

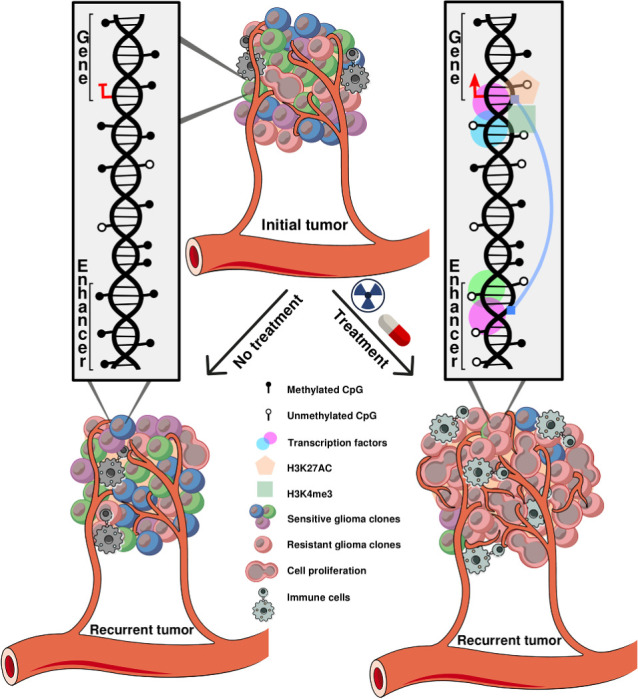
Standard treatments are related to loss of DNA methylation in IDHmut glioma, resulting in epigenetic activation of genes associated with tumor progression and alterations in the microenvironment that resemble treatment-naïve IDHwt glioma.
Abstract
Tumor adaptation or selection is thought to underlie therapy resistance in glioma. To investigate longitudinal epigenetic evolution of gliomas in response to therapeutic pressure, we performed an epigenomic analysis of 132 matched initial and recurrent tumors from patients with IDH-wildtype (IDHwt) and IDH-mutant (IDHmut) glioma. IDHwt gliomas showed a stable epigenome over time with relatively low levels of global methylation. The epigenome of IDHmut gliomas showed initial high levels of genome-wide DNA methylation that was progressively reduced to levels similar to those of IDHwt tumors. Integration of epigenomics, gene expression, and functional genomics identified HOXD13 as a master regulator of IDHmut astrocytoma evolution. Furthermore, relapse of IDHmut tumors was accompanied by histologic progression that was associated with survival, as validated in an independent cohort. Finally, the initial cell composition of the tumor microenvironment varied between IDHwt and IDHmut tumors and changed differentially following treatment, suggesting increased neoangiogenesis and T-cell infiltration upon treatment of IDHmut gliomas. This study provides one of the largest cohorts of paired longitudinal glioma samples with epigenomic, transcriptomic, and genomic profiling and suggests that treatment of IDHmut glioma is associated with epigenomic evolution toward an IDHwt-like phenotype.
Significance:
Standard treatments are related to loss of DNA methylation in IDHmut glioma, resulting in epigenetic activation of genes associated with tumor progression and alterations in the microenvironment that resemble treatment-naïve IDHwt glioma.
Graphical Abstract
Introduction
Despite advances in our biological understanding, molecular classification and surgical techniques, management of diffuse gliomas of adulthood remains challenging making it an incurable disease (1, 2). Compared with gliomas of the same grade that carry intact isocitrate dehydrogenase (IDH) 1 and 2 genes, gliomas with IDH mutations exhibit a less aggressive clinical course that has led to their separation as distinct tumor types in the 2016 World Health Organization (WHO) classification of Tumors of the Central Nervous System (CNS; ref. 3). On the basis of the revised 2021 WHO Classification (4), IDH-mutant (IDHmut) tumors now comprise two distinct tumor types, namely “oligodendroglioma, IDH-mutant and 1p/19q-codeleted, CNS WHO grade 2 or 3,” and “astrocytoma, IDH-mutant, CNS WHO grade 2, 3, or 4.” Yet, there is controversy on the morphologic criteria used to distinguish CNS WHO grades 2 and 3, and homozygous CDKN2A loss, a signature lesion of CNS WHO grade 4 among IDHmut astrocytomas, which is currently the only diagnostic molecular marker in these tumors (5–7). Thus, additional molecular characterization is needed to establish which of these tumors will rapidly progress and which will remain quiescent for several years with or without adequate therapy (2).
Epigenetics play a vital role in stratifying CNS tumors and gliomas into clinically relevant subtypes (8, 9). Studies enlightened that oncometabolite 2-hydroxyglutarate produced by IDHmut leads glioma cells to encompass subtypes with different DNA methylation patterns, named glioma CpG island methylator phenotype (GCIMP), which is associated with distinct chromatin remodeling processes and better clinical outcome than gliomas not carrying this phenotype (1, 10, 11). Further investigation revealed a subset of IDH-mutant gliomas that presented with a lower degree of DNA methylation and poorer outcome, named GCIMP-low, distinct from the previously described highly methylated tumors that have a better outcome, now renamed as GCIMP-high (9). However, the longitudinal trajectory of evolution of the glioma epigenome remains incompletely characterized and it is unknown whether the epigenetic changes marking glioma progression occur in concert with other molecular and biological changes (genome, transcriptome, immune cell infiltrates, etc.). Even less clear is the impact exerted by standard-of-care treatment on the epigenetic evolution of glioma, especially when considering the frequent transition to more aggressive forms of the disease at recurrence (12–15).
Current treatments for gliomas include surgery followed by radiotherapy and/or alkylating chemotherapy (e.g., temozolomide). Recent studies have revealed fundamental molecular genetic changes associated with glioma treatment including the development of a hypermutation phenotype (12, 16), increase in small deletion burden and acquisition of CDKN2A homozygous deletions associated with radiotherapy and acquired aneuploidy associated with cell cycle–related genes and overall poorer outcome (17, 18). Interestingly, not all temozolomide-treated gliomas develop a hypermutator status, which challenges the possible mechanisms driving this temozolomide treatment–induced molecular phenotype (19, 20).
In this study, by leveraging the Glioma Longitudinal AnalySiS (GLASS) international consortium (17, 21, 22), we analyze an epigenetic cohort of 132 patients with glioma with matched initial and first recurrent tumors, and include additional molecular data and clinical data to characterize the evolution of both IDH-wildtype (IDHwt) and IDHmut gliomas. This is the largest cohort of paired initial and recurrent glioma samples profiled with epigenomics, transcriptomics, and genomics, that we know of being used in the literature. Also, the GLASS in the Netherlands (GLASS-NL), a collaboration of several centers in the Netherlands treating patients with glioma, was included in this study to evaluate the effects of treatment in the epigenome of gliomas in an independent cohort. This consortium has collected material from 100 astrocytoma, IDHmut patients who underwent at least two surgical resections (surgical interval > 6 months). Our study aimed at identifying key master regulators of tumor progression, identifying changes in the tumor microenvironment and epigenetic drivers of glioma evasion to treatment and examining differences in these processes between IDHwt and IDHmut gliomas to derive better informed tailored treatments.
Materials and Methods
Biospecimens/GLASS datasets
Datasets added to GLASS came from both published and unpublished sources. The GLASS epigenomic cohort consists of 354 DNA methylation samples (total of 143 patients; 132 of them with high-quality molecular data from at least two-time points that were used in this study) profiled by either Illumina 450K or EPIC BeadChip methylation arrays and described below. For those same patients, we also profiled DNA sequencing data from 64 patients, whole-genome sequencing (WGS) or whole-exome sequencing (WXS); and RNA sequencing from 54 patients, available through the GLASS consortium, resulting in the largest cohort of matched glioma samples profiled with epigenomics, transcriptomics and genomics platforms (Supplementary Tables S1 and S2).
Newly generated DNA methylation data was collected from four different institutions: Henry Ford Hospital (N = 103), University of Leeds (United Kingdom; N = 8), Chinese University of Hong Kong (N = 6), and Luxembourg Institute of Health (N = 54). The DNA was extracted at each institution. New DNA methylation data from Henry Ford Hospital and from Chinese University of Hong Kong was generated at the University of Southern California. Briefly, the DNA was bisulfite-converted (Zymo EZ DNA methylation Kit; Zymo Research) and profiled using an Illumina Human EPIC array (EPIC). For the Luxembourg Institute of Health samples, DNA methylation data was generated by the Illumina EPIC array at the Helmholtz Zentrum München (Research Unit of Molecular Epidemiology, Institute of Epidemiology, German Research Center for Environmental Health, Neuherberg, Germany) or by the Laboratoire National de Santé (Neuropathology Unit, National Center of Pathology, Dudelange, Luxembourg). Samples from University of Leeds were profiled locally using Illumina 450K BeadChip methylation arrays. The raw DNA methylation intensity data files (IDAT) were processed with the minfi package (23). We performed noob (normal-exponential convolution using out-of-band probes) background correction (24) and dye bias correction using the minfi package (v 1.36.0; ref. 23). The DNA methylation value for each locus is presented as a β value [β = (M/(M+U)] in which M and U indicate the mean methylated and unmethylated signal intensities for each locus, respectively. β-Values range from zero to one, with scores of zero indicating no DNA methylation and scores of one indicating complete DNA methylation. A detection P value also accompanies each data point and compares the signal intensity difference between the analytic probes and a set of negative control probes on the array. Any data point with a corresponding P value greater than 1E-4 is deemed not to be statistically significantly different from background and was thus masked as “NA”. All processed data files that were used in our analysis can be found at Synapse (https://www.synapse.org/glass).
The raw DNA methylation IDAT files from public datasets were accessed and processed as described for the GLASS datasets above. Sample ID and tissue source site from our entire longitudinal glioma cohort are listed in Supplementary Table S1.
The generation and processing of gene expression data was described in previous GLASS publication (22). RNA expression data used in this study was downloaded from the GLASS Synapse portal and the transcripts per million (TPM) data matrix was filtered for selected protein coding genes only. Next, batch effects due to the different Aliquot Batches were corrected using the COMBAT algorithm with aliquots as covariates (25). Whole-exome and/or whole-genome sequencing data were generated and processed as described during creation of the initial GLASS dataset (17).
Quality control
DNA methylation quality control was performed using the entire GLASS epigenetic samples to ensure the identity check of samples matched to their corresponding patient. The DNA methylation signals of probes querying high-frequency SNPs were used to calculate a pairwise agreement score across samples (26). Only samples that passed our pairwise agreement score cutoff were kept in the GLASS epigenetic cohort (N = 354).
Data analysis
Data visualization and data analysis were performed using R version 4.1.0 software packages (www.r-project.org) and Bioconductor (27). Unless specified, all statistical tests were performed using two-tailed tests and significance was obtained with FDR < 5%. Differential analysis between paired initial and first recurrent samples was performed using paired tests; other analyses were performed using independent-samples tests (type of test is indicated in each method's subsection).
Classification of longitudinal gliomas
Longitudinal glioma samples were classified as either IDHwt (Classic-like, Mesenchymal-like, LGm6) or IDHmut (Codel, GCIMP-high, and GCIMP-low) DNA methylation subtypes using the CpG methylation signatures and method previously defined by our group (9).
Our cohort was also classified into the Pan-CNS DNA methylation-based classification (8) by uploading idat files into the portal https://www.molecularneuropathology.org/mnp. Some samples could not be assigned to a Pan-CNS DNA methylation-based subtype due to the rigid calibrated classifier score cutoff, therefore their subtype is not available in Supplementary Table S1.
In addition, the samples were classified into the recent transcriptomic pathway-based classification of glioblastomas (28) using MWW-GST on the basis of the highest positive normalized enrichment score (29).
Estimation of 1p/19q codeletion status, IDH mutation, and MGMT promoter methylation using DNA methylation data
The molecular status of 1p/19q codeletion status and the IDH mutation were determined according to The Cancer Genome Atlas (TCGA) molecular subtypes (9). The status of the MGMT promoter methylation was defined using data from microarray as described in ref. 30. For some patients, the status of IDH, 1p/19q codeletion and MGMT promoter methylation were also provided by the original tissue source institution and this information is provided in Supplementary Table S1 (Columns “idh_status,” “codel_status,” and “mgmt_methylation,” respectively). There are a few cases with inconsistent information among the different methodologies. As all methodologies are well established, we used the status defined by DNA methylation microarray as the information is available for all samples.
Estimation of tumor purity
Tumor purity was estimated using the package InfiniumPurify (31) and we used combined normal samples from different tissue types to construct a panel of normal methylomes as reference, according to ref. 32.
Hypermutation status in gliomas
Patients with known temozolomide treatment status after initial surgery and known genomic data for both initial and recurrent samples were classified in hypermutator and nonhypermutator phenotype. Hypermutation was defined for all recurrent tumors that had received temozolomide after initial surgery and had more than 10 mutations per megabase sequenced, as described previously (17).
Chromatin immunoprecipitation sequencing
Sample selection
Seventeen fresh-frozen GCIMP tumor samples (nine primary GCIMP-high samples, four primary GCIMP-low, and three recurrent GCIMP-low tumors) were collected from Hermelin Brain Tumor Center (HBTC) tumor bank at Henry Ford Hospital (HFH), with extensive clinical follow-up, to profile H3K27ac and H3K4me3. These samples were selected on the basis of data availability, such as RNA sequencing, high tumor purity evaluated by neuropathologists, and sufficient amount of tissue.
Sample preparation
About 100 to 400 mg of fresh frozen tissues by sample were cut. Then, tissue samples were sent to Active Motif for the following steps, according to their protocol: samples were cross-linked for 10 minutes by adding fresh formaldehyde directly to the culture medium at a final concentration of 1%. Using 10X (1.15 mol/L) glycine for 5 minutes at room temperature to quench the reaction. Chromatin from fixed cells were sonicated using a Bioruptor Pico (Diagenode, catalog no. B01060001) with 30 seconds on/30 seconds off cycles to produce fragments between 200 and 500 base pairs. For immunoprecipitation, 100 grams of sonicated chromatin were used and 10 grams (10%) were saved as an input control. To probe for active enhancers, samples were incubated at 4°C overnight with an H3K27ac antibody (Active Motif, catalog no. 39133) or an IgG control (Sigma, catalog no. R9133). As a secondary, protein A/G magnetic beads (Pierce, catalog no. 88802) were added to the samples prior to an additional incubation for 2 hours at 4°C. The beads were then washed with a series of salt buffers before elution. The immunoprecipitated and input control DNA were purified using A QIAprep Spin Miniprep Kit (Qiagen, catalog no. 27104). Finally, the samples were single-end sequenced with read lengths of 75 bp each and an average coverage of approximately 100×.
Data processing
First, FastQC (version 0.11.5) was used to do quality control checks by each sample on the raw sequence data, followed by MultiQC (version 1.4; ref. 33) to combine all reports into a single report by experiment. All the samples, from both chromatin immunoprecipitation sequencing (ChIP-seq)-specific antibodies to H3K27ac and H3K4me3, showed average Phred scores above 30, low level of duplication, and no adapter sequence content. The software used to map the sequence files to the most recent reference genome (hg38) was bwa-mem (version 0.7.15). The output of this tool is a SAM file. After the alignment, SAM files were converted to BAM, filtered to only include reads with mapping quality greater than 30 and, finally, sorted using SAMtools (version 1.3.1; ref. 34). Duplicated reads were tagged and removed using the picard MarkDuplicates tool (version 2.7.1).
ChIP peaks calling
After mapping reads, peaks were called, by sample, to identify regions of ChIP enrichment (FDR ≤ 0.01) in gliomas compared to control (input) using MACS2 (version 2.1.1; ref. 35). The output of this tool contains the genomic location of each peak, followed by the absolute peak summit position, pileup height, fold enrichment over the control (input), log10 transformed P value and FDR.
Differentially bound peaks
At the beginning of all analyses, we compared nine GCIMP-high primary samples versus four GCIMP-low primary samples (only primary status) to identify which differentially bound peaks are specific for each GCIMP condition, regardless of tumor recurrence status. R/Bioconductor package DiffBind (36) was used. This tool allows the user to input peak calling files from MACS2 and is composed of several steps. First, DiffBind reads in the files and associated metadata and then detects common peaks across all the samples toward creating a single set of binding site intervals. Next, DiffBind counts the number of reads that overlap each binding site interval, by sample, using sequence read files. To do the differential analysis, DiffBind divides the samples by group according to the metadata provided by the user and then compares the groups by performing differential binding affinity analysis using DESeq2, by default. Finally, each peak is assigned with a fold change (FC), P value, and FDR representing the confidence in which they are differentially bound. Differentially bound peaks identified by DiffBind (version 3.4.11) were then assigned to discrete categories based on genomic position, using gencode (version 39; ref. 37) as reference for gene location: promoter [2,000-bp window surrounding known transcription start site (TSS)] or intergenic (nonpromoter) regions.
Identification of the putative master epigenetic regulator gene
We used RNA sequencing data TCGA GCIMP-high and GCIMP-low samples and performed a paired Wilcoxon rank-sum test followed by FDR to identify differentially expressed genes (FDR < 0.05). On the basis of the FC between GCIMP-high and GCIMP-low, genes with FDR < 0.05 were classified into upregulated and downregulated when FC > 0 and FC < 0, respectively. Integration between transcriptomic and epigenomic data was initially performed by mapping the RNA sequencing results to active TSS regions obtained from H3K27ac and H3K4me3 data. As a preliminary result, we characterized the regions with gains and losses of epigenetic biomarkers in GCIMP-low and we were able to relate these marks to gene expression level. To visualize the distribution of the H3K27ac and H3K4me3 peaks in the regulatory regions of the gene identified as a putative epigenetic regulator of the progression from GCIMP-high to GCIMP-low, we used Integrative Genomics Viewer (IGV; version 2.15.2).
Functional validation – putative epigenetic master regulator gene
CRISPR - HOXD13 KO
Knockout of the HOXD13 target gene was performed using the CRISPR methodology. The experiment using the IDHwt cell line (HF3016) derived from a patient and expressing high levels of HOXD13, was conducted in six biological replicates: Negative control (represented by Control 1 and Control 2), HOXD13 KO 1 (represented by 1a and 1b), and HOXD13 K0 2 (represented by 2a and 2b). The construction HOXD13 KO 1 was built using gRNA VSGHSOH-28531007 lot# V19080103 and HOXD14 KO 2 was constructed using gRNA VSGHSOH-28552455 lot# V19080103, both obtained from ORIGENE.
In compliance with institutional regulations, tissue sample freshly resected from glioblastoma patient HF3016 was enzymatically dissociated and cultured in serum-free neurosphere media consisting of DMEM/F12 media (Invitrogen), N2 supplement (Gibco), and 0.5 mg/mL BSA, supplemented with growth factors 20 ng/mL EGF and 20 ng/mL bFGF (PeproTech; NMGF), to select for cancer stem cells and used prior to achieving passage 20. A subset of media samples was tested and identified to be absent of Mycoplasma contamination (ATCC Universal Mycoplasma Detection Kit 30–1012K). The identity of cell lines was confirmed by comparing the genotype with the patient germline using short tandem repeat (STR) analysis. The experiment was carried out for 8 days. On day 1 and day 2, the neurospheres were decanted and dissociated into single cells in DPBS (Mg and Ca-free). The single cells were counted using a hemocytometer and Trypan Blue. Next, the total amount of cells was resuspended in NMGF medium supplemented with 2% FBS to achieve 4×105 cells/mL. The viral suspension was prepared in a microtube to achieve the indicated multiplicity of infection (MOI) of 1. The equation to calculate a volume of lentiviral stock for a given MOI was: V = MOI × CN/VT × 1,000, where: V = volume of lentiviral stock in μL; MOI = desired multiplicity of infection; CN = number of cells in the well at transduction; VT = viral titer in TU/mL (indicated in the Certificate of Analysis) and multiplied by 1,000 to convert the volume from mL to μL.
Next, the solution was carefully mixed by pipetting up and down and placed in a viral tissue culture incubator for 6 hours. Next, we added 2% FBS NMGF medium to the wells and the plate was incubated in a viral tissue culture incubator for 48 hours. On day 3, the cells were observed under microscope and from day 4 to day 8 the cell morphology was recorded by performing a partial change of antibiotic-containing medium every 2–3 days. Surviving cells were harvested from each well with TrypLe express at 37°C, transferred to 15 mL tube, and resuspended in NMGF containing no FBS and no antibiotic.
Cell proliferation assay
The cell proliferation assay related to the HOXD13 KO was performed in four different time points: days 0, 3, 6, and 9. The experiment was conducted using 1,000 cells/well of black bottom transparent 96-well plate per time point. In total, we used 6 wells/cell line/day in 100 μL of medium. The measurement of cell viability (per time point) was performed adding 100 μL of CellTiter-Glo Reagent (substrate prediluted in buffer) to the cell culture medium, mixing the contents for 2 minutes on an orbital shaker to induce cell lysis and stabilizing the luminescent signal keeping the plate at room temperature for 10 minutes. The luminescence was recorded using a cell imaging multimode microplate reader (Cytation 3).
Quantification of the relative HOXD13 expression levels
We analyzed the quantification of HOXD13 expression using the real-time PCR (qPCR) technique. Total RNA was isolated from 1 well of 6-well plate cells on 50%–80% confluence using RNeasy total RNA isolation kit from Qiagen (#217004). DNA-se digestion step (Dnase I Qiagen) was incorporated into the RNA isolation process to eliminate potential genomic DNA contamination. Reverse transcription of about 0.5 mg/rxt total RNA was performed using Thermo Fisher Scientific Superscript III RT kit/oligodt priming, according to the manufacturer. The c-DNA obtained was diluted 1/20 and used 8 mL/rxt in real-time PCR (about 7ng/rxt if we consider RT 100% efficient). For real-time PCR, we used SYBR Green I dye detection, a highly specific double-stranded DNA-binding dye, which allows the detection of product accumulation during PCR, including nonspecific reaction products (primer dimmers). We designed (vector NTI software) highly specific primers for genes of interest: I (housekeeping gene) = TAAGTGGGATCGAGACATGTAAGC; HOXD13 forward (fw) = TAAGTGGGATCGAGACATGTAAGC, HOXD13 reverse (rev) = CTAGAGCTACCTGTGGAGCA.
Longitudinal DNA methylation changes of IDHmut-noncodel gliomas
We selected IDHmut noncodel initial and first recurrent pairs with available DNA methylation data (N = 59) and performed a paired Wilcoxon rank-sum test followed by FDR to identify differentially methylated probes (FDR < 0.05). On the basis of the mean DNA methylation difference between initial and first recurrent tumors, the CpG probes with FDR < 0.05 were classified into hypomethylated and hypermethylated. Only hypomethylated probes were selected for further analysis (81,958 hypomethylated probes).
To integrate the data, we combined the DNA methylation and the corresponding gene expression of 11 IDHmut noncodel samples with both DNA methylation and RNA expression data available and mapped each differentially methylated CpG probe to the nearest 20 genes (10 upstream and 10 downstream genes, independent of the distance). We then calculated the Spearman rank correlation between DNA methylation and gene expression data with the associated P value corrected by FDR. Pathway analysis was performed using Reactome (38). Finally, we searched for previously identified/validated HOXD13 targets at ChEA (39) and CISTROME (40) databases.
DNA methylation changes associated with treatment
Only patients with known temozolomide and RT treatment status after initial surgery were included in the analysis. In total, we identified 6 patients who received both temozolomide and RT, 12 patients who received only temozolomide, 18 patients who received RT only and 33 patients who did not receive additional treatment besides surgery. We used the Kruskal–Wallis test by ranks followed by multiple testing corrections using the Benjamini and Hochberg (BH) method for FDR estimation (41) to identify differentially methylated sites between these four groups at first recurrence. To define differentially methylated CpG probes, we selected probes with FDR < 0.01 and absolute mean DNA methylation difference between each group >20%. Known motif discovery analysis was conducted using HOMER as previously described and hypomethylated CpG probes overlapping the top DNA motif were selected for further investigation. To understand the biological context of these probes, we integrated the DNA methylation at these CpGs with the 20 nearest genes and the Mann–Whitney U test was used to test the null hypothesis that overall gene expression in the treated group (temozolomide only, RT only, or combination of temozolomide and RT) is greater than that in the untreated group. The P value was corrected for multiple hypotheses using FDR. The CpG-gene pairs with FDR < 0.01 were then sent to our collaborators who are part of the GLASS-NL consortium for validation of our results.
DNA methylation and gene expression data from the GLASS-NL cohort
The GLASS-NL consortium has collected material from 100 IDHmut astrocytoma (1p19q non codeleted) patients who underwent at least two surgical resections. Material for analysis had to be available for both resections, and the surgical interval between resections was >6 months. Detailed clinical data, imaging, and treatment data of patients was collected within the consortium. All institutions obtained ethics approval from their institutional review boards or ethics review committees before initiation of the project. All patients provided written informed consent according to local and national guidelines.
DNA and RNA were isolated from formalin-fixed paraffin-embedded (FFPE) tumor samples as previously described (42). Evaluation of the area with highest tumor content was done by the pathologist (P. Wesseling) on a hematoxylin and eosin–stained section. Macrodissection of the marked area was then done on 10–20 10-μm consecutive slides. DNA and RNA extraction was performed using the QIAamp DNA FFPE and RNeasy FFPE kit respectively (both Qiagen). DNA methylation profiling was performed with the Infinium MethylationEPIC BeadChip according to the manufacturer's instructions making use of the Infinium FFPE DNA Restoration Kit. RNA sequencing was done by Genomescan and data processing, alignment, and further analysis of read counts was done as described previously (43).
Deconvolution analysis
We first constructed a signature matrix from reference DNA methylation profiles of pure flow-sorted populations of cells from the literature. This signature matrix represents a set of differentially methylated CpGs selected and weighted to reflect specificity for a given cell type and is used as the basis of cell deconvolution by methylCIBERSORT. Our final signature matrix consisted of 10 cell types: CD19+ cells (B cells; N = 6), CD8+ T cells (N = 6), CD56+ (natural killer cells; N = 6), and neutrophils (N = 12) were from the FlowSorted.Blood.450k Bioconductor package version 1.30.0 (44). CD4+ effector T cells (N = 6) and Tregs (N = 4) were from ref. 45, accessed through the MethylCIBERSORT R package (46). Vascular endothelial cells’ (N = 2) data were from ref. 47. Monocyte-derived macrophage (N = 4) data were from ref. 48. Neuron (N = 31) and glia cells (N = 31) were from ref. 49. The MethylCIBERSORT R package was used to derive the DNA methylation signature for the deconvolution and the signature matrix was exported and uploaded to the CIBERSORTx portal to be deconvoluted using 1,000 permutations without quantile normalization.
Validation of tumor cell composition
Sections of formalin-fixed, paraffin-embedded human glioma surgical samples were deparaffinized with xylene and rehydrated through graded alcohol into deionized H20. Antigens were unmasked by incubation for 45 minutes at 95°C in Diva Decloaker (Biocare, DV2004) using Biocare's Decloaking chamber, and sections were stained with the antibodies listed on the Reporting Summary, visualized with intelliPATH FLX DAB Chromogen Kit (Biocare, IPK5010) and counterstained with intelliPATH Hematoxylin (Biocare, IPCS5006L).
Images of the tissue sections stained by IHC using CD163, CD31, and CD8 antibodies were captured by an Olympus IX70 microscope and a digital camera. For quantitative analysis, we selected eight representative areas in each section. Images of the representative areas were captured at a 10× magnification. For CD163 and CD31 stainings, individual cells per area were identified by strong brown stain and counted by using ImageJ (NIH, Bethesda, MD) by an algorithm to evaluate staining using hematoxylin and DAB staining specific built-in color deconvolution plug-in. For CD8 immunostaining, positive cells identified in each area by strong brown stain were manually counted. The cell counting was repeated three times. All images were analyzed in a blinded fashion.
Data availability
The newly generated data analyzed in this study were obtained from GLASS at https://www.synapse.org/glass. All the processed molecular data accompanied by the corresponding clinical data for the GLASS consortium is available on Synapse.
Raw DNA and RNA sequencing data generation and data processing were described in previous GLASS publications (17, 22). Newly generated epigenomic data, including DNA methylation array idat files, can be accessed in Gene Expression Omnibus (GEO) at GSE248471.
Public data included in the GLASS cohort were downloaded from TCGA/GDC (https://portal.gdc.cancer.gov) and in the European Genome-Phenome Archive (EGA) at EGAS00001001255 (50), EGAS00001001854 (51), and EGAS00001001588 (15, 50–52).
DNA methylation processed data used in the deconvolution analysis was downloaded from GEO at GSE66351 (neuron and glia; ref. 49), GSE35069 (CD19+, CD8+ T, CD56+, neutrophils; ref. 44), GSE49667 (CD4+ effector T cells and Tregs; ref. 45), GSE122126 (vascular endothelial cells; ref. 47), and GSE118696 (monocyte-derived macrophage; ref. 48).
Results
Molecular evolution of matched initial and recurrent gliomas
The GLASS-international DNA methylation cohort consists of 132 patients with high-quality molecular data from at least two-time points, resulting in a total of 354 samples profiled by either Illumina 450K or EPIC BeadChip methylation arrays (Supplementary Tables S1 and S2). A few patients have multiple fragments from the same tumor profiled by DNA methylation and were excluded from most of the analyses whenever we found heterogeneity. We selected 132 patients with available initial and first recurrence tumors for further analysis (Supplementary Table S2). The patients at initial diagnosis represented the three major glioma subtypes, which here were defined by DNA methylation signatures (9): IDHmut and 1p/19q-codeleted oligodendroglioma (IDHmut-codel; n = 13); IDHmut astrocytoma without 1p/19q codeletion (IDHmut-noncodel; n = 59); and IDHwt glioblastoma (n = 60). Among the 132 patients with profiled DNA methylation, 54 patients had RNA sequencing data, 64 had DNA sequencing genomic data, either whole-genome sequencing (WGS) or whole-exome sequencing (WXS), and 49 had all three molecular data sets (Fig. 1A, patient level; Supplementary Fig. S1A, sample level).
Figure 1.

Epigenomic evolution of matched initial and recurrent gliomas. A, Venn diagram of number of patients who had DNA methylation, genomic (WGS/WXS), and/or RNA sequencing profiling. B, Clinical and molecular overview of matched initial and first recurrent DNA methylation cohort. Each column represents a single patient (N = 132) at two separate time points grouped by IDH status and ordered by increase of gain of DNA methylation at recurrent tumor from left to right. The gain of DNA methylation represents the percentage of probes that showed an increase of DNA methylation at recurrence. Top plot shows the surgical interval of each patient. C, Frequency of patients with hypermutator tumors that switched or retained molecular subtype at recurrence. Patients are distinguished by IDH status.
To investigate the temporal differences, we evaluated the most relevant molecular and clinical features in gliomas. Our cohort includes the previously described DNA methylation-based glioma TCGA subtypes: Three IDHmut-specific DNA methylation subtypes (Codel, GCIMP-high, and GCIMP-low) and three IDHwt-specific subtypes (Classic-like, Mesenchymal-like, LGm6; ref. 9; Fig. 1B) and we used this molecular classification throughout the analyses in this study. Over time the majority of patients retained the original subtype, with only a minor fraction of patients switching subtypes. Specifically, 20% (14/71) of IDHmut and 27% (16/60) of IDHwt tumors switched subtypes. Among IDHmut cases that switched subtypes, tumors of 10 patients (10/14, 71%) switched from GCIMP-high to GCIMP-low, a subtype associated with worse overall survival (time from initial surgery to death or last follow-up; Supplementary Table S3A). Patient GLSS-SF-0001 showed spatial subtyping heterogeneity at recurrence (two fragments showed transition to GCIMP-low subtype and one fragment retained the GCIMP-high phenotype) and was excluded from this statistic. Among IDHwt cases that switched subtype, tumors of 8 patients (8/16, 50%) switched from classic-like or LGm6 to the mesenchymal-like subtype. Conversely, 7 patients (7/16, 44%) switched from mesenchymal to classic-like or LGm6. Less frequent subtype shifts were also observed. We tested whether tumor purity, which was estimated by DNA methylation, affects subtype switches in our bulk samples and we did not find purity differences between initial and recurrent tumors from patients that switched versus those that did not switch subtype (Supplementary Fig. S1B). The only subtype change affected by tumor purity was switches to LGm6, which showed lower tumor purity at recurrence (Supplementary Fig. S1B; t test, initial vs. recurrent P = 0.021), corroborating with a previous study that reported the tumor microenvironment contributing to LGm6 subtype assignment (53).
A Pan-CNS DNA methylation-based classification (8) and a recent pathway-based classification of glioblastomas (28) were also assigned to our cohort (Fig. 1B). On the basis of the Pan-CNS DNA methylation–based classification, 20 IDHmut astrocytomas (20/49, 41%) progressed to high-grade astrocytoma upon first recurrence; whereas the IDHwt cases, tumors of 7 patients (7/21, 33%) switched from mesenchymal to RTK II, 7 cases (7/17, 41%) switched from RTK II to mesenchymal and 3 (3/9, 33%) from RTK I to mesenchymal. Patient GLSS-SF-0017 showed spatial heterogeneity at recurrence and patient GLSS-SF-0018 showed heterogeneity at initial tumor and were excluded from this statistic.
When the patients were stratified according to the genome-wide gain or loss of DNA methylation groups upon first recurrence, patients with IDHmut gliomas showed a higher proportion of samples losing DNA methylation than patients with IDHwt gliomas (39% (28/72) vs. 8% (5/60); Fisher test, P = 0.0006; Fig. 1B). Interestingly, 3 of 5 (60%) of IDHmut tumors that switched TCGA subtypes and had genomic data available evidenced hypermutator phenotype at first recurrence (Fig. 1C; Supplementary Table S3B). Two of these switched from GCIMP-high to GCIMP-low. The tumor of one patient switched from IDHmut Codel to GCIMP-high (noncodel subtype). In contrast, only 1 of 23 (4%) IDHmut patients that retained their subtype became hypermutator at recurrence (Fisher test, P = 0.01). All switches in IDHmut tumors were toward a more aggressive phenotype (e.g., GCIMP-low and/or grade 4), suggesting an association between DNA methylation change, tumor progression, and hypermutation acquisition.
Master regulators associated with IDHmut glioma progression
To further investigate the changes in the epigenome that occur over time in gliomas, we compared the genome-wide DNA methylation characteristics of the initial compared with first recurrent tumor samples, stratified by IDH status (Supplementary Fig. S1C). IDHwt gliomas showed a more stable epigenome over time (i.e., zero CpG probes presented a differentially methylated mean difference greater than 15%; N = 60), while the epigenome of IDHmut gliomas showed genome-wide loss of DNA methylation (674 CpG probes with DNA methylation difference >15%; N = 72) throughout the disease evolution (Supplementary Fig. S1C). IDHmut patients that progressed from GCIMP-high to GCIMP-low showed the most prominent loss of DNA methylation, particularly at intergenic regions, reinforcing the association between the loss of DNA methylation and tumor progression in this subtype (Supplementary Fig. S1D–S1F), confirming previous findings of our group and others (15, 54). Compared with patients with tumors that remained GCIMP-high at recurrence, those with recurrent GCIMP-low more often had histologically higher-grade astrocytoma, were less often managed by a watch-and-wait strategy and exhibited inferior survival (Supplementary Fig. S1G; log-rank P = 0. 06; Supplementary Table S3A).
Next, we investigated the impact of the epigenomic changes on the transcriptional landscape of GCIMP-high and GCIMP-low tumors using an integrative approach that combines epigenome and transcriptome data to define master regulators (MR). We profiled 9 IDHmut GCIMP-high and 4 GCIMP-low tumors at diagnosis with ChIP-seq for the H3K27Ac and H3K4me3 active promoter marks. We compared GCIMP-high and GCIMP-low tumors and identified an increase of both H3K27Ac and H3K4me3 peaks at known TSS with corresponding increase in gene expression levels in GCIMP-low (Fig. 2A; fold change > 0 representing loss in GCIMP-low; fold change < 0 representing gain in GCIMP-low; and P < 0.05). Among the 45 activated genes enriched by epigenetic peaks in GCIMP-low, we identified several members of the HOX and FOX family of transcription factors (Fig. 2A), known to play a role in cancer (55–58). Notably, we identified HOXD13, a member of the HOX transcription factor family previously associated with the development and maintenance of brain cancer (59, 60). This gene also showed an increase of H3K27Ac and H3K4me3 peaks in matched recurrent GCIMP-low (N = 3) that progressed from GCIMP-high (N = 3; Fig. 2B), suggesting a role in glioma progression. We next expanded our analysis and identified the overexpression of HOXD13 in the matched recurrent IDHmut noncodel GLASS transcriptomic cohort (N = 24 paired samples; t test, P = 0.0019; Fig. 2C). HOXD13 was shown to contribute to glioma progression by regulating tumor invasion, growth, and cell stemness (60). Here we show that recurrent IDHmut tumors have higher stemness activity than their corresponding initial tumors (Fig. 2D), as defined by their degree of undifferentiation based on a pluripotent stemness epigenomic signature described in our previous study (61). To evaluate the oncogenic role of HOXD13 in glioma, we used CRISPR to knockout its expression in vitro using two distinct constructions (Fig. 2E) in a patient-derived IDHwt cell line expressing high levels of HOXD13. It is important to note, that we did not observe expression of HOXD13 in a primary IDHmut cell line and because recurrent IDHmut resembles the epigenetic and clinical phenotype of IDHwt, HOXD13 knockout resulted in a decrease of cell proliferation in a time-dependent manner in a glioma cell line, reinforcing its role in glioma evolution (Fig. 2F).
Figure 2.

Master regulators associated with IDHmut glioma recurrence/progression. A, Starburst plot of all H3K27ac and H3K4me3 peaks overlapping known transcription start sites. Significant gains or losses of H3K27ac (y-axis) and H3K4me3 (x-axis) are highlighted. Each dot indicates a gene and the shape indicates the expression difference between GCIMP-low versus GCIMP-high. Triangles, upregulated genes; squares, downregulated genes. If a gene is enriched for both H3K27ac and H3K4me3, this implies active TSS and the associated gene expression is defined as upregulated (turquoise, top right corner). If a gene is depleted for both H3K4me3 and H3K27ac, this implies weak or quiescent expression of the associated gene (green, bottom left corner). Gray, not significant gene promoter enrichment. Only the most significant upregulated genes are labeled in this plot. B, Genome browser representation focused on the HOXD family. The region related to HOXD13 gene (hg18.chr2: 176,092,721–176,095,944) is highlighted in turquoise. HOXD13 is more enriched by the H3K27ac and H3K4me3 peaks in the GCIMP-Low (recurrent) samples (n = 3) than in their corresponding GCIMP-High (primary) pairs (n = 3), top and bottom graph, respectively. C, HOXD13 expression level by RNA sequencing of GLASS IDHmut samples stratified by initial/recurrent and codel status (N = 11 codels and 26 noncodels patients). Each box represents quartiles, and the center line represents the median of each group. The whiskers represent absolute range. D, Stemness activity in the GLASS IDHmut samples stratified by initial/recurrent and codel status. Each box represents quartiles, and the center line represents the median of each group. The whiskers represent absolute range. E, Quantification analysis of the relative HOXD13 expression levels (2-ΔΔCt) between samples: control, n = 2; HOXD13 1 sgRNA HOXD13–07, n = 2; HOXD13 2 sgRNA HOXD13–55, n = 2 biological replicates. The boxplots represent the analysis of three technical replicates for each sample. F, Proliferation growth curve over 9 days of analysis (control, N = 2; HOXD13 1 sgRNA HOXD13–07, N = 2; HOXD13 2 sgRNA HOXD13–55, N = 2 biological replicates). **, P < 0.01
To further explore the epigenomic context of the DNA methylation changes associated with the progression of IDHmut-noncodel gliomas, we compared initial and first recurrent tumors by DNA methylation (N = 59 paired samples) and identified a total of 81,958 differentially methylated probes between the time-separated groups (paired Wilcoxon rank-sum test, FDR < 0.05; Supplementary Table S4). We searched for nearby genes using a set of 11 IDHmut-noncodel samples with both DNA methylation and RNA expression data available and identified 1,080 genes with anticorrelated expression, considered potential epigenetically regulated genes. A pathway-based analysis of these genes resulted in the enrichment of cell cycle and proliferation-related activities (Supplementary Table S4B). Interestingly, of those, 83 genes have been previously experimentally identified by ChIP-seq as HOXD13 target in human or mouse, according to the ChEA (39) and CISTROME (40) databases and are upregulated in our IDHmut-noncodel transcriptomic cohort (N = 23 paired samples; Supplementary Table S4C), while no changes were observed in the IDHmut-codel cohort (data not shown). Some of these HOXD13 targets are well-established oncogenes related to cell proliferation: Centromere protein F (CENPF) has been described to enhances the progression of adrenocortical carcinoma (62) and was associated to poor prognosis in breast cancer (63) and gliomas (64); Proliferating cell nuclear antigen (PCNA) has been reported as a prognostic indicator in gliomas (65, 66) and has been tested as a potential target to inhibit tumor cell proliferation (67); Homeobox protein A7 (HOXA7) has been reported to promote tumor growth and metastasis in liver cancer (68).
Our study revealed that IDHmut gliomas exhibit a more dynamic epigenome, characterized by significant loss of DNA methylation during disease progression and recurrence. This epigenetic landscape is accompanied by the epigenetic activation of HOXD13 and other oncogenes at recurrence. In contrast, the epigenome of IDHwt gliomas appears to be relatively preserved longitudinally, with minimal changes observed in DNA methylation patterns. These findings underscore the distinct molecular characteristics between IDHmut and IDHwt gliomas, suggesting a potential role for epigenetic alterations in driving disease progression in the former.
DNA methylation loss associated with recurrent IDHmut gliomas after standard treatment
It has been shown that treatment (radiotherapy and alkylating chemotherapy) improves the progression-free survival of IDHmut gliomas (69). However, recurrence of IDHmut lower-grade glioma is frequently associated with progression to higher histological grades. Treatment of IDHmut lower-grade gliomas with temozolomide and/or radiotherapy has been linked to many genomic alterations, such as a hypermutator phenotype (12, 20), and a strong tendency towards aneuploidy and a specific radiotherapy-associated deletion signature by genetic analysis (18). Herein, we observe that treatment is associated with epigenomic changes. We sought to identify the DNA methylation changes triggered at recurrence by the different treatment choices made in our cohort. Towards this goal, we divided our cohort into 4 groups: patients who received temozolomide only (N = 12), radiotherapy only (N = 18), the combination of radiotherapy and temozolomide (RT+TMZ; N = 6) and patients who did not receive additional treatment after the first surgery but were managed by a watch-and-wait approach (N = 33; Supplementary Tables S3C and S3D and S5). The methylome analysis of first recurrent IDHmut gliomas across the 4 groups defined 620 DMP (Kruskal–Wallis test by ranks, FDR < 0.01 and absolute DNA methylation difference > 20%; Fig. 3A; Supplementary Table S6). Upon first investigation, we determined that these CpGs were associated with consistent loss of DNA methylation in patients who received any treatment besides surgery after initial diagnosis (temozolomide only, radiotherapy only, or RT+TMZ) compared to their initial counterparts. On the other hand, the methylome of the recurrent sample of patients who did not receive treatment resembled the initial tumor (Fig. 3A). There were no CpG probes that distinguished the different groups that received any specific type of treatment (temozolomide only, radiotherapy only, or RT+TMZ), suggesting that the introduction of any treatment regimen involving TMZ and/or RT after initial surgery is associated to decrease of DNA methylation levels of treatment-naïve gliomas. The loss of DNA methylation after treatment is not affected by tumor purity (Supplementary Fig. S1H; t test, initial vs. recurrent P = 0.51 and 0.95 in the treated and untreated groups, respectively). The interrogation of the same CpGs corroborated this observation in IDHwt initial and recurrent gliomas, which showed a similar DNA hypomethylation profile to the IDHmut treatment arm (temozolomide only, radiotherapy only, or RT+TMZ; Fig. 3A). To evaluate whether the observed decrease of DNA methylation in our discovery cohort (GLASS-International; this study) is consistent in an independent cohort, we sought to validate our findings in a yet unpublished dataset of paired glioma samples from the GLASS in the Netherlands consortium (GLASS-NL; validation; ref. 21). The validation cohort consists of 36 treated paired glioma samples and 64 untreated paired samples, all of which are IDHmut astrocytomas at diagnosis. The loss of DNA methylation pattern upon treatment after initial surgery was confirmed in the validation cohort (Fig. 3B and C). Because the GLASS-NL cohort was comprised of only IDHmut astrocytomas, we repeated our discovery test on only the IDHmut astrocytomas of the GLASS-International cohort (excluding oligodendroglioma samples), separated by treatment group and selected a new set of DNA hypomethylated CpG probes (N = 981) in the treated samples (FDR < 0.01 and absolute DNA methylation difference > 25%). Sixty-one percent (381/620) of the previously described CpG probe list overlapped with this new signature showing consistent DNA methylation changes. Again, the loss of DNA methylation pattern after treatment was confirmed in the validation cohort (Supplementary Fig. S2A; Supplementary Table S5).
Figure 3.

DNA methylation loss associates with malignant transformation of glioma after standard treatment. A, Heat map of DNA methylation data. Hierarchical clustering analysis of 620 CpG probes that are associated with different treatment strategies in IDHmut paired glioma samples. Columns represent glioma samples; rows represent CpG probes. Samples were stratified and clustered on the basis of IDH mutation status and initial/recurrent status and CpGs were ordered using hierarchical clustering methods. Nonneoplastic brain samples are represented on the left of the heat map. DNA methylation β values range from 0 (low) to 1 (high). Additional tracks are included at the top of the heat maps to identify each sample membership within separate cluster analysis. B, Heat map of DNA methylation data in the validation cohort - GLASS-NL, showing the same 620 CpG probes of A. C, Boxplot of the average DNA methylation β value of the 620 CpG probes from A, in IDHmut samples. Samples are stratified by initial/recurrent status and by treated/nontreated status. Left, GLASS-International samples; right, GLASS-NL samples. Each box represents quartiles, and the center line represents the median of each group. The whiskers represent absolute range. D, Evolution of tumor histology (2021 WHO classification) from initial to recurrent samples after treatment compared with nontreated gliomas. E, Scatter plot of mean DNA methylation of CpG probes and mean gene expression of the epigenetically regulated genes after treatment (Supplementary Table S5). Each dot is a sample.
By comparing different clinical and molecular features of these tumors, we observed a significant enrichment of progression to IDHmut astrocytoma CNS WHO grade 4 in the treatment versus the nontreatment groups in the discovery set (Fisher test, P = 0.03; Fig. 3D) but not with the validation datasets (Fisher test, P = 0.3; Supplementary Fig. S2B). In addition, we noticed that 34% (10/29) of GCIMP-high tumors progressed to GCIMP-low in the treatment group versus 4% (1/27) in the nontreatment group (Fisher test, P = 0.005). The same was observed with the molecular Pan-CNS classification (8), in which 68% (15/22) of IDHmut astrocytomas progressed to high-grade astrocytoma in the treatment group versus 23% (6/26) in the nontreatment group (Fisher test, P = 0.003; Supplementary Table S1). As expected, only in the treatment group, we observed hypermutator samples (N = 5; Fig. 3A). To consider whether the selection of the treatment regimen might have been biased by the age at diagnosis, we performed an ANOVA on age at the initial tumor resection and we did not find an age difference across the four groups (ANOVA F-test, P = 0.59). To explore the biological implications of these epigenomic changes, we searched for association between DNA methylation, DNA sequence motif and gene expression. We identified 24 distal CpG-gene pairs (FDR < 0.01) enriched for the NEUROD1 motif resulting in 18 unique genes potentially regulated by DNA methylation after treatment (Fig. 3E; Supplementary Fig. S2C; Supplementary Table S5D), which included known oncogenes and cancer-related genes such as MYB, previously reported to control cell survival and proliferation (70–72), and RSPO4, an agonist of Wnt/B-catenin pathway and known to play a role in multiple cancers (73).
Tumor microenvironment changes and clinical implications of treatment in IDHmut gliomas revealed by DNA methylation
As the observed changes of DNA methylation may not only be driven by tumor cell-intrinsic-events but may also underscore changes in the methylome associated with nontumor cell populations present in the glioma tumor microenvironment (TME), we applied a methylation-specific approach for the deconvolution of nontumor cells to the longitudinal glioma cohort. MethylCIBERSORT (46) uses genome-wide DNA methylation data to deconvolute cell types from individual tumor samples. Our estimated cell populations consisted of 10 cell types: glia, neuron, endothelial cells (CD31+), B cells (CD19+), CD4 effector T cells, CD8+ T cells, T regs, natural killer cells (CD56+), macrophages, and neutrophils. The detailed description is given in Materials and Methods. Compared with untreated recurrencies, recurrent IDHmut tumors that were treated after surgery were marked by increased infiltration of endothelial cells (CD31+) and CD8 T lymphocytes (CD8+; Fig. 4A; Supplementary Table S6), suggesting that treatment might impact the tumor microenvironment and most notably angiogenesis. Our in silico cell fraction estimation was validated by IHC staining in representative IDHmut samples that received treatment after initial surgery (Fig. 4B). As a further validation, the relative proportion of nontumor cells estimated by methylCIBERSORT was significantly correlated with the glioma cell compartments estimated by gene expression for the GLASS-international transcriptomic cohort (Supplementary Fig. S3A; ref. 22). Additional changes in the TME were also observed within IDHmut and IDHwt longitudinally, particularly a significant increase in macrophage in recurrent IDHwt tumors (Supplementary Fig. S3B–S3D).
Figure 4.

Tumor microenvironment and clinical implications of treatment in IDH-mutant gliomas. A, CD31 and CD8 proportions (range scaled from 0 to 100%) in samples originating from IDHmut matched initial and recurrent tumors in treated and nontreated patients. Each box in A and B represents quartiles, and the center line represents the median of each group. The whiskers represent absolute range. B, Illustrative immunohistochemical stainings for two marker proteins (CD31 and CD8) in an individual patient showing change of levels of tumor-infiltrating immune cells between initial (left) and recurrent (right) tumors. CD8 stainings are shown in two different magnifications. Boxplots represent the number of CD31- (top) and CD8-positive cells (bottom) counted per area for individual patients. C and D, Overall survival and surgical interval analysis of IDHmut gliomas for the GLASS International (C) and GLASS-NL (D) cohorts.
We also investigated whether TME differs spatially in patients with multiple fragments available. We found that macrophages, neutrophils, and natural killer cells are the cell types that vary the most across spatially distinct samples, although most cell fractions are conserved in space (Supplementary Fig. S4A). Furthermore, the 3 tumors that showed spatial subtyping heterogeneity also showed TME and purity heterogeneity (GLSS-SF-0001, GLSS-SF-0017, GLSS-SF-0018), although we did not observe consistent changes (Supplementary Fig. S4A). A previous report showed relative DNA methylation subtype stability across glioma specimens from the same patient, especially for IDH mutants, as we show here in ref. 74.
To evaluate the clinical implications of these findings, we next assessed the clinical follow-up of the entire IDHmut GLASS-International cohort (including patients that do not have DNA methylation data) divided into treatment (N = 73) or nontreatment (N = 37) groups. First, although the overall survival medians are close, in univariate analysis, patients receiving treatment after initial surgery had a worse survival than patients who did not receive treatment beyond surgery (log-rank P = 0.03, Fig. 4C, left). However, patients left untreated after initial surgery had a progression-free interval (PFI) of 27 months that was significantly worse compared to a PFI of 40.5 months in the treatment group (log-rank P = 0.009; Fig. 4C, middle), thus confirming the results of EORTC 22845 (75). When we focused on the survival interval from second surgery to the last follow-up, we found a markedly worse survival in previously treated patients (log-rank P = 0.0001; Fig. 4C, right). The same associations of treatment and clinical implications were observed in the validation GLASS-NL cohort (Fig. 4D) and when we evaluated only grade 2 patients (Supplementary Fig. S4B).
Discussion
This work reports and analyzes the largest cohort of matched glioma samples profiled with epigenomics, transcriptomics, and genomics platforms to uncover the diverse molecular routes that drive treatment-resistant gliomas during progression. By applying an integrative molecular approach, we highlight the critical epigenetic mechanisms by which gliomas evade treatment, in addition to known transcriptomic and genetic evolution mechanisms. We also identified epigenomic changes that may be useful as biomarkers for continuous monitoring of disease progression and treatment response prediction.
Our cohort consisted of the three major glioma subtypes (IDHmut-noncodel, IDHmut-codel and IDHwt) and revealed key evolutionary differences across these subgroups. Earlier work from us and others described the GCIMP glioma phenotype in IDHmut glioma characterized by higher levels of DNA methylation likely as a direct result of the epigenetic effects of the oncometabolite 2-hydroxyglutarate that is generated by mutant IDH enzymes (10, 11). These tumors exhibited favorable clinical outcomes compared with IDHwt gliomas, which have lower DNA methylation levels (10). Indeed, in our previous work, we reported that the extent of genome-wide DNA methylation showed broad positive correlation with clinical outcome for IDHwt glioma, now defined as “molecular GBM'' given their aggressive behavior independent of histological grading (4), with these tumors having the lowest levels of DNA methylation genome-wide. Here we confirm and extend these notions to tumor progression. In particular, we found that IDHwt gliomas presented an initial low genome-wide DNA methylation, which shows minimal changes during the course of the disease. This is consistent with the malignant state of IDHwt gliomas at diagnosis and which does not appear to progress during treatment.
Conversely, we uncovered pronounced epigenetic changes in IDHmut gliomas, which invariably converged toward lower DNA methylation levels in recurrent treated tumors compared to untreated neoplasms. In the most extreme cases, the treatment-induced evolution of IDHmut glioma resulted in a state of DNA hypomethylation comparable to IDHwt gliomas. Thus, the epigenetic trajectory of glioma progression was associated with progressively lower levels of DNA methylation in IDHmut tumors or was essentially moot in the case of IDHwt gliomas as glioma initiation in the absence of IDH mutations coincided with the lowest possible levels of DNA methylation.
Following diagnosis and surgery, IDHmut glioma patients may or may not undergo treatment with radiation or alkylating chemotherapy or both (76, 77). A recognized phenomenon after using the alkylating agent temozolomide for glioma therapy is the acquisition of a hypermutator phenotype (12, 17). Here, we found that treatment with radiation and/or chemotherapy individually or combined is associated with progressive loss of DNA methylation at recurrence. These findings were confirmed in an independent cohort.
The treatment-associated epigenetic drift towards the hypomethylated state parallels the histopathological shift from a lower- to a higher-grade phenotype. Altogether, these results may indicate that the initial treatment with radiotherapy and/or temozolomide triggered a more aggressive evolution at the time of the tumor recurrence, which compromised the survival probability of these patients at that recurrence. However, the retrospective and the nonrandomized nature of our cohort leaves open the possibility that, regardless of methylation status, the primary tumors undergoing treatment were in fact naturally more aggressive than those left untreated, thus explaining the worse survival.
More specifically, in our limited and retrospective subset, the introduction of treatment after initial surgery in IDHmut gliomas is associated with a significant delay in tumor progression. However, the time from second surgery to progression or death is significantly shorter when compared with untreated patients, which is likely associated with the loss of DNA methylation and activation of associated genes that we uncovered. These findings remain consistent with previous observations from large clinical trials that reported the beneficial role of chemotherapy and radiotherapy for the survival of patients (75). Further clinical and experimental studies will be needed, using our findings as a starting point, to elucidate the mechanisms and potential causal effect of treatment-associated molecular changes.
The therapy-associated changes of the epigenetic evolution of IDHmut glioma were also mirrored by specific changes in the TME at recurrence. At recurrence, CD8 and endothelial cell–related signatures were elevated in treated IDHmut gliomas compared to untreated tumors. Together, these findings indicate that the epigenomic and genomic changes associated with more aggressive histotypes of IDHmut gliomas at recurrence coincided with specific changes of the TME (neoangiogenesis and changes in T-cell composition), indicating the convergence of IDHmut glioma evolution toward features more typical to the most aggressive IDHwt subtype.
Having recognized the significance of epigenetic changes in IDHmut noncodel glioma evolution, our study specifically focused on this subtype to comprehensively analyze ChIP-seq, transcriptome, and methylome data. Our aim was to identify the distinctive features associated with the transition toward DNA methylation loss and subsequent transcriptional activation of key drivers of disease progression, referred to as master regulators (MR). Through the integration of histone marks and transcriptome analyses, we successfully identified and validated HOXD13 as a candidate MR driving the progression of IDHmut astrocytomas. To establish its role, we performed in vitro editing experiments, which confirmed the involvement of HOXD13 in the activation of proliferation and stemness during recurrence compared with the initial tumor state. However, it is worth noting that the limited availability of patient-derived IDHmut cell lines, consequently these cells are very challenging for genetic manipulation (e.g., CRISPR-CAS9; ref. 78). Furthermore, the few IDHmut glioma cell lines have exclusively been generated from primary glioma classified into the codel or G-CIMP-high state, rather than the G-CIMP-low phenotype of interest in our study. In addition, these cell lines predominantly originate from the initial tumor stage, where HOXD13 expression levels are low. Consequently, utilizing IDHmut cell lines as a suitable model was not feasible for our investigation. Ideally, an ideal model would involve a recurrent patient-derived cell line specifically exhibiting the G-CIMP-low transition. Nevertheless, our validation experiments, despite the limitations of the available cell line models, provide support for the role of HOXD13 as a master regulator in the progression of IDHmut glioma. Furthermore, our findings indicate that IDHmut noncodel tumors that progress towards the G-CIMP-low phenotype share molecular similarities with IDHwt tumors, including low methylation levels, as well as clinical outcomes associated with poorer prognosis. Therefore, we believe that the validation experiments conducted in this study contribute to establishing HOXD13 as a crucial regulator of glioma progression in the IDHmut context.
In summary, we found loss of DNA methylation associated with standard treatment in IDHmut tumors, which results in epigenetic activation of genes associated with early tumor progression and alterations in the TME contexture toward angiogenesis and a T-cell composition that resembles a treatment-naïve IDHwt glioma. In untreated IDHmut patients the epigenome does not change significantly, and tumors progress later than the treated ones, either spontaneously or after subsequent treatment. Further studies are needed to elucidate the mechanisms and potential causal effect of treatment-associated molecular changes.
Supplementary Material
Supp Figures
Table S1: GLASS surgery-level clinical and molecular characteristics (N=354)
Table S2: List of 132 pairs (ID of Initial and Recurrent samples)
Table S3A/B/C/D: Clinical summary of GLASS epigenomic samples S3A: Clinical summary of IDHmut patients who switched from GCIMP-high to GCIMP-low S3B: Clinical summary of IDHmut and IDHwt patients with known hypermutator status S3C: Clinical summary of IDHmut and IDHwt patients with known treatment information S3D: Clinical summary of IDHmut patients with known treatment information
Table S4: Master regulators - list of samples and features (Related to Figure 2) S4A: List of sample profiled for ChIP-seq S4B: Results of H3K4me3 differential analysis S4C: Results of H3K27Ac differential analysis S4D: Results of gene expression differential analysis S4E: List of IDHmut non-codels samples profiled for both gene expression and DNA methylation S4F: List of hypomethylated probes between IDHmut non-codel initial and first recurrent pairs S4G: List of anticorrelated genes S4H: List of HOXD13 predicted targets
Table S5: Treatment-related probes and samples (Related to Figure 3) S5A: List of 69 IDHmut pairs with treatment information (ID of Initial and Recurrent samples and group assignment) S5B: List of differentially methylated probes associated with treatment in IDHmut gliomas S5C: List of differentially methylated probes associated with treatment in IDHmut astrocytomas S5D: List of CpG-gene pairs (epigenetic regulation associated with treatment)
Table S6: Deconvolution cell proportions and probe-signature matrix S6A: Methylcibersort cell proportions S6B: Methylcibersort cell reference mixture
List of collaborator authors
Supplemental Figures Legends
Acknowledgments
This work is supported by the NIH under grant numbers R01CA222146 and R01CA270365 (to H. Noushmehr), R01CA222146 (to I. Datta, L.M. Poisson), R01NS096236 (to E.G. Van Meir), R01NS117666 (to E.G. Van Meir), R01NS042645 (to S. Bakas), P50CA190991–07 (to M. Khasraw), R21 NS114873 and Cancer Center Support Grant P30 CA034196 (to R.G. Verhaak); the Department of Defense grant number CA170278 (T.S. Sabedot., L.M. Poisson, H. Noushmehr); grants #2018/00583–0 and #2019/14928–1, São Paulo Research Foundation (FAPESP; to T.M. Malta); Leeds Cares grant (9R11/14–11) and the Sidney Driscol Neuroscience Foundation Contribution to Brain Tumour Northwest (to L.F. Stead); University of Colorado Department of Neurosurgery Nervous System Biorepository (to D.R. Ormond); FNR CORE C20/BM/14646004 (GLASS-LUX) and FNRS-Televie TETHER (to S.P. Niclou); FNRS-Televie TETHER (to A.-C. Hau ). F.S. Varn is supported by the JAX Scholar Program and a postdoctoral fellowship from The Jane Coffin Childs Memorial Fund for Medical Research; GLASS-NL was supported by the Dutch Cancer Society KWF grant number 11026 (to B. Ylstra, B.A. Westerman, P. Wesseling, M.C.M. Kouwenhoven, W. Vallentgoed, P.J. French, M.J. van den Bent, J.M. Niers, M. Smits). A list of all contributors to the GLASS Consortium appears in the Supplementary Appendix.
Footnotes
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
Contributor Information
Consortium The GLASS:
Adelheid Woehrer, Allison K Lowman, Ana C deCarvalho, Ana Valeria Castro, Andrea Transou, Andrew R Brodbelt, Ann-Christin Hau, Anna Lasorella, Anna Golebiewska, Annemiek Walenkamp, Annette M Molinaro, Antonio Iavarone, Azzam Ismail, Bart A Westerman, Bauke Ylstra, Christoph Bock, D. Ryan Ormond, Daniel J Brat, Emre Kocakavuk, Erwin G Van Meir, Floris P Barthel, Frederick S Varn, Fulvio D'Angelo, Gaetano Finocchiaro, Ganesh Rao, Gelareh Zadeh, Guido Reifenberger, Ho Keu ngNg, Hoon Kim, Houtan Noushmehr, Hrvoje Miletic, Hui K Gan, Indrani Datta, Jack Rock, James M Snyder, Jason T Huse, Jennifer M Connelly, Jill S Barnholtz-Sloan, Johanna M Niers, John F deGroot, Kadir C Akdemir, Kasthuri S Kannan, Keith L Ligon, Kenneth Aldape, Ketan R Bulsara, Kevin C Johnson, Kristin D Alfaro, Laila M Poisson, Luciano Garofano, Lucy F Stead, MacLean P Nasrallah, Marion Smits, Martin J van den Bent, Mathilde CM Kouwenhoven, Michael Weller, Mohammad Hasanain, Mustafa Khasraw, Peter V Gould, Peter A Sillevis Smitt, Peter S LaViolette, Philip D Tatman, Pieter Wesseling, Pim J French, Rameen Beroukhim, Roel G.W. Verhaak, Simona Migliozzi, Simone P Niclou, Spyridon Bakas, Steven Kalkanis, Sun Ha Paek, Susan C Short, Tabatabai Ghazaleh, Tathiane M Malta, Thais S Sabedot, Tobias Weiss, Tobias Walbert, Ujjwal Baid, Wies Vallentgoed, and W. K. Alfred Yung
Authors' Disclosures
I. Datta reports grants from NIH/NCI during the conduct of the study. W.R. Vallentgoed reports grants from KWF Dutch Cancer Society during the conduct of the study. S. Bakas reports grants from NIH/NCI/ITCR (award number U01-CA242871) during the conduct of the study. J.S. Barnholtz-Sloan reports other support from NCI/NIH during the conduct of the study. M. Khasraw reports relationships outside the scope of the work in this article (research funding to institutions: BMS, AbbVie, Daiichi Sankyo, Immvira Therapeutics, BioNTech, Celldex, Astellas, Personalis, CNS Pharmaceuticals, and Immorna Therapeutics. Honoraria: JAX Lab for Genomic Research, Johnson and Johnson, Voyager Therapeutics, George Clinical, AnHeart Therapeutics, Novocure, Servier, and Stemline Menarini). S.P. Niclou reports grants from FNR and from LIH during the conduct of the study. M. Smits reports grants from Medical Delta and from Dutch Cancer Society KWF during the conduct of the study; personal fees from GE Healthcare, Bracco, AuntMinnie, and Fondazione Internazionale Menarini outside the submitted work. M.J. van den Bent reports grants from Dutch Cancer Society during the conduct of the study and personal fees from Servier, Boehringer-Ingelheim, Carthera, Genenta, Fore Biotherapeutics, Hoffman la Roche, Incyte Corporation, and AstraZeneca outside the submitted work. E.G. Van Meir reports grants from the NIH during the conduct of the study and patents issued, licensed, and with royalties paid from OncoSpherix, LLC. for an anticancer drug. T. Weiss reports personal fees from Philogen outside the submitted work. M. Weller reports grants from Versameb and Quercis, and personal fees from Bayer, Curevac, Medac, Neurosense, Novocure, Novartis, Orbus, Philogen, Roche, and Servier outside the submitted work. P.J. French reports grants from KWF during the conduct of the study and grants from Strijd van Salland, KWF, Brain Tumour Charity, and Hersentumorfonds outside the submitted work. L.M. Poisson reports grants from NIH/NCI during the conduct of the study. R.G. Verhaak reports grants and other support from Boundless Bio during the conduct of the study. No other disclosures were reported.
Authors' Contributions
T.M. Malta: Conceptualization, data curation, formal analysis, validation, investigation, visualization, writing–original draft, writing–review and editing. T.S. Sabedot: Data curation, formal analysis, validation, visualization, writing–original draft, writing–review and editing. N.S. Morosini: Formal analysis, validation, visualization. I. Datta: Formal analysis. L. Garofano: Formal analysis, methodology. W. Vallentgoed: Formal analysis, validation, visualization. F.S. Varn: Data curation, formal analysis. K. Aldape: Resources. F. D'Angelo: Formal analysis. S. Bakas: Resources. J.S. Barnholtz-Sloan: Resources. H.K. Gan: Resources. M. Hasanain: Validation, methodology. A.-C. Hau: Data curation. K.C. Johnson: Data curation, formal analysis. S. Cazacu: Formal analysis. A.C. deCarvalho: Resources, formal analysis, validation. M. Khasraw: Resources, investigation. E. Kocakavuk: Data curation. M.C.M. Kouwenhoven: Resources, investigation. S. Migliozzi: Data curation, validation. S.P. Niclou: Resources, data curation, investigation. J.M. Niers: Investigation. D.R. Ormond: Investigation. S.H. Paek: Resources. G. Reifenberger: Resources. P.A. Sillevis Smitt: Investigation. M. Smits: Investigation. L.F. Stead: Resources, investigation. M.J. van den Bent: Resources, investigation, writing–review and editing. E.G. Van Meir: Resources. A. Walenkamp: Investigation. T. Weiss: Investigation. M. Weller: Investigation, writing–review and editing. B.A. Westerman: Investigation. B. Ylstra: Investigation. P. Wesseling: Resources, investigation. A. Lasorella: Resources, formal analysis, validation. P.J. French: Resources, investigation, writing–review and editing. L.M. Poisson: Resources, data curation, investigation. The GLASS Consortium: Resources. R.G.W. Verhaak: Conceptualization, resources, data curation, supervision, funding acquisition, investigation, writing–original draft. A. Iavarone: Conceptualization, resources, data curation, supervision, funding acquisition, investigation, writing–review and editing. H. Noushmehr: Conceptualization, resources, data curation, supervision, funding acquisition, investigation, writing–original draft, writing–review and editing.
References
- 1. Malta TM, de Souza CF, Sabedot TS, Silva TC, Mosella MS, Kalkanis SN, et al. Glioma CpG island methylator phenotype (G-CIMP): biological and clinical implications. Neuro Oncol 2018;20:608–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Weller M, van den Bent M, Preusser M, Le Rhun E, Tonn JC, Minniti G, et al. EANO guidelines on the diagnosis and treatment of diffuse gliomas of adulthood. Nat Rev Clin Oncol 2021;18:170–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, et al. The 2016 world health organization classification of tumors of the central nervous system: a summary. Acta Neuropathol 2016;131:803–20. [DOI] [PubMed] [Google Scholar]
- 4. Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D, et al. The 2021 WHO classification of tumors of the central nervous system: a summary. Neuro Oncol 2021;23:1231–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shirahata M, Ono T, Stichel D, Schrimpf D, Reuss DE, Sahm F, et al. Novel, improved grading system(s) for IDH-mutant astrocytic gliomas. Acta Neuropathol 2018;136:153–66. [DOI] [PubMed] [Google Scholar]
- 6. Brat DJ, Aldape K, Colman H, Figrarella-Branger D, Fuller GN, Giannini C, et al. cIMPACT-NOW update 5: recommended grading criteria and terminologies for IDH-mutant astrocytomas. Acta Neuropathol 2020;139:603–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tesileanu CMS, van den Bent MJ, Sanson M, Wick W, Brandes AA, Clement PM, et al. Prognostic significance of genome-wide DNA methylation profiles within the randomized, phase 3, EORTC CATNON trial on non-1p/19q deleted anaplastic glioma. Neuro Oncol 2021;23:1547–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Capper D, Jones DTW, Sill M, Hovestadt V, Schrimpf D, Sturm D, et al. DNA methylation-based classification of central nervous system tumours. Nature 2018;555:469–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ceccarelli M, Barthel FP, Malta TM, Sabedot TS, Salama SR, Murray BA, et al. Molecular profiling reveals biologically discrete subsets and pathways of progression in diffuse glioma. Cell 2016;164:550–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Noushmehr H, Weisenberger DJ, Diefes K, Phillips HS, Pujara K, Berman BP, et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell 2010;17:510–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Turcan S, Rohle D, Goenka A, Walsh LA, Fang F, Yilmaz E, et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 2012;483:479–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Johnson BE, Mazor T, Hong C, Barnes M, Aihara K, McLean CY, et al. Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science 2014;343:189–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Klughammer J, Kiesel B, Roetzer T, Fortelny N, Nemc A, Nenning K-H, et al. The DNA methylation landscape of glioblastoma disease progression shows extensive heterogeneity in time and space. Nat Med 2018;24:1611–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kim H, Zheng S, Amini SS, Virk SM, Mikkelsen T, Brat DJ, et al. Whole-genome and multisector exome sequencing of primary and post-treatment glioblastoma reveals patterns of tumor evolution. Genome Res 2015;25:316–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. de Souza CF, Sabedot TS, Malta TM, Stetson L, Morozova O, Sokolov A, et al. A distinct DNA methylation shift in a subset of glioma CpG island methylator phenotypes during tumor recurrence. Cell Rep 2018;23:637–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hunter C, Smith R, Cahill DP, Stephens P, Stevens C, Teague J, et al. A hypermutation phenotype and somatic MSH6 mutations in recurrent human malignant gliomas after alkylator chemotherapy. Cancer Res 2006;66:3987–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Barthel FP, Johnson KC, Varn FS, Moskalik AD, Tanner G, Kocakavuk E, et al. Longitudinal molecular trajectories of diffuse glioma in adults. Nature 2019;576:112–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kocakavuk E, Anderson KJ, Varn FS, Johnson KC, Amin SB, Sulman EP, et al. Radiotherapy is associated with a deletion signature that contributes to poor outcomes in patients with cancer. Nat Genet 2021;53:1088–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mathur R, Zhang Y, Grimmer MR, Hong C, Zhang M, Bollam S, et al. MGMT promoter methylation level in newly diagnosed low-grade glioma is a predictor of hypermutation at recurrence. Neuro Oncol 2020;22:1580–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Touat M, Li YY, Boynton AN, Spurr LF, Iorgulescu JB, Bohrson CL, et al. Mechanisms and therapeutic implications of hypermutation in gliomas. Nature 2020;580:517–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. GLASS Consortium. Glioma through the looking GLASS: molecular evolution of diffuse gliomas and the glioma longitudinal analysis consortium. Neuro Oncol 2018;20:873–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Varn FS, Johnson KC, Martinek J, Huse JT, Nasrallah MP, Wesseling P, et al. Glioma progression is shaped by genetic evolution and microenvironment interactions. Cell 2022;185:2184–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fortin J-P, Triche TJ, Hansen KD. Preprocessing, normalization and integration of the Illumina HumanMethylationEPIC array with minfi. Bioinformatics 2017;33:558–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Triche TJ, Weisenberger DJ, Van Den Berg D, Laird PW, Siegmund KD. Low-level processing of illumina infinium DNA methylation BeadArrays. Nucleic Acids Res 2013;41:e90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Leek JT, Johnson WE, Parker HS, Jaffe AE, Storey JD. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 2012;28:882–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Heiss JA, Just AC. Identifying mislabeled and contaminated DNA methylation microarray data: an extended quality control toolset with examples from GEO. Clin Epigenetics 2018;10:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol 2004;5:R80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Garofano L, Migliozzi S, Oh YT, D'Angelo F, Najac RD, Ko A, et al. Pathway-based classification of glioblastoma uncovers a mitochondrial subtype with therapeutic vulnerabilities. Nat Cancer 2021;2:141–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Frattini V, Pagnotta SM, Tala, Fan JJ, Russo MV, Lee SB, et al. A metabolic function of FGFR3-TACC3 gene fusions in cancer. Nature 2018;553:222–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bady P, Sciuscio D, Diserens A-C, Bloch J, van den Bent MJ, Marosi C, et al. MGMT methylation analysis of glioblastoma on the infinium methylation BeadChip identifies two distinct CpG regions associated with gene silencing and outcome, yielding a prediction model for comparisons across datasets, tumor grades, and CIMP-status. Acta Neuropathol 2012;124:547–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Qin Y, Feng H, Chen M, Wu H, Zheng X. InfiniumPurify: an R package for estimating and accounting for tumor purity in cancer methylation research. Genes Dis 2018;5:43–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zheng X, Zhang N, Wu H-J, Wu H. Estimating and accounting for tumor purity in the analysis of DNA methylation data from cancer studies. Genome Biol 2017;18:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ewels P, Magnusson M, Lundin S, Käller M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016;32:3047–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/map format and SAMtools. Bioinformatics 2009;25:2078–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol 2008;9:R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ross-Innes CS, Stark R, Teschendorff AE, Holmes KA, Ali HR, Dunning MJ, et al. Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature 2012;481:389–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Navarro Gonzalez J, Zweig AS, Speir ML, Schmelter D, Rosenbloom KR, Raney BJ, et al. The UCSC genome browser database: 2021 update. Nucleic Acids Res 2021;49:D1046–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gillespie M, Jassal B, Stephan R, Milacic M, Rothfels K, Senff-Ribeiro A, et al. The reactome pathway knowledgebase 2022. Nucleic Acids Res 2022;50:D687–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lachmann A, Xu H, Krishnan J, Berger SI, Mazloom AR, Ma'ayan A. ChEA: transcription factor regulation inferred from integrating genome-wide ChIP-X experiments. Bioinformatics 2010;26:2438–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zheng R, Wan C, Mei S, Qin Q, Wu Q, Sun H, et al. Cistrome data browser: expanded datasets and new tools for gene regulatory analysis. Nucleic Acids Res 2019;47:D729–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Benjamini Y, Drai D, Elmer G, Kafkafi N, Golani I. Controlling the false discovery rate in behavior genetics research. Behav Brain Res 2001;125:279–84. [DOI] [PubMed] [Google Scholar]
- 42. Draaisma K, Chatzipli A, Taphoorn M, Kerkhof M, Weyerbrock A, Sanson M, et al. Molecular evolution of IDH wild-type glioblastomas treated with standard of care affects survival and design of precision medicine trials: a report from the EORTC 1542 study. J Clin Oncol 2020;38:81–99. [DOI] [PubMed] [Google Scholar]
- 43. Hoogstrate Y, Vallentgoed W, Kros JM, de Heer I, de Wit M, Eoli M, et al. EGFR mutations are associated with response to depatux-m in combination with temozolomide and result in a receptor that is hypersensitive to ligand. Neurooncol Adv 2020;2:vdz051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Reinius LE, Acevedo N, Joerink M, Pershagen G, Dahlén S-E, Greco D, et al. Differential DNA methylation in purified human blood cells: implications for cell lineage and studies on disease susceptibility. PLoS One 2012;7:e41361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhang Y, Maksimovic J, Naselli G, Qian J, Chopin M, Blewitt ME, et al. Genome-wide DNA methylation analysis identifies hypomethylated genes regulated by FOXP3 in human regulatory T cells. Blood 2013;122:2823–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chakravarthy A, Furness A, Joshi K, Ghorani E, Ford K, Ward MJ, et al. Pan-cancer deconvolution of tumour composition using DNA methylation. Nat Commun 2018;9:3220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Moss J, Magenheim J, Neiman D, Zemmour H, Loyfer N, Korach A, et al. Comprehensive human cell-type methylation atlas reveals origins of circulating cell-free DNA in health and disease. Nat Commun 2018;9:5068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Dekkers KF, Neele AE, Jukema JW, Heijmans BT, de Winther MPJ. Human monocyte-to-macrophage differentiation involves highly localized gain and loss of DNA methylation at transcription factor binding sites. Epigenetics Chromatin 2019;12:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gasparoni G, Bultmann S, Lutsik P, Kraus TFJ, Sordon S, Vlcek J, et al. DNA methylation analysis on purified neurons and glia dissects age and Alzheimer's disease-specific changes in the human cortex. Epigenetics Chromatin 2018;11:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mazor T, Pankov A, Johnson BE, Hong C, Hamilton EG, Bell RJA, et al. DNA methylation and somatic mutations converge on the cell cycle and define similar evolutionary histories in brain tumors. Cancer Cell 2015;28:307–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Mazor T, Chesnelong C, Pankov A, Jalbert LE, Hong C, Hayes J, et al. Clonal expansion and epigenetic reprogramming following deletion or amplification of mutant IDH1. Proc Natl Acad Sci U S A 2017;114:10743–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bai H, Harmancı AS, Erson-Omay EZ, Li J, Coşkun S, Simon M, et al. Integrated genomic characterization of IDH1-mutant glioma malignant progression. Nat Genet 2016;48:59–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Chaligne R, Gaiti F, Silverbush D, Schiffman JS, Weisman HR, Kluegel L, et al. Epigenetic encoding, heritability and plasticity of glioma transcriptional cell states. Nat Genet 2021;53:1469–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Nomura M, Saito K, Aihara K, Nagae G, Yamamoto S, Tatsuno K, et al. DNA demethylation is associated with malignant progression of lower-grade gliomas. Sci Rep 2019;9:1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Myatt SS, Lam EW-F. The emerging roles of forkhead box (Fox) proteins in cancer. Nat Rev Cancer 2007;7:847–59. [DOI] [PubMed] [Google Scholar]
- 56. Bach D-H, Long NP, Luu T-T-T, Anh NH, Kwon SW, Lee SK. The dominant role of forkhead box proteins in cancer. Int J Mol Sci 2018;19:3279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Bhatlekar S, Fields JZ, Boman BM. HOX genes and their role in the development of human cancers. J Mol Med 2014;92:811–23. [DOI] [PubMed] [Google Scholar]
- 58. Paço A, Aparecida de Bessa Garcia S, Leitão Castro J, Costa-Pinto AR, Freitas R. Roles of the HOX proteins in cancer invasion and metastasis. Cancers (Basel) 2020;13:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Abdel-Fattah R, Xiao A, Bomgardner D, Pease CS, Lopes MBS, Hussaini IM. Differential expression of HOX genes in neoplastic and non-neoplastic human astrocytes. J Pathol 2006;209:15–24. [DOI] [PubMed] [Google Scholar]
- 60. Zhang J, Deng M, Tong H, Xue W, Guo Y, Wang J, et al. A novel miR-7156–3p-HOXD13 axis modulates glioma progression by regulating tumor cell stemness. Int J Biol Sci 2020;16:3200–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Malta TM, Sokolov A, Gentles AJ, Burzykowski T, Poisson L, Weinstein JN, et al. Machine learning identifies stemness features associated with oncogenic dedifferentiation. Cell 2018;173:338–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Huang Y-G, Li D, Wang L, Su X-M, Tang X-B. CENPF/CDK1 signaling pathway enhances the progression of adrenocortical carcinoma by regulating the G2–M-phase cell cycle. J Transl Med 2022;20:78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Sun J, Huang J, Lan J, Zhou K, Gao Y, Song Z, et al. Overexpression of CENPF correlates with poor prognosis and tumor bone metastasis in breast cancer. Cancer Cell Int 2019;19:264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Zhang M, Zhang Q, Bai J, Zhao Z, Zhang J. Transcriptome analysis revealed CENPF associated with glioma prognosis. Math Biosci Eng 2021;18:2077–96. [DOI] [PubMed] [Google Scholar]
- 65. Korkolopoulou P, Christodoulou P, Lekka-Katsouli I, Kouzelis K, Papanikolaou A, Panayotides I, et al. Prognostic significance of proliferating cell nuclear antigen (PCNA) expression in gliomas. Histopathology 1994;25:349–55. [DOI] [PubMed] [Google Scholar]
- 66. Vigliani MC, Chiò A, Pezzulo T, Soffietti R, Giordana MT, Schiffer D. Proliferating cell nuclear antigen (PCNA) in low-grade astrocytomas: its prognostic significance. Tumori 1994;80:295–300. [DOI] [PubMed] [Google Scholar]
- 67. Lv Q, Zhang J, Yi Y, Huang Y, Wang Y, Wang Y, et al. Proliferating cell nuclear antigen has an association with prognosis and risks factors of cancer patients: a systematic review. Mol Neurobiol 2016;53:6209–17. [DOI] [PubMed] [Google Scholar]
- 68. Tang B, Qi G, Sun X, Tang F, Yuan S, Wang Z, et al. HOXA7 plays a critical role in metastasis of liver cancer associated with activation of Snail. Mol Cancer 2016;15:57. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 69. Bell EH, Zhang P, Shaw EG, Buckner JC, Barger GR, Bullard DE, et al. Comprehensive genomic analysis in NRG oncology/RTOG 9802: a phase III trial of radiation versus radiation plus procarbazine, lomustine (CCNU), and vincristine in high-risk low-grade glioma. J Clin Oncol 2020;38:3407–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Malaterre J, Mantamadiotis T, Dworkin S, Lightowler S, Yang Q, Ransome MI, et al. c-Myb is required for neural progenitor cell proliferation and maintenance of the neural stem cell niche in adult brain. Stem Cells 2008;26:173–81. [DOI] [PubMed] [Google Scholar]
- 71. Ramsay RG, Gonda TJ. MYB function in normal and cancer cells. Nat Rev Cancer 2008;8:523–34. [DOI] [PubMed] [Google Scholar]
- 72. Cicirò Y, Sala A. MYB oncoproteins: emerging players and potential therapeutic targets in human cancer. Oncogenesis 2021;10:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Chartier C, Raval J, Axelrod F, Bond C, Cain J, Dee-Hoskins C, et al. Therapeutic targeting of tumor-derived r-spondin attenuates β-catenin signaling and tumorigenesis in multiple cancer types. Cancer Res 2016;76:713–23. [DOI] [PubMed] [Google Scholar]
- 74. Verburg N, Barthel FP, Anderson KJ, Johnson KC, Koopman T, Yaqub MM, et al. Spatial concordance of DNA methylation classification in diffuse glioma. Neuro Oncol 2021;23:2054–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. van den Bent MJ, Afra D, de Witte O, Ben Hassel M, Schraub S, Hoang-Xuan K, et al. Long-term efficacy of early versus delayed radiotherapy for low-grade astrocytoma and oligodendroglioma in adults: the EORTC 22845 randomised trial. Lancet 2005;366:985–90. [DOI] [PubMed] [Google Scholar]
- 76. Baumert BG, Hegi ME, van den Bent MJ, von Deimling A, Gorlia T, Hoang-Xuan K, et al. Temozolomide chemotherapy versus radiotherapy in high-risk low-grade glioma (EORTC 22033–26033): a randomised, open-label, phase 3 intergroup study. Lancet Oncol 2016;17:1521–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Buckner JC, Shaw EG, Pugh SL, Chakravarti A, Gilbert MR, Barger GR, et al. Radiation plus procarbazine, CCNU, and vincristine in low-grade glioma. N Engl J Med 2016;374:1344–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Mehrjardi NZ, Hänggi D, Kahlert UD. Current biomarker-associated procedures of cancer modeling-a reference in the context of IDH1 mutant glioma. Cell Death Dis 2020;11:998. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supp Figures
Table S1: GLASS surgery-level clinical and molecular characteristics (N=354)
Table S2: List of 132 pairs (ID of Initial and Recurrent samples)
Table S3A/B/C/D: Clinical summary of GLASS epigenomic samples S3A: Clinical summary of IDHmut patients who switched from GCIMP-high to GCIMP-low S3B: Clinical summary of IDHmut and IDHwt patients with known hypermutator status S3C: Clinical summary of IDHmut and IDHwt patients with known treatment information S3D: Clinical summary of IDHmut patients with known treatment information
Table S4: Master regulators - list of samples and features (Related to Figure 2) S4A: List of sample profiled for ChIP-seq S4B: Results of H3K4me3 differential analysis S4C: Results of H3K27Ac differential analysis S4D: Results of gene expression differential analysis S4E: List of IDHmut non-codels samples profiled for both gene expression and DNA methylation S4F: List of hypomethylated probes between IDHmut non-codel initial and first recurrent pairs S4G: List of anticorrelated genes S4H: List of HOXD13 predicted targets
Table S5: Treatment-related probes and samples (Related to Figure 3) S5A: List of 69 IDHmut pairs with treatment information (ID of Initial and Recurrent samples and group assignment) S5B: List of differentially methylated probes associated with treatment in IDHmut gliomas S5C: List of differentially methylated probes associated with treatment in IDHmut astrocytomas S5D: List of CpG-gene pairs (epigenetic regulation associated with treatment)
Table S6: Deconvolution cell proportions and probe-signature matrix S6A: Methylcibersort cell proportions S6B: Methylcibersort cell reference mixture
List of collaborator authors
Supplemental Figures Legends
Data Availability Statement
The newly generated data analyzed in this study were obtained from GLASS at https://www.synapse.org/glass. All the processed molecular data accompanied by the corresponding clinical data for the GLASS consortium is available on Synapse.
Raw DNA and RNA sequencing data generation and data processing were described in previous GLASS publications (17, 22). Newly generated epigenomic data, including DNA methylation array idat files, can be accessed in Gene Expression Omnibus (GEO) at GSE248471.
Public data included in the GLASS cohort were downloaded from TCGA/GDC (https://portal.gdc.cancer.gov) and in the European Genome-Phenome Archive (EGA) at EGAS00001001255 (50), EGAS00001001854 (51), and EGAS00001001588 (15, 50–52).
DNA methylation processed data used in the deconvolution analysis was downloaded from GEO at GSE66351 (neuron and glia; ref. 49), GSE35069 (CD19+, CD8+ T, CD56+, neutrophils; ref. 44), GSE49667 (CD4+ effector T cells and Tregs; ref. 45), GSE122126 (vascular endothelial cells; ref. 47), and GSE118696 (monocyte-derived macrophage; ref. 48).