

Abstract
Synthetic chemistry plays an indispensable role in drug discovery, contributing to hit compounds identification, lead compounds optimization, candidate drugs preparation, and so on. As Nobel Prize laureate James Black emphasized, “the most fruitful basis for the discovery of a new drug is to start with an old drug”1. Late-stage modification or functionalization of drugs, natural products and bioactive compounds have garnered significant interest due to its ability to introduce diverse elements into bioactive compounds promptly. Such modifications alter the chemical space and physiochemical properties of these compounds, ultimately influencing their potency and druggability. To enrich a toolbox of chemical modification methods for drug discovery, this review focuses on the incorporation of halogen, oxygen, and nitrogen—the ubiquitous elements in pharmacophore components of the marketed drugs—through late-stage modification in recent two decades, and discusses the state and challenges faced in these fields. We also emphasize that increasing cooperation between chemists and pharmacists may be conducive to the rapid discovery of new activities of the functionalized molecules. Ultimately, we hope this review would serve as a valuable resource, facilitating the application of late-stage modification in the construction of novel molecules and inspiring innovative concepts for designing and building new drugs.
Key words: Late-stage modification, Drug space, Halogenation, Oxygenation, Nitrogenation, Synthetic chemistry
Graphical abstract
This article reviews the recent strategies applied to late-stage modification of bioactive compounds and tries to provide a toolbox for efficient molecule generation in drug discovery.

1. Introduction
The development of drug discovery has been accelerated by the emergence of new strategies and technologies, such as combinatorial chemistry, large compound libraries, high-throughput screening, cheminformatics, omics, and artificial intelligence2, 3, 4, 5, 6. However, organic synthesis remains a rate-limiting factor in drug discovery, despite decades of groundbreaking research in academia7, 8, 9. Recently, late-stage modification (LSM), or called late-stage functionalization (LSF) of drugs, natural products and bioactive compounds has attracted more and more attention from pharmaceutical chemists. As Nobel Prize winner James Black highlighted, “the most fruitful basis for the discovery of a new drug is to start with an old drug”1. In the field of pharmaceutical science, LSM enables the rapid generation of efficient tool molecules for the investigation of structure–activity relationship (SAR) and optimization of druggability by modifying numerous analogs of bioactive compounds and natural products without resorting to de novo synthesis10,11.
LSM of bioactive compounds offers the opportunity to introduce new effects that were either designed or previously unanticipated. This approach can modulate the physicochemical properties, such as solubility, acidity coefficient (pKa), and oil-water partition coefficient (logP), by incorporating different polarity groups into bioactive compounds. Moreover, adjusting the shape and size of substituent groups can optimize their fit within target cavities. Halogen, oxygen, and nitrogen are common elements in pharmacophore groups and are frequently found in marketed drugs. Particularly, the groups containing halogen, oxygen, and nitrogen elements can form halogen and hydrogen bonds with amino acid residues, and may enhance binding affinity and retention time with targets12. Analysis of US Food and Drug Administration (FDA) approved drugs from 2015 to 2020, reveals that nitrogen or oxygen elements are present in most drugs, while nearly half of them also contain halogen elements13. In this review, we will expound on the importance of introducing halogen, oxygen, and nitrogen elements here individually, emphasizing their potential for influencing potency and druggability with target molecules. While other functionalization of bioactive compounds are the same important in LSM, such as methylation14, 15, 16, trifluoromethylation17, 18, 19, and so on, some excellent articles have summarized this field well.
For instance, metabolic instability mediated by P450 enzymes poses a significant challenge in drug discovery12. Thus, blocking susceptible metabolic sites of bioactive compounds with fluorine atoms has emerged as an effective strategy in the early stage of drug discovery20, 21, 22. Ibuprofen is a famous non-steroidal anti-inflammatory drug, that works as an inhibitor of cyclooxygenases. When introducing a fluorine atom on the active benzyl site, its metabolic stability will be increased. The clearance was decreased from 19 to 12 μg/(min·mg) protein in human microsome and from 71 to 39 μg/(min·mg) protein in rat microsome (Fig. 1)23. Terfenadine, a non-sedating antihistamine withdrawn due to high cardiotoxicity, underwent a remarkable transformation. Its carboxylic acid metabolite, Fexofenadine24,25, was proven safe after oxygen atom incorporation, paving the way for its emergence as a second-generation antihistamine (Fig. 1)26,27. There was a typical case to show the application of introducing nitrogen elements in drug discovery. Pioglitazone is a strong and selective PPARγ agonist, which was approved for diabetes by FDA in 199928, and applied to treat other metabolic diseases in clinical later29,30. In 2013, Baran introduced azide group with a linker into this drug, and then the new derivative could occur click reaction and be applied in other situations (Fig. 1)31.
Figure 1.

Influences after introducing ‘X’, ‘O’, and ‘N’ into bioactive compounds. Therefore, LSM has been a convenient and powerful strategy for incorporating ‘X’, ‘O’, and ‘N’ into bioactive compounds. This review would explore the role of LSM in the generation of new molecules from the insight of pharmaceutical science. Specifically, late-stage halogenation, oxygenation, and nitrogenation strategies will be expounded along the logic of conversion of functional groups.
2. Halogenation of bioactive compounds
The inclusion of halogen atoms in a molecule can significantly alter its properties. The varying sizes and electronegativities of different halogens result in distinct changes in the physicochemical characteristics of bioactive compounds32. Introducing halogen atoms would raise the logP and reduce the solubility of benzene (Table 1), consequently influencing the pharmacokinetic properties and binding affinity of halogenated bioactive compounds. For example, Deschloroketamine, which is an analog of Ketamine without chlorine, has a lower logP than Fluoroketamine, Ketamine, and Bromoketamine33. And when a halogen atom was removed in a non-nucleoside reverse transcriptase inhibitor (NNRTI) presented by Anderson, its solubility was enhanced obviously34.
Table 1.
Selected properties of substrate and product in halogenation.
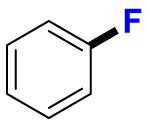
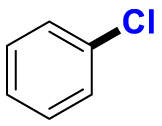
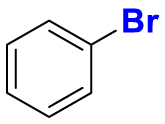
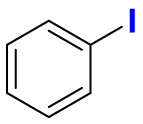
2.1. Fluorination of bioactive compounds
The incorporation of fluorine atoms into bioactive compounds has been a prominent strategy in drug discovery, with several approved drugs containing fluorine atoms. Such as Tivicay40,41, Verzenio42, Isentress43,44, and so on, contain at least one fluorine atom among the top 200 small molecule drugs sold45, 46, 47. This part explores the effects of introducing fluorine elements on the characteristics and potency of these molecules. The primary goal was to prevent in vivo metabolism by incorporating fluorine atoms into drug candidates47,48. The similarity in size between fluorine (atomic radius 0.42 Å), hydrogen (atomic radius 0.53 Å), and oxygen atoms (atomic radius 0.48 Å) allows for seamless replacement49, blocking active sites without significant steric effects50,51. Moreover, the introduction of fluorine atoms can predictably enhance potency, primarily through the formation of new hydrogen bonds and the electron-withdrawing effect52,53. These interactions improve binding affinity with targets and enhance the π‒π stacking effect of benzene rings52,53. Additionally, the aryl C–F moiety serves as a hydrophobic isostere, mimicking the pyridone carbonyl moiety and improving bioavailability54.
The incorporation of fluorine atoms into bioactive compounds also has significant effects on their physicochemical properties, such as lipophilicity and membrane permeability55,56. Moreover, different moieties containing fluorine atoms can influence membrane permeability in distinct ways, with aryl, vinyl, and alkyl fluorine playing varying roles48,57. While the proximal fluorine atoms might change the logP of molecules containing amine groups, due to the interaction of fluorine and the nearby N–H58,59.
Therefore, the strategic introduction of fluorine atoms into bioactive compounds in late-stage by rational design may increase the potency and improve the pharmacokinetic properties, thus facilitating drug discovery and development. Furthermore, trifluoromethylation is also a fantastic strategy for introducing fluorine atoms into bioactive compounds, there were several good reviews described this field from different perspectives17, 18, 19.
2.1.1. Conversion of C–H bond to C–F bond
The development of C–H bond functionalization based on transition-metal catalysis has opened up new possibilities for the fluorination of aromatic bioactive molecules. This review discusses notable examples of C–H bond fluorination strategies reported in recent two decades. Wang and coworkers successfully fluorinated an azo-macrocyclic compound through aryl‒Cu(III) complex intermediate (Scheme 1a)60. Daugulis reported site-selective sp3 C–H arylation directed by 8-animoquinoline and picolinic acid auxiliaries in 200561. In 2013, this strategy was developed for o-fluorination of benzoic acid and some heterocyclic carboxylic acids with high selectivity of monofluorination and diflourination in different conditions62. Picolinamides, a PARP inhibitor, could be fluorinated with moderate yield under this condition (Scheme 1b)62. Xu and coworkers demonstrated an o-fluorination strategy for phenols, where the directing group pyridine could be removed after modification of molecules. Intriguingly, the pesticide Diflufenican and bioactive 2-phenoxyl nicotinic acid derivatives could be regioselective and late-stage modified smoothly, even with the presence of other potential sites (Scheme 1c)63. In 2018, Xu group presented a nitrate-promoted Pd-catalysis method for the fluorination of three drug derivatives (Scheme 1d)64. Although the presented examples are limited due to the existence of directing groups that restrict substrate structures, they highlight the potential of C–H bond fluorination in the synthesis of bioactive compounds.
Scheme 1.

Directed fluorination of arenes.
In addition to directed fluorination strategies, non-directed methods have also been explored for the modification of aromatic bioactive molecules. In 2013, Hartwig reported that the 2-fluorination of a broad range of substituted pyridines occurs with AgF2, and some medicinally relevant compounds can also be fluorinated using this method (Scheme 2a)65. In mechanism, the Ag atom is firstly coordinated with the nitrogen atom of pyridine, then the [Ag]-F bond is added to the π system of pyridine, and a second equivalent of AgF2 is abstracted from the hydrogen-atom to form product (Scheme 2a)65. Subsequently, they added nucleophilic reagents and bases into this system to further convert to 2-fluoropyridines66. Notably, Ritter's group has made a breakthrough in direct aromatic C–H fluorination with a broad substrate scope. The aromatic rings with all kinds of electron-donating groups and a few electron-withdrawing groups could be fluorinated by designed palladium catalyst, including some bioactive compounds. This reaction would be a practical tool for LSM, while overcoming its limitation of regioselectivity in the future (Scheme 2b)67. Li and Nicewicz utilized organic photoredox catalysis to introduce 18F atoms into aromatic molecules with excellent substrate tolerance, and the steric effects might be the main factor to influence site-selectivity. Excitingly, numerous bioactive compounds were fluorinated through this method within a short time, and might be a great supplement for PET (Scheme 2c)68. The main challenge in non-directed strategies is achieving selectivity, as current approaches rely on the electronic properties of substituents on aromatic rings, but precise control remains elusive.
Scheme 2.

Non-directed fluorination of arenes.
Although directed aryl fluorination has made significant progress in recent twenty years, challenges remain for chemists due to the strong metal–fluorine bond hindering reductive elimination69, 70, 71 and the limited substrate scope imposed by directing groups. Conversely, non-directed fluorination of arenes, particularly heteroaromatics, often lacks regioselectivity. Despite these challenges, many commercially available small molecule drugs feature aryl fluoride moieties, such as Invega72, Lynparza73, and Erleada74. The development of improved aryl fluorination methods could enable the late-stage fluorination of more bioactive compounds, leading to their rapid progression as potential drug candidates.
No-aromatic fluorination has also been developed by chemists over the past two decades. In 2012, Groves and coworkers reported the aliphatic C–H bond fluorination strategy catalyzed by manganese porphyrin for the first time. The secondary carbon could be fluorinated more easily than primary and tertiary carbons slightly, and a kind of androgen was selectively fluorinated on A-ring under this condition (Scheme 3a)75. Tan and coworkers presented a new strategy of fluorination for unactivated secondary alkyl C–H bonds via photocatalysis, and (+)-Sclareolide, a terpenoid from plants, was subjected to this fluorination method (Scheme 3b)76. Lectka demonstrated a photocatalytic approach that selectively fluorinates peptides containing phenylalanine-like residues77, and in another work the bioactive polycyclic terpenoid derivatives with α,β-unsaturated ketones could be regioselectively fluorinated via 1,5-hydrogen atom transfer process (Scheme 3c)78. In 2018, Xu, Luo and coworkers used acetone oxime ether as a removable directing group to achieve the fluorination of the β-C position of hydroxyl groups, facilitating the conversion of steroid molecules into the desired products (Scheme 3d)79. In 2022, Alexanian and coworkers developed an aliphatic C–H functionalization strategy, including fluorination, through radical chain transfer mechanism. Many kinds of C–H bonds could be functionalized in mild to good yields, and some bioactive compounds could be fluorinated with good regioselectivity (Scheme 3e)80.
Scheme 3.

Direct fluorination of alkanes.
As we know, the methylene C–H bonds of alkyl bioactive molecules are often indistinguishable to fluorination reagents, unless the directing group takes effect. In addition, chirality is also an important factor in drugs. Enhancing the stereoselective fluorination of aliphatic C–H bonds, particularly those in aliphatic heterocycles, would greatly facilitate the application of late-stage fluorination in drug discovery. This would enable the selective introduction of fluorine atoms while preserving the desired stereochemistry of bioactive compounds.
While common aliphatic C–H bonds are challenging to modify, the benzyl and allyl C–H bonds fluorination was developed leading to the emergence of several new approaches. Doyle successfully achieved allyl fluorination, demonstrating its applicability to a steroid derivative (Scheme 4a)81. Additionally, Doyle and coworkers developed another general strategy for benzyl C–H fluorination through photocatalysis, enabling smooth modification of various bioactive compounds and drug derivatives (Scheme 4b)82. Britton revealed a metal-free fluorination method for benzyl C–H bonds, offering potential applications in late-stage functionalization of bioactive molecules and positron emission tomography (PET) (Scheme 4c)83. Benzyl and allyl C–H bonds are appropriate fluorination sites in bioactive compounds, potentially improving their pharmacokinetic properties by blocking potential metabolic positions.
Scheme 4.

Fluorination of benzyl and allyl C–H bonds.
2.1.2. Conversion of C‒X to C–F bonds
Besides the conversion of C–H bonds to C–F bonds, some works on C‒X (X = B, OTf, Cl, Br, I, Si, S, Sn) to C–F bonds have been published in the last two decades. Ritter and coworkers fluorinated aryl stannanes with silver catalyst using commercial fluorination reagent Select-F84. By first converting hydroxyl groups to tin groups, they successfully transferred several pharmaceutically active molecules from hydroxyl to fluoride derivatives (Scheme 5a)84. Ritter also developed a silver-catalyzed fluorinate ion method for fluorination of many stannanes derived from complex small molecules, including quinine, taxol and rifamycin S (Scheme 5b)85. Then, they combined photoredox catalysis with transition metal catalysis to enable site-selectivity fluorination of aryl sulfonium salts, which came from C–H bond thianthrenation of arenes. This approach allowed for late-stage modification of numerous drugs and their derivatives (Scheme 5c)86,87.
Scheme 5.

Conversion of C‒X to C–F.
In 2009, Buchwald used palladium-catalyst to convert aryl triflates to aryl fluorides (Scheme 5d)88. This reaction could proceed at room temperature attributed to their new ligand AlPhos of the system89. Furthermore, they expanded their substrates to aryl halides (Br and I) in 2014, and the vascular disorder drug Nicergoline could be fluorinated at late-stage to provide an analog (Scheme 5e)90.
Hartwig and coworkers have contributed to this field as well. They smoothly converted aryl trisiloxanes to aryl fluorides mediated by copper (Scheme 5f)91,92. Aggarwal and coworkers reported the transformation from alkyl boronates to alkyl fluorides in 201593. Scott, Sanford and coworkers completed radio fluorination of pharmaceutically relevant scaffolds via organoboronate intermediates94.
Nicewicz and Li cooperated to accomplish the photocatalytic method of converting C‒X bonds to C–F bonds, where X can be Cl, Br, I, NO2 and OTf groups. This approach allows for the late-stage fluorination of potential bioactive compounds with a wide substrate scope (Scheme 5g)95. Unlike C–H fluorination, the conversion of C‒X bonds to C–F bonds is distinct because the ‘X’ group occupies specific positions within the molecules. This position fixed ‘X’ group enables the introduction of fluorine atoms without the need for challenging C–H activations.
2.1.3. Conversion of C–O to C–F bonds
The development of deoxyfluorination reagents has provided new strategies for converting C–OH or C‒OR into C–F bonds, enabling late-stage modification of drug molecules96. Various methods have been established and applied to enhance the properties of pharmaceutical compounds. In 2008, Fasan combined CYP450-catalyzed oxygenation with deoxyfluorination to improve the blood‒brain barrier (BBB) crossing potential97 and metabolic stability23 of Ibuprofen methyl ester (Scheme 6a). In 2011, Ritter group created a new deoxyfluorination reagent PhenoFluor and accomplished the direct transformation from phenol to aryl fluorides98. Subsequently, this commercial reagent was applied to late-stage fluorination of alcohol, and so many drugs performed well under this condition (Scheme 6b)99. In 2015, they further upgraded the reagent to PhenoFluorMix, which facilitated the deoxyfluorination of phenols100. In 2019, Ritter and coworkers utilized PhenoFluorMix to deoxyfluorinate sterides favorably101. Nicewicz and Li developed photocatalysis for late-stage deoxyfluorination and used it for 18F isotope labeling of Estrone (Scheme 6c)102. Moreover, many scientists have made contributions to this field103, 104, 105, 106, 107. Deoxyfluorination has emerged as an effective approach for introducing fluorine atoms into bioactive molecules using readily available alcohols and phenols as starting materials.
Scheme 6.

Conversion of C–O to C–F.
2.1.4. Other conversions
Besides the methods mentioned above, several other functional group transformations have been developed for late-stage fluorination of bioactive compounds. Decarboxylative fluorination has been well developed over past decades, and Li group used silver-catalyst and Selectfluor to decarboxylative fluorinate dehydrolithocholic acid derivatives (Scheme 7a)108. In 2018, Renaud and coworkers designed a new generation of radical fluorination agents called NFASs (Scheme 7b)109, which have lower N–F bond dissociation energies and can be operated under milder conditions compared to the first (F2) and second (NFSI and Selectfluor) generation agents. NFASs could hydroxyfluoride the carbon–carbon double bonds under 60 °C, and a derivative of steroids underwent well. Additionally, more reactions transfer different functional groups to fluorine through other mechanisms (Scheme 7c)110, 111, 112. These advancements signify a promising future for easier late-stage fluorination of bioactive compounds, as an increasing number of novel fluorinating methods continue to emerge.
Scheme 7.

Other fluorination strategies.
Bioactive compounds labeled with 18F could be used as tracers for non-invasive visualization and quantification of molecular interactions, receptor binding, metabolism, and other dynamic processes in living organisms. PET is a significate application of fluorination using 18F tracers, and so many works have been reported over the past decades113, 114, 115, 116, 117.
Despite the development of numerous late-stage fluorination strategies, several challenges in this field remain unresolved. One major challenge in nucleophilic fluorination is the weak nucleophilicity of fluorine anions in the presence of hydrogen bond donors, which are common pharmacophore features in bioactive molecules. Oppositely, unproductive side reactions might occur without hydrogen bond donors due to the fluorine's basicity. Electrophilic fluorination using reagents like NFSI and Select-F offers high reactivity but comes with drawbacks such as high cost and poor atom economy. Overcoming this obstacle through the development of new general transition-metal catalytic methods would greatly benefit late-stage fluorination strategies.
For medicinal chemistry, selective late-stage fluorination of bioactive molecules is essential for rational design of SAR and optimization of pharmacokinetics. However, achieving regioselectivity without directing groups presents a significant challenge that hinders these goals. The use of directing groups, on the other hand, restricts the substrate scope, posing an additional obstacle. While various fluorination methods through functional group conversion have been developed, they often require pre-functionalization and cannot accurately modify bioactive compounds. Consequently, there remains a lack of practical and widely applicable late-stage fluorination reactions for medicinal chemistry. Furthermore, the purification of C–H fluorinated products from starting materials continues to be problematic45,118. Addressing these challenges and advancing fluorination strategies would greatly benefit medicinal chemistry, enabling more practical and efficient late-stage modifications.
2.2. Chlorination of bioactive compounds
The introduction of chlorine atoms into bioactive compounds, as exemplified by Xarelto119,120, Jardiance121, and Vraylar122, among the top 200 marketed small molecule drugs, can significantly alter their bioactivity spectrum and pharmacokinetic properties. The larger atomic radius of chlorine (atomic radius 0.79 Å) compared to hydrogen (atomic radius 0.53 Å) can result in different steric effects49, potentially influencing the binding affinity of bioactive compounds. Furthermore, heavy halogens like chlorine, bromine, and iodine can form a unique region called the σ-hole in the positive outer region along the covalent bond123. This leads to the formation of halogen bonds124, a non-covalent interaction, with nucleophilic groups such as oxygen, sp3-hybridized nitrogen, aromatic rings, or sulfur125, 126, 127. These interactions can significantly improve binding affinity128. Additionally, the introduction of chlorine atoms can slightly increase molecular lipophilicity and membrane permeability (Table 1)36,38,39,129,130, thus impacting the absorption and distribution of bioactive compounds. Moreover, chlorine incorporation serves as an effective method to block metabolic sites, similar to fluorine incorporation131.
2.2.1. Chlorination of arenes
Chlorination strategies of arenes have been well developed and applied to late-stage modification of bioactive compounds for drug discovery. In 2014, Baran group invented a new chlorinating reagent called “Palau'chlor”, which is practical and reactive with broad substrate scope and high regioselectivity. Clotrimazole could be selectively chlorinated with good yield132. In 2016, Trauner group used N-chlorosuccinimide (NCS) as the chlorine source, and Pd(OAc)2 catalyzed o-dichlorination of azobenzene. Interestingly, the introduction of chlorine atoms prevents the switch of benzene rings, resulting in the formation of photoswitches (Scheme 8a)133. Regioselective chlorination of phenol derivatives was achieved in the presence of pyridine directing groups catalyzed by palladium, demonstrating the versatility of this methodology. Similar conditions were successfully applied to diflufenican and estrone derivatives134. In 2019, Jiao group developed an electrocatalytic strategy for arenes chlorination with 1,2-dichloroethane as a chlorine source, and bioactive compounds were chlorinated successfully (Scheme 8b)135. Next year, they reported DMSO-catalyzed chlorination of bioactive molecules using NCS, demonstrating the compatibility of this mild reaction condition with numerous natural products and drugs (Scheme 8c)136. Notably, the chlorination of gemfibrozil, a lipid-lowering drug, was shown to decrease its solubility in water137, and potentially enhance its antiandrogen activity138. In 2021, they also developed alternative system in which DMSO was replaced by nitroxides appeared and expanded the substrate scope of the past work139. Taran also announced the selective chlorination of iminosydnones for the fast release of pro-drugs under mild conditions140.
Scheme 8.

Chlorination of arenes.
Anilines are the common structure in pharmaceutical compounds, a general chlorinating method for different substituted anilines was revealed by Huang, Feng, Chen and coworkers using photo-organo co-catalysis with good selectivity (Scheme 8d)141. McNally and Paton reported that the C–H bond of aromatic bioactive compounds could be transferred to triaryl phosphorus group, and then converted to chlorine. This method enables the chlorination of pyridines and other drugs containing pyridine building blocks (Scheme 8e)142. Functional groups transformation was also applied in this field, Cornella group converted aminoheterocycles into heterocyclic chlorides (Scheme 8f)143. The anticancer drug Dabrafenib could become a proteolysis-targeting chimeras (PROTAC) binder after this transformation144. Recently, Studer and coworkers make a breakthrough in meta-functionalization of pyridines, quinolines, and isoquinolines. They proposed a mechanism that dearomatization of the substrates occurred after reacting with dimethyl acetylenedicarboxylate (DMAD) and methyl pyruvate (MP), followed by rearomatization and functionalization. Among the process, meta-halogenation of bioactive compounds could be completed with medium to good yields (Scheme 8g)145.
2.2.2. C(sp3)‒H bond chlorination
There have been some efficient works on the C(sp3)‒H chlorination in recent years. Enzyme engineering is a hot research topic for chlorination. Buller engineered a halogenase WelO5∗ for chlorination of martinelline-derived fragment and soraphens selectively (Scheme 9a)146,147. Meanwhile, Hoebenreich engineered Wi-WelO15 to chlorinate a kind of alkaloid148. Selective chlorination of tertiary C(sp3)‒H bond was achieved using a combination of copper and hypervalent iodine reagents in mild condition. The different substrates with various types of groups could be tolerated in this system with good selectivity. And many bioactive compounds were suitably chlorinated in excellent yields (Scheme 9b)149. A method of photocatalytic C(sp3)‒H chlorination was put forward, and 5α-cholestane could bear the condition well150. A new copper catalytic strategy could be used to chlorinate benzyl, allylic and γ-carbonyl hydrogen, and several drugs and natural products were chlorinated in medium to excellent yields (Scheme 9c)151.
Scheme 9.

Chlorination of alkanes.
While the chlorination strategy has been well-established, the concept of late-stage modification in chlorination remains relatively unexplored. To the best of our knowledge, there are quite a few drugs containing chlorine atoms, and the positions and quantities of substituents are key determinants of their biological activity. Hence, it is imperative to develop selective chlorination methods that allow precise control over regioselectivity. Moreover, expanding the substrate scope of these strategies, particularly for chlorination of electron-deficient aromatic rings, remains an unsolved challenge152, 153, 154, 155. This necessitates the exploration of new chlorination reactions that exhibit both regioselectivity and broad substrate compatibility, which can significantly contribute to the rapid advancement of medicinal chemistry and drug discovery.
2.3. Bromination of bioactive compounds
Opsumit156, one of the top 200 small molecule drugs for pulmonary arterial hypertension, contains two bromine atoms in different aromatic rings. The introduction of bromine atoms may have a similar impact on bioactive compounds with the introduction of chlorine atoms to a certain extent. The larger radius of bromine atoms (0.94 Å)49 contributes to enhanced hydrophobic effects and van der Waals interactions, potentially improving binding affinity in bioactive compounds. However, it may also introduce steric clash effects and reduce binding affinity. The strategies developed are usually suitable for chlorination, bromination, and even iodination, so we would just expound these works in a certain chapter without repetition.
2.3.1. Bromination of arenes
Bromination of bioactive molecules often accompanies chlorination due to the suitability of developed methods for both halogenations, and occasionally iodination. In 2015, Jiao group reported a general method for oxidative bromination and iodination of electron-rich arenes and heteroarenes, however, this strategy could not complete the bromination and iodination of electron-deficient aromatic rings. Even though, many drugs could be brominated under mild oxidation reagent DMSO and relatively low temperature (Scheme 10a)157. Lou group used this strategy to brominate Marchantin C, and obtained a series of analogs with anticancer activity 2–6 fold higher158. In 2022, Jiao and coworkers made a breakthrough in bromination of electron-deficient aromatic rings. They reported an efficient catalyst m-NBSA, and it could promote electrophilic halogenation of arenes with electron-withdrawing substituents. And the bromination of lots of bioactive molecules proceeded under this condition smoothly (Scheme 10b)152. Panda developed a DMSO promoting bromination of many arenes including bioactive compounds159. Furthermore, Koley brominated aminoquinolines including drug analogs without metal, oxidant or additive under a mild condition160. Correa created a bromination method with click reaction at ortho-position of phenylacetylene, potentially applicable in biological studies (Scheme 10c)161.
Scheme 10.

Bromination of arenes.
Electrochemistry was also used for the bromination of drug molecules by Liu, Rivera and coworkers in 2017162. Lewis pioneered the directed evolution of flavin-dependent halogenases, providing a powerful tool for late-stage bromination of bioactive compounds. And different halogenases could be applied to brominate bioactive compounds selectively (Scheme 10d)163. Chen reported a tungstate-catalyzed biomimetic oxidative halogenation of arenes, enabling the bromination of select bioactive molecules164. Porphyrins and their analogs are significant for organisms in different parts. Shinokubo selectively realized the bromination of porphyrins and analogs on different aromatic rings165. Thibaudeau revealed a strategy for the bromination of aniline building blocks166. In 2021, Sharma group developed a selective para-bromination method for the N-substituted site of the aromatic ring, which has been successfully applied in drug modification167. Lately, McNally group developed an excellent strategy for halogenation of the 3-position of pyridines using a ring-opening and ring-closing process via an NTf-Zincke imine intermediate. Many bioactive molecules can undergo late-stage modifications through this reaction (Scheme 10e)168.
2.3.2. Other bromination
There are a few strategies developed for alkyl bromination, offering opportunities to modify bioactive compounds. In 2019, Jiang group reported that visible-light-promoted oxidative halogenation of alkynes could get dibrominated alkanes. This method could provide several late-stage dibrominated drug derivatives (Scheme 11)169.
Scheme 11.

Other bromination reactions.
The bromination methods used for bioactive compounds modification faced similar problems with chlorination, including regioselectivity and substrate scope. While bromine is a superior halogen bond donor compared to chlorine, its introduction can affect the effectiveness of ligands. Nevertheless, a significant drawback of introducing bromine atoms is the reduction in solubility for molecules (Table 1), which may prevent bioactive compounds from becoming drug candidates.
3. Oxygenation of bioactive compounds
The introduction of oxygen functionality into bioactive compounds can lead to various changes in their properties. For physicochemical properties, incorporation of different groups containing oxygen atoms generally decreases the compound's logP and improves its solubility, except for the carbonyl group (Table 2). Moreover, the binding affinity between the compound and its target may increase due to the formation of new interactions, such as hydrogen bonds and salt bridges. In some drugs, the removal of oxygen-containing groups may lead to the loss of their biological activities170, 171, 172. Furthermore, introduction of oxygen atoms may alter the pharmacokinetic properties of bioactive compounds, for example, introduction of a hydroxyl group may accelerate the metabolic rate of the drug173.
Table 2.
Selected properties of substrate and product in oxygenation.
| Substrate | Product | Substrate/Product |
|
|---|---|---|---|
| LogPa | Sw (mg/L)a | ||
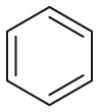 |
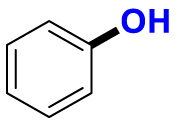 |
2.13/1.64 | 1789/76,500 |
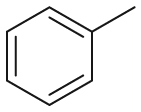 |
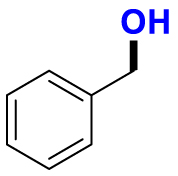 |
2.52/1.02 | 517/38,000 |
|
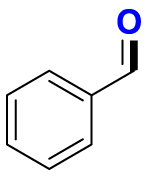 |
2.52/5 | 517/410 |
|
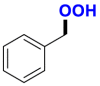 |
2.52/1.45c | 517/19,000b |
 |
 |
2.95/1.59 | 300/2000b |
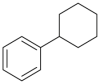 |
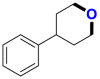 |
4.69c/2.49c | 2.9b/810b |
Data from references38,39,174,175; logP: octan-1-ol/H2O partition coefficient; Sw: aqueous solubility at 25 °C, mg/L.
Predicted Sw at pH 7, 25 °C by Scifinder (https://origin-scifinder.cas.org/scifinder/view/scifinder/scifinderExplore.jsf).
Predicted logP at 25 °C by Scifinder (https://origin-scifinder.cas.org/scifinder/view/scifinder/scifinderExplore.jsf).
Indeed, oxygenation reactions have been extensively developed, encompassing various transformations such as C–H bond oxygenation176, 177, 178, oxygen insertion179, conversion of functional groups to hydroxyls180, 181, 182, 183, 184, 185, hydroperoxidation186, 187, 188 and oxygenation through C–C/C=C bond cleavage189, 190, 191, some of which are applied to late-stage modification of bioactive molecules.
3.1. Hydroxylation of bioactive compounds
Hydroxyl groups frequently appear in marketed drugs, and they play crucial roles in the pharmacodynamic and pharmacokinetic properties of drugs192. These groups not only contribute to the formation of hydrogen bonds, which can increase the biological activity of drugs193,194, but they can also enhance the metabolism of bioactive compounds by serving as prodrugs195,196. Moreover, hydroxyl groups can also be a synthetic handle for the rapid synthesis of new analogs197, as well as a link position for PROTAC, among others.
3.1.1. C–H bond hydroxylation of arenes
Phenols are significant building blocks in drugs, and introducing hydroxyl in bioactive molecules is a convenient way to seek a molecule with better activity176. As the well-sold drug Advair198, which treats asthma, contains phenolic hydroxyl groups.
In 2012, Rao and coworkers announced that Pd-catalyzed carbonyl directed o-hydroxylation of arenes, demonstrating good selectivity and moderate yields in the hydroxylation of Ibuprofen ethyl ester199. They later replaced the Pd catalyst with Ru or Rh catalysts in a similar system, successfully hydroxylating Fenofibrate under this condition (Scheme 12a)200. Siegel developed a metal-free C–H bond oxidation strategy for the preparation of tocopherol and its derivatives, as well as late-stage functionalization of natural product clovanemagnolol precursor201. Moreover, Jiao group reported oxime ether directed C–H bond hydroxylation of arenes, and a nonsteroidal anti-inflammatory drug Zaltoprofen could be modified under this condition (Scheme 12b)202. Ritter group reported a mild method whereby bioactive compounds could be converted to corresponding phenols via methanesulfonate intermediates203. Ritter and coworkers revealed another strategy for hydroxylation of arenes through aryl sulfonium salts intermediates, and the substrates were hydroxylated at electron-rich positions. Many drugs could be late-stage hydroxylated well in mild condition (Scheme 12c)204. Through the same aryl sulfonium salts intermediates, Patureau uses UV-light and TEMPO to hydroxylate arenes205.
Scheme 12.

Hydroxylation of arenes.
In addition, Yu and coworkers developed a palladium-catalyzed hydroxylation method with molecular oxygen as an oxygen source. This method enabled the smooth modification of drugs and peptides with excellent site-selectivity (Scheme 12d)206. Han group reported an arene C–H hydroxylation method using an iron catalyst. This transformation from arenes to phenols exhibited remarkable selectivity even in the absence of directing groups (Scheme 12e)207. Recently, Correa revealed a Ru-catalyzed method by which many Tyr-containing peptides could be hydroxylated208. This innovative approach provides a means to introduce hydroxyl groups selectively into peptide structures.
3.1.2. C(sp3)‒H bond hydroxylation
There were a number of strategies for late-stage C(sp3)‒H hydroxylation of molecules. In 2009, Sherman group engineered P450 mono-oxygenase with an amino-sugar-derived anchoring group to achieve the hydroxylation of many complex molecules209. In 2012, Fasan and coworkers achieved the selective hydroxylation of artemisinin via modified P450 catalysis210. These P450 engineering techniques have been applied to develop methods for the late-stage hydroxylation of bioactive compounds (Scheme 13a)211, 212, 213, 214, 215. Furthermore, Lei developed a site-selective and metal-free aliphatic C–H hydroxylation method of cholesterol in total synthesis216. In 2007, White group reported an iron-catalysis method for hydroxylation of aliphatic C–H bond in which bioactive molecules could be hydroxylated217,218. They also developed a series of oxidative diversification methods for amino acids and peptides catalyzed by iron, including hydroxylation219. In 2019, they achieved chemoselective tertiary C–H hydroxylation using Mn(PDP)/chloroacetic acid catalyst for late-stage functionalization (Scheme 13b)220. Stoltz disclosed functionalization of natural product core cyanthiwigin, with hydroxylation occurring at positions C12 and C15 under different conditions respectively (Scheme 13c)221. Ritter group reported a selective benzyl hydroxylation method similar to their previous work for arenes, tolerating different groups and complex molecules (Scheme 13d)222. Notably, hydroxylation of celecoxib led to the formation of its metabolite with lower activity223,224. Baran revealed an electrochemical C–H oxidation strategy, the selectivity of this method could be controlled by using different condition. And hydroxylation of some bioactive compounds could bear this condition (Scheme 13e)225.
Scheme 13.

Hydroxylation of alkanes.
3.1.3. Other hydroxylation reactions
There were some late-stage hydroxylation strategies of bioactive compounds through functional group transformation. Maloney, Fier and coworkers reported two approaches that converted aromatic halides to phenols under mild condition (Scheme 14a)226,227. Shi group transferred aryl borides to phenols with trichloroacetonitrile as an activator under blue LED, and steride derivatives underwent this condition228. Another work of converting aryl borides to phenols was reported by Maurya, and many bioactive molecules could be late-stage hydroxylated (Scheme 14b)229. Jiao and coworkers used DMSO as an oxygen source to transfer simple organobromides or olefins to bromohydrins, and the hydroxybromination of (+)-δ-tocopherol derivative underwent to produce the corresponding product (Scheme 14c)230. They also reported another DMSO involving a unique transformation introducing hydroxyl into cyclohexanones with aromatization. A cholesterol analog was smoothly converted to the corresponding catechol231.
Scheme 14.

Other hydroxylation reactions.
The precise introduction of hydroxyl groups into bioactive compounds remains a formidable challenge due to the structural similarity between methylene C–H and methyl C–H232. However, the position of hydroxyl groups is critical for investigating the SAR and druggability of bioactive compounds in medicinal chemistry and drug discovery. Therefore, the accurate prediction of hydroxylation sites in bioactive molecules holds significant significance. The achievement of selectivity in late-stage hydroxylation reactions would mark a breakthrough, especially in the context of bioactive compounds containing aryl rings or aliphatic chains.
3.2. Carbonylation of bioactive compounds
Aldehyde groups are well-known structural alerts in pharmaceutical science due to their high reactivity and rapid metabolism. However, it is noteworthy that certain marketed drugs, including Streptomycin and Spiramycin233,234, contain aldehyde groups, suggesting that their presence can be safely designed into bioactive compounds235. Other carbonyl groups, such as ketones, esters, and amides, predominantly appear as linkers or skeletons within bioactive molecules.
3.2.1. C(sp3)‒H bond oxygenation
C(sp3)‒H bond oxygenation is another way to introduce oxygen element into bioactive compounds. Although achieving selective C–H bond oxidation remains a challenge, significant progress has been made in recent years. In 2013, White group developed an electrophilic, bulky catalyst Fe(S,S)-PDP, and used it to promote oxidation of methylenes selectively236. Then, they revealed another oxidation method for methylenes catalyzed by manganese which is coordinated by similar ligands previously used with moderate to good chemo-selectivity (Scheme 15a)237. Bryliakov and colleagues applied this catalyst in the C–H oxidation of (−)-ambroxide238. Recently, Li group utilized this type of manganese catalyst to complete highly selective benzyl oxidation with a wide substrate scope239. In 2016, Maes developed an aerobic oxidation method using iron or copper catalysts for the oxidation of (aryl) (heteroaryl)methanes, and the natural product Papaverine could be modified to corresponding ketone under this condition240. Talbot and Burley developed a metal-free method that used iodine as oxidant agent to transfer methylenes near nitrogen atoms to carbonyl groups, and several industrially relevant drug scaffolds could be selectively oxidized (Scheme 15b)241. Zhao reported an oxidation reaction of tertiary amines at two positions with PIDA/I2 system, and Obscurine worked well under this condition242. In 2019, a graphitic carbon nitride based heterogeneous photocatalyst was used to facilitate methylenes oxidizing, and Corydaline, Indoprofen, and Indobufen could be generated using this method243. In the same year, Han and coworkers developed a significant protocol, in which the toluene motif could be selectively converted to benzaldehyde, and many bioactive compounds containing toluene structure were oxidized to corresponding oxygenation products smoothly (Scheme 15c)244. Then in 2020, Chand and coworkers revealed another strategy to selectively oxidize C(sp3)‒H bonds to carbonyls by molybdenum catalysis245. Costas, Stefano, Olivo and coworkers developed a strategy for predicting reaction sites and late-stage oxidizing bioactive molecules (Scheme 15d)246. Barham applied flow chemistry to accomplish the oxidation of N–CH3 commonly found in various bioactive compounds, expanding the synthetic possibilities for these molecules (Scheme 15e)247. Similar to hydroxylation, there is still a lack of universal regioselective strategies for conversion of C(sp3)‒H to carbonyl groups.
Scheme 15.

C(sp3)‒H bonds oxidation of bioactive compounds.
3.3. Peroxidation of bioactive compounds
Peroxidation represents an alternative approach for incorporating oxygen elements into small molecules. While its utilization in the modification of bioactive compounds remains limited, it bears significance in certain cases. Notably, Artemisinin, a renowned drug employed in malaria treatment, features a peroxide group as its key pharmacophore. The breakdown of the peroxide bridge and the production of hydroperoxides may be one of the explanations for its antimalarial activity248, 249, 250. Meanwhile, many works have been focused on the development of antimalarial drugs containing peroxide groups251, 252, 253.
In 2006, Iskra group converted ketones to gem-dihydroperoxides under catalysis of iodine, and androstane-3,17-dione could be transferred to corresponding product with good yield (Scheme 16a)254. Su and coworkers revealed a singlet oxygen-mediated strategy for selective C(sp3)‒H hydroperoxidation (Scheme 16b)255. In 2020, Xing and coworkers employed photocatalysis for the hydroperoxidation of benzylic C(sp3)‒H bonds. This method was applied to the late-stage modification of celecoxib, an anti-inflammatory drug, resulting in the desired product (Scheme 16c)256.
Scheme 16.

Peroxidation of bioactive compounds.
The significance of the peroxide group in antimalarial drugs has been well-established, and its application will continue to expand to other diseases with the development of synthetic chemistry and pharmaceutical science. However, the construction of peroxide bridges in late-stage modifications remains a challenge for chemists.
3.4. Oxygenation with C–C/C=C bond cleavage
Carbon–carbon bonds cleavage is a suitable procedure to introduce oxygen atoms. Beller and coworkers realized a manganese and cobalt co-catalyzed the C(sp3)‒C(sp3) bond oxygenation under high pressure. Linezolid, a kind of prescription antibiotic, was successfully converted into open-ring oxidative product (Scheme 17a)257. Han group presented an iron-catalyzed oxidative C–C bond cleavage strategy for converting allylarenes to aryl aldehydes. This method enabled the smooth modification of various bioactive compounds containing the corresponding structures (Scheme 17b)258. Jiao group broke C=C double bonds to introduce oxygen into small molecules to form cyclic imides. This method could be applied to synthesize succinimide-containing medicines and drug analogs (Scheme 17c)259. In 2021, Zografos revealed a work about epoxidation of alkenes by introducing molecular oxygen. And a number of bioactive molecules could be late-stage modified to epoxides with medium to excellent yields (Scheme 17d)260.
Scheme 17.

Oxygenation through carbon–carbon/carbon = carbon bonds cleavage.
An aliphatic ring containing one or two oxygen atoms is common in marketed drugs, such as Lexapro261 and Cialis262,263. Changing the carbon atom to oxygen atom selectively in late-stage will be more convenient than de novo synthesis using the substrate containing the moiety. While there have been remarkable advancements in oxygenation methods involving C–C/C=C bond cleavage, certain deficiencies still exist, especially regarding regioselective C–C/C=C bond cleavage in complex bioactive molecules. In this context, skeleton editing has emerged as a valuable approach for C–C/C=C bond cleavage and is expected to become a practical tool in this field.
4. Nitrogenation of bioactive compounds
Of the versatile role of nitrogen elements among drugs, 182 drugs contain at least one nitrogen atom among the top 200 small molecule drugs by retail sales in 2021264. Nitrogen can appear in various forms, including primary amines, amides, N-heterocycles, and so on, which are common pharmacophore groups in bioactive compounds. Late-stage nitrogenation offers a means to modify the structure and physicochemical properties of bioactive compounds, which in turn can have a substantial impact on their pharmacodynamics and pharmacokinetics. For example, the introduction of amino groups into the benzene ring reduces logP and improves water solubility, while construction triazole in phenylacetylene does the opposite (Table 3). These structural and physicochemical changes are of great importance in medicinal derivatization. Leonori and coworkers applied their late-stage C–H amination method in microscale parallel reactor, and efficiently enriched derivative library of natural products and peptides265. Inspired by adequate combination of virtual screening and large-scale convergent synthesis266, it can be speculated that tighter collaboration between chemists and pharmacists will stimulate the potential of late-stage modification into hit/lead exploration. Late-stage nitrogenation serves not only in the investigation of SAR and structural modification but also in optimizing druggability, as well as the development of novel molecular probes/antibody conjugated drugs (ADC)/drug-delivery conjugates/PROTACs. Some potential applications for in situ proteome profiling have been proposed before267. Hence, installing such nitrogen-bearing functional moieties to bioactive compounds in a minimally impacted way is of great importance. Considering the numerous synthetic approaches in these filed, the latest advances focusing on late-stage nitrogenation of bioactive compounds, related skeleton-editing methodologies and specific N,O-difunctionalization approaches are discussed below.
Table 3.
Selected properties of substrate and product in nitrogenation.
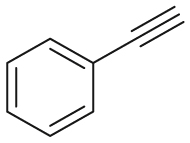
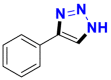
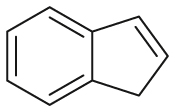
| Substrate | Product | Substrate/Product |
|
|---|---|---|---|
| LogPa | Sw (mg/L)a | ||
|
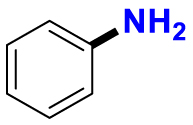 |
2.13/0.90 | 1789/34,100 |
 |
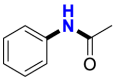 |
1.58/1.00 | 5500/4900b |
 |
 |
2.70c/2.90c | 430b/200b |
 |
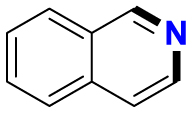 |
3.04c/2.08 | 280b/8100b |
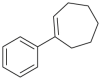 |
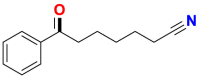 |
4.85c/2.16c | 1.6b/180b |
Data from references36,268, 269, 270, 271, 272, 273; logP: octan-1-ol/H2O partition coefficient; Sw: aqueous solubility at 25 °C, mg/L.
Predicted Sw at pH 7, 25 °C by Scifinder (https://origin-scifinder.cas.org/scifinder/view/scifinder/scifinderExplore.jsf).
Predicted logP at 25 °C by Scifinder (https://origin-scifinder.cas.org/scifinder/view/scifinder/scifinderExplore.jsf).
4.1. Primary amination of bioactive compounds
Despite the common increase of hydrogen bond donors, introduction of primary amines brings about different properties changes counting on substrate types274. Compared with parent arenes, primary amination will augment electronic density of conjugated ligands, and enhance their π‒π/cation‒π interaction with electron-deficient target pockets275. As for alkyl substrates, primary amination provides a potential positive electronic center276, which will benefit receptor-ligand electrostatic interactions as well as compound dissolution.
As the ubiquity of primary amines across bioactive compounds, their synthesis method counts a great deal. As for anilines, electrophilic aromatic substitution (SEAr) is one of the most practical, atom-economic reactions in functionalizing arenes. The traditional SEAr approach in aniline synthesis involves the nitration of arenes followed by reduction. But that seems unsuitable for late-stage application due to its poor functional group tolerance, bad reliance on protective group manipulations, and highly acidic reaction conditions.
Transition-metal-catalyzed arenes C–H bond activation is an important approach for direct amination. Yu and Dai initially explored an oxazoline-based directing group within copper-mediated late-stage heterocyclic functionalization, and then combined it with oximes in primary amination of telmisartan (Scheme 18a)277,278. Furthermore, Chang and Baik reported the usage of aqueous ammonia as a nitrogen source in copper-catalyzed aryl C–H bonds amination through a disproportionation pathway279. After simple transformation of the oxazoline-derived directing group, Bexarotene and Probenecid ortho-amination derivatives can be easily constructed (Scheme 18b). Applying the innate carboxyl in benzoic acid as directing group, Matute and Johansson got rapid access to various drug amination derivatives via iridium-catalyzed arenes C–H bond activation under room temperature (Scheme 18c)280. Besides, Falck and colleagues uncovered a meta-selective arene amination approach through a Fe=N participated SEAr mechanism and utilized it to late-stage modification (Scheme 18d)281.
Scheme 18.

Transition-metal-catalyzed arenes C–H bond activation.
Over the past decade, direct C–H bond electrophilic amination has emerged as an efficient method, including newly developed electrophilic aminating reagents utilization, such as hydroxylamine-O-sulfonic acid (HOSA)282, novel integration with electrochemistry or photochemistry, and so on. For instance, Yoshida group coupled anodic oxidation with nucleophilic pyridine attack, among which the first-step one-electron oxidation lays foundation for aromatic C–H functionalization (Scheme 19a)283. The amino group is introduced at site with the largest coefficient of the LUMO in cation intermediate. Unlike that, Nicewicz284, Tung and Wu285 utilized photocatalytic approach to activate arenes substrates. Nicewicz converted diverse protected phenols, haloarenes and nitrogen heteroaromatics to corresponding anilines through an organic photooxidant and nitroxyl radical catalyst system (Scheme 19b)284. Except for monosubstituted arenes, other various substrates such as protected phenols, haloarenes, and nitrogen heteroaromatics were also applicable to this method. Tung and Wu developed hydrogen-evolution cross-coupling strategy for amination and hydroxylation of arenes. Their exquisite dual catalyst system, comprising photocatalysis and cobalt catalysis, accomplished atom-economic amination of arenes with no need of any oxidant (Scheme 19c)285.
Scheme 19.

Direct C–H bond amination via electrochemistry and photochemistry strategies.
Elevating reactivity of electrophilic aminating reagents through attaching more electron-deficient leaving groups could achieve direct amination under mild conditions. Vedejs group applied O-di(p-methoxyphenyl)phosphinylhydroxylamine as NH2+ equivalent for direct amination of stabilized sodium and potassium enolates (Scheme 20a)286. Falck presented dirhodium-catalyzed C–H arene amination with hydroxylamines as nitrogen sources282. Morandi and coworkers developed the Minisci protocol with MsONH3OTf as aminating reagent287. The protonated state increases reagent electrophilicity and the deactivating ammonium substituent prevents over-amination. This method directly constructed primary anilines in Flurbiprofen, 17β-estradiol-3-methyl ether, and dextromethorphan with good selectivity on the more electron-rich aromatic site (Scheme 20b). As they descried in substrate table, this method was compatible with unprotected amines and hydroxyl groups, and its application in more complex bioactive substrates is expected. Ritter and coworkers realized hexafluoroisopropanol (HFIP)-aided radical aromatic C–H bond amination that provided free anilines in a single step288. The solvent HFIP, known for strong polarity and hydrogen bond-donating ability but weak nucleophilicity, expanded substrate scope across more electron-poor arenes, such as nitrobenzene, by lowering the lowest unoccupied molecular orbital (LUMO) of MsONH3OTf to increase overall electrophilicity. The late-stage functionalization of Moclobemide and Rufinamide revealed its potential for drug modification (Scheme 20c). Jiao group developed various RCO2NH3OTf as new redox-active aminating reagents and introduced primary amines to β-d-galactopyranoside, 18-crown-6 derivatives affording single regioisomers (Scheme 20d)289. Some unprotected hydroxyl-substituted substrates also worked well. Moreover, a Ti-mediated electrophilic amination method was disclosed by Sanford with hydroxylamine as a nitrogen source (Scheme 20e)290. Several heteroaromatic substrates like thiophene could also be transformed with modest regioselectivity. Additionally, a one-pot pyridination-aminolysis approach has been discovered by Ritter group291. With aid of visible light, the pyridinium radical cation generated in situ from N-OTf 2-ethlpyridine effectively aminated bioactive arenes, which enabled quick access to the broad-spectrum antimicrobial Sulfamethoxazole (Scheme 20f). There was high compatibility with heterocyclic arenes like pyrrole and isoxazole in this method, which implies great potential in late-stage pharmacophores derivatization.
Scheme 20.

Direct C–H amination by electrophilic aminating reagents.
Radosevich group disclosed the primary amination of aryl boronic acids through capturing newly-generated intermediate oxazaphosphirane292. They coupled Nef decomposition with PIII/PV=O redox cycling to sustainably produce active intermediates (Scheme 21a). Besides improving the electrophilicity of N sources, enhancing nucleophilicity of aromatic arenes has also been investigated. Kürti and Ess combined aryl Grignard reagents with N–H oxaziridine for direct primary amination of pharmaceutically relevant estradiol, terpenoid, and carbohydrate derivatives (Scheme 21b)293. Chang group came up with C2-selective pyrimidines amination in an enthalpy-driven manner, during which the C2/C4 selectivity was accomplished through nucleophilic substitution of hydrogen (SNH) process, and applied it to fast synthesis of the anticancer drug Dabrafenib (Scheme 21c)294.
Scheme 21.

Primary amination of prepared aryl reagents.
The site-selectivity of late-stage amination methods is highly desired, as the position of amino groups significantly impacts the activity and metabolic feature of bioactive compounds. While there have been advancements in reactant activation methods, achieving precise site-selectivity in amination reactions remains challenging, particularly for heteroaromatic substrates.
Transition-metal-catalyzed cross-coupling of amines and aryl halides is a classical method to construct primary (hetero)arylamine. Ma and coworkers developed Ullmann-type cross-coupling of (hetero)aryl halides to afford various amination products, which has been widely used in industrial and academic pharmaceutical synthesis295, 296, 297, 298. For instance, their CuI/BPMPO system295, 296, 297, 298 and Cu2O/MNBO system295, 296, 297, 298 enable versatile primary amines construction from (hetero)aryl chlorides and bromides, respectively (Scheme 22a). And Chen simplified the synthesis of retinoic acid receptor α/β (RARα/β) agonist Tamibarotene with a non-pressurized Ullmann-type l-proline/DMSO system (Scheme 22b)299. Besides, Buchwald–Hartwig amination is also universal across drug discovery300. As selected example, Pyke group applied sequential Buchwald–Hartwig amination to synthesize ligands of Tec Src homology 3 (SH3) domain, which could be useful in cancer and osteoporosis drug development. In the second Buchwald–Hartwig reaction, they utilized lithium bis(trimethylsilyl)amide (LiHMDS, or LHMDS) as an ammonia equivalent for primary aniline formation (Scheme 22c)301.
Scheme 22.

Cross-coupling of aryl halides for primary anilines formation.
Since reductive amination of unsaturated bonds is highly atom-economic in primary amination, Jagadeesh and Beller discovered Ni(OTf)2-Triphos catalyzed hydrogenative coupling of nitriles with 15N-isotope labeling ammonia/NH4OAc, accessing to 15N-labeled drug-derived primary amines which could be useful for specific metabolites identification (Scheme 23a)302. They also developed a cobalt-based homogeneous catalyst and disclosed its application in reducing carbonyl compounds with gaseous ammonia and hydrogen (Scheme 23b)303. Notably, Leonori reported a tandem method for non-canonical aniline synthesis, consisting of condensation between amines and carbonyls before progressively dehydroaromatization of newly formed cyclohexene304. This protocol successfully modified commercial medicines and natural products with aniline substructure (Scheme 23c). Li group converted various naturally bioactive phenols into corresponding anilines by use of hydrazine as nitrogen source. According to their proposed mechanism, in situ generated palladium hydride reduces phenol to cyclohexanone intermediate, which subsequently undergoes hydrazine condensation, dehydrogenative rearomatization, and finally reductive N–N bond cleavage (Scheme 23d)305. As a common bioactive motif, amino acid bearing chiral α-primary amine carboxylic acid, thus the prevention of undesired self-coupling is always achieved by passively introducing protecting groups. However, these extra protecting-deprotecting procedures inevitably lead to efficiency loss. Struggling with this dilemma, Zhou's work realized direct asymmetric amino acids synthesis from diazoesters and ammonia with only catalytic copper306. They solved both the metal poison problem and enantioselective carbene insertion problem with Tp∗Cu–HBD complex (Scheme 23e).
Scheme 23.

Late-stage nucleophilic primary amination.
Additionally, late-stage dealkylating C–C bond amination is an effective route for actualizing reversal of hydrophobic to hydrophilic properties (Table 3). Site-direct C–C bond primary amination is an emerging strategy for substituted anilines synthesis. In this field, Jiao and coworkers developed dealkylating C–C bond amination via C(sp2) –C(sp3) bond307 and C(sp2) –C(sp2) bond308 cleavage from alkylarenes and styrenes respectively, involving newly generated benzyl azide intermediate followed by rearrangement under acidic conditions (Scheme 24a). Furthermore, the easy preparation of phenylalanine derivatives might help to accelerate the development of chemokine receptor CCR3 antagonists309. And aza-Hock rearrangement C–C bond amination, starting from benzyl alcohols via hydroxylamine intermediates, has been realized by Hashmi and coworkers (Scheme 24b)310.
Scheme 24.

Late-stage dealkylating C–C bond amination.
While there are diverse methods for primary amine synthesis, it is still underdeveloped in direct nitrogenation to construct α-fully substituted primary amines. Since sterically hindered primary amines are popular among pharmaceutical agents311, 312, 313, 314, developing such late-stage approach would help to introduce extra hydrogen bond attachment and potential positively charged site.
4.2. Amidation of bioactive compounds
Since amide is an important building block among peptides, chemical probes, drugs, and natural products, it plays an increasingly prominent role in drug design and development. Among the top 200 small molecule drug retail sales in 2021, 120 drugs contain at least one amide264. Compared with parent carbonyl compounds, newly-generated amides provide extra hydrogen bond acceptors and donors, and one-atom linker extension implies more flexible molecular conformations.
Chang and Baik developed two-step carboxylic acid amination to afford γ-lactams through intramolecular C–H insertion315,316. 1,4,2-dioxazol-5-ones prepared from carboxylic acids, proceeded decarboxylation after coordinating with Ir catalyst, and the desired γ-lactams were generated through enantioselective C–H insertion of metal nitrenes (Scheme 25a). Furthermore, Hong and Chang elaborated on a stepwise lactone-to-NH lactam replacement approach for easy bio-isosteres conversion317. Lactone is a common biological interaction motif but well-known for its metabolic instability, and this defect might be ameliorated by lactone/lactam transformation while retaining bioactive hydrogen bond interaction. They advanced the previous method315,316 by combining reductive C–O bond cleavage of lactones (Scheme 25b). The late-stage conversion of steroid bromodomain inhibitor to its lactam derivatives implied that favorable binding mode of products might emerge from the newly-introduced free NH. Chen and Ma reported an amine-boranes involved photoredox amidation reaction, in which photocatalyst fac-Ir(ppy)3 assisted acyl radical intermediate formation and the radical subsequently generated amide‒borane complex before its conversion to desired amide (Scheme 25c)318.
Scheme 25.

Late-stage amide formation from carboxylic acids.
Apart from carboxylic acid, other carbonyl compounds can also be used for late-stage amide construction. Beckmann rearrangement of newly synthesized oximes is a common stepwise method to construct amides from ketones and aldehydes (Scheme 26)319, 320, 321. For attaining late-stage application of Beckmann rearrangement, many researchers modified the traditional harsh conditions by employing various promoters322,323. The groups Lambert324 and Guo325 developed geminal dihalides-activated Beckmann rearrangement, and applied it to late-stage modification of steroids. Lambert and coworkers proposed a self-propagating mechanism, in which a formerly generated nitrilium ion might alkylate the latter oxime to proceed rearrangement (Scheme 26a)324. Guo and colleagues similarly applied dichloroimidazolidineddiones (DCIDs) in similar fashion for Pregnenolone derivation (Scheme 26b)325. Hall group developed a boronic acid/perfluoropinacol system for mildly converting Pregnenolone oxime to corresponding amide, without protecting alcohol (Scheme 26c)326. McLaughlin and Brennan disclosed calcium-catalyzed Beckmann rearrangement, in which the transient [HO– Ca2+ PF6‒] attacked the nitrilium to accelerate following rearrangement (Scheme 26d)323. These achieved Prasterone/Pregnenolone-derived amides could also be used as cytotoxic agents on different cancer cell lines327,328.
Scheme 26.

Late-stage stepwise amide synthesis by Beckmann rearrangement.
In addition, late-stage Beckmann rearrangement of cyclic nature products will provide unnatural skeletal types for activity screening in the early stage of drug discovery. Hyodo group actualized direct NH insertion to ketones via transoximation/Beckmann rearrangement, and replaced explosive O-protected hydroxylamines with O-protected oximed (Scheme 27a)329. Osborn applied Beckmann rearrangement to afford glycoside-derived lactams from oxime precursors (Scheme 27b)330. Zhang accessed poly-heterocyclic skeletal types from β-caryophyllene (Scheme 27c)331.
Scheme 27.

One-pot amide synthesis and Beckmann rearrangement in natural product derivatization.
In the field of Schmidt-type transformation332, Aubé and Tantillo synthesized a series of fused lactams and bridged lactams in a regioselective way by utilizing stabilized cation‒π/cation‒n interaction (Scheme 28a)333, 334, 335, 336, 337. Jiao group described a copper-catalyzed aerobic oxidative system for converting ketones to primary amides338. Looking for more benign nitrogen sources, they further developed cascade activation of nitromethane for preparing amides and nitriles. In this approach, the active N-donor species are generated through successive activation by triflic anhydride/formic acid/acetic acids (Scheme 28b)339.
Scheme 28.

Schmidt-type amide synthesis.
In the field of amide synthesis, since α-substituted aliphatic chiral amides are common substructures across bioactive compounds like Aliskiren340, 341, 342, 343 and Cebulactam A344, 345, 346, efficient α-enantiopure amides synthesis methods and late-stage application require further development.
4.3. Late-stage introduction of triazole and tetrazole
As representative products of "click chemistry", triazole and tetrazole are widely used across every stage in drug discovery, such as labeling biomacromolecules with fluorescent probes for target identification347 and mechanism of action study348, and rapid preparation of ADC drugs (Scheme 29a)31.
Scheme 29.

Late-stage introduction of triazole and tetrazole in drug discovery.
Triazole and tetrazole are more than connection units in bioconjugation, and they can also serve as critical pharmacophores and bio-isosteres in place of carboxylic acids, aromatic rings, and double bonds349, 350, 351, 352. Besides that, according to their specific steric properties, they could be applied as effective amide surrogates but have fixed configurations and increased metabolic stability353. Moody group applied late-stage Chan-Lam amination to effective construction of diverse triazole-contained integrin inhibitors with various substituents for idiopathic pulmonary fibrosis (IPF) drug SAR study (Scheme 29b)354. Recently Glorius and Fridman developed steroid-based and ergosterol-based triazole cationic amphiphiles for antifungal bioactive compounds optimization (Scheme 29c)355. Furthermore, Moss and Wang found that the reduction of amyloid-β protein (Aβ) plateau significantly increased about 2-fold when introduced tetrazole into the cyano position of the parent compound, which implied the newly introduced tetrazole structure might occupy an essential binding site (Scheme 29d)356. Maurya and coworkers conjugated Ciprofloxacin with various bioactive molecules in CuI-nanoparticle-catalyzed click reaction. And they evaluated the antibacterial activity of these pharmaceutical conjugates, further broadened the chemical space of lead discovery (Scheme 29e)357. Besides, NH-triazole is also a useful handle for further structural modification and biological optimization.
Since azide is one of the prototypical triazole precursors, in-situ azide generation will simplify triazole introduction. Abell and Sirinivasan successfully converted arenes to N-aryl-1,2,3-triazoles through a one-pot tandem reaction, which was initiated from C–H bond borylation, followed by copper-catalyzed azidation and azide–alkyne click reaction at the end358. This approach was applied to equip (±)-α-tocopherol nicotinate, nicotine, and resveratrol with triazole (Scheme 30a). Metzger group synthesized various tetrazole analogs of natural fatty acids from corresponding fatty nitriles, and provided potential homoprostanoids due to structure similarity (Scheme 30b)359. The group of Seo afforded triazole- and tetrazole-decorated-side chain peptoids via nucleophilic substitution and subsequent [3 + 2]-cycloaddition on solid support (Scheme 30c)360. Jiao and coworkers accessed triazole equipped Estradiol derivatives through copper-catalyzed nitrogenation of alkynes. In this approach, the key intermediated is generated in situ by Pummerer-type rearrangement between the N donor DPPA and the solvent DMSO (Scheme 30d)361. While in Gazvoda's work, they obtained 4-substituted-1H-1,2,3-triazoles derivatives through terminal alkyne coupling with newly formed hydrazoic acids (Scheme 30e)362. They avoided direct usage of hazardous hydrazoic acid by in situ generations from sodium azide under formic acid conditions. In addition, the formic acid also served as a reductant for regeneration of Cu(I) species. Except for classical [3 + 2] cycloaddition, other innovative late-stage triazole synthesis methods have also been reported. Bi group used primary amines as substrate, developed revised [4 + 1] annulation with perfluoroalkyl N-mesylhydrazones (PFHZ-Ms)363 or difluoroacetaldehyde N-tosylhydrazones (DFHZ-Tfs)364 accessing to regioselective preparation of substituted 1,2,3-triazole products (Scheme 30f). The reported mechanism involved twice HF elimination and N,N-diisopropylethylamine aided sulfonic acid release before intramolecular cyclization.
Scheme 30.

Late-stage introduction of triazole and tetrazole.
Due to their special conformation, atropisomeric 1,2,3-triazoles are useful pharmacophores in medicinal chemistry365, 366, 367, but the late-stage introduction is rarely studied. Direct cycloaddition reaction might be stuck by decreased reactivity of sterically substituted internal alkynes, therefore novel stereo-controlled synthetic protocols are highly required.
4.4. Late-stage skeletal editing with nitrogenation
Compared to branches modification, direct skeletal editing will completely reverse physicochemical and pharmaceutical properties, further expanding drug chemical space for phenotypic and target screening in the early stage of drug discovery368. We herein introduce researches focusing on late-stage skeletal editing with Nitrogenation. Levin and colleagues developed osmium nitrides as N-reagents and applied them to construct isoquinolines from indenes (Scheme 31a)369, which could simplify the synthetic routes of isoquinoline scaffold and facilitate its application in antifungal studies370. Morandi group disclosed the skeletal editing reaction of indoles to access corresponding aromatic ring nitrogen incorporation products via nitrene insertion (Scheme 31b)371. Diverse indole-contained sterically constrained substrates like five- or six-membered fused indole rings were evaluated with this method, affording corresponding quinazoline and quinoxaline scaffolds with good yield. Besides, they also accomplished nitrogen atom insertion into indenes in a similar way and successfully achieved the 15N-labled Papaverine probe (Scheme 31c)372. Cheng group developed electrochemical method for direct ammonia insertion, accessing diverse substituted quinolines and pyridines (Scheme 31d)373. Wei and colleagues directly converted arylcycloalkenes into corresponding N-heterocycles through Cobalt-catalyzed nitrogen atom insertion, through which the aziridine intermediate underwent oxidative ring-opening and dehydrogenation process before performing product (Scheme 31e)374.
Scheme 31.

Late-stage skeleton N atom introduction.
Dong and Liu realized oxygen-to-nitrogen editing through zinc/nickel tandem catalyzed boron insertion of C–O bond in strained cyclic ethers (Scheme 31f)375. According to the proposed cleavage-then-rebound pathway, the Ni-catalyzed C–Br/B–Br coupling is the rate-determining step in boron intermediate production375. Other annular skeletons like lactams can be achieved through insertion and substitution strategy either. Since Schmidt reaction and Beckmann rearrangement are classical ring expansion methods for cyclic ketones accessing lactams339,376, 377, 378, Wahl group explored skeletal editing reaction of cyclobutanones toward γ-lactams via Aza-Beayer-Villiger mechanism and applied it to late-stage skeleton editing (Scheme 31g)379,380. They took use of the inherent ring strain of hemiaminal intermediate to promote leaving group mediated rearrangement under room temperature, which enabled convenient synthesis of phosphordiesterase-4 (PDE-4) inhibitor Rolipram.
Since scaffold hopping is an effective approach to discovering new skeleton compounds, skeletal editing may become a novel method for this field without de novo synthesis. As seen the skeleton nitrogenation is fairly terse and efficient, but its late-stage application is rarely reported, which partly attribute to limited functional group compatibility and uncontrollable reaction selectivity. As more catalysts are discovered and more skeletal editing methods are used for LSF, skeletal editing will hopefully improve the pace and quality of bioactive compounds modification and synthesis in medicinal chemistry.
4.5. Late-stage N,O-incorporation via C–C bond cleavage
In light of the importance of oxygenation and nitrogenation in bioactive derivatization, specific combination methodologies of the N,O-incorporation, which provide a shortcut to equip versatile pharmacophores and biological handles, will be discussed below. In the field of oxo nitriles synthesis, Jiao group discovered TEMPO-catalyzed aerobic oxygenation and nitrogenation system, through which they simultaneously introduced carbonyl and cyano groups via C=C double-bond cleavage. In this process, TEMPO-induced azido free radical attacks substrate alkene before terminating with molecular oxygen to form peroxide radical intermediate (Scheme 32a)381. Besides, Shi and Jiao also developed a photoinduced C(sp2)‒C(sp3) bond cleavage approach to building cyano-containing ketones with DMSO as an oxidant382. Liu and coworkers disclosed visible-light-promoted selective cleavage of arene vicinal C(sp3)-C(sp3) bond, and the subsequent deconstructive nitrogenation successfully remodel bioactive steroid skeleton (Scheme 32b)383.
Scheme 32.

Direct synthesis of oxo nitriles via N,O-incorporation.
Concerning direct synthesis of β-azido alcohols, Jiao and colleagues reported Mn-catalyzed hydroxyazidation of olefins, with air as oxygen source and azidotrimethylsilane (TMSN3) as nitrogen source (Scheme 33a)384. Furthermore, with promotion of visible light, Lu group developed a metal-free aerobic hydroxyazidation of alkenes (Scheme 33b)385. Vankar and coworkers disclosed a TEMPO-PIFA-TMSN3 system, through which they obtained 1-azido-2-deoxysugars from glycals with water as oxygen source (Scheme 33c)386.
Scheme 33.

Direct synthesis of β-azido alcohols via N,O-incorporation.
Besides above nitrogentation approaches, Baran, Zhu, and other scientists developed diverse methods for equipping nitrogen-bearing drug-like fragments such as imides387,388, and morpholines389, 390, 391, which could effectively diversify bioactive molecules with broad substrate scope and good regioselectivity. Chiral chromanols especially chiral 3-amino-4-chromanols are fairly important structural units in bioactive compounds and drugs392, 393, 394, and useful synthons for the synthesis of therapeutic compounds395. Direct enantioselective construction of chiral 3-amino-4-chromanols is still in high demand.
5. Conclusions
Since late-stage modification has become a convenient and efficient strategy for the derivatization of bioactive compounds and natural products, it plays an increasingly important role in drug discovery. However, the disconnection of reported methodologies with their applications to modify bioactive compounds is a loss for pharmaceutical research and medicinal chemistry. Here we highlighted the latest progress on late-stage halogenation, oxygenation and nitrogenation methods, which enrich the toolbox for bioactive compounds and natural products modification. Despite the rapid evolution of synthetic methodology in every aspect, there are still some common difficulties and challenges to be solved before its widespread late-stage application. From a pharmaceutical science perspective, due to the particularity of bioactive compounds, desired approaches aimed at late-stage derivatization should meet the following requirements: multifunctional group toleration, predictable site/enantio-selectivity, broad substrate scope, and transformation effectiveness.
Firstly, LSM should demonstrate tolerance towards multifunctional groups commonly found in bioactive compounds, such as hydroxyl, carbonyl, carboxyl, nitro, cyano, amino, amide, heterocycles, α,β-unsaturated ketones, and more13. Recent advancements in catalysts and new reactions have contributed to addressing these challenges under mild conditions. For instance, naturally occurring bioactive peptides396,397 often contain a variety of above functional groups, and modifying these peptides with unnatural peptides can enhance their druggability and therapeutic potential398, 399, 400. In recent years, significant advancements have been made in LSM of short peptides, with numerous chemical137,401, 402, 403, enzymatic307,400, or photocatalytic404 approaches. Some strategies have successfully achieved the C–F, C–Cl, C–O, and C–N bond formations in amino acids and short peptides using C–H activation approaches143,402,404. However, challenges remain in achieving late-stage halogenation, oxygenation, and nitrogenation of naturally occurring bioactive peptides, especially those with unprotected carboxyl, hydroxyl, and amino groups, thus milder and more compatible conditions are needed. Additionally, providing details on chemoselectivity attempts and incompatible functional groups can assist medicinal chemists in adopting newly discovered methods.
Secondly, predictable chemo-, regio-, and enantio-selectivity are crucial factors during the transition from hit to lead compounds. Pharmaceutical chemists often perform SAR studies by introducing various substituents at specific positions of hit compounds using convergent synthetic routes. For instance, naturally occurring bioactive saccharides405 and peptides406 play vital roles in living cells and have potential therapeutic applications with subtle modification407,408. Chemists have made significant progress in selectively functionalizing the β-, γ-, and δ-positions of amino acids and short peptides using directing group strategies. However, these approaches are seldom utilized in bioactive peptides LSM due to limitations in suitable directing groups or amino acids137,402. Similarly, chemists have devised various methods, such as transition-metal catalysis, enzyme catalysis, organocatalysis, and photoredox catalysis for the selective modification of mono- and oligosaccharides409,410. Yet, achieving selective LSM of bioactive saccharides remains a challenge. Enzymatic catalysis, such as glycosidic bond formation, shows promise as it holds the potential for directional evolutionary advancements to address this issue. Further exploration of enzymatic approaches may lead to breakthroughs in achieving selective LSM of bioactive saccharides. While some reactions can generate multiple isomers and derivatives quickly291, efforts are required to identify the specific bioactive structure. Biocatalytic transformations are well-known for their high selectivity compared to other catalysts, but few enzymatic late-stage methods have been disclosed mainly because of the innately narrow substrate scope411. Ongoing efforts like molecular biology, bioinformatics, protein engineering412 and (meta-)genome mining413 hold promise for expanding the scope of enzymatic late-stage methods.
Thirdly, broad substrate scope is essential due to the diversity of targets and binding pockets for bioactive molecules. These molecules encompass various scaffolds, including benzene rings, steroids, N-heterocycles414, O-heterocycles415, and other heterocycles416. For example, the functionalization of N-heterocycles and heteroaromatics remains facing huge challenges in synthetic chemistry7,9. In our opinion, while it is difficult for a single reaction to be compatible with multiple scaffolds, achieving multiple substrate compatibility while ensuring site-selectivity is feasible and necessary for LSM in pharmaceutical science.
Fourthly, transformation efficiency is crucial as bioactive compounds are valuable and hard-earned resources, whether obtained through chemical synthesis or natural extraction. It is essential to avoid introducing additional auxiliary groups, such as directing groups and protecting groups, to simplify the reaction process and achieve atom economy.
In addition to the method itself, the new molecules generated through LSM are also a valuable resource for drug screening based on target and phenotype. However, there exists a disconnect between the development of synthetic methods and the evaluation of product bioactivity, making it difficult for drug development researchers to recognize the practical significance of specific transformations. Therefore, cooperation between chemists and pharmacists, along with the establishment of compound resource-sharing platforms, can facilitate the rapid discovery of the potential applications of these generated molecules.
While it may be challenging to satisfy all the aforementioned requirements simultaneously, controlling functional group toleration and site/enantio-selectivity to a large extent will enable wide applicability of LSM methods, even within a limited range of substrates and transformation effectiveness. Meanwhile, the progressive integration of multiple technologies can expedite the development process. For instance, the rapid development of computational chemistry and artificial intelligence in reaction designing and synthesis planning have rescued chemists from verbose route exploring417,418. While multi-parameter optimization is the specialty of automation technology, high-throughput screening of reaction systems based on existing methods in biologically friendly environments might accelerate target reaction disclosure419,420. Ultimately, the combination of various technology-assisted synthetic chemistry approaches will gradually overcome the outlined challenges, and the late-stage modification will greatly facilitate the discovery and development of the next generation of medicines.
Author contributions
Ning Jiao conceived the project and directed the research. Tongyu Huo, Xinyi Zhao, Xiaodong Dou, and Ning Jiao wrote the paper. Zengrui Cheng, Jialiang Wei, and Minghui Zhu discussed the text the citation.
Conflicts of interest
The authors declare no conflicts of interest.
Acknowledgments
Financial support from the National Key R&D Program of China (No. 2021YFA1501700), the National Natural Science Foundation of China (Nos. 22293014, 22131002, 22161142019, 81821004), the Ministry of Science and Technology of China Changping Laboratory, Peking University Special Fund for COVID-19, and the New Cornerstone Science Foundation through the XPLORER PRIZE are greatly appreciated.
Footnotes
Peer review under the responsibility of Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences.
Contributor Information
Xiaodong Dou, Email: xddou@pku.edu.cn.
Ning Jiao, Email: jiaoning@pku.edu.cn.
References
- 1.Raju T.N.K. The nobel chronicles. Lancet. 2000;356:436. doi: 10.1016/S0140-6736(05)73588-1. [DOI] [PubMed] [Google Scholar]
- 2.Guillemard L., Kaplaneris N., Ackermann L., Johansson M.J. Late-stage C–H functionalization offers new opportunities in drug discovery. Nat Rev Chem. 2021;5:522–545. doi: 10.1038/s41570-021-00300-6. [DOI] [PubMed] [Google Scholar]
- 3.Lasso J.D., Castillo Pazos D.J., Li C.J. Green chemistry meets medicinal chemistry: a perspective on modern metal-free late-stage functionalization reactions. Chem Soc Rev. 2021;50:10955–10982. doi: 10.1039/d1cs00380a. [DOI] [PubMed] [Google Scholar]
- 4.Lombardino J.G., Lowe J.A., 3rd The role of the medicinal chemist in drug discovery—then and now. Nat Rev Drug Discov. 2004;3:853–862. doi: 10.1038/nrd1523. [DOI] [PubMed] [Google Scholar]
- 5.Moir M., Danon J.J., Reekie T.A., Kassiou M. An overview of late-stage functionalization in today's drug discovery. Expet Opin Drug Discov. 2019;14:1137–1149. doi: 10.1080/17460441.2019.1653850. [DOI] [PubMed] [Google Scholar]
- 6.Wencel Delord J., Glorius F. C‒H bond activation enables the rapid construction and late-stage diversification of functional molecules. Nat Chem. 2013;5:369–375. doi: 10.1038/nchem.1607. [DOI] [PubMed] [Google Scholar]
- 7.Blakemore D.C., Castro L., Churcher I., Rees D.C., Thomas A.W., Wilson D.M., et al. Organic synthesis provides opportunities to transform drug discovery. Nat Chem. 2018;10:383–394. doi: 10.1038/s41557-018-0021-z. [DOI] [PubMed] [Google Scholar]
- 8.Jana R., Begam H.M., Dinda E. The emergence of the C‒H functionalization strategy in medicinal chemistry and drug discovery. Chem Commun. 2021;57:10842–10866. doi: 10.1039/d1cc04083a. [DOI] [PubMed] [Google Scholar]
- 9.Campos K.R., Coleman P.J., Alvarez J.C., Dreher S.D., Garbaccio R.M., Terrett N.K., et al. The importance of synthetic chemistry in the pharmaceutical industry. Science. 2019;363 doi: 10.1126/science.aat0805. [DOI] [PubMed] [Google Scholar]
- 10.Börgel J., Ritter T. Late-stage functionalization. Chem. 2020;6:1877–1887. [Google Scholar]
- 11.Zhang L., Ritter T. A perspective on late-stage aromatic C‒H bond functionalization. J Am Chem Soc. 2022;144:2399–2414. doi: 10.1021/jacs.1c10783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Purser S., Moore P.R., Swallow S., Gouverneur V. Fluorine in medicinal chemistry. Chem Soc Rev. 2008;37:320–330. doi: 10.1039/b610213c. [DOI] [PubMed] [Google Scholar]
- 13.Bhutani P., Joshi G., Raja N., Bachhav N., Rajanna P.K., Bhutani H., et al. U.S. FDA approved drugs from 2015-June 2020: a perspective. J Med Chem. 2021;64:2339–2381. doi: 10.1021/acs.jmedchem.0c01786. [DOI] [PubMed] [Google Scholar]
- 14.Aynetdinova D., Callens M.C., Hicks H.B., Poh C.Y.X., Shennan B.D.A., Boyd A.M., et al. Installing the “magic methyl”―C‒H methylation in synthesis. Chem Soc Rev. 2021;50:5517–5563. doi: 10.1039/d0cs00973c. [DOI] [PubMed] [Google Scholar]
- 15.Castellino N.J., Montgomery A.P., Danon J.J., Kassiou M. Late-stage functionalization for improving drug-like molecular properties. Chem Rev. 2023;123:8127–8153. doi: 10.1021/acs.chemrev.2c00797. [DOI] [PubMed] [Google Scholar]
- 16.Novaes L.F.T., Ho J.S.K., Mao K., Liu K., Tanwar M., Neurock M., et al. Exploring electrochemical C(sp3)–H oxidation for the late-stage methylation of complex molecules. J Am Chem Soc. 2022;144:1187–1197. doi: 10.1021/jacs.1c09412. [DOI] [PubMed] [Google Scholar]
- 17.Shang W., Sun H., Chen W., Liu J. Diversification of pharmaceutical molecules via late-stage C(sp2)–H functionalization. Green Synth Catal. 2023;4:104–123. [Google Scholar]
- 18.Li H.P., He X.H., Peng C., Li J.L., Han B. A straightforward access to trifluoromethylated natural products through late-stage functionalization. Nat Prod Rep. 2023;40:988–1021. doi: 10.1039/d2np00056c. [DOI] [PubMed] [Google Scholar]
- 19.Clarke S.L., McGlacken G.P. Methyl fluorosulfonyldifluoroacetate (MFSDA): an underutilised reagent for trifluoromethylation. Chem Eur J. 2017;23:1219–1230. doi: 10.1002/chem.201602511. [DOI] [PubMed] [Google Scholar]
- 20.Bohm H.J., Banner D., Bendels S., Kansy M., Kuhn B., Muller K., et al. Fluorine in medicinal chemistry. Chembiochem. 2004;5:637–643. doi: 10.1002/cbic.200301023. [DOI] [PubMed] [Google Scholar]
- 21.Müller K., Faeh C., Diederich F. Fluorine in pharmaceuticals: looking beyond intuition. Science. 2007;317:1881. doi: 10.1126/science.1131943. [DOI] [PubMed] [Google Scholar]
- 22.Park B.K., Kitteringham N.R., O'Neill P.M. Metabolism of fluorine-containing drugs. Annu Rev Pharmacol Toxicol. 2001;41:443–470. doi: 10.1146/annurev.pharmtox.41.1.443. [DOI] [PubMed] [Google Scholar]
- 23.Nodwell M.B., Bagai A., Halperin S.D., Martin R.E., Knust H., Britton R. Direct photocatalytic fluorination of benzylic C–H bonds with N-fluorobenzenesulfonimide. Chem Commun. 2015;51:11783–11786. doi: 10.1039/c5cc04058b. [DOI] [PubMed] [Google Scholar]
- 24.Mazier C., Jaouen M., Sari M.A., Buisson D. Microbial oxidation of terfenadine and ebastine into fexofenadine and carebastine. Bioorg Med Chem Lett. 2004;14:5423–5426. doi: 10.1016/j.bmcl.2004.07.076. [DOI] [PubMed] [Google Scholar]
- 25.El Ouarradi A., Salard Arnaud I., Buisson D. Biooxidation of methyl group: application to the preparation of alcohol and acid metabolites of terfenadine, ebastine and analogues. Tetrahedron. 2008;64:11738–11744. [Google Scholar]
- 26.Genovino J., Sames D., Hamann L.G., Toure B.B. Accessing drug metabolites via transition-metal catalyzed C‒H oxidation: the liver as synthetic inspiration. Angew Chem Int Ed Engl. 2016;55:14218–14238. doi: 10.1002/anie.201602644. [DOI] [PubMed] [Google Scholar]
- 27.Bernstein D.I., Schoenwetter W.F., Nathan R.A., Storms W., Ahlbrandt R., Mason J. Efficacy and safety of Fexofenadine hydrochloride for treatment of seasonal allergic rhinitis. Ann Allergy Asthma Immunol. 1997;79:443–448. doi: 10.1016/S1081-1206(10)63041-4. [DOI] [PubMed] [Google Scholar]
- 28.Miller J.L. FDA approves pioglitazone for diabetes. Am J Health Syst Pharm. 1999;56:1698. doi: 10.1093/ajhp/56.17.1698. [DOI] [PubMed] [Google Scholar]
- 29.Musso G., Cassader M., Paschetta E., Gambino R. Thiazolidinediones and advanced liver fibrosis in nonalcoholic steatohepatitis: a meta-analysis. JAMA Intern Med. 2017;177:633–640. doi: 10.1001/jamainternmed.2016.9607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cusi K., Orsak B., Bril F., Lomonaco R., Hecht J., Ortiz Lopez C., et al. Long-term pioglitazone treatment for patients with nonalcoholic steatohepatitis and prediabetes or type 2 diabetes mellitus. Ann Intern Med. 2016;165:305–315. doi: 10.7326/M15-1774. [DOI] [PubMed] [Google Scholar]
- 31.Zhou Q., Gui J., Pan C.M., Albone E., Cheng X., Suh E.M., et al. Bioconjugation by native chemical tagging of C‒H bonds. J Am Chem Soc. 2013;135:12994–12997. doi: 10.1021/ja407739y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maria Faisca Phillips A., Pombeiro A.J.L. Recent developments in enantioselective organocatalytic cascade reactions for the construction of halogenated ring systems. Eur J Org Chem. 2021;2021:3938–3969. [Google Scholar]
- 33.Wang P.F., Neiner A., Lane T.R., Zorn K.M., Ekins S., Kharasch E.D. Halogen substitution influences ketamine metabolism by cytochrome P450 2B6: in vitro and computational approaches. Mol Pharm. 2019;16:898–906. doi: 10.1021/acs.molpharmaceut.8b01214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Frey K.M., Puleo D.E., Spasov K.A., Bollini M., Jorgensen W.L., Anderson K.S. Structure-based evaluation of non-nucleoside inhibitors with improved potency and solubility that target HIV reverse transcriptase variants. J Med Chem. 2015;58:2737–2745. doi: 10.1021/jm501908a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cheng Y.Y., Yuan H. Quantitative study of electrostatic and steric effects on physicochemical property and biological activity. J Mol Graphics Modell. 2006;24:219–226. doi: 10.1016/j.jmgm.2005.08.005. [DOI] [PubMed] [Google Scholar]
- 36.Baduru K.K., Trapp S., Burken J.G. Direct measurement of voc diffusivities in tree tissues: impacts on tree-based phytoremediation and plant contamination. Environ Sci Technol. 2008;42:1268–1275. doi: 10.1021/es071552l. [DOI] [PubMed] [Google Scholar]
- 37.Jiang Y., Liu J., Hu Y., Fujita T. Novel topological index for research on structure-property relationships of complex organic compounds. J Comput Chem. 2003;24:842–849. doi: 10.1002/jcc.10237. [DOI] [PubMed] [Google Scholar]
- 38.von der Ohe P.C., Kühne R., Ebert R.U., Altenburger R., Liess M., Schüürmann G. Structural alerts―a new classification model to discriminate excess toxicity from narcotic effect levels of organic compounds in the acute daphnid assay. Chem Res Toxicol. 2005;18:536–555. doi: 10.1021/tx0497954. [DOI] [PubMed] [Google Scholar]
- 39.Chiou C.T., Schmedding D.W., Manes M. Improved prediction of octanol−water partition coefficients from liquid−solute water solubilities and molar volumes. Environ Sci Technol. 2005;39:8840–8846. doi: 10.1021/es050729d. [DOI] [PubMed] [Google Scholar]
- 40.Dolutegravir (Tivicay) for HIV JAMA. 2014;312:428–429. doi: 10.1001/jama.2014.8406. [DOI] [PubMed] [Google Scholar]
- 41.Rhyne D.N., Byrd E.S., Klibanov O.M. Dolutegravir (Tivicay) for HIV infection. Nurse Pract Am J Prim Health Care. 2014;39:11–15. doi: 10.1097/01.NPR.0000444657.88872.7f. [DOI] [PubMed] [Google Scholar]
- 42.Kim E.S. Abemaciclib: first global approval. Drugs. 2017;77:2063–2070. doi: 10.1007/s40265-017-0840-z. [DOI] [PubMed] [Google Scholar]
- 43.Deeks E.D. Raltegravir once-daily tablet: a review in HIV-1 infection. Drugs. 2017;77:1789–1795. doi: 10.1007/s40265-017-0827-9. [DOI] [PubMed] [Google Scholar]
- 44.Sayana S., Khanlou H. Raltegravir: the first in a new class of integrase inhibitors for the treatment of HIV. Expert Rev Anti Infect Ther. 2008;6:419–426. doi: 10.1586/14787210.6.4.419. [DOI] [PubMed] [Google Scholar]
- 45.Cheng Q., Ritter T. New directions in C‒H fluorination. Trends Chem. 2019;1:461–470. [Google Scholar]
- 46.Champagne P.A., Desroches J., Hamel J.D., Vandamme M., Paquin J.F. Monofluorination of organic compounds: 10 years of innovation. Chem Rev. 2015;115:9073–9174. doi: 10.1021/cr500706a. [DOI] [PubMed] [Google Scholar]
- 47.Richardson P. Fluorination methods for drug discovery and development. Expet Opin Drug Discov. 2016;11:983–999. doi: 10.1080/17460441.2016.1223037. [DOI] [PubMed] [Google Scholar]
- 48.Smart B.E. Fluorine substituent effects (on bioactivity) J Fluor Chem. 2001;109:3–11. [Google Scholar]
- 49.Clementi E., Raimondi D.L. Atomic screening constants from SCF functions. J Chem Phys. 1963;38:2686–2689. [Google Scholar]
- 50.Schlosser M., Michel D. About the “physiological size” of fluorine substituents: comparison of sensorially active compounds with fluorine and methyl substituted analogues. Tetrahedron. 1996;52:99–108. [Google Scholar]
- 51.Richardson P. Applications of fluorine to the construction of bioisosteric elements for the purposes of novel drug discovery. Expet Opin Drug Discov. 2021;16:1261–1286. doi: 10.1080/17460441.2021.1933427. [DOI] [PubMed] [Google Scholar]
- 52.Kaan H.Y., Ulaganathan V., Rath O., Prokopcova H., Dallinger D., Kappe C.O., et al. Structural basis for inhibition of Eg5 by dihydropyrimidines: stereoselectivity of antimitotic inhibitors enastron, dimethylenastron and fluorastrol. J Med Chem. 2010;53:5676–5683. doi: 10.1021/jm100421n. [DOI] [PubMed] [Google Scholar]
- 53.Parlow J.J., Kurumbail R.G., Stegeman R.A., Stevens A.M., Stallings W.C., South M.S. Synthesis and X-ray crystal structures of substituted fluorobenzene and benzoquinone inhibitors of the tissue factor VIIa complex. Bioorg Med Chem Lett. 2003;13:3721–3725. doi: 10.1016/j.bmcl.2003.08.002. [DOI] [PubMed] [Google Scholar]
- 54.Parlow J.J., Stevens A.M., Stegeman R.A., Stallings W.C., Kurumbail R.G., South M.S. Synthesis and crystal structures of substituted benzenes and benzoquinones as tissue factor VIIa inhibitors. J Med Chem. 2003;46:4297–4312. doi: 10.1021/jm030233b. [DOI] [PubMed] [Google Scholar]
- 55.Morgenthaler M., Schweizer E., Hoffmann Roder A., Benini F., Martin R.E., Jaeschke G., et al. Predicting and tuning physicochemical properties in lead optimization: amine basicities. ChemMedChem. 2007;2:1100–1115. doi: 10.1002/cmdc.200700059. [DOI] [PubMed] [Google Scholar]
- 56.Landry M.L., Crawford J.J. LogD contributions of substituents commonly used in medicinal chemistry. ACS Med Chem Lett. 2020;11:72–76. doi: 10.1021/acsmedchemlett.9b00489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Huchet Q.A., Kuhn B., Wagner B., Fischer H., Kansy M., Zimmerli D., et al. On the polarity of partially fluorinated methyl groups. J Fluor Chem. 2013;152:119–128. [Google Scholar]
- 58.Swahn B.M., Kolmodin K., Karlstrom S., von Berg S., Soderman P., Holenz J., et al. Design and synthesis of beta-site amyloid precursor protein cleaving enzyme (BACE1) inhibitors with in vivo brain reduction of beta-amyloid peptides. J Med Chem. 2012;55:9346–9361. doi: 10.1021/jm3009025. [DOI] [PubMed] [Google Scholar]
- 59.Ettorre A., D'Andrea P., Mauro S., Porcelloni M., Rossi C., Altamura M., et al. hNK2 receptor antagonists. The use of intramolecular hydrogen bonding to increase solubility and membrane permeability. Bioorg Med Chem Lett. 2011;21:1807–1809. doi: 10.1016/j.bmcl.2011.01.074. [DOI] [PubMed] [Google Scholar]
- 60.Yao B., Wang Z.L., Zhang H., Wang D.X., Zhao L., Wang M.X. Cu(ClO4)2-mediated arene C‒H bond halogenations of azacalixar-omatics using alkali metal halides as halogen sources. J Org Chem. 2012;77:3336–3340. doi: 10.1021/jo300152u. [DOI] [PubMed] [Google Scholar]
- 61.Zaitsev V.G., Shabashov D., Daugulis O. Highly regioselective arylation of sp3 C‒H bonds catalyzed by palladium acetate. J Am Chem Soc. 2005;127:13154–13155. doi: 10.1021/ja054549f. [DOI] [PubMed] [Google Scholar]
- 62.Truong T., Klimovica K., Daugulis O. Copper-catalyzed, directing group-assisted fluorination of arene and heteroarene C‒H bonds. J Am Chem Soc. 2013;135:9342–9345. doi: 10.1021/ja4047125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lou S.J., Chen Q., Wang Y.F., Xu D.Q., Du X.H., He J.Q., et al. Selective C–H bond fluorination of phenols with a removable di-recting group: late-stage fluorination of 2-phenoxyl nicotinate derivatives. ACS Catal. 2015;5:2846–2849. [Google Scholar]
- 64.Ning X.Q., Lou S.J., Mao Y.J., Xu Z.Y., Xu D.Q. Nitrate-promoted selective C‒H fluorination of benzamides and benzeneacetamides. Org Lett. 2018;20:2445–2448. doi: 10.1021/acs.orglett.8b00793. [DOI] [PubMed] [Google Scholar]
- 65.Fier P.S., Hartwig J.F. Selective C‒H fluorination of pyridines and diazines inspired by a classic amination reaction. Science. 2013;342:956–960. doi: 10.1126/science.1243759. [DOI] [PubMed] [Google Scholar]
- 66.Fier P.S., Hartwig J.F. Synthesis and late-stage functionalization of complex molecules through C‒H fluorination and nucleophilic aromatic substitution. J Am Chem Soc. 2014;136:10139–10147. doi: 10.1021/ja5049303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yamamoto K., Li J., Garber J.A.O., Rolfes J.D., Boursalian G.B., Borghs J.C., et al. Palladium-catalysed electrophilic aromatic C‒H fluorination. Nature. 2018;554:511–514. doi: 10.1038/nature25749. [DOI] [PubMed] [Google Scholar]
- 68.Chen W., Huang Z., Tay N.E.S., Giglio B., Wang M., Wang H., et al. Direct arene C–H fluorination with 18F−via organic photoredox catalysis. Science. 2019;364:1170–1174. doi: 10.1126/science.aav7019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Racowski J.M., Gary J.B., Sanford M.S. Carbon(sp3)‒fluorine bond-forming reductive elimination from palladium(IV) complexes. Angew Chem Int Ed Engl. 2012;51:3414–3417. doi: 10.1002/anie.201107816. [DOI] [PubMed] [Google Scholar]
- 70.Mankad N.P., Toste F.D. C(sp3)‒F reductive elimination from alkylgold(III) fluoride complexes. Chem Sci. 2012;3:72–76. doi: 10.1039/C1SC00515D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Li Y., Wu Y., Li G.S., Wang X.S. Palladium-catalyzed C‒F bond formation via directed C‒H activation. Adv Synth Catal. 2014;356:1412–1418. [Google Scholar]
- 72.News in brief. Expert Rev Neurother. 2009;9:1459–1461. [Google Scholar]
- 73.Griguolo G., Dieci M.V., Guarneri V., Conte P. Olaparib for the treatment of breast cancer. Expert Rev Anticancer Ther. 2018;18:519–530. doi: 10.1080/14737140.2018.1458613. [DOI] [PubMed] [Google Scholar]
- 74.Al Salama Z.T. Apalutamide: first global approval. Drugs. 2018;78:699–705. doi: 10.1007/s40265-018-0900-z. [DOI] [PubMed] [Google Scholar]
- 75.Liu W., Huang X., Cheng M.J., Nielsen R.J., Goddard W.A., Groves J.T. Oxidative aliphatic C‒H fluorination with fluoride ion catalyzed by a manganese porphyrin. Science. 2012;337:1322–1325. doi: 10.1126/science.1222327. [DOI] [PubMed] [Google Scholar]
- 76.Kee C.W., Chin K.F., Wong M.W., Tan C.H. C‒H selective fluorination of alkyl C‒H bonds via photocatalysis. Chem Commun. 2014;50:8211–8214. doi: 10.1039/c4cc01848f. [DOI] [PubMed] [Google Scholar]
- 77.Bume D.D., Pitts C.R., Jokhai R.T., Lectka T. Direct, visible light-sensitized benzylic C‒H fluorination of peptides using dibenzosuberenone: selectivity for phenylalanine-like residues. Tetrahedron. 2016;72:6031–6036. [Google Scholar]
- 78.Pitts C.R., Bume D.D., Harry S.A., Siegler M.A., Lectka T. Multiple enone-directed reactivity modes lead to the selective photochemical fluorination of polycyclic terpenoid derivatives. J Am Chem Soc. 2017;139:2208–2211. doi: 10.1021/jacs.7b00335. [DOI] [PubMed] [Google Scholar]
- 79.Mao Y.J., Lou S.J., Hao H.Y., Xu D.Q. Selective C(sp3)-H and C(sp2)-H fluorination of alcohols using practical auxiliaries. Angew Chem Int Ed Engl. 2018;57:14085–14089. doi: 10.1002/anie.201808021. [DOI] [PubMed] [Google Scholar]
- 80.Fazekas T.J., Alty J.W., Neidhart E.K., Miller A.S., Leibfarth F.A., Alexanian E.J. Diversification of aliphatic C–H bonds in small molecules and polyolefins through radical chain transfer. Science. 2022;375:545–550. doi: 10.1126/science.abh4308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Braun M.G., Doyle A.G. Palladium-catalyzed allylic C‒H fluorination. J Am Chem Soc. 2013;135:12990–12993. doi: 10.1021/ja407223g. [DOI] [PubMed] [Google Scholar]
- 82.Leibler I.N., Tekle Smith M.A., Doyle A.G. A general strategy for C(sp3)-H functionalization with nucleophiles using methyl radical as a hydrogen atom abstractor. Nat Commun. 2021;12:6950. doi: 10.1038/s41467-021-27165-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Meanwell M., Adluri B.S., Yuan Z., Newton J., Prevost P., Nodwell M.B., et al. ??? Direct heterobenzylic fluorination, difluorination and trifluoromethylthiolation with dibenzenesulfonamide derivatives. Chem Sci. 2018;9:5608–5613. doi: 10.1039/c8sc01221k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Furuya T., Strom A.E., Ritter T. Silver-mediated fluorination of functionalized aryl stannanes. J Am Chem Soc. 2009;131:1662–1663. doi: 10.1021/ja8086664. [DOI] [PubMed] [Google Scholar]
- 85.Tang P., Furuya T., Ritter T. Silver-catalyzed late-stage fluorination. J Am Chem Soc. 2010;132:12150–12154. doi: 10.1021/ja105834t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Berger F., Plutschack M.B., Riegger J., Yu W., Speicher S., Ho M., et al. Site-selective and versatile aromatic C‒H functionalization by thianthrenation. Nature. 2019;567:223–228. doi: 10.1038/s41586-019-0982-0. [DOI] [PubMed] [Google Scholar]
- 87.Li J., Chen J., Sang R., Ham W.S., Plutschack M.B., Berger F., et al. Photoredox catalysis with aryl sulfonium salts enables site-selective late-stage fluorination. Nat Chem. 2020;12:56–62. doi: 10.1038/s41557-019-0353-3. [DOI] [PubMed] [Google Scholar]
- 88.Watson D.A., Su M., Teverovskiy G., Zhang Y., García-Fortanet J., Kinzel T., et al. Formation of ArF from LPdAr(F): catalytic conversion of aryl tri-flates to aryl fluorides. Science. 2009;325:1661–1664. doi: 10.1126/science.1178239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sather A.C., Lee H.G., De La Rosa V.Y., Yang Y., Muller P., Buchwald S.L. A fluorinated ligand enables room-temperature and regioselec-tive Pd-catalyzed fluorination of aryl triflates and bromides. J Am Chem Soc. 2015;137:13433–13438. doi: 10.1021/jacs.5b09308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lee H.G., Milner P.J., Buchwald S.L. Pd-catalyzed nucleophilic fluorination of aryl bromides. J Am Chem Soc. 2014;136:3792–3795. doi: 10.1021/ja5009739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Fier P.S., Luo J., Hartwig J.F. Copper-mediated fluorination of arylboronate esters. Identification of a copper(III) fluoride complex. J Am Chem Soc. 2013;135:2552–2559. doi: 10.1021/ja310909q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Dorel R., Boehm P., Schwinger D.P., Hartwig J.F. Copper-mediated fluorination of aryl trisiloxanes with nucleo-philic fluoride. Chem Eur J. 2020;26:1759–1762. doi: 10.1002/chem.201905040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sandford C., Rasappan R., Aggarwal V.K. Synthesis of enantioenriched alkylfluorides by the fluorination of boronate complexes. J Am Chem Soc. 2015;137:10100–10103. doi: 10.1021/jacs.5b05848. [DOI] [PubMed] [Google Scholar]
- 94.Wright J.S., Sharninghausen L.S., Preshlock S., Brooks A.F., Sanford M.S., Scott P.J.H. Sequential Ir/Cu-mediated method for the meta-selective C‒H radiofluorination of (hetero)arenes. J Am Chem Soc. 2021;143:6915–6921. doi: 10.1021/jacs.1c00523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Chen W., Wang H., Tay N.E.S., Pistritto V.A., Li K.P., Zhang T., et al. Arene radiofluorination enabled by photoredox-mediated halide interconversion. Nat Chem. 2022;14:216–223. doi: 10.1038/s41557-021-00835-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Aggarwal T., Sushmita, Verma A.K. Achievements in fluorination using variable reagents through a deoxyfluorination reaction. Org Chem Front. 2021;8:6452–6468. [Google Scholar]
- 97.Rentmeister A., Arnold F.H., Fasan R. Chemo-enzymatic fluorination of unactivated organic compounds. Nat Chem Biol. 2009;5:26–28. doi: 10.1038/nchembio.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Tang P., Wang W., Ritter T. Deoxyfluorination of phenols. J Am Chem Soc. 2011;133:11482–11484. doi: 10.1021/ja2048072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Sladojevich F., Arlow S.I., Tang P., Ritter T. Late-stage deoxyfluorination of alcohols with PhenoFluor. J Am Chem Soc. 2013;135:2470–2473. doi: 10.1021/ja3125405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Fujimoto T., Ritter T. PhenoFluorMix: practical chemoselective deoxyfluorination of phenols. Org Lett. 2015;17:544–547. doi: 10.1021/ol5035518. [DOI] [PubMed] [Google Scholar]
- 101.Chen J. Late-stage deoxyfluorination of phenols with PhenoFluorMix. Org Synth. 2019;96:16–35. [Google Scholar]
- 102.Tay N.E.S., Chen W., Levens A., Pistritto V.A., Huang Z., Wu Z., et al. 19F- and 18F- arene deoxyfluorination via organic photoredox-catalysed polarity-reversed nucleophilic aromatic substitution. Nat Catal. 2020;3:734–742. doi: 10.1038/s41929-020-0495-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Schimler S.D., Cismesia M.A., Hanley P.S., Froese R.D., Jansma M.J., Bland D.C., et al. Nucleophilic deoxyfluorination of phenols via aryl fluorosulfonate intermediates. J Am Chem Soc. 2017;139:1452–1455. doi: 10.1021/jacs.6b12911. [DOI] [PubMed] [Google Scholar]
- 104.Laine D., Denavit V., Lessard O., Carrier L., Fecteau C.E., Johnson P.A., et al. Fluorine effect in nucleophilic fluorination at C4 of 1,6-anhydro-2,3-dideoxy-2,3-difluoro-beta-d-hexopyranose. Beilstein J Org Chem. 2020;16:2880–2887. doi: 10.3762/bjoc.16.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zhang W., Gu Y.C., Lin J.H., Xiao J.C. Dehydroxylative fluorination of tertiary alcohols. Org Lett. 2020;22:6642–6646. doi: 10.1021/acs.orglett.0c02438. [DOI] [PubMed] [Google Scholar]
- 106.Hamala V., Cervenkova Stastna L., Kurfirt M., Curinova P., Balouch M., Hrstka R., et al. The effect of deoxyfluorination and O-acylation on the cytotoxicity of N-acetyl-d-gluco- and d-galactosamine hemiacetals. Org Biomol Chem. 2021;19:4497–4506. doi: 10.1039/d1ob00497b. [DOI] [PubMed] [Google Scholar]
- 107.Ma J., Xu W., Xie J. Predictable site-selective radical fluorination of tertiary ethers. Sci China Chem. 2019;63:187–191. [Google Scholar]
- 108.Yin F., Wang Z., Li Z., Li C. Silver-catalyzed decarboxylative fluorination of aliphatic carboxylic acids in aqueous solution. J Am Chem Soc. 2012;134:10401–10404. doi: 10.1021/ja3048255. [DOI] [PubMed] [Google Scholar]
- 109.Meyer D., Jangra H., Walther F., Zipse H., Renaud P. A third generation of radical fluorinating agents based on N-fluoro-N-arylsulfonamides. Nat Commun. 2018;9:4888. doi: 10.1038/s41467-018-07196-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Thompson S., Lee S.J., Jackson I.M., Ichiishi N., Brooks A.F., Sanford M.S., et al. Synthesis of [18F]-gamma-fluoro-alpha,beta,-unsaturated esters and ketones via vinylogous 18F-fluorination of alpha-diazoacetates with [18F] AgF. Synthesis. 2019;51:4401–4407. doi: 10.1055/s-0039-1690012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Yoo W.J., Kondo J., Rodriguez-Santamaria J.A., Nguyen T.V.Q., Kobayashi S. Efficient synthesis of alpha-trifluoromethyl carboxylic acids and esters through fluorocarboxylation of gem-difluoroalkenes. Angew Chem Int Ed Engl. 2019;58:6772–6775. doi: 10.1002/anie.201902779. [DOI] [PubMed] [Google Scholar]
- 112.Hafliger J., Livingstone K., Daniliuc C.G., Gilmour R. Difluorination of alpha-(bromomethyl)styrenes via I(I)/I(III) catalysis: facile access to electrophilic linchpins for drug discovery. Chem Sci. 2021;12:6148–6152. doi: 10.1039/d1sc01132d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Lee E., Kamlet A.S., Powers D.C., Neumann C.N., Boursalian G.B., Furuya T., et al. A fluoride-derived electrophilic late-stage fluorination reagent for PET imaging. Science. 2011;334:639–642. doi: 10.1126/science.1212625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Preshlock S., Tredwell M., Gouverneur V. 18F‒ labeling of arenes and heteroarenes for applications in positron emission tomography. Chem Rev. 2016;116:719–766. doi: 10.1021/acs.chemrev.5b00493. [DOI] [PubMed] [Google Scholar]
- 115.Halder R., Ritter T. 18F-Fluorination: challenge and opportunity for organic chemists. J Org Chem. 2021;86:13873–13884. doi: 10.1021/acs.joc.1c01474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Wang Y., Lin Q., Shi H., Cheng D. Fluorine-18: radiochemistry and target-specific PET molecular probes design. Front Chem. 2022;10 doi: 10.3389/fchem.2022.884517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Daly B., Ling J., de Silva A.P. Current developments in fluorescent PET (photoinduced electron transfer) sensors and switches. Chem Soc Rev. 2015;44:4203–4211. doi: 10.1039/c4cs00334a. [DOI] [PubMed] [Google Scholar]
- 118.Campbell M.G., Ritter T. Modern carbon-fluorine bond forming reactions for aryl fluoride synthesis. Chem Rev. 2015;115:612–633. doi: 10.1021/cr500366b. [DOI] [PubMed] [Google Scholar]
- 119.Rosencher N., Arnaout L., Chabbouh T., Bellamy L. Rivaroxaban (Xarelto®) : efficacité et tolerance. Ann Fr Anesth Reanim. 2008;27:S22–S27. doi: 10.1016/S0750-7658(08)75143-8. [DOI] [PubMed] [Google Scholar]
- 120.FDA approves novel, dual targeted treatment for type 2 diabetes mellitus. Nurse Pract Am J Prim Health Care. 2022;47:48. [Google Scholar]
- 121.Shafiq A., Mahboob E., Samad M.A., Ur Rehman M.H., Tharwani Z.H. The dual role of empagliflozin: cardio renal protection in T2DM patients. Ann Med Surg (Lond) 2022;81 doi: 10.1016/j.amsu.2022.104555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.McCormack P.L. Cariprazine: first global approval. Drugs. 2015;75:2035–2043. doi: 10.1007/s40265-015-0494-7. [DOI] [PubMed] [Google Scholar]
- 123.Kolar M.H., Hobza P. Computer modeling of halogen bonds and other sigma-hole interactions. Chem Rev. 2016;116:5155–5187. doi: 10.1021/acs.chemrev.5b00560. [DOI] [PubMed] [Google Scholar]
- 124.Shinada N.K., de Brevern A.G., Schmidtke P. Halogens in protein‒ligand binding mechanism: a structural perspective. J Med Chem. 2019;62:9341–9356. doi: 10.1021/acs.jmedchem.8b01453. [DOI] [PubMed] [Google Scholar]
- 125.Wilcken R., Zimmermann M.O., Lange A., Joerger A.C., Boeckler F.M. Principles and applications of halogen bonding in medicinal chemistry and chemical biology. J Med Chem. 2013;56:1363–1388. doi: 10.1021/jm3012068. [DOI] [PubMed] [Google Scholar]
- 126.Scholfield M.R., Ford M.C., Vander Zanden C.M., Billman M.M., Ho P.S., Rappe A.K. Force field model of periodic trends in biomolecular halogen bonds. J Phys Chem B. 2015;119:9140–9149. doi: 10.1021/jp509003r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Parisini E., Metrangolo P., Pilati T., Resnati G., Terraneo G. Halogen bonding in halocarbon‒protein complexes: a structural survey. Chem Soc Rev. 2011;40:2267–2278. doi: 10.1039/c0cs00177e. [DOI] [PubMed] [Google Scholar]
- 128.Maignan S., Guilloteau J.P., Choi Sledeski Y.M., Becker M.R., Ewing W.R., Pauls H.W., et al. Molecular structures of human factor Xa complexed with ketopiperazine inhibitors: preference for a neutral group in the S1 pocket. J Med Chem. 2003;46:685–690. doi: 10.1021/jm0203837. [DOI] [PubMed] [Google Scholar]
- 129.Cheng Y.Y., Yuan H. Quantitative study of electrostatic and steric effects on physicochemical property and biological activity. J Mol Graphics Modell. 2006;24:219–226. doi: 10.1016/j.jmgm.2005.08.005. [DOI] [PubMed] [Google Scholar]
- 130.Jiang Y.R., Liu J.Y., Hu Y.H., Fujita T. Novel topological index for research on structure‒property relationships of complex organic compounds. J Comput Chem. 2003;24:842–849. doi: 10.1002/jcc.10237. [DOI] [PubMed] [Google Scholar]
- 131.Naumann K. Influence of chlorine substituents on biological activity of chemicals: a review. Pest Manag Sci. 2000;56:3–21. [Google Scholar]
- 132.Rodriguez R.A., Pan C.M., Yabe Y., Kawamata Y., Eastgate M.D., Baran P.S. Palau’chlor: a practical and reactive chlorinating reagent. J Am Chem Soc. 2014;136:6908–6911. doi: 10.1021/ja5031744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Konrad D.B., Frank J.A., Trauner D. Synthesis of redshifted azobenzene photoswitches by late-stage functionalization. Chem Eur J. 2016;22:4364–4368. doi: 10.1002/chem.201505061. [DOI] [PubMed] [Google Scholar]
- 134.Gao C., Li H., Liu M., Ding J., Huang X., Wu H., et al. Regioselective C‒H chlorination: towards the sequential difunctionalization of phenol derivatives and late-stage chlorination of bioactive compounds. RSC Adv. 2017;7:46636–46643. [Google Scholar]
- 135.Liang Y., Lin F., Adeli Y., Jin R., Jiao N. Efficient electrocatalysis for the preparation of (hetero)aryl chlo-rides and vinyl chloride with 1,2-dichloroethane. Angew Chem Int Ed Engl. 2019;58:4566–4570. doi: 10.1002/anie.201814570. [DOI] [PubMed] [Google Scholar]
- 136.Song S., Li X., Wei J., Wang W., Zhang Y., Ai L., et al. DMSO-catalysed late-stage chlorination of (hetero)arenes. Nat Catal. 2019;3:107–115. [Google Scholar]
- 137.Andrzejczyk N.E., Greer J.B., Nelson E., Zhang J., Rimoldi J.M., Gadepalli R.S.V., et al. Novel disinfection byproducts formed from the pharmaceutical gemfibrozil are bioaccumulative and elicit increased toxicity relative to the parent compound in marine polychaetes (neanthes arenaceodentata) Environ Sci Technol. 2020;54:11127–11136. doi: 10.1021/acs.est.0c01080. [DOI] [PubMed] [Google Scholar]
- 138.Bulloch D.N., Lavado R., Forsgren K.L., Beni S., Schlenk D., Larive C.K. Analytical and biological characterization of halogenated gemfibrozil produced through chlorination of wastewater. Environ Sci Technol. 2012;46:5583–5589. doi: 10.1021/es3006173. [DOI] [PubMed] [Google Scholar]
- 139.Wang W., Li X., Yang X., Ai L., Gong Z., Jiao N., et al. Oxoammonium salts are catalysing efficient and selective halogenation of olefins, alkynes and aromatics. Nat Commun. 2021;12:3873. doi: 10.1038/s41467-021-24174-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Feng M., Madegard L., Riomet M., Louis M., Champagne P.A., Pieters G., et al. Selective chlorination of iminosydnones for fast release of amide, sulfonamide and urea-containing drugs. Chem Commun. 2022;58:8500–8503. doi: 10.1039/d2cc02784d. [DOI] [PubMed] [Google Scholar]
- 141.Xie W., Wang M., Yang S., Chen Y., Feng J., Huang Y. C‒H chlorination of (hetero)anilines via photo/organo co-catalysis. Org Biomol Chem. 2022;20:5319–5324. doi: 10.1039/d2ob00834c. [DOI] [PubMed] [Google Scholar]
- 142.Levy J.N., Alegre-Requena J.V., Liu R., Paton R.S., McNally A. Selective halogenation of pyridines using designed phosphine reagents. J Am Chem Soc. 2020;142:11295–11305. doi: 10.1021/jacs.0c04674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Ghiazza C., Faber T., Gomez-Palomino A., Cornella J. Deaminative chlorination of aminoheterocycles. Nat Chem. 2022;14:78–84. doi: 10.1038/s41557-021-00812-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Posternak G., Tang X., Maisonneuve P., Jin T., Lavoie H., Daou S., et al. Functional characterization of a PROTAC directed against BRAF mutant V600E. Nat Chem Biol. 2020;16:1170–1178. doi: 10.1038/s41589-020-0609-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Cao H., Cheng Q., Studer A. Radical and ionic meta-C–H functionalization of pyridines, quinolines, and isoquinolines. Science. 2022;378:779–785. doi: 10.1126/science.ade6029. [DOI] [PubMed] [Google Scholar]
- 146.Hayashi T., Ligibel M., Sager E., Voss M., Hunziker J., Schroer K., et al. Evolved aliphatic halogenases enable regiocomplementary C‒H functionalization of a pharmaceutically relevant compound. Angew Chem Int Ed Engl. 2019;58:18535–18539. doi: 10.1002/anie.201907245. [DOI] [PubMed] [Google Scholar]
- 147.Buchler J., Malca S.H., Patsch D., Voss M., Turner N.J., Bornscheuer U.T., et al. Algorithm-aided engineering of aliphatic halogenase WelO5∗ for the asymmetric late-stage functionalization of soraphens. Nat Commun. 2022;13:371. doi: 10.1038/s41467-022-27999-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Duewel S., Schmermund L., Faber T., Harms K., Srinivasan V., Meggers E., et al. Directed evolution of an FeII-dependent halogenase for asymmet-ric C(sp3)–H chlorination. ACS Catal. 2019;10:1272–1277. [Google Scholar]
- 149.Fawcett A., Keller M.J., Herrera Z., Hartwig J.F. Site selective chlorination of C(sp3)-H bonds suitable for late-stage functionalization. Angew Chem Int Ed Engl. 2021;60:8276–8283. doi: 10.1002/anie.202016548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.McMillan A.J., Sienkowska M., Di Lorenzo P., Gransbury G.K., Chilton N.F., Salamone M., et al. Practical and selective sp3 C‒H bond chlorination via aminium radicals. Angew Chem Int Ed Engl. 2021;60:7132–7139. doi: 10.1002/anie.202100030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Jin J., Zhao Y., Kyne S.H., Farshadfar K., Ariafard A., Chan P.W.H. Copper(I)-catalysed site-selective C(sp3)-H bond chlorination of ketones, (E)-enones and alkylbenzenes by dichloramine-T. Nat Commun. 2021;12:4065. doi: 10.1038/s41467-021-23988-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Wang W., Yang X., Dai R., Yan Z., Wei J., Dou X., et al. Catalytic electrophilic halogenation of arenes with electron-withdrawing substituents. J Am Chem Soc. 2022;144:13415–13425. doi: 10.1021/jacs.2c06440. [DOI] [PubMed] [Google Scholar]
- 153.Djerassi C. Brominations with N-bromosuccinimide and related compounds. the Wohl-Ziegler reaction. Chem Rev. 1948;43:271–317. doi: 10.1021/cr60135a004. [DOI] [PubMed] [Google Scholar]
- 154.Prakash G.K.S., Mathew T., Hoole D., Esteves P.M., Wang Q., Rasul G., et al. N-Halosuccinimide/BF3−H2O, efficient electrophilic halogenating systems for aromatics. J Am Chem Soc. 2004;126:15770–15776. doi: 10.1021/ja0465247. [DOI] [PubMed] [Google Scholar]
- 155.Rozen S., Brand M., Lidor R. Aromatic bromination using bromine fluoride with no Friedel-Crafts catalyst. J Org Chem. 1988;53:5545–5547. [Google Scholar]
- 156.Patel T., McKeage K. Macitentan: first global approval. Drugs. 2014;74:127–133. doi: 10.1007/s40265-013-0156-6. [DOI] [PubMed] [Google Scholar]
- 157.Song S., Sun X., Li X., Yuan Y., Jiao N. Efficient and practical oxidative bromination and iodination of arenes and heteroarenes with dmso and hydrogen halide: a mild protocol for late-stage functionalization. Org Lett. 2015;17:2886–2889. doi: 10.1021/acs.orglett.5b00932. [DOI] [PubMed] [Google Scholar]
- 158.Sun B., Li L., Hu Q.W., Xie F., Zheng H.B., Niu H.M., et al. Design, synthesis and biological evaluation of novel macrocyclic bisbibenzyl analogues as tubulin polymerization inhibitors. Eur J Med Chem. 2016;121:484–499. doi: 10.1016/j.ejmech.2016.06.007. [DOI] [PubMed] [Google Scholar]
- 159.Dalai P.G., Palit K., Panda N. Generation of dimethyl sulfoxide coordinated thermally stable halogen cation pools for C‒H halogenation. Adv Synth Catal. 2022;364:1031–1038. [Google Scholar]
- 160.Dutta H.S., Khan B., Khan A.A., Raziullah, Ahmad A., Kant R., et al. Metal-free, oxidant-free, site-selective C‒H halogenations to ami-noquinolines at room temperature using N-halosaccharins. ChemistrySelect. 2017;2:6488–6492. [Google Scholar]
- 161.Goitia A., Gomez-Bengoa E., Correa A. Selective C(sp2)–H halogenation of "click" 4-aryl-1,2,3-triazoles. Org Lett. 2017;19:962–965. doi: 10.1021/acs.orglett.7b00275. [DOI] [PubMed] [Google Scholar]
- 162.Tan Z., Liu Y., Helmy R., Rivera N.R., Hesk D., Tyagarajan S., et al. Electrochemical bromination of late stage intermediates and drug molecules. Tetrahedron Lett. 2017;58:3014–3018. [Google Scholar]
- 163.Fisher B.F., Snodgrass H.M., Jones K.A., Andorfer M.C., Lewis J.C. Site-selective C‒H halogenation using flavin-dependent halogenases identified via family-wide activity profiling. ACS Cent Sci. 2019;5:1844–1856. doi: 10.1021/acscentsci.9b00835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Ma Z., Lu H., Liao K., Chen Z. Tungstate-catalyzed biomimetic oxidative halogenation of (hetero)arene under mild condition. iScience. 2020;23 doi: 10.1016/j.isci.2020.101072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Longevial J.F., Miyagawa K., Shinokubo H. Site-selective halogenation on meso-mesityl substituents of 10,20-dimesityl-5,15-diazaporphyrins with an AuX3/AgOTf combination. Dalton Trans. 2020;49:14786–14789. doi: 10.1039/d0dt02727h. [DOI] [PubMed] [Google Scholar]
- 166.Mamontov A., Martin-Mingot A., Metayer B., Karam O., Zunino F., Bouazza F., et al. Complementary site-selective halogenation of nitrogen-containing (hetero)aromatics with superacids. Chem Eur J. 2020;26:10411–10416. doi: 10.1002/chem.202000902. [DOI] [PubMed] [Google Scholar]
- 167.Gupta S.S., Manisha Kumar R., Dhiman A.K., Sharma U. Predictable site-selective functionalization: promoter group assisted para-halogenation of N-substituted (hetero)aromatics under metal-free condition. Org Biomol Chem. 2021;19:9675–9687. doi: 10.1039/d1ob02000e. [DOI] [PubMed] [Google Scholar]
- 168.Boyle B.T., Levy J.N., de Lescure L., Paton R.S., McNally A. Halogenation of the 3-position of pyridines through Zincke imine intermediates. Science. 2022;378:773–779. doi: 10.1126/science.add8980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Li Y., Mou T., Lu L., Jiang X. Visible-light-promoted oxidative halogenation of alkynes. Chem Commun. 2019;55:14299–14302. doi: 10.1039/c9cc07655g. [DOI] [PubMed] [Google Scholar]
- 170.Weber J.M., Leung J.O., Swanson S.J., Idler K.B., McAlpine J.B. An erythromycin derivative produced by targeted gene disruption in Saccharopolyspora erythraea. Science. 1991;252:114–117. doi: 10.1126/science.2011746. [DOI] [PubMed] [Google Scholar]
- 171.Zambias R.A., Hammond M.L., Heck J.V., Bartizal K., Trainor C., Abruzzo G., et al. Preparation and structure‒activity relationships of simplified analogs of the antifungal agent cilofungin: a total synthesis approach. J Med Chem. 1992;35:2843–2855. doi: 10.1021/jm00093a018. [DOI] [PubMed] [Google Scholar]
- 172.Xu L., Liu H., Hong A., Vivian R., Murray B.P., Callebaut C., et al. Structure‒activity relationships of diamine inhibitors of cytochrome P450 (CYP) 3A as novel pharmacoenhancers. Part II: P2/P3 region and discovery of cobicistat (GS-9350) Bioorg Med Chem Lett. 2014;24:995–999. doi: 10.1016/j.bmcl.2013.12.057. [DOI] [PubMed] [Google Scholar]
- 173.Davies N.M. Clinical pharmacokinetics of ibuprofen. Clin Pharmacokinet. 1998;34:101–154. doi: 10.2165/00003088-199834020-00002. [DOI] [PubMed] [Google Scholar]
- 174.Sanderson H., Thomsen M. Comparative analysis of pharmaceuticals versus industrial chemicals acute aquatic toxicity classification according to the United Nations classification system for chemicals. Assessment of the (Q)SAR predictability of pharmaceuticals acute aquatic toxicity and their predominant acute toxic mode-of-action. Toxicol Lett. 2009;187:84–93. doi: 10.1016/j.toxlet.2009.02.003. [DOI] [PubMed] [Google Scholar]
- 175.Kulikov A.U., Galat M.N. Comparison of C18 silica bonded phases selectivity in micellar liquid chromatography. J Separ Sci. 2009;32:1340–1350. doi: 10.1002/jssc.200800716. [DOI] [PubMed] [Google Scholar]
- 176.Ottenbacher R.V., Talsi E.P., Bryliakov K.P. Recent progress in catalytic oxygenation of aromatic C‒H groups with the environmentally benign oxidants H2O2 and O2. Appl Organomet Chem. 2020;34 [Google Scholar]
- 177.Fessner N.D. P450 monooxygenases enable rapid late-stage diversification of natural products via C‒H bond activation. ChemCatChem. 2019;11:2226–2242. doi: 10.1002/cctc.201801829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.White M.C., Zhao J. Aliphatic C‒H oxidations for late-stage functionalization. J Am Chem Soc. 2018;140:13988–14009. doi: 10.1021/jacs.8b05195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Alford J.S., Abascal N.C., Shugrue C.R., Colvin S.M., Romney D.K., Miller S.J. Aspartyl oxidation catalysts that dial in functional group selectivity, along with regio- and stereoselectivity. ACS Cent Sci. 2016;2:733–739. doi: 10.1021/acscentsci.6b00237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Xia S., Gan L., Wang K., Li Z., Ma D. Copper-catalyzed hydroxylation of (hetero)aryl halides under mild conditions. J Am Chem Soc. 2016;138:13493–13496. doi: 10.1021/jacs.6b08114. [DOI] [PubMed] [Google Scholar]
- 181.Ma D., Jiang Y., Maurer S., Liu W., Zhang X. An efficient synthesis of phenol via CuI/8-hydroxyquinoline-catalyzed hydroxylation of aryl halides and potassium hydroxide. Synlett. 2010;2010:976–978. [Google Scholar]
- 182.Gallon B.J., Kojima R.W., Kaner R.B., Diaconescu P.L. Palladium nanoparticles supported on polyaniline nanofibers as a semi-heterogeneous catalyst in water. Angew Chem Int Ed. 2007;46:7251–7254. doi: 10.1002/anie.200701389. [DOI] [PubMed] [Google Scholar]
- 183.Schulz T., Torborg C., Schäffner B., Huang J., Zapf A., Kadyrov R., et al. Practical imidazole-based phosphine ligands for selective palladium-catalyzed hydroxylation of aryl halides. Angew Chem Int Ed. 2009;48:918–921. doi: 10.1002/anie.200804898. [DOI] [PubMed] [Google Scholar]
- 184.Anderson K.W., Ikawa T., Tundel R.E., Buchwald S.L. The selective reaction of aryl halides with KOH: synthesis of phenols, aromatic ethers, and benzofurans. J Am Chem Soc. 2006;128:10694–10695. doi: 10.1021/ja0639719. [DOI] [PubMed] [Google Scholar]
- 185.Cheung C.W., Buchwald S.L. Palladium-catalyzed hydroxylation of aryl and heteroaryl halides enabled by the use of a palladacycle precatalyst. J Org Chem. 2014;79:5351–5358. doi: 10.1021/jo500662s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Hu X., Maimone T.J. Four-step synthesis of the antimalarial cardamom peroxide via an oxygen stitching strategy. J Am Chem Soc. 2014;136:5287–5290. doi: 10.1021/ja502208z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Xuan J., Zhu A., Ma B., Ding H. Diastereoselective synthesis of the hydroperoxide-keto form of (+/‒)-Steenkrotin B. Org Lett. 2018;20:4153–4156. doi: 10.1021/acs.orglett.8b01875. [DOI] [PubMed] [Google Scholar]
- 188.Guo W., Liu Y., Li C. Asymmetric catalytic 1,2-hydroperoxidation of Isatin-derived ketimine with hydrogen peroxide in the crowding environment of PEGs. Org Lett. 2017;19:1044–1047. doi: 10.1021/acs.orglett.7b00032. [DOI] [PubMed] [Google Scholar]
- 189.Joarder D., Gayen S., Sarkar R., Bhattacharya R., Roy S., Maiti D.K. (Ar-tpy)Ru(II)(ACN)3: a water-soluble catalyst for aldehyde amidation, olefin oxo-scissoring, and alkyne oxygenation. J Org Chem. 2019;84:8468–8480. doi: 10.1021/acs.joc.9b00487. [DOI] [PubMed] [Google Scholar]
- 190.Liu J., Wen X., Qin C., Li X., Luo X., Sun A., et al. Oxygenation of simple olefins through selective allylic C‒C bond cleavage: a direct approach to cinnamyl aldehydes. Angew Chem Int Ed Engl. 2017;56:11940–11944. doi: 10.1002/anie.201705671. [DOI] [PubMed] [Google Scholar]
- 191.Wan J.P., Gao Y., Wei L. Recent advances in transition-metal-free oxygenation of alkene C=C double bonds for carbonyl generation. Chem Asian J. 2016;11:2092–2102. doi: 10.1002/asia.201600671. [DOI] [PubMed] [Google Scholar]
- 192.Cramer J., Sager C.P., Ernst B. Hydroxyl groups in synthetic and natural-product-derived therapeutics: a perspective on a common functional group. J Med Chem. 2019;62:8915–8930. doi: 10.1021/acs.jmedchem.9b00179. [DOI] [PubMed] [Google Scholar]
- 193.Stokker G.E., Hoffman W.F., Alberts A.W., Cragoe E.J., Jr., Deana A.A., Gilfillan J.L., et al. 3-Hydroxy-3-methylglutaryl-coenzyme a reductase inhibitors. I. Structural modification of 5-substituted 3,5-dihydroxypentanoic acids and their lactone derivatives. J Med Chem. 1985;28:347–358. doi: 10.1021/jm00381a014. [DOI] [PubMed] [Google Scholar]
- 194.Roth B.D. 1 the discovery and development of atorvastatin, a potent novel hypolipidemic agent. Prog Med Chem. 2002;40:1–22. doi: 10.1016/s0079-6468(08)70080-8. [DOI] [PubMed] [Google Scholar]
- 195.Sica D.A., Gehr T.W.B., Ghosh S. Clinical pharmacokinetics of losartan. Clin Pharmacokinet. 2005;44:797–814. doi: 10.2165/00003088-200544080-00003. [DOI] [PubMed] [Google Scholar]
- 196.Copeland R.A., Pompliano D.L., Meek T.D. Drug–target residence time and its implications for lead optimization. Nat Rev Drug Discov. 2006;5:730–739. doi: 10.1038/nrd2082. [DOI] [PubMed] [Google Scholar]
- 197.Charlton S.N., Hayes M.A. Oxygenating biocatalysts for hydroxyl functionalisation in drug discovery and development. ChemMedChem. 2022;17 doi: 10.1002/cmdc.202200115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Nelson H.S. Advair: combination treatment with fluticasone propionate/salmeterol in the treatment of asthma. J Allergy Clin Immunol. 2001;107:397–416. doi: 10.1067/mai.2001.112939. [DOI] [PubMed] [Google Scholar]
- 199.Shan G., Yang X., Ma L., Rao Y. Pd-catalyzed C‒H oxygenation with TFA/TFAA: expedient access to oxygen-containing heterocycles and late-stage drug modification. Angew Chem Int Ed Engl. 2012;51:13070–13074. doi: 10.1002/anie.201207458. [DOI] [PubMed] [Google Scholar]
- 200.Shan G., Han X., Lin Y., Yu S., Rao Y. Broadening the catalyst and reaction scope of regio- and chemoselective C‒H oxygenation: a convenient and scalable approach to 2-acylphenols by intriguing Rh(II) and Ru(II) catalysis. Org Biomol Chem. 2013;11:2318–2322. doi: 10.1039/c3ob27457h. [DOI] [PubMed] [Google Scholar]
- 201.Yuan C., Liang Y., Hernandez T., Berriochoa A., Houk K.N., Siegel D. Metal-free oxidation of aromatic carbon-hydrogen bonds through a reverse-rebound mechanism. Nature. 2013;499:192–196. doi: 10.1038/nature12284. [DOI] [PubMed] [Google Scholar]
- 202.Liang Y.F., Wang X., Yuan Y., Liang Y., Li X., Jiao N. Ligand-promoted Pd-catalyzed oxime ether directed C‒H hydroxylation of arenes. ACS Catal. 2015;5:6148–6152. [Google Scholar]
- 203.Borgel J., Tanwar L., Berger F., Ritter T. Late-stage aromatic C‒H oxygenation. J Am Chem Soc. 2018;140:16026–16031. doi: 10.1021/jacs.8b09208. [DOI] [PubMed] [Google Scholar]
- 204.Sang R., Korkis S.E., Su W., Ye F., Engl P.S., Berger F., et al. Site-selective C‒H oxygenation via aryl sulfonium salts. Angew Chem Int Ed. 2019;58:16161–16166. doi: 10.1002/anie.201908718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Zhao Y., Yu C., Liang W., Atodiresei I.L., Patureau F.W. TEMPO-mediated late stage photochemical hydroxylation of biaryl sulfonium salts. Chem Commun. 2022;58:2846–2849. doi: 10.1039/d1cc07057f. [DOI] [PubMed] [Google Scholar]
- 206.Li Z., Wang Z., Chekshin N., Qian S., Qiao J.X., Cheng P.T., et al. A tautomeric ligand enables directed C‒H hydroxylation with molecular oxygen. Science. 2021;372:1452–1457. doi: 10.1126/science.abg2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Cheng L., Wang H., Cai H., Zhang J., Gong X., Han W. Iron-catalyzed arene C‒H hydroxylation. Science. 2021;374:77–81. doi: 10.1126/science.abj0731. [DOI] [PubMed] [Google Scholar]
- 208.Andrade Sampedro P., Matxain J.M., Correa A. Ru-catalyzed C‒H hydroxylation of tyrosine-containing di- and tripeptides toward the assembly of L-DOPA derivatives. Adv Synth Catal. 2022;364:2072–2079. [Google Scholar]
- 209.Li S., Chaulagain M.R., Knauff A.R., Podust L.M., Montgomery J., Sherman D.H. Selective oxidation of carbolide C‒H bonds by an engineered macrolide P450 mono-oxygenase. Proc Natl Acad Sci USA. 2009;106:18463–18468. doi: 10.1073/pnas.0907203106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Zhang K., Shafer B.M., Demars M.D., 2nd, Stern H.A., Fasan R. Controlled oxidation of remote sp3 C‒H bonds in artemisinin via P450 catalysts with fine-tuned regio- and stereoselectivity. J Am Chem Soc. 2012;134:18695–18704. doi: 10.1021/ja3073462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Kolev J.N., O'Dwyer K.M., Jordan C.T., Fasan R. Discovery of potent parthenolide-based antileukemic agents enabled by late-stage P450-mediated C‒H functionalization. ACS Chem Biol. 2014;9:164–173. doi: 10.1021/cb400626w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Le-Huu P., Heidt T., Claasen B., Laschat S., Urlacher V.B. Chemo-, regio-, and stereoselective oxidation of the monocyclic diterpenoid β-cembrenediol by P450 BM3. ACS Catal. 2015;5:1772–1780. [Google Scholar]
- 213.Espinoza R.V., Haatveit K.C., Grossman S.W., Tan J.Y., McGlade C.A., Khatri Y., et al. Engineering P450 TamI as an iterative biocatalyst for selective late-stage C‒H functionalization and epoxidation of Tirandamycin antibiotics. ACS Catal. 2021;11:8304–8316. doi: 10.1021/acscatal.1c01460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Espinoza R.V., Maskeri M.A., Turlik A., Nangia A., Khatri Y., Montgomery J., et al. Epoxidation and late-stage C‒H functionalization by P450 TamI are mediated by variant heme-iron oxidizing species. ACS Catal. 2022;12:3731–3742. [Google Scholar]
- 215.Alwaseem H., Giovani S., Crotti M., Welle K., Jordan C.T., Ghaemmaghami S., et al. Comprehensive structure-activity profiling of micheliolide and its targeted proteome in leukemia cells via probe-guided late-stage C‒H functionalization. ACS Cent Sci. 2021;7:841–857. doi: 10.1021/acscentsci.0c01624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Liu W., Li X., Chen J., Li T., Dong M., Lei X. Site-selective and metal-free aliphatic C‒H oxidation enabled synthesis of [5,24,25-D3]-(25S)-Δ7-Dafachronic acid. Chem Eur J. 2015;21:5345–5349. doi: 10.1002/chem.201500324. [DOI] [PubMed] [Google Scholar]
- 217.Chen M.S., White M.C. A predictably selective aliphatic C‒H oxidation reaction for complex molecule synthesis. Science. 2007;318:783–787. doi: 10.1126/science.1148597. [DOI] [PubMed] [Google Scholar]
- 218.Nanjo T., de Lucca E.C., Jr., White M.C. Remote, late-stage oxidation of aliphatic C‒H bonds in amide-containing molecules. J Am Chem Soc. 2017;139:14586–14591. doi: 10.1021/jacs.7b07665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Osberger T.J., Rogness D.C., Kohrt J.T., Stepan A.F., White M.C. Oxidative diversification of amino acids and peptides by small-molecule iron catalysis. Nature. 2016;537:214–219. doi: 10.1038/nature18941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Chambers R.K., Zhao J., Delaney C.P., White M.C. Chemoselective tertiary C‒H hydroxylation for late-stage functionalization with Mn(PDP)/Chloroacetic acid catalysis. Adv Synth Catal. 2020;362:417–423. doi: 10.1002/adsc.201901472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Kim K.E., Adams A.M., Chiappini N.D., Du Bois J., Stoltz B.M. Cyanthiwigin natural product core as a complex molecular scaffold for comparative late-stage C‒H functionalization studies. J Org Chem. 2018;83:3023–3033. doi: 10.1021/acs.joc.7b03291. [DOI] [PubMed] [Google Scholar]
- 222.Tanwar L., Borgel J., Ritter T. Synthesis of benzylic alcohols by C‒H oxidation. J Am Chem Soc. 2019;141:17983–17988. doi: 10.1021/jacs.9b09496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Zheng X., Cui X.X., Gao Z., Zhao Y., Shi Y., Huang M.T., et al. Inhibitory effect of dietary atorvastatin and celecoxib together with voluntary running wheel exercise on the progression of androgen-dependent LNCaP prostate tumors to androgen independence. Exp Ther Med. 2011;2:221–228. doi: 10.3892/etm.2011.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Penning T.D., Talley J.J., Bertenshaw S.R., Carter J.S., Collins P.W., Docter S., et al. Synthesis and biological evaluation of the 1,5-diarylpyrazole class of cyclooxygenase-2 inhibitors: identification of 4-[5-(4-methylphenyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl]benzenesulfonamide (SC-58635, Celecoxib) J Med Chem. 1997;40:1347–1365. doi: 10.1021/jm960803q. [DOI] [PubMed] [Google Scholar]
- 225.Saito M., Kawamata Y., Meanwell M., Navratil R., Chiodi D., Carlson E., et al. N-Ammonium ylide mediators for electrochemical C‒H oxidation. J Am Chem Soc. 2021;143:7859–7867. doi: 10.1021/jacs.1c03780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Fier P.S., Maloney K.M. Reagent design and ligand evolution for the development of a mild copper-catalyzed hydroxylation reaction. Org Lett. 2017;19:3033–3036. doi: 10.1021/acs.orglett.7b01403. [DOI] [PubMed] [Google Scholar]
- 227.Fier P.S., Maloney K.M. Synthesis of complex phenols enabled by a rationally designed hydroxide surrogate. Angew Chem Int Ed Engl. 2017;56:4478–4482. doi: 10.1002/anie.201700244. [DOI] [PubMed] [Google Scholar]
- 228.Fang Y., Zhao R., Yao Y., Liu Y., Chang D., Yao M., et al. Trichloroacetonitrile as an efficient activating agent for the ipso-hydroxylation of arylboronic acids to phenolic compounds. Org Biomol Chem. 2019;17:7558–7563. doi: 10.1039/c9ob01568j. [DOI] [PubMed] [Google Scholar]
- 229.Upadhyay R., Singh D., Maurya S.K. Highly efficient heterogeneous V2O5@TiO2 catalyzed the rapid transformation of boronic acids to phenols. Eur J Org Chem. 2021;2021:3925–3931. [Google Scholar]
- 230.Song S., Huang X., Liang Y.F., Tang C., Li X., Jiao N. From simple organobromides or olefins to highly value-added bromohydrins: a versatile performance of dimethyl sulfoxide. Green Chem. 2015;17:2727–2731. [Google Scholar]
- 231.Liang Y.F., Li X., Wang X., Zou M., Tang C., Liang Y., et al. Conversion of simple cyclohexanones into catechols. J Am Chem Soc. 2016;138:12271–12277. doi: 10.1021/jacs.6b07269. [DOI] [PubMed] [Google Scholar]
- 232.Hartwig J.F., Larsen M.A. Undirected, homogeneous C‒H bond functionalization: challenges and opportunities. ACS Cent Sci. 2016;2:281–292. doi: 10.1021/acscentsci.6b00032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Schatz A., Bugle E., Waksman S.A. Streptomycin, a substance exhibiting antibiotic activity against gram-positive and gram-negative bacteria. Proc Soc Exp Biol Med. 1944;55:66–69. doi: 10.1097/01.blo.0000175887.98112.fe. [DOI] [PubMed] [Google Scholar]
- 234.Umbreit Wayne W., Tonhazy N.E. The action of streptomycin III. J Bacteriol. 1949;58:769–776. doi: 10.1128/jb.58.6.769-776.1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Gampe C., Verma V.A. Curse or cure? A perspective on the developability of aldehydes as active pharmaceutical ingredients. J Med Chem. 2020;63:14357–14381. doi: 10.1021/acs.jmedchem.0c01177. [DOI] [PubMed] [Google Scholar]
- 236.Chen M.S., White M.C. Combined effects on selectivity in Fe-catalyzed methylene oxidation. Science. 2010;327:566–571. doi: 10.1126/science.1183602. [DOI] [PubMed] [Google Scholar]
- 237.Zhao J., Nanjo T., de Lucca E.C., Jr., White M.C. Chemoselective methylene oxidation in aromatic molecules. Nat Chem. 2019;11:213–221. doi: 10.1038/s41557-018-0175-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Ottenbacher R.V., Samsonenko D.G., Nefedov A.A., Talsi E.P., Bryliakov K.P. Mn aminopyridine oxidase mimics: switching between biosynthetic-like and xenobiotic regioselectivity in C‒H oxidation of (‒)-ambroxide. J Catal. 2021;399:224–229. [Google Scholar]
- 239.Zhou J., Jia M., Song M., Huang Z., Steiner A., An Q., et al. Chemoselective oxyfunctionalization of functionalized benzylic compounds with a manganese catalyst. Angew Chem Int Ed Engl. 2022;61 doi: 10.1002/anie.202205983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Sterckx H., De Houwer J., Mensch C., Herrebout W., Tehrani K.A., Maes B.U. Base metal-catalyzed benzylic oxidation of (aryl)(heteroaryl)methanes with molecular oxygen. Beilstein J Org Chem. 2016;12:144–153. doi: 10.3762/bjoc.12.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Griffiths R.J., Burley G.A., Talbot E.P. Transition-metal-free amine oxidation: a chemoselective strategy for the late-stage formation of lactams. Org Lett. 2017;19:870–873. doi: 10.1021/acs.orglett.7b00021. [DOI] [PubMed] [Google Scholar]
- 242.Zhu Y., Shao L.D., Deng Z.T., Bao Y., Shi X., Zhao Q.S. PIDA/I2-mediated alpha- and beta-C(sp3)–H bond dual functionalization of tertiary amines. J Org Chem. 2018;83:10166–10174. doi: 10.1021/acs.joc.8b01424. [DOI] [PubMed] [Google Scholar]
- 243.Geng P., Tang Y., Pan G., Wang W., Hu J., Cai Y. Ag-C3N4-based heterogeneous photocatalyst for visible light mediated aerobic benzylic C‒H oxygenations. Green Chem. 2019;21:6116–6122. [Google Scholar]
- 244.Hu P., Tan M., Cheng L., Zhao H., Feng R., Gu W.J., et al. Bio-inspired iron-catalyzed oxidation of alkylarenes enables late-stage oxidation of complex methylarenes to arylaldehydes. Nat Commun. 2019;10:2425. doi: 10.1038/s41467-019-10414-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Thiruvengetam P., Chand D.K. Controlled and predictably selective oxidation of activated and unactivated C(sp3)-H bonds catalyzed by a molybdenum-based metallomicellar catalyst in water. J Org Chem. 2022;87:4061–4077. doi: 10.1021/acs.joc.1c02855. [DOI] [PubMed] [Google Scholar]
- 246.Olivo G., Capocasa G., Ticconi B., Lanzalunga O., Di Stefano S., Costas M. Predictable selectivity in remote C‒H oxidation of steroids: analysis of substrate binding mode. Angew Chem Int Ed Engl. 2020;59:12703–12708. doi: 10.1002/anie.202003078. [DOI] [PubMed] [Google Scholar]
- 247.Mandigma M.J.P., Zurauskas J., MacGregor C.I., Edwards L.J., Shahin A., d'Heureuse L., et al. An organophotocatalytic late-stage N-CH3 oxidation of trialkylamines to N-formamides with O2 in continuous flow. Chem Sci. 2022;13:1912–1924. doi: 10.1039/d1sc05840a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Robert A., Dechy-Cabaret O., Cazelles J., Meunier B. From mechanistic studies on artemisinin derivatives to new modular antimalarial drugs. Acc Chem Res. 2002;35:167–174. doi: 10.1021/ar990164o. [DOI] [PubMed] [Google Scholar]
- 249.Wu Y. How might qinghaosu (artemisinin) and related compounds kill the intraerythrocytic malaria parasite? A chemist's view. Acc Chem Res. 2002;35:255–259. doi: 10.1021/ar000080b. [DOI] [PubMed] [Google Scholar]
- 250.Posner G.H., O'Neill P.M. Knowledge of the proposed chemical mechanism of action and cytochrome P450 metabolism of antimalarial trioxanes like artemisinin allows rational design of new antimalarial peroxides. Acc Chem Res. 2004;37:397–404. doi: 10.1021/ar020227u. [DOI] [PubMed] [Google Scholar]
- 251.Nicolaou K.C., Lu M., Totokotsopoulos S., Heretsch P., Giguere D., Sun Y.P., et al. Synthesis and biological evaluation of epidithio-, epitetrathio-, and bis-(methylthio)diketopiperazines: synthetic methodology, enantioselective total synthesis of epicoccin G, 8,8′-epi-ent-rostratin B, gliotoxin, gliotoxin G, emethallicin E, and haematocin and discovery of new antiviral and antimalarial agents. J Am Chem Soc. 2012;134:17320–17332. doi: 10.1021/ja308429f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Opsenica I., Opsenica D., Smith K.S., Milhous W.K., Šolaja B.A. Chemical stability of the peroxide bond enables diversified synthesis of potent tetraoxane antimalarials. J Med Chem. 2008;51:2261–2266. doi: 10.1021/jm701417a. [DOI] [PubMed] [Google Scholar]
- 253.Griesbeck A.G., Neudörfl J., Hörauf A., Specht S., Raabe A. Antimalarial peroxide dyads from natural artemisinin and hydroxyalkylated 1,2,4-trioxanes. J Med Chem. 2009;52:3420–3423. doi: 10.1021/jm9002523. [DOI] [PubMed] [Google Scholar]
- 254.Žmitek K., Zupan M., Stavber S., Iskra J. Iodine as a catalyst for efficient conversion of ketones to gem-dihydroperoxides by aqueous hydrogen peroxide. Org Lett. 2006;8:2491–2494. doi: 10.1021/ol060590r. [DOI] [PubMed] [Google Scholar]
- 255.Sagadevan A., Hwang K.C., Su M.D. Singlet oxygen-mediated selective C‒H bond hydroperoxidation of ethereal hydrocarbons. Nat Commun. 2017;8:1812. doi: 10.1038/s41467-017-01906-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Inoa J., Patel M., Dominici G., Eldabagh R., Patel A., Lee J., et al. Benzylic hydroperoxidation via visible-light-induced C(sp3)-H activation. J Org Chem. 2020;85:6181–6187. doi: 10.1021/acs.joc.0c00385. [DOI] [PubMed] [Google Scholar]
- 257.Leonard D.K., Li W., Junge K., Beller M. Improved bimetallic cobalt-manganese catalysts for selective oxidative cleavage of Morpholine derivatives. ACS Catal. 2019;9:11125–11129. [Google Scholar]
- 258.Liu B., Cheng L., Hu P., Xu F., Li D., Gu W.J., et al. Iron-catalyzed oxidative C‒C(vinyl) sigma-bond cleavage of allylarenes to aryl aldehydes at room temperature with ambient air. Chem Commun. 2019;55:4817–4820. doi: 10.1039/c9cc01995b. [DOI] [PubMed] [Google Scholar]
- 259.Li J., Wei J., Zhu B., Wang T., Jiao N. Cu-catalyzed oxygenation of alkene-tethered amides with O2via unactivated C=C bond cleavage: a direct approach to cyclic imides. Chem Sci. 2019;10:9099–9103. doi: 10.1039/c9sc03175h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Petsi M., Orfanidou M., Zografos A.L. Organocatalytic epoxidation and allylic oxidation of alkenes by molecular oxygen. Green Chem. 2021;23:9172–9178. [Google Scholar]
- 261.Wang X., Fan Y., Li G., Li H. The efficacy of escitalopram in major depressive disorder: a multicenter randomized, placebo-controlled double-blind study. Int Clin Psychopharmacol. 2021;36:133–139. doi: 10.1097/YIC.0000000000000350. [DOI] [PubMed] [Google Scholar]
- 262.Rosenzweig E.B. Tadalafil for the treatment of pulmonary arterial hypertension. Expet Opin Pharmacother. 2010;11:127–132. doi: 10.1517/14656560903413542. [DOI] [PubMed] [Google Scholar]
- 263.Arif S.A., Poon H. Tadalafil: a long-acting phosphodiesterase-5 inhibitor for the treatment of pulmonary arterial hypertension. Clin Therapeut. 2011;33:993–1004. doi: 10.1016/j.clinthera.2011.06.008. [DOI] [PubMed] [Google Scholar]
- 264.McGrath N.A., Brichacek M., Njardarson J.T. A graphical journey of innovative organic architectures that have improved our lives. J Chem Educ. 2010;87:1348–1349. [Google Scholar]
- 265.Ruffoni A., Juliá F., Svejstrup T.D., McMillan A.J., Douglas J.J., Leonori D. Practical and regioselective amination of arenes using alkyl amines. Nat Chem. 2019;11:426–433. doi: 10.1038/s41557-019-0254-5. [DOI] [PubMed] [Google Scholar]
- 266.Kaplan A.L., Confair D.N., Kim K., Barros-Álvarez X., Rodriguiz R.M., Yang Y., et al. Bespoke library docking for 5-HT2A receptor agonists with antidepressant activity. Nature. 2022;610:582–591. doi: 10.1038/s41586-022-05258-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Lim Y.J., Kuang Y., Wu J., Yao S.Q. Late-stage C(sp2)−H functionalization: a powerful toolkit to arm natural products for in situ proteome profiling? Chem Eur J. 2020;27:3575–3580. doi: 10.1002/chem.202004373. [DOI] [PubMed] [Google Scholar]
- 268.Ruan X., Zhu L., Chen B. Adsorptive characteristics of the siloxane surfaces of reduced-charge bentonites saturated with tetramethylammonium cation. Environ Sci Technol. 2008;42:7911–7917. doi: 10.1021/es801034h. [DOI] [PubMed] [Google Scholar]
- 269.Harden J., Jewell A., Donaldson F.P., Nyman M.C. Benzidine transformation processes in natural sediments. Environ Toxicol Chem. 2006;25:1969–1974. doi: 10.1897/05-274r.1. [DOI] [PubMed] [Google Scholar]
- 270.Liu X., Hefesha H., Scriba G., Fahr A. Retention behavior of neutral and positively and negatively charged solutes on an immobilized-artificial-membrane (IAM) stationary phase. Helv Chim Acta. 2008;91:1505–1512. [Google Scholar]
- 271.Bengtson G., Panek D., Fritsch D. Hydrogenation of acetophenone in a pervaporative catalytic membrane reactor with online mass spectrometric monitoring. J Membr Sci. 2007;293:29–35. [Google Scholar]
- 272.Chen Z., Weber S.G. High-throughput method for lipophilicity measurement. Anal Chem. 2006;79:1043–1049. doi: 10.1021/ac061649a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Bi E., Schmidt T.C., Haderlein S.B. Sorption of heterocyclic organic compounds to reference soils: column studies for process identification. Environ Sci Technol. 2006;40:5962–5970. doi: 10.1021/es060470e. [DOI] [PubMed] [Google Scholar]
- 274.Richter M.F., Drown B.S., Riley A.P., Garcia A., Shirai T., Svec R.L., et al. Predictive compound accumulation rules yield a broad-spectrum antibiotic. Nature. 2017;545:299–304. doi: 10.1038/nature22308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Chiellini G., Nesi G., Digiacomo M., Malvasi R., Espinoza S., Sabatini M., et al. Design, synthesis, and evaluation of thyronamine analogues as novel potent mouse trace amine associated receptor 1 (mTAAR1) agonists. J Med Chem. 2015;58:5096–5107. doi: 10.1021/acs.jmedchem.5b00526. [DOI] [PubMed] [Google Scholar]
- 276.Kuhn B., Guba W., Hert J., Banner D., Bissantz C., Ceccarelli S., et al. A real-world perspective on molecular design. J Med Chem. 2016;59:4087–4102. doi: 10.1021/acs.jmedchem.5b01875. [DOI] [PubMed] [Google Scholar]
- 277.Shang M., Wang M.M., Saint-Denis T.G., Li M.H., Dai H.X., Yu J.Q. Copper-mediated late-stage functionalization of heterocycle-containing molecules. Angew Chem Int Ed. 2017;56:5317–5321. doi: 10.1002/anie.201611287. [DOI] [PubMed] [Google Scholar]
- 278.Xu L.L., Wang X., Ma B., Yin M.X., Lin H.X., Dai H.X., et al. Copper mediated C‒H amination with oximes: en route to primary anilines. Chem Sci. 2018;9:5160–5164. doi: 10.1039/c8sc01256c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Kim H., Heo J., Kim J., Baik M.H., Chang S. Copper-mediated amination of aryl C‒H bonds with the direct use of aqueous ammonia via a disproportionation pathway. J Am Chem Soc. 2018;140:14350–14356. doi: 10.1021/jacs.8b08826. [DOI] [PubMed] [Google Scholar]
- 280.Weis E., Johansson M.J., Martín-Matute B. Late-stage amination of drug-like benzoic acids: access to anilines and drug conjugates through directed iridium-catalyzed C‒H activation. Chem Eur J. 2021;27:18188–18200. doi: 10.1002/chem.202103510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Anugu R.R., Munnuri S., Falck J.R. Picolinate-directed arene meta-C‒H amination via FeCl3 catalysis. J Am Chem Soc. 2020;142:5266–5271. doi: 10.1021/jacs.9b13753. [DOI] [PubMed] [Google Scholar]
- 282.Paudyal M.P., Adebesin A.M., Burt S.R., Ess D.H., Ma Z., Kurti L., et al. Dirhodium-catalyzed C‒H arene amination using hydroxylamines. Science. 2016;353:1144–1147. doi: 10.1126/science.aaf8713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Morofuji T., Shimizu A., Yoshida J. Electrochemical C‒H amination: synthesis of aromatic primary amines via N-arylpyridinium ions. J Am Chem Soc. 2013;135:5000–5003. doi: 10.1021/ja402083e. [DOI] [PubMed] [Google Scholar]
- 284.Romero N.A., Margrey K.A., Tay N.E., Nicewicz D.A. Site-selective arene C‒H amination via photoredox catalysis. Science. 2015;349:1326–1330. doi: 10.1126/science.aac9895. [DOI] [PubMed] [Google Scholar]
- 285.Zheng Y.W., Chen B., Ye P., Feng K., Wang W., Meng Q.Y., et al. Photocatalytic hydrogen-evolution cross-couplings: benzene C‒H amination and hydroxylation. J Am Chem Soc. 2016;138:10080–10083. doi: 10.1021/jacs.6b05498. [DOI] [PubMed] [Google Scholar]
- 286.Smulik J.A., Vedejs E. Improved reagent for electrophilic amination of stabilized carbanions. Org Lett. 2003;5:4187–4190. doi: 10.1021/ol035629w. [DOI] [PubMed] [Google Scholar]
- 287.Legnani L., Prina Cerai G., Morandi B. Direct and practical synthesis of primary anilines through iron-catalyzed C‒H bond amination. ACS Catal. 2016;6:8162–8165. [Google Scholar]
- 288.D'Amato E.M., Börgel J., Ritter T. Aromatic C–H amination in hexafluoroisopropanol. Chem Sci. 2019;10:2424–2428. doi: 10.1039/c8sc04966a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Liu J., Wu K., Shen T., Liang Y., Zou M., Zhu Y., et al. Fe-catalyzed amination of (hetero)arenes with a redox-active aminating reagent under mild conditions. Chem Eur J. 2017;23:563–567. doi: 10.1002/chem.201605476. [DOI] [PubMed] [Google Scholar]
- 290.See Y.Y., Sanford M.S. C–H amination of arenes with hydroxylamine. Org Lett. 2020;22:2931–2934. doi: 10.1021/acs.orglett.0c00598. [DOI] [PubMed] [Google Scholar]
- 291.Ham W.S., Hillenbrand J., Jacq J., Genicot C., Ritter T. Divergent late-stage (hetero)aryl C‒H amination by the pyridinium radical cation. Angew Chem Int Ed. 2019;58:532–536. doi: 10.1002/anie.201810262. [DOI] [PubMed] [Google Scholar]
- 292.Hong S.Y., Radosevich A.T. Chemoselective primary amination of aryl boronic acids by PIII/PV=O-catalysis: synthetic capture of the transient Nef inter-mediate HNO. J Am Chem Soc. 2022;144:8902–8907. doi: 10.1021/jacs.2c02922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Gao H., Zhou Z., Kwon D.H., Coombs J., Jones S., Behnke N.E., et al. Rapid heteroatom transfer to arylmetals utilizing multifunctional reagent scaffolds. Nat Chem. 2016;9:681–688. doi: 10.1038/nchem.2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Ham W.S., Choi H., Zhang J., Kim D., Chang S. C2-selective, functional-group-divergent amination of pyrimidines by enthalpy-controlled nucleophilic functionalization. J Am Chem Soc. 2022;144:2885–2892. doi: 10.1021/jacs.1c13373. [DOI] [PubMed] [Google Scholar]
- 295.Ma D., Cai Q. Copper/amino acid catalyzed cross-couplings of aryl and vinyl halides with nucleophiles. Acc Chem Res. 2008;41:1450–1460. doi: 10.1021/ar8000298. [DOI] [PubMed] [Google Scholar]
- 296.Yang Q., Zhao Y., Ma D. Cu-mediated Ullmann-type cross-coupling and industrial applications in route design, process development, and scale-up of pharmaceutical and agrochemical processes. Org Process Res Dev. 2022;26:1690–1750. [Google Scholar]
- 297.Fan M., Zhou W., Jiang Y., Ma D. Assembly of primary (hetero)arylamines via CuI/oxalic diamide-catalyzed coupling of aryl chlorides and ammonia. Org Lett. 2015;17:5934–5937. doi: 10.1021/acs.orglett.5b03230. [DOI] [PubMed] [Google Scholar]
- 298.Gao J., Bhunia S., Wang K., Gan L., Xia S., Ma D. Discovery of N-(naphthalen-1-yl)-N′-alkyl oxalamide ligands enables Cu-catalyzed aryl amination with high turnovers. Org Lett. 2017;19:2809–2812. doi: 10.1021/acs.orglett.7b00901. [DOI] [PubMed] [Google Scholar]
- 299.Bao X., Qiao X., Bao C., Liu Y., Zhao X., Lu Y., et al. Synthesis of tamibarotene via ullmann-type coupling. Org Process Res Dev. 2017;21:748–753. [Google Scholar]
- 300.Forero Cortés P.A., Haydl A.M. The 25th anniversary of the buchwald–hartwig amination: development, applications, and outlook. Org Process Res Dev. 2019;23:1478–1483. [Google Scholar]
- 301.Smith J.A., Jones R.K., Booker G.W., Pyke S.M. Sequential and selective buchwald−hartwig amination reactions for the controlled functionalization of 6-bromo-2-chloroquinoline: synthesis of Ligands for the Tec Src homology 3 domain. J Org Chem. 2008;73:8880–8892. doi: 10.1021/jo801808r. [DOI] [PubMed] [Google Scholar]
- 302.Chandrashekhar V.G., Baumann W., Beller M., Jagadeesh R.V. Nickel-catalyzed hydrogenative coupling of nitriles and amines for general amine synthesis. Science. 2022;376:1433–1441. doi: 10.1126/science.abn7565. [DOI] [PubMed] [Google Scholar]
- 303.Murugesan K., Wei Z., Chandrashekhar V.G., Neumann H., Spannenberg A., Jiao H., et al. Homogeneous cobalt-catalyzed reductive amination for synthesis of functionalized primary amines. Nat Commun. 2019;10:5443. doi: 10.1038/s41467-019-13351-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Dighe S U., Juliá F., Luridiana A., Douglas J.J., Leonori D. A photochemical dehydrogenative strategy for aniline synthesis. Nature. 2020;584:75–81. doi: 10.1038/s41586-020-2539-7. [DOI] [PubMed] [Google Scholar]
- 305.Qiu Z., Lv L., Li J., Li C., Li C. Direct conversion of phenols into primary anilines with hydrazine catalyzed by palladium. Chem Sci. 2019;10:4775–4781. doi: 10.1039/c9sc00595a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Li M., Pan J., Zhou Q. Enantioselective synthesis of amino acids from ammonia. Nat Catal. 2022;5:571–577. [Google Scholar]
- 307.Liu J., Qiu X., Huang X., Luo X., Zhang C., Wei J., et al. From alkylarenes to anilines via site-directed carbon-carbon amination. Nat Chem. 2018;11:71–77. doi: 10.1038/s41557-018-0156-y. [DOI] [PubMed] [Google Scholar]
- 308.Liu J., Pan J., Luo X., Qiu X., Zhang C., Jiao N. Selective dealkenylative functionalization of styrenes via C‒C bond cleavage. Research. 2020;2020:1–9. doi: 10.34133/2020/7947029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.CCR-3 receptor antagonists. Dhanak D, Widdowson KL, White JR, inventors. SmithKline Beecham Corp, assignee. US6420424B1, 1999 Apr 27.
- 310.Wang T., Stein P.M., Shi H., Hu C., Rudolph M., Hashmi A.S.K. Hydroxylamine-mediated C‒C amination via an aza-hock rearrangement. Nat Commun. 2021;12:1–8. doi: 10.1038/s41467-021-27271-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Rajapakse H.A., Nantermet P.G., Selnick H.G., Munshi S., McGaughey G.B., Lindsley S.R., et al. Discovery of oxadiazoyl tertiary carbinamine inhibitors of β-secretase (BACE-1) J Med Chem. 2006;49:7270–7273. doi: 10.1021/jm061046r. [DOI] [PubMed] [Google Scholar]
- 312.Caldwell J.J., Davies T.G., Donald A., McHardy T., Rowlands M.G., Aherne G.W., et al. Identification of 4-(4-aminopiperidin-1-yl)-7H-pyrrolo[2,3-d]pyrimidines as selective inhibitors of protein kinase b through fragment elaboration. J Med Chem. 2008;51:2147–2157. doi: 10.1021/jm701437d. [DOI] [PubMed] [Google Scholar]
- 313.McHardy T., Caldwell J.J., Cheung K.M., Hunter L.J., Taylor K., Rowlands M., et al. Discovery of 4-amino-1-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)piperidine-4-carboxamides as selective, orally active inhibitors of protein kinase b (akt) J Med Chem. 2010;53:2239–2249. doi: 10.1021/jm901788j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Kolocouris A., Tzitzoglaki C., Johnson F.B., Zell R., Wright A.K., Cross T.A., et al. Aminoadamantanes with persistent in vitro efficacy against H1N1 (2009) influenza A. J Med Chem. 2014;57:4629–4639. doi: 10.1021/jm500598u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Hong S.Y., Park Y., Hwang Y., Kim Y.B., Baik M.H., Chang S. Selective formation of gamma-lactams via C‒H amidation enabled by tailored iridium catalysts. Science. 2018;359:1016–1021. doi: 10.1126/science.aap7503. [DOI] [PubMed] [Google Scholar]
- 316.Park Y., Chang S. Asymmetric formation of γ-lactams via C‒H amidation enabled by chiral hydrogen-bond-donor catalysts. Nat Catal. 2019;2:219–227. [Google Scholar]
- 317.Jung H., Chang S., Hong S. Strategic approach to the metamorphosis of γ-lactones to NH γ-lactams via reductive cleavage and C‒H amidation. Org Lett. 2019;21:7099–7103. doi: 10.1021/acs.orglett.9b02673. [DOI] [PubMed] [Google Scholar]
- 318.Miao Y., Kang J., Ma Y., Chen X. Visible light-mediated synthesis of amides from carboxylic acids and amine-boranes. Green Chem. 2021;23:3595–3599. [Google Scholar]
- 319.Crochet P., Cadierno V. Catalytic synthesis of amides via aldoximes rearrangement. Chem Commun. 2015;51:2495–2505. doi: 10.1039/c4cc08684h. [DOI] [PubMed] [Google Scholar]
- 320.Reddy T.N., Beatriz A., Rao V.J., de Lima D.P. Carbonyl compounds' journey to amide bond formation. Chem Asian J. 2019;14:344–388. doi: 10.1002/asia.201801560. [DOI] [PubMed] [Google Scholar]
- 321.Kaur K., Srivastava S. Beckmann rearrangement catalysis: a review of recent advances. New J Chem. 2020;44:18530–18572. [Google Scholar]
- 322.Furuya Y., Ishihara K., Yamamoto H. Cyanuric chloride as a mild and active Beckmann rearrangement catalyst. J Am Chem Soc. 2005;127:11240–11241. doi: 10.1021/ja053441x. [DOI] [PubMed] [Google Scholar]
- 323.Kiely-Collins H.J., Sechi I., Brennan P.E., McLaughlin M.G. Mild, calcium catalysed Beckmann rearrangements. Chem Commun. 2018;54:654–657. doi: 10.1039/c7cc09491d. [DOI] [PubMed] [Google Scholar]
- 324.Vanos C.M., Lambert T.H. Cyclopropenium-activated Beckmann rearrangement. Catalysis versus self-propagation in reported organocatalytic Beckmann rearrangements. Chem Sci. 2010;1:705–708. [Google Scholar]
- 325.Gao Y., Liu J., Li Z., Guo T., Xu S., Zhu H., et al. Dichloroimidazolidinedione-activated Beckmann rearrangement of ketoximes for accessing amides and lactams. J Org Chem. 2018;83:2040–2049. doi: 10.1021/acs.joc.7b02983. [DOI] [PubMed] [Google Scholar]
- 326.Mo X., Morgan T.D.R., Ang H.T., Hall D.G. Scope and mechanism of a true organocatalytic beckmann rearrangement with a boronic acid/perfluoropinacol system under ambient conditions. J Am Chem Soc. 2018;140:5264–5271. doi: 10.1021/jacs.8b01618. [DOI] [PubMed] [Google Scholar]
- 327.Huang Y., Cui J., Jia L., Gan C., Song H., Zeng C., et al. Synthesis and evaluation of some 17-acetamidoandrostane and N,N-dimethyl-7-deoxycholic amide derivatives as cytotoxic agents: structure/activity studies. Molecules. 2013;18:7436–7447. doi: 10.3390/molecules18077436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Huang Y., Cui J., Zhong Z., Gan C., Zhang W., Song H. Synthesis and cytotoxicity of 17a-aza-D-homo-androster-17-one derivatives. Bioorg Med Chem Lett. 2011;21:3641–3643. doi: 10.1016/j.bmcl.2011.04.093. [DOI] [PubMed] [Google Scholar]
- 329.Hyodo K., Hasegawa G., Oishi N., Kuroda K., Uchida K. Direct and catalytic amide synthesis from ketones via transoximation and Beckmann rearrangement under mild conditions. J Org Chem. 2018;83:13080–13087. doi: 10.1021/acs.joc.8b01810. [DOI] [PubMed] [Google Scholar]
- 330.Benning R.K., Osborn H.M.I., Turkson A. Regioselective Beckmann rearrangements of furanoside and pyranoside-derived oximes. Tetrahedron: Asymmetry. 2011;22:109–116. [Google Scholar]
- 331.Ma S., Yu J., Fan H., Li Z., Zhang A., Zhang Q. Exploring sesquiterpene alkaloid-like scaffolds via Beckmann-transannular remodelling of beta-caryophyllene. RSC Adv. 2017;7:40510–40516. [Google Scholar]
- 332.Szostak M., Aubé J. Chemistry of bridged lactams and related heterocycles. Chem Rev. 2013;113:5701–5765. doi: 10.1021/cr4000144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Yao L., Aubé J. Cation‒π control of regiochemistry of intramolecular Schmidt reactions en route to bridged bicyclic lactams. J Am Chem Soc. 2007;129:2766–2767. doi: 10.1021/ja068919r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Szostak M., Yao L., Aubé J. Cation-n control of regiochemistry of intramolecular Schmidt Reactions en route to bridged bicyclic lactams. Org Lett. 2009;11:4386–4389. doi: 10.1021/ol901771b. [DOI] [PubMed] [Google Scholar]
- 335.Szostak M., Aubé J. Synthesis, structural analysis, and reactivity of bridged orthoamides by intramolecular Schmidt reaction. J Am Chem Soc. 2010;132:2530–2531. doi: 10.1021/ja910654t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336.Szostak M., Yao L., Aubé J. Synthesis of medium-bridged twisted lactams via cation‒π control of the regiochemistry of the intramolecular Schmidt reaction. J Org Chem. 2010;75:1235–1243. doi: 10.1021/jo902574m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337.Gutierrez O., Aubé J., Tantillo D.J. Mechanism of the acid-promoted intramolecular Schmidt reaction: theoretical assessment of the importance of lone pair-cation, cation‒π, and steric effects in controlling regioselectivity. J Org Chem. 2011;77:640–647. doi: 10.1021/jo202338m. [DOI] [PubMed] [Google Scholar]
- 338.Tang C., Jiao N. Copper-catalyzed aerobic oxidative C‒C bond cleavage for C‒N bond formation: from ketones to amides. Angew Chem Int Ed. 2014;53:6528–6532. doi: 10.1002/anie.201403528. [DOI] [PubMed] [Google Scholar]
- 339.Liu J., Zhang C., Zhang Z., Wen X., Dou X., Wei J., et al. Nitromethane as a nitrogen donor in Schmidt-type formation of amides and nitriles. Science. 2020;367:281–285. doi: 10.1126/science.aay9501. [DOI] [PubMed] [Google Scholar]
- 340.Bokuda K., Morimoto S., Seki Y., Yatabe M., Watanabe D., Yatabe J., et al. Greater reductions in plasma aldosterone with aliskiren in hypertensive patients with higher soluble (Pro)renin receptor level. Hypertens Res. 2018;41:435–443. doi: 10.1038/s41440-018-0037-1. [DOI] [PubMed] [Google Scholar]
- 341.Shen J., Feng W., Wang Y., Zhao Q., Flavorta B.L., Lu J. Efficacy and safety of aliskiren combination therapy: a protocol for an umbrella review. BMJ Open. 2021;11 doi: 10.1136/bmjopen-2020-043807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342.Savoia C., De Ciuceis C., Paini A., Carletti R., Arrabito E., Nicoletti C., et al. Effect of direct renin inhibition on vascular function after long-term treatment with aliskiren in hypertensive and diabetic patients. J Hypertens. 2021;39:169–180. doi: 10.1097/HJH.0000000000002595. [DOI] [PubMed] [Google Scholar]
- 343.Mahfoz A.M., Gawish A.Y. Insight into the hepatoprotective, hypolipidemic, and antidiabetic impacts of aliskiren in streptozotocin-induced diabetic liver disease in mice. Diabetol Metab Syndrome. 2022;14:163. doi: 10.1186/s13098-022-00935-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344.Pimentel Elardo S.M., Gulder T.A.M., Hentschel U., Bringmann G. Cebulactams A1 and A2, new macrolactams isolated from Saccharopolyspora cebuensis, the first obligate marine strain of the genus Saccharopolyspora. Tetrahedron Lett. 2008;49:6889–6892. [Google Scholar]
- 345.Yang S., Xi Y., Chen J., Yang Z. Asymmetric total synthesis of (−)-cebulactam A1. Org Chem Front. 2014;1:91–99. [Google Scholar]
- 346.Xie C., Niu S., Xia J., Peng K., Zhang G., Yang X. Saccharopolytide A, a new cyclic tetrapeptide with rare 4-hydroxy-proline moieties from the deep-sea derived actinomycete Saccharopolyspora cebuensis MCCC 1A09850. Nat Prod Res. 2017;32:1627–1631. doi: 10.1080/14786419.2017.1392956. [DOI] [PubMed] [Google Scholar]
- 347.Pang Z., Schafroth M.A., Ogasawara D., Wang Y., Nudell V., Lal N.K., et al. In situ identification of cellular drug targets in mammalian tissue. Cell. 2022;185:1793–1805. doi: 10.1016/j.cell.2022.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348.Liechti G.W., Kuru E., Hall E., Kalinda A., Brun Y.V., VanNieuwenhze M., et al. A new metabolic cell-wall labelling method reveals peptidoglycan in Chlamydia trachomatis. Nature. 2013;506:507–510. doi: 10.1038/nature12892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Dheer D., Singh V., Shankar R. Medicinal attributes of 1,2,3-triazoles: current developments. Bioorg Chem. 2017;71:30–54. doi: 10.1016/j.bioorg.2017.01.010. [DOI] [PubMed] [Google Scholar]
- 350.Bonandi E., Christodoulou M.S., Fumagalli G., Perdicchia D., Rastelli G., Passarella D. The 1,2,3-triazole ring as a bioisostere in medicinal chemistry. Drug Discov Today. 2017;22:1572–1581. doi: 10.1016/j.drudis.2017.05.014. [DOI] [PubMed] [Google Scholar]
- 351.Zou Y., Liu L., Liu J., Liu G. Bioisosteres in drug discovery: focus on tetrazole. Future Med Chem. 2020;12:91–93. doi: 10.4155/fmc-2019-0288. [DOI] [PubMed] [Google Scholar]
- 352.Herr R.J. 5-Substituted-1H-tetrazoles as carboxylic acid isosteres: medicinal chemistry and synthetic methods. Bioorg Med Chem Lett. 2002;10:3379–3393. doi: 10.1016/s0968-0896(02)00239-0. [DOI] [PubMed] [Google Scholar]
- 353.Kumari S., Carmona A.V., Tiwari A.K., Trippier P.C. Amide bond bioisosteres: strategies, synthesis, and successes. J Med Chem. 2020;63:12290–12358. doi: 10.1021/acs.jmedchem.0c00530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354.Robinson H., Oatley S.A., Rowedder J.E., Slade P., Macdonald S.J.F., Argent S.P., et al. Late-stage functionalization by Chan-Lam amination: rapid access to potent and selective integrin inhibitors. Chem Eur J. 2020;26:7678–7684. doi: 10.1002/chem.202001059. [DOI] [PubMed] [Google Scholar]
- 355.Wegner T., Elias R., Roling L., Raj N., Gerke V., Fridman M., et al. Cationic, steroid-based imidazolium amphiphiles show tunable backbone-dependent membrane selectivity in fungi. ACS Infect Dis. 2022;8:1815–1822. doi: 10.1021/acsinfecdis.2c00164. [DOI] [PubMed] [Google Scholar]
- 356.Soto-Ortega D.D., Murphy B.P., Gonzalez-Velasquez F.J., Wilson K.A., Xie F., Wang Q., et al. Inhibition of amyloid-β aggregation by coumarin analogs can be manipulated by functionalization of the aromatic center. Bioorg Med Chem. 2011;19:2596–2602. doi: 10.1016/j.bmc.2011.03.010. [DOI] [PubMed] [Google Scholar]
- 357.Upadhyay R., Kumar R., Jangra M., Rana R., Nayal O.S., Nandanwar H., et al. Synthesis of bioactive complex small molecule–Ciprofloxacin conjugates and evaluation of their antibacterial activity. ACS Comb Sci. 2020;22:440–445. doi: 10.1021/acscombsci.0c00060. [DOI] [PubMed] [Google Scholar]
- 358.Srinivasan R., Coyne A.G., Abell C. Regioselective conversion of arenes to N-aryl-1,2,3-triazoles using C‒H borylation. Chem Eur J. 2014;20:11680–11684. doi: 10.1002/chem.201403021. [DOI] [PubMed] [Google Scholar]
- 359.Fürmeier S., Metzger Jürgen O. Synthesis of new heterocyclic fatty compounds. Eur J Org Chem. 2003;2003:885–893. [Google Scholar]
- 360.Lee Y.J., Kang D., Seo J. Facile method for the synthesis of triazole- and tetrazole-containing peptoids on a solid support. Tetrahedron Lett. 2018;59:3311–3316. [Google Scholar]
- 361.Shen T., Huang X., Liang Y., Jiao N. Cu-catalyzed transformation of alkynes and alkenes with azide and dimethyl sulfoxide reagents. Org Lett. 2015;17:6186–6189. doi: 10.1021/acs.orglett.5b03179. [DOI] [PubMed] [Google Scholar]
- 362.Jankovič D., Virant M., Gazvoda M. Copper-catalyzed azide-alkyne cycloaddition of hydrazoic acid formed in situ from sodium azide affords 4-monosubstituted-1,2,3-triazoles. J Org Chem. 2022;87:4018–4028. doi: 10.1021/acs.joc.1c02775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363.Ning Y., Wang H., Sivaguru P., Li S., Zanoni G., Nolan S.P., et al. Defluorinative [4+1] annulation of perfluoroalkyl N-mesylhydrazones with primary amines provides 5-fluoroalkyl 1,2,3-triazoles. Green Chem. 2021;23:7976–7981. [Google Scholar]
- 364.Wang H., Ning Y., Sivaguru P., Zanoni G., Bi X. [4+1] Annulation of in situ generated azoalkenes with amines: a powerful approach to access 1-substituted 1,2,3-triazoles. Chin Chem Lett. 2022;33:1550–1554. [Google Scholar]
- 365.Goyard D., Chajistamatiou A.S., Sotiropoulou A.I., Chrysina E.D., Praly J.P., Vidal S. Efficient atropodiastereoselective access to 5,5′-bis-1,2,3-triazoles: studies on 1-glucosylated 5-halogeno 1,2,3-triazoles and their 5-substituted derivatives as glycogen phosphorylase inhibitors. Chem Eur J. 2014;20:5423–5432. doi: 10.1002/chem.201304989. [DOI] [PubMed] [Google Scholar]
- 366.Taddei M., Ferrini S., Giannotti L., Corsi M., Manetti F., Giannini G., et al. Synthesis and evaluation of new Hsp90 inhibitors based on a 1,4,5-trisubstituted 1,2,3-triazole scaffold. J Med Chem. 2014;57:2258–2274. doi: 10.1021/jm401536b. [DOI] [PubMed] [Google Scholar]
- 367.Mohamed H.A., Bekheit M.S., Ewies E.F., Awad H.M., Betz R., Hosten E.C., et al. Design of new hybrids indole/phthalimide/oxadiazole-1,2,3 triazole agents and their anticancer properties. J Mol Struct. 2023;1274 [Google Scholar]
- 368.Jurczyk J., Woo J., Kim S.F., Dherange B.D., Sarpong R., Levin M.D. Single-atom logic for heterocycle editing. Nat Synth. 2022;1:352–364. doi: 10.1038/s44160-022-00052-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369.Kelly P.Q., Filatov A.S., Levin M.D. A synthetic cycle for heteroarene synthesis by nitride insertion∗∗. Angew Chem Int Ed. 2022;61 doi: 10.1002/anie.202213041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370.Pedras M., Abdoli A., Sarma Mamillapalle V. Inhibitors of the detoxifying enzyme of the phytoalexin brassinin based on quinoline and isoquinoline scaffolds. Molecules. 2017;22:1345–1360. doi: 10.3390/molecules22081345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371.Reisenbauer J.C., Green O., Franchino A., Finkelstein P., Morandi B. Late-stage diversification of indole skeletons through nitrogen atom insertion. Science. 2022;377:1104–1109. doi: 10.1126/science.add1383. [DOI] [PubMed] [Google Scholar]
- 372.Finkelstein P., Reisenbauer J.C., Botlik B.B., Green O., Florin A., Morandi B. Nitrogen atom insertion into indenes to access isoquinolines. Chem Sci. 2023;14:2954–2959. doi: 10.1039/d2sc06952k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 373.Liu S., Cheng X. Insertion of ammonia into alkenes to build aromatic N-heterocycles. Nat Commun. 2022;13:425. doi: 10.1038/s41467-022-28099-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 374.Wang J., Lu H., He Y., Jing C., Wei H. Cobalt-catalyzed nitrogen atom insertion in arylcycloalkenes. J Am Chem Soc. 2022;144:22433–22439. doi: 10.1021/jacs.2c10570. [DOI] [PubMed] [Google Scholar]
- 375.Lyu H., Kevlishvili I., Yu X., Liu P., Dong G. Boron insertion into alkyl ether bonds via zinc/nickel tandem catalysis. Science. 2021;372:175–182. doi: 10.1126/science.abg5526. [DOI] [PubMed] [Google Scholar]
- 376.De Luca L., Giacomelli G., Porcheddu A. Beckmann Rearrangement of oximes under very mild conditions. J Org Chem. 2002;67:6272–6274. doi: 10.1021/jo025960d. [DOI] [PubMed] [Google Scholar]
- 377.Sahasrabudhe K., Gracias V., Furness K., Smith B.T., Katz C.E., Reddy D.S., et al. Asymmetric Schmidt reaction of hydroxyalkyl azides with ketones. J Am Chem Soc. 2003;125:7914–7922. doi: 10.1021/ja0348896. [DOI] [PubMed] [Google Scholar]
- 378.Cochran J.E., Waal N. Photochemical rearrangement of chiral oxaziridines in continuous flow: application toward the scale-up of a chiral bicyclic lactam. Org Process Res Dev. 2016;20:1533–1539. [Google Scholar]
- 379.Sietmann J., Ong M., Mück Lichtenfeld C., Daniliuc C.G., Wahl J.M. Desymmetrization of prochiral cyclobutanones via nitrogen insertion: a concise route to chiral γ-lactams. Angew Chem Int Ed. 2021;60:9719–9723. doi: 10.1002/anie.202100642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 380.Ong M., Arnold M., Walz A.W., Wahl J.M. Stereospecific nitrogen insertion using amino diphenylphosphinates: an Aza-Baeyer-Villiger Rearrangemen. Org Lett. 2022;24:6171–6175. doi: 10.1021/acs.orglett.2c02361. [DOI] [PubMed] [Google Scholar]
- 381.Wang T., Jiao N. TEMPO-catalyzed aerobic oxygenation and nitrogenation of olefins via C=C double-bond cleavage. J Am Chem Soc. 2013;135:11692–11695. doi: 10.1021/ja403824y. [DOI] [PubMed] [Google Scholar]
- 382.Zhao B., Tan H., Chen C., Jiao N., Shi Z. Photoinduced C‒C bond cleavage and oxidation of cycloketoxime esters. Chin J Chem. 2018;36:995–999. [Google Scholar]
- 383.Wang Y., Wang N., Zhao J., Sun M., You H., Fang F., et al. Visible-light-promoted site-specific and diverse functionalization of a c(sp3)–c(sp3) bond adjacent to an arene. ACS Catal. 2020;10:6603–6612. [Google Scholar]
- 384.Sun X., Li X., Song S., Zhu Y., Liang Y., Jiao N. Mn-catalyzed highly efficient aerobic oxidative hydroxyazidation of olefins: a direct approach to β-azido alcohols. J Am Chem Soc. 2015;137:6059–6066. doi: 10.1021/jacs.5b02347. [DOI] [PubMed] [Google Scholar]
- 385.Yang B., Lu Z. Visible-light-promoted metal-free aerobic hydroxyazidation of alkenes. ACS Catal. 2017;7:8362–8365. [Google Scholar]
- 386.Chennaiah A., Vankar Y.D. One-step TEMPO-catalyzed and water-mediated stereoselective conversion of glycals into 2-azido-2-deoxysugars with a pifa–trimethylsilyl azide reagent system. Org Lett. 2018;20:2611–2614. doi: 10.1021/acs.orglett.8b00814. [DOI] [PubMed] [Google Scholar]
- 387.Foo K., Sella E., Thomé I., Eastgate M.D., Baran P.S. A Mild, ferrocene-catalyzed C–H imidation of (hetero)arenes. J Am Chem Soc. 2014;136:5279–5282. doi: 10.1021/ja501879c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 388.Rit R.K., Shankar M., Sahoo A.K. C–H imidation: a distinct perspective of C–N bond formation. Org Biomol Chem. 2017;15:1282–1293. doi: 10.1039/c6ob02162j. [DOI] [PubMed] [Google Scholar]
- 389.Kourounakis A.P., Xanthopoulos D., Tzara A. Morpholine as a privileged structure: a review on the medicinal chemistry and pharmacological activity of morpholine containing bioactive molecules. Med Res Rev. 2019;40:709–752. doi: 10.1002/med.21634. [DOI] [PubMed] [Google Scholar]
- 390.Tzara A., Xanthopoulos D., Kourounakis A.P. Morpholine as a scaffold in medicinal chemistry: an update on synthetic strategies. ChemMedChem. 2020;15:392–403. doi: 10.1002/cmdc.201900682. [DOI] [PubMed] [Google Scholar]
- 391.He Y., Wu H., Wang Q., Zhu J. Catalytic enantioselective synthesis of morpholinones enabled by aza-benzilic ester rearrangement. J Am Chem Soc. 2021;143:7320–7325. doi: 10.1021/jacs.1c03915. [DOI] [PubMed] [Google Scholar]
- 392.DeWald H.A., Heffner T.G., Jaen J.C., Lustgarten D.M., McPhail A.T., Meltzer L.T., et al. Synthesis and dopamine agonist properties of (+‒)-trans-3,4,4a,10b-tetrahydro-4-propyl-2H,5H-[1]benzopyrano [4,3-b]-1,4-oxazin-9-ol and its enantiomers. J Med Chem. 1990;33:445–450. doi: 10.1021/jm00163a068. [DOI] [PubMed] [Google Scholar]
- 393.Pontillo J., Wu D., Ching B., Hudson S., Genicot M.J., Gao Y., et al. Synthesis and structure‒activity relationships of selective norepinephrine reuptake inhibitors (sNRI) with improved pharmaceutical characteristics. Bioorg Med Chem Lett. 2008;18:6151–6155. doi: 10.1016/j.bmcl.2008.10.013. [DOI] [PubMed] [Google Scholar]
- 394.Battiti F.O., Cemaj S.L., Guerrero A.M., Shaik A.B., Lam J., Rais R., et al. The significance of chirality in drug design and synthesis of bitopic ligands as d3 receptor (D3R) selective agonists. J Med Chem. 2019;62:6287–6314. doi: 10.1021/acs.jmedchem.9b00702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 395.Harris P.A., Berger S.B., Jeong J.U., Nagilla R., Bandyopadhyay D., Campobasso N., et al. Discovery of a first-in-class receptor interacting protein 1 (RIP1) kinase specific clinical candidate (GSK2982772) for the treatment of inflammatory diseases. J Med Chem. 2017;60:1247–1261. doi: 10.1021/acs.jmedchem.6b01751. [DOI] [PubMed] [Google Scholar]
- 396.Niu X., Wu M., Li G., Zhou X., Cao W., Zhai W., et al. Identification and optimization of peptide inhibitors to block VISTA/PSGL-1 interaction for cancer immunotherapy. Acta Pharm Sin B. 2023;13:4511–4522. doi: 10.1016/j.apsb.2023.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 397.Cong W., Shen H., Liao X., Zheng M., Kong X., Wang Z., et al. Discovery of an orally effective double-stapled peptide for reducing ovariectomy-induced bone loss in mice. Acta Pharm Sin B. 2023;13:3770–3781. doi: 10.1016/j.apsb.2023.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 398.Muratspahić E., White A.M., Ciotu C.I., Hochrainer N., Tomašević N., Koehbach J., et al. Development of a selective peptide κ-opioid receptor antagonist by late-stage functionalization with cysteine staples. J Med Chem. 2023;66:11843–11854. doi: 10.1021/acs.jmedchem.3c00426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 399.Coskun T., Urva S., Roell W.C., Qu H., Loghin C., Moyers J.S., et al. LY3437943, a novel triple glucagon, GIP, and GLP-1 receptor agonist for glycemic control and weight loss: from discovery to clinical proof of concept. Cell Metabol. 2022;34:1234–1247. doi: 10.1016/j.cmet.2022.07.013. [DOI] [PubMed] [Google Scholar]
- 400.Alexander A.K., Elshahawi S.I. Promiscuous enzymes for residue-specific peptide and protein late-stage functionalization. Chembiochem. 2023;24 doi: 10.1002/cbic.202300372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 401.Kaplaneris N., Rogge T., Yin R., Wang H., Sirvinskaite G., Ackermann L. Late-stage diversification through manganese-catalyzed C−H activation: access to acyclic, hybrid, and stapled peptides. Angew Chem Int Ed. 2019;58:3476–3480. doi: 10.1002/anie.201812705. [DOI] [PubMed] [Google Scholar]
- 402.Shabani S., Wu Y., Ryan H.G., Hutton C.A. Progress and perspectives on directing group-assisted palladium-catalysed C–H functionalisation of amino acids and peptides. Chem Soc Rev. 2021;50:9278–9343. doi: 10.1039/d0cs01441a. [DOI] [PubMed] [Google Scholar]
- 403.Allouche E.M.D., Grinhagena E., Waser J. Hypervalent iodine-mediated late-stage peptide and protein functionalization. Angew Chem Int Ed. 2021;61 doi: 10.1002/anie.202112287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 404.Bellotti P., Huang H.M., Faber T., Glorius F. Photocatalytic late-stage C–H functionalization. Chem Rev. 2023;123:4237–4352. doi: 10.1021/acs.chemrev.2c00478. [DOI] [PubMed] [Google Scholar]
- 405.Gao X., Sun C., Yu Z., Cang J., Tian X., Huo X., et al. Correlation analysis between the chemical contents and bioactivity for the quality control of alismatis rhizoma. Acta Pharm Sin B. 2018;8:242–251. doi: 10.1016/j.apsb.2017.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 406.Zhu Q., Chen Z., Paul P.K., Lu Y., Wu W., Qi J. Oral delivery of proteins and peptides: challenges, status quo and future perspectives. Acta Pharm Sin B. 2021;11:2416–2448. doi: 10.1016/j.apsb.2021.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 407.Gattringer J., Gruber C.W., Hellinger R. Peptide modulators of cell migration: overview, applications and future development. Drug Discov Today. 2023;28 doi: 10.1016/j.drudis.2023.103554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 408.Rodriguez J., O'Neill S., Walczak M.A. Constrained saccharides: a review of structure, biology, and synthesis. Nat Prod Rep. 2018;35:220–229. doi: 10.1039/c7np00050b. [DOI] [PubMed] [Google Scholar]
- 409.Dimakos V., Taylor M.S. Site-selective functionalization of hydroxyl groups in carbohydrate derivatives. Chem Rev. 2018;118:11457–11517. doi: 10.1021/acs.chemrev.8b00442. [DOI] [PubMed] [Google Scholar]
- 410.Witte M.D., Minnaard A.J. Site-selective modification of (oligo)saccharides. ACS Catal. 2022;12:12195–12205. doi: 10.1021/acscatal.2c03876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 411.Romero E., Jones B.S., Hogg B.N., Rué Casamajo A., Hayes M.A., Flitsch S.L., et al. Enzymatic late-stage modifications: better late than never. Angew Chem Int Ed. 2021;60:16824–16855. doi: 10.1002/anie.202014931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 412.Batista V.F., Galman J.L., A G., Pinto D.C., Silva A.M.S., Turner N.J. Monoamine oxidase: tunable activity for amine resolution and functionalization. ACS Catal. 2018;8:11889–11907. [Google Scholar]
- 413.Yang M., Fehl C., Lees K.V., Lim E.K., Offen W.A., Davies G.J., et al. Functional and informatics analysis enables glycosyltransferase activity prediction. Nat Chem Biol. 2018;14:1109–1117. doi: 10.1038/s41589-018-0154-9. [DOI] [PubMed] [Google Scholar]
- 414.Vitaku E., Smith D.T., Njardarson J.T. Analysis of the structural diversity, substitution patterns, and frequency of nitrogen heterocycles among U.S. FDA approved pharmaceuticals. J Med Chem. 2014;57:10257–10274. doi: 10.1021/jm501100b. [DOI] [PubMed] [Google Scholar]
- 415.Delost M.D., Smith D.T., Anderson B.J., Njardarson J.T. From oxiranes to oligomers: architectures of U.S. FDA approved pharmaceuticals containing oxygen heterocycles. J Med Chem. 2018;61:10996–11020. doi: 10.1021/acs.jmedchem.8b00876. [DOI] [PubMed] [Google Scholar]
- 416.Taylor R.D., MacCoss M., Lawson A.D. Combining molecular scaffolds from FDA approved drugs: application to drug discovery. J Med Chem. 2017;60:1638–1647. doi: 10.1021/acs.jmedchem.6b01367. [DOI] [PubMed] [Google Scholar]
- 417.Segler M.H.S., Preuss M., Waller M.P. Planning chemical syntheses with deep neural networks and symbolic AI. Nature. 2018;555:604–610. doi: 10.1038/nature25978. [DOI] [PubMed] [Google Scholar]
- 418.Klucznik T., Mikulak-Klucznik B., McCormack M.P., Lima H., Szymkuć S., Bhowmick M., et al. Efficient syntheses of diverse, medicinally relevant targets planned by computer and executed in the laboratory. Chem. 2018;4:522–532. [Google Scholar]
- 419.Granda J.M., Donina L., Dragone V., Long D.L., Cronin L. Controlling an organic synthesis robot with machine learning to search for new reactivity. Nature. 2018;559:377–381. doi: 10.1038/s41586-018-0307-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 420.Angello N.H., Rathore V., Beker W., Wołos A., Jira E.R., Roszak R., et al. Closed-loop optimization of general reaction conditions for heteroaryl Suzuki‒Miyaura coupling. Science. 2022;378:399–405. doi: 10.1126/science.adc8743. [DOI] [PubMed] [Google Scholar]