

Abstract
Modular functionalization enables versatile exploration of chemical space and has been broadly applied in structure–activity relationship (SAR) studies of aromatic scaffolds during drug discovery. Recently, the bicyclo[1.1.1]pentane (BCP) motif has increasingly received attention as a bioisosteric replacement of benzene rings due to its ability to improve the physicochemical properties of prospective drug candidates, but studying the SARs of C2-substituted BCPs has been heavily restricted by the need for multistep de novo synthesis of each analogue of interest. Here we report a programmable bis-functionalization strategy to enable late-stage sequential derivatization of BCP bis-boronates, opening up opportunities to explore the SARs of drug candidates possessing multisubstituted BCP motifs. Our approach capitalizes on the inherent chemoselectivity exhibited by BCP bis-boronates, enabling highly selective activation and functionalization of bridgehead (C3)-boronic pinacol esters (Bpin), leaving the C2-Bpin intact and primed for subsequent derivatization. These selective transformations of both BCP bridgehead (C3) and bridge (C2) positions enable access to C1, C2-disubstituted and C1, C2, C3-trisubstituted BCPs that encompass previously unexplored chemical space.
Infinite chemical space provides boundless opportunities for medicinal chemists, within which three-dimensional scaffolds with drug-like properties are particularly sought after1–3. The past decade has witnessed growing interest in the chemical space of bicyclo[1.1.1] pentane (BCP), a class of motifs that are considered medicinally relevant as a three-dimensional bioisosteric replacement of aromatic moieties (Fig. 1a)4–11. Such consideration was driven by its potential to improve physicochemical, pharmacological and toxicological properties of drug candidates6,10,11, with bridgehead (C1 and C3)-substituted BCPs widely recognized as saturated bioisosteres for mono- and para-substituted benzenes in synthetic chemistry12–25. Analogously, it has been hypothesized that C1, C2-disubstituted BCPs could represent bioisosteres of ortho-and meta-substituted benzenes10,26–28. However, despite the recent reports from Baran et al.28, Ma et al.29,30, our group31 and others32–34, efficient synthesis of bridge-substituted BCPs still proves challenging and the practical applications of such BCPs in medicinal chemistry remain heavily constrained. In contrast to C(sp2) cross-coupling chemistry, which enables rapid diversification of arenes35, derivatization of the C(sp3)-rich bioisostere BCPs requires multistep de novo synthesis (Fig. 1b)31. Such constraints represent the biggest bottleneck to access and evaluate these promising compounds in structure–activity relationship (SAR) studies. Thus, development of a modular and programmable strategy to access C1, C2-disubstituted and C1, C2, C3-trisubstituted BCPs should enable more comprehensive SAR campaigns and chemical space exploration.
Fig. 1 |. Significance, challenges and strategy for accessing multi-substituted BCPs.
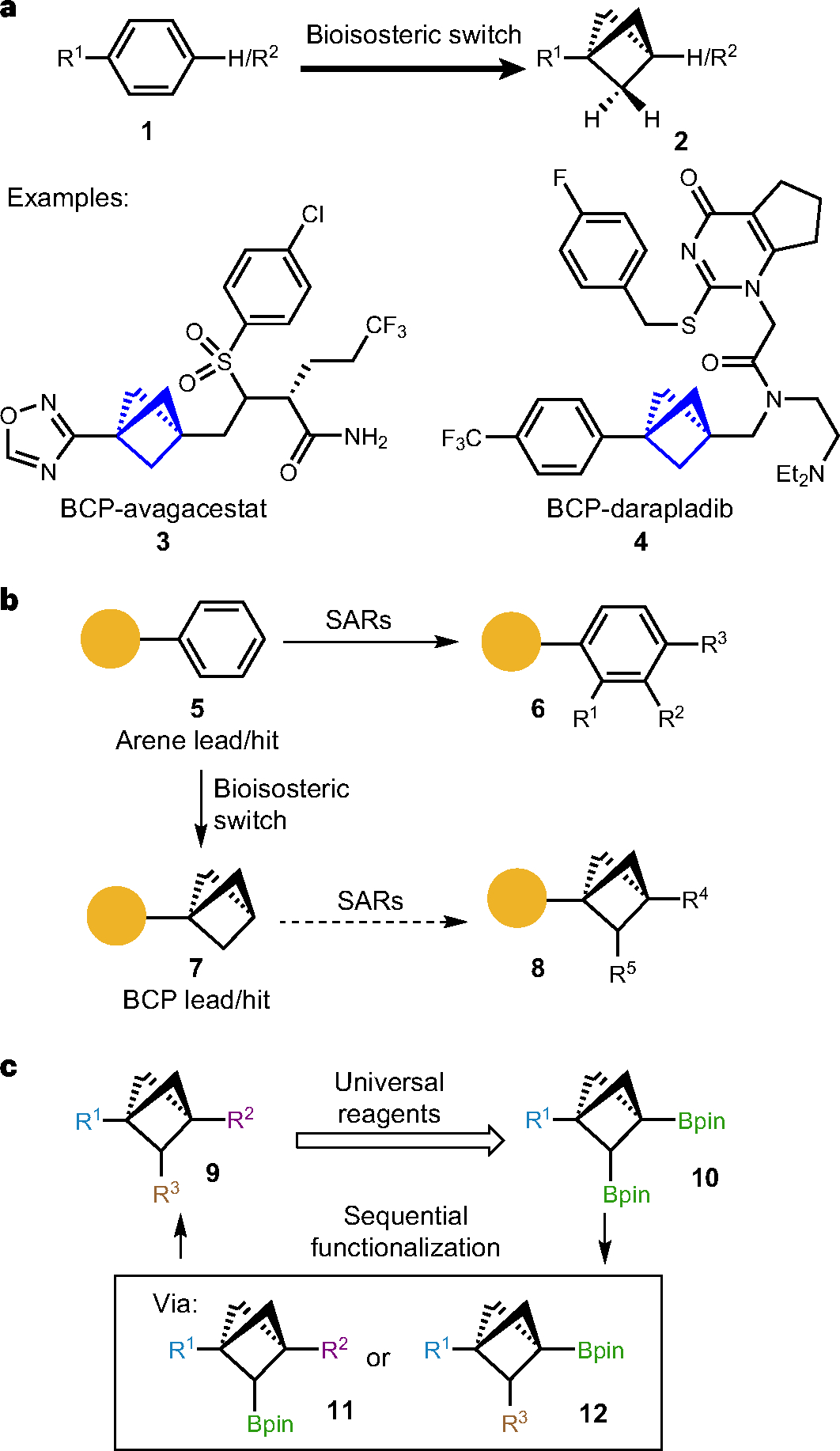
a, Significance of bicyclo[1.1.1]pentanes as a 3D bioisostere for arenes in medicinal chemistry. Bicyclo[1.1.1]pentanes has been applied as benzene 3D-surrogates in drug discovery owing to their metabolic stability from their high BDE and kinetically inhibited HAT process. Several BCP derivatives of drug molecules such as BCP-avagacestat and BCP-darapladib have demonstrated their potential to improve physicochemical, pharmacological (ADME) and toxicological (safety) properties. b, Synthetic challenges in SAR studies with a BCP core structure. Owing to well-established C(sp2)–C(sp2) cross-coupling reactions and abundant available building blocks, it is synthetically accessible to obtain diverse derivatives of arene lead compounds for further SAR studies; however, derivatization of the C(sp3)-rich bioisostere BCPs has been synthetically challenging due to non-programmable synthetic route, requirement of de novo synthesis and inaccessible chemical space. c, Programmable and sequential functionalization of bridge-substituted BCPs. To access multisubstituted BCPs (9), a sequential functionalization strategy from BCP bis-boronate (10) was proposed taking advantage of different reactivity of two boronates. Diversification of R1, chemoselectivity of two boronates and reactivity of boronic esters are main challenges in this strategy.
The inert C–H bonds within BCPs not only provide metabolic stability for this three-dimensional scaffold36–38, but also create the synthetic hurdles to generally access bridge (C2)-substituted derivatives via C–H functionalization27. Despite our earlier success in multisubstituted BCP syntheses via intramolecular coupling strategy31, the installation of the bridge-substitution group before cyclization substantially increased the synthetic steps required to access each analogue. To address these issues, our goal was to identify a late-stage BCP intermediate with two functional handles that could be selectively and rapidly modified to pursue divergent SAR exploration. To that end, our group previously reported31 the preparation of two C1-alkyl BCPs bearing Bpin substituents at both C2 and C3. Inspired by breakthrough reports by Morken39–41, Aggarwal42,43 and others44,45, we postulated that these BCP bis-boronates could be selectively and sequentially functionalized (Fig. 1c) and this synthetic approach would provide rapid access to bridge-substituted C1, C2-di- and C1, C2, C3-tri-substituted BCPs. Here we describe a strategy that leverages the endogenous properties of BCP bis-boronates to enable the modular and efficient synthesis of multisubstituted BCPs via sequential functionalization.
Results and discussion
The striking reactivity difference between C2- and C3-Bpin units was initially observed in the boronate ligand exchange reaction with BCP 13 (Fig. 2a). Under treatment of 1,8-diaminonaphthalene in toluene, selective formation of C3-Bdan-substituted product 15 was identified by NMR analysis and confirmed by single-crystal X-ray diffraction. In comparison, the C2-cyclopentyl-substituted BCP 14 afforded much lower yield of the corresponding product (16, 7%). In a similar vein, this chemoselectivity was also observed in the hydrazone coupling46. The bridgehead C3-Bpin in BCP bis-boronates displayed an enhanced reactivity and underwent coupling with sulfonyl hydrazone 17, whereas C2-Bpin in bis-boronates and BCP 14 remained mostly unreacted (Fig. 2a). We initially hypothesized that—as noted by Morken39,40, and others42,43,44,45—selective activation at the C3 position in this bis-boronate system is probably due to intramolecular coordination (for example, an oxygen lone pair–boron interaction) of the Bpin at the C2 position; however, careful analysis of B3–C3–C1 angles of these BCP X-ray crystal structures (see Fig. 3 and ‘X-ray crystallographic data for BCP compounds’ section in the Supplementary Information) revealed no obvious initial interaction between the two adjacent Bpin groups in the BCP scaffolds, presumably because the strained bicyclic scaffold restricts the spatial orientations of these groups (Fig. 2a).
Fig. 2 |. Preliminary chemoselectivity of BCP bis-boronates and theoretical explanation.
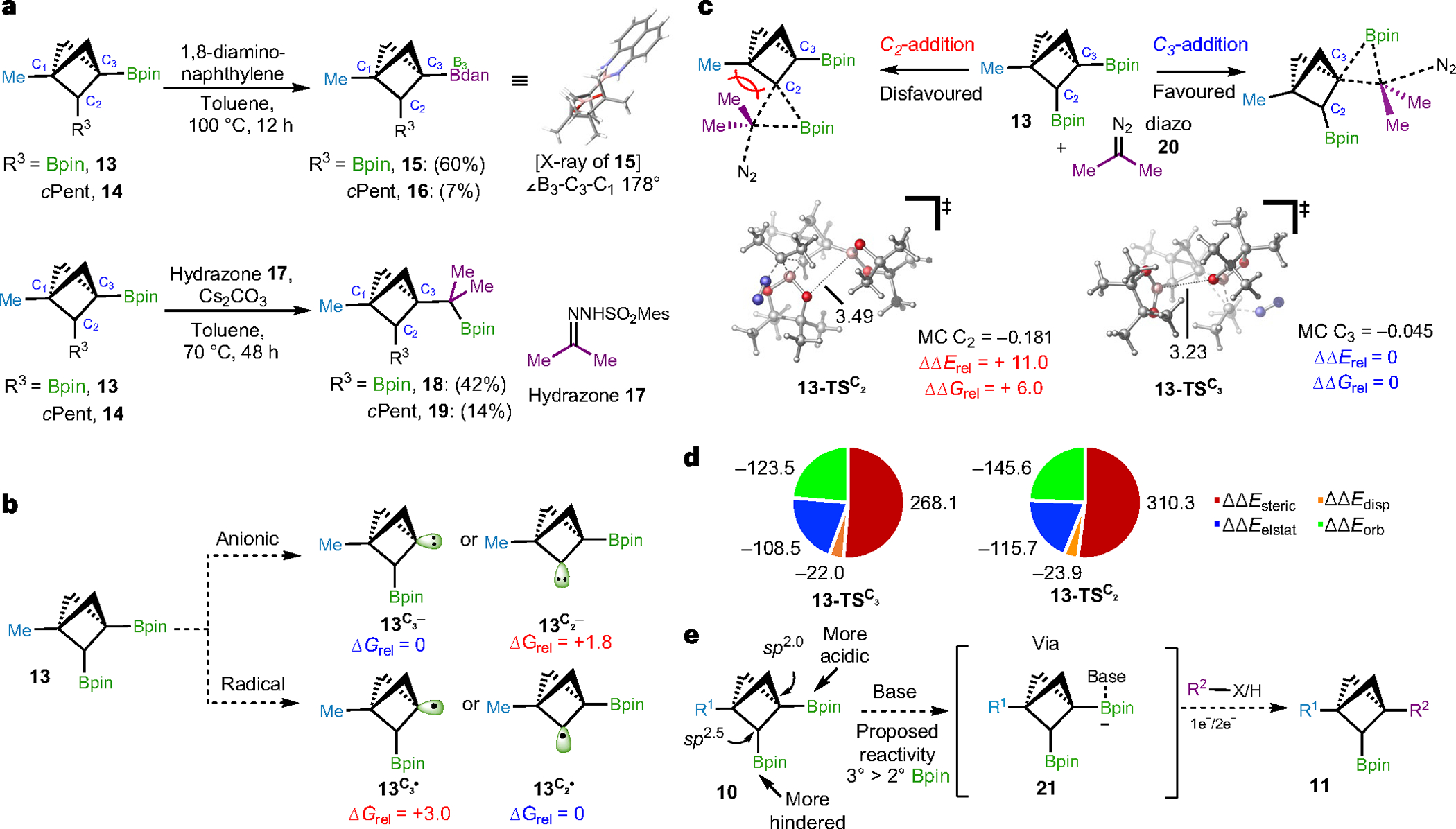
a, Preliminary results for BCP bis-boronates reactivities. In the ligand-exchange and hydrazone coupling reactions, the bridgehead C3-Bpin in BCP bis-boronates presented an increased reactivity and underwent the two transformations, whereas C2-Bpin in bis-boronates and C2-alkyl BCP boronates remained mostly intact. b, Energetic comparison between BCP C3− and C2-anionic and radical intermediates suggest an anion-mediated transformation at the bridgehead position (C3−). c, Concerted addition/migration/N2 extrusion hydrazone coupling at the C3− and C2− positions of BCP bis-boronates show substantial preference for reactivity at C3−. d, Energy decomposition analysis of the concerted transition states in hydrazone coupling reveal that steric interactions greatly impact the selectivity. e, Working hypothesis for selective BCP bis-boronates functionalizations. See Supplementary Information for computational methods and details. Energy values in b, c and d are in kcal mol−1. cPent, cyclopentyl.
Fig. 3 |. Scope and synthesis of BCP bis-boronates.
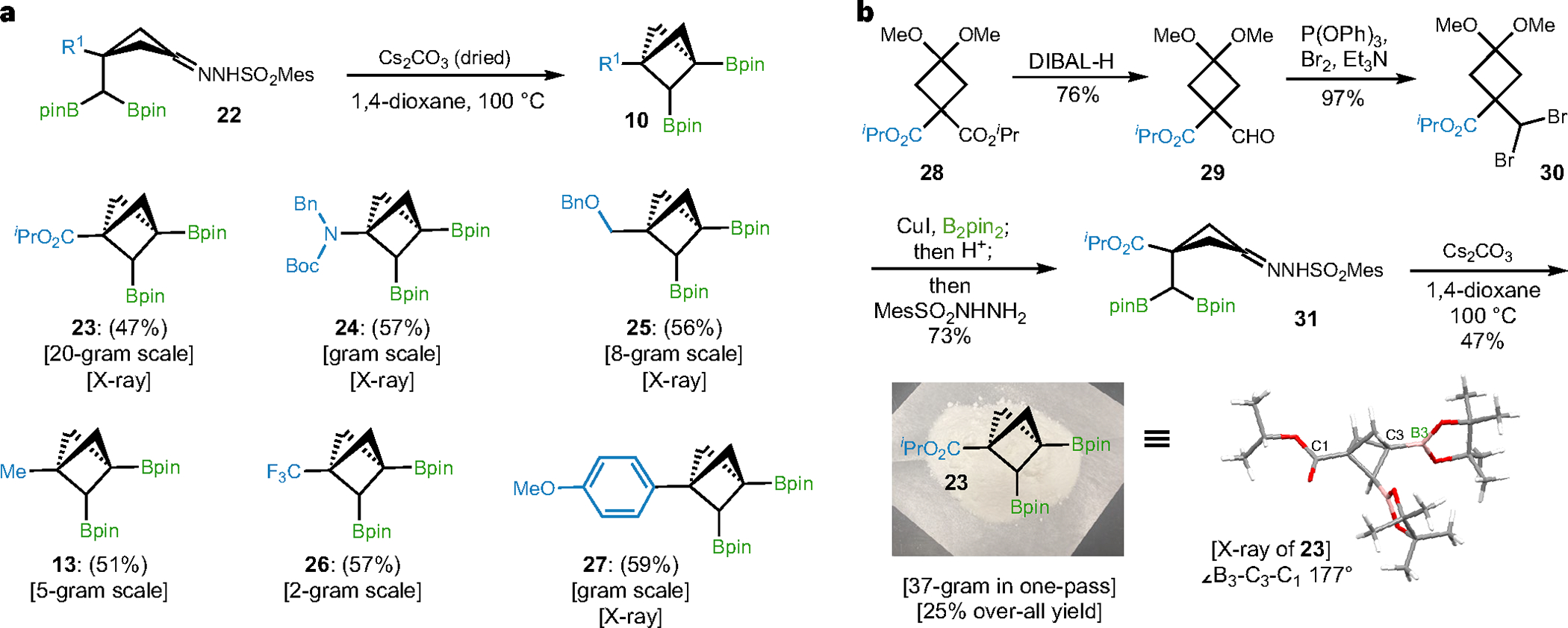
a, Substrate scope of BCP bis-boronates (13, 23–27). Six C1-substituted BCP bis boronates (esters, amino, alkyls, CF3, aryl) were obtained via intramolecular coupling of sulfonyl hydrazones and boronic esters. b, Representative synthesis route towards BCP 23 via (1) DIBAL reduction; (2) dibromination; (3) diborylation/hydrolysis/ hydrazine condensation; and (4) intramolecular coupling. See ‘Synthetic details and optimization of intramolecular coupling’ section in the Supplementary Information for more details.
Quantum mechanical calculations
We turned to quantum mechanical calculations to gain further insights into the selective reactivity of BCP bis-boronates. First, analysis of the lowest energy conformations of BCP bis-boronate 13 found no apparent Bpin–Bpin non-covalent interactions, consistent with the crystal structures (as determined by natural bond orbital analysis and O–B distances; see Supplementary Information for details). Comparison between the putative anionic intermediates generated revealed an appoximately tenfold thermodynamic preference for the C3 (bridgehead) 3° anionic intermediate (ΔΔGrel 13C2−/C3− = +1.8 kcal mol−1; Fig. 2b). In contrast, DFT calculations show an energetic preference for the formation of the 2° alkyl radical, which might occur via C2-Bpin homolysis (ΔΔGrel 13C2⋅/C3⋅ = −3.0 kcal mol−1; Fig. 2b). Such results can be rationalized by the higher s-orbital-character of the C3 carbon (behaving as sp2) compared with the C2 carbon (sp2.5)37,38, leading to a higher electronegativity at the bridgehead position and an increased Lewis acidity of the attached Bpin. Given that exclusive reactivity at the bridgehead position was observed for all transformations (see below), these calculations support an anion-mediated C–B activation pathway (Fig. 2c).
Further study probed into the bond-forming step, in which we modelled the hydrazone coupling reaction46 between bis-boronate 13 and a diazo compound 20 presumably generated from the corresponding sulfonyl hydrazone under basic conditions (Fig. 2c). In agreement with the synthetic outcomes (Fig. 2a), these calculations highlighted a strong preference for activation and functionalization of the C3 carbon versus C2-Bpin functionalization. Specifically, DFT calculations identified an energetically much lower concerted addition/migration/N2 extrusion transition state that will lead to the corresponding C–C coupling at the C3- than at the C2-carbon (13–TSC3 versus 13–TSC2, ΔΔGrel = +6.0 kcal mol–1). Moreover, the resulting C3-product is also more thermodynamically favourable than the C2-coupling product by similar energy (see Supplementary Information). Inspection of the Mulliken charges (MC) of the transition states (Fig. 2c) revealed a greater buildup of negative charge in 13–TSC2 (MC C2 = –0.181; MC C3 = –0.045). Further energy decomposition analysis of 13–TSC2 and 13–TSC3 (Fig. 2d) suggested that despite greater non-covalent electrostatic, orbital, and dispersion stabilizing interactions in 13–TSC2, much more substantial steric interactions were observed in 13–TSC2 (ΔΔEsteric13–TSC2 = +310.3 kcal mol−1, ΔΔEsteric13–TSC3 = +268.1 kcal mol−1), overall resulting in a higher barrier via the C2 transition state (ΔΔErel = +11.0 kcal mol−1; Fig. 2c)47.
Overall, we attribute the selectivity to (1) greater buildup of negative charge and (2) less destabilizing steric interactions between the BCP scaffold and pinacol ligand in the transition states at the C3 position (see Supplementary Information for further details). Taking advantage of the higher reactivity of C3-Bpin, we envisioned a sequential functionalization protocol of BCP bis-boronates, where C3- and C2-Bpin can be transformed into multiple functional groups and access divergent C1, C2, C3-tri-substituted BCPs (Fig. 2e).
Synthesis of BCP bis-boronates
The advantages of this strategy to access bridge-substituted BCPs not only draws from its modularity, but also is reinforced by the synthetic efficacy and practicality in the preparation of various BCP bis-boronates. In our previous study31, the alkyl substituted BCP bis-boronate (13) was synthesized from readily accessible aldehyde via diborylation of its derived sulfonyl hydrazones48 and intramolecular coupling31 on gram scale and in moderate yield. Bicyclo[1.1.1] pentanes 24 and 25 were prepared via similar routes (Fig. 3a); however, the carboxylate ester-, trifluoromethyl- and aryl-substituted BCP bis-boronates were found unobtainable by the challenging access to geminal bisBpin precursors (yield < 5%) and the diminished yield in intramolecular coupling. These BCPs (23, 26, 27) were successfully accessed (Fig. 3a) through crucial modifications including the diborylation of germinal dibromo compounds49 and intramolecular coupling under dry conditions (pre-preparation of sulfonylhydrazone, dry solvent and base). As an example, synthesis of ester C1-substituted BCP 23 (Fig. 3b) starts from an affordable cyclobutyl diester 28. The cyclobutyl sulfonylhydrazone 31 was generated in high yields through a sequence of DIBAL-H reduction, dibromination and diborylation. The following cyclization mediated by anhydrous Cs2CO3 robustly afforded 23 on 37-gram scale in a single pass without deterioration of yield. Notably, all the synthesized BCP bis-boronates are stable crystalline solids and can be stored at −20 °C without any noticeable degradation for several months.
C3-functionalization of BCP bis-boronates
With the knowledge gained from computational results and a variety of BCP bis-boronates prepared, we sought to investigate selective C3-Bpin deborylative functionalization strategies (Table 1). Consistent with theoretical modelling, BCP bis-boronates underwent selective alkylation at the bridgehead (C3) Bpin with sulfonylhydrazones31 (18, 32–35), leaving the resultant alkyl Bpins intact. It is worth noting that other non-BCP alkyl Bpins were generally incompatible with this hydrazone coupling46.
Table 1 |.
Selective C3-Bpin functionalization of BCP bis-boronates
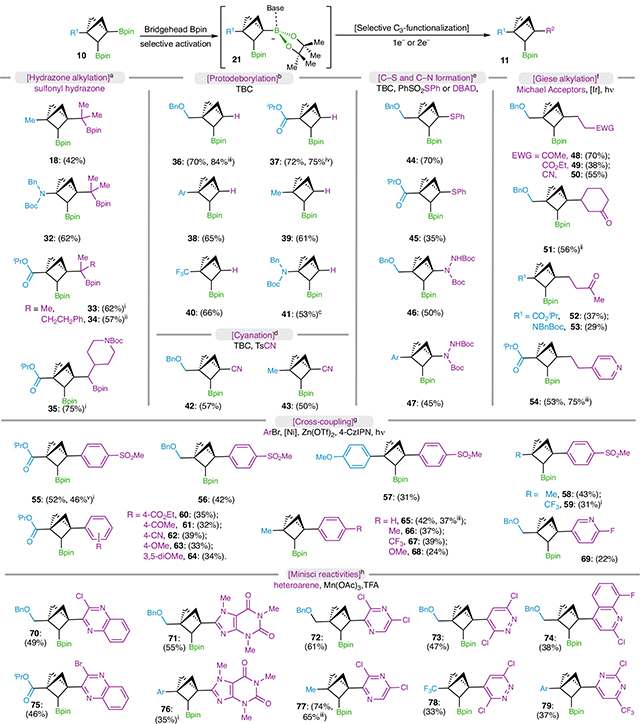
|
Reaction conditions with BCP BisBpin as the Limiting reagent at 0.05–0.2mmol scale:
sulfonyl hydrazone (2.0 equiv.), Cs2CO3 (3.0 equiv.), toluene (0.2M), 70°C, 18–48h
TBC (2.5 equiv.), toluene (0.2M), argon or air atmosphere, 100°C, 2–12h
4-CzlPN (5mol%), MeOBcat (30 mol%), acetone/MeOH (1:1, 0.1 M), blue LEDs, r.t., 2 h
TsCN (2.0 equiv.), TBC (20mol%), toluene (0.2M), 18h
PhSO2SPh or DBAD (2.0 equiv.), TBC (20mol%), toluene (0.2M), 70°C or 100 °C, 18–24h
[Ir] (5mol%), DMAP (30mol%), Michael acceptor (2.0 equiv.), acetone/MeOH (1:1, 0.1M), blue LEDs, r.t., 24h
4-CzlPN (2–5mol%), [Ni] (10–20mol%), ArBr (3.0 equiv.), Zn(OTf)2 (2.0 equiv.), DMAP (4.0 equiv.), DMA (0.2M), blue LEDs, r.t., 24–60h
heteroarene (3.0 equiv.), Mn(OAc)3·2H2O (2.5 equiv.), TFA (2.5 equiv.), AcOH/H2O (1:1, 0.1M), 50°C, 18h.
The structure was verified by X-ray experiments
d.r. =1:1
2.0 mmol scale
10mmol scale
1.0 mmol scale. [Ir], (Ir[dF(CF3)ppy]2(dtbbpy))PF6, [Ni], Ni(dtbbpy)Cl2 or Ni(cod)2+dtbbpy.
EWG, electron-withdrawing group; Bcat, boronic acid catechol ester; ppy, 2-phenylpyridine; dtbbpy, 4,4-di-tert-butyl-2,2-dipyridyl; cod, 1,5-cyclooctadiene; DMA, dimethylacetamide.
Furthermore, our study continued with C3-protodeborylation of BCP bis-boronates to afford C2-Bpin-substituted BCPs, which provides a modular tool to access a library of diverse 1,2-disubstituted BCPs, a class of potential bioisosteres for ortho- and meta-disubstituted arenes. Inspired by Renaud’s studies50,51, BCP bis-boronates (13, 25, 27) underwent effective protodeborylation with tert-butyl catechol (TBC) upon heating under argon atmosphere and C2-Bpin-disubstituted BCPs were afforded with good yields (36, 38–39). Other BCPs containing electron-withdrawing groups (23, 26) at the C1 position featured lower reactivities, and their protodeborylation was successfully promoted by air (37, 40); however, this TBC strategy only led to decomposition of starting material in preparation of nitrogen C3-substituted BCP (41), which was only made possible with a photoinduced protodeborylation approach18.
Enabled by the TBC strategy, the C3-centred radicals formed in situ can also react with tosyl cyanide (TsCN), PhSO2SPh and di-tert-butyl azodicarboxylate (DBAD) to afford nitrile- (42, 43), thioether- (44, 45) and hydrazide- (46, 47) containing BCPs in moderate yields. C3-selective Giese alkylation of BCPs (48–54) was achieved with Ley’s conditions52,53 via the bridgehead C3 radical from photoinduced oxidation of a Bpin-DMAP (4-dimethylaminopyridine) ‘ate’ complex.
Assessment of nickel-catalysed cross-coupling started with aryl bromides—an abundant class of building blocks—yet all of the previously reported conditions failed to afford the desired product54–58. Extensive screening efforts identified a photosensitized method with 4-CzlPN as the photosensitizer and Zn(OTf)2 as a key Lewis acid additive, where arylbromoides with diverse electronic properties were well tolerated (55–69; see Supplementary Information for more details).
Minisci reactivities59 could also be achieved with selective introduction of a series of heteroarenes including quinoxaline (70, 75), caffeine (71, 76), pyrazine (72, 77), pyridazine (73, 78), quinoline (74) and pyrimidine (79), and moderate to good yields of the products were afforded.
Late-stage C2-functionalization of BCP boronates
With several scalable preparations of BCP C2-Bpins (36 and 37, 54–55, 65, 77), further derivatizations of such BCPs were explored to demonstrate broad applications of a sequential functionalization strategy for accessing C1, C2-di- (Fig. 4a) and C1, C2, C3-tri-substituted BCPs (Fig. 4b) at a late stage. Successful transformation of Bpin group in 36 to the more stable trifluoroborate salt (80, 73%) and the boronic acid (81, 68%) created further functionalization opportunities. In comparison with other non-BCP secondary boronic species, BCP C2-boronates were found to exhibit inferior reactivity owing to a combination of specific hybridization38 at the C2 bridge position and steric hinderance36. Fortunately, attempts to leverage BCP 36 in C–O and C–S formation proved successful, as demonstrated in oxidation (82) and Renaud’s radical trapping51 (83). Nickel-catalysed ‘ate’ complex coupling (84)42 with PhLi and Aggarwal’s arylation (85)60 from boronate 36, Molander’s photo-induced cross-coupling (86)55–58 and Minisci-type heteroarylation (87)59 from trifluorobororate salt 80 enables the installation of aryl and heteroaryl groups at the bridge (C2) position. tert-Butyl-catechol-mediated C–N formation affords hydrazine (88)51 in 95% yield and amination with nitroarene via Radosevich’s protocol affords anilines (89, 90)61 in moderate yields. Sulfonyl hydrazone coupling (91)46 of boronic acid (81), Giese-type alkylation (92) and cyanation (93) via ‘ate’ complex42 using PhLi from boronate 36, and Matteson homologation (94)62 afford alkylation, cyanation and CH2 insertion product on the BCP motif in moderate to high yields.
Fig. 4 |. Syntheses of structurally divergent BCP compounds.
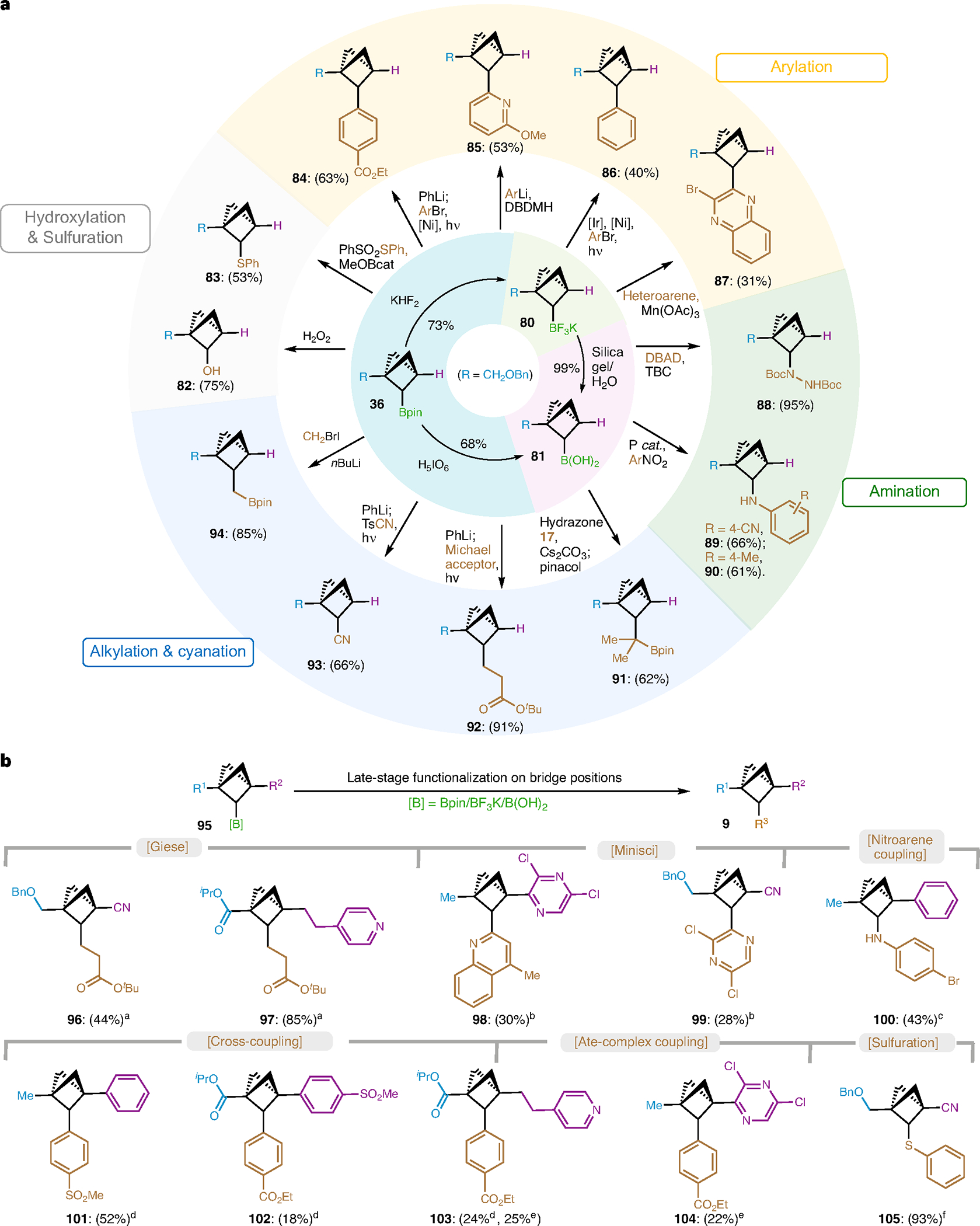
a, Functionalization of C2-Bpin as a versatile building block towards C1, C2-disubstituted BCPs. b, Late-stage functionalization to access C1, C2, C3-trisubstituted BCPs. Reaction conditions: aBCP C2-Bpin (1.0 equiv.), PhLi (1.2 equiv.) THF (0.2 M), −78 °C to r.t., 1 h; then 4-CzlPN (5 mol%), tert-butyl acrylate (2.0 equiv.), THF/MeCN (0.1 M), blue LEDs, 15 h. bBCP C2-BF3K (1.0 equiv.), heteroarene (3.0 equiv.), Mn(OAc)3 (2.5 equiv.), TFA (2.5 equiv.), AcOH/H2O (1:1, 0.1 M), 50 °C, 18 h. cBCP C2-B(OH)2 (1.0 equiv.), ArNO2 (1.0 equiv.), 1,2,2,3,4,4-hexamethyl-phosphetane 1-oxide (15 mol%), PhSiH3 (2.0 equiv.), m-xylene (0.5 M), 120 °C, 8 h. dBCP C2-BF3K (1.0 equiv.), [Ir] (5 mol%), Ni(dtbbpy) Cl2 (20 mol%), ArBr (5.0 equiv.), Cs2CO3 (6.0 equiv.), dioxane or THF (0.1 M), blue LEDs, 24 h. eBCP C2-Bpin (1.0 equiv.), PhLi (1.2 equiv.) THF (0.2 M), −78 °C to r.t., 1 h; then 4-CzlPN (5 mol%), Ni(dtbbpy)Cl2 (10 mol%), ArBr (3.0 equiv.), THF/DMA (0.1 M), blue LEDs, 15 h. fBCP C2-Bpin (1.0 equiv.), PhSO2SPh (4.0 equiv.), MeOBcat (1.0 equiv.), TBC (0.3 equiv.), toluene (0.2 M), 80 °C, 24 h. See the ‘Experimental procedures and characterization data of substrates’ section in Supplementary Information for details.
To further exploit the potential of BCP bis-boronates as a versatile building block, a series of structurally diverse trisubstituted BCPs were accessed through a C3–C2 functionalization sequence. Representatives of bridgehead disubstituted BCP C2-Bpin (42, 54, 55, 65, 77) enabled employment of Bpin functionalization strategies, including Giese-type alkylation (96, 97), Minisci-type heteroarylation (98, 99), amination (100) arylation (101, 102, 103, 104) and sulfuration (105).
Alternatively, to first achieve C2-Bpin functionalization, C3-Bpin was transformed into N-methylimidodiacetic boronic acid ester (BMIDA) or tetramethyl N-methyliminodiacetic boronic acid ester (BTIDA) (109, 110)63,64, followed by selective functionalization of C2-Bpin via oxidation (111) or Giese-type alkylation (112) with the C3-boronates remaining intact (Extended Data Fig. 1).
Furthermore, the medicinal chemistry rationale for pursuing poly-substituted BCPs was showcased via the improvement in physicochemical and ADME properties of biologically relevant compounds. The BCP match-pair 115 of arene 116 a p-38 kinase inhibitor65, was prepared via the sequential functionalization strategy from 43, and physicochemical and ADME properties for both analogues profiled (Extended Data Fig. 2). The data highlights that the theoretical improvement in Fsp3 (fraction of sp3 carbon atoms) values by introducing the BCP also results in a substantial improvement in measured solubility (>10-fold) and reduction of lipophilicity, while retaining excellent permeability.
Bicyclo[1.1.1]pentane derivatives feature characteristic three-dimensional topology and represent a C(sp3)-rich chemical space. In a comparative study we enumerated libraries of trisubstituted BCP derivatives (1,2,4- and 1,3,4-trisubstituted arenes) with substituents selected from synthetically accessible moieties demonstrated earlier (with protecting groups removed; see Supplementary Information for more details). These compounds were evaluated with principal moments of inertia (PMI) metrics, where shape diversity of a given library can be assessed via computation of principal moments of inertia for each molecule (see Supplementary Information for more details)66. In contrast to arene compounds, trisubstituted BCP compounds featured much greater structural diversity as they occupied much broader space (Fig. 5a). More importantly, BCP compounds exhibited higher three-dimensionality as they extend into the sphere-like space, whereas the corresponding arenes are heavily concentrated along the ‘rod-disk’ axis.
Fig. 5 |. PMIs between substituted BCPs and arenes.
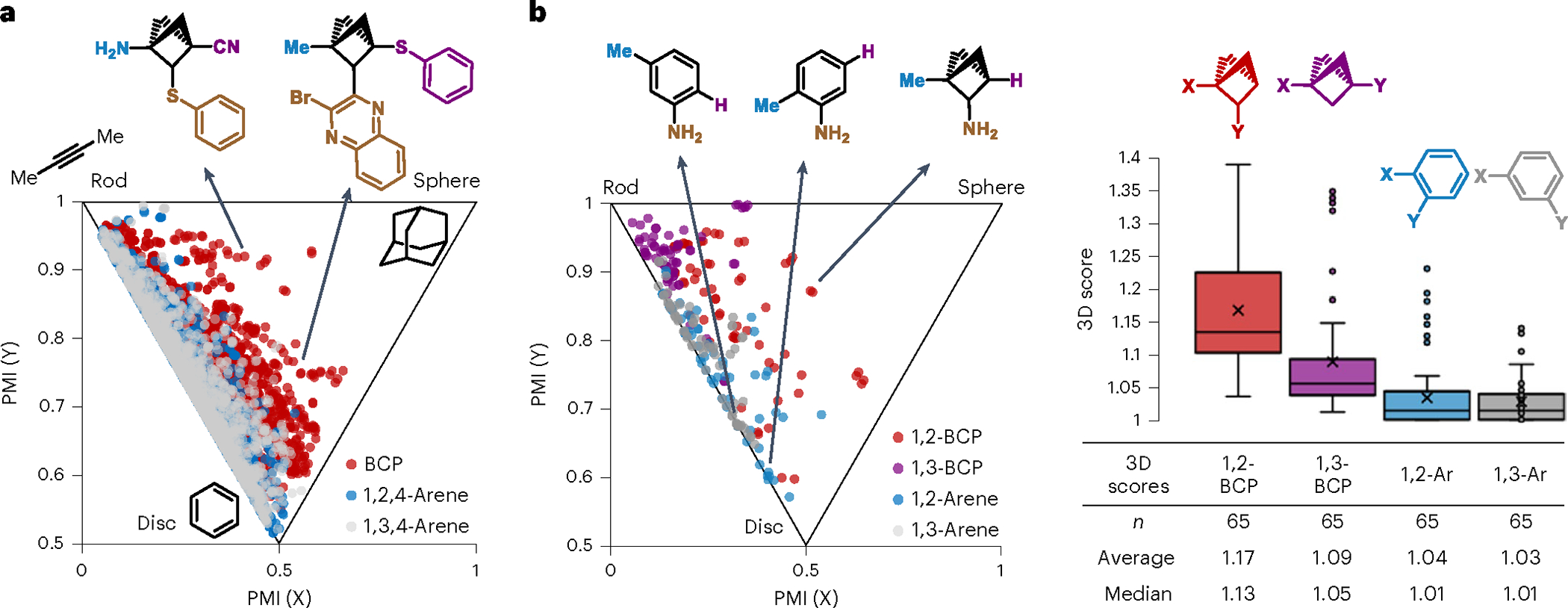
a, PMI analysis of trisubstituted BCP and arene chemical space. b, PMI and 3D scores comparison between selected disubstituted BCP and arenes. 3D scores were presented as box-whisker plots ranging between 1.00 and 2.00. The lines are the median, the average is marked by crosses, and outliers are shown as dots. See Methods and Source Data Fig. 5 for details (quartiles, boundaries and so on) on the box-whisker plots.
Further study focused on a selection of difunctionalized BCP compounds and arenes sharing the same set of substituents (Fig. 5b; see the Supplementary Information for details). Analysis of these compounds indicated that C1, C2-difunctionalized BCP compounds are more three-dimensional than 1, 2-and 1,3-difunctionalized arenes and C1, C3-disubstituted BCP compounds. Calculation of 3D scores67, that is, the sum of PMI (X) and PMI (Y), quantitatively demonstrated the merit of this C2-functionalization strategy, as the C1, C2-difunctionalized BCP compounds scored broader and overall higher (average = 1.17, median = 1.13; Fig. 5b) than C1, C2-BCP compounds (average = 1.09, median = 1.05) and arenes (average ≤ 1.04, median ≤ 1.01) in the box-whisker plot. These results together suggested that this BCP C2-functionalization strategy represent a unique entry to a novel three-dimensional chemical space.
In summary we developed a selective and sequential bis-functionalization strategy of BCP bis-boronates. Computational studies shed insights into the differentiated reactivities between C2- and C3-Bpins and such results proved consistent with synthetic outcomes. Combination of the scalable, crystalline BCP bis-boronates and enabling Bpin functionalization strategies allowed the access to an array of novel, diverse di- and tri-substituted BCP compounds. Systematic investigation of large and yet unexplored chemical space of C2-substitution on BCPs demonstrated their higher shape diversity and three-dimensionality than conventional arene derivatives. Further application of this approach should help assemble molecular libraries with more three-dimensional structures and create a broader avenue to SAR studies68–71.
Online content
Any methods, additional references, Nature Portfolio reporting summaries, source data, extended data, Supplementary Information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41557-023-01342-7.
Methods
General procedure for intramolecular coupling to access BCP bis-boronates
A one-necked (24/40 joint) round-bottomed flask, equipped with a teflon-coated magnetic stir bar, was flame-dried under vacuum and then cooled to 23 °C under an atmosphere of argon. Then the flask was charged with sulfonyl hydrazone (1.0 equiv.) and dried cesium carbonate (3.0 equiv.). (Note: cesium carbonate was dried at 120 °C under vacuum for 18 h.). After being evacuated and backfilled with argon from a balloon three times, dioxane (0.2 M) was added into the flask and the reaction mixture was allowed to stir at 100 °C. After it was confirmed that the starting material, sulfonyl hydrazone, was consumed through TLC analysis, the reaction was cooled to room temperature, filtered through Celite, washed with excess hexanes, and concentrated to remove excess solvents. The crude reaction was purified through flash chromatography (hexanes: ethyl acetate, 20:1 to 10:1) on silica gel to afford the title compound, which was further purified through recrystallization in hexanes at −40 °C, affording the product with >99% purity as white solids31.
Recrystallization procedure.
The product after chromatography was dissolved in hexanes (around 1.0 ml g−1) at room temperature and then cooled to −40 °C. After the solution of the product was slowly stirred at −40 °C for 1 h, the suspension was filtered and the white solid was washed with cooled hexanes quickly and dried under vacuum for 1 h.
General procedure A for hydrazone coupling of BCP bis-boronates
A screw-capped 13 × 100 mm Pyrex culture tube was charged with cesium carbonate (3.0 equiv.), 2-mesitylsulfonyl hydrazone (2.0 equiv.) and BCP bis-boronate (1.0 equiv.). Then the tube was evacuated and backfilled with argon for three times, followed by addition of toluene (0.1 M) via a syringe. After stirring for at 70 °C for 18–48 h, the reaction mixture was cooled to room temperature. The suspended solution was then filtered over celite and washed with diethyl ether. The solvent was removed under high vacuum, and the crude residue was purified by chromatography on silica gel.
The procedure was applied in the synthesis of compounds 18 and 32–35.
General procedure B for deborylation of BCP bis-boronates
A screw-capped 13 × 100 mm Pyrex culture tube or a flame-dried 100 ml Pyrex flask was charged with BCP bisboronate (1.0 equiv.) and tert-butyl catechol (2.5 equiv.). The tube or the flask was then evacuated and backfilled with argon or air for three times, followed by addition of toluene (0.1 M) via a syringe. After stirring for at 100 °C for 2–12 h when it is confirmed that the starting material was consumed totally, the reaction mixture was cooled to room temperature. Next, the solvent was removed under high vacuum, and the crude residue was purified by chromatography on silica gel.
The procedure was applied in the synthesis of compounds 36–41.
General procedures C for cyanation of BCP bis-boronates
A flame-dried screw-capped 13 × 100 mm Pyrex culture tube was charged with BCP bisboronate (1.0 equiv.), para-toluenesulfonyl cyanide (2.0 equiv.) and tert-butyl catechol (0.25 equiv.). Then the tube or the flask was evacuated and backfilled with argon for three times, followed by addition of toluene (0.2 M) via a syringe. After stirring for at 70 °C for 18–24 hours when it is confirmed that the starting material was consumed totally, the reaction mixture was cooled to room temperature. Next, the solvent was removed under high vacuum, and the crude residue was purified by chromatography on silica gel.
The procedure was applied in the synthesis of compounds 42 and 43.
General procedures D for C–S and C–N formation of BCP bis-boronates
A flame-dried screw-capped 13 × 100 mm Pyrex culture tube was charged with BCP bisboronate (1.0 equiv.), radical trapping reagent (2.0 equiv., PhSO2SPh or DBAD) and tert-butyl catechol (0.25 equiv.). Then the tube or the flask was evacuated and backfilled with argon for three times, followed by addition of toluene (0.2 M) via a syringe. After stirring for at 70 °C or 100 °C for 18–24 h when it is confirmed that the starting material was consumed totally, the reaction mixture was cooled to room temperature. Next, the solvent was removed under high vacuum, and the crude residue was purified by chromatography on silica gel.
The procedure was applied in the synthesis of compounds 44–47.
General procedure E for Giese-type reaction of BCP bis-boronates
A screw-capped 13 × 100 mm Pyrex culture tube was charged with (Ir[dF(CF3)ppy]2(dtbpy))PF6 (5 mol%), DMAP (30 mol%), and BCP bisboronate (1.0 equiv.). Then the tube or the flask was evacuated and backfilled with argon for three times, followed by addition of Michael acceptor (2.0 equiv.) and methanol/acetone (0.1 M, 1:1) solvent via a syringe. Then the headspace of the tube was purged with a gentle stream of argon for approximately 10 s. After stirring in a 450 nm photoreactor for 24 h when it is confirmed that the starting material was consumed totally, the reaction mixture was concentrated under high vacuum, and the crude residue was purified by chromatography on silica gel.
The procedure was applied in the synthesis of compounds 48–54.
General procedure F for cross-coupling of BCP bis-boronates General procedure F1.
A flame-dried screw-capped 13 × 100 mm Pyrex culture tube was charged with BCP bisboronate (1.0 equiv.), aryl bromide (3.0 equiv.), 4-CzlPN (5 mol%), Ni(dtbbpy)Cl2 (10 mol%), Zn(OTf)2 (2.0 equiv.) and DMAP (4.0 equiv.). Then the tube was evacuated and backfilled with argon three times, followed by addition of DMA (0.2 M) solvent via a syringe. The headspace of the tube was then purged with a gentle stream of argon for approximately 10 s and the reaction was allowed to stir in a 450 nm PennPhD integrated photoreactor (M2) for 24–60 h. After it is confirmed that the starting material was consumed totally, the reaction mixture quenched with water, extracted with ethyl acetate or diethyl ether, washed with brine, dried by Na2SO4 and concentrated under high vacuum. The crude residue was purified by chromatography on silica gel.
The procedure was applied in the synthesis of compounds 56 and 69.
General procedure F2.
Preparation of [Ni] catalyst: a flame-dried screw-capped 13 × 100 mm Pyrex culture tube was charged with Ni(cod)2 (0.2 mmol, 55 mg) and dtbbpy (0.24 mmol, 64.4 mg). Then the tube was evacuated and backfilled with argon three times, followed by addition of DMA (4.0 ml) solvent via a syringe. The tube was then sonicated for 20 min to dissolve the catalyst.
A flame-dried screw-capped 13 × 100 mm Pyrex culture tube was charged with BCP bisboronate (1.0 equiv.), aryl bromide (3.0 equiv.), Zn(OTf)2 (2.0 equiv.) and DMAP (4.0 equiv.). Then the tube was evacuated and backfilled with argon for three times, followed by addition of 4-CzlPN solution (0.02 equiv., 0.02 M) in DMA and [Ni] catalyst solution (0.2 equiv., 0.05 M) in DMA via a syringe. The headspace of the tube was then purged with a gentle stream of argon for approximately 10 s and the reaction was allowed to stir in a 450 nm PennPhD integrated photoreactor (M2) for 24–60 h. After it is confirmed that the starting material was consumed totally, the reaction mixture quenched with water, extracted with ethyl acetate or diethyl ether, washed with brine, dried by Na2SO4 and concentrated under high vacuum. The crude residue was purified by chromatography on silica gel.
The procedure was applied in the synthesis of compounds 55, 57–68.
General procedure G for Minisci reaction of BCP bis-boronates
A screw-capped 13 × 100 mm Pyrex culture tube was charged with BCP bisboronate (1.0 equiv.), heteroarene (3.0 equiv.) and Mn(OAc)3 (2.5 equiv.). Then the tube or the flask was evacuated and backfilled with argon for three times, followed by addition of acetic acid/water (0.1 M, 1:1) solvent via a syringe. Trifluoroacetic acid (5.0 equiv.) was then added into the reaction. The headspace of the tube was then purged with a gentle stream of argon for approximately 10 s and the reaction was stirred at 50 °C for 18 h. After it was confirmed that the starting material was completely consumed, the reaction mixture was concentrated under high vacuum to remove excess acetic acid, quenched with Na2CO3 solution, extracted with ethyl acetate, dried with Na2SO4 and concentrated under high vacuum. The crude residue was purified by chromatography on silica gel.
The procedure was applied in the synthesis of compounds 70–79.
Data availability
Experimental data as well as characterization data for all new compounds prepared in the course of these studies are provided in the Supplementary Information. Crystallographic data for the structures reported in this Article have been deposited at the Cambridge Crystallographic Data Centre under deposition nos. CCDC 2158998 (15), 2159002 (23), 2158995 (24), 2159016 (25), 2159001 (27), 2162135 (33), 2160325 (55), 2160336 (59) and 2162136 (76), see the ‘X-ray crystallographic data’ section in the Supplementary Information. Copies of the data can be obtained free of charge via https://www.ccdc.cam.ac.uk/structures/. Source Data are provided with this paper.
Extended Data
Extended Data Fig. 1 |. Selective functionalization of C2-boronate.
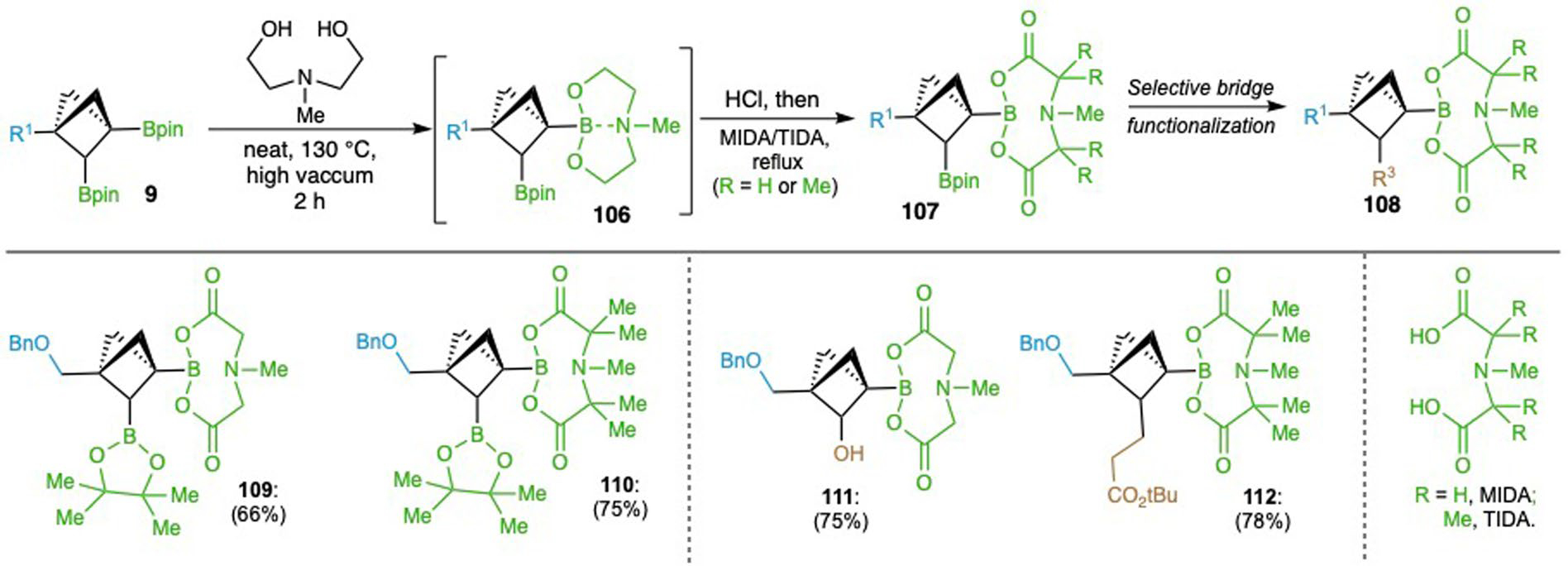
To achieve C2-functionalization with C3-boron retained, BCP bis-boronate 23 was firstly transformed into C3-BMIDA and BTIDA esters (109, 110), followed by oxidation and Giese-type alkylation to afford C2-functionalized products (111,112).
Extended Data Fig. 2 |. Synthesis and ADME study of p-38 kinase inhibitor 116 and its BCP analogue 115.
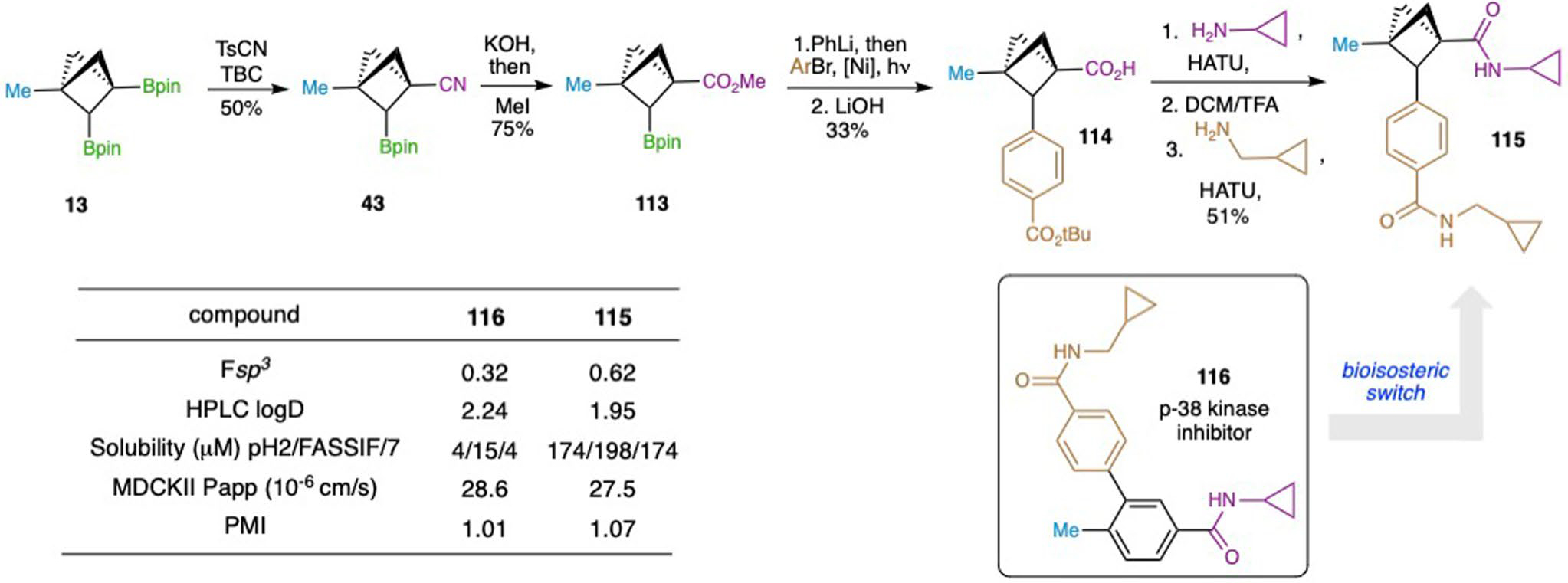
The BCP analogue 115 of arene 116, a p-38 kinase inhibitor65, was prepared via the selective functionalization sequence: 1) cyanation; 2) hydrolysis and esterification; 3) arylation; 4) hydrolyses and amide couplings. The physicochemical and ADME properties for both compounds were profiled. Fsp3, the fraction of sp3 carbon atoms; Log D, distribution coefficient; Solubility, high-throughput equilibrium solubility; MDCKII, Madin-Darby canine kidney cells; Papp, apparent permeability.
Supplementary Material
Acknowledgements
Financial support for this work was provided by the National Science Foundation (CAREER CHE-2143925 to T.Q.), National Institutes of Health (R01GM141088 to T.Q. and R35GM137797 to O.G.), Camille and Henry Dreyfus Foundation (to O.G.) and UT Southwestern Eugene McDermott Scholarship (to T.Q.). Preliminary results were made possible by the support of Welch Foundation (I-2010–20190330 to T.Q.) and American Chemistry Society Petroleum Research Fund (62223-DNI1 to T. Q.). UT Southwestern Amgen Scholars program supported a fellowship to J.B.W. We thank F. Lin (UTSW) for assistance with NMR spectroscopy; H. Baniasadi (UTSW) for HRMS; and V. Lynch (UT-Austin) for X-ray crystallographic analysis. We gratefully acknowledge the Texas A&M University HPRC resources (https://hprc.tamu.edu), UMD Deepthought2, MARCC/BlueCrab HPC clusters and XSEDE (CHE160082 and CHE160053) for computational resources. We thank the Chen, Tambar, Ready, De Brabander, Smith and Falck groups (UTSW) for generous access to equipment, and helpful discussions. We are grateful to S. W. Krska, X. Ma, and D. Levorse (Merck & Co., Inc.) for feedback on this manuscript and assistance with ADME profiling.
Footnotes
Competing interests
The authors declare the following competing financial interest(s): T.Q., Y.Y. and J.T. from UT Southwestern Medical Center are listed as inventors on US patent application no. 63/146,266, which covers the ‘synthesis of BCP bis-boronates’ in the manuscript, and on the US provisional application no. 63/321,700, which covers ‘C3-functionalization of BCP bis-boronates’ and ‘late-stage C2-functionalization of BCP boronates’ in the manuscript. The remaining authors declare no competing interests.
Additional information
Extended data is available for this paper at https://doi.org/10.1038/s41557-023-01342-7.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41557-023-01342-7.
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
References
- 1.Lovering F, Bikker J & Humblet C Escape from flatland: increasing saturation as an approach to improving clinical success. J. Med. Chem. 52, 6752–6756 (2009). [DOI] [PubMed] [Google Scholar]
- 2.Talele TT Opportunities for tapping into three-dimensional chemical space through a quaternary carbon. J. Med. Chem. 63, 13291–13315 (2020). [DOI] [PubMed] [Google Scholar]
- 3.Blakemore DC et al. Organic synthesis provides opportunities to transform drug discovery. Nat. Chem. 10, 383–394 (2018). [DOI] [PubMed] [Google Scholar]
- 4.Costantino G et al. Synthesis and biological evaluation of 2-(3′-(1H-tetrazol-5-yl)bicyclo[1.1.1]pent-1-yl)glycine (S-TBPG), a novel mGlu1 receptor antagonist. Bioorg. Med. Chem. 9, 221–227 (2001). [DOI] [PubMed] [Google Scholar]
- 5.Mikhailiuk PK et al. Conformationally rigid trifluoromethyl-substituted α-amino acid designed for peptide structure analysis by solid-state 19F NMR spectroscopy. Angew. Chem. Int. Ed. 45, 5659–5661 (2006). [DOI] [PubMed] [Google Scholar]
- 6.Stepan AF et al. Application of the bicyclo[1.1.1]pentane motif as a nonclassical phenyl ring bioisostere in the design of a potent and orally active γ-secretase inhibitor. J. Med. Chem. 55, 3414–3424 (2012). [DOI] [PubMed] [Google Scholar]
- 7.Westphal MV, Wolfstaedter BT, Plancher J, Gatfield J & Carreira EM Evaluation of tert-butyl isosteres: case studies of physicochemical and pharmacokinetic properties, efficacies, and activities. ChemMedChem 10, 461–469 (2015). [DOI] [PubMed] [Google Scholar]
- 8.Measom ND et al. Investigation of a bicyclo[1.1.1]pentane as a phenyl replacement within an LpPLA2 Inhibitor. ACS Med. Chem. Lett. 8, 43–48 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Auberson YP et al. Improving nonspecific binding and solubility: bicycloalkyl groups and cubanes as para-phenyl bioisosteres. ChemMedChem 12, 590–598 (2017). [DOI] [PubMed] [Google Scholar]
- 10.Mikhaliuk PK Saturated bioisoteres of benzene: where to go next? Org. Biomol. Chem. 17, 2839–2849 (2019). [DOI] [PubMed] [Google Scholar]
- 11.Subbaiah MAM & Meanwell NA Bioisosteres of the phenyl ring: recent strategic applications in lead optimization and drug design. J. Med. Chem. 64, 14046–14128 (2021). [DOI] [PubMed] [Google Scholar]
- 12.Gianatassio R et al. Strain-release amination. Science 351, 241–246 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Caputo DFJ et al. Synthesis and applications of highly functionalized 1-halo-3-substituted bicyclo[1.1.1]pentanes. Chem. Sci. 9, 5295–5300 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nugent J et al. A general route to bicyclo[1.1.1]pentanes through photoredox catalysis. ACS Catal. 9, 9568–9574 (2019). [Google Scholar]
- 15.Zhang X et al. Copper-mediated synthesis of drug-like bicyclopentanes. Nature 580, 220–226 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Trongsiriwat N et al. Reactions of 2-aryl-1,3-dithianes and [1.1.1] propellane. Angew. Chem. Int. Ed. 58, 13416–13420 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yu S, Jing C, Noble A & Aggarwal VK 1,3-Difunctionalizations of [1.1.1]-propellane via 1,2-metalate rearrangements of boronate complexes. Angew. Chem. Int. Ed. 59, 3917–3921 (2020). [DOI] [PubMed] [Google Scholar]
- 18.Kim JH, Ruffoni A, Al-Faiyz YSS, Sheikh N & Leonori D Divergent strain-release amino-functionalization of [1.1.1] propellane with electrophilic nitrogen-radicals. Angew. Chem. Int. Ed. 59, 8225–8231 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Garlets ZJ et al. Enantioselective C–H functionalization of bicyclo[1.1.1]pentanes. Nat. Catal. 3, 351–357 (2020). [Google Scholar]
- 20.Harmata AS, Spiller TE, Sowden MJ & Stephenson CRJ Photochemical formal (4 + 2)-cycloaddition of imine-substituted bicyclo[1.1.1]pentanes and alkenes. J. Am. Chem. Soc. 143, 21223–21228 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yu S, Jing C, Noble A & Aggarwal VK Iridium-catalyzed enantioselective synthesis of α-chiral bicyclo[1.1.1]pentanes by 1,3-difunctionalization of [1.1.1]propellane. Org. Lett. 22, 5650–5655 (2020). [DOI] [PubMed] [Google Scholar]
- 22.Polites VC, Badir SO, Keess S, Jolit A & Molander GA Nickel-catalyzed decarboxylative cross-coupling of bicyclo[1.1.1]pentyl radicals enabled by electron donor-acceptor complex photoactivation. Org. Lett. 23, 4828–4833 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nugent J, Sterling AJ, Frank N, Mousseau JJ & Anderson EA Synthesis of α-quaternary bicyclo[1.1.1]pentanes through synergistic organophotoredox and hydrogen atom transfer catalysis. Org. Lett. 23, 8628–8633 (2021). [DOI] [PubMed] [Google Scholar]
- 24.Wong MLJ, Sterling AJ, Mousseau JJ, Duarte F & Anderson EA Direct catalytic asymmetric synthesis of α-chiral bicyclo[1.1.1]pentanes. Nat. Commun. 12, 1644 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pickford HD et al. Twofold radical-based synthesis of N,C-difunctionalized bicyclo[1.1.1]pentanes. J. Am. Chem. Soc. 143, 9729–9736 (2021). [DOI] [PubMed] [Google Scholar]
- 26.Denisenko A, Garbuz P, Shishkina SV, Voloshchuk NM & Mikhailiuk PK Saturated bioisosteres of ortho-substituted benzenes. Angew. Chem. Int. Ed. 59, 20515–20521 (2020). [DOI] [PubMed] [Google Scholar]
- 27.Anderson JM, Measom ND, Murphy JA & Poole DL Bridge functionalisation of bicyclo[1.1.1]pentane derivatives. Angew. Chem. Int. Ed. 60, 24754–24769 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhao J-X et al. 1,2-Difunctionalized bicyclo[1.1.1]pentanes: long-sought-after mimetics for ortho/meta-substituted arenes. Proc. Natl Acad. Sci. USA 118, e2108881118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ma X, Han Y & Bennett DJ Selective synthesis of 1-dialkylamino-2-alkylbicyclo-[1.1.1]pentanes. Org. Lett. 22, 9133–9138 (2020). [DOI] [PubMed] [Google Scholar]
- 30.Ma X, Sloman DL, Han Y & Bennett DJ A selective synthesis of 2,2-difluorobicyclo[1.1.1]pentane analogues:’BCP-F2’. Org. Lett. 21, 7199–7203 (2019). [DOI] [PubMed] [Google Scholar]
- 31.Yang Y et al. An intramolecular coupling approach to alkyl bioisosteres for the synthesis of multisubstituted bicycloalkyl boronates. Nat. Chem. 13, 950–955 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Applequist DE, Renken TL & Wheeler JW Polar substituent effects in 1,3-disubstituted bicyclo[1.1.1]pentanes. J. Org. Chem. 47, 4985–4995 (1982). [Google Scholar]
- 33.Bychek RM et al. Difluoro-substituted bicyclo[1.1.1]pentanes for medicinal chemistry: design, synthesis, and characterization. J. Org. Chem. 84, 15106–15117 (2019). [DOI] [PubMed] [Google Scholar]
- 34.Ryan EM, Amber LT & Edward AA Synthesis and applications of polysubstituted bicyclo[1.1.0]butanes. J. Am. Chem. Soc. 143, 21246–21251 (2021). [DOI] [PubMed] [Google Scholar]
- 35.Buskes MJ & Blanco MJ Impact of cross-coupling reactions in drug discovery and development. Molecules 25, 3493–3514 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wiberg KB & Williams VZ Bicyclo[1.1.1]pentane derivatives. J. Org. Chem. 35, 369–373 (1970). [Google Scholar]
- 37.Levin MD, Kaszynski P & Michl J Bicyclo[1.1.1]pentanes, [n] saffanes, [1.1.1]propellanes, and tricyclo[2.1.0.02,5]pentanes. Chem. Rev. 100, 169–223 (2000). [DOI] [PubMed] [Google Scholar]
- 38.Jarret RM & Cusumano L 13C–13C coupling in [1.1.1]propellane. Tetrahedron Lett. 31, 171–174 (1990). [Google Scholar]
- 39.Mlynarski SN, Schuster CH & Morken JP Asymmetric synthesis from terminal alkenes by cascades of diboration and cross-coupling. Nature 505, 386–390 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Blaisdell TP & Morken JP Hydroxyl-directed cross-coupling: a scalable synthesis of debromohamigeran E and other targets of interest. J. Am. Chem. Soc. 137, 8712 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu X, Sun C, Mlynarski S & Morken JP Synthesis and stereochemical assignment of arenolide. Org. Lett. 20, 1898 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kaiser D, Noble A, Fasano V & Aggarwal VK 1,2-Boron shifts of β-boryl radicals generated from bis-boronic esters using photoredox catalysis. J. Am. Chem. Soc. 141, 14104–14109 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fawcett A et al. Regio- and stereoselective homologation of 1,2-bis(boronic esters): stereocontrolled synthesis of 1,3-diols and Sch725674. Angew. Chem., Int. Ed. 55, 14663–14667 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nóvoa L, Trulli L, Parra A & Tortosa M Stereoselective diboration of spirocyclo-butenes: a platform for the synthesis of spirocycles with orthogonal exit vectors. Angew. Chem. Int. Ed. 60, 11763–11768 (2021). [DOI] [PubMed] [Google Scholar]
- 45.Crudden CM et al. Iterative protecting group-free cross-coupling leading to chiral multiply arylated structures. Nat. Commun. 7, 11065–11071 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang Y et al. Practical and modular construction of C(sp3)–rich alkyl boron compounds. J. Am. Soc. Chem. 143, 471–480 (2021). [DOI] [PubMed] [Google Scholar]
- 47.Qi X, Kohler DG, Hull KL & Liu P Energy decomposition analyses reveal the origins of catalyst and nucleophile effects on regioselectivity in nucleopalladation of alkenes. J. Am. Chem. Soc. 141, 11892–11904 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li H, Wang L, Zhang Y & Wang J Transition-metal-free synthesis of pinacol alkylboronates from tosylhydrazones. Angew. Chem. Int. Ed. 51, 2943–2946 (2012). [DOI] [PubMed] [Google Scholar]
- 49.Yang CT, Zhang ZQ, Liu YC & Liu L Copper-catalyzed cross-coupling reaction of organoboron compounds with primary alkyl halides and pseudohalides†. Angew. Chem. Int. Ed. 50, 3904–3907 (2011). [DOI] [PubMed] [Google Scholar]
- 50.Pozzi D, Scanlan EM & Renaud P A mild radical procedure for the reduction of B-alkylcatecholboranes to alkanes. J. Am. Chem. Soc. 127, 14204–14205 (2005). [DOI] [PubMed] [Google Scholar]
- 51.André-Joyaux E, Kuzovlev A, Tappin ND & Renaud P A general approach to deboronative radical chain reaction with pinacol alkylboronic esters. Angew. Chem. Int. Ed. 59, 13859–13864 (2020). [DOI] [PubMed] [Google Scholar]
- 52.Lima F et al. A Lewis base catalysis approach for the photoredox activation of boronic acids and esters. Angew. Chem. Int. Ed. 56, 15136–15140 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lima F et al. Organic photocatalysis for the radical couplings of boronic acid derivatives in batch and flow. Chem. Commun. 54, 5606–5609 (2018). [DOI] [PubMed] [Google Scholar]
- 54.Lima F et al. Visible light activation of boronic esters enables efficient photoredox C(sp2)–C(sp3) cross-couplings in flow. Angew. Chem. Int. Ed. 55, 14085–14089 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tellis JC, Primer DN & Molander GA Single-electron transmetalation in organoboron cross-coupling by photoredox/nickel dual catalysis. Science 345, 433–436 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gutierrez O, Tellis JC, Primer DN, Molander GA & Kozlowski MC Nickel-catalyzed cross-coupling of photoredox-generated radicals: uncovering a general manifold for stereoconvergence in nickel-catalyzed cross-couplings. J. Am. Chem. Soc. 137, 4896–4899 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Primer DN & Molander GA Enabling the cross-coupling of tertiary organoboron nucleophiles through radical-mediated alkyl transfer. J. Am. Chem. Soc. 139, 9847–9850 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yuan M, Song Z, Badir SO, Molander GA & Gutierrez O On the nature of C(sp3)–C(sp2) bond formation in nickel-catalyzed tertiary radical cross-couplings: a case study of Ni/photoredox catalytic crosscoupling of alkyl radicals and aryl halides. J. Am. Chem. Soc. 142, 7225–7234 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Molander GA, Colombel V & Braz VA Direct alkylation of heteroaryls using potassium alkyl- and alkoxymethyltrifluoroborates. Org. Lett. 13, 1852–1855 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Odachowski M et al. Development of enantiospecific coupling of secondary and tertiary boronic esters with aromatic compounds. J. Am. Chem. Soc. 138, 9521–9532 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nykaza TV et al. Intermolecular reductive C–N cross coupling of nitroarenes and boronic acids by PIII/PV=O catalysis. J. Am. Chem. Soc. 140, 15200–15205 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sadhu KM & Matteson DS (Chloromethyl)lithium: efficient generation and capture by boronic esters and a simple preparation of diisopropyl (chloromethyl)boronate. Organometallics 4, 1687–1689 (1985). [Google Scholar]
- 63.Li J, Grillo AS & Burke MD From synthesis to function via iterative assembly of N-methyliminodiacetic acid boronate building blocks. Acc. Chem. Res. 48, 2297–2307 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Blair DJ et al. Automated iterative Csp3–C bond formation. Nature 604, 92–97 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Angell RM et al. Biphenyl amide p38 kinase inhibitors 4: DFG-in and DFG-out binding modes. Bioorg. Med. Chem. Lett. 18, 4433–4437 (2008). [DOI] [PubMed] [Google Scholar]
- 66.Sauer WH & Schwarz MK Molecular shape diversity of combinatorial libraries: a prerequisite for broad bioactivity. J. Chem. Inf. Comput. Sci. 43, 987–1003 (2003). [DOI] [PubMed] [Google Scholar]
- 67. Prosser KE, Stokes RW & Cohen SM Evaluation of 3-dimensionality in approved and experimental drug space. ACS Med. Chem. Lett. 11, 1292–1298 (2020). During manuscript editing process after it is accepted, several publications on synthesis of bridge-substituted BCPs and BCP boronates were reported.
- 68.Dong W et al. Exploiting the sp2 character of bicyclo[1.1.1] pentyl radicals in the transition-metal-free multi-component difunctionalization of [1.1.1]propellane. Nat. Chem. 14, 1068–1077 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yu IF et al. Catalytic undirected borylation of tertiary C–H bonds in bicyclo[1.1.1]pentanes and bicyclo[2.1.1]hexanes. Nat. Chem. 15, 685–693 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wright BA et al. Skeletal editing approach to bridge-functionalized bicyclo[1.1.1] pentanes from azabicyclo[2.1.1] hexanes. J. Am. Chem. Soc. 145, 0960–10966 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Garry OL et al. Rapid access to 2-substituted bicyclo[1.1.1] pentanes. J. Am. Chem. Soc. 145, 3092–3100 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Experimental data as well as characterization data for all new compounds prepared in the course of these studies are provided in the Supplementary Information. Crystallographic data for the structures reported in this Article have been deposited at the Cambridge Crystallographic Data Centre under deposition nos. CCDC 2158998 (15), 2159002 (23), 2158995 (24), 2159016 (25), 2159001 (27), 2162135 (33), 2160325 (55), 2160336 (59) and 2162136 (76), see the ‘X-ray crystallographic data’ section in the Supplementary Information. Copies of the data can be obtained free of charge via https://www.ccdc.cam.ac.uk/structures/. Source Data are provided with this paper.