

Abstract
Atherosclerosis is a leading cause of death in the world. A significant body of evidence suggests that inflammation and various players are implicated and have pivotal roles in the formation of atherosclerotic plaques. Toll-like receptor 4 (TLR4) is linked with different stages of atherosclerosis. This receptor is highly expressed in the endothelial cells (ECs) and atherosclerotic plaques. TLR4 activation can lead to the production of inflammatory cytokines and related responses. Lectin-like oxidized low-density lipoprotein-1 (LOX-1), an integral membrane glycoprotein with widespread expression on the ECs, is involved in atherosclerosis and has some common pathways with TLR4 in atherosclerotic lesions. In addition, proprotein convertase subtilisin/kexin type9 (PCSK9), which is a regulatory enzyme with different roles in cholesterol uptake, is implicated in atherosclerosis. At present, TLR4, PCSK9, and LOX-1 are increasingly acknowledged as key players in the pathogenesis of atherosclerotic cardiovascular diseases. Herein, we presented the current evidence on the structure, functions, and roles of TLR4, PCSK9, and LOX-1 in atherosclerosis.
1. Introduction
1.1. Atherosclerosis
Atherosclerosis is one of the most common causes of cardiovascular diseases (CVDs) worldwide and a common cause of death in the United States of America, Europe, and Japan [1, 2]. Smoking, hypertension, dyslipidemia, and diabetes mellitus are the major risk factors for atherosclerosis [3]. Endothelial dysfunction is a hallmark in the pathogenesis of atherosclerosis. One of the early stages of atherosclerosis is vascular wall damage, accompanied by changes in arterial permeability [4]. Disruption in arterial permeability results in loss of vascular polarity and makes the endothelial cells (ECs) spindle-shaped; this process is the reason why endothelial dysfunction and plaque formation usually occur in areas with stressful hemodynamic states such as arteries with high curvature or arterial branches with turbulent blood flow [5, 6] Furthermore, adhesion molecules like vascular cell adhesion molecule-1 (VCAM-1), intercellular adhesion molecule-1 (ICAM-1), E-selectin, and P-selectin facilitate the diapedesis of leukocytes and the inflammatory response [7–10].
1.2. Lipid Accumulation
During the early steps in atherosclerotic plaque formation, the LDL particles undergo oxidation, which is influenced by reactive oxygen species (ROS). The LDL particles will subsequently bind to scavenger receptors (SRs) on macrophages to produce foam cells with abundant amounts of lipid reserves [5, 8, 11–13]. The LDL uptake into the macrophages can be done by micropinocytosis or by phagocytosed crystals [11]. The formation of fatty streaks from the accumulation of LDL-oxidized particles in foam cells is a characteristic of the early stages of atherosclerosis [8, 9] (Figure 1).
Figure 1.
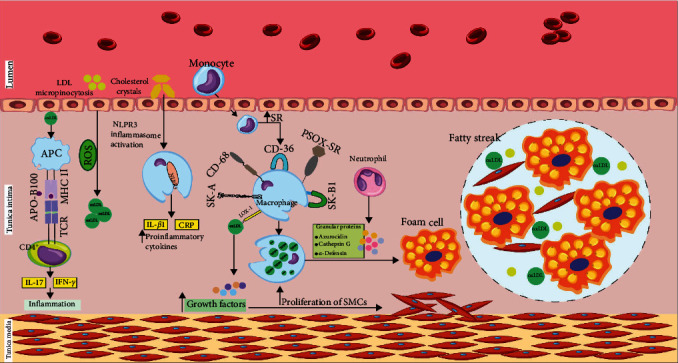
Different players in atherosclerosis. Increased LDL enters the cell via micropinocytosis and is converted into oxLDL. Additionally, after digesting oxLDL by APC, TCRs detect APO-B100 as an autoantigen by MHC2, leading to the release of IL-17 and IFN-γ. Absorbed cholesterol crystals can activate NOD, LRR, and pyrin domain-containing protein 3 (NLRP3) inflammasome, causing the production of IL-1β. Furthermore, monocytes pass through the endothelium and differentiate into scavenger receptors, including SK-A, SK-B1, CD36, CD68, LOX-1, and SR-PSOX. Macrophages will be enabled to absorb more oxLDL. Meanwhile, granular proteins such as azurocidin, cathepsin G, and α-defensin produced by neutrophils, facilitate the conversion of macrophages to foam cells. Additionally, the binding of oxLDL to LOX-1 leads to the release of growth factors that can increase the proliferation of smooth muscle cells. In summary, the aggregation of foam cells can create fatty streaks, which is the initial step in the formation of atherosclerotic plaque. LDL, low-density lipoprotein; oxLDL, oxidized low-density lipoprotein; APC, antigen-presenting cell; TCRs, T-cell receptors; APO-B100, apolipoprotein B-100; MHC2, major histocompatibility complex class II; IL-17, interleukin-17; IFN-γ, interferon-gamma; NLRP3, NOD-, LRR-, and pyrin domain-containing protein 3; IL-1β, interleukin-1β; SK-A, scavenger receptor class A; SK-B1, scavenger receptor class B type 1; CD36, cluster of differentiation 36; CD68, cluster of differentiation 68; LOX-1, lectin-like oxidized low-density lipoprotein receptor-1; SR-PSOX, scavenger receptor that binds phosphatidylserine and oxidized lipoproteins.
As monocytes cROS the endothelium, they become tissue macrophages, resulting in the differentiation of a series of SRs such as SK-A, SK-B1, CD36, CD68, lectin-like oxidized low-density lipoprotein-1 (LOX-1), and SR-PSOX, which enable them to pick up the oxidized LDL and phosphatidylserine particles [7, 8, 10, 11]. Cholesterol particles adsorbed by activating the NLRP3 (NOD-, leucine-rich repeat (LRR)-, and pyrin domain-containing protein 3) nuclear pathway cause the production of pro-inflammatory cytokines like IL-1β and C-reactive protein (CRP), accompanied by a gradual increase in the levels of prostaglandins, matrix metalloproteinases (MMP), nitric oxide species (NOS), and ROS [11, 14, 15]. After digestion of LDL oxidized particles by antigen-presenting cell (APC), the Apo-B100 component is detected as an autoantigen by the MHC-II molecule via T-cell receptor (TCR) of CD4+ T lymphocytes [6, 11, 14]. Furthermore, neutrophils, under innate immunity, can exacerbate the atherosclerosis process by producing and secreting granular proteins such as azurocidin, cathepsin G, and α-defensin; these cells can also facilitate the conversion of macrophages to foam cells [7]. In advanced atherosclerotic lesions, mast cells secret histamine, serotonin, leukotriene, thromboxane, serine protease, and other eicosanoids, which may lead to plaque rupture [7, 14], especially in the presence of metalloproteinases [16]. Growth factors are secreted from the endothelium, smooth muscle cells (SMCs), and macrophages in response to OxLDL and LOX-1 interaction, which may lead to smooth muscle proliferation, extracellular matrix production, and the formation of new arteries [17]. It is important to note that growth factors are released under specific circumstances, particularly when levels of oxidative stress are low [18]. These new and unstable arteries increase the risk of intraplaque bleeding and subsequent atherosclerotic plaque rupture [5, 6, 8, 10, 19].
1.3. Complications of Atherosclerotic Plaques
In general, plaques may go into a nonobstructive, asymptomatic, and prolonged state over several months to years, which may progress toward plaque rupture or chronic vascular occlusion [14, 20]. In the extracellular matrix of the plaque, there is a balance between reconstruction and degradation depending on factors such as necrotic nucleus size, temperature, or pH of the environment tissue [5]. In general, it is estimated that up to 65% of plaques ultimately lead to cap rupture, 30%–35% lead to superficial erosion, while only 2%–7% of them remain stable and lead to calcification [10].
Some plaques only lead to erosion or localized damage to the endothelium without the plaque rupturing. Areas with a high risk of erosion have high levels of smooth muscle tissues and proteoglycans and low levels of macrophages. Since surface vulnerability increases endothelial destruction, there may be an increase in aggregation of the platelets in the eroded areas, which may eventually lead to thrombosis formation [5, 14].
2. Toll-Like Receptor 4 (TLR4)
2.1. Structure, Function, and Signaling
TLRs were first discovered in the dorsal–ventral development of drosophila in 1997 [21]. TLR4 is a type 1 transmembrane protein that contains an LRR extracellular domain and a carboxy-terminal intracellular domain similar to the intracellular domain of the interleukin 1 receptor [22]. As shown in Table 1, TLR4, similar to the other TLRs in its family, can recognize different types of exogenous pathogen-associated molecular patterns, such as lipopolysaccharide (LPS). Likewise, several endogenous ligands for TLR4 have also been discovered, including domain A fibronectin (Fibronectin-EDA) and heat shock proteins.
Table 1.
Potential ligands for TLR4.
| Endogenous ligands | Exogenous ligands |
|---|---|
| Fibrinogen/fibrin | Fusion protein (RSV) |
| Heat shock proteins | LPS |
| Minimally modified LDL | Lipoteichoic acids |
| OxLDL | Taxol |
| Heparan sulfate | Mannuronic acid polymers |
Palsson-McDermmot and O'Neill [23] explained the downstream signaling cascade of TLRs entirely in 2008. TLR4 expressed on the surface of hematopoietic and nonhematopoietic cells such as ECs is in noncovalent association with myeloid differentiation 2 (MD2). This association is required for ligand-induced activation and forms the TLR4/MD2 receptor complex. Upon LPS recognition that leads to several molecular interactions, including LPS-binding protein (LBP), CD14, MD-2, and TLR4 and oligomerization of TLR4, downstream adaptors such as myeloid differentiation factor 88 (MyD88) are recruited through interactions with the Toll-interleukin-1 receptor (TIR) domains [24]. However, TLR4 mainly acts through MyD88-dependent and MyD88-independent pathways. In response to MyD88 binding, IL-1R-associated kinase 1 (IRAK1) is activated due to phosphorylation of IRAK1 by IL-1R-associated kinase 4 (IRAK4). This allows tumor necrosis factor-associated receptor 6 (TRAF6) to bind the phosphorylated IRAK4-IRAK1 complex. The second complex, TAK1-binding protein 1 (TAB1), TAK1-binding protein 2/3 (TAB2/3), is activated when IRAK1–TRAF6 dissociates from the TLR4 (Figure 2). When TAK1 is activated, it stimulates the inhibitor of nuclear factor-κB (IkB) kinase complex (IKK complex), which then phosphorylates IkB proteins, which causes its degradation. After that, due to the translocation of NF-κB to the nucleus, the production of several pro-inflammatory cytokines is promoted. In the independent myD88 pathway, upon activating TLR4, TIR domain-containing adapter molecules (TRIF or TICAM-1) bind to the intracellular TIR domain. TRIF activates IFN-related factor 3 (IRF3), which then activates the transcription of target genes like interferons [22, 23].
Figure 2.
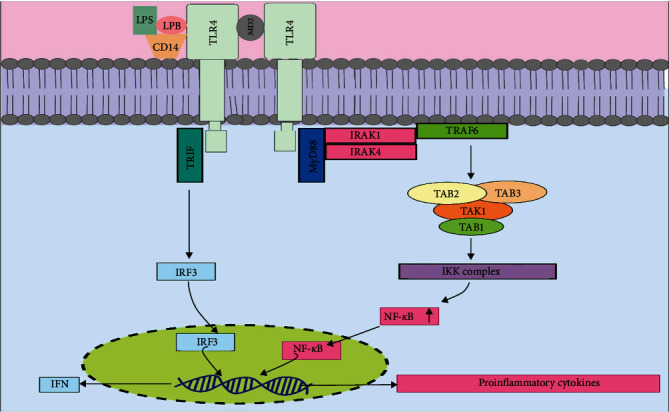
TLR4 signaling cascade. Circulating LBP recognizes LPS in the plasma and brings it to CD14. This aids the loading of LPS onto the LPS receptor complex, which is composed of dimerized TLR4 receptors and two molecules of the extracellular adapter MD-2. Subsequent signals activated by TLR4 can be subdivided into those dependent on MyD88, which occur early, and those independent of MyD88, which occur later and use the adapters TRIF and TRAM. LPS signaling leads to the early activation of NF-κB, IRF3, and MAPK kinase pathways, which are mediated by MyD88. After the subsequent activation and phosphorylation of IRAK, TRAF6 becomes activated, which gives rise to the expression of numerous pro-inflammatory genes. As a later response to LPS, TLR4 gives rise to the activation of TRAF6 and TBK1, an event mediated by the adapters; TRIF and TRAM. LPS, lipopolysaccharide; LBP, LPS-binding protein; MyD88, myeloid differentiation factor 88; IRAK, IL-1R-associated kinase; TLR, Toll-like receptor; TRAF, tumor necrosis factor receptor-associated factor; TRIF, Toll/IL-1R domain-containing adaptor-inducing IFN-β; IKK, inhibitor of nuclear factor-κB (IκB) kinase.
2.2. TLR4 and Atherosclerosis
The critical role of TLRs in atherosclerosis is well-documented. In addition to its role in pathogen recognition, TLR4 is expressed by a variety of cells in atherosclerotic lesions. Although TLR4 is expressed at low levels by ECs in normal arteries, Edfeldt et al. [25] reported increased expression of TLR4 on the ECs of human atherosclerotic lesions. Vascular smooth muscle cells (VSMCs) that reside in the media of healthy adult arteries and regulate vascular tone seem to upregulate TLR4 expression in human atherosclerotic vessels [26]. It was found that TLR4 is expressed in adventitial fibroblasts at the site of the formation of intimal lesions [27]. Dendritic cells also mediate the immunity-related processes of atherogenesis development through cell–cell contact with innate and adaptive immune cells [28]. Platelets are involved in the atherosclerosis process. Activated platelets can release platelet microparticles, which are highly procoagulant [29, 30]. Additionally, TLR4 is expressed on platelets, and activation of platelets by LPS triggers coagulation via TLR4 [31, 32]. High mobility group box 1 protein (HMGB1) is involved in the activation of platelets and plays a role in coagulant dysfunction during hemorrhagic shock and resuscitation [33]. Ahrens et al. [34] showed that in human coronary artery thrombi, the level of HMGB1 expression was increased. A growing body of evidence has shown that the formation of foamy macrophage cells through the interaction between activated monocytes and oxidized LDL (oxLDL) plays the main role in the development and progression of atherosclerosis. These lipid-laden foamy macrophages form the basis of the primary lesion. However, TLR4 also plays a significant part in this process by affecting the oxLDL-induced differentiation of macrophages to foam cells alongside the induction of inflammatory cytokine expression in VSMCs [35].
In the early stage of atherogenesis, activation of ECs and their overexpression of adhesive molecules leads to the rolling of circulating monocytes along the vascular surface and subsequent adherence at the site of activation. However, the LPS-induced activation of TLR4 on macrophages initiates a signal cascade that leads to the production of ROS and cytokines [25]. Methe et al. [36] found that acute myocardial infarction (MI) and unstable angina are associated with enhanced expression and signaling events downstream of TLR4 in circulating monocytes. Satoh et al. [37] reported a strong association between activation of TLR4 and heart failure following MI. The role of TLR4 in atherosclerosis is also supported by several loss-of-function animal models. According to Coenen et al.'s [38] study, deficiency of TLR4 expression in mice macrophages could reduce atherosclerotic lesion size under fed low-fat diets. Zeng et al. [39] found that treatment of high-fat fed ApoE−/− mice with intermittent hypoxia triggered the activation of pro-inflammatory TLR4/NF-κB signaling, leading to accelerated growth and vulnerability of atherosclerotic plaque. Malgor et al. [40] also reported an overexpression of Wnt5a in coincident with TLR4 and TLR2 in an advanced stage of atherosclerosis.
Several studies have demonstrated the role of TLR4 in plaque rupture. Activated macrophage cells within the plaque degrade extracellular matrix by secretion of MMP and proteolytic enzymes, which lead to plaque rupture. Recognition of LPS by TLR4 induces the expression of MMP9 in human macrophages, which degrades the collagen of fibrous caps [41]. Induction of apoptotic molecules such as Fas–Fas ligand is another important event in plaque rupture [42]. Destabilization of plaque may also occur through the induction and activation of proteolytic enzymes via TLR4 in macrophages. Proteolytic enzymes are capable of degrading the components of the extracellular matrix and predispose plaque to rupture [43]. Together, these results suggested a pivotal role for TLR4 in atherosclerosis progression. Our previous investigations have provided evidence for TLR4 role in human atherosclerosis and associated complications [44–46]. We showed that there is an increase in monocyte expression of TLR4 in patients with chronic coronary syndrome (CCS) who underwent percutaneous coronary intervention (PCI) [45]. In another study, we found that thrombolytic (fibrinolytic) therapies caused a more increase in monocyte expression of TLR4 expression and function compared to PCI in patients with acute coronary syndromes (ACS) [44]. Moreover, it was shown that 100 mg hydrocortisone prior to PCI was effective to cause a reduction in TLR4 expression and function in patients with CCS [47].
3. LOX-1
3.1. Structure, Function, and Signaling
LOX-1 (also known as SR-E1) is a type II integral membrane glycoprotein that is encoded by the lectin-like oxidized low-density lipoprotein receptor 1 (OLR1) gene located on chromosome 12. This receptor belongs to the C-type lectin family and is the major receptor of oxLDL. LOX-1 consists of four domains, including a short N-terminal cytoplasmic domain, a transmembrane domain, an extracellular stalk region (neck domain), and a C-type extracellular lectin-like domain (which is responsible for binding to ligands, especially oxLDL) at the C-terminus [48]. Several studies indicate the expression of LOX-1 in different types of cells like ECs, SMCs, macrophages/monocytes, platelets, fibroblasts, cardiomyocytes, airway epithelial cells, renal, and neuronal tissues [49–55]. The ligands that have been introduced for this receptor include modified lipoproteins (oxLDL, acetylated LDL, and hypochlorite-modified high-density lipoprotein), bacteria, apoptotic cells, advanced glycation end-products (AGEs), activated platelets, polyinosinic acid, carrageenan, phosphatidylserine, phosphatidylinositol, and CRP [55–60]. Importantly, LOX-1 is also expressed in atheroma-derived of human and animal atherosclerotic lesions. The expression of LOX-1 is induced by several pro-inflammatory cytokines (TNF-α, IL-1, IFN-γ), CRP, LPS, modified lipoproteins, hypertension-related stimuli (angiotensin II, endothelin-1, and fluid shear stress), hyperglycemic stimuli (high glucose and AGEs), IL-6, and some other stimuli-like homocysteine and free radicals [48, 61–63]. Pathological conditions such as diabetes mellitus, hypertension, hyperlipidemia, myocardial ischemia, and atherosclerosis are associated with an induction in LOX-1 expression. LOX-1 is involved in OxLDLs/LDLs transcytosis, leading to the macrophages transformation to foam cells and proliferation of SMCs [64].
3.2. Influence of LOX-1 on ECs
ECs activation by the LOX-1/oxLDL axis, which tends to endothelial dysfunction, is a hallmark of atherosclerosis which leads to the reduced endothelium-dependent relaxation, increased monocyte adhesion to ECs, facilitates foam cell formation, and apoptosis of ECs [65].
Endothelial dysfunction is partially a consequence of oxLDL/LOX-1 interaction. Several signaling pathways play a part in this process. One pathway involves the production of ROS caused by increased activity of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase. ROS, especially superoxide, impairs endothelial NO synthase (eNOS), which is responsible for producing nitric oxide using L-arginine and oxygen [66]. Meanwhile, LOX-1 directly increases L-arginase-1, which metabolizes arginine into ornithine and urea. As a result, there is a further decrease in NO levels and increased levels of inactivated NO. Finally, depletion of NO can lead to EC impairment [67]. Another pathway is related to EC apoptosis, which can enhance SMC proliferation and coagulation. OxLDL and LOX-1 can work together to activate the apoptosis pathway and deactivate the anti-apoptosis pathway; for example, they can increase caspase-3 and caspase-9 and decrease Bcl2 (an antiapoptotic protein) [68]. Lastly, oxLDL has a destructive effect on endothelial progenitor cells (EPCs), which disturbs the migration and proliferation of EPCs [69], and the resulting EPC dysfunction may play an important role in atherogenesis.
Leukocyte adhesion to the ECs is a crucial step in the development of atherosclerosis. Li and Mehta [70] demonstrated that incubation of human coronary artery ECs (HCAECs) with oxLDL results in the upregulation of monocyte chemoattractant protein-1 (MCP-1) expression, beside, using a human LOX-1 antisense RNA could inhibit this response. It suggests that LOX-1 is a key factor in ox-LDL–mediated monocyte adhesion to HCAECs [70]. Moreover, the binding of oxLDL to LOX-1 activates the NF-κB signaling pathway and promotes monocyte adhesion to ECs [61].
3.3. LOX-1 and Atherosclerosis
LOX-1 is a cell surface SR that participates in the binding, endocytosis, and proteolytic degradation of oxLDL and also mediates the induction of endothelial dysfunction, vascular inflammation, foam cell formation, and collagen deposition, resulting in atherosclerosis [71]. Previous studies have shown that LOX-1 is overexpressed in atherosclerotic lesions. Inoue et al. [72] showed the overexpression of LOX-1 in atherosclerosis in a mice model. Expression of LOX-1 is mainly regulated through a feed-forward system stimulated by oxLDL, a major component of atherosclerosis. Several signaling pathways are induced by LOX-1, which leads to the activation of protein kinase, transcription factors, and regulation of apoptotic and antiapoptotic genes; the final result is the development of atheroma. In pro-inflammatory states, there is an increase in the expression of LOX-1 up to 40% [73]. Internalization of oxLDL into ECs by LOX-1 increases in the macrophages. In this process, calpains, which is a calcium-dependent protease, have a crucial role in macrophage migration [74]. Wang et al. [75] showed that macrophage migration is associated with upregulation of LOX-1 and calpain-2 and downregulation of calpain-1, the same as oxLDL. As a result, macrophages remain in the intima layer of arteries and then transform into foam cells, promoting the development of plaques. Eto et al. [76] demonstrated that after vascular injury, the expression of LOX-1 increases [76]. Overproduction of oxLDL is responsible for the upregulation of LOX-1 on SMCs, which triggers pro-apoptotic pathways in vascular SMCs [77]. Exposure to high levels of oxLDL can induce upregulation of pro-apoptotic protein Bcl-2-associated X protein (Bax) and also downregulation of antiapoptotic B-cell lymphoma 2 (Bcl- 2). This process may have an impact on vulnerability and rupture of the atherosclerotic lesions [78].
OxLDL/LOX-1 activation and enhancement of NADPH oxidase may lead to the increased production and activation of mitogen-activated protein kinase (MAPK) and transcription factors (such as NF-κB), which can elevate the production of ROS and reduce the production of NO, ultimately leading to apoptosis and autophagy (autophagy refers to the destruction of cytoplasmic components by lysosomes, which differs from endocytic degradation by extracellular proteins and plasma membranes and is performed by the autophagosome [79]). Ding et al. [80] showed the dose-dependent VSMC's behavior in response to oxLDL level; a modest concentration (20–40 μg/ml) caused autophagy and apoptosis, and a higher concentration (60–100 μg/ml) caused apoptosis and declined autophagy. In vivo, it has been demonstrated that deletion of LOX-1 in low-density lipoprotein receptor (LDLR)/LOX-1 double-knockout mice alleviated autophagy [81]. Noncoding RNA, specifically microRNA, negatively modulates gene expression via binding to their mRNA [82]. Hsa-let-7g is a microRNA that can diminish the expression of LOX-1 and ROS formation in VSMCs [80]. It can also cause an overexpression of autophagy markers (beclin-1, LC3, and Atg5). Hence, there is a similar effect between hsa-let-7g and LOX-1 antibody.
Colocalization of LOX-1 and oxLDL within SMCs of human restenotic plaques suggests that LOX-1 has an effect on oxLDL-dependent VSMC proliferation and restenosis [76]. VSMC proliferation is involved in atherogenesis through vascular remodeling and subsequent lesion formation [83–85]. NF-κB- and JNK-signaling pathways are involved in VSMC proliferation, as well [74]. NO is an antioxidant that inhibits VSMC proliferation by reducing the ubiquitin-conjugating enzyme UbcH10 level, which is responsible for the ubiquitination of cell cycle protein. This reduction leads to G0/G1 cell cycle arrest, which in turn inhibits VSMC proliferation [86]. As a result of increased ROS production and decreased NO production Ang2 (a LOX-1 and VSMC proliferation inducer) can be upregulated [87, 88].
3.4. Influence of LOX-1 on Platelets
LOX-1 is expressed on the surface of human platelets in an activation-dependent manner. The binding of oxLDL to platelets induces thrombus formation by contributing to the ADP-induced activation of fibrinogen receptors such as alpha (IIb) beta (3) and alpha (2) beta (1) integrins [65].
Different pro-inflammatory cytokines like platelet factor 4 (PF4 or CXCL4) and growth factors like platelet-derived growth factor (PDGF) are secreted from the activated platelets [89, 90]. PF4 released from activated platelets facilitates the uptake of oxLDL into the macrophages, which may lead to foam cell formation [91]. PDGF released from activated platelets causes the proliferation and migration of VSMCs [90]. The binding of activated platelets to endothelial surface LOX-1 causes the secretion of endothelin-1, which induces vascular constriction and endothelial dysfunction [92]. Also, the formation of ROS and, subsequently, the inactivation of NO are the next events. As a result, it seems that LOX-1 induces atherosclerosis through binding to oxLDL and activated platelets. It has been demonstrated that aspirin and statins reduce the expression of LOX-1 in platelets [65]. Additionally, LOX-1 and ox-LDL interaction may cause destabilization of plaque through the release of the extracellular MMP inducer CD147 [93].
4. Proprotein Convertase Subtilisin/Kexin Type 9 (PCSK9)
4.1. Structure, Function, and Signaling
PCSK9, a key protein in lipid metabolism, is the ninth member of the proprotein convertase family and is encoded by the PCSK9 gene in humans on chromosome 1 [94, 95]. PCSK9 is abundantly expressed in the cells of arterial walls, such as endothelium, SMCs, and macrophages, which can regulate vascular homeostasis and atherosclerosis [96–98]. PCSK9 is primarily biosynthesized in the hepatocytes, where it binds LDLR; in addition, it is also expressed in many other tissues, including the kidney, small intestine, lung, pancreas, and brain [99]. In hepatocytes, PCSK9 transports the immature LDLR made in the endoplasmic reticulum to the Golgi membrane, which is glucosidated in the Golgi and then converts to its mature form. Mature LDLR that reaches the cellular level can now be linked to circulating LDL [100–102]. Emma et al. [103] showed that there is a negative correlation between PCSK9 level and liver damage, which means PCSK9 may play a protective role against liver damage. They also showed that PCSK9 levels decreased in hepatic steatosis [103]. In addition, circulating PCSK9 can interfere with the metabolism of triglycerides in heart cells, skeletal muscles, and adipose tissues by degrading very low-density lipoprotein receptors (VLDLR) and apolipoprotein E 2 receptors (ApoER2 or LRP8) [101, 104]. In the atherosclerotic plaque, degradation of LRP-1 (lipoprotein-related protein-1), in which PCSK 9 increases the degradation of this protein, leads to increased expression of tissue factor by ECs and increased pro-inflammatory response by macrophages [105, 106]. The use of NADPH oxidase inhibitors or NF-κB knockout in EC cells has been shown to reduce the production of ROS and LOX-1 and reduce PCSK9 expression [80, 97, 107]. Molecular mechanisms dependent on the regulatory effects of PCSK9 involved in the proliferation and migration of SMCs include the effects of this enzyme on LDLR, LRP-1, VLDLR, and CD36 [108–110]. PCSK9 can proliferate SMCs by mammalian targets of rapamycin [111]. Moreover, PCSK9 is involved in the synthesis of cytokines as well as pro-inflammatory triggers (LPS, ox-LDL, IL-6, IL-1β, TNFα, and INF-γ). In addition, PCSK9 in macrophages can modulate oxLDL uptake and increase foam cells by regulating LOX-1, CD36, and SRA [98, 112–114]. In addition to its role in the degradation of LDL receptors, PCSK9 is associated with an increased risk of coronary artery disease [115]. Based on observational epidemiological studies, plasma levels of CRP are associated with an increased risk of subsequent coronary artery disease [116]. According to a large prospective multicenter study of patients with ACS, those with higher levels of circulating PCSK9 suffer a greater degree of acute-phase inflammation as measured by hs-CRP [117]. Dwivedi et al. [118] observed a relationship between PCSK9 and inflammation. In a mouse sepsis model, PCSK9 overexpression exacerbated lung and liver inflammation, whereas PCSK9 deficiency reduced levels of IL-6 in the blood and reduced organ inflammation. Additionally, some human studies have shown that patients with a PCSK9 LOF allele have significantly lower plasma levels of pro-inflammatory cytokines like TNF-α, IL-6, IL-8, and MCP-1 compared with those with a GOF allele [119].
Many polycystic ovary syndrome (PCOS) patients are obese and suffer from atherogenic dyslipidemia, leading to a higher risk of CVD. Recent investigations have elucidated that PCSK9 directly affected ovarian lipid metabolism in PCOS mice. Wang et al. [120] showed that PCSK9 inhibition by alirocumab partly improved lipid profiles and the morphology and function of the ovary in PCOS mice, including dysfunctions associated with endocrine function, follicular growth, and ovulation [112].
4.2. PCSK9 and Atherosclerosis
Although the exact role of PCSK9 in the formation of atherosclerotic plaque is unclear, several studies have provided strong evidence for PCSK9 blockade in ischemic heart disease and the development of atherosclerotic plaque through an inflammation-mediated process. The expression of PCSK9 in ECs and SMCs is triggered in pro-inflammatory conditions comprised of ox-LDL, TNF-α, IL-1β, and LPS [80]. Boyd et al. [121] have found that elevated plasma levels of PCSK9 are associated with systemic inflammatory response syndrome and sepsis. Denis et al. [122] also reported a positive correlation between PCSK9 and atherosclerosis. According to Cheng et al. [123], PCSK9 levels are linearly correlated with the fraction and amount of necrotic core tissue in coronary atherosclerosis, independently of serum LDL cholesterol levels. Notably, PCSK9 inhibitors, either as fully human monoclonal antibodies (evolocumab and alirocumab) or as humanized monoclonal antibodies (bocosizumab), effectively lower LDL-C levels [124]. Treatment with these medicines could significantly reduce major adverse cardiovascular events. In the FOURIER (Further Cardiovascular Outcomes Research with PCSK9 Inhibition in Subjects with Elevated Risk) trial, evolocumab significantly reduced LDL-C levels by 15% for patients with atherosclerotic CVD and LDL-C > 70 mg/dL on statin therapy (HR 0.85; 95% CI, 0.79–0.92; P < 0.001) the risk of the primary endpoint (a composite of CV death, MI, stroke, hospitalization for unstable angina, or coronary revascularization) and by 20% (HR 0.80; 95% CI, 0.73–0.88; P < 0.001) the risk of the secondary endpoint (a composite of CV death, MI, or stroke) after a median follow-up of 2.2 years [125]. In addition, based on the results of the ODYSSEY OUTCOMES trial, alirocumab therapy reduced the incidence of ACS (the incidence of the primary endpoint (a composite of death from CHD, nonfatal MI, fatal or nonfatal stroke, unstable angina requiring hospitalization)) by 15% in patients on high-intensity statin therapy (HR 0.85; 95% CI, 0.78–0.93; P < 0.001). The greatest absolute reduction was observed in the subgroup of patients with the highest baseline LDL-C levels (100 mg/dL) [126]. A study by Bonaca MP indicated that the presence of PCSK9 antibodies was more prevalent in patients with higher CV risk. In fact, patients with peripheral artery disease (PAD) had a greater benefit from evolocumab therapy than patients without PAD [127].
Recent studies have proved that decreasing PCSK9 expression via endogenous RNA interference is a promising therapeutic approach to acutely reducing LDLc and have paved the way for the development of novel PCSK9 lowering agents for the management of severe hypercholesterolemia.
As the first-in-class cholesterol-lowering small interfering RNA (siRNA), Inclisiran (Leqvio®; Novartis) targets triantennary N-acetylgalactosamine carbohydrates (GalNAc). This agent has been approved in the EU in December 2020 as a possible treatment for adults with primary hypercholesterolemia (heterozygous familial and nonfamilial) and mixed dyslipidemia [128].
4.3. Possible Cross-Talks between LOX-1, TLR4, and PCSK9
As previously mentioned, NO overproduction can lead to endothelial dysfunction, which is the initial step toward atherosclerosis [129]. OxLDL interferes in modulating the eNOS/inducible nitric oxide synthase (iNOS) machinery. As the oxLDL level rises, so does HMGB1. HMGB1 is a nonhistone DNA-binding protein expressed in most cells, mainly in the nucleus and as a structural component of chromatin. Among the important roles of this protein can be mentioned the participation in the process of DNA replication, recombination, transcription, and repair [130, 131]. HMGB1 can act as a cytokine by being expressed on the plasma membrane or secreted in the extracellular environment and interacts with TLR 2, TLR4, and TLR9 [132]. The interaction of HMGB1 with TLR4 is involved in inducing the release of cytokines such as TNFα, IL-1, IL-6, and IL-8 by activated macrophages through the NF-κB pathway [133]. In normal aorta, HMGB1 is expressed in ECs, SMCs, and in CD68 positive macrophages, but in abnormal conditions and in atherosclerotic lesions, HMGB1 expression is increased. Also, strong expression of HMGB1 has been observed in the areas near the necrotic core of atherosclerotic lesions [134]. HMGB1 can activate vascular ECs and thereby lead to the expression and secretion of ICAM-1, VCAM-1, colony-stimulating factor granulocyte, RAGE, and TNFα [135, 136]. In response to cellular stress, HMGB1 is released into the extracellular space, studies showed that suppression of HMGB1 leads to reduced LDL transcytosis in human coronary artery EC and vice versa. Studies have shown that this protein plays a role in autophagy and the inhibition of inflammatory nucleosomes, leading to a reduction in inflammation [137, 138].
As shown in Figure 3, HMGB1 is involved in TLR4/Caveolin-1 expression pathway in ECs [139]. This pathway downregulates eNOS activity. One important part of TLR4 activation is Caveolin-1 Tyr14 phosphorylation [140]. Furthermore, there is a positive feedback between TLR4 and LOX-1 [141]. Therefore, when TLR4 is activated (by modulating HMGB1), the LOX-1 level also increases. The high level of LOX-1 increases the NF-κB pathway in the nucleus [67]. NF-κB signaling can affect iNOS and causes vascular damage due to an increase in iNOS level (which leads to increased EC apoptosis) and an eNOS level decrease (which leads to reduced protective autophagy); endothelial dysfunction can be initiated [142]. NADPH Oxidase is a key mediator of the LOX-1-PCSK9 axis. In addition, there is a strong correlation between the intracellular ROS concentration and PCSK9 expression [107]. After attaching oxLDL to LOX-1, ROS synthesis is promoted due to the activation of NADPH oxidase. A rise in intracellular ROS leads to upregulation of PCSK9 through a rise in TNF-α and reducing eNOS level [143, 144]. Increasing PCSK9 levels cause the degradation of LDLR and consequently leads to LOX-1 upregulation [145]. As mentioned earlier, the reduction in eNOS levels reduces protective autophagy. The combination of increased iNOS and reduced eNOS leads to endothelial dysfunction. More interestingly, RIF and MyD88 are selective adapter molecules involved in TLR4 signaling. According to a study by Liu et al. [146], although the TLR4-MyD88-NF-κB pathway plays an important role in regulating PCSK9, TLR4-TRIF does not. Through the TLR4-MyD88-NF-κB pathway, NF-κB translocation causes the expression of pro-inflammatory genes such as IL-1β, IL-18, MCP-1, IL-6, TNFα, IL-12, IFNγ, and GM-CSF, leading to an upregulation of PCSK9 [146].
Figure 3.
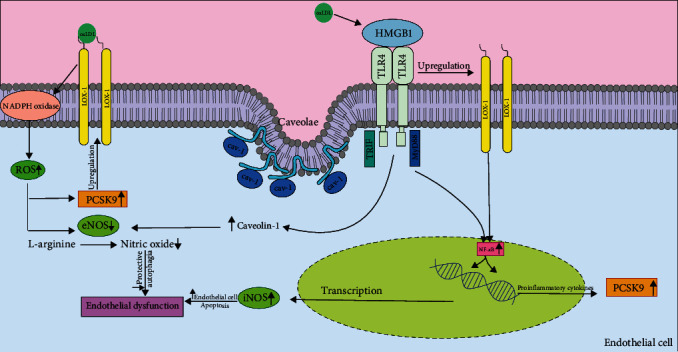
LOX-1, PCSK9, and TLR4 functions in EC. Overall, activated NF-κB upregulates the expression of inflammatory cytokines, such as IL-6, IL-1, and TNF-α. As shown, HMGB1 activity causes a decrease in the activity of eNOS. NADPH oxidase is a key mediator of the LOX-1-PCSK9 axis. Upon its activation, there will be an increase in ROS production, which is an inducer for PCSK9 production and further impairment of the function of endothelial cells. ROS, reactive oxygen species; HMGB1, high mobility group box 1 protein; TLR4, Toll-like receptor 4; eNOS, endothelial nitric oxide synthase.
5. Conclusion
TLR4, LOX-1, and PCSK9 have distinctive roles in atherosclerosis development. Data from clinical and experimental investigations indicate that their inhibitions can be effective to slow the progression of atherosclerosis. Inflammation has a well-distinguished position in atherothrombosis. At present, it is not clearly known how and when to diminish it. Moreover, it is not still conclusive which pharmaceutical interventions are preferred choices. Of note, current findings by important studies like FOURIER, ODYSSEY, and CANTOS are to pave the way for future research and a more robust understanding about lowering lipid levels and also inflammation inhibition.
Acknowledgments
The authors acknowledge funding received from the European Society of Cardiology in the form of an ESC Research Grant.
Data Availability
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
Conflicts of Interest
The authors declare that there are no conflicts of interest.
Authors' Contributions
The study concept and design were done by Seyedeh Zahra Banihashemian and Bahador Bagheri. Acquisition of data was done by Zahra Khatibiyan Feyzabadi, Ahmad Nouri, Ali Azadfallah, Mahyar Mahdizade Ari, and Parisa Alavi Toosi. Drafting of the Manuscript was done by Maral Hemmati, Mahboubeh Darban, Zahra Khatibiyan Feyzabadi, Ahmad Nouri, Ali Azadfallah, and Parisa Alavi Toosi. Critical revision of the manuscript for important intellectual content was done by Seyedeh Zahra Banihashemian and Bahador Bagheri. Study supervision was done by Seyedeh Zahra Banihashemian and Bahador Bagheri. All authors are responsible for drafting and revising the article and contributed equally.
References
- 1.Organization WH. Classification of atherosclerotic lesions. World Health Organization Technical Report Series . 1985;57(143):1–20. [PubMed] [Google Scholar]
- 2.Moran A. E., Forouzanfar M. H., Roth G. A., et al. Temporal trends in ischemic heart disease mortality in 21 world regions, 1980 to 2010: the global burden of disease 2010 study. Circulation . 2014;129(14):1483–1492. doi: 10.1161/CIRCULATIONAHA.113.004042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Criqui M. H. Epidemiology of atherosclerosis: an updated overview. The American Journal of Cardiology . 1986;57(5):C18–C23. doi: 10.1016/0002-9149(86)91022-2. [DOI] [PubMed] [Google Scholar]
- 4.Gimbrone M. A., Jr, García-Cardeña G. Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circulation Research . 2016;118(4):620–636. doi: 10.1161/CIRCRESAHA.115.306301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stefanadis C., Antoniou C. K., Tsiachris D., Pietri P. Coronary atherosclerotic vulnerable plaque: current perspectives. Journal of the American Heart Association . 2017;6(3) doi: 10.1161/JAHA.117.005543.e005543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sterpetti A. V. Inflammatory cytokines and atherosclerotic plaque progression. Therapeutic implications. Current Atherosclerosis Reports . 2020;22 doi: 10.1007/s11883-020-00891-3.75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moriya J. Critical roles of inflammation in atherosclerosis. Journal of Cardiology . 2019;73(1):22–27. doi: 10.1016/j.jjcc.2018.05.010. [DOI] [PubMed] [Google Scholar]
- 8.Bobryshev Y. V., Ivanova E. A., Chistiakov D. A., Nikiforov N. G., Orekhov A. N. Macrophages and their role in atherosclerosis: pathophysiology and transcriptome analysis. BioMed Research International . 2016;2016:13. doi: 10.1155/2016/9582430.9582430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bergheanu S. C., Bodde M. C., Jukema J. W. Pathophysiology and treatment of atherosclerosis: current view and future perspective on lipoprotein modification treatment. Netherlands Heart Journal . 2017;25:231–242. doi: 10.1007/s12471-017-0959-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rehberger Likozar A., Zavrtanik M., Šebeštjen M. Lipoprotein(a) in atherosclerosis: from pathophysiology to clinical relevance and treatment options. Annals of Medicine . 2020;52(5):162–177. doi: 10.1080/07853890.2020.1775287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wolf D., Ley K. Immunity and inflammation in atherosclerosis. Circulation Research . 2019;124:315–327. doi: 10.1161/CIRCRESAHA.118.313591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lu H., Daugherty A. Atherosclerosis. Arteriosclerosis, Thrombosis, and Vascular Biology . 2015;35(3):485–491. doi: 10.1161/ATVBAHA.115.305380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aziz M., Yadav K. S. Pathogenesis of atherosclerosis. Medical & Clinical Reviews . 2016;2(3):1–6. [Google Scholar]
- 14.Hansson G. K., Libby P., Tabas I. Inflammation and plaque vulnerability. Journal of Internal Medicine . 2015;278(5):483–493. doi: 10.1111/joim.12406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Duewell P., Kono H., Rayner K. J., et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature . 2010;464(7293):1357–1361. doi: 10.1038/nature08938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Newby A. C. Metalloproteinases and vulnerable atherosclerotic plaques. Trends in Cardiovascular Medicine . 2007;17(8):253–258. doi: 10.1016/j.tcm.2007.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ros R. Atherosclerosis—an inflammatory disease. The New England Journal of Medicine . 1999;340:115–126. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- 18.Dandapat A., Hu C., Sun L., Mehta J. L. Small concentrations of oxLDL induce capillary tube formation from endothelial cells via LOX-1-dependent redox-sensitive pathway. Arteriosclerosis, Thrombosis, and Vascular Biology . 2007;27(11):2435–2442. doi: 10.1161/ATVBAHA.107.152272. [DOI] [PubMed] [Google Scholar]
- 19.Camaré C., Pucelle M., Nègre-Salvayre A., Salvayre R. Angiogenesis in the atherosclerotic plaque. Redox Biology . 2017;12:18–34. doi: 10.1016/j.redox.2017.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ahmadi A., Argulian E., Leipsic J., Newby D. E., Narula J. From subclinical atherosclerosis to plaque progression and acute coronary events: JACC state-of-the-art review. Journal of the American College of Cardiology . 2019;74(12):1608–1617. doi: 10.1016/j.jacc.2019.08.012. [DOI] [PubMed] [Google Scholar]
- 21.Hashimoto C., Hudson K. L., Anderson K. V. The Toll gene of drosophila, required for dorsal–ventral embryonic polarity, appears to encode a transmembrane protein. Cell . 1988;52(2):269–279. doi: 10.1016/0092-8674(88)90516-8. [DOI] [PubMed] [Google Scholar]
- 22.Rock F. L., Hardiman G., Timans J. C., Kastelein R. A., Bazan J. F. A family of human receptors structurally related to Drosophila toll. Proceedings of the National Academy of Sciences . 1998;95(2):588–593. doi: 10.1073/pnas.95.2.588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pålsson-McDermott E. M., O’Neill L. A. J. Signal transduction by the lipopolysaccharide receptor, Toll-like receptor-4. Immunology . 2004;113(2):153–162. doi: 10.1111/j.1365-2567.2004.01976.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lu Y.-C., Yeh W.-C., Ohashi P. S. LPS/TLR4 signal transduction pathway. Cytokine . 2008;42(2):145–151. doi: 10.1016/j.cyto.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 25.Edfeldt K., Swedenborg J., Hansson G. K., Yan Z.-Q. Expression of Toll-like receptors in human atherosclerotic lesions: a possible pathway for plaque activation. Circulation . 2002;105(10):1158–1161. doi: 10.1161/circ.105.10.1158. [DOI] [PubMed] [Google Scholar]
- 26.Stoll L. L., Denning G. M., Li W.-G., et al. Regulation of endotoxin-induced proinflammatory activation in human coronary artery cells: expression of functional membrane-bound CD14 by human coronary artery smooth muscle cells. The Journal of Immunology . 2004;173(2):1336–1343. doi: 10.4049/jimmunol.173.2.1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vink A., Schoneveld A. H., van der Meer J. J., et al. In vivo evidence for a role of Toll-like receptor 4 in the development of intimal lesions. Circulation . 2002;106(15):1985–1990. doi: 10.1161/01.CIR.0000032146.75113.EE. [DOI] [PubMed] [Google Scholar]
- 28.Bobryshev Y. V., Lord R. S. Structural heterogeneity and contacting interactions of vascular dendritic cells in early atherosclerotic lesions of the human aorta. Journal of Submicroscopic Cytology and Pathology . 1996;28(1):49–60. [PubMed] [Google Scholar]
- 29.Suades R., Padró T., Vilahur G., Badimon L. Circulating and platelet-derived microparticles in human blood enhance thrombosis on atherosclerotic plaques. Thrombosis and Haemostasis . 2012;108(6):1208–1219. doi: 10.1160/TH12-07-0486. [DOI] [PubMed] [Google Scholar]
- 30.Wang Z. T., Wang Z., Hu Y. W. Possible roles of platelet-derived microparticles in atherosclerosis. Atherosclerosis . 2016;248:10–16. doi: 10.1016/j.atherosclerosis.2016.03.004. [DOI] [PubMed] [Google Scholar]
- 31.Shiraki R., Inoue N., Kawasaki S., et al. Expression of Toll-like receptors on human platelets. Thrombosis Research . 2004;113(6):379–385. doi: 10.1016/j.thromres.2004.03.023. [DOI] [PubMed] [Google Scholar]
- 32.Rondina M. T., Schwertz H., Harris E. S., et al. The septic milieu triggers expression of spliced tissue factor mRNA in human platelets. Journal of Thrombosis and Haemostasis . 2011;9(4):748–758. doi: 10.1111/j.1538-7836.2011.04208.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ding N., Chen G., Hoffman R., et al. Toll-like receptor 4 regulates platelet function and contributes to coagulation abnormality and organ injury in hemorrhagic shock and resuscitation. Circulation: Cardiovascular Genetics . 2014;7(5):615–624. doi: 10.1161/CIRCGENETICS.113.000398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ahrens I., Chen Y.-C., Topcic D., et al. HMGB1 binds to activated platelets via the receptor for advanced glycation end products and is present in platelet rich human coronary artery thrombi. Thrombosis and Haemostasis . 2015;114(5):994–1003. doi: 10.1160/TH14-12-1073. [DOI] [PubMed] [Google Scholar]
- 35.Yang K., Zhang X. J., Cao L. J., et al. Toll-like receptor 4 mediates inflammatory cytokine secretion in smooth muscle cells induced by oxidized low-density lipoprotein. PLoS ONE . 2014;9(4) doi: 10.1371/journal.pone.0095935.e95935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Methe H., Kim J.-O., Kofler S., Weis M., Nabauer M., Koglin J. Expansion of circulating Toll-like receptor 4–positive monocytes in patients with acute coronary syndrome. Circulation . 2005;111(20):2654–2661. doi: 10.1161/CIRCULATIONAHA.104.498865. [DOI] [PubMed] [Google Scholar]
- 37.Satoh M., Shimoda Y., Maesawa C., et al. Activated Toll-like receptor 4 in monocytes is associated with heart failure after acute myocardial infarction. International Journal of Cardiology . 2006;109(2):226–234. doi: 10.1016/j.ijcard.2005.06.023. [DOI] [PubMed] [Google Scholar]
- 38.Coenen K. R., Gruen M. L., Lee-Young R. S., Puglisi M. J., Wasserman D. H., Hasty A. H. Impact of macrophage Toll-like receptor 4 deficiency on macrophage infiltration into adipose tissue and the artery wall in mice. Diabetologia . 2009;52(2):318–328. doi: 10.1007/s00125-008-1221-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zeng X., Guo R., Dong M., Zheng J., Lin H., Lu H. Contribution of TLR4 signaling in intermittent hypoxia-mediated atherosclerosis progression. Journal of Translational Medicine . 2018;16(1):1–11. doi: 10.1186/s12967-018-1479-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Malgor R., Bhatt P. M., Connolly B. A., et al. Wnt5a, TLR2 and TLR4 are elevated in advanced human atherosclerotic lesions. Inflammation Research . 2014;63(4):277–285. doi: 10.1007/s00011-013-0697-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Grenier D., Grignon L. Response of human macrophage-like cells to stimulation by Fusobacterium nucleatum ssp. nucleatum lipopolysaccharide. Oral Microbiology and Immunology . 2006;21(3):190–196. doi: 10.1111/j.1399-302X.2006.00278.x. [DOI] [PubMed] [Google Scholar]
- 42.Li H., Sun B. Toll-like receptor 4 in atherosclerosis. Journal of Cellular and Molecular Medicine . 2007;11(1):88–95. doi: 10.1111/j.1582-4934.2007.00011.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shah P. K., Galis Z. S. Matrix metalloproteinase hypothesis of plaque rupture: players keep piling up but questions remain. Circulation . 2001;104(16):1878–1880. doi: 10.1161/circ.104.16.1878. [DOI] [PubMed] [Google Scholar]
- 44.Garjani A., Sohrabi B., Movassaghpour A. A., et al. Thrombolytic therapy up-regulates inflammatory mediators compared to percutaneous coronary intervention (PCI) Iranian Journal of Allergy, Asthma, and Immunology . 2016;15(4):257–263. [PubMed] [Google Scholar]
- 45.Bagheri B., Sohrabi B., Movassaghpur A., et al. Monocyte expression of Toll-like receptor-4 in patients with stable angina undergoing percutanoeus coronary intervention. Iranian Journal of Immunology . 2012;9(3):149–158. doi: 10.22034/iji.2012.16867. [DOI] [PubMed] [Google Scholar]
- 46.Shokri M., Bagheri B., Garjani A., et al. Everolimus-eluting stents reduce monocyte expression of Toll-like receptor 4. Advanced Pharmaceutical Bulletin . 2015;5(5):643–647. doi: 10.15171/apb.2015.087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bagheri B., Sohrabi B., Movassaghpour A. A., et al. Hydrocortisone reduces Toll-like receptor 4 expression on peripheral CD14+ monocytes in patients undergoing percutaneous coronary intervention. Iranian Biomedical Journal . 2014;18(2):76–81. doi: 10.6091/ibj.1275.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pirillo A., Norata G. D., Catapano A. L. OxLDL, and atherosclerosis. Mediators of Inflammation . 2013;2013:12. doi: 10.1155/2013/152786.152786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dieudonné A., Torres D., Blanchard S., et al. Scavenger receptors in human airway epithelial cells: role in response to double-stranded RNA. PLoS ONE . 2012;7(8) doi: 10.1371/journal.pone.0041952.e41952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Draude G., Hrboticky N., Lorenz R. L. The expression of the lectin-like oxidized low-density lipoprotein receptor (LOX-1) on human vascular smooth muscle cells and monocytes and its down-regulation by lovastatin. Biochemical Pharmacology . 1999;57(4):383–386. doi: 10.1016/s0006-2952(98)00313-x. [DOI] [PubMed] [Google Scholar]
- 51.Chen M., Kakutani M., Naruko T., et al. Activation-dependent surface expression of LOX-1 in human platelets. Biochemical and Biophysical Research Communications . 2001;282(1):153–158. doi: 10.1006/bbrc.2001.4516. [DOI] [PubMed] [Google Scholar]
- 52.Kataoka H., Kume N., Miyamoto S., et al. Oxidized LDL modulates Bax/Bcl-2 through the lectinlike Ox-LDL receptor-1 in vascular smooth muscle cells. Arteriosclerosis, Thrombosis, and Vascular Biology . 2001;21(6):955–960. doi: 10.1161/01.atv.21.6.955. [DOI] [PubMed] [Google Scholar]
- 53.Yoshida H., Kondratenko N., Green S., Steinberg D., Quehenberger O. Identification of the lectin-like receptor for oxidized low-density lipoprotein in human macrophages and its potential role as a scavenger receptor. Biochemical Journal . 1998;334(1):9–13. doi: 10.1042/bj3340009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen H. X., Sheng W. B. Application of low power laser irradiation and olfactory ensheathing cell transplantation in treatment of spinal cord injury. Chinese Journal of Tissue Engineering Research . 2008;12(51):10179–10183. [Google Scholar]
- 55.Jin P., Cong S. LOX-1 and atherosclerotic-related diseases. Clinica Chimica Acta . 2019;491:24–29. doi: 10.1016/j.cca.2019.01.006. [DOI] [PubMed] [Google Scholar]
- 56.Oka K., Sawamura T., Kikuta K., et al. Lectin-like oxidized low-density lipoprotein receptor 1 mediates phagocytosis of aged/apoptotic cells in endothelial cells. Proceedings of the National Academy of Sciences . 1998);95(16):9535–9540. doi: 10.1073/pnas.95.16.9535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen M., Masaki T., Sawamura T. LOX-1, the receptor for oxidized low-density lipoprotein identified from endothelial cells: implications in endothelial dysfunction and atherosclerosis. Pharmacology & Therapeutics . 2002;95(1):89–100. doi: 10.1016/s0163-7258(02)00236-x. [DOI] [PubMed] [Google Scholar]
- 58.Shimaoka T., Kume N., Minami M., et al. LOX-1 supports adhesion of Gram-positive and Gram-negative bacteria. The Journal of Immunology . 2001;166(8):5108–5114. doi: 10.4049/jimmunol.166.8.5108. [DOI] [PubMed] [Google Scholar]
- 59.Murphy J. E., Tacon D., Tedbury P. R., et al. LOX-1 scavenger receptor mediates calcium-dependent recognition of phosphatidylserine and apoptotic cells. Biochemical Journal . 2006;393(1):107–115. doi: 10.1042/BJ20051166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shih H. H., Zhang S., Cao W., et al. CRP is a novel ligand for the oxidized LDL receptor LOX-1. Heart and Circulatory Physiology . 2009;296(5):H1643–H1650. doi: 10.1152/ajpheart.00938.2008. [DOI] [PubMed] [Google Scholar]
- 61.Xu S., Ogura S., Chen J., Little P. J., Moss J., Liu P. LOX-1 in atherosclerosis: biological functions and pharmacological modifiers. Cellular and Molecular Life Sciences . 2013;70:2859–2872. doi: 10.1007/s00018-012-1194-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pirillo A., Uboldi P., Ferri N., Corsini A., Kuhn H., Catapano A. L. Upregulation of lectin-like oxidized low density lipoprotein receptor 1 (LOX-1) expression in human endothelial cells by modified high density lipoproteins. Biochemical and Biophysical Research Communications . 2012;428(2):230–233. doi: 10.1016/j.bbrc.2012.10.020. [DOI] [PubMed] [Google Scholar]
- 63.Lubrano V., Ballardin M., Longo V., Paolini M., Scarpato R. Hydroperoxides and cytokines as biomarkers in detecting atherosclerosis predisposition in cigarette smokers. Modern Research in Inflammation . 2012;01(1):11–17. doi: 10.4236/mri.2012.11002. [DOI] [Google Scholar]
- 64.Luchetti F., Crinelli R., Nasoni M. G., et al. LDL receptors, caveolae and cholesterol in endothelial dysfunction: oxLDLs accomplices or victims? British Journal of Pharmacology . 2021;178(16):3104–3114. doi: 10.1111/bph.15272. [DOI] [PubMed] [Google Scholar]
- 65.Kattoor A. J., Goel A., Mehta J. L. LOX-1: regulation, signaling and its role in atherosclerosis. Antioxidants . 2019;8(7) doi: 10.3390/antiox8070218.218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Getz G. S., Reardon C. A. Arginine/arginase NO NO NO. Arteriosclerosis, Thrombosis, and Vascular Biology . 2006;26(2):237–239. doi: 10.1161/01.ATV.0000202014.54609.9d. [DOI] [PubMed] [Google Scholar]
- 67.Akhmedov A., Rozenberg I., Paneni F., et al. Endothelial overexpression of LOX-1 increases plaque formation and promotes atherosclerosis in vivo. European Heart Journal . 2014;35(40):2839–2848. doi: 10.1093/eurheartj/eht532. [DOI] [PubMed] [Google Scholar]
- 68.Li D., Mehta J. L. Intracellular signaling of LOX-1 in endothelial cell apoptosis. Circulation Research . 2009;104(5):566–568. doi: 10.1161/CIRCRESAHA.109.194209. [DOI] [PubMed] [Google Scholar]
- 69.Ji K.-T., Qian L., Nan J.-L., et al. Ox-LDL induces dysfunction of endothelial progenitor cells via activation of NF-κB. BioMed Research International . 2015;2015:8. doi: 10.1155/2015/175291.175291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Li D., Mehta J. L. Antisense to LOX-1 inhibits oxidized LDL–mediated upregulation of monocyte chemoattractant protein-1 and monocyte adhesion to human coronary artery endothelial cells. Circulation . 2000;101(25):2889–2895. doi: 10.1161/01.CIR.101.25.2889. [DOI] [PubMed] [Google Scholar]
- 71.Chui P. C., Guan H.-P., Lehrke M., Lazar M. A. PPARγ regulates adipocyte cholesterol metabolism via oxidized LDL receptor 1. Journal of Clinical Investigation . 2005;115(8):2244–2256. doi: 10.1172/JCI24130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Inoue K., Arai Y., Kurihara H., Kita T., Sawamura T. Overexpression of lectin-like oxidized low-density lipoprotein receptor-1 induces intramyocardial vasculopathy in apolipoprotein E–null mice. Circulation Research . 2005;97(2):176–184. doi: 10.1161/01.RES.0000174286.73200.d4. [DOI] [PubMed] [Google Scholar]
- 73.Schaeffer D. F., Riazy M., Parhar K. S., et al. LOX-1 augments oxLDL uptake by lysoPC-stimulated murine macrophages but is not required for oxLDL clearance from plasma. Journal of Lipid Research . 2009;50(8):1676–1684. doi: 10.1194/jlr.M900167-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Barreto J., Karathanasis S. K., Remaley A., Sposito A. C. Role of LOX-1 (lectin-like oxidized low-density lipoprotein receptor 1) as a cardiovascular risk predictor: mechanistic insight and potential clinical use. Arteriosclerosis, Thrombosis, and Vascular Biology . 2021;41(1):153–166. doi: 10.1161/ATVBAHA.120.315421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wang X., Ding Z., Lin J., Guo Z., Mehta J. L. LOX-1 in macrophage migration in response to ox-LDL and the involvement of calpains. Biochemical and Biophysical Research Communications . 2015;467(1):135–139. doi: 10.1016/j.bbrc.2015.09.100. [DOI] [PubMed] [Google Scholar]
- 76.Eto H., Miyata M., Kume N., et al. Expression of lectin-like oxidized LDL receptor-1 in smooth muscle cells after vascular injury. Biochemical and Biophysical Research Communications . 2006;341(2):591–598. doi: 10.1016/j.bbrc.2005.12.211. [DOI] [PubMed] [Google Scholar]
- 77.Sun Y., Chen X. Ox-LDL-induced LOX-1 expression in vascular smooth muscle cells: role of reactive oxygen species. Fundamental & Clinical Pharmacology . 2011;25(5):572–579. doi: 10.1111/j.1472-8206.2010.00885.x. [DOI] [PubMed] [Google Scholar]
- 78.Clarke M. C. H., Figg N., Maguire J. J., et al. Apoptosis of vascular smooth muscle cells induces features of plaque vulnerability in atherosclerosis. Nature Medicine . 2006;12:1075–1080. doi: 10.1038/nm1459. [DOI] [PubMed] [Google Scholar]
- 79.Mizushima N. Autophagy: process and function. Genes & Development . 2007;21(22):2861–2873. doi: 10.1101/gad.1599207. [DOI] [PubMed] [Google Scholar]
- 80.Ding Z., Liu S., Wang X., et al. Cross-talk between LOX-1 and PCSK9 in vascular tissues. Cardiovascular Research . 2015;107(4):556–567. doi: 10.1093/cvr/cvv178. [DOI] [PubMed] [Google Scholar]
- 81.Ding Z., Liu S., Wang X., Khaidakov M., Dai Y., Mehta J. L. Oxidant stress in mitochondrial DNA damage, autophagy and inflammation in atherosclerosis. Scientific Reports . 2013;3 doi: 10.1038/srep01077.1077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhang W.-F., Zhu T.-T., Xiong Y.-W., et al. Negative feedback regulation between microRNA let-7g and LOX-1 mediated hypoxia-induced PASMCs proliferation. Biochemical and Biophysical Research Communications . 2017;488(4):655–663. doi: 10.1016/j.bbrc.2017.01.073. [DOI] [PubMed] [Google Scholar]
- 83.Rudijanto A. The role of vascular smooth muscle cells on the pathogenesis of atherosclerosis. Acta Medica Indonesiana . 2007;39(2):86–93. [PubMed] [Google Scholar]
- 84.Willis A. I., Pierre-Paul D., Sumpio B. E., Gahtan V. Vascular smooth muscle cell migration: current research and clinical implications. Vascular and Endovascular Surgery . 2004;38(1):11–23. doi: 10.1177/153857440403800102. [DOI] [PubMed] [Google Scholar]
- 85.Newby A. C., Zaltsman A. B. Molecular mechanisms in intimal hyperplasia. The Journal of Pathology . 2000;190(3):300–309. doi: 10.1002/(SICI)1096-9896(200002)190:3<300::AID-PATH596>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 86.Tsihlis N. D., Oustwani C. S., Vavra A. K., Jiang Q., Keefer L. K., Kibbe M. R. Nitric oxide inhibits vascular smooth muscle cell proliferation and neointimal hyperplasia by increasing the ubiquitination and degradation of UbcH10. Cell Biochemistry and Biophysics . 2011;60:89–97. doi: 10.1007/s12013-011-9179-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Li F. F., Shang X. K., Du X. L., Chen S. Rapamycin treatment attenuates angiotensin II-induced abdominal aortic aneurysm formation via VSMC phenotypic modulation and down-regulation of ERK1/2 activity. Current Medical Science . 2018;38:93–100. doi: 10.1007/s11596-018-1851-z. [DOI] [PubMed] [Google Scholar]
- 88.Dudzinski D. M., Igarashi J., Greif D., Michel T. The regulation and pharmacology of endothelial nitric oxide synthase. Annual Review of Pharmacology and Toxicology . 2006;46:235–276. doi: 10.1146/annurev.pharmtox.44.101802.121844. [DOI] [PubMed] [Google Scholar]
- 89.Lindemann S., Krämer B., Seizer P., Gawaz M. Platelets, inflammation and atherosclerosis. Journal of Thrombosis and Haemostasis . 2007;5(Suppl 1):203–211. doi: 10.1111/j.1538-7836.2007.02517.x. [DOI] [PubMed] [Google Scholar]
- 90.Ros R. The pathogenesis of atherosclerosis: a perspective for the 1990s. Nature . 1993;362:801–809. doi: 10.1038/362801a0. [DOI] [PubMed] [Google Scholar]
- 91.Nassar T., Sachais B. S., Akkawi S., et al. Platelet factor 4 enhances the binding of oxidized low-density lipoprotein to vascular wall cells. Journal of Biological Chemistry . 2003;278(8):6187–6193. doi: 10.1074/jbc.M208894200. [DOI] [PubMed] [Google Scholar]
- 92.Kakutani M., Masaki T., Sawamura T. A platelet-endothelium interaction mediated by lectin-like oxidized low-density lipoprotein receptor-1. Proceedings of the National Academy of Sciences . 2000;97(1):360–364. doi: 10.1073/pnas.97.1.360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yang S.-H., Li Y.-T., Du D.-Y. Oxidized low-density lipoprotein-induced CD147 expression and its inhibition by high-density lipoprotein on platelets in vitro. Thrombosis Research . 2013;132(6):702–711. doi: 10.1016/j.thromres.2013.10.003. [DOI] [PubMed] [Google Scholar]
- 94.O’Connell E. M., Lohoff F. W. Proprotein convertase subtilisin/kexin type 9 (PCSK9) in the brain and relevance for neuropsychiatric disorders. Frontiers in Neuroscience . 2020;14 doi: 10.3389/fnins.2020.00609.609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Seidah N. G., Benjannet S., Wickham L., et al. The secretory proprotein convertase neural apoptosis-regulated convertase 1 (NARC-1): liver regeneration and neuronal differentiation. Proceedings of the National Academy of Sciences . 2003;100(3):928–933. doi: 10.1073/pnas.0335507100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ferri N., Tibolla G., Pirillo A., et al. Proprotein convertase subtilisin kexin type 9 (PCSK9) secreted by cultured smooth muscle cells reduces macrophages LDLR levels. Atherosclerosis . 2012;220(2):381–386. doi: 10.1016/j.atherosclerosis.2011.11.026. [DOI] [PubMed] [Google Scholar]
- 97.Wu C.-Y., Tang Z.-H., Jiang L., Li X.-F., Jiang Z.-S., Liu L.-S. PCSK9 siRNA inhibits HUVEC apoptosis induced by ox-LDL via Bcl/Bax-caspase9-caspase3 pathway. Molecular and Cellular Biochemistry . 2012;359:347–358. doi: 10.1007/s11010-011-1028-6. [DOI] [PubMed] [Google Scholar]
- 98.Giunzioni I., Tavori H., Covarrubias R., et al. Local effects of human PCSK9 on the atherosclerotic lesion. The Journal of Pathology . 2016;238(1):52–62. doi: 10.1002/path.4630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Sun H. L., Wu Y. R., Song F. F., et al. Role of PCSK9 in the development of mouse periodontitis before and after treatment: a double-edged sword. The Journal of Infectious Diseases . 2018;217(4):667–680. doi: 10.1093/infdis/jix574. [DOI] [PubMed] [Google Scholar]
- 100.Strøm T. B., Tveten K., Leren T. P. PCSK9 acts as a chaperone for the LDL receptor in the endoplasmic reticulum. Biochemical Journal . 2014;457(1):99–105. doi: 10.1042/BJ20130930. [DOI] [PubMed] [Google Scholar]
- 101.Poirier S., Mayer G., Poupon V., et al. Dissection of the endogenous cellular pathways of PCSK9-induced low density lipoprotein receptor degradation: evidence for an intracellular route. Journal of Biological Chemistry . 2009;284(42):28856–28864. doi: 10.1074/jbc.M109.037085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Schulz R., Schlüter K.-D., Laufs U. Molecular and cellular function of the proprotein convertase subtilisin/kexin type 9 (PCSK9) Basic Research in Cardiology . 2015;110 doi: 10.1007/s00395-015-0463-z.4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Emma M. R., Giannitrapani L., Cabibi D., et al. Hepatic and circulating levels of PCSK9 in morbidly obese patients: relation with severity of liver steatosis. Biochimica et Biophysica Acta (BBA)—Molecular and Cell Biology of Lipids . 2020;1865(12) doi: 10.1016/j.bbalip.2020.158792.158792 [DOI] [PubMed] [Google Scholar]
- 104.Poirier S., Mayer G., Benjannet S., et al. The proprotein convertase PCSK9 induces the degradation of low density lipoprotein receptor (LDLR) and its closest family members VLDLR and ApoER2. Journal of Biological Chemistry . 2008;283(4):2363–2372. doi: 10.1074/jbc.M708098200. [DOI] [PubMed] [Google Scholar]
- 105.Wang M., Li Y.-F., Guo Y.-G., Chen M.-M., Jiang Z.-L., Song J.-Y. Positive correlation between plasma PCSK9 and tissue factors levels in patients with angiographically diagnosed coronary artery disease and diabetes mellitus. Journal of Geriatric Cardiology . 2016;13(4):312–315. doi: 10.11909/j.issn.1671-5411.2016.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Overton C. D., Yancey P. G., Major A. S., Linton M. F., Fazio S. Deletion of macrophage LDL receptor-related protein increases atherogenesis in the mouse. Circulation Research . 2007;100(5):670–677. doi: 10.1161/01.RES.0000260204.40510.aa. [DOI] [PubMed] [Google Scholar]
- 107.Ding Z., Liu S., Wang X., et al. Hemodynamic shear stress via ROS modulates PCSK9 expression in human vascular endothelial and smooth muscle cells and along the mouse aorta. Antioxidants & Redox Signaling . 2015;22(9):760–771. doi: 10.1089/ars.2014.6054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Canuel M., Sun X., Asselin M.-C., Paramithiotis E., Prat A., Seidah N. G. Proprotein convertase subtilisin/kexin type 9 (PCSK9) can mediate degradation of the low density lipoprotein receptor-related protein 1 (LRP-1) PloS ONE . 2013;8(5) doi: 10.1371/journal.pone.0064145.e64145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ferri N., Marchianò S., Tibolla G., et al. PCSK9 knock-out mice are protected from neointimal formation in response to perivascular carotid collar placement. Atherosclerosis . 2016;253:214–224. doi: 10.1016/j.atherosclerosis.2016.07.910. [DOI] [PubMed] [Google Scholar]
- 110.Navarro-Lérida I., Sánchez-Perales S., Calvo M., et al. A palmitoylation switch mechanism regulates Rac1 function and membrane organization. The EMBO Journal . 2012;31(3):534–551. doi: 10.1038/emboj.2011.446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ding Z., Liu S., Wang X., et al. CROS-talk between PCSK9 and damaged mtDNA in vascular smooth muscle cells: role in apoptosis. Antioxidants & Redox Signaling . 2016;25(18):997–1008. doi: 10.1089/ars.2016.6631. [DOI] [PubMed] [Google Scholar]
- 112.Mehta J. L. Oxidized or native low-density lipoprotein cholesterol: which is more important in atherogenesis? Journal of the American College of Cardiology . 2006;48(5):980–982. doi: 10.1016/j.jacc.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 113.Ding Z., Wang X., Liu S., et al. NLRP3 inflammasome via IL-1β regulates PCSK9 secretion. Theranostics . 2020;10(16):7100–7110. doi: 10.7150/thno.45939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Tang Z.-H., Peng J., Ren Z., et al. New role of PCSK9 in atherosclerotic inflammation promotion involving the TLR4/NF-κB pathway. Atherosclerosis . 2017;262:113–122. doi: 10.1016/j.atherosclerosis.2017.04.023. [DOI] [PubMed] [Google Scholar]
- 115.Zamarrón-Licona E., Rodríguez-Pérez J. M., Posadas-Sánchez R., et al. Variants of PCSK9 gene are associated with subclinical atherosclerosis and cardiometabolic parameters in Mexicans. The GEA project. Diagnostics . 2021;11(5) doi: 10.3390/diagnostics11050774.774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.The Emerging Risk Factors Collaboration. C-reactive protein concentration and risk of coronary heart disease, stroke, and mortality: an individual participant meta-analysis. The Lancet . 2010;375(9709):132–140. doi: 10.1016/S0140-6736(09)61717-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Gencer B., Montecucco F., Nanchen D., et al. Prognostic value of PCSK9 levels in patients with acute coronary syndromes. European Heart Journal . 2016;37(6):546–553. doi: 10.1093/eurheartj/ehv637. [DOI] [PubMed] [Google Scholar]
- 118.Dwivedi D. J., Grin P. M., Khan M., et al. Differential expression of PCSK9 modulates infection, inflammation, and coagulation in a murine model of sepsis. Shock . 2016;46(6):672–680. doi: 10.1097/SHK.0000000000000682. [DOI] [PubMed] [Google Scholar]
- 119.Walley K. R., Thain K. R., Russell J. A., et al. PCSK9 is a critical regulator of the innate immune response and septic shock outcome. Science Translational Medicine . 2014;6(258) doi: 10.1126/scitranslmed.3008782.258ra143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Wang M., Zhao D., Xu L., et al. Role of PCSK9 in lipid metabolic disorders and ovarian dysfunction in polycystic ovary syndrome. Metabolism Clinical and Experimental . 2019;94:47–58. doi: 10.1016/j.metabol.2019.02.002. [DOI] [PubMed] [Google Scholar]
- 121.Boyd J. H., Fjell C. D., Russell J. A., Sirounis D., Cirstea M. S., Walley K. R. Increased plasma PCSK9 levels are associated with reduced endotoxin clearance and the development of acute organ failures during sepsis. Journal of Innate Immunity . 2016;8(2):211–220. doi: 10.1159/000442976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Denis M., Marcinkiewicz J., Zaid A., et al. Gene inactivation of proprotein convertase subtilisin/kexin type 9 reduces atherosclerosis in mice. Circulation . 2012;125(7):894–901. doi: 10.1161/CIRCULATIONAHA.111.057406. [DOI] [PubMed] [Google Scholar]
- 123.Cheng J. M., Oemrawsingh R. M., Garcia-Garcia H. M., et al. PCSK9 in relation to coronary plaque inflammation: results of the ATHEROREMO-IVUS study. Atherosclerosis . 2016;248:117–122. doi: 10.1016/j.atherosclerosis.2016.03.010. [DOI] [PubMed] [Google Scholar]
- 124.Alkhalil M. Proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors, reality or dream in managing patients with cardiovascular disease. Current Drug Metabolism . 2019;20(1):72–82. doi: 10.2174/1389200219666180816141827. [DOI] [PubMed] [Google Scholar]
- 125.Sabatine M. S., Giugliano R. P., Keech A. C., et al. Evolocumab and clinical outcomes in patients with cardiovascular disease. New England Journal of Medicine . 2017;376(18):1713–1722. doi: 10.1056/NEJMoa1615664. [DOI] [PubMed] [Google Scholar]
- 126.Schwartz G. G., Steg P. G., Szarek M., et al. Alirocumab and cardiovascular outcomes after acute coronary syndrome. New England Journal of Medicine . 2018;379(22):2097–2107. doi: 10.1056/NEJMoa1801174. [DOI] [PubMed] [Google Scholar]
- 127.Bonaca M. P., Nault P., Giugliano R. P., et al. Low-density lipoprotein cholesterol lowering with evolocumab and outcomes in patients with peripheral artery disease: insights from the FOURIER trial (further cardiovascular outcomes research with PCSK9 inhibition in subjects with elevated risk) Circulation . 2018;137(4):338–350. doi: 10.1161/CIRCULATIONAHA.117.032235. [DOI] [PubMed] [Google Scholar]
- 128.Lamb Y. N. Inclisiran: first approval. Drugs . 2021;81:389–395. doi: 10.1007/s40265-021-01473-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Vanhoutte P. M. Endothelial dysfunction: the first step toward coronary arteriosclerosis. Circulation Journal . 2009;73(4):595–601. doi: 10.1253/circj.cj-08-1169. [DOI] [PubMed] [Google Scholar]
- 130.Einck L., Bustin M. The intracellular distribution and function of the high mobility group chromosomal proteins. Experimental Cell Research . 1985;156(2):295–310. doi: 10.1016/0014-4827(85)90539-7. [DOI] [PubMed] [Google Scholar]
- 131.Abdulahad D. A., Westra J., Limburg P. C., Kallenberg C. G. M., Bijl M. HMGB1 in systemic lupus erythematosus: Its role in cutaneous lesions development. Autoimmunity Reviews . 2010;9(10):661–665. doi: 10.1016/j.autrev.2010.05.015. [DOI] [PubMed] [Google Scholar]
- 132.Yu M., Wang H., Ding A., et al. HMGB1 signals through Toll-like receptor (TLR) 4 and TLR2. Shock . 006;26(2):174–179. doi: 10.1097/01.shk.0000225404.51320.82. [DOI] [PubMed] [Google Scholar]
- 133.Park J. S., Svetkauskaite D., He Q., et al. Involvement of Toll-like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. Journal of Biological Chemistry . 2004;279(9):7370–7377. doi: 10.1074/jbc.M306793200. [DOI] [PubMed] [Google Scholar]
- 134.Kalinina N., Agrotis A., Antropova Y., et al. Increased expression of the DNA-binding cytokine HMGB1 in human atherosclerotic lesions: role of activated macrophages and cytokines. Arteriosclerosis, Thrombosis, and Vascular Biology . 2004;24(12):2320–2325. doi: 10.1161/01.ATV.0000145573.36113.8a. [DOI] [PubMed] [Google Scholar]
- 135.Inoue K., Kawahara K., Biswas K. K., et al. HMGB1 expression by activated vascular smooth muscle cells in advanced human atherosclerosis plaques. Cardiovascular Pathology . 2007;16(3):136–143. doi: 10.1016/j.carpath.2006.11.006. [DOI] [PubMed] [Google Scholar]
- 136.Porto A., Palumbo R., Pieroni M., et al. Smooth muscle cells in human atherosclerotic plaques secrete and proliferate in response to high mobility group box 1 protein. The FASEB Journal . 2006;20(14):2565–2566. doi: 10.1096/fj.06-5867fje. [DOI] [PubMed] [Google Scholar]
- 137.Banerjee S., de Freitas A., Friggeri A., Zmijewski J. W., Liu G., Abraham E. Intracellular HMGB1 negatively regulates efferocytosis. The Journal of Immunology . 2011;187(9):4686–4694. doi: 10.4049/jimmunol.1101500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Haraba R., Suica V. I., Uyy E., Ivan L., Antohe F. Hyperlipidemia stimulates the extracellular release of the nuclear high mobility group box 1 protein. Cell and Tissue Research . 2011;346:361–368. doi: 10.1007/s00441-011-1277-4. [DOI] [PubMed] [Google Scholar]
- 139.Tian K., Ogura S., Little P. J., Xu S.-W., Sawamura T. Targeting LOX-1 in atherosclerosis and vasculopathy: current knowledge and future perspectives. Annals of the New York Academy of Sciences . 2019;1443(1):34–53. doi: 10.1111/nyas.13984. [DOI] [PubMed] [Google Scholar]
- 140.Jiao H., Zhang Y., Yan Z., et al. Caveolin-1 Tyr14 phosphorylation induces interaction with TLR4 in endothelial cells and mediates MyD88-dependent signaling and sepsis-induced lung inflammation. The Journal of Immunology . 2013;191(12):6191–6199. doi: 10.4049/jimmunol.1300873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Ding Z., Liu S., Wang X., et al. Lectin-like ox-LDL receptor-1 (LOX-1)–Toll-like receptor 4 (TLR4) interaction and autophagy in CATH.a differentiated cells exposed to angiotensin II. Molecular Neurobiology . 2015;51:623–632. doi: 10.1007/s12035-014-8756-z. [DOI] [PubMed] [Google Scholar]
- 142.Gliozzi M., Scicchitano M., Bosco F., et al. Modulation of nitric oxide synthases by oxidized LDLs: role in vascular inflammation and atherosclerosis development. International Journal of Molecular Sciences . 2019;20(13) doi: 10.3390/ijms20133294.3294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Tam J., Thankam F., Agrawal D. K., Radwan M. M. Critical role of LOX-1-PCSK9 axis in the pathogenesis of atheroma formation and its instability. Heart, Lung and Circulation . 2021;30(10):1456–1466. doi: 10.1016/j.hlc.2021.05.085. [DOI] [PubMed] [Google Scholar]
- 144.Faria A. M., Papadimitriou A., Silva K. C., Lopes de Faria J. M., Lopes de Faria J. B. Uncoupling endothelial nitric oxide synthase is ameliorated by green tea in experimental diabetes by re-establishing tetrahydrobiopterin levels. Diabetes . 2012;61(7):1838–1847. doi: 10.2337/db11-1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Tavori H., Fan D., Blakemore J. L., et al. Serum proprotein convertase subtilisin/kexin type 9 and cell surface low-density lipoprotein receptor: evidence for a reciprocal regulation. Circulation . 2013;127(24):2403–2413. doi: 10.1161/CIRCULATIONAHA.113.001592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Liu S., Deng X., Zhang P., et al. Blood flow patterns regulate PCSK9 secretion via MyD88-mediated pro-inflammatory cytokines. Cardiovascular Research . 2020;116(10):1721–1732. doi: 10.1093/cvr/cvz262. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.