

Abstract
BACKGROUND:
Polygenicity and genetic heterogeneity pose great challenges for studying psychiatric conditions. Genetically informed approaches have been implemented in neuroimaging studies to address this issue. However, the effects on functional connectivity of rare and common genetic risks for psychiatric disorders are largely unknown. Our objectives were to estimate and compare the effect sizes on brain connectivity of psychiatric genomic risk factors with various levels of complexity: oligogenic copy number variants (CNVs), multigenic CNVs, and polygenic risk scores (PRSs) as well as idiopathic psychiatric conditions and traits.
METHODS:
Resting-state functional magnetic resonance imaging data were processed using the same pipeline across 9 datasets. Twenty-nine connectome-wide association studies were performed to characterize the effects of 15 CNVs (1003 carriers), 7 PRSs, 4 idiopathic psychiatric conditions (1022 individuals with autism, schizophrenia, bipolar conditions, or attention-deficit/hyperactivity disorder), and 2 traits (31,424 unaffected control subjects).
RESULTS:
Effect sizes on connectivity were largest for psychiatric CNVs (estimates: 0.2–0.65 score), followed by psychiatric conditions (0.15–0.42), neuroticism and fluid intelligence (0.02–0.03), and PRSs (0.01–0.02). Effect sizes of CNVs on connectivity were correlated to their effects on cognition and risk for disease (r = 0.9, p = 5.93 × 10−6). However, effect sizes of CNVs adjusted for the number of genes significantly decreased from small oligogenic to large multigenic CNVs (r = −0.88, p = 8.78 × 10−6). PRSs had disproportionately low effect sizes on connectivity compared with CNVs conferring similar risk for disease.
CONCLUSIONS:
Heterogeneity and polygenicity affect our ability to detect brain connectivity alterations underlying psychiatric manifestations.
Polygenicity and genetic heterogeneity pose great challenges for studying mechanisms and risk underlying psychiatric conditions (1). Rare copy number variants (CNVs), as well as common variants, confer risk for neurodevelopmental and psychiatric disorders such as autism spectrum disorder and schizophrenia (SZ). CNVs that increase risk for autism and/or SZ also decrease IQ (2,3). Their effect sizes range from large to mild (e.g., 22q11.2 and 15q11.2 deletions decrease IQ by 29 and 3 points, respectively, and increase risk for SZ with odd ratios of 23 and 1.9, respectively) (Table 1) (4,5). Effect sizes of CNVs on cognition and risk for neurodevelopmental and psychiatric disorders are positively correlated to the number of genes they contain. To account for the fact that not all genes contribute equally to the impact of CNVs, we developed a CNV severity score, which is the sum of genes encompassed in CNVs, weighted by the sensitivity of each gene to loss of function (6). Both the number of genes included in a CNV and its severity score are measures of the level of multigenicity of a CNV. This severity score can predict the effect size of CNVs on cognition with close to 80% accuracy (7,8). Furthermore, the mean effect size on cognitive ability of one point of this severity score is similar for benign oligogenic and deleterious multigenic CNVs, which suggests that effect sizes of large CNVs are the additive effects of many individual genes with small effects.
Table 1.
Demographics: CNVs
| CNV (hg19) | Number of Genes / Well-Known Gene | Status | Ascertainment, n | Age, Years, Mean (SD) | Sex, F/M, n | Cohorts | Previously Published | |||
|---|---|---|---|---|---|---|---|---|---|---|
| Clinical | Unselected | IQ Loss | OR ASD | OR SZ | ||||||
| Psychiatric CNVs | ||||||||||
| 1q21.1 1: 146.53–147.39 |
7 / CHD1L | DEL | 15 | 10 | 44.4 (19) | 12/13 | UKBB, MRG, Cardiff, SFARI | 15 | 3.2 | 6.4 |
| DUP | 6 | 13 | 50.9 (19) | 13/6 | 25 | 5.3 | 2.9 | |||
| 22q11.2 22: 19.04–21.47 |
49 / TBX1 | DEL | 43 | 0 | 16.9 (7) | 19/24 | UCLA | 28.8 | 32.3 | 23 |
| DUP | 12 | 10 | 39.4 (23) | 12/10 | UCLA, UKBB, Cardiff, MRG | 8.3 | 2 | 0.2 | ||
| 16p11.2 16: 29.65–30.20 |
27 / KCTD13 | DEL | 28 | 4 | 21.7 (20) | 13/19 | SFARI, MRG, UKBB | 26 | 14.3 | 1.1 |
| DUP | 29 | 6 | 34.1 (19) | 14/21 | 11 | 10.5 | 11.7 | |||
| 15q11.2 15: 22.81–23.09 |
4 / CYFIP1 | DEL | 0 | 103 | 64.3 (7) | 55/48 | UKBB | 3 | 1.3 | 1.9 |
| Nonpsychiatric CNVs | ||||||||||
| 15q11.2 15: 22.81–23.09 |
4 / CYFIP1 | DUP | 0 | 136 | 63.7 (7) | 76/60 | UKBB | 0.9 | 1 | 1 |
| 15q13.3 15: 31.08–32.46 |
5 / CHRNA7 | DUP | 0 | 190 | 64.4 (7) | 100/90 | 0.9 | 0.7 | 1.24 | |
| 2q13 2: 110.86–110.98 |
3 / NPHP1 | DEL | 0 | 183 | 63.1 (7) | 110/73 | – | 1 | – | |
| DUP | 0 | 88 | 64.7 (8) | 43/45 | – | 1 | – | |||
| 16p13.11 16: 15.51–16.29 |
6 / MYH11 | DUP | 0 | 40 | 64.7 (6) | 21/19 | 2 | 1 | 1.5 | |
| 13q12.12 13: 23.56–24.88 |
5 / SPATA13 | DEL | 0 | 22 | 63.5 (6) | 12/9 | 2.1 | – | – | |
| DUP | 0 | 20 | 60.8 (7) | 10/10 | 0.6 | – | – | |||
| TAR 1: 145.39–147.39 |
15 / RBM8 | DUP | 0 | 29 | 59.8 (7) | 14/15 | 2.4 | – | 1 | |
The cohort column provides the cohorts used to perform case-control studies for each of the 29 CWASs. IQ loss indicates mean decrease in IQ points associated with each CNV (7,8). ORs for the enrichment of CNVs in autism and schizophrenia were previously published (4,5,9,11,26,27,52,65–70). ORs for the enrichment of CNVs in ADHD were not available. The nonpsychiatric CNVs were defined as variants without any previous association with psychiatric conditions in large case-control studies (4,26–28); and detailed information relative to diagnosis, IQ, and motion are available in the Supplement. Information relative to scanning sites, motion, and diagnoses are also available in eTables 2–4 in the Supplement. All sites scanned control subjects. The table shows the numbers of CNV carriers (DEL, DUP), individuals with idiopathic psychiatric conditions (SZ, ASD, ADHD, BIP), and control subjects after MRI quality control. Chromosome number and coordinates are presented in megabases (Hg19).
ADHD, attention-deficit/hyperactivity disorder; ASD, autism spectrum disorder; BIP, bipolar disorder; Chr, chromosome; CNV, copy number variant; CWAS, connectome-wide association study; DEL, deletion; DUP, duplication; F, female; M, male; MRG, Montreal rare genomic disorder; MRI, magnetic resonance imaging; OR, odds ratio; SFARI, Simons Foundation Autism Research Initiative; SZ, schizophrenia; UCLA, University of California, Los Angeles; UKBB, UK Biobank.
Similarly, for common variants, polygenic risk scores (PRSs) are additive models developed to estimate the aggregate effects of thousands of single nucleotide polymorphisms (SNPs) with very small individual effects (9,10). The risk for SZ ranges from odds ratio of 3.3 to 4.6 (9) when comparing individuals in the bottom and top deciles of PRS-SZ, similar or higher than the risk conferred by some oligogenic CNVs such as 1q21.1 and 15q11.2 deletions. For PRS-IQ, contrasting bottom and top deciles shows moderate to large effect sizes of approximately 9 to 12 points of IQ (11), which is similar to several CNVs associated with neurodevelopmental and psychiatric disorders (e.g., 16p11.2 duplication) (Table 1).
Because cognition is thought to be subserved by largescale brain networks (12), it is reasonable to hypothesize that the effects of genetic variants on cognition and behavior (8,13) are mediated by brain structure and networks (14,15). The organization of such networks can be inferred using resting-state functional magnetic resonance imaging (rs-fMRI) (16,17). Functional connectivity (FC) has gained traction in the past decade, characterizing increasingly reproducible patterns of alterations associated with psychiatric conditions (18). However, these studies reported small effect sizes, which appeared discordant with the severity of autism and attentiondeficit/hyperactivity disorder (ADHD) (19,20). Genetically informed approaches have been introduced with the hope of focusing on a specific biological risk. Effects of CNVs on FC have been investigated at only 2 genomic loci in humans (16p11.2 and 22q11.2) (14,21), demonstrating robust effects. Little is known about the effect sizes on FC of psychiatric PRS (22,23). While PRSs and CNVs can have similar effect sizes on psychiatric risk, the effects on connectivity of these 2 classes of variants with vastly different levels of genomic complexity have never been compared.
Our aims were to 1) estimate and compare the effects of oligogenic (e.g., 4 protein-coding genes for 15q11.2 CNVs) and multigenic (e.g., 49 protein-coding genes for 22q11.2 CNVs) CNVs (24) and PRSs for psychiatric conditions on brain connectivity; 2) characterize the relation between effect sizes of genomic variants on cognition/behavior and connectivity; and 3) test the relation between the level of multigenicity (measured by the number of genes and the severity score) and effect sizes of CNVs on connectivity.
To this end, we analyzed rs-fMRI data in 33,452 individuals and performed 29 connectome-wide association studies (CWASs) for 15 CNVs, 7 PRSs, 4 idiopathic conditions, inflammatory bowel disease, and 2 traits (fluid intelligence and neuroticism).
METHODS AND MATERIALS
The selection process for the CNVs, PRSs, and psychiatric conditions and traits are detailed in the Supplementary Materials and Methods.
Cohorts
Our analysis included 33,452 individuals from 9 datasets (Figure 1 and Tables 1 and 2). Each study of the corresponding dataset was approved by the research ethics review boards of the respective institutions. This project was approved by the research ethics review board at the Centre Hospitalier Universitaire Sainte-Justine.
Figure 1.
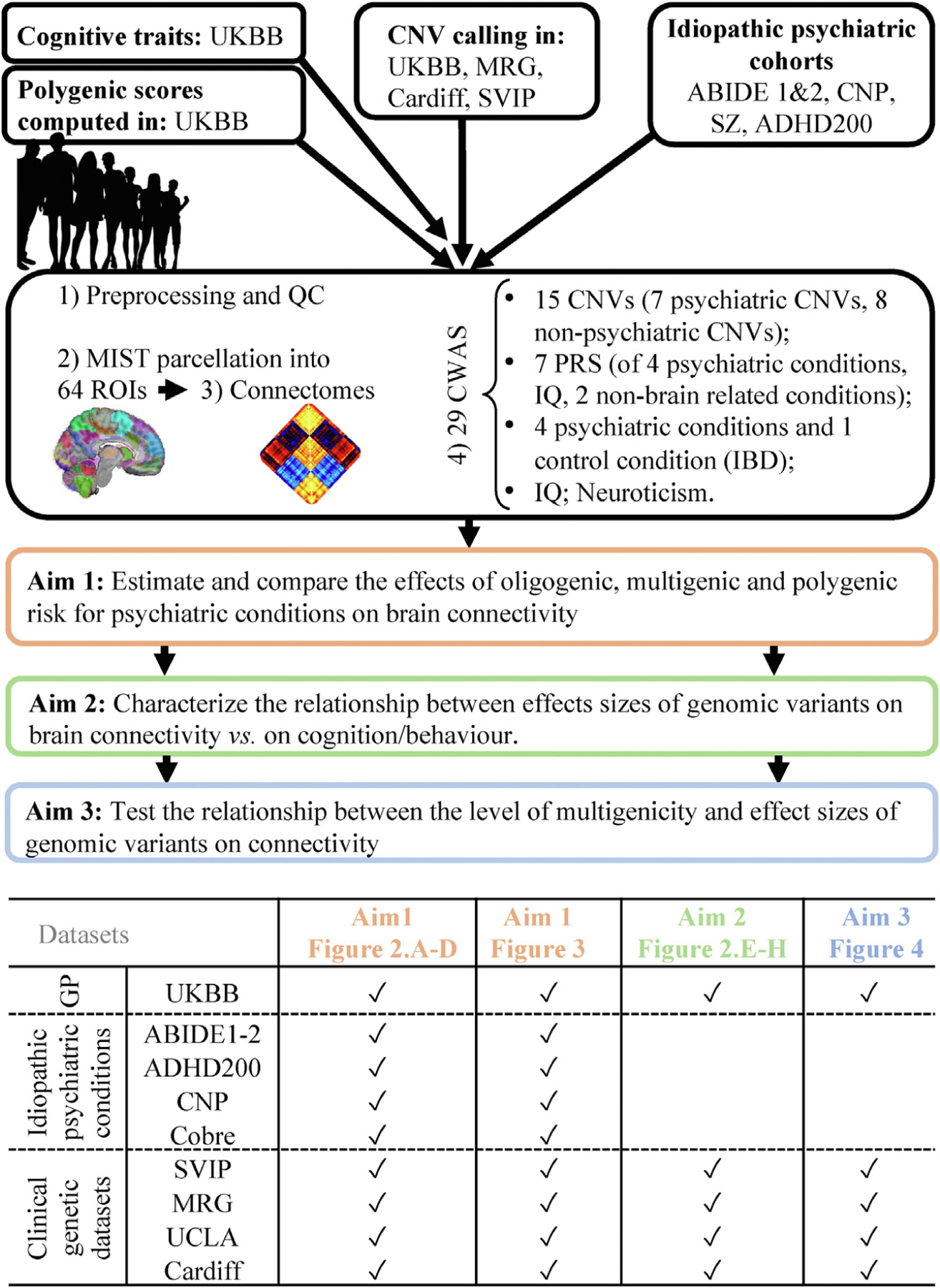
Method flowchart. ABIDE, Autism Brain Imaging Data Exchange; ADHD, attention-deficit/hyperactivity disorder; CNP, Consortium for Neuro-psychiatric Phenomics; CNV, copy number variant; CWAS, connectome-wide association study; GP, general population; IBD, inflammatory bowel disease; MIST, Multiresolution Intrinsic Segmentation Template; MRG, Montreal rare genomic disorder family project; PRS, polygenic risk score; ROI, region of interest; QC, quality control; SVIP, Simons Variation in Individuals Project; SZ, schizophrenia; UCLA, University of California, Los Angeles; UKBB, UK Biobank.
Table 2.
Demographics: Conditions, Polygenic Scores, and Traits
| Conditions, Polygenic Scores, and Traits | Ascertainment, n | Age, Years, Mean (SD) | Sex, F/M, n | Cohorts | Previously Published | |||
|---|---|---|---|---|---|---|---|---|
| Clinical | Unselected | IQ Loss | OR ASD | OR SZ | ||||
| Idiopathic Psychiatric Conditions | ||||||||
| SZ | 283 | 0 | 33.9 (9.2) | 73/210 | Montreal-SZ CNP | – | – | – |
| BIP | 44 | 0 | 35 (9) | 20/24 | CNP | – | – | – |
| ASD | 472 | 0 | 14.9 (6) | 0/472 | ABIDE1, ABIDE2 | – | – | – |
| ADHD | 223 | 0 | 14.8 (9.5) | 66/157 | ADHD-200 CNP | – | – | – |
| Nonpsychiatric Condition | ||||||||
| IBD | 0 | 287 | 64.7 (7.5) | 144/143 | UKBB | – | – | – |
| Polygenic Scores | ||||||||
| Autism | 0 | 29460 | 64.2 (7.5) | 15840/13620 | UKBB | – | 2.7 | - |
| Schizophrenia | – | – | 3.5 | |||||
| BIP | – | – | – | |||||
| Cross-disorder | – | – | – | |||||
| IQ | 9–12 | – | – | |||||
| Nonpsychiatric conditions: LDL, CKD | – | – | – | |||||
| Traits | ||||||||
| Fluid intelligence | 0 | 27522 | 64 (7.5) | 14777/12745 | UKBB | – | – | – |
| Neuroticism | 0 | 24025 | 64 (7.5) | 12723/11302 | UKBB | – | – | – |
| Controls | ||||||||
| UKBB | 0 | 30185 | 64.1 (7.5) | 16260/13925 | UKBB | – | – | – |
| SFARI | 0 | 84 | 26.7 (15) | 35/49 | SFARI | – | – | – |
| MRG | 0 | 39 | 34 (16) | 25/14 | MRG | – | – | – |
| Cardiff | 0 | 8 | 39.8 (4) | 4/4 | Cardiff | – | – | – |
| UCLA | 0 | 43 | 13 (4.6) | 22/21 | UCLA | – | – | – |
| Psychiatric cohorts | 0 | 1066 | 20 (11) | 244/822 | – | – | – | – |
The cohort column provides the cohorts used to perform case-control studies for each of the 29 CWASs. IQ loss indicates mean decrease in IQ points associated with each CNV (7,8). ORs for the enrichment of PRSs in autism and schizophrenia were previously published (4,5,9,11,26,27,52,65–70). Detailed information relative to diagnosis, IQ, and motion are available in the Supplement. Information relative to scanning sites, motion, and diagnoses are also available in eTables 2–4 in the Supplement. All sites scanned control subjects. The table shows the numbers of CNV carriers (DEL, DUP), individuals with idiopathic psychiatric conditions (SZ, ASD, ADHD, BIP), and control subjects after MRI quality control.
ABIDE, Autism Brain Imaging Data Exchange; ADHD, attention-deficit/hyperactivity disorder; ASD, autism spectrum disorder; BIP, bipolar disorder; CKD, chronic kidney disease; CNP, Consortium for Neuropsychiatric Phenomics; CNV, copy number variant; CWAS, connectome-wide association study; DEL, deletion; DUP, duplication; F, female; IBD, inflammatory bowel disease; LDL, low-density lipoprotein; M, male; MRG, Montreal rare genomic disorder; MRI, magnetic resonance imaging; OR, odds ratio; PRS, polygenic risk score; SFARI, Simons Foundation Autism Research Initiative; SZ, schizophrenia; UCLA, University of California, Los Angeles; UKBB, UK Biobank.
Clinical Genetic Datasets.
We used 4 genetically informed CNV datasets, which were recruited based on the presence of a CNV associated with neurodevelopmental and psychiatric disorders, regardless of symptomatology (detailed in Supplementary Materials and Methods). Of note, the term oligogenic refers to CNVs containing 1 gene < CNV ≤5 genes, while multigenic CNVs contain more than 5 genes. These categories are descriptive, and cutoffs are descriptive. None of the analyses rely on these categories.
These 4 datasets included the Simons Variation in Individuals Project (SVIP for 16p11.2 and 1q21.1 CNVs) (25) and the University of California, Los Angeles 22q11.2 CNV project (UCLA). fMRI data have not yet been published for the Montreal rare genomic disorder family project (MRG, Canada) (8) and the Define Neuropsychiatric-CNVs Project (Cardiff, United Kingdom).
Unselected Population.
CNVs associated with neurodevelopmental and psychiatric disorders and nonpsychiatric CNVs were also identified in the UK Biobank (UKBB) dataset (25) (Supplementary Materials and Methods). Nonpsychiatric CNVs were defined as variants without any previous association with a psychiatric condition in large case-control studies (4,26–28).
Idiopathic Psychiatric Conditions Cohorts.
We used the Autism Brain Imaging Data Exchange 1 (ABIDE1) (29), Autism Brain Imaging Data Exchange 2 (ABIDE2) (30), ADHD-200 (31), the Consortium for Neuropsychiatric Phenomics (CNP) (32), and an aggregate dataset of 10 SZ studies (14,33); collectively, these datasets include individuals with idiopathic autism, ADHD, SZ, and bipolar disorder (BIP), as well as their respective control subjects. Psychiatric assessments are detailed in Supplementary Materials and Methods.
CNV Calling and PRS Computation
CNVs were identified in the UKBB using PennCNV (34) and QuantiSNP (35) following previously published methods (7) (Supplementary Materials and Methods).
We computed 7 PRSs for individuals of European ancestry in the UKBB using Bayesian regression and continuous shrinkage priors (36) (Table 2; Supplementary Materials and Methods; and eTable 1 in the Supplement).
rs-fMRI Preprocessing
All datasets were preprocessed using the same parameters of Neuroimaging Analysis Kit (37). Preprocessed data were visually controlled for quality of the coregistration, head motion, and related artifacts (Supplementary Materials and Methods).
Computing Connectomes
We segmented the brain into 64 functional seed-based regions and 12 networks defined by the Multiresolution Intrinsic Segmentation Template (MIST) brain parcellation (38). FC was computed as the temporal pairwise Pearson’s correlation between the average time series of the 64 seed-based regions, and then Fisher- transformed. The connectome of each individual encompassed 2080 connectivity values: (63 × 64)/2 = 2016 region-to-region connectivity + 64 within seed-based region connectivity. We chose the 64 parcel atlas of the MIST parcellation (https://simexp.github.io/multiscale_dashboard/index.html) because it falls within the range of network resolution previously identified to be maximally sensitive to FC alterations in neurodevelopmental and psychiatric disorders such as autism (39). We corrected for multiple comparisons using a false discovery rate (FDR) strategy (40).
Connectome-wide Association Studies
We performed 29 CWASs using one of the two approaches:
Contrasting cases and their respective controls for 7 CNVs associated with neurodevelopmental and psychiatric disorders and 8 nonpsychiatric CNVs (Tables 1 and 2), 4 idiopathic psychiatric disorder cohorts (autism, SZ, BIP, and ADHD), and 1 non–brain-related condition (inflammatory bowel disease). Control subjects refer to 1) individuals without a CNV for analysis investigating the effect of CNVs and 2) individuals without a diagnosis in analyses investigating effects of psychiatric conditions.
Investigating the linear effects of 7 continuous PRSs: autism, BIP, SZ, cross-disorder (ADHD, autism, BIP, SZ, anorexia nervosa, major depression, obsessive-compulsive disorder, and Tourette syndrome), and IQ, as well as 2 non–brain-related control traits (low-density lipoprotein and chronic kidney disease) and 2 continuous traits provided by the UKBB (neuroticism and fluid intelligence).
FC was -scored based on the variance of the pooled control subjects used for each CWAS (Cohorts column in Tables 1 and 2). They were conducted by linear regression, in which -scored FC values were the dependent variables and genetic or diagnostic status or traits were the explanatory variables. PRSs and traits were normalized within the UKBB sample.
It was previously demonstrated that global signal–adjusted (GSA)–FC profiles show stronger correlations with cognition (41) and reduce confounding effects in multisite studies (42). We therefore used GSA-FC profiles for this study. Global effect sizes obtained without GSA are available in eTable 5 in the Supplement.
Models were adjusted for sex, scanning site, head motion, age, and global signal (= GSA) defined as the mean of all 2080 Fisher’s values (42). FC profiles were defined as the 2080 values of 2080 connections.
This linear regression was applied for each of the 2080 functional connections. Because all raw connectomes were normalized on the variance of the control subjects, regression estimates can be interpreted as scores. We corrected for multiple testing using FDR (q < .05) as well as a permutation procedure (see Supplementary Materials and Methods). Effect size of genetic risk, conditions, and traits on connectivity was defined as the top decile of the 2080 absolute values. Sensitivity analyses using a cross-validation approach (43) ensured that effect sizes were stable across the different sample sizes investigated in the study.
Multiple Testing
Within each independent variable (15 CNVs, 7 PRSs, 4 conditions, and 2 traits), we corrected for the number of tests (2080 connections) using the Benjamini-Hochberg FDR correction at a threshold of q < .05 (40,44). We also computed an empirical p value by conducting a permutation test, shuffling the genetic or clinical status labels of the individuals included in each CWAS (5000 permutations). We estimated the empirical p value by calculating the frequency of obtaining an effect size equal to or greater than the original observation (45).
Estimating Effect Sizes Using Cross-validation
We generated effect sizes for each sample using K-fold cross-validation with 2-, 5-, and 10-fold cross-validation (43). For each genetic risk, condition, and trait, we split the sample into K segments (for case-control analyses, segments are stratified accordingly); then, for K iterations, we held out a segment as a training sample to generate a betamap and identify the connections with effect size estimates in the top decile. On the remaining independent test group, we extracted the top decile connections and computed their mean effect sizes. The overall effect size was computed as the mean of K estimates (eFigure 1 in the Supplement and the Supplement).
Bootstrap Procedure to Estimate 95th Confidence Intervals of Effect Size Ratios
We identified the 95% confidence intervals for the ratios of effect sizes using a bootstrap procedure (46). First, for each sample, we generated the actual betamap and identified the top decile connections and their mean X. Then, for 5000 iterations, we resampled with replacement of the same number of subjects (for case-control analyses, the resampling was performed separately in each group), generated a resampled betamap, and took the mean of the identified connections to form a distribution (x_1, x_2, ...x_5000). To generate a distribution of ratios for a given pair X_1, X_2 (where X_1 > X_2), we took the ratios of the bootstrap distributions (x_1_1/x_2_1, x_1_2/x_2_2, ,…x_1_5000/x_2_5000).
Sum of Genes and CNV Severity Score
The CNV severity score was previously published and is an additive model (7,8). It is the sum of genes included in a CNV, and each gene is weighted by its sensitivity to loss function, which is measured by the loss of function observed/expected upper bound fraction (LOEUF) score, which is available for each coding gene (47). Smaller values of LOEUF represent genes with highest sensitivity to loss of function (more severe genes); therefore, the inverse of LOEUF is used in the additive model:
This severity score is predictive of CNV effect size on cognition (7,8) and risk for psychiatric conditions (3,48).
As a sensitivity analysis, we computed a CNV severity score based on the probability of being loss of function intolerant (49), which is another constraint score with a binary distribution (>0.8 for intolerant genes and close to 0 for all other genes). As a result, this score only takes into account the contribution of intolerant genes.
RESULTS
Effects of Genetic Risk Factors and Psychiatric Conditions on Brain Connectivity
All 7 CNVs were associated with neurodevelopmental and psychiatric disorders, and none of the 9 nonpsychiatric CNVs significantly altered functional connections (FDR, 2080 connections, q < .05) (Table 3). Empirical p-value analyses (pval effect)–performing contrasts in 5000 randomly sampled groups–found the same level of significance compared with the FDR procedure (Table 3).
Table 3.
Connectome-wide Association Study Summary
| CNV/Conditions/Traits | Global Signal Adjustment | |||||
|---|---|---|---|---|---|---|
| Connections | Values | Top-decile Values | p-Value Effect | |||
| Positive | Negative | Minimum | Maximum | |||
| Psychiatric CNVs | ||||||
| 1q21.1 DEL | 1 | 11 | −1.07 | 0.62 | 0.44 | .002 |
| 1q21.1 DUP | 4 | 0 | −0.62 | 0.84 | 0.48 | .002 |
| 15q11.2 DEL | 1 | 0 | −0.29 | 0.36 | 0.2 | .01 |
| 16p11.2 DEL | 124 | 149 | −0.98 | 1.67 | 0.57 | <2 × 10−4 |
| 16p11.2 DUP | 4 | 3 | −1.04 | 0.55 | 0.38 | .002 |
| 22q11.2 DEL | 4 | 13 | −1.48 | 1 | 0.65 | <2 × 10−4 |
| 22q11.2 DUP | 0 | 2 | −0.78 | 0.69 | 0.43 | .04 |
| Nonpsychiatric CNVs | ||||||
| TAR DUP | 0 | 0 | −0.48 | 0.51 | 0.28 | ns |
| 2q13 DEL | 0 | 0 | −0.15 | 0.19 | 0.11 | ns |
| 2q13 DUP | 0 | 0 | −0.34 | 0.26 | 0.18 | ns |
| 13q12.12 DEL | 0 | 0 | −0.54 | 0.5 | 0.34 | ns |
| 13q12.12 DUP | 0 | 0 | −0.53 | 0.48 | 0.31 | ns |
| 15q11.2 DUP | 0 | 0 | −0.24 | 0.24 | 0.16 | .04 |
| 15q13.3 DUP | 0 | 0 | −0.20 | 0.18 | 0.11 | ns |
| 16p13.11 DUP | 0 | 0 | −0.42 | 0.40 | 0.26 | ns |
| Polygenic Score | ||||||
| Cross-disorder | 23 | 22 | −0.02 | 0.03 | 0.01 | <2 × 10−4 |
| Autism | 3 | 1 | −0.02 | 0.02 | 0.01 | .04 |
| Schizophrenia | 30 | 27 | −0.02 | 0.03 | 0.01 | <2 × 10−4 |
| Bipolar disorder | 16 | 2 | −0.02 | 0.03 | 0.01 | .002 |
| IQ | 74 | 42 | −0.02 | 0.02 | 0.01 | <3 × 10−4 |
| Control PRSs | ||||||
| Low-density lipoprotein | 0 | 0 | −0.02 | 0.02 | 0.009 | ns |
| Chronic kidney disease | 0 | 0 | −0.02 | 0.02 | 0.01 | ns |
| Psychiatric Conditions | ||||||
| Autism | 51 | 55 | −0.26 | 0.36 | 0.16 | <2 × 10−4 |
| Schizophrenia | 221 | 258 | −0.41 | 0.51 | 0.30 | <2 × 10−4 |
| Bipolar disorder | 33 | 24 | −0.66 | 0.65 | 0.43 | <2 × 10−4 |
| ADHD | 0 | 0 | −0.22 | 0.22 | 0.15 | <2 × 10−4 |
| Inflammatory bowel disease | 0 | 0 | −0.16 | 0.16 | 0.11 | ns |
| Traits | ||||||
| Fluid intelligence | 311 | 281 | −0.04 | 0.04 | 0.02 | <2 × 10−4 |
| Neuroticism | 208 | 208 | −0.03 | 0.04 | 0.02 | <2 × 10−4 |
The number of significantly altered connections (false discovery rate–corrected) for each CWAS (n = 29) (eTable 5 in the Supplement). Control PRSs are the non–brain-related PRSs. Connections are the number of positive or negative connections surviving false discovery rate correction. Minimum-maximum are -scored values. Top-decile values are the effect size of genetic risk, conditions, and traits on connectivity defined as the top decile of the 2080 absolute values. The p-value effect is the empirical p value obtained by conducting a permutation test, shuffling the genetic or clinical status labels of the individuals included in each CWAS (5000 permutations). We estimated the empirical p value by calculating the frequency of obtaining an effect size equal to or greater than the original observation.
ADHD, attention-deficit/hyperactivity disorder; CNV, copy number variant; CWAS, connectome-wide association study; DEL, deletion; DUP, duplication; ns, nonsignificant; PRS, polygenic risk score.
The previously published 22q11.2 deletion FC profile showed the largest effects (mean of brainwide estimates in the top decile = 0.65), followed by the 16p11.2 deletion, which also showed large effects on FC profiles (0.57). The 22q11.2 and 16p11.2 FC profiles were robust and correlated (r = 0.7 and 0.83) to previously published profiles that were based on smaller samples (14). 1q21.1 deletion and duplication FC profiles showed moderate to large effects on FC. 15q11.2 deletion showed the mildest effects among CNVs associated with neurodevelopmental and psychiatric disorders. Individual CNV FC profiles in 3-dimensional maps showing effect sizes for each of the 64 functional regions are available at https://claramoreau9.github.io/Braimaps_Github.html.
All brain-related PRSs (SZ, BIP, autism, cross-disorder, and IQ) altered FC profiles. The non–brain-related PRSs (low-density lipoprotein, chronic kidney disease) showed no significant effects (Table 3).
Individuals diagnosed with idiopathic SZ, BIP, and autism but not ADHD had significantly altered FC compared with control subjects. SZ, ADHD, and autism FC profiles were previously published (14), but we recomputed them with additional individuals. Correlations between new and previously published profiles were 0.95, 0.70, and 0.86, respectively.
Effect sizes were largest for CNVs associated with neurodevelopmental and psychiatric disorders, followed by psychiatric conditions, fluid intelligence, neuroticism, and PRS (Figure 2A–D). Effect sizes of deletions were on average 1.3-fold larger than their reciprocal duplications. Effect sizes for a change in 1 SD of a cognitive trait or a PRS were on average one order of magnitude smaller than those associated with CNVs (Figure 2E). To test the relation between genome-wide association study (GWAS) sample size and the effect of PRS on FC, we compared the PRS-SZ based on the most recent GWAS in 76,755 subjects with SZ (50) to the one based on an older GWAS computed with 23,585 subjects with SZ (51). The 2 FC profiles associated with the 2 PRS-SZ were correlated (r = 0.89), but the number of significant connections was higher for the FC profile based on the larger SZ-GWAS. The effect size (for 1 SD of PRS) was also larger, with the top decile of values increasing from 0.0138 (95% CI, 0.011–0.016) to 0.016 (95% CI, 0.013–0.019).
Figure 2.
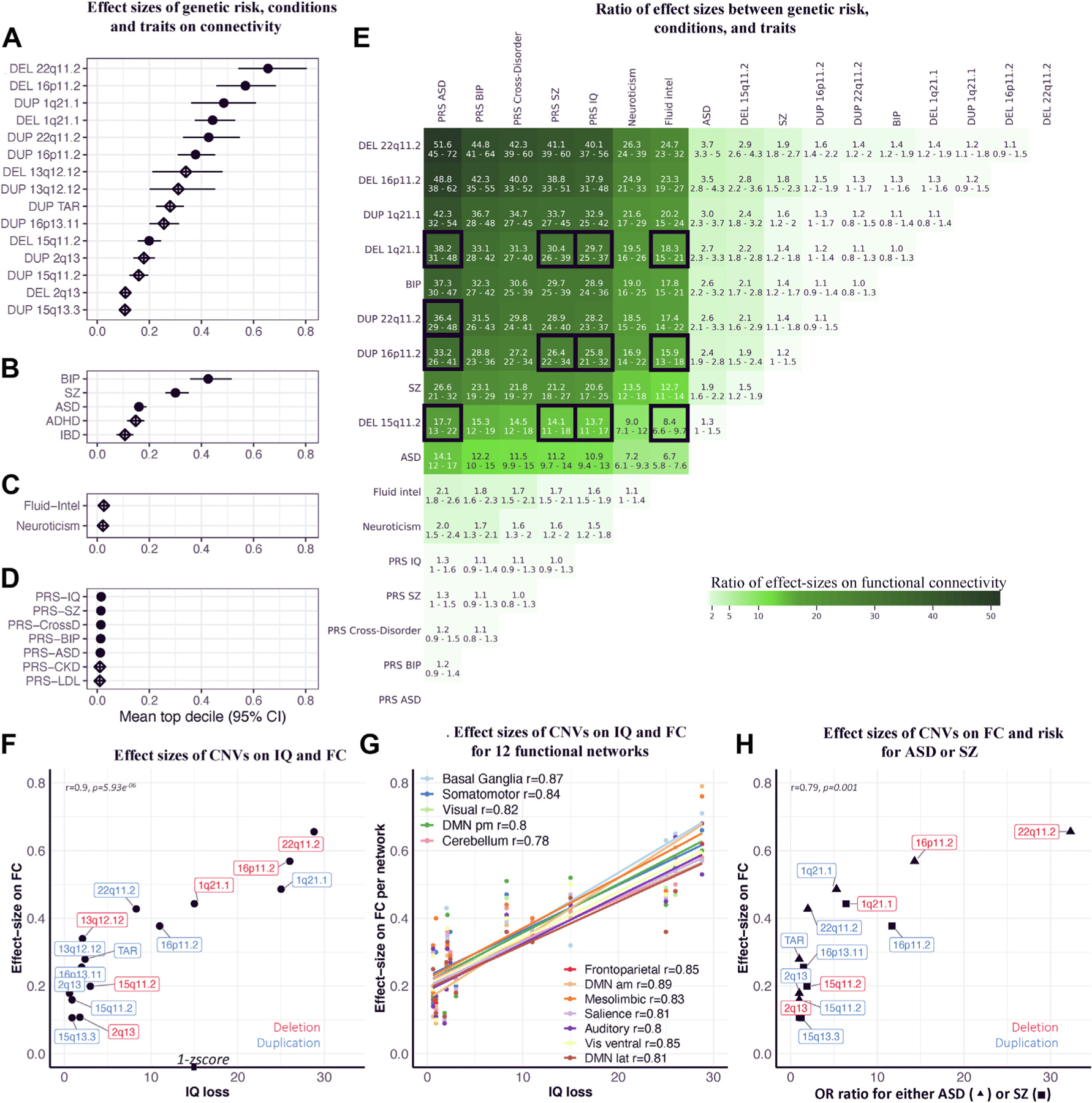
Relation between effect sizes of CNVs on cognition and connectivity. (A–D) Effect size of genetic risk, conditions, and traits on FC. Effect sizes of (A) CNVs, (B) idiopathic psychiatric conditions, (C) traits, and (D) PRSs on FC. Each dot (or diamond) is the mean of estimates in the top decile (the 208 connections with the highest estimates). x-axis values represent the effect sizes -scored on the variance of the control group. Full dots represent significant effect sizes (the intersection of FC profiles with altered connections surviving false discovery rate and empirical p values using 5000 permutation tests q < 0.05) (Table 3), and empty diamonds are nonsignificant effect sizes. (E) Ratio of effect sizes between genetic risk, conditions, and traits. Ratios are only computed for groups that have significant effect sizes on FC. The ratio is the line (numerator) divided by the column (denominator). The 95% confidence interval for each ratio was computed using a bootstrap procedure (43,46) (see Methods and Materials). Boxes with black borders highlight CNVs and PRSs that should have similar effects on connectivity because they are matched for effect size on cognition or risk for disease. We also highlight CNVs that have effect sizes on cognitive ability equal or smaller than 1 score to highlight the discordance with effect of 1 score of fluid intelligence on connectivity. (F) Effect sizes of CNVs on IQ and FC. We used previously published effect sizes of CNVs on IQ (7). The x-axis indicates decrease in IQ associated with each CNV. The y-axis indicates effect sizes of CNVs on FC (top decile of estimates). (G) Effect sizes of CNVs on IQ and FC for 12 functional networks. The x-axis indicates decrease in IQ associated with each CNV (7). The y-axis indicates effect sizes of CNVs on FC for 12 functional networks (mean of the top decile of network-wide estimates) (For a representation of each network individually, see eFigure 5 in the Supplement). (H) Effect sizes of CNVs on FC and risk for autism or SZ. Correlation between previously published effect sizes of CNVs on autism or SZ risk (4,26,28,52) and their effect sizes on FC. We used the highest risk conferred by each CNV for either autism (26,28) or SZ (4,52). The x-axis indicates odd ratios for autism (▲) or SZ (■). The y-axis indicates effect sizes on FC (top decile of estimates). ADHD, attention-deficit/hyperactivity disorder; am, anteromedial; ASD, autism spectrum disorder; BIP, bipolar disorder; CKD, chronic kidney disease; CNV, copy number variant; CrossD, cross-disorder; DEL, deletion; DMN, default mode network; DUP, duplication; FC, functional connectivity; FP, frontoparietal network; IBD, inflammatory bowel disease; intel, intelligence; lat, lateral; LDL, low-density lipoprotein; OR, odds ratio; pm, posteromedial; PRS, polygenic risk score; SZ, schizophrenia; Vis, visual.
Sensitivity analyses showed that effect size estimates were robust to several cross-validations as well as the effects of sex, pooled or matched control subjects, clinical or unselected ascertainment, and medication (Supplementary Results).
Relation Between Effect Sizes of CNVs on Connectivity and Cognition or Risk for Neurodevelopmental and Psychiatric Disorders
We observed a correlation between the effect size of CNVs on FC and their previously reported effect size on cognitive ability (7) (r = 0.9, p = 5.93 × 10−6), but effects of CNVs on FC were systematically smaller than their effect on cognitive ability (Figure 2F). Effect size on FC was also correlated with previously reported general risk for neurodevelopmental and psychiatric disorders, i.e., the highest risk conferred by each CNV for either autism (26,28) or SZ (4,52) (r = 0.79, p = .001) (Figure 2H). As expected, this correlation was weaker for autism (r = 0.75) and SZ (r = 0.6) risk separately because some CNVs confer a high risk for autism but not SZ and vice versa (eFigure 4 in the Supplement). The correlation with cognitive ability was similar across all 12 networks (Figure 2G; eFigure 5 in the Supplement).
Most Networks Are Affected by Genetic Risk and Conditions
Genetic risk, conditions, and traits affected connections that were distributed across all functional brain networks (Figure 3). However, basal ganglia–thalamus and somatomotor networks exhibited overconnectivity across most genetic risks and conditions (sum values: 2.1 and 1.1, respectively). In contrast, limbic and auditory networks were predominantly underconnected (sum values: −2.7, −1.3, respectively).
Figure 3.
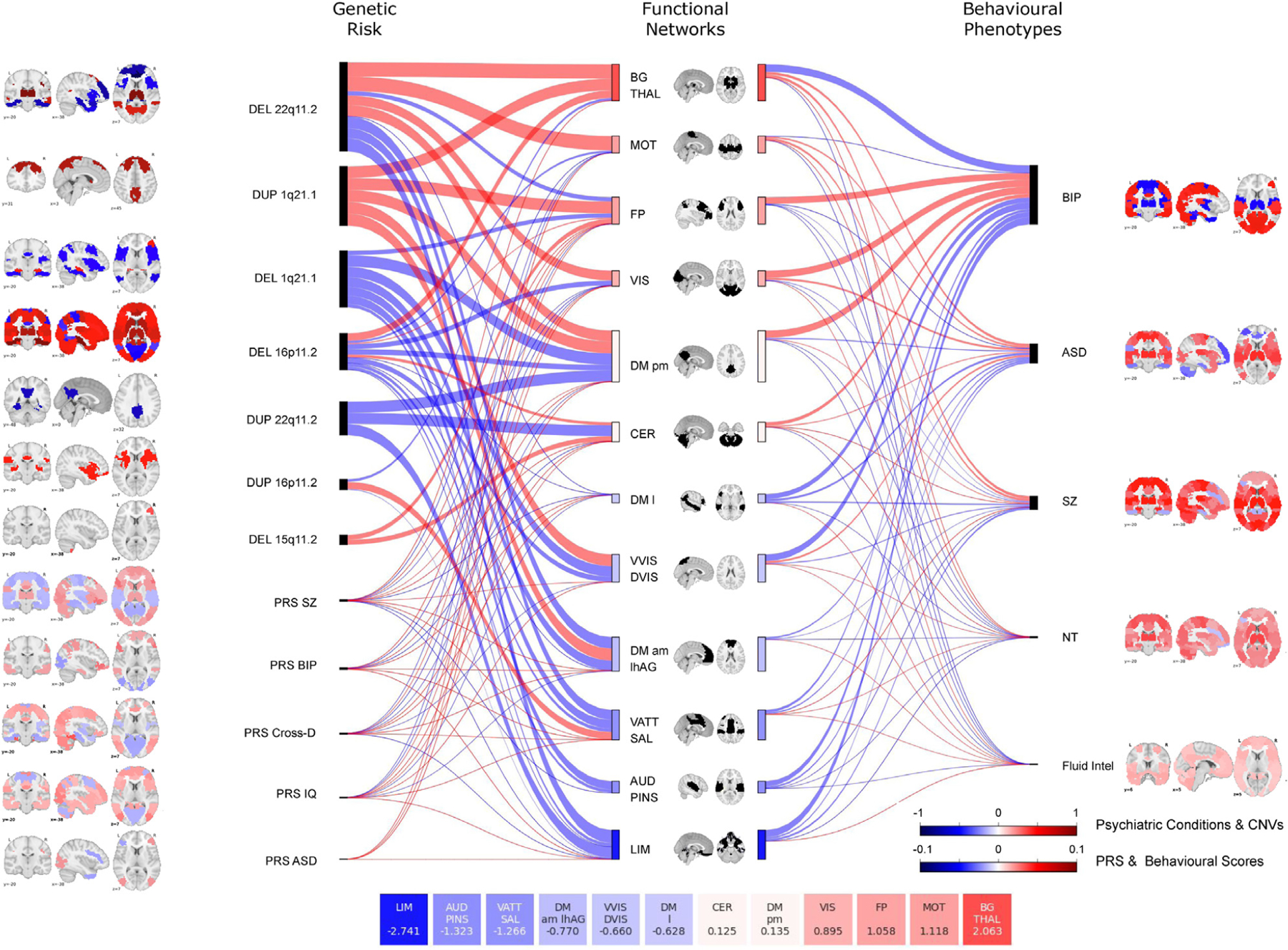
Similarities at the network level across genetic risks, psychiatric conditions, and traits. Sankey plot shows effect sizes across 12 networks for genetic risk (left) and conditions, and traits (right). The thickness of the connecting lines represents these effect sizes, which were defined as the mean value of all significant connections within the network and between the network and the other 11 networks. The length and color of rectangles on either side of each network in the middle of the Sankey plot represents the sum of effect sizes across all genetic risks, conditions, and traits for that particular network. For each network, effect size values are summarized in the 12 boxes (bottom of the figure). Brain maps represent the maximum estimate value for each functional region (Table 3). Red indicates overconnectivity; blue indicates underconnectivity. Color bars represent the value. ASD, autism spectrum disorder; AUD PINS, auditory network and posterior insula; BG Thal, basal ganglia thalamus; BIP, bipolar disorder; CER, cerebellum; Cross-D, cross-disorder; CNV, copy number variant; DEL, deletion; DM am lhAG, default mode network anteromedial and left angular gyrus; DM l, default mode network lateral; DM pm, default mode network posteromedial; DUP, duplication; Fluid intel, fluid intelligence; FP, frontoparietal network; LIM, limbic network; MOT, somatomotor network; NT, neuroticism; PRS, polygenic risk score; SZ, schizophrenia; VATT SAL, ventral attentional and salience network; VIS, visual network; VVIS DVIS, ventral and dorsal visual network.
Effect Sizes of Individual Genes Within CNVs Decrease as CNVs Increase in Number of Genes
We first asked if there was a relation between the number of genes in a CNV (Figure 4A) and its effect size on FC. This was the case (r = 0.72, p = .002) (Figure 4B), and the relation was similar when genes were weighted by their sensitivity to gene dosage (severity score, r = 0.76, p = .0009) (Figure 4C).
Figure 4.
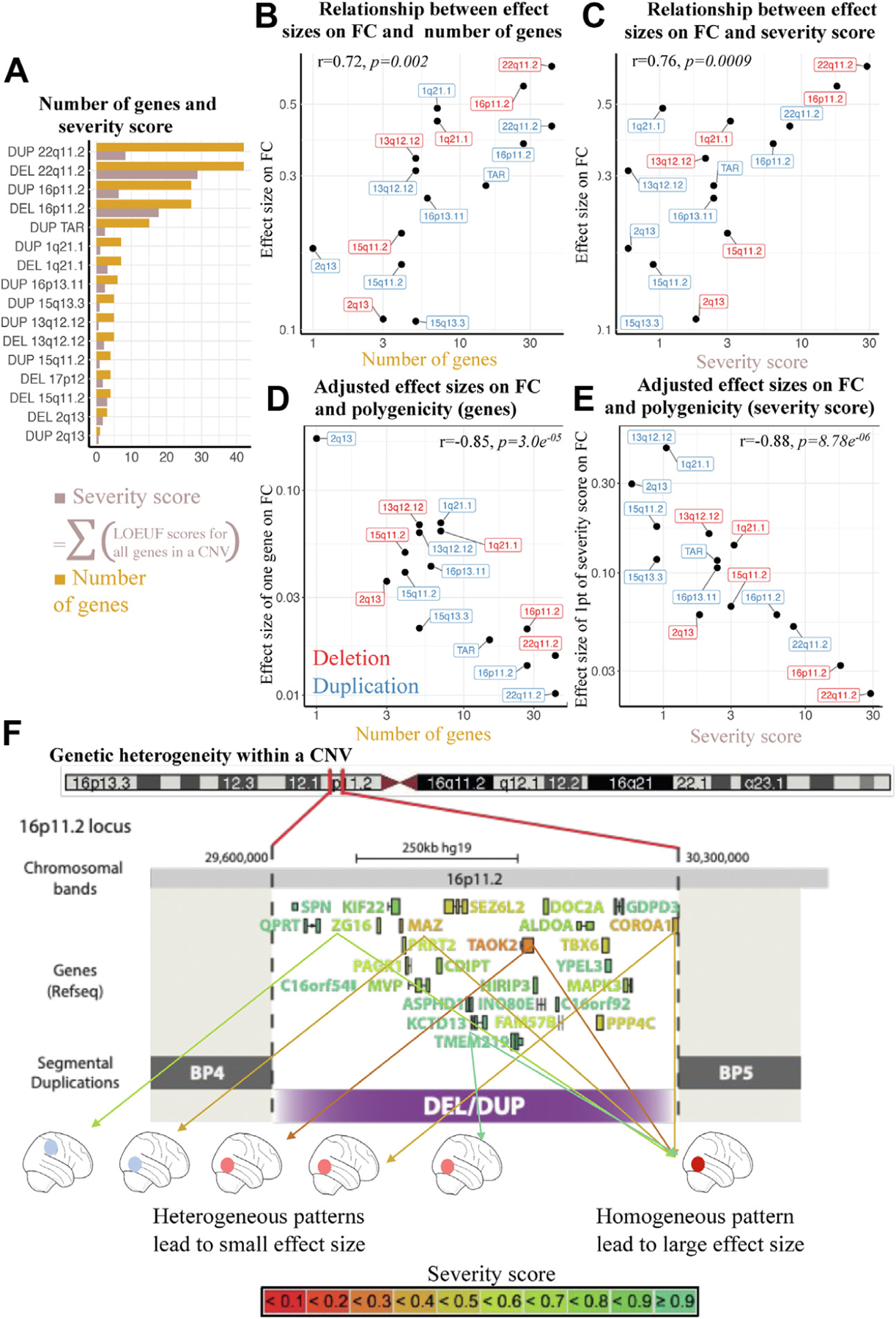
Relation between multigenicity and connectivity. (A) Number of genes and severity score. Bar plot showing for each CNV, the number of genes encompassed, and the sum of genes weighted by their intolerance score (sum of 1/LOEUF). The sum of 1/LOEUF values of all genes encompassed in a CNV is highly predictive of the effect size of CNVs on cognitive ability. (B) Relation between effect sizes on FC and number of genes. The y-axis indicates effect size of CNVs on FC. The x-axis indicates number of genes in each CNV. (C) Relation between effect sizes on FC and severity score. The y-axis indicates effect size of CNVs on FC. The x-axis indicates severity score for each CNV. (D) Adjusted effect sizes on FC and multigenicity (genes). The y-axis indicates mean effect of 1 gene on FC (CNV effect sizes on FC adjusted for number of genes). The x-axis indicates number of genes in each CNV. (E) Adjusted effect sizes on FC and multigenicity (severity score). The y-axis indicates mean effect on FC of one point of severity score (CNV effect sizes adjusted by the severity score). The x-axis indicates severity score for each CNV. (F) Genetic heterogeneity within a CNV. Genes encompassed in the 16p11.2 CNV are color-coded based on their LOEUF score. Two scenarios are represented. Right, genes converge on shared brain patterns: the effect size of the CNV increases linearly with the number of intolerant genes and is large. Left, genes within a CNV are associated with distinct patterns: the resulting effect size is weaker. BP, breakpoint; CNV, copy number variant; DEL, deletion; DUP, duplication; FC, functional connectivity; LOEUF, loss of function observed/expected upper bound fraction; PRS, polygenic risk score.
To investigate the effects of multigenicity on connectivity, we computed for each CNV an effect size adjusted for gene content (effect size divided by the number of genes included in the CNV). We observed that the adjusted effect size of CNVs significantly decreased as CNVs increased in number of genes (r = −0.85, p = 3 × 10−5) (Figure 4D). Using the severity score showed the same phenomenon (r = −0.88, p = 8.8 × 10−6) (Figure 4E). In other words, compared with small oligogenic CNVs, large multigenic CNVs have smaller effects on FC than expected based on the number of genes they contain. We performed the same analysis using probability of being loss of function intolerant (instead of LOEUF) to test the assumption that only a few intolerant genes contribute to the CNV-associated FC alterations (eFigure 6 in the Supplement). The same decrease in effect size was observed as the number of intolerant genes increased in CNVs.
In contrast, there was no relation between the severity score and its adjusted effect size on IQ (r = 0.30, p = .25) (eFigure 7 in the Supplement).
To further investigate the effect of multigenicity on FC, we examined PRS and CNVs with similar effects (previously published) (Table 3) on cognitive ability and risk for autism and SZ. PRS effect sizes on connectivity were disproportionately lower (between 38- and 13-fold lower) than those observed for the selected CNVs (1q21.1 deletions, 16p11.2, and 22q11.2 duplications and 15q11.2 deletions) (Figure 2E).
DISCUSSION
Main Findings
In this large rs-fMRI dataset, we demonstrated that most rare and common genetic risks for neurodevelopmental and psychiatric disorders affect FC, but effect sizes vary over an order of magnitude across variants. We showed that the effect sizes of CNVs on FC were correlated with their previously reported effects on cognitive ability and risk for autism and SZ connectome-wide. This relation was observed across all brain networks, which is consistent with the fact that fluid intelligence is thought to be subserved by networks widely distributed across the brain (53). Whether these associations across all networks are causal of IQ decrease in CNV carriers remains an open question.
Multigenicity had a profound impact on FC signals; as CNVs increased in size and number of genes, effect size of CNVs adjusted for gene content (number of genes and severity score) rapidly decreased. In line with this observation, PRS had minute effects on FC, and the latter was disproportionately low compared with that observed for CNVs with similar effect sizes on IQ and risk for autism or SZ.
Similar Effect Sizes Across Functional and Structural MRI
Effect sizes on FC across neurodevelopmental CNVs and psychiatric conditions are consistent with those reported for structural MRI measures (54,55) even when much larger samples are investigated (Cohen’s d = −1 and d = 0.6 for cortical surface and thickness, respectively, in n = 475 carriers of the 22q11.2 deletion) (56). For autism (57) and SZ (58), previously reported effect sizes for cortical thickness (Cohen’s d = 0.21 and 0.5, respectively) were also similar to those observed in our study for FC (54). To date, the only effect sizes reported for PRS were for SZ ( for cortical surface and thickness) and are consistent with the very small effects in this study (59).
Even Small Levels of Multigenicity Increase Heterogeneity at the FC Level
We observed that the effect size on FC of one gene (the CNV-adjusted effect size) declines (by an order of magnitude) for increasingly multigenic CNVs. In other words, a gene would contribute to a smaller FC effect in a multigenic CNV compared with a gene with the same severity score (sensitivity to gene dosage) encompassed in a small oligogenic one. Multigenic CNVs may therefore represent heterogeneous combinations of relatively distinct FC profiles associated with each dosage-sensitive gene (Figure 4F). This suggests that genes within a CNV or a polygenic score may cancel out each other’s effects on FC, leading to weaker effect sizes.
This effect of multigenicity may not be restricted to FC. As an example, Down syndrome, which encompasses more than 200 protein-coding genes (60), has an extreme effect size on cognition (a mean decrease of 3.3 SD) (8,61) but has been associated with smaller effect sizes on MRI structural measures (below 1.65 Cohen’s d) (62,63). Based on our observations, genetic effects on rs-fMRI would be best observed (with the largest effects) in the context of monogenic variants such as FMR1, NRXN1, or CHD8. This would likely apply to other brain modalities.
Why Are Polygenic Scores Associated With Such Small Effect Sizes?
The microscopic effect sizes associated with autism-PRS, SZ-PRS, and intelligence-PRS are possibly related to extreme levels of heterogeneity. Our comparison of 2 PRS-SZ based on GWASs of different sample sizes suggests that further increasing the GWAS sample size will improve the detection of significant connections altered by PRS but will not substantially increase the effect size of psychiatric PRS on rsfMRI.
The CNV-PRS discordance is striking for PRS-IQ, which has been associated with moderate to large effects on cognitive ability. There are infinite combinations of different common variants that would lead to the same PRS. This may explain why PRS shows minimal convergence on a particular connectivity pattern. Of note, both traits (fluid intelligence and neuroticism) also showed similar effect sizes, suggesting comparable levels of heterogeneity.
However, alternative interpretations are possible. Current PRSs may be vastly improved when larger GWASs will be available. Some of the CNVs investigated have small sample sizes, which leads to inflated effect sizes. FC may not represent a relevant intermediate phenotype for genetic risk or cognitive traits.
Limitations
Genetic heterogeneity is only one of the plausible interpretations that may explain observations such as the survivor effect where large deleterious multigenic CNVs are only observed in resilient individuals with disproportionally low alterations at the brain connectivity level. Similarly, bias toward less individuals, who have a higher probability of completing the MRI scan coils contributes as well to this drop in effect size. An alternative interpretation that may explain why effect sizes of CNVs on FC < cognition is that FC is a noisy metric, and noise could be increased in individuals with neurodevelopmental conditions (i.e., head movement despite being carefully adjusted for).
For PRSs, the portability across populations is poor, which may contribute to the small effects of PRSs on FC. Finally, CNVs including intolerant genes are under negative selection, whereas this is likely not the case for psychiatric PRSs, which are the sum of many variants that are individually frequent. This fundamental difference may contribute to differences in FC effect sizes observed for a PRS and a CNV matched for the level of disease risk.
Confounding factors including sex bias and age differences may have influenced some of the results. However, carefully conducted sensitivity analyses provided similar results (Supplemental Results). Larger samples will be required to detect potential interactions between genetic risk and age or sex.
Conclusions
Polygenicity may predominantly result in polyconnectivity, a scenario in which thousands of autism or SZ genomic risk variants lead to a diverse set of connectivity patterns associated with the conditions. Future studies will require both indepth partitionings of polygenic scores and clustering of rare variants–based on relevant gene functions–to obtain mechanistically coherent subgroups of individuals.
Supplementary Material
KEY RESOURCES TABLE
| Resource Type | Specific Reagent or Resource | Source or Reference | Identifiers | Additional Information |
|---|---|---|---|---|
| Add additional rows as needed for each resource type | Include species and sex when applicable. | Include name of manufacturer, company, repository, individual, or research lab. Include PMID or DOI for references; use “this paper” if new. | Include catalog numbers, stock numbers, database IDs or accession numbers, and/or RRIDs. RRIDs are highly encouraged; search for RRIDs at https://scicrunch.org/resources. | Include any additional information or notes if necessary. |
| Antibody | NA | NA | NA | NA |
| Bacterial or Viral Strain | NA | NA | NA | NA |
| Biological Sample | NA | NA | NA | NA |
| Cell Line | NA | NA | NA | NA |
| Chemical Compound or Drug | NA | NA | NA | NA |
| Commercial Assay Or Kit | NA | NA | NA | NA |
| Deposited Data; Public Database | NA | NA | NA | NA |
| Genetic Reagent | NA | NA | NA | NA |
| Organism/Strain | NA | NA | NA | NA |
| Peptide, Recombinant Protein | NA | NA | NA | NA |
| Recombinant DNA | NA | NA | NA | NA |
| Sequence-Based Reagent | NA | NA | NA | NA |
| Software; Algorithm | NeuroImaging Analysis Kit, Scikit-Learn, R, Python | https://github.com/claramoreau9/NeuropsychiatricCNVs_Connectivity | NA | NA |
| Transfected Construct | NA | NA | NA | NA |
| Other |
ACKNOWLEDGMENTS AND DISCLOSURES
This research was supported by Compute Canada (ID 3037 and gsf-624), the Brain Canada Multi-Investigator Research Initiative (MIRI), Canada First Research Excellence Fund, Institute of Data Valorization, and Healthy Brain, Healthy Lives (to SJ). SJ is a recipient of a Canada Research Chair in neurodevelopmental disorders, and a chair from the Jeanne et Jean Louis Levesque Foundation. This work was supported by a grant from the Brain Canada MIRI (to SJ) and a grant from The Canadian Institutes of Health Research (Grant No. CIHR 400528 [to SJ]). CAM and TB are supported by AIMS-2-TRIALS, which received support from the Innovative Medicines Initiative 2 Joint undertaking under grant agreement (Grant No. 777394). The Cardiff Copy Number Variant cohort was supported by the Wellcome Trust Strategic Award DEFINE and the National Centre for Mental Health with funds from Health and Care Research Wales (code 100202/Z/12/Z). Data from the UCLA cohort provided by CEB (participants with 22q11.2 deletions or duplications and control subjects) was supported through grants from the National Institutes of Health (NIH) (Grant No. U54EB020403), the National Institute of Mental Health (Grant Nos. R01MH085953, R01MH100900, 1U01MH119736, and R21MH116473), and the Simons Foundation (SFARI Explorer Award). Finally, data from another study were obtained through the OpenFMRI project (http://openfmri.org) from the Consortium for Neuropsychiatric Phenomics (CNP), which was supported by NIH Roadmap for Medical Research grants (Grant Nos. UL1-DE019580, RL1MH083268, RL1MH083269, RL1DA024853, RL1MH083270, RL1LM009833, PL1MH083271, and PL1NS062410). PB is a fellow (Chercheur boursier Junior 2) of the Fonds de recherche du Québec - Santé. Data preprocessing and analyses were supported in part by the Courtois foundation (to PB). This work was supported by Simons Foundation (Grant Nos. SFARI219193 and SFARI274424). PMT was funded in part by the U.S. NIH grants (Grant Nos. R01MH116147, P41EB015922, R01MH111671, and U01 AG068057). PT received the Canadian Institute of Health Research Scholarship.
CAM, SJ, and PB designed the overall study and drafted the manuscript. CAM, AH, and SGWU processed 90% of all the fMRI data and performed all imaging analyses. GH, J-LM, and EAD performed the CNV calling. PO preprocessed the schizophrenia data. HS performed the UKBB fMRI preprocessing. LMS and LA computed the polygenic scores. AL gave feedback on the statistics used in this manuscript. TB and TR gave feedback on the analyses using polygenic risk and LOEUF scores. TB, PMT, DCG, and CEB contributed to the interpretation of the data and reviewed the manuscript. KK, KJ, C-OM, PT, NY, EAD, and SL recruited/scanned patients for the Montreal rare genomic disorder family dataset. CEB provided the UCLA 22q.11.2 fMRI data. DEJL, MJO, MBMvdB, JH, and AIS provided the Cardiff CNV fMRI data. All authors provided feedback on the manuscript.
We thank all of the families at the participating Simons Variation in Individuals Project (SVIP) sites, as well as the Simons VIP Consortium. We appreciate obtaining access to imaging and phenotypic data on the SFARI Base. Approved researchers can obtain the Simons VIP population dataset described in this study by applying at https://base.sfari.org. We are grateful to all families who participated in the 16p11.2 European Consortium.
Data from UK Biobank were downloaded under the application 40980 and can be accessed via their standard data access procedure (see http://www.ukbiobank.ac.uk/register-apply). UK Biobank CNVs were called using the pipeline developed in Jacquemont Lab and described in https://github.com/labjacquemont/MIND-GENESPARALLELCNV. The final CNV calls are available from UK Biobank returned datasets (Return ID: 3104, https://biobank.ndph.ox.ac.uk/ukb/dset.cgi?id=3104). BIDE1, ABIDE2, COBRE, ADHD-200, CNP, and 16p11.2 SVIP data are publicly available at: http://fcon_1000.projects.nitrc.org/indi/abide/abide_I.html, http://fcon_1000.projects.nitrc.org/indi/abide/abide_II.html, http://schizconnect.org/queries/new, http://fcon_1000.projects.nitrc.org/indi/adhd200/, https://www.openfmri.org/dataset/ds000030/, and https://www.sfari.org/funded-project/simons-variation-in-individuals-project-simons-vip/. The 22q11.2 UCLA raw data are currently available by request from the principal investigator (PI). Raw imaging data for the Montreal rare genomic disorder family dataset are going to be available on the LORIS platform in 2023. The Cardiff raw data are not publicly available yet; contact the principal investigator for further information. All processed connectomes are available through a request to the corresponding authors. Code for all analyses and visualizations, values, and p values for the 29 functional connectivity profiles are available online through the GitHub platform with Jupyter notebook: https://github.com/claramoreau9/NeuropsychiatricCNVs_Connectivity.
PMT received partial research grant support from Biogen, Inc., for research unrelated to this study. MJO, HH, and MBMvdB have a research grant from Takeda Pharmaceuticals outside the scope of this work. JH is a founding director of the company Meomics (unrelated to this work).
Footnotes
All other authors report no biomedical financial interests or potential conflicts of interest.
Supplementary material cited in this article is available online at https://doi.org/10.1016/j.biopsych.2022.08.024.
Contributor Information
Clara A. Moreau, Human Genetics and Cognitive Functions, Institut Pasteur, Université Paris Cité, Paris, France; Sainte-Justine Research Center, University of Montréal, Montréal, Canada; Centre de Recherche de l’Institut Universitaire de Gériatrie de Montréal, Montreal, Canada
Annabelle Harvey, Sainte-Justine Research Center, University of Montréal, Montréal, Canada; Centre de Recherche de l’Institut Universitaire de Gériatrie de Montréal, Montreal, Canada.
Kuldeep Kumar, Sainte-Justine Research Center, University of Montréal, Montréal, Canada.
Guillaume Huguet, Sainte-Justine Research Center, University of Montréal, Montréal, Canada.
Sebastian G.W. Urchs, Centre de Recherche de l’Institut Universitaire de Gériatrie de Montréal, Montreal, Canada; Montreal Neurological Institute, McGill University, Montreal, Canada
Elise A. Douard, Sainte-Justine Research Center, University of Montréal, Montréal, Canada
Laura M. Schultz, Lifespan Brain Institute, Children’s Hospital of Philadelphia, University of Pennsylvania, Philadelphia, Pennsylvania
Hanad Sharmarke, Centre de Recherche de l’Institut Universitaire de Gériatrie de Montréal, Montreal, Canada.
Khadije Jizi, Sainte-Justine Research Center, University of Montréal, Montréal, Canada.
Charles-Olivier Martin, Sainte-Justine Research Center, University of Montréal, Montréal, Canada.
Nadine Younis, Sainte-Justine Research Center, University of Montréal, Montréal, Canada.
Petra Tamer, Sainte-Justine Research Center, University of Montréal, Montréal, Canada.
Thomas Rolland, Human Genetics and Cognitive Functions, Institut Pasteur, Université Paris Cité, Paris, France.
Jean-Louis Martineau, Sainte-Justine Research Center, University of Montréal, Montréal, Canada.
Pierre Orban, Centre de Recherche de l’Institut Universitaire en Santé Mentale de Montréal, Montréal, Canada; Département de Psychiatrie et d’Addictologie, Université de Montréal, Montréal, Canada.
Ana Isabel Silva, Neuroscience and Mental Health Research Institute, Cardiff University, Cardiff, United Kingdom; MRC Centre for Neuropsychiatric Genetics and Genomics, Cardiff University, Cardiff, United Kingdom; School for Mental Health and Neuroscience, Faculty of Health, Medicine and Life Sciences, Maastricht University, The Netherlands.
Jeremy Hall, Neuroscience and Mental Health Research Institute, Cardiff University, Cardiff, United Kingdom; MRC Centre for Neuropsychiatric Genetics and Genomics, Cardiff University, Cardiff, United Kingdom.
Marianne B.M. van den Bree, Neuroscience and Mental Health Research Institute, Cardiff University, Cardiff, United Kingdom; MRC Centre for Neuropsychiatric Genetics and Genomics, Cardiff University, Cardiff, United Kingdom
Michael J. Owen, Neuroscience and Mental Health Research Institute, Cardiff University, Cardiff, United Kingdom; MRC Centre for Neuropsychiatric Genetics and Genomics, Cardiff University, Cardiff, United Kingdom
David E.J. Linden, MRC Centre for Neuropsychiatric Genetics and Genomics, Cardiff University, Cardiff, United Kingdom; School for Mental Health and Neuroscience, Faculty of Health, Medicine and Life Sciences, Maastricht University, The Netherlands
Aurelie Labbe, Département des Sciences de la Décision, HEC, Québec, Montréal, Canada.
Sarah Lippé, Sainte-Justine Research Center, University of Montréal, Montréal, Canada.
Carrie E. Bearden, Integrative Center for Neurogenetics, Semel Institute for Neuroscience and Human Behavior, Departments of Psychiatry and Biobehavioral Sciences and Psychology, University of California, Los Angeles, Los Angeles, California
Laura Almasy, Department of Biomedical and Health Informatics, Children’s Hospital of Philadelphia, Pennsylvania; Department of Genetics, University of Pennsylvania, Philadelphia, Pennsylvania; Lifespan Brain Institute, Children’s Hospital of Philadelphia, University of Pennsylvania, Philadelphia, Pennsylvania.
David C. Glahn, Harvard Medical School, Department of Psychiatry, Boston, Massachusetts; Boston Children’s Hospital, Tommy Fuss Center for Neuropsychiatric Disease Research, Boston, Massachusetts
Paul M. Thompson, Imaging Genetics Center, Stevens Institute for Neuroimaging and Informatics, Keck USC School of Medicine, Marina del Rey, California
Thomas Bourgeron, Human Genetics and Cognitive Functions, Institut Pasteur, Université Paris Cité, Paris, France.
Pierre Bellec, Centre de Recherche de l’Institut Universitaire de Gériatrie de Montréal, Montreal, Canada.
Sebastien Jacquemont, Sainte-Justine Research Center, University of Montréal, Montréal, Canada.
REFERENCES
- 1.Lee PH, Feng Y-CA, Smoller JW (2021): Pleiotropy and cross-disorder genetics among psychiatric disorders. Biol Psychiatry 89:20–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stefansson H, Meyer-Lindenberg A, Steinberg S, Magnusdottir B, Morgen K, Arnarsdottir S, et al. (2014): CNVs conferring risk of autism or schizophrenia affect cognition in controls. Nature 505:361–366. [DOI] [PubMed] [Google Scholar]
- 3.Douard E, Zeribi A, Schramm C, Tamer P, Loum MA, Nowak S, et al. (2021): Effect sizes of deletions and duplications on autism risk across the genome. Am J Psychiatry 178:87–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marshall CR, Howrigan DP, Merico D, Thiruvahindrapuram B, Wu W, Greer DS, et al. (2017): Contribution of copy number variants to schizophrenia from a genome-wide study of 41,321 subjects. Nat Genet 49:27–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Davies RW, Fiksinski AM, Breetvelt EJ, Williams NM, Hooper SR, Monfeuga T, et al. (2020): Using common genetic variation to examine phenotypic expression and risk prediction in 22q11.2 deletion syndrome. Nat Med 26:1912–1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Collins RL, Glessner JT, Porcu E, Niestroj L-M, Ulirsch J, Kellaris G, et al. (2021): A cross-disorder dosage sensitivity map of the human genome. Cell 185:3041–3055.e25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huguet G, Schramm C, Douard E, Jiang L, Labbe A, Tihy F, et al. (2018): Measuring and estimating the effect sizes of copy number variants on general intelligence in community-based samples. JAMA Psychiatry 75:447–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huguet G, Schramm C, Douard E, Tamer P, Main A, Monin P, et al. (2021): Genome-wide analysis of gene dosage in 24,092 individuals estimates that 10,000 genes modulate cognitive ability. Mol Psychiatry 26:2663–2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zheutlin AB, Dennis J, Karlsson Linnér R, Moscati A, Restrepo N, Straub P, et al. (2019): Penetrance and pleiotropy of polygenic risk scores for schizophrenia in 106,160 patients across four health care systems. Am J Psychiatry 176:846–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Murray GK, Lin T, Austin J, McGrath JJ, Hickie IB, Wray NR (2021): Could polygenic risk scores be useful in psychiatry?: A review. JAMA Psychiatry 78:210–219. [DOI] [PubMed] [Google Scholar]
- 11.Richards AL, Pardiñas AF, Frizzati A, Tansey KE, Lynham AJ, Holmans P, et al. (2020): The relationship between polygenic risk scores and cognition in schizophrenia. Schizophr Bull 46:336–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sydnor VJ, Larsen B, Bassett DS, Alexander-Bloch A, Fair DA, Liston C, et al. (2021): Neurodevelopment of the association cortices: Patterns, mechanisms, and implications for psychopathology. Neuron 109:2820–2846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sniekers S, Stringer S, Watanabe K, Jansen PR, Coleman JRI, Krapohl E, et al. (2017): Genome-wide association meta-analysis of 78, 308 individuals identifies new loci and genes influencing human intelligence. Nat Genet 49:1107–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moreau CA, Urchs SGW, Kuldeep K, Orban P, Schramm C, Dumas G, et al. (2020): Mutations associated with neuropsychiatric conditions delineate functional brain connectivity dimensions contributing to autism and schizophrenia. Nat Commun 11:5272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grasby KL, Jahanshad N, Painter JN, Colodro-Conde L, Bralten J, Hibar DP, et al. (2020): The genetic architecture of the human cerebral cortex. Science 367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Biswal B, Yetkin FZ, Haughton VM, Hyde JS (1995): Functional connectivity in the motor cortex of resting human brain using echo-planar MRI. Magn Reson Med 34:537–541. [DOI] [PubMed] [Google Scholar]
- 17.van den Heuvel MP, Hulshoff Pol HE (2010): Exploring the brain network: A review on resting-state fMRI functional connectivity. Eur Neuropsychopharmacol 20:519–534. [DOI] [PubMed] [Google Scholar]
- 18.Holiga Š, Hipp JF, Chatham CH, Garces P, Spooren W, D’Ardhuy XL, et al. (2019): Patients with autism spectrum disorders display reproducible functional connectivity alterations. Sci Transl Med 11: eaat9223. [DOI] [PubMed] [Google Scholar]
- 19.Moreau CA, Raznahan A, Bellec P, Chakravarty M, Thompson PM, Jacquemont S (2021): Dissecting autism and schizophrenia through neuroimaging genomics. Brain 144:1943–1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bernanke J, Luna A, Chang L, Bruno E, Dworkin J, Posner J (2022): Structural brain measures among children with and without ADHD in the Adolescent Brain and Cognitive Development Study cohort: A cross-sectional US population-based study. Lancet Psychiatry 9:222–231. [DOI] [PubMed] [Google Scholar]
- 21.Bertero A, Liska A, Pagani M, Parolisi R, Masferrer ME, Gritti M, et al. (2018): Autism-associated 16p11.2 microdeletion impairs prefrontal functional connectivity in mouse and human. Brain 141:2055–2065. [DOI] [PubMed] [Google Scholar]
- 22.Cao H, Zhou H, Cannon TD (2021): Functional connectome-wide associations of schizophrenia polygenic risk. Mol Psychiatry 26:2553–2561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alnæs D, Kaufmann T, van der Meer D, Córdova-Palomera A, Rokicki J, Moberget T, et al. (2019): Brain heterogeneity in schizophrenia and its association with polygenic risk. JAMA Psychiatry 76:739–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Morrow BE, McDonald-McGinn DM, Emanuel BS, Vermeesch JR, Scambler PJ (2018): Molecular genetics of 22q11.2 deletion syndrome. Am J Med Genet A 176:2070–2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sudlow C, Gallacher J, Allen N, Beral V, Burton P, Danesh J, et al. (2015): UK Biobank: An open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med 12:e1001779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sanders SJ, He X, Willsey AJ, Ercan-Sencicek AG, Samocha KE, Cicek AE, et al. (2015): Insights into autism spectrum disorder genomic architecture and biology from 71 risk loci. Neuron 87:1215–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jønch AE, Douard E, Moreau C, Van Dijck A, Passeggeri M, Kooy F, et al. (2019): Estimating the effect size of the 15Q11.2 BP1-BP2 deletion and its contribution to neurodevelopmental symptoms: Recommendations for practice. J Med Genet 56:701–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moreno-De-Luca D, Sanders SJ, Willsey AJ, Mulle JG, Lowe JK, Geschwind DH, et al. (2013): Using large clinical data sets to infer pathogenicity for rare copy number variants in autism cohorts. Mol Psychiatry 18:1090–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Di Martino A, Yan CG, Li Q, Denio E, Castellanos FX, Alaerts K, et al. (2014): The autism brain imaging data exchange: Towards a largescale evaluation of the intrinsic brain architecture in autism. Mol Psychiatry 19:659–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Di Martino A, O’Connor D, Chen B, Alaerts K, Anderson JS, Assaf M, et al. (2017): Enhancing studies of the connectome in autism using the autism brain imaging data exchange II. Sci Data 4:170010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.HD-200 Consortium (2012): The ADHD-200 consortium: a model to advance the translational potential of neuroimaging in clinical neuroscience. Front Syst Neurosci 6:62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Poldrack RA, Congdon E, Triplett W, Gorgolewski KJ, Karlsgodt KH, Mumford JA, et al. (2016): A phenome-wide examination of neural and cognitive function. Sci Data 3:160110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Orban P, Desseilles M, Mendrek A, Bourque J, Bellec P, Stip E (2017): Altered brain connectivity in patients with schizophrenia is consistent across cognitive contexts. J Psychiatry Neurosci 42:17–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang K, Li M, Hadley D, Liu R, Glessner J, Grant SFA, et al. (2007): PennCNV: An integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res 17:1665–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Colella S, Yau C, Taylor JM, Mirza G, Butler H, Clouston P, et al. (2007): QuantiSNP: An Objective Bayes Hidden-Markov Model to detect and accurately map copy number variation using SNP genotyping data. Nucleic Acids Res 35:2013–2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ge T, Chen CY, Ni Y, Feng Y-CA, Smoller JW (2019): Polygenic prediction via Bayesian regression and continuous shrinkage priors. Nat Commun 10:1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bellec P, Carbonell FM, Perlbarg V, Lepage C, Lyttelton O, Fonov V, et al. (2011). In: A neuroimaging analysis kit for MATLAB and Octave: Proceedings of the 17th International Conference on Functional Mapping of the Human Brain, 2735–2746. [Google Scholar]
- 38.Urchs S, Armoza J, Benhajali Y, St-Aubin J, Orban P, Bellec P (2017): MIST: A multi-resolution parcellation of functional brain networks. MNI Open Res 1:3. [Google Scholar]
- 39.Abraham A, Milham MP, Di Martino A, Craddock RC, Samaras D, Thirion B, Varoquaux G (2017): Deriving reproducible biomarkers from multi-site resting-state data: An autism-based example. Neuroimage 147:736–745. [DOI] [PubMed] [Google Scholar]
- 40.Benjamini Y, Hochberg Y (1995): Controlling the false discovery rate: A practical and powerful approach to multiple testing. J R Stat Soc B 57:289–300. [Google Scholar]
- 41.Li J, Kong R, Liégeois R, Orban C, Tan Y, Sun N, et al. (2019): Global signal regression strengthens association between resting-state functional connectivity and behavior. Neuroimage 196:126–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yan CG, Craddock RC, Zuo XN, Zang YF, Milham MP (2013): Standardizing the intrinsic brain: Towards robust measurement of inter-individual variation in 1000 functional connectomes. Neuroimage 80:246–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Varoquaux G (2018): Cross-validation failure: Small sample sizes lead to large error bars. Neuroimage 180:68–77. [DOI] [PubMed] [Google Scholar]
- 44.Bellec P, Benhajali Y, Carbonell F, Dansereau C, Albouy G, Pelland M, et al. (2015): Impact of the resolution of brain parcels on connectome-wide association studies in fMRI. Neuroimage 123:212–228. [DOI] [PubMed] [Google Scholar]
- 45.Phipson B, Smyth GK (2010): Permutation P-values should never be zero: Calculating exact P-values when permutations are randomly drawn. Stat Appl Genet Mol Biol 9:Article39. [DOI] [PubMed] [Google Scholar]
- 46.Efron B, Tibshirani RJ. Google. An introduction to the bootstrap Available at: https://playgoogle.com/store/books/details?id=gLlpIUxRntoC. Accessed October 3, 2021.
- 47.Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, et al. (2020): The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581:434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wainberg M, Merico D, Huguet G, Zarrei M, Jacquemont S, Scherer SW, Tripathy SJ (2022): Deletion of loss-of-function-intolerant genes and risk of 5 psychiatric disorders. JAMA Psychiatry 79:78–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. (2016): Analysis of protein-coding genetic variation in 60,706 humans. Nature 536:285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Trubetskoy V, Pardiñas AF, Qi T, Panagiotaropoulou G, Awasthi S, Bigdeli TB, et al. (2022): Mapping genomic loci implicates genes and synaptic biology in schizophrenia. Nature 604:502–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bipolar Disorder and Schizophrenia Working Group of the Psychiatric Genomics Consortium (2018): Genomic dissection of bipolar disorder and schizophrenia, including 28 subphenotypes. Cell 173:1705–1715. e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kirov G, Rees E, Walters JTR, Escott-Price V, Georgieva L, Richards AL, et al. (2014): The penetrance of copy number variations for schizophrenia and developmental delay. Biol Psychiatry 75:378–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Assem M, Glasser MF, Van Essen DC, Duncan J (2020): A domain-general cognitive core defined in multimodally parcellated human cortex. Cereb Cortex 30:4361–4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Moreau CA, Ching CR, Kumar K, Jacquemont S, Bearden CE (2021): Structural and functional brain alterations revealed by neuroimaging in CNV carriers. Curr Opin Genet Dev 68:88–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Modenato C, Kumar K, Moreau C, Martin-Brevet S, Huguet G, Schramm C, et al. (2021): Effects of eight neuropsychiatric copy number variants on human brain structure. Transl Psychiatry 11:399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sun D, Ching CRK, Lin A, Forsyth JK, Kushan L, Vajdi A, et al. (2020): Large-scale mapping of cortical alterations in 22q11.2 deletion syndrome: Convergence with idiopathic psychosis and effects of deletion size. Mol Psychiatry 25:1822–1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.van Rooij D, Anagnostou E, Arango C, Auzias G, Behrmann M, Busatto GF, et al. (2018): Cortical and subcortical brain morphometry differences between patients with autism spectrum disorder and healthy individuals across the lifespan: Results from the ENIGMA ASD working group. Am J Psychiatry 175:359–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.van Erp TGM, Walton E, Hibar DP, Schmaal L, Jiang W, Glahn DC, et al. (2018): Cortical brain abnormalities in 4474 individuals with schizophrenia and 5098 control subjects via the Enhancing Neuro Imaging Genetics through Meta Analysis (ENIGMA) consortium. Biol Psychiatry 84:644–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhu X, Ward J, Cullen B, Lyall DM, Strawbridge RJ, Smith DJ, Lyall LM (2021): Polygenic risk for schizophrenia, brain structure, and environmental risk in UK Biobank. Schizophr Bull Open 2. [Google Scholar]
- 60.Antonarakis SE, Skotko BG, Rafii MS, Strydom A, Pape SE, Bianchi DW, et al. (2020): Down syndrome. Nat Rev Dis Primers 6:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Capone GT, Grados MA, Kaufmann WE, Bernad-Ripoll S, Jewell A (2005): Down syndrome and comorbid autism-spectrum disorder: Characterization using the aberrant behavior checklist. Am J Med Genet A 134:373–380. [DOI] [PubMed] [Google Scholar]
- 62.Modenato C, Martin-Brevet S, Moreau CA, Rodriguez-Herreros B, Kumar K, Draganski B, et al. (2021): Lessons learned from neuroimaging studies of copy number variants: A systematic review. Biol Psychiatry 90:596–610. [DOI] [PubMed] [Google Scholar]
- 63.Bletsch A, Mann C, Andrews DS, Daly E, Tan GMY, Murphy DGM, Ecker C (2018): Down syndrome is accompanied by significantly reduced cortical grey-white matter tissue contrast. Hum Brain Mapp 39:4043–4054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Moreau CA, Kumar K, Harvey A, Huguet G, Urchs S, Douard EA, et al. (2021): Atlas of functional connectivity relationships across rare and common genetic variants, traits, and psychiatric conditions. medRxiv 10.1101/2021.05.21.21257604. [DOI]
- 65.Kendall KM, Rees E, Escott-Price V, Einon M, Thomas R, Hewitt J, et al. (2017): Cognitive performance among carriers of pathogenic copy number variants: Analysis of 152,000 UK Biobank subjects. Biol Psychiatry 82:103–110. [DOI] [PubMed] [Google Scholar]
- 66.Malhotra D, Sebat J (2012): CNVs: Harbingers of a rare variant revolution in psychiatric genetics. Cell 148:1223–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rees E, Kirov G, Sanders A, Walters JTR, Chambert KD, Shi J, et al. (2014): Evidence that duplications of 22q11.2 protect against schizophrenia. Mol Psychiatry 19:37–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bernier R, Steinman KJ, Reilly B, Wallace AS, Sherr EH, Pojman N, et al. (2016): Clinical phenotype of the recurrent 1q21.1 copy-number variant. Genet Med 18:341–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.D’Angelo D, Lebon S, Chen Q, Martin-Brevet S, Snyder LG, Hippolyte L, et al. (2016): Defining the effect of the 16p11.2 duplication on cognition, behavior, and medical comorbidities. JAMA Psychiatry 73:20–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rees E, Kendall K, Pardiñas AF, Legge SE, Pocklington A, Escott-Price V, et al. (2016): Analysis of intellectual disability copy number variants for association with schizophrenia. JAMA Psychiatry 73:963–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.