeneity in cardiac excitation-contraction coupling. Sci Adv. 2015;1(1):e1400205. doi: 10.1126/sciadv.1400205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kuster DW, et al. Altered C10 domain in cardiac myosin binding protein-C results in hypertrophic cardiomyopathy. Cardiovasc Res. 2019;115(14):1986–1997. doi: 10.1093/cvr/cvz111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Barefield D (2014) Haploinsufficiency of cardiac myosin binding protein-C in the development of hypertrophic cardiomyopathy
- 83.Harris SP, Lyons RG, Bezold KL. In the thick of it: HCM-causing mutations in myosin binding proteins of the thick filament. Circ Res. 2011;108(6):751–764. doi: 10.1161/CIRCRESAHA.110.231670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Harris SP, et al. Hypertrophic cardiomyopathy in cardiac myosin binding protein-C knockout mice. Circ Res. 2002;90(5):594–601. doi: 10.1161/01.RES.0000012222.70819.64. [DOI] [PubMed] [Google Scholar]
- 85.Rosas PC et al (2019) Cardiac myosin binding protein-C phosphorylation mitigates age-related cardiac dysfunction: hope for better aging? JACC 4(7):817–830 [DOI] [PMC free article] [PubMed]
- 86.Bull M, et al. Alternative splicing of titin restores diastolic function in an HFpEF-like genetic murine model (Ttn ΔIAjxn) Circ Res. 2016;119(6):764–772. doi: 10.1161/CIRCRESAHA.116.308904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Itoh-Satoh M, et al. Titin mutations as the molecular basis for dilated cardiomyopathy. Biochem Biophys Res Commun. 2002;291(2):385–393. doi: 10.1006/bbrc.2002.6448. [DOI] [PubMed] [Google Scholar]
- 88.Slater RE, Strom JG, Granzier H. Effect of exercise on passive myocardial stiffness in mice with diastolic dysfunction. J Mol Cell Cardiol. 2017;108:24–33. doi: 10.1016/j.yjmcc.2017.04.006. [DOI] [PubMed] [Google Scholar]
- 89.Soetkamp D, et al. Myofilament phosphorylation in stem cell treated diastolic heart failure. Circ Res. 2021;129(12):1125–1140. doi: 10.1161/CIRCRESAHA.119.316311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hopf A-E, et al. Diabetes-induced cardiomyocyte passive stiffening is caused by impaired insulin-dependent titin modification and can be modulated by neuregulin-1. Circ Res. 2018;123(3):342–355. doi: 10.1161/CIRCRESAHA.117.312166. [DOI] [PubMed] [Google Scholar]
- 91.Vafiadaki E, Arvanitis DA, Sanoudou D. Muscle LIM protein: master regulator of cardiac and skeletal muscle functions. Gene. 2015;566(1):1–7. doi: 10.1016/j.gene.2015.04.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Chang Y, et al. Zebrafish cysteine and glycine-rich protein 3 is essential for mechanical stability in skeletal muscles. Biochem Biophys Res Commun. 2019;511(3):604–611. doi: 10.1016/j.bbrc.2019.02.115. [DOI] [PubMed] [Google Scholar]
- 93.Knöll R, et al. Telethonin deficiency is associated with maladaptation to biomechanical stress in the mammalian heart. Circ Res. 2011;109(7):758–769. doi: 10.1161/CIRCRESAHA.111.245787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hayashi T, et al. Tcap gene mutations in hypertrophic cardiomyopathy and dilated cardiomyopathy. J Am Coll Cardiol. 2004;44(11):2192–2201. doi: 10.1016/j.jacc.2004.08.058. [DOI] [PubMed] [Google Scholar]
- 95.Chen SN et al (2008) Pathogenesis of hypertrophic cardiomyopathy caused by the myozenin 2 mutations involves calcineurin-dependent and -independent mechanisms. Am Heart Assoc
- 96.Xu W, Baribault H, Adamson ED. Vinculin knockout results in heart and brain defects during embryonic development. Development. 1998;125(2):327–337. doi: 10.1242/dev.125.2.327. [DOI] [PubMed] [Google Scholar]
- 97.Zemljic-Harpf A, Manso AM, Ross RS. Vinculin and talin: focus on the myocardium. J Investig Med. 2009;57(8):849–855. doi: 10.2310/JIM.0b013e3181c5e074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Tangney JR et al (2013) Novel role for vinculin in ventricular myocyte mechanics and dysfunction. Biophysical J 104(7):1623–1633 [DOI] [PMC free article] [PubMed]
- 99.Noll NA, Lal H, Merryman WD. Mouse models of heart failure with preserved or reduced ejection fraction. Am J Pathol. 2020;190(8):1596–1608. doi: 10.1016/j.ajpath.2020.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Roh J, et al. Heart failure with preserved ejection fraction: heterogeneous syndrome, diverse preclinical models. Circ Res. 2022;130(12):1906–1925. doi: 10.1161/CIRCRESAHA.122.320257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kasner M, et al. Diastolic tissue Doppler indexes correlate with the degree of collagen expression and cross-linking in heart failure and normal ejection fraction. J Am Coll Cardiol. 2011;57(8):977–985. doi: 10.1016/j.jacc.2010.10.024. [DOI] [PubMed] [Google Scholar]
- 102.López B, et al. Myocardial collagen cross-linking is associated with heart failure hospitalization in patients with hypertensive heart failure. J Am Coll Cardiol. 2016;67(3):251–260. doi: 10.1016/j.jacc.2015.10.063. [DOI] [PubMed] [Google Scholar]
- 103.Leite-Moreira AF. Current perspectives in diastolic dysfunction and diastolic heart failure. Heart. 2006;92(5):712–718. doi: 10.1136/hrt.2005.062950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ouzounian M, Lee DS, Liu PP. Diastolic heart failure: mechanisms and controversies. Nat Clin Pract Cardiovasc Med. 2008;5(7):375–386. doi: 10.1038/ncpcardio1245. [DOI] [PubMed] [Google Scholar]
- 105.González A et al (2019) The complex dynamics of myocardial interstitial fibrosis in heart failure. Focus on collagen cross-linking. Biochim Biophys Acta Mol Cell Res 1866(9):1421–1432 [DOI] [PubMed]
- 106.Bowers SL, Meng Q, Molkentin JD. Fibroblasts orchestrate cellular crosstalk in the heart through the ECM. Nature Cardiovascular Research. 2022;1(4):312–321. doi: 10.1038/s44161-022-00043-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Fan D, et al. Cardiac fibroblasts, fibrosis and extracellular matrix remodeling in heart disease. Fibrogenesis & tissue repair. 2012;5:1–13. doi: 10.1186/1755-1536-5-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Zile MR et al (201) Plasma biomarkers that reflect determinants of matrix composition identify the presence of left ventricular hypertrophy and diastolic heart failure. Circulation 4(3):246–256 [DOI] [PMC free article] [PubMed]
- 109.Zile MR, et al. Myocardial stiffness in patients with heart failure and a preserved ejection fraction: contributions of collagen and titin. Circulation. 2015;131(14):1247–1259. doi: 10.1161/CIRCULATIONAHA.114.013215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Patel RB, Shah SJ, Inciardi RM. Collagen homeostasis of the left atrium: an emerging treatment target to prevent heart failure? Eur J Heart Fail. 2022;24(2):332. doi: 10.1002/ejhf.2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kolijn D, et al. Enhanced cardiomyocyte function in hypertensive rats with diastolic dysfunction and human heart failure patients after acute treatment with soluble guanylyl cyclase (sGC) activator. Front Physiol. 2020;11:345. doi: 10.3389/fphys.2020.00345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.González A, et al. New targets to treat the structural remodeling of the myocardium. J Am Coll Cardiol. 2011;58(18):1833–1843. doi: 10.1016/j.jacc.2011.06.058. [DOI] [PubMed] [Google Scholar]
- 113.van Heerebeek L, et al. Low myocardial protein kinase G activity in heart failure with preserved ejection fraction. Circulation. 2012;126(7):830–839. doi: 10.1161/CIRCULATIONAHA.111.076075. [DOI] [PubMed] [Google Scholar]
- 114.Zhao W, et al. Effects of cardiomyocyte-specific deletion of STAT3–a murine model of heart failure with preserved ejection fraction. Frontiers in Cardiovascular Medicine. 2020;7:613123. doi: 10.3389/fcvm.2020.613123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kolijn D, et al. Empagliflozin improves endothelial and cardiomyocyte function in human heart failure with preserved ejection fraction via reduced pro-inflammatory-oxidative pathways and protein kinase Gα oxidation. Cardiovasc Res. 2021;117(2):495–507. doi: 10.1093/cvr/cvaa123. [DOI] [PubMed] [Google Scholar]
- 116.Pérez-Torres I, et al. Nitrosative stress and its association with cardiometabolic disorders. Molecules. 2020;25(11):2555. doi: 10.3390/molecules25112555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Breitkreuz M, Hamdani N. A change of heart: oxidative stress in governing muscle function? Biophys Rev. 2015;7:321–341. doi: 10.1007/s12551-015-0175-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Townsend DM, et al. Nitrosative stress–induced S-glutathionylation of protein disulfide isomerase leads to activation of the unfolded protein response. Can Res. 2009;69(19):7626–7634. doi: 10.1158/0008-5472.CAN-09-0493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Uehara T, et al. S-Nitrosylated protein-disulphide isomerase links protein misfolding to neurodegeneration. Nature. 2006;441(7092):513–517. doi: 10.1038/nature04782. [DOI] [PubMed] [Google Scholar]
- 120.Nasoni M et al (2023) When nitrosative stress hits the endoplasmic reticulum: possible implications in oxLDL/oxysterols-induced endothelial dysfunction. Free Radic Biol Med [DOI] [PubMed]
- 121.Gu Z, Nakamura T, Lipton SA. Redox reactions induced by nitrosative stress mediate protein misfolding and mitochondrial dysfunction in neurodegenerative diseases. Mol Neurobiol. 2010;41:55–72. doi: 10.1007/s12035-010-8113-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Nakato R, et al. Regulation of the unfolded protein response via S-nitrosylation of sensors of endoplasmic reticulum stress. Sci Rep. 2015;5(1):14812. doi: 10.1038/srep14812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Failure E, On DH (1998) How to diagnose diastolic heart failure. Eur Heart J 19(7):990–1003 [DOI] [PubMed]
- 124.From AM, Scott CG, Chen HH. The development of heart failure in patients with diabetes mellitus and pre-clinical diastolic dysfunction a population-based study. J Am Coll Cardiol. 2010;55(4):300–305. doi: 10.1016/j.jacc.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Levy D, et al. The progression from hypertension to congestive heart failure. JAMA. 1996;275(20):1557–1562. doi: 10.1001/jama.1996.03530440037034. [DOI] [PubMed] [Google Scholar]
- 126.Pinho-Gomes AC, Rahimi K. Management of blood pressure in heart failure. Heart. 2019;105(8):589–595. doi: 10.1136/heartjnl-2018-314438. [DOI] [PubMed] [Google Scholar]
- 127.Hirota Y. A clinical study of left ventricular relaxation. Circulation. 1980;62(4):756–763. doi: 10.1161/01.CIR.62.4.756. [DOI] [PubMed] [Google Scholar]
- 128.Little WC, Downes TR. Clinical evaluation of left ventricular diastolic performance. Prog Cardiovasc Dis. 1990;32(4):273–290. doi: 10.1016/0033-0620(90)90017-V. [DOI] [PubMed] [Google Scholar]
- 129.Husain S, et al. Invasive monitoring in patients with heart failure. Curr Cardiol Rep. 2009;11(3):159–166. doi: 10.1007/s11886-009-0024-x. [DOI] [PubMed] [Google Scholar]
- 130.Cao P, et al. Chapter II: diagnostic methods. Eur J Vasc Endovasc Surg. 2011;42:S13–S32. doi: 10.1016/S1078-5884(11)60010-5. [DOI] [PubMed] [Google Scholar]
- 131.Kirchhof P, et al. 2016 ESC Guidelines for the management of atrial fibrillation developed in collaboration with EACTS. Europace. 2016;18(11):1609–1678. doi: 10.1093/europace/euw295. [DOI] [PubMed] [Google Scholar]
- 132.Oh JK, et al. Diastolic heart failure can be diagnosed by comprehensive two-dimensional and Doppler echocardiography. J Am Coll Cardiol. 2006;47(3):500–506. doi: 10.1016/j.jacc.2005.09.032. [DOI] [PubMed] [Google Scholar]
- 133.Borlaug BA, Kass DA. Mechanisms of diastolic dysfunction in heart failure. Trends Cardiovasc Med. 2006;16(8):273–279. doi: 10.1016/j.tcm.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 134.Gaasch WH, Zile MR. Left ventricular diastolic dysfunction and diastolic heart failure. Annu Rev Med. 2004;55:373–394. doi: 10.1146/annurev.med.55.091902.104417. [DOI] [PubMed] [Google Scholar]
- 135.Douglas PS (2003) The left atrium: a biomarker of chronicdiastolic dysfunction andcardiovascular disease risk. American College of Cardiology Foundation Washington, DC, (p. 1206–1207) [DOI] [PubMed]
- 136.Tschöpe C, et al. The role of NT-proBNP in the diagnostics of isolated diastolic dysfunction: correlation with echocardiographic and invasive measurements. Eur Heart J. 2005;26(21):2277–2284. doi: 10.1093/eurheartj/ehi406. [DOI] [PubMed] [Google Scholar]
- 137.Watanabe S, et al. Myocardial stiffness is an important determinant of the plasma brain natriuretic peptide concentration in patients with both diastolic and systolic heart failure. Eur Heart J. 2006;27(7):832–838. doi: 10.1093/eurheartj/ehi772. [DOI] [PubMed] [Google Scholar]
- 138.Paelinck BP, et al. Assessment of diastolic function by cardiovascular magnetic resonance. Am Heart J. 2002;144(2):198–205. doi: 10.1067/mhj.2002.123316. [DOI] [PubMed] [Google Scholar]
- 139.Ibrahim EH, et al. Diastolic cardiac function by MRI-imaging capabilities and clinical applications. Tomography. 2021;7(4):893–914. doi: 10.3390/tomography7040075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Cremer PC et al (2022) Effect of mavacamten on diastolic function in patients with obstructive hypertrophic cardiomyopathy: insights from the VALOR-HCM study. Circulation 146(Suppl_1):A12577–A12577 [DOI] [PubMed]
- 141.Olivotto I, et al. Mavacamten for treatment of symptomatic obstructive hypertrophic cardiomyopathy (EXPLORER-HCM): a randomised, double-blind, placebo-controlled, phase 3 trial. The Lancet. 2020;396(10253):759–769. doi: 10.1016/S0140-6736(20)31792-X. [DOI] [PubMed] [Google Scholar]
- 142.Raake P, et al. Gene therapy targets in heart failure: the path to translation. Clin Pharmacol Ther. 2011;90(4):542–553. doi: 10.1038/clpt.2011.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Anderson RL, et al. Deciphering the super relaxed state of human β-cardiac myosin and the mode of action of mavacamten from myosin molecules to muscle fibers. Proc Natl Acad Sci. 2018;115(35):E8143–E8152. doi: 10.1073/pnas.1809540115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Halas M, et al. Effects of sarcomere activators and inhibitors targeting myosin cross-bridges on Ca2+-activation of mature and immature mouse cardiac myofilaments. Mol Pharmacol. 2022;101(5):286–299. doi: 10.1124/molpharm.121.000420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Anker SD, et al. Empagliflozin in heart failure with a preserved ejection fraction. N Engl J Med. 2021;385(16):1451–1461. doi: 10.1056/NEJMoa2107038. [DOI] [PubMed] [Google Scholar]
- 146.Nassif ME, et al. Empagliflozin effects on pulmonary artery pressure in patients with heart failure: results from the EMBRACE-HF trial. Circulation. 2021;143(17):1673–1686. doi: 10.1161/CIRCULATIONAHA.120.052503. [DOI] [PubMed] [Google Scholar]
- 147.Solomon SD, et al. Dapagliflozin in heart failure with mildly reduced or preserved ejection fraction. N Engl J Med. 2022;387(12):1089–1098. doi: 10.1056/NEJMoa2206286. [DOI] [PubMed] [Google Scholar]
- 148.Pitt B, et al. Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med. 2003;348(14):1309–1321. doi: 10.1056/NEJMoa030207. [DOI] [PubMed] [Google Scholar]
- 149.Izawa H, et al. Mineralocorticoid receptor antagonism ameliorates left ventricular diastolic dysfunction and myocardial fibrosis in mildly symptomatic patients with idiopathic dilated cardiomyopathy: a pilot study. Circulation. 2005;112(19):2940–2945. doi: 10.1161/CIRCULATIONAHA.105.571653. [DOI] [PubMed] [Google Scholar]
- 150.Pitt B, et al. Spironolactone for heart failure with preserved ejection fraction. N Engl J Med. 2014;370(15):1383–1392. doi: 10.1056/NEJMoa1313731. [DOI] [PubMed] [Google Scholar]
- 151.McMurray J, O’Connor C. Lessons from the TOPCAT trial. N Engl J Med. 2014;370(15):1453–1454. doi: 10.1056/NEJMe1401231. [DOI] [PubMed] [Google Scholar]
- 152.Ryba DM et al (2019) Sphingosine-1-phosphate receptor modulator, FTY720, improves diastolic dysfunction and partially reverses atrial remodeling in a Tm-E180G mouse model linked to hypertrophic cardiomyopathy. Circulation 12(11):e005835 [DOI] [PMC free article] [PubMed]
- 153.Rosas PC, Solaro RJ (2022) Implications of S-glutathionylation of sarcomere proteins in cardiac disorders, therapies, and diagnosis. Front Cardiovasc Med 9 [DOI] [PMC free article] [PubMed]