

Abstract
While our understanding of the molecular biology of Alzheimer’s disease (AD) has grown, the etiology of the disease, especially the involvement of peripheral infection, remains a challenge. In this study, we hypothesize that peripheral infection represents a risk factor for AD pathology. To test our hypothesis, APP/PS1 mice underwent cecal ligation and puncture (CLP) surgery to develop a polymicrobial infection or non-CLP surgery. Mice were euthanized at 3, 30, and 120 days after surgery to evaluate the inflammatory mediators, glial cell markers, amyloid burden, gut microbiome, gut morphology, and short-chain fatty acids (SCFAs) levels. The novel object recognition (NOR) task was performed 30 and 120 days after the surgery, and sepsis accelerated the cognitive decline in APP/PS1 mice at both time points. At 120 days, the insoluble Aβ increased in the sepsis group, and sepsis modulated the cytokines/chemokines, decreasing the cytokines associated with brain homeostasis IL-10 and IL-13 and increasing the eotaxin known to influence cognitive function. At 120 days, we found an increased density of IBA-1-positive microglia in the vicinity of Aβ dense-core plaques, compared with the control group confirming the predictable clustering of reactive glia around dense-core plaques within 15 μm near Aβ deposits in the brain. In the gut, sepsis negatively modulated the α- and β-diversity indices evaluated by 16S rRNA sequencing, decreased the levels of SCFAs, and significantly affected ileum and colon morphology in CLP mice. Our data suggest that sepsis-induced peripheral infection accelerates cognitive decline and AD pathology in the AD mouse model.
INTRODUCTION
According to the 2019 Global Burden of Disease Study, the number of persons with dementia will rise from 57.4 million to 152.8 million cases by 2050 [1]. Alzheimer’s disease (AD) constitutes a major subset of the total prevalence of dementia, and it is expected that by 2050, 106.8 million people worldwide will be living with AD [2]. Most AD cases are sporadic, but a small minority of cases are linked with mutations in the amyloid precursor protein (APP) or other proteins assisting in its proteolytic processing [3]. Sporadic AD is frequently caused by aging, hereditary, and environmental factors. Other risk factors associated with AD include chronic cerebral hypoperfusion [4], atherosclerosis [5], chronic inflammation [6, 7], cardiovascular diseases [7], traumatic brain injury (TBI) [8], and infection [9–11], among many others [12].
Regardless of progress in understanding the molecular biology of AD, the specific events dictating its etiology have been a subject of debate [13–15]. Along this line, there is growing evidence that amyloid-beta (Aβ) peptides have antibacterial and antiviral properties [16, 17] as part of an innate immune response in the brain [17]. However, the significance of microbial infection in promoting AD pathology has recently gained attention in the scientific literature [16–18]. In this sense, a potential risk factor for AD could comprise microbial infections, including chronic spirochetes brain infection [19], human immunodeficiency virus (HIV) [20], herpes simplex virus type (HSV) [11], HSV-1 [21], human herpesvirus 6 A (HHV)-6A, and HHV-7 [10] with increased risks of developing dementia. The peripheral infection has been previously noted to trigger the host immune response by increasing the levels of pro-inflammatory mediators, including acute phase proteins, cytokines, and chemokines in the bloodstream, and affecting the gut and blood-brain barrier (BBB) permeabilities [14, 22, 23], all individually acknowledged as AD risk factors. As a result of bacterial sepsis, the compromised BBB facilitates the entry of peripheral inflammatory mediators and immune cells into the brain, either triggering or exacerbating glial cell activation and neuroinflammation in Wistar rats [22, 23] and increasing fibrillar amyloid plaque formation in the hippocampus of APP/PS1 mice [24].
Our previous publications demonstrate an association between infection and AD changes in experimental non transgenic rodent models [22, 25, 26]. Here, APP/PS1 mice were subjected to cecal ligation and puncture (CLP) surgery to develop mild-grade sepsis (medium ligation). Mice were euthanized at different time points after the procedure (3, 30, and 120 days) to evaluate sepsis’s effect on AD pathology in the short, intermediate, and long-term. Prior to the experimental endpoint, experimental and control mice were evaluated for cognitive decline. We next evaluated the gut microbiome, gut morphology, and short-chain fatty acids (SCFAs) to explore how peripheral infection modulates the gut-brain axis and potentially affects AD pathology. In addition, we also studied glial activation, inflammatory markers, and Aβ burden in the brain to understand whether sepsis-induced peripheral infection influences AD hallmarks.
MATERIAL AND METHODS
Transgenic mice and housing
APP/PS1 mice overexpress the human APP harboring the Swedish double mutation and presenilin-1 with the delta-E9 mutation [27]. These animals mimic AD pathology by developing Aβ aggregates at ~4 months old [27]. The Animal Welfare Committee (AWC) of the Center for Laboratory Animal Medicine and Care (CLAMC), UTHealth at Houston, TX, USA, permitted the experimental procedures for this study (IACUC Number: AWC-18–0124). Animals were selected randomly in a blinded manner for different time points. For more details, see Supplementary Methods.
Experimental design
The time point and brain structures investigated in the experimental design of this study were determined considering the previous works [22, 28, 29] as described in the Supplementary Methods.
CLP and non-CLP surgeries
Polymicrobial sepsis was performed in APP/PS1 mice by the CLP or sham surgeries as described in previous studies [30]. For more details, see Supplementary Methods.
Novel object recognition (NOR) task
The mice were submitted to the NOR task before euthanizing on day 30 and day 120 after CLP or sham surgeries in accordance with protocol [31], detailed in the Supplementary Methods.
Brain collection and total protein concentration estimation
Brain collection and total protein quantification were performed according to the standard protocols [32] described in the Supplementary Methods.
Quantification of inflammatory cytokines and chemokines by multiplex assay
For the quantification of cytokines and chemokines levels, multiplex fluorescent immunoassay kits were used according to the manufacturer’s instructions [32] and details provided in the Supplementary Methods.
Immunofluorescence
An immunofluorescence (IF) assay was performed using specific antibodies from the saggital brain sections. Analyses of Aβ plaque-associated microglia were made using previously published reports [32] described in the Supplementary Methods.
Soluble and insoluble Aβ levels using Milliplex
Soluble and insoluble Aβ levels were determined in accordance with protocol [33] described in the Supplementary Methods.
Fecal collection and 16S rRNA sequencing
Fecal sample collection and DNA extraction were performed as described in the Supplementary Methods. The 16S rRNA gene sequencing data, taxonomic annotations, and α- and β-diversity calculation was performed according to standard protocols [34]. Details are given in Supplementary Methods.
Short-chain fatty acids (SCFAs) quantification
The SCFAs extraction procedure was performed as described by Zhao et al. [35], and details are provided in the Supplementary Methods.
Gut histology
Intestinal tissues (ileum and colon) was processed for histological studies as described in the Supplementary Methods.
Gut mucus lectin staining
Intestinal sections (ileum, cecum, and colon) were obtained for the mucus lectin staining following a specific protocol described in the Supplementary Methods.
Statistical analyses
A detailed description of the statistical analyses is provided in the Supplementary Methods.
RESULTS
The timeframe and experimental design
Male, 50-day-old APP/PS1 mice were euthanized at 3, 30, and 120 days time points to investigate the short, intermediate, and long-term effects of sepsis in the AD mouse model. A schematic representation of the experimental design is given in Supplementary Fig. 1. The behavioral tasks were assessed for mice euthanized 30 and 120 days after the surgeries. The mice from CLP and non-CLP groups were followed for ten days, and mortality was recorded. Interestingly, the APP/PS1 mice subjected to sepsis had an 87.88% survival rate compared with wild-type mice, with a 56% survival rate.
Polymicrobial sepsis accelerates cognitive impairment in APP/PS1 mice
We evaluated non-spatial memory in intermediate and long-term time point groups using the NOR task to verify if sepsis predisposes to cognitive impairment (Fig. 1B–E). We found no differences between the groups on exploratory behavior (Fig. 1B, D). This parameter was evaluated by the recognition index, which was significantly reduced in sepsis survivors mice as compared to the control group at 30 (Fig. 1C, t = 2.552 and df = 18, p < 0.05), n = 10 and 120 days (Fig. 1E, t = 2.257 and df = 12, p < 0.05), n = 7 after sepsis induction. Despite being free from infection, post-septic mice spent less time with the new object reflecting cognitive impairments at both time points. Of note, previous studies reported cognitive impairment in APP/PS1 mice aged 7 to 8 months [36, 37], suggesting that sepsis accelerates the decline in cognitive functions.
Fig. 1. Cognitive impairment evaluated by novel object recognition (NOR) task in APP/PS1 mice challenged with CLP.
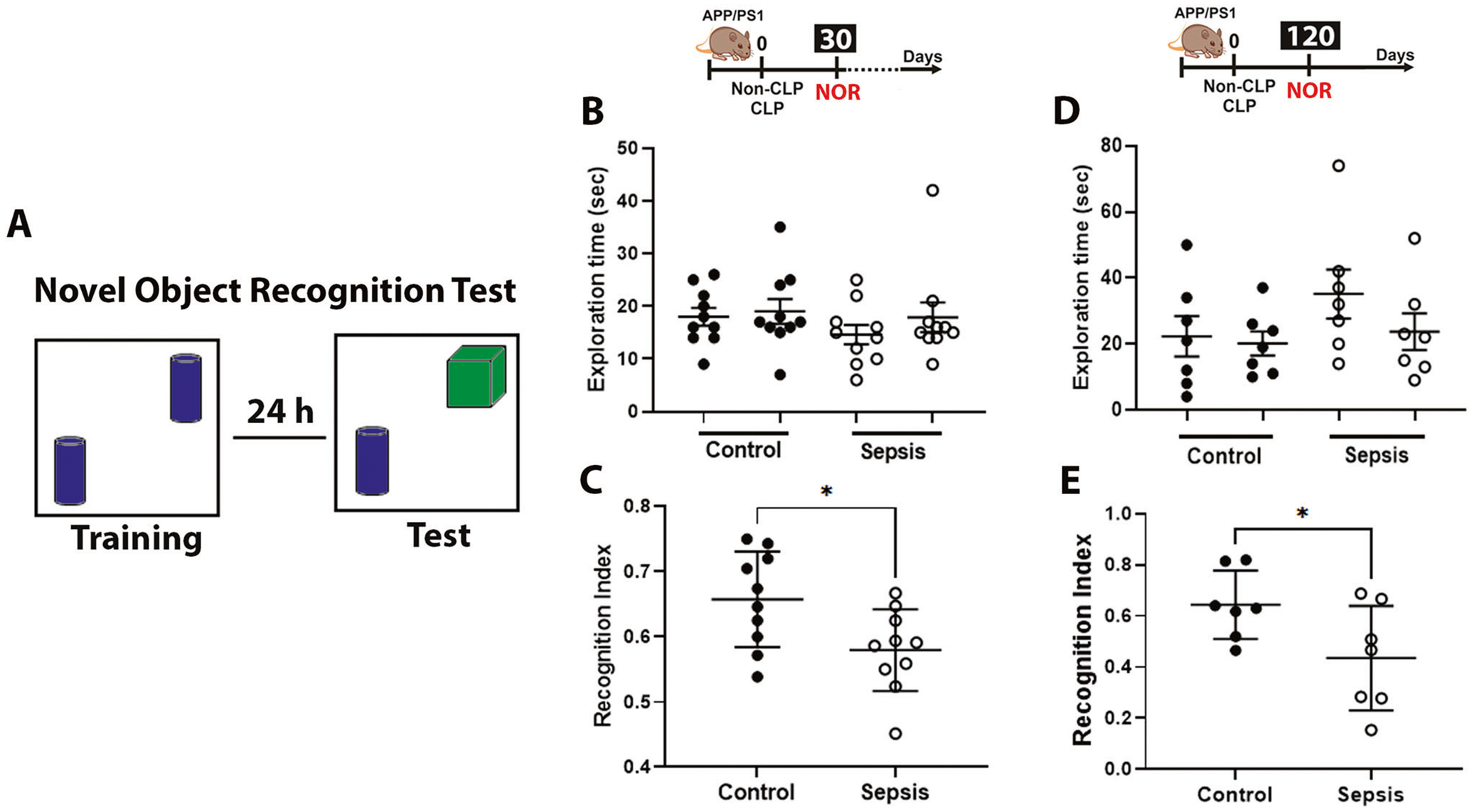
A Schematic representation of the NOR task. B, D Exploration time at 30 and 120 after CLP and non-CLP surgery. No difference between the control and sepsis groups was observed in the locomotor activity. C, E Recognition index, as explained in Materials and Methods. The results are expressed as the mean ± SEM for n = 7–10. Unpaired Student’s t-tests analyzed the differences between control and sepsis at each time point. *p < 0.05 compared to controls.
Polymicrobial sepsis increases the levels of insoluble Aβ1–40 and Aβ1–42 in the APP/PS1 mice brain
The levels of aqueous insoluble and soluble Aβ1–40 and Aβ1–42 evaluated by Milliplex did not change at three days (Fig. 2A–D, p ≥ 0.05) and 30 days after sepsis (Fig. 2E–H, p ≥ 0.05). Notably, insoluble Aβ1–40 and Aβ1–42 concentrations were increased in the brain of APP/PS1 mice at 120 days after sepsis (Fig. 2I, J, t = 2.983, df = 14, p < 0.01 and t = 2.474, df = 14, p < 0.05, respectively), indicating a deleterious long-term effect of sepsis in brain pathology. However, the soluble Aβ1–40 and Aβ1–42 levels did not change at this specific time for post-septic mice compared to controls (Fig. 2K, L, p ≥ 0.05, respectively, n = 6–8). Our findings imply a progressive shift of brain Aβ1–40 and Aβ1–42 from soluble to insoluble pools at more prolonged periods, which are known to play mechanistic roles in the onset and progression of AD pathology [38].
Fig. 2. Effect of polymicrobial sepsis on the insoluble and soluble fraction of Aβ1–40 and Aβ1–42 in the APP/PS1 mice brain.
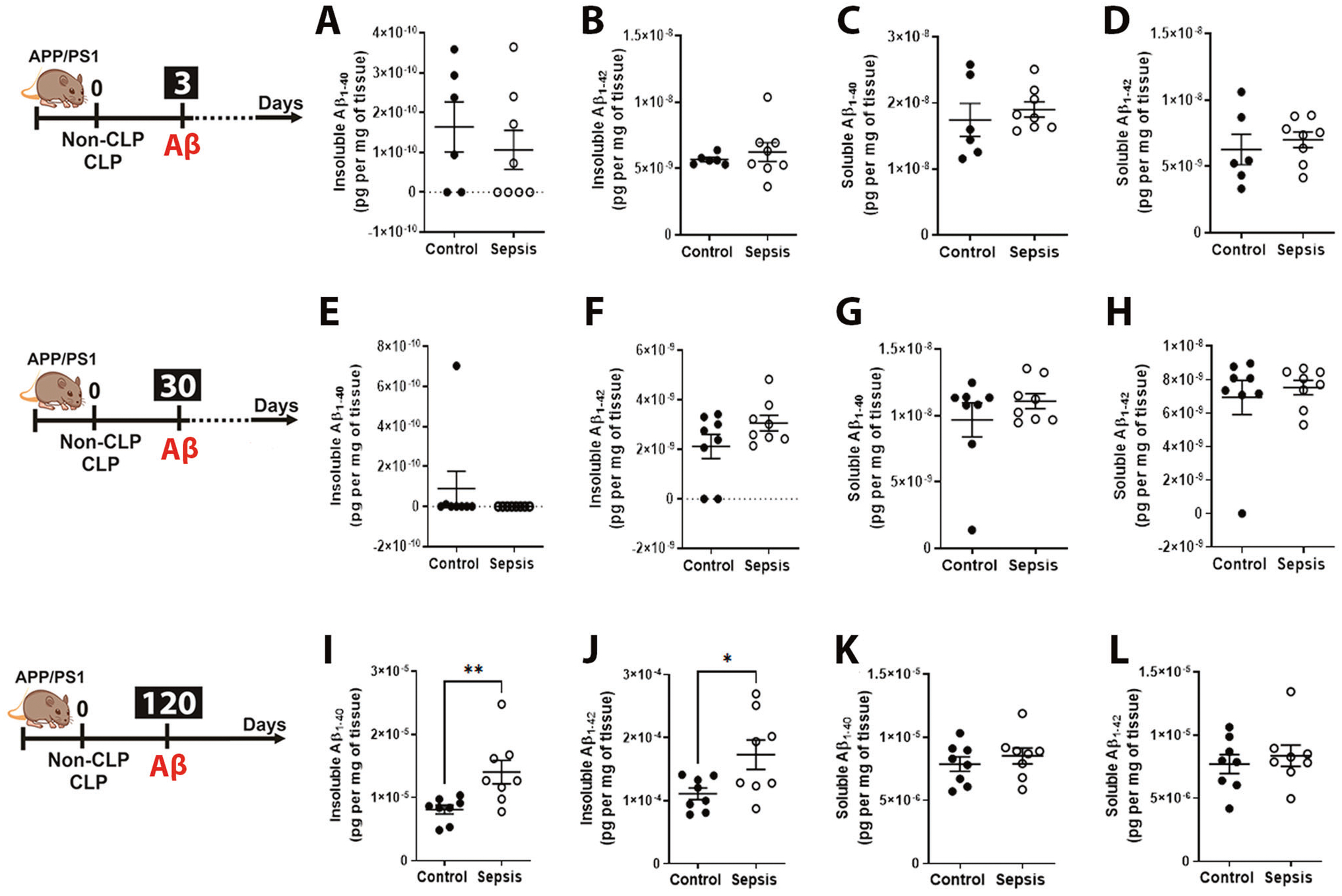
Using ELISA, aqueous soluble and insoluble Aβ1–40 and Aβ1–42 were measured. A–D Insoluble and soluble Aβ1–40 and Aβ1–42 after 3 days. E–H Insoluble and soluble Aβ1–40 and Aβ1–42 after 30 days. I–L Insoluble and soluble Aβ1–40 and Aβ1–42 after 120 days. The results are expressed as the mean ± SEM for n = 6–8. Unpaired Student’s t-tests was used to analyze the differences between control and sepsis groups at each time point. *p < 0.05 and **p < 0.01 compared to controls.
Polymicrobial sepsis modifies the microglia cell markers and Aβ burden in the APP/PS1 mice brain
To evaluate the dynamic changes of microglial activation after sepsis, we compared immunofluorescence staining for IBA-1 in PFC and hippocampal (Fig. 3) regions between CLP and non-CLP groups. At three days after the sepsis induction, IBA-1 increased in the PFC (Fig. 3B, C, t = 6.467, df = 6, p < 0.001) and in the hippocampus (Fig. 3P, Q, t = 3.456, df = 6, p < 0.05). At 30 days after sepsis, IBA-1 increased in the PFC (Fig. 3F, G, t = 2.468, df = 6, p < 0.05); on the contrary, IBA-1 did not change in the hippocampus in the CLP-group compared to the control group (Fig. 3T, U, p ≥ 0.05). At 120 days after sepsis induction, IBA-1 staining did not change between groups in the PFC and hippocampus (Fig. 3L, M, Z and A1, p ≥ 0.05). However, we found an upsurge in the deposition of Aβ burden in the hippocampus but not in PFC of 120 days post-septic mice compared to the age-matched controls (Fig. 3Z and B1, t = 2.494, df = 6, p < 0.05). Our results corroborate the previous finding where 2-month-old APP/PS1 subjected to CLP displayed elevated amyloid plaques two months after CLP [24]. Further, a three-dimensional (3D) reconstruction of 4G8 positive amyloid plaques was generated for a longer time to investigate the number of plaque-associated microglia. Utilizing the distance transformation tool in the IMARIS software, a 3D region of interest (ROI) was created with a 15 μm radius from the edge of the plaque. A surface rendering of the IBA-1 positive cell was generated for all IBA-1 positive staining in this 3D plaque-associated ROI to evaluate the number of plaque-associated microglia. Quantifying the volume of IBA-1 positive staining showed a significantly increased volume of IBA-1 positive staining surrounding plaque in sepsis mice compared to the control mice. As shown in figure 3D1, at 120 days, we found a significantly increased density of IBA-1-positive microglia in the vicinity of Aβ dense-core plaques, compared with the control group confirming the predictable clustering of reactive glia round dense-core plaques within 15 μm near Aβ deposits in the brain. (Fig. 3D1, t = 2.490, df = 6, p < 0.05).
Fig. 3. Effect of polymicrobial sepsis on microglial, astroglial activation, and Aβ burden in the APP/PS1 mice PFC and hippocampus.
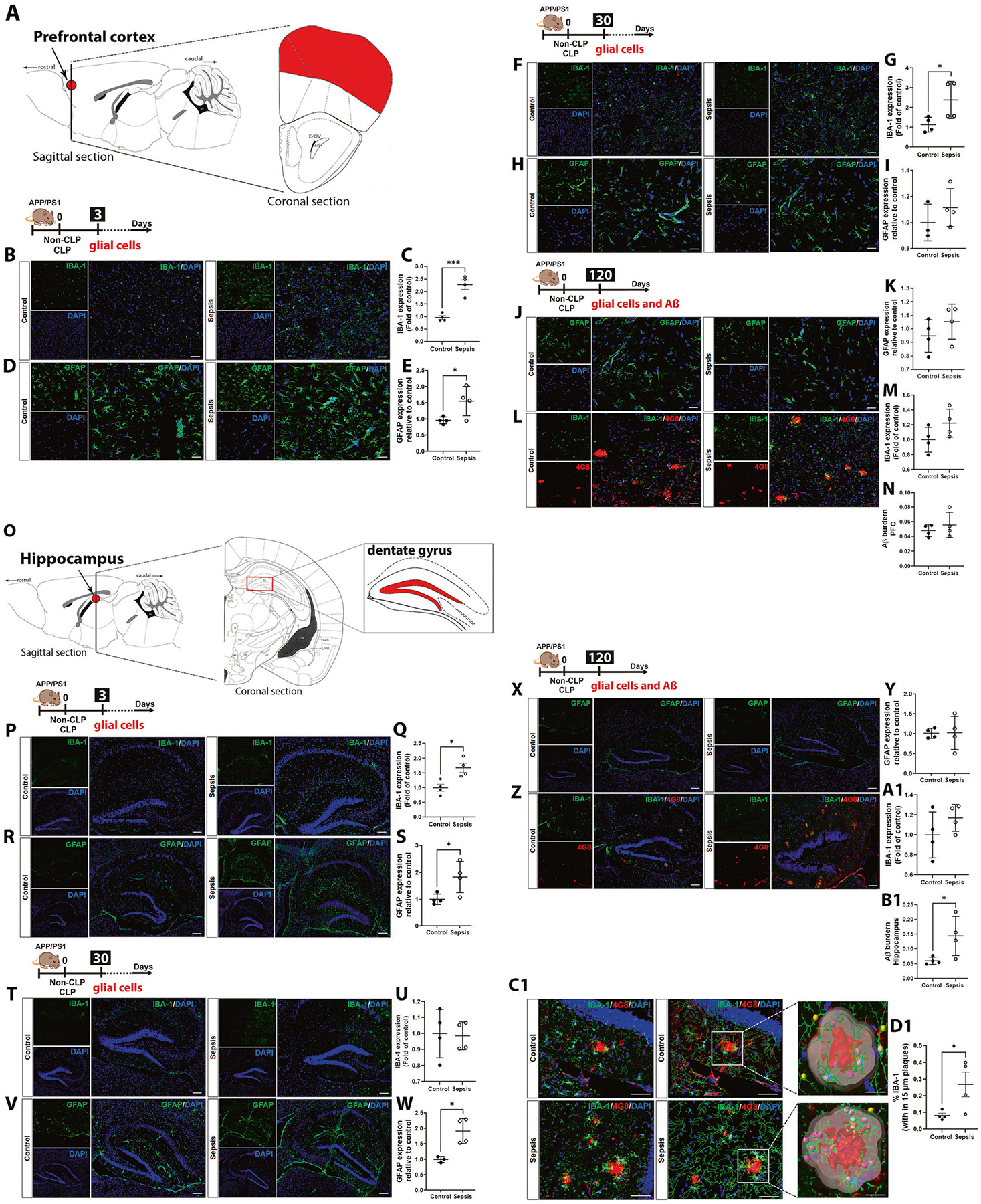
Effect of sepsis on microglial activation at 3, 30, and 120-time points after CLP and non-CLP surgery evaluated by immunofluorescence. A Schematic representation indicates the PFC. B Representative PFC images of IBA-1 immunostaining after three days (magnification, ×20; scale bar = 50 μm). C Quantification of PFC IBA-1 immunostaining after three days. Activation of microglia was observed in sepsis mice in PFC three days after sepsis induction. D Representative PFC images of GFAP immunostaining after three days (magnification, ×40; scale bar = 30 μm). E Quantification of PFC GFAP immunostaining after three days. Astrogliosis was observed in sepsis mice in PFC three days after sepsis induction. F Representative PFC images of IBA-1 immunostaining after 30 days (magnification, ×20; scale bar = 50 μm). G Quantification of PFC IBA-1 immunostaining after 30 days. Microgliosis was observed in sepsis mice in PFC thirty days after sepsis induction. H Representative PFC images of GFAP immunostaining after 30 days (magnification, ×40; scale bar = 30 μm). I Quantification of PFC GFAP immunostaining after 30 days. J Representative PFC images of GFAP immunostaining after 120 days (magnification, ×40; scale bar = 30 μm). K Quantification of PFC GFAP immunostaining after 120 days. L Representative PFC images of IBA-1 and 4G8 immunostaining after 120 days (magnification, ×20; scale bar = 50 μm). M Quantification of PFC IBA-1 immunostaining after 120 days. N Quantification of Aβ burden in PFC by 4G8 immunostaining after 120 days. O Schematic representation indicates the hippocampus highlighting the dentate gyrus. P Representative hippocampal dentate gyrus images of IBA-1 immunostaining after three days (magnification, ×10, scale bar = 150 μm). Q Quantification of hippocampal IBA-1 immunostaining. Activation of microglia was observed in sepsis mice in the hippocampus three days after sepsis induction. R Representative hippocampal dentate gyrus images of GFAP immunostaining after three days (magnification, ×10; scale bar = 150 μm). S Quantification of hippocampal GFAP immunostaining after three days. Astrogliosis was observed in the PFC and hippocampus of sepsis mice three days after sepsis induction. T Representative hippocampal dentate gyrus images of IBA-1 immunostaining after 30 days (magnification, ×10; scale bar = 150 μm). U Quantification of hippocampal IBA-1 immunostaining after 30 days. V Representative hippocampal dentate gyrus images of GFAP immunostaining after 30 days (magnification, ×10; scale bar = 150 μm). W Quantification of hippocampal GFAP immunostaining after 30 days. X Representative hippocampal dentate gyrus images of GFAP immunostaining after 120 days (magnification, ×10; scale bar = 150 μm). Y Quantification of hippocampal GFAP immunostaining after 120 days. Z Representative hippocampal dentate gyrus images of IBA-1 and 4G8 immunostaining after 120 days (magnification, ×10; scale bar = 150 μm). A1 Quantification of hippocampal IBA-1 immunostaining after 120 days. B1 Quantification of Aβ burden in the hippocampus by 4G8 immunostaining after 120 days. C1 Representative images of Imaris 3D reconstruction of plaques (magnification, ×40; scale bar = 20 μm). D1 Quantification of plaque-associated microglia within a 15 μm radius of plaques. Immunofluorescence investigated the effect of sepsis on astroglial and microglial expression at 3, 30, and 120-time points after CLP and non-CLP surgery. The results are expressed as the mean ± SEM for n = 4. Unpaired Student’s t-tests analyzed the differences between control and sepsis at each time point. *p < 0.05 and ***p < 0.001 compared to controls.
Polymicrobial sepsis modulates astrocytes in the APP/PS1 mice brain
At three days after sepsis induction, GFAP increased in the PFC (Fig. 3D, E, t = 2.561, df = 6, p < 0.05) and hippocampus (Fig. 3R, S, t = 2.706, df = 6, p < 0.05). Thirty days after CLP, the GFAP did not change in the PFC (Fig. 3H, I, p ≥ 0.05) but increased in the hippocampus (Fig. 3V, W, t = 3.729, df = 5, p < 0.05). At 120 days after sepsis induction, the GFAP expression did not change in PFC and hippocampus (Fig. 3J, K, X, Y, p ≥ 0.05), n = 3–4. In agreement with the previous publication [39], astrocyte markers exacerbate in the first hours after the insult via inflammation and cytokine production. However, 30 days after the CLP surgery, the astroglial marker remains activated and concomitant with IL-1α, TNF-α, eotaxin, and IFN-γ levels in the hippocampus, an essential area of the brain associated with memory [40].
Polymicrobial sepsis modulates the cytokines and chemokines in the APP/PS1 mice plasma and brain
To determine the significant relationship between peripheral infection/inflammation and AD pathology, we evaluated 23 cytokines/chemokines in plasma and the most critical areas in the brain associated with sepsis [28] and AD [41, 42]. In plasma, as expected, at three days, the sepsis group demonstrated significant escalation of IFN-γ (t = 5.159, df = 9, p < 0.001), IL-5 (t = 3.761, df = 9, p < 0.01), IL-9 (t = 8.801, df = 6, p < 0.001), IL-12 (p40) (t = 4.914, df = 9, p < 0.001), IL-17A (t = 2.955, df = 4, p < 0.05), KC (t = 2.327, df = 8, p < 0.05), and TNF-α (t = 2.833, df = 8, p < 0.05) as compared to the control group. At 30 and 120 days after sepsis, the levels of RANTES (t = 4.170, df = 9, p < 0.01) reduced and TNF-α (t = 3.084, df = 8, p < 0.05) increased respectively in sepsis mice (Supplementary Fig. 2). After three days, sepsis increased the levels of eotaxin (t = 2.264, df = 10), IFN-γ (t = 5.539, df = 10, p < 0.01), IL-6 (t = 2.430, df = 10), IL-12 (p70) (t = 2.274, df = 10), and MCP-1 (t = 2.253, df = 10, p < 0.05) in PFC (Fig. 4A) and increased the levels of eotaxin (t = 2.264, df = 10), G-CSF (t = 2.522, df = 6), IFN-γ (t = 3.234, df = 6), IL-1α (t = 2.727, df = 7), IL-4 (t = 2.326, df = 8), KC (t = 2.773, df = 8), and TNF-α (t = 2.259, df = 10) in the hippocampus (Fig. 4B, p < 0.05). At 30 days after sepsis induction, IFN-γ (t = 3.195, df = 10, p < 0.01) and IL-1α (t = 2.414, df = 10, p < 0.05) increased; IL-6 (t = 2.462, df = 10, p < 0.05), IL-13 (t = 3.348, df = 10, p < 0.01), and IL-17α (t = 2.356, df = 10, p < 0.05) decreased in the PFC (Fig. 4C). In the hippocampus, eotaxin (t = 2.282, df = 9), IFN-γ (t = 2.154, df = 6), IL-1α (t = 2.376, df = 6), IL-12 (p40) (t = 3.228, df = 8), and TNF-α (t = 2.569, df = 6) levels increased compared to the control group (Fig. 4D, p < 0.05). At 120 days after sepsis, eotaxin (t = 2.603, df = 10), KC (t = 2.905, df = 10), MIP-1α (t = 2.789, df = 6), and RANTES (t = 3.132, df = 10) increased in the PFC (Fig. 4E, p < 0.05), whereas in the hippocampus, IL-6 (t = 2.725, df = 7), IL-10 (t = 2.764, df = 7), and IL-13 (t = 3.227, df = 5) decreased (Fig. 4F, p < 0.05), n = 6.
Fig. 4. Assessment of immune activation by measuring the levels of cytokines and chemokines in the APP/PS1 mice brain after CLP challenge.
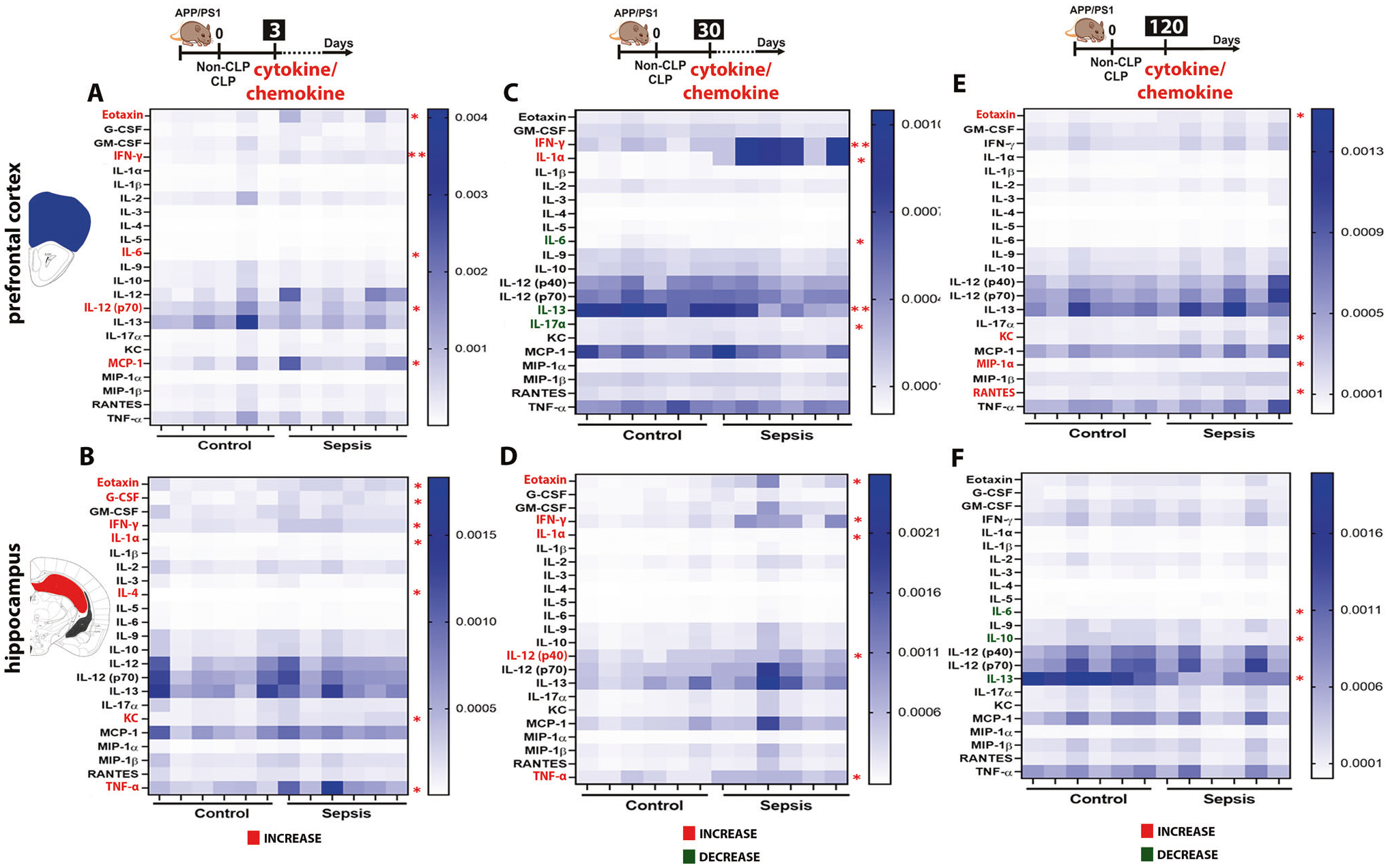
Evaluation of brain immune modulation at 3, 30, and 120-time points after CLP and non-CLP surgery in PFC and hippocampus using mouse cytokine 23-plex assay. The cytokines and chemokines include interleukin (IL)-1α, IL-1β, IL-2, IL-3, IL-4, IL-5, IL-6, IL-9, IL-10, IL-12 (p40), IL-12 (p70), IL-13, IL-17A, eotaxin, granulocyte colony-stimulating factor (G-CSF), granulocyte-macrophage colony-stimulating factor (GM-CSF), interferon (IFN)-γ, keratinocytes-derived chemokine (KC), monocyte chemoattractant protein (MCP)-1, macrophage inflammatory protein (MIP)-1α, MIP-1β, regulated upon activation, normal T cell expressed and presumably secreted (RANTES), and tumor necrosis factor (TNF)-α, the unit of expression given as pg/μg of protein. A Heatmap of the PFC cytokine/chemokine levels after three days. Sepsis increased the levels of eotaxin, IL-6, IL-12, MCP-1, and IFN-γ in the sepsis group compared to the control. B Heatmap of the hippocampus cytokine/chemokine levels after three days. The sepsis group had elevated levels of eotaxin, G-CSF, IFN-γ, IL-1α, IL-4, KC, and TNF-α compared to the control group. C Heatmap of the PFC cytokine/chemokine levels after 30 days. IFN-γ and IL-1α increased; IL-6 and IL-17α decreased in the sepsis group compared to the control group. D Heatmap of the hippocampus cytokine/chemokine levels after 30 days. Eotaxin, IFN-γ, IL-1α, IL-12, and TNF-α levels increased compared to the control group. E Heatmap of the PFC cytokine/chemokine levels after 120 days. Increased levels of eotaxin, KC, MIP-1 α, and RANTES in the sepsis group compared to the control group. F Heatmap of the hippocampus cytokine/chemokine levels after 120 days. Sepsis decreased IL-6, IL-10, and IL-13 in survivors’ mice. The results are expressed as the mean ± SEM for n = 6. Unpaired Student’s t-tests analyzed the differences between control and sepsis at each time point. *p < 0.05 and **p < 0.01 compared to controls.
Sepsis changes the microbiome profile of APP/PS1 mice
Gut microbiome alterations have been reported in AD and sepsis [43, 44]. At three days after sepsis, the indexes did not change in the sepsis group (Fig. 5A, B, p ≥ 0.05), but at 30 and 120 days, the α-diversity decreased as measured by observed OTUs statistical analysis (Fig. 5A, p < 0.01 and p < 0.05, respectively) and also when using the Shannon statistical method (Fig. 5B, p < 0.001 and p < 0.05, respectively). The β-diversity variance between groups did not change three days after sepsis (Fig. 5C, F, p ≥ 0.05, unweighted and weighted, respectively). At 30 days after sepsis, the β-diversity significantly differed between the groups (Fig. 5D, unweighted, p = 0.004, R2: 0.259, and Fig. 5G weighted p = 0.004, R2: 0.341). Similarly, at 120 days after sepsis, the β-diversity significantly differed between the groups (Fig. 5E, unweighted, p = 0.009, R2: 0.2, and Fig. 5H, weighted p = 0.006, R2: 0.216). In summary, these data demonstrate that CLP significantly leads to dysbiosis in the gut.
Fig. 5. Analysis of gut microbiome profile and the relative abundance of phyla and class level difference after polymicrobial infection as measured in the feces of APP/PS1 mice.
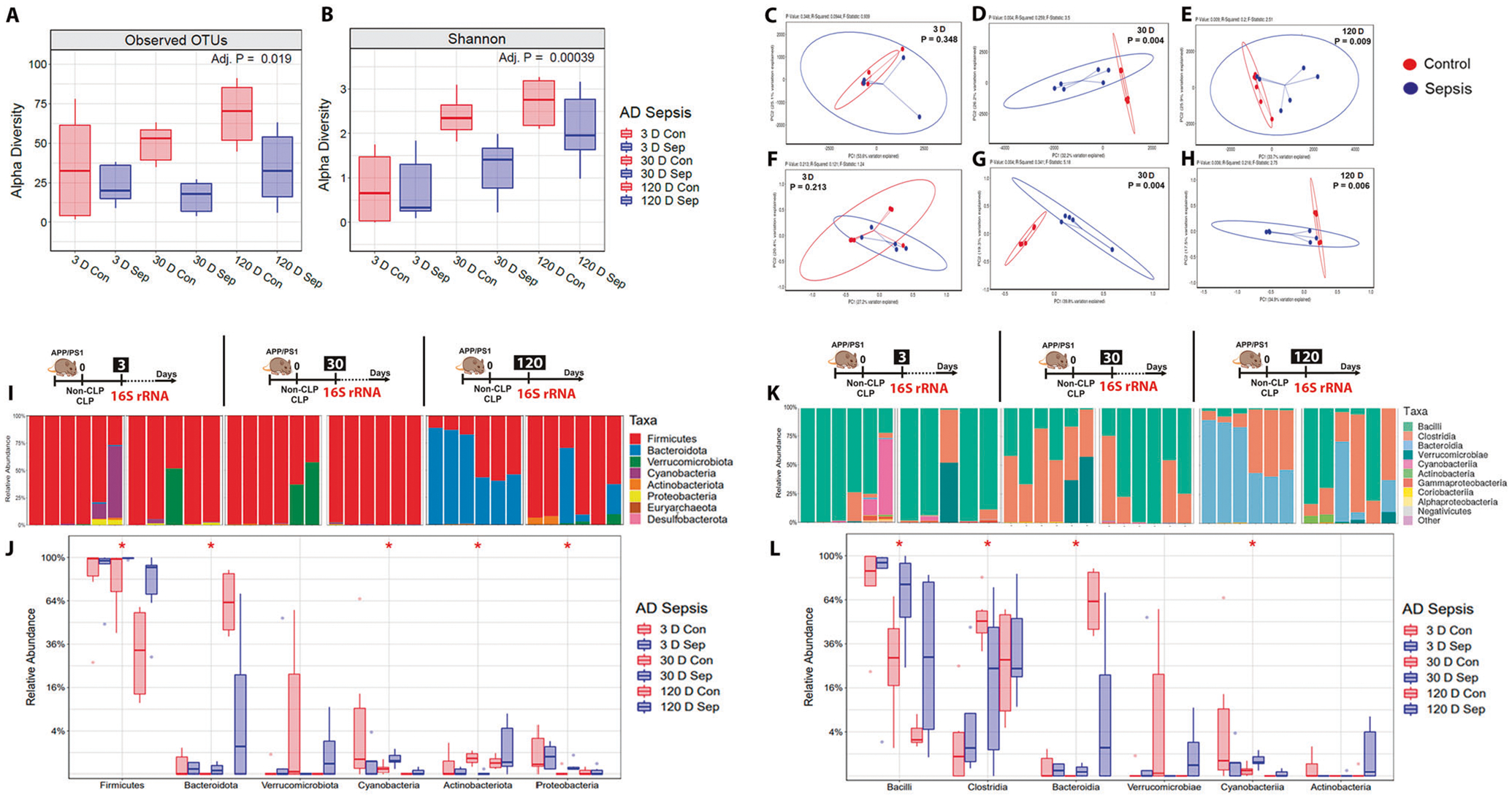
Using 16S rRNA sequencing, the α-diversity (microbiota richness) and β-diversity (difference in global microbiota composition between samples) were determined at 3, 30, and 120-time points after CLP and non-CLP surgery. A α-diversity: Observed Operational Taxonomic Units (OTUs). B α-diversity: Shannon-diversity index. At 30 and 120 days after sepsis induction, there was a significant reduction in the α-diversity difference in sepsis-surviving mice compared to the control group. C and F β-diversity at three days: Principal coordinate analysis using unweighted and weighed UniFrac. D and G β-diversity at 30 days: Principal coordinate analysis using unweighted and weighted UniFrac, p = 0.004. E and H β-diversity at 120 days: Principal coordinate analysis using unweighted UniFrac, p = 0.009 and weighted UniFrac, p = 0.006, n = 6. The 16S rRNA sequencing was used to measure the relative abundance of the phyla and class at 3, 30, and 120-time points after CLP and non-CLP surgery. I Relative abundance stacked bar plots of taxa from CLP or non-CLP-treated mice at the phylum level. J Quantification of taxa at the phylum level. No change was found at 3 and 120 days, p ≥ 0.05. At 30 days, the phylum Actinobacteria decreased, and the phylum Bacteriodetes, Cyanobacteria, Firmicutes, and Proteobacteria increased in the sepsis group compared with the control group, p < 0.05. K Relative abundance stacked bar plots of taxa from at the class level. L Quantification of taxa at the class level. Similarly, there was no change at the class level in 3 and 120 days, p ≥ 0.05). The class Bacilli, Bacteroidia, and Cyanobacteria increased, but the class Clostridia decreased in the sepsis group at the intermediate time point, p < 0.05, n = 6. For 16S rRNA sequencing, Kruskal–Wallis test with post hoc Benjamini-Hochberg correction was used for statistical analysis.
Sepsis changes the relative abundance of phyla and class in the feces of APP/PS1 mice
Several studies support a link between gut microbiota and brain amyloidosis [43, 45, 46]. The relative phyla abundance is demonstrated in Fig. 5I–L. At 3 and 120 days, the relative phyla abundance did not change (Fig. 5I, J, p ≥ 0.05). At 30 days, the phylum Actinobacteriota decreased, and the phylum Bacteroidota, Cyanobacteria, Firmicutes, and Proteobacteria increased in the CLP group compared with the control group (Fig. 5I, J, p < 0.05). The relative abundance of the class did not change at 3 and 120 days after sepsis (Fig. 5K, L, p ≥ 0.05). At 30 days after the surgeries, the class Bacilli, Bacteroidota, and Cyanobacteria increased, but the class Clostridia decreased in the sepsis group (Fig. 5K, L, p < 0.05). The genus did not modify between the groups at all three-time points (p ≥ 0.05, data not shown) n = 5–6.
Short-chain fatty acids (SCFAs) levels decreased in APP/PS1 mice subjected to sepsis
We evaluated fecal SCFAs to identify if infection influences their production in the gut. The concentration of acetic acid (Fig. 6A, t = 2.281, df = 9, p < 0.05) decreased three days after sepsis. At 30 days after sepsis, the acetic (t = 3.534, df = 9), butyric (t = 2.337, df = 10), and isovaleric acid (t = 2.482, df = 9) levels decreased in the feces of the CLP-group compared to the control group (Fig. 6A, C, E, p < 0.01, p < 0.05, and p < 0.05, respectively). At 120 days after sepsis, the isobutyric acid levels decreased in the feces of the sepsis group (Fig. 6B, t = 3.150, df = 9, p < 0.05). The propionic, valeric, and hexanoic acid levels did not change between the groups (Fig. 6D, F, G, p ≥ 0.05). Acetate has been recognized as an inhibitor of inflammatory responses in different models [23, 47]. Nonetheless, its levels decreased in the CLP group. Besides, as expected, the acetate-producer Clostridia class was significantly reduced in the CLP group.
Fig. 6. Cecum feces SCFAs levels and morphometric measurement of intestinal villus length and crypt depth after polymicrobial sepsis in APP/PS1 mice.
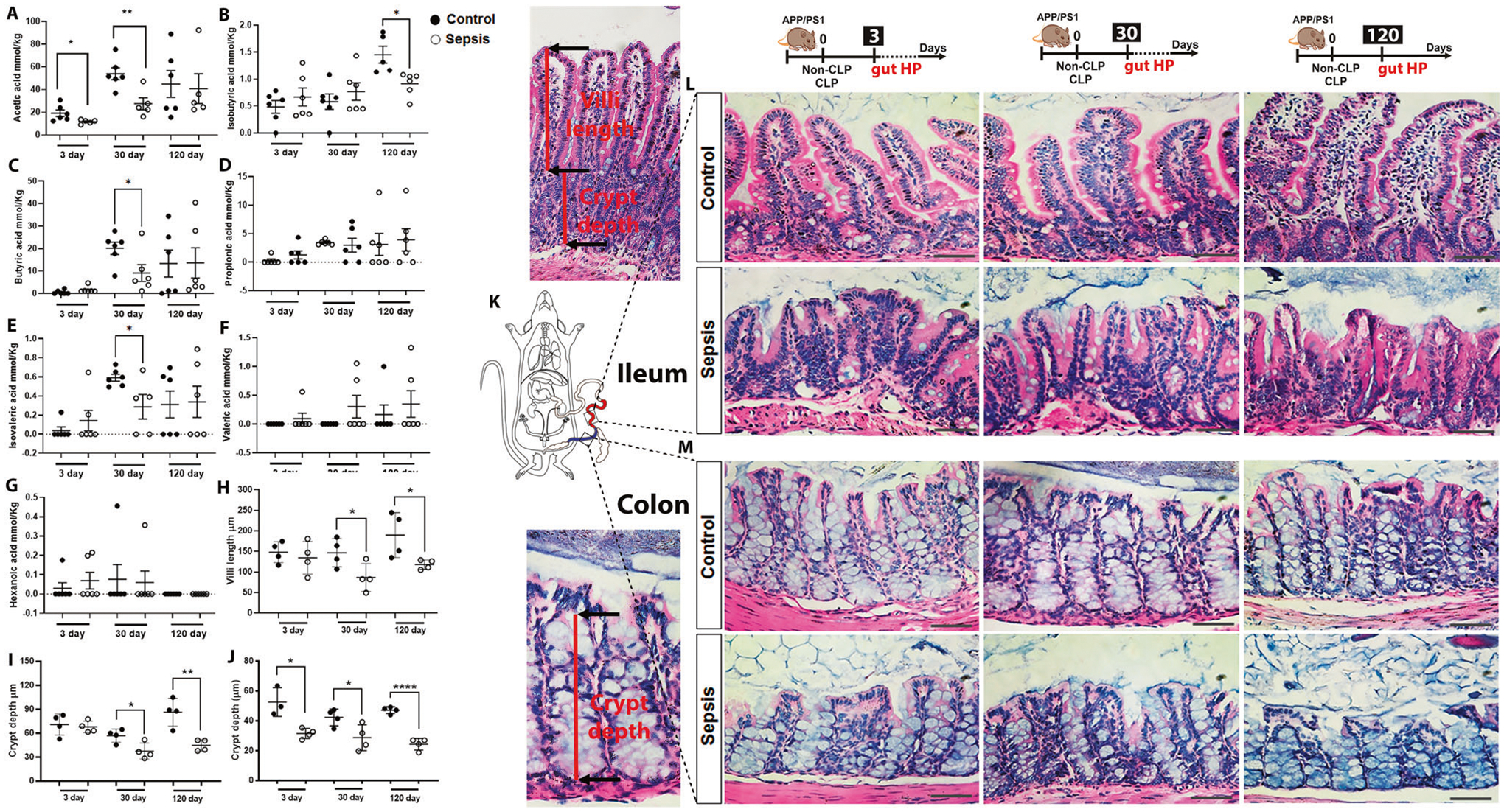
Detection of cecum feces SCFAs levels using GC-MS at 3, 30, and 120-time points after CLP and non-CLP surgery in APP/PS1 mice. A Acetic acid. B Isobutyric acid. C Butyric acid. D Propionic acid. E Isovaleric acid. F Valeric acid. G Hexanoic acid. The results are expressed as the mean ± SEM for n = 6. Intestinal architecture in the small intestine and colon was examined after hematoxylin and eosin (H&E) staining at 3, 30, and 120-time points after CLP and non-CLP surgery in APP/PS1 mice. H Ileum villi length at 3, 30, and 120 days after CLP and non-CLP surgery in APP/PS1 mice. I Ileum crypt depth at 3, 30, and days CLP and non-CLP surgery in APP/PS1 mice. J Colon crypt depth 3, 30, and 120 days CLP and non-CLP surgery in APP/PS1 mice. K Schematic representation indicates the ileum and colon region investigated in this experiment. L Representative images of gut ileum at 3, 30, and 120 days after sepsis and control group, magnification ×40. M Representative images of the gut colon at 3, 30, and 120 days in sepsis and control group, magnification ×40. The results are expressed as the mean ± SEM for n = 4. Unpaired Student’s t-tests analyzed the differences between control and sepsis at each time point. Scale bar = 100 μm. *p < 0.05 and **p < 0.01 compared to controls.
Sepsis decreases the ileum villus length, crypt depth, and colon crypt depth and fucosylated goblet cells of APP/PS1 mice
We first measured ileal villus length, crypt depth, and colon crypt depth to evaluate how sepsis affects gut tissue integrity. The ileal villus length and crypt depth did not change in the sepsis group compared to the control group 3 days after the CLP surgery (Fig. 6H, I, p ≥ 0.05). At 30 days after sepsis induction, the APP/PS1 mice reduced the villi length (t = 2.491, df = 6, p < 0.05) and crypt depth (t = 2.954, df = 6, p < 0.05) along with increased inflammation including leukocyte infiltration (Fig. 6H, I). At 120 days after sepsis induction, the APP/PS1 mice reduced the villi length (t = 2.582, df = 6, p < 0.05) and crypt depth (t = 4.433, df = 6, p < 0.01) (Fig. 6H, I). The colon crypt depth significantly reduced at 3 (t = 2.870, df = 6, p < 0.05), 30 (t = 2.643, df = 6, p < 0.05), and 120 days (t = 10.14, df = 6, p < 0.0001) after sepsis (Fig. 6M, J). The ileal fucosylated goblet cells of the post-septic mice showed a significant increase in the 30 days group (Supplementary Fig. 3A, B, t = 2.635, df = 6, p < 0.05), whereas the fucosylated goblet cells in the colon decreased at 120 days after sepsis (Supplementary Fig. 3E, F, t = 2.449, df = 6, p < 0.05).
Sepsis did not change intestinal Aβ deposition
To test the hypothesis of whether intestinal Aβ deposition occurs before cerebral Aβ deposition in APP/PS1 mice, we used a 4G8 antibody to stain the gut ileal sections. We found no specific signal for 4G8 staining in control and sepsis groups at any time tested. Supplementary Fig. 4 shows the DAB and immunofluorescence 4G8 staining in the gut ileum and brain of APP/PS1.
DISCUSSION
The present study investigated the role of peripheral infection and inflammation in the progression of AD pathogenesis. Peripheral infection triggered by CLP surgery in mice generated polymicrobial sepsis [30], followed by a severely dysregulated immune response. Recent reports show that survivors of infection-related hospitalizations suffer long-term cognitive dysfunction [48, 49]. Also, in a cohort study including 20,698 individuals residents of a nursing home, infection-related hospitalization was associated with immediate and persistent cognitive decline, with the most significant increase in cognitive Function Scale scores among older residents, those with AD and related dementias (ADRD) diagnosis, and the those who had experienced sepsis [50]. Different mechanisms connect microbial infections to AD pathophysiology, including but not limited to Aβ misfolding [51], Aβ antimicrobial properties [17, 26, 52], and neuroinflammation [17, 53, 54]. An in vivo experiment using the intracerebral inoculation of Salmonella Typhimurium in a transgenic 5XFAD mouse model provided more support for infection involvement in AD pathogenesis. In the above study, the 5XFAD mice had higher brain amyloid deposition co-localized with the invading bacteria compared to the control group, and the survival rate increased in the mice overexpressing human Aβ compared to nontransgenic mice [52]. Similarly, we demonstrated that APP/PS1 mice subjected to sepsis had a higher survival rate, 87.88%, relative to wild-type C57BL/6 mice, 56%, and Wistar rats, 58.33%, subjected to sepsis induction, suggesting possible protection by Aβ, a possible antimicrobial peptide (AMP), against infection [26]. Our research team also reported an association between infection and AD, including high levels of Aβ production, higher Ser-202-phosphorylated Tau, and RAGE expression followed by cognitive impairment in Wistar rats subjected to sepsis [22]. In addition, in Wistar rats, the experimental induction of pneumococcal meningitis, with intracisternal inoculation of the S. pneumococcus, enhanced the levels of Aβ1–42, microglial cell activation, and memory impairment [25].
The APP/PS1 mice in the current study showed cognitive impairments at 30- and 120 days following CLP surgery compared to their respective non-CLP surgery groups. APP/PS1 mice do not exhibit cognitive impairment before around eight months old [36, 37]; however, our study shows that CLP mice showed cognitive impairment at 2.6 and 5.6-month-old after exposure to peripheral infection, indicating that sepsis accelerates cognitive impairment in a mouse model of AD. In addition, the APP/PS1 mice subjected to sepsis presented higher levels of insoluble Aβ1–40 and Aβ1–42 in the brain than the control group. Sepsis was induced before the Aβ plaque was formed, and microbial infection induced by CLP surgery may initiate the early Aβ misfolding events. Our results are supported by evidence showing that infection-induced peripheral or systemic inflammation could be a risk factor in AD progression [55].
We next evaluated the glial cell markers to understand the effect of peripheral infection/inflammation in the brain. We found that microglial cells were activated a few hours after sepsis induction in the PFC and hippocampus. However, only microglia cells in the PFC remained active 30 days following CLP surgery. Astrocyte reactivity started a few hours after sepsis induction (3 days) and remained activated in the hippocampus 30 days after CLP surgery. We also found an increased association between the IBA1-positive microglial cells concerning the nearest dense-core Aβ plaques in post-septic APP/PS1 mice, demonstrating that sepsis makes the microglia more active in detecting Aβ plaques. Sepsis accelerated the Aβ burden in the hippocampus, an important brain area associated with learning and memory. Our results support previous findings that bacterial sepsis increased the hippocampal fibrillar Aβ plaques load in APP/PS1 mice [24], and Aβ levels are increased in the Wistar rats’ PFC and hippocampus [22]. The increased Aβ pathology in the hippocampus is a hypothesis to explain why cognitive impairment is commonly observed in sepsis survivors. Whether the increase in Aβ lesions observed in APP/PS1 subjected to sepsis is a specific event or a secondary outcome due to damage in this particular brain region will be evaluated in future studies.
We also evaluated a profile of cytokines and chemokines in the PFC and hippocampus, the essential areas associated with AD pathophysiology, and in the plasma. As expected, pro-inflammatory cytokines/chemokines increased three days after CLP surgery compared to non-CLP surgery. The 30 day time point did not present any difference between the groups; however, at 120 days, the plasma levels of TNF-α increased, and the mice also presented Aβ deposition. Several pro-inflammatory cytokines increased three days after sepsis induction in the PFC and hippocampus, including IFN-γ, and IL-6, accompanied by elevated glial cell reactivity. Neuroinflammation was expected since the first few hours following sepsis are critical in the immune system’s attempt to combat infection or reduce inflammation. However, systemic inflammatory mediators are known to influence the BBB, increasing its permeability [14]. Experimental peripheral infection increased the BBB permeability a few hours after CLP surgery [56], presenting a BBB pathophysiology similar to experimental pneumococcal meningitis in which the bacteria is introduced into the fourth ventricle [57]. Compared to cognitively healthy participants, BBB in mild cognitive impairment (MCI) patients was more permeable to small molecules like water but not to larger molecules like albumin. BBB water permeability was associated with AD biomarkers in the CSF, particularly Aβ [58]. At 30 days after sepsis induction, the IFN-γ and IL-1α levels remained elevated in the PFC, and IFN-γ, IL-1α, eotaxin, TNF-α, and IL-12 remained elevated in the hippocampus. However, the blood culture of the sepsis survival mice was negative, demonstrating no more infection in those animals and suggesting that sepsis induces a chronic neuroinflammatory state that may be favorable for further AD changes. Another critical data is decreased IL-13 levels in the PFC at 30 and 120 days after sepsis induction. IL-13 is an important cytokine produced by innate lymphoid cells and associated with increasing neuronal survival [59, 60], reducing neuroinflammation, and promoting recovery in a mouse model of TBI [61]. The administration of IL-13 also reduced lesion volume and induced anti-inflammatory microglial and macrophage phenotypes, providing neuroprotection in an experimental model of ischemic stroke [62]. In IL-13-deficient mice, the working and reference memories were impaired, both required for efficient complex learning [63]. Considering this, the consistently decreased levels of IL-13 in APP/PS1 mice subjected to sepsis suggest that peripheral infection/inflammation affects brain-protective mechanisms in the short and long-term. Eotaxin is another chemokine associated with AD, as it negatively correlates with neurogenesis and cognitive function [64]. Our study showed APP/PS1 mice had increased eotaxin levels at 3, 30, and 120 days after CLP surgery. Previous research has found that circulating eotaxin levels are higher in AD patients [65], and higher plasma eotaxin levels have been associated with increased long-term forgetting [66]. Considering changes in eotaxin in our study and the eotaxin are higher in neuroinflammation and neurodegenerative disorders [67], using eotaxin modulators may help lessen the neurologic complications after sepsis.
The gut microbiota imbalance has been connected to AD pathogenesis in preclinical and clinical studies [68, 69]. We evaluated gut microbiota composition to understand the microbiota-gut-brain axis on the peripheral infection/inflammation associated with AD. The gut microbiota and their metabolites were analyzed at different time points after sepsis in APP/PS1 mice; however, 30 days after CLP surgery was a defining moment of significant changes. Our results demonstrated that sepsis profoundly altered the α- and β-diversity 30 and 120 days after the CLP surgery. Sepsis decreased Clostridia abundance, which could contribute to the reduction in SCFAs at 30 days. Clostridium was reported to be a major acetate and butyrate-producing bacteria (1), and Proteobacteria, considered a microbial signature of dysbiosis [70] increased in the CLP group, demonstrating that sepsis profoundly modulates the gut microbiota in APP/PS1 mice. Also, sepsis increased the phyla Bacteroidota, Cyanobacteria, and Proteobacteria, as well as the classes Bacilli, Bacteroidia, and Cyanobacteria, 30 days after CLP surgery. Most research has shown that the Bacteroidetes phylum has pro-inflammatory properties due to endotoxins and cytokine production [71], and Proteobacteria is associated with bacterial dysbiosis induced by inflammation in the gut [72].
In congruence with the above data, the ileum villi length and crypt depth decreased at 30 and 120 days, and colon crypt depth decreased at 3, 30, and 120 days in the sepsis group compared to the control group. Butyric, acetic, and propionic acids are the most significant SCFAs generated by bacteria, and they play essential roles in intestinal homeostasis, circadian rhythm, neuroimmune function, and behavior [13, 14, 73]. The acetic acid decreased at three and 30 days; butyric and isobutyric acid levels subsequently decreased at 30 and 120 days. In addition, SCFAs are a significant energy source for intestinal epithelial cells, which have been shown to improve gut barrier function [73]. Similarly, we found that loss of fucosylated mucus from intestinal goblet cells at the chronic time validated that persisting inflammation would cause loss of protective mucus barrier and accelerated infection [74]. Gut epithelial dysfunction is an age-related regenerative decline [75], demonstrating that sepsis-induced peripheral infection accelerated the dysfunction of intestinal epithelial cells in APP/PS1 mice. In the brain, SCFAs, especially butyric acid, binds to the (aryl hydrocarbon receptor) AHR on the microglia and astrocyte cells blocking nuclear factor kappa-B (NF-κB), decreasing inflammation, neurotoxicity, and immune cell recruitment [76]. Signals from host-microbe interaction were also detected in amnestic mild cognitive impairment and AD, with lower levels of acetic, propionic, butyric, and isovaleric acids, among others, compared to healthy individuals [46]. Our results suggest that infection/inflammation can promote gut dysfunction and neuroinflammation, contributing to the pathogenesis of AD.
CONCLUSION
Considering the global incidence of sepsis and the expected increase in ADRD cases, the current study strengthens the evidence for an association between acute systemic inflammation followed by peripheral infection as an inductor of gut dyshomeostasis, neuroinflammation, and the deposition of Aβ plaques in the brain. The peripheral infection accelerated the Aβ burden with co-localized microglia cells in the brain and an early cognitive decline in APP/PS1 mouse model. Moreover, sepsis triggered long-term gut dysbiosis, reflected by decreased microbial diversity and SCFA levels, changing the gut morphology without the Aβ deposition. Our findings indicate that peripheral infection and inflammation accelerate and exacerbate AD pathology. Mechanistic and neurocognitive studies will be needed to understand the causes and consequences of sepsis-accelerated AD neuropathology.
Supplementary Material
ACKNOWLEDGEMENTS
We acknowledge MD Anderson Advanced Microscopy Core Facility, NIH grant (S10RR029552) and Microbiome Insights Inc., Vancouver, Canada (SCFAs analysis), and the infrastructure and support of the Alkek Center for Metagenomics and Microbiome Research – CMMR (16S rRNA sequencing). This work was supported by startup funds from The University of Texas Health Science Center at Houston to RM and TB, Alzheimer’s Association® AARGDNTF-19–619645 and TARCC 2022–24 to TB, NIH/NIA grant 1RF1AG072491 to RM and TB, and FAPESP grant 21/06496–4 to CHRC.
Footnotes
COMPETING INTERESTS
The authors declare no competing interests.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41380-023-02172-2.
DATA AVAILABILITY
The datasets during and/or analyzed during the current study are available from the corresponding author upon reasonable request.
REFERENCES
- 1.GBD 2019 Dementia Forecasting Collaborators. Estimation of the global prevalence of dementia in 2019 and forecasted prevalence in 2050: an analysis for the Global Burden of Disease Study 2019. Lancet Public Health. 2022;7:e105–e125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brookmeyer R, Johnson E, Ziegler-Graham K, Arrighi HM. Forecasting the global burden of Alzheimer’s disease. Alzheimer’s Dement. 2007;3:186–91. [DOI] [PubMed] [Google Scholar]
- 3.Bateman RJ, Aisen PS, De Strooper B, Fox NC, Lemere CA, Ringman JM, et al. Autosomal-dominant Alzheimer’s disease: a review and proposal for the prevention of Alzheimer’s disease. Alzheimer’s Res Therap. 2011;3:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Duncombe J, Kitamura A, Hase Y, Ihara M, Kalaria RN, Horsburgh K. Chronic cerebral hypoperfusion: a key mechanism leading to vascular cognitive impairment and dementia. Closing the translational gap between rodent models and human vascular cognitive impairment and dementia. Clin Sci. 2017;131:2451–68. [DOI] [PubMed] [Google Scholar]
- 5.Liao J, Chen G, Liu X, Wei ZZ, Yu SP, Chen Q, et al. C/EBPβ/AEP signaling couples atherosclerosis to the pathogenesis of Alzheimer’s disease. Mol Psychiat. 2022;27:3034–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Licastro F Special Issue Editorial: “Infections, Inflammation and Neurodegeneration in Alzheimer Disease” Infections, Neuronal Senescence, and Dementia. Int J Mol Sci. 2022;23:5865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jiang M, Zhang X, Yan X, Mizutani S, Kashiwazaki H, Ni J, et al. GSK3β is involved in promoting Alzheimer’s disease pathologies following chronic systemic exposure to Porphyromonas gingivalis lipopolysaccharide in amyloid precursor protein(NL-F/NL-F) knock-in mice. Brain behav Immun. 2021;98:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ackermans NL, Varghese M, Williams TM, Grimaldi N, Selmanovic E, Alipour A, et al. Evidence of traumatic brain injury in headbutting bovids. Acta Neuropathol. 2022;144:5–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Iwashyna TJ, Ely EW, Smith DM, Langa KM. Long-term cognitive impairment and functional disability among survivors of severe sepsis. JAMA. 2010;304:1787–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Readhead B, Haure-Mirande JV, Funk CC, Richards MA, Shannon P, Haroutunian V, et al. Multiscale Analysis of Independent Alzheimer’s Cohorts Finds Disruption of Molecular, Genetic, and Clinical Networks by Human Herpesvirus. Neuron. 2018;99:64–82.e67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tzeng NS, Chung CH, Lin FH, Chiang CP, Yeh CB, Huang SY, et al. Anti-herpetic Medications and Reduced Risk of Dementia in Patients with Herpes Simplex Virus Infections-a Nationwide, Population-Based Cohort Study in Taiwan. Neurotherapeutics. 2018;15:417–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moreno-Gonzalez I, Morales R, Baglietto-Vargas D, Sanchez-Varo R. Editorial: Risk Factors for Alzheimer’s Disease. Front Agin Neurosci. 2020;12:124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Doifode T, Giridharan VV, Generoso JS, Bhatti G, Collodel A, Schulz PE, et al. The impact of the microbiota-gut-brain axis on Alzheimer’s disease pathophysiology. Pharmacol Res. 2021;164:105314. [DOI] [PubMed] [Google Scholar]
- 14.Barichello T, Generoso JS, Collodel A, Petronilho F, Dal-Pizzol F. The blood-brain barrier dysfunction in sepsis. Tissue Barriers. 2021;9:1840912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Licinio J, Wong ML. Molecular Psychiatry special issue: advances in Alzheimer’s disease. Mol Psychiat. 2021;26:5467–70. [DOI] [PubMed] [Google Scholar]
- 16.Moir RD, Lathe R, Tanzi RE. The antimicrobial protection hypothesis of Alzheimer’s disease. Alzheimer’s & Dement. 2018;14:1602–14. [DOI] [PubMed] [Google Scholar]
- 17.Hur JY, Frost GR, Wu X, Crump C, Pan SJ, Wong E, et al. The innate immunity protein IFITM3 modulates γ-secretase in Alzheimer’s disease. Nature. 2020;586:735–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Holmes C, Cotterell D. Role of infection in the pathogenesis of Alzheimer’s disease: implications for treatment. CNS Drugs. 2009;23:993–1002. [DOI] [PubMed] [Google Scholar]
- 19.Miklossy J Chronic inflammation and amyloidogenesis in Alzheimer’s disease - role of Spirochetes. J Alzheimer’s Dis. 2008;13:381–91. [DOI] [PubMed] [Google Scholar]
- 20.Grant I, Franklin DR Jr, Deutsch R, Woods SP, Vaida F, Ellis RJ, et al. Asymptomatic HIV-associated neurocognitive impairment increases risk for symptomatic decline. Neurology. 2014;82:2055–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Itzhaki RF, Wozniak MA. Herpes simplex virus type 1 in Alzheimer’s disease: the enemy within. J Alzheimer’s Dis. 2008;13:393–405. [DOI] [PubMed] [Google Scholar]
- 22.Gasparotto J, Girardi CS, Somensi N, Ribeiro CT, Moreira JCF, Michels M, et al. Receptor for advanced glycation end products mediates sepsis-triggered amyloid-β accumulation, Tau phosphorylation, and cognitive impairment. J Biol Chem. 2018;293:226–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Giridharan VV, Generoso JS, Lence L, Candiotto G, Streck E, Petronilho F, et al. A crosstalk between gut and brain in sepsis-induced cognitive decline. J Neuroinflamm. 2022;19:114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Basak JM, Ferreiro A, Cohen LS, Sheehan PW, Nadarajah CJ, Kanan MF, et al. Bacterial sepsis increases hippocampal fibrillar amyloid plaque load and neuroinflammation in a mouse model of Alzheimer’s disease. Neurobiol Dis. 2021;152:105292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Giridharan VV, Generoso JS, Collodel A, Dominguini D, Faller CJ, Tardin F, et al. Receptor for Advanced Glycation End Products (RAGE) Mediates Cognitive Impairment Triggered by Pneumococcal Meningitis. Neurotherapeutics. 2021;18:640–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barichello T, Giridharan VV, Comim CM, Morales R. What is the role of microbial infection in Alzheimer’s disease? Revista brasileira de psiquiatria (Sao Paulo, Brazil : 1999) 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Garcia-Alloza M, Robbins EM, Zhang-Nunes SX, Purcell SM, Betensky RA, Raju S, et al. Characterization of amyloid deposition in the APPswe/PS1dE9 mouse model of Alzheimer disease. Neurobiol Dis. 2006;24:516–24. [DOI] [PubMed] [Google Scholar]
- 28.Heming N, Mazeraud A, Verdonk F, Bozza FA, Chrétien F, Sharshar T. Neuroanatomy of sepsis-associated encephalopathy. Critic Care. 2017;21:65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ehler J, Barrett LK, Taylor V, Groves M, Scaravilli F, Wittstock M, et al. Translational evidence for two distinct patterns of neuroaxonal injury in sepsis: a longitudinal, prospective translational study. Critic Care. 2017;21:262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rittirsch D, Huber-Lang MS, Flierl MA, Ward PA. Immunodesign of experimental sepsis by cecal ligation and puncture. Nat Protoc. 2009;4:31–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Idris N, Neill J, Grayson B, Bang-Andersen B, Witten LM, Brennum LT, et al. Sertindole improves sub-chronic PCP-induced reversal learning and episodic memory deficits in rodents: involvement of 5-HT(6) and 5-HT (2 A) receptor mechanisms. Psychopharmacology. 2010;208:23–36. [DOI] [PubMed] [Google Scholar]
- 32.Scaini G, Fries GR, Valvassori SS, Zeni CP, Zunta-Soares G, Berk M, et al. Perturbations in the apoptotic pathway and mitochondrial network dynamics in peripheral blood mononuclear cells from bipolar disorder patients. Transl Psychiat. 2017;7:e1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kawarabayashi T, Younkin LH, Saido TC, Shoji M, Ashe KH, Younkin SG. Age-dependent changes in brain, CSF, and plasma amyloid (beta) protein in the Tg2576 transgenic mouse model of Alzheimer’s disease. J Neurosci. 2001;21:372–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nature Biotechnol. 2019;37:852–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhao G, Nyman M, Jönsson JA. Rapid determination of short-chain fatty acids in colonic contents and faeces of humans and rats by acidified water-extraction and direct-injection gas chromatography. Biomed Chromatogr 2006;20:674–82. [DOI] [PubMed] [Google Scholar]
- 36.Radde R, Bolmont T, Kaeser SA, Coomaraswamy J, Lindau D, Stoltze L, et al. Abeta42-driven cerebral amyloidosis in transgenic mice reveals early and robust pathology. EMBO Rep. 2006;7:940–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Serneels L, Van Biervliet J, Craessaerts K, Dejaegere T, Horré K, Van Houtvin T, et al. gamma-Secretase heterogeneity in the Aph1 subunit: relevance for Alzheimer’s disease. Science. 2009;324:639–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang J, Dickson DW, Trojanowski JQ, Lee VM. The levels of soluble versus insoluble brain Abeta distinguish Alzheimer’s disease from normal and pathologic aging. Exp Neurol. 1999;158:328–37. [DOI] [PubMed] [Google Scholar]
- 39.Sofroniew MV, Vinters HV. Astrocytes: biology and pathology. Acta Neuropathol. 2010;119:7–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bird CM, Burgess N. The hippocampus and memory: insights from spatial processing. Nature Rev Neurosci. 2008;9:182–94. [DOI] [PubMed] [Google Scholar]
- 41.Sun Q, Zhang J, Li A, Yao M, Liu G, Chen S, et al. Acetylcholine deficiency disrupts extratelencephalic projection neurons in the prefrontal cortex in a mouse model of Alzheimer’s disease. Nature Commun. 2022;13:998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhurakovskaya E, Ishchenko I, Gureviciene I, Aliev R, Gröhn O, Tanila H. Impaired hippocampal-cortical coupling but preserved local synchrony during sleep in APP/PS1 mice modeling Alzheimer’s disease. Sci Rep. 2019;9:5380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vogt NM, Kerby RL, Dill-McFarland KA, Harding SJ, Merluzzi AP, Johnson SC, et al. Gut microbiome alterations in Alzheimer’s disease. Sci Reports. 2017;7:13537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Adelman MW, Woodworth MH, Langelier C, Busch LM, Kempker JA, Kraft CS, et al. The gut microbiome’s role in the development, maintenance, and outcomes of sepsis. Critic Care. 2020;24:278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu P, Wu L, Peng G, Han Y, Tang R, Ge J, et al. Altered microbiomes distinguish Alzheimer’s disease from amnestic mild cognitive impairment and health in a Chinese cohort. Brain Behav Immun. 2019;80:633–43. [DOI] [PubMed] [Google Scholar]
- 46.Wu L, Han Y, Zheng Z, Peng G, Liu P, Yue S, et al. Altered Gut Microbial Metabolites in Amnestic Mild Cognitive Impairment and Alzheimer’s Disease: Signals in Host-Microbe Interplay. Nutrients. 2021;13:228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wen C, Xie T, Pan K, Deng Y, Zhao Z, Li N, et al. Acetate attenuates perioperative neurocognitive disorders in aged mice. Aging. 2020;12:3862–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yende S, Austin S, Rhodes A, Finfer S, Opal S, Thompson T, et al. Long-Term Quality of Life Among Survivors of Severe Sepsis: Analyses of Two International Trials. Critic Care Med. 2016;44:1461–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Buchman TG, Simpson SQ, Sciarretta KL, Finne KP, Sowers N, Collier M, et al. Sepsis Among Medicare Beneficiaries: 1. The Burdens of Sepsis, 2012–2018. Critic Care Med. 2020;48:276–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gracner T, Agarwal M, Murali KP, Stone PW, Larson EL, Furuya EY, et al. Association of Infection-Related Hospitalization With Cognitive Impairment Among Nursing Home Residents. JAMA Network Open. 2021;4:e217528–e217528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hanseeuw BJ, Betensky RA, Jacobs HIL, Schultz AP, Sepulcre J, Becker JA, et al. Association of Amyloid and Tau With Cognition in Preclinical Alzheimer Disease: A Longitudinal Study. JAMA Neurol. 2019;76:915–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kumar DK, Choi SH, Washicosky KJ, Eimer WA, Tucker S, Ghofrani J, et al. Amyloid-β peptide protects against microbial infection in mouse and worm models of Alzheimer’s disease. Sci Transl Med. 2016;8:340ra372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Licinio J, Wong M-L. Advances in Molecular Psychiatry – March 2023: mitochondrial function, stress, neuroinflammation – bipolar disorder, psychosis, and Alzheimer’s disease. Mol Psychiat. 2023;28:968–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Haage V, De Jager PL. Neuroimmune contributions to Alzheimer’s disease: a focus on human data. Mol Psychiat. 2022;27:3164–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Giridharan VV, Masud F, Petronilho F, Dal-Pizzol F, Barichello T. Infection-Induced Systemic Inflammation Is a Potential Driver of Alzheimer’s Disease Progression. Front Agin Neurosci. 2019;11:122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Comim CM, Vilela MC, Constantino LS, Petronilho F, Vuolo F, Lacerda-Queiroz N, et al. Traffic of leukocytes and cytokine up-regulation in the central nervous system in sepsis. Intens Care Med. 2011;37:711–8. [DOI] [PubMed] [Google Scholar]
- 57.Barichello T, Generoso JS, Silvestre C, Costa CS, Carrodore MM, Cipriano AL, et al. Circulating concentrations, cerebral output of the CINC-1 and blood–brain barrier disruption in Wistar rats after pneumococcal meningitis induction. Eur J Clin Microbiol Infect Dis. 2012;31:2005–9. [DOI] [PubMed] [Google Scholar]
- 58.Lin Z, Sur S, Liu P, Li Y, Jiang D, Hou X, et al. Blood-Brain Barrier Breakdown in Relationship to Alzheimer and Vascular Disease. Annal Neurol. 2021;90:227–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shin WH, Lee DY, Park KW, Kim SU, Yang MS, Joe EH, et al. Microglia expressing interleukin-13 undergo cell death and contribute to neuronal survival in vivo. Glia. 2004;46:142–52. [DOI] [PubMed] [Google Scholar]
- 60.Barichello T The role of innate lymphoid cells (ILCs) in mental health. Discov Mental Health. 2022;2:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Miao W, Zhao Y, Huang Y, Chen D, Luo C, Su W, et al. IL-13 Ameliorates Neuroinflammation and Promotes Functional Recovery after Traumatic Brain Injury. J Immunol. 2020;204:1486–98. [DOI] [PubMed] [Google Scholar]
- 62.Kolosowska N, Keuters MH, Wojciechowski S, Keksa-Goldsteine V, Laine M, Malm T, et al. Peripheral Administration of IL-13 Induces Anti-inflammatory Microglial/Macrophage Responses and Provides Neuroprotection in Ischemic Stroke. Neurotherapeutics. 2019;16:1304–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Brombacher TM, Nono JK, De Gouveia KS, Makena N, Darby M, Womersley J, et al. IL-13-Mediated Regulation of Learning and Memory. J Immunol. 2017;198:2681–8. [DOI] [PubMed] [Google Scholar]
- 64.Villeda SA, Luo J, Mosher KI, Zou B, Britschgi M, Bieri G, et al. The ageing systemic milieu negatively regulates neurogenesis and cognitive function. Nature. 2011;477:90–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Choi C, Jeong JH, Jang JS, Choi K, Lee J, Kwon J, et al. Multiplex analysis of cytokines in the serum and cerebrospinal fluid of patients with Alzheimer’s disease by color-coded bead technology. J Clin Neurol. 2008;4:84–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kong Y, Li HD, Wang D, Gao X, Yang C, Li M, et al. Group 2 innate lymphoid cells suppress the pathology of neuromyelitis optica spectrum disorder. FASEB J. 2021;35:e21856. [DOI] [PubMed] [Google Scholar]
- 67.Nazarinia D, Behzadifard M, Gholampour J, Karimi R, Gholampour M. Eotaxin-1 (CCL11) in neuroinflammatory disorders and possible role in COVID-19 neurologic complications. Acta Neurol Belg. 2022;122:865–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhang L, Wang Y, Xiayu X, Shi C, Chen W, Song N, et al. Altered Gut Microbiota in a Mouse Model of Alzheimer’s Disease. J Alzheimer’s Dis. 2017;60:1241–57. [DOI] [PubMed] [Google Scholar]
- 69.Marizzoni M, Cattaneo A, Mirabelli P, Festari C, Lopizzo N, Nicolosi V, et al. Short-Chain Fatty Acids and Lipopolysaccharide as Mediators Between Gut Dysbiosis and Amyloid Pathology in Alzheimer’s Disease. J Alzheimer’s Dis. 2020;78:683–97. [DOI] [PubMed] [Google Scholar]
- 70.Shin NR, Whon TW, Bae JW. Proteobacteria: microbial signature of dysbiosis in gut microbiota. Trend Biotechnol. 2015;33:496–503. [DOI] [PubMed] [Google Scholar]
- 71.Stojanov S, Berlec A, Štrukelj B. The Influence of Probiotics on the Firmicutes/Bacteroidetes Ratio in the Treatment of Obesity and Inflammatory Bowel disease. Microorganisms. 2020;8:1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Carvalho FA, Koren O, Goodrich JK, Johansson ME, Nalbantoglu I, Aitken JD, et al. Transient inability to manage proteobacteria promotes chronic gut inflammation in TLR5-deficient mice. Cell Host Microbe. 2012;12:139–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cryan JF, O’Riordan KJ, Sandhu K, Peterson V, Dinan TG. The gut microbiome in neurological disorders. Lancet Neurol. 2020;19:179–94. [DOI] [PubMed] [Google Scholar]
- 74.Ganesh BP, Klopfleisch R, Loh G, Blaut M. Commensal Akkermansia muciniphila exacerbates gut inflammation in Salmonella Typhimurium-infected gnotobiotic mice. PloS One. 2013;8:e74963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jasper H Intestinal Stem Cell Aging: Origins and Interventions. Annu Rev Physiol. 2020;82:203–26. [DOI] [PubMed] [Google Scholar]
- 76.Rothhammer V, Quintana FJ. The aryl hydrocarbon receptor: an environmental sensor integrating immune responses in health and disease. Nature Rev Immunol. 2019;19:184–97. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets during and/or analyzed during the current study are available from the corresponding author upon reasonable request.