

Significance
CAR T cell therapy, a transformative treatment for blood cancers, is limited by T cell dysfunction, especially in chronic lymphocytic leukemia (CLL). Our study reveals a pivotal role for the human and great ape-specific p53 isoform, Δ133p53α, in enhancing the therapeutic efficacy of CAR T cells. Expressing Δ133p53α in CAR T cells improves their metabolic robustness, enabling superior antitumor function particularly under high tumor burden conditions. Importantly, this modification enhanced the effectiveness of CAR T cells from CLL patients who previously did not benefit from CAR T cell therapy. Our findings underscore the potential of taking the approach to modulating the p53 pathway to improve CAR T cell treatments.
Keywords: T cell, senescence, cancer therapy
Abstract
Chimeric antigen receptor (CAR) T cell dysfunction is a major barrier to achieving lasting remission in hematologic cancers, especially in chronic lymphocytic leukemia (CLL). We have shown previously that Δ133p53α, an endogenous isoform of the human TP53 gene, decreases in expression with age in human T cells, and that reconstitution of Δ133p53α in poorly functional T cells can rescue proliferation [A. M. Mondal et al., J. Clin. Invest. 123, 5247–5257 (2013)]. Although Δ133p53α lacks a transactivation domain, it can form heterooligomers with full-length p53 and modulate the p53-mediated stress response [I. Horikawa et al., Cell Death Differ. 24, 1017–1028 (2017)]. Here, we show that constitutive expression of Δ133p53α potentiates the anti-tumor activity of CD19-directed CAR T cells and limits dysfunction under conditions of high tumor burden and metabolic stress. We demonstrate that Δ133p53α-expressing CAR T cells exhibit a robust metabolic phenotype, maintaining the ability to execute effector functions and continue proliferating under nutrient-limiting conditions, in part due to upregulation of critical biosynthetic processes and improved mitochondrial function. Importantly, we show that our strategy to constitutively express Δ133p53α improves the anti-tumor efficacy of CAR T cells generated from CLL patients that previously failed CAR T cell therapy. More broadly, our results point to the potential role of the p53-mediated stress response in limiting the prolonged antitumor functions required for complete tumor clearance in patients with high disease burden, suggesting that modulation of the p53 signaling network with Δ133p53α may represent a translationally viable strategy for improving CAR T cell therapy.
The introduction of chimeric antigen receptor (CAR) T cell therapy has revolutionized the treatment landscape for patients with relapsed or refractory (r/r) hematologic malignancies (1–4). Nonetheless, T cell dysfunction is a critical limitation for a subset of hematologic patients that either do not respond to CAR T cell therapy, or experience relapse in the absence of antigen loss (5–7). For instance, in chronic lymphocytic leukemia (CLL), the complete response (CR) rate to CAR T cell therapy is estimated at 25%, compared with 44% in r/r non-Hodgkin lymphoma (NHL) and 77% in r/r B cell acute lymphoblastic leukemia (B-ALL) (8). Furthermore, in B-ALL, roughly 20% of CAR T cell patients experience relapse without the loss of CD19 antigen, highlighting the importance of T cell dysfunction even in the indication for which CAR T cell therapy is most effective (5, 9). In CLL, CAR T cell dysfunction is particularly challenging due to a combination of factors that impair baseline T cell function including patient age, treatment regimen, tumor microenvironment (TME), and metabolic impairments (10, 11). Given the availability of validated CAR targets, hematologic malignancies represent a useful context for developing strategies to mitigate CAR T cell dysfunction, with findings potentially useful in the context of solid tumors, where a lack of validated CAR targets has complicated progress (12–14).
The human TP53 gene encodes the transcription factor p53α, also referred to as full-length p53 (p53FL), and gives rise to at least 11 additional protein isoforms through usage of alternative promoters and splicing (15). These protein isoforms of p53, alongside p53 family members p63 and p73, contribute to the diverse, context-dependent p53-mediated transcriptional responses, which can influence nearly all fundamental processes in the cell, including proliferation, metabolism, stemness, and survival (16, 17). Indeed, decades of intense investigation—and more publications than any gene in history—have revealed that p53 is more than just the “guardian of the genome,” as it also mediates several pro-survival and pro-proliferative transcriptional programs in response to milder forms of cellular stress (18, 19).
The p53 isoform Δ133p53α, only expressed in humans and great apes, lacks the first 132 residues found in p53FL containing both transactivation domains, the proline-rich domain, and loop 1 of the DNA binding domain (Fig. 1A). Nonetheless, Δ133p53α plays an important role in modulating the transcriptional activity of p53, as well as p53 signaling network members p63 and p73, via interaction with each through an oligomerization domain (20–22). Although the mechanistic details are not yet fully elucidated, there is evidence that Δ133p53 selectively inhibits transcriptional activity at specific p53FL response elements in a dominant-negative manner, with a net bias toward pro-survival cell fates in appropriate cellular contexts (15, 23–25). This isoform-mediated regulation of p53’s function as a transcription factor is thought to contribute to the complex “decision-making” in which p53 can initiate an appropriate stress response to a wide variety of different forms and intensities of cellular stress (16–18). Across several cell types, it has been shown that Δ133p53α-mediated modulation of the p53 signaling network leads to upregulated DNA repair, improved glucose uptake, enhanced proliferation, as well as reduced senescence without evidence for increased risk of transformation (15, 21, 26–30).
Fig. 1.
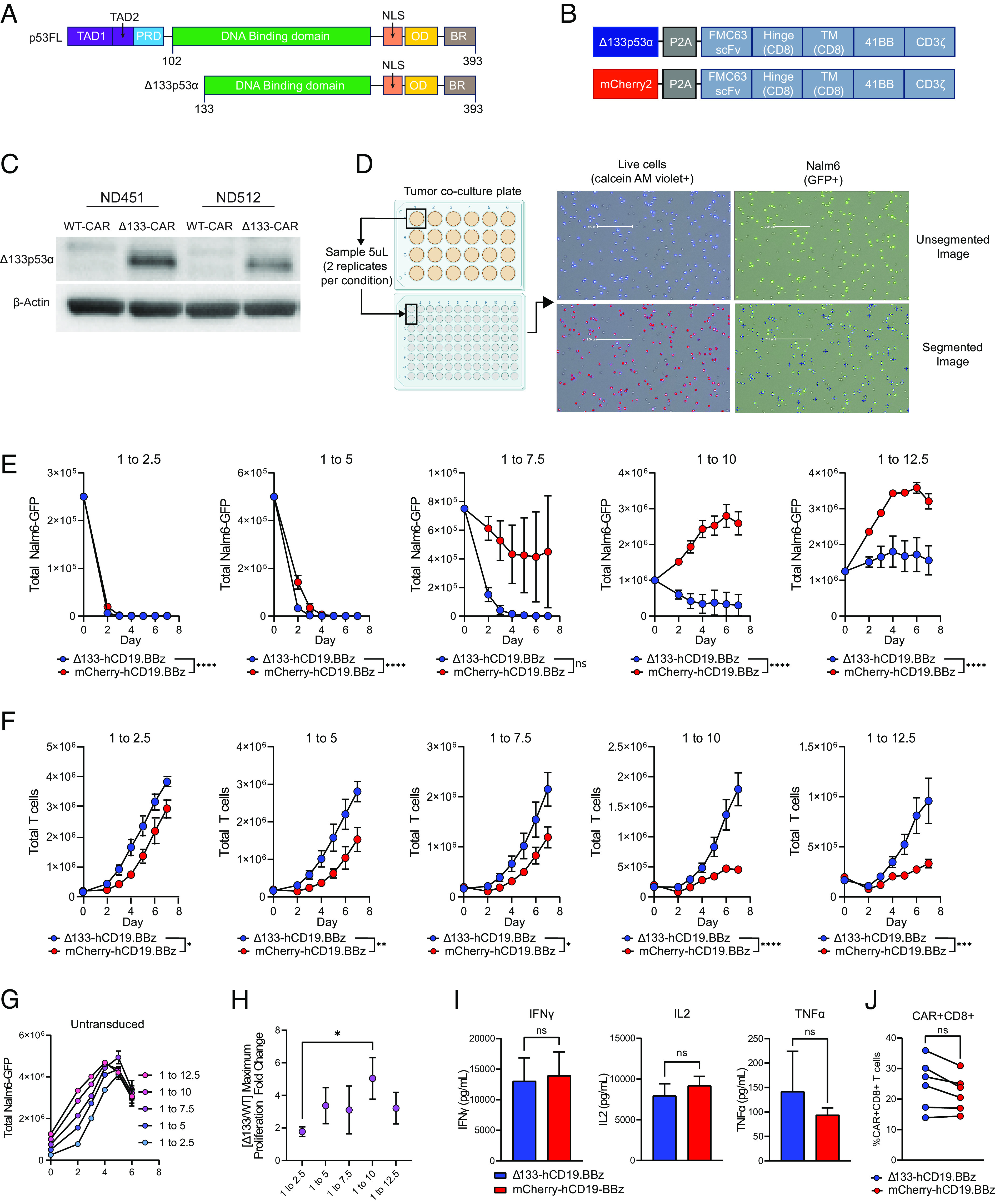
Δ133p53α-CAR T cells improve in vitro cytotoxicity and proliferation during high tumor burden conditions. (A) Schematic of human full-length p53 (p53FL) and Δ133p53α. TAD, transactivation domain; PRD, proline-rich domain; NLS, nuclear localization signal; OD, oligomerization domain; BR, basic region. (B) CAR expression vectors for Δ133p53α-expressing and WT, mCherry-expressing CARs. TM, transmembrane domain; WT, wild type. (C) Western blot for Δ133p53α expression in Δ133p53α CAR T cells and WT CAR T cells at day 10 of primary expansion from two normal donors. Δ133, Δ133p53α. (D) Schematic outlining in vitro co-culture assay setup and accompanying tumor and T cell measurements. Representative images show GFP+ tumor cells (Nalm6-GFP) and corresponding segmented images used for quantification by Celigo software. Live cells are identified by detection of fluorescent calcein AM violet, with total T cells being calculated by subtracting the number of GFP+ cells from the total number of live cells. Representative images showing segmentation and counting performed by Celigo software are shown. (E) Tumor measurements over time from in vitro co-culture assay described in D from six separate normal donors. Co-cultures were run at 5 effector to target ratios, indicated by the number over each graph. Data are mean ± SEM. Statistical significance was assessed using a two-way ANOVA, with reported values representing the interacting term of CAR treatment over time. (F) T cell proliferation during tumor co-culture from the same experiments shown in E. Data are from replicate experiments with six healthy donors. Statistical significance was assessed using a two-way ANOVA, with reported values representing the interacting term of CAR treatment over time. (G) Representative untransduced T cells co-cultured with Nalm6-GFP tumor cells, included as a negative control. (H) Ratio between maximum proliferation of Δ133p53α-CARs and wild-type CARs at each effector to target ratio. Maximum proliferation for each condition was determined as the maximum fold-change from baseline CAR T cells. Statistical significance was calculated using multiple t tests. (I) Cytokine secretion at 24-h time point of in vitro co-culture at 1 to 7.5 effector to target ratio. (J) Proportion of CAR+CD8+ T cells in each comparator group for each healthy donor used for data generated in E–H. Statistical significance for I and J was assessed using paired t tests. ns = not significant, *P 0.05, **P 0.01, ***P 0.001, ****P 0.0001.
We have previously shown that Δ133p53α expression decreases with age in human T cells and that ectopic expression of Δ133p53α enhances the proliferative capacity of a near-senescent CD28-CD57+ T cell subset (31). Given the association between age and baseline impairment of T cell function, as well as the overlap between p53-activating stresses and factors known to contribute to T cell dysfunction, we hypothesized that constitutive expression of Δ133p53α would improve CAR T cell therapy through modulation of the p53-mediated stress response (32, 33).
Here, we demonstrate that constitutive expression of Δ133p53α improves the antitumor function of CD19-directed CAR T cells manufactured from both healthy normal donors and from CLL patients that failed to respond to CAR T cell therapy during clinical trials. We observe that ectopic expression of Δ133p53α improves antitumor function in part through modulation of p53FL transcriptional activity and improved metabolic function. These results inform the development of improved CAR T cell therapies and point to the importance of the p53 signaling network as a regulator of prolonged CAR T cell responses.
Results
CAR T Cells that Constitutively Express Δ133p53α Demonstrate Enhanced In Vitro Tumor Clearance during High Tumor Burden Conditions.
To test the hypothesis that constitutive expression of Δ133p53α (Fig. 1A) can improve chimeric antigen receptor (CAR) T cell function, we co-expressed Δ133p53α alongside the well-characterized, humanized CD19-directed CAR with a 4-1BB co-stimulatory domain (hCD19.BBz) using a P2A self-cleaving linker (Fig. 1B). We evaluated the impact of Δ133p53α expression both upstream and downstream of the P2A linker to account for any potential impact of the residual 2A peptide linker post-cleavage and moved forward with Δ133p53α expressed upstream of the P2A linker due to observation of a more consistent improvement in antitumor function over matched CAR controls (SI Appendix, Fig. S1 A–C). To eliminate any impact of construct size on the level of surface CAR expression, we designed our wild-type (WT) CAR construct to express the similarly sized fluorescent protein, mCherry2, in the same position as Δ133p53α (Fig. 1B). Using this construct, we observed stable expression of ectopic Δ133p53α (Fig. 1C). No significant differences were observed during CAR manufacturing between Δ133p53α-expressing CARs and WT CARs in proliferation, cell volume, or surface CAR MFI (SI Appendix, Fig. S2 A–C).
To simultaneously evaluate the cytotoxicity and proliferative capacity of Δ133p53α-expressing CARs, we developed an in vitro co-culture assay capable of accurately quantitating tumor cell clearance and T cell proliferation simultaneously. To do so, we used a Celigo Image cytometer to measure absolute counts of a B-ALL cell line, Nalm6, engineered to express green fluorescent protein (GFP) and the total number of live cells via calcein AM violet, through direct sub-sampling from each co-culture condition at each time point (Fig. 1D).
Using this assay, we measured tumor clearance and proliferation of CAR T cells generated from six healthy donors at five effector to target (E:T) ratios (i.e. CAR+ T cells to tumor cells), ranging from 1 to 2.5 up to 1 to 12.5. Interestingly, we observed a consistent pattern across donors in which differences in tumor clearance became more pronounced as tumor burden increased from the lowest tumor dose (1 to 2.5) until the second highest tumor dose (1 to 10), with the Δ133p53α-expressing CAR T cells (referred to as Δ133-CARs) exhibiting significantly more tumor clearance than wild-type (WT) controls (Fig. 1 E and G). A similar pattern was observed for CAR T cell proliferation (Fig. 1F), with the largest relative differences in maximum proliferation between the Δ133-CAR and WT CAR observed at a 1 to 10 E:T ratio (Fig. 1H). At a 1 to 12.5 tumor dose, we observe that Δ133-CARs have robust anti-tumor effects, whereas WT CARs fail to kill tumor and allow tumor proliferation kinetics similar to non-transduced controls. Donor-to-donor differences in antitumor function are responsible for the variance observed at a 1 to 7.5 in WT CARs and a 1 to 12.5 for the Δ133-CARs. Individual tumor clearance and proliferation curves for each donor are presented in SI Appendix, Figs. S3 and S4. Given the role of Δ133p53α in modulating the p53-mediated stress response, our results point to the possibility that increased stress associated with higher tumor burden potentiates the effects of Δ133p53α.
Minimal differences were observed in cytokine production after 24-h co-culture with Nalm6 at a 1 to 7.5 E:T ratio (Fig. 1I). The improved tumor clearance and proliferation of Δ133-CARs could not be attributed to baseline differences in %CAR+CD8+ (Fig. 1J) or initial %CAR+ T cells, as each co-culture is normalized for an equivalent number of input-CAR+ T cells.
Δ133-CAR T Cells Exhibit Superior Tumor Clearance in a Nalm6 Xenograft Mouse Model.
To evaluate the in vivo functionality of Δ133-CAR T cells, we used NOD/SCID/IL2Rγ−/− (NSG) mice engrafted with Nalm6 leukemia cells engineered to express both GFP and luciferase and then compared tumor clearance between Δ133- and WT CARs from three normal healthy donors. Based on in vitro findings which the improvements conferred by Δ133p53α were most significant under high tumor load, we used an intentionally high starting tumor burden (Fig. 2A). Under these conditions, we observed a consistent improvement in tumor clearance across donors, with Δ133-CARs driving a reduction in mean tumor luminescence approaching background levels by day 21 (Fig. 2 B and C). Differences in engraftment were observed at day 12 (Fig. 2D). Improvements in tumor clearance corresponded with improvements in survival, with significantly more mice administered Δ133-CARs alive at the final time point compared with WT CARs (Fig. 2E).
Fig. 2.
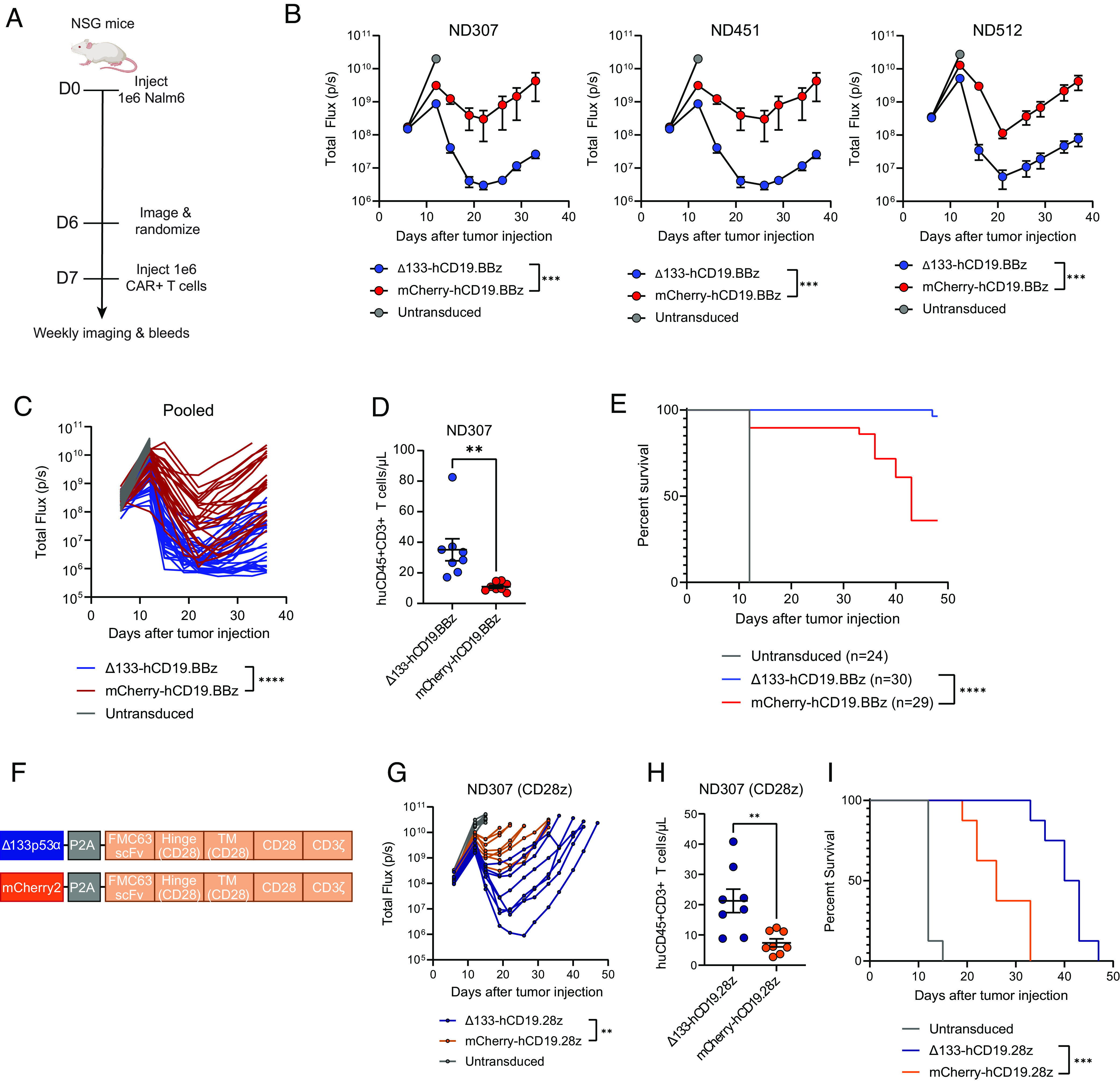
Δ133-CAR T cells exhibit superior tumor clearance in a B cell leukemia xenograft mouse model. (A) NSG mice were injected intravenously with 1 × 106 Nalm6 leukemia cells transduced with GFP-P2A-CBG on day 0. Mice were imaged on day 6 and randomized into three groups, including both treatment groups and an untransduced T cell control. Either 1 × 106 freshly thawed CAR+ T cells or a matched total number of untransduced T cells were injected intravenously on day 7. (B) Tumor progression for mice administered CARs from three healthy donors was monitored using bioluminescent imaging. Data are mean ± SEM of n = 8 to 11 mice depending on the donor. In each graph, data are reported up to the timepoint when the first mouse was lost in the WT or Δ133-CAR group. Statistical significance was determined via two-way ANOVA and is based on the CAR treatment X time interaction term. (C) Time course of tumor progression in individual mice across three experiments shown in B. Statistical significance was determined via two-way ANOVA and is based on the CAR treatment X time interaction term. (D) T cell engraftment on day 12 after injection. Statistical significance was calculated using an unpaired t test. Data not shown for ND451 and ND512 due to sample mishandling preventing measurement at day 12. (E) Kaplan–Meier curves of mice pooled from all experiments showing survival of each group over time. Data were analyzed using a log rank Mantel–Cox test. (F) CD19-directed CAR expression vector with CD28 co-stimulatory domain used in G–I. (G) Tumor progression for mice administered hCD19.28z CARs. Data represent tumor progression for each individual mouse. Statistical significance was determined via two-way ANOVA and is based on the CAR treatment X time interaction term. (H) T cell engraftment at initial bleed measurement. Statistical significance was calculated using an unpaired t test. (I) Kaplan–Meier curves showing survival of each group of mice over time. Data were analyzed using a log rank Mantel–Cox test. ns = not significant, *P 0.05, **P 0.01, ***P 0.001, ****P 0.0001.
Given the consistency of functional improvements observed with Δ133p53α alongside the 4-1BB costimulatory domain both in vitro and in vivo, we next evaluated whether the benefits would extend to a CAR with a CD28 costimulatory domain. To test this, we generated matching constructs with Δ133p53α and mCherry2 alongside a humanized CD19-directed CAR with a CD28 co-stimulatory domain (hCD19.28z) (Fig. 2F). Again, antitumor function was improved by Δ133p53α, albeit with reduced overall potency in both groups compared with the 41BB-containing CAR (Fig. 2G). T cell engraftment at day 12 with the hCD19.28z constructs showed that Δ133-CARs have more than double the mean expansion compared with WT 28z CARs (Fig. 2H). Differences in tumor clearance and engraftment were reflected in survival differences which again were in favor of the Δ133p53α-expressing hCD19.28z CAR (Fig. 2I).
Δ133-CAR T Cells Are Characterized by Enhanced Metabolic Capacity and Reduced Transcription of p53 Target Genes.
To gain insight into the mechanism by which Δ133p53α confers improved antitumor function, we performed bulk RNAseq on CAR T cells isolated before and during in vitro co-culture with Nalm6-GFP tumor cells at a 1 to 10 effector to target ratio, from three healthy normal donors (Fig. 3A). We employed the in vitro assay detailed in Fig. 1D, with a modification to use a 300 μL media feed after day 3 measurement instead of the standard 600uL. This change was based on a prior observation indicating that restriction of nutrient availability accentuates differences in tumor clearance between Δ133p53α and WT CAR T cells.
Fig. 3.
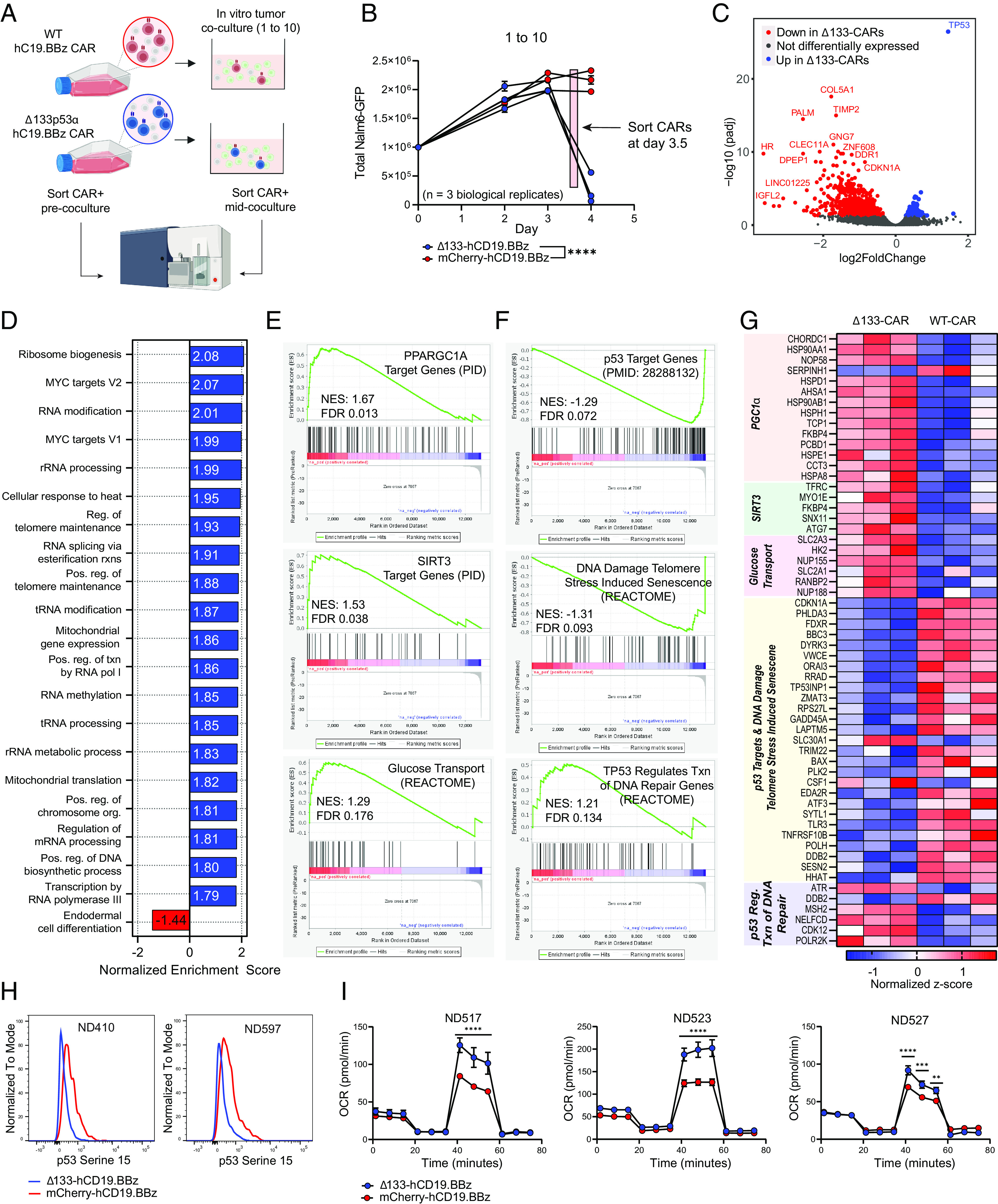
Transcriptomic analysis of Δ133-CAR T cells under stress reveals differences in transcription of p53 targets and key metabolic genes. (A) Schematic of sample generation for RNAseq analysis. CAR+ T cells were isolated prior to co-culture (day 0) and again mid-coculture, approximately 8 h prior to day 4 measurement. CAR T cells from the same three healthy donors were used for both isolations. (B) Tumor co-culture experiment used to generate CARs for RNAseq analysis. An effector to target ratio of 1 to 10 was used. Each data point represents the mean ± SEM of two technical replicates for each healthy donor. Statistical significance was determined via two-way ANOVA and is based on the CAR treatment X time interaction term. (C) Volcano plot showing differentially expressed genes in WT CAR T cells and Δ133-CAR T cells isolated mid-coculture. All genes with an adjusted P-value ≤ 0.05 are colored either red or blue, if upregulated in WT or Δ133-CARs, respectively. (D) Summary of results for GSEA of all Hallmark and GO Biological Process gene sets. For Δ133-CARs, only the top 20 of the 80 gene sets with FDR < 0.1, ranked by normalized enrichment score are shown. (E) Additional gene sets identified during targeted GSEA for metabolic gene sets relevant for CAR T cell function. (F) Additional gene sets identified during targeted GSEA for p53 transcriptional activity. (G) Heatmap of individual genes from gene sets in E and F with adjusted P-values ≤ 0.05. Corresponding gene sets for each gene are highlighted in vertical text. (H) Phospho-p53 (Serine 15) staining of CAR T cells on day 3 of tumor co-culture for two healthy donors measured by flow cytometry. (I) Oxygen consumption rate (OCR) as measured by Seahorse extracellular flux analysis. Each of the three healthy donors was evaluated following tumor co-culture. Data points for each time series represent mean oxygen consumption rate ± SEM for eight replicates. Statistical significance was determined using multiple unpaired t tests for each time point. ns = not significant, *P 0.05, **P 0.01, ***P 0.001, ****P 0.0001.
Comparison of differentially expressed genes between Δ133p53α and WT CARs pre-coculture revealed minimal differences, with the top hit, TP53, due to ectopic expression of the Δ133p53α transgene (SI Appendix, Fig. S5A). The low amount of differentially expressed genes is consistent with the lack of functional differences observed during CAR manufacture and under unstressed conditions; however, this may also reflect the relatively low transcriptional activity of rested CAR T cells.
For the Δ133p53α and WT CARs isolated during tumor co-culture (Fig. 3B), 99 of the top 100 most differentially expressed genes were downregulated in the Δ133-CARs cells (Fig. 3C) (SI Appendix, Table S1). In contrast to individual genes, gene set enrichment analysis (GSEA) of all Hallmark and gene ontology biological process (GO: BP) gene sets revealed 80 separate gene sets upregulated in the Δ133-CARs with a false discovery rate (FDR) ≤0.1, compared with only 1 gene set enriched in the WT CARs (SI Appendix, Table S2). A summary of the top 20 gene sets enriched in the Δ133-CARs sorted by normalized enrichment scores (NES), as well as the 1 gene set enriched in the WT CARs, is shown in Fig. 3D. In the Δ133-CARs, which demonstrated significantly enhanced anti-tumor function both in vitro and in vivo, we observed an enrichment of several gene sets associated with increased biosynthetic processes, metabolic processes, and maintenance of cellular integrity, including ribosome biogenesis, mitochondrial translation, and regulation of telomere maintenance. Consistent with our functional data, these findings suggest that Δ133-CARs generate enhanced anti-tumor activity in part due to robust upregulation of metabolic and biosynthetic processes.
Based on these findings, we performed GSEA on additional gene sets related to essential metabolic processes in T cells. Further supporting our initial unbiased GSEA findings, we identified additional gene sets associated with upregulation of targets of the transcription factors PGC1α and SIRT3, as well as an enrichment of glucose transport (GLUT3) and metabolism (HK2) (Fig. 3E). That HK11 localizes to the outer mitochondrial membrane, via voltage-dependent anion channel (VDAC), and initiates glycolytic reprogramming implicates its importance for activation, proliferation, and effector function in this context. We previously showed how glycolytic metabolism is indispensable for human T cell proliferation (34), and others have shown how glycolysis supports IFNγ production in effector T cells (35). Unlike other scenarios, in which there can be a trade-off between glycolytic versus oxidative function, Δ133-CARs maintain a hybrid metabolic state exemplified by superior glycolytic and mitochondrial features compared to WT CARs. PGC1α has been shown to be critical for regulation of mitochondrial biogenesis, which has been identified as an important limiting factor in CAR T cell therapy for CLL (7, 36, 37).
Given the role of Δ133p53α as a modulator of p53 transcription, we looked at additional gene sets related to p53-specific functions, including a set of validated p53 target genes identified by a meta-analysis of 16 high-throughput datasets (38). Notably, Δ133p53α-CAR T cells had reduced expression of p53 target genes and genes associated with stress-induced senescence from DNA damage to telomeres, including canonical senescence and apoptosis genes CDKN1A, PHLDA3, ZMAT3, and GADD45A (Fig. 3 F and G). Despite the reduced transcription of several canonical p53 target genes, Δ133p53α-CAR T cells had increased expression of genes associated with p53-mediated DNA repair, consistent with previous findings that Δ133p53α enhances DNA repair and contributes to genome stability (23, 26, 28, 30, 39, 40). Although we observe an overall reduction in expression of p53 target genes in the Δ133-CARs, it is important to emphasize that this doesn’t represent inhibition of p53 target gene expression, but rather a reduction in activation, as there is an enrichment of p53 target genes when comparing Δ133-CARs mid-coculture to Δ133-CARs pre-coculture (SI Appendix, Fig. S5B).
To determine whether this suggested difference in p53 activation is reflected at the protein level, we measured phosphorylation of p53 at serine 15, a critical posttranslational modification driving p53 accumulation and activation (41–44). As anticipated from our GSEA, p53-S15 levels were higher in WT CARs than Δ133-CARs (Fig. 3H). We’ve replicated this observation in multiple contexts and have found that Δ133-CAR T cells have consistently lower p53-S15 phosphorylation, both with CD19.BBz CAR T cells and CD19.28z CAR T cells (SI Appendix, Fig. S6).
To validate the suggested enhanced metabolic function from our RNAseq data, we performed Seahorse extracellular flux analysis to assess the spare respiratory capacity of WT and Δ133p53α-expressing CAR T cells after tumor co-culture. Consistent with the enrichment in PGC1α transcriptional targets, Δ133-CAR T cells exhibit increased mitochondrial spare respiratory capacity, indicating a greater capacity for energy production while under increased energy demand (Fig. 3I), providing a potential explanation for enhanced anti-tumor function (36).
Constitutive Expression of Δ133p53α Enhances CAR T Cell Function in CAR-T Refractory Leukemia Patients.
Given the observed metabolic and functional enhancements in Δ133p53α-expressing CAR T cells, we hypothesized that Δ133p53α would improve the anti-tumor activity of CAR T cells generated from chronic lymphocytic leukemia (CLL) patients, where responses to CAR T cell therapy have been suboptimal, and specifically linked to impaired mitochondrial function. To evaluate this hypothesis, we obtained apheresis product that was cryopreserved prior to original clinical manufacture for patients during clinical trial NCT01029366. We manufactured Δ133- and corresponding WT CAR T cells from one responding patient and two non-responding patients, with no notable differences during primary expansion in proliferation, cell volume, or transduction efficiency (Fig. 4A and SI Appendix, Fig. S7 A–E). To be consistent with the current clinical manufacturing protocol for CLL patients, all CLL patient expansions were performed in culture media supplemented with interleukin 7 (IL7) and interleukin 15 (IL15). Consequently, we observed an overall greater level of proliferation across both groups compared with typical normal donor expansions.
Fig. 4.
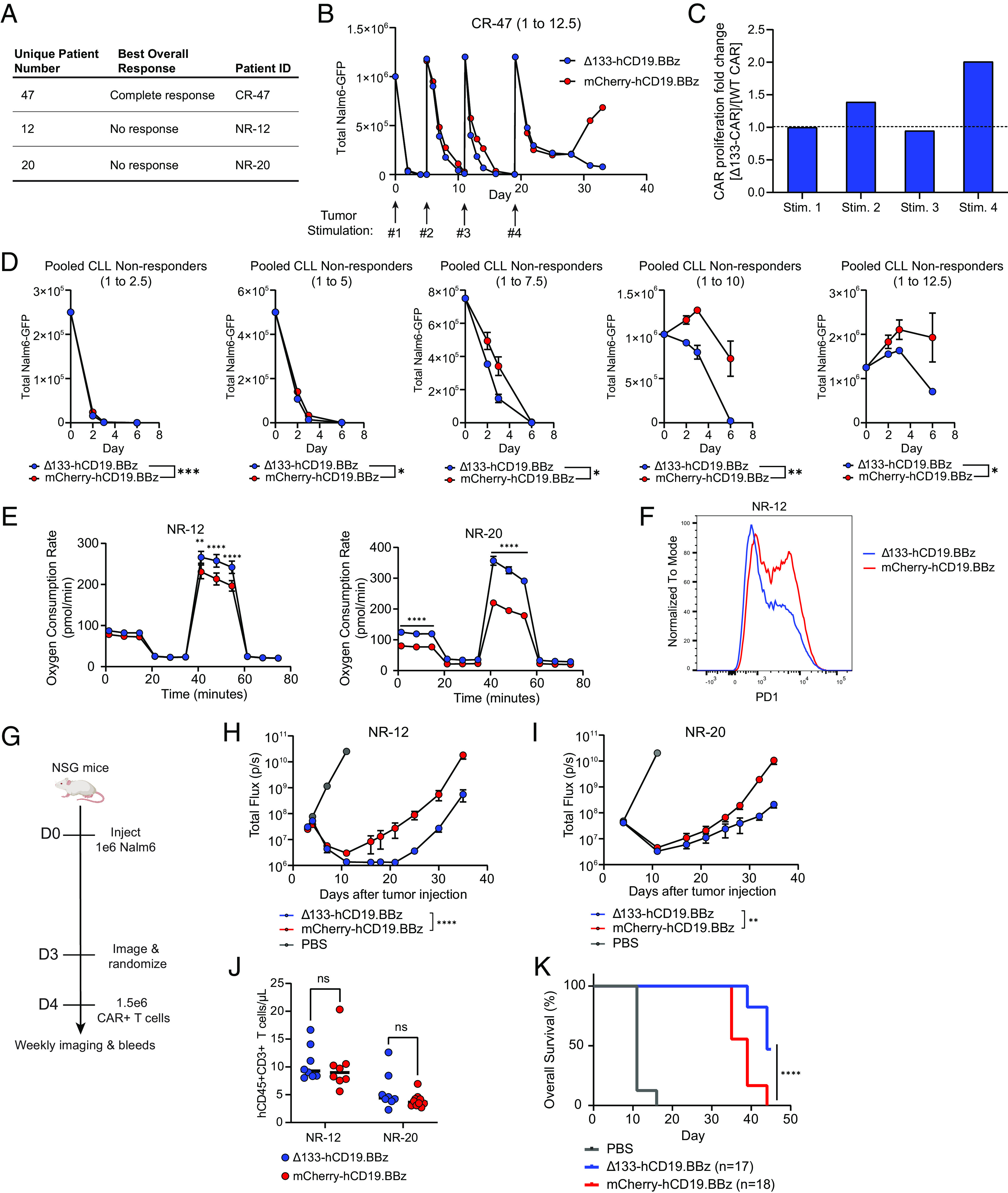
Δ133p53α expression in non-responding leukemia patient CAR T cells improves in vitro and in vivo efficacy. (A) Table outlining CLL patient T cells used for CAR manufacturing alongside clinical response to initial CD19-directed CAR T cell therapy. (B) In vitro tumor co-culture data for CR-47. Data shown are for an effector to target ratio of 1 to 12.5, with the timing of fresh tumor stimulations indicated by arrows. For each tumor restimulation, the number of CARs was normalized between treatment groups prior to tumor addition. Data are mean ± SEM of two technical replicates. (C) Ratio of population doublings between Δ133-CARs and WT CARs at the end of each tumor stimulation for CR-47. (D) Pooled tumor measurements over time from in vitro co-culture from NR-12 and NR-20. Co-cultures were run at 5 effector to target ratios, as indicated by the number over each graph. Data are mean ± SEM. Statistical significance was assessed using a two-way ANOVA, with reported values representing the interacting term of CAR treatment over time. (E) Oxygen consumption rate as measured by Seahorse extracellular flux analysis following co-culture at 1 to 4 E:T ratio. Data points for each time series represent mean oxygen consumption rate ± SEM for eight replicates. Statistical significance was determined using multiple unpaired t tests for each time point. (F) Surface PD1 staining of CAR T cells from NR-12 on day 5 of tumor co-culture at an effector to target ratio of 1 to 8 measured by flow cytometry. (G) NSG mice were injected intravenously with 1 × 106 Nalm6 leukemia cells transduced with GFP-P2A-CBG on day 0. Mice were imaged on day 3 and randomized into three groups, including both treatment groups and a PBS control. Then, 1.5 × 106 freshly thawed CAR+ T cells were injected intravenously on day 4. (H) Tumor progression for mice administered CARs from NR-12 monitored using bioluminescent imaging. Data are mean ± SEM of n = 8 mice. Statistical significance was determined via two-way ANOVA and is based on the CAR treatment X time interaction term. (I) Tumor progression for mice administered CARs from patient NR-20 monitored using bioluminescent imaging. Data are mean ± SEM of n = 8 mice. Statistical significance was determined via two-way ANOVA and is based on the CAR treatment X time interaction term. (J) T cell engraftment 1-week post-CAR injection for NR-12 and NR-20. Statistical significance was determined using a paired t test. (K) Kaplan–Meier curves showing survival of mice from both NR-12 and NR-20 over time. Data were analyzed using a log rank Mantel–Cox test. ns = not significant, *P 0.05, **P 0.01, ***P 0.001, ****P 0.0001.
Using apheresis product from complete responder (CR) patient 47, referred to as CR-47, Δ133p53α conferred an improvement in both in vitro tumor clearance and proliferation (Fig. 4 B and C). However, observation of differences in tumor clearance required multiple tumor stimulations, unlike previous in vitro co-cultures with normal donors in which Δ133-CARs demonstrated clear improvements following one tumor stimulation. Furthermore, notable differences in tumor clearance were only present at a 1 to 12.5 effector to target ratio. We attribute the overall increase in tumor clearance capacity to the use of supplemental IL7 and IL15, both of which have been shown to potently impact T cell persistence, differentiation status, and metabolism (45–47). Despite the overlap in functional benefits with IL7 and IL15, constitutive expression of Δ133p53α nonetheless confers improvements in both tumor clearance and proliferation in CARs generated from a CR CLL patient, with a reduction in p53-S15 expression consistent with what was observed in normal donor CARs (SI Appendix, Fig. S8).
We next evaluated the impact of constitutive expression of Δ133p53α in CAR T cells generated from the cryopreserved apheresis product of two patients who did not respond to CAR T cell therapy. Similar to CR-47, CAR T cells manufactured from non-responding (NR) patients 12 (NR-12) and 20 (NR-20), did not exhibit differences in proliferation or cell volume between groups, but were again more proliferative than typical normal donor expansions, albeit to a lesser degree than CR-47 (SI Appendix, Fig. S7 A and B). Unlike CR-47, and more in line with normal donor in vitro co-cultures, we observed differences during in vitro tumor clearance following one tumor stimulation, with Δ133-CARs demonstrating improved tumor clearance under high tumor burden (Fig. 4D and SI Appendix, Fig. S9 A and B). Mirroring earlier findings in healthy donors, Δ133-CAR T cells exhibited increased mitochondrial spare respiratory capacity following in vitro co-culture, with especially pronounced differences between groups for CAR T cells from NR-20 (Fig. 4E). Although differences were less significant than in normal donor-derived CARs, Δ133-CARs from NR-12 and NR-20 were more proliferative than WT CARs (SI Appendix, Fig. S9 C and D). Meaningful differences in exhaustion markers were never observed in healthy donors; however, we observed a reduction in PD1 expression for NR-12 in the Δ133-CAR T cells following co-culture at a 1 to 8 E:T ratio (Fig. 4F).
Finally, we evaluated CLL-derived CAR T cells in vivo from NR-12 and NR-20 in an NSG mouse model which was titrated to account for the reduced anticipated potency of CLL-derived CARs. Following intravenous injection of NSG mice with 1 × 106 Nalm6 leukemia cells engineered to express GFP and luciferase, 1.5 × 106 CAR+ T cells were injected intravenously on day 4 (Fig. 4G). Δ133-CAR T cells derived from both patients demonstrated improved tumor clearance, again reducing tumor burden near background levels despite minimal differences in engraftment (Fig. 4 H–J). In addition to enhanced tumor clearance, Δ133-CAR T cells improved survival, with approximately 50% of mice alive at the final timepoint, compared with the loss of all mice by day 43 in the control group (Fig. 4K).
Discussion
The inability of T cells to maintain effector functions during antitumor responses is an important limitation for CAR T cell therapy (48–50). Here, we demonstrate that constitutive expression of Δ133p53α may help to address this limitation as Δ133p53α-expressing CAR T cells exhibit enhanced antitumor activity both in vitro and in vivo, particularly under high tumor burden conditions. Based on previous findings in which Δ133p53α improved the performance of poorly functional T cells from aged donors, it was unclear whether constitutive expression of Δ133p53α would improve the function of CARs derived from healthy donors (31). However, we observed a marked improvement in antitumor function in CAR T cells generated from young, healthy donors, demonstrating that the benefits of Δ133p53α are not limited to addressing functional limitations present in aged T cells. The broader utility of Δ133p53α is further illustrated by improvements in spare respiratory capacity and corresponding functional enhancements under nutrient-limiting conditions, where Δ133p53α-expressing CAR T cells not only clear significantly more tumor, but also display increased proliferation.
Nonetheless, we observe that Δ133p53α also confers an improvement in antitumor function in CAR T cells generated from CLL patients with advanced age that were heavily pretreated and did not previously respond to CAR T cell therapy. Consistent with normal donor data, we observe an improvement in antitumor function in vitro and in vivo, as well as an increase in spare respiratory capacity. This is notable as mitochondrial function has been shown to be an important predictor of efficacy for CLL patients that respond to CAR T cell therapy (7, 10). Improvements were observed in Δ133-CARs despite the use of expansion media supplemented with IL7 and IL15, which would be expected to provide a set of benefits that at least partially overlaps with constitutive expression of Δ133p53α (45–47). Given the potential translational relevance of Δ133p53α in the context of CLL, it will be important to perform follow-up experiments using CLL tumor cell lines, which have been shown to directly cause dysfunction through multiple mechanisms, including impaired immune synapse formation (10, 11, 51).
A consistent observation throughout multiple contexts is that the functional benefits conferred by Δ133p53α appear to be stress dependent. During primary expansion and low tumor conditions, Δ133p53α CAR T cells demonstrate minimal differences compared with wild-type controls, consistent with the view that Δ133p53α functions primarily by modulation of the p53-mediated stress response (15, 20, 24, 39). Further corroborating the stress-dependent nature of Δ133p53α is the proportional improvement in tumor clearance as tumor burden increases, suggesting that stress potentiates the effects of Δ133p53α. In addition to excess antigen, CARs are subject to nutrient depletion and low pH during in vitro co-culture, with co-culture media appearing bright yellow as early as day 3, attributable to the pH indicator phenol red. An important aspect of our in vitro assay is that different E:T ratios are achieved by varying the amount of tumor against a constant number of CARs. As a result, the lowest E:T ratio has the highest total concentration of rapidly proliferating cells (i.e., CAR T cells and tumor cells), thus coupling maximal nutrient depletion and antigen exposure. The requirement of increased tumor burden for Δ133-CARs to demonstrate functional improvements suggests that some degree of p53 signaling network activation is required for Δ133p53α to impact CAR T cell function, which is consistent with the lack of transactivation domains on Δ133p53α and its general nature as an inhibitory p53 isoform.
Under these stress-inducing conditions, Δ133-CARs display enhanced expression of several transcriptional programs that drive proliferation and biosynthetic processes critical to supporting effector function. From the initial unbiased GSEA of all Hallmark and Gene Ontology (GO) gene sets, the most enriched gene sets were in the Δ133-CARs and involved upregulation of myc targets, upregulation of mitochondrial gene expression and translation, and upregulation of ribosomal biogenesis. Subsequent targeted GSEA performed on gene sets specific to T cell metabolism further support the enhanced metabolic profile of Δ133-CARs, with upregulation of targets of the transcription factor PCG-1α, critical for mitochondrial biogenesis, and upregulation of genes associated with increased glucose transport (36, 37, 52). Given the competition for nutrients between CAR T cells and tumor cells during an antitumor response, these molecular changes in the Δ133-CARs are highly advantageous toward maintaining prolonged effector functions in such a metabolically demanding context (53).
Alongside these metabolic improvements, Δ133-CARs have reduced activation of p53 and reduced transcription of critical p53 target genes. Δ133-CARs have reduced expression of canonical senescence and apoptosis genes including CDKN1A, PHLDA3, ZMAT3, and BAX. In contrast, Δ133-CARs have an enrichment in DNA repair genes regulated by p53. This is consistent with prior findings in which Δ133p53α biases p53-mediated transcription toward genes associated with DNA repair, rather than broad inhibition of p53 (26, 40). Importantly, Δ133-CARs do not completely inhibit p53FL transcriptional activity, as demonstrated by the maintenance of transcription at several validated p53 target genes. Although further inquiry is needed to confirm our findings, our results suggest that Δ133-CARs do not simply inhibit p53 but rather redirect p53 transcription.
Interestingly, the improvements in antitumor function conferred by Δ133p53α in CD19-directed CARs may extend to other forms of adoptive cell therapy, as another group has shown that Δ133p53α improves the fitness of T cells engineered to express a tumor-antigen specific T cell receptor (TCR) (54). Given Δ133p53α’s modulation of the ubiquitous p53-mediated stress response, these results warrant investigation of Δ133p53α expression in additional forms of cell therapy, including engineered tumor-infiltrating lymphocytes (TILs) and other immune cells, such as natural killer (NK) cells and macrophages.
More broadly, the improved performance of Δ133-CAR T cells indirectly suggests that p53 may be a critical regulator of CAR T cell dysfunction during antitumor responses. Given p53’s role as the master regulator of homeostasis and the enormous homeostatic demands of tumor clearance, this appears to be a reasonable and intuitive takeaway. Furthermore, many canonical p53-activating stresses such as nutrient depletion, replication stress, and hypoxia overlap with drivers of T cell dysfunction, with many of the functional and phenotypic characteristics of dysfunctional T cells also under the control of p53 including metabolism, differentiation, and proliferation (13, 16–18, 49). Yet despite this alignment, limited evidence from over a decade of intense investigation into understanding T cell dysfunction points to p53 as a key mediator of T cell dysfunction. However, an important caveat around this relative lack of evidence is that much of our understanding of T cell dysfunction has been informed using transcriptomic and genetic tools not equipped to identify proteins like p53 which are regulated post-translationally and are not amenable to genetic deletion (55–58). These observations, taken together, present a compelling case for a reassessment of p53’s role in T cell dysfunction.
A potential limitation of our study is that we have not addressed the role of Δ133p53α beyond hematologic malignancies. Further studies will be required to determine the role of Δ133p53α in modulating CAR T cell antitumor responses in the solid tumor microenvironment. Another area for additional investigation is around the safety of constitutive expression of Δ133p53α to determine whether there are increased risks for CAR T cell transformation. Importantly, previous work has shown that overexpression of Δ133p53α extends the replicative lifespan without evidence for immortalization or malignant transformation in normal human fibroblasts, CD8+ T cells, astrocytes, and epithelial cells (21, 27, 29, 31). Furthermore, we have not observed any evidence of transformation in our extensive series of experiments with T cells from healthy donors or leukemia patients. Nonetheless, more targeted evaluation of this risk will be needed for this strategy to be translated into patients.
In summary, our findings point to the potential for using the p53 isoform Δ133p53α to improve CAR T cell antitumor function through the modulation of the p53 signaling network. Constitutive expression of Δ133p53α may represent a viable translational strategy for improving CAR T cell therapy in CLL and in solid tumor indication and warrants further investigation.
Materials and Methods
Plasmid Design and Construction.
All plasmids used in this study were designed using Snapgene (Dotmatics) and generated via cloning of gene fragments (Genscript) into pTRPE (PMID: 27332733), a third-generation self-inactivating lentiviral vector. Plasmids were sequence verified using Sanger sequencing (Genewiz). All CAR constructs used a CD8 leader sequence, (GGGGS)3 linker, CD8 hinge, and CD8 transmembrane domain and were expressed under the control of an EF1α promoter.
Primary Human T Cells from Healthy Donors and CLL Patients.
Deidentified human T cells were obtained from peripheral blood mononuclear cells (PBMCs) of healthy donors through the University of Pennsylvania Human Immunology Core. To obtain T cells from CLL patients who enrolled in single-agent CTL019 therapy, CD4 and CD8 T cells were isolated from cryopreserved apheresis that was collected prior to initial clinical CAR manufacture. All subjects provided written informed consent according to the Declaration of Helsinki and the International Conference on Harmonization Guidelines for Good Clinical Practice.
Tumor Cell Lines.
NALM6 leukemic cells were acquired from the American Type Culture Collection (ATCC), routinely authenticated by the University of Arizona Genetics Core, and confirmed negative for Mycoplasma. NALM6 cells were transduced with a lentiviral vector encoding click beetle green (CBG) luciferase and GFP under and elongation factor 1 alpha (EF1α) promoter, separated by a P2A bicistronic linker. A fluorescence-activated cell sorter (FACS) was then used to collect ~100% GFP-positive cells, which were then expanded and cryopreserved for future use. CD19 and GFP positivity was routinely checked prior to use for in vitro and in vivo experiments by flow cytometry. NALM6 cells were maintained in culture with R10 medium: RPMI 1640 (Gibco) supplemented with 10% heat-inactivated fetal bovine serum (FBS; Seradigm), 2% 1M Hepes buffer solution (Gibco), 1% 100× GlutaMAX (Gibco), and 1% penicillin (10,000 U/mL) + streptomycin (10,000 µg/mL).
Vector Production, T Cell Isolation, and Generation of CAR T Cells.
Lentiviral vectors and human CAR T cells were produced as previously described (4, 59). For WT control CARs, the fluorescent protein mCherry2 was used, as it is a brighter version of mCherry (60). Human T cells were stimulated with anti-human CD3/CD28 antibody-coated magnetic beads (Dynabeads, Gibco) at a bead-to-T cell ratio of 3:1. Twenty-four hours after bead stimulation, T cells were lentivirally transduced to express the constructs shown in Fig. 1B, using a multiplicity of infection (MOI) of 4. At day 5 of T cell expansion, magnetic beads were removed from culture and cells were kept in culture to continue expanding until resting down to a volume of ~350 fL prior to cryopreservation. For cryopreservation of CAR T cells, CAR T cells were resuspended in a freezing medium consisting of 90% heat-inactivated FBS (Seradigm) and 10% dimethyl sulfoxide (Sigma-Aldrich), subaliquotted into 1 mL cryovials, and then placed in controlled rate freezing containers at −80 °C. CAR expression was assessed prior to cryopreservation using an anti-idiotype antibody. Expansion of CAR T cells from healthy human donors was performed in R10 medium (see above), whereas CAR T cells derived from CLL patient samples were performed using an alternative medium to be consistent with current clinical protocols: Optimizer T cell Expansion Basal Medium (Gibco) supplemented with 2.5% CTS OptMizer Supplement (Gibco), 5% Human AB serum (Valley Biomedical), 1% 100× GlutaMax (Gibco), 5 ng/mL recombinant human IL7 (Miltenyi), and 5 ng/mL recombinant human IL15.
Western Blot.
Western blotting was used to assess ectopic expression of Δ133p53α. Lysates from CAR T cells isolated at the end of primary expansion were collected using ice-cold RIPA buffer (Cell Signaling Technology) supplemented with protease inhibitor (Sigma-Aldrich) for 30 min on ice, with periodic vortexing, followed by centrifugation at 15,000 rpm for 15 min at 4 °C. Protein concentrations of each whole cell lysate were determined using a DC protein quantification assay (BioRad). Lysates were diluted to 20 to 40 µg/sample with loading buffer and were treated with 2-mercaptoethanol and incubated at 95 °C for 5 min. Samples were run on a 10% tris-glycine mini protein gel (Thermo) at 200 V for approximately 1 h. Transfer was performed onto a polyvinylidene difluoride (PVDF) membrane in 1× tris-glycine transfer buffer (Novex). The primary antibody used for detection of Δ133p53α (SAPU; provided by JC Bourdon) was applied at a 1:5,000 dilution in blocking buffer at 4 °C overnight with gentle rocking. Secondary antibody, donkey anti-sheep IgG-HRP (Jackson ImmunoResearch) was applied at a 1:40,000 dilution and incubated at room temperature for 30 min with gentle rocking. All steps and washes were performed in the following buffer: 10 mM Tris-HCl pH 7.5, 150 mM NaCl, 0.05% Tween 20. Quantitative image analysis was performed using Image Lab Software (Bio-Rad).
In Vitro Cytokine Secretion Assay.
CAR T cells were co-cultured for 24 h with Nalm6 leukemia cells at a 1:7.5 effector to target ratio. Supernatants were measured for concentration of the cytokines IL-2, TNFα, and IFNγ using the human T helper 1 (TH1)/TH2 cytokine cytometric bead array kit II for human T cells (BD Biosciences) according to manufacturer’s instructions. Sample data were acquired using a Fortessa LSR II flow cytometer (BD Biosciences) with FACSDiva software (BD Biosciences). Data were analyzed using the BD CBA analysis software (BD Biosciences).
Flow Cytometry and Antibodies.
For measurement of surface markers, cells were stained with either fixable live/dead violet or near-infrared stain (Thermo) in PBS depending on the panel, followed by surface staining in PBS containing 2.5% heat-inactivated FBS (Seradigm). CD19 CAR staining was performed using an AF647 or APC conjugated anti-idiotype antibody directed at the FMC63 scFv. CD8 staining was performed using anti-CD8 APC-H7 (BD) and finally PD1 staining was performed using anti-PD1 BV711 (BioLegend). Measurement of intracellular markers was performed by first staining cells with fixable live/dead near-infrared stain (Thermo). After washing, cells were fixed using 1.5% paraformaldehyde at room temperature for 10 min. Washed cells were then resuspended in cold methanol and stained on ice for 15 min. Cells were then either stored at −80 °C for temporary storage to enable batch analysis or were immediately washed and resuspended in PBS with 2.5% FBS (Seradigm) prior to intracellular staining. Phosphorylated p53 serine 15 was stained using phospho-p53 Ser15 antibody conjugated to either PE or AF488 depending on the panel (Cell Signaling Technology). All data were collected by a Fortessa LSR II cytometer (BD Biosciences) equipped with FACSDiva software. Analysis was performed using FlowJo version 10 (BD Biosciences).
In Vitro Tumor Co-culture.
In vitro tumor clearance was assessed using a Celigo Image Cytometer (Nexcelom). CAR T cells were co-cultured with NALM6 cells engineered to express GFP in 24-well cell culture plates, at five different effector-to-target (E:T) ratios: 1 to 2.5, 1 to 5, 1 to 7.5, 1 to 10, and 1 to 12.5. To set up the assay, a master mix is made for each E:T, with a fixed concentration of CAR T cells of 1 × 105/mL (normalized for CAR transduction %) across all E:T and a final concentration of either 2.5 × 105, 5 × 105, 7.5 × 105, 1 × 106, 1.25 × 106/mL NALM6. Each master mix is then dispensed into a 24-well plate with 1mL per well. To count tumor cells and T cells, 5 µL of a well-mixed cell suspension is transferred into a compatible 96-well clear-bottomed plate containing 95 µL PBS. Next, 100 µL of 2 µM calcein AM violet (Thermo) in PBS is added to each well (200 µL total: 5 µL cell suspension + 95 µL PBS + 100 µL 2 µM calcein AM violet), mixed, and then incubated at room temperature for 25 min protected from light. Following incubation, the 96-well clear-bottomed plate is spun for 1 min at 100 g and then each well is imaged and quantitated using a Celigo Image Cytometer using brightfield, alongside the blue and green channels to image all live cells and tumor cells, respectively. Live T cell counts are determined by subtracting the number of GFP+ cells from the total number of live cells. For each condition, two replicate measurements are made. Following day 3 sampling, 600 µL of pre-warmed R10 is added to each well.
B-ALL Xenograft Model.
The University of Pennsylvania Institutional Animal Care and Use Committee (IACUC) approved all animal experiments and all animal procedures were performed in the animal facility at the University of Pennsylvania in accordance with all Federal and Institutional requirements. Six- to eight-week-old male NOD/scid/IL2rg−/− (NSG) mice bred in the vivarium at the University of Pennsylvania and maintained under pathogen-free conditions, were used for all in vivo experiments. For assessment of CAR T cells generated from healthy normal donors, mice were injected via tail vein with 1 × 106 NALM6 cells engineered to express luciferase in 200 µL PBS. Seven days after tumor injection, mice were randomized based on tumor burden and then injected via tail vein with 1 × 106 CAR-expressing T cells (CAR+) or with an equivalent number of non-transduced T cells in 200 µL PBS. For assessment of CAR T cells generated from CLL patient samples, mice were injected via tail vein with 1 × 106 NALM6 cells (luciferase positive). Four days after tumor injection, mice were randomized based on tumor burden and then injected via tail vein with 1.5 × 106 CAR-expressing T cells (CAR+) in 200 µL PBS or with 200 µL PBS alone, as sample limitations preventing the use of a non-transduced control group. Tumor burden was measured through weekly bioluminescent imaging, with imaging data analyzed using Living Image version 4.4 (Caliper LifeSciences, PerkinElmer). For peripheral blood human T cell counts, retro-orbital bleeds were performed to collect 50 µL of blood for staining and lysis in TrucountTM tubes (BD). Throughout the study, mice were observed for indications of graft-versus-host disease (GVHD) and any signs of overt toxicity, such as body weight loss exceeding 20%, fur loss, diarrhea, conjunctivitis, and hind limb paralysis. Mice were euthanized once total flux exceeded 1 × 1010 photons per second, with one set of exceptions: due to the use of a high starting tumor burden prior to CAR T cell injection, several mice in the ND451 and ND512 experiments were above the 1 × 1010 cutoff at the first time point following CAR injection (D12). Rather than euthanize in accordance with the requirements of the Animal Welfare Act and Regulations, all affected mice following the first tumor measurement, an exception was made to raise the cutoff to 2 × 1010, such that an additional 7 mice were kept alive for ND451 (3 in the Δ133-CAR group; 4 in the WT CAR group) and an additional 8 mice for ND512 (all 8 mice were in the WT CAR group). No other exceptions were made.
Bulk RNA-seq.
Cell pellets were collected following CAR isolation and snap frozen on dry ice, with all subsequent steps performed by Azenta Life Sciences. Purity and integrity of extracted RNA was determined using Nanodrop 2000 and TapeStation. RNA libraries were prepared by isolating polyA-tailed mRNA from total RNA, with ERCC RNA spike-ins added to serve as internal controls. Sequencing was performed on an Illumina HiSeq 2500 with 150-bp paired-end reads. Differential gene expression between Δ133- and WT-CARs was evaluated using DESeq2, with p-values calculated using the Wald test. Adjusted P-values were determined using the Benjamini–Hochberg method.
Seahorse Extracellular Flux Analysis.
CAR T cell oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) were measured using a Seahorse XF96 Extracellular Flux Analyzer according to the manufacturer’s instructions. CAR T cells were collected following Nalm6 coculture, checked for purity, washed, and resuspended in Seahorse EF medium (Agilent) at a concentration of 1 × 106 live CAR T cells/mL, with 175 µL of cell suspension added to each well. OCR and ECAR were measured under basal conditions, followed by sequential injections of oligomycin (1 µM), fluoro-carbonylcyanidephenylhydrazone (FCCP, 1 µM), and rotenone/antimycin A (500 nM), per manufacturer instructions.
Statistical Analysis and Figure Illustrations.
Statistical analysis and graphing were performed with Prism version 10 (Graphpad Software). The statistical tests used for each analysis are listed in the corresponding figure legends. Illustrations in Figs. 2A, 3A, and 4G were created with BioRender.com.
Supplementary Material
Appendix 01 (PDF)
Acknowledgments
This work was supported by T32 predoctoral training grant T32GM008076 (C.R.), National Cancer Institute grant number 5T32CA009140 (P.C.R.), NIH RO-1 CA278837 (R.O.C.), and the Ludwig Research Foundation–Princeton Branch (R.O.C.). We would like to thank M. Angela Aznar and Jesse Rodriguez for technical input as well as Carolyn Shaw and Yujie Ma for logistical support. We also acknowledge the resources provided by Human Immunology Core, Flow and Cell Sorting Facility, Stem Cell and Xenograft Core at the University of Pennsylvania.
Author contributions
C.R., I.H., P.C.R., J.S., R.M.Y., and C.H.J. designed research; C.R., L.C., A.R.K., D.G., T.D., N.W., P.C.R., D.B., J.S., and R.S.O. performed research; I.H., M.S., and C.C.H. contributed new reagents/analytic tools; C.R., I.H., J.S., R.S.O., R.M.Y., and C.C.H. analyzed data; and C.R., R.M.Y., and C.H.J. wrote the paper.
Competing interests
C.H.J. is an inventor of patents related to the CAR therapy product which is the subject of this paper, as well as other CAR therapy products, and may be eligible to receive a select portion of royalties paid from Kite to the University of Pennsylvania. C.H.J. is a scientific co-founder and holds equity in Capstan Therapeutics, Dispatch Biotherapeutics and Bluewhale Bio. C.H.J. serves on the board of AC Immune and is a scientific advisor to BluesphereBio, Cabaletta, Carisma, Cartography, Cellares, Cellcarta, Celldex, Danaher, Decheng, ImmuneSensor, Kite, Marble Therapeutics, Poseida, Verismo, Viracta, and WIRB-Copernicus group. C.H.J., R.M.Y., and J.S. are inventors on patents and/or patent applications licensed to Novartis Institutes of Biomedical Research and Kite and may receive license revenue from such licenses. R.O.C. is an inventor on patents licensed to Novartis of Biomedical Research and has equity in Nucleus Biologics and Stoic Bio. R.O.C. is also a scientific advisor to Nucleus Biologics. C.R. consults for Marble Therapeutics and is a scientific advisor to Boston Labs.
Footnotes
Reviewers: J.A.B., Sonoma Biotherapeutics; and A.J.L., Institute for Advanced Study.
Data, Materials, and Software Availability
RNA-seq data were deposited onto Gene Expression Omnibus (GEO) accession number GSE248382 (61). All other data are included in the manuscript and/or SI Appendix.
Supporting Information
References
- 1.Kalos M., et al. , T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci. Transl. Med. 3, 95ra73 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maude S. L., et al. , Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 371, 1507–1517 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Melenhorst J. J., et al. , Decade-long leukaemia remissions with persistence of CD4+ CAR T cells. Nature 602, 503–509 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Porter D. L., et al. , Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci. Trans. Med. 7, 303ra139 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lamble A. J., et al. , Preinfusion factors impacting relapse immunophenotype following CD19 CAR T cells. Blood Adv. 7, 575–585 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mehta P. H., et al. , T cell fitness and autologous CAR T cell therapy in haematologic malignancy. Front. Immunol. 12, 780442 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fraietta J. A., et al. , Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat. Med. 24, 563–571 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grigor E. J. M., et al. , Risks and benefits of chimeric antigen receptor T-Cell (CAR-T) therapy in cancer: A systematic review and meta-analysis. Transfus. Med. Rev. 33, 98–110 (2019). [DOI] [PubMed] [Google Scholar]
- 9.Zhang X., Zhu L., Zhang H., Chen S., Xiao Y., CAR-T cell therapy in hematological malignancies: Current opportunities and challenges. Front. Immunol. 13, 927153 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.van Bruggen J. A. C., et al. , Chronic lymphocytic leukemia cells impair mitochondrial fitness in CD8(+) T cells and impede CAR T-cell efficacy. Blood 134, 44–58 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhao Z., Gregoire C., Oliveira B., Chung K., Melenhorst J. J., Challenges and opportunities of CAR T-cell therapies for CLL. Semin. Hematol. 60, 25–33 (2023). [DOI] [PubMed] [Google Scholar]
- 12.Finck A. V., Blanchard T., Roselle C. P., Golinelli G., June C. H., Engineered cellular immunotherapies in cancer and beyond. Nat. Med. 28, 678–689 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Newick K., O’Brien S., Moon E., Albelda S. M., CAR T cell therapy for solid tumors. Annu. Rev. Med. 68, 139–152 (2017). [DOI] [PubMed] [Google Scholar]
- 14.Maher J., Davies D. M., CAR based immunotherapy of solid tumours—A clinically based review of target antigens. Biology 12, 287 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Joruiz S. M., Bourdon J. C., p53 Isoforms: Key regulators of the cell fate decision. Cold Spring Harb. Perspect. Med. 6, a026039 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aylon Y., Oren M., The paradox of p53: What, how, and why? Cold Spring Harb. Perspect. Med. 6, a026328 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mehta S., et al. , Adaptive homeostasis and the p53 isoform network. EMBO Rep. 22, e53085 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kastenhuber E. R., Lowe S. W., Putting p53 in context. Cell 170, 1062–1078 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kruiswijk F., Labuschagne C. F., Vousden K. H., p53 in survival, death and metabolic health: A lifeguard with a licence to kill. Nat. Rev. Mol. Cell Biol. 16, 393–405 (2015). [DOI] [PubMed] [Google Scholar]
- 20.Bourdon J. C., et al. , p53 isoforms can regulate p53 transcriptional activity. Genes Dev. 19, 2122–2137 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fujita K., et al. , p53 isoforms Delta133p53 and p53beta are endogenous regulators of replicative cellular senescence. Nat. Cell Biol. 11, 1135–1142 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Khoury M. P., Bourdon J. C., The isoforms of the p53 protein. Cold Spring Harb. Perspect. Biol. 2, a000927 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Horikawa I., et al. , Delta133p53 represses p53-inducible senescence genes and enhances the generation of human induced pluripotent stem cells. Cell Death Differ. 24, 1017–1028 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Khoury M. P., Bourdon J. C., p53 isoforms: An intracellular microprocessor? Genes Cancer 2, 453–465 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aoubala M., et al. , p53 directly transactivates Δ133p53α, regulating cell fate outcome in response to DNA damage. Cell Death Differentiation 18, 248–258 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Steffens Reinhardt L., Groen K., Newton C., Avery-Kiejda K. A., The role of truncated p53 isoforms in the DNA damage response. Biochim. Biophys. Acta Rev. Cancer 1878, 188882 (2023). [DOI] [PubMed] [Google Scholar]
- 27.Turnquist C., et al. , p53 isoforms regulate astrocyte-mediated neuroprotection and neurodegeneration. Cell Death Differ. 23, 1515–1528 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Horikawa I., Harris C. C., Delta133p53: A p53 isoform enriched in human pluripotent stem cells. Cell Cycle 16, 1631–1632 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mondal A. M., et al. , Delta133p53alpha, a natural p53 isoform, contributes to conditional reprogramming and long-term proliferation of primary epithelial cells. Cell Death Dis. 9, 750 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.von Muhlinen N., et al. , p53 isoforms regulate premature aging in human cells. Oncogene 37, 2379–2393 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mondal A. M., et al. , p53 isoforms regulate aging- and tumor-associated replicative senescence in T lymphocytes. J. Clin. Invest. 123, 5247–5257 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Minato N., Hattori M., Hamazaki Y., Physiology and pathology of T-cell aging. Int. Immunol. 32, 223–231 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Levine A. J., p53: 800 million years of evolution and 40 years of discovery. Nat. Rev. Cancer 20, 471–480 (2020). [DOI] [PubMed] [Google Scholar]
- 34.O’Connor R. S., et al. , The CPT1a inhibitor, etomoxir induces severe oxidative stress at commonly used concentrations. Sci. Rep. 8, 6289 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chang C. H., et al. , Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell 153, 1239–1251 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Scharping N. E., et al. , The tumor microenvironment represses t cell mitochondrial biogenesis to drive intratumoral T cell metabolic insufficiency and dysfunction. Immunity 45, 374–388 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang L., et al. , Mitochondria dysfunction in CD8+ T cells as an important contributing factor for cancer development and a potential target for cancer treatment: A review. J. Exp. Clin. Cancer Res. 41, 227 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fischer M., Census and evaluation of p53 target genes. Oncogene 36, 3943–3956 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Joruiz S. M., Beck J. A., Horikawa I., Harris C. C., The Δ133p53 Isoforms, Tuners of the p53 Pathway. Cancers 12, 3422 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gong L., et al. , p53 isoform Δ113p53/Δ133p53 promotes DNA double-strand break repair to protect cell from death and senescence in response to DNA damage. Cell Res. 25, 351–369 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Loughery J., Cox M., Smith L. M., Meek D. W., Critical role for p53-serine 15 phosphorylation in stimulating transactivation at p53-responsive promoters. Nucleic Acids Res. 42, 7666–7680 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jones R. G., et al. , AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol. Cell 18, 283–293 (2005). [DOI] [PubMed] [Google Scholar]
- 43.She Q.-B., Chen N., Dong Z., ERKs and p38 kinase phosphorylate p53 protein at serine 15 in response to UV radiation. J. Biol. Chem. 275, 20444–20449 (2000). [DOI] [PubMed] [Google Scholar]
- 44.Shieh S.-Y., Ikeda M., Taya Y., Prives C., DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell 91, 325–334 (1997). [DOI] [PubMed] [Google Scholar]
- 45.Giuffrida L., et al. , IL-15 preconditioning augments CAR T cell responses to checkpoint blockade for improved treatment of solid tumors. Mol. Therapy 28, 2379–2393 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xu Y., et al. , Closely related T-memory stem cells correlate with in vivo expansion of CAR.CD19-T cells and are preserved by IL-7 and IL-15. Blood 123, 3750–3759 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhou J., et al. , Chimeric antigen receptor T (CAR-T) cells expanded with IL-7/IL-15 mediate superior antitumor effects. Protein Cell 10, 764–769 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.June C. H., O’Connor R. S., Kawalekar O. U., Ghassemi S., Milone M. C., CAR T cell immunotherapy for human cancer. Science 359, 1361–1365 (2018). [DOI] [PubMed] [Google Scholar]
- 49.Philip M., Schietinger A., CD8+ T cell differentiation and dysfunction in cancer. Nat. Rev. Immunol. 22, 209–223 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rafiq S., Hackett C. S., Brentjens R. J., Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat. Rev. Clin. Oncol. 17, 147–167 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Roessner P. M., Seiffert M., T-cells in chronic lymphocytic leukemia: Guardians or drivers of disease? Leukemia 34, 2012–2024 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Scharping N. E., et al. , Mitochondrial stress induced by continuous stimulation under hypoxia rapidly drives T cell exhaustion. Nat. Immunol. 22, 205–215 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Klein Geltink R. I., Kyle R. L., Pearce E. L., Unraveling the complex interplay between T cell metabolism and function. Ann. Rev. Immunol. 36, 461–488 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Legscha K. J., et al. , Delta133p53alpha enhances metabolic and cellular fitness of TCR-engineered T cells and promotes superior antitumor immunity. J. Immunother. Cancer 9, e001846 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jiménez A., et al. , Time-series transcriptomics and proteomics reveal alternative modes to decode p53 oscillations. Mol. Syst. Biol. 18, e10588 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kastan M. B., Onyekwere O., Sidransky D., Vogelstein B., Craig R. W., Participation of p53 protein in the cellular response to DNA damage. Cancer Res. 51, 6304–6311 (1991). [PubMed] [Google Scholar]
- 57.Kubbutat M. H. G., Jones S. N., Vousden K. H., Regulation of p53 stability by Mdm2. Nature 387, 299–303 (1997). [DOI] [PubMed] [Google Scholar]
- 58.Lakin N. D., Jackson S. P., Regulation of p53 in response to DNA damage. Oncogene 18, 7644–7655 (1999). [DOI] [PubMed] [Google Scholar]
- 59.Posey A. D. Jr., et al. , Engineered CAR T cells targeting the cancer-associated Tn-glycoform of the membrane mucin MUC1 control adenocarcinoma. Immunity 44, 1444–1454 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shen Y., Chen Y., Wu J., Shaner N. C., Campbell R. E., Engineering of mCherry variants with long Stokes shift, red-shifted fluorescence, and low cytotoxicity. PLoS One 12, e0171257 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Roselle C., Chen L., Young R. M., June C. H., Enhancing chimeric antigen receptor T cell therapy by modulating the p53 signaling network with Δ133p53α. Gene Expression Omnibus (GEO). https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE248382. Deposited 21 November 2023.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Data Availability Statement
RNA-seq data were deposited onto Gene Expression Omnibus (GEO) accession number GSE248382 (61). All other data are included in the manuscript and/or SI Appendix.