

Significance
Invariant natural killer T (iNKT) cells are innate-like T lymphocytes that participate in the immune response to microbial pathogens by bridging innate and acquired immunity. Among several effector subsets of iNKT cells, follicular helper NKT (NKTFH) cells contribute to helping antibody production. However, the mechanism of NKTFH differentiation remains elusive. This study shows that interleukin-27, a cytokine derived from Gr-1+ monocytes and macrophages, is important for NKTFH differentiation via supporting the energetic demand by stimulating mitochondrial metabolism. Furthermore, our data demonstrate that NKTFH cells are important for protecting against Streptococcus pneumoniae infection through stimulating IgG production by vaccination. Our results on the mechanisms of iNKT cell–mediated vaccination provide insights into an important and broadly applicable vaccination strategy.
Keywords: iNKT, follicular helper, IL-27, Streptococcus pneumoniae, mitochondrial metabolism
Abstract
Invariant natural killer T (iNKT) cells are innate-like T lymphocytes that express an invariant T cell receptor α chain and contribute to bridging innate and acquired immunity with rapid production of large amounts of cytokines after stimulation. Among effecter subsets of iNKT cells, follicular helper NKT (NKTFH) cells are specialized to help B cells. However, the mechanisms of NKTFH cell differentiation remain to be elucidated. In this report, we studied the mechanism of NKTFH cell differentiation induced by pneumococcal surface protein A and α-galactosylceramide (P/A) vaccination. We found that Gr-1+ cells helped iNKT cell proliferation and NKTFH cell differentiation in the spleen by producing interleukin-27 (IL-27) in the early phase after vaccination. The neutralization of IL-27 impaired NKTFH cell differentiation, which resulted in compromised antibody production and diminished protection against Streptococcus pneumoniae infection by the P/A vaccine. Our data indicated that Gr-1+ cell–derived IL-27 stimulated mitochondrial metabolism, meeting the energic demand required for iNKT cells to differentiate into NKTFH cells. Interestingly, Gr-1+ cell–derived IL-27 was induced by iNKT cells via interferon-γ production. Collectively, our findings suggest that optimizing the metabolism of iNKT cells was essential for acquiring specific effector functions, and they provide beneficial knowledge on iNKT cell–mediated vaccination-mediated therapeutic strategies.
Bridging innate and adaptive immunity is imperative for effective protection against microbial infections. Invariant natural killer T (iNKT) cells are a unique subset of T lymphocytes with a memory-like phenotype, and expression of an invariant T cell receptor (TCR) α chain composed of a Vα14-Jα18 (Trav11-Traj18) rearrangement in mice (1–5). iNKT cells recognize glycolipid antigens presented by CD1d, a nonclassical MHC class I molecule (6). Following TCR stimulation by a potent synthetic glycolipid antigen α-galactosylceramide (α-GalCer), iNKT cells proliferate very rapidly and produce a large amount of cytokines, such as interferon-γ (IFNγ), interleukin-4 (IL-4), and interleukin-17 (7). As a result, iNKT cells play a key role in modulating immune responses such as infection control, autoimmunity, and antitumor immunity.
iNKT cells differentiate into effector cells during their development in the thymus, leading to the emergence of distinct subsets termed NKT1, NKT2, and NKT17 cells (3, 8–11). On the other hand, the differentiation of iNKT cells into other functional subsets, such as follicular helper NKT (NKTFH) cells, occurs in the periphery following TCR stimulation by α-GalCer (12, 13). NKTFH cells exhibit a specific phenotype and functions similar to follicular helper T (TFH) cells, including expression of the chemokine receptor CXCR5, the inhibitory receptor PD-1, and the transcription factor Bcl-6, in addition to secreting interleukin-21 (IL-21). Consistent with these phenotypic features, previous reports have suggested that NKTFH cells support germinal center (GC) formation and antibody production, which is dependent on IL-21 (12, 13). However, the mechanistic understanding remains very limited as to how iNKT cells acquire a follicular helper phenotype.
An effective immune response is critically dependent on energy metabolism (14). The cellular metabolism of T cells plays pivotal and distinct roles in their proliferation and acquisition of effector functions (15, 16). Upon antigen recognition, T cells switch metabolism from oxidative phosphorylation (OXPHOS) and fatty acid oxidation to aerobic glycolysis. In contrast, iNKT cells exhibit basally low glycolysis and mainly rely on OXPHOS, even upon activation (17, 18). After stimulation, augmentation of OXPHOS in activated iNKT cells allows for efficient generation of a high amount of adenosine triphosphate (ATP), which drives their massive proliferation and cytokine production. Recently, the importance of OXPHOS rather than glycolysis in the metabolic regulation of TFH cell differentiation has been reported (19). However, it remains to be explored which of these metabolic pathways are critical for the differentiation of NKTFH cells.
In this report, we studied the mechanism of NKTFH cell differentiation by using α-GalCer and pneumococcal surface protein A (PspA), which is a vaccine candidate against Streptococcus pneumoniae, the leading cause of community-acquired pneumonia (20–26). We found that Gr-1+CD11b+ cells (Gr-1+ cells) played an important role in the proliferation of iNKT cells and differentiation into NKTFH cells in the spleen after PspA/α-GalCer (P/A) vaccination. We examined the mechanism of Gr-1+ cell–mediated NKTFH differentiation and the metabolic regulation of iNKT cells. We also have elucidated how Gr-1+ cells are stimulated after glycolipid-mediated activation of iNKT cells. Furthermore, we have addressed whether the differentiation into NKTFH cells is necessary for vaccine-elicited protection against S. pneumoniae infection.
Results
Gr-1+ Cells Support iNKT Cell Proliferation and NKTFH Cell Differentiation.
To investigate the mechanism underlying NKTFH cell differentiation, we first assessed the kinetics of the splenic iNKT cell population in mice after vaccination with a mixture of α-GalCer and recombinant PspA protein (P/A). Following immunization, there was a transient increase in the number of iNKT cells in the spleen (Fig. 1A). The incorporation of 5-Ethynyl-2′-deoxyuridine (EdU) suggested that iNKT cells proliferated, perhaps in the spleen (SI Appendix, Fig. S1A), although migration of iNKT cells from other tissues may also have occurred. A previous report has shown that iNKT cells can differentiate into either follicular or nonfollicular phenotype cells after α-GalCer stimulation, and the former was defined as NKTFH cells and the latter as effector NKT (NKTeff) cells (27–29). These subsets have distinct transcriptional gene programs (27). Consistent with the previous report, we observed CXCR5+ PD-1+ iNKT (NKTFH) cells in the spleen after P/A immunization (Fig. 1B). The number of iNKT cells reached a peak at day 3 (Fig. 1A), when the cells were incorporating EdU (SI Appendix, Fig. S1A), and then declined. The percentage of NKTFH cells reached a later peak at day 5 (Fig. 1C). Our data suggested that NKTFH cells may be derived from proliferating iNKT cells, with higher EdU incorporation as compared to CXCR5− PD-1− iNKT (NKTeff) cells (Fig. 1D). The NKTFH cells induced by P/A immunization had the capacity to produce Il4 and Il21 transcripts, as measured using cytokine gene reporter mice analyzed ex vivo without stimulation, indicating their potential role in initiating GC formation and efficient antibody production (Fig. 1E) (30).
Fig. 1.

Gr-1+ cells play a crucial role in proliferation of iNKT cells and differentiation of NKTFH cells. (A) Kinetics of the absolute number of iNKT cells after P/A immunization. (B) Flow cytometry plots showing NKTFH cells on day 5 after P/A immunization. (C) Kinetics of frequency of iNKT cells and NKTFH as a percentage of iNKT cells after P/A immunization. (D) Percentage incorporating EdU of naive iNKT cells, NKTeff cells, and NKTFH cells on day 3 after P/A immunization. (E) Flow cytometry plots and percentage of IL-4 and IL-21 produced by NKTFH cells on day 5 after P/A immunization. (F) Confocal immunofluorescence microscopy of the spleen on day 3 after P/A immunization. (G) Flow cytometry plots and absolute numbers of NKTFH cells from mice on day 5 after P/A immunization treated with anti-Gr-1 antibody or isotype control. (H) Flow cytometry plots and absolute numbers of NKTFH cells from mice on day 5 after P/A immunization treated with anti-Gr-1 antibody or isotype control at indicated time points. Isotype control was administered on day 1 after P/A immunization. Data are pooled from two experiments (A and C) with two to seven mice per group and are representative of two independent experiments. Data in (D, E, G, and H) with three to five mice per group. Statistical analyses were performed using one-way ANOVA with Tukey’s correction (D, G, and H); *P < 0.05, ***P < 0.001, ****P < 0.0001; ns, not significant.
As α-GalCer induces strong TCR stimulation of iNKT cells, robust TCR signaling is expected to be essential for generating NKTFH cells. During thymic development, iNKT cells are believed to receive strong TCR signaling by intrinsic ligands (31). Although it is not possible to precisely compare these intrinsic signals to those mediated by α-GalCer, unlike other iNKT subsets such as NKT1, NKT2, and NKT17 cells, NKTFH cells are not observed in the thymus (3, 8–11). Therefore, we hypothesized that certain cells in the periphery support differentiation of iNKT cells into the follicular phenotype. To identify these “helper cells,” we initially analyzed the localization of iNKT cells in naïve mice. Consistent with a prior report, iNKT cells predominantly localized in the red pulp (SI Appendix, Fig. S1B) (32). Notably, some iNKT cells colocalized with Gr-1+ cells, in the red pulp following P/A immunization (Fig. 1F). To examine the involvement of Gr-1+ cells in NKTFH cell differentiation, we administrated an anti-Gr-1 antibody for specific cell depletion (SI Appendix, Fig. S1C). Injection of anti-Gr-1 antibody 1 d after P/A immunization significantly decreased NKTFH cell differentiation on day 5 (Fig. 1G). This decrease was accompanied by a reduction in the total number of iNKT cells (SI Appendix, Fig. S1D). As NKTFH cells were more proliferative (Fig. 1D), the reduction of NKTFH cells could have been due to a blockade of iNKT cell proliferation and NKTFH differentiation, although effects on cell survival were not ruled out.
Next, to elucidate the timing of Gr-1+ cell involvement in iNKT cell proliferation, anti-Gr-1 antibody was administrated at various time points (SI Appendix, Fig. S1E). Injection of anti-Gr-1 antibody, 1 d before and after P/A immunization, significantly decreased NKTFH cells, unlike 3 d after P/A immunization, which had no effect (Fig. 1H). These results suggested that Gr-1+ cells regulated iNKT cell proliferation and NKTFH cell differentiation in the early phase after P/A immunization.
Gr-1+ Cell–Derived IL-27 Regulated NKTFH Cell Differentiation.
To identify the key attribute of Gr-1+ cells that influenced NKTFH cell differentiation, we isolated splenic Gr-1+ cells before and on days 1 and 3 after P/A immunization (SI Appendix, Fig. S2A) and performed RNA sequence (RNA-seq) analysis. Principle component analysis (PCA) revealed that Gr-1+ cells on day 1 after P/A immunization showed a unique gene expression profile compared with those before immunization or on day 3 after P/A immunization (Fig. 2A). Considering the significant decrease of NKTFH cells by the depletion of Gr-1+ cells one day after P/A immunization (Fig. 1H), we inferred that Gr-1+ cells present on day 1 following P/A immunization played a crucial role in both iNKT cell proliferation and NKTFH cell differentiation. Among the cytokine-related gene transcripts whose expression was increased on day 1 and decreased on day 3 after P/A immunization (Fig. 2B), our initial focus was directed to the Il27a gene encoding the IL-27p28 protein, because this cytokine has previously been indicated to play a role in the induction of TFH cell differentiation (33, 34). However, the mechanism of IL-27-mediated TFH cell differentiation remains to be elucidated. IL-27 is a member of the IL-6/IL-12 heterodimeric cytokine family and is composed of IL-27p28 and Ebi3 (Epstein-Barr virus induced 3) protein (35). We confirmed that splenic iNKT cells expressed the IL-27 receptor α chain on the cell surface (SI Appendix, Fig. S2B), and the phosphorylation of STAT1 and STAT3, downstream of the IL-27 receptor, was observed with recombinant IL-27 stimulation of iNKT cells (SI Appendix, Fig. S2C). Because Gr-1+ cells transiently expressed mRNA of Il27 and EBi3 on day 1 following P/A immunization (Fig. 2C), we attempted to detect IL-27 protein in Gr-1+ cells by using an in vivo-Golgi stop method modified from a published protocol (36). One day after P/A immunization, mice received an intravenous injection of monensin, an inhibitor of intracellular protein transport used for enhanced intracellular detection of proteins, including cytokines, and about 18 h later, we analyzed IL-27-producing cells with intracellular cytokine staining. IL-27p28 was detected in approximately 20% of Gr-1+ cells on day 1 after P/A immunization. In contrast, naive Gr-1+ cells did not produce this protein (Fig. 2D). Although a previous report shows that several immune cells produce IL-27 in a context-dependent manner (35), our findings suggested that Gr-1+ cells in the spleen were the primary producers of IL-27 (Fig. 2E).
Fig. 2.
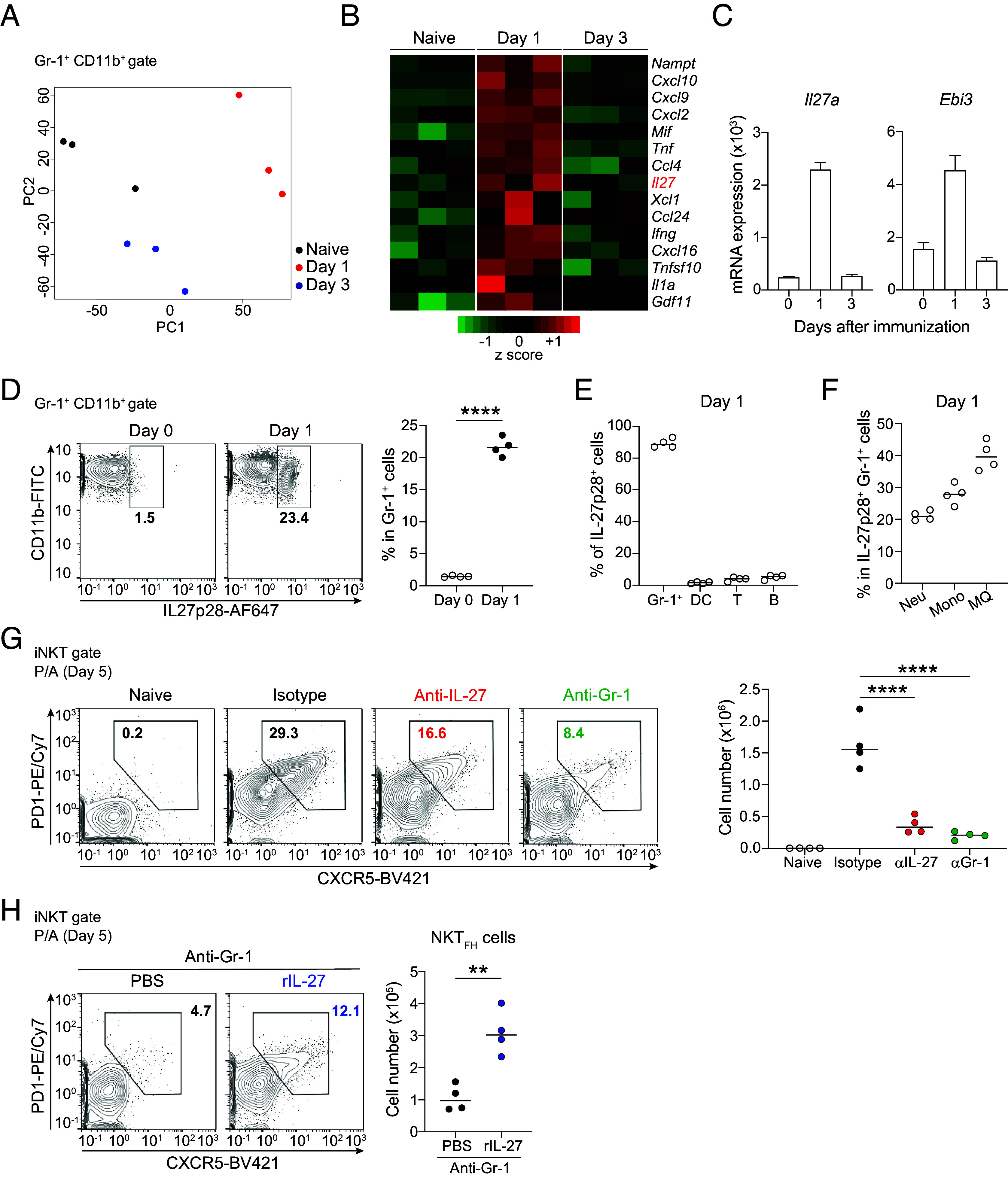
IL-27 derived from Gr-1+ cells is required for maximal proliferation of iNKT cells and differentiation of NKTFH cells. (A) PCA plot of RNA-seq data from naive Gr-1+ cells, Gr-1+ cells on day 1, and Gr-1+ cells on day 3 after P/A immunization. Each symbol represents the analysis of cells from a single mouse. (B) Heat map of genes differentially expressed in experimental replicates between naive Gr-1+ cells, Gr-1+ cells on day 1, and Gr-1+ cells on day 3 after P/A immunization. (C) Il27A and Ebi3 mRNA expression in naive Gr-1+ cells, Gr-1+ cells on day 1, and Gr-1+ cells on day 3 after P/A immunization. (D) Flow cytometry plots and frequency of IL-27 p28 expression in naive Gr-1+ cells and Gr-1+ cells on day 1 after P/A immunization. (E) Frequency of IL-27 p28-producing cells on day 1 after P/A immunization. (DC: dendritic cell, T: T cell, B: B cell). (F) Frequency of percentage of IL-27 p28-producing Gr-1+ cells on day 1 after P/A immunization. (Neu: neutrophil, Mono: monocyte, MQ: macrophage). (G) Flow cytometry plots and absolute numbers of NKTFH cells from naive mice, mice on day 5 after P/A immunization treated with anti-IL-27 antibody, anti-Gr-1 antibody, or isotype control. (H) Flow cytometry plots and absolute numbers of NKTFH cells from Gr-1+ cell–depleted mice on day 5 after P/A immunization treated with or without rIL-27. Data are representative of two independent experiments (C–H) with four mice per group (D–H), and error bars represent mean ± SD. Statistical analyses were performed using unpaired Student’s t test (D and H) or one-way ANOVA with Tukey’s correction (G); **P < 0.01, ****P < 0.0001.
We gated myeloid cell subsets (SI Appendix, Fig. S2D), and among Gr-1+ cells, macrophages and monocytes were suggested to be primary sources of IL-27 in the early phase of P/A immunization, although more than 20% of neutrophils also produced IL-27 (Fig. 2F). Injection of anti-Gr-1 antibody depleted not only monocytes and macrophages but also neutrophils (SI Appendix, Fig. S2 E and F). Thus, to assess the primary sources of IL-27, we used an anti-Ly-6G antibody, which depleted only neutrophils (SI Appendix, Fig. S2G), and thereby tested the role of neutrophils in NKTFH cell differentiation. Even in the absence of neutrophils (SI Appendix, Fig. S2H), the number of NKTFH cells was unreduced (SI Appendix, Fig. S2I). These results suggested that monocytes and macrophages play an important role in NKTFH differentiation following this immunization.
We then analyzed the functional role of IL-27 in the proliferation and differentiation of iNKT cells by administrating neutralizing IL-27 antibodies into mice (SI Appendix, Fig. S3A). Treatment with anti-IL-27 antibodies impaired iNKT cell proliferation and NKTFH cell differentiation on day 5 after P/A immunization, with the decreased number of NKTFH cells comparable to the decrease in mice treated with anti-Gr-1 antibodies (Fig. 2G and SI Appendix, Fig. S3B). To confirm Gr-1+ cell–derived IL-27 is necessary for optimal NKTFH cell differentiation, recombinant IL-27 protein (rIL-27) was intravenously (i.v.) injected into the anti-Gr-1 antibody-treated mice after P/A immunization (SI Appendix, Fig. S3C). The treatment with rIL-27 partially rescued iNKT cell proliferation and NKTFH cell differentiation (Fig. 2H and SI Appendix, Fig. S3D). Therefore, these results suggested that IL-27 was involved in the differentiation of NKTFH cells accompanied by iNKT cell proliferation.
IL-27 Supports Mitochondrial Metabolism for the Bioenergetic Demand of iNKT Cells.
To explore the role of IL-27 in iNKT cells, we performed RNA sequence analysis of iNKT cells sorted from spleens of naïve, P/A immunized isotype control-treated, and P/A immunized anti-IL27 antibody-treated mice (SI Appendix, Fig. S4A). We identified 60 genes whose expression was increased after P/A immunization and decreased by IL-27 neutralization (Fig. 3A). Further gene enrichment analysis revealed that the up-regulated genes were associated with mitochondrial biogenesis in iNKT cells (Fig. 3B). Intriguingly, the transcript levels of genes categorized under “flavin adenine dinucleotide binding” and “mitochondrial inner membrane” were particularly enriched in the up-regulated gene set after P/A immunization and were decreased following IL-27 neutralization (Fig. 3C).
Fig. 3.

IL-27 supports metabolic adaptation of iNKT cells by enhancing the mitochondrial metabolism. (A) Venn diagram analysis of differences in gene expression comparing isotype control/naive >2.0 increase and isotype control/anti-IL-27 antibody >2.0 expression increase. (B) Enrichment scores of isotype control/naive >2.0 population and isotype control/anti-IL-27 antibody >2.0 population. (C) Heat map of genes differentially expressed between iNKT cells from naive mice, mice on day 3 after P/A immunization treated with anti-IL-27 antibody, or isotype control. (D) gMFI of iNKT cells from mice on day 3 after P/A immunization treated with anti-IL-27 antibody or isotype control showing mitochondrial mass, potential, and ROS measured by staining with MitoTracker, TMRM, and MitoSox, respectively. (E) gMFI of iNKT cells and CD4+T cells cultured with and without rIL-27 showing mitochondrial mass, potential, and ROS measured by staining with MitoTracker, TMRM, and MitoSox. (F) TEM showing the mitochondrial morphology of iNKT cells cultured with and without rIL-27. (G) The number and area of mitochondria of iNKT cells cultured with and without rIL-27. (H) Intracellular levels of ATP in iNKT cells cultured with and without rIL-27. Data are representative of two independent experiments (D, E, and H) with three to four mice per group and error bars represent mean ± SD. Representative TEM images are shown (F). (Scale bar, 2.0 μm.) The number and area of mitochondria were quantified from more than 50 cells per group (G). Statistical analyses were performed using unpaired Student’s t test (D, E, G, and H) *P < 0.05, **P < 0.01, ***P < 0.001; ns, not significant.
Recent studies have demonstrated that iNKT cells heavily rely on mitochondrial functions, especially OXPHOS, to fulfill their metabolic demands for cell proliferation and cytokine expression (17, 18). We observed that NKTFH cells showed a greater proliferation rate than NKTeff cells (Fig. 1D), and that neutralization of IL-27 diminished EdU incorporation by iNKT cells (SI Appendix, Fig. S4B). Therefore, we postulated that IL-27 plays a crucial role in optimizing the bioenergetic state of iNKT cells, which subsequently promoted their proliferation and differentiation into NKTFH cells. To assess the impact of IL-27 on mitochondrial metabolism in iNKT cells, we analyzed mitochondrial mass, ΔΨm, and mitochondrial reactive oxygen species (ROS) in iNKT cells following IL-27 neutralization. Three days posttreatment, the mean fluorescence intensity (MFI) of mitochondrial mass, ΔΨm, and mitochondrial ROS in iNKT cells from anti-IL-27 antibody-treated mice were significantly reduced in comparison to their control counterparts (Fig. 3D). To determine whether this metabolic change was specific to iNKT cells, we employed a competitive culture system utilizing isolated CD8− B220− splenic fractions, predominantly consisting of CD4+ T cells with approximately 1% of iNKT cells. Following 2 d of stimulation with plate-bound CD3ε antibody plus soluble CD28 antibody with rIL-27, we observed that iNKT cells cultured with rIL-27 significantly increased mitochondrial function, while CD4+ T cells in the same cultures did not (Fig. 3E). Consistently, the expressions of mitochondrial biogenesis-related genes were increased in iNKT cells treated with rIL-27 (SI Appendix, Fig. S4C). Thus, IL-27 induced cell type–specific effects on mitochondrial metabolism. This interpretation is supported by a previous report indicating that IL-27 selectively augments mitochondrial spare respiratory capacity in vaccine-elicited CD8+ T cells (37).
To confirm alterations in the mitochondrial potential of rIL-27-treated iNKT cells, we performed morphological analysis using transmission electron microscopy (TEM) (Fig. 3F). Although the number of mitochondria slightly decreased in iNKT cells following rIL-27 treatment, the change was not significant (Fig. 3G). On the other hand, the area of mitochondria was significantly increased in iNKT cells (Fig. 3G) but not in CD4+ T cells (SI Appendix, Fig. S4D), implying increased mitochondrial fusion in iNKT cells. These observations were consistent with the increased mitochondrial membrane potential (Fig. 3E). While mitochondrial fission is correlated with facilitated aerobic glycolysis, such as that observed in effector T cells, mitochondrial fusion is observed in resting or memory T cells that rely on OXPHOS for bioenergetic demands (38). Therefore, TEM analysis data revealed that IL-27 facilitated mitochondrial potential likely with mitochondrial biogenesis and fusion. Consistent with the facilitated mitochondrial capacity, iNKT cells cultured with IL-27 expressed higher levels of intracellular ATP, suggesting that IL-27 facilitated OXPHOS in iNKT cells (Fig. 3H). Collectively, these results indicated that IL-27 plays a pivotal role in regulating mitochondrial potential through the modulation of mitochondrial biogenesis-related genes, thereby facilitating the bioenergetic demands needed for iNKT cell proliferation and the subsequent differentiation into NKTFH cells.
iNKT Cells Induce IL-27-Producing Gr-1+ Cells via IFNγ Production.
IL-27-producing Gr-1+ cells were induced 1 d after P/A immunization, but not in the naive condition (Fig. 2 B–D). Since there was no difference in the number of Gr-1+ cells between day 0 and day 1 after P/A immunization (SI Appendix, Fig. S5A), these findings led us to postulate that a reprogramming factor transforms Gr-1+ cells into IL-27 producers. To investigate this possibility, we conducted a gene set enrichment analysis (GSEA) using the gene sets of Gr-1+ cells on day 0 and day 1 after P/A immunization (Fig. 2B). The results revealed that the signature genes for the response to IFNγ were significantly increased in Gr-1+ cells on day 1 after P/A immunization (Fig. 4A). This result is consistent with the fact that α-GalCer administration induces IFNγ from iNKT cells through strong TCR stimulation. Indeed, our data showed that approximately 60% of iNKT cells intensively produced IFNγ without restimulation 2 h after P/A immunization (Fig. 4B).
Fig. 4.

IFNγ from activated iNKT cells reprograms Gr-1+ cells to produce IL-27. (A) GSEA of response to IFNγ genes comparing naive Gr-1+ cells and Gr-1+ cells on day 1 after P/A immunization. (B) Flow cytometry plots and frequency of IFNγ producing iNKT cells from naive mice and 2 h after P/A immunization. (C) Il27a and Ebi3 mRNA expression in Gr-1+ cells cultured with and without IFNγ for 3 h. (D) Flow cytometry plots and absolute numbers of Gr-1+ cells on day 1 after P/A immunization treated with anti-IFNγ antibody or isotype control. (E) Absolute numbers of NKTFH cells from mice on day 5 after P/A immunization treated with anti-IFNγ antibody, anti-Gr-1 antibody, or isotype control. Data are representative of two independent experiments (B–E) with four mice per group (B, D, and E) and error bars represent mean ± SD. Statistical analyses were performed using unpaired Student’s t test (B) or one-way ANOVA with Tukey’s correction (D and E); *P < 0.05, ***P < 0.001, ****P < 0.0001.
To assess the potential for reprograming with IFNγ, we cultured isolated Gr-1+ cells with IFNγ. Short-term cultivation with IFNγ resulted in increased gene expression of Il27a and Ebi3 in isolated Gr-1+cells (Fig. 4C). In addition, in vivo treatment with anti-IFNγ antibody significantly reduced IL-27 producing Gr-1+ cells (Fig. 4D). Thus, IFNγ had the potential to reprogram gene expression in Gr-1+ cells to promote IL-27 production. Importantly, treatment with an anti-IFNγ antibody significantly reduced the cell number of NKTFH cells, similar to treatment with an anti-Gr-1 antibody (Fig. 4E). These results suggested that iNKT cell–derived IFNγ reprogramed gene expression in Gr-1+ cells toward an NKTFH cell phenotype.
Consistent with a previous report (39), P/A immunization also induced TCR downregulation/reappearance in vivo and abolished secondly responsiveness of iNKT cells to α-GalCer similarly to α-GalCer alone (SI Appendix, Fig. S5 B and C). However, our data suggested that iNKT cell proliferation and NKTFH differentiation via IL-27 was not hindered as long as initial IFNγ production is induced by P/A immunization (Figs. 2G and 4 B and E).
IL-27 Confers iNKT-Mediated Protection against Systemic Bacterial Infection.
Previous reports have indicated that NKTFH cells serve as helper cells for B cells (12, 13). Therefore, we sought to evaluate whether P/A immunization induced the production of PspA-specific antibodies. Two weeks postimmunization, we detected PspA-specific IgG in the sera of immunized mice (Fig. 5A), but not in mice immunized with α-GalCer alone (SI Appendix, Fig. S6A). Notably, no specific antibody response was observed in mice deficient in iNKT cells (Jα18KO mice) (SI Appendix, Fig. S6B), indicating that IgG antibody production was dependent on iNKT cells for P/A immunization. Furthermore, ELISA with urea-wash demonstrated that IgG produced by the P/A vaccine exhibited a high degree of avidity toward the PspA antigen (Fig. 5B). Collectively, these results suggested that B cell affinity maturation may have occurred in the response to PspA, although iNKT cells can not recognize this antigen. NKTFH cells are thought to function as helper cells for antibody production by supporting the generation of GC B cells. In this regard, wild-type mice exhibited the generation of PspA-reactive GC B cells on day 13 following P/A immunization, whereas Jα18KO mice failed to do so (SI Appendix, Fig. S6C). The generation of PspA-specific GC B cells was similarly compromised upon IL-27 neutralization (SI Appendix, Fig. S6D), indicating that IL-27 also played a vital role in the iNKT cell–mediated GC response, likely through its promotion of NKTFH cell differentiation (Fig. 2).
Fig. 5.

P/A immunization confers long-term protection against S. pneumoniae systemic infection. (A) PspA-specific antibody titers determined by ELISA on day 14 after P/A immunization. (B) Urea wash analysis of PspA-specific antibody titers determined by ELISA on day 14 after P/A immunization. (C) Survival curve after S. pneumoniae (BG7322) systemic infection comparing P/A and P/Veh immunization. (D) PspA-specific antibody titers determined by ELISA on day 14 after P/A immunization treated with anti-IL-27 antibody or isotype control. (E) Avidity of PspA-specific antibody titers determined by ELISA on day 14 after P/A immunization in mice treated with anti-IL-27 antibody or isotype control. (F) Survival curve after S. pneumoniae (BG7322) systemic infection comparing P/Veh, P/A immunization with anti-IL-27 antibody, and P/A immunization with isotype control. Data are representative of two independent experiments with six to eight mice per group, and error bars represent mean ± SD. Statistical analyses were performed using unpaired Student’s t test (A, B, D, and E) or log-rank tests (C and F); **P < 0.01, ***P < 0.001, ****P < 0.0001.
Pathogen-specific antibodies are crucial for host protection against systemic bacterial infections such as bacteremia. Therefore, we investigated whether iNKT cell–mediated antibody production could protect against systemic S. pneumoniae infection. After 3 wk of immunization, mice were intraperitoneally (i.p.) infected with S. pneumoniae and the survival of infected mice was monitored (SI Appendix, Fig. S6E). Consistent with IgG production (Fig. 5 A and B), P/A-immunized mice survived for over 10 d, while all mice in the vehicle group succumbed by day 3 (Fig. 5C). Notably, this protection was not observed in Jα18KO mice (SI Appendix, Fig. S6F), which is consistent with the impaired PspA-specific IgG production in these mice.
A recent work finds that administration of 7DW8-5, a synthetic analog of α-GalCer, induces protection against respiratory pathogens without vaccine antigens (40). Therefore, we assessed whether the protection induced by P/A vaccine was based on the antibody response to the vaccine antigen, PspA. α-GalCer alone treatment did not protect mice from lethal systemic S. pneumoniae infection (SI Appendix, Fig. S6G), suggesting that PspA-specific antibody production mediated by iNKT cells is important for protection against S. pneumoniae.
Furthermore, our findings demonstrated that IL-27 neutralization (SI Appendix, Fig. S6H) significantly reduced the PspA-specific IgG titer in immunized mice (Fig. 5D), in agreement with the diminished PspA-specific GC B cells (SI Appendix, Fig. S6D). Although the generation of GC B cells was entirely disrupted by IL-27 neutralization, the decrease in IgG titer was less pronounced than anticipated. To further investigate potential differences, we compared the avidity of antibodies using the same methods as before (Fig. 5B). We found that PspA-specific IgG had lower avidity upon IL-27 neutralization (Fig. 5E). Consequently, IL-27 neutralization not only reduced the production of antigen-specific IgG but also hindered the acquisition of avidity, which might have resulted from the attenuation of the GC reaction. In line with the decrease in serum titer and avidity, the survival rate of anti-IL-27 antibody-treated mice (SI Appendix, Fig. S6H) decreased by approximately 60% on day 5 compared to control mice (Fig. 5F). These findings indicated that IL-27-dependent iNKT cell proliferation and differentiation into NKTFH cells was important for eliciting vaccine-induced protection against systemic S. pneumoniae infection.
Discussion
Previous reports show that iNKT cells have the potential to differentiate into NKTFH cells, which express IL-21 and surface molecules such as PD-1 and CXCR5 similar to CD4+ TFH cells (12, 13). However, the differentiation machinery underlying this process has not been fully assessed. In the present study, we demonstrated that IL-27 supported the bioenergetic requirements of peripheral iNKT cell proliferation followed by differentiation into NKTFH cells and that this machinery is essential for producing high-affinity antigen-specific antibodies and eliciting protection against S. pneumoniae infection by a P/A vaccine.
Upon activation, T cells augment their active glucose catabolism including aerobic glycolysis (15). On the other hand, iNKT cells rely more on OXPHOS for energetic demands, instead of aerobic glycolysis (17, 18). Indeed, iNKT cells exhibit lower glucose uptake than T cells, and OXPHOS inhibition impairs cytokine production and proliferation of iNKT cells (17). After P/A immunization, the number of iNKT cells transiently increased and then was reduced. This reduction might have resulted from inadequate meeting of energetic demands in these lymphocytes. iNKT cells rapidly produce abundant cytokines following TCR stimulation, which requires a relatively high energy input (18). NKTFH cells were induced after the population expansion of iNKT cells, and their proliferation rate was high (Fig. 1D), suggesting that NKTFH cell differentiation may require much energy. Therefore, iNKT cells, which are less dependent on glycolysis, might differentiate into the follicular helper subset by maximizing OXPHOS with enhanced ATP production. Our data revealed that IL-27 neutralization inhibited the enhanced mitochondrial function in iNKT cells (Fig. 3D). A recent report shows that IL-27 supports mitochondrial function in subunit vaccine-elicited CD8+ T cells by regulating Krebs cycle enzymes (37). Our results indicated that IL-27 treatment facilitated ATP production by increasing mitochondrial mass and membrane potential in iNKT cells (Fig. 3 E–H). Notably, IL-27 treatment did not affect mitochondria in CD4+ T cells (Fig. 3E), suggesting there is T cell–type specificity in the effects of IL-27. Further studies are necessary to elucidate how IL-27 signaling specifically regulates mitochondrial function in different cell types.
In our study, Gr-1+ cells were the major producers of IL-27 (Fig. 2 D and E), and monocytes and macrophages were most frequent among IL-27-producing Gr-1+ cells (Fig. 2F). Given that macrophages are required for IL-4 production by iNKT cells following influenza virus infection (30), it is plausible that they possess specialized histological and functional features that enable them to initiate iNKT cell activation. Indeed, Gr-1+ cells, including macrophages, were abundant in the red pulp of the spleen where many iNKT cells were localized (Fig. 1F). This histological proximity likely confers advantages to Gr-1+ cells in acquiring the ability to produce IL-27, which occurs in a manner dependent on IFNγ, a cytokine predominantly produced by activated iNKT cells after P/A immunization. It is known that iNKT cell activation may alter many cell types, here the effects on Gr-1+ cells were critical. Gr-1+ cell depletion had a more profound effect on iNKT cell differentiation than the neutralization of IL-27 (Fig. 2G), suggesting there are other factors that influence the communication between Gr-1+ cells and iNKT cells. Furthermore, the gene expression profile of Gr-1+ cells was dramatically changed on day 1 after P/A immunization, suggesting that they have additional functions beyond modulating iNKT cell response.
Because iNKT cells are capable of enhancing immune responses, they hold significant potential for broad applications, including antitumor responses and vaccination against bacterial and viral infections (41–43). iNKT cell agonists, such as α-GalCer, have been proposed as potential vaccine adjuvants (42, 44). Our findings also indicated that iNKT cell–mediated vaccination is useful for protection against systemic S. pneumoniae infection by inducing antigen-specific IgG production (Fig. 5). However, we did not address all aspects of the detailed mechanisms of antigen-specific IgG production and protection against S. pneumoniae infection mediated by NKTFH cells in this study. Further studies are necessary to clarify the functions of NKTFH cells induced by P/A immunization.
Our study provided insights into iNKT cell–specific energetic regulation, which is essential for the acquisition of iNKT cell helper function in the periphery. We expect that this knowledge will pave the way for the development of therapies and vaccinations that harness the iNKT cell response.
Materials and Methods
Mice.
C57BL/6 mice were obtained from CREA Inc. IL-21-hCD2 and IL-4-hCD2 reporter mice were kindly provided by Masato Kubo at Tokyo University of Science (45). Jα18KO (Traj18−/−) mice were kindly provided by Masaru Taniguchi at RIKEN Center for Integrative Medical Science (46). Animal experiments were approved by the Jikei University Animal Care and Use Committee. Unless otherwise indicated, age-matched mice (6 to 12 wk) were used for experiments. All mice were maintained under specific pathogen-free conditions in accordance with the Jikei University guidelines.
Mouse Treatments.
For the P/A immunization, 2 μg of α-GalCer (KRN7000) was mixed with 2 μg recombinant PspA protein in PBS (refer to P/A) followed by intraperitoneal injection. To deplete Gr-1+cells, mice were injected i.p. with 100 μg of anti-Gr-1 antibody (RB8-6C5, provided by Kazuyoshi Kawakami, Tohoku University) at 24 h after P/A immunization. To deplete neutrophils or to neutralize IL-27, mice were i.p. with 500 μg of anti-Ly-6G antibody (1A8; BioXcell) or i.p. with 200 μg of anti-IL-27 antibody (MM-27.7B1; BioXcell), respectively, at 6 h before and 48 h after P/A immunization. For the experiments in Fig. 2H, mice were injected i.v. with 1 μg of recombinant IL-27 at 24 h and 48 h after P/A immunization. To neutralize IFNγ, mice were injected i.p. with 200 μg of anti-IFNγ antibody (R4-6A2; in house) at 24 h, 6 h before, and 24 h after P/A immunization.
Cell Sorting.
Splenocytes were collected and washed through a mesh followed by a short red blood cell lysis with Ammonium-Chloride-Potassium Lysing Buffer (Thermo). For depletion of B cells and CD8+ T cells, cells were incubated with an anti-CD16/CD32 antibody (2.4G2; TONBO Biosciences) followed by biotinylated anti-CD8α (53-6-7) and anti-B220 (RA3-6B2) antibodies (BioLegend). Cells were subsequently incubated with streptavidin nanobeads (BioLegend) and separated using a LS column (Miltenyi Biotec) in the magnetic field of a MACS Separator (Miltenyi Biotec). The negative fraction was collected, counted, and used for in vitro iNKT cell stimulation. For sorting of iNKT cells, the negative fraction cells were stained with anti-TCRβ antibody (H57-597; BioLegend), CD1d tetramers loaded with α-GalCer (MBL), and anti-CD19 antibody (6D5; BioLegend) followed by cell sorting (FACSAria III; BD). For sorting of Gr-1+ cells, splenocytes were incubated with PE-labeled anti-Gr-1 antibody (RB6-8C5; BioLegend) and anti-PE Microbeads (Miltenyi Biotec) and then separated positively using a LS column followed by sorting after staining with an anti-CD11b antibody (M1/70; BioLegend).
In Vitro iNKT Cell Stimulation.
Isolated cells were cultivated in RPMI-1640 medium (Wako) supplemented with 10% (volume/volume) heat-inactivated FBS, 10 mM HEPES pH7.5, 50 μM 2-mercaptoethanol (Gibco), and 1% (volume/volume) penicillin and streptomycin (Sigma). CD8 negative B220 negative splenic cells, sorted iNKT and CD4+ T cells were activated for the indicated times in plates coated with 2.5 μg/mL of anti-CD3ε, and in the presence of 1 μg/mL anti-CD28. rIL-2 and rIL-27 were added at a concentration of 10 ng/mL. For IL-27-induced STAT1/3 phosphorylation, sorted iNKT cells were incubated with or without rIL-27 (10 ng/mL). To assess hyporesponsiveness of iNKT cells by P/A immunization, mice were killed at the indicated time points, and splenocytes were cultured with αGC (100 ng/mL). After 3 d, culture supernatants were evaluated for IFNγ by ELISA.
Flow Cytometry.
Mouse spleen cells were incubated with anti-CD16/CD32 antibody (2.4G2, 1:500) (TONBO Biosciences) and stained with the following antibodies: anti-TCRβ-FITC (H57-597; 1:100), anti-CD3ε-APC/Cy7 (145-2C11; 1:200), anti-B220-biotin, APC/Cy7 (RA3-6B2; 1:200), anti-CD8-biotin (53-6.7; 1:200), anti-CD138–BV421(281-2; 1:200), anti-CD38-PE/Cy7 (90; 1:500), anti-GL7-PerCP/Cy5.5(GL-7; 1:200), anti-CD169-FITC (3D6.112; 1:100), anti-IgM-FITC (RMM-1; 1:200), anti-IgD-FITC (11-26c.2a; 1:200), anti-Gr-1-BV421 (RB6-8C5; 1:200), anti-Ly6G-PE (1A8; 1:200), anti-Ly6c-FITC (HK1.4; 1:200), anti-CD11b-APC (M1/70; 1:200), anti-CD11c-PE/Cy7 (N418; 1:200), anti-F4/80-PE (BM8; 1:200), anti-IL-27p28-AF647 (MM27-7B1; 1:100), anti-IFNγ-AF647 (XMG1.2), anti-IL-4-AF488 (11B11), human CD2-APC (RPA-2.10) antibodies and Rat IgG1 isotype control-AF488, AF647 (RTK2071), Mouse IgG2a isotype control-AF647 (MOPC-173; 1:100) were purchased from BioLegend. Anti-IL-27Rα-PE (2918; 1:100), anti-pSTAT-1-AF488 (4a; 1:100), anti-pSTAT3-AF488 (4/P-STAT3; 1:100), anti-PD-1-PE/Cy7 (J43; 1:200) and anti-CXCR5-biotin (2GD; 1:100) antibodies and Rat IgG2a isotype control-PE (RTK2758; 1:100) were purchased from BD Bioscience. To exclude dead cells in the analysis, we stained spleen cells with Fixable Viability Dye (eBioscience) or propidium iodide (TOYOBO). For iNKT cell staining, PE-labeled CD1d tetramers loaded with α-GalCer (MBL) and BV421-labeled CD1d tetramers loaded with PBS57 (kindly provided by the Tetramer Core Facility of the US NIH) were used. For intracellular staining, cells were incubated in a fixation/permeabilization buffer (BD Bioscience) and then incubated with antibodies. Data were analyzed using FlowJo software (BD Bioscience).
Measurement of Mitochondrial Parameters.
To measure mitochondrial parameters, cells were incubated with potentiometric dye MitoTracker (2 nM, Invitrogen), TMRM (500 nM; Invitrogen), and MitoSox (5 μM, Invitrogen) for 30 min except for MitoSox for 10 min at 37 °C in RPMI-1640 without glucose media (Wako) for flow cytometry.
ATP Assays.
ATP levels were measured using CellTiter-Glo Luminescent Cell Viability reagent (Promega) according to the manufacturer’s instructions.
EdU Staining.
At one day before immunization, day 1, and day 2 after P/A immunization, we i.p. injected mice with 1.0 mg EdU (TCI). At day 3, we killed mice and performed staining of EdU using a Click-iT Plus EdU Alexa 488 Flow Cytometry Assay Kit (Invitrogen).
In Vivo Intracellular Cytokine Synthesis Assay.
We measured IL-27-producing cells in vivo on the basis of a previously described protocol with modifications (36). Briefly, at day 1 after P/A immunization, we i.v. injected mice with 200 μL of a PBS solution containing 1.7 μg monensin (Invitrogen) 16 h before harvesting.
Infection.
Mice were infected with S. pneumoniae (Serotype 6A, BG7322). Bacterial suspensions were diluted in PBS, and 200 μL was administered i.p. into the mice. Body weights of mice were recorded daily before and after S. pneumoniae infection.
TEM.
In vitro–stimulated iNKT cells and CD4+ T cells, with or without rIL-27, were harvested after 2-d culture by centrifugation at 400 g 4 °C for 10 min and fixed with 2.5% glutaraldehyde. The fixed cells were analyzed using a TEM H-7500 microscope (Hitachi, Ltd., Tokyo, Japan), and images were captured. Data were analyzed with ImageJ.
Real-Time PCR.
Total cellular RNA from ex vivo–sorted or in vitro–cultured cells was extracted with TRIzol reagent (Thermo). The extracted RNA was reverse-transcribed into cDNA using SuperScript IV VILO Master Mix (Thermo Fisher), and the resulting cDNA was amplified by RT-PCR. Ct values were quantified using a StepOnePlus Real-Time PCR system (Applied Biosystems) with the default fast mode setting (stage 1; 95 °C for 20 s, stage 2; 40 cycles at 95 °C for 1 s, and 60.0 °C for 20 s). Each PCR was run in triplicate, and relative gene expression was quantified using the 2-ΔΔCT method and normalized to Rpl13a expression for iNKT cells and Hprt expression for Gr-1+ cells. The primer pairs and universal probe (Roche) are listed in SI Appendix, Table S1.
3’mRNA-seq Library Preparation.
TRIzol reagent (Thermo Fisher) was used for the extraction of total cellular RNA, and the Quantus Fluorometer (Promega) was used for determination of RNA concentrations. A total of 500 ng of RNA was used for the 3′mRNA library preparation with QuantSeq 3′mRNA-seq Library Prep Kit FWD (LEXOGEN), according to the manufacturer’s protocol. After the PCR step, the size distribution and yield of the library were determined by the D1000 high-sensitivity tape station (Agilent) or Agilent High Sensitivity DNA kit on the bioanalyzer (Agilent). The pooled libraries were loaded on the Illumina NextSeq 500 platform and analyzed by 75-bp single reads.
Analysis of RNA-seq Data.
Adaptor sequences were trimmed from the raw RNA-seq reads with fastp (47). Trimmed reads of each sample were mapped to the reference mouse genome mm10 by using STAR (48) and normalized to 1 million reads in the original library. Genes with an average of 10 or more reads in either group were subjected for further analysis. PCA analysis and heatmap were depicted with R software (https://cran.r-project.org/) and amap. GSEA was performed to determine the statistical significance of the enrichment of known transcriptional signatures in a ranked list of genes (49, 50).
ELISA.
To measure the relative amount of anti-PspA IgG in the serum of mice, plates were coated with 0.5 μg/mL recombinant PspA overnight at 4 °C. After blocking with Block Ace (KAC), serially diluted mouse serum was added and incubated for 1 h at RT. Antibody was detected using 80 ng/mL peroxidase-conjugated goat anti-mouse IgG (Jackson) for 1 h at RT and visualized using TMB substrate (Thermo). For measuring avidity, plates were incubated for 15 min at RT with PBS containing 7M urea after washing of serially diluted serum. Data showed that loss of avidity was determined by subtracting 100 from the percentage of titer with urea to titer without urea (Fig. 5E). The amount of IFNγ was measured by the IFN-gamma ELISA Kit (R&D) according to the manufacturer’s instructions.
Confocal Fluorescence Microscopy.
For immunofluorescent staining, spleen samples were fixed in 4% paraformaldehyde and equilibrated in 30% sucrose/PBS. Cryostat sections of the spleen were stained and mounted with fluorescent mounting medium (Dako). Histological analyses were carried out with a confocal laser microscope (LSM880, Carl Zeiss). Analysis of iNKT cell localization was performed as previously described (32). Streptavidin, Alexa647 conjugate (Invitrogen), APC-labeled CD1d tetramers loaded with α-GalCer (MBL), biotinylated-goat anti-streptavidin antibody (Vector Lab.), and monoclonal antibodies against Gr-1 (RB6-8C5), CD11b (M1/70), and CD169 (3D6.112) (BioLegend) were used. Data were analyzed with ImageJ.
Supplementary Material
Appendix 01 (PDF)
Acknowledgments
We are grateful to Dr. Masaru Taniguchi (RIKEN Center for Integrative Medical Science) for kindly providing Jα18KO (Traj18−/−) mice and to the NIH Tetramer Core Facility for kindly providing PBS57 loaded CD1d tetramers. We thank Naoko Toda, Satomi Yamada, Izumi Kinoshita, and Mieko Utsugi for their expert technical assistance. We thank Dr. Toshiaki Tachibana, Emi Kikuchi, and Yuki Takemura for acquisition of TEM analysis. We thank Dr. Shinya Sugimoto for helpful discussions. This work was supported by the Ministry of Education, Culture, Sports, Science and Technology, Japan: Grants-in-Aid for Scientific Research (B) #19H03705, #22H03123, (C) #21K07085, Grants-in-Aid for Early-Career Scientists #19K16704, Scientific Grants from Terumo Life Science Foundation, Senshin Medical Research Foundation, Daiichi Sankyo Foundation of Life Science, Astellas Foundation for Research on Metabolic Disorders, and The Jikei University Graduate Research Fund.
Author contributions
Y. Kamii, K.H., and Y. Kinjo designed research; Y. Kamii, K.H., T.K., A.C., T.I., and Y.E. performed research; M.S., Y.A., T.O., M. Kubo, K.Y., K. Kawakami, K.O., J.A., and K. Kuwano contributed new reagents/analytic tools; Y. Kamii, K.H., T.K., and Y.E. analyzed data; M.S., Y.A., T.O., M. Kubo, K.Y., K. Kawakami, K.O., J.A., K. Kuwano, M. Kronenberg, and Y.E. discussions/suggestions; Y. Kinjo supervised this study; and Y. Kamii, K.H., M. Kronenberg, Y.E., and Y. Kinjo wrote the paper.
Competing interests
The authors declare no competing interest.
Footnotes
This article is a PNAS Direct Submission.
Data, Materials, and Software Availability
All study data are included in the article and/or SI Appendix. The RNA-seq data have been deposited in the Gene Expression Omnibus at NCBI (https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE254186 (51).
Supporting Information
References
- 1.Brennan P. J., Brigl M., Brenner M. B., Invariant natural killer T cells: An innate activation scheme linked to diverse effector functions. Nat. Rev. Immunol. 13, 101–117 (2013). [DOI] [PubMed] [Google Scholar]
- 2.Brigl M., Brenner M. B., CD1: Antigen presentation and T cell function. Annu. Rev. Immunol. 22, 817–890 (2004). [DOI] [PubMed] [Google Scholar]
- 3.Gapin L., Development of invariant natural killer T cells. Curr. Opin. Immunol. 39, 68–74 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kronenberg M., Toward an understanding of NKT cell biology: Progress and paradoxes. Annu. Rev. Immunol. 23, 877–900 (2005). [DOI] [PubMed] [Google Scholar]
- 5.Taniguchi M., Harada M., Kojo S., Nakayama T., Wakao H., The regulatory role of Valpha14 NKT cells in innate and acquired immune response. Annu. Rev. Immunol. 21, 483–513 (2003). [DOI] [PubMed] [Google Scholar]
- 6.Godfrey D. I., MacDonald H. R., Kronenberg M., Smyth M. J., Van Kaer L., NKT cells: What’s in a name? Nat. Rev. Immunol. 4, 231–237 (2004). [DOI] [PubMed] [Google Scholar]
- 7.Salio M., Silk J. D., Jones E. Y., Cerundolo V., Biology of CD1- and MR1-restricted T cells. Annu. Rev. Immunol. 32, 323–366 (2014). [DOI] [PubMed] [Google Scholar]
- 8.Godfrey D. I., Stankovic S., Baxter A. G., Raising the NKT cell family. Nat. Immunol. 11, 197–206 (2010). [DOI] [PubMed] [Google Scholar]
- 9.Constantinides M. G., Bendelac A., Transcriptional regulation of the NKT cell lineage. Curr. Opin. Immunol. 25, 161–167 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jameson S. C., Lee Y. J., Hogquist K. A., Innate memory T cells. Adv. Immunol. 126, 173–213 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kwon D. I., Lee Y. J., Lineage differentiation program of invariant natural killer T cells. Immune Netw. 17, 365–377 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chang P. P., et al. , Identification of Bcl-6-dependent follicular helper NKT cells that provide cognate help for B cell responses. Nat. Immunol. 13, 35–43 (2011). [DOI] [PubMed] [Google Scholar]
- 13.King I. L., et al. , Invariant natural killer T cells direct B cell responses to cognate lipid antigen in an IL-21-dependent manner. Nat. Immunol. 13, 44–50 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hotamisligil G. S., Foundations of immunometabolism and implications for metabolic health and disease. Immunity 47, 406–420 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Buck M. D., O’Sullivan D., Pearce E. L., T cell metabolism drives immunity. J. Exp. Med. 212, 1345–1360 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Endo Y., Kanno T., Nakajima T., Fatty acid metabolism in T-cell function and differentiation. Int. Immunol. 34, 579–587 (2022). [DOI] [PubMed] [Google Scholar]
- 17.Kumar A., et al. , Enhanced oxidative phosphorylation in NKT cells is essential for their survival and function. Proc. Natl. Acad. Sci. U.S.A. 116, 7439–7448 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weng X., et al. , Mitochondrial metabolism is essential for invariant natural killer T cell development and function. Proc. Natl. Acad. Sci. U.S.A. 118, e2021385118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ray J. P., et al. , The interleukin-2-mTORc1 kinase axis defines the signaling, differentiation, and metabolism of T helper 1 and follicular B helper T cells. Immunity 43, 690–702 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Briles D. E., et al. , Intranasal immunization of mice with a mixture of the pneumococcal proteins PsaA and PspA is highly protective against nasopharyngeal carriage of Streptococcus pneumoniae. Infect. Immun. 68, 796–800 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chang B., et al. , Distribution and variation of serotypes and pneumococcal surface protein A clades of Streptococcus pneumoniae strains isolated from adult patients with invasive pneumococcal disease in Japan. Front. Cell. Infect. Microbiol. 11, 617573 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Crain M. J., et al. , Pneumococcal surface protein A (PspA) is serologically highly variable and is expressed by all clinically important capsular serotypes of Streptococcus pneumoniae. Infect. Immun. 58, 3293–3299 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McDaniel L. S., Sheffield J. S., Delucchi P., Briles D. E., PspA, a surface protein of Streptococcus pneumoniae, is capable of eliciting protection against pneumococci of more than one capsular type. Infect. Immun. 59, 222–228 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nakahashi-Ouchida R., et al. , A nanogel-based trivalent PspA nasal vaccine protects macaques from intratracheal challenge with pneumococci. Vaccine 39, 3353–3364 (2021). [DOI] [PubMed] [Google Scholar]
- 25.Orihuela C. J., et al. , Microarray analysis of pneumococcal gene expression during invasive disease. Infect. Immun. 72, 5582–5596 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Piao Z., et al. , Protective properties of a fusion pneumococcal surface protein A (PspA) vaccine against pneumococcal challenge by five different PspA clades in mice. Vaccine 32, 5607–5613 (2014). [DOI] [PubMed] [Google Scholar]
- 27.Murray M. P., et al. , Transcriptome and chromatin landscape of iNKT cells are shaped by subset differentiation and antigen exposure. Nat. Commun. 12, 1446 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shimizu K., et al. , Eomes transcription factor is required for the development and differentiation of invariant NKT cells. Commun. Biol. 2, 150 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shimizu K., et al. , KLRG+ invariant natural killer T cells are long-lived effectors. Proc. Natl. Acad. Sci. U.S.A. 111, 12474–12479 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gaya M., et al. , Initiation of antiviral B Cell immunity relies on innate signals from spatially positioned NKT cells. Cell 172, 517–533.e20 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moran A. E., et al. , T cell receptor signal strength in Treg and iNKT cell development demonstrated by a novel fluorescent reporter mouse. J. Exp. Med. 208, 1279–1289 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee Y. J., et al. , Tissue-specific distribution of iNKT cells impacts their cytokine response. Immunity 43, 566–578 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Batten M., et al. , IL-27 supports germinal center function by enhancing IL-21 production and the function of T follicular helper cells. J. Exp. Med. 207, 2895–2906 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gringhuis S. I., et al. , Fucose-based PAMPs prime dendritic cells for follicular T helper cell polarization via DC-SIGN-dependent IL-27 production. Nat. Commun. 5, 5074 (2014). [DOI] [PubMed] [Google Scholar]
- 35.Yoshida H., Hunter C. A., The immunobiology of interleukin-27. Annu. Rev. Immunol. 33, 417–443 (2015). [DOI] [PubMed] [Google Scholar]
- 36.Liu F., Whitton J. L., Cutting edge: Re-evaluating the in vivo cytokine responses of CD8+ T cells during primary and secondary viral infections J. Immunol. 174, 5936–5940 (2005). [DOI] [PubMed] [Google Scholar]
- 37.Klarquist J., et al. , Clonal expansion of vaccine-elicited T cells is independent of aerobic glycolysis. Sci. Immunol. 3, eaas9822 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Buck M. D., et al. , Mitochondrial dynamics controls T cell fate through metabolic programming. Cell 166, 63–76 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Parekh V. V., et al. , Glycolipid antigen induces long-term natural killer T cell anergy in mice. J. Clin. Invest. 115, 2572–2583 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tsuji M., et al. , An immunostimulatory glycolipid that blocks SARS-CoV-2, RSV, and influenza infections in vivo. Nat. Commun. 14, 3959 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Doherty D. G., Melo A. M., Moreno-Olivera A., Solomos A. C., Activation and regulation of B cell responses by invariant natural killer T cells. Front. Immunol. 9, 1360 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kinjo Y., et al. , Functions of CD1d-restricted invariant natural killer T Cells in antimicrobial immunity and potential applications for infection control. Front. Immunol. 9, 1266 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Webb T. J., Yuan W., Meyer E., Dellabona P., Editorial: NKT cells in cancer immunotherapy. Front. Immunol. 11, 1314 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fujii S. I., Yamasaki S., Sato Y., Shimizu K., Vaccine designs utilizing invariant NKT-licensed antigen-presenting cells provide NKT or T cell help for B cell responses. Front. Immunol. 9, 1267 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Harada Y., et al. , The 3’ enhancer CNS2 is a critical regulator of interleukin-4-mediated humoral immunity in follicular helper T cells. Immunity 36, 188–200 (2012). [DOI] [PubMed] [Google Scholar]
- 46.Dashtsoodol N., et al. , Generation of novel Traj18-deficient mice lacking Valpha14 natural killer T cells with an undisturbed T cell receptor alpha-chain repertoire. PLoS One 11, e0153347 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen S., Zhou Y., Chen Y., Gu J., fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dobin A., Gingeras T. R., Mapping RNA-seq reads with STAR. Curr. Protoc. Bioinformatics 51, 11 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mootha V. K., et al. , PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 34, 267–273 (2003). [DOI] [PubMed] [Google Scholar]
- 50.Subramanian A., et al. , Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kamii Y., et al. , IL-27 regulates the differentiation of follicular helper NKT cells via metabolic adaptation of mitochondria. Gene Expression Omnibus at NCBI. https://www.ncbi.nlm.nih.gov/geo/. Deposited 25 January 2024. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Data Availability Statement
All study data are included in the article and/or SI Appendix. The RNA-seq data have been deposited in the Gene Expression Omnibus at NCBI (https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE254186 (51).