

Abstract
Idiopathic pulmonary fibrosis (IPF) is characterized by progressive, often fatal loss of lung function due to overactive collagen production and tissue scarring. Patients with IPF have a sevenfold-increased risk of developing lung cancer. The COVID-19 pandemic has increased the number of patients with lung diseases, and infection can worsen prognoses for those with chronic lung diseases and disease-associated cancer. Understanding the molecular pathogenesis of IPF-associated lung cancer is imperative for identifying diagnostic biomarkers and targeted therapies that will facilitate prevention of IPF and progression to lung cancer. To understand how IPF-associated fibroblast activation, matrix remodeling, epithelial-to-mesenchymal transition (EMT), and immune modulation influences lung cancer predisposition, we developed a mouse model to recapitulate the molecular pathogenesis of pulmonary fibrosis–associated lung cancer using the bleomycin and Lewis lung carcinoma models. We demonstrate that development of pulmonary fibrosis–associated lung cancer is likely linked to increased abundance of tumor-associated macrophages and a unique gene signature that supports an immune-suppressive microenvironment through secreted factors. Not surprisingly, preexisting fibrosis provides a pre-metastatic niche and results in augmented tumor growth, and tumors associated with bleomycin-induced fibrosis are characterized by a dramatic loss of cytokeratin expression, indicative of EMT.
Implications:
This characterization of tumors associated with lung diseases provides new therapeutic targets that may aid in the development of treatment paradigms for lung cancer patients with preexisting pulmonary diseases.
Introduction
Idiopathic pulmonary fibrosis (IPF) is a debilitating and fatal lung disease with a median survival of 3 to 4 years following diagnosis (1, 2). The approximately 3 million people worldwide affected by IPF exhibit signs of permanent lung scarring and suffer respiratory and other organ failure related to hypoxia (3). The global COVID-19 pandemic has intensified the seriousness of pulmonary fibrosis because lung infections worsen fibrosis in the lung (4); furthermore, the effects of IPF on lung function increase patients’ risk of developing COVID-19, while those recovering from severe COVID-19 are at high risk for developing pulmonary fibrosis (5, 6). While many genetic factors, along with environmental factors such as smoking and other forms of oxidative damage are linked to IPF, its main cause remains largely unknown, thus preventing optimal treatment and diagnosis (7). Treatment options remain limited and include anti-inflammatory therapy, transplant, palliation, or clinical trial recruitment (8). Novel anti-fibrotic agents like pirfenidone and nintedanib slow disease progression (9, 10) and temporarily alleviate fibrotic scarring, but have not yet extended median survival rates of patients with IPF.
While IPF is a devastating, often fatal inflammatory lung disease, lung cancer remains the leading cause of cancer-related mortality worldwide (11). With the emergence of immune checkpoint therapies and KRAS targeted therapies, great strides are being made in the treatment of lung cancer. However, an estimated 127,070 Americans will die of lung cancer in 2023, accounting for ∼21% of all cancer-related deaths in the United States (12). Epidemiologic evidence indicates that IPF increases the risk of lung cancer by sevenfold (13) and thus, about 22% of patients with IPF develop lung cancer and exhibit a worse prognosis with poorer survival compared with the already dismal survival of patients with IPF (14–17). Cigarette smoke, chronic lung diseases like chronic obstructive pulmonary disease, fibrotic disorders, and recently, COVID, increase the risk for lung cancer in patients with IPF (18, 19). As no guidelines exist to screen patients with IPF for lung cancer, and common therapeutic options for lung cancer often exacerbate IPF, a critical need remains to identify the mechanistic link between these diseases, and in turn, illuminate the molecular underpinnings of IPF-associated lung cancer (IPF-LC) and the potential drivers that lead to lung cancer in patients with IPF. To our knowledge, there is currently only one preclinical model that mimics the pathobiology of pulmonary fibrosis–associated lung cancer, described most recently in a study of tumor-associated fibrosis by Herzog and colleagues (20).
To gain a mechanistic understanding of the pathobiology of IPF-LC and identify and evaluate new therapeutic strategies, we developed a novel mouse model that combines the bleomycin-induced fibrosis model with the Lewis lung carcinoma model in a syngeneic background (21, 22). Previous work has shown that mouse models of bleomycin-induced pulmonary fibrosis do not uniformly recapitulate human disease (23), but because our model is characterized by some aspects associated specifically with IPF, we discuss this disease throughout the paper. Because the tumor cells (LLC-1) are immunologically compatible with the host, the role of the immune response, which is a key contributor to cancer progression and metastasis, can be studied. This new murine model allowed us to interrogate the role of the tumor microenvironment (TME) in the context of fibrosis, lung cancer, or pulmonary fibrosis–associated lung cancer and provided mechanistic insight into the pathobiology of IPF and importantly, new therapeutic biomarkers and targets.
Materials and Methods
Animals
Female and male 8- to 10-week-old C57BL/6-albino mice (The Jackson Laboratory, strain/stock: 000058 B6(Cg)-Tyrc-2J/J, RRID: IMSR_JAX:000058) were maintained in accordance with the University of Michigan's Institutional Animal Care and Use Committee guidelines approved protocol (IACUC PRO 00010349). C57B/L6-albino mice were randomized into four different experimental cohorts: (i) control mice not receiving bleomycin or LLC-1 Luciferase expressing cells (LLC-1 Luc), (ii) mice that received two doses of 0.5 mg/kg and 1 mg/kg bleomycin in 50 μL saline [double oropharyngeal aspiration (OA) schedule at day 0 and day 4] to induce fibrosis, (iii) mice wherein LLC-1 Luc cells were injected intravenously through the tail vein at a concentration of 1 × 106 cells per mouse in 100 μL saline, and (iv) mice where LLC-1 Luc cells were implanted intravenously into bleomycin preconditioned mice (OA). Because the bleomycin model exhibits two distinct phases, the inflammatory (2 weeks) and the fibrotic (2 weeks) phase, LLC-1 Luc cells were injected 2 weeks post bleomycin administration during the fibrotic phase. We found that a double OA schedule of bleomycin at day 0 and day 4 produced the most consistent induction of lung fibrosis as previously described (24, 25).
Transgenic mouse model
Transgenic CCSP-rtTA mice (26) were first crossed with transgenic TRE-TGFα mice (27). The double transgenic offspring (CCSPrtTA+, TRE-TGFa+) of those was crossed to the transgenic TRE-KrasG12D model (28) to obtain four experimental groups: controls, IPF (CCSPrtTA+, TRE-TGFa+, TRE-KrasG12D-), LC (CCSPrtTA+, TRE-TGFa-, TRE-KrasG12D+), and IPF-LC (CCSPrtTA+, TRE-TGFa+, TRE-KrasG12D+). To induce IPF, LC or IPF-LC, mice were given doxycycline (Sigma, catalog number D9891) containing water at a concentration of 0.5 mg/mL at study start and when mice were 6 weeks and older. Two different ‘control groups’ were used for this study to control for the effects of doxycycline on lung tissue and for leakiness of the TetO inducible genes. Single transgenic control mice were given doxycycline in the drinking water at the same dose as experimental cohorts and triple transgenics were given regular drinking water.
Bleomycin
Bleomycin was obtained through the University of Michigan Health Services Pharmacy (Meitheal Pharmaceuticals, catalog no. 71288–106–10). Bleomycin powder was resuspended in 1xPBS at a concentration of 1 U/mL (equal to 1 mg/mL) and stored at −20°C. A dose of 0.5 mg/kg was given by OA at the beginning of the study (day 0) and 1 mg/kg four days later (day 4).
Cell lines
The murine Lewis lung carcinoma-1 (LLC-1) cell line was obtained from ATCC in 2018 (ATCC, catalog no. CRL-1642, RRID:CVCL_4358) and maintained in DMEM with 10% FBS and 1% Pen/Strep. LLC-1 cells were infected with lentivirus FUGW (29) to express firefly luciferase and GFP to obtain LLC-1 Luciferase expressing cells for bioluminescence imaging (BLI; LLC-1 Luc). LLC-1 cells bear a heterozygous KrasG12C mutation (25). All cell lines were regularly checked for mycoplasma contamination every other week using MycoAlert Mycoplasma Detection Kit (Lonza). Cell lines were cultured for a period of ∼ 2 months, after which they were replaced by new thaws. Cells were purchased from ATCC for this study specifically and not additionally authenticated by STR.
BLI
In vivo BLI was performed under anesthesia using the IVIS Spectrum In Vivo Imaging System (PerkinElmer) according to the manufacturer's protocol. In short, mice received an intraperitoneal injection of 100 μL D-luciferin (stock of 40 mg/mL, Promega, E160). Mice were allowed to move freely for 5 minutes post injection prior to initiating anesthesia via isoflurane. Mice were imaged 5 minutes after isoflurane administration (10 minutes post injection). Imaging occurred at indicated timepoints postimplantation of LLC-1 Luc cells. Quantification of total flux (photons per second, p/s/cm3) using a standardized circular region of interest spanning the entire lung was performed at each timepoint using IVIS Spectrum In Vivo Imaging Software.
Microcomputed tomography
Microcomputed tomography (Micro-CT) imaging was performed similarly to methods described by Bowman and colleagues (30). In brief, imaging was performed at indicated timepoints using Bruker SkyScan 1176 System with the following parameters: 60 kilovolt peaks (kVp) with a 1 mm aluminum filter, 168 μA, 158-ms exposure, 0.5° rotation step over 360°, and a reconstructed voxel size of 35 μm.
Multiplex immunofluorescence and multispectral imaging
Multiplex immunofluorescence (mIF) was performed as previously described (31, 32) on lung sections for a minimum of 3 mice per experimental group. Antibodies used are shown in Supplementary Table S1. Images of immunofluorescence antibody-stained murine lung sections were acquired using the Mantra Quantitative Pathology Workstation (PerkinElmer) and captured at 20x magnification. All cube filters were used for each image capture (DAPI, CY3, CY5, CY7, Texas Red, Qdot) and the saturation protection feature was used. After image acquisition, analyses were performed using inForm Cell Analysis software (PerkinElmer) as previously described (31, 32). Basic phenotypes were composed of the following: Arg1, CK19, DAPI, CD3+, F4/80, and CD8+. The scoring feature was used to determine the appropriate range of the mean signal intensity of each stain within the cytoplasm for CD8+ and nucleus for DAPI. These scoring ranges were used to make secondary phenotypes in R: macrophages, T cells, epithelial cells, and ‘other.’ Final phenotypes, including primary (CK19, F4/80, and CD3) and secondary phenotypes (macrophages and CD8+ T cells) made in R were quantitatively analyzed using a program designed in R. The mean numbers of cells per high powered field at 20x magnification of five fields per mouse were plotted on the y-axis. A minimum of 3 mice per experimental group were used for mIF.
Histology and IHC
Murine lungs were perfused in 1x PBS then fixed in formalin and stored in 70% ethanol before embedding in paraffin. The top half of the right lung lobe was used for histology and sent to the University of Michigan Histology Core (https://www.pathology.med.umich.edu/core-resources/research-histology-core) for paraffin embedding, sectioning, hematoxylin and eosin (H&E) and Masson Trichrome staining. Additional sections were hybridized with antibodies against Ki67, CD3+, CD8+, and CK7, performed by the core facility. In brief, for H&E staining, sections were deparaffinized in xylene, re-hydrated in ethanol, and briefly washed in distilled water. Staining was performed with Harris hematoxylin solution, and counterstained with eosin-phloxine solution, prior to mounting with xylene based mounting medium. For Masson Trichrome staining, Bouin's solution was preheated for 1 hour at 56°C to 60°C. Sections were deparaffinized and hydrated in deionized water. Sections were then incubated in Bouin's solution for 1 hour in oven. Slides and lids were removed from the oven, allowed to cool for 10 minutes, then washed in tap water until their yellow color disappeared (approx. 10 minutes). Slides were placed in Weigerts Iron Hematoxylin solution for 10 minutes and rinsed in running tap water for 5 minutes. Rinse was repeated in deionized water. Subsequently, slides were blotted to remove excess water and stained in Biebrich Scarlet-Acid Fuchsin for 10 minutes, then rinsed in deionized water until clear. Slides were again blotted to remove excess water and incubated in phosphomolybdic-phosphotungstic acid for 15 minutes. Slides were blotted, incubated in aniline blue for 8 minutes and rinsed in deionized water until clear. Slides were dehydrated in 70% ethanol, 95% ethanol and 100% ethanol for 10 seconds each with agitation prior to mounting with xylene based mounting medium. Quantification of trichome staining was performed using ImageJ software (ImageJ, RRID:SCR_003070).
Evaluation of lung lesion size and number
H&E-stained sections of the murine lungs were scanned using the Nikon Supercool Scan 5000 at 1X magnification. Evaluation of lung lesion number and size was blinded and performed on at least three sections per experimental group by three different readers. The number of lesions was counted, and the size measured with a ruler in cm on 1X magnified scans. The number of lesions and lesion size was graphed for each group and statistical significance was determined by one-way ANOVA using GraphPad Prism (GraphPad Prism, RRID:SCR_002798).
Cytometry by time-of-flight
Murine lung tissues from all four experimental cohorts were placed into DMEM Complete after lung perfusion with 1xPBS. Subsequently, lung tissues were mechanically minced and enzymatically digested with collagenase P (1 mg/mL DMEM) and subsequently filtered through a 40-μm mesh to obtain single cells. Up to 1 × 107 cells were stained with Cell-ID cisplatin (1.67 μmol/L) for 5 minutes at room temperature, followed by a ‘Fix and Perm-Sensitive Surface Epitopes and Nuclear Antigen Staining’ protocol according to the manufacturer's instructions (Fluidigm) for mouse samples (33). Briefly, after quenching cisplatin reaction with 5X volume of MaxPar cell staining buffer, cells were centrifuged at 300 × g for 5 minutes. Up to 3 million cells per sample were stained with cell surface antibody cocktail (see Supplementary Table S2) in 100 μL volume of MaxPar cell staining buffer for 30 minutes at room temperature. Cells were subsequently washed twice in 1 mL MaxPar cell staining buffer and then fixed in 1.6% freshly made formaldehyde solution for 10 minutes at room temperature. After fixation, cells were washed once in 1 mL MaxPar cell staining buffer and permeabilized with 1 mL nuclear antigen staining buffer for 20 minutes at room temperature. Cells were then washed twice with 1 mL nuclear antigen staining perm and centrifuged in between at 800 × g for 5 minutes, followed by staining with intracellular antibody cocktail (see Supplementary Table S2) in 50 μL volume of nuclear antigen staining perm for 45 minutes at room temperature. Subsequently, cells were washed with 2 mL nuclear antigen staining perm followed by a wash with 2 mL MaxPar cell staining buffer, resuspended in 2 mL cell intercalation solution (125 nmol/L Cell-ID Intercalator-Ir in Maxpar fix and perm buffer) and shipped to the Flow Cytometry Core at the University of Rochester Medical Center, where sample preparation was finalized, and Mass Cytometer analysis performed on a CYTOF2. Raw FCS files were analyzed using the Premium Cytobank software (cytobank.org) and FlowSOM-viSNE (34) or viSNE as previously described (33). Doublets were excluded from the analysis by using event length in conjunction with the DNA intercalator (Iridium). We analyzed only live cells for CyTOF analysis in Cytobank by selecting cells negative for the Cisplatin live/dead stain as recommended by the manufacturer (35). Only immune cells (CD45+ cells) were analyzed and quantified by gating on CD45+. The marker negative population represents nonimmune cells that are present in the tissue, which likely includes tumor cells. Statistical analyses were performed in GraphPad Prism using paired t tests with statistical significances established with a P value less than 0.05.
Flow cytometry
Lung tissue was collected in 10 mL of FACS buffer (2% FBS, 500 mL PBS, 5 mmol/L EDTA) and pushed through 70-μm cell strainer with syringe plunger. Red cell lysis was performed in 10 mL ACK (Ammonium-Chloride-Potassium) lysing buffer (Invitrogen, catalog no. 11814389001) following the manufacturer's instructions. Cells were washed and centrifuged and 500,000 cells were stained in 200 μL 1x PBS plus 0.5 μg of indicated antibody for 30 minutes in the dark: APC anti-mouse CD274 (B7-H1, PD-L1; BioLegend, catalog no. 124311 (also 124312), RRID:AB_10612935) or appropriate IgG isotype. After incubation 500 μL of the FACS buffer was added to the antibody staining solution and centrifuged for 1,800 RPM for 5 minutes. Supernatant was removed and the process repeated for a total of two washes. After the second wash, the pellet was resuspended in 500 μL of FACS buffer and cells were analyzed using Accuri Flow Cytometer. Data was analyzed using Accuri software and data graphed using GraphPad Prism.
Bulk RNA sequencing
RNA was isolated from lung tissue using the Qiagen RNA isolation kit according to the manufacturer's protocol. RNA was stored in 30 μL of RNase/DNase free water and quantified using the NanoDrop 2000c (Thermo Fisher). 30 μg of RNA was sent to the University of Michigan Advanced Genomics Core for RNA Quant Sequencing. Total RNA quality was assessed using the Tapestation 4200 (Agilent). 500 ng of DNase-treated total RNA was used to generate QuantSeq 3′ mRNA FWD (Lexogen). Pooled libraries were subjected to 100 bp paired-end sequencing according to the manufacturer's protocol (Illumina NextSeq550). Bcl2fastq2 Conversion Software (Illumina) was used to generate de-multiplexed Fastq files. Reads were trimmed for adapters and quality using Trim Galore 0.5.0 (Trim Galore, RRID:SCR_011847). Trimmed reads were aligned to the mm10 reference using STAR 2.6.0a (STAR, RRID:SCR_004463). Annotations for mm10 were from UCSC and obtained from iGenomes (Illumina). Alignments were collapsed to UMIs using collapse_UMI_bam from Lexogen. Gene counting was carried out using Rsubread/featureCounts. DESeq2 1.38.3 (DESeq2, RRID:SCR_015687; ref. 36) was used for differential gene expression analysis and genes with adjusted P value < 0.05 and fold-change > 1.5 (log2 fold-change > 0.58) in either direction were considered significant. Genes uniquely regulated in the IPF-LC group were selected if they were significantly up- or downregulated in the IPF-LC group when compared with genes in the control group. The same genes had to show no statistically significant regulation in the LC or IPF group when compared with the control groups. In other words, these genes were unique to the IPF-LC group when compared with normal tissue and to both IPF and lung cancer tissues.
Data availability
The datasets supporting the current study are available from the corresponding author upon request and available in the NIH Gene Expression Omnibus (GEO) database (RRID:SCR_005012). RNA raw data files and analysis was deposited with GEO accession number GSE224134. Material or data that require a Material Transfer Agreement (MTA) can be provided by the University of Michigan pending scientific review and the execution of an MTA negotiated by the university's Office of Technology Transfer. Requests for data that require an MTA should be submitted to the corresponding author, Dr. Stefanie Galban, sgalban@umich.edu.
Results
Preexisting pulmonary fibrosis results in augmented tumor growth
To establish a new IPF-LC mouse model, we induced lung fibrosis by intratracheal delivery of bleomycin and pulmonary lesions through intravenous injection of the murine, heterozygous KrasG12C mutant Lewis lung carcinoma cell line (LLC-1) in a syngeneic C57BL/6 background (Fig. 1A). Two distinct phases for bleomycin-induced IPF, an inflammatory (0–2 weeks) and a fibrotic (2–4 weeks) phase, have previously been described (24). This guided our decision to inject LLC-1 Luc cells 2 weeks post instillation at the beginning of the fibrotic phase. We optimized the bleomycin OA schedule and confirmed a previously reported benefit of double administration on day 0 and 4 to obtain the most consistent induction of lung fibrosis (37). Development of pulmonary fibrosis and lesions were assessed by micro-CT and bioluminescence (Fig. 1B–D), respectively, as well as by histologic analysis at the end of the study (Fig. 1E). As expected, lung metastases were detected in the LC group (‘LC’), which received LLC-1 Luc cells alone, and in the IPF-LC group, where lung fibrosis was induced prior to LC initiation (‘IPF-LC’). Bioluminescence activity increased significantly over time in the IPF-LC group when compared with the LC group, indicative of more aggressive tumor seeding and growth in the lungs due to pulmonary fibrosis (Fig. 1C). This was confirmed by histologic analysis at the end of the study, when both the number and size of the pulmonary lesions were significantly augmented in the IPF-LC group versus the LC group (Fig. 1F and G). Statistical analysis between groups to assess the correlation between lung cancer and fibrosis revealed that mice with fibrosis experienced a higher tumor burden and larger lung tumors when compared with mice with no fibrosis (Fig. 1F and G). As expected, we also observed pulmonary fibrosis, determined by trichome staining of collagenous connective tissue fibers, in both bleomycin-induced fibrosis mice (‘IPF’) and the IPF-LC group (Fig. 1H). Interestingly, LLC-1 Luc cells were preferentially detected around the airways and the lung periphery, correlating with deposition of collagenous fibers post bleomycin administration in these areas (Fig. 1E). In summary, we have developed a murine model using bleomycin and LC cells that recapitulates the pathobiology of IPF-LC and have detected an increased tumor load in fibrotic lungs.
Figure 1.

Modeling pathogenesis of IPF-LC in a murine model. A, Schematic indicating experimental outline: C57BL/6-albino mice were randomized into four experimental cohorts for all comparative studies: control, IPF, LC, and IPF-LC. Mice in the control group received appropriate vehicles (control), mice in the IPF group received 0.5 mg/kg and 1 mg/kg of bleomycin on day 0 and day 4, respectively, by OA. Mice in the LC group received LLC-1 Luc cells (1 × 106 cells/100 μL) by intravenous injection into the tail vein (intravenously) on day 14 and mice in the IPF-LC group received 0.5 mg/kg and 1 mg/kg of bleomycin (OA) on day 0 and day 4, respectively, and LLC-1 Luc cells (intravenously) on day 14. Timepoints for micro-CT and BLI or tissue collection are denoted by a yellow star, a purple circle, and a red arrow respectively. B, Representative micro-CT images from all groups at study endpoint. Tumor lesions in LC group and IPF-LC group are indicated by red arrows and circled in pink. C, Representative BLI images from mice in LC and IPF-LC groups at week 1 and 3 (day 21 and day 28) post LLC-1 Luc injection. D, Quantification of bioluminescence at indicated timepoints of mice in the LC (n = 4) and IPF-LC (n = 4) groups. Data is presented ± SEM and statistics were performed using one-way ANOVA with post hoc Tukey (Honestly Significant Difference (HSD)) test with statistical significance denoted as: **, P ≤ 0.01. E, Representative images of gross morphology, H&E and trichome staining of lung images and lung sections acquired at study endpoint at 5 weeks post first bleomycin injection. Black arrows indicate tumors on H&E-stained lung sections of mice in LC and IPF-LC groups. F, Quantification of the number of tumors in the LC and IPF-LC groups counted on 1x H&E-stained lung sections presented ± SEM. Statistical test was performed with one-way ANOVA with post hoc Tukey HSD test and significance indicated as: *, P ≤ 0.05. G, Quantification of the average tumor size in the lung in LC and IPF-LC groups measured on 1x H&E-stained lung sections. Data is represented ±SEM and statistical significance determined using one-way ANOVA with post hoc Tukey HSD test and significance of *, P ≤ 0.05. H, Quantification of collagen in trichrome staining of lung sections. No statistical difference was determined between groups.
Induction of fibrosis in a genetically engineered model of lung cancer
To develop a more physiologically relevant and spontaneous model of IPF-LC we used two previously described inducible transgenic models where lung cancer-associated KrasG12D and fibrosis-associated TGFα expression is directed to the lung through the control of reverse tetracycline transactivator by the Clara cell secretory protein promoter Scgb1a1 (CCSP; refs. 27, 38; Supplementary Fig. S1A). The reverse tetracycline transactivator is expressed in club cells (rtTA), and in the presence of doxycycline, results in expression of KrasG12D and/or TGFα in these cells, thus inducing lung cancer and/or fibrosis. We generated four experimental cohorts: (i) control mice lacking CCSPrtTA, KrasG12D, and TGFα, (ii) LC-positive mice with CCSPrtTA and KrasG12D but no TGFα transgene, (iii) IPF-positive mice with CCSPrtTA and TGFα but no KrasG12D transgene, and (iv) IPF-LC mice that were positive for all three transgenes. All mice were administered doxycycline twice a week at a concentration of 0.5 mg/mL in drinking water for the duration of the 20-week experiment. As expected, expression of TGFα in the lung induced fibrosis in both the IPF and IPF-LC groups and expression of KrasG12D resulted in lung cancer lesions in the LC group. However, fibrosis progressed quickly and resulted in mortality prior to the detection of lung tumors in the IPF-LC group. Statistical analysis revealed significant differences in weight changes between the IPF or IPF-LC groups when compared with control or LC groups, with IPF and IPF-LC mice exhibiting a marked loss of body weight (Supplementary Fig. S1B). In addition, the IPF and IPF-LC groups exhibited significantly accelerated mortality compared with other groups, with only the LC and control groups surviving until the end of the 20-week study period (Supplementary Fig. S1C). Because no difference in survival was observed between the IPF and IPF-LC groups, no molecular profiling was performed. In summary, this spontaneous genetically engineered model of IPF-LC was found to be inadequate to determine the impact of fibrosis on tumor growth and aggressiveness. Future modification of this model where expression of TGFα and KrasG12D are controlled independently of each other will be necessary to answer this question.
Tumor progression of pulmonary lesions is associated with increased tumor-associated macrophages (TAM) and decreased T-cell infiltration. The tumor immune microenvironment plays a pivotal role in tumor initiation and progression by creating an immune suppressive, tumor promoting or tumor killing environment. To study the infiltrating immune cells in IPF-LC presumably contributing to tumor initiation and promotion, we used a previously described tissue staining method to phenotype tumor epithelial cells (CK19), macrophages (F4/80), Arginase 1 expressing immune suppressive TAMs, and T cells (CD3, CD8, CD4; ref. 31). Multiplex fluorescent IHC was performed on murine lung sections of all four groups (control, IPF, LC and IPF-LC; Fig. 2A). Increase in macrophage abundance or infiltration as determined by F4/80 marker expression was observed in the lungs of IPF-LC mice when compared with LC mice, indicating their contribution to accelerated tumor growth in this group (Fig. 2B). Arginase 1 positive cells, a phenotypic marker for TAMs often associated with M2 polarized immune suppressive macrophages, were detected in the IPF-LC group with a statistically significant difference to the LC group (Fig. 2C). Interestingly, a significant decrease in Cytokeratin 19 (CK19) staining of epithelial cells was observed in IPF-LC when compared with LC tumors (Fig. 2D). This may indicate a TGF-dependent transition that results in a reduction of Cytokeratin 19 expression, as previously described in lung epithelium (39). When querying all T cells (CD3+), no statistical difference between the LC and IPF-LC groups was observed (Fig. 2E), but a trend towards decreased T cell abundance was observed. CD8+ cytotoxic T cells, however, were statistically significantly downregulated in the IPF-LC group when compared with the LC group (Fig. 2F), which would support a phenotype of more rapid tumor promotion observed in the IPF-LC group and an immune suppressive TME. To further assess proliferation and differences in T cell abundance, additional histologic analyses were performed on lung sections from all experimental cohorts (control, IPF, LC, and IPF-LC). Sections were stained for proliferation marker Ki67, but no statistical difference was observed between tumor bearing mice in the LC and IPF-LC groups (Fig. 2G and H). CD3+ T cells and CD8+ cytotoxic T cells were reduced in IPF-LC lung lesions and limited to the periphery of the tumor when compared with tumors in the LC group (Fig. 2I and J). In summary, we observed an increase in macrophages with Arginase 1 expressing TAMs predominantly enriched in lungs from IPF-LC mice and a decrease in cytotoxic T cells. This finding indicates macrophage polarization to an M2 phenotype, which corroborates with our findings of increased tumor growth in lungs of IPF-LC mice.
Figure 2.

Tumor progression of pulmonary lesions is associated with increased macrophage infiltration and decrease of cytotoxic T cells. A, mIF performed on lung section from at least 3 mice per group. Representative composite images of immunofluorescence (left) are shown at 4x magnification and antibody stains are shown in corresponding colors: Arg1 (orange), CK19 (light blue), DAPI (dark blue), CD3+ (green), F4/80 (pink), CD8+ (yellow). Representative images of computational phenotype are depicted on the right with cell phenotypes in corresponding colors as indicated: macrophages (orange), T cells (green), epithelial (pink), other (grey). B–F, Quantification of F4/80+ macrophages, Arg1+ F4/80 macrophages, and CK19 epithelial, all T cells (CD3) and CD8+ T cells abundance of quantified composite images. Data is represented ± SEM and statistical significance determined by one-way ANOVA with post hoc Fisher least significant difference (LSD) test and denoted as: *, P < 0.05; ***, P < 0.005; or ns, no statistical difference. G, Representative images (20x) of lung section in each group at endpoint stained with Ki-67, CD3+, and CD8+. H–J, Quantification of Ki67, CD3+ T cells and CD8+ T cells counted on lung section stained with corresponding antibody. Data represented ± SEM and statistical significance determined with one-way ANOVA with post hoc Tukey HSD test and significance of: **, P < 0.01; ***, P < 0.001; or ns, not statistically different.
Detection of a unique TME in IPF-LC
To study the relative abundance of immune cells in IPF-LC, lungs from control mice, mice treated with bleomycin (IPF), mice injected with LLC-1 Luc cells (LC), or both (IPF-LC) were obtained at study end point, homogenized, and suspended as single cells for CyTOF analysis (Fig. 3A). Only immune cells (CD45+ cells) were specifically quantified and categorized into different immune cell clusters based on their immune marker expression and previously described naming convention (ref. 40; Fig. 3A). The marker negative (pink) population represents nonimmune cells that are present in the tissue and likely include tumor cells. Confirming our previous observations by mIF, certain macrophages and TAMs were found to be elevated in the IPF-LC group when compared with lungs from IPF or LC mice (Fig. 3B and C). Specifically, macrophages expressing CD274 marker showed a trend toward higher abundance in the IPF-LC group. Immune cells expressing CD11c, but low levels of CD206, here termed TAMs, were significantly more abundant in lungs from IPF-LC mice. However, CD45+, CD3+, Ly6C high myeloid-derived suppressor cells (MDSC), likely tumor-associated neutrophils (TAN), as well as MDSCs expressing Ly6G high showed a downward trend (Fig. 3D; Supplementary Fig. S2). No statistically significant difference in overall B, CD4+ T cell or APC/dendritic cell numbers was determined (see Supplementary Fig. S2). Interestingly, flow cytometry of dissociated lung sections indicated an increase in CD274 marker expression, supporting an immune suppressive microenvironment (Fig. 3E and F). However, CD274 expression on all cells assessed by flow cytometry showed a relatively high expression in control, IPF and IPF-LC groups; this differed from CyTOF analysis, likely indicating CD274 expression on nonimmune cells, which has been described for AT2 cells (41). In summary, our findings suggest TME differences between the IPF-LC and the LC groups, suggestive of an immune suppressive and tumor promoting niche for lung cancer progression and metastases in the IPF-LC group.
Figure 3.

Increased TAMs and CD274 expression in IPF-LC. A, CyTOF of 3 lungs per experimental group was performed at study endpoint (5 weeks post initial bleomycin injection). Representative tSNE plots of CyTOF analysis with specific immune cell clusters depicted and circled in corresponding colors. B–D, Quantification of abundance of cell-specific expression markers for macrophages (CD45+, CD274+, CD11b+, CD206+, CD11c high), TAMs (CD45+, CD206 low, CD11c+), and MDSCs (CD45+, CD3+, Ly6C high), using FlowSOM-viSNE. Data ±SEM with statistical analysis using one-way ANOVA with post hoc Fisher least significant difference (LSD) Test (*, P ≤ 0.05; ns, no statistical difference). E and F, Flow cytometry of lung tissue obtained from 2 mice per group at endpoint stained for CD274 marker expression using anti-CD274-APC. Statistical significance was determined using one-way ANOVA with post hoc Fisher LSD test (*, P ≤ 0.05).
Identification of a unique gene signature of IPF-LC
To gain mechanistic insight into the molecular underpinnings of IPF-LC we performed bulk RNA sequencing (RNA-seq) analysis on lung tissue obtained from control, IPF, LC and IPF-LC mice. Sequencing was performed on lung tissue from 3 mice per group and principal component analysis (see Supplementary Fig. S3) and an unbiased cluster analysis of the sequencing data was performed. A heat map of gene log2 fold changes for the indicated comparisons shows distinct patterns between the groups when compared with control or each other (Fig. 4A). Genes with statistical significance are highlighted with intense blue or red colors based on whether they were down- or upregulated, respectively. We then compared IPF-LC with IPF and LC to expand our initial heat map and found the log2 fold change to be lower for both up- and downregulated genes when comparing these groups than when comparing IPF-LC, IPF, and LC to control (Fig. 4A). To depict overlap and shared up- and downregulated genes, as well as independently regulated genes in the different groups, we used Venn Diagrams (Fig. 4B and C). Interestingly, we identified unique gene signatures for IPF-LC and LC groups, but not for IPF, along with overlap between IPF-LC and LC groups (Fig. 4B and C). Next, we focused on genes uniquely modulated in the IPF-LC group to better understand the molecular pathobiology of IPF-LC. The log2 fold changes for the top 25 up- and downregulated protein-coding genes in IPF-LC are shown in a heat map comparing how the genes are expressed in LC, IPF and IPF-LC groups versus control (Fig. 4D). In summary, we have identified uniquely regulated genes in the IPF-LC groups, which may provide insight into the pathobiology of IPF-LC.
Figure 4.

RNA-seq identifies genes uniquely regulated in IPF-LC. Lung tissue from 3 mice per group was used at endpoint for bulk RNA-seq to evaluate differential gene expression in experimental groups. A, Heat map of gene log2 fold changes for the indicated comparisons. B and C, The intersections of genes significantly differentially expressed in IPF-LC, IPF, and LC (vs. control) are represented by Venn diagrams for upregulated (B) and downregulated (C) genes. D, Heat map of log2 fold changes for the top 25 up- and downregulated protein-coding genes in IPF-LC only. Genes with adjusted P values < 0.05 and fold change > 1.5 were considered significantly differentially expressed.
Pathway analysis identifies immune suppressive, secreted factors uniquely regulated in IPF-LC
To better understand the molecular pathobiology of IPF-LC and identify signaling nodes regulated in this disease, we performed GSEA Hallmark pathway analysis using all genes uniquely regulated in IPF-LC. Interestingly, but not surprisingly, we found that myc, oxidative phosphorylation, DNA repair, E2F targets and mTOR signaling pathways were upregulated, and identified IFNγ response, IFNα response, and inflammatory response among the downregulated pathways (Fig. 5A). Querying genes in the three Hallmark pathways for IFNγ, IFNα, and inflammatory responses in the four experimental groups, we found their expression to be significantly reduced in the IPF-LC group when compared with control (Fig. 5B). This was validated for a select group of genes by qRT-PCR (Fig. 5C). In summary, signaling pathway analysis and confirmatory qRT-PCR identified genes within inflammatory and IFN response pathways to be downregulated upon tumor progression in the IPF-LC group, providing evidence of an immune suppressive TME.
Figure 5.

Pathway analysis identifies secreted factors uniquely regulated in IPF-LC. A, Enrichment of mSigDB Hallmark gene sets (FDR < 0.05) for all IPF-LC-regulated genes (red: upregulated; blue: downregulated). B, Selected genes, which appear in three indicated Hallmark gene sets depicted in (A) differentially expressed in IPF-LC versus control. Heat map of log10 normalized counts (z-score) for these selected genes in all groups: control, IPF, LC, IPF-LC. Symbols in legend indicate in which Hallmark gene sets they appeared (IFNγ response, IFNα response, inflammatory response). C, qRT-PCR of selected genes downregulated in the IPF-LC group, normalized to cyclophilin as housekeeping gene and fold change calculated compared with control. Significance was determined using unpaired Student t test of RNA obtained from lung tissue of 3 mice per control and IPF-LC group. Statistical significance: *, P < 0.05; ****, P < 0.005.
Loss of cytokeratin 7 marks epithelial-to-mesenchymal transition in IPF-LC
Epithelial-to-mesenchymal transition (EMT), the process by which epithelial cells lose their epithelial abilities and become mesenchymal cells and increase their expression of cancer stemness genes, is observed in many malignancies, including lung cancer, when tumors progress. We queried known EMT gene expression to identify a potential EMT in the IPF-LC group denoting more aggressive tumor growth. Genes that are typically expressed in epithelial cell compartments, like Cytokeratin 7/Krt7, Mucin 1/Muc1 and E Cadherin/Cdh1, were downregulated in the IPF-LC group, whereas genes that denote a more mesenchymal phenotype, like Vimentin/Vim, N-Cadherin/Cdh2 and fibroblast-specific protein-1 (also called S100A4), were upregulated (Fig. 6A). Next, we performed IHC on lung tissue obtained from the four different experimental groups and found that Cytokeratin 7 expression (CK7) was completely lost in the IPF-LC group (Fig. 6B), indicating a mesenchymal, more aggressive, and potentially invasive and metastatic phenotype. In summary, the progression of lung cancer observed in IPF-LC may in part be due to EMT.
Figure 6.
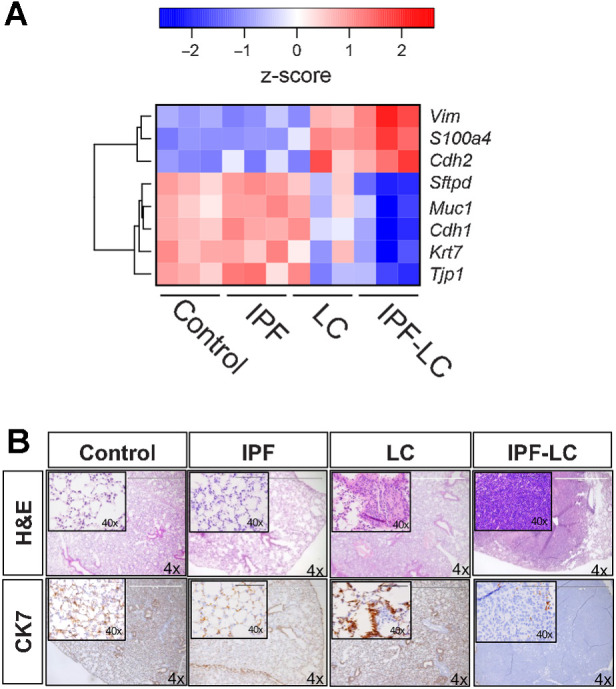
Pronounced EMT in IPF-LC. RNA-seq data as described in Fig. 4 was used to query genes important in EMT. A, Heat map of log10 normalized counts (z-score) for selected mesenchymal and epithelial marker genes in all groups: control, IPF, LC, IPF-LC. B, Representative images (4x; 40x inset) of lung section in each group stained with H&E (top) and Cytokeratin 7 (bottom).
Discussion
In this study we developed and explored a new mouse model for IPF-LC to gain mechanistic insight into the link between IPF and lung cancer and identify potential therapeutic targets to develop future treatment options for patients with IPF-LC. Our model combined the commonly used bleomycin model for IPF with the Lewis lung adenocarcinoma (LLC-1 Luc) model to improve understanding of the lung microenvironment in the development of IPF-LC. We identified an increase in immune cell infiltration that likely contributes to a tumor promoting and immune suppressive microenvironment in IPF-LC, but not in lung cancer or IPF alone. Our findings indicate that macrophages, particularly tumor-associated Arginase 1 positive macrophages, may communicate with T cells to produce a tumor promoting environment that results in growth and proliferation as assessed by preclinical CT and BLI and histologic assessment. Furthermore, we identified a unique gene signature in IPF-LC that provides biological insight into the pathobiology of this disease and may provide future biomarkers and therapeutic targets.
While our findings provide an opportunity for future mechanistic work and testing of therapeutic interventions, we acknowledge that this study and mouse model have some limitations. In bleomycin-induced pulmonary fibrosis models, the rapid onset of disease and apparent resolution within a few weeks is not a faithful representation of human pulmonary fibrosis, which develops over many years and does not resolve on its own (42). Moreover, this model recapitulates only some aspects of IPF (43). The combination of the bleomycin model with the LLC-1 lung tumor metastasis model, which bears the KrasG12C mutation, has previously been explored (44), but has not provided insight into the composition of the tumor microenvironmental or transcriptomic changes in IPF-LC. Further, the LLC-1 model only recapitulates lung cancer with the KrasG12C mutation, but not with other Kras or co-occurring mutations in other oncogenes or tumor suppressors that may affect the development of an immune suppressive, tumor promoting microenvironment differently (25, 45). Supporting the significance of developing and characterizing models for lung cancer associated with lung disease, Herzog and colleagues recently developed a bleomycin-induced lung fibrosis model in combination with a genetically engineered mouse model of NSCLC and showed that fibrosis exacerbated lung cancer progression (20), which could be halted when TGFβ signaling was inhibited (20).
TAMs affect patient responses to chemo-, immuno-, and radiotherapies (46) and TAM abundance correlates with aggressiveness and metastatic potential of tumors (47). It remains to be investigated whether the IPF-induced lung microenvironment results in a polarization of lung resident alveolar macrophages repolarized by disseminated tumor cells that seed in the lung, or in a recruitment of distant, bone marrow derived macrophages that infiltrate the lung to provide a pre-metastatic niche. TAMs provide viable targets for immune therapies (48), and targeted therapies to inhibit or reprogram TAMs that may be used in combination with other therapies are being developed to increase prognosis in cancer patients (49). Our data indicate that preexisting lung disease, like fibrosis, increases the infiltration or polarization of macrophages, specifically M2-like TAMs that express Arginase 1. As such, these TAMs may contribute to the high mortality rates and the low 5-year survival observed in patients with IPF-LC compared with lung cancer patients.
Our data indicate that macrophages and TAMs, but not Ly6C MDSCs, were more abundant in the IPF-LC group. Because MDSC are known to regulate a plethora of immune modulatory processes, including T-cell migration, T-cell death, tumor neovascularization and tumor growth, we were surprised to notice a downward trend of MDSCs in the IPF-LC group. Thus, future studies will include a detailed characterization of tumors associated with IPF to evaluate the role of MDSCs, or TANs, in tumor progression in this group. Furthermore, the identification of TAMs and MDSCs as potential key players in modulating an immunosuppressive, tumor-promoting microenvironment in IPF-LC may provide future opportunities for therapeutic intervention, as it is being explored in other malignancies (50).
In addition, our observation of decreased CD8+ cytotoxic T cells and CD4+ helper T cells, as well as antigen presenting dendritic cells in the IPF-LC compared with the LC group, further supports a more immunosuppressive TME in IPF-LC tumors, as both lymphocytes are required for cell-mediated immune responses in cancer (51). However, disparities were observed among the methods used when analyzing CD8+ or CD4+ T cells, specifically with IHC, mIF or CyTOF. Future studies are needed to discern the role of various T cells in lung cancer associated with lung disease, as CD4+ T helper cells play an important role in maintaining effective antitumor immunity (52) and can differentiate into effector cells needed in the antitumor response. In addition, CD4+ T helper cells primarily mediate antitumor immunity by assisting CD8+ cytotoxic T cells and antibody responses by providing stimulus for priming CD8+ T cells (53). Similarly, the antitumorigenic role of CD8+ cytotoxic T cells, their interaction with CD4+ T cells and other immune cells, and their association with better patient outcome has been well established, and our findings of reduced CD8+ and CD4+ T cell abundance in IPF-LC was therefore not surprising (54–60).
Along with key immune cells that may be contributors to an immune suppressive and thereby tumor proliferative microenvironment in IPF-LC, we also identified genes and signaling pathways that are uniquely modulated in IPF-LC, providing further insight into its pathogenesis. While the significant overlap in gene regulation between the LC and IPF-LC groups that we identified was expected, given the significant seeding and proliferation of tumor cells in both, we were surprised that we were unable to identify genes uniquely regulated in the IPF group. The relatively low dose of bleomycin and fast resolution of the fibrosis phenotype in the bleomycin-induced model may explain these findings. Nevertheless, identification of the unique gene signature detected in IPF-LC provides insight into the signaling nodes regulated in IPF-LC and may provide future therapeutic targets. In fact, the downregulation of the inflammatory response and IFNγ and IFNα-mediated signaling of CD40 is interesting, as downregulation of CD40 has been shown to contribute to accumulation of MDSC in other cancers (61), while downregulation of granzyme A may indicate lack of cytotoxic T cells, which typically produce granzymes to kill aberrant cells, thereby promoting tumor growth (62). These findings corroborate with our data indicating reduction in CD8+ T cells and increased abundance of M2-like TAMs.
Finally, transcriptomic analysis demonstrated that IPF-LC is accompanied by an EMT, which corroborated with a remarkable reduction in cytokeratin 7 staining in IPF-LC, thereby likely contributing to a more aggressive tumor promoting and immune suppressive microenvironment (63). This may indicate that lung disease, especially fibrosis, caused by environmental factors, infections, or genetic predisposition or induced by treatment, may trigger EMT in pre-cancerous cells and/or provide a niche for circulating cancer cells. As bleomycin is used in the treatment of several types of malignancies, including certain types of lymphoma, long-term adverse effects should be considered (64).
In summary, our findings provide rationale for targeting disease associated immune cells and signaling pathways to develop future therapeutic regimens for IPF-LC and the IPF-LC model provides an opportunity to interrogate and therapeutically modulate both the TME and signaling pathways regulated in IPF-LC.
Supplementary Material
Supplemental Figure S1. Transgenic mouse IPF-LC model development.
Supplemental Figure S2. Expression of immune cell surface markers by CyTOF.
Supplemental Figure S3. PCA biplot (PC1 and PC2) of VST-normalized gene counts for the top 500 most variable genes.
Supplemental Table S1. Stains and antibodies used for mIHC.
Supplemental Table S2. Antibody panel used for CyTOF analysis.
Acknowledgments
This work was supported by the University of Michigan Rogel Cancer Center “First and Goal” funding (to S. Galban). Flow cytometry work was performed at the University of Michigan Flow Cytometry Core, which is supported by the NCI of the NIH grants P30 CA046592 and P30 CA04659229. Histology was performed at the Rogel Cancer Center Tissue and Molecular Pathology Shared Resource, which is funded by NCI grant P30 CA04659229. Multiplex immunohistochemistry (mIHC) was performed by the Frankel Lab and supported by the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) of the NIH grant R01 DK128102 (to T.L. Frankel). CyTOF was performed at the Flow Cytometry Core at the University of Rochester Medical Center, with guidance and analysis provided by the Pasca di Magliano Lab, which is supported by NCI grants R01 CA260752, R01 CA271510, U01 CA224145, U01 CA274154, and U54 CA274371 (to M. Pasca di Magliano). The authors wish to thank Michelle Paulsen of the Ljungman Lab for assisting with Bru Sequencing.
Footnotes
Note: Supplementary data for this article are available at Molecular Cancer Research Online (http://mcr.aacrjournals.org/).
Authors' Disclosures
C.J. Galbán reports other support from Imbio, LLC outside the submitted work. T.L. Frankel reports grants from NIH during the conduct of the study. S. Galban reports grants from University of Michigan Rogel Cancer Center during the conduct of the study. No disclosures were reported by the other authors.
Authors' Contributions
I. Barravecchia: Data curation, formal analysis, investigation, writing–review and editing. J.M. Lee: Data curation, methodology, writing–original draft. J. Manassa: Writing–original draft. B. Magnuson: Data curation, software, formal analysis, validation, methodology, writing–original draft, writing–review and editing. S.F. Ferris: Data curation, investigation. S. Cavanaugh: Investigation. N.G. Steele: Data curation, formal analysis. C.E. Espinoza: Investigation. C.J. Galban: Data curation, supervision, investigation. N. Ramnath: Writing–review and editing. T.L. Frankel: Resources, data curation, writing–review and editing. M. Pasca di Magliano: Resources, data curation, writing–review and editing. S. Galban: Conceptualization, resources, data curation, software, formal analysis, supervision, funding acquisition, validation, visualization, methodology, writing–original draft, project administration, writing–review and editing.
References
- 1. Raghu G, Chen S-Y, Yeh W-S, Maroni B, Li Q, Lee Y-C, et al. Idiopathic pulmonary fibrosis in US Medicare beneficiaries aged 65 years and older: incidence, prevalence, and survival, 2001–11. Lancet Respir Med 2014;2:566–72. [DOI] [PubMed] [Google Scholar]
- 2. Cottin V, Richeldi L. Neglected evidence in idiopathic pulmonary fibrosis and the importance of early diagnosis and treatment. Eur Respir Rev 2014;23:106–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nalysnyk L, Cid-Ruzafa J, Rotella P, Esser D. Incidence and prevalence of idiopathic pulmonary fibrosis: review of the literature. Eur Respir Rev 2012;21:355–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lechowicz K, Drożdżal S, Machaj F, Rosik J, Szostak B, Zegan-Barańska M, et al. COVID-19: the potential treatment of pulmonary fibrosis associated with SARS-CoV-2 infection. J Clin Med 2020;9:1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ojo AS, Balogun SA, Williams OT, Ojo OS. Pulmonary fibrosis in COVID-19 survivors: predictive factors and risk reduction strategies. Pulm Med 2020;2020:6175964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. McGroder CF, Zhang D, Choudhury MA, Salvatore MM, D'Souza BM, Hoffman EA, et al. Pulmonary fibrosis 4 months after COVID-19 is associated with severity of illness and blood leucocyte telomere length. Thorax 2021;76:1242–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Oh CK, Murray LA, Molfino NA. Smoking and idiopathic pulmonary fibrosis. Pulm Med 2012, 2012. 808260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.du Bois RM. Strategies for treating idiopathic pulmonary fibrosis. Nat Rev Drug Discov 2010;9:129–40. [DOI] [PubMed] [Google Scholar]
- 9. Taniguchi H, Ebina M, Kondoh Y, Ogura T, Azuma A, Suga M, et al. Pirfenidone in idiopathic pulmonary fibrosis. Eur Respir J 2010;35:821–9. [DOI] [PubMed] [Google Scholar]
- 10. Wollin L, Maillet I, Quesniaux V, Holweg A, Ryffel B. Antifibrotic and anti-inflammatory activity of the tyrosine kinase inhibitor nintedanib in experimental models of lung fibrosis. J Pharmacol Exp Ther 2014;349:209–20. [DOI] [PubMed] [Google Scholar]
- 11. Global Burden of Disease Cancer Collaboration, Fitzmaurice C, Dicker D, Pain A, Hamavid H, Moradi-Lakeh M, MacIntyre MF. et al. The global burden of cancer 2013. JAMA Oncol 2015;1:505–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Siegel RL, Miller KD, Wagle NS, Jemal A. Cancer statistics, 2023. CA Cancer J Clin 2023;73:17–48. [DOI] [PubMed] [Google Scholar]
- 13. Hubbard R, Venn A, Lewis S, Britton J. Lung cancer and cryptogenic fibrosing alveolitis. A population-based cohort study. Am J Respir Crit Care Med 2000;161:5–8. [DOI] [PubMed] [Google Scholar]
- 14. Le Jeune I, Gribbin J, West J, Smith C, Cullinan P, Hubbard R. The incidence of cancer in patients with idiopathic pulmonary fibrosis and sarcoidosis in the UK. Respir Med 2007;101:2534–40. [DOI] [PubMed] [Google Scholar]
- 15. Ozawa Y, Suda T, Naito T, Enomoto N, Hashimoto D, Fujisawa T, et al. Cumulative incidence of and predictive factors for lung cancer in IPF. respirol 2009;14:723–8. [DOI] [PubMed] [Google Scholar]
- 16. Tomassetti S, Gurioli C, Ryu JH, Decker PA, Ravaglia C, Tantalocco P, et al. The impact of lung cancer on survival of idiopathic pulmonary fibrosis. Chest 2015;147:157–64. [DOI] [PubMed] [Google Scholar]
- 17. Yoon JH, Nouraie M, Chen X, Zou RH, Sellares J, Veraldi KL, et al. Characteristics of lung cancer among patients with idiopathic pulmonary fibrosis and interstitial lung disease - analysis of institutional and population data. Respir Res 2018;19:195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kato E, Takayanagi N, Takaku Y, Kagiyama N, Kanauchi T, Ishiguro T, et al. Incidence and predictive factors of lung cancer in patients with idiopathic pulmonary fibrosis. ERJ Open Res 2018;4:00111–2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Houghton AM. Mechanistic links between COPD and lung cancer. Nat Rev Cancer 2013;13:233–45. [DOI] [PubMed] [Google Scholar]
- 20. Herzog BH, Baer JM, Borcherding N, Kingston NL, Belle JI, Knolhoff BL, et al. Tumor-associated fibrosis impairs immune surveillance and response to immune checkpoint blockade in non–small cell lung cancer. Sci Transl Med 15:eadh8005. [DOI] [PubMed] [Google Scholar]
- 21. Liu T, De Los Santos FG, Phan SH. The bleomycin model of pulmonary fibrosis. Methods Mol Biol 2017;1627:27–42. [DOI] [PubMed] [Google Scholar]
- 22. Sugiura K, Stock CC. Studies in a tumor spectrum. III. The effect of phosphoramides on the growth of a variety of mouse and rat tumors. Cancer Res 1955;15:38–51. [PubMed] [Google Scholar]
- 23. Gul A, Yang F, Xie C, Du W, Mohammadtursun N, Wang B, et al. Pulmonary fibrosis model of mice induced by different administration methods of bleomycin. BMC Pulmonary Medicine 2023;23:91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Izbicki G, Segel MJ, Christensen TG, Conner MW, Breuer R. Time course of bleomycin-induced lung fibrosis. Int J Exp Pathol 2002;83:111–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Agalioti T, Giannou AD, Krontira AC, Kanellakis NI, Kati D, Vreka M, et al. Mutant KRAS promotes malignant pleural effusion formation. Nat Commun 2017;8:15205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tichelaar JW, Lu W, Whitsett JA. Conditional expression of fibroblast growth factor-7 in the developing and mature lung. J Biol Chem 2000;275:11858–64. [DOI] [PubMed] [Google Scholar]
- 27. Hardie WD, Le Cras TD, Jiang K, Tichelaar JW, Azhar M, Korfhagen TR. Conditional expression of transforming growth factor-alpha in adult mouse lung causes pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol 2004;286:L741–9. [DOI] [PubMed] [Google Scholar]
- 28. Collins MA, Bednar F, Zhang Y, Brisset J-C, Galbán S, Galbán CJ, et al. Oncogenic Kras is required for both the initiation and maintenance of pancreatic cancer in mice. J Clin Invest 2012;122:639–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Smith MCP, Luker KE, Garbow JR, Prior JL, Jackson E, Piwnica-Worms D, et al. CXCR4 regulates growth of both primary and metastatic breast cancer. Cancer Res 2004;64:8604–12. [DOI] [PubMed] [Google Scholar]
- 30. Bowman BM, Sebolt KA, Hoff BA, Boes JL, Daniels DL, Heist KA, et al. Phosphorylation of FADD by the kinase CK1alpha promotes KRASG12D-induced lung cancer. Sci Signal 2015;8:ra9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lazarus J, Maj T, Smith JJ, Perusina Lanfranca M, Rao A, D'Angelica MI, et al. Spatial and phenotypic immune profiling of metastatic colon cancer. JCI Insight 2018;3:e121932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. McGue JJ, Edwards JJ, Griffith BD, Frankel TL. Multiplex fluorescent immunohistochemistry for preservation of tumor microenvironment architecture and spatial relationship of cells in tumor tissues. Methods Mol Biol 2023;2660:235–46. [DOI] [PubMed] [Google Scholar]
- 33. Steele NG, Carpenter ES, Kemp SB, Sirihorachai VR, The S, Delrosario L, et al. Multimodal mapping of the tumor and peripheral blood immune landscape in human pancreatic cancer. Nat Cancer 2020;1:1097–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Van Gassen S, Callebaut B, Van Helden MJ, Lambrecht BN, Demeester P, Dhaene T, et al. FlowSOM: Using self-organizing maps for visualization and interpretation of cytometry data. Cytometry A 2015;87:636–45. [DOI] [PubMed] [Google Scholar]
- 35. Iyer A, Hamers AAJ, Pillai AB. CyTOF for the Masses. Front Immunol 2022;13:815828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ruscitti F, Ravanetti F, Essers J, Ridwan Y, Belenkov S, Vos W, et al. Longitudinal assessment of bleomycin-induced lung fibrosis by micro-CT correlates with histological evaluation in mice. Multidiscip Respir Med 2017;12:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Fisher GH, Wellen SL, Klimstra D, Lenczowski JM, Tichelaar JW, Lizak MJ, et al. Induction and apoptotic regression of lung adenocarcinomas by regulation of a K-Ras transgene in the presence and absence of tumor suppressor genes. Genes Dev 2001;15:3249–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Xu G-P, Li Q-Q, Cao X-X, Chen Q, Zhao Z-H, Diao Z-Q, et al. The effect of TGFβ1 and Smad7 gene transfer on the phenotypic changes of rat alveolar epithelial cells. Cell Mol Biol Lett 2007;12:457–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhang Y, Lazarus J, Steele NG, Yan W, Lee H-J, Nwosu ZC, et al. Regulatory T-cell depletion alters the tumor microenvironment and accelerates pancreatic carcinogenesis. Cancer Discov 2020;10:422–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ahmadvand N, Carraro G, Jones MR, Shalashova I, Noori A, Wilhelm J, et al. Cell-surface programmed death ligand-1 expression identifies a sub-population of distal epithelial cells enriched in idiopathic pulmonary fibrosis. Cells 2022;11:1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chua F, Gauldie J, Laurent GJ. Pulmonary fibrosis: searching for model answers. Am J Respir Cell Mol Biol 2005;33:9–13. [DOI] [PubMed] [Google Scholar]
- 43. Usuki J, Fukuda Y. Evolution of three patterns of intra-alveolar fibrosis produced by bleomycin in rats. Pathol Int 1995;45:552–64. [DOI] [PubMed] [Google Scholar]
- 44. Orr FW, Adamson IY, Young L. Promotion of pulmonary metastasis in mice by bleomycin-induced endothelial injury. Cancer Res 1986;46:891–7. [PubMed] [Google Scholar]
- 45. El-Telbany A, Ma PC. Cancer genes in lung cancer: racial disparities: are there any? Genes Cancer 2012;3:467–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P. Tumor-associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol 2017;14:399–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Nardin A, Abastado JP. Macrophages and cancer. Front Biosci 2008;13:3494–505. [DOI] [PubMed] [Google Scholar]
- 48. Anfray C, Ummarino A, Torres Andón F, Allavena P. Current strategies to target tumor-associated macrophages to improve antitumor immune responses. Cells 2019;9:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhang S-Y, Song X-Y, Li Y, Ye L-L, Zhou Q, Yang W-B. Tumor-associated macrophages: a promising target for a cancer immunotherapeutic strategy. Pharmacol Res 2020:105111. [DOI] [PubMed] [Google Scholar]
- 50. Law AMK, Valdes-Mora F, Gallego-Ortega D. Myeloid-derived suppressor cells as a therapeutic target for cancer. Cells 2020;9:561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Rad FR, Ajdary S, Omranipour R, Alimohammadian MH, Hassan ZM. Comparative analysis of CD4 +and CD8+T cells in tumor tissues, lymph nodes and the peripheral blood from patients with breast cancer. Iran Biomed J 2015;19:35–44. [Google Scholar]
- 52. Hung K, Hayashi R, Lafond-Walker A, Lowenstein C, Pardoll D, Levitsky H. The central role of CD4(+) T cells in the antitumor immune response. J Exp Med 1998;188:2357–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Topalian SL, Taube JM, Anders RA, Pardoll DM. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat Rev Cancer 2016;16:275–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Shimizu S, Hiratsuka H, Koike K, Tsuchihashi K, Sonoda T, Ogi K, et al. Tumor-infiltrating CD8(+) T-cell density is an independent prognostic marker for oral squamous cell carcinoma. Cancer Med 2019;8:80–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Shinagawa N, Yamazaki K, Tamura Y, Imai A, Kikuchi E, Yokouchi H, et al. Immunotherapy with dendritic cells pulsed with tumor-derived gp96 against murine lung cancer is effective through immune response of CD8+cytotoxic T lymphocytes and natural killer cells. Cancer Immunol Immunother 2008;57:165–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Dranoff G. Cytokines in cancer pathogenesis and cancer therapy. Nat Rev Cancer 2004;4:11–22. [DOI] [PubMed] [Google Scholar]
- 57. Curtsinger JM, Lins DC, Johnson CM, Mescher MF. Signal 3 tolerant CD8 T cells degranulate in response to antigen but lack granzyme B to mediate cytolysis. J Immunol 2005;175:4392–9. [DOI] [PubMed] [Google Scholar]
- 58. Trapani JA, Sutton VR. Granzyme B: pro-apoptotic, antiviral and antitumor functions. Curr Opin Immunol 2003;15:533–43. [DOI] [PubMed] [Google Scholar]
- 59. Slifka MK, Whitton JL. Antigen-specific regulation of T cell-mediated cytokine production. Immunity 2000;12:451–7. [DOI] [PubMed] [Google Scholar]
- 60. Russell JH, Ley TJ. Lymphocyte-mediated cytotoxicity. Annu Rev Immunol 2002;20:323–70. [DOI] [PubMed] [Google Scholar]
- 61. Shen J, Chen X, Wang Z, Zhang G, Chen W. Downregulation of CD40 expression contributes to the accumulation of myeloid-derived suppressor cells in gastric tumors. Oncol Lett 2014;8:775–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Cullen SP, Brunet M, Martin SJ. Granzymes in cancer and immunity. Cell Death Differ 2010;17:616–23. [DOI] [PubMed] [Google Scholar]
- 63. Bartis D, Mise N, Mahida RY, Eickelberg O, Thickett DR. Epithelial–mesenchymal transition in lung development and disease: does it exist and is it important? Thorax 2014;69:760–5. [DOI] [PubMed] [Google Scholar]
- 64. Sleijfer S. Bleomycin-induced pneumonitis. Chest 2001;120:617–24. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure S1. Transgenic mouse IPF-LC model development.
Supplemental Figure S2. Expression of immune cell surface markers by CyTOF.
Supplemental Figure S3. PCA biplot (PC1 and PC2) of VST-normalized gene counts for the top 500 most variable genes.
Supplemental Table S1. Stains and antibodies used for mIHC.
Supplemental Table S2. Antibody panel used for CyTOF analysis.
Data Availability Statement
The datasets supporting the current study are available from the corresponding author upon request and available in the NIH Gene Expression Omnibus (GEO) database (RRID:SCR_005012). RNA raw data files and analysis was deposited with GEO accession number GSE224134. Material or data that require a Material Transfer Agreement (MTA) can be provided by the University of Michigan pending scientific review and the execution of an MTA negotiated by the university's Office of Technology Transfer. Requests for data that require an MTA should be submitted to the corresponding author, Dr. Stefanie Galban, sgalban@umich.edu.