

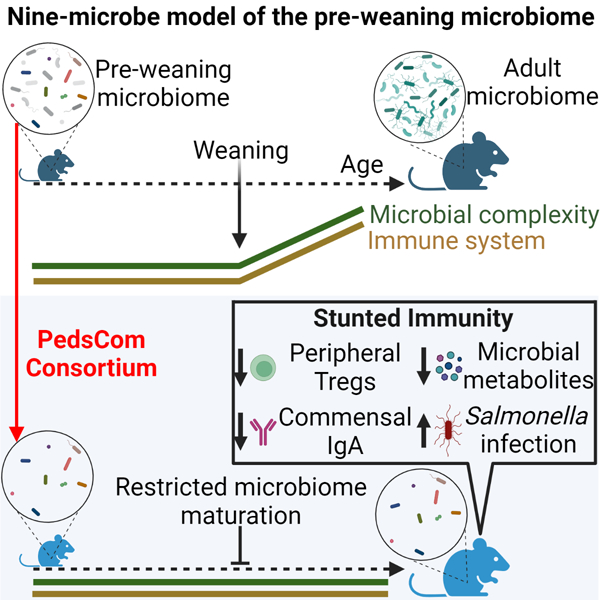
Summary
Disruptions to the intestinal microbiome during weaning lead to negative effects on host immune function. However, the critical host-microbe interactions during weaning required for immune system development remain poorly understood. We find that restricting microbiome maturation during weaning stunts immune system development and increases susceptibility to enteric infection. We developed a gnotobiotic mouse model of the early-life microbiome, Pediatric Community (PedsCom). These mice develop fewer peripheral regulatory T cells and less IgA, hallmarks of microbiota-driven immune system development. Further, adult PedsCom mice retain high susceptibility to Salmonella infection, characteristic of young mice and children. Altogether, our work illustrates how the post-weaning transition in microbiome composition contributes to normal immune maturation and protection from infection. Accurate modelling of the pre-weaning microbiome provides a window into the microbial requirements for healthy development and suggests an opportunity to design microbial interventions at weaning to improve immune development in human infants.
Keywords: Microbiome, Immune system development, Gnotobiotic
Graphical Abstract
One Sentence Summary:
Arresting microbiome development stunts immune ontogeny
eTOC blurb
The transition from a pre-weaning intestinal microbiome to an adult-like community is a critical period in host development. Lubin et al. explore the consequences of restricting the intestinal microbiome to a pre-weaning configuration on normal immune system maturation, major microbially-derived metabolite generation and susceptibility to enteric infection.
Introduction
Bacteria colonize the intestinal tract at birth, support nutrient metabolism and shape immune system development and function.1 The early-life microbiome is compositionally distinct from that of older individuals2, and these differences have been implicated in increased susceptibility to enteric pathogens in infants.3 This increased susceptibility to enteric infections has long been thought to be due to immune system immaturity4, but has been more recently linked to the immature state of the pediatric microbiome.3 In humans, the microbiome rapidly shifts its composition, diversity and function from birth to three years of age, after which the microbiome stabilizes and resembles that of an adult microbiome.2 In mice, the developmental progression is more compressed with young mice initiating solid food around days 12–14, fully weaned by day 21, and developing a microbiome that resembles an adult microbiome by day 28.5 The compositional and functional differences of the early-life microbiome are particularly evident in the pre-weaning stage of life when the infant is solely nourished by maternal milk.6,7
Perturbations in the composition of the early-life microbiome around weaning are linked to an increased risk for developing autoimmunity8, allergies9 and obesity10 later in life. Remarkably, the consequences of early-life perturbations persist even if the microbiome is restored to a state resembling healthy subjects8,9,11, suggesting that there is a window of opportunity that is critical for host-microbe interactions during ontogeny.12 Notably, a specific population of regulatory T cells (Tregs), which develop in the gut around weaning, regulate long term immune system health.11 The initiation of Treg development is coincident with a spike in the expression of pro-inflammatory cytokines tumor necrosis factor alpha (TNF-α) and interferon gamma (IFNγ) in the gut, referred to as the ‘weaning reaction’. This process requires the presence of microbes that expand at weaning, and preventing the weaning reaction during ontogeny leads to long-term defects in regulation of inflammatory responses and gut barrier dysfunction.11 Further mechanistic studies are required to understand and leverage the pre-weaning microbiome to support immune system development and health later in life.
Gnotobiotic models, which enable the commensal microbiome to be rationally designed and manipulated, are valuable tools for the study of host-microbiota interactions. Studies of gnotobiotic mice demonstrate that specific commensal microbes modulate immune system function by inducing the secretion of antimicrobial peptides13 and development of innate lymphoid cells14, peripheral regulatory T cells (pTregs)15,16, and IgA production.17 Gnotobiotic mice designed to model adult microbiomes [e.g., altered Schaedler flora18 (ASF) and Oligo-Mouse-Microbiota-1219 (Oligo-MM12)] reduce the highly complex and diverse adult microbial communities from hundreds of members to a small, defined consortium, which facilitates the study of dynamic and complex interactions in fine detail. Such simplified communities can also serve as a platform for the introduction of additional microbes to identify novel functions such as specific systemic antibody targeting of commensals during homeostasis20, the ability of pathogenic microbes to alter immune responses to established commensals 21,22, and the ability of specific commensals to prevent disease.19,23 The recent development of microbial communities19,24 that recapitulate and standardize the intestinal microbiome of adult mice highlights the utility of rigorously designed gnotobiotic communities. However, to date, no published consortium has been designed to specifically model the distinct function of the pre-weaning microbiome.
To experimentally probe the impact of the pre-weaning microbiome on immune system function and response to enteric infection, we developed a tractable gnotobiotic model that mirrors the early-life resident microbiota. Using extensive culture conditions in parallel with metagenomic sequencing, we rationally designed a nine-member microbial consortium that we call PedsCom, for Pediatric Community, which represents >90% of the microbial reads present in the intestines of pre-weaning mice. PedsCom is vertically transmitted from dam to pup. However, unlike the major taxonomic shifts that characterize normal development of the microbiome during weaning, the relative abundances of PedsCom bacteria remain largely unchanged during the transition from milk to a fiber-rich solid food diet. Thus, adult PedsCom mice retain an intestinal microbiota with a pre-weaning configuration. We found that restricting the intestinal microbiome to a pre-weaning state led to a stunting of immune system development and increased susceptibility to neonatal-associated infection.
Results
Features of pre-weaning microbiomes and construction of a defined consortium functionally similar to infant microbiomes
We first sought to evaluate the development of the intestinal microbiome in our specific pathogen free (SPF) NOD mouse colony from birth to adulthood to determine a pre-weaning timepoint to build a defined consortium. We tracked fecal microbes at days 7, 14, 18, 28, and 42 post birth and assessed microbial composition and diversity by 16S rRNA gene amplicon sequencing of the V4 region. After quality control filtering, the library consisted of 4,241,547 reads clustered into 509 amplicon sequence variants (ASVs) by the DADA2 pipeline.25 The fecal microbiomes from 7- and 14-day old pre-weaning mice that are exclusively breast-fed were highly similar and dominated by Lactobacillus. During the transition from milk to a solid food diet on days 14–18, there was a dramatic shift in the fecal microbiome composition, which by day 18 resembles an adult microbiome (day 42) (Fig. 1A). Thus, fecal microbiota derived from 14-day-old mice retain the characteristics of pre-weaning microbiomes yet are poised to undergo rapid maturation during weaning.
Figure 1. Features of pre-weaning (early-life) microbiomes and construction of a defined consortium functionally similar to infant microbiomes.
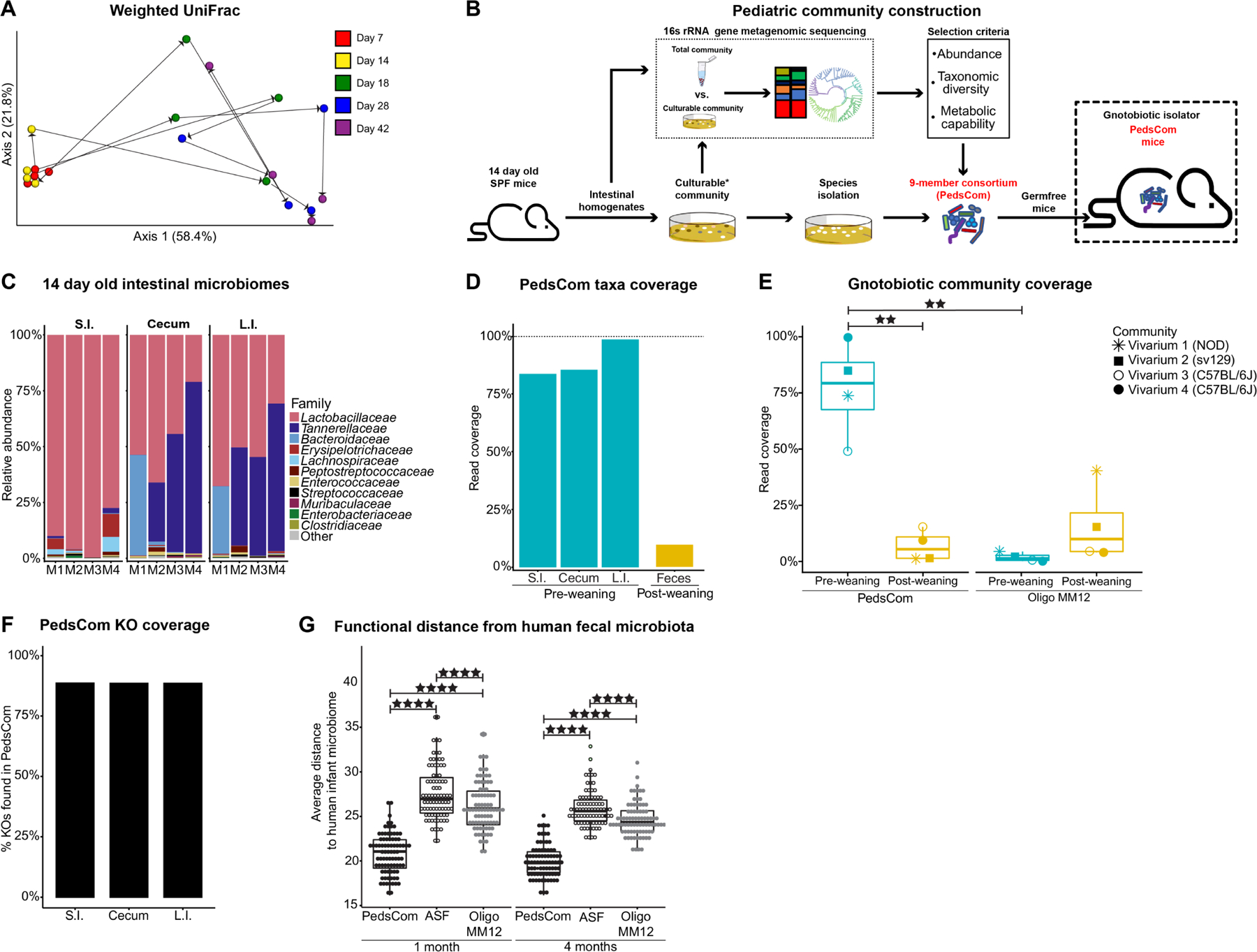
A. Weighted UniFrac Principal Coordinates Analysis (PCoA) of fecal microbiota from SPF NOD mice beta-diversity during ontogeny. Microbial community determined by 16S rRNA gene sequencing. Representative litter shown (N = 4). B. Schematic of pediatric community construction. C. Intestinal microbiomes of 14-day old donor mice. Family level relative abundance determined by 16S rRNA gene sequencing (N = 4, representative data from two litters). D. Read coverage of PedsCom taxa per organ site (N = 4). Median read coverage of PedsCom taxa in the fecal microbiomes of 10-week-old adult SPF NOD mice (N = 19). E. Median read coverage of Oligo-MM12 and PedsCom taxa in the fecal microbiomes of pre- and post-weaning SPF NOD (vivarium 1, N = 171), sv129 (vivarium 2, N = 42) or C57BL/6J (vivarium 3, N = 25, vivarium 4, N = 22) mice housed in different facilities. F. Percent of KEGG orthologues (KOs) represented by PedsCom consortium in the SPF microbial community of the small intestine, cecum, and large intestine of day 14 donor mice. G. Euclidean distance comparison of human infant fecal microbiome (N = 84) determined by shotgun metagenomics to the predicated functions of PedsCom, Altered Schaedler Flora (ASF) and Oligo-MM12. Data presented with box and whisker plot. Mann-Whitney-Wilcoxon test **p<0.01, ****p<0.0001. SPF = Specific pathogen free, S.I. = small intestine, L.I. = large intestine. Teal and yellow colors indicate pre- and post-weaning timepoints, respectively. See also Figure S1.
We extensively cultured intestinal microbiota from 14-day-old SPF Eα16/NOD mice, generating a culture collection of microbes from the pre-weaning period (Fig. 1B). In parallel to this culture approach, the bacterial composition of the pre-weaning intestinal microbiome was determined by 16S rRNA gene amplicon sequencing of small intestinal, cecal and large intestinal homogenates. After processing, the total library contained 1,311,028 reads from 12 intestinal samples and 67 cultured samples. The small intestine was dominated by Lactobacillus spp., which transitioned to a Bacteroidetes-dominant (Tannerellaceae - Parabacteroides, Bacteroidaceae - Bacteroides) microbiome in the cecum and large intestine (Fig. 1C). There was no significant difference in alpha diversity between intestinal sites (Fig. S1A). This pattern differs from what is observed in adult mice, in which diversity increases along the gastrointestinal (G.I.) tract.26,27 We cultured microbes that account for >99% of the total sequenced reads and 82% of the amplicon sequence variants (ASVs) (Fig. S1B). Overall, the top 20 ASVs present at each site represented >97% of the read abundance. This low bacterial diversity is a hallmark of the early-life intestinal microbiomes of mice and humans.6 Altogether, the low diversity of pre-weaning microbiomes and relative evenness of microbial diversity along the G.I. tract provided an opportunity to construct a defined consortium that would closely model the pre-weaning microbiome using a limited number of species.
We hypothesized that a defined consortium composed of microbes isolated from pre-weaning mice would effectively colonize the gastrointestinal tract of germfree C57BL/6 mice, be vertically transmitted from dam to offspring, and recapitulate the functions of early-life intestinal microbiomes in progeny. We selected nine isolates designated PedsCom (Table 1) to 1) maximize the taxonomic diversity of the complete community across all three intestinal sites, 2) attain ≥ 80% of the predicted metabolic functions of the complete community across all three intestinal sites, and 3) limit the number of microbes to less than ten for ease of use. These nine bacteria represent a remarkably high proportion of the microbial sequences present in the small intestine, cecum and large intestine (83%, 85% and 98%, respectively) but only 9.6% of the relative abundance of conventionally raised adult fecal microbiomes (Fig. 1D). To assess the generalizability of the PedsCom consortium across widely used mouse strains and under different housing conditions, we compared the relative abundance of the nine PedsCom taxa in pre- (≤14 days-old) and post-weaning (>14 days-old) SPF mice from the original colony (donor) from which PedsCom was derived (vivarium 1 = ARC Philadelphia, USA) and three additional vivaria [vivarium 2 = CTRB (Philadelphia, USA), vivarium 3 (Aachen, Germany) and vivarium 4 (Alabama, USA) and two additional genetic backgrounds (vivarium 2 = sv129, vivaria 3 and 4 = C57BL/6). The PedsCom taxa were detected at strikingly higher abundances in pre-weaning mice when compared to post-weaning timepoints (79.3% vs 5.4%) (Fig. 1E).
Table 1.
Summary of PedsCom consortia.
| Isolate | Phylum | Class | Order | Family | Genus | Species | Contigs | Plasmids | Genome (MB) | % GC content | PATRIC CDS | KO |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PC20 | Firmicutes | Bacilli | Bacillales | Staphylococcaceae | Staphylococcus | xylosus | 1 | 0 | 2.83 | 32.8 | 2582 | 1563 |
| PC04 | sciuri | 1 | 0 | 2.84 | 32.6 | 2766 | 1644 | |||||
| PC15 | Lactobacillales | Enterococcaceae | Enterococcus | faecalis | 4 | 1 | 2.88 | 37.5 | 2756 | 1466 | ||
| PC39 | Lactobacillaceae | Lactobacillus | murinus | 1 | 0 | 2.48 | 39.7 | 2468 | 1197 | |||
| PC38 | johnsonii | 7 | 0 | 1.92 | 34.6 | 1817 | 984 | |||||
| PC18 | Clostridia | Clostridiales | Lachnospiraceae | Anaerostipes | sp. | 4 | 2 | 3.39 | 43.5 | 3820 | 1605 | |
| PC17 | Clostridiaceae | Clostridium | intestinale | 1 | 0 | 4.69 | 30.4 | 4669 | 2108 | |||
| PC08 | Proteobacteria | Gammaproteobacteria | Enterobacterales | Enterobacteriaceae | Kosakonia | cowanii | 1 | 2 | 4.99 | 55.8 | 4800 | 2886 |
| PC19 | Bacteroidetes | Bacteroidia | Bacteroidales | Tannerellaceae | Parabacteroides | distasonis | 1 | 0 | 5.33 | 45.1 | 4614 | 1535 |
It is important to note that defined gnotobiotic communities invariably lack low-abundance species that may provide unique and important functions. To determine the functional coverage of the PedsCom consortium to the complete pre-weaning microbiome, molecular functions [Kyoto Encyclopedia of Genes and Genomes (KEGG) Orthology groups; (KOs)]28 were predicted by PICRUSt2 analysis.29 The PedsCom consortium provides ~90% of the KOs detected in the pre-weaning SI, cecum, and LI communities (Fig.1F). We conclude that the PedsCom consortium faithfully recapitulates the composition and function of pre-weaning microbes across multiple vivaria and strains.
We next compared the taxonomy of PedsCom to Oligo-MM12, a recently designed gnotobiotic consortium of intestinal microbes derived from adult mice.19 PedsCom is composed of a larger proportion of isolates from the class Bacilli (Enterococcus faecalis, Lactobacillus johnsonii, Lactobacillus murinus, Staphylococcus sciuri, and Staphylococcus xylosus) compared to Oligo-MM12. The increase in Bacilli mirrors the increased prevalence of this class in infants compared to adults.6,7,30 Clostridium intestinale is a member of Clostridiaceae (clostridium cluster I), a family that is strongly associated with the early-life microbiome and is absent in Oligo-MM12.31 In PedsCom, the Lachnospiraceae lineage of Clostridia (cluster XIVa) is represented by Anaerostipes sp. PC18; an isolate closely related to Anaerostipes caccae (average nucleotide identity: 92.16%). The Bacteroidetes lineage is represented by Parabacteroides distasonis, while Oligo-MM12 contains Bacteroides caecimuris and Muribaculum intestinale. Muribaculaceae species are a dominant member of the adult mouse microbiome, positively associated with dietary fiber fermentation.32,33 Lastly, PedsCom contains an Enterobacteriaceae isolate, Kosakonia cowanii, a family of microbes absent in Oligo-MM12. Enterobacteriaceae is a critical component of modeling the pre-weaning microbiome with levels reaching ~20% relative abundance in human neonates.34 These differences in consortium design were evident when comparing the relative abundance of Oligo-MM12 taxa to pre- and post-weaning microbes at 4 different animal facilities (Fig. 1E). Oligo-MM12 taxa are much less abundant in pre-weaning SPF mice than PedsCom taxa (1.4% vs. 79.3%) and trend higher in post-weaning mice (1.4% vs. 10.0%). We conclude that in comparison to Oligo-MM12, the PedsCom consortium more faithfully recapitulates the composition of the pre-weaning microbiome.
To determine if the taxa from pre-weaning communities that were not included in PedsCom contain relevant functions for the pre-weaning microbiome, we compared the abundance and functions of the “non-PedsCom” taxa in pre- and post-weaning mice (Fig. S1C). The 20 most abundant taxa are dominated by Firmicutes of the Clostridia and Erysipelotrichia classes (11/20) (Fig. S1D). Furthermore, there is considerable functional overlap between the 9 PedsCom microbes and non-PedsCom taxa, with only 485 of the 2,981 unique KOs not detected in PedsCom. These missing KOs fall primarily in the membrane transport, glycan biosynthesis and metabolism, and general metabolism KEGG superpathways, including a trend toward complex carbohydrate metabolism that is associated with a solid food diet (Fig. S1E). Additionally, only five of the 78 taxa analyzed were enriched in the pre-weaning SPF mice microbiome (Fig. S1F). We conclude that non-PedsCom taxa are predominantly adult-associated microbes that are unlikely to provide key pre-weaning metabolic functions.
Based on the taxonomic associations of PedsCom members with the early-life microbiome, we hypothesized that PedsCom is more functionally similar to human early-life microbiomes than consortia derived from adult mice like Oligo-MM12 and Altered Schaedler Flora (ASF).18 Like Oligo-MM12, ASF is an eight-member consortium commonly used in gnotobiotic studies, which also lacks the Bacilli dominance, cluster I Clostridia, and Enterobacteriaceae representation found in the PedsCom consortium. We performed whole genome sequencing on each PedsCom isolate with the Oxford Nanopore and Illumina MiSeq platforms and the combined reads were assembled with the Unicycler pipeline.35 Genomes were annotated using the RAST algorithm through the PATRIC webserver.36,37 In aggregate, the genes of the PedsCom consortium consisted of a total of 30,292 coding sequences (CDS) that were assigned to 14,988 KOs. To assess whether PedsCom faithfully recapitulates the metabolic function of neonatal microbiomes, the functional potential of PedsCom, Oligo-MM12 and ASF were compared to human neonatal microbiomes. The KO abundances of the three consortia were compared to a published metagenomic data set of fecal microbiomes from 84 human infants at one and four months of age.38 The PedsCom community was more similar to the human infant fecal microbiota at both one and four months of age than either ASF or Oligo-MM12 (Fig. 1G). This similarity indicated that the functional repertoire of PedsCom more closely resembles that of pre-weaning age microbiomes of human infants, although it is important to note the considerable diversity in the observed neonatal microbiome means that PedsCom is only one possible early-life microbial consortium.
Community dynamics of PedsCom consortium is restricted during weaning
We next investigated whether the nine-member PedsCom consortium stably colonizes germfree mice and measured the effect of weaning on community composition. To generate gnotobiotic PedsCom mice, 1×109 colony-forming units (CFUs) of each PedsCom isolate were orally gavaged into 6-week-old female C57BL/6 germfree (GF) mice (n=3). All nine microbes were recovered from the fecal microbiome one day post-gavage. From day 2 to day 28, the fecal microbiomes were dominated by P. distasonis and K. cowanii. The species L. johnsonii, S. xylosus and S. sciuri were below 0.1% relative abundance one day post inoculation, and undetectable in the fecal microbiome by day seven. In contrast, C. intestinale, which was present at low abundance on day 1 (0.029%), increased 10-fold by day 7 (0.31%) and 60-fold to 1.74% by day 21. Overall, 7 of the 9 member PedsCom consortium of microbes were detected in the initial survey of the fecal microbiomes of germfree mice gavaged as adults, and these microbes formed a stable community that resembles a normal pre-weaning microbiome (Fig. 2A). The two microbes that appeared to be lost (S. xylosus and S. sciuri) were detected in progeny born to these adult PedsCom founders indicating successful colonization and vertical transmission of all nine PedsCom microbes.
Figure 2. Community dynamics of PedsCom consortium is restricted during weaning.
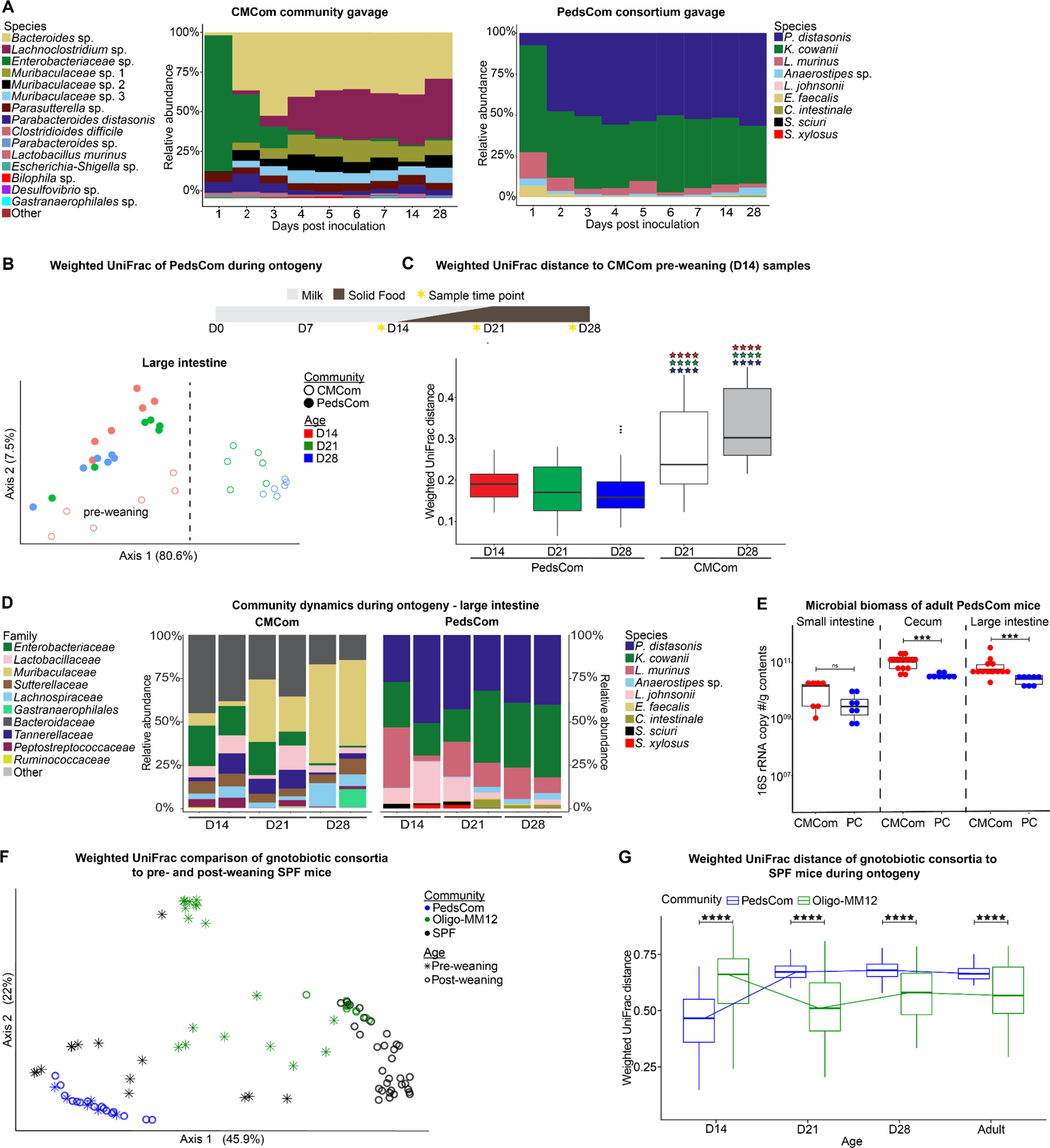
A. Representative species level fecal microbiota of ex-germfree female C57BL/6J mice gavaged with PedsCom consortium (N = 6 mice, 2 independent experiments) or CMCom community (N = 3 mice). B. Weighted UniFrac Principal Coordinates Analysis (PCoA) of the large intestine microbiota of PedsCom and CMCom mice at 14, 21 and 28 days old. Dashed line highlights separation of CMCom pre-weaning and post-weaning samples. N = 6 samples per timepoint. C. Median weighted UniFrac distance comparison of day 14 CMCom large intestine microbiota (pre-weaning) to all PedsCom and post-weaning (D21, D28) CMCom microbiota. Color of stars indicates PedsCom timepoint compared (D14 = red, D21 = green, D28 = blue). N = 6 samples per timepoint. Data represented with box and whisker plot. Mann-Whitney-Wilcoxon test ****p<0.0001. D. Species and family level 16S rRNA gene relative abundance of microbiota from PedsCom and CMCom mice during ontogeny in the large intestine. Two representative litters for each community and timepoint. E. Intestinal biomass of adult CMCom and PedsCom mice (16S rRNA gene copies per gram intestinal contents; PedsCom - N = 8 per tissue, CMCom – S.I. N = 7, Cecum and L.I, N = 13). F. Weighted UniFrac PCoA of the large intestine microbiota of pre- and post-weaning SPF sv129 mice from vivarium 2 (N = 42) to PedsCom (N = 22) and Oligo-MM12 (N = 51). G. Median weighted UniFrac distance of pre-weaning (day 14) and post-weaning (≥ day 21) SPF vivarium 2 mice to all PedsCom and Oligo-MM12 timepoints. Data presented with box and whisker plot. Mann-Whitney-Wilcoxon test **p<0.01, ***p<0.001. See also Figure S2–3.
To generate a control line of mice harboring an undefined, complex community, the cecal contents of a conventionally housed 6-week-old adult Eα16/NOD mouse was gavaged into female GF mice (n=3) and maintained in a separate gnotobiotic isolator. We designated this closed but complex adult community as the Complex Mature Community, CMCom. The fecal microbial communities from CMCom gavaged mice exhibited substantial changes in relative abundance during the first week post-colonization (Fig. 2A). Enterobacteriaceae initially dominated the fecal microbial community with >80% relative abundance by day 1, but by the end of the first week, microbes from the Bacteroidetes and Firmicutes became the dominant taxa. This pattern recapitulated normal taxonomic shifts during development of the microbiome although at a much-accelerated time scale.
We anticipated that the PedsCom consortium would be resistant to diet-induced changes in relative abundance at weaning since the predominant microbes pre-weaning would likely be inefficient users of complex carbohydrates present in a solid food diet. In contrast, we predicted that microbes present in the pre-weaning microbiome of CMCom would be displaced by adult-associated microbiota during the transition to solid food. Indeed, we observed age-determined taxonomic progression in CMCom intestinal microbiota, demonstrating a developmental arc of community composition from days 14 to 28 (Fig. 2B and Fig. S2A,B), which corresponds with a significant increase in beta-diversity community distances in pre-weaning (day 14) to post-weaning intestinal communities (days 21 and 28) (Fig. 2C). Major taxonomic shifts in CMCom included the expected drop in relative abundance of Enterobacteriaceae and Lactobacillaceae through the weaning period (Fig. 2D and S2C,D). Additionally, Bacteroidetes families Bacteroidaceae and Tannerellaceae were replaced with members of the Muribaculaceae lineage in CMCom mice during weaning.
As predicted, we found no clear separation of intestinal samples by age in PedsCom mice by weighted UniFrac analysis (Fig. 2B and Fig. S2A,B). In fact, PedsCom samples from all age groups clustered more closely with 14-day old microbiomes of CMCom mice, demonstrating persistent similarity to the pre-weaning microbiome (Fig. 2C). Further supporting this observation, the relative abundances of dominant PedsCom members remained remarkably stable in the small intestine, cecum, and large intestine during the transition from a milk-based diet to a solid food diet [P. distasonis (36.04% to 34.46%), K. cowanii (22.03% to 32.45%), and L. murinus (13.96% to 18.92%)] (Fig. 2D and S2C,D). Modest shifts in the relative abundances of taxa in the large intestines included increases in Anaerostipes (0.49% to 1.57%), C. intestinale (0.01% to 2.73%) and reduction of L. johnsonii (1.97% to 0.04%), S. xylosus, and S. sciuri (0.02% to undetectable) by week four.
CMCom and PedsCom had several taxa in common (e.g., P. distasonis, L. murinus, E. faecalis) in the cecum and large intestine, especially at earlier time points. In CMCom, these taxa decreased in abundance and were replaced by Muribaculaceae, Bacteroidaceae and Lachnospiraceae during the transition to solid food from 14 to 28 days old. In contrast, relative abundances of these bacteria in PedsCom mice remained unchanged into adulthood (Fig. 2D and Fig. S2E). We also observed significantly lower microbial biomass in the cecum (3.4 × 1010 vs. 1.0 × 1011) and large intestine (2.6 × 1010 vs. 5.6 × 1010) of PedsCom mice compared to CMCom (Fig. 2E), which is consistent with lower microbial biomass in pre-weaning versus adult mice.39
We next compared the microbial dynamics of PedsCom and Oligo-MM12 to SPF mice from multiple vivaria. We predicted that PedsCom mice would resemble the community composition of pre-weaning mice more closely than Oligo-MM12 and importantly that Oligo-MM12 would develop diet-induced changes in relative abundance at weaning similar to SPF mice. PedsCom samples at all ages are significantly closer to 14-day-old SPF mice than Oligo-MM12 mice (Fig. 2F,G). This relationship shifts post-weaning, with Oligo-MM12 mice becoming more similar to mice older than 21-days-old. This shift in community similarity highlights the early-life association of the PedsCom taxa compared to the adult-derived Oligo-MM12 community. The similarity of PedsCom to pre-weaning SPF communities and diet-induced shifts in Oligo-MM12 was also present in mice from vivarium 3 (Fig. S3), demonstrating that the PedsCom consortium of microbes recapitulates the microbiome features of the pre-weaning mice across multiple genetic backgrounds and housed in different facilities.
Taken together, the PedsCom nine-member consortium of microbes derived from the pre-weaning microbiota is vertically transmitted from dam to pups and maintains a pre-weaning configuration through the weaning period into adulthood. This ‘locking in’ of the PedsCom community provides a unique opportunity to investigate the immunologic and physiologic impacts of progression from a pre-weaning to adult microbiome.
Restriction of intestinal microbiome maturation stunts cellular and humoral immune system development
We used the PedsCom model to test whether components of the immune system that depend on normal microbiome maturation are impacted by locking in a pre-weaning intestinal community. We hypothesized that arresting microbiome maturation at weaning would stunt the development of those components of the immune system that develop during and soon after weaning. We focused on CD4+Foxp3+RORγ+ peripheral Tregs (pTregs) which are induced by specific commensal microbes and food antigens during weaning and are required to maintain intestinal homeostasis15,16,40 (Fig. 3A). As expected, pTreg proportions increased in CMCom mice from 14–28 days old, (0% to 30.4%, large intestine) with a sharp rise occurring post-weaning.15,16 In contrast, pTregs proportions were considerably lower but not absent in PedsCom mice (0% to 11.6%, large intestine) (Fig. 3B). The partial induction of pTregs in PedsCom mice suggests that radical shifts of abundance or exogenous introduction of microbes colonizing the gut is not absolutely required to induce this cell population at weaning. Interestingly, though the proportion of pTregs continues to increase in PedsCom mice into adulthood, the populations remained significantly lower than those of CMCom (Fig. 3C). In addition, we compared the induction of pTregs by PedsCom to Oligo-MM12 and found that Oligo-MM12 colonization induced significantly more pTregs in the cecum and large intestine (Fig. 3C). This finding suggests that the lack of pTreg induction by PedsCom is more likely due to the composition of this community than its lower complexity compared to SPF microbiomes.
Figure 3. Restriction of intestinal microbiome maturation stunts cellular and humoral immune system development.
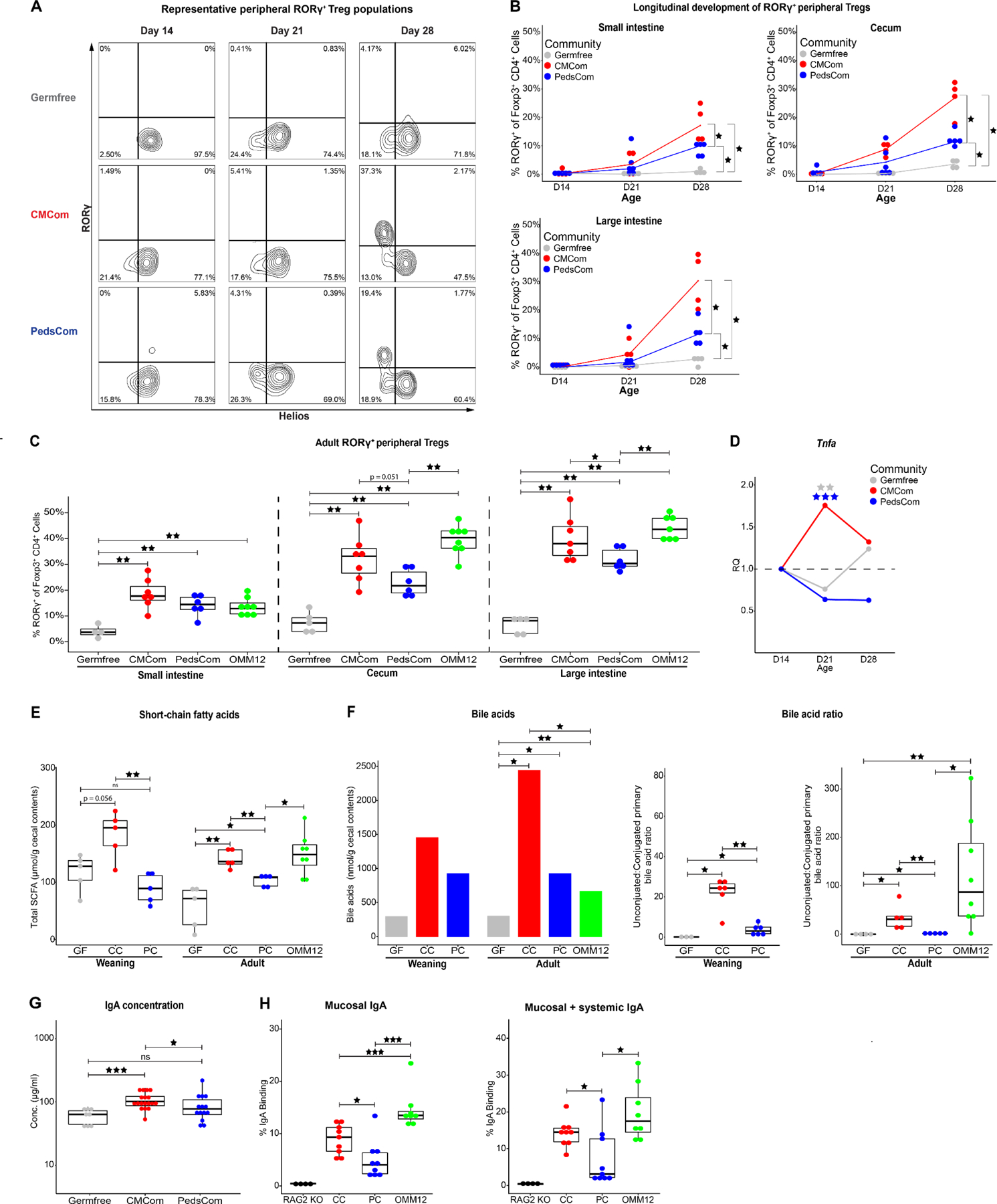
A. Representative flow cytometry plots of CD4+Foxp3+RORγ+Helios− peripheral Treg (pTreg) cell populations. Cells were gated on CD45+TCRβ+CD19−CD8−CD4+Foxp3+. B. Percentage of pTregs in the small intestine, cecum and large intestine lamina propria of germfree, PedsCom and CMCom mice during ontogeny. N = 3 for day 21 germfree cecum, N ≥ 4 per other communities and timepoints. Lines represent median value for each community. Mann-Whitney-Wilcoxon test *p<0.05, **p<0.01, ***p<0.001. C. Percentage of pTregs in the small intestine, cecum and large intestine lamina propria of germfree, CMCom, PedsCom and Oligo-MM12 7-12-week-old adult mice. N ≥ 4 for germfree tissues, N ≥ 6 per other communities and timepoints. D. Tumor necrosis factor alpha (TNFα) transcript levels in distal small intestinal tissue of germfree, PedsCom and CMCom mice during weaning. Relative quantitation (RQ) represents the fold change at day 21 and day 28 relative to mean baseline transcript levels at day 14 in each community. Day 21 represents pooled data of mice sacrificed at day 21 or day 22 post birth. Germfree (N: D14 = 3, D21 = 7, D28 = 8), PedsCom (N: D14 = 4, D21 = 7, D28 = 4), CMCom (N: D14 = 7, D21 = 13, D28 = 5). Independent sample t-test **p<0.01, ***p<0.001. E. Median cecal short-chain fatty acid concentration in germfree, CMCom and PedsCom mice at weaning (day 21) and Oligo-MM12 mice in adulthood (8–14 weeks old). N ≥ 5 per timepoint, per group. GF = germfree, CC = CMCom, PC = PedsCom, OMM12 = Oligo-MM12. F. Median cecal primary bile acid concentration in germfree, CMCom, PedsCom and Oligo-MM12 at weaning [day 21 (N: germfree = 3, CMCom and PedsCom = 6)] and adulthood [8–14 weeks old (N: germfree = 4, CMCom, PedsCom = 5, Oligo-MM12 = 8)] (left panel). The median molar ratios of primary deconjugated to primary conjugated bile acid in each study group. G. Serum IgA concentration in germfree, PedsCom and CMCom 6-12-week-old adult mice (N: germfree = 8, CMCom = 18, PedsCom = 14) (middle and right panels). H. Percent of mucosal and systemic IgA binding to PedsCom, CMCom and Oligo-MM12 fecal microbes from 7-10-week-old mice. (N: RAG2−/− = 4, CMCom, PedsCom = 9, Oligo-MM12 = 8). Mann-Whitney-Wilcoxon test *p<0.05, **p<0.01, ***p<0.001. Data in C and E-H presented with box and whisker plots. See also Figure S4–6.
We also analyzed other lymphoid cell populations in the lamina propria of the small intestine, cecum, large intestine and the spleen. The overall percentage of αβ, γδ, CD4+, CD8+ and Foxp3−RORγ+ (Th17) T cells and B cells were similar between PedsCom and CMCom mice at weaning (Fig. S4) and adulthood (Fig. S5).
The development of pTregs in gut tissues coincides with a spike in pro-inflammatory cytokines in the gut known as the ‘weaning reaction’.11 Consistent with conventionally housed mice, Tnfa gene expression was elevated at day 21 in CMCom but not PedsCom relative to day 14 levels (Fig. 3D). These data indicate that the PedsCom microbes do not induce the characteristic Tnfa inflammatory response at weaning. Short chain fatty acids (SCFAs) in the gut at weaning are capable of inducing the Tnfa-associated weaning reaction in germfree mice and importantly increasing pTreg proportions.11 We investigated whether the amount of SCFAs associated with PedsCom colonization could explain the stunting of pTreg development. Targeted metabolomic analysis of PedsCom cecal contents revealed significantly lower SCFA levels at weaning (day 21) compared to CMCom mice (Fig. 3E). PedsCom SCFA levels remained significantly lower into adulthood when compared to CMCom and Oligo-MM12 mice (Fig. 3E). The higher levels of total SCFAs in CMCom and Oligo-MM12 mice were primarily driven by acetate (Fig. S6A,B). Butyrate, which is capable of inducing pTreg generation16, was also significantly higher in CMCom adults than PedsCom (Fig. S6B). Similarly, PedsCom cecal samples collected at Zeitgeber time (ZT)1500 (corresponding to 10pm), which is the peak of intestinal SCFA levels41, had lower concentrations of SCFA compared to CMCom (Fig. S6C,D). Thus, PedsCom mice have lower levels of immunostimulatory SCFAs compared to a complex microbial community or an adult-like gnotobiotic community.
Since bacterial colonization increases bile acid levels in the gut42 and bacterial deconjugation of primary bile acids supports the generation of pTregs43, we investigated whether differences in the bile acid profile of PedsCom, Oligo-MM12, CMCom, and germfree mice might contribute to the low levels of pTregs in PedsCom mice. As expected, primary bile acid levels in the cecum were significantly higher in the adult colonized groups compared to germfree controls (Fig. 3F). In PedsCom colonized mice there was a trend toward lower concentrations of primary bile acids compared to CMCom during weaning and adulthood. There were no significant differences in bile acid levels in adult PedsCom and Oligo-MM12 (Fig. 3F). In addition to overall lower concentrations of primary bile acids in PedsCom mice, the ratio of deconjugated to conjugated bile acids was highly skewed towards deconjugated molecules in Oligo-MM12 and CMCom samples, which is consistent with bile acid profiles in conventionally housed mice43,44 (Fig. 3F). This skewing is in contrast to PedsCom mice that had significantly reduced molar ratios of deconjugated:conjugated bile acids when compared to CMCom mice at weaning (3:1 vs. 24:1) and Oligo-MM12 and CMCom mice in adulthood (1:1 vs. 87:1 vs. 31:1). These results indicate that the PedsCom consortium has defects in stimulating host bile acid production and a reduced capability for bile acid deconjugation. Taken together, reduced levels of bile acids and SCFAs in PedsCom mice indicate a defect in the level of microbially-derived metabolites associated with pTreg generation.
Germfree mice have less serum IgA and more serum IgE than conventionally housed mice, due to a lack of early-life commensal stimulation.45 These effects become evident soon after weaning when maternal IgA wains and nascent endogenous IgA becomes the dominant mucosal antibody.46 Serum IgA concentration (78.0 μg/ml) were significantly lower in PedsCom mice compared to CMCom mice (101.7 μg/ml) and similar to germfree mice (63.8 μg/ml) (Fig. 3G). Notably, IgE concentration was significantly higher in PedsCom mice compared to CMCom controls (30.0 ng/ml vs. 13.7 ng/ml) (Fig. S6E). Diminished serum IgA and elevated serum IgE are both consistent with decreased microbial stimulation of the immune system during development.
Since PedsCom colonization leads to reduced systemic IgA compared to CMCom, we hypothesized that PedsCom microbes may also induce less secretory IgA capable of binding to these commensal microbes. Acetate levels regulate secretory IgA production in the host47 and lower levels of microbial metabolites such as acetate suggest that PedsCom mice may have altered commensal IgA targeting. Indeed, microbial flow cytometry (mFLOW) analysis of fecal microbiota showed that IgA binds to a lower percentage of the fecal microbes in PedsCom compared to Oligo-MM12 and CMCom mice (4.1% vs. 13.5 vs. 9.3%) (Fig. 3H and S6F). Further, fecal microbiota incubated with autologous serum antibodies also exhibited less binding of IgA to the PedsCom microbes compared to Oligo-MM12 and CMCom mice (Fig. 3H). In summary, lower serum IgA levels in PedsCom mice correspond to less mucosal IgA targeting of the PedsCom consortium members compared to adult-associated commensals. Reduced intestinal pTreg populations and commensal-mediated antibody levels indicate that the restriction of intestinal microbiota in PedsCom mice to this pre-weaning configuration stunts these key components of intestinal and systemic immune development.
Introduction of adult-associated taxa to PedsCom weanlings restores peripheral Treg development
Since restriction of the mouse intestinal microbiome to a pre-weaning configuration with PedsCom leads to a stunting of cellular and humoral immunity, we investigated whether introduction of adult-associated taxa at weaning would rescue key aspects of microbiome and immune system development in PedsCom mice. We hypothesized that colonization with taxa abundant in CMCom adult mice would lead to microbiome maturation in PedsCom mice, increase SCFA and bile acid levels, and subsequently induce higher levels of pTregs. 21-day-old PedsCom mice were co-housed with an adult CMCom female donor mouse for 2 days and then analyzed 14 days later (Fig. 4A). This brief exposure to CMCom microbiota reduced the relative abundance of PedsCom isolates by ~95% (Fig. 4B), equivalent to the abundance of PedsCom taxa in the adult CMCom donor. We found that the newly acquired taxa also reduced absolute abundances of PedsCom isolates (Fig. 4C). Absolute abundance in five out of nine PedsCom isolates decreased (P. distasonis, K. cowanii, L. murinus, E. faecalis, and C. intestinale) (Fig. 4D). To further investigate the microbes transferred to PedsCom from co-housed mice, we analyzed the top 10 ASVs acquired from the adult CMCom donor (Table 2). These ASVs were primarily of the Muribaculaceae family, which are common commensals in adult murine intestinal microbiomes. We conclude that introduced adult-associated microbes effectively rescue the locked-in, pre-weaning microbiome in PedsCom mice.
Figure 4. Introduction of adult-associated taxa to PedsCom weanlings restores peripheral Treg development.
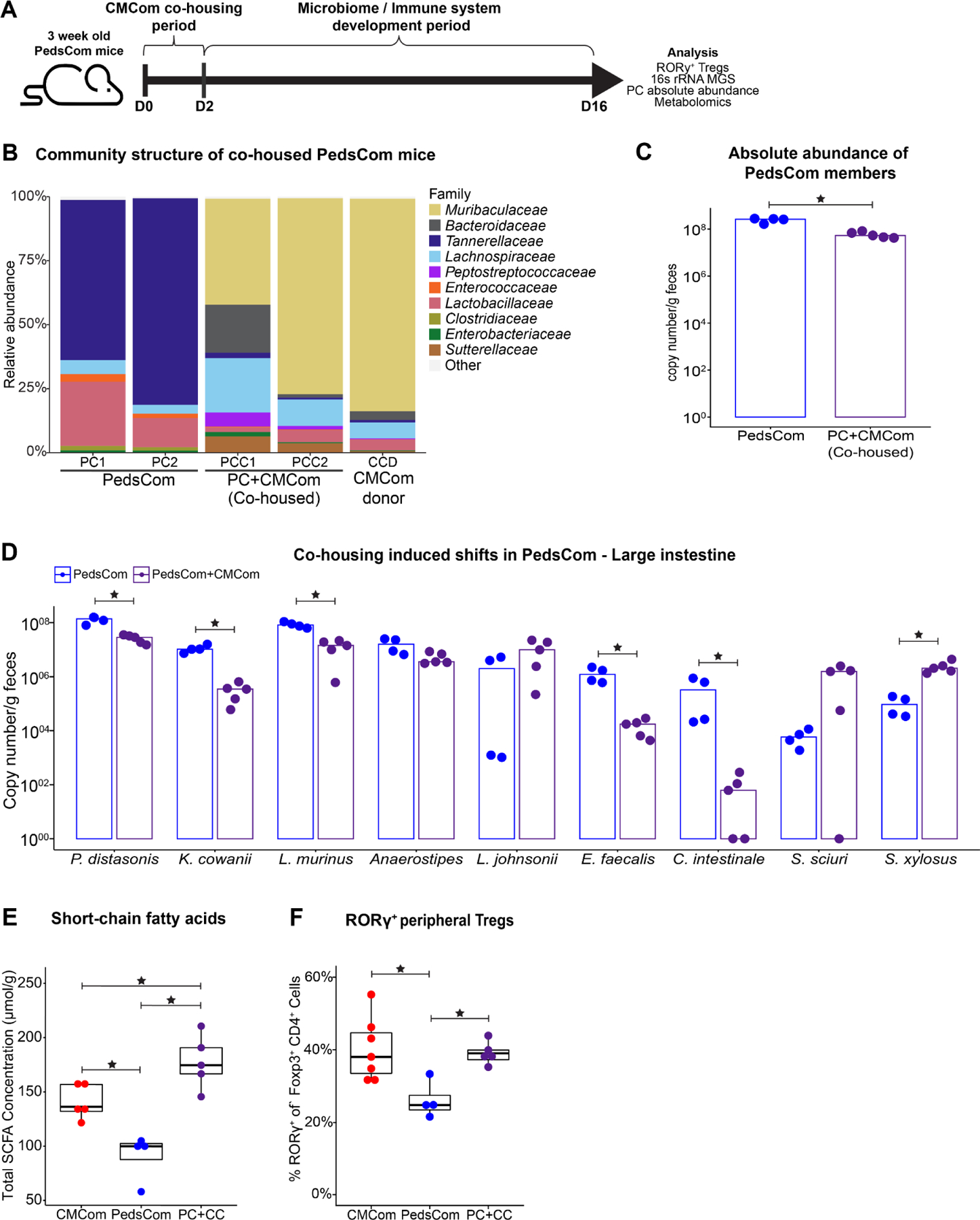
A. Schematic of PedsCom and CMCom co-housing experiment. B. Comparison of the large intestine microbiomes of PedsCom co-housed mice to CMCom donor. Family level relative abundance determined by 16S rRNA gene sequencing. Representative data from two PedsCom and two PedsCom co-housed with one CMCom mice shown. C. Median absolute abundance of PedsCom isolates in fecal samples from PedsCom and PedsCom co-housed mice by rpoB gene qPCR analysis. D. Median absolute abundance of individual PedsCom members in fecal samples from PedsCom and PedsCom co-housed mice. E. Cecal short-chain fatty acid concentration in CMCom, PedsCom and PedsCom co-housed mice. F. Percent of pTregs in the cecum and large intestine lamina propria of CMCom, PedsCom and PedsCom co-housed adult mice. CMCom N = 7, PedsCom mice N = 4. Data presented with box and whisker plot. (N ≥ 4). Mann-Whitney-Wilcoxon tests *p<0.05. See also Figure S6.
Table 2.
Top 10 ASVs acquired by PedsCom mice during co-housing with adult CMCom mice.
| Average relative abundance of top 10 transferred ASVs | Top BLAST hit of representative sequence | |||||
|---|---|---|---|---|---|---|
| ASV | Donor | Co-Housed | ASV ID (Phylum:Family) | Species | Strain | % ID |
| ASV01 | 24.53% | 19.73% | Bacteroidota:Muribaculaceae | Muribaculum gordoncarteri | TLL-A4 | 85.66 |
| ASV02 | 12.16% | 9.09% | Bacteroidota:Muribaculaceae | Muribaculum gordoncarteri | TLL-A4 | 86.89 |
| ASV03 | 4.72% | 9.49% | Firmicutes:Lachnospiraceae | Enterocloster clostridioformis | YL32 | 100 |
| ASV04 | 25.64% | 8.31% | Bacteroidota:Muribaculaceae | Duncaniella freteri | TLL-A205 | 92.15 |
| ASV05 | 11.34% | 6.05% | Bacteroidota:Muribaculaceae | Muribaculum gordoncarteri | TLL-A4 | 85.66 |
| ASV06 | 0.49% | 3.96% | Proteobacteria:Sutterellaceae | Parasutterella sp. | DSM108034 | 100 |
| ASV07 | 1.04% | 4.00% | Bacteroidota:Bacteroidaceae | Bacteroides caecimuris | I48 | 100 |
| ASV08 | 0.67% | 2.54% | Bacteroidota:Muribaculaceae | Muribaculum intestinale | YL7/YL27 | 85.83 |
| ASV09 | 0.78% | 2.67% | Cyanobacteria:Gastranaerophilales | Candidatus Melainabacteria | MEL.A1 | 88.4 |
| ASV10 | 1.07% | 2.25% | Firmicutes:Lachnospiraceae | Enterocloster clostridioformis | YL32 | 100 |
We predicted that the addition of adult-associated species to PedsCom mice would lead to increases in microbial metabolites and immune system maturation. As expected, total SCFA levels were significantly higher in PedsCom co-housed mice compared to controls (Fig. 4E). This increase was primarily driven by higher acetic and isobutyric acid concentrations in PedsCom co-housed mice to levels comparable to CMCom controls (Fig. S6G). However, total primary bile acid levels and the molar ratio of unconjugated to conjugated bile acids remained lower in PedsCom co-housed mice compared to CMCom controls. (Fig. S6H,I). Co-housing PedsCom weanlings with a CMCom adult mouse completely rescued colonic pTreg levels [PedsCom co-housed (39%) vs CMCom (38%), p = 0.88] (Fig. 4F). In addition, we found a trend towards increased mucosal and serum IgA binding to PedsCom co-housed fecal samples (Fig. S6J). Thus, cohousing of PedsCom mice led to the development of adult-like microbiome, a partial rescue of weaning-associated microbial metabolites and complete restoration of colonic pTregs.
The metabolic profiling of control and co-housed PedsCom mice suggests that lower levels of SCFAs, rather than BAs, may account for the lower levels of pTregs in PedsCom mice. To test this hypothesis, we supplemented the drinking water of weaning age PedsCom mice with SCFAs (acetate, propionate, butyrate) for three weeks. We found no significant differences in cecal, colonic, or splenic pTreg proportions in SCFA supplemented PedsCom mice compared to controls (Fig. S6K). Taken together, these studies suggest that additional adult-associated microbial metabolites are required for immune system maturation in PedsCom mice.
PedsCom mice are highly susceptible to Salmonella infection
Since PedsCom mice have a locked-in, pre-weaning microbiome and defects in immune system development, we next assessed the response of PedsCom mice to Salmonella infection, a leading cause of pediatric mortality48, for which susceptibility is linked to features of pediatric intestinal microbiota.3,49 To determine if adult mice harboring PedsCom microbes maintain the high susceptibility of neonatal mice to enteric pathogens, we orally challenged PedsCom, germfree and CMCom adult mice with 5×108 CFUs of wild-type S. enterica s. Typhimurium SL1344 (Salmonella). Germfree mice were highly susceptible to Salmonella infection with 100% mortality occurring within 3 days (Fig. 5A). CMCom mice were resistant with 100% survival 7 days post infection (d.p.i.). PedsCom mice, however, were more susceptible than CMCom to infection with only 50% survival at day 7 post infection, and all PedsCom mice succumbed to infection by day 12. The early onset of mortality of PedsCom-colonized adult mice compared to CMCom suggests that the PedsCom mice lack protective mechanisms found in mice colonized with adult intestinal communities.
Figure 5. Increased susceptibility of PedsCom gnotobiotic mice to Salmonella infection.
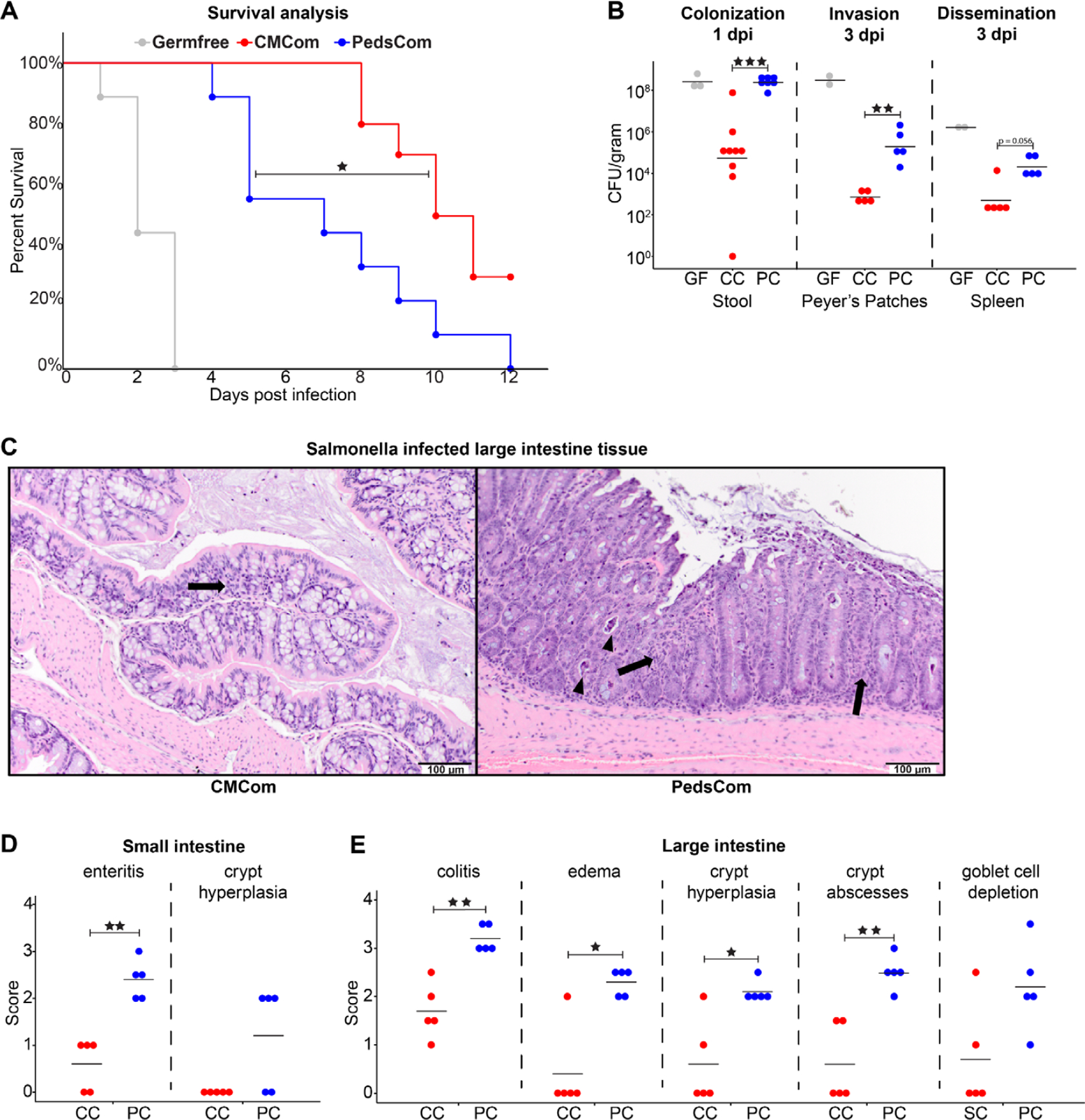
A. Percent survival of adult PedsCom, CMCom and GF mice infected with Salmonella enterica s. Typhimurium SL1344. Adult mice were 10–17 weeks of age. Germfree (N = 9), PedsCom (N = 9), and CMCom (N = 10). Log-rank Mantel-Cox test *p<0.05. Data from two independent experiments. B. Salmonella burden 1 day post infection in feces [Germfree (N = 3), PedsCom (N = 9), and CMCom (N = 7)], Peyer’s patches and spleen 3 days post infection [Germfree (N = 2), PedsCom and CMCom (N = 5)]. CFU per gram feces or indicated tissue. GF = Germfree, CC = CMCom, PC = PedsCom. C. Pathologic features of CMCom and PedsCom large intestine tissue 3 days post Salmonella infection. Arrows indicate minor inflammatory infiltrates within the mucosa and arrowheads indicate crypt abscesses. (H&E staining, 200x magnification, scale bar: 100 μm). D and E. Pathological scores of small and large intestine tissue 3 days post Salmonella infection in PedsCom and CMCom mice (N = 5 per tissue). Median values shown. Mann-Whitney-Wilcoxon test *p<0.05, **p<0.01, ***p<0.001.
To investigate the pathogenesis of Salmonella infection in PedsCom mice, we compared Salmonella burden in the gut, invasion into Peyer’s patches, and systemic dissemination to the spleen between the three groups. We found a ~3 log increase in CFUs per gram of feces one day post infection (d.p.i.) in PedsCom mice compared to CMCom controls (Fig. 5B) indicating increased Salmonella burden in the distal intestines of PedsCom mice. The increased bacterial burden of PedsCom mice relative to CMCom is consistent with studies in young SPF mice and studies of transplanted complex neonatal and pre-weaning microbiota into adult germfree mice.3 Salmonella rapidly colonizes germfree mice to high levels, leading to catastrophic loss of epithelial barrier function, ulceration of the gut and rapid death.50 In contrast, while Salmonella burden in PedsCom mice was comparable to germfree controls, a large discrepancy in survival existed between these two groups, indicating that high bacterial levels in the distal intestines alone do not fully account for differences in survival. Salmonella burden in the Peyer’s patches of PedsCom mice was significantly higher compared to CMCom mice and trended higher in the spleen of PedsCom mice (Fig. 5B). Thus, PedsCom mice are more susceptible to pathogen invasion of the Peyer’s patches and subsequent systemic invasion than CMCom mice, likely explaining the increased mortality. However, compared to germfree controls, PedsCom partially prevents invasion of Peyer’s patches and systemic spread, despite similar intestinal density of Salmonella.
To assess the level of intestinal damage caused by Salmonella infection in PedsCom and CMCom, we scored fixed tissue sections from the small intestine and large intestine for the degree of pathology. In uninfected PedsCom and CMCom mice, we found no obvious differences in intestinal tissue morphology. The most common lesions in infected mice included inflammation characterized by inflammatory cells (predominantly neutrophils, lymphocytes, and histiocytes) expanding in the lamina propria of the mucosa and in the most severely affected animals, effacing and replacing crypts (Fig. 5C). Secondary changes included attenuation of the mucosa, crypt hyperplasia, edema, crypt abscesses (degenerate leukocytes and necrotic debris within crypt lumens), and necrotic debris in the lumen of the intestine. As expected, the large intestine exhibited more severe damage than the small intestine.51 Small intestine histology revealed significantly worse enteritis in all PedsCom mice (Fig. 5D). We also found significantly higher colitis, edema, crypt hyperplasia scores, and an increased frequency of crypt abscesses in infected PedsCom large intestines (Fig. 5E). Overall, these data demonstrate that adult PedsCom mice retain high susceptibility to Salmonella infection characteristic of pre-weaning mice, providing evidence for a critical role of intestinal microbiome maturation in the resistance to this enteric infection.
Discussion
In this study, we developed a microbial community, PedsCom, that recapitulates the relative abundances (>90%), taxonomic diversity and functional capability of pre-weaning intestinal microbiomes. Using these mice, we made two important findings. First, PedsCom retains the characteristics of a pre-weaning microbiome into adulthood. Second, restriction of the intestinal microbiome to a pre-weaning configuration leads to stunting of immune system development.
The first finding of a locked-in microbiota suggests that the predominant microbes at pre-weaning are resistant to diet-induced shifts in relative abundance associated with intestinal microbiome maturation. A large increase of Clostridia at weaning is a hallmark of microbiome maturation. However, the persistence of the most abundant members of PedsCom through the weaning period suggests that the species selected for this consortium accurately recapitulate the functional limitations of pre-weaning associated microbes. PedsCom Clostridia may be less efficient utilizers of complex carbohydrates, which are introduced during the transition to solid food. Deficiencies in complex carbohydrate metabolism of pre-weaning microbes would allow for exogenous or low-abundance microbes to take advantage of this new nutrient source to successfully engraft during weaning. For example, members of the family Muribaculaceae (formerly known as S24-7) are enriched with glycoside hydrolases responsible for the breakdown of starch as well as host-indigestible plant cell wall components, arabinan, xylan, and pectin.52 The Muribaculaceae are found at 2% relative abundance in the pre-weaning (day 14) large intestine of CMCom mice but rise sharply during weaning to 36% by day 21 before stabilizing at 50% of the community in adult mice. It is possible that the weaning period is a window for host-intrinsic events that facilitate the introduction of new microbes and maturation of the microbiome. With this in mind, we speculate that the weaning period may be an ideal time to introduce probiotics, which may better engraft and influence immune system development.
Restriction of the intestinal microbiome to a pre-weaning configuration leads to stunting of immune system development as illustrated by 1) decreased pTreg expansion in intestinal tissues, 2) loss of the Tnfa-associated weaning reaction, 3) lower serum IgA, 4) lower fecal IgA binding to commensal microbiota, 5) elevated serum IgE, and 6) increased susceptibility to Salmonella infection. To begin to dissect the molecular mechanisms underpinning the stunted immune system development in PedsCom mice, we investigated whether alterations in microbially-derived metabolites could explain the defects in immune system maturation in PedsCom mice. The weaning reaction11, intestinal Treg induction53–55 and secretory IgA47 are all influenced by microbially-derived SCFAs. Though the PedsCom consortium induces some pTregs and produces some SCFAs, it is likely that the lack of clostridial species acquired post-weaning reduces total SCFA generation, thereby limiting full pTreg expansion. This relationship is illustrated by the post-weaning expansion in SPF mice of Clostridia of cluster XIVa and IV, which includes commensal microbes capable of metabolizing complex carbohydrates to SCFAs and thereby inducing colonic Tregs.56,57 Predicted functional analyses of PedsCom confirm that transporters for complex-carbohydrate derived oligosaccharides are absent, which may account for the lower levels of SCFA in PedsCom mice. Further, acetate stimulates germinal center B cells in the colon leading to increased secretory IgA in conventionally housed mice.47 The decreased acetate levels in PedsCom relative to Oligo-MM12 and CMCom suggested that the reduction in systemic IgA levels and secretory IgA binding in fecal samples might be rescued by SCFA supplementation, yet this was not the case in our experiments. The lack of expansion of pTregs and IgA binding to commensal microbes in PedsCom mice following SCFA supplementation suggests the picture is more complex and may involve multiple microbial metabolites and perhaps additional microbial antigens to fully support immune maturation.
Additionally, since bacterial deconjugation of primary bile acids also plays an important role in the generation of pTregs43, the reduction of intestinal pTreg proportions in PedsCom mice may be driven by the reduction in the levels of deconjugated bile acids. Unexpectedly, predicted bile salt hydrolase (BSH) genes are present in each PedsCom member’s genome except K. cowanii, which would suggest that bile acid deconjugation should be widespread. However, the abundance of BSH genes increases in the murine intestinal microbiome post-weaning5, suggesting that adult-associated microbes contribute to increasing bile acid deconjugation in the gut. Secondary bile acids are also reported to induce pTregs.43 However, the hydroxysteroid dehydrogenases responsible for secondary bile acid production are limited to a small number of clostridial species58, and no appreciable levels of secondary bile acids were detected in either PedsCom or CMCom samples, which suggests that the lack of these metabolites does not explain the difference in pTreg abundances. Additionally, we found that ‘maturing’ the PedsCom community with adult-associated microbes leads to increased levels of SCFAs and pTreg proportions, without changes in the bile acid profile. Overall, the reductions in intestinal SCFAs in PedsCom mice highlight that microbes that expand or are acquired during the weaning period provide microbial antigens and produce intestinal metabolites that drive cellular and humoral immune system maturation.
The increased susceptibility of adult PedsCom mice to Salmonella infection could be due to stunting of the immune system, or reduced competition with the specific constituent species of the consortium. Compared to CMCom controls, Salmonella causes severe intestinal epithelial damage and immune cell infiltration in PedsCom mice, quickly becoming a systemic infection. Disease progression in PedsCom mice closely mirrors non-Typhoidal Salmonella models in neonatal SPF mice59, suggesting that the loss of protection conferred by adult-associated microbes largely contributed to Salmonella susceptibility in PedsCom adults. It is also possible that immunological defects associated with the disruption of the weaning reaction11 and decreased pTreg levels contributed to increased systemic infection in PedsCom mice. However, in light of the ~1000x higher Salmonella burden in PedsCom compared to CMCom controls, we suspect that this increased level of Salmonella is more likely the primary driver of increased susceptibility of PedsCom.
It is also possible that the low complexity of PedsCom more than the specific features of the pre-weaning consortium is responsible for the increased susceptibility to Salmonella infection. This complexity-versus-composition question has been debated for other gnotobiotic communities. For example, ASF colonized adult mice are highly susceptible to Salmonella infection, with the low complexity of the consortium described as the cause.60 However, Brugiroux et. al.19 showed that the addition of five supplementary microbes from ASF to Oligo-MM12 (now a 17-member consortium) did not alter Salmonella burden compared to Oligo-MM12 alone. In contrast, the addition of three facultative anaerobes which can compete for oxygen with S. enterica s. Typhimurium, reduced Salmonella burden in Oligo-MM12 mice. This indicates that the features of microbes introduced, as opposed to absolute numbers, are responsible for reducing Salmonella burden. The ability of certain microbes to restrict Salmonella infection and not others highlights what one would intuitively expect; that just increasing the number of species present will not necessarily decrease pathogen burden. The PedsCom consortium lacks several carbohydrate transporters, which could lead to increased susceptibility to Salmonella colonization. These include transporters for the monosaccharides utilized by S. enterica s. Typhimurium (D-xylose, myo-inositol)61,62 and oligosaccharides linked to complex carbohydrate fermentation in adult-associated species63, likely leading to a reduced bacterial biomass and subsequently increased susceptibility to infection. It is likely that the susceptibility to Salmonella infections in adult PedsCom mice is due to both the lack of adult-associated taxa to effectively occupy available niches and compete for resources and stunted immune system development leading to a pro-inflammatory environment during infection.
Our study has defined a consortium (PedsCom) as a model for investigating the unique function of the pre-weaning intestinal microbiome and its impact on host functions. Critically, PedsCom’s resistance to weaning-associated microbial shifts and stunted immune system maturation allows it to serve as a model to elucidate the specific components of the developing immune system that require transition from a pre-weaning to an adult-associated microbiome. Accurate modelling of the pre-weaning microbiome provides an exciting opportunity to design microbial interventions at weaning to improve immune system development and long-term health.
Limitations of the Study
This study provides compelling evidence that restricting the intestinal microbiome to a pre-weaning configuration impairs the development of the immune system and resistance to Salmonella infection. The next steps involve identifying the specific molecular mechanism by which PedsCom community composition and function leads to stunted immunity (e.g., decreased pTregs) and increased susceptibility to Salmonella infection. Co-housing PedsCom mice with a complex mature community recovered pTregs and SCFAs levels. However, supplementation of SCFAs alone did not rescue pTreg levels, which led us to conclude that additional bacterial-derived metabolites and/or bacterial antigens are required, a finding which is consistent with the growing number of microbial-derived metabolites that impact immune system development. Additionally, since PedsCom mice are several orders of magnitude more colonized with Salmonella than control mice, the degree to which defects in immunity contribute to the increased susceptibility of PedsCom to Salmonella remains an open question.
STAR Methods
Resource Availability
Lead Contact
Further information and request for resources and reagents should be directed to and will be fulfilled by the lead contact, Michael Silverman (silvermam1@chop.edu)
Materials Availability
The members of the PedsCom consortium are available upon request.
Data and Code Availability
Whole genome sequences of PedsCom isolates and 16S rRNA gene metagenomic datasets generated in this study have been deposited to NCBI BioProject: PRJNA934396 and are publicly available as of the date of publication.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental Models and Subject Details
Mice
Specific pathogen free NOD and Eα16/NOD8 mice were bred and housed in Abramson Research Center (ARC) vivarium at Children’s Hospital of Philadelphia (CHOP). Specific pathogen free sv129 mice were housed in Colket Translational Research Building (CTRB) vivarium at CHOP. CHOP housed mice were fed LabDiet 5058 (Cat# 0007689, MO, USA) ad libitum. Germfree and gnotobiotic C57BL/6J mice were derived and housed in the Hill Pavilion gnotobiotic mouse facility, University of Pennsylvania. Germfree and gnotobiotic mice are housed in flexible film isolators [Class Biologically Clean (CBClean), WI, USA]. Hill housed mice were fed autoclaved LabDiet 5021 (Cat# 0006540) ad libitum. Germfree and gnotobiotic mice were caged on autoclaved Beta-chip hardwood bedding (Nepco, NY, USA). Sterility checks were regularly performed on isolators each month and additionally prior to any transfer of animals. Freshly collected pellets were cultured on brain heart infusion (BHI) (Oxoid, UK), NB1, and Sabouraud media for 65–70 hours at 37°C under aerobic and anaerobic conditions with positive and negative control samples. Isolator sterility was confirmed externally every 3–4 months by Charles River Laboratories (NJ, USA). For all experiments, mice of both sexes between 2–17 weeks of age were used and randomly assigned to experimental groups. Animal studies were performed under protocols approved by the Institutional Animal Care and Use Committee (IACUC) at CHOP and the University of Pennsylvania.
Bacterial strain isolation and gavage conditions
Whole small intestine, cecum, and large intestine tissues were collected from 14-day old Eα16/NOD mice and homogenized under anaerobic conditions (90% N2, 5% CO2, 5% H2) in sterile, reduced 0.1% L-cysteine, phosphate buffered saline (rPBS). A sample of the organ homogenates was collected and stored at −80°C for sequencing. Serial dilutions of homogenates were plated on four bacterial growth media: Tryptic Soy Agar (Becton-Dickinson [BD], MD, USA), Chocolate Agar (BD), Laked Blood Agar with 100 mg/L kanamycin, 7.5 mg/L vancomycin (BD), and Yeast Casitone Fatty Acid (YCFA) prepared as previously described.64 Plates were cultured under three different growth conditions: anaerobic at 37°C for five days, ambient air at 37°C for three days, ambient air supplemented with 5% CO2 at 37°C for three days. Colony isolates were restreaked for purity and preliminary identification performed by 16S rRNA gene Sanger sequencing. Isolates were stored at −80°C in 25% glycerol, BHI media. Whole plate scrapings were collected for sequencing. PedsCom bacteria were grown under the following conditions: Clostridium intestinale PC17, Anaerostipes sp. PC18, Parabacteroides distasonis PC19 – anaerobic at 37°C, BHI supplemented with hemin (5 mg/L) and vitamin K (0.5 mg/L) (BD); Lactobacillus johnsonii PC38, Lactobacillus murinus PC39 – anaerobic at 37°C, de Man Rogosa Sharpe (MRS) media (Sigma, MO, USA); Staphylococcus sciuri PC04, Kosakonia cowanii PC08, Enterococcus faecalis PC15, Staphylococcus xylosus PC20 – ambient air at 37°C, BHI. Six-to-eight-week old Germfree C57BL/6J mice were inoculated with 100 μl of a pooled sample of 1×109 colony forming units (CFU) of each isolate in rPBS to generate the PedsCom gnotobiotic line. To generate the CMCom gnotobiotic line, whole cecal contents of a six-week-old female SPF NOD/Eα16 mouse was collected anaerobically in rPBS and 100 μl of the slurry was gavaged into six-to-eight-week-old germfree C57L/6J mice. Oligo-MM12 isolates were obtained from German Collection of Microorganisms and Cell Cultures (DSMZ). Oligo-MM12 isolates were grown under the following conditions: Enterococcus faecalis KB1 – ambient air at 37°C, BHI; Limosilactobacillus reuteri I49 – anaerobic at 37°C, MRS; Bacteroides caecimuris I48 – anaerobic at 37°C, chopped meat broth (Anaerobe Systems, CA, USA) supplemented with hemin (5 mg/L) and vitamin K (0.5 mg/L); Clostridium innocuum I46, Enterocloster clostridioformis YL32, Muribaculum intestinale YL27, Blautia coccoides YL58 – anaerobic at 37°C, BHI supplemented with hemin (5 mg/L) and vitamin K (0.5 mg/L); Flavonifractor plautii YL31, Turicimonas muris YL45, Bifidobacterium animals YL2, Acutalibacter muris KB18, Akkermansia muciniphila YL44 – anaerobic at 37°C, Anaerobic Akkermansia Medium supplemented with 0.5 g/L N-acetylglucosamine (Sigma).65 Oligo-MM12 gnotobiotic mice were generated by gavage of 21-day-old germfree C57BL/6J mice as previously described.65 Briefly, individual cultures of Oligo-MM12 isolates were diluted to matching OD600 values, combined and aliquoted in 10% glycerol, and stored at −80°C. Recipients of Oligo-MM12 were gavaged twice with 100 μl of the cultured microbes three days apart.
Methods Details
Sample collection and DNA isolation
Fecal pellets or intestinal contents were collected in sterile microcentrifuge tubes and stored at −80°C. Bacterial DNA was isolated from samples using the DNAeasy PowerSoil kit (Qiagen, Germany) according to manufacturer instructions.
16S rRNA gene metagenomic analysis and functional prediction
The V4 variable region of the 16S rRNA gene was sequenced by the PennCHOP microbiome program sequencing core on the Illumina MiSeq platform as previously described.66 Publicly available 16S rRNA gene metagenomic datasets were obtained for Oligo-MM12 and SPF mouse fecal microbiomes during ontogeny.5,67,68 Sequenced libraries were processed using QIIME2 (ver. 2020.6)69 and de noised and clustered into amplicon sequence variants (ASVs) with Deblur.70 Taxonomic classification was performed with SILVA 99% rRNA gene reference database (ver. 138). Taxa bar plots, alpha diversity, and beta-diversity analysis were performed with the R package phyloseq.71 Metabolic function predictions was conducted by analyzing 16S rRNA gene metagenomic sequence using PICRUSt2.29 PICRUSt2 annotated KEGG orthologs were analyzed using the BURRITO web server.72 Differential abundance analyses were performed on 16s rRNA gene ASVs merged at 97% identity cutoff with the ANCOM methodology.73
Whole genome sequencing
Bacterial genomic DNA was isolated using the Blood and Cell Culture DNA kit and 500/G genomic tips (Qiagen) according to manufacturer instructions. DNA purity was assessed with NanoDrop 2000 UV-Vis Spectrophotometer (ThermoFisher, MA, USA) and long-read sequencing was performed using the RBK-004 rapid barcoding kit and MinION sequencer (Oxford Nanopore Technologies, UK) according to manufacturer’s instructions. Short reads were sequenced by the PennCHOP microbiome program sequencing core using the Nextera XT library preparation kit (Illumina, CA, USA) and the Illumina HiSeq 2500. Contaminating reads were identified using Mash screen.74 Long and short read sequences were assembled using the unicycler pipeline.35 Assembled genomes were annotated using the RAST tool kit (RASTtk) on the Pathosystems Resource Integration Center (PATRIC) webserver.36,37 Average nucleotide identity (ANI) was calculated using the ANI Calculator program with the OrthoANIu algorithm.75
Predicted metabolic function
Publicly available assembled genome files of ASF76 and Oligo-MM1277 isolates were obtained from PATRIC. Tables of KEGG orthologs (KO) for each consortium were generated by genome annotation through the KEGG automatic annotation server (KAAS).28 Analyses of predicted metabolic function were conducted using the MicrobiomeAnalyst R package.78 Publicly available shotgun metagenomic datasets were obtained for human infant fecal microbiomes for functional comparison to consortia.38
Lymphoid cell isolation and flow cytometry
Mouse small intestine, cecum, and colon samples were separated, cleaned of fat, flushed with ice cold PBS and vertically incised. Cleaned tissues were incubated for 15 min in 30 ml of intraepithelial cell dissociation medium (RPMI, 0.02% dithiothreitol, 1 mM EDTA, 1.3% FBS) with stirring at 37°C. Tissues were then minced and incubated for 35–45 min in 25 ml of digestion medium [RPMI, 0.5 mg/ml dispase (Gibco, Life Technologies, CA, US), 1.5 mg/ml collagenase type II (Gibco), 4% FBS] with stirring at 37°C. Peyer’s patches from the small intestine and the cecal patch was removed before these tissues were used for the preparation of the lamina propria cell suspensions. Splenocytes were processed in parallel by mechanical disruption and incubation in ACK lysis buffer (Gibco) for 1 min at room temperature for use as internal processing and staining controls. Cell populations were stained for surface markers in 4% FBS RPMI for 15 min on ice, in the dark. Cell populations were fixed and permeabilized overnight at 4°C in permeabilization buffer (eBioscience, MA, USA). Permeabilized cell populations were stained for intracellular markers for 50 min, in the dark, at room temperature. Stained cell populations were analyzed on the LSRFortessa (BD), and data analysis was performed on FlowJo v10 software (BD). Fluorophore-conjugated antibodies and dilutions used in staining panel: BV510-CD45 1:200 (clone 30F11; Biolegend, CA, USA), PE-Cy7-TCRβ 1:100 (clone H57-597; Biolegend), APC-Cy7-CD19 1:100 (clone 6D5; Biolegend), AF700-CD8 1:100 (clone 53–6.7; Biolegend), FITC-CD4 1:100 (clone Rm4-5; Biolegend), APC-Foxp3 1:100 (FJK-16s; eBioscience), PE- RORγ 1:100 (clone B2D; eBioscience), Pacific Blue-Helios 1:33 (clone 22F6; Biolegend).
Reverse transcription quantitative real-time PCR
Two-millimeter tissue sections were excised 5 cm from the distal end of the small intestine (ileum) and stored in RNAlater (ThermoFisher) overnight at 4°C, then at −80°C for long-term storage. RNA was extracted from tissue samples using the E.Z.N.A. total RNA kit I (Omega Bio-tek, GA, USA) according to manufacturer instructions. cDNA synthesis was performed using the High-Capacity RNA-to-cDNA Kit (Thermofisher) with 2 μg of input RNA. Data acquisition was performed on the 7500 Fast Real-time PCR system using PowerUp SYBR Green PCR master mix (Thermofisher). Cycle threshold (Ct) values of Tnfa were normalized to mean Ct values of Gapdh endogenous controls. Data analysis was performed using the 7500 software v2.3 (Thermofisher). The 2−ΔΔCt method was used to analyze the relative fold-change of Tnfa at 21 and 28-days-old, with baseline levels set at 14-days old in each community. Murine Tnfa and Gapdh primer pairs were obtained from the PrimerBank database.79
Targeted metabolomics analysis
Mouse cecal contents were flash frozen on dry ice and stored at −80°C prior to analysis. Short-chain fatty acids were analyzed by the PennCHOP microbiome core on an Acquity uPLC System with a photodiode array detector and a HSS T3 1.8 μm 2.1×150 mm column (Waters, UK). The flow rate was set to 0.25 ml/min with a 5 μl injection volume and column temperature of 40°C, the sample temperature was 4°C. The samples were run with a gradient of 100% eluent A (100 mM sodium phosphate monobasic) for 5 min to 70% eluent B (methanol) for 5–22 min to 100% eluent A for 3 min. The photodiode array is set to read absorbance at 215 nm with 4.8 nm resolution. Short-chain fatty acids assayed: acetic acid, propionic acid, isobutyric acid, butyric acid, isovaleric acid, valeric acid, isocaproic acid, caproic acid and heptanoic acid. Samples are quantified against standard curves of at least five points run in triplicate. Limit of detection was 5 μmol/g cecal contents. Bile acids were analyzed on an Acquity uPLC System with a Cortecs UPLC C-18+ 1.6 mm 2.1 × 50 mm column with a QDa single quadrupole mass detector (Waters). The flow rate was set to 0.8 ml/min with a 4 μl injection volume and column temperature of 30°C, the sample temperature was 4°C. The samples were run with a gradient of 70% eluent A (0.1% formic acid in water) for 2.5 min to 100% eluent B (0.1% formic acid in acetonitrile) for 0.6 min to 70% eluent A for 0.9 min. The mass detection channels are: +357.35 for chenodeoxycholic acid and deoxycholic acid; +359.25 for lithocholic acid; −407.5 for cholic, alphamuricholic, betamuricholic, gamma muricholic, and omegamuricholic acids; −432.5 for glycolithocholic acid; −448.5 for glycochenodeoxycholic and glycodeoxycholic acids; −464.5 for glycocholic acid; −482.5 for taurolithocholic acid; −498.5 for taurochenodeoxycholic and taurodeoxycholic acids; and −514.4 for taurocholic acid. Samples were quantified against standard curves of at least five points run in triplicate. Limit of detection was 0.3 nmol/g cecal contents.
Serum ELISA
Euthanized mice were bled orbitally, and the serum was isolated by a 15 min incubation of blood at room temperature followed by centrifugation at 587 x g for 15 minutes at 4°C. Isolated serum was aliquoted and stored at −20°C. Serum ELISA analysis was performed according to standard protocols. Briefly, 96-well MaxiSorp Immuno plates (ThermoFisher) were coated with unconjugated capture polyclonal antibody goat anti-mouse IgA, IgG1, IgG2b, IgG2c, IgG3, IgM, IgE (Bethyl laboratories, MA, USA) in coating buffer (2.93 g/L NaHCO3, 1.59 g/L Na2CO3, pH 9.6) overnight at 4°C. Coated plates were washed with wash buffer (0.1% Tween-20 PBS) and incubated at room temperature with blocking buffer (2% BSA PBS) for one hour. Serum samples were diluted in blocking buffer and incubated on plates for 1.5 hours. Plates were washed and HRP-conjugated detection antibodies were incubated with the plates at room temperature for one hour. Plates were developed with the OptEIA TMB substrate reagent set (BD).
Microbial flow cytometry
Fecal pellets were homogenized in 1 mL PBS, 1% BSA and centrifuged at 100 x g for 15 minutes to pellet solid material. Fecal supernatants were transferred to sterile microcentrifuge tubes and centrifuged at 8000 x g for five minutes to pellet the bacterial fraction and resuspended to an OD600 of 0.1 in PBS, 1% BSA. Bacteria were fixed with 4% paraformaldehyde for 20 minutes at room temperature. Bacteria were washed and resuspended in blocking buffer (PBS, 1% BSA, 20% normal rat serum) and incubated for 20 minutes at 4°C. To determine the precent of fecal microbes bound by secreted IgA, the processed fecal samples were incubated with PE-conjugated anti-mouse IgA antibodies (clone mA-6E1; eBioscience) diluted 1:25 in PBS 1% BSA for 20 minutes at 4°C, washed and incubated in SytoBC nucleic acid stain (ThermoFisher) diluted 1:500 in TBS, for 15 minutes at room temperature. To determine the percentage of fecal microbes bound by systemic immunoglobulins, the processed fecal samples were incubated with autologous diluted serum and stained with anti-mouse PE-IgA as previously described. Flow cytometry analysis on stained bacteria populations was performed on the LSRFortessa and data analysis was performed on FlowJo ver. 10 software. SytoBC and isotype-stained controls were used to define the gates for Ig-coated microbes.
Absolute quantitation of intestinal microbiota
Bacterial biomass was determined by 16S rRNA gene copies per gram cecal contents using the EUB 1114F, 1221R universal primer set.80 Data acquisition was performed on the CFX Opus 96 PCR system (Bio-Rad, CA, USA) using SYBR Green PCR master mix (Thermofisher). TaqMan custom primer and probe sets were designed to target the DNA-directed RNA polymerase subunit beta (rpoB) gene for each PedsCom member. Primer and probe sets were generated using the PrimerQuest Tool (Integrated DNA Technologies, IA, USA) and compared with the Multiple Primer Analyzer tool (Thermofisher) and are available in Table S1. Absolute quantification of each PedsCom member was determined by rpoB gene copies per gram feces. Data acquisition was performed on the CFX Opus 96 PCR system using TaqMan Multiplex master mix (Thermofisher). Data analysis was performed on CFX Maestro ver. 2.3 (Bio-Rad).
Co-housing experiments
Weaning age (21-day-old) PedsCom littermates were co-housed with an adult female CMCom mouse for two days after which the adult was removed and sacrificed for microbiome analysis. PedsCom mice co-housed with a germfree adult female mouse served as controls. Experiments were performed 16 days post co-housing.
Short-chain fatty acid supplementation
Weaning age (21-day-old) PedsCom littermates were separated and housed under sterile conditions. Mice were treated with either mix of short-chain fatty acids [sodium acetate (150mM), sodium propionate (150mM) (Sigma), sodium butyrate (100mM) (Thermofisher)], or pH and sodium matched control drinking water for three weeks.55 The drinking water solutions were sterile filtered prior to use and replaced every seven days. Following the treatment period, mice were sacrificed and the effect of short-chain fatty acid supplementation on intestinal peripheral Treg proportions was assessed.
Salmonella infection model
A streptomycin resistant mutant of Salmonella enterica s. Typhimurium SL1344 (Salmonella) was cultured with Luria-Bertani (LB) media in ambient air at 37°C. Cultures of Salmonella were resuspended in sterile PBS and 10-17-week-old mice were infected with 5×108 CFU by oral gavage. Salmonella colonization was enumerated 24 hours post infection (h.p.i) by serial dilution of fecal homogenates on LB with 100 μg/ml streptomycin. Cohorts of infected mice were tracked longitudinally for mortality rates. A separate cohort was sacrificed 36 h.p.i. and the Peyer’s patches, spleen and liver were collected, homogenized in sterile PBS and plated on LB with 100 μg/ml streptomycin for Salmonella enumeration by CFU/g tissue. Small intestine and large intestine tissue samples were collected for histopathological assessment.
Histopathological assessment
Small intestine and large intestine tissue samples from Salmonella infected mice and healthy controls were fixed in 10% phosphate-buffered formalin for >24 hours and routinely processed and embedded in paraffin blocks (FFPE). FFPE tissue blocks were routinely sectioned and stained with hematoxylin and eosin (H&E) on glass slides. Histopathological examination was performed blinded to experimental groups by a veterinary pathologist using light microscopy. Samples of small intestine and large intestine were evaluated unpaired as an additional layer of blind evaluation. Detailed assessment of lesions included descriptive evaluation of tissues with morphologic diagnoses as well as semiquantitative scoring of the degrees of inflammation and associated findings, including degree of edema, crypt hyperplasia, goblet cell loss, and frequency of crypt abscesses in each tissue. Scores for each parameter were assigned from 0 (unremarkable) to 4 (severe) at half point intervals.
Quantification and Statistical Analysis
Statistical analysis
Log-rank Mantel-Cox tests were performed with Prism 8 (GraphPad Software Inc). RT-qPCR statistical analysis was performed using independent sample t-tests on mean values with the ggpubr package. All other statistical analyses were performed using non-parametric Mann-Whitney-Wilcoxon tests on median values by either base R or the ggpubr package. P values <0.05 were considered statistically significant. Outliers were determined by z-score analysis.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-mouse CD45 BV510 (30F11) | Biolegend | Cat# 103137; RRID:AB_2561392 |
| Anti-mouse TCRb PE-Cy7 (H57-597) | Biolegend | Cat# 109223; RRID:AB_1027654 |
| Anti-mouse CD19 APC-Cy7 (6D5) | Biolegend | Cat# 115530; RRID:AB_830707 |
| Anti-mouse CD8 AF700 (53-6.7) | Biolegend | Cat# 100730; RRID:AB_493703 |
| Anti-mouse CD4 FITC (Rm4-5) | Biolegend | Cat# 100510; RRID:AB_312713 |
| Anti-mouse FoxP3 APC (FJK-16s) | Thermo Fisher Scientific | Cat# 17-5773-82; RRID:AB_469457 |
| Anti-mouse RORγ PE (B2D) | Thermo Fisher Scientific | Cat# 12-6981-82; RRID:AB_10807092 |
| Anti-mouse Helios Pacific Blue (22F6) | Biolegend | Cat# 137220; RRID:AB_10690535 |
| Anti-mouse IgA Unconjugated | Bethyl Laboratories | Cat# A90-103A; RRID:AB_67136 |
| Anti-mouse IgG1 Unconjugated | Bethyl Laboratories | Cat# A90-105A; RRID:AB_67147 |
| Anti-mouse IgG2b Unconjugated | Bethyl Laboratories | Cat# A90-109A; RRID:AB_67157 |
| Anti-mouse IgG2c Unconjugated | Bethyl Laboratories | Cat# A90-136A; RRID:AB_67162 |
| Anti-mouse IgE Unconjugated | Bethyl Laboratories | Cat# A90-115A; RRID:AB_67142 |
| Anti-mouse IgA HRP-conjugated | Bethyl Laboratories | Cat# A90-103P; RRID:AB_67140 |
| Anti-mouse IgG1 HRP-conjugated | Bethyl Laboratories | Cat# A90-105P; RRID:AB_67150 |
| Anti-mouse IgG2b HRP-conjugated | Bethyl Laboratories | Cat# A90-109P; RRID:AB_67160 |
| Anti-mouse IgG2c HRP-conjugated | Bethyl Laboratories | Cat# A90-136P; RRID:AB_67165 |
| Anti-mouse IgE HRP-conjugated | Bethyl Laboratories | Cat# A90-115P; RRID:AB_67145 |
| Anti-mouse IgA PE (mA-6E1) | Thermo Fisher Scientific | Cat# 12-4204-82; RRID:AB_465917 |
| Bacterial and virus strains | ||
| Staphylococcus xylosus PC20 | This study | N/A |
| Staphylococcus sciuri PC04 | This study | N/A |
| Enterococcus faecalis PC15 | This study | N/A |
| Lactobacillus murinus PC39 | This study | N/A |
| Anaerostipes sp. PC18 | This study | N/A |
| Lactobacillus johnsonii PC38 | This study | N/A |
| Clostridium intestinale PC17 | This study | N/A |
| Kosakonia cowanii PC08 | This study | N/A |
| Parabacteroides distasonis PC19 | This study | N/A |
| Bifidobacterium animalis subsp. animalis YL2 | DSMZ | DSM No: 26074 |
| Muribaculum intestinale YL27 | DSMZ | DSM No: 28989 |
| Flavonifractor plautii YL31 | DSMZ | DSM No: 26117 |
| Enterocloster clostridioformis YL32 | DSMZ | DSM No: 26114 |
| Akkermansia muciniphila YL44 | DSMZ | DSM No: 26127 |
| Turicimonas muris YL45 | DSMZ | DSM No: 26109 |
| Blautia coccoides YL58 | DSMZ | DSM No: 26115 |
| Clostridium innocuum I46 | DSMZ | DSM No: 26113 |
| Bacteroides caecimuris I48 | DSMZ | DSM No: 26085 |
| Limosilactobacillus reuteri I49 | DSMZ | DSM No: 32035 |
| Enterococcus faecalis KB1 | DSMZ | DSM No: 32036 |
| Acutalibacter muris KB18 | DSMZ | DSM No: 26090 |
| Salmonella enterica s. Typhimurium SL1344 (StrepR) | Brodsky lab strain collection | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| RNAlater | ThermoFisher Scientific | Cat# AM7020 |
| Sodium acetate | Sigma-Aldrich | Cat# S5636 |
| Sodium propionate | Sigma-Aldrich | Cat# P1880 |
| Sodium butyrate | ThermoFisher Scientific | Cat# A11079.22 |
| Critical commercial assays | ||
| DNAeasy Powersoil kit | Qiagen | Cat# 12888-100 |
| Blood and Cell Culture DNA kit | Qiagen | Cat# 19060 |
| Nanopore Rapid Barcoding Kit | Oxford Nanopore Technologies | Cat# RBK-004 |
| Nextera XT library preparation kit | Illumina | Cat# FC-131-1024 |
| E.Z.N.A. total RNA kit I | Omega Bio-tek | Cat# R6834-01 |
| High-Capacity RNA-to-cDNA kit | ThermoFisher Scientific | Cat# 4387406 |
| PowerUp SYBR Green PCR master mix | ThermoFisher Scientific | Cat# A25742 |
| TaqMan multiplex master mix | ThermoFisher Scientific | Cat# 4461882 |
| Deposited data | ||
| 16S rRNA gene metagenomics: PedsCom and CMCom intestinal samples | This study | BioProject: PRJNA934396 |
| 16S rRNA gene metagenomics: SPF NOD and sv129 mouse feces (vivarium 1 + 2) | This study | BioProject: PRJNA934396 |
| 16S rRNA gene metagenomics: Oligo-MM12 C57BL/6J mouse feces | (Wyss et al., 2020) | https://doi.org/10.6084/m9.figshare.c.4763306.v1 |
| 16S rRNA gene metagenomics: SPF C57BL/6J mouse feces (vivarium 3) | (van Best et al., 2020) | ENA: ERP116798 |
| 16S rRNA gene metagenomics: SPF C57BL/6J mouse feces (vivarium 4) | (Kemp et al., 2021) | SRA: SRP247412 |
| Shotgun metagenomics: Human infant feces | (Bittinger et al., 2020) | SRA: SRP217052 |
| Whole genome sequences of PedsCom isolates | This Study | BioProject: PRJNA934396 |
| Whole genome sequences of Oligo-MM12 isolates | (Garzetti et al., 2017) | BioProject: PRJNA289613 |
| Whole genome sequences of Altered-Schaedler Flora isolates | (Wannemuehler et al., 2014) | BioProject: PRJNA176000, PRJNA176001, PRJNA176002, PRJNA176003, PRJNA176004, PRJNA213740, PRJNA213743, PRJNA175999 |
| Experimental models: Organisms/strains | ||
| Mouse: NOD | Jackson laboratories | RRID:MGI:2159775 |
| Mouse: C57BL/6J | Jackson laboratories | RRID:IMSR_JAX:000664 |
| Mouse: sv129 | Jackson laboratories | RRID:IMSR_JAX:000691 |
| Oligonucleotides | ||
| Primer: murine Tnfa: Forward: CTGAACTTCGGGGTGATCGG |
(Wang et al., 2012) | PrimerBank ID: 133892368c2 |
| Primer: murine Tnfa: Reverse: GGCTTGTCACTCGAATTTTGAGA | (Wang et al., 2012) | PrimerBank ID: 133892368c2 |
| Primer: murine Gapdh: Forward: AGGTCGGTGTGAACGGATTTG | (Wang et al., 2012) | PrimerBank ID: 6679937a1 |
| Primer: murine Gapdh: Reverse: TGTAGACCATGTAGTTGAGGTCA | (Wang et al., 2012) | PrimerBank ID: 6679937a1 |
| Primer: EUB1114 Forward: CGGCAACGAGCGCAACCC | (Denman and McSweeney, 2006) | N/A |
| Primer: EUB1221 Reverse: CCATTGTAGCACGTGTGTAGCC | (Denman and McSweeney, 2006) | N/A |
| PedsCom TaqMan primer and probe sets | This study | See table S1 for sequences |
| Software and algorithms | ||
| QIIME2 ver. 2020.6 | (Bolyen et al., 2019) | https://qiime2.org/ |
| Deblur | (Amir et al., 2017) | https://github.com/biocore/deblur |
| Phyloseq | (McMurdie and Holmes, 2013) | https://joey711.github.io/phyloseq/ |
| PICRUSt2 | (Douglas et al., 2020) | https://github.com/picrust/picrust2 |
| BURRITO | (McNally et al., 2018) | https://github.com/borenstein-lab/burrito |
| ANCOM | (Mandal et al., 2015) | https://github.com/FrederickHuangLin/ANCOMBC |
| Mash screen | (Ondov et al., 2019) | https://github.com/marbl/Mash |
| Unicycler pipeline | (Wick et al., 2017) | https://github.com/rrwick/Unicycler |
| RASTtk | (Brettin et al., 2015) | https://www.bv-brc.org/remote |
| KEGG automatic annotation server | (Moriya et al., 2007) | https://www.genome.jp/kegg/kaas/ |
| MicrobiomeAnalyst | (Chong et al., 2020) | https://www.microbiomeanalyst.ca/ |
| FlowJo v10 | Becton-Dickinson | https://www.flowjo.com/ |
| 7500 software ver. 2.3 | ThermoFisher Scientific | N/A |
| CFX Maestro ver. 2.3 | Bio-Rad | N/A |
| Prism 8 | GraphPad Software Inc. | https://www.graphpad.com/scientific-software/prism/ |
| ggpubr | CRAN | https://rpkgs.datanovia.com/ggpubr/ |
| Other | ||
| Illumina MiSeq platform | lllumina | Cat# SY-410-1003 |
| minION sequencer | Oxford Nanopore Technologies | Cat# MIN-101B |
| Illumina HiSeq 2500 platform | Illumina | Cat# SY-401-2501 |
| LSR Fortessa | Becton-Dickinson | Cat# 649225B1 |
| 7500 Fast Real-time PCR system | ThermoFisher Scientific | Cat# 4351106 |
| CFX Opus 96 PCR system | Bio-Rad | Cat# 12011319 |
| Acquity uPLC Core System | Waters | Cat# 176015000 |
| Acquity QDa detector | Waters | Cat# 176003208 |
| HSS T3 1.8 µm 2.1 × 150 mm column | Waters | Cat# 186009635 |
| Cortecs UPLC C-18+ 1.6 mm 2.1 × 50 mm column | Waters | Cat# 186007114 |
Highlights.
Develop a bacterial consortium to model the murine pre-weaning intestinal microbiome
Restricted intestinal microbiome maturation leads to stunted immune system development
Restricted microbiome development increases susceptibility to Salmonella infection
Acknowledgements
The authors thank Kyle Bittinger, Ceylan Tanes, William Bailis, Matthew Weitzman, Laurence Eisenlohr, Kathryn Knoop, Neil Surana and Joseph Zackular for helpful discussions. The authors also thank Dmitri Kobuley and Michelle Albright of the PennCHOP microbiome program gnotobiotic mouse facility at Hill Pavilion and Elliot Friedman and Dylan Curry of the PennCHOP microbiome microbial culture and metabolomics core. The graphical abstract was made with BioRender. This work was supported by National Institutes of Health grants R21AI146629 (M.A.S.), R01AI137526-01 (P.J.P.), Juvenile Diabetes Research Foundation grant 5-CDA-2020-946-S-B (M.A.S.), National Institutes of Health T32 Postdoctoral Program in Clinical Pharmacology T32GM008562 (J.L.) and the Children’s Hospital of Philadelphia/University of Pennsylvania Postdoctoral Research Fellowship for Academic Diversity (J.L.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
We declare no financial conflict of interest.
Inclusion and Diversity
One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in their field of research or within their geographical location. One or more of the authors of this paper self-identifies as a gender minority in their field of research. One or more of the authors of this paper self-identifies as a member of the LGBTQIA+ community. One or more of the authors of this paper received support from a program designed to increase minority representation in their field of research.
References
- 1.Gensollen T, Iyer SS, Kasper DL, and Blumberg RS (2016). How colonization by microbiota in early life shapes the immune system. Science 352, 539–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominguez-Bello MG, Contreras M, Magris M, Hidalgo G, Baldassano RN, Anokhin AP, et al. (2012). Human gut microbiome viewed across age and geography. Nature 486, 222–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kim Y-G, Sakamoto K, Seo S-U, Pickard JM, Gillilland MG, Pudlo NA, Hoostal M, Li X, Wang TD, Feehley T, et al. (2017). Neonatal acquisition of Clostridia species protects against colonization by bacterial pathogens. Science 356, 315–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wynn JL, and Levy O (2010). Role of Innate Host Defenses in Susceptibility to Early Onset Neonatal Sepsis. Clin. Perinatol 37, 307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van Best N, Rolle-Kampczyk U, Schaap FG, Basic M, Olde Damink SWM, Bleich A, Savelkoul PHM, von Bergen M, Penders J, and Hornef MW (2020). Bile acids drive the newborn’s gut microbiota maturation. Nat. Commun 11, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Koenig JE, Spor A, Scalfone N, Fricker AD, Stombaugh J, Knight R, Angenent LT, and Ley RE (2011). Succession of microbial consortia in the developing infant gut microbiome. Proc. Natl. Acad. Sci 108, 4578–4585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hasegawa M, Osaka T, Tawaratsumida K, Yamazaki T, Tada H, Chen GY, Tsuneda S, Núñez G, and Inohara N (2010). Transitions in Oral and Intestinal Microflora Composition and Innate Immune Receptor-Dependent Stimulation during Mouse Development. Infect. Immun 78, 639–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Silverman M, Kua L, Tanca A, Pala M, Palomba A, Tanes C, Bittinger K, Uzzau S, Benoist C, and Mathis D (2017). Protective major histocompatibility complex allele prevents type 1 diabetes by shaping the intestinal microbiota early in ontogeny. Proc. Natl. Acad. Sci. U. S. A 114, 9671–9676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Galazzo G, van Best N, Bervoets L, Dapaah IO, Savelkoul PH, Hornef MW, Hutton EK, Morrison K, Holloway AC, McDonald H, et al. (2020). Development of the Microbiota and Associations With Birth Mode, Diet, and Atopic Disorders in a Longitudinal Analysis of Stool Samples, Collected From Infancy Through Early Childhood. Gastroenterology 158, 1584–1596. [DOI] [PubMed] [Google Scholar]
- 10.Miller SA, Wu RKS, and Oremus M (2018). The association between antibiotic use in infancy and childhood overweight or obesity: a systematic review and meta-analysis. Obes. Rev 19, 1463–1475. [DOI] [PubMed] [Google Scholar]
- 11.Al Nabhani Z, Dulauroy S, Marques R, Cousu C, Al Bounny S, Déjardin F, Sparwasser T, Bérard M, Cerf-Bensussan N, and Eberl G (2019). A Weaning Reaction to Microbiota Is Required for Resistance to Immunopathologies in the Adult. Immunity 50, 1276–1288.e5. [DOI] [PubMed] [Google Scholar]
- 12.Hornef MW, and Torow N (2020). ‘Layered immunity’ and the ‘neonatal window of opportunity’ – timed succession of non‐redundant phases to establish mucosal host–microbial homeostasis after birth. Immunology 159, 15–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Natividad JMM, Hayes CL, Motta JP, Jury J, Galipeau HJ, Philip V, Garcia-Rodenas CL, Kiyama H, Bercik P, and Verdú EF (2013). Differential Induction of Antimicrobial REGIII by the Intestinal Microbiota and Bifidobacterium breve NCC2950. Appl. Environ. Microbiol 79, 7745–7754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Atarashi K, Tanoue T, Ando M, Kamada N, Nagano Y, Narushima S, Suda W, Imaoka A, Setoyama H, Nagamori T, et al. (2015). Th17 Cell Induction by Adhesion of Microbes to Intestinal Epithelial Cells. Cell 163, 367–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sefik E, Geva-Zatorsky N, Oh S, Konnikova L, Zemmour D, McGuire AM, Burzyn D, Ortiz-Lopez A, Lobera M, Yang J, et al. (2015). Individual intestinal symbionts induce a distinct population of ROR + regulatory T cells. Science 349, 993–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ohnmacht C, Park J-H, Cording S, Wing JB, Atarashi K, Obata Y, Gaboriau-Routhiau V, Marques R, Dulauroy S, Fedoseeva M, et al. (2015). The microbiota regulates type 2 immunity through RORγt+ T cells. Science 349, 989–993. [DOI] [PubMed] [Google Scholar]
- 17.Hapfelmeier S, Lawson MAE, Slack E, Kirundi JK, Stoel M, Heikenwalder M, Cahenzli J, Velykoredko Y, Balmer ML, Endt K, et al. (2010). Reversible microbial colonization of germ-free mice reveals the dynamics of IgA immune responses. Science 328, 1705–1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dewhirst FE, Chien CC, Paster BJ, Ericson RL, Orcutt RP, Schauer DB, and Fox JG (1999). Phylogeny of the defined murine microbiota: altered Schaedler flora. Appl. Environ. Microbiol 65, 3287–3292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brugiroux S, Beutler M, Pfann C, Garzetti D, Ruscheweyh H-J, Ring D, Diehl M, Herp S, Lötscher Y, Hussain S, et al. (2016). Genome-guided design of a defined mouse microbiota that confers colonization resistance against Salmonella enterica serovar Typhimurium. Nat. Microbiol 2, 16215. [DOI] [PubMed] [Google Scholar]
- 20.Ansaldo E, Slayden LC, Ching KL, Koch MA, Wolf NK, Plichta DR, Brown EM, Graham DB, Xavier RJ, Moon JJ, et al. (2019). Akkermansia muciniphila induces intestinal adaptive immune responses during homeostasis. Science 364, 1179–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gomes-Neto JC, Kittana H, Mantz S, Segura Munoz RR, Schmaltz RJ, Bindels LB, Clarke J, Hostetter JM, Benson AK, Walter J, et al. (2017). A gut pathobiont synergizes with the microbiota to instigate inflammatory disease marked by immunoreactivity against other symbionts but not itself. Sci. Rep 7, 17707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hand TW, Dos Santos LM, Bouladoux N, Molloy MJ, Pagán AJ, Pepper M, Maynard CL, Elson CO, and Belkaid Y (2012). Acute Gastrointestinal Infection Induces Long-Lived Microbiota-Specific T Cell Responses. Science 337, 1553–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bolsega S, Basic M, Smoczek A, Buettner M, Eberl C, Ahrens D, Odum KA, Stecher B, and Bleich A (2019). Composition of the Intestinal Microbiota Determines the Outcome of Virus-Triggered Colitis in Mice. Front. Immunol 10, 1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Darnaud M, De Vadder F, Bogeat P, Boucinha L, Bulteau AL, Bunescu A, Couturier C, Delgado A, Dugua H, Elie C, et al. (2021). A standardized gnotobiotic mouse model harboring a minimal 15-member mouse gut microbiota recapitulates SOPF/SPF phenotypes. Nat. Commun 12, 6686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, and Holmes SP (2016). DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gu S, Chen D, Zhang J-N, Lv X, Wang K, Duan L-P, Nie Y, and Wu X-L (2013). Bacterial Community Mapping of the Mouse Gastrointestinal Tract. PLoS One 8, 74957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yuan C, Graham M, Staley C, and Subramanian S (2020). Mucosal Microbiota and Metabolome along the Intestinal Tract Reveal a Location-Specific Relationship. mSystems 5, e00055–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moriya Y, Itoh M, Okuda S, Yoshizawa AC, and Kanehisa M (2007). KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res 35, W182–W185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Douglas GM, Maffei VJ, Zaneveld JR, Yurgel SN, Brown JR, Taylor CM, Huttenhower C, and Langille MGI (2020). PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol 38, 685–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Palmer C, Bik EM, DiGiulio DB, Relman DA, and Brown PO (2007). Development of the Human Infant Intestinal Microbiota. PLOS Biol 5, e177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yassour M, Vatanen T, Siljander H, Hämäläinen A-M, Härkönen T, Ryhänen SJ, Franzosa EA, Vlamakis H, Huttenhower C, Gevers D, et al. (2016). Natural history of the infant gut microbiome and impact of antibiotic treatments on strain-level diversity and stability. Sci. Transl. Med 8, 343ra81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lagkouvardos I, Lesker TR, Hitch TCA, Gálvez EJC, Smit N, Neuhaus K, Wang J, Baines JF, Abt B, Stecher B, et al. (2019). Sequence and cultivation study of Muribaculaceae reveals novel species, host preference, and functional potential of this yet undescribed family. Microbiome 7, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu H, Liao C, Wu L, Tang J, Chen J, Lei C, Zheng L, Zhang C, Liu YY, Xavier J, et al. (2022). Ecological dynamics of the gut microbiome in response to dietary fiber. ISME J 16, 2040–2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Niu J, Xu L, Qian Y, Sun Z, Yu D, Huang J, Zhou X, Wang Y, Zhang T, Ren R, et al. (2020). Evolution of the Gut Microbiome in Early Childhood: A Cross-Sectional Study of Chinese Children. Front. Microbiol 11, 439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wick RR, Judd LM, Gorrie CL, and Holt KE (2017). Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLOS Comput. Biol 13, e1005595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brettin T, Davis JJ, Disz T, Edwards RA, Gerdes S, Olsen GJ, Olson R, Overbeek R, Parrello B, Pusch GD, et al. (2015). RASTtk: A modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci. Rep 5, 8365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Davis JJ, Wattam AR, Aziz RK, Brettin T, Butler R, Butler RM, Chlenski P, Conrad N, Dickerman A, Dietrich EM, et al. (2020). The PATRIC Bioinformatics Resource Center: expanding data and analysis capabilities. Nucleic Acids Res 48, D606–D612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bittinger K, Zhao C, Li Y, Ford E, Friedman ES, Ni J, Kulkarni CV, Cai J, Tian Y, Liu Q, et al. (2020). Bacterial colonization reprograms the neonatal gut metabolome. Nat. Microbiol 56 5, 838–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Knoop KA, Gustafsson JK, McDonald KG, Kulkarni D, Coughlin PE, McCrate S, Kim D, Hsieh C-S, Hogan SP, Elson CO, et al. (2017). Microbial Antigen Encounter During a Pre-weaning Interval is Critical for Tolerance to Gut Bacteria. Sci. Immunol 2. [DOI] [PMC free article] [PubMed]
- 40.Kim KS, Hong SW, Han D, Yi J, Jung J, Yang BG, Lee JY, Lee M, and Surh CD (2016). Dietary antigens limit mucosal immunity by inducing regulatory T cells in the small intestine. Science 351, 858–863. [DOI] [PubMed] [Google Scholar]
- 41.Tahara Y, Yamazaki M, Sukigara H, Motohashi H, Sasaki H, Miyakawa H, Haraguchi A, Ikeda Y, Fukuda S, and Shibata S (2018). Gut Microbiota-Derived Short Chain Fatty Acids Induce Circadian Clock Entrainment in Mouse Peripheral Tissue. Sci. Rep 8, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sayin SI, Wahlström A, Felin J, Jäntti S, Marschall HU, Bamberg K, Angelin B, Hyötyläinen T, Orešič M, and Bäckhed F (2013). Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR antagonist. Cell Metab 17, 225–235. [DOI] [PubMed] [Google Scholar]
- 43.Song X, Sun X, Oh SF, Wu M, Zhang Y, Zheng W, Geva-Zatorsky N, Jupp R, Mathis D, Benoist C, et al. (2019). Microbial bile acid metabolites modulate gut RORγ+ regulatory T cell homeostasis. Nature 557, 410–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sayin SI, Wahlström A, Felin J, Jäntti S, Marschall HU, Bamberg K, Angelin B, Hyötyläinen T, Orešič M, and Bäckhed F (2013). Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR antagonist. Cell Metab 17, 225–235. [DOI] [PubMed] [Google Scholar]
- 45.Cahenzli J, Köller Y, Wyss M, Geuking MB, and McCoy KD (2013). Intestinal microbial diversity during early-life colonization shapes long-term IgE levels. Cell Host Microbe 14, 559–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rogier EW, Frantz AL, Bruno MEC, Wedlund L, Cohen DA, Stromberg AJ, and Kaetzel CS (2014). Secretory antibodies in breast milk promote long-term intestinal homeostasis by regulating the gut microbiota and host gene expression. Proc. Natl. Acad. Sci. U. S. A 111, 3074–3079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Takeuchi T, Miyauchi E, Kanaya T, Kato T, Nakanishi Y, Watanabe T, Kitami T, Taida T, Sasaki T, Negishi H, et al. (2021). Acetate differentially regulates IgA reactivity to commensal bacteria. Nature 595, 560–564. [DOI] [PubMed] [Google Scholar]
- 48.Lanata CF, Fischer-Walker CL, Olascoaga AC, Torres CX, Aryee MJ, Black RE, and UNICEF, for the C.H.E.R.G. of the W.H.O. and (2013). Global Causes of Diarrheal Disease Mortality in Children <5 Years of Age: A Systematic Review. PLoS One 8, 72788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dupont A, Sommer F, Zhang K, Repnik U, Basic M, Bleich A, Kühnel M, Bäckhed F, Litvak Y, Fulde M, et al. (2016). Age-Dependent Susceptibility to Enteropathogenic Escherichia coli (EPEC) Infection in Mice. PLOS Pathog 12, e1005616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stecher B, Macpherson AJ, Hapfelmeier S, Kremer M, Stallmach T, and Hardt WD (2005). Comparison of Salmonella enterica serovar typhimurium colitis in germfree mice and mice pretreated with streptomycin. Infect. Immun 73, 3228–3241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Barthel M, Hapfelmeier S, Quintanilla-Martínez L, Kremer M, Rohde M, Hogardt M, Pfeffer K, Rüssmann H, and Hardt WD (2003). Pretreatment of Mice with Streptomycin Provides a Salmonella enterica Serovar Typhimurium Colitis Model That Allows Analysis of Both Pathogen and Host. Infect. Immun 71, 2839–2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ormerod KL, Wood DLA, Lachner N, Gellatly SL, Daly JN, Parsons JD, Dal’Molin CGO, Palfreyman RW, Nielsen LK, Cooper MA, et al. (2016). Genomic characterization of the uncultured Bacteroidales family S24-7 inhabiting the guts of homeothermic animals. Microbiome 4, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Arpaia N, Campbell C, Fan X, Dikiy S, Van Der Veeken J, Deroos P, Liu H, Cross JR, Pfeffer K, Coffer PJ, et al. (2013). Metabolites produced by commensal bacteria promote peripheral regulatory T cell generation HHS Public Access. Nature 504, 451–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, Takahashi D, Nakanishi Y, Uetake C, Kato K, Kato T, et al. (2013). Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 504, 446–450. [DOI] [PubMed] [Google Scholar]
- 55.Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly-Y M, Glickman JN, and Garrett WS (2013). The microbial metabolites, short chain fatty acids, regulate colonic Treg cell homeostasis. Science 341, 569–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Atarashi K, Tanoue T, Oshima K, Suda W, Nagano Y, Nishikawa H, Fukuda S, Saito T, Narushima S, Hase K, et al. (2013). Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature 500, 232–236. [DOI] [PubMed] [Google Scholar]
- 57.Atarashi K, Tanoue T, Shima T, Imaoka A, Kuwahara T, Momose Y, Cheng G, Yamasaki S, Saito T, Ohba Y, et al. (2011). Induction of colonic regulatory T cells by indigenous Clostridium species. Science 331, 337–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ridlon JM, Kang DJ, and Hylemon PB (2006). Bile salt biotransformations by human intestinal bacteria. J. Lipid Res 47, 241–259. [DOI] [PubMed] [Google Scholar]
- 59.Burns-Guydish SM, Olomu IN, Zhao H, Wong RJ, Stevenson DK, and Contag CH (2005). Monitoring Age-Related Susceptibility of Young Mice To Oral Salmonella enterica Serovar Typhimurium Infection Using an In Vivo Murine Model. Pediatr. Res 58, 153–158. [DOI] [PubMed] [Google Scholar]
- 60.Stecher B, Chaffron S, Käppeli R, Hapfelmeier S, Freedrich S, Weber TC, Kirundi J, Suar M, McCoy KD, Von Mering C, et al. (2010). Like Will to Like: Abundances of Closely Related Species Can Predict Susceptibility to Intestinal Colonization by Pathogenic and Commensal Bacteria. PLOS Pathog 6, e1000711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Seif Y, Kavvas E, Lachance JC, Yurkovich JT, Nuccio SP, Fang X, Catoiu E, Raffatellu M, Palsson BO, and Monk JM (2018). Genome-scale metabolic reconstructions of multiple Salmonella strains reveal serovar-specific metabolic traits. Nat. Commun 9, 3771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kröger C, Stolz J, and Fuchs TM (2010). Myo-Inositol transport by Salmonella enterica serovar Typhimurium. Microbiology 156, 128–138. [DOI] [PubMed] [Google Scholar]
- 63.Bäckhed F, Roswall J, Peng Y, Feng Q, Jia H, Kovatcheva-Datchary P, Li Y, Xia Y, Xie H, Zhong H, et al. (2015). Dynamics and Stabilization of the Human Gut Microbiome during the First Year of Life. Cell Host Microbe 17, 690–703. [DOI] [PubMed] [Google Scholar]
- 64.Duncan SH, Barcenilla A, Stewart CS, Pryde SE, and Flint HJ (2002). Acetate utilization and butyryl coenzyme A (CoA): Acetate-CoA transferase in butyrate-producing bacteria from the human large intestine. Appl. Environ. Microbiol 68, 5186–5190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Eberl C, Ring D, Münch PC, Beutler M, Basic M, Slack EC, Schwarzer M, Srutkova D, Lange A, Frick JS, et al. (2020). Reproducible Colonization of Germ-Free Mice With the Oligo-Mouse-Microbiota in Different Animal Facilities. Front. Microbiol 10, 2999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Huntley J, Fierer N, Owens SM, Betley J, Fraser L, Bauer M, et al. (2012). Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J 6, 1621–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wyss M, Brown K, Thomson CA, Koegler M, Terra F, Fan V, Ronchi F, Bihan D, Lewis I, Geuking MB, et al. (2020). Using Precisely Defined in vivo Microbiotas to Understand Microbial Regulation of IgE. Front. Immunol 10, 3107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kemp KM, Colson J, Lorenz RG, Maynard CL, and Pollock JS (2021). Early life stress in mice alters gut microbiota independent of maternal microbiota inheritance. Am. J. Physiol. - Regul. Integr. Comp. Physiol 320, R663–R674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, Alexander H, Alm EJ, Arumugam M, Asnicar F, et al. (2019). Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol 37, 852–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Amir A, McDonald D, Navas-Molina JA, Kopylova E, Morton JT, Zech Xu Z, Kightley EP, Thompson LR, Hyde ER, Gonzalez A, et al. (2017). Deblur Rapidly Resolves Single-Nucleotide Community Sequence Patterns. mSystems 2. [DOI] [PMC free article] [PubMed]
- 71.McMurdie PJ, and Holmes S (2013). phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS One 8, e61217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.McNally CP, Eng A, Noecker C, Gagne-Maynard WC, and Borenstein E (2018). BURRITO: An Interactive Multi-Omic Tool for Visualizing Taxa-Function Relationships in Microbiome Data. Front. Microbiol 9, 365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mandal S, Van Treuren W, White RA, Eggesbø M, Knight R, and Peddada SD (2015). Analysis of composition of microbiomes: a novel method for studying microbial composition. Microb. Ecol. Health Dis 26, 27663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ondov BD, Starrett GJ, Sappington A, Kostic A, Koren S, Buck CB, and Phillippy AM (2019). Mash Screen: high-throughput sequence containment estimation for genome discovery. Genome Biol 20, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yoon SH, Ha S. min, Lim J, Kwon S, and Chun J (2017). A large-scale evaluation of algorithms to calculate average nucleotide identity. Antonie van Leeuwenhoek, Int. J. Gen. Mol. Microbiol 110, 1281–1286. [DOI] [PubMed] [Google Scholar]
- 76.Wannemuehler MJ, Overstreet AM, Ward DV, and Phillips GJ (2014). Draft Genome Sequences of the Altered Schaedler Flora, a Defined Bacterial Community from Gnotobiotic Mice. Genome Announc 2, 287–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Garzetti D, Brugiroux S, Bunk B, Pukall R, McCoy KD, Macpherson AJ, and Stecher B (2017). High-Quality Whole-Genome Sequences of the Oligo-Mouse-Microbiota Bacterial Community. Genome Announc 5, e00758–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chong J, Liu P, Zhou G, and Xia J (2020). Using MicrobiomeAnalyst for comprehensive statistical, functional, and meta-analysis of microbiome data. Nat. Protoc 15, 799–821. [DOI] [PubMed] [Google Scholar]
- 79.Wang X, Spandidos A, Wang H, and Seed B (2012). PrimerBank: a PCR primer database for quantitative gene expression analysis, 2012 update. Nucleic Acids Res 40, D1144–D1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Denman SE, and McSweeney CS (2006). Development of a real-time PCR assay for monitoring anaerobic fungal and cellulolytic bacterial populations within the rumen. FEMS Microbiol. Ecol 58, 572–582. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Whole genome sequences of PedsCom isolates and 16S rRNA gene metagenomic datasets generated in this study have been deposited to NCBI BioProject: PRJNA934396 and are publicly available as of the date of publication.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.