

Summary
Despite advances in treatment, lung cancer survival rates remain low. A better understanding of the cellular heterogeneity and interplay of cancer-associated fibroblasts (CAFs) within the tumor microenvironment will support the development of personalized therapies. We report a spatially resolved single-cell imaging mass cytometry (IMC) analysis of CAFs in a non-small cell lung cancer cohort of 1,070 patients. We identify four prognostic patient groups based on 11 CAF phenotypes with distinct spatial distributions and show that CAFs are independent prognostic factors for patient survival. The presence of tumor-like CAFs is strongly correlated with poor prognosis. In contrast, inflammatory CAFs and interferon-response CAFs are associated with inflamed tumor microenvironments and higher patient survival. High density of matrix CAFs is correlated with low immune infiltration and is negatively correlated with patient survival. In summary, our data identify phenotypic and spatial features of CAFs that are associated with patient outcome in NSCLC.
Keywords: Cancer-associated fibroblasts, Imaging mass cytometry, Single cell, Spatial analysis, Non-small cell lung cancer
Graphical abstract
Highlights
-
•
Imaging mass cytometry analysis of CAF heterogeneity in over 1,000 patients
-
•
Definition of good and poor prognostic CAF phenotypes
-
•
Poor prognostic CAFs are associated with metastasis and chemoresistance
-
•
CAF spatial distribution correlates with immune infiltration and prognosis
Cords et al. use multiplexed imaging mass cytometry to analyze a cohort of >1,000 NSCLC patients and show that CAF phenotypes with varying spatial location are associated with different states of the tumor microenvironment and are an independent factor for patient prognosis.
Introduction
Lung cancer is the deadliest cancer worldwide, accounting for 18% of all cancer-related deaths.1 The most prevalent type of lung cancer is non-small cell lung cancer (NSCLC), with an estimated 5-year survival of 10–23%,1,2 and with lung adenocarcinoma (LUAD) and lung squamous cell carcinoma (LUSC) subtypes accounting for more than 70% of all NSCLC cases.3 Lung cancer is often diagnosed at an advanced stage, and mortality drops to 5.5% for patients with distant metastases at diagnosis.4 Thus, there is an urgent need to identify effective therapeutic targets for NSCLC.
Immune checkpoint inhibitor therapy has increased the overall survival of NSCLC patients, showing the potential to target the tumor microenvironment (TME) in this disease,5,6 as also seen for other indications.7,8,9,10,11 Cancer-associated fibroblasts (CAFs) are a heterogeneous set of cells that are the main constituents of the tumor-surrounding stroma. CAFs drive fibrosis in the TME, causing increased extracellular matrix (ECM) deposition and remodeling, which can actively hamper the anti-tumor immune response by excluding immune cells.12 In many tumor types, including NSCLC, CAFs impact treatment efficiency; GPR77+ and CD10+ CAFs are associated with tumor stemness and chemoresistance,13 whereas LRRC15+ CAFs have tumor-promoting and immunosuppressive activity.14 In NSCLC, the stromal compartment is also important for cancer progression and therapeutic success but is not well understood.15
The effects of CAFs on tumor progression are likely to be multifactorial.16 CAFs exert their tumor-promoting effects through modulation of inflammation,10 T cell exclusion via altered matrix production,17 or promotion of cell survival.13,18 Certain CAF subtypes also display tumor inhibitory functions and have been linked with improved therapy outcome.18,19,20 For instance, the dense collagen matrix produced by CAFs can restrict tumor growth and metastasis.19,21,22 On the other hand, stromal cell depletion has also been linked to tumor outgrowth despite enhanced immune cell infiltration.19,21,22 Due to their tumor promoting and inhibiting properties, CAFs have recently moved into the spotlight as potential therapeutic targets, but clinical trials broadly targeting the tumor stroma have so far not succeeded.23,24,25,26 The full extent of CAF heterogeneity and its relationship to tumor biology and clinical outcome needs further study.
We and others have previously proposed a CAF classification system based on single-cell RNA sequencing (scRNA-seq) data from breast, lung, colon, head and neck cancer, and pancreatic ductal adenocarcinoma tumors.17,27,28,29,30,31,32,33,34,35,36,37 The 9 CAF types we annotated in this previous work are tumor type independent, recapitulate CAF types identified in molecular biology studies27,36,38,39,40 and are present in NSCLC.30 Here, we used IMC41 to characterize heterogeneous CAF phenotypes and the interactions of these cells with tumor and immune cells in NSCLC.42,43,44 We identified most of our previously annotated CAF types in this dataset, showed that CAF phenotypes were a strong prognostic factor for NSCLC, and identified specific CAF phenotypes associated with good and poor patient prognosis. Further, we discovered that different CAF types varied in their spatial distribution in the TME. Our data lay the foundation for future mechanistic studies to identify CAF phenotypes that causally affect tumor growth or improve immune infiltration.
Results
IMC analysis of non-small cell lung cancer samples
We investigated the phenotypic heterogeneity of CAFs in NSCLC. The tumor microarray (TMA) was generated from samples from a patient cohort (n = 1,070) with at least 15 years follow-up (Table 1).42,43,44 The samples included 618 pathologist-classified LUADs and 401 LUSCs; the remaining (n = 51) were mixed or undefined. Most cancers were stage 1–2, 60 patients had stage 4 tumors. 82 patients who received neoadjuvant therapy (were excluded from survival analyses; all other patient samples (n = 987) were treatment naive. 730 patients were smokers at diagnosis; 26 patients identified as former smokers and 86 as never smokers. The smoking status of 228 patients and ethnicity of all patients were unknown. Lymph node metastases (TNM: N1-N3) were found in 503 patients and distant metastases (TNM: M1 and M2) in 90 patients at diagnosis. Of all patients, 480 experienced relapses within the 15 years of follow-up. We analyzed two cores for 85% (n = 909) and one core for 15% (n = 161) of patients; all cores were sampled from the tumor invasive front. For patients with two cores, the distribution of all cell types except hypoxic tCAFs was the same in both cores (STAR Methods), which were pooled for analysis. We also acquired images of 92 cores from normal lungs and patients with other pulmonary morbidities as staining controls. In total, 2,070 cores were analyzed from 1,070 subjects.
Table 1.
Clinical cohort description
| Sex | |
| Male | 710 (66.4%) |
| Female | 348 (32.5%) |
| Missing | 12 (1.1%) |
| Age | |
| Mean (SD) | 64.0 (9.96) |
| Median [Min, Max] | 64.5 [18.0, 87.0] |
| Missing | 12 (1.1%) |
| Tumor type | |
| Adenocarcinoma | 618 (57.8%) |
| Squamous cell carcinoma | 401 (37.5%) |
| Adeno squamous cell carcinoma | 32 (3.0%) |
| Large cell carcinoma | 9 (0.8%) |
| NSCLC | 2 (0.2%) |
| Grade | |
| 1 | 62 (5.8%) |
| 2 | 481 (45.0%) |
| 3 | 515 (48.1%) |
| Missing | 12 (1.1%) |
| Size [cm] | |
| Mean (SD) | 4.04 (2.26) |
| Median [Min, Max] | 3.50 [0.300, 16.0] |
| Missing | 12 (1.1%) |
| T | |
| 1 | 131 (12.2%) |
| 2 | 129 (12.1%) |
| 3 | 381 (35.6%) |
| 4 | 118 (11.0%) |
| 5 | 215 (20.1%) |
| 6 | 84 (7.9%) |
| Missing | 12 (1.1%) |
| N | |
| 0 | 555 (51.9%) |
| 1 | 268 (25.0%) |
| 2 | 220 (20.6%) |
| 3 | 14 (1.3%) |
| Missing | 13 (1.2%) |
| M | |
| 0 | 967 (90.4%) |
| 1 | 71 (6.6%) |
| 2 | 19 (1.8%) |
| Missing | 13 (1.2%) |
| Stage | |
| 1 (IA) | 182 (17.0%) |
| 2 (IB) | 191 (17.9%) |
| 3 (IIA) | 191 (17.9%) |
| 4 (IIB) | 113 (10.6%) |
| 5 (IIIA) | 289 (27.0%) |
| 6 (IIIB) | 32 (3.0%) |
| 7 (IV) | 60 (5.6%) |
| Missing | 12 (1.1%) |
| Chemotherapy (neoadjuvant) | |
| No neoadjuvant chemotherapy | 961 (89.8%) |
| Neoadjuvant chemotherapy | 78 (7.3%) |
| Missing | 31 (2.9%) |
| Radiotherapy (neoadjuvant) | |
| No neoadjuvant radiotherapy | 1035 (96.7%) |
| Neoadjuvant radiotherapy | 4 (0.4%) |
| Missing | 31 (2.9%) |
| Relapse | |
| No relapse | 555 (51.9%) |
| Relapse | 480 (44.9%) |
| Missing | 35 (3.3%) |
| Overall survival | |
| Mean (SD) | 1490 (1190) |
| Median [Min, Max] | 1230 [0, 5730] |
| Missing | 2 (0.2%) |
| Disease free survival | |
| Mean (SD) | 1430 (1220) |
| Median [Min, Max] | 1060 [0, 5100] |
| Missing | 138 (12.9%) |
| Smoking status | |
| No | 86 (8.0%) |
| Currently | 730 (68.2%) |
| Former | 26 (2.4%) |
| Unknown | 197 (18.4%) |
| Missing | 31 (2.9%) |
| Chemotherapy (adjuvant) | |
| Yes | 138 |
| No | 901 |
| Missing | 31 |
Table 1. Cohort description including number and percentage information on the distribution of sex, age, tumor type, tumor grade, tumor size, TNM classification, tumor stage, neoadjuvant and adjuvant chemotherapy, neoadjuvant radiotherapy, relapse, and smoking status as well as overall and disease-free survival. Categories are printed bold.
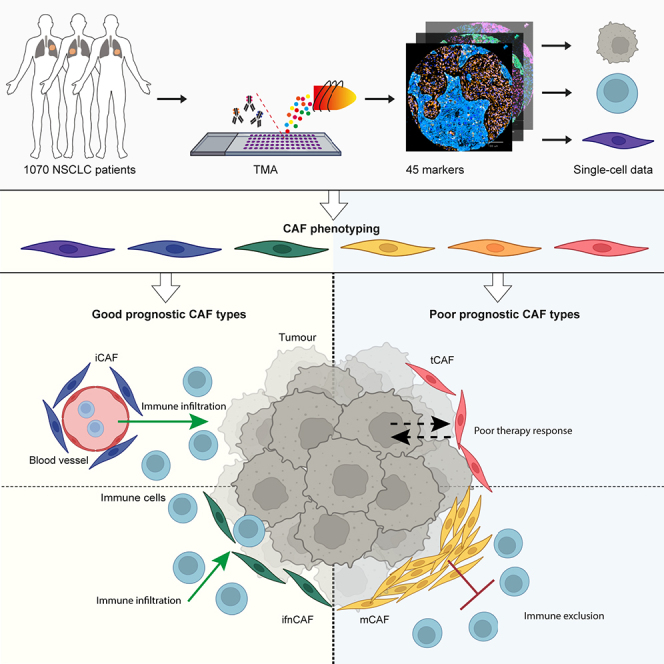
We stained the TMA with 45 metal-labeled antibodies, performed IMC analysis, and quantified marker expression in the resulting highly multiplexed images at single-cell resolution (Figure 1A). These data were then used for cellular phenotyping and correlation of cell types and spatial structures with clinical parameters such as survival (Figure 1B). The antibodies were selected to ensure a comprehensive analysis of CAFs and of other cells of the TME such as endothelial cells, epithelial cells, and immune cells including myeloid cells, neutrophils, B cells, and multiple subsets of T cells (Figures 1C and S1A; Table S1). Marker selection for the identification of CAF subsets was based on our previously established CAF panel for IMC analysis of CAFs37 (STAR Methods) which was also validated against scRNA-seq data from NSCLC.30
Figure 1.

Overview of experimental and analytical set-up
(A) Experimental set-up. NSCLC samples (2070 cores, 1,070 patients) and controls were stained with metal-tagged antibodies and imaged using IMC. Cell and tumor-stroma masks were generated.
(B) Analytical setup. Single-cell data were used to identify cell phenotypes, and to evaluate correlations between cell types and clinical data. CAF based patient stratifications were used for survival and TMA composition analyses. Finally, spatial properties of CAFs and immune cell types were analyzed and integrated with clinical data.
(C) Markers in the antibody panel.
Cell masks were generated using ilastik45 training and CellProfiler46 (Figures 2A and S1B‒S1E), using a strategy we previously described and validated for fibroblasts (STAR Methods).37 We also generated tumor-stroma masks (Figures 2A and S1B‒S1E), which showed the expected average marker (SMA and panCK) expression per cell (Figures 2A and S1E).
Figure 2.

Cell phenotypes and distribution in NSCLC
(A) Exemplary LUAD (left) and LUSC cores (right) after cell segmentation based on panCK as a tumor marker and SMA as a stromal marker. Top to bottom, cells colored by average expression of SMA and panCK; cell assignment based on the generated tumor-stroma masks; cell category assignment; and the binned distance of all non-tumor cells to the nearest tumor-stroma border (30 μm bins [red, orange, yellow, green, blue, purple] from tumor-stroma interface; gray: tumor).
(B) Heatmap of marker expression (normalized from 0 to 1) for all cell phenotypes (labeled cell types). The bar chart above the heatmap shows the number of cells (square root for visualization) of each cell phenotype per tumor type.
(C) Mean proportions of the indicated cell categories per patient, annotated for clinical features. Dist. Met.: distant metastasis; LN met.: lymph node metastasis; Neoad.: neoadjuvant therapy.
(D) tSNE of a subset of 1,000 cells per image colored by cell category.
(E) Proportions of cell categories for all cells measured. For D, E, colors as in B.
For cell phenotyping, all cells were first split into tumor (n = 2,979,712) and non-tumor cells (n = 3,004,742) using a Gaussian mixture model based on the expression of panCK. The non-tumor cells were further clustered into immune cells (n = 1,179,114), endothelial cells (n = 150,062), and CAFs (n = 1,086,678). Immune cells were then categorized into myeloid cells (HLA-DR+ and CD68+), neutrophils (MPO+), B cells (CD20+), and T cells (CD3+). T cells were further clustered into those expressing CD4 and those expressing CD8 and then both types were further grouped into IDO+, PD-1+, TCF1/7+ and dividing Ki-67+. CD4+/FOXP3+ cells were identified as regulatory T cells. Endothelial cells were divided into blood endothelial cells (CD31+/vWF+/LYVE-1-/CD146+), lymphatic endothelial cells (LYVE-1+/CD146+), and high endothelial venules (PNAd+). Tumor cells were further clustered into non-hypoxic (panCK+/CAIX−) and hypoxic (panCK+/CAIX+) cells (Figure 2B). CAFs were identified as cells expressing vimentin, SMA, FAP, and PDGFR-β and lacking panCK expression. All cores showed varying proportions of tumor, stromal, and immune cells (Figure 2C). Roughly 50% of cells were classified as tumor cells, 20% as stromal cells, and 20% as immune cells; about 10% of cells could not be assigned with our panel (Figures 2D and 2E). We observed no correlation between compositions of these general cell types and clinical parameters such as grade, metastasis, or relapse (Figure 2C).
CAF phenotypes in NSCLC
In order to understand whether CAFs are associated with different states of the TME and with tumor progression in NSCLC, we first analyzed CAF phenotypes in our cohort. The 1,086,678 CAFs were clustered using FLOWSOM47 to identify subtypes (Figures S2A and S2B). We previously classified CAF subtypes based on scRNA-seq data from breast, lung, colon, head and neck cancer, and pancreatic ductal adenocarcinoma, and validated an IMC panel to identify these CAF types in tumor tissue samples.37
Most of these previously defined CAF types were also present in the NSCLC cohort analyzed here, including ifnCAFs (IDO+), tCAFs (CD10+/CD73+), hypoxic tCAFs (CAIX+/CD10+), iCAFs (CD34+/CD248+), vCAF (CD146+/CD34-), dCAFs (Ki-67+), SMA CAFs (SMA+/FAP−/MMP−/Collagen−), and hypoxic CAFs (CAIX+). We also identified PDPN CAFs (PDPN+), which were unique to this NSCLC dataset, and split collagen-expressing CAFs into mCAFs (MMP11+/SMA+/Collagen+), as in our previous scRNA-seq study, and collagen CAFs (Collagen+/FN+/MMP11-/SMA−) (STAR Methods for technical differences with our previous work) (Figures 3A–3D). SMA CAFs made up the biggest proportion of CAFs in our NSCLC dataset, followed by mCAFs, vCAFs, and iCAFs; all CAF types were present in LUAD and LUSC samples (Figure 3A, top). Lacking a suitable antibody for the pericyte marker RGS5, we were not able to distinguish pericytes from fibroblasts. This is especially relevant for SMA CAFs and vCAFs, as their characteristic markers (SMA, CD146, and PDGFR-β, respectively) are abundant around vessels. Visual assessment however showed that SMA CAFs as well as PDGFR-β+ CAFs did not exclusively cluster around vessel structures, indicating that these cells are not solely pericytes or smooth muscle vascular cells (Figure 3D). However, as we cannot define pericytes with our current marker panel, they are most likely merged into other CAF phenotypes near vessel cells, especially CD146+ vCAFs.
Figure 3.

CAF phenotypes in NSCLC
(A) Heatmap of mean marker expression (z-scaled from −2 to 3) for all CAF phenotypes identified after FLOWSOM clustering. The bar chart above the heatmap shows the number (square root for visualization) of CAFs per tumor type for each cluster.
(B) UMAP of a random subset (n = 200,000 cells) of CAFs showing the mean intensity per cell (scaled from 0 to 1) for the indicated markers.
(C) UMAP of a random subset (n = 200,000 cells) of CAFs colored by subtype.
(D) Images showing all CAFs (left) and SMA CAFs (right); CAFs are shown with white masks overlaid on the indicated markers.
(E) Mean proportions (left plot) and absolute numbers (right plot) of each CAF type per patient are shown. Color bar (right) indicates CAF types. The hierarchical clustering tree is colored (different color scale from CAF types) by four meta-clustered patient groups (1, 2, 3, 4).
(F) Heatmap (z-scaled expression) showing enrichment of each CAF type per patient group as defined in E.
CAFs showed different enrichment patterns between LUAD and LUSC. dCAFs, tCAFs, iCAFs, vCAFs, and SMA CAFs were more abundant in LUAD, while hypoxic tCAFs, PDPN CAFs, and mCAFs were enriched in LUSC (Figure S3A). We found in addition minor differences between different growth patterns of LUAD, with mCAFs and collagen CAFs enriched in solid and micropapillary tumors, consistent with the relatively high stroma content of these tumor types (Figure S3B). Patients clustered into four patient groups based on proportions of CAF types (Figures 3E, 3F, and S3C), with group 1 enriched in SMA CAFs, group 2 in matrix and collagen CAFs, group 3 in several CAF types including tCAFs, hypoxic CAFs, hypoxic tCAFs and vCAFs, and group 4 in iCAFs and ifnCAFs.
CAF phenotypes are an independent prognostic factor in NSCLC
We next asked whether the CAF types and compositions in a tumor were associated with patient outcome or clinical presentation. A comparison between patients with overall survival longer or shorter than the median (3 years in this cohort) identified CAF phenotypes associated with longer survival (ifnCAFs and iCAFs), whereas all other CAF types (e.g., hypoxic tCAFs, tCAFs) were associated with shorter survival (Figure 4A). Similarly, patients who relapsed within the follow-up period of 15 years had tumors enriched for hypoxic tCAFs, hypoxic CAFs, PDPN CAFs, collagen CAFs, tCAFs, and dCAFs (all p < 0.05; Figure S3D), again suggesting that these CAF types are associated with poor patient outcome. Analysis of the association between CAF types and relapse as well as sex generally supported these observations (Figure S3D). Differential analyses conducted separately for LUAD and LUSC tumors showed similar enrichments to the joint analysis (Figures S3E and S3F).
Figure 4.

Association of CAF types with clinical parameters
(A) Differential abundance testing of all cell types between patient groups defined by long (left) and short (right) overall survival, split by median. p Values < 0.05 are shown in blue, p values > 0.05 in grey.
(B) Kaplan-Meier overall survival curves for patient groups (PG) defined by CAF composition (PG 1–4). The table shows patient numbers according to tumor type and p values for comparison between groups (∗p < 0.05, ∗∗∗p < 0.001).
(C) CoxPH-model for patient groups, corrected for tumor type and grade. Patient group 1 serves as a reference. p Values < 0.05 are shown in blue, p values > 0.05 in grey.
(D and E) Kaplan-Meier (plots for overall survival comparing patients stratified as high and low based on the median proportion of indicated CAF types. Good prognosis CAFs are in D, poor prognosis CAFs in E. All comparisons with significant differences (log rank testing p < 0.05) are shown.
(F) Lasso-regressed CoxPH model including mean CAF type proportions per patient, patient stratification into high and low for each CAF type (by median proportion), and all clinical data.
We next studied whether the CAF groups were associated with distinct survival. CAF groups 1 and 4 were enriched for good prognosis CAFs and groups 2 and 3 with poor-prognosis CAFs; these groups indeed showed a significant difference in overall survival (Figure 4B). While the patient groups did not have the same distribution of LUAD and LUSC tumors (Figure 4B), the differences in prognosis between these groups remained in a Cox proportional hazard (CoxPH) model corrected for tumor type (Figure 4C), suggesting that these effects are not explained by tumor type alone. This model was also corrected for grade, which in LUAD is correlated with histological growth patterns, again suggesting that the prognostic differences in the patient groups are independent of LUAD subtypes. Analysis of patients stratified as high and low for some individual CAF types also showed associations with survival, with high proportions of SMA CAFs, ifnCAFs, and iCAFs (only in LUSC) associated with longer survival (p < 0.05) (Figure 4D), and high proportions of hypoxic CAFs, collagen CAFs, and tCAFs (only in LUAD) associated with shorter survival (p < 0.05) (Figure 4E). High proportions of collagen CAFs, ifnCAFs, and iCAFs and low proportions of vCAFs were also associated with disease-free survival (Figure S4A). Stratifying patients by the median density of the CAF types gave similar results (Figures S4B and S4C). Interestingly, high mCAF density was associated with significantly lower overall survival in LUSC (p = 0.025, Figure S4B).
A lasso regressed CoxPH model for overall survival considering all CAF types and all clinical factors selected the proportions of tCAFs and hypoxic tCAFs as independent predictors of poor prognosis (Figure 4F). ifnCAF, dCAF, and SMA CAF proportions were predictors of good prognosis, albeit with low hazard ratios (Figure 4F). As expected, other prognostic factors included tumor grade, stage, and metastasis. When density (rather than proportion) of all cell types and clinical parameters were analyzed, hypoxic tCAFs and tCAFs, as well as vCAFs, hypoxic CAFs in general, and mCAFs were predictors of poor prognosis (Figure S4D). Other than the mCAF low stratification, no CAF type was selected as a predictor of good outcome, but clinical features (M0, low stage) were, as expected (Figure S4D). Overall, our differential abundance analysis suggests that specific CAF types and compositions are associated with poor prognosis (tCAFs, hypoxic tCAFs, collagen CAF, and mCAFs) or better prognosis (ifnCAFs, iCAFs, and SMA CAFs) in NSCLC.
CAF types are associated with chemoresistance and metastasis
To gain further insight into potential mechanisms that underlie the prognostic value of specific CAF types, we investigated their relationship with chemoresistance and metastasis as clinically relevant drivers of poor patient outcome in NSCLC. We first asked whether chemotherapy affected CAF type distribution in NSCLC tumors by comparing the 74 patients who had received neoadjuvant chemotherapy with a matched untreated control group (n = 72, STAR Methods). The neoadjuvant-treated group was significantly enriched in dCAFs in both LUAD and LUSC (Figures 5A and 5B, p ≤ 0.001 each), as were SMA CAFs in neoadjuvant-treated LUSC (Figure 5B, p = 0.023), suggesting a reactive remodeling stroma after therapy. In LUAD, treatment naive patients were enriched in ifnCAFs and tCAFs (p = 0.01 and 0.011) (Figure 5A). We observed no other differences between the two groups.
Figure 5.

CAF types are associated with chemoresistance and metastasis
(A and B) Boxplots showing the average patient proportions for the indicated CAF types comparing patients that received neoadjuvant therapy versus a matched treatment-naïve cohort for LUAD (A) and LUSC (B). Exact p values are indicated.
(C and D) Differential abundance testing of all CAF types comparing patients receiving adjuvant chemotherapy who did or did not relapse, in LUAD (C) and LUSC (D). p Values < 0.05 are shown in blue, p values > 0.05 in grey.
(E and F) Differential abundance testing of all CAF types comparing patients with (N1/2) and without (N0) lymph node at diagnosis, in LUAD (E) and in LUSC (F). p Values < 0.05 are shown in blue, p values > 0.05 in grey.
(G) Boxplot showing the average proportions per patient of the indicated CAF types in matched patient groups with and without distant metastases at diagnosis. Exact p values are indicated.
Next, we examined tumors of patients treated with chemotherapy after surgery (n = 138 patients) and asked whether the distribution of CAF types was associated with relapse and therefore potentially with resistance to treatment. We detected a significant enrichment of ifnCAFs in LUAD and LUSC patients who did not experience relapse (n = 65 patients) in comparison to those who did (n = 68 patients) (Figures 5C and 5D). Associations with relapse were different for the two tumor types, with dCAFs, collagen CAFs, and tCAFs being significantly associated with relapse in LUAD (Figure 5C) and PDPN CAFs, hypoxic tCAFs and hypoxic CAFs in LUSC (p < 0.05) (Figure 5D). Notably, the CAF type associations with relapse in this smaller, chemotherapy-treated subset were similar to our observations in the entire cohort, suggesting that these associations are independent of treatment.
We next sought to investigate whether CAF types in the primary tumor were correlated with the occurrence of metastasis. We first compared CAF-type abundance in patients with (n = 503) and without (n = 555) lymph node metastasis. Interestingly, we observed that positive-prognostic iCAFs and SMA CAFs were enriched in patients with no dissemination to lymph nodes, while negative-prognostic tCAFs, hypoxic CAFs and hypoxic tCAFs showed the opposite effect (Figures 5E and 5F). Next, we compared patients with distant metastasis at the time of diagnosis (n = 90) to a matched control group (n = 90, STAR Methods). We compared LUSC and LUAD samples together, given the relatively small sample numbers, and found that higher proportions of tCAFs were associated with distant metastasis at the time of diagnosis, while dCAFs showed the opposite effect (Figure 5G, p < 0.05 in both cases).
Taken together, these results are consistent with our identification of tCAFs and hypoxic tCAFs as poor prognostic factors and of ifnCAFs, iCAFs, SMA CAFs, and dCAFs as good prognostic factors in NSCLC.
CAF types are associated with distinct TME compositions
We next sought to understand whether the prognostic CAF types we have defined are associated with distinct immune compositions. We first analyzed the proportions and the densities of all cell types across the cohort. Neither CAF-type proportions nor densities were strongly correlated with those of other cells of the TME, but a few clusters that included CAFs were detected (Figures S5A and S5B). ifnCAFs formed part of an immune cell cluster that also included B cells, lymphatic endothelial cells, and IDO+ T cells (Figure S5A, cluster 1). vCAFs and iCAFs were weakly positively correlated with endothelial cells (lymphatic and blood) (Figure S5A, cluster 2); the same was found when cell type densities were analyzed (Figure S5B).
When we analyzed only CAF types, we saw no strong correlations in the data, except that the proportions of mCAFs and SMA CAFs in individual patient images were strongly anti-correlated (p < 0.001; Figure S5A); we observed the opposite effect in an analysis of cell density (Figure S5B), most probably reflecting the high density of both of these CAF types in the TME. In addition, proportions of iCAFs were anti-correlated with those of SMA CAFs and mCAFs (p < 0.001), and proportions of ifnCAFs were positively correlated with those of iCAFs and tCAFs (p < 0.001; Figure S5A).
iCAFs were significantly, if weakly, anti-correlated with all hypoxic cells (LUAD r = −0.14, p = 0.00033/LUSC: r = −0.16, p = 0.0012) and were enriched in normoxic patches (p < 0.05, Figures S5C‒S5H), in contrast to recent findings in pancreatic cancer,48,49 which, however, defined iCAFs using a different marker (IL6) than in this study. Indeed, all of the CAF types were enriched in normoxic patches in our lung dataset, with the exception of CAIX+ hypoxic cells as expected (Figures S5G‒S5H). Endothelial cells (marking blood vessels) were enriched in normoxic patches, supporting our definition of CAIX+ cells as genuinely hypoxic.
In the absence of strong correlations at the level of individual CAF phenotypes, we investigated the TME composition of the prognostic patient groups that reflect different CAF compositions. We found that the poor prognostic patient groups (2, 3) were enriched in hypoxic tumor cells as well as neutrophils but depleted of immune cell types (Figures 6A and S6). In contrast, the two good prognostic patient groups (1, 4) showed an overall immune cell-enriched phenotype with little to no hypoxia suggesting a more inflamed TME state in these groups (Figures 6A and S6). Analysis at the level of individual CAF types for patients stratified as “high” or “low” also showed similar trends (Figures S7A and S7B). The exception was TMEs high in tCAFs and PDPN CAFs, which were enriched in PD-1+/CD4+ T cells, IDO+ T cells, neutrophils, regulatory T cells, and myeloid cells, indicative of immune exhaustion (Figure S7A). Taken together, these data show that CAF types and prognostic patient groups defined by CAF composition are associated with distinct TME states. Poor prognosis groups and their CAF types are found in tumors enriched for exhausted immune cells and hypoxia, and, in contrast, good prognosis patient groups and their CAF types present with a more inflammatory TME.
Figure 6.

The spatial relationship of CAFs with the tumor microenvironment
(A) Heatmap showing enrichment or depletion of cell types between patient groups defined by CAF composition (1, 2, 3, and 4).
(B) Density curves showing the distance per cell of the indicated CAF types to the closest tumor-stroma border for LUAD (left) and LUSC (right).
(C) Neighborhood analysis heatmap showing nearest 15 neighbors of each cell within a radius of 20 μm (to cell, Y axis; from cell, X axis). Plotted are mean significances over all images, split by LUAD (left) and LUSC (right). Positive/negative (red/blue) mean scores indicate higher/lower cellular interactions compared to a random null-distribution.
(D) Images with mask overlays (white) of SMA CAFs (SMA+), mCAFs (MMP11+), collagen CAFs (Collagen I/Fibronectin+), vCAFs (CD146+), iCAFs (CD34+), tCAFs (CD10+), hypoxic CAFs (CAIX+). The general stroma marker SMA is in red in all cases except for images showing SMA CAFs and hypoxic CAFs. Pan cytokeratin is in magenta in all cases except for the image showing hypoxic CAFs (lower right), all CAFs are panCK−; DNA is in blue. Scalebars are in μm.
CAFs have defined spatial distributions within the TME
Since CAF types are associated with TMEs with distinct characteristics, we analyzed the spatial distributions of cell types in NSCLC TMEs. We defined tumor-stroma masks, ensured that the basic cell types were indeed localized in the expected compartments, and calculated the distances of each cell to the closest tumor-stroma interface. We saw striking differences in the spatial distributions of different CAF types (Figure 6B). SMA CAFs and mCAFs were almost exclusively in the stromal compartment, whereas hypoxic tCAFs were almost exclusively in the tumor compartment. Further, tCAFs and iCAFs were generally found in the tumor compartment, whereas ifnCAFs accumulated at the tumor-stroma interface. These results were independent of tumor type (Figure 6B).
Next, we assessed the pairwise spatial interaction between CAFs and all other cells and their association with distinct structures within the TME. A neighborhood analysis over a radius of 20 μm (STAR Methods) revealed that SMA CAFs, mCAFs, and vCAFs were spatially near each other and that vCAFs and iCAFs were near blood and lymphatic endothelial cells, independent of tumor type (Figures 6C and 6D). The latter observation likely explains the presence of iCAFs in the tumor compartment as a result of vessels being incorporated into tumor masks (Figure 6C). ifnCAFs were near IDO+/CD4+ and IDO+/CD8+ T cells, likely a result of IFN stimulation from surrounding cells; however, only ifnCAFs and IDO+/CD8+ T cells showed positive interaction scores with tumor cells. Poor-prognosis tCAFs did not show strong spatial interactions with other cell types but had a slight tendency to neighbor tumor cells and ifnCAFs. All hypoxic cells (hypoxic tCAFs, hypoxic CAFs, and tumor cells) clustered spatially in a hypoxic neighborhood that included collagen CAFs to some extent tCAFs. Collagen CAFs also showed strong interactions with mCAFs. SMA CAFs and mCAFs showed the most cellular interactions, likely reflecting their high abundance, whereas dCAFs and PDPN CAFs showed no strong interactions, consistent with their relative sparsity within the TME. Splitting the neighborhood analyses by tumor type (LUAD vs. LUSC) showed similar patterns (Figure 6C). We did not find any major differences for cell-to-cell interactions when comparing additional clinical parameters or our prognostic patient groups (data not shown).
It is known that fibrosis and a dense stroma can prevent immune cells from reaching the tumor, resulting in poor patient prognosis.50,51 Our data revealed that mCAFs are a dominant constituent of the tumor stroma (Figure 3E). Further, high mCAF density was a predictor of poor overall survival in LUSC (Figure S4B) and the mCAF-enriched patient group 2 was immune cell depleted compared to the other patient groups (Figure 6A). We therefore analyzed the effects of mCAF density on the spatial distribution of immune cells, by comparing cellular interactions in LUSC patients stratified by median mCAF density (STAR Methods, Figure 7A).52 This revealed that all immune cells were enriched spatially near tumor cells in patients stratified as mCAF density low (Figure 7A). Further, all immune cells, including CD4+ and CD8+ T cells, B cells, myeloid cells, and neutrophils were found significantly more often near the tumor-stroma interface (−30 μm–0 μm) or invading the tumor (0 μm–30 μm) in mCAF-low patients compared to mCAF-high patients (p < 0.05); with the reverse effect seen further away from the tumor (−90 to −30 μm) (p < 0.05) (Figure 7B). This effect was also visible in images of mCAF density high (Figure 7C) and low samples (Figure 7D). None of the other CAF types showed this effect. Our data thus show that the density of mCAFs might contribute toward poor patient prognosis by driving fibrosis and thus affecting immune cell access to the tumor in NSCLC.
Figure 7.

High mCAF density blocks out immune cells
(A) SpicyR analysis of cell pairs in patients stratified by median mCAF density. Positive scores (red) indicate higher cellular interactions between cell pairs in patients with low mCAF density, negative scores (blue) indicate higher cellular interactions in patient with high mCAF density; white indicates no difference.
(B) Proportions of B cells (upper) and CD4+ T cells (lower) in patients stratified by mCAF density, within bins of 30 μm from the tumor-stroma border (∗p values < 0.05).
(C and D) Images showing the indicated markers for a mCAF density high image (C) and a mCAF density low image (D), together with the corresponding masks colored by tumor cells (magenta), immune cells (yellow) and mCAFs (cyan). Scale bar, 150 μm.
In summary, we have shown that fibroblast phenotypes in the TME are a strong prognostic factor for NSCLC patient outcome. Our data provides evidence for both good- and poor-prognosis CAF types. The presence of tCAFs was strongly correlated with poor prognosis and associated with tumors characterized by hypoxic and immune-exhausted microenvironments. In contrast, iCAFs that express CD34 and CD248 as well as ifnCAFs were associated with inflamed tumor microenvironments and higher overall patient survival.
Discussion
Understanding cellular processes and interactions in the TME and how they are connected to disease progression and patient outcome is of key importance for patient stratification, therapy development, and ultimately precision oncology applications. Studies of stromal cells are particularly crucial. The first experimental therapies broadly targeting the tumor stroma have not been successful in clinical studies, suggesting that more precise stromal targets are needed.53 Here, we identified prognostic cellular factors within the stroma of NSCLC, a disease with poor overall survival and moderate therapeutic response rates. We identified individual CAF types in LUAD and LUSC and studied the composition and spatial organization of these cells within the TME and how these relate to clinical parameters including survival.
Our defined CAF types share markers with CAF types defined by others. CD34+ iCAFs have been previously described as Pi16+ CAFs,17,35 as were cells similar to our ifnCAFs,34 typically as a subpopulation of a broader iCAF or myofibroblast group17,36,54,55; the same is also true for CCL19/CCL21+ rCAFs.17,56 We note that iCAFs are frequently described as IL6+ CAFs, but we did not include IL-6 in our marker panel due to the lack of a suitable antibody and therefore cannot describe IL-6 expression in our CD34+ iCAFs. Previous work has defined inflammatory CAFs with different markers.21,39,55,57 Also, matrix-associated CAFs have been described in the literature as having high expression of MMP11 and collagens.17,36,54,55 In our study, we distinguished between mCAFs (FAP+ MMP11+) and collagen CAFs (FAP+ MMP11-), with the latter cells likely to be a subgroup within the diverse group of matrix-associated CAFs,17,34 but that express matrix deposition and remodeling factors other than MMP11.17,36,37,54,55 CD10/CD73+ CAFs, which we have named tCAFs, have also been previously described, in all cases specifically identified as panCK-negative fibroblasts17 and thus distinguishing them from epithelial populations that are also known to express CD10.58 While CD10 is generally used as a marker for alveolar fibroblasts of the lung, high CD10 expression in NSCLC CAFs has been associated with chemoresistance and cancer stemness.58 Finally, our vCAFs are similar to the MHY11+aSMA+ CAFs described by Grout et al.17 as well as with previously identified vascular or vessel CAFs.36 Taken together, our analyses show that the commonly used stratification of CAFs into iCAFs, myofibroblasts and apCAFs can be further refined into smaller, functionally relevant groups. Our study unfortunately does not identify MHC-II+ antigen-presenting CAFs due to widespread expression of this marker.38
We showed that patients can be classified into good and poor prognosis groups based on their CAF composition, independent of tumor type (i.e., LUAD or LUSC), and with individual CAF types also being strong independent prognostic factors in NSCLC. Good prognostic patient groups 1 and 4 were enriched in SMA CAFs, iCAFs, and ifnCAFs, and showed also an immune-enriched microenvironment. Since SMA CAFs and iCAFs are, in addition, spatially near vessel structures, this suggests that these CAF types actively promote immune cell infiltration. ifnCAFs may be an indirect marker for an ongoing immune response; they have been previously associated with inflammation and IFN expression and we previously showed that they strongly express the IFN stimulated chemokines CXCL9, CXCL10, and CXCL11. Further, ifnCAFs are close to tumor cells, suggesting that the ongoing inflammation may directly affect these cells. However, the effect of interferons on tumor cells is complex and may be pro- or anti-tumor.59,60 Intriguingly, ifnCAFs were enriched in the tumors of non-relapsing patients both in the whole cohort and in the subset of patients who received chemotherapy post-surgery, suggesting a tumor-suppressing role of ifnCAFs through interferon signaling cues.
In contrast, tCAFs and mCAFs (among others) were enriched in poor-prognosis patient groups 2 and 3 and were found in hypoxic TMEs enriched in poor prognostic neutrophils. tCAFs and hypoxic tCAFs were further enriched in patients with distant metastases at the time of diagnosis, suggesting the hypothesis that a role of these cell types in metastasis is the reason for their poor prognostic effect. In our previous study, tCAFs also showed enrichment of TGF-β signaling, glycolysis, stress response and angiogenesis in a functional enrichment analysis, further pointing toward pro-tumourigenic functions. Since early micrometastasis is a driver of poor patient prognosis,61 it is conceivable that tCAFs could drive early metastatic dissemination and hence worsen prognosis. This is consistent with their close spatial interaction with tumor cells. We interestingly also observed that tCAFs were depleted in patients post neoadjuvant therapy, which may suggest that tCAFs are either targeted by chemotherapy or that their phenotype depends on close interactions with tumor cells which are depleted by chemotherapy. In contrast, tCAFs and hypoxic tCAFs were enriched in relapsing patients, including those who relapsed after chemotherapy, in line with the known link between stromal CD10 expression and chemoresistance in lung cancer.13
Interestingly, our spatial analysis of CAF types in NSCLC revealed no significant differences between LUAD and LUSC as well as between the different growth patterns of LUAD (data not shown). It further showed good correspondence with the spatial CAF-type distributions we detected in our multiplexed imaging study of breast cancer.37 This suggests that the architecture and spatial distribution of CAF types within the tumor stroma might be conserved across cancer types. It is also conceivable that the different CAF types in cancer are the result of their spatial proximity to different structures (vessels, stroma, and tumor) and therefore of their interaction with different cell types. Mechanistic studies using co-cultures would be useful to investigate CAF plasticity further.
Poor-prognostic tCAFs, as well as good-prognostic and pro-inflammatory ifnCAFs, are both located near tumor cells, suggesting juxtracrine or paracrine effects between CAFs and tumor cells. tCAFs express CD73, a 5′-nucleotidase that dephosphorylates AMP to adenosine. An increase of available adenosine in the extracellular matrix facilitates growth, migration, survival, and adhesion of tumor cells.13,62,63,64 Adenosine also has immunosuppressive functions and has been associated with T cell exhaustion and thus could indirectly drive tumor growth by facilitating immune escape.65 tCAFs may therefore promote tumor growth by making adenosine available in the tumor-surrounding stroma. Furthermore, it has previously been shown that tumor cells can reprogram nearby stromal cells for their own metabolic benefit.66,67 Based on the physical proximity of tumor cells and tCAFs it could be speculated that this mechanism may be at play. For good-prognostic ifnCAFs, which are also located near tumor cells, expression of IDO suggests that these cells may be reacting to ongoing inflammation, a hypothesis supported by immune cell enrichment in patients with high proportions of ifnCAFs.68 However, ifnCAF high patients were also significantly enriched for hypoxic tCAFs, myeloid cells and potentially immunosuppressed IDO+ T cells.69,70 This, and their close interaction with tumor cells stands in conflict with their overall apparent good prognostic function. Functional experiments are needed to fully understand the mechanisms of CAF heterogeneity and the synergistic effect between CAFs, tumor cells and inflammatory processes within the TME.71 Still, CAFs might serve as a marker for the state of the TME.
We hypothesize that our data may have implications for anti-CAF directed therapy, which has so far not been successful.72 This may be due to targeting of all CAFs, which we would predict to reduce therapeutic efficiency given the mixed prognostic effects we have observed for different CAF types. It would potentially be beneficial, and has been hypothesized before,23 to target or eliminate poor prognostic CAFs while supporting good prognostic types. Poor prognostic tCAFs and hypoxic tCAFs, which we see are correlated with chemoresistance and metastasis, could be potential cellular targets for precision medicine. Further, if our hypothesis is correct that the spatial proximity of tCAFs with tumor cells is functionally important, a mode of action for potential treatments might lie in interrupting the molecular interactions between these cell types to potentially slow tumor growth and decrease metastasis and chemoresistance. Nevertheless, further experiments will be needed to strengthen the case for these CAF types playing a causal role in tumor growth.
We showed that mCAF density has a negative effect on immune cell infiltration in NSCLC, whereas other CAF types did not play this role. Thus, a dense mCAF barrier may exclude immune cells from the tumor, hamper the anti-immune response and negatively impact patient survival. This finding might suggest that approaches that disrupt the tumor-surrounding stroma may allow more immune cells to reach the tumor and yield higher success rates for immunotherapy. However, previously reported negative effects of stroma depletion must be considered in such settings.19,22 In general, a balance between the tumor promoting and suppressing effects of fibrosis must be found if these cells are to be effective drug targets.
More generally, our findings highlight the importance of understanding CAF heterogeneity to enable development of therapies that inhibit functions of poor-prognosis CAFs or that support the good-prognosis ones. Previous work in pancreatic ductal adenocarcinoma73 and breast cancer34 suggests that there are links between specific CAF types and patient response to therapy. It will therefore be of interest to analyze in more detail how the stromal compartment changes after different types of therapy and how our defined CAF types relate to therapeutic response. The data we provide here on treatment-naïve patients can serve as a reference in that regard.
Limitations of the study
Our study has limitations: First, despite the highly multiplexed nature of IMC, there may be CAF types that are not captured by our 45-antibody panel. For instance, due to the lack of a high-quality antibody for pericyte marker RGS5,74 we cannot distinguish pericytes from vCAFs. We mitigated this effect by careful antibody panel design informed by intensive literature research and by our own and public scRNA-seq data.30,37 Second, the simplification of fibroblasts to their nuclear core during segmentation meant that we were unable to investigate interactions that occur at the edges of the cells. Third, only one or two cores were imaged per patient, and all cores were taken from the invasive front. Thus, CAF features and heterogeneity that we describe may be unique to this region of NSCLC tumors. Finally, our study is descriptive and investigated correlation rather than causation, thus, our derived hypotheses are speculative. Nevertheless, this study is the largest multiplex imaging analysis of NSCLC and fibroblasts to date and should serve as a reference and resource for future work.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-Vimentin; Clone (EPR3776); Lot(GR286525-2) | Abcam | Cat#ab193555; RRID: AB_2814713 |
| Rabbit polyclonal anti-p75 (CD271); Clone (polyclonal_ANT-007); Lot(ANT007AN0702) | alomone labs | Cat#ANT-007; RRID: AB_2039968 |
| Rabbit polyclonal anti-Collagen I; Clone (Polyclonal_Collagen I); Lot(GR3271183-3) | Abcam | Cat#ab34710; RRID: AB_731684 |
| Mouse monoclonal anti-Fibronectin; Clone (10/Fibronectin); Lot(6251888) | BD Biosciences | Cat#610078; RRID: AB_397486 |
| Rat monoclonal anti-Podoplanin; Clone (NC-08); Lot(B260834) | Biolegend | Cat#337002; RRID: AB_1595511 |
| Mouse monoclonal anti-pan Cytokeratin; Clone (AE1); Lot(3252910) | Millipore | Cat#MAB1612; RRID: AB_2132794 |
| Goat polyclonal anti-Carbonic Anhydrase IX; Clone (polyclonal_CA9_AF2188); Lot(VNQ0319011) | R&D Systems | Cat#AF2188; RRID: AB_416562 |
| Rabbit monoclonal anti-Histone H3; Clone (D1H2); Lot(15) | Cell Signal Technology | Cat#4499BF; RRID: AB_10544537 |
| Rabbit polyclonal anti-CD248; Clone (Polyclonal_Proteintech); Lot(00019535) | Proteintech | Cat#18160; RRID: AB_10858230 |
| Mouse monoclonal anti-CXCL-12; Clone (79018); Lot(JOJ0519031) | R&D Systems | Cat#MAB350-100; RRID: AB_2088149 |
| Goat polyclonal anti-LYVE-1; Clone (Polyclonal_LYVE-1); Lot(KPY0119052) | R&D Systems | Cat#AF2089; RRID: AB_355144 |
| Goat polyclonal anti-CCL21 ; Clone (Polyclonal_CCL21 / 6Ckine); Lot(AYJ218071) | R&D Systems | Cat#AF366; RRID: AB_355327 |
| Mouse monoclonal anti-Cadherin-11; Clone (283416); Lot(VLT0219091) | R&D Systems | Cat#MAB1790; RRID: AB_2076970 |
| Mouse monoclonal anti-CD45RA; Clone (HI100); Lot(B291512) | Biolegend | Cat#304102; RRID: AB_314406 |
| Rabbit monoclonal anti-Caveolin-1; Clone (D46G3); Lot(7) | Cell Signal Technology | Cat#3267BF; RRID: AB_2275453 |
| Mouse monoclonal anti-FOXP3; Clone (236A/E7); Lot(2129676) | eBioscience | Cat#14-4777-82; RRID: AB_467556 |
| Rabbit monoclonal anti-MMP9; Clone (D6O3H); Lot(4) | Cell Signal Technology | Cat#A0398; RRID: AB_2798289 |
| Rabbit monoclonal anti-CD140b/PDGFRb; Clone (Y92); Lot(GR296584-4) | Abcam | Cat#ab215978; RRID: AB_2894841 |
| Mouse monoclonal anti-CD8a; Clone (C8/144B); Lot(2132595) | eBioscience | Cat#14-0085-82; RRID: AB_11150240 |
| Rabbit polyclonal anti-Myeloperoxidase; Clone (Polyclonal MPO); Lot(20071134) | Dako | Cat#AB_2335676; RRID: AB_2335676 |
| Mouse monoclonal anti-CD15; Clone (HI98); Lot(B291510) | Biolegend | Cat#301902; RRID: AB_314194 |
| Rabbit monoclonal anti-CD4; Clone (EPR6855); Lot(GR3215375-27) | Abcam | Cat#ab181724; RRID: AB_2864377 |
| Rabbit monoclonal anti-Indoleamine 2- 3-dioxygenase; Clone (SP260); Lot(GR3259345-2) | Abcam | Cat#ab245737; RRID: AB_2894840 |
| Rabbit monoclonal anti-CD10; Clone (E5P7S); Lot(2) | Cell Signal Technology | Cat#65534; RRID: AB_2894842 |
| Goat polyclonal anti-CD146; Clone (Polyclonal_MCAM/CD146_RnD_AF932); Lot(ECL0319041) | R&D Systems | Cat#AF932; RRID: AB_355721 |
| Mouse monoclonal anti-SMA; Clone (1A4); Lot(2183900) | eBioscience | Cat#14-9760-82; RRID: AB_2572996 |
| Rabbit monoclonal anti-CD73; Clone (D7F9A); Lot(2) | Cell Signal Technology | Cat#13160BF; RRID: AB_2716625 |
| Rat monoclonal anti-PNAd; Clone (MECA-79); Lot(B257139) | Biolegend | Cat#120802; RRID: AB_493555 |
| Rabbit monoclonal anti-VCAM-1; Clone (EPR5047); Lot(GR3255420-3) | Abcam | Cat#ab215380; RRID: AB_2894839 |
| Rabbit monoclonal anti-CD34; Clone (EP373Y); Lot(GR3271518-1) | Abcam | Cat#ab198395; RRID: AB_2889381 |
| Rabbit polyclonal anti-vWF; Clone (poly vwf); Lot(3322998) | Millipore | Cat#AB7356; RRID: AB_92216 |
| Mouse monoclonal anti-HLA-DR; Clone (TAL 1B5); Lot(GR3306331-1) | Abcam | Cat#ab20181; RRID: AB_445401 |
| Rabbit polyclonal anti-CD3; Clone (polyclonal_A0452); Lot(20073981) | Dako | Cat#A0452; RRID: AB_2335677 |
| Rabbit polyclonal anti-MMP11; Clone (polyclonal); Lot(GR111256-10) | Abcam | Cat#ab53143; RRID: AB_2042361 |
| Mouse monoclonal anti-Keratin Epithelial; Clone (AE3); Lot(3255457) | Millipore | Cat#MAB1611; RRID: AB_2134409 |
| Mouse monoclonal anti-CD20; Clone (L26); Lot(2172592) | eBioscience | Cat#14-0202-82; RRID: AB_10734340 |
| Rabbit monoclonal anti-CD31; Clone (EPR3094); Lot(GR3229164-1) | Abcam | Cat#ab207090; RRID: AB_2889382 |
| Rabbit monoclonal anti-FSP1 / S100A4; Clone (EPR2761(2)); Lot(GR317174-3) | Abcam | Cat#136784; RRID: AB_10807552 |
| Sheep polyclonal anti-FAP; Clone (polyclonal_FAP); Lot(ZKW0619101) | R&D Systems | Cat#AF3715; RRID: AB_2102369 |
| Mouse monoclonal anti-CD68; Clone (KP1); Lot(2162103) | eBioscience | Cat#14-0688-82; RRID: AB_11151139 |
| Rabbit monoclonal anti-CD279 (PD-1); Clone (D4W2J); Lot(6) | Cell Signal Technology | Cat# I44:I47; RRID: AB_2864408 |
| Rabbit monoclonal anti-TCF1/TCF7; Clone (C63D9); Lot(10) | Cell Signal Technology | Cat#2203; RRID: AB_2534154 |
| Mouse monoclonal anti-CD45RO; Clone (UCHL1); Lot(B291511) | Biolegend | Cat#304202; RRID: AB_314418 |
| Rabbit monoclonal anti-K-Cadherin ; Clone (D3T3I); Lot(2) | Cell Signal Technology | Cat#48111; RRID: AB_2799334 |
| Mouse monoclonal anti-Ki-67; Clone (B56); Lot(8239549) | BD Biosciences | Cat#556003; RRID: AB_396287 |
| Biological samples | ||
| Human NSCLC TMA sections | University Hospital Zurich, Zurich, Switzerland | https://www.usz.ch/ |
| Chemicals, peptides, and recombinant proteins | ||
| Iridium intercalator | Fluidigm | Cat#201192B |
| Critical commercial assays | ||
| MaxPar X8 Multimetal Labeling Kit | Fluidigm | Cat#201300 |
| Deposited data | ||
| IMC images | This paper | Zenodo: https://doi.org/10.5281/zenodo.7961844 |
| Clinical data | This paper | Zenodo: |
| Software and algorithms | ||
| CellProfiler v3.1.9 | McQuin et al.46 | http://cellprofiler.org |
| HistoCAT | Schapiro et al.75 | https://github.com/BodenmillerGroup/histoCAT/releases |
| Ilastik v.1.3.3 | Berg et al.45 | https://www.ilastik.org/news/2019/09/30/ilastik-1.3.3 |
| Python v 3.4.3 | Python Software Foundation | https://www.python.org |
| R v4.1 | R Development Core Team | https://www.r-project.org |
| Bodenmiller lab imctools | Bodenmiller lab | https://github.com/BodenmillerGroup/imctools |
| Bodenmiller lab segmentation pipeline | Bodenmiller lab | https://github.com/BodenmillerGroup/ImcSegmentationPipeline |
| Bodenmiller CellProfiler plugins | Bodenmiller lab | https://github.com/BodenmillerGroup/ImcPluginsCP |
| Bodenmiller lab spillover compensation | Bodenmiller lab | https://github.com/BodenmillerGroup/cyTOFcompensation |
| imcRtools v1.3.7 | Windhager et al.76 | https://doi.org/10.1038/s41596-023-00881-0. |
| Rphenoannoy v0.1.0 | Levine JH et al.77 | https://doi.org/10.1016/j.cell.2015.05.047 |
| FLOWSOM v2.2.0 | Van et al.47 | https://onlinelibrary.wiley.com/doi/full/10.1002/cyto.a.22625. |
| Analysis code | This paper | https://doi.org/10.5281/zenodo.7981268 |
Resource availability
Lead contact
Please direct all questions regarding this manuscript to Bernd Bodenmiller (bernd.bodenmiller@uzh.ch).
Material availability
No new materials were generated within the scope of this project.
Data and code availability
All data have been deposited at zenodo under https://doi.org/10.5281/zenodo.7961844 and are publicly available as of the date of publication. DOIs are listed in the key resources table.
All original code has been deposited at GitHub and is publicly available as of the date of publication. DOIs are listed in the key resources table.
Experimental model and study participant details
The TMA cohort used in this study is an existing clinical sample cohort that was kindly provided by the Pathology Department of the University Hospital Zurich (USZ).42,43,44 All samples were collected with approval of the Ethics Committee of Zurich #2019-01597 and Stv. 29-2009. Information regarding the ethnicity of the patients as well as information on the mutation status of the patients is not available for this cohort. Information about sex and age are provided as part of the anonymised clinical data, information on ancestry, race, ethnicity and the socioeconomic status was not collected as part of this cohort and is thus not available. The influence of sex as a co-founding factor on the results was analysed.
Method details
Antibody panel, staining, and imaging
Samples were first deparaffinised using HistoClear and then rehydrated with a graded ethanol series (100%, 96%, 90%, 80%, 70%). For antigen retrieval, the slides were incubated in heat-induced epitope retrieval (Tris-EDTA) buffer at pH 9.2 at 95°C for 30 minutes. Afterwards, the slides were blocked with 3% BSA (Bovine serum albumin) in Tris buffered saline with Tween (TBS-T) (TBS plus 0.1% Tween) for 45 minutes and were then stained with the antibody panel diluted in TBS with 0.1% Tween (Table S1) overnight at 4°C. After washing in TBS (Trizma and NaCl at pH 7.6) for 30 minutes, the samples were stained with a DNA intercalator (CellID™ Iridium Intercalator, diluted 1:100 in TBS). Finally, the slides were dipped into double distilled H2O and dried with pressured air. Slides were analysed by IMC over a period of 7 weeks. The samples were laser-ablated at 400 Hz using a Hyperion imaging mass cytometry system coupled to a Helios time-of-flight mass cytometer with a resolution of 1 μm. We tuned the machine between each TMA slide to ensure machine stability. Images of each slide were checked for their staining using histoCAT.75 We acquired data on 2,070 cores; two cores were acquired in two steps due to technical issues (88A_4,2 and 88A_5,2). 193Ir and 191Ir and Histone were used for segmentation but excluded from subsequent clustering analyses. Cadherin-6 was excluded from all analyses as it did not show any specific staining pattern. All antibodies were selected based on our previous study of CAF heterogeneity including a scRNA-seq dataset of NSCLC.37 There, we first defined different CAF types based on unsupervised clustering of transcriptomic data. Based on these results, we selected a subset of markers that allowed identification of these different CAF types using IMC. We evaluated their specificity by re-clustering our scRNA-seq data using only this subset of markers. We were able to identify all of our CAF types with our IMC marker subset as detected using all transcriptomic data available.37
Segmentation pipeline
The raw data was converted to tiff files and segmented using a pipeline that includes CellProfiler (3.1.9)46 and ilastik.45 Cells were segmented with ilastik using iridium for nuclear staining, and fibroblasts were reduced to their nuclear cores based on the iridium signal and treating SMA signal as background. We previously validated this approach for fibroblast segmentation in a study on a breast cancer cohort, where we showed that this segmentation revealed the same CAF types as detected with scRNA-seq as well as when using automated Mesmer based78 nuclear segmentation.37 Our previous analyses showed that this segmentation approach recapitulates CAF heterogeneity and allows us to investigate CAF type heterogeneity. Tumour-stroma masks were also generated with ilastik using SMA as the marker for stroma identification and panCK as the marker for tumour identification. The tumour-stroma masks were then used together with the single-cell masks to calculate the mean intensity of each marker per cell and to assign cells as either stroma or tumour based on spatial location. Compensation was done using the measured compensation slide that contained pure spots of each used metal-tagged antibody to correct for spill over between channels. The raw counts were then censored using a 99th-percentile cut-off and transformed using arc-sinh transformation with a cofactor of 1. For visualisation, normalised counts based on the arc-sinh transformed counts from 0 to 1 are shown. All analyses were carried out using the arc-sinh transformed values.
Cell phenotype clustering
All analyses were performed using R (version 4.1.2). All cells were first split into tumour and non-tumour cells using a Gaussian mixture model based on the mean cellular expression of panCK. This was done for each TMA block, and all tumour (n = 2,855,748) and non-tumour (n = 3,128,955) cells were merged. The non-tumour cells were subclustered using a combination of Rphenograph (Rphenoannoy) and FLOWSOM.47,77 Both the non-tumour and the tumour cell subsets were clustered again using all markers to guarantee purity of non-tumour cells from tumour cells. Tumour cells identified at later clustering stages were merged back with the previously defined tumour cells. The final dataset contained 2,979,712 tumour cells, 1,179,114 immune cells, 1,086,678 fibroblasts, 150,062 vessel cells (both blood and lymphatic endothelial cells), 588,888 undefined (other) cells; 148 cells that originated from the removed images are not included in the final dataset.
Non-tumour cells were clustered (Rphenograph, k = 30) and split into tumour, vessel, immune, and stromal cells. The tumour cells were merged into the tumour cell subset. Immune were clustered with all markers. The immune cells were then clustered using FLOWSOM (k_max = 40, som = 25) to exclude T cells. Cells that expressed CD20 and CD3 were integrated into the T cell subset. T cells were then clustered with Rphenograph (k = 20, cluster 17) first including CD20, which resulted in a fraction of 16,825 CD20+ B cells that were merged back into the immune cell subset. Subsequently, 391,422 T cells were clustered using only T cell markers (CD3, CD4, CD8, Ki-67, PD-1, TCF-1/7, IDO, FOXP3)) and grouped into CD4, CD4 T reg, IDO CD4, IDO CD8, Ki-67 CD8, Ki-67 CD4, TCF1/7 CD4, TCF1/7 CD8, PD-1 CD4, CD8. Immune cells excluding T cells were clustered (Rphenograph, k = 12) and categorised as myeloid cells (HLA-DR+/CD68+), neutrophils (MPO+/MMP9+), and B cells (CD20+).
Stromal cells were clustered over all markers, and 5,612 tumour and 16,641 immune cells were excluded. Clustering over all CAF and vessel markers including CD31, vWF, LYVE-1, and PNAd was used to subset 508,786 endothelial and lymphatic cells (Rphenograph, k = 20). These were clustered (using SMA, CD146, FAP, VCAM1, podoplanin, p75 / CD271, vimentin, PNAd, LYVE-1, CD140b, CD34, vWF/CD31, CXCL12, CCL21) to exclude 384,307 CAFs that were merged back into the CAF subset. Vessel cells (n = 150,062) were split into high endothelial venules (PNAd+/LYVE-1+/CCL21+), lymphatic endothelial cells (LYVE-1+ /CCL21+), and endothelial cells (CD146+/CD31+/vWF+/CCL21-). CAFs (n = 1,086,918) were over-clustered using FLOWSOM (k_max: 45, som = 40, markers displayed in heatmap, Figure 2A) and annotated based on marker expression as: mCAFs (FAP+/MMP11+/Collagen+), iCAFs (CD34+/CD248+), tCAFs (CD10+/CD73+), hypoxic tCAFs (CD10+/CAIX+), hypoxic CAFs (CAIX+), IFNCAFs (IDO+), vCAFs (CD146+/CD34-), dCAFs (Ki-67+), PDPN CAFs (PDPN+), and SMA CAFs (SMA+). Some differences were observed between the CAF types detected in this dataset and the CAF types identified in the breast cancer IMC analysis.37 In the breast cancer dataset, we had previously identified mCAFs as PDPNlow/collagen-fibronectinhigh/FAP+ cells that did not express CD10, CD34, or CD146. In that analysis we did not stain for MMP-11 due to the lack of an antibody. Now that we had access to an anti-MMP-11 antibody, we identified mCAFs (MMP11+) as well as Collagen CAFs (MMP-/Collagen+). In the original breast cancer publication these are likely grouped together as mCAFs. Due to the absence of CXCL13 in this panel, we did not identify CXCL13+ CAFs. CCL21+ CAFs were also absent, which is not surprising given their rarity and association with TLS. We also identified PDPN CAFs in this dataset. The final tumour cell subset (n = 2,979,716) was divided into hypoxic and non-hypoxic cells using a Gaussian mixture model based on panCK expression.
The final cell classification into tumour, stroma as well as cell categorisation into tumour, immune, CAF, vessel and other was evaluated by comparing the cell type distribution within the given tumour-stroma masks (Table S2; Figures S1B‒S1E). 95% of all tumour cells were found in the tumour compartment and over 80% of all CAFs were found in the stromal compartment. The remaining CAFs were mostly found at the edge of the tumour masks (within the first 10 μm, Table S3).
Quantification and statistical analysis
Data exclusion and subsetting
When analysing individual cell categories (e.g., CAFs only), the images with less than 100 cells (lowest 5-10%) were excluded from all CAF-only analyses, resulting in 1025 patients in total. Proportions were calculated per cell category and overall densities were calculated over the entire tumour core, if not otherwise indicated, as the area sum of the stromal and tumour masks. When analysing correlations between patients for all cell types, the same threshold (5-10%) was applied to exclude low cell number images, resulting in a total of 1070 patients. Two cores were available for 85% of the patients. We ran a paired t-test analysis comparing the distribution of all cell types between the two cores for each patient. Apart from hypoxic CAFs (p = 0.005717), the p-values for all paired cell type analyses were not significant (p > 0.05), suggesting that the different cell types come from the same overall distribution. For patients with two cores, proportions were thus calculated for the respective cell type category for both cores together. For density calculations, the densities were first calculated per core before the mean of the two cores was calculated, similar to the publication by Fischer et al.79
Analysis of matched cohorts
Matched control cohorts were generated from subsets of the dataset. For the comparison of neoadjuvant chemotherapy treated versus treatment naive patients were matched based on tumour type, stage, sex, grade. For the comparison of distant metastasis and the absence thereof, patients were matched by tumour type, TNM (T only), grade, absence of neoadjuvant treatment and sex. We do not have any information about the development of distant metastasis after diagnosis. Where possible, cell numbers per patient were also roughly matched if more than one control candidate was available.
Statistical analyses
The Mann-Whitney-U test was used for two-group comparisons and the Kruskal-Wallis test was used for multiple group comparisons. Differential abundance tests were carried out using the edgeR80 and diffcyt81 packages. We calculated Kaplan-Meier curves for survival analyses and tested differences between groups by using log rank test. In case the median survival difference was only significant for the respective CAF type in one tumour type (LUAD, LUSC) the respective tumour type is indicated in the plot. For all other analyses survival differences were significant in both tumour types and the analysis is shown for the whole cohort. Pearson correlation was used for evaluation of cell type proportions and densities. The Bonferroni post-hoc test was used in our analyses to account for multiple testing. The non-lasso regressed CoxPH models was corrected for tumour type and grade. Differential abundance and survival analyses were carried out for all patients pooled and split by tumour type (LUAD, LUSC) respectively. Non-significant results are not being shown.
Spatial analysis
We used the histoCAT neighbourhood analysis included in the imcRtools package.75,80 The maximal radius per cell was set to 20 μm, and we analysed the 15 nearest neighbours of each cell. The spicyR package was used to compare the cellular neighbours between patient groups stratified based on CAF density.52
Hypoxia patch detection analysis
In order to assess the abundance of endothelial cells as an orthogonal measure of hypoxia in CAIX-defined hypoxic versus normoxic areas, we used patch detection based on cells defined as hypoxic. A spatial graph using expansion with a threshold of 15 was calculated based on the coordinates of the centroids of each cell. We defined hypoxic and normoxic ‘patches’ using graph-based expansion from all hypoxic (CAIX+) cells (graph threshold: 15, expansion: 5), requiring a minimum of 10 hypoxic cells to define a patch. Note that these hypoxic patches could also contain normoxic (CAIX-) cells. All regions outside of hypoxic patches were then designated as normoxic patches.
Acknowledgments
B.B. was funded by two SNSF project grants (#310030_205007: Analysis of breast tumor ecosystem properties for precision medicine approaches and #316030_213512: Cellular-resolution high-performance mass spectrometric imaging of biological samples), an NIH grant (UC4 DK108132), the CRUK IMAXT Grand Challenge, and the European Research Council (ERC) under the European Union's Horizon 2020 Program under the ERC grant agreement no. 866074 (“Precision Motifs”). B.B. and L.C. were founded by an SNSF SINERGIA project grant (#177208: Defining the identity and differentiation pathways of the immune-stimulating fibroblastic tumor-stroma). We would like to thank the entire consortium of the SNSF SINERGIA grant for the fruitful discussions and especially the Ludewig lab for providing test sample tissues. The graphical abstract was created with BioRender (biorender.com).
Author contributions
L.C. and B.B. conceived the study. L.C. designed the experiments, developed the IMC antibody panel, generated the IMC dataset, analyzed all data, and wrote the manuscript with inputs from N.dS. and B.B. S.E. helped with the IMC antibody panel design and antibody testing. M.H., J.H.R., and H.M. provided the samples as well as the clinical pathological data and gave clinical input. M.H. specified the growth pattern of all LUAD patients. N.dS. supervised writing and revising the manuscript. B.B. supervised the work and provided input at all stages.
Declaration of interests
B.B. has co-founded Navignostics, a spin-off company of the University of Zurich, and is one of its shareholders and a board member.
Published: January 18, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ccell.2023.12.021.
Supplemental information
References
- 1.Sung H., Ferlay J., Siegel R.L., Laversanne M., Soerjomataram I., Jemal A., Bray F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA. Cancer J. Clin. 2021;71:209–249. doi: 10.3322/caac.21660. [DOI] [PubMed] [Google Scholar]
- 2.Schabath M.B., Cote M.L. Cancer progress and priorities: Lung cancer. Cancer Epidemiol. Biomarkers Prev. 2019;28:1563–1579. doi: 10.1158/1055-9965.EPI-19-0221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pikor L.A., Ramnarine V.R., Lam S., Lam W.L. Genetic alterations defining NSCLC subtypes and their therapeutic implications. Lung Cancer. 2013;82:179–189. doi: 10.1016/j.lungcan.2013.07.025. [DOI] [PubMed] [Google Scholar]
- 4.Garon E.B., Hellmann M.D., Rizvi N.A., Carcereny E., Leighl N.B., Ahn M.J., Eder J.P., Balmanoukian A.S., Aggarwal C., Horn L., et al. Five-year overall survival for patients with advanced non‒small-cell lung cancer treated with pembrolizumab: results from the phase I KEYNOTE-001 study. J. Clin. Oncol. 2019;37:2518–2527. doi: 10.1200/JCO.19.00934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Robert C. A decade of immune-checkpoint inhibitors in cancer therapy. Nat. Commun. 2020;11:3801. doi: 10.1038/s41467-020-17670-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sahai E., Astsaturov I., Cukierman E., DeNardo D.G., Egeblad M., Evans R.M., Fearon D., Greten F.R., Hingorani S.R., Hunter T., et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer. 2020;20:174–186. doi: 10.1038/s41568-019-0238-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hanahan D., Weinberg R.A. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 8.Turley S.J., Cremasco V., Astarita J.L. Immunological hallmarks of stromal cells in the tumour microenvironment. Nat. Rev. Immunol. 2015;15:669–682. doi: 10.1038/nri3902. [DOI] [PubMed] [Google Scholar]
- 9.Smyth M.J., Ngiow S.F., Ribas A., Teng M.W.L. Combination cancer immunotherapies tailored to the tumour microenvironment. Nat. Rev. Clin. Oncol. 2016;13:143–158. doi: 10.1038/nrclinonc.2015.209. [DOI] [PubMed] [Google Scholar]
- 10.Altorki N.K., Markowitz G.J., Gao D., Port J.L., Saxena A., Stiles B., McGraw T., Mittal V. The lung microenvironment: an important regulator of tumour growth and metastasis. Nat. Rev. Cancer. 2019;19:9–31. doi: 10.1038/s41568-018-0081-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Finak G., Bertos N., Pepin F., Sadekova S., Souleimanova M., Zhao H., Chen H., Omeroglu G., Meterissian S., Omeroglu A., et al. Stromal gene expression predicts clinical outcome in breast cancer. Nat. Med. 2008;14:518–527. doi: 10.1038/nm1764. [DOI] [PubMed] [Google Scholar]
- 12.Bruni D., Angell H.K., Galon J. The immune contexture and Immunoscore in cancer prognosis and therapeutic efficacy. Nat. Rev. Cancer. 2020;20:662–680. doi: 10.1038/s41568-020-0285-7. [DOI] [PubMed] [Google Scholar]
- 13.Su S., Chen J., Yao H., Liu J., Yu S., Lao L., Wang M., Luo M., Xing Y., Chen F., et al. CD10+GPR77+ Cancer-Associated Fibroblasts Promote Cancer Formation and Chemoresistance by Sustaining Cancer Stemness. Cell. 2018;172:841–856.e16. doi: 10.1016/j.cell.2018.01.009. [DOI] [PubMed] [Google Scholar]
- 14.Krishnamurty A.T., Shyer J.A., Thai M., Gandham V., Buechler M.B., Yang Y.A., Pradhan R.N., Wang A.W., Sanchez P.L., Qu Y., et al. LRRC15+ myofibroblasts dictate the stromal setpoint to suppress tumour immunity. Nature. 2022;611:148–154. doi: 10.1038/s41586-022-05272-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Genova C., Dellepiane C., Carrega P., Sommariva S., Ferlazzo G., Pronzato P., Gangemi R., Filaci G., Coco S., Croce M. Therapeutic Implications of Tumor Microenvironment in Lung Cancer: Focus on Immune Checkpoint Blockade. Front. Immunol. 2021;12:799455. doi: 10.3389/fimmu.2021.799455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Caligiuri G., Tuveson D.A. Activated fibroblasts in cancer: Perspectives and challenges. Cancer Cell. 2023;41:434–449. doi: 10.1016/j.ccell.2023.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grout J.A., Sirven P., Leader A.M., Maskey S., Hector E., Puisieux I., Steffan F., Cheng E., Tung N., Maurin M., et al. Spatial Positioning and Matrix Programs of Cancer-Associated Fibroblasts Promote T-cell Exclusion in Human Lung Tumors. Cancer Discov. 2022;12:2606–2625. doi: 10.1158/2159-8290.CD-21-1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ogawa Y., Masugi Y., Abe T., Yamazaki K., Ueno A., Fujii-Nishimura Y., Hori S., Yagi H., Abe Y., Kitago M., Sakamoto M. Three distinct stroma types in human pancreatic cancer identified by image analysis of fibroblast subpopulations and collagen. Clin. Cancer Res. 2021;27:107–119. doi: 10.1158/1078-0432.CCR-20-2298. [DOI] [PubMed] [Google Scholar]
- 19.Özdemir B.C., Pentcheva-Hoang T., Carstens J.L., Zheng X., Wu C.C., Simpson T.R., Laklai H., Sugimoto H., Kahlert C., Novitskiy S.V., et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell. 2014;25:719–734. doi: 10.1016/j.ccr.2014.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rhim A.D., Oberstein P.E., Thomas D.H., Mirek E.T., Palermo C.F., Sastra S.A., Dekleva E.N., Saunders T., Becerra C.P., Tattersall I.W., et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell. 2014;25:735–747. doi: 10.1016/j.ccr.2014.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bhattacharjee S., Hamberger F., Ravichandra A., Miller M., Nair A., Affo S., Filliol A., Chin L., Savage T.M., Yin D., et al. Tumor restriction by type I collagen opposes tumor-promoting effects of cancer-associated fibroblasts. J. Clin. Invest. 2021;131 doi: 10.1172/JCI146987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen Y., Kim J., Yang S., Wang H., Wu C.J., Sugimoto H., LeBleu V.S., Kalluri R. Type I collagen deletion in αSMA+ myofibroblasts augments immune suppression and accelerates progression of pancreatic cancer. Cancer Cell. 2021;39:548–565.e6. doi: 10.1016/j.ccell.2021.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen Y., McAndrews K.M., Kalluri R. Clinical and therapeutic relevance of cancer-associated fibroblasts. Nat. Rev. Clin. Oncol. 2021;18:792–804. doi: 10.1038/s41571-021-00546-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Valkenburg K.C., De Groot A.E., Pienta K.J. Targeting the tumour stroma to improve cancer therapy. Nat. Rev. Clin. Oncol. 2018;15:366–381. doi: 10.1038/s41571-018-0007-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bughda R., Dimou P., D’Souza R.R., Klampatsa A. Fibroblast Activation Protein (FAP)-Targeted CAR-T Cells: Launching an Attack on Tumor Stroma. ImmunoTargets Ther. 2021;10:313–323. doi: 10.2147/ITT.S291767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brünker P., Wartha K., Friess T., Grau-Richards S., Waldhauer I., Koller C.F., Weiser B., Majety M., Runza V., Niu H., et al. RG7386, a novel tetravalent FAP-DR5 antibody, effectively triggers FAP-dependent, avidity-driven DR5 hyperclustering and tumor cell apoptosis. Mol. Cancer Ther. 2016;15:946–957. doi: 10.1158/1535-7163.MCT-15-0647. [DOI] [PubMed] [Google Scholar]
- 27.Chen Z., Zhou L., Liu L., Hou Y., Xiong M., Yang Y., Hu J., Chen K. Single-cell RNA sequencing highlights the role of inflammatory cancer-associated fibroblasts in bladder urothelial carcinoma. Nat. Commun. 2020;11:5077. doi: 10.1038/s41467-020-18916-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Puram S.V., Tirosh I., Parikh A.S., Patel A.P., Yizhak K., Gillespie S., Rodman C., Luo C.L., Mroz E.A., Emerick K.S., et al. Single-cell transcriptomic analysis of primary and metastatic tumor ecosystems in head and neck cancer. Cell. 2017;171:1611–1624.e24. doi: 10.1016/j.cell.2017.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee H.-O., Hong Y., Etlioglu H.E., Cho Y.B., Pomella V., Van den Bosch B., Vanhecke J., Verbandt S., Hong H., Min J.W., et al. Lineage-dependent gene expression programs influence the immune landscape of colorectal cancer. Nat. Genet. 2020;52:594–603. doi: 10.1038/s41588-020-0636-z. [DOI] [PubMed] [Google Scholar]
- 30.Lambrechts D., Wauters E., Boeckx B., Aibar S., Nittner D., Burton O., Bassez A., Decaluwé H., Pircher A., Van den Eynde K., et al. Phenotype molding of stromal cells in the lung tumor microenvironment. Nat. Med. 2018;24:1277–1289. doi: 10.1038/s41591-018-0096-5. [DOI] [PubMed] [Google Scholar]
- 31.Tietscher S., Wagner J., Anzeneder T., Langwieder C., Rees M., Sobottka B., de Souza N., Bodenmiller B. A comprehensive single-cell map of T cell exhaustion-associated immune environments in human breast cancer. Nat. Commun. 2023;14:98. doi: 10.1038/s41467-022-35238-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Friedman G., Levi-Galibov O., David E., Bornstein C., Giladi A., Dadiani M., Mayo A., Halperin C., Pevsner-Fischer M., Lavon H., et al. Cancer-associated fibroblast compositions change with breast cancer progression linking the ratio of S100A4+ and PDPN+ CAFs to clinical outcome. Nat. Cancer. 2020;1:692–708. doi: 10.1038/s43018-020-0082-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Costa A., Kieffer Y., Scholer-Dahirel A., Pelon F., Bourachot B., Cardon M., Sirven P., Magagna I., Fuhrmann L., Bernard C., et al. Fibroblast Heterogeneity and Immunosuppressive Environment in Human Breast Cancer. Cancer Cell. 2018;33:463–479.e10. doi: 10.1016/j.ccell.2018.01.011. [DOI] [PubMed] [Google Scholar]
- 34.Kieffer Y., Hocine H.R., Gentric G., Pelon F., Bernard C., Bourachot B., Lameiras S., Albergante L., Bonneau C., Guyard A., et al. Single-cell analysis reveals fibroblast clusters linked to immunotherapy resistance in cancer. Cancer Discov. 2020;10:1330–1351. doi: 10.1158/2159-8290.CD-19-1384. [DOI] [PubMed] [Google Scholar]
- 35.Buechler M.B., Pradhan R.N., Krishnamurty A.T., Cox C., Calviello A.K., Wang A.W., Yang Y.A., Tam L., Caothien R., Roose-Girma M., et al. Cross-tissue organization of the fibroblast lineage. Nature. 2021;593:575–579. doi: 10.1038/s41586-021-03549-5. [DOI] [PubMed] [Google Scholar]
- 36.Bartoschek M., Oskolkov N., Bocci M., Lövrot J., Larsson C., Sommarin M., Madsen C.D., Lindgren D., Pekar G., Karlsson G., et al. Spatially and functionally distinct subclasses of breast cancer-associated fibroblasts revealed by single cell RNA sequencing. Nat. Commun. 2018;9:5150. doi: 10.1038/s41467-018-07582-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cords L., Tietscher S., Anzeneder T., Langwieder C., Rees M., de Souza N., Bodenmiller B. Cancer-associated fibroblast classification in single-cell and spatial proteomics data. Nat. Commun. 2023;14:4294. doi: 10.1038/s41467-023-39762-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Elyada E., Bolisetty M., Laise P., Flynn W.F., Courtois E.T., Burkhart R.A., Teinor J.A., Belleau P., Biffi G., Lucito M.S., et al. Cross-species single-cell analysis of pancreatic ductal adenocarcinoma reveals antigen-presenting cancer-associated fibroblasts. Cancer Discov. 2019;9:1102–1123. doi: 10.1158/2159-8290.CD-19-0094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Öhlund D., Handly-Santana A., Biffi G., Elyada E., Almeida A.S., Ponz-Sarvise M., Corbo V., Oni T.E., Hearn S.A., Lee E.J., et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017;214:579–596. doi: 10.1084/jem.20162024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wu S.Z., Roden D.L., Wang C., Holliday H., Harvey K., Cazet A.S., Murphy K.J., Pereira B., Al-Eryani G., Bartonicek N., et al. Stromal cell diversity associated with immune evasion in human triple-negative breast cancer. EMBO J. 2020;39:e104063. doi: 10.15252/embj.2019104063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Giesen C., Wang H.A.O., Schapiro D., Zivanovic N., Jacobs A., Hattendorf B., Schüffler P.J., Grolimund D., Buhmann J.M., Brandt S., et al. Highly multiplexed imaging of tumor tissues with subcellular resolution by mass cytometry. Nat. Methods. 2014;11:417–422. doi: 10.1038/nmeth.2869. [DOI] [PubMed] [Google Scholar]
- 42.Gachechiladze M., Škarda J., Kolek V., Grygárková I., Langová K., Bouchal J., Kolář Z., Baty F., Stahel R., Weder W., et al. Prognostic and predictive value of loss of nuclear RAD51 immunoreactivity in resected non-small cell lung cancer patients. Lung Cancer. 2017;105:31–38. doi: 10.1016/j.lungcan.2017.01.009. [DOI] [PubMed] [Google Scholar]
- 43.Tischler V., Pfeifer M., Hausladen S., Schirmer U., Bonde A.K., Kristiansen G., Sos M.L., Weder W., Moch H., Altevogt P., Soltermann A. L1CAM protein expression is associated with poor prognosis in non-small cell lung cancer. Mol. Cancer. 2011;10:127. doi: 10.1186/1476-4598-10-127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Soltermann A., Tischler V., Arbogast S., Braun J., Probst-Hensch N., Weder W., Moch H., Kristiansen G. Prognostic significance of epithelial-mesenchymal and mesenchymal- epithelial transition protein expression in non-small cell lung cancer. Clin. Cancer Res. 2008;14:7430–7437. doi: 10.1158/1078-0432.CCR-08-0935. [DOI] [PubMed] [Google Scholar]
- 45.Berg S., Kutra D., Kroeger T., Straehle C.N., Kausler B.X., Haubold C., Schiegg M., Ales J., Beier T., Rudy M., et al. Ilastik: interactive machine learning for (bio) image analysis. Nat. Methods. 2019;16:1226–1232. doi: 10.1038/s41592-019-0582-9. [DOI] [PubMed] [Google Scholar]
- 46.McQuin C., Goodman A., Chernyshev V., Kamentsky L., Cimini B.A., Karhohs K.W., Doan M., Ding L., Rafelski S.M., Thirstrup D., et al. CellProfiler 3.0: Next-generation image processing for biology. PLoS Biol. 2018;16 doi: 10.1371/journal.pbio.2005970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Van Gassen S., Callebaut B., Van Helden M.J., Lambrecht B.N., Demeester P., Dhaene T., Saeys Y. FlowSOM: Using self-organizing maps for visualization and interpretation of cytometry data. Cytometry A. 2015;87:636–645. doi: 10.1002/cyto.a.22625. [DOI] [PubMed] [Google Scholar]
- 48.Schwörer S., Cimino F.V., Ros M., Tsanov K.M., Ng C., Lowe S.W., Carmona-Fontaine C., Thompson C.B. Hypoxia Potentiates the Inflammatory Fibroblast Phenotype Promoted by Pancreatic Cancer Cell-Derived Cytokines. Cancer Res. 2023;83:1596–1610. doi: 10.1158/0008-5472.CAN-22-2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Garcia Garcia C.J., Huang Y., Fuentes N.R., Turner M.C., Monberg M.E., Lin D., Nguyen N.D., Fujimoto T.N., Zhao J., Lee J.J., et al. Stromal HIF2 Regulates Immune Suppression in the Pancreatic Cancer Microenvironment. Gastroenterology. 2022;162:2018–2031. doi: 10.1053/j.gastro.2022.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lodyga M., Hinz B. TGF-β1 - A truly transforming growth factor in fibrosis and immunity. Semin. Cell Dev. Biol. 2020;101:123–139. doi: 10.1016/j.semcdb.2019.12.010. [DOI] [PubMed] [Google Scholar]
- 51.Cremasco V., Astarita J.L., Grauel A.L., Keerthivasan S., MacIsaac K., Woodruff M.C., Wu M., Spel L., Santoro S., Amoozgar Z., et al. FAP delineates heterogeneous and functionally divergent stromal cells in immune-excluded breast tumors. Cancer Immunol. Res. 2018;6:1472–1485. doi: 10.1158/2326-6066.CIR-18-0098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Canete N.P., Iyengar S.S., Ormerod J.T., Baharlou H., Harman A.N., Patrick E. spicyR: spatial analysis of in situ cytometry data in R. Bioinformatics. 2022;38:3099–3105. doi: 10.1093/bioinformatics/btac268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bejarano L., Jordāo M.J.C., Joyce J.A. Therapeutic Targeting of the Tumor Microenvironment. Cancer Discov. 2021;11:933–959. doi: 10.1158/2159-8290.CD-20-1808. [DOI] [PubMed] [Google Scholar]
- 54.Zhang M., Yang H., Wan L., Wang Z., Wang H., Ge C., Liu Y., Hao Y., Zhang D., Shi G., et al. Single-cell transcriptomic architecture and intercellular crosstalk of human intrahepatic cholangiocarcinoma. J. Hepatol. 2020;73:1118–1130. doi: 10.1016/j.jhep.2020.05.039. [DOI] [PubMed] [Google Scholar]
- 55.Yu Z., Zhang J., Zhang Q., Wei S., Shi R., Zhao R., An L., Grose R., Feng D., Wang H. Single-cell sequencing reveals the heterogeneity and intratumoral crosstalk in human endometrial cancer. Cell Prolif. 2022;55:e13249. doi: 10.1111/cpr.13249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Foster D.S., Januszyk M., Delitto D., Yost K.E., Griffin M., Guo J., Guardino N., Delitto A.E., Chinta M., Burcham A.R., et al. Multiomic analysis reveals conservation of cancer-associated fibroblast phenotypes across species and tissue of origin. Cancer Cell. 2022;40:1392–1406.e7. doi: 10.1016/j.ccell.2022.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Werba G., Weissinger D., Kawaler E.A., Zhao E., Kalfakakou D., Dhara S., Wang L., Lim H.B., Oh G., Jing X., et al. Single-cell RNA sequencing reveals the effects of chemotherapy on human pancreatic adenocarcinoma and its tumor microenvironment. Nat. Commun. 2023;14:3912. doi: 10.1038/s41467-023-39680-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Keller P.J., Arendt L.M., Skibinski A., Logvinenko T., Klebba I., Dong S., Smith A.E., Prat A., Perou C.M., Gilmore H., et al. Defining the cellular precursors to human breast cancer. Proc. Natl. Acad. Sci. USA. 2012;109:2772–2777. doi: 10.1073/pnas.1017626108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cheon H., Wang Y., Wightman S.M., Jackson M.W., Stark G.R. How cancer cells make and respond to interferon-I. Trends Cancer. 2023;9:83–92. doi: 10.1016/j.trecan.2022.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Benveniste E.N., Qin H. Type I interferons as anti-inflammatory mediators. Sci. STKE. 2007;2007:pe70. doi: 10.1126/stke.4162007pe70. [DOI] [PubMed] [Google Scholar]
- 61.Al Bakir M., Huebner A., Martínez-Ruiz C., Grigoriadis K., Watkins T.B.K., Pich O., Moore D.A., Veeriah S., Ward S., Laycock J., et al. The evolution of non-small cell lung cancer metastases in TRACERx. Nature. 2023;616:534–542. doi: 10.1038/s41586-023-05729-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Stagg J., Divisekera U., McLaughlin N., Sharkey J., Pommey S., Denoyer D., Dwyer K.M., Smyth M.J. Anti-CD73 antibody therapy inhibits breast tumor growth and metastasis. Proc. Natl. Acad. Sci. USA. 2010;107:1547–1552. doi: 10.1073/pnas.0908801107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Turcotte M., Spring K., Pommey S., Chouinard G., Cousineau I., George J., Chen G.M., Gendoo D.M.A., Haibe-Kains B., Karn T., et al. CD73 is associated with poor prognosis in high-grade serous ovarian cancer. Cancer Res. 2015;75:4494–4503. doi: 10.1158/0008-5472.CAN-14-3569. [DOI] [PubMed] [Google Scholar]
- 64.Wang L., Zhou X., Zhou T., Ma D., Chen S., Zhi X., Yin L., Shao Z., Ou Z., Zhou P. Ecto-5′-nucleotidase promotes invasion, migration and adhesion of human breast cancer cells. J. Cancer Res. Clin. Oncol. 2008;134:365–372. doi: 10.1007/s00432-007-0292-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vijayan D., Young A., Teng M.W.L., Smyth M.J. Targeting immunosuppressive adenosine in cancer. Nat. Rev. Cancer. 2017;17:709–724. doi: 10.1038/nrc.2017.86. [DOI] [PubMed] [Google Scholar]
- 66.Fu Y., Liu S., Yin S., Niu W., Xiong W., Tan M., Li G., Zhou M. The reverse Warburg effect is likely to be an Achilles’ heel of cancer that can be exploited for cancer therapy. Oncotarget. 2017;8:57813–57825. doi: 10.18632/oncotarget.18175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pavlides S., Whitaker-Menezes D., Castello-Cros R., Flomenberg N., Witkiewicz A.K., Frank P.G., Casimiro M.C., Wang C., Fortina P., Addya S., et al. The reverse Warburg effect: Aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle. 2009;8:3984–4001. doi: 10.4161/cc.8.23.10238. [DOI] [PubMed] [Google Scholar]
- 68.Dragica J., Song M., Wang L. Vol. 8. BioMed Central Ltd unless otherwise stated. Part of Springer Nature; 2020. (Roles of IFN-γ in Tumor Progression and Regression a Review Biomarker Research). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Uyttenhove C., Pilotte L., Théate I., Stroobant V., Colau D., Parmentier N., Boon T., Van den Eynde B.J. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat. Med. 2003;9:1269–1274. doi: 10.1038/nm934. [DOI] [PubMed] [Google Scholar]
- 70.Zimmermann J.A., Hettiaratchi M.H., McDevitt T.C. Enhanced Immunosuppression of T Cells by Sustained Presentation of Bioactive Interferon-γ Within Three-Dimensional Mesenchymal Stem Cell Constructs. Stem Cells Transl. Med. 2017;6:223–237. doi: 10.5966/sctm.2016-0044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hu H., Piotrowska Z., Hare P.J., Chen H., Mulvey H.E., Mayfield A., Noeen S., Kattermann K., Greenberg M., Williams A., et al. Three subtypes of lung cancer fibroblasts define distinct therapeutic paradigms. Cancer Cell. 2021;39:1531–1547.e10. doi: 10.1016/j.ccell.2021.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yang D., Liu J., Qian H., Zhuang Q. Q. Cancer-associated fibroblasts: from basic science to anticancer therapy. Exp. Mol. Med. 2023;55:1322–1332. doi: 10.1038/s12276-023-01013-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dominguez C.X., Müller S., Keerthivasan S., Koeppen H., Hung J., Gierke S., Breart B., Foreman O., Bainbridge T.W., Castiglioni A., et al. Single-cell RNA sequencing reveals stromal evolution into LRRC15+ myofibroblasts as a determinant of patient response to cancer immunotherapy. Cancer Discov. 2020;10:232–253. doi: 10.1158/2159-8290.CD-19-0644. [DOI] [PubMed] [Google Scholar]
- 74.Bondjers C., Kalén M., Hellström M., Scheidl S.J., Abramsson A., Renner O., Lindahl P., Cho H., Kehrl J., Betsholtz C. Transcription profiling of platelet-derived growth factor-B-deficient mouse embryos identifies RGS5 as a novel marker for pericytes and vascular smooth muscle cells. Am. J. Pathol. 2003;162:721–729. doi: 10.1016/S0002-9440(10)63868-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Schapiro D., Jackson H.W., Raghuraman S., Fischer J.R., Zanotelli V.R.T., Schulz D., Giesen C., Catena R., Varga Z., Bodenmiller B. histoCAT: analysis of cell phenotypes and interactions in multiplex image cytometry data. Nat. Methods. 2017;14:873–876. doi: 10.1038/nmeth.4391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Windhager J., Zanotelli V.R.T., Schulz D., Meyer L., Daniel M., Bodenmiller B., Eling N. An end-to-end workflow for multiplexed image processing and analysis. Nat. Protoc. 2023;18:3565–3613. doi: 10.1038/s41596-023-00881-0. [DOI] [PubMed] [Google Scholar]
- 77.Levine J.H., Simonds E.F., Bendall S.C., Davis K.L., Amir E.a.D., Tadmor M.D., Litvin O., Fienberg H.G., Jager A., Zunder E.R., et al. Data-Driven Phenotypic Dissection of AML Reveals Progenitor-like Cells that Correlate with Prognosis. Cell. 2015;162:184–197. doi: 10.1016/j.cell.2015.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Greenwald N.F., Miller G., Moen E., Kong A., Kagel A., Dougherty T., Fullaway C.C., McIntosh B.J., Leow K.X., Schwartz M.S., et al. Whole-cell segmentation of tissue images with human-level performance using large-scale data annotation and deep learning. Nat. Biotechnol. 2022;40:555–565. doi: 10.1038/s41587-021-01094-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Jackson H.W., Fischer J.R., Zanotelli V.R.T., Ali H.R., Mechera R., Soysal S.D., Moch H., Muenst S., Varga Z., Weber W.P., Bodenmiller B. The single-cell pathology landscape of breast cancer. Nature. 2020;578:615–620. doi: 10.1038/s41586-019-1876-x. [DOI] [PubMed] [Google Scholar]
- 80.Robinson M.D., McCarthy D.J., Smyth G.K. A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Weber L.M., Nowicka M., Soneson C., Robinson M.D. Differential discovery in high-dimensional cytometry via high-resolution clustering. Commun. Biol. 2019;2:183. doi: 10.1038/s42003-019-0415-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data have been deposited at zenodo under https://doi.org/10.5281/zenodo.7961844 and are publicly available as of the date of publication. DOIs are listed in the key resources table.
All original code has been deposited at GitHub and is publicly available as of the date of publication. DOIs are listed in the key resources table.