

Abstract

Herein, we report the preparation and characterization of the Group 13 metal complexes of a tripodal tris(nitroxide)-based ligand, designated (TriNOx3–)M (M = Al (1), Ga (2), In (3)). Complexes 1 and 2 both activate the O–H bond of a range of alcohols spanning a ∼10 pKa unit range via an element-ligand cooperative pathway to afford the zwitterionic complexes (HTriNOx2–)M–OR. Structures of these alcohol adduct products are discussed. We demonstrate that the thermodynamic and kinetic aspects of the reactions are both influenced by the identity of the metal, with 1 having higher reaction equilibrium constants and proceeding at a faster rate relative to 2 for any given alcohol. These parameters are also influenced by the pKa of the alcohol, with more acidic alcohols reacting both to more completion and faster than their less acidic counterparts. Possible mechanistic pathways for the O–H activation are discussed.
Short abstract
Aluminum and gallium complexes supporting a tripodal tris(nitroxide) ligand exhibit reversible, element-ligand cooperative O−H bond activation of various alkyl and aryl alcohols. The equilibrium position of the reactions are affected by both the identity of the group 13 element and the pKa of the alcohol, and the aluminum complex reacts both faster and more fully with an alcohol relative to its gallium counterpart
Introduction
The development of transition-metal coordination complexes designed to undergo the metal–ligand cooperative (MLC) breaking of chemical bonds is an exciting and rich field of research.1,2 Of particular importance have been the development of systems for the splitting of polar H–X (X = heteroatom) bonds, which serves as an important step to introducing these reagents into catalytic processes without formal metal-based oxidative addition. This breadth of success is in contrast to the element-ligand cooperative (ELC) chemistry of the main-group elements, which has been much less developed.3 We have been investigating the coordination chemistry of aluminum and other group 13 metal complexes supporting redox-active and/or noninnocent ligand frameworks,4,5 and specifically, have an interest in understanding the role that the complexes play in ELC chemistry.
There is precedent for ELC chemistry between aluminum and H–X bonds (Scheme 1). The Berben group has shown that their bis(imino)pyridine aluminum hydride complex (PhI2P2–)AlH undergoes ELC chemistry with select anilines6 and alcohols7 to form the (PhHI2P1–)Al(X)H complexes, where X represents an alkoxo or amido ligand that is installed at the metal ion while the ligand is protonated. They have advanced this chemistry to develop catalytic systems for the dehydrocoupling of amines,6 the dehydrogenation of formic acid,8 and carbonyl transfer hydrogenation.9
Scheme 1. Examples of ELC Reactivity for Aluminum and Gallium Complexes with Polar H–X Bonds, Including This Work (Bottom).
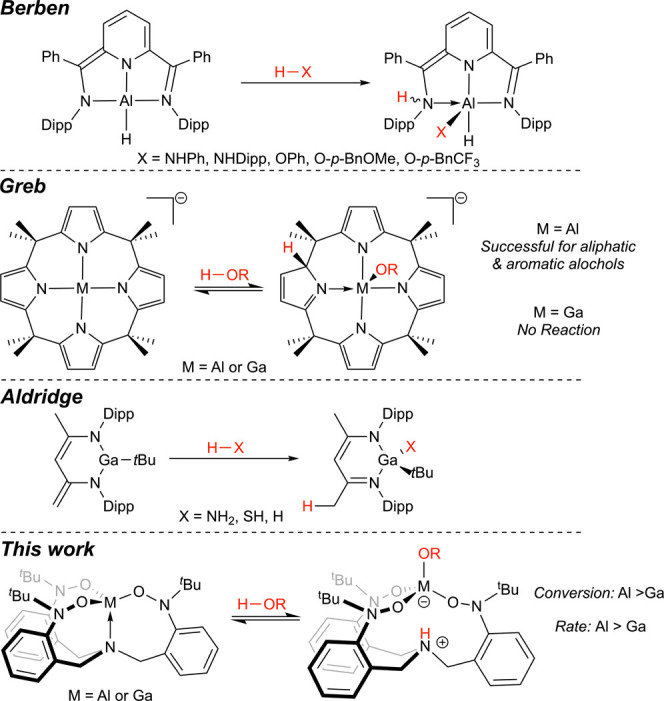
Recently, the Greb group reported the preparation of a methylcalix[4]pyrrolato aluminate complex10 and described its ELC chemistry,11 including its reactivity with alcohols.12 The complex reacts with a variety of aliphatic and aromatic alcohols via a reversible ELC process that involves dearomatization/rearomatization of the calix[4]pyrrole ligand to facilitate the protonation/deprotonation step. Although also shown to undergo ELC with carbon dioxide, the corresponding calix[4]pyrrolato gallate complex does not react directly with i-PrOH, which the authors attributed to differences in Lewis acidity between the metal ions.13
The Aldridge group has demonstrated ELC chemistry for their β-diketiminato galium complex (Dipp2Nacnac’)Ga(tBu), which reacts with a range of H–X bonds to give the [(Dipp2Nacnac)Ga(tBu)X] (X = NH2, SH, H) complexes.14 The reactivity was applicable to H–X bonds of varying polarities and laid the basis for the catalytic reduction of carbon dioxide to MeOBpin by HBpin.
We have reported the synthesis of the (TriNOx3–)Al (1, TriNOx3– = [{(2-tBuNO)C6H4CH2}3N]3–) complex and showed that it is an effective catalyst for the hydroboration reaction of carbonyl compounds to their boronic esters with HBpin.15 Complex 1 combines a Lewis acidic aluminum ion along with several basic sites within the TriNOx3– ligand framework, and we proposed an ELC pathway involving synergistic activation of both the carbonyl (at the Al3+ ion) and borane (at a nitrogen atom of a N–O arm of the TriNOx3– ligand) for the hydroboration reaction. In our exploration of synthetic routes to 1, we discovered that the TriNOxH3 ligand precursor reacts incompletely with trimethylaluminum at room temperature to give the complex (HTriNOx2–)AlMe, which when heated undergoes a third deprotonation to liberate methane and give 1. We postulated that reaction of 1 with a polar H–X reagent would result in the analogous (HTriNOx2–)AlX complexes via an ELC pathway, where the Lewis acidic aluminum would accept the electrons from the heteroatom X and the TriNOx3– ligand would accept the proton.
Herein, we report the synthesis and characterization of the aluminum and gallium (TriNOx3–)M complexes and discuss their reactivity with alcohols to give the (HTriNOx2–)M–OR complexes. Unlike with Greb’s gallate complex, our gallium system undergoes ELC directly with alcohols and we are able to compare and contrast reactivity between the two metal ions across a range of protic substrates. We show that the aluminum complex reacts both faster and with greater completion with a given alcohol relative to the same reaction with gallium and develop a mechanistic description supported by a kinetic analysis.
Results and Discussion
Synthesis and Characterization of (TriNOx)M Complexes
The (TriNOx3–)M (M = Al (1); Ga (2)) complexes are most easily prepared from the reaction between {M(NMe2)3}2 with two equivalents of TriNOxH3 ligand precursor in toluene (Scheme 2). After heating the reaction mixtures for 12 h at 50 °C, complexes 1 and 2 can be cleanly isolated from the reactions following the removal of volatiles, giving off-white solids in average yields of 80 and 70%, respectively.16 This synthetic route is an improvement over our previously reported preparation of 1-py (py = pyridine) in which salt metathesis was used to install the TriNOx3– ligand.15 The salt metathesis method was unsuccessful in the preparation of 2 as the isolation of pure (TriNOx3–)Ga from the reaction byproducts was always complicated by partial decomposition of the complex into some unknown material. 1 and 2 are both indefinitely stable in the solid state when stored under a nitrogen atmosphere at −80 °C, although they do decompose if stored in a glovebox over the course of weeks to months if care is not taken to protect the complexes from volatile reagents, regardless of the temperature at which they are stored.171 and 2 are soluble in hydrocarbon solvents such as toluene and benzene as well as in more polar solvents such as tetrahydrofuran, chloroform, and methylene chloride but have minimal solubility in acetonitrile, pentane, or hexanes.
Scheme 2. Synthesis of the (TriNOx3–)M Complexes 1–3 and Solid-State Structure of the (TriNOx3–)Ga (2) Complex.

Ellipsoids are projected at the 50% probability, and H atoms are omitted for clarity. One of the ligand arms is depicted using a wireframe model. R1 = 0.0637.
We also prepared the (TriNOx3–)In (3) complex through the reaction of {In(NEt2)3}2 with two equivalents of TriNOxH3 in a toluene/pyridine mixture at room temperature (Scheme 2). Following evaporation of the solvents, complex 3 was collected in 56% yield after purification by precipitation from a concentrated pyridine solution at −25 °C. 3 is much less soluble than its aluminum and gallium counterparts; it has very limited solubility even in polar solvents like THF or dichloromethane and gives homogeneous solutions only in boiling pyridine. Based on this limited solubility, complex 3 was not included in the alcohol reactivity studies carried out with the other (TriNOx)M complexes (vide infra).
Complexes 1–3 were readily characterized by 1H and 13C NMR spectroscopies. All of the complexes exhibit a single resonance in their 1H NMR spectrum assignable to the tBu groups of the ligand as well as four sets of aromatic resonances that each integrate to three protons, indicating 3-fold symmetry of the tripodal ligand when bound to the group 13 metal ions. In all cases, the protons of the bridgehead CH2 groups in 1–3 are diastereotopic and are assignable as two doublets (J = 11–12 Hz) in the 1H NMR spectrum, each integrating to three protons. The 13C{1H} NMR spectra for the complexes each have six unique aromatic resonances along with signatures for both the tBu substituents and methylene bridgehead carbons of the TriNOx3– ligand.
Single crystals of the (TriNOx3–)Ga complex were grown from a concentrated THF solution layered with hexanes at −25 °C, allowing for the characterization of 2 by X-ray crystallography. The molecule lies on a 3̅ rotary inversion axis that passes through the gallium ion and basal nitrogen atom (N(2)), and there are multiple types of disorder resulting in a total of four superimposed molecules in the asymmetric unit. A representation of one of these molecules is shown in Scheme 2. Full details of the various disorders and their modeling are available in the Supporting Information. The gallium ion in 2 sits within the ligand core and is coordinated by all three oxygen atoms of the nitroxide groups and the bridgehead nitrogen in a tetrahedral geometry (τ4 = 0.94). The Ga–O (1.854(9) Å) distances in 2 are comparable to the Ga–O distances in the others structurally characterized, 4-coordinate gallium ions supporting a NOOO primary coordination sphere,18 although our Ga–N (2.22(2) Å) is somewhat longer than the Ga–N distances for the same comparison group.
We were surprised by the absence of coordinated Lewis base at the gallium ion in the solid-state structure of 2 given that 1 crystallizes as its base adduct with pyridine.15 With this in mind, we used the Gutmann–Beckett method19,20 to evaluate the Lewis acidity of 1 and 2 in solution. Attempts to collect similar data for complex 3 were inhibited by lack of solubility of 3 in either CDCl3 or CD2Cl2. The difference in 31P chemical shift (Δδ) of Et3PO·(TriNOx)Al and Et3PO measured in C6D6 is 19.7 ppm. In contrast, we do not observe a Δδ between free Et3PO and the 2/Et3PO mixture, suggesting that the gallium ion does not coordinate the Lewis base in solution. We carried out the analogous experiments using Et3PS in place of Et3PO. In this case, we do not observe a Δδ between the Et3PS and its mixture with either 1 or 2, suggesting that the lack of Lewis acidity of 2 is not solely due to a mismatch in the hardness of Et3PO. At this point, we are unsure where the difference in Lewis acidity originates, but we do suggest that this difference plays a fundamental role in the reactivity of 1 and 2 with alcohols, concepts which we explore in more detail throughout the remainder of this paper.
Reactivity of the (TriNOx3–)M Complexes with ROH
We investigated the reactivity of the (TriNOx3–)M complexes 1 and 2 with various alcohols (Scheme 3). (TriNOx3–)Al was reacted with tert-butanol (t-BuOH) in toluene at room temperature to give the Zwitterion (HTriNOx2–)Al–OtBu (4) in which the O–H bond of the alcohol has reacted to install a tert-butoxy ligand at the metal ion and protonate the bridgehead nitrogen of the TriNOx3– ligand. Using one equivalent of t-BuOH gives 4 in 86% conversion after 24 h,21 but the reaction can be pushed to completion by increasing the amount of alcohol to three equivalents. Using these conditions, 4 was isolated in 80% yield after 12 h following removal of volatiles. In contrast, the reaction between (TriNOx3–)Ga (2) and t-BuOH is much less successful. Reaction of 2 with three equivalents of t-BuOH in toluene gives (HTriNOx2–)Ga–OtBu (5) in only 21% conversion after 24 h. The conversion improves only slightly with increasing the equivalents of t-BuOH (25% conversion with 6 equiv) and stirring 2 in neat t-BuOH results in decomposition of the complex into free TriNOxH3 ligand precursor as evidenced by 1H NMR spectrocopy.22
Scheme 3. Reactivity of the (TriNOx3–)M Complexes 1 (M = Al) and 2 (M = Ga) with Alcohols to Give the Complexes (HTriNOx2–)M–OR 4–9.

We next carried out the reaction of the (TriNOx3–)M complexes with phenol (PhOH). The 1:1 reaction between 1 or 2 and phenol in toluene at room temperature results in the formation of the alcohol adduct complexes (HTriNOx2–)Al–OPh (8) and (HTriNOx2–)Ga–OPh (9), respectively. These reactions occur much faster than the reactions between 1 and 2 with t-BuOH, as evidenced by the generation of opaque reaction solitons within minutes in the former. The products 8 and 9 were isolated in 82% and 75% yield, respectively, after the removal of volatiles from the crude reactions after 12 h.
Finally, we investigated the reactivity of 1 and 2 with benzyl alcohol (BnOH). The reaction between 1 with a stoichiometric amount of BnOH in toluene gives (HTriNOx2–)Al–OBn (6) in 100% conversion after 12 h at room temperature. Following workup, 6 was isolated in 82% yield. Conversely, the reaction of 2 with BnOH gives (HTriNOx2–)Ga–OBn (7) in only 56% conversion under identical conditions, which is increased only slightly (to 61%) when three equivalents of benzyl alcohol are used. Increasing the amount of alcohol further results in the consumption of the starting material, but free TriNOxH3 is also produced along with 7. Collectively, these results suggest that the identity of the metal ion and the specific alcohol both influence the thermodynamic and kinetic parameters of the reaction between (TriNOx3–)M and ROH. In particular, reactions with 1 proceed both faster and in higher conversion relative to the reaction of 2 with the same alcohol. Additionally, for a given metal, as the alcohol becomes more acidic the reaction proceeds both faster and in higher conversion. We more fully examine these dependencies below.
Complexes 4, 6, 8, and 9 are stable for the period of weeks if stored in the solid state under a nitrogen atmosphere at −25 °C, but are not stable in the long term under these conditions. All of the complexes are partially soluble in toluene and soluble in both chloroform and methylene chloride. Complexes 4 and 6 are both also soluble in benzene. Neither 8 or 9 are readily soluble in THF and require several hours of stirring to achieve a homogeneous solution. However, the complexes are not stable in THF and solubilization is always accompanied by partial decomposition of the complexes as judged by the presence of free TriNOxH3 in the 1H NMR spectra of their solutions. We suspect that none of the complexes 4, 6, 8, and 9 have long-term stability in THF solution, although we have not rigorously tested this hypothesis. The complexes are not stable pyridine, resulting in partial decomposition of the complexes into mixtures of (TriNOx3–)M and what we suspect is [(TriNOx3–)M–OR][py–H].
Complexes 4, 6, 8, and 9 were characterized by 1H and 13C NMR spectroscopies. In all cases, the 1H NMR spectra of the complexes exhibit a single resonance assignable to the protons of the tBu groups of the tripodal ligand. These signals come at chemical shifts ∼0.5 ppm upfield relative to the resonance for the ligand tBu groups in the 1H NMR spectra of 1 and 2 and suggest pseudo-C3 symmetry of the (HTriNOx2–)M–OR complexes in solution. The spectra also all display a broad singlet at ∼11 ppm that can be assigned to the N–H proton of the complexes. The diastereotopic protons of the CH2 groups in the (HTriNOx2–)M–OR complexes appear as a doublet (J = 12 Hz) and a doublet-of-doublets (J ∼ 12 Hz; 8–12 Hz), the latter splitting pattern of which arises from coupling between the methylene proton with its diastereotopic partner, and the newly formed N–H proton on the bridgehead nitrogen atom. The 13C NMR spectra for the complexes each have six unique aromatic resonances assignable to the HTriNOx2– ligand along with signatures for both the ligand tBu substituents and methylene carbons. The NMR signatures of the various apical groups of the (HTriNOx2–)M–OR complexes are also observed in the 1H and 13C NMR spectra complexes 4, 6, 8, and 9. For example, the 1H NMR spectra of 4 has a signal at δ 1.81 ppm assignable to the tBu group the tert-butoxy ligand, and the CH2 of the benzyloxy ligand appears as a set of diastereotopic protons in the δ 5.5–5.6 ppm range of in the 1H NMR spectrum of 6.
Solid-state structures of complexes 4, 6, 8, and 9 were obtained by single-crystal X-ray diffraction. Representations of the molecules are shown in Figure 1 and details regarding the collection and refinement of the data sets can be found in the Supporting Information. The complexes are all similar in structure, with the metal ion coordinated by four alkoxide ligands in a tetrahedral geometry (τ4 = 0.93–0.95). The average Al–O distance between the aluminum ion and the oxygen atoms of the TriNOx ligand in 4, 6, and 8 is 1.777(3) Å. The similar set of Ga–O distances in 9 are longer (1.855(6) Å), as is expected with the increased size of the metal ion. In all the complexes, the M(OR)4 fragment sits at the top of the bonding pocket of the HTriNOx2– ligand, with the protonated bridgehead N–H sitting at the bottom and pointing into the ligand. The N–H participates in hydrogen bonding with one of the oxygen atoms of a nitroxide group (O---Have = 1.862 Å for 4, 6, and 8; O---H = 1.902 Å for 9). The average N–O distance across all four complexes is 1.45 Å, which is in the range of analogous metrics observed in other metal complexes of the TriNOx ligand.23−29 The Al–O bond distances for the Al–OtBu (1.7240(10) Å for 4), Al–OBn (1.7446(8) Å for 6), and Al–OPh (1.7453(12) Å for 8) interactions are all in the range of the other structurally characterized terminal Al–OR bonds of their respective types.30−32 Similarly, at 1.852(4) Å the Ga–O bond distance for the Ga–OPh interaction in 9 is in the range of other structurally characterized terminal Ga–OPh bonds.33
Figure 1.

Solid state structures of the (HTriNOx2–)M–OR compounds 4, 6, 8, and 9. Ellipsoids are projected at 30% probability and the tert-butyl groups of the TriNOx ligand are shown in wireframe for clarity. With the exception of the N–H, hydrogen atoms have been omitted for clarity. (i) (HTriNOx2–)Al–OtBu (4), R1 = 0.0427; τ4[Al(1)] = 0.95; O(1)---H(4), 1.842 Å. (ii) (HTriNOx2–)Al–OBn (6), R1 = 0.0371; τ4[Al(1)] = 0.91; O(1)---H(4), 1.853 Å. (iii) (HTriNOx2–)Al–OPh (8), R1 = 0.0612; τ4[Al(1)] = 0.94; O(1)---H(4), 1.891 Å. (iv) (HTriNOx2–)Ga–OPh (9), R1 = 0.0694; τ4[Ga(1)] = 0.93, O(1)---H(4), 1.902 Å.
The VT-NMR spectra of 4 were collected over the 293–353 K temperature range and demonstrate the reversibility of the alcohol addition reaction (Figure 2i). As the temperature of a sample of 4 is increased from 293 K, 1 and t-BuOH are formed at the expense of 4. By 323 K, the 1:4 ratio is 0.09:0.91, determined via a comparison of the integrations of the bridgehead protons of the two metal complexes. Increasing the temperature to 353 K further increases the 1:4 ratio to 0.46:0.54. When returned to room temperature, the liberated t-BuOH adds to the (TriNOx3–)Al complex to reform the alcohol adduct 4, giving a final 1:4 ratio of 0.03:0.97. The reaction is clean with 4, 1, and t-BuOH being the only species observed in the spectra over the temperature range investigated and with no noticeable generation of free TriNOxH3.
Figure 2.

(i) Diastereotopic proton region of the 1H NMR spectra of the 4 complex over the temperature range 293–353 K; (ii) dependence of the ln(Keq) on temperature for the reaction 1 + t-BuOH ⇄ 4.
The enthalpy and entropy of the forward 1 + t-BuOH ⇄ 4 reaction were determined from the temperature dependence of the equilibrium constant for the Keq for the reaction at various temperatures (Figure 2ii). The reaction is exothermic with a ΔH of −225 kJ/mol, which agrees with the VT-NMR experiment of 4, which showed increasing amounts of 1 as T increases. The ΔS is large (−181 J/mol K), which we attribute to the forward reaction generating the more ordered (HTriNOx2–)Al–OtBu adduct. Additionally, the creation of charge separation in the (HTriNOx2–)Al–OtBu complex would also be expected to result in more organization of the solvent and hence a large negative entropy.
The VT-NMR spectra of both 6 and 8 were also collected (see the Supporting Information). Complex 6 behaves similarly to 4, although the 1:6 ratio is smaller relative to the 1:4 ratio across every temperature examined with a final 1:6 ratio of 0.11:0.89 at 353 K. On return to room temperature, 6 is fully reformed with no trace of 1. The phenoxide complex 8 is stable in solution, with no appearance of 1 across the 293–353 K temperature range. The expected Al–O bond strengths in the three complexes 4, 6, and 8 should be Al–OPh < Al–OBn < Al–OtBu, as supported by the Al–O bond lengths observed in the solid-state structures. The observed thermal stabilities of the complexes as gauged by the VT-NMR experiments thus suggest that it is a competition between the Al–OR and H–OR bond strengths that determines the equilibrium.
We further explored these concepts by carrying out the reaction of 1 and 2 with a broader range of alcohols. With the exception of t-BuOH, 1 reacts to completion to give the (HTriNOx2–)Al–OR products with the majority of the alcohols we studied, and we highlight the reaction with both i-PrOH (Figure S19) and 9-fluorenemethanol (Figure S20) as specific examples. The position of the 1 + t-BuOH ⇄ 4 is solvent-dependent, with equilibrium lying further toward 4 in C6D6 relative to CDCl3 (see Figures S17 and S18). Interestingly, the 2 + t-BuOH ⇄ 5 equilibrium seems less solvent-dependent, with a conversion of ∼20% in either C6D6 or CDCl3.
The reactions with 1 give higher conversions in comparison to the reaction of the same alcohol with 2 across the range of alcohols studied. We attribute this difference to the bonding preferences for aluminum versus gallium. The RO– ligands in the (HTriNOx2–)M–OR complexes are classified as hard according to Pearson’s theory of hard/soft acids and bases;34,35 as such, a given alkoxide should form a stronger bond with the harder aluminum ion relative to with the softer gallium ion, resulting in the (HTriNOx2–)Al–OR products being favored over their (HTriNOx2–)Ga–OR counterparts. To support this reasoning, we investigated the 1:1 reaction of 1 and 2 with t-butylmercaptan (t-BuSH, pKa = 17.9), which would incorporate the softer tBuS– anion in the presumptive (HTriNOx2–)M–StBu products (Figure S30). In the case of 1, no adduct product is observed in the 1H NMR spectra of its reaction with t-BuSH after 24 h, although the resonances for 1 are all broadened in the presence of the thiol.36 Conversely, the reaction between the t-BuSH and 2 gives (HTriNOx2–)Ga–StBu in 82% conversion under identical reaction conditions.
The full data for the gallium series shows a correlation between the pKa of the alcohol37 and the reaction Keq, such that more acidic alcohols favor the formation of the (HTriNOx2–)Ga–OR products (Table 1, Figure 3). We would expect to see a similar trend for the (HTriNOx2–)Al–OR complexes, but in these cases, the majority of the reactions go to completion. There is no correlation between the reaction Keq and size of the alcohol as judged by the A-value of the alkoxide ligand (see the Supporting Information). However, there is clearly an upper limit to the size of alkoxide that can be accommodated, since neither 1 or 2 react with 2,4,6-tri-tert-butylphenol even though the alcohol pKa (22.8) would suggest a quantitative reaction in both cases.
Table 1. Percent Conversion and Keq Values of Reactions of Various Alcohols with Complex 2.

| entry | R | pKaa | % conversionc | Keqd |
|---|---|---|---|---|
| 1 | tBu | 32.2 | 21 | 0.049e |
| 2 | iPr | 30.3 | 42 | 0.32 |
| 3 | 1-adamantyl | 29.9b | 36 | 0.20 |
| 4 | Bn | 28.3b | 38 | 0.16e |
| 5 | 9-MeFl | 28.0b | 84 | 5.0 |
| 6 | HCCCH2 | 26.3b | 93 | 64 |
| 7 | CF3CH2 | 23.5 | 91 | 150 |
| 8 | CCl3CH2 | 22.2b | 98 | 250 |
In DMSO as reported in Hans Reich’s Bordwell pKa table.38
Approximated using G4. See the Supporting Information for details.
Determined from the ratio of 2:(HTriNOx2–)Ga–OR in the 1H NMR spectra of a 1:1 mixture of 2 and ROH.
Calculated using concentrations of each species determined from integration of the 1H NMR spectra of a mixture of a 1:1 mixture of 2:ROH against internal standard.
Value determined in C6D6.
Figure 3.
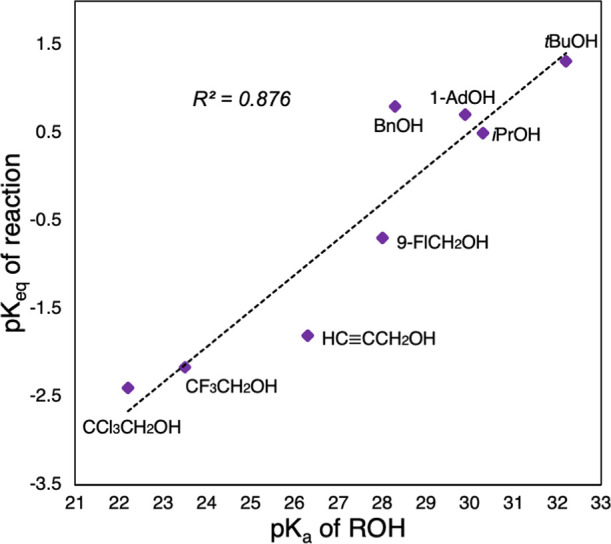
Plot of the reaction pKeq versus ROH pKa for the 1:1 reaction of (TriNOx3–)Ga (2) with various alcohols in CDCl3.
Kinetic Analysis and Mechanistic Considerations
Our initial observations on the reactivity of the (TriNOx3–)M complexes with alcohols suggested that the kinetic parameters of the reactions are also influenced by the identities of both the metal ion and the reacting alcohol. To more fully explore these observations, we monitored the conversion over time for the reaction of 1 and 2 with t-BuOH and i-PrOH to give the (HTriNOx2–)M–OR alcohol adducts. The reaction of t-BuOH proceeds with a rate constant an order of magnitude greater with 1 (k = 0.008 ± 0.002 min–1) relative to its reaction with 2 (0.0005 ± 0.0004 min–1) (Figure 4). A similar difference in rate was observed in the reactions of i-PrOH with 1 and 2 (see Supporting Information). In this case, i-PrOH reacts with 1 faster than our experimental capabilities to accurately determine a reaction rate, although our data suggests a rate constant of k ∼ 0.4 min–1.39 We were able to determine the rate constant for the reaction of 2 with i-PrOH (k = 0.009 ± 0.002 min–1), which when directly compared to the value for the reaction between 2 and t-BuOH suggests that more acidic alcohols result in faster reaction rates. The reactions between MeOH with 1 and 2 are both too fast to extract reliable rate constants, although the data clearly shows that the reaction with 1 is significantly faster than that with 2 and as predicted based on its lower pKa is the fastest reacting alcohol studied for either metal complex.
Figure 4.

Reaction of 1 and 2 with t-BuOH in C6D6 at 20 °C: (i) Concentration of products over time for the two reactions. (ii) Initial rate data for the reaction of 1 with t-BuOH. (iii) Initial rate data for the reaction of 2 with t-BuOH. Replicate trials are represented by blue, black, and red lines.
We make the following mechanistic proposals based on this kinetic investigation (Scheme 4). First, we think that it is unlikely that the reaction proceeds through direct interaction between the M–N bond of the (TriNOx3–)M complexes with the O–H bond of the alcohol given that the M–N bond sits within the ligand pocket. Instead, we propose that the reaction first involves the formation of a Lewis acid–base adduct between the (TriNOx3–)M complex and alcohol (Pathway I). This is expected to be less favorable for 2 relative to 1 given their relative Lewis acidities (vide supra) which results in faster reaction rates for 1 versus 2 for any given alcohol. Upon coordination with the metal, the alcohol proton becomes more acidic and transfers to an oxygen atom of one of the N–O arms of the TriNOx ligand, installing the alkoxo ligand and protonating the ligand backbone. We expect this step to be rate-limiting and dependent on the pKa of the reacting alcohol. A second proton transfer to the basal nitrogen commensurate with breaking the M–N bond generates the (HTriNOx2–)M–OR products.
Scheme 4. Mechanistic Proposals for the Reaction of 1 (M = Al) and 2 (M = Ga) with Alcohol to Give (HTriNOx2–)M–OR.

We have also considered a mechanism where the alcohol protonates the TriNOx ligand to generate the ion pair [(HTriNOx2–)M][−OR] as the initial step of the reaction (Pathway II). It is not clear which site on the TriNOx ligand would be protonated, although our previous study on the reactivity of 1-py with MeOTf suggests that the nitrogen atoms of the nitroxide arms are the most basic sites in the (TriNOx3–)Al complex.15 After this protonation step, coordination of the alkoxide anion to the metal ion installs the alkoxo ligand, which we expect to simultaneously weaken the M–N interaction. The final step involves proton transfer to the basal nitrogen commensurate with fully breaking the M–N bond to give the (HTriNOx2–)M–OR products. The rate dependence on the alcohol pKa is highlighted in this mechanistic pathway, although it is less obvious why 1 would react an order of magnitude faster than 2. However, Pathway II offers a reasonable route to explain how the (HTriNOx2–)Ga–OR complexes are formed without precoordination of the alcohol to the gallium ion in 2, which we have evaluated as lacking Lewis acidity. This is also in agreement with the chemistry reported by the Greb group, who showed enhanced ELC between their calix[4]pyrrolato gallate complex with i-PrOH with preprotonation of the ligand framework.13 At this point, it is unclear whether one or both pathways are operative, and the specific nature of the proton transfer steps involved in either pathway is not fully elucidated. With these qualifiers, it is tempting to hypothesize that the two complexes 1 and 2 may proceed through different pathways, especially given the difference in rate constants between the complexes. We are currently investigating the mechanistic details more fully.
Experimental Section
Physical Measurements
All NMR spectra were recorded using a Bruker 400 MHz spectrometer (399.78 MHz for 1H, 100.52 MHz for 13C) at ambient temperature unless otherwise specified. Chemical shifts were referenced to residual solvent. s = singlet, bs = broad singlet, d = doublet, t = triplet, dd = doublet of doublets, m = multiplet. CHN analyses were performed at the CENTC Elemental Analysis Facility at the University of Rochester (for 4, 6, 8, and 9) or at the Midwest Microlab (for 2 and 3).
Safety Statement
No uncommon hazards are noted.
Preparation of Compounds
All reactions and manipulations were performed under an inert atmosphere (N2) using standard Schlenk techniques or in a Vacuum Atmospheres, Inc. NextGen glovebox equipped with oxygen and moisture purifier systems. Glassware was dried overnight at 160 °C before use. C6D6, CDCl3, THF-d8, and pyridine-d5 were degassed and stored over 4 Å molecular sieves prior to use. Tetrahydrofuran, toluene, dichloromethane, hexane, and pentane were sparged for 20 min with dry argon and dried using a commercial two-column solvent purification system comprising of two columns packed with neutral alumina (for tetrahydrofuran and dichloromethane) or Q5 reactant then neutral alumina (for hexanes, toluene, and pentane). Anhydrous benzene and pyridine were further dried over 4 Å molecular sieves prior to use. The (TriNOx)H3 ligand precursor,23 [M(NMe2)3]2 (M = Al, Ga),40 and [In(NEt2)3]241 starting materials were prepared according to literature procedures. All other reagents were purchased from commercial sources and used as received.
General Protocol for the Synthesis of (TriNOx3–)M (1, M = Al; 2, M = Ga)
TriNOxH3 (0.50 g, 0.91 mmol) was loaded into a round-bottom Schlenk flask equipped with a stir bar and dissolved in toluene (30 mL). [M(NMe2)3]2 (0.41 mmol) was then added to the reaction, and the flask was sealed, removed from the glovebox, and heated at 50 °C. After 12 h, the reaction was removed from heat, cooled to room temperature, and brought back into the glovebox where volatiles were removed from the reaction mixture under reduced pressure. The resulting material was triturated with pentane (3 × 10 mL) to give 1 or 2 as off-white solids.
Characterization data for 1: Yield = 0.42 g, 0.73 mmol (80%).161H NMR (CDCl3): δ 7.57 (d, J = 8 Hz, 3H), 7.27 (m, 6H), 7.06 (t, J = 7 Hz, 3H), 4.47 (d, J = 12 Hz, 3H, NCH2), 3.05 (d, J = 12 Hz, 3H, NCH2), 1.27 (s, 27H, C(CH3)3). 13C{1H} NMR (CDCl3): 152.4, 133.0, 132.1, 129.1, 128.2, 124.1, 61.5, 58.2, 27.8. The elemental purity of 1 has previously been confirmed as the 1-py adduct.15
Characterization data for 2. Yield: 0.39 g, 0.64 mmol (70%).161H NMR (C6D6): δ 7.63 (d, J = 8 Hz, 3H), 7.04 (m, 6H), 6.90 (t, J = 7 Hz, 3H), 4.87 (d, J = 12 Hz, 3H, NCH2), 2.84 (d, J = 12 Hz, 3H, NCH2), 1.40 (s, 27H, C(CH3)3). 13C{1H} NMR (C6D6): δ 152.6, 133.1, 132.2, 129.4, 124.9, 124.4, 62.2, 58.2, 27.7. Anal. Calcd for C33H45GaN4O3: C, 64.40; H, 7.37; N, 9.10. Found: C, 63.77; H, 7.46; N, 9.01. Crystals suitable for X-ray diffraction were obtained from a saturated THF solution layered with hexane at −25 °C.
Synthesis of (TriNOx3–)In (3)
[In(NEt2)3]2 (0.21 g, 0.32 mmol) was added to a round-bottom Schlenk flask equipped with a stir bar and dissolved in toluene (∼25 mL). TriNOxH3 (0.35 g, 0.64 mmol) was separately dissolved in toluene (∼25 mL) and transferred to the Schlenk flask. The reaction was allowed to stir at room temperature for 12 h after which volatiles were removed from the heterogeneous reaction mixture under reduced pressure. The crude reaction mixture was dissolved in boiling pyridine (∼20 mL), and the resulting solution was allowed to slowly cool to −25 °C. After 24 h, the resultant white powder was collected over a medium-porosity frit, washed with cold pyridine followed by hexane and then dried under vacuum to give 3 as a white powder. Yield: 0.12 g, 0.18 mmol (56%). 1H NMR (py-d5): δ 7.84 (d, J = 8 Hz, 3H), 7.41 (m, 6H), 7.24 (t, J = 7 Hz, 3H), 5.14 (d, J = 11 Hz, 3H, NCH2), 2.58 (d, J = 11 Hz, 3H, NCH2), 0.99 (s, 27H, C(CH3)3); 13C{1H} NMR (py-d5): δ 153.0, 134.2, 132.8, 129.0, 127.9, 124.9, 60.3, 59.7, 26.3. Anal. Calcd for C33H45InN4O3: C, 60.00; H, 6.87; N, 8.48. Found: C, 59.67; H, 6.70; N, 8.27.
Synthesis of (HTriNOx2–)Al–OtBu (4)
tert-Butanol (38.8 mg, 0.52 mmol) was dissolved in toluene (∼1 mL) and added to a stirring toluene (10 mL) solution of 1 (100 mg, 0.18 mmol) in a vial. The homogeneous mixture was allowed to stir at room temperature for 12 h, after which volatiles were removed from the reaction mixture via vacuum evaporation to give 3 as a white solid. Yield: 89 mg, 0.14 mmol (81% yield). 1H NMR (C6D6): δ 10.94 (bs, 1H), 7.82 (d, J = 8.0 Hz, 3H), 6.89 (t, J = 7.4, 3H), 6.70 (d, J = 6.4 Hz, 3H), 4.70 (d, J = 12.0 Hz, 3H), 2.24 (dd, J1 = 12.0 Hz, J2 = 10.0 Hz, 3H), 1.81 (s, 9H, OC(CH3)3), 1.00 (s, 27H, C(CH3)3).4213C{1H} NMR (C6D6): δ 154.3, 132.1, 131.7, 129.5, 126.1, 124.3, 67.8, 61.5, 57.1, 34.6, 26.7. Anal. Calcd for C37H55AlN4O4·(CH2Cl2)1.5: C, 59.80; H, 7.43; N, 7.25. Found: C, 59.90; H, 7.19; N, 7.42. Crystals suitable for X-ray diffraction were obtained from a saturated THF solution layered with hexane at −25 °C.
Synthesis of (HTriNOx2–)Al–OBn (6)
Benzyl alcohol (19.0 mg, 0.176 mmol) was added as a solution in toluene (1 mL) to a stirring toluene (10 mL) solution of 1 (100 mg, 0.18 mmol) in a vial. The reaction was allowed to stir at room temperature for ∼12 h. Volatiles were subsequently removed from the reaction via vacuum evaporation to give 6 as a white powder. Yield: 100 mg, 0.15 mmol (82% yield). 1H NMR (C6D6): δ 11.03 (bs, 1H), 7.93 (d, J = 8 Hz, 2H), 7.85 (d, J = 8 Hz, 3H), 7.67 (m, 1H), 7.38 (t, J = 8 Hz, 3H), 6.90 (m, 5H), 6.71 (d, J = 8 Hz, 3H), 5.59 (d, J = 12 Hz, 1H), 5.51 (d, J = 12 Hz, 1H), 4.71 (d, J = 12 Hz, 3H), 2.25 (dd, J1 = 12 Hz, J2 = 8 Hz, 3H), 0.94 (s, 27H, C(CH3)3). 13C{1H} NMR (CDCl3): δ 153.3, 132.1, 131.1, 129.3, 129.0, 127.2, 126.4, 125.7, 125.0, 124.7, 65.2, 61.4, 57.5, 26.3. Anal. Calcd for C40H53AlN4O4·(CH2Cl2): C, 64.39; H, 7.11; N, 7.33. Found: C, 64.59; H, 7.10; N, 6.77. Crystals suitable for X-ray diffraction were obtained from a saturated THF solution layered with hexane at −25 °C.
Synthesis of (HTriNOx2–)Al–OPh (8)
Phenol (16.5 mg, 0.175 mmol) was added as a solid to a toluene (10 mL) solution of 1 (100 mg, 0.175 mmol) stirring in a vial. The reaction was allowed to stir at room temperature for ∼12 h after which volatiles were subsequently removed from the reaction under reduced pressure to give 8 as a white powder. Yield: 100 mg, 0.15 mmol (82%). 1H NMR (CDCl3):43 δ 10.94 (bs, 1H), 7.66 (d, J = 8 Hz, 3H), 7.30 (t, J = 8 Hz, 3H), 7.05 (m, 7H), 6.96 (d, J = 8 Hz, 2H), 6.59 (t, J = 8 Hz, 2H), 4.95 (d, J = 12 Hz, 3H), 3.10 (dd, J1 = 12 Hz, J2 = 10 Hz, 3H), 0.64 (s, 27H, C(CH3)3). 13C{1H} NMR (CDCl3): δ 153.3, 132.2, 131.1, 129.4, 128.3, 125.6, 124.8, 121.7, 120.6, 116.3, 61.6, 57.5, 26.1. Anal. Calcd for C39H51AlN4O4·(CH2Cl2)0.5: C, 66.98; H, 7.26; N, 7.91. Found: C, 66.10; H, 7.28; N, 7.46. Crystals suitable for X-ray diffraction were obtained from a saturated THF solution layered with hexane at −25 °C.
Synthesis of (HTriNOx2–)Ga–OPh (9)
Phenol (15.4 mg, 0.164 mmol) was added as a solid to a stirring toluene (∼10 mL) solution of 2 (100 mg, 0.164 mmol) in a vial. The reaction was allowed to stir at room temperature for ∼12 h after which volatiles were removed from the reaction under vacuum to give 9 as a white powder. Yield: 85 mg, 0.12 mmol (75%). 1H NMR (CDCl3):43 δ 10.91(bs, 1H, NH), 7.67 (d, J = 8 Hz, 3H), 7.30 (t, J = 8 Hz, 3H), 7.04 (m, 9H), 6.59 (t, J = 8 Hz, 2H), 4.95 (d, J = 12 Hz, 3H), 3.09 (dd, J1 = 12 Hz, J2 = 12 Hz, 3H), 0.66 (s, 27H, C(CH3)3). 13C{1H} NMR (CDCl3): δ 162.8, 153.1, 132.2, 130.9, 129.4, 128.4, 125.4, 124.8, 120.2, 116.3, 61.8, 57.5, 26.2. Anal. Calcd for C39H51GaN4O4·(CH2Cl2)0.75: C, 61.82; H, 6.72; N, 7.25. Found: C, 61.87; H, 6.66; N, 7.05. Crystals suitable for X-ray diffraction were obtained from a saturated THF solution layered with hexane at −25 °C.
Conclusions
In summary, we have shown that the O–H bond of alcohols can be cleaved via an ELC pathway by the tripodal complexes (TriNOx3–)Al and (TriNOx3–)Ga. The thermodynamic and kinetic aspects of the reactions are both influenced by the identity of the metal, with 1 having higher reaction equilibrium constants and proceeding at a faster rate relative to 2 for any given alcohol. These parameters are also influenced by the pKa of the alcohol, with more acidic alcohols reacting both to more completion and faster than their less acidic counterparts. We expect this knowledge to lay the groundwork for the ELC of other polar H–X bonds, an area we are actively exploring. Additionally, we are currently trying to better understand the differences in Lewis acidity between 1 and 2, especially in how these differences result in divergent reactivity.
Acknowledgments
This work was funded by the National Science Foundation (CHE-1664902) and the Dreyfus Foundation (TH-19-021). J.S.S. was supported by both a Eugene M. Lang and Robert. F. Pasternack Summer Research Fellowship. M.L.M. was supported through a Frances Velay Women’s Science Research Fellowship. V.W.G. was supported through a Robert F. Pasternack Research Fellowship. A.R.C. was supported through a James H. Scheuer Summer Internship in Environmental Studies.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.inorgchem.3c02902.
1H and 13C{1H} NMR spectra for all compounds, experimental protocols for the Guttmann, VT-NMR, van’t Hoff, and Keq experiments, 1H NMR spectra used to determine the Keq values, details regarding collection of the kinetic data, and details regarding the calculations of alcohol pKa and R-group A-factors (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Khusnutdinova J. R.; Milstein D. Metal–Ligand Cooperation. Angew. Chem., Int. Ed. 2015, 54 (42), 12236–12273. 10.1002/anie.201503873. [DOI] [PubMed] [Google Scholar]
- Elsby M. R.; Baker R. T. Strategies and Mechanisms of Metal–Ligand Cooperativity in First-Row Transition Metal Complex Catalysts. Chem. Soc. Rev. 2020, 49 (24), 8933–8987. 10.1039/D0CS00509F. [DOI] [PubMed] [Google Scholar]
- Greb L.; Ebner F.; Ginzburg Y.; Sigmund L. M. Element-Ligand Cooperativity with p-Block Elements. Eur. J. Inorg. Chem. 2020, 2020 (32), 3030–3047. 10.1002/ejic.202000449. [DOI] [Google Scholar]
- Berben L. A. Catalysis by Aluminum(III) Complexes of Non-Innocent Ligands. Chem. – Eur. J. 2015, 21 (7), 2734–2742. 10.1002/chem.201405400. [DOI] [PubMed] [Google Scholar]
- Gahlaut P. S.; Yadav K.; Gautam D.; Jana B. Recent Developments in the Syntheses of Aluminum Complexes Based on Redox-Active Ligands. J. Organomet. Chem. 2022, 963, 122298 10.1016/j.jorganchem.2022.122298. [DOI] [Google Scholar]
- Myers T. W.; Berben L. A. Aluminum–Ligand Cooperative N–H Bond Activation and an Example of Dehydrogenative Coupling. J. Am. Chem. Soc. 2013, 135 (27), 9988–9990. 10.1021/ja4032874. [DOI] [PubMed] [Google Scholar]
- Sherbow T. J.; Carr C. R.; Saisu T.; Fettinger J. C.; Berben L. A. Insight into Varied Reaction Pathways for O–H and N–H Bond Activation by Bis(Imino)Pyridine Complexes of Al(III). Organometallics 2016, 35 (1), 9–14. 10.1021/acs.organomet.5b00743. [DOI] [Google Scholar]
- Myers T. W.; Berben L. A. Aluminium–Ligand Cooperation Promotes Selective Dehydrogenation of Formic Acid to H2 and CO2. Chem. Sci. 2014, 5 (7), 2771–2777. 10.1039/C4SC01035C. [DOI] [Google Scholar]
- Carr C. R.; Vesto J. I.; Xing X.; Fettinger J. C.; Berben L. A. Aluminum-Ligand Cooperative O–H Bond Activation Initiates Catalytic Transfer Hydrogenation. ChemCatChem. 2022, 14 (13), e202101869 10.1002/cctc.202101869. [DOI] [Google Scholar]
- Ebner F.; Wadepohl H.; Greb L. Calix[4]Pyrrole Aluminate: A Planar Tetracoordinate Aluminum(III) Anion and Its Unusual Lewis Acidity. J. Am. Chem. Soc. 2019, 141 (45), 18009–18012. 10.1021/jacs.9b10628. [DOI] [PubMed] [Google Scholar]
- Ebner F.; Sigmund L. M.; Greb L. Metal–Ligand Cooperativity of the Calix[4]Pyrrolato Aluminate: Triggerable C–C Bond Formation and Rate Control in Catalysis. Angew. Chem., Int. Ed. 2020, 59 (39), 17118–17124. 10.1002/anie.202007717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigmund L. M.; Greb L. Reversible OH-Bond Activation and Amphoterism by Metal–Ligand Cooperativity of Calix[4]Pyrrolato Aluminate. Chem. Sci. 2020, 11 (35), 9611–9616. 10.1039/D0SC03602A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigmund L. M.; Engels E.; Richert N.; Greb L. Calix[4]Pyrrolato Gallate: Square Planar-Coordinated Gallium(III) and Its Metal–Ligand Cooperative Reactivity with CO2 and Alcohols. Chem. Sci. 2022, 13 (37), 11215–11220. 10.1039/D2SC03054C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdalla J. A. B.; Riddlestone I. M.; Tirfoin R.; Aldridge S. Cooperative Bond Activation and Catalytic Reduction of Carbon Dioxide at a Group 13 Metal Center. Angew. Chem. Int. Ed 2015, 54 (17), 5098–5102. 10.1002/anie.201500570. [DOI] [PubMed] [Google Scholar]
- Woodside A. J.; Smith M. A.; Herb T. M.; Manor B. C.; Carroll P. J.; Rablen P. R.; Graves C. R. Synthesis and Characterization of a Tripodal tris(Nitroxide) Aluminum Complex and its Catalytic Activity toward Carbonyl Hydroboration. Organometallics 2019, 38 (5), 1017–1020. 10.1021/acs.organomet.8b00933. [DOI] [Google Scholar]
- These yields are calculated from the average of five (for 1) and three (for 2) reactions conducted over the course of Summer 2022.
- We have not rigorously identified the decomposition products, although it appears that 1 and 2 both react to give (HTriNOx2–)M-X (X = monoanionic ligand) materials similar in structure to the alcohol adduct products discussed in this manuscript.
- a Atwood J. L.; Lee F. C.; Raston C. L.; Robinson K. D. Bimetallic Aluminium and Gallium Derivatives of 1,1,1,5,5,5-Hexafluoropentane-2,4-Dione via Selective Metallation/Hydrometallation. J. Chem. Soc., Dalton Trans. 1994, 13, 2019–2021. Ga-N: 2.024 Å. Ga-O: 1.806, 1.798, 1.795 Å; Ga-N: 2.033 Å. Ga-O: 1.794, 1.805, 1.784 Å 10.1039/dt9940002019. [DOI] [Google Scholar]; b Valet M.; Hoffman D. M. Synthesis of Homoleptic Gallium Alkoxide Complexes and the Chemical Vapor Deposition of Gallium Oxide Films. Chem. Mater. 2001, 13 (6), 2135–2143. Ga-N: 2.022 Å. Ga-O: 1.822, 1.806, 1.799 Å 10.1021/cm0014177. [DOI] [Google Scholar]; c Mîinea L.; Suh S.; Bott S. G.; Liu J.-R.; Chu W.-K.; Hoffman D. M. Synthesis of Aluminium and Gallium Fluoroalkoxide Compounds and the Low Pressure Metal-Organic Chemical Vapor Deposition of Gallium Oxide Films. J. Mater. Chem. 1999, 9 (4), 929–935. 10.1039/a808460b. [DOI] [Google Scholar]; Ga-N: 1.924 Å. Ga-O: 1.803, 1.801, 1.811 Å.
- Mayer U.; Gutmann V.; Gerger W. The Acceptor Number — A Quantitative Empirical Parameter for the Electrophilic Properties of Solvents. Monatshefte Für Chem. Chem. Mon. 1975, 106 (6), 1235–1257. 10.1007/BF00913599. [DOI] [Google Scholar]
- Beckett M. A.; Brassington D. S.; Coles S. J.; Hursthouse M. B. Lewis Acidity of Tris(Pentafluorophenyl)Borane: Crystal and Molecular Structure of B(C6F5)3·OPEt3. Inorg. Chem. Commun. 2000, 3 (10), 530–533. 10.1016/S1387-7003(00)00129-5. [DOI] [Google Scholar]
- Please note, the equilibrium position for the 1 + t-BuOH ⇄ 4 reaction is dependent on solvent and the conversion values reported here are those in C6D6.
- We also attempted to prepare the (HTriNOx2–)Ga–OtBu complex via salt metathesis through the reaction of a stoichiometric amount of potassium tert-butoxide with (HTriNOx2-)GaCl. The reaction was unsuccessful, giving 2 as the major species after the reaction.
- Bogart J. A.; Lippincott C. A.; Carroll P. J.; Schelter E. J. An Operationally Simple Method for Separating the Rare-Earth Elements Neodymium and Dysprosium. Angew. Chem., Int. Ed. 2015, 54 (28), 8222–8225. 10.1002/anie.201501659. [DOI] [PubMed] [Google Scholar]
- Bogart J. A.; Cole B. E.; Boreen M. A.; Lippincott C. A.; Manor B. C.; Carroll P. J.; Schelter E. J. Accomplishing Simple, Solubility-Based Separations of Rare Earth Elements with Complexes Bearing Size-Sensitive Molecular Apertures. Proc. Natl. Acad. Sci. U. S. A. 2016, 113 (52), 14887–14892. 10.1073/pnas.1612628113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogart J. A.; Lippincott C. A.; Carroll P. J.; Booth C. H.; Schelter E. J. Controlled Redox Chemistry at Cerium within a Tripodal Nitroxide Ligand Framework. Chem. - Eur. J. 2015, 21 (49), 17850–17859. 10.1002/chem.201502952. [DOI] [PubMed] [Google Scholar]
- Wilson H. H.; Yu X.; Cheisson T.; Smith P. W.; Pandey P.; Carroll P. J.; Minasian S. G.; Autschbach J.; Schelter E. J. Synthesis and Characterization of a Bridging Cerium(IV) Nitride Complex. J. Am. Chem. Soc. 2023, 145 (2), 781–786. 10.1021/jacs.2c12145. [DOI] [PubMed] [Google Scholar]
- Lapsheva E. N.; Cheisson T.; Lamsfus C. Á.; Carroll P. J.; Gau M. R.; Maron L.; Schelter E. J. Reactivity of Ce(IV) Imido Compounds with Heteroallenes. Chem. Commun. 2020, 56 (35), 4781–4784. 10.1039/C9CC10052K. [DOI] [PubMed] [Google Scholar]
- Cheisson T.; Kersey K. D.; Mahieu N.; McSkimming A.; Gau M. R.; Carroll P. J.; Schelter E. J. Multiple Bonding in Lanthanides and Actinides: Direct Comparison of Covalency in Thorium(IV)- and Cerium(IV)-Imido Complexes. J. Am. Chem. Soc. 2019, 141 (23), 9185–9190. 10.1021/jacs.9b04061. [DOI] [PubMed] [Google Scholar]
- Boreen M. A.; Bogart J. A.; Carroll P. J.; Schelter E. J. Rearrangement in a Tripodal Nitroxide Ligand To Modulate the Reactivity of a Ti–F Bond. Inorg. Chem. 2015, 54 (19), 9588–9593. 10.1021/acs.inorgchem.5b01687. [DOI] [PubMed] [Google Scholar]
- For representative Al–O bond distances in complexes with a four-coordinate Al(III) ion and with a terminal Al–OBn group, see:; a Keyes L. K.; Todd A. D. K.; Giffin N. A.; Veinot A. J.; Hendsbee A. D.; Robertson K. N.; Geier S. J.; Masuda J. D. Reaction of Sterically Encumbered Phenols, TEMPO-H, and Organocarbonyl Insertion Reactions with L–AlH2 (L = HC(MeCNDipp)2, Dipp = 2,6-Diisopropylphenyl). RSC Adv. 2017, 7 (59), 37315–37323. 10.1039/C7RA06526D. [DOI] [Google Scholar]; b Peddarao T.; Sarkar N.; Nembenna S. Mono- and Bimetallic Aluminum Alkyl, Alkoxide, Halide and Hydride Complexes of a Bulky Conjugated bis-Guanidinate(CBG) Ligand and Aluminum Alkyls as Precatalysts for Carbonyl Hydroboration. Inorg. Chem. 2020, 59 (7), 4693–4702. 10.1021/acs.inorgchem.9b03778. [DOI] [PubMed] [Google Scholar]
- For representative Al–O bond distances in complexes with a four-coordinate Al(III) ion and with a terminal Al–OPh group, see:; a Brown R. K.; Hooper T. N.; Rekhroukh F.; White A. J. P.; Costa P. J.; Crimmin M. R. Alumination of Aryl Methyl Ethers: Switching between sp2 and sp3 C–O Bond Functionalisation with Pd-Catalysis. Chem. Commun. 2021, 57 (88), 11673–11676. 10.1039/D1CC05408B. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Zaworotko M. J.; Kerr C. R.; Atwood J. L. Reaction of the Phenoxide Ion with Trimethylaluminum. Isolation and Crystal Structure of [K.Cntdot.Dibenzo-18-Crown-6][Al2Me6OPh] and K[AlMe2(OPh)2]. Organometallics 1985, 4 (2), 238–241. 10.1021/om00121a005. [DOI] [Google Scholar]; c Cole M. L.; Hibbs D. E.; Jones C.; Junk P. C.; Smithies N. A. Imidazolium Formation from the Reaction of N-Heterocyclic Carbene Stabilised Group 13 Trihydride Complexes with Organic Acids. Inorg. Chim. Acta 2005, 358 (1), 102–108. 10.1016/j.ica.2004.07.036. [DOI] [Google Scholar]; d Ogrin D.; Poppel L. H. van; Bott S. G.; Barron A. R. Aluminium Alkyl and Aryloxide Complexes of Pyrazine and Bipyridines: Synthesis and Structure. Dalton Trans. 2004, 21, 3689–3694. 10.1039/B410662H. [DOI] [PubMed] [Google Scholar]; e Knabel K.; Krossing I.; Nöth H.; Schwenk-Kircher H.; Schmidt-Amelunxen M.; Seifert T. The Aluminum–Nitrogen Bond in Monomeric Bis(Amino)Alanes: A Systematic Experimental Study of bis(Tetramethylpiperidino)Alanes and Quantum Mechanical Calculations on the Model System (H2N)2AlY. Eur. J. Inorg. Chem. 1998, 1998 (8), 1095–1114. 10.1002/(SICI)1099-0682(199808)1998:8<1095::AID-EJIC1095>3.0.CO;2-D. [DOI] [Google Scholar]; f Dagorne S.; Le Bideau F.; Welter R.; Bellemin-Laponnaz S.; Maisse-François A. Well-Defined Cationic Alkyl– and Alkoxide–Aluminum Complexes and Their Reactivity with ε-Caprolactone and Lactides. Chem. – Eur. J. 2007, 13 (11), 3202–3217. 10.1002/Chem.200601112. [DOI] [PubMed] [Google Scholar]
- For representative Al–O bond distances in complexes with a four-coordinate Al(III) ion and with a terminal Al–OtBu group, see:; a Gianopoulos C. G.; Kumar N.; Zhao Y.; Jia L.; Kirschbaum K.; Mason M. R. Aluminum Alkoxide, Amide and Halide Complexes Supported by a Bulky Dipyrromethene Ligand: Synthesis, Characterization, and Preliminary ε-Caprolactone Polymerization Activity. Dalton Trans. 2016, 45 (35), 13787–13797. 10.1039/C6DT02449A. [DOI] [PubMed] [Google Scholar]; b Liu L. L.; Zhou J.; Cao L. L.; Stephan D. W. Phosphaaluminirenes: Synthons for Main Group Heterocycles. J. Am. Chem. Soc. 2019, 141 (42), 16971–16982. 10.1021/jacs.9b09330. [DOI] [PubMed] [Google Scholar]; c Veith M.; Frères J.; König P.; Schütt O.; Huch V.; Blin J. Nanoscaled Sn and Pb Particles Aligned in Al2O3 Tubes Obtained from Molecular Precursors. Eur. J. Inorg. Chem. 2005, 2005 (18), 3699–3710. 10.1002/ejic.200500368. [DOI] [Google Scholar]; d Kiriratnikom J.; Chotchatchawankul S.; Haesuwannakij S.; Kiatisevi S.; Phomphrai K. Synthesis and Characterization of Neutral and Cationic Aluminum Complexes Supported by a Furfuryl-Containing Aminophenolate Ligand for Ring-Opening Polymerization of ε-Caprolactone. New J. Chem. 2018, 42 (11), 8374–8383. 10.1039/C8NJ00937F. [DOI] [Google Scholar]; e Chisholm M. H.; DiStasi V. F.; Streib W. E. Kinetic Control in the Alcoholysis Reaction Involving Hexakisdimethylamidodialuminium and tert-Butanol. Preparation, Crystal and Molecular Structures of Al(OBut)3(HNMe2) and Al2(NMe2)(OBut)5. Polyhedron 1990, 9 (2), 253–255. 10.1016/S0277-5387(00)80577-X. [DOI] [Google Scholar]; f Asada T.; Hoshimoto Y.; Ogoshi S. Rotation-Triggered Transmetalation on a Heterobimetallic Cu/Al N-Phosphine-Oxide-Substituted Imidazolylidene Complex. J. Am. Chem. Soc. 2020, 142 (21), 9772–9784. 10.1021/jacs.0c03252. [DOI] [PubMed] [Google Scholar]; g Lin C.-Y.; Lee H. M.; Huang J.-H. Synthesis of Aluminum Alkoxide and Bis-Alkoxide Compounds Containing Bidentate Pyrrolyl Ligands. J. Organomet. Chem. 2007, 692 (17), 3718–3722. 10.1016/j.jorganchem.2007.05.013. [DOI] [Google Scholar]; h Uhl W.; Jana B. Reactions of β-Diketiminatoaluminum Hydrides with tert-Butyl Hydrogenperoxide – Facile Formation of Dialuminoxanes Containing Al–O–Al Groups. J. Organomet. Chem. 2009, 694 (7), 1101–1106. 10.1016/j.jorganchem.2008.09.036. [DOI] [Google Scholar]
- a van Poppel L. H.; Bott S. G.; Barron A. R. Molecular Structures of Ga(tBu)2(OPh)(Pyz)·PhOH and [(tBu)2Ga(H2O)(μ-OH)Ga(tBu)2]2(μ-OC6H4O)·4(2-Mepy): Intra- and Inter-Molecular Hydrogen Bonding to Gallium Aryloxides. Polyhedron 2002, 21 (19), 1877–1882. 10.1016/S0277-5387(02)01066-5. [DOI] [Google Scholar]; b van Poppel L. H.; Bott S. G.; Barron A. R. 1,4-Dioxobenzene Compounds of Gallium: Reversible Binding of Pyridines to [{(tBu)2Ga}2(μ-OC6H4O)]n in the Solid State. J. Am. Chem. Soc. 2003, 125 (36), 11006–11017. 10.1021/ja0208714. [DOI] [PubMed] [Google Scholar]; c Linti G.; Frey R.; Köstler W.; Urban H. On the Chemistry of Gallium, 8. Synthesis and Structure of [tris(trimethylsilyl)Silyl]-Substituted Gallanes and Gallates. Chem. Ber. 1996, 129 (5), 561–569. 10.1002/cber.19961290514. [DOI] [Google Scholar]; d Uzelac M.; Kennedy A. R.; Hevia E. Trans-Metal-Trapping Meets Frustrated-Lewis-Pair Chemistry: Ga(CH2SiMe3)3-Induced C–H Functionalizations. Inorg. Chem. 2017, 56 (15), 8615–8626. 10.1021/acs.inorgchem.7b00549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson R. G. Hard and Soft Acids and Bases. J. Am. Chem. Soc. 1963, 85 (22), 3533–3539. 10.1021/ja00905a001. [DOI] [Google Scholar]
- Pearson R. G. Absolute Electronegativity and Hardness: Application to Inorganic Chemistry. Inorg. Chem. 1988, 27 (4), 734–740. 10.1021/ic00277a030. [DOI] [Google Scholar]
- The Δν1/2 for the C(CH3)3 resonance of 1 is ∼3 Hz while the Δν1/2 for the C(CH3)3 resonance of 1 in the presence of t-BuSH is ∼21 Hz. We hypothesize that that this broadening comes from the 1 + t-BuSH ⇄ [1-H]+ + t-BuS– equilibrium, where the TriNOx ligand is protonated without coordination of the thiolate anion to the complex.
- Where available, experimentally determined pKa values (in DMSO) are used. For those alcohols without experimentally determined DMSO pKa values, we developed a correlation curve that provided an approximate linear relationship between the experimentally determined DMSO pKa values of alcohols from the literature and calculated pKa values of the same alcohols predicted by DFT. The resulting linear fit was then used to predict a DMSO pKa value for alcohols not listed in the Bordwell pKa tables. See the Supporting Information for additional details.
- Reich H.Bordwell pKa Table. https://organicchemistrydata.org/hansreich/resources/pka/.
- This approximation is obtained from the linear regression between the first data point at t = 2 min and the origin.
- Waggoner K. M.; Olmstead M. M.; Power P. P. Structural and Spectroscopic Characterization of the Compounds [Al(NMe2)3]2, [Ga(NMe2)3]2, [(Me2N)2Al{μ-N(H)1-Ad}]2 (1-Ad = 1-Adamantanyl) and [{Me(μ-NPh2)Al}2NPh(μ-C6H4)]. Polyhedron 1990, 9 (2), 257–263. 10.1016/S0277-5387(00)80578-1. [DOI] [Google Scholar]
- Rossetto G.; Brianese N.; Camporese A.; Porchia M.; Zanella P.; Bertoncello R. Synthesis and Characterization of Hexakis(Diethylamido)Diindium(III) and Bis-Cyclopentadienyl(Diethylamido)Indium(III). Main Group Met. Chem. 1991, 14, 113–122. [Google Scholar]
- One of the aryl signals is obscured by the signal for the residual protons of the C6D6 solvent. Taking the 1H NMR spectra in THF-d8 shows the four expected aryl signals. See Figure S7.
- Complexes 8 and 9 are not readily soluble in C6D6 and require prolonged stirring to go into THF-d8, resulting in partial decomposition of the complexes as judged by free TriNOxH3 in the resultant NMR spectra of the solutions. The complexes are solubilized in CDCl3 with gentle heating.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.