

Abstract
RNA editing in Trypanosoma brucei mitochondria produces mature mRNAs by a series of enzyme-catalyzed reactions that specifically insert or delete uridylates in association with a macromolecular complex. Using a mitochondrial fraction enriched for in vitro RNA editing activity, we produced several monoclonal antibodies that are specific for a 21-kDa guide RNA (gRNA) binding protein initially identified by UV cross-linking. Immunofluorescence studies localize the protein to the mitochondrion, with a preference for the kinetoplast. The antibodies cause a supershift of previously identified gRNA-specific ribonucleoprotein complexes and immunoprecipitate in vitro RNA editing activities that insert and delete uridylates. The immunoprecipitated material also contains gRNA-specific endoribonuclease, terminal uridylyltransferase, and RNA ligase activities as well as gRNA and both edited and unedited mRNA. The immunoprecipitate contains numerous proteins, of which the 21-kDa protein, a 90-kDa protein, and novel 55- and 16-kDa proteins can be UV cross-linked to gRNA. These studies indicate that the 21-kDa protein associates with the ribonucleoprotein complex (or complexes) that catalyze RNA editing.
RNA editing produces mature mRNAs in the mitochondria of trypanosomatids by guide RNA (gRNA)-directed posttranscriptional insertion and deletion of uridylates (U’s) (2). This process can be so extensive that most of the coding sequence, as well as the initiation and termination codons, results from RNA editing (1, 11, 27, 28, 30). Stage-specific RNA editing appears to regulate mitochondrial respiration in the different life stages of African trypanosomes (9, 29). The mRNAs for components of respiratory complex I are preferentially edited in the mammalian stage of the life cycle, where the trypanosomes lack cytochromes, rely on glycolysis for energy production, and utilize complex I and alternate oxidase for terminal respiration. In contrast, the invertebrate stage predominantly utilizes cytochrome-mediated oxidative phosphorylation for energy generation while editing cytochrome mRNAs only in this stage.
The edited mRNA sequence is specified by trans-acting small RNA molecules called gRNAs, which are complementary to their edited cognate mRNAs (4). The gRNAs have three distinct regions. A 5- to 15-nucleotide (nt) region at the 5′ end of gRNAs is complementary to the sequence of its cognate preedited mRNA that is immediately 3′ to the region that will be edited. Formation of a duplex between these regions, called the anchor duplex, is an essential prelude to editing. A 55- to 70-nt guiding region, immediately 3′ to the anchor region, contains the sequence information that can specify the insertion or deletion of U’s at 1 to 20 internucleotide sites in the pre-mRNA. The third region contains the 3′ end of the gRNA, which has a 5- to 24-nt oligo(U) tail that is added posttranscriptionally, presumably by a terminal uridylyltransferase (TUTase) activity. The function of the oligo(U) tail is unknown, but it may enhance the interaction between the gRNA and the region of the RNA that is 5′ of the editing site, which tends to be purine rich (5, 25). While gRNAs have distinct sequences, they appear to form similar structures in vitro with 5′ and 3′ stem-loops (12, 24).
In vitro studies indicate that RNA editing occurs by a series of enzymatically catalyzed steps (13, 25, 26). These studies suggest that editing is initiated by endoribonucleolytic cleavage of the pre-mRNA at a site that is 5′ to the anchor duplex. Following cleavage, U’s are added or removed at the 3′ end of the 5′ cleavage product by one or more enzymes that are yet to be fully characterized. The 5′ cleavage product with the added or removed U’s is ligated with the 3′ cleavage product by an RNA ligase to produce the edited product. The mechanism by which the number of U’s is specified by the gRNA is not yet determined. Chimeric RNA molecules in which gRNAs are covalently linked at their 3′ end to the 3′ portion of a pre-RNA are also detected both in vivo and in vitro (6, 13, 20). These molecules do not appear to be editing intermediates as previously proposed but rather seem to be aberrant by-products of the editing reaction (13, 25).
In vitro editing activity requires Trypanosoma brucei mitochondrial extract in addition to pre-mRNA, gRNA, divalent cations, ATP, and UTP (for insertion) (13, 26). The in vitro RNA editing activities that insert or delete U’s sediment at ∼20S in isokinetic glycerol gradients, indicating that editing occurs in association with a multicomponent macromolecular complex (8). The 20S fraction also contains the gRNA-specific endoribonuclease activity as well as TUTase and RNA ligase. However, substantial TUTase and RNA ligase activities also sediment at ∼40S, suggesting that multiple forms of the editing complex may exist (8, 18). Such a complex (or complexes) would be predicted to contain multiple molecules to account for the several catalytic activities and other functions such as RNA binding, positioning, translocation, and unwinding. Four RNP complexes (G1 to G4) that form with mitochondrial extract and gRNA have also been visualized on native polyacrylamide gels (10, 19). These complexes were found to be gRNA specific, since homologous and heterologous gRNA prevent their formation whereas non-gRNA transcripts do not. The complexes contain protein since both sodium dodecyl sulfate (SDS) and proteinase K prevent their formation.
While editing occurs in association with an RNP complex, the components of that complex remain largely unknown and hence uncharacterized. Several candidate protein components have been identified by their ability to UV cross-link specifically with gRNA (15, 16, 19). In T. brucei, these include 21- and 90-kDa proteins that are highly specific for gRNA, based on RNA competition experiments (19). The 21-kDa protein (gBP21) has been recently purified, and its gene has been cloned and sequenced (14). The sequence predicts an arginine-rich protein, and the recombinant protein binds gRNA with high affinity. UV cross-linking of the 90-kDa protein requires that the gRNAs have an oligo(U) tail, while the 21-kDa protein binds to the 3′ stem-loop of gRNA (12, 19).
The development of an in vitro assay for RNA editing (26) permitted the enrichment of editing activity by biochemical techniques. Material enriched for RNA editing activity was used to produce a panel of 81 independent monoclonal antibodies (MAbs), of which six are specific for a 21-kDa protein that binds gRNA. The MAbs alter the mobility on native polyacrylamide gels of gRNA-specific RNP complexes G1 and G2, and they immunoprecipitate the in vitro RNA editing activity as well as all catalytic activities and RNAs associated with editing. The immunoprecipitate contains multiple proteins, including some with mobilities similar to those of the eight major proteins reported to have been found in a fraction enriched for editing activity by biochemical techniques (21) as well as proteins that specifically cross-link with gRNA (19). The data presented here indicate that gBP21 can associate with RNP complexes that are capable of RNA editing in vitro. The possible significance of this association is discussed.
MATERIALS AND METHODS
Antigen preparation and MAb production.
T. brucei procyclic and bloodstream forms (EATRO 164) were grown in vitro as previously described (31). Subcellular fractionation, storage of mitochondrial vesicles, and preparation of mitochondrial lysate that is active for in vitro RNA editing were performed as described by Corell et al. (8). The lysate was fractionated by slowly adding solid (NH4)2SO4 at 4°C to a concentration of 30% with stirring for 1 h. The supernatant from centrifugation at 15,000 rpm for 30 min at 4°C in a Beckman JA-20 rotor contained the in vitro RNA editing activity (assay described below). The supernatant was raised to 45% (NH4)2SO4 at 4°C with stirring for 1 h and recentrifuged. The pellet, which contained the in vitro RNA editing activity, was resuspended in 500 μl of HHE (20 mM HEPES [pH 7.9], 50 mM KCl, 10 mM magnesium acetate, 0.5 mM dithiothreitol) with 1 mM ATP and layered on a 11.5-ml 10 to 40% glycerol gradient. Gradients were centrifuged at 38,000 rpm in a Beckman SW40 rotor for 5 h at 4°C and fractions of 500 μl were collected. Fractions containing RNA editing activity were concentrated by adding (NH4)2SO4 to 70% as described above to produce the antigen for MAb production, which was performed by standard protocols (7). Five female BALB/c mice were injected subcutaneously with ∼15 μg of antigen in Freund’s complete adjuvant and again 14 days later with antigen in Freund’s incomplete adjuvant. The mice were injected intraperitoneally with antigen in incomplete Freund’s adjuvant on day 35 and then both intraperitoneally and intravenously with antigen in incomplete Freund’s adjuvant on day 56. Three days later, splenocytes from the best responder were fused with the SP2/O myeloma cell line and grown in 96-well plates. Supernatants were screened by enzyme-linked immunosorbent assay (ELISA) for antibody against the immunogen, using plates coated with immunogen (10 μg/ml) and using goat anti-mouse immunoglobulin G (IgG) conjugated with alkaline phosphatase (Bio-Rad) as the secondary antibody. Clones from positive wells were made by limiting dilution and rescreened by ELISA to yield a total of 81 clones.
Plasmids and RNA.
gRNA expression plasmids for gA6[14]wt and gA6[14]Δ16G, as well as mRNA substrates A6short/Tag.1 and A6-eEs1, were constructed as previously described (13, 26). gRNAs and mRNA substrates were transcribed by using T7 RNA polymerase and purified on a 9% (wt/vol) denaturing polyacrylamide gel, followed by visualization by UV shadowing. Uniformly labeled transcripts were transcribed in the presence of [α-32P]UTP (800 Ci/mmol) and purified as described above except that the transcripts were visualized by autoradiography. mRNA substrates were labeled by ligation of [32P]pCp to the 3′ end of the transcript as previously described (25), purified on a 9% (wt/vol) denaturing polyacrylamide gel, and visualized by autoradiography.
Western blot analysis.
A 600-ng aliquot of total mitochondrial lysate was separated by polyacrylamide gel electrophoresis on an SDS–12% (wt/vol) polyacrylamide gel, followed by transfer to nitrocellulose by using standard protocols (7). The filters were blocked for 1 h in phosphate-buffered saline (PBS; 4.3 mM Na2HPO4, 1.4 mM KH2PO4, 137 mM NaCl, 2.7 mM KCl [pH 7.3]) containing 10% (wt/vol) nonfat milk and 0.2% (vol/vol) Tween 20. Tissue culture supernatants diluted 1:100 were incubated with the filter overnight at 4°C with gentle agitation. After several washes in PBS, goat anti-mouse antibody conjugated to horseradish peroxidase (Bio-Rad) diluted 1:5,000 was added, and the mixture was incubated at room temperature for 2 h. The Amersham ECL kit was used to visualize the MAb protein target, following the manufacturer’s protocol.
Indirect immunofluorescence.
Procyclic trypanosomes grown to mid-logarithmic phase were washed in TDB (20 mM Na2HPO4, 2 mM NaH2PO4, 5 mM KCl, 80 mM NaCl, 1 mM MgSO4, 10 mM glucose [pH 7.4]), resuspended to 5 × 106 cells/ml in TDB, and spotted onto printed microscope slides. The parasites were allowed to adhere to the slide for 15 min in a moist chamber and were then fixed for 10 min at room temperature with 1.5% (vol/vol) formaldehyde in PBS. Bloodstream-form parasites harvested at the peak of parasitemia were washed with TDB, fixed in suspension with 1.5% (vol/vol) formaldehyde in PBS at 2 × 107 cells/ml, and then spotted onto slides pretreated with 3% bovine serum albumin in PBS supplemented with 0.02% (wt/vol) NaN3. Cells were permeabilized by treatment for 10 min with 0.1% (vol/vol) Triton X-100 in PBS, washed with PBS, and then blocked for 1 h with 3% (wt/vol) bovine serum albumin in PBS supplemented with 0.02% (wt/vol) NaN3. The cells were incubated with the undiluted hybridoma supernatants for 1 h in a moist chamber, washed extensively with PBS, and incubated for 1 h with fluorescein isothiocyanate-conjugated goat anti-mouse antibody (Dianova) diluted 1:200 in PBS. The fixed cells were treated for 5 min with 4′,6-diamidino-2-phenylindole (DAPI; 0.8 μg/μl in PBS) to stain DNA. The cells were washed with PBS and mounted with ProLong antifade medium (Molecular Probes) according to the manufacturer’s specifications. Fluorescence was observed with a Nikon fluorescence microscope equipped with the appropriate filters.
Gel retardation assay.
Ten femtomoles of radiolabeled gRNA was incubated with mitochondrial lysate (5 μg) in 15 μl of binding buffer (20 mM HEPES [pH 7.9], 50 mM KCl, 10 mM magnesium acetate, 1 mM EDTA, 0.5 mM dithiothreitol, 750 ng of yeast RNA per μl) for 30 min at 27°C. Immediately after incubation, complexes were separated on a 4% (wt/vol) nondenaturing polyacrylamide gel (acrylamide/methylene bisacrylamide ratio of 80:1) in 50 mM Tris-glycine (pH 8.8) at 5 to 10 V/cm. After electrophoresis, gels were fixed in 10% (vol/vol) methanol–10% (vol/vol) acetic acid for 30 min, dried, and subjected to autoradiography. Supershift experiments included ∼500 ng of MAb in the reaction mixture.
Immunoprecipitation.
Immunomagnetic beads (Dynabeads M-450; Dynal) coated with goat anti-mouse IgG were coupled with purified MAbs at a concentration of 1.5 μg of MAb/mg of immunomagnetic beads. Coupling was performed in PBS for 30 min at 4°C with bidirectional mixing. After incubation, a magnet was applied for 2 min, followed by three to four 1-min washes with PBS. In experiments where MAbs were cross-linked to the immunomagnetic beads, 1.2 × 106 MAb-specific beads were incubated in 10 ml of 0.2 M triethanolamine (pH 9.0) at 23°C with 52 mg of dimethyl pimelimidate dihydrochloride (Sigma) for 45 min with mixing. After incubation, the beads were incubated in an additional 10 ml of 0.2 M triethanolamine (pH 9.0) at 23°C for 2 h and then washed with either PBS or HHE. Immunoprecipitation of mitochondrial proteins was performed by incubating crude mitochondrial lysate with MAb-specific immunomagnetic beads at a mitochondrial protein/MAb ratio of 10:1 (vol/vol). The mixture was allowed to incubate at 4°C with bidirectional mixing for 2 h. After incubation, the beads were washed five times (1 min each) with cold HHE. The final washed beads were resuspended in the amount of HHE appropriate for the experimental design being used. For SDS-PAGE analysis of proteins, MAb-specific immunomagnetic beads prepared as described above were incubated after the final wash with elution buffer (25 mM glycine [pH 2.5], 150 mM KCl, 1% [wt/vol] SDS) for 10 min at 4°C with bidirectional mixing. The eluted proteins were then dialyzed against HHE, separated by SDS-PAGE on a 10% (wt/vol) gel, and detected by silver staining.
In vitro RNA editing, TUTase, and RNA ligase assays.
The in vitro deletion assay was modified from previously published protocols (13, 25). Three milligrams of MAb-specific immunomagnetic beads used in immunoprecipitation reactions as described above was resuspended in 60 μl of assay buffer (HHE with 5 mM CaCl2, 1 U of RNasin, 1 mM ATP, 750 ng of Torula yeast RNA per μl, 0.25 pmol of gA6[14]Δ16G, and 2.5 pmol of [32P]pCp-labeled A6short/Tag.1) and incubated at 27°C for 1 h with mixing. The in vitro insertion assay was essentially the same as the deletion assay except that (i) [32P]pCp-labeled A6-eES1 in combination with gA6[14]wt was used instead of A6short/Tag.1 with gA6[14]Δ16G and (ii) 1 mM UTP was included in the reaction mixture. After incubation, 40 μl of Tris-EDTA was added, followed by phenol-chloroform extraction. The RNA was then precipitated by adding 5 μg of glycogen, 12 μl of 3 M sodium acetate, and 300 μl of cold ethanol. The pellet was then washed with 70% (vol/vol) ethanol and resuspended in 5 μl of diethylpyrocarbonate-treated H2O and 5 μl of formamide dye (95% [vol/vol] formamide, 0.09% [wt/vol] bromophenol blue, 0.09% [wt/vol] xylene cyanol FF). The samples were then loaded onto a 9% (wt/vol) denaturing polyacrylamide gel. After electrophoresis, the gels were dried and the RNA was visualized by autoradiography.
TUTase assays were modified from previously reported protocols (8). MAb-specific immunomagnetic beads (1.5 mg) from immunoprecipitation reactions described above were resuspended in 20 μl of HHE with 1 mM ATP, 1 μg of yeast tRNA, and 5 μCi of [α-32P]UTP (800 Ci/mmol). After incubation for 30 min at 27°C, a 5-μl aliquot of the reaction mixture was spotted onto duplicate fiberglass disks, followed by four 15-min washes in cold 10% (wt/vol) trichloroacetic acid with 50 mM sodium pyrophosphate on ice. Incorporated radioactivity was quantified by liquid scintillation counting.
RNA ligase activity was assessed by self-adenylylation of the enzyme (22). MAb-specific immunomagnetic beads (1.5 mg) from immunoprecipitation reactions described above were resuspended in 20 μl of HHE with 10% (vol/vol) dimethyl sulfoxide, 1 μg of yeast tRNA, and 5 μCi of [α-32P]ATP (3,000 Ci/mmol). The reaction mixture was incubated at 27°C for 30 min, and then 6.6 μl of 3× SDS-PAGE loading dye (150 mM Tris-HCl [pH 6.8], 300 mM dithiothreitol, 6% [wt/vol] SDS, 0.3% [wt/vol] bromophenol blue, 30% [vol/vol] glycerol) was added. Proteins were separated by SDS-PAGE on a 10% gel. The gel was dried, and proteins were visualized by autoradiography.
RNA isolation and analysis.
Immunoprecipitation of associated RNA was performed with MAb-specific magnetic beads prepared as described above. Mitochondrial extract was mixed with the MAb-specific beads at a ratio of 4:1 (vol/vol) for 4 h at 4°C. The RNA was extracted from the immunoprecipitated material with an equal volume of phenol-chloroform and then precipitated with ethanol. The RNA was then separated on either a 2% (vol/vol) formaldehyde-containing agarose gel or a 6% (wt/vol) polyacrylamide–7 M urea gel and then transferred to nitrocellulose as previously described (23). The membranes were prehybridized in 5× SSPE (50 mM NaH2PO4 [pH 7.0], 40 mM NaOH, 900 mM NaCl, 5 mM EDTA)–1% (wt/vol) SDS–10× Denhardt’s solution–50 μg of denatured sheared salmon sperm DNA per ml for 5 h at the hybridization temperatures listed below. The membranes were hybridized overnight in 5× SSPE with 32P 5′ end-labeled oligonucleotide probes MURF4.1 (CCTTTCTCCTTCATTTCCTCTCCTGTCTCCTTCTCTTCCGCCC) at 50°C, MURF4.14 (CACAATAATACATACATAATAACAAACGCAACC) at 50°C, and gA6-1 (CACTGTCAAAATCTGATTCGTTATCGGAG) at 55°C. The membranes were washed sequentially for 1, 2, and 3 min in 5× SSPE–1% (wt/vol) SDS at room temperature and for 6 min in 1× SSPE–1% (wt/vol) SDS at the hybridization temperature. The RNA was visualized by autoradiography.
UV cross-linking.
The gRNA-specific cross-linking to proteins in crude mitochondrial extract was performed as previously described (19) except that the buffer used was HHE. In experiments where UV cross-linking was performed after immunoprecipitation (see Fig. 9), MAb-specific immunomagnetic beads (1.5 mg) from immunoprecipitation reactions described above were resuspended in 15 μl of HHE containing 10 fmol of radiolabeled gRNA (see above) and 250 ng of yeast RNA per μl as a competitor. The reaction mixture was incubated for 20 min at rom temperature and then subjected to UV cross-linking as previously described (19). Following UV cross-linking, 1.5 μl of RNase A (10-mg/ml stock) was added, and the reaction mixture was incubated for 15 min at 37°C. After incubation, 3.5 μl of 3× SDS-PAGE loading dye was added. Proteins were then separated on an SDS–12% polyacrylamide gel; the gel was dried and subjected to autoradiography.
FIG. 9.
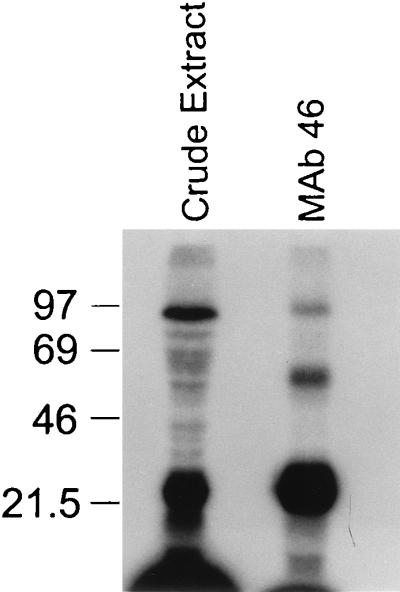
Proteins in the immunoprecipitate that UV cross-link with gRNA. Uniformly labeled gRNA was added to crude mitochondrial extract and to material immunoprecipitated with MAb 46, UV cross-linked, treated with RNase A, and separated on an SDS–12% polyacrylamide gel, followed by visualization of the proteins by autoradiography as described in Materials and Methods. The cross-linked 21-kDa protein as well as proteins of ∼55, ∼16, and ∼90 kDa are evident. Sizes are indicated in kilodaltons at the left.
RESULTS
Production and identification of anti-gBP21 MAbs.
Precipitation with (NH4)2SO4 and glycerol gradient sedimentation allowed substantial enrichment of the in vitro RNA editing activity (Fig. 1). All detectable in vitro RNA editing activity precipitates at between 30 and 45% (NH4)2SO4 (Fig. 1A), which represents ∼10% of the protein in total mitochondrial extract. The bulk of the precipitated activity sediments at ∼20S (fractions 16 to 18) in 10 to 40% (vol/vol) glycerol gradients (Fig. 1B), indicating that the activity is associated with complexes. (NH4)2SO4 precipitation did not affect the sedimentation of editing complexes compared to gradients for which (NH4)2SO4 precipitation was not performed (data not shown) (8). This ∼20S fraction from the 30 to 45% (NH4)2SO4 fraction was used to raise a panel of MAbs as described in Materials and Methods. Eighty-one independent MAbs that showed a variety of protein targets based on Western blot analysis were obtained (data not shown). Of these, six were found to be specific for a ∼21-kDa protein by Western blot analysis, as illustrated by assays using MAb 56, the antibody used most extensively in this study (Fig. 2A). We examined the possibility that this was the ∼21-kDa protein previously identified as a gRNA-specific UV-cross-linking protein that is present in mitochondrial extracts (19). To do this, we incubated uniformly labeled gRNA with mitochondrial extract, UV cross-linked the RNA to the protein, and treated the preparation with RNase A to remove the non-cross-linked portion of the RNA as described in Materials and Methods. This treatment labeled several proteins, as we previously had found (Fig. 2B, crude extract) (19). However, all six MAbs immunoprecipitated the labeled ∼21-kDa protein when coupled with magnetic beads (Fig. 2B [data not shown for two of the MAbs]). Magnetic beads alone or coupled with a control MAb (anti-T. brucei dihydrolipoyl transacylase MAb ODB2) did not immunoprecipitate labeled proteins. Similarly, experiments using an RNA transcript of equal size derived from pBluescript (Stratagene) did not immunoprecipitate any UV-cross-linked proteins (data not shown). In addition, the MAbs reacted with recombinant gBP21 protein in Western blot analysis (Fig. 2C; data for MAb 56 shown). Thus, these six MAbs are specific for gBP21, and this protein is present in fractions enriched for in vitro RNA editing activity. Further Western blot analysis determined that only four of the six MAb cell lines (MAb lines 46, 56, 61, and 67) produced antibodies in high quantities. Consequently, these four MAbs were used in subsequent experiments.
FIG. 1.

Antigen for MAb production. The in vitro RNA editing activity was enriched by subcellular fractionation of mitochondria followed by (NH4)2SO4 precipitation and glycerol gradient fractionation to prepare immunogen used for the production of MAbs (see Materials and Methods). (A) In vitro editing assay (U deletion) of fractions from sequential steps of (NH4)2SO4 precipitation of crude mitochondrial lysate, showing editing activity in the 45% pellet. The product formed by the deletion of three U’s from editing site 1 of the input RNA (A6short/Tag.1) is indicated. (B) In vitro RNA editing assay (U deletion) of the 20S fraction from glycerol gradient sedimentation of the 45% (NH4)2SO4 precipitate showing the editing activity.
FIG. 2.

Identification of target protein. (A) Western blot analysis of crude mitochondrial lysate separated by SDS-PAGE on a 12% (wt/vol) gel. MAb 56 (and five other MAbs not shown) recognized an ∼21-kDa protein. (B) Immunoprecipitation of gRNA UV-cross-linked proteins. Proteins that cross-link with uniformly labeled gRNA in crude mitochondrial lysate and immunoprecipitate with the MAbs specific to gBP21 after treatment with RNase A were separated by SDS-PAGE on a 12% (wt/vol) gel and visualized by autoradiography as described in Materials and Methods. The four MAbs shown (and two others not shown) recognize an ∼21-kDa UV-cross-linking protein. (C) Western blot analysis showing that the MAbs (MAb 56 shown) recognize recombinant gBP21. Time points refer to sampling intervals after induction of culture (see Materials and Methods). Sizes are indicated in kilodaltons at the left.
Immunofluorescence analysis with MAb 56 shows distinct staining of the mitochondrion, most intensely in the kinetoplast (Fig. 3A [DAPI] and 3B [MAb 56]). The bloodstream forms show the typical single tubular mitochondrion (Fig. 3C), while the procyclic forms have a network of interconnected tubules (Fig. 3B). Thus, gBP21 appears to be exclusively localized in the mitochondrion and preferentially located in the kinetoplast region.
FIG. 3.

Immunofluorescence with MAbs specific to gBP21. Procyclic and bloodstream forms of T. brucei were fixed and stained with MAbs against gBP21 and with DAPI (see Materials and Methods). (A) DAPI staining showing the nucleus and smaller kinetoplast of procyclic T. brucei. (B) Procyclic T. brucei after incubation with MAb 56 and development with a goat anti-mouse antibody conjugated with fluorescein isothiocyanate. The kinetoplast is evident as an intensely staining spot in the network of mitochondrial tubules. (C) Bloodstream T. brucei examined as in panel B, showing the single tubular mitochondrion.
Anti-gBP21 MAbs recognize complexes that are associated with editing.
The anti-gBP21 MAbs supershift gRNA-specific complexes that are visualized in 4% (wt/vol) nondenaturing polyacrylamide gels (Fig. 4). The G1, G2, and G3 complexes that were previously shown to be specific for gRNA (10, 19) are shown in Fig. 4 (No MAb). The G4 complex is not resolved in these gels. The G1 and G2 complexes, which were shown to contain a ∼21-kDa protein that UV cross-links to gRNA (19), supershift upon the inclusion of anti-gBP21 MAbs (Fig. 4). Supershifting of the G3 complex was not detected. As with the formation of the RNP complexes, the supershift is not affected by the addition of heterologous RNA transcripts (pBluescript) as a competitor of both the formation of the RNP complexes and the subsequent supershift (data not shown). In addition, SDS and proteinase K digestion ablates both the formation of the RNP complex and the subsequent supershift (data not shown). The amount of G1 and G2 that is supershifted is dependent on the amount of MAb added to the reaction and thus is probably not due to differences between the MAbs (data not shown). As seen in Fig. 4 (MAb 61), all of G1 and G2 can be supershifted by the MAbs. These studies indicate that the G1 and G2 RNP complexes contain gBP21 and that all of the labeled gRNA is associated with the gBP21 protein. They also imply that while G3 contains gRNA, the specificity for gRNA does not appear to entail gBP21 or its state is such that anti-gBP21 MAb cannot produce a detectable mobility shift.
FIG. 4.

Supershift of gRNA-specific RNP complexes G1 and G2. Specific complexes that form with uniformly labeled gRNA were separated on a 4% (wt/vol) nondenaturing polyacrylamide gel and then autoradiographed. The lane marked as containing no MAb shows gRNA-specific RNP complexes that form in the absence of the MAbs. Lanes in which MAbs specific for gBP21 were added to the reaction mixture (see Materials and Methods) show a supershift of RNP complexes G1 and G2.
The anti-gBP21 MAbs can immunoprecipitate in vitro RNA editing activity.
Immunomagnetic beads coupled with either of four anti-gBP21 MAbs immunoprecipitated both deletion and insertion editing activities (Fig. 5). As shown in Fig. 5A, the four MAbs (MAbs 46, 56, 61, and 67) precipitate the activity that specifically deletes four U’s from the synthetic mRNA substrate. The gRNA/pre-mRNA chimeras that form in vitro (as well as in vivo) and which are thought to be nonproductive by-products of editing are also observed (13, 25). In addition, the specific 3′ cleavage product of the pre-mRNA is produced. This finding demonstrates that the gRNA-specific endoribonuclease is also present in the immunoprecipitated material. By inference, the material contains a 3′ exoribonuclease activity that removes the U’s from the 3′ end of the 5′ cleavage product of the pre-mRNA, although the reverse activity of the enzyme that adds U’s could also be responsible. A control MAb (ODB2) attached to immunomagnetic beads and immunomagnetic beads alone do not immunoprecipitate activities responsible for production of edited RNA, chimeras, or the endoribonuclease activity. Similarly, the anti-gBP21 MAbs immunoprecipitate the activity that specifically inserts two U’s into pre-mRNA (Fig. 5B [results for MAb 61 not shown]). The immunoprecipitate produces edited RNA, gRNA/pre-mRNA chimeras, and the 3′ cleavage product of the pre-mRNA that is the result of gRNA-specific endoribonucleolytic cleavage. Micrococcal nuclease treatment of mitochondrial extracts before immunoprecipitation abolished the ability of the MAbs to immunoprecipitate both deletion and insertion type editing, suggesting a requirement for an RNA component (data not shown). The editing activity is in excess relative to the MAb in these experiments since the extract incubated with the immunomagnetic beads is sufficient for hundreds of in vitro editing assays. Attempts to deplete the extract of editing activity were inconclusive since while repeated immunoprecipitations resulted in abolishment of activity in the supernatant, the need to use less supernatant and to handle the material more may alone have resulted in the abolishment of activity in the supernatant. Thus, it is unclear if gBP21 is associated with all complexes that are capable of editing.
FIG. 5.

Immunoprecipitation of in vitro editing activity. Material that immunoprecipitates with MAbs against gBP21 was tested for its ability to support in vitro RNA editing that deletes (A) or inserts (B) U’s. The right gel in panel B shows a longer separation of the RNA molecules that increases the resolution of the edited product from the input RNA. Input RNA, 3′ cleavage product, chimeras, and edited product are diagramed to the right of the corresponding RNA molecule. Editing products were identified by their comigration with known editing products. Editing activity is immunoprecipitated with magnetic beads coupled with MAbs specific for gBP21 but not with immunomagnetic beads alone (No MAb) or with immunomagnetic beads coupled to a control antibody (ODB2).
In addition to the endo- and exoribonuclease activities, the anti-gBP21 MAbs immunoprecipitate the 3′-terminal TUTase and RNA ligase activities (Fig. 6) that are implied by the cleavage/ligation model for RNA editing (30). The TUTase activity, which may add U’s to the 3′ end of gRNA, is present in abundance in the material immunoprecipitated with each of the anti-gBP21 MAbs but absent from immunoprecipitates resulting from the use of uncoated beads or beads coated with control antibody ODB2 (Fig. 6A). Similarly, RNA ligase, as measured by self-adenylylation, is also immunoprecipitated by the four anti-gBP21 MAbs but not in the control immunoprecipitations (Fig. 6B). Thus, the anti-gBP21 MAbs immunoprecipitate all of the individual activities that are expected to be associated with RNA editing as well as the overall in vitro RNA editing activity itself. As found with in vitro RNA editing activity, micrococcal nuclease treatment of extracts before immunoprecipitation abolishes the ability of the MAbs to immunoprecipitate TUTase or the self-adenylylatable proteins (data not shown).
FIG. 6.

Immunoprecipitation of TUTase and [α-32P]ATP self-adenylylatable proteins (RNA ligase). (A) TUTase activity, as monitored by the addition of [α-32P]UTP to yeast tRNA (counts per minute are shown), immunoprecipitates with the anti-gBP21 MAbs but not with immunomagnetic beads alone (No MAb) or immunomagnetic beads coupled with control MAb ODB2. (B) Anti-gBP21 MAbs immunoprecipitate the [α-32P]ATP self-adenylylatable proteins (RNA ligase) while immunomagnetic beads and the control antibody (ODB2) do not.
Northern blot analyses of the material that is immunoprecipitated by the anti-gBP21 MAbs reveal the presence of gRNA (as expected since the MAbs are against a gRNA binding protein) as well as preedited, partially edited, and edited mRNAs (Fig. 7). A probe that is specific for gA6[14] gRNA reveals the presence of endogenous gRNA (Fig. 7A) (3). Hybridization with the MURF4.1 oligonucleotide probe, which is complementary to 3′ A6 mRNA sequence that is not edited, reveals preedited A6 mRNA as well as partially edited A6 mRNA in the immunoprecipitated material (Fig. 7B) (3). The MURF4.14 oligonucleotide probe, which is complementary to 5′ edited A6 sequence, reveals fully and partially edited A6 mRNA (Fig. 7C) (3). Control MAb ODB2 as well as immunomagnetic beads alone do not immunoprecipitate either gRNA or mRNA (data not shown). Thus, the material that is immunoprecipitated by antibodies specific for gBP21 contains all activities and RNAs that are expected to be part of the complex that catalyzes RNA editing.
FIG. 7.

Immunoprecipitation of gRNA and mRNA. Northern blots of immunoprecipitated RNA were probed with oligonucleotide probes for gA6[14]gRNA (A), MURF4.1 (which is complementary to 3′ A6 mRNA sequence which does not get edited; this probe reveals preedited as well as partially edited A6 mRNA) (B), and MURF4.14 (which is complementary to 5′ edited A6 sequence; this probe reveals fully and partially edited A6 mRNA) (C).
Protein profile of immunoprecipitated proteins.
SDS-PAGE analysis of the material immunoprecipitated by the anti-gBP21 antibody shows a complex pattern of proteins upon silver staining. However, multiple experiments reveal a consistent pattern of 13 major proteins with apparent molecular masses of 18, 24, 25, 28, 30, 32, 45, 47, 50, 52, 64, 65, and 69 kDa (Fig. 8). This pattern, which has remained consistent throughout multiple experiments, is not seen when immunoprecipitation experiments are performed with immunomagnetic beads alone or when MAb ODB2 is used.
FIG. 8.
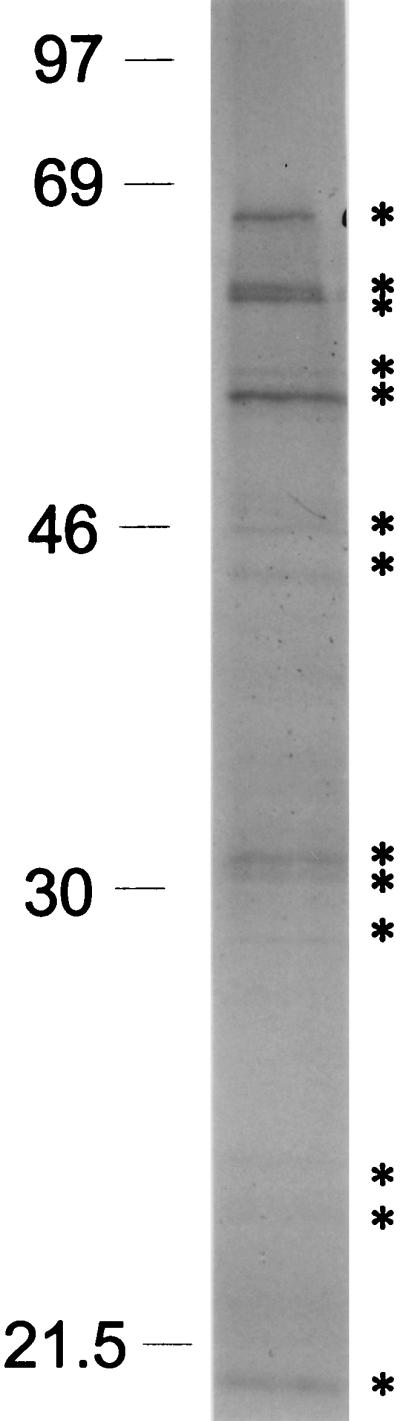
SDS-PAGE analysis of immunoprecipitated proteins. Proteins present in the material immunoprecipitated from crude mitochondrial extract with gBP21-specific MAb 56 were separated by SDS-PAGE on a 10% (wt/vol) gel and silver stained. Thirteen prominent bands are seen; sizes of standards are indicated in kilodaltons at the left.
Immunoprecipitation of gRNA binding proteins.
We examined proteins in the immunoprecipitated material that UV cross-link to gRNA since several proteins have been implicated in RNA editing by their ability to UV cross-link to gRNA. In these experiments, immunoprecipitation with anti-gBP21 MAb 46 was followed by UV cross-linking to added uniformly labeled gRNA and RNase A treatment (see Materials and Methods). As expected, the 21-kDa gBP21 protein is prominently seen in addition to a 90-kDa protein, which may be the same protein that we previously observed to require the gRNA U tail for UV cross-linking (19). Furthermore, a ∼55-kDa and a ∼16-kDa protein, neither of which has previously been described, were also observed (Fig. 9). This finding suggests that these proteins are closely associated with gRNA in RNA editing complexes.
DISCUSSION
This study reports that MAbs specific for the gRNA binding protein gBP21 can immunoprecipitate in vitro RNA editing activity and its component activities of gRNA-specific endoribonuclease, TUTase, and RNA ligase (protein self-adenylylation). In addition, gRNA and preedited, partially edited, and fully edited mRNAs are also immunoprecipitated. This finding suggests that the antibodies immunoprecipitate functional editing complexes by virtue of their association with the gBP21 protein. The MAbs are specific for gBP21 since they reveal a single ∼21-kDa protein in Western blot analyses of samples from isolated T. brucei mitochondria (Fig. 2A) and from Escherichia coli expressing recombinant gBP21 (Fig. 2C) and since they immunoprecipitate a ∼21-kDa protein that specifically cross-links with gRNA (Fig. 2B). The recovery of six of 81 MAbs specific for gBP21 shows that this protein is present in the 20S fraction from the 30 to 45% (NH4)2SO4 precipitate and suggests that it may be abundant in this fraction and/or highly immunogenic.
The immunoprecipitation of editing activity and its associated activities appears to be specific but does not definitively demonstrate a functional association of gBP21 with the editing complex. The specificity is evident from the results that MAbs specific for other mitochondrial components or non-T. brucei antigens as well as immunomagnetic beads to which additional antibodies are not coupled do not precipitate the activities. The inability to immunoprecipitate the activities after treatment of mitochondrial extract with (and subsequent to inactivation of) micrococcal nuclease suggests a functional association between gBP21 and the editing complex which involves RNA. However, although the addition of EGTA does not inhibit the editing activity (and other activities), the editing activity is not detectable in either the immunoprecipitate or supernatant after micrococcal nuclease treatment and inactivation. In contrast, TUTase and the self-adenylylatable proteins are seen in the supernatant after treatment. The loss of in vitro RNA editing activity may have been the consequence of the manipulations, dilution, or dissociation. Thus, a more direct assessment is needed to ascertain the functional association of gBP21 with the complex. Nevertheless, the immunoprecipitation of RNA editing activities that insert and delete U’s by the anti-gBP21 MAbs indicates that gBP21 is associated with complexes that perform both types of RNA editing, which implies that they may be similar.
The coprecipitation of in vitro RNA editing activities that insert and delete U’s, the component activities of endoribonuclease, TUTase, and RNA ligase, as well as gRNA and mRNA, suggests that all of these activities and molecules might be assembled within a multicomponent macromolecular complex. This is consistent with the current view that editing occurs by a series of enzyme catalyzed steps (4, 13, 25). According to this model, editing is initiated by an endoribonucleolytic cleavage at a site that is directed by the gRNA (25). This gRNA-dependent endoribonuclease sediments at ∼20S, as do RNA editing complexes (8), while gRNA-independent endoribonuclease activity remains at the top of glycerol gradients. The relationship between the two endoribonuclease activities is unclear; they may be distinct enzymes or simply differ in their association with the complex (17). Nevertheless, gRNA-dependent endoribonuclease is immunoprecipitated by anti-gBP21 MAbs (Fig. 5). The MAbs also immunoprecipitate activities that add or remove U’s at the 3′ end of the 5′ cleavage product of pre-mRNA, since RNA edited by U insertion or deletion is produced by the immunoprecipitate. TUTase activity, detected as the addition of U’s to yeast tRNA, is also immunoprecipitated by the anti-gBP21 MAbs (Fig. 6A) and may be responsible for the U insertion. It is unclear if this enzyme is also responsible for the posttranscriptional addition of U’s to the 3′ end of gRNA or if there are multiple U-addition enzymes. Removal of U’s during RNA editing may conceivably occur by the reverse activity of the same enzyme that adds U’s. However, one study (21) suggests that, at least for TUTase, this may not be the case since UMP and not UTP is released upon U removal, although these studies did not examine authentic sites that are edited. The anti-gBP21 MAbs also immunoprecipitate the 57- and (to a lesser extent) 50-kDa self-adenylylatable proteins (Fig. 6B). These proteins are probably RNA ligases since they accumulate when incubated with nonligatable substrates but deadenylylate and release AMP upon incubation with ligatable substrates (22). Thus, they may be responsible for the RNA ligase activity that catalyzes the final step in a single round of RNA editing. The requirement for ATP α-β bond hydrolysis for in vitro RNA editing may reflect the ATP requirement of RNA ligase, although other steps in editing may also require ATP hydrolysis (22, 26).
The in vitro RNA editing activity as well as the associated gRNA-dependent endoribonuclease and RNA editing-associated U addition and deletion activities sediment with a broad profile centered at ∼20S in glycerol gradients (8). While TUTase and the 50- and 57-kDa self-adenylylatable proteins also sediment with a peak in this region of the gradient, a second peak of TUTase and the self-adenylylatable proteins, which is more prominent in some strains and studies, is centered at ∼40S (8, 18). In addition, gRNA and preedited mRNA sediment near 30S, while partially and fully edited mRNAs sediment with a broad profile nearer 40S (8, 18). Perhaps the greater apparent sedimentation coefficient reflects the increased mRNA size due to RNA editing and/or gRNA or protein accumulation during editing. One possibility, which we favor, is that complexes sedimenting at ∼20S can edit exogenously added RNAs whereas those sedimenting down to ∼40S are associated with endogenous RNA and thus do not edit exogenous RNA (30). An alternative suggestion is that the ∼20S and ∼40S peaks represent different complexes (18). Western blot analysis shows that most gBP21 protein is well above the 20S fraction in glycerol gradients, indicating that the majority of it is not (or not stably) associated with the editing complexes (data not shown). However, G1 and G2 gRNA-specific complexes that contain gBP21 which form in vitro (19) can be quantitatively supershifted by the anti-gBP21 MAbs, while the G3 complexes that contain a 90-kDa protein but not gBP21 are not (Fig. 4). Thus, gBP21 may be in substantial excess relative to editing complexes and/or it does not remain associated with the editing complex during the entire editing process. One possibility is that gBP21 plays a role in the association of the gRNA with the editing complex. Perhaps G1 and G2 represent early steps in the association of gRNA with a complex that associates the gRNA with the editing complex, and the G3 (and perhaps G4) complex represents a later step in the assembly of the complex. The localization of gBP21 throughout the mitochondrion with a greater concentration in the kinetoplast might imply a functional partitioning (Fig. 3). Perhaps the kinetoplast, which is the site of preedited RNA and gRNA transcription, is also a site of assembly of the editing complexes.
Studies of gRNA structure (12, 24) indicate that gRNAs assume a double stem-loop structure with the anchor sequence contained within the 5′ stem and the 3′ U tail extending from and not included within the 3′ stem. In vitro footprint analysis shows that gBP21 protects a substantial part of the 3′ stem-loop structure (12). This might leave the 5′ region free to form a duplex with the pre-mRNA. UV cross-linking to gRNA with the 90-kDa protein, which is U tail dependent (19), sediments at 10S to 20S on glycerol gradients after incubation with mitochondrial extract, as does UV-cross-linked gBP21 (8). However, the UV-cross-linked 90-kDa protein is primarily seen in the ∼40S fraction, while most UV-cross-linked 21-kDa protein is in a 10S fraction if the extract is fractionated before UV cross-linking. Perhaps there is a greater affinity or stability of the association of the gRNA with the editing complex than for gBP21, and most 90-kDa protein remains associated with a complex whereas most 21-kDa protein is free in solution. Elucidation of the roles of these proteins and the other proteins associated with the editing complex will await other studies, including gene knockout experiments.
The composition of the editing complex(es) is uncertain. In addition to gRNA and pre-mRNA, the complex(es) likely contains multiple proteins, and the possibility that other RNAs are present has not yet been excluded. UV cross-linking has identified numerous potential protein constituents of the editing complex, including gBP21 and the 90-kDa U-tail binding protein which become conspicuous in competition experiments (19) and 55- and 16-kDa proteins revealed upon anti-gBP21 MAb enrichment (Fig. 9). Complexes enriched by immunoaffinity purification consistently contain proteins with approximate apparent molecular masses of 18, 24, 25, 28, 30, 32, 45, 47, 50, 52, 64, 65, and 69 kDa (Fig. 8). These may include the proteins of 16, 21, 55, and 90 kDa, given the inaccuracy of measurements, especially in comparisons of silver-stained proteins with those radiolabeled by RNA cross-linking and with the variation in silver staining intensities seen between proteins. Complexes with RNA editing and associated activities prepared by biochemical techniques have fewer prominent proteins. Those prepared by the Sollner-Webb group (21) have major proteins with apparent molecular masses of 21, 45, 50, 55, 58, 66, 90, and 95 kDa. These protein profiles are quite similar although not identical. In any event, a demonstration of a specific association between one or more of the proteins is needed before it can be concluded that these proteins are components of the editing complex.
ACKNOWLEDGMENTS
This work was supported by NIH grant GM42188 to K.D.S., who is also a Burroughs Wellcome Scholar of Molecular Parasitology. T.E.A. was supported by NIH postdoctoral fellowship 1F32AI09206-01; S.H. and H.U.G. were supported by funding from the Deutsche Forschungsgemeinschaft.
We thank Reza Salavati and Rob Igo for critical reading of the manuscript and members of the Stuart laboratory for many helpful discussions.
REFERENCES
- 1.Benne R. RNA editing in trypanosomes. Eur J Biochem. 1994;221:9–23. doi: 10.1111/j.1432-1033.1994.tb18710.x. [DOI] [PubMed] [Google Scholar]
- 2.Benne R, van den Burg J, Brakenhoff J P J, Sloof P, Van Boom J H, Tromp M C. Major transcripts of the frameshifted coxII gene from trypanosome mitochondria contain four nucleotides that are not encoded in the DNA. Cell. 1986;46:819–826. doi: 10.1016/0092-8674(86)90063-2. [DOI] [PubMed] [Google Scholar]
- 3.Bhat G J, Koslowsky D J, Feagin J E, Smiley B L, Stuart K. An extensively edited mitochondrial transcript in kinetoplastids encodes a protein homologous to ATPase subunit 6. Cell. 1990;61:885–894. doi: 10.1016/0092-8674(90)90199-o. [DOI] [PubMed] [Google Scholar]
- 4.Blum B, Bakalara N, Simpson L. A model for RNA editing in kinetoplastid mitochondria: “guide” RNA molecules transcribed from maxicircle DNA provide the edited information. Cell. 1990;60:189–198. doi: 10.1016/0092-8674(90)90735-w. [DOI] [PubMed] [Google Scholar]
- 5.Blum B, Simpson L. Guide RNAs in kinetoplastid mitochondria have a nonencoded 3′ oligo(U) tail involved in recognition of the preedited region. Cell. 1990;62:391–397. doi: 10.1016/0092-8674(90)90375-o. [DOI] [PubMed] [Google Scholar]
- 6.Blum B, Simpson L. Formation of guide RNA/messenger RNA chimeric molecules in vitro, the initial step of RNA editing, is dependent on an anchor sequence. Proc Natl Acad Sci USA. 1992;89:11944–11948. doi: 10.1073/pnas.89.24.11944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Coligan E, Kruibsbeek A M, Margulies D H, Shevach E M, Strober W. Current protocols in immunology. New York, N.Y: John Wiley & Sons, Inc.; 1991. [Google Scholar]
- 8.Corell R A, Read L K, Riley G R, Nellissery J K, Allen T, Kable M L, Wachal M D, Seiwert S, Myler P J, Stuart K D. Complexes from Trypanosoma brucei that exhibit deletion editing and other editing-associated properties. Mol Cell Biol. 1996;16:1410–1418. doi: 10.1128/mcb.16.4.1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Feagin J E, Stuart K. Development aspects of uridine addition within mitochondrial transcripts of Trypanosoma brucei. Mol Cell Biol. 1988;8:1259–1265. doi: 10.1128/mcb.8.3.1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Göringer H U, Koslowsky D J, Morales T H, Stuart K D. The formation of mitochondrial ribonucleoprotein complexes involving guide RNA molecules in Trypanosoma brucei. Proc Natl Acad Sci USA. 1994;91:1776–1780. doi: 10.1073/pnas.91.5.1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hajduk S L, Harris M E, Pollard V W. RNA editing in kinetoplastid mitochondria. FASEB J. 1993;7:54–63. doi: 10.1096/fasebj.7.1.8422975. [DOI] [PubMed] [Google Scholar]
- 12.Hermann T, Schmid B, Heumann H, Göringer H U. A three-dimensional working model for a guide RNA from Trypanosoma brucei. Nucleic Acids Res. 1997;25:2311–2318. doi: 10.1093/nar/25.12.2311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kable M L, Seiwert S D, Heidmann S, Stuart K. RNA editing: a mechanism for gRNA-specified uridylate insertion into precursor mRNA. Science. 1996;273:1189–1195. doi: 10.1126/science.273.5279.1189. [DOI] [PubMed] [Google Scholar]
- 14.Köller J, Müller U, Schmid B, Missel A, Kruft V, Stuart K, Göringer H U. Trypanosoma brucei gBP21: an arginine rich mitochondrial protein that binds to guide RNA with high affinity. J Biol Chem. 1997;272:3749–3757. doi: 10.1074/jbc.272.6.3749. [DOI] [PubMed] [Google Scholar]
- 15.Köller J, Nörskau G, Paul A S, Stuart K, Göringer H U. Different Trypanosoma brucei guide RNA molecules associate with an identical complement of mitochondrial proteins in vitro. Nucleic Acids Res. 1994;22:1988–1995. doi: 10.1093/nar/22.11.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leegwater P, Speijer D, Benne R. Identification by UV cross-linking of oligo(U)-binding proteins in mitochondria of the insect trypanosomatid Crithidia fasciculata. Eur J Biochem. 1995;227:780–786. doi: 10.1111/j.1432-1033.1995.tb20201.x. [DOI] [PubMed] [Google Scholar]
- 17.Piller K J, Rusché L N, Cruz-Reyes J, Sollner-Webb B. Resolution of the RNA editing gRNA-directed endonuclease from two other endonucleases of Trypanosoma brucei mitochondria. RNA. 1997;3:279–290. [PMC free article] [PubMed] [Google Scholar]
- 18.Pollard V W, Harris M E, Hajduk S L. Native mRNA editing complexes from Trypanosoma brucei mitochondria. EMBO J. 1992;11:4429–4438. doi: 10.1002/j.1460-2075.1992.tb05543.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Read L K, Göringer H U, Stuart K. Assembly of mitochondrial ribonucleoprotein complexes involves specific guide RNA (gRNA)-binding proteins and gRNA domains but does not require preedited mRNA. Mol Cell Biol. 1994;14:2629–2639. doi: 10.1128/mcb.14.4.2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Riley G R, Myler P J, Stuart K. Quantification of RNA editing substrates, products and potential intermediates: implication for development regulation. Nucleic Acids Res. 1995;23:708–712. doi: 10.1093/nar/23.4.708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rusché L N, Cruz-Reyes J, Piller K J, Sollner-Webb B. Purification of a functional enzymatic editing complex from Trypanosoma brucei mitochondria. EMBO J. 1997;16:4069–4081. doi: 10.1093/emboj/16.13.4069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sabatini R, Hajduk S L. RNA ligase and its involvement in guide RNA/mRNA chimera formation. Evidence for a cleavage-ligation mechanism of Trypanosoma brucei mRNA editing. J Biol Chem. 1995;270:7233–7240. doi: 10.1074/jbc.270.13.7233. [DOI] [PubMed] [Google Scholar]
- 23.Sambrook J, Fritsch T, Maniatis T. Molecular cloning: a laboratory manual. 2nd ed. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- 24.Schmid B, Riley G R, Stuart K, Göringer H U. The secondary structure of guide RNA molecules from Trypanosoma brucei. Nucleic Acids Res. 1995;23:3093–3102. doi: 10.1093/nar/23.16.3093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Seiwert S D, Heidmann S, Stuart K. Direct visualization of uridylate deletion in vitro suggests a mechanism for kinestoplastid RNA editing. Cell. 1996;84:1–20. doi: 10.1016/s0092-8674(00)81062-4. [DOI] [PubMed] [Google Scholar]
- 26.Seiwert S D, Stuart K. RNA editing: transfer of genetic information from gRNA to precursor mRNA in vitro. Science. 1994;266:114–117. doi: 10.1126/science.7524149. [DOI] [PubMed] [Google Scholar]
- 27.Simpson L, Maslov D A, Blum B. RNA editing in Leishmania mitochondria. In: Benne R, editor. RNA editing: the alteration of protein coding sequences of RNA. Chichester, England: Ellis Horwood Ltd.; 1993. pp. 53–86. [Google Scholar]
- 28.Sollner-Webb B. RNA editing. Curr Opin Cell Biol. 1991;3:1056–1061. doi: 10.1016/0955-0674(91)90129-m. [DOI] [PubMed] [Google Scholar]
- 29.Stuart K. RNA editing in mitochondria of African trypanosomes. In: Benne R, editor. RNA editing: the alteration of protein coding sequences of RNA. Chichester, England: Ellis Harwood Ltd.; 1993. pp. 26–52. [Google Scholar]
- 30.Stuart K, Allen T E, Heidmann S, Seiwert S D. RNA editing in kinetoplastid protozoa. Microbiol Mol Biol Rev. 1997;61:105–120. doi: 10.1128/mmbr.61.1.105-120.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stuart K, Gobright E, Jenni L, Milhausen M, Thomashow L, Agabian N. The IsTaR 1 serodeme of Trypanosoma brucei: development of a new serodeme. J Parasitol. 1984;70:747–754. [PubMed] [Google Scholar]