

Abstract
Mechanical forces influence different cell types in our bodies. Among the earliest forces experienced in mammals is blood movement in the vascular system. Blood flow starts at the embryonic stage and ceases when the heart stops. Blood flow exposes endothelial cells (ECs) that line all blood vessels to hemodynamic forces. ECs detect these mechanical forces (mechanosensing) through mechanosensors, thus triggering physiological responses such as changes in vascular diameter. In this review, we focus on endothelial mechanosensing and on how different ion channels, receptors, and membrane structures detect forces and mediate intricate mechanotransduction responses. We further highlight that these responses often reflect collaborative efforts involving several mechanosensors and mechanotransducers. We close with a consideration of current knowledge regarding the dysregulation of endothelial mechanosensing during disease. Because hemodynamic disruptions are hallmarks of cardiovascular disease, studying endothelial mechanosensing holds great promise for advancing our understanding of vascular physiology and pathophysiology.
Keywords: endothelial cells, shear stress, mechanosensing, mechanosensors, ion channels, mechanotransduction, Piezo1, G protein–coupled receptors
1. INTRODUCTION
1.1. Making Sense of Senses
The way we sense external cues, known as exteroception, has intrigued humankind for millennia. Exteroception is sculpted by our senses. The last decades have intriguingly witnessed major developments in our understanding of how we see, hear, smell, and taste. While much remained unknown about our sense of touch, the discoveries of fundamental mechanosensitive proteins in recent years have advanced our understanding of how mechanical forces are converted into cellular electrical signals (1). Venturing beyond mechanical exteroception—sensitivity to forces in the external environment—the mammalian body intriguingly experiences inherent internal forces. Sensing these internal forces, known as mechanical interoception, is facilitated by mechanosensors that sense forces so that mechanical signals are translated into downstream signaling mechanisms that affect cellular and tissue function.
1.2. Cardiovascular Mechanosensation
Under physiological conditions, a myriad of mechanical forces is exerted on different cell types within the body. The mammalian heart, for example, starts beating at the embryonic stage. Blood flow in the developing vasculature represents one of the earliest forces experienced before birth. Throughout life, the vascular system is exposed to pressure forces due to intravascular blood pressure, as well as frictional forces due to blood flow–mediated shear stress (2) (Figure 1). Depending on their anatomical context and physiological environment, some vascular cells are more likely to sense and respond to certain mechanical forces. The arterial wall is composed of smooth muscle cells (SMCs) that circumferentially surround the blood vessel wall and endothelial cells (ECs) that line the lumen of all vessels. SMCs are predominantly sensitive to intravascular pressure, and ECs are most affected by shear stress (2). SMCs and ECs are equipped with mechanosensors that sense forces (mechanosensing) so that these stimuli are translated into cellular responses (mechanotransduction).
Figure 1:
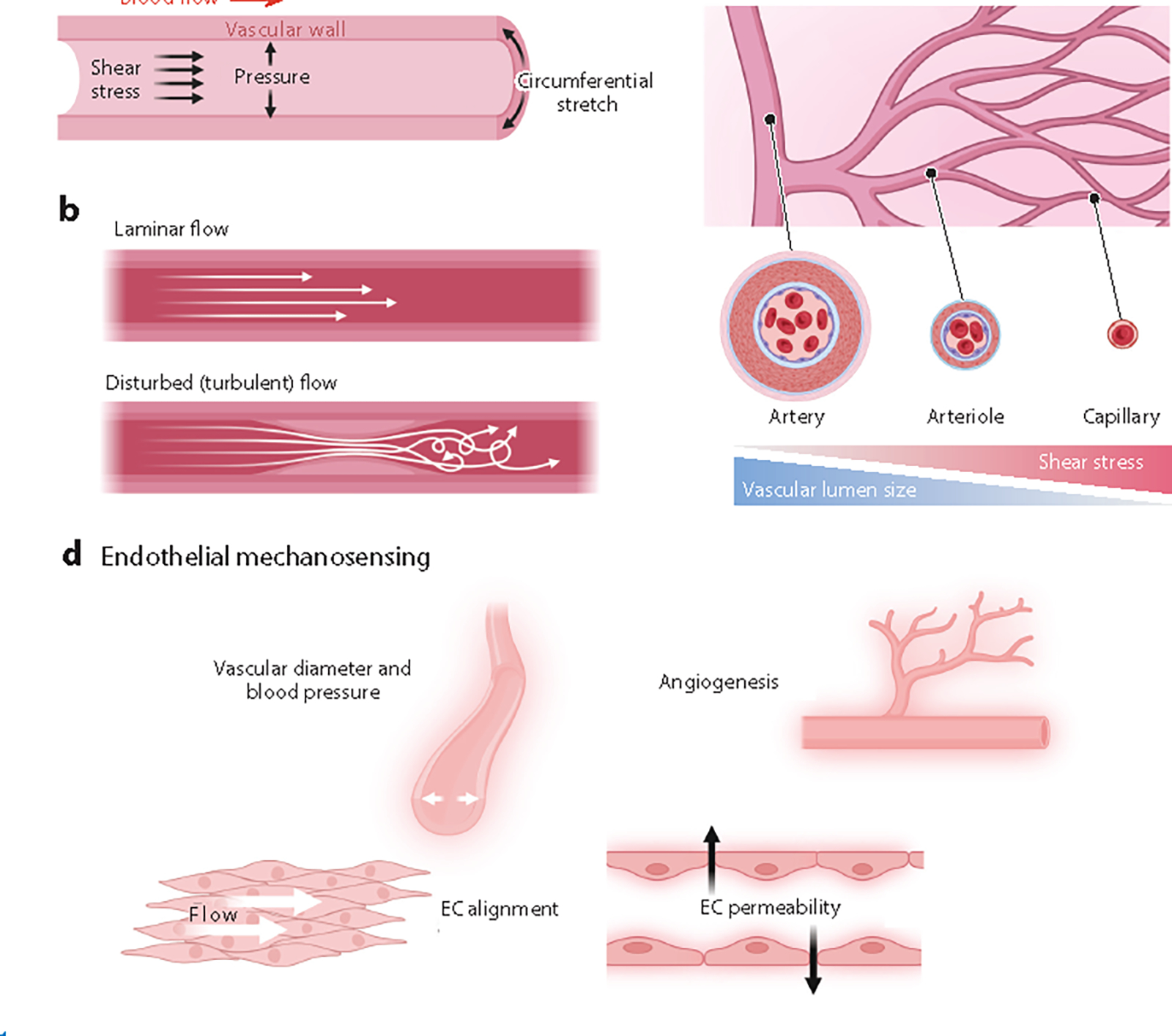
Hemodynamic forces and endothelial mechanosensing. (a) Blood movement in the vasculature generates hemodynamic forces. Shear stress is triggered by blood flow and represents the frictional force parallel to the vascular wall. Hydrostatic pressure is the perpendicular force exerted on the vascular wall, and circumferential stretch reflects the vessel wall stretching in the circumferential direction. (b) Blood flow patterns can be either laminar or disturbed, and these patterns are linked to atheroprotection or atherogenesis, respectively. (c) As blood flows from larger arteries to smaller arterioles and then to capillaries, the vascular lumen size decreases and the shear stress onto endothelial cells (ECs) increases. (d) Key physiological responses to endothelial mechanosensing. Figure adapted from images created with BioRender.com.
Cardiovascular diseases are the leading cause of death worldwide. Hallmarks defining cardiovascular disease include disruptions in hemodynamic forces. For example, intravascular pressure is elevated during hypertension, and blood flow is disturbed during atherosclerosis. These disruptions profoundly impact mechanosensing and therefore mechanotransduction and vascular function (3). Given the huge implications of endothelial function in regulating cardiovascular homeostasis (Figure 1), this article focuses on how ECs respond to shear stress. The main aim of this article is not to provide a systematic review. Rather, we highlight the indispensability of vascular endothelial mechanosensing for cardiovascular health and how the ability of ECs to sense mechanical cues maintains blood flow. We also discuss endothelial dysfunction in pathological contexts during which hemodynamic forces are disrupted. The goal is that this understanding will establish a foundation for identifying novel targets for therapeutic interventions.
2. THE ENDOTHELIUM AND FORCES
2.1. Endothelial Cells
The endothelium is fundamental for vascular health. ECs maintain selective permeability that regulates the transfer of ions, nutrients, gases, immune cells, and other substances. The luminal endothelial membrane faces the vascular lumen and is directly exposed to blood constituents. The basolateral surface, on the other hand, is typically separated from surrounding tissues by a self-secreted glycoprotein basement membrane (4). Noteworthy, significant heterogeneity between ECs in different vascular beds has been observed (5). EC functions vary in different environments, and the endothelial morphology can be altered to suit function. For instance, cardiac ECs are characterized by high metabolic rates and minimal transendothelial transport (6). Blood–brain barrier ECs are equipped with specialized tight junctions and selective barriers. In distinction, ECs in the liver vasculature are fenestrated (7). Not only are different vessels lined with unique ECs, but they are also subjected to unique mechanical stresses. As an example, vascular ECs in the heart and skeletal muscles experience muscle contraction–associated forces that are absent in the brain.
2.2. Hemodynamic Forces
The mechanical forces generated from blood movement in the cardiovascular system are collectively known as hemodynamic forces. These forces include pressure and shear stress. Pressure develops when fluid exerts hydrostatic forces perpendicular to the vessel wall (8). Shear stress, on the other hand, is the tangential frictional force exerted by flow over the endothelial surface of the blood vessel (Figure 1). Intravascular pressure depends on cardiac output and vessel compliance and can be described with the bulk flow law (9), an equation analogous to Ohm’s law in the context of fluid mechanics, where the pressure gradient is equal to flow multiplied by resistance :
| 1. |
Pressure increases when blood flow increases (more cardiac output) or when vascular resistance to flow increases during vasoconstriction. Other factors such as viscosity and vascular diameter affect vascular resistance, which in turn affects blood flow (9–11). This is described by the formula for the Poiseuille resistance:
| 2. |
Equation 2 relates to the vascular resistance to flow , where represents the viscosity of blood, denotes the length of the vessel, and is the radius of the blood vessel. This formula indicates that resistance is inversely proportional to the fourth power of the radius , highlighting the profound impact of even a small change in vascular diameter on vascular resistance. Another important aspect of hemodynamics is the pattern of fluid flow. Factors affecting flow patterns are tied with the Reynolds number :
| 3. |
In this equation, is fluid density, is velocity, is vascular diameter, and is viscosity (9). dictates whether blood flow is laminar or turbulent (Figure 1). Laminar flow (low ) is linear, where friction decreases blood velocity closest to the wall. Conversely, turbulent flow (high ) represents a disruption in the laminar flow and can result from differences in the fluid’s speed and direction.
As mentioned earlier, the primary force that the endothelium is exposed to is shear stress. The magnitude of shear stress at the vascular wall can be expressed as a function of flow using Poiseuille’s law:
| 4. |
where shear stress magnitude is proportional to blood viscosity and blood flow and inversely proportional to the third power of the internal vascular radius (10). To summarize, several hemodynamic forces are generated within the cardiovascular system; shear stress is dictated by blood flow and is the force with the most influence on ECs (Figure 1).
2.3. Sensing Shear Stress
The endothelium plays a pivotal role in vascular physiology through mechanisms that ultimately cause vasodilation. These mechanisms canonically involve potent vasodilator nitric oxide (NO) release and/or membrane potential hyperpolarization. Synthesized by and released from ECs, NO diffuses to neighboring vascular SMCs and activates soluble guanylyl cyclase, resulting in cyclic guanosine monophosphate (cGMP) production and signaling. The latter signaling activates protein kinase G, which phosphorylates several proteins, leading to decreased SMC calcium ion (Ca2+) concentration levels, SMC relaxation and, ultimately, vasodilation. ECs additionally transduce membrane potential (Vm) hyperpolarization through the activation of K+ channels. Given that ECs are coupled electrically to SMCs, hyperpolarizing signals are transmitted to SMCs, leading to Ca2+ channel deactivation, a decrease in SMC Ca2+ levels and, eventually, vasodilation. Therefore, ECs serve as a central hub orchestrating the modulation of vascular tone and blood vessel diameter.
ECs are constantly exposed to changes in shear stress and blood pressure, which impact their function. In response to these mechanical forces, blood vessels constrict or dilate to maintain proper blood flow and tissue perfusion (12, 13). Vasodilation causes shear stress to return to baseline levels, demonstrating the crucial role of the endothelium in not only mechanosensing, but also maintaining shear stress magnitude. This feedback mechanism is especially important in smaller blood vessels such as capillaries. Because red blood cells exhibit non-Newtonian features when flowing through capillaries (14), blood viscosity increases during vasoconstriction and therefore enhances shear stress.
Endothelial dysfunction and presumably defective mechanosensation are significant contributors to vascular diseases involving hemodynamic disruptions, including hypertension and atherosclerosis (15). On the mechanistic level, one can speculate that defective EC mechanosensing leads to defects in vascular diameter control, reduced vasodilator bioavailability, and elevations in reactive oxygen species and proinflammatory factors (15). Re-envisioning cardiovascular disease through the defective-mechanosensing lens is an area for future research.
The strategic location of ECs as the innermost layer of blood vessels allows them to sense hemodynamic forces through subcellular compartments or protein complexes collectively called mechanosensors (Figure 2). Examples of mechanosensors include ion channels, G protein–coupled receptors (GPCRs), caveolae, tyrosine kinase receptors, glycocalyx, primary cilia, junctional complexes, integrins, and the cytoskeleton (16, 17). Following mechanosensing, downstream signaling (mechanotransduction) allows ECs to change their morphology, gene expression, and cellular behavior and to release vasodilators and inflammatory mediators that facilitate communication between ECs and SMCs (17, 18). Mechanotransduction mechanisms ultimately affect vascular function and blood flow control (Figure 1). In the next section, we cover some key endothelial mechanosensors and the consequences of their activation.
Figure 2:

Mechanosensors in endothelial cells. Endothelial cells lining the inside of blood vessels are constantly exposed to vascular mechanical forces. Mechanosensing these forces is feasible through various mechanosensors. Examples of these mechanosensors include ion channels (Piezo1, TRPV4, Kir2.1, ENaC, TREK-1), receptors (GPCR, receptor tyrosine kinase), and membrane structures (caveolae, PECAM-1, PlexinD1, glycocalyx, cadherin, integrin, cilia). Abbreviations: ENaC, epithelial sodium channel; GPCR, G protein–coupled receptor; Kir2.1, inwardly rectifying K+ channel 2.1; PECAM-1, platelet endothelial cell adhesion molecule-1; TRPV4, transient receptor potential vanilloid 4 channel. Figure adapted from images created with BioRender.com.
3. MECHANOSENSORS
Since the discovery that ECs sense shear stress (19), studies have focused on understanding the mechanisms behind mechanosensing. It is evident that ECs are equipped with dedicated mechanosensors that detect and respond to stimuli exerted by blood flow (Figure 2). Once triggered, mechanosensors initiate signaling pathways in ECs or adjacent cells. In this section, we cover some categories of endothelial mechanosensors: ion channels, GPCRs, and membrane structures. Additional mechanosensing mechanisms, including but not limited to TRPC6 (transient receptor potential canonical 6), TRPM4 (transient receptor potential melastatin 4), ASIC (acid-sensing ion channel), TRPV1 (transient receptor potential vanilloid 1), TRPP1/TRPP2 (transient receptor potential polycystic 1 and 2), P2X (ionotropic purinergic receptor), Ca2+-activated Cl− channels, TMEM63 (transmembrane 63), primary cilia, and tyrosine kinases, have been reviewed in recent articles (20, 21).
3.1. Ion Channels
Some classes of ion channels are ligand-gated or voltage-activated, and some others are mechanically activated. Mechanosensitive ion channels are directly activated by physiological physical forces. Due to the vital location of ECs, how the endothelium senses and responds to blood flow and associated mechanical forces has intrigued scientists for decades. The first evidence for an endothelial mechanosensitive channel that mediates mechanically induced cation influx dates to over three decades ago (22). This pioneering discovery was followed shortly by the discovery of shear stress–activated K+ currents in ECs (23).
The activity of endothelial mechanosensitive channels changes directly in response to shear stress, whereas the activity of other channels could be indirectly altered by forces. Several criteria need to be met for an ion channel to be considered a true mechanosensor (24, 25). First, overexpression of the channel in a null cell must confer mechanosensitivity, and deleting or inhibiting the channel must abolish the mechanoresponse. Second, message and protein expression of the channel must be evident in the mechanosensitive cell—the EC in this context. Third, the purported mechanosensitive channel must have a direct role in ion permeation in response to force. Fourth, channel expression must be indispensable for mechanosensing, rather than being a downstream target of another mechanosensing mechanism. Last, site-directed mutagenesis of the ion channel must alter the mechanoresponse.
Here, we cover Piezo1, transient receptor potential vanilloid 4 channel (TRPV4), epithelial sodium channel (ENaC), inwardly rectifying K+ channel 2.1 (Kir2.1), and TREK-1 channels, where the evidence of physiological involvement in endothelial mechanosensing and mechanotransduction is more substantiated. Additional endothelial ion channels purported to be mechanosensors are reviewed in depth elsewhere (21).
3.1.1. Piezo1.
Piezo1 is a bona fide mechanosensitive ion channel that fulfils the criteria of a true mechanosensor (24, 25). The functional expression of the Piezo1 channel in ECs is well established (17, 26–28). Inspired by the embryonic lethality of genetic deletion of endothelial Piezo1, the Beech and the Patapoutian laboratories (17, 28) were the first to demonstrate that Piezo1 is a mechanosensor involved in EC alignment during vascular development. Piezo1 is a nonselective cation channel that is slightly more permeable to Ca2+ than to Na+. Electrophysiological and imaging studies in ECs from different vascular beds revealed that Piezo1 activation leads to cationic currents and intracellular Ca2+ signals (17, 26, 27). Piezo1 activation can be achieved experimentally through mechanical (e.g., shear stress) or chemical activation (e.g., Yoda1, a selective Piezo1 activator) (25). The effects of Yoda1 in ECs mimicked shear stress–induced responses and were attenuated by mechano-gated channel blockers (26, 27). A limitation in Piezo1 research, however, is the lack of specific and selective inhibitors.
Endothelial Ca2+ signaling plays an important role during vascular development. Earlier efforts following the discovery of Piezo1 in ECs were directed at understanding the impact of Piezo1-mediated Ca2+ transients on vascular development. The activation of Ca2+-dependent enzymes is vital for the spatial organization and alignment of ECs during development. Li et al. (17) found that the Ca2+-activated protease calpain, which cleaves focal adhesion proteins required for cellular orientation, is upregulated upon Piezo1 activation and is important during vascular development. Ranade et al. (28) found that Piezo1 activation is critical for stress fiber orientation and EC alignment. Piezo1 additionally activates several Ca2+-dependent endothelial metalloproteinases involved in angiogenesis (29). In brain endothelial tip cells, Piezo1-mediated Ca2+ events modulate pathfinding and cerebrovascular patterning (30). Collectively, studies have demonstrated that Piezo1-mediated Ca2+ signaling is essential for vascular development, patterning, and angiogenesis.
Ca2+ transients trigger downstream signaling and facilitate Ca2+-dependent endothelial processes that influence vascular diameter control. A central mechanism activated by Ca2+ signaling is the synthesis of the potent vasodilator NO. Studies have demonstrated that flow-mediated vasodilation is attributed to Piezo1-mediated Ca2+ influx, Ca2+-dependent activation of endothelial NO synthase (eNOS), NO generation and, ultimately, SMC relaxation (Figure 3). Further, loss of NO-mediated vasodilation in EC-specific Piezo1 knockout mice has been proposed to cause hypertension (27). Other studies, however, found no effect of genetic Piezo1 deletion on blood pressure (31). Nonetheless, Piezo1-mediated vasodilation in different vascular beds is mediated through NO production (27, 32–34). Endothelial Piezo1 has been shown to influence GPCR activity. Laminar flow activates Piezo1, enabling the release of adenosine triphosphate (ATP), which then activates purinergic Gq/11-PCRs to ultimately enhance NO production (35). GqPCR activity is notably an important regulator of ion channels in ECs (36, 37) (Figure 3), but whether GqPCR signaling affects endothelial Piezo1 activity is unclear.
Figure 3:

Flexibility, diversity, and cross talk between endothelial mechanosensors and mechanotransducers. (a) Example flow chart showing how different mechanosensors ultimately trigger vasodilation. Mechanotransduction occurs via one or both major pathways: NO production and release and/or EC Vm hyperpolarization. (b) Exemplary cross talk between EC mechanosensors downstream of mechano-GqPCR activation. Mechanical activation of GqPCR evokes vasodilation via NO release or hyperpolarization. GqPCR activation can directly activate PI3K/Akt, leading to eNOS activation and NO synthesis. On the other hand, GqPCR canonically facilitates PLC activity and the hydrolysis of PIP2 into DAG and IP3. The three metabolites (PIP2, DAG, and IP3) modulate the activity of mechanosensitive ion channels or their downstream effectors. PIP2 depletion inhibits Kir2.1 and TREK-1 channels but disinhibits TRPV4 activity. DAG/PKC signaling facilitates TRPV4 activity evoking Ca2+ transients that facilitate eNOS activity and/or IK/SK Ca2+-activated K+ channels; both pathways ultimately lead to vasodilation. IP3/IP3R signaling facilitates Ca2+ release from the endoplasmic reticulum, and these Ca2+ transients enhance eNOS and IK/SK activities. Abbreviations: Akt, protein kinase B; B2R, bradykinin 2 receptor; Ca2+, calcium ion; DAG, diacylglycerol; EC, endothelial cell; ENaC, epithelial sodium channel; eNOS, endothelial NO synthase; GPR68, G protein-coupled receptor 68; GqPCR, Gq protein–coupled receptor; H1R, histamine H1 receptor; IK/SK, intermediate-conductance/small-conductance; IP3, inositol 1,4,5-trisphosphate; IP3R, IP3 receptor; Kir2.1, inwardly rectifying K+ channel 2.1; NO, nitric oxide; PECAM-1, platelet endothelial cell adhesion molecule-1; PI3K, phosphatidylinositol 3-kinase; PIP2, phosphatidylinositol 4,5-bisphosphate; PKC, protein kinase C; PLC, phospholipase C; S1PR1, sphingosine-1-phosphate receptor 1; TRPV4, transient receptor potential vanilloid 4 channel; VE-cadherin, vascular endothelial-cadherin; VEGFR2, vascular endothelial growth factor receptor 2; Vm, membrane potential. Figure adapted from images created with BioRender.com.
The cation influx associated with Piezo1 activation depolarizes cells, including ECs (31, 38). ECs are structurally and electrically connected to SMCs by myo-endothelial projections and gap junctions (39, 40). This electrical coupling facilitates the communication between ECs and adjacent SMCs and presumably allows the spread of Piezo1-mediated electrical signals from ECs to SMCs. There is evidence suggesting that Piezo1-induced EC depolarization spreads to SMCs and activates voltage-gated Ca2+ channels, causing mesenteric artery vasoconstriction (31), which explains the observations of flow-mediated vasoconstriction instead of vasodilation and the lack of this response in EC Piezo1 knockout mice. These findings, however, contrast with others demonstrating flow-induced vasodilation that was NO dependent and attenuated in EC Piezo1 knockout mice (27). These discrepancies await further exploration and could be partly explained by distinctive expression patterns in different artery branches or differences in mouse models. Additionally, it is unclear whether Piezo1 stimulates Ca2+-activated ion channels in ECs. If so, Piezo1 activity could hyperpolarize or depolarize EC Vm by activating Ca2+-activated K+ or Cl− channels, respectively—a hypothesis that awaits experimental testing. In summary, Piezo1 is a key mechanosensing mechanism in ECs. In a later section of this review (Section 4.3), we discuss crucial gaps in our understanding of the physiological roles of Piezo1, and we speculate that Piezo1 function is dictated by the vascular bed type (peripheral versus central) and size (macro- versus microcirculation).
3.1.2. TRPV4.
TRPV4 was first discovered in the endothelium over two decades ago (41). TRPV4 has been identified since then in ECs from different vascular beds, such as mesenteric resistance arteries, pulmonary arteries, carotid arteries, skeletal muscle arterioles, and brain capillaries (42–45). TRPV4 is a nonselective cation channel that is more permeable to Ca2+ than Na+ (46, 47). The endothelial TRPV4 channel contributes to several vascular functions including vascular tone regulation, EC orientation, angiogenesis, and permeability (45, 48).
Studies showed that the TRPV4 channel is sensitive to forces and that EC TRPV4 is crucial for shear stress detection (42–44). Whether TRPV4 is a true mechanosensor has, however, been controversial. Direct channel activation by mechanical force has not been robustly demonstrated, suggesting that TRPV4 may not be a bona fide mechanosensor (47, 49–51). Additionally, the fact that TRPV4 can be activated physiologically by endogenous substances released in response to shear stress [e.g., arachidonic acid (AA) and epoxyeicosatrienoic acids (EETs)] could explain how TRPV4 activity increases with force (42, 52, 53). Along the same lines, recent observations in cultured ECs demonstrated that Piezo1 activation triggers the production of AA metabolites, ultimately enhancing TRPV4 activity (54). These findings suggest that Piezo1 is the mechanosensor and TRPV4 is functionally coupled downstream to amplify and sustain mechanically induced Ca2+ influx.
Endothelial TRPV4 activation leads to vasodilation through NO release and/or through altering EC Vm. These two mechanisms are not necessarily mutually exclusive and could act synergistically (Figure 3). Highly localized Ca2+ events upon TRPV4 channel activation, known as TRPV4 sparklets, are coupled to (a) eNOS activation, NO production, and vasodilation or (b) the activation of Ca2+-activated K+ channels and downstream EC hyperpolarization (42–44, 55,56)(Figure 3). Genetic ablation of TRPV4 leads to blunted endothelial Ca2+ responses and reduced NO release (57). Amplification of TRPV4-mediated Ca2+ signaling occurs through inositol 1,4,5-trisphosphate receptor (IP3R) sensitization (58).
TRPV4 channels interact with a plethora of proteins and molecules, some of which are directly affected by mechanical forces. Like several other TRP channels, TRPV4 is activated downstream of GqPCR stimulation. Endothelial GqPCR activation enhances phospholipase C (PLC) activity and the hydrolysis of phosphatidylinositol 4,5-bisphosphate (PIP2) into IP3—that causes Ca2+ release from intracellular stores—and diacylglycerol (DAG), which activates protein kinase C (PKC). EC GqPCR signaling activates TRPV4 channels in PKC- or PIP2-dependent manners (36,59), and IP3/IP3R signaling amplifies TRPV4 activity (58). As some GPCRs are mechanosensitive (60) (discussed below), their activation by forces could enhance EC TRPV4 activity, leading to the perception that TRPV4s are essential mechanosensors (Figure 3). It is noteworthy that GqPCR signaling and downstream ion channel regulation (e.g., TRPV4 and Piezo1) could be a key intersection point between different mechanosensing mechanisms. The scaffolding protein caveolin-1 (discussed below) interacts with TRPV4 and colocalizes with gap junction proteins to facilitate electrical coupling. Caveolin-1 is indeed required for endothelial Ca2+ influx and subsequent vasodilation (61, 62). Altogether, evidence supports that caveolae are crucial microdomains involved in TRPV4 activity and vasodilation. Other TRP channels are crucial for EC signaling besides mechanosensing and flow-mediated vasodilation. Cerebral endothelial transient receptor potential ankyrin 1 channel (TRPA1), for example, is involved in neurovascular coupling, and TRPA1 activation by reactive oxygen species leads to vasodilation (63, 64).
3.1.3. Kir2.1.
One of the earliest attempts to identify EC mechanosensors discovered a K+ channel sensitive to forces (23). Using cultured aortic ECs, the authors identified a Kir channel that was shear stress activated. They proposed that this channel contributes to flow-mediated hyperpolarization and vascular tone regulation (23). This work kick-started the search for the role of Kir channels in mechanosensation and blood flow control. The Kir2.1 channel is expressed in ECs from different vascular beds (65–67), sensitive to extracellular K+ concentration, and activated by hyperpolarization (68). Flow activates Kir2.1, in a shear stress magnitude–and oscillation frequency–dependent manner, causing K+ efflux and hyperpolarization. Additional to Vm modulation, Kir2.1 activity has been linked to NO production (66, 69, 70) (Figure 3). Ahn et al. (66) suggested that EC Kir2.1 is crucial for flow-induced phosphorylation and activation of eNOS and Akt, as well as subsequent NO generation, presumably in a Ca2+-independent manner.
Shear stress increases Kir2.1 activity, but the channel might not be the primary mechanosensor. Using cell-attached electrophysiology, shear activated Kir channels, even though the cell-attached membrane were not always directly exposed to shear stress (71). Upstream mechanosensing mechanisms are likely involved in the response of Kir2.1 to shear stress. One speculation is that shear stress alters plasma membrane lipids and fluidity that in turn affect Kir2.1 activity. Kir2.1 is indeed a key regulatory target of phosphoinositides (e.g., PIP2) and cholesterol (37, 72).
3.1.4. ENaC.
ENaC was originally thought, as the name implies, to be exclusively expressed in epithelial cells. Accumulating evidence, however, demonstrated that ENaC is functionally expressed in ECs of various vascular tissues (73–75). There is evidence that the endothelial ENaC channel is directly activated by laminar flow. Mechanosensitivity of ENaC depends on the interaction between the extracellular matrix and α-ENaC extracellular glycosylated asparagines, an interaction that underlies the responsiveness of endothelial ENaC to shear stress (74, 76). The endothelial glycocalyx is also involved in the ability of endothelial ENaC to facilitate flow-mediated vasodilation, and EC ENaC deletion eliminates shear stress–mediated relaxation (75, 77, 78). Several modulators regulate endothelial ENaC activation in response to shear stress; some of these also affect other purported mechanosensors. EETs, for example, activate TRPV4 but inhibit ENaC (74). These studies signify a role for EC ENaC in mechanosensing and mechanotransduction.
There is evidence that an increase in ENaC channel density and/or activity [ENaC gain-of-function (GOF)] stiffens ECs and causes vasoconstriction. ENaC GOF due to increased channel expression has been observed during hypertension (79, 80). The mechanisms by which ENaC GOF affects endothelial function and shear stress–mediated vasodilation involve reduced NO production. The loss of NO results from enhanced Na+ influx, which attenuates the driving force for the uptake of l-arginine (NO precursor) by cationic amino acid transporters. Alternatively, NO loss can occur via the negative modulation of phosphoinositide 3-kinase (PI3K)/Akt-pathway that is crucial for NO synthesis (81,82). To summarize, while normal endothelial ENaC facilitates shear stress–mediated vasodilation, enhanced ENaC activity is linked to endothelial dysfunction. The divergent impact of ENaC on vascular function could be explained by vascular bed heterogeneity and whether ENaC activity levels are physiological or pathologically enhanced (80, 83). In summary, ENaC involvement in endothelial mechanosensing is quite complex and requires additional investigations.
3.1.5. TREK-1.
Members of the two-pore domain K+ (K2P) channel superfamily are proposed endothelial mechanosensors. K2P channels are expressed in different vascular beds where they regulate vascular function (84). TRAAK, TREK-1, and TREK-2 are members of the K2P channel family, and the TREK-1 channel has been extensively studied in ECs (85, 86). TREK-1 responds directly to both positive and negative pressure in lipid bilayers, and the channel’s mechanosensitivity is preserved in different electrophysiological configurations (85, 87). TREK-1 is directly gated by membrane tension; plasma membrane deformation relieves lipid block of ion conduction and therefore enhances channel activity, leading to hyperpolarizing currents (85, 88–90). In addition to altering EC Vm, TREK-1 is implicated in endothelium-dependent, NO-mediated vasodilation (91) (Figure 3).
In line with being mechanosensitive, the TREK-1 channel is sensitive to osmotic pressure and activated by ischemia-associated swelling. The cerebrovascular TREK-1 channel is, therefore, protective during ischemia via hyperpolarizing ECs and vasodilation (87, 92, 93). One speculation is that TREK-1-mediated hyperpolarization synergizes with other hyperpolarizing channels or mechanosensors (e.g., Kir2.1) or antagonizes depolarizing channels (e.g., Piezo1 or TRPV4). The TREK-1 channel is modulated by GPCRs, some of which are mechanosensors (85, 86, 88, 93–95) (Figure 3). PIP2 enhances TREK-1 activity, and cyclic adenosine monophosphate/protein kinase A (cAMP/PKA) suppresses the channel. TREK-1 activity is therefore inhibited by Gs-PCR (increased inhibitory PKA) and Gq-PCR (decreased stimulatory PIP2) but not by Gi-PCR (decreased PKA) signaling (94, 95). Collectively, endothelial TREK-1’s role in mechanosensing is likely affected by the other mechanosensitive GPCRs present.
3.1.6. Perspectives on ion channels as mechanosensors.
We covered candidate endothelial mechanosensitive ion channels, and the evidence among these is most solid for Piezo1 to be a true mechanosensor. Piezo1 is expressed in almost all vascular beds studied to date, but the functions of endothelial Piezo1 remain understudied. This reflects the relatively recent discovery of Piezo1 and the lack of specific/selective inhibitors. As discussed above, different mechanosensors work together, and Piezo1 could be a missing link in these mechanisms. We expect that the near future will witness advances in our understanding of mechanosensors in general and of Piezo1 as a dominant channel mechanosensor in ECs.
3.2. G Protein–Coupled Receptors
Canonical activation of a GPCR is initiated when an extracellular ligand binds to the receptor, triggering second messenger cascades involving Ca2+ signals, cyclic nucleotides, or β-arrestin. Some GPCRs, however, mediate mechanochemical signal transduction independent of ligand binding (96, 97). Mechanisms underlying GPCR mechanosensitivity are not fully understood but likely involve changes in membrane fluidity that allow GPCRs to adopt an active conformation even in the absence of a receptor agonist (98–100). It has been additionally shown that helix 8, a conserved helical motif located immediately after transmembrane segment 7, endows GPCRs with mechanosensitivity. Structural and functional analyses revealed that mechanical stimulation elongates helix 8 and therefore activates GPCR (101).
3.2.1. GPR68.
G protein–coupled receptor 68 (GPR68) is an endothelial Gq/11-coupled receptor that mediates flow-induced vasodilation by enhancing NO production (Figure 3). Deletion of GPR68 impaired flow-mediated dilation of mesenteric arteries but did not affect blood pressure regulation (60). GPR68 possesses a helix 8 structure, which could explain how the receptor responds to shear stress in ECs (101). Contradicting evidence using cultured cells, however, showed that flow-induced GPR68 activation persisted despite the deletion of helix 8 or the pharmacological inhibition of G protein signaling (102). The latter is consistent with findings that shear stress induces Gq/11 activation independently of GPCR activation in cultured ECs (103).
3.2.2. Bradykinin 2 receptor.
Another endothelial mechanosensor is the Gq/11-coupled bradykinin 2 (B2) receptors (97). Shear stress, hypo-osmotic stimulation, or a change in the plasma membrane fluidity activates the B2 receptor, leading to an increase in intracellular [Ca2+]. Endothelial B2 receptor–mediated Ca2+ signaling leads to vasodilation by increasing NO production or promoting Ca2+-dependent hyperpolarization (97, 104, 105) (Figure 3). Further, the endothelial B2 receptor enhances the production of AA, which upregulates Ca2+-influx via TRPV4 (52), suggesting potential cross talk between the B2 receptor and other candidate mechanosensors.
3.2.3. Sphingosine-1-phosphate receptor 1 and histamine H1 receptor.
It has been shown that shear stress activates sphingosine-1-phosphate receptor 1 (S1PR1) and histamine H1 receptor (H1R) in ECs. S1PR1 activation initiates NO production, leading to vasodilation. S1PR1 is also crucial for vascular development and blood pressure homeostasis (106, 107). Studies collectively suggest that S1PR1 plays a key role in an endothelial mechanosensory pathway, more like a mechanotransducer than a sensor. H1R possesses helix 8 and was therefore reported to be an endothelial mechanosensing GPCR capable of facilitating NO signaling and vasodilation (101).
3.2.4. Perspectives on mechanosensitive GPCRs.
Endothelial GPCRs functioning as purported mechano-GPCRs include GPR68, B2 receptor, S1PR1, and H1R. Among these, the mechanosensing evidence suggests that GPR68 could play the dominant role. However, expression at the messenger RNA (mRNA) level is most evident for S1PR1 and rather low for other candidates (108). Further, some purported mechanosensitive Gq/11-coupled receptors lack evidence of direct activation by mechanical forces when expressed in heterologous expression systems, corroborating the idea that GPCRs are indirectly involved in mechanosensation (109). Downstream effects of GPCR signaling overlap with and modulate other mechanosensitive ion channels; the complement of EC receptors could therefore coordinate and regulate the activity of mechanosensitive ion channels (110) (Figure 3). This suggests that mechanosensors are functionally connected and cooperative and could therefore compensate for one another.
3.3. Membrane Structures as Mechanosensors
There are multiple embedded protein microdomains in the EC plasma membrane. These microdomains confer unique mechanical properties to different regions and therefore contribute to EC heterogeneity. When ECs are exposed to shear stress forces, plasma membrane regions with higher mechanical resistance concentrate forces (e.g., focal adhesion sites). The latter leads to the creation of areas of high strain or stiffness that are capable of mechanosensing. Additional membrane structures that are candidate mechanosensors include the glycocalyx and caveolae (Figure 2).
3.3.1. Glycocalyx.
The endothelial glycocalyx is a gel-like layer that lines the luminal surface of all ECs. Composed of proteoglycans, glycosaminoglycans, glycoproteins, and adsorbed plasma proteins (111), it was initially considered to be solely a structural barrier. Recent research, however, suggests that the glycocalyx serves as an important mechanosensing function (112–114). Functioning as the interface between flowing blood and ECs, the glycocalyx converts mechanical stimuli into intracellular signals. The glycocalyx interacts with mechanosensitive ion channels like ENaC (76) and contributes to shear-induced signaling pathways involving EC junctional proteins [e.g., platelet endothelial cell adhesion molecule-1 (PECAM-1)] (115). The glycocalyx is additionally a key player in shear stress–mediated proteoglycan distribution and caveolin-1 expression (116, 117).
There is an established link between glycocalyx and NO production. Degrading specific glycocalyx components in bovine aortic ECs compromised shear-induced, but not GPCR-mediated, NO synthesis, highlighting unique mechanosensing and transduction pathways (118). Along the same lines, depletion of glycocalyx components, such as heparan sulfate, hyaluronic acid, and sialic acids, blocks shear stress–induced NO production but does not affect shear stress–induced prostaglandin I2 production or agonist-induced NO production (119–123). Interestingly, some studies demonstrated that an intact glycocalyx might not be necessary for conduit artery endothelium-dependent vasodilation in vivo, suggesting tissue-specific differences in glycocalyx mechanotransduction (124).
Pathological conditions characterized by atheroprone blood flow, such as atherosclerosis, disrupt the glycocalyx (125) and impair endothelial mechanosensing capabilities (116). Loss or shedding of the glycocalyx diminishes the endothelial response to mechanical forces, leading to endothelial dysfunction, increased vascular permeability, inflammation, disrupted cytoskeletal remodeling, and disrupted EC alignment (125–127). To summarize, the glycocalyx is a complex and dynamic structure that functions as an endothelial mechanosensing system. The roles of the glycocalyx in regulating EC functions in response to flow, particularly in vivo, are not fully understood and warrant further research.
3.3.2. PECAM-1 and PlexinD1.
PECAM-1, also known as CD31, is a cell-adhesion molecule and an EC mechanosensor. PECAM-1 acts cooperatively with different junctional proteins. A junctional mechanosensory complex composed of PECAM-1, vascular endothelial (VE) cadherin, and vascular endothelial growth factor receptor 2 (VEGFR2) is crucial for endothelial mechanosensing and alignment (128). Flow increases the force on PECAM-1, initiating a signaling cascade that leads to NO production (129, 130). When exposed to shear stress, the tyrosine residues at the cytoplasmic tail of PECAM-1 become phosphorylated, creating a docking site for signaling molecules such as PI3K (131–133). These signaling molecules modulate downstream effectors, including the activation of mitogen-activated protein kinase/extracellular signal-regulated kinase 1/2 (ERK) and PI3K/Akt, both of which activate downstream eNOS and NO production (129, 134, 135) (Figure 3). PECAM-1 facilitates eNOS activation, not only by enhancing its activity, but also through physical interactions at EC–EC junctions. An association between PECAM-1 and eNOS is crucial during abrupt changes in flow (136, 137). In addition to NO release, PECAM-1 is involved in the release of other vasodilatory factors (e.g., cyclooxygenase products) in response to shear stress (138). Collectively, PECAM-1 is indispensable in mediating shear stress–induced vasodilation.
While flow triggers increase tension on PECAM-1, data suggest that an upstream mechanosensor could initiate this signaling cascade (139), with implications for whether PECAM-1 itself is a mechanosensor. The fact that tyrosine kinase activation and subsequent phosphorylation of PECAM-1 are required for PECAM-1/eNOS/NO signal transduction may suggest that PECAM-1 is not the primary mechanosensing mechanism. Further, previous reports demonstrated that shear stress could stimulate the phosphorylation of endothelial ERK and Akt despite the absence of PECAM-1 (140). To summarize, a junctional complex of PECAM-1, VE-cadherin, and VEGFR2 plays a crucial role in EC mechanosensation and/or mechanotransduction.
PlexinD1 is a transmembrane receptor implicated in different cellular functions, such as migration and proliferation. Endothelial PlexinD1 has been identified as a mechanosensor acting upstream of the junctional PECAM-1/VEGFR2/VE-cadherin complex and integrins (141). PlexinD1 responds to shear stress by forming a multiprotein complex with VEGFR2 and facilitating the phosphorylation of Akt, ERK, and eNOS (Figure 3). In addition to accelerating eNOS activation, PlexinD1 modulates integrin-mediated cell adhesion, cytoskeleton rearrangements, and cell motility (142, 143). Because PlexinD1 acts upstream of other proposed mechanosensors, its activation is likely necessary and sufficient to mediate shear stress–mediated Akt/ERK responses (141).
3.3.3. Caveolae.
Caveolae are specialized membrane invaginations enriched with cholesterol, caveolin, and other signaling molecules. They are involved in several vascular cellular functions, and endothelial caveolae are likely involved in mechanosensing. Caveolae respond to mechanical perturbations by rapidly and reversibly disassembling, thereby releasing membrane areas and molecules as they flatten out. In response to tension, caveolae release free caveolins, the building block of caveolae, therefore acting as a membrane tension buffer (144, 145). Released caveolins interact with other signaling molecules to initiate mechanosensing signals.
Laminar shear stress increases caveolin-1 at the plasma membrane, alters ERK and Akt signaling patterns, and enhances eNOS accumulation and activation within the caveolae (146, 147) (Figure 3). Studies have further shown that caveolae interact with focal adhesion molecules and that this interaction is involved in flow-induced responses (148,149). Further, caveolae couple mechanical stress to integrin activation and therefore regulate the early steps of the mechanosensing response (150). These effects support the involvement of EC caveolae in mechanosensing.
Caveolae are key regulators of other proteins and signaling molecules, such as those involved in EC–Ca2+ signaling. Examples include candidate mechanosensors such as GPCRs and TRPV4, both of which are involved in endothelial-dependent, shear stress–induced vasodilation (61, 62, 151). It has been further demonstrated that caveolae are involved in flow-dependent ATP release from ECs, which in turn engages purinergic signaling to amplify Ca2+ signaling (152). Whether the endothelial Piezo1 channel is regulated by caveolin-1 and whether Piezo1 is preferentially localized in EC caveolae remain unknown. In conclusion, it is evident that caveolae play an important role in facilitating endothelial mechanosensing as well as several mechanotransduction processes.
3.3.4. Integrins.
Integrins connect the extracellular matrix and the actin cytoskeleton via multiprotein focal adhesion complexes. Although integrins are proposed to be sensitive to shear forces or cytoskeletal tension changes (153–155), they can be viewed as a point of integration for different shear-induced signaling cascades. PI3K, for example, is activated by shear stress through integrins, but it can also be activated downstream of the PECAM-1 pathway (132, 156). The response of integrins to shear stress is also modulated by other mechanosensors, such as PlexinD1 (141) and caveolin (150). Whether they are directly or indirectly sensitive to shear stress, integrins are involved in several mechanosensing pathways contributing to ECs’ capacity to sense and respond to shear stress.
4. ENDOTHELIAL MECHANOSENSING IN CONTEXT
Endothelial mechanosensing is undoubtedly indispensable for vascular function. Our understanding of mechanosensing mechanisms has profoundly grown over the past two decades. Important in this context is realizing that EC mechanosensing is quite far from being a one-size-fits-all system.
4.1. Diversity Is the Theme
Sensing hemodynamic forces entails several factors: (a) mechanosensors, some working in tandem and others as potential backup or opposing mechanisms; (b) signaling proteins, as regulators or amplifiers; and (c) multiprotein complexes that function as integrated units. Consequent endothelial mechanotransduction mechanisms involve rapid responses, such as acute changes in vascular diameter, or slow responses including changes in the EC cytoskeleton or permeability. Mechanosensing and mechanotransduction pathways are therefore complex and diverse. Functional responses vary between species, vascular beds, or locations within vascular networks. Mutations, upregulation, or downregulation of mechanosensors or their downstream transduction activities further feed into such functional diversity.
4.2. Mechanosensors Team Up Together
When considering mechanosensing and mechanotransduction, the physiology is complex and at times puzzling. The solution demands a holistic approach, which realizes that independent mechanosensors have common downstream effectors. Mechanosensing additionally transduces signaling through molecules that amplify or suppress the activity of other mechanosensors. The approach also recognizes the physical association or colocalization of mechanosensors and mechanotransducers. Here, we briefly discuss these considerations.
A common feature of mechanosensing is the ability of different mechanosensors to trigger the same effector. For example, the Akt-eNOS-NO pathway, NO generation, and subsequent vasodilation represent a prominent endothelial signaling cascade that is influenced by different mechanosensors including ion channels, receptors, or membrane structures (27, 66, 81, 105, 129, 141, 157) (Figure 3). This indicates a common and conserved function (NO synthesis) despite the diversity of the sensor. Among ion channel mechanosensors, arterial EC Piezo1 and Kir2.1 channels mediate shear stress–induced NO release, despite one channel being depolarizing (Piezo1:Ca2+/Na+ influx) and the other hyperpolarizing (Kir2.1:K+ efflux)(27,66). Other studies linked flow-mediated activation of the TRP polycystin (PKD2) channel to eNOS activation and K+ channel–mediated hyperpolarization (158). These studies and other similar observations highlight endothelial signaling flexibility. ECs utilize different toolkits to ensure robust signaling and mechanotransduction trajectories in response to hemodynamic forces.
Colocalization of endothelial membrane structures, such as caveolae, with mechanosensitive ion channels or GPCRs facilitates mechanosensing (61, 62, 159). Many of these molecular players are implicated in Akt-NO signaling, supporting the cross talk between caveolae and other mechanosensors. Additionally, eNOS itself associates with caveolin-1 (160), and hypercholesterolemia disrupts caveolin-1/eNOS heterocomplexes, leading to endothelial dysfunction (151, 161). These observations indicate that distinct mechanosensors and regulators colocalize, allowing them to serve endothelial function. Disruption of these complexes interferes with EC mechanosensing and could underlie pathophysiology. We anticipate that future efforts to address mechanosensor colocalization will greatly benefit from new technical advances such as superresolution microscopy.
Several mechanosensing mechanisms intersect with one another. Activation of an EC mechanosensor is often linked to changes in metabolite levels. Shear stress activates GqPCRs and consequently enhances the generation of DAG and IP3 and the depletion of PIP2. Changes in intracellular metabolite levels have significant impacts on mechanosensitive ion channels. DAG enhances arterial EC TRPV4 activity. PIP2 depletion enhances TRPV4 and TREK-1 activities and suppresses Kir2.1 activity. Piezo1 activation can enhance AA production, and AA activates TRPV4 and TREK-1 and their downstream responses (36, 37, 54, 59, 87, 94). The intersections between different metabolic pathways are therefore important elements in mechanosensing (Figure 3).
4.3. Mechanosensitive Piezo1 and Vascular Function
Endothelial Piezo1 channel expression and function were discovered less than a decade ago (17, 28). Yet, evidence implicating Piezo1 as a key vascular mechanosensor has evolved. Earlier, we discussed Piezo1 as a mechanosensing mechanism from a general standpoint (Section 3.1.1). Here, we speculate on diverse physiological contexts in which Piezo1 engages in vascular function and blood flow control.
4.3.1. Piezo1 and peripheral vascular function.
Genetic deletion of Piezo1 from the vascular endothelium is embryonically lethal. Research has explored Piezo1 function in the peripheral vasculature, demonstrating that the EC Piezo1 channel is involved in vascular development, angiogenesis, flow-mediated vasodilation, vascular permeability, leukocyte extravasation, and exercise capacity (17, 27–29, 31, 33, 162–164). There are reports that EC Piezo1 activation induces vasodilation, vasoconstriction, or no detectable effect on vascular diameter (27, 31–34). Some studies implicated EC Piezo1 in blood pressure regulation, but others reached conflicting conclusions that Piezo1 deletion has no overt effect on blood pressure. One study suggested a role for vascular SMC Piezo1 in arterial remodeling during hypertension but no effect on vascular tone or blood pressure (165). Contradictory findings could be explained by Piezo1 triggering unique downstream signaling events in different blood vessels or by differences in regulatory mechanisms. For instance, larger arteries are exposed to shear stress levels that differ from those imposed onto arteriolar and capillary ECs. Additionally, flow pulsatility—present in arteries but almost absent in capillaries—could be a major determinant of downstream signaling following EC Piezo1 activation. Elucidating the mechanisms governing peripheral EC Piezo1 function requires further research.
4.3.2. Piezo1 and cerebral blood flow control.
Compared to the evolving understanding of peripheral endothelial Piezo1 expression and function, the current knowledge of Piezo1 in the central nervous system endothelium is still in its infancy. Within the different cell types in the brain, Piezo1 is most highly expressed in ECs. Single-cell RNA sequencing studies demonstrated that mural vascular cells (SMCs and pericytes), glia, microglia, and neurons show little or no Piezo 1 expression (108, 166, 167). The cerebral circulation is a unique system, not only because of its anatomical location, but also due to a quite distinct blood delivery system. Neurons in the brain rely on an on-demand delivery strategy in which active neurons signal to the vasculature to increase regional cerebral blood flow (CBF)—a phenomenon termed functional hyperemia (168). This is unique from peripheral organs in which changes in blood flow are more homogeneous. The increase in CBF during functional hyperemia delivers O2 and nutrients to active neurons and simultaneously elevates frictional forces imposed onto ECs of small cerebral arterioles and capillaries.
Brain capillary ECs express functional Piezo1 channels that respond to flow and pressure changes by mediating cation influx and Ca2+ transients (26). As neural activity is associated with increased blood flow, such increases are likely the physiological trigger for Piezo1. Additionally, blood cells traveling through small capillaries impose forces distinct from those in larger vessels due to the non-Newtonian nature of blood (14). The exact role of Piezo1 in CBF regulation remains unknown. Because brain capillary Piezo1 channels mediate Ca2+ transients, channel activation likely generates NO, leading to localized vasodilation and increased perfusion (27, 56). ECs are also electrically coupled to one another and overlying mural cells, so that a change in EC Vm, triggered by Piezo1 activation, could affect surrounding cells (Figure 3). Further work is needed to fully understand the role of Piezo1 in CBF control. An important consideration, however, is that not all hemodynamic triggers are the same. A local change in red blood cell flux promotes a spatially restricted mechanical stimulus that differs spatiotemporally and in magnitude from large-scale hemodynamic changes in active brain regions (e.g., motor cortex during movement). The uniqueness of these scenarios emphasizes the importance of understanding the context of mechanosensing by Piezo1 when considering its possible role in CBF control. In other words, central nervous system EC Piezo1 activation should not be viewed as a one-size-fits-all system. Our laboratory is actively trying to bridge the knowledge gaps regarding the role of Piezo1 in central endothelial mechanosensing.
Decoding the role of EC mechanosensing in CBF control, via Piezo1 or other mechanosensors, will raise many new questions. What determines the set point of mechanosensing; is it simply a function of mechanosensor activity, expression levels, or the mechanical forces experienced by ECs? How is mechanosensing altered in diseases with hemodynamic disruptions (e.g., hypertension)? Is mechanosensing compromised in cerebrovascular and neurodegenerative diseases, such as stroke and Alzheimer’s disease? If so, can it be therapeutically targeted? Is endothelial mechanosensing plastic? Is it modified by use or disease? Are human mutations risk factors for cardiovascular diseases? Do externally assisted hemodynamics—like those in patients with artificial hearts—affect mechanosensing mechanisms and CBF? We anticipate that revealing Piezo1-mediated mechanisms that govern the precise tuning of blood flow to neural demands will advance our understanding of mechanosensing and CBF regulation and could provide new therapeutic targets for improving blood perfusion in disease.
5. ENDOTHELIAL MECHANOSENSING IN DISEASE
Mechanosensing involves a force and a sensor. When either or both are altered, disrupted mechanosensing and/or mechanotransduction ensue, leading to endothelial dysfunction. To that end, hemodynamic disruption and changes in blood flow patterns are hallmarks of vascular diseases such as hypertension and atherosclerosis. Disruptions also occur during less clear scenarios such as in collateral vessels and during ischemia-reperfusion (Figure 4). On the other hand, mechanosensors are liable to post-translational modifications and mutations that affect their function. Here, we discuss pathologies associated with altered mechanosensing. We further consider untapped areas of mechanosensing research.
Figure 4:
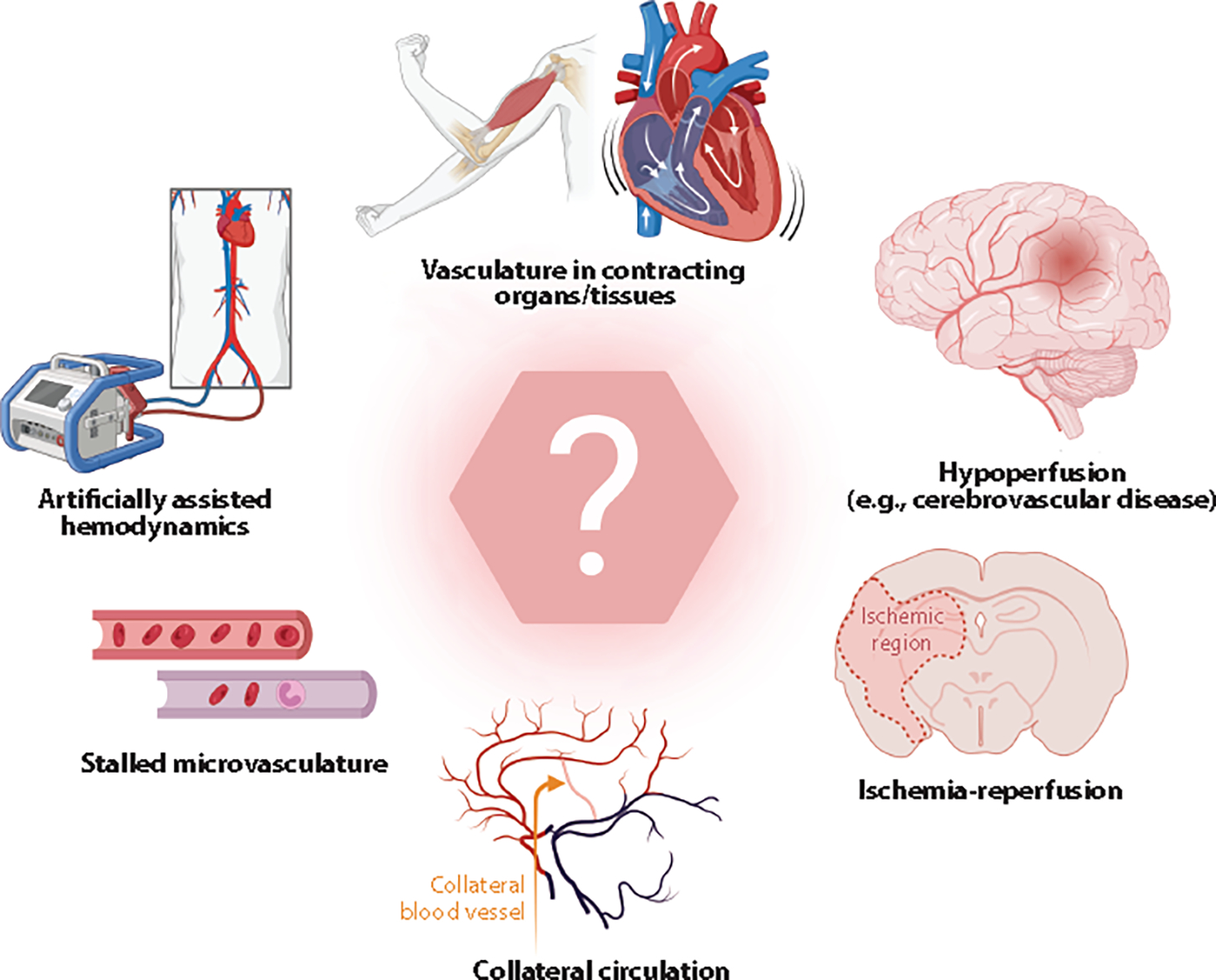
Less-understood examples where endothelial mechanosensing is likely engaged. The question mark symbolizes untapped areas where endothelial mechanosensing is understudied and therefore less clear. Figure adapted from images created with BioRender.com.
5.1. Flow Pattern Determines Endothelial Cell Signaling
Hemodynamic disruptions can trigger pathological outcomes rather than physiological responses. Atherosclerosis is a prominent example of altered forces imposed onto ECs. Atheroprotective or atherogenic endothelial signaling characterizes vascular regions depending on blood flow patterns (35). Blood vessels experiencing high, laminar flow display EC alignment in the flow direction, anti-inflammatory phenotype, low oxidative stress, cell turnover, and permeability (2). In distinction, vascular regions with low or oscillatory flow, such as arterial bifurcations, are characterized by an inflammatory phenotype, high EC proliferation, apoptosis, increased permeability, EC misalignment, enhanced proinflammatory gene expression, and extracellular matrix synthesis (2, 169–171). This demonstrates that different hemodynamic profiles shift endothelial signaling and could therefore protect against or aggravate vascular disease.
Shear stress and flow patterns dictate inflammatory signaling in ECs. Disturbed flow activates an inflammatory phenotype characterized by the upregulation of reactive oxygen species production, oxidative stress, cytokine expression, alterations in the extracellular matrix, and sustained changes in inflammatory gene expression (hours to days), even in atherosclerosis-resistant animal models (2, 172–174). These changes evoked by disturbed flow result in a constantly activated inflammatory phenotype, persistent inflammation, and blood vessel remodeling. In contrast, laminar flow only temporarily activates endothelial inflammatory signaling, which declines rapidly (175).
5.2. Hypertension Influences Endothelial Cell Signaling and Mechanosensors
Chronic hypertension is characterized by major disruptions of hemodynamic forces (176) that are experienced by ECs and likely impact mechanosensing pathways. Hypertension increases oxidative stress, reactive oxygen species generation, and inflammation. Blood flow disturbances are additional risk factors for the formation of atherosclerotic plaques (2, 169, 172). It has been amply reported that hypertension associates with altered expression and/or function of mechanosensors and mechanotransducers. Reactive oxygen species generated during hypertension interact with and eliminate NO and therefore inhibit vasodilation (177). Animal studies further demonstrated that the activity of mechanosensitive channels is affected by disease. Whether the altered activity of a mechanosensor is a cause or an outcome of changes in forces remains unclear. Hypertension affects vascular TRPV4, Kir2.1, and Piezo1 channels, among many others (59, 165, 178). Human mutations in genes encoding mechanosensors (e.g., PIEZO1) are common and could be risk factors in cardiovascular disease (179). Collectively, disrupted hemodynamics and dysregulated mechanosensing are intimately involved in vascular dysfunction and disease.
5.3. Untapped Areas
We listed two examples where mechanosensing is likely involved in cardiovascular disease. These examples only scratch the surface of possibilities and scenarios in which altered mechanosensing could be overlooked (Figure 4). For instance, a unique feature of capillaries is that they temporarily undergo a no-flow phenomenon, known as capillary stalling. This stalling would present a situation where forces/mechanosensing would be silenced. Recovery of flow in stalled capillaries could influence, or be influenced by, mechanosensing mechanisms (e.g., Piezo1). Along the lines of dramatic changes from no flow to flow, collateral vessels in the brain normally experience very low to no flow. Collateral vessels could become patent after stroke to salvage brain tissues, begging the question of whether stop-start of mechanosensing occurs, and if so, whether it affects the trajectory of vascular disease. Ischemia-reperfusion injury (e.g., heart or kidney) is another example of the stop-start mechanosensing scenario.
Hemorrhage (e.g., hemorrhagic stroke) evokes abnormal mechanical forces due, in part, to blood cells leaking out of a burst region. Traumatic brain injury represents an acute physical impact that could cause large mechanosensing and signaling events. Cardiac arrhythmias and artificially assisted hemodynamics alter flow patterns and pulsatility, and it is unknown whether these changes affect mechanosensing and vascular function. Chronic hypoperfusion of the brain during neurodegenerative diseases could also affect mechanosensors chronically and their ability to regulate cerebrovascular function. To summarize, there are many untapped areas that would benefit from being viewed through a mechanosensing lens.
6. CONCLUDING REMARKS
Mechanosensing by the endothelium is crucial for cardiovascular function and health. Evidence from the last years portrays a complex picture of the mechanisms involved. Endothelial mechanosensors not only detect hemodynamic forces, but they also team up with one another and/or enable common signal transduction pathways that ultimately influence vascular function. Mechanosensing, as the name implies, encompasses a sensing element and a mechanical force element. Both elements are disrupted during diseases such as hypertension, atherosclerosis, or stroke. Better understanding of endothelial mechanosensing has the potential to advance our ability to therapeutically target cardiovascular disease.
It is also our vision that the field will benefit from a holistic view that captures different elements involved in endothelial mechanosensing, whether it be a true mechanosensor or not. We also believe that it is crucial to reconceptualize our views of different vascular pathologies to encompass alterations in hemodynamic forces, vascular wall structure, and mechanosensing mechanisms.
ACKNOWLEDGMENTS
The authors thank Manuel Navedo and Thomas Longden for the feedback on the manuscript. This work was supported by grants from the National Institutes of Health (NIH) National Heart, Lung, and Blood Institute (R01HL169681), National Institute of General Medical Sciences (P20GM135007), National Institute of Aging (R21AG082193), and National Institute of Neurological Disorders and Stroke (R01NS119971), as well as the American Heart Association (20CDA35310097), the Totman Medical Research Trust, the Larner College of Medicine at the University of Vermont, and the Cardiovascular Research Institute of Vermont.
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Abraira VE, Ginty DD. 2013. The sensory neurons of touch. Neuron 79(4):618–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hahn C, Schwartz MA. 2009. Mechanotransduction in vascular physiology and atherogenesis. Nat. Rev. 10(1):53–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cecchi E, Giglioli C, Valente S, Lazzeri C, Gensini GF, et al. 2011. Role of hemodynamic shear stress in cardiovascular disease. Atherosclerosis 214(2):249–56 [DOI] [PubMed] [Google Scholar]
- 4.Kubota Y, Kleinman HK, Martin GR, Lawley TJ. 1988. Role of laminin and basement membrane in the morphological differentiation of human endothelial cells into capillary-like structures. J. Cell Biol. 107(4):1589–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Przysinda A, Feng W, Li G. 2020. Diversity of organism-wide and organ-specific endothelial cells. Curr. Cardiol. Rep. 22:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eelen G, de Zeeuw P, Treps L, Harjes U, Wong BW, Carmeliet P. 2018. Endothelial cell metabolism. Physiol. Rev. 98(1):3–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dalal PJ, Muller WA, Sullivan DP. 2020. Endothelial cell calcium signaling during barrier function and inflammation. Am. J. Pathol. 190(3):535–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Paszkowiak JJ, Dardik A, Haven N. 2003. Basic science review arterial wall shear stress: observations from the bench to the bedside. Vasc. Endovasc. Surg. 37(1):47–57 [DOI] [PubMed] [Google Scholar]
- 9.Belloni FL. 1999. Teaching the principles of hemodynamics. Adv. Physiol. Educ. 277(6):S187–202 [DOI] [PubMed] [Google Scholar]
- 10.Langille B 1993. Remodeling of developing and mature arteries: endothelium, smooth muscle, and matrix. J. Cardiovasc. Pharmacol. 21:S11–17 [DOI] [PubMed] [Google Scholar]
- 11.Badeer HS. 2001. Hemodynamics for medical students. Adv. Physiol. Educ. 25(1):44–52 [DOI] [PubMed] [Google Scholar]
- 12.Pyke KE, Tschakovsky ME. 2005. The relationship between shear stress and flow-mediated dilatation: implications for the assessment of endothelial function. J. Physiol. 568(Part 2):357–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Davies PF. 1995. Flow-mediated endothelial mechanotransduction. Physiol. Rev. 75(3):519–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Drew PJ. 2022. Neurovascular coupling: motive unknown. Trends Neurosci. 45(11):809–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Godo S, Shimokawa H. 2017. Endothelial functions. Arterioscler. Thromb. Vasc. Biol. 37(9):e108–14 [DOI] [PubMed] [Google Scholar]
- 16.Ando J, Yamamoto K. 2009. Vascular mechanobiology endothelial cell responses to fluid shear stress. Circ. J. 73:1983–92 [DOI] [PubMed] [Google Scholar]
- 17.Li J, Hou B, Tumova S, Muraki K, Bruns A, et al. 2014. Piezo1 integration of vascular architecture with physiological force. Nature 515(7526):279–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Malek AM, Alper SL, Izumo S. 1999. Hemodynamic shear stress and its role in atherosclerosis. JAMA 282(21):2035–42 [DOI] [PubMed] [Google Scholar]
- 19.Dewey CF,Bussolari SR,Gimbrone MA,Davies PF. 1981.The dynamic response of vascular endothelial cells to fluid shear stress. J. Biomech. Eng. 103(3):177–85 [DOI] [PubMed] [Google Scholar]
- 20.Fang Y, Wu D, Birukov KG. 2019. Mechanosensing and mechanoregulation of endothelial cell functions. Compr. Physiol. 9(2):873–904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Davis MJ,Earley S,Li Y-S, Chien S. 2023.Vascular mechanotransduction.Physiol. Rev.103(2):1247–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lansman JB, Hallam TJ, Rink TJ. 1987. Single stretch-activated ion channels in vascular endothelial cells as mechanotransducers? Nature 325(6107):811–13 [DOI] [PubMed] [Google Scholar]
- 23.Olesen ren- P, ClaphamH DE, Davies PF. 1988. Haemodynamic shear stress activates a K+ current in vascular endothelial cells. Nature 331(6152):168–70 [DOI] [PubMed] [Google Scholar]
- 24.Árnadóttir J, Chalfie M. 2010. Eukaryotic mechanosensitive channels. Annu. Rev. Biophys. 39:111–37 [DOI] [PubMed] [Google Scholar]
- 25.Syeda R, Florendo MN, Cox CD, Kefauver JM, Santos JS, et al. 2016. Piezo1 channels are inherently mechanosensitive. Cell Rep. 17(7):1739–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Harraz OF, Klug NR, Senatore AJ, Hill-Eubanks DC, Nelson MT. 2022. Piezo1 is a mechanosensor channel in central nervous system capillaries. Circ. Res. 130(10):1531–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang SP, Chennupati R, Kaur H, Iring A, Wettschureck N, Offermanns S. 2016. Endothelial cation channel PIEZO1 controls blood pressure by mediating flow-induced ATP release. J. Clin. Investig. 126(12):4527–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ranade SS, Qiu Z, Woo SH, Hur SS, Murthy SE, et al. 2014. Piezo1, a mechanically activated ion channel, is required for vascular development in mice. PNAS 111(28):10347–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kang H, Hong Z, Zhong M, Klomp J, Bayless KJ, et al. 2019. Piezo1 mediates angiogenesis through activation of MT1-MMP signaling. Am. J. Physiol. Cell Physiol. 316(1):C92–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu T, Du X, Zhang B, Zi H, Yan Y, et al. 2020. Piezo1-mediated Ca2+ activities regulate brain vascular pathfinding during development. Neuron 108(1):180–192.e5 [DOI] [PubMed] [Google Scholar]
- 31.Rode B, Shi J, Endesh N, Drinkhill MJ, Webster PJ, et al. 2017. Piezo1 channels sense whole body physical activity to reset cardiovascular homeostasis and enhance performance. Nat. Commun. 8(1):350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lhomme A, Gilbert G, Pele T, Deweirdt J, Henrion D, et al. 2019. Stretch-activated Piezo1 channel in endothelial cells relaxes mouse intrapulmonary arteries. Am. J. Respir. Cell Mol. Biol. 60(6):650–58 [DOI] [PubMed] [Google Scholar]
- 33.John L, Ko NL, Gokin A, Gokina N, Mandalà M, Osol G. 2018. The Piezo1 cation channel mediates uterine artery shear stress mechanotransduction and vasodilation during rat pregnancy. Am. J. Physiol. Heart Circ. Physiol. 315(4):H1019–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Evans EL, Cuthbertson K, Endesh N, Rode B, Blythe NM, et al. 2018. Yoda1 analogue (Dooku1) which antagonizes Yoda1-evoked activation of Piezo1 and aortic relaxation. Br. J. Pharmacol. 175(10):1744–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Albarrán-Juárez J, Iring A, Wang S, Joseph S, Grimm M, et al. 2018. Piezo1 and Gq/G11 promote endothelial inflammation depending on flow pattern and integrin activation. J. Exp. Med. 215(10):2655–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Harraz OF,Longden TA,Hill-Eubanks D,Nelson MT. 2018.PIP2 depletion promotes TRPV4 channel activity in mouse brain capillary endothelial cells. eLife 7:e38689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Harraz OF, Longden TA, Dabertrand F, Hill-Eubanks D, Nelson MT. 2018. Endothelial GqPCR activity controls capillary electrical signaling and brain blood flow through PIP2 depletion. PNAS 115(15):E3569–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ye Y, Barghouth M, Dou H, Luan C, Wang Y, et al. 2022. A critical role of the mechanosensor PIEZO1 in glucose-induced insulin secretion in pancreatic β-cells. Nat. Commun. 13:4237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu D, Minami M, Kawamura H, Puro D. 2006. Electrotonic transmission within pericyte-containing retinal microvessels. Microcirculation 13(5):353–63 [DOI] [PubMed] [Google Scholar]
- 40.Yamamoto Y, Imaeda K, Suzuki H. 1999. Endothelium-dependent hyperpolarization and intercellular electrical coupling in guinea-pig mesenteric arterioles. J. Physiol. 514(Part 2):505–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Watanabe H, Vriens J, Suh SH, Benham CD, Droogmans G, Nilius B. 2002. Heat-evoked activation of TRPV4 channels in a HEK293 cell expression system and in native mouse aorta endothelial cells. J. Biol. Chem. 277(49):47044–51 [DOI] [PubMed] [Google Scholar]
- 42.Köhler R, Heyken W-T, Heinau P, Schubert R, Si H, et al. 2006. Evidence for a functional role of endothelial transient receptor potential V4 in shear stress-induced vasodilatation. Arterioscler. Thromb. Vasc. Biol. 26(7):1495–502 [DOI] [PubMed] [Google Scholar]
- 43.Hartmannsgruber V, Heyken WT, Kacik M, Kaistha A, Grgic I, et al. 2007. Arterial response to shear stress critically depends on endothelial TRPV4 expression. PLOS ONE 2(9):e827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mendoza SA,Fang J,Gutterman DD,Wilcox DA,Bubolz AH,et al.2010.TRPV4-mediated endothelial Ca2+ influx and vasodilation in response to shear stress. Am. J. Physiol. Heart Circ. Physiol. 298:466–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Earley S, Brayden JE. 2015. Transient receptor channels in the vasculature. Physiol. Rev. 95:645–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Strotmann R, Harteneck C, Nunnenmacher K, Schultz G, Plant TD. 2000. OTRPC4, a nonselective cation channel that confers sensitivity to extracellular osmolarity. Nat. Cell Biol. 2(10):695–702 [DOI] [PubMed] [Google Scholar]
- 47.Liedtke W, Choe Y, Martí-Renom MA, Bell AM, Denis CS, et al. 2000. Vanilloid receptor-related osmotically activated channel (VR-OAC), a candidate vertebrate osmoreceptor. Cell 103(3):525–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.White JPM, Cibelli M, Urban L, Nilius B, McGeown JG, Nagy I. 2016. TRPV4: molecular conductor of a diverse orchestra. Physiol. Rev. 96(3):911–73 [DOI] [PubMed] [Google Scholar]
- 49.Loukin S, Zhou X, Su Z, Saimi Y, Kung C. 2010. Wild-type and brachyolmia-causing mutant TRPV4 channels respond directly to stretch force. J. Biol. Chem. 285(35):27176–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Servin-Vences MR,Moroni M,Lewin GR,Poole K. 2017.Direct measurement of TRPV4 and PIEZO1 activity reveals multiple mechanotransduction pathways in chondrocytes. eLife 6:e21074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nikolaev YA, Cox CD, Ridone P, Rohde PR, Cordero-Morales JF, et al. 2019. Mammalian TRP ion channels are insensitive to membrane stretch. J. Cell Sci. 132:jcs238360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Watanabe H, Vriens J, Prenen J, Droogmans G, Voets T, Nillus B. 2003. Anandamide and arachidonic acid use epoxyeicosatrienoic acids to activate TRPV4 channels. Nature 424(6947):434–38 [DOI] [PubMed] [Google Scholar]
- 53.Loot AE, Popp R, Fisslthaler B, Vriens J, Nilius B, Fleming I. 2008.Role of cytochrome P450-dependent transient receptor potential V4 activation in flow-induced vasodilatation. Cardiovasc. Res. 80(3):445–52 [DOI] [PubMed] [Google Scholar]
- 54.Swain SM, Liddle RA. 2021. Piezo1 acts upstream of TRPV4 to induce pathological changes in endothelial cells due to shear stress. J. Biol. Chem. 296:100171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sonkusare SK, Bonev AD, Ledoux J, Liedtke W, Kotlikoff MI, et al. 2012. Elementary Ca2+ signals through endothelial TRPV4 channels regulate vascular function. Science 336(6081):597–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Longden TA, Mughal A, Hennig GW, Harraz OF, Shui B, et al. 2021. Local IP3 receptor-mediated Ca2+ signals compound to direct blood flow in brain capillaries. Sci. Adv. 7(30):eabh0101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.McFarland SJ, Weber DS, Choi C-S, Lin MT, Taylor MS. 2020. Ablation of endothelial TRPV4 channels alters the dynamic Ca2+ signaling profile in mouse carotid arteries. Int. J. Mol. Sci. 21(6):2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Heathcote HR, Lee MD, Zhang X, Saunter CD, Wilson C, McCarron JG. 2019. Endothelial TRPV4 channels modulate vascular tone by Ca2+-induced Ca2+ release at inositol 1,4,5-trisphosphate receptors. Br. J. Pharmacol. 176(17):3297–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sonkusare SK, Dalsgaard T, Bonev AD, Hill-Eubanks DC, Kotlikoff MI, et al. 2014. AKAP150-dependent cooperative TRPV4 channel gating is central to endothelium-dependent vasodilation and is disrupted in hypertension. Sci. Signal. 7(333):ra66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Xu J, Mathur J, Vessières E, Hammack S, Nonomura K, et al. 2018. GPR68 senses flow and is essential for vascular physiology. Cell 173(3):762–75.e16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rath G, Saliez J, Behets G, Romero-Perez M, Leon-Gomez E, et al. 2012. Vascular hypoxic pre-conditioning relies on TRPV4-dependent calcium influx and proper intercellular gap junctions communication. Arterioscler. Thromb. Vasc. Biol. 32(9):2241–49 [DOI] [PubMed] [Google Scholar]
- 62.Saliez J, Bouzin C, Rath G, Ghisdal P, Desjardins F, et al. 2008. Role of caveolar compartmentation in endothelium-derived hyperpolarizing factor-mediated relaxation. Circulation 117(8):1065–74 [DOI] [PubMed] [Google Scholar]
- 63.Thakore P,Alvarado MG,Ali S,Mughal A,Pires PW,et al.2021.Brain endothelial cell TRPA1 channel sinitiate neurovascular coupling. eLife 10:e63040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sullivan MN,Gonzales A,Pires PW,Leo MD,Gonzales AL,et al.2015.Localized TRPA1 channel Ca2+ signals stimulated by reactive oxygen species promote cerebral artery dilation. Sci. Signal. 8(358):ra2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Longden TA, Dabertrand F, Koide M, Gonzales AL, Tykocki NR, et al. 2017. Capillary K+-sensing initiates retrograde hyperpolarization to locally increase cerebral blood flow. Nat. Neurosci. 20:717–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ahn SJ, Fancher IS, Bian JT, Zhang CX, Schwab S, et al. 2017. Inwardly rectifying K+ channels are major contributors to flow-induced vasodilatation in resistance arteries. J. Physiol. 595(7):2339–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zaritsky JJ, Eckman DM, Wellman GC, Nelson MT, Schwarz TL. 2000. Targeted disruption of Kir2.1 and Kir2.2 genes reveals the essential role of the inwardly rectifying K+ current in K+-mediated vasodilation. Circ. Res. 87(2):160–66 [DOI] [PubMed] [Google Scholar]
- 68.Hibino H, Inanobe A, Furutani K, Murakami S, Findlay I, Kurachi Y. 2010. Inwardly rectifying potassium channels: their structure, function, and physiological roles. Physiol. Rev. 90(1):291–366 [DOI] [PubMed] [Google Scholar]
- 69.Hoger JH, Ilyin VI, Forsyth S, Hoger A. 2002. Shear stress regulates the endothelial Kir2.1 ion channel. PNAS 99(11):7780–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lieu DK, Pappone PA, Barakat AI. 2004. Differential membrane potential and ion current responses to different types of shear stress in vascular endothelial cells. Am. J. Physiol. Cell Physiol. 286(6):1367–75 [DOI] [PubMed] [Google Scholar]
- 71.Jacobs ER, Cheliakine C, Gebremedhin D, Birks EK, Davies PF, Harder DR. 1995. Shear activated channels in cell-attached patches of cultured bovine aortic endothelial cells. Pflügers Arch.431(1):129–31 [DOI] [PubMed] [Google Scholar]
- 72.Romanenko VG, Fang Y, Byfield F, Travis AJ, Vandenberg CA, et al. 2004. Cholesterol sensitivity and lipid raft targeting of Kir2.1 channels. Biophys. J. 87(6):3850–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kusche-Vihrog K, Callies C, Fels J, Oberleithner H. 2010. The epithelial sodium channel (ENaC): Mediator of the aldosterone response in the vascular endothelium? Steroids 75(8–9):544–49 [DOI] [PubMed] [Google Scholar]
- 74.Wang S, Meng F, Mohan S, Champaneri B, Gu Y. 2009. Functional ENaC channels expressed in endothelial cells: a new candidate for mediating shear force. Microcirculation 16(3):276–87 [DOI] [PubMed] [Google Scholar]
- 75.Sternak M, Bar A, Adamski MG, Mohaissen T, Marczyk B, et al. 2018. The deletion of endothelial sodium channel a (αENaC) impairs endothelium-dependent vasodilation and endothelial barrier integrity in endotoxemia in vivo. Front. Pharmacol. 9:178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Knoepp F, Ashley Z, Barth D, Baldin JP, Jennings M, et al. 2020. Shear force sensing of epithelial Na+ channel (ENaC) relies on N-glycosylated asparagines in the palm and knuckle domains of αENaC. PNAS 117(1):717–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tarjus A, Maase M, Jeggle P, Martinez-Martinez E, Fassot C, et al. 2017. The endothelial αENaC contributes to vascular endothelial function in vivo. PLOS ONE 12(9):e0185319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cosgun ZC, Sternak M, Fels B, Bar A, Kwiatkowski G, et al. 2022. Rapid shear stress-dependent ENaC membrane insertion is mediated by the endothelial glycocalyx and the mineralocorticoid receptor. Cell. Mol. Life Sci. 79:235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Jeggle P, Callies C, Tarjus A, Fassot C, Fels J, et al. 2013. Epithelial sodium channels tiffens the vascular endothelium in vitro and in Liddle mice. Hypertension 61(5):1053–59 [DOI] [PubMed] [Google Scholar]
- 80.Zhang J, Yuan HK, Chen S, Zhang ZR. 2022. Detrimental or beneficial: role of endothelial ENaC in vascular function. J. Cell Physiol. 237(1):29–48 [DOI] [PubMed] [Google Scholar]
- 81.Pérez FR, Venegas F, González M, Andrés S, Vallejos C, et al. 2009. Endothelial epithelial sodium channel inhibition activates endothelial nitric oxide synthase via phosphoinositide 3-kinase/Akt in small-diameter mesenteric arteries. Hypertension 53(6):1000–7 [DOI] [PubMed] [Google Scholar]
- 82.Guo D, Liang S, Wang S, Tang C, Yao B, et al. 2016. Role of epithelial Na+ channels in endothelial function. J. Cell Sci. 129(2):290–97 [DOI] [PubMed] [Google Scholar]
- 83.Ashley Z, Mugloo S, McDonald FJ, Fronius M. 2018. Epithelial Na+ channel differentially contributes to shear stress-mediated vascular responsiveness in carotid and mesenteric arteries from mice. Am. J. Physiol. Heart Circ. Physiol. 314(5):H1022–32 [DOI] [PubMed] [Google Scholar]
- 84.Gurney A, Manoury B. 2008. Two-pore potassium channels in the cardiovascular system. Eur. Biophys. J. 38(3):305–18 [DOI] [PubMed] [Google Scholar]
- 85.Brohawn SG, Su Z, MacKinnon R. 2014.Mechanosensitivity is mediated directly by the lipid membrane in TRAAK and TREK1 K+ channels. PNAS 111(9):3614–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lesage F, Terrenoire C, Romey G, Lazdunski M. 2000.Human TREK2, a 2P domain mechano-sensitive K+ channel with multiple regulations by polyunsaturated fatty acids, lysophospholipids, and Gs, Gi, and Gq protein-coupled receptors. J. Biol. Chem. 275(37):28398–405 [DOI] [PubMed] [Google Scholar]
- 87.Patel AJ, Honoré E, Maingret F, Lesage F, Fink M, et al.1998.A mammalian two pore domain mechano gated S-like K+ channel. EMBO J. 17(15):4283–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Brohawn SG. 2015. How ion channels sense mechanical force: insights from mechanosensitive K2P channels TRAAK, TREK1, and TREK2. Ann. N. Y. Acad. Sci. 1352(1):20–32 [DOI] [PubMed] [Google Scholar]
- 89.Honoré E, Patel AJ, Chemin J, Suchyna T, Sachs F. 2006. Desensitization of mechano-gated K2P channels. PNAS 103(18):6859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Schmidpeter PAM, Petroff JT 2nd, Khajoueinejad L, Wague A, Frankfater C, et al. 2023. Membrane phospholipids control gating of the mechanosensitive potassium leak channel TREK1. Nat. Commun. 14:1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Garry A, Fromy B, Blondeau N, Henrion D, Brau F, et al. 2007. Altered acetylcholine, bradykinin and cutaneous pressure-induced vasodilation in mice lacking the TREK1 potassium channel: the endothelial link. EMBO Rep. 8(4):354–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Rao AM, Hatcher JF, Kindy MS, Dempsey RJ. 1999. Arachidonic acid and leukotriene C4: role in transient cerebral ischemia of gerbils. Neurochem. Res. 24(10):1225–32 [DOI] [PubMed] [Google Scholar]
- 93.Maingret F, Patel AJ, Lesage F, Lazdunski M, Honoré E. 1999. Mechano- or acid stimulation, two interactive modes of activation of the TREK-1 potassium channel. J. Biol. Chem. 274(38):26691–96 [DOI] [PubMed] [Google Scholar]
- 94.Chemin J, Patel AJ, Duprat F, Lauritzen I, Lazdunski M, Honoré E. 2005. A phospholipid sensor controls mechanogating of the K+ channel TREK-1. EMBO J. 24(1):44–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Mathie A 2007. Neuronal two-pore-domain potassium channels and their regulation by G protein-coupled receptors. J. Physiol. 578(2):377–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Takada Y, Kato C, Kondo S, Korenaga R, Ando J. 1997.Cloning of cDNAs encoding G protein-coupled receptor expressed in human endothelial cells exposed to fluid shear stress. Biochem. Biophys. Res. Commun. 240(3):737–41 [DOI] [PubMed] [Google Scholar]
- 97.Chachisvilis M, Zhang YL, Frangos JA. 2006. G protein-coupled receptors sense fluid shear stress in endothelial cells. PNAS 103(42):15463–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Gudi S, Nolan JP, Frangos JA. 1998. Modulation of GTPase activity of G proteins by fluid shear stress and phospholipid composition. PNAS 95(5):2515–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Yamamoto K,Ando J. 2015.Vascular endothelial cell membranes differentiate between stretch and shear stress through transitions in their lipid phases. Am. J. Physiol. Heart Circ. Physiol. 309(7):H1178–85 [DOI] [PubMed] [Google Scholar]
- 100.Yamamoto K, Ando J. 2013. Endothelial cell and model membranes respond to shear stress by rapidly decreasing the order of their lipid phases. J Cell Sci. 126(5):1227–34 [DOI] [PubMed] [Google Scholar]
- 101.Erdogmus S, Storch U, Danner L, Becker J, Winter M, et al. 2019. Helix 8 is the essential structural motif of mechanosensitive GPCRs. Nat. Commun. 10:5784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ozkan AD, Gettas T, Sogata A, Phaychanpheng W, Zhou M, Lacroix JJ. 2021. Mechanical and chemical activation of GPR68 probed with a genetically encoded fluorescent reporter. J. Cell Sci. 134(16):jcs255455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.dela Paz NG, Melchior B, Frangos JA. 2017. Shear stress induces Gαq/11 activation independently of G protein-coupled receptor activation in endothelial cells. Am. J. Physiol. Cell Physiol. 312(4):C428–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Liao JK, Homcy CJ. 1993. The G proteins of the Gαi and Gαq family couple the bradykinin receptor to the release of endothelium-derived relaxing factor. J. Clin. Investig. 92(5):2168–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Hu Y, Chen M, Wang M, Li X. 2022.Flow-mediated vasodilation through mechanosensitive G protein-coupled receptors in endothelial cells. Trends Cardiovasc. Med. 32(2):61–70 [DOI] [PubMed] [Google Scholar]
- 106.Jung B, Obinata H, Galvani S, Mendelson K, Ding B Sen, et al. 2012. Flow-regulated endothelial S1P receptor-1 signaling sustains vascular development. Dev. Cell 23(3):600–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Cantalupo A, Gargiulo A, Dautaj E, Liu C, Zhang Y, et al. 2017. S1PR1 (sphingosine-1-phosphate receptor 1) signaling regulates blood flow and pressure. Hypertension 70(2):426–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Vanlandewijck M, He L, Mäe MA, Andrae J, Ando K, et al. 2018. A molecular atlas of cell types and zonation in the brain vasculature. Nature 554(7693):475–80 [DOI] [PubMed] [Google Scholar]
- 109.Marullo S, Doly S, Saha K, Enslen H, Scott MGH, Coureuil M. 2020. Mechanical GPCR activation by traction forces exerted on receptor N-glycans. ACS Pharmacol. Transl. Sci. 3(2):171–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Harraz OF,Hill-Eubanks D,Nelson MT. 2020.PIP2: a critical regulator of vascular ion channels hiding in plain sight. PNAS 117(34):20378–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Reitsma S, Slaaf DW, Vink H, van Zandvoort MAMJ, oude Egbrink. 2007. The endothelial glycocalyx: composition, functions, and visualization. Pflügers Arch. 454(3):345–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Tarbell JM, Simon SI, Curry F- RE. 2014. Mechanosensing at the vascular interface. Annu. Rev. Biomed. Eng. 16:505–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zeng Y, Zhang XF, Fu BM, Tarbell JM. 2018. The role of endothelial surface glycocalyx in mechanosensing and transduction. Mol. Cell. Tissue Eng. Vasc. Syst. 1097:1–27 [DOI] [PubMed] [Google Scholar]
- 114.Weinbaum S, Tarbell JM, Damiano ER. 2007. The structure and function of the endothelial glycocalyx layer. Annu. Rev. Biomed. Eng. 9:121–67 [DOI] [PubMed] [Google Scholar]
- 115.Nikmanesh M, Shi Z-D, Tarbell JM. 2012. Heparan sulfate proteoglycan mediates shear stress-induced endothelial gene expression in mouse embryonic stem cell-derived endothelial cells. Biotechnol. Bioeng. 109(2):583–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Harding IC, Mitra R, Mensah SA, Herman IM, Ebong EE. 2018. Pro-atherosclerotic disturbed flow disrupts caveolin-1 expression, localization, and function via glycocalyx degradation. J. Transl. Med. 16(1):364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ebong EE, Lopez-Quintero SV, Rizzo V, Spray DC, Tarbell JM. 2014.Shear-induced endothelial NOS activation and remodeling via heparan sulfate, glypican-1, and syndecan-1. Integr. Biol. 6(3):338–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Florian JA, Kosky JR, Ainslie K, Pang Z, Dull RO, Tarbell JM. 2003. Heparan sulfate proteoglycan is a mechanosensor on endothelial cells. Circ. Res. 93(10):e136–42 [DOI] [PubMed] [Google Scholar]
- 119.Yen W, Cai B, Yang J, Zhang L, Zeng M, et al. 2015. Endothelial surface glycocalyx can regulate flow-induced nitric oxide production in microvessels in vivo. PLOS ONE 10(1):e0117133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Mochizuki S, Vink H, Hiramatsu O, Kajita T, Shigeto F, et al. 2003. Role of hyaluronic acid glycosaminoglycans in shear-induced endothelium-derived nitric oxide release. Am. J. Physiol. Heart Circ. Physiol. 285(2):H722–26 [DOI] [PubMed] [Google Scholar]
- 121.Hecker M, Mulsch A, Bassenge E, Busse R. 1993. Vasoconstriction and increased flow: two principal mechanisms of shear stress-dependent endothelial autacoid release. Am. J. Physiol. Heart Circ. Physiol. 265(3):H828–33 [DOI] [PubMed] [Google Scholar]
- 122.Kumagai R, Lu X, Kassab GS. 2009. Role of glycocalyx in flow-induced production of nitric oxide and reactive oxygen species. Free Radic Biol. Med. 47(5):600–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Pahakis MY, Kosky JR, Dull RO, Tarbell JM. 2007. The role of endothelial glycocalyx components in mechanotransduction of fluid shear stress. Biochem. Biophys. Res. Commun. 355(1):228–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Ruane-O’Hora T, Ahmeda A, Markos F. 2020. The vascular glycocalyx is not a mechanosensor in conduit arteries in the anesthetized pig. PeerJ 8:e8725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Cancel LM, Ebong EE, Mensah S, Hirschberg C, Tarbell JM. 2016. Endothelial glycocalyx, apoptosis and inflammation in an atherosclerotic mouse model. Atherosclerosis 252:136–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Thi MM, Tarbell JM, Weinbaum S, Spray DC. 2004. The role of the glycocalyx in reorganization of the actin cytoskeleton under fluid shear stress: a “bumper-car” model. PNAS 101(47):16483–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Moon JJ, Matsumoto M, Patel S, Lee L, Guan J-L, Li S. 2005. Role of cell surface heparan sulfate proteoglycans in endothelial cell migration and mechanotransduction. J. Cell Physiol. 203(1):166–76 [DOI] [PubMed] [Google Scholar]
- 128.Liu Y, Collins C, Kiosses WB, Murray AM, Joshi M, et al. 2013. A novel pathway spatiotemporally activates Rac1 and redox signaling in response to fluid shear stress. J. Cell Biol. 201(6):863–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Fleming I, Fisslthaler B, Dixit M, Busse R. 2005. Role of PECAM-1 in the shear-stress-induced activation of Akt and the endothelial nitric oxide synthase (eNOS) in endothelial cells. J. Cell Sci. 118:4103–11 [DOI] [PubMed] [Google Scholar]
- 130.Bagi Z, Frangos JA, Yeh JC, White CR, Kaley G, Koller A. 2005. PECAM-1 mediates NO-dependent dilation of arterioles to high temporal gradients of shearstress.Arterioscler. Thromb. Vasc. Biol.25(8):1590–95 [DOI] [PubMed] [Google Scholar]
- 131.Masuda M, Osawa M, Shigematsu H, Harada N, Fujiwara K. 1997. Platelet endothelial cell adhesion molecule-1 is a major SH-PTP2 binding protein in vascular endothelial cells. FEBS Lett. 408(3):331–36 [DOI] [PubMed] [Google Scholar]
- 132.Tzima E, Irani-Tehrani M, Kiosses WB, Dejana E, Schultz DA, et al. 2005. A mechanosensory complex that mediates the endothelial cell response to fluid shear stress. Nature 437(7057):426–31 [DOI] [PubMed] [Google Scholar]
- 133.Chiu YJ, McBeath E, Fujiwara K. 2008. Mechanotransduction in an extracted cell model: Fyn drives stretch- and flow-elicited PECAM-1 phosphorylation. J. Cell Biol. 182(4):753–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Osawa M, Masuda M, Kusano KI, Fujiwara K. 2002. Evidence for a role of platelet endothelial cell adhesion molecule-1 in endothelial cell mechanosignal transduction: Is it a mechanoresponsive molecule? J. Cell Biol. 158(4):773–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Tai LK, Zheng Q, Pan S, Jin ZG, Berk BC. 2005. Flow activates ERK1/2 and endothelial nitric oxide synthase via a pathway involving PECAM1, SHP2, and Tie2. J. Biol. Chem. 280(33):29620–24 [DOI] [PubMed] [Google Scholar]
- 136.Govers R, Bevers L, De Bree P, Rabelink TJ. 2002. Endothelial nitric oxide synthase activity is linked to its presence at cell-cell contacts. Biochem. J. 361(Part 2):193–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Dusserre N, L’Heureux N, Bell KS, Stevens HY, Yeh J, et al. 2004. PECAM-1 interacts with nitric oxide synthase in human endothelial cells: Implication for flow-induced nitric oxide synthase activation. Arterioscler. Thromb. Vasc. Biol. 24(10):1796–802 [DOI] [PubMed] [Google Scholar]
- 138.Liu Y, Bubolz AH, Shi Y, Newman PJ, Newman DK, Gutterman DD. 2006. Peroxynitrite reduces the endothelium-derived hyperpolarizing factor component of coronary flow-mediated dilation in PECAM-1-knockout mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 290(1):57–65 [DOI] [PubMed] [Google Scholar]
- 139.Conway DE, Breckenridge MT, Hinde E, Gratton E, Chen CS, Schwartz MA. 2013. Fluid shear stress on endothelial cells modulates mechanical tension across VE-cadherin and PECAM-1. Curr. Biol. 23(11):1024–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Sumpio BE, Yun S, Cordova AC, Haga M, Zhang J, et al. 2005. MAPKs (ERK1/2, p38) and AKT can be phosphorylated by shear stress independently of platelet endothelial cell adhesion molecule-1 (CD31) in vascular endothelial cells. J. Biol. Chem. 280(12):11185–91 [DOI] [PubMed] [Google Scholar]
- 141.Mehta V, Pang KL, Rozbesky D, Nather K, Keen A, et al. 2020. The guidance receptor plexin D1 is a mechanosensor in endothelial cells. Nature 578(7794):290–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Uesugi K, Oinuma I, Katoh H, Negishi M. 2009. Different requirement for Rnd GTPases of R-RasGAP activity of plexin-C1 and plexin-D1. J. Biol. Chem. 284(11):6743–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Sandri C, Caccavari F, Valdembri D, Camillo C, Veltel S, et al. 2012. The R-Ras/RIN2/Rab5 complex controls endothelial cell adhesion and morphogenesis via active integrin endocytosis and Rac signaling. Cell Res. 22(10):1479–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Sinha B, Köster D, Ruez R, Gonnord P, Bastiani M, et al. 2011. Cells respond to mechanical stress by rapid disassembly of caveolae. Cell 144(3):402–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Yu J, Bergaya S, Murata T, Alp IF, Bauer MP, et al. 2006. Direct evidence for the role of caveolin-1 and caveolae in mechanotransduction and remodeling of blood vessels. J. Clin. Investig. 116(5):1284–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Rizzo V,Sung A,Oh P,Schnitzer JE. 1998.Rapid mechanotransduction in situ at the luminal cell surface of vascular endothelium and its caveolae. J. Biol. Chem. 273(41):26323–29 [DOI] [PubMed] [Google Scholar]
- 147.Rizzo V, McIntosh DP, Oh P, Schnitzer JE. 1998. In situ flow activates endothelial nitric oxide synthase in luminal caveolae of endothelium with rapid caveolin dissociation and calmodulin association. J. Biol. Chem. 273(52):34724–29 [DOI] [PubMed] [Google Scholar]
- 148.Sonveaux P, Martinive P, DeWever J, Batova Z, Daneau G, et al. 2004. Caveolin-1 expression is critical for vascular endothelial growth factor-induced ischemic hindlimb collateralization and nitric oxide-mediated angiogenesis. Circ. Res. 95(2):154–61 [DOI] [PubMed] [Google Scholar]
- 149.Radel C, Rizzo V. 2005. Integrin mechanotransduction stimulates caveolin-1 phosphorylation and recruitment of Csk to mediate actin reorganization. Am. J. Physiol. Heart Circ. Physiol. 288(2):936–45 [DOI] [PubMed] [Google Scholar]
- 150.Lolo FN, Pavón DM, Grande-García A, Elósegui-Artola A, Segatori VI, et al. 2022. Caveolae couple mechanical stress to integrin recycling and activation. eLife 11:e82348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Frank PG, Woodman SE, Park DS, Lisanti MP. 2003. Caveolin, caveolae, and endothelial cell function. Arterioscler. Thromb. Vasc. Biol. 23(7):1161–68 [DOI] [PubMed] [Google Scholar]
- 152.Yamamoto K, Furuya K, Nakamura M, Kobatake E, Sokabe M, Ando J. 2011. Visualization of flow-induced ATP release and triggering of Ca2+ waves at caveolae in vascular endothelial cells. J. Cell Sci. 124(Part 20):3477–83 [DOI] [PubMed] [Google Scholar]
- 153.Wang N, Butler JP, Ingber DE. 1993. Mechanotransduction across the cell surface and through the cytoskeleton. Science 260(5111):1124–27 [DOI] [PubMed] [Google Scholar]
- 154.Matthews BD, Overby DR, Mannix R, Ingber DE. 2006. Cellular adaptation to mechanical stress: role of integrins, Rho, cytoskeletal tension and mechanosensitive ion channels. J. Cell Sci. 119(3):508–18 [DOI] [PubMed] [Google Scholar]
- 155.Choquet D, Felsenfeld DP, Sheetz MP. 1997. Extracellular matrix rigidity causes strengthening of integrin-cytoskeleton linkages. Cell 88(1):39–48 [DOI] [PubMed] [Google Scholar]
- 156.Collins C, Guilluy C, Welch C, O’Brien ET, Hahn K,et al.2012.Localized tensional forces on PECAM-1 elicit a global mechanotransduction response via the integrin-RhoA pathway. Curr. Biol. 22:2087–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Boyd NL, Park H, Yi H, Boo YC, Sorescu GP, et al. 2003. Chronic shear induces caveolae formation and alters ERK and Akt responses in endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 285(3):1113–22 [DOI] [PubMed] [Google Scholar]
- 158.Mackay CE, Leo MD, Fernández-Peña C, Hasan R, Yin W, et al. 2020. Intravascular flow stimulates PKD2 (Polycystin-2) channels in endothelial cells to reduce blood pressure. eLife 9:e56655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Lisanti MP, Scherer PE, Vidugiriene J, Tang ZL, Hermanowski-Vosatka A, et al.1994.Characterization of caveolin-rich membrane domains isolated from an endothelial-rich source: implications for human disease. J. Cell Biol. 126(1):111–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Rizzo V, Morton C, DePaola N, Schnitzer JE, Davies PF. 2003. Recruitment of endothelial caveolae into mechanotransduction pathways by flow conditioning in vitro. Am. J. Physiol. Heart Circ. Physiol. 285(4):1720–29 [DOI] [PubMed] [Google Scholar]
- 161.Feron O, Dessy C, Moniotte S, Desager JP, Balligand JL. 1999. Hypercholesterolemia decreases nitric oxide production by promoting the interaction of caveolin and endothelial nitric oxide synthase. J. Clin. Investig. 103(6):897–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Friedrich EE, Hong Z, Xiong S, Zhong M, Di A, et al. 2019. Endothelial cell Piezo1 mediates pressure-induced lung vascular hyperpermeability via disruption of adherens junctions. PNAS 116(26):12980–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Bartoli F, Debant M,Chuntharpursat-Bon E, Evans EL, Musialowski KE, et al. 2022. Endothelial Piezo1 sustains muscle capillary density and contributes to physical activity. J. Clin. Investig. 132(5):e141775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Wang SP, Wang B, Shi Y, Möller T, Stegmeyer RI, et al. 2022. Mechanosensation by endothelial PIEZO1 is required for leukocyte diapedesis. Blood 140(3):171–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Retailleau K, Duprat F, Arhatte M, Ranade SS, Peyronnet R, et al. 2015. Piezo1 in smooth muscle cells is involved in hypertension-dependent arterial remodeling. Cell Rep. 13(6):1161–71 [DOI] [PubMed] [Google Scholar]
- 166.Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, et al. 2014. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex.J. Neurosci.34(36):11929–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Saunders A, Macosko EZ, Wysoker A, Goldman M, Krienen FM, et al. 2018. Molecular diversity and specializations among the cells of the adult mouse brain. Cell 174(4):1015–30.e16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Iadecola C 2017. The neurovascular unit coming of age: a journey through neurovascular coupling in health and disease. Neuron 96(1):17–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Cheng C, Tempel D, Van Haperen R, Van Der Baan A, Grosveld F, et al. 2006. Atherosclerotic lesion size and vulnerability are determined by patterns of fluid shear stress. Circulation 113(23):2744–53 [DOI] [PubMed] [Google Scholar]
- 170.Brooks AR, Lelkes PI, Rubanyi GM. 2004. Gene expression profiling of vascular endothelial cells exposed to fluid mechanical forces: relevance for focal susceptibility to atherosclerosis. Endothelium 11(1):45–57 [DOI] [PubMed] [Google Scholar]
- 171.Mohan S, Mohan N, Sprague EA. 1997. Differential activation of NF-κB in human aortic endothelial cells conditioned to specific flow environments. Am. J. Physiol. Cell Physiol. 273(2):C572–78 [DOI] [PubMed] [Google Scholar]
- 172.Jongstra-Bilen J, Haidari M, Zhu SN, Chen M, Guha D, Cybulsky MI. 2006. Low-grade chronic inflammation in regions of the normal mouse arterial intima predisposed to atherosclerosis. J. Exp. Med. 203(9):2073–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Orr AW, Sanders JM, Bevard M, Coleman E, Sarembock IJ, Schwartz MA. 2005. The subendothelial extracellular matrix modulates NF-κB activation by flow a potential role in atherosclerosis. J. Cell Biol. 169(1):191–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Thoumine O, Nerem RM, Girard FR. 1995. Oscillatory shear stress and hydrostatic pressure modulate cell-matrix attachment proteins in cultured endothelial cells. In Vitro Cell. Dev. Biol. Anim. 31:45–54 [DOI] [PubMed] [Google Scholar]
- 175.Hsieh H-J, Cheng C-C, Wu S-T, Chiu J-J, Wung B-S, Wang DL. 1998. Increase of reactive oxygen species (ROS) in endothelial cells by shear flow and involvement of ROS in shear-induced c-fos expression. J. Cell Physiol. 175:156–62 [DOI] [PubMed] [Google Scholar]
- 176.Mayet J, Hughes A. 2003. Cardiac and vascular pathophysiology in hypertension. Heart 89(9):1104–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Harrison DG, Widder J, Grumbach I, Chen W, Weber M, Searles C. 2006. Endothelial mechanotransduction, nitric oxide and vascular inflammation. J. Intern. Med. 259(4):351–63 [DOI] [PubMed] [Google Scholar]
- 178.Koide M, Harraz OF, Dabertrand F, Longden TA, Ferris HR, et al. 2021. Differential restoration of functional hyperemia by antihypertensive drug classes in hypertension-related cerebral small vessel disease. J. Clin. Investig. 131(18):e149029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Ma S, Cahalan S, LaMonte G, Grubaugh ND, Zeng W, et al. 2018. Common PIEZO1 allele in African populations causes RBC dehydration and attenuates Plasmodium infection. Cell 173(2):443–55.e12 [DOI] [PMC free article] [PubMed] [Google Scholar]