

Abstract
Spatial transcriptomics technologies have shed light on the complexities of tissue structures by accurately mapping spatial microenvironments. Nonetheless, a myriad of methods, especially those utilized in platforms like Visium, often relinquish spatial details owing to intrinsic resolution limitations. In response, we introduce TransformerST, an innovative, unsupervised model anchored in the Transformer architecture, which operates independently of references, thereby ensuring cost-efficiency by circumventing the need for single-cell RNA sequencing. TransformerST not only elevates Visium data from a multicellular level to a single-cell granularity but also showcases adaptability across diverse spatial transcriptomics platforms. By employing a vision transformer-based encoder, it discerns latent image-gene expression co-representations and is further enhanced by spatial correlations, derived from an adaptive graph Transformer module. The sophisticated cross-scale graph network, utilized in super-resolution, significantly boosts the model’s accuracy, unveiling complex structure–functional relationships within histology images. Empirical evaluations validate its adeptness in revealing tissue subtleties at the single-cell scale. Crucially, TransformerST adeptly navigates through image-gene co-representation, maximizing the synergistic utility of gene expression and histology images, thereby emerging as a pioneering tool in spatial transcriptomics. It not only enhances resolution to a single-cell level but also introduces a novel approach that optimally utilizes histology images alongside gene expression, providing a refined lens for investigating spatial transcriptomics.
Keywords: spatial transcriptomics, single-cell RNA-seq, graph transformer
INTRODUCTION
Understanding the tissue structures at the spot and single-cell resolution helps to extract fine-grained information for tissue microenvironment detection. How tissue heterogeneity shapes the structure–function interactions at enhanced resolution remains an open question in current spatial transcriptomics (ST) analysis. Contemporary ST technologies facilitate the inference of large-scale structural connectivity and the delineation of spatial heterogeneity patterns inherent in disease pathology. [1, 2]. ST methods can be generally grouped into two main categories: methods based on fluorescence in situ hybridization or sequencing, such as seqFISH [3, 4], seqFISH+ [5], MERFISH [6, 7], STARmap [8] and FISSEQ [9], have the capability to attain single-cell resolution. However, these technologies measure gene expression with low throughput and less sensitivity. The in situ capturing-based approach forms the second category, comprising methods such as ST [10], SLIDE-seq [11], SLIDE-seqV2 [12], HDST [13] and 10x Visium. These techniques are designed for high-throughput gene expression analysis while maintaining the integrity of spatial patterns. The primary limitation of in situ capturing methods, a category of barcoding-based ST, is their limited spatial resolution. This is particularly evident in widely used technologies like Visium, where the resolution is typically constrained to 20 to 100 cells per barcode. Such a resolution makes it challenging to profile spatial neighborhoods in detail. While other protocols like Slide-seq, PIXEL-seq, Seq-Scope and Stereo-seq, as well as microfluidics-based barcoding methods like DBIT-seq, achieve higher resolutions ranging from 10  m to approximately 500 nm per barcode, they are not inherently ‘cell aware.’ The barcodes in these methods are randomly distributed relative to cell positions, often overlapping cell–cell boundaries, which complicates the association of a spatial barcode with a specific cell. Furthermore, these methods generally exhibit lower sensitivity, mainly due to the requirement for in situ reverse transcription, and the cost per sample is often higher than other methods. Despite these challenges, barcoding methods, including Visium, offer significant advantages in throughput, as the acquisition time does not increase with the sample size or the number of features detected, allowing for the parallel processing of multiple samples and bulk sequencing. Prominent technologies offer spot measurements with diameters of 100
m to approximately 500 nm per barcode, they are not inherently ‘cell aware.’ The barcodes in these methods are randomly distributed relative to cell positions, often overlapping cell–cell boundaries, which complicates the association of a spatial barcode with a specific cell. Furthermore, these methods generally exhibit lower sensitivity, mainly due to the requirement for in situ reverse transcription, and the cost per sample is often higher than other methods. Despite these challenges, barcoding methods, including Visium, offer significant advantages in throughput, as the acquisition time does not increase with the sample size or the number of features detected, allowing for the parallel processing of multiple samples and bulk sequencing. Prominent technologies offer spot measurements with diameters of 100  m in the ST platform and 55
m in the ST platform and 55  m in the Visium platform. Given the constrained resolution of existing ST technologies, there is a pressing need for advanced data analysis techniques to uncover the intricate tissue heterogeneities in tumor microenvironments, brain disorders and embryonic development [1, 14, 15].
m in the Visium platform. Given the constrained resolution of existing ST technologies, there is a pressing need for advanced data analysis techniques to uncover the intricate tissue heterogeneities in tumor microenvironments, brain disorders and embryonic development [1, 14, 15].
Traditional approaches to ST analysis fall short in seamlessly integrating original gene expression, spatial relationships and histology images due to the following limitations. (1) A majority of the current methods employ dimension-reduction techniques to mitigate computational demands. Yet, these reduced features often compromise the heterogeneity of gene expression in certain tissues. (2) Several workflows, like Seurat [16], are tailored for single-cell RNA sequencing (scRNA-seq) analysis, which can inadvertently distort the spatial relationships. (3) To the best of our knowledge, minimal efforts have been directed toward examining the heterogeneity across tissue structures at both spot and enhanced resolutions. Several approaches such as RCTD [17], stereoscope [18], SPOTlight [19], SpatialDWLS [20] and cell2location [21] have been developed to integrate scRNA-seq with ST, enhancing the resolution of spatial gene expression. However, such methods hinge on the availability of appropriate single-cell references. In many cases, the acquisition of appropriate single-cell references is impeded by financial limitations, technical obstacles and biological factors [22, 23]. Some deconvolution methods use public scRNAseq references such as Human Cell Atlas [24], BRAIN Initiative Cell Census Network (BICCN) [25] and Human BioMolecular Atlas [26] to solve the problem, but the batch effects and tissue heterogeneity in samples may result in incomplete cell types. Moreover, single-cell references and ST are affected by different perturbations, which may affect the deconvolution accuracy [27].
Prior ST analysis techniques, particularly those utilizing Visium technology, were unable to elevate gene expression to single-cell resolution without relying on scRNA-seq data. BayesSpace [28] employs a Bayesian prior to investigate the neighborhood structure, enhancing the resolution to a subspot level, which remains less refined than single-cell resolution. However, the high computational complexity and lack of flexibility hinder its application in multimodal ST data analysis. CCST [29] leverages graph convolutional networks to integrate gene expression with overarching spatial information. SpaGCN [30] combines gene expression, spatial information and histology image through a graph convolution model. Importantly, many current methods, including BayesSpace, CCST and SpaGCN, depend on principle component analysis (PCA) to isolate highly variable features. This approach falls short when it comes to uncovering nonlinear relationships. As detailed in [31], STAGATE utilizes an adaptive graph attention autoencoder to discern spatial domains. It achieves better performance for the identification of tissue types and highly expressed gene patterns. However, the utility of STAGATE is limited to spot resolution analysis. ConST [32] is a cutting-edge ST data analysis framework that uses contrastive learning techniques to effectively process and integrate multi-modal ST data. DeepST combines the capabilities of a graph neural network (GNN) autoencoder with a denoising autoencoder to craft an enriched latent representation of augmented ST data. Moreover, as detailed in [33], DeepST employs domain adversarial neural networks to synchronize ST data from different batches, thereby elevating the depth and accuracy of ST analysis. StLearn, as referenced in [34], employs a deep learning approach tailored for the image domain and relies on linear PCA for extracting features from spatial gene expression. However, its limited focus on gene expression and spatial relationships potentially constrains its efficacy across diverse platforms. STdeconvolve [35] utilizes latent Dirichlet allocation to deconvolve the cell type proportions within each multi-cellular pixel. As highlighted in [35], STdeconvolve might struggle to distinguish specific cell types in the absence of highly co-expressed genes unique to each type. Furthermore, it lacks the capability to pinpoint the exact location of individual cell types within each multi-cellular pixel. BLEEP [36] is a novel approach that leverages contrastive learning to generate a low-dimensional joint embedding space from a reference dataset, utilizing paired image and gene expression profiles at micrometer resolution to accurately impute gene expression in diverse image patches. TCGN [37] is an innovative model combining convolutional layers, transformer encoders and GNNs to efficiently and accurately estimate gene expressions from H&E-stained pathological slides, making it a significant advancement in precision health applications. SpatialPCA [38] is an innovative dimension reduction technique tailored for ST. It effectively extracts significant biological signals from data, maintains spatial correlation structures and facilitates advanced analyses such as identifying spatial domains, inferring developmental trajectories and constructing detailed spatial maps, thereby uncovering essential molecular and immunological patterns in intricate tissue contexts. Vesalius [39] is a cutting-edge tool designed for ST data, utilizing image processing technology to decode tissue anatomy, uniquely identifying regions comprising multiple cell types, and effectively revealing tissue structures and cell-specific gene expression patterns in high-resolution datasets, including mouse brain, embryo, liver and colon.
Current methodologies in ST analysis often underutilize the rich information embedded within histology images when combined with gene expression data. Predominant methods, such as SpaGCN, typically leverage merely the spatial location of each spot in constructing graphs, thereby neglecting the intricate textural features present within the histology images. This oversight potentially omits valuable contextual data regarding cellular structures, tissue architectures and localized expression patterns, which could otherwise enhance the granularity and accuracy of spatial gene expression mappings. The nuanced visual details within histology images, such as cellular alignments, tissue morphologies and pathological markers, can provide an additional layer of data that, when effectively integrated with ST, could unveil deeper insights into spatially resolved biological phenomena and disease progressions. Thus, there is a compelling need for the development of advanced analytical methods that holistically integrate both the spatial coordinates and the detailed textural features of histology images to fully harness the synergistic potential of combining these data with spatial gene expression. While our study focuses on ST, it is contextualized by advancements in deep learning for drug discovery as demonstrated in the works on multimodal representation learning and interaction prediction in TripletMultiDTI [40], drug combination studies using transformers in DeepTraSynergy [41] and compound–protein interaction prediction enhancements in DeepCompoundNet [42].
To address existing challenges, we developed TransformerST, an innovative Transformer-based framework crafted to correlate the heterogeneity of local gene expression properties with various tissues in histology images, concurrently unveiling the dependency of structural relationships at a single-cell resolution (Figure 1). TransformerST encompasses three pivotal components: a vision transformer, an adaptive graph Transformer model fortified with multi-head attention and a cross-scale model dedicated to super-resolved gene expression reconstruction. The initial component effectively incorporates vision transformer structures, adeptly capturing genuine local gene expression patterns in tandem with histology visuals. This model takes in a co-representation of image and gene, sourced from the histology images, and amalgamates both local and overarching gene expressions within each spot, culminating in the formation of a spot-to-spot correlation graph. The adaptive graph transformer approach identifies tissue types by amalgamating spatial gene expression, spatial relationships and histology images, while also employing an adaptive parameter learning step to more astutely explore the relationship between spatial gene features and graph neighboring dependence. Lastly, the super-resolved resolution is enhanced through the cross-scale internal GNN, which recovers more detailed tissue structures in histology images at a single-cell resolution. The proposed approach offers the subsequent benefits.
Figure 1.

Schematic representation of TransformerST. A, The VisionTransformer encoder amalgamates spatial gene expression, spatial location and histology image, facilitating the exploration of image-gene expression co-expression. B, The Adaptive Graph Transformer model is employed to harness spatial neighboring dependence, enabling the association of spatial gene expression patterns at the original resolution. C, The Cross-scale Internal Graph Network is utilized for the super-resolved reconstruction of gene expression, taking concatenated embedding and histology image as inputs to elevate gene expression from multicellular to single-cell resolution.
The proposed approach sheds light on the dynamic structural–functional relationships in ST at a single-cell resolution. While the incorporation of scRNA-seq data is prevalent in deconvolution studies [17, 19, 43], it may introduce bias when single-cell measurements are not available for real-world applications. The proposed method can infer the tissue microenvironment at both spot and single-cell resolution without relying on scRNA-seq data. Our method can produce gene expression data for each pixel in histology images, achieving a resolution higher than that of single-cell sequencing. However, the resolution of the enhanced spatial gene expression hinges on the ST technology, which can span from subcellular to single-cell or multi-cell levels. Additionally, the enhanced resolution is influenced by the quality of the image captured. For instance, when supplied with a high-quality histology image coupled with Visium data, our proposed method has the capacity to generate single-cell resolution gene expression data.
The proposed approach enables the integration of heterogeneous spatial gene expression with histology images using multimodal data. While most of the existing methods utilize linear PCA for feature extraction, the proposed method learns and reconstructs the original expressive gene pattern with a large number of highly variable genes (HVGs). The proposed method provides a novel pipeline for tissue type identification, spatial-resolved gene reconstruction and gene expression prediction from histology images (if available). It can be easily transferred to different ST platforms, such as STomics or 10x Visium.
The proposed method is assessed to investigate the pertinence of various tissue types. This method represents the first attempt to reconstruct gene expression at a single-cell resolution without employing scRNA-seq as a reference. Experimental outcomes, derived from various ST datasets, highlight the robustness and effectiveness of our proposed approach, outshining contemporary methods in terms of representation quality.
While TransformerST is equipped for transcriptomics deconvolution, our main emphasis is on clustering and super-resolution. Unlike deconvolution, which estimates cell type proportions per spot, our super-resolution pinpoints both location and gene expression for each cell. Coupled with the clustering task at both the spot and single-cell levels, TransformerST is adept at analyzing the cell type for each individual cell.
RESULTS
Overview of the proposed method and evaluations. Our proposed methodology for analyzing spatial transcriptomic data across multiple tissues addresses the limitations of existing techniques, many of which depend on the availability of scRNA-seq data to enhance resolution. This requirement is not always feasible, especially in tissues such as the lung. In contrast, our method is engineered to function without the need for a single-cell reference, significantly broadening its versatility. This expands its applicability in ST studies considerably. Crucially, our approach is tailored to enhance the resolution of spatial transcriptomic data up to the granularity of individual cells, even in the absence of a single-cell reference. This facilitates the categorization of cell types at both the original and single-cell resolutions. Compared with existing methods, TransformerST stands out for its efficiency, requiring significantly less computational time for both clustering and super-resolution tasks (Table 1 and Table 2). In Table 3, we evaluate our method, TransformerST, in comparison with state-of-the-art methods in ST. This comparison, which highlights the varying capacities of different methods to handle a range of tasks in ST analysis, is thoroughly detailed in Section S2 of the Supplementary Material. While our clustering approach is similar to existing methods, our proposed method is unique in its ability to identify tissue type at both spot and enhanced resolution. To the best of our understanding, this represents the inaugural approach to attain single-cell resolution in ST without resorting to single-cell reference datasets. While BayesSpace [28] and STdeconvolve [35] have shown incremental enhancements in ST data resolution, they have not reached the granularity of single-cell resolution.
Table 1.
Computational time for tissue type identification with LIBD human dorsolateral prefrontal cortex
| Method | Runtime/mins | GPU/CPU |
|---|---|---|
| TransformerST-3000 HVGs | 6.5 | GPU |
| TransformerST-200 PCA | 3 | GPU |
| BayesSpace | 21 | CPU |
| stLearn | 0.5 | GPU |
| SpaGCN | 2 | GPU |
| CCST | 3 | GPU |
| STAGATE | 7 | GPU |
| Gitto | 17 | CPU |
Table 2.
Computational time for super-resolved gene expression reconstruction with IDC sample
| Method | Runtime/mins | GPU/CPU |
|---|---|---|
| TransformerST-3000 HVGs | 29 | GPU |
| BayesSpace | 200 | CPU |
| STdeconvolve | 54 | CPU |
Table 3.
Comparison between TransformerST with baselines
| Methods | Objective | Super-resolution | Reference-free | Histology image |
|---|---|---|---|---|
| TransformerST | Clustering, super-resolution | Single-cell | Yes | Yes |
| SpaGCN | Clustering | Original | No | Yes |
| BayesSpace | Clustering, super-resolution | Multi-cellular | Yes | No |
| CCST | Clustering | Original | No | No |
| STAGATE | Clustering | Original | No | No |
| DeepST | Clustering | Original | No | Yes |
| stLearn | Clustering | Original | No | No |
| STdeconvolve | Deconvolution | Multi-cellular | Yes | No |
To showcase the strength of the proposed method, we evaluated its performance with several publicly available datasets. In tissue identification experiments at original resolution, we showed the spot resolution clustering results with human dorsolateral prefrontal cortex data (DLPFC). We additionally validated TransformerST using our in-house mouse lung data, which were generated with the 10x Visium platform. A portion of this in-house data features fluorescence staining as an alternative method for obtaining ground truth information (Figure 2 and Figure 3). TransformerST outperforms several state-of-the-art approaches such as stLearn [34], Mclust, Kmeans, Louvain, Giotto, BayesSpace [28], CCST [29], STAGATE [31] and SpaGCN [30]. To evaluate the super-resolution performance of TransformerST, we used three data from different ST platforms. Specifically, we used the melanoma data from the ST platform to evaluate the super-resolution performance at subspot resolution when the histology image is missing (Figure 4). We demonstrated the improved resolution performance at the single-cell level using invasive ductal carcinoma (IDC) samples that were human epidermal growth factor receptor 2 amplified (HER+), obtained through the 10x Visium platform (Figure 5). The IDC was manually annotated by a pathologist to exclude the overexposed regions.
Figure 2.

Tissue identification in human dorsolateral prefrontal cortex Visium data at spot resolution. The ARI is used to evaluate the similarity between cluster labels acquired by each method against manual annotations. A, Tissue types assignments by different spatial clustering methods for sample 151672. B, Tissue types assignments by different spatial clustering methods for sample 151508. C, Summary of all 12 samples’ clustering accuracy.
Figure 3.
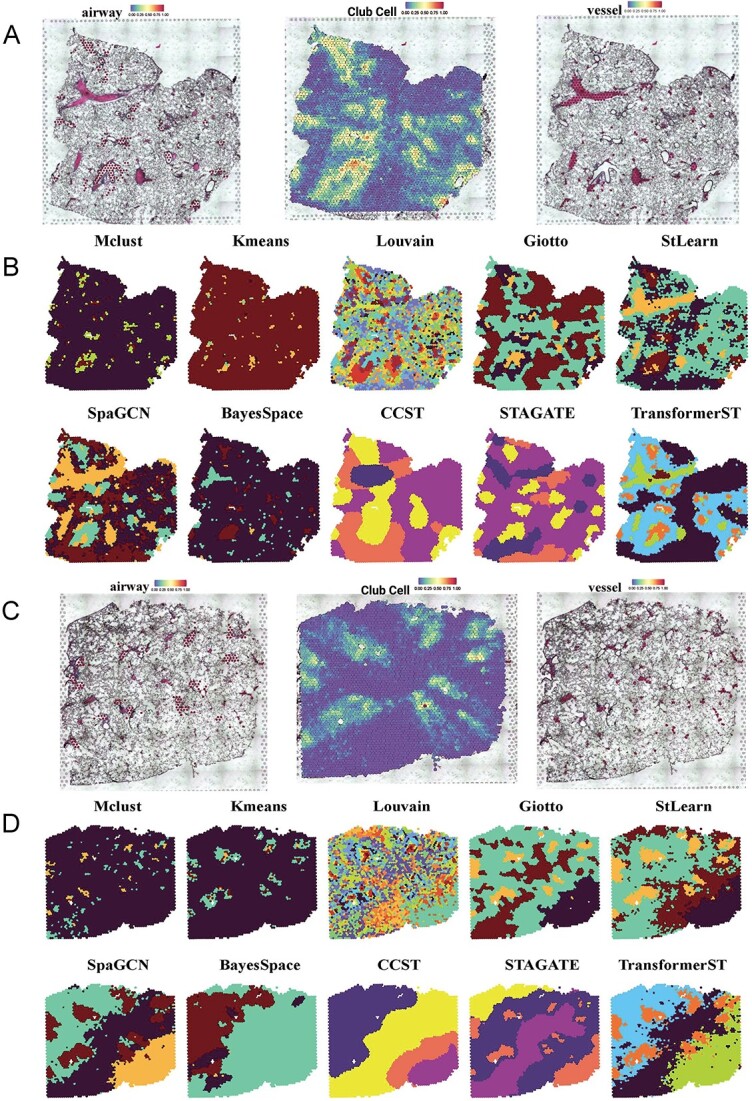
Tissue identification in mouse lung Visium data at spot resolution. A, Manual annotations of airways (left) and blood vessels (right) of the first slice. Pathologists identified regions of significant regions according to the histology image. Airways were defined in line with the proportion of club cells (middle) within each slice. B, Tissue types assignments by different spatial clustering methods for the first sample. C, Manual annotations of airways (left) and blood vessels (right) of the second slice. D, Tissue types assignments by different spatial clustering methods for the second sample.
Figure 4.

Tissue identification with super-resolved gene expression in melanoma ST data. A, Tissue type assignments by different spatial clustering methods for melanoma sample. B, Enhanced subspot tissue identification of melanoma sample with BayesSpace, STdeconvolve and TransformerST.
Figure 5.

Tissue identification with super-resolved gene expression in IDC Visium data. A, Tissue type assignments by different spatial clustering methods for IDC sample. The pathologist annotated different regions in different colors (carcinoma in situ outlined in red, invasive carcinoma (IC) in Blue, Nontumor in Brown, benign hyperplasia in yellow and unclassified tumor in orange). B, Histology imaging of tissue. C, Cell type proportion of IDC sample with ST-deconvolve. D, Enhanced super-resolved tissue identification of IDC sample with BayesSpace at subspot resolution. E, Enhanced super-resolved tissue identification of IDC sample with TransformerST at single-cell resolution.
Moreover, our research, as detailed in Section S4 and depicted in Supplementary Figure 3, employed the 36 tissue sections from the HER2+ breast cancer dataset [44] to evaluate the effectiveness of TransformerST in gene expression prediction and super-resolution. Utilizing a leave-one-out evaluation method (36 fold), we trained the clustering and super-resolution model on 32 sections, with the remaining section used for evaluation. This singular experimental approach effectively showcased TransformerST’s capabilities in predicting gene expression and achieving super-resolution at the single-cell level.
Subsequently, in the supplementary material detailed in Section S5 and visualized in Supplementary Figure 4, we explored the accuracy of detecting spatial variable genes (SVGs) and meta-genes using DLPFC and IDC samples. Our proposed method notably reduces computational complexity and more efficiently reconstructs enhanced gene expression at a single-cell resolution. The SVGs and meta-genes identified by our approach demonstrate superior biological interpretability. Additionally, we compared TransformerST with SpatialPCA and Vesalius using Moran’s I and Geary’s C statistical tests to further underscore TransformerST’s performance in capturing spatial gene expression patterns.
We employed the Xenium in situ data from a human breast cancer block in a simulation experiment, demonstrating our method’s effectiveness in clustering and super-resolution. These findings are elaborated in Supplementary Section S6 and depicted in Supplementary Figure 5.
It should be noted that all the baseline methods were applied with the default parameters. Besides the experiments described in the manuscript, we also employed data from Stereo-seq sourced from mouse olfactory bulb and mouse lung tissues [45]. The results of these additional analyses can be found in Section S3 of the supplementary material.
Tissue type identification at original resolution
Tissue identification in human dorsolateral prefrontal cortex Visium data. The LIBD recently procured data for the human DLPFC using the 10x Visium technique. This comprehensive dataset includes 12 tissue samples, with each one having manual annotations distinguishing six cortical layers and the white matter. The annotations, as detailed in the original research by [46], offer a foundation for assessing the efficacy of identifying tissue types at the granularity of individual spots. We evaluated the tissue type identification of TransformerST compared with StLearn, Mclust, Kmeans, Louvain, Giotto, BayesSpace, CCST, STAGATE, DeepST, conST and SpaGCN. We employed the adjusted Rand index (ARI) as a metric to measure the congruence between the actual annotations and the outcomes of our clustering approach [44].
The clustering accuracy (ARI) of sections 151672 and 151508 are shown in Figure 2A and Figure 2B. Compared with the baseline methods, TransformerST could learn the dynamic graph representation between spatial gene expression and spatial neighbors. Specifically, the proposed method was implemented using the top 3000 HVGs; other comparison methods, such as BayesSpace and SpaGCN, used 15 PCs from the top 3000 HVGs. Gitto, CCST, STAGATE, ConST, DeepST and StLearn used the recommended parameters in the previous papers. The proposed method could use the highly expressive gene and spatial dependence of neighboring embedding to achieve the highest tissue identification performance of both samples. In our analysis focused on section 151672 of the human DLPFC dataset, Figure 2A shows methods such as TransformerST, Gitto, STAGATE, ConST, DeepST, and SpaGCN effectively highlight spatial gene expression patterns that closely match manual annotations. Among these, TransformerST achieves the highest Adjusted Rand Index (ARI) of 0.687, indicating superior alignment, followed by Gitto with an ARI of 0.573, STAGATE at 0.561, ConST at 0.544, and SpaGCN at 0.565. The visual difference among these results is not significant. BayesSpace, Mclust, DeepST and CCST also provided decent results (ARI is 0.439 for BayesSpace, 0.479 for Mclust, 0.45 for DeepST and 0.427 for CCST) and outperformed Louvain, StLearn and Kmeans. In Figure 2B, for section 151508, TransformerST had the highest clustering accuracy and provided distinct layers of clusters (ARI is 0.592). CCST and STAGATE outperformed other methods but provided a worse performance than TransformerST.
The remaining clustering results with all 12 DLPFC samples are shown in Figure 2C. TransformerST achieved the best performance with a mean ARI (0.564). Compared with the second performer STAGATE with mean ARI (0.502), TransformerST increased the tissue identification performance by 12.4%. The difference between BayesSpace, CCST, DeepST, ConST and SpaGCN is not significant. Additionally, the runtime of TransformerST at spot resolution is comparable with other clustering methods for spot-level annotation, which uses 6.5 min with 3000 HVGs and 3 min for 200PCs. (Table 1). These results further demonstrate the superiority of TransformerST in exploring spatial expression patterns and provide clear cluster differences between brain layers.
Tissue identification in mouse lung Visium data at spot resolution. To further assess the performance of TransformerST in tissue identification, we performed Visium experiments on slices of mouse lung tissues [47]. Single-cell suspension processed side-by-side was subjected to a scRNA-seq experiment and utilized to deconvolute the Visium data.
A pathologist subsequently pinpointed areas of interest, such as airways and blood vessels, based on the histological images provided [47]. Airways were delineated based on the proportion of club cells deconvoluted within each tissue section. In the study by [47], a pathologist manually determined the thresholds for each tissue section to align the chosen spots with the histological representation of the airways. Spots were identified as airways when the percentage of club cells exceeded the set threshold (top 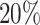 for slice A1, top
for slice A1, top 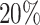 for slice A2, top
for slice A2, top 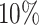 for slice A3 and top
for slice A3 and top 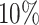 for slice A4). Blood vessels were identified based on their correspondence with the vascular regions depicted in the histological images. We employed a random trees pixel classifier in QuPath (version 0.2.3), set at a downsample rate of 16, to predict the likelihood of blood vessels presence within each spot across all tissue slices. All the training samples of the random trees pixel classifier came from the manual annotation of slice A1. Then, the pathologist [47] used the threshold 0.5 to select the blood vessels (Figure 3A and Figure 3C).
for slice A4). Blood vessels were identified based on their correspondence with the vascular regions depicted in the histological images. We employed a random trees pixel classifier in QuPath (version 0.2.3), set at a downsample rate of 16, to predict the likelihood of blood vessels presence within each spot across all tissue slices. All the training samples of the random trees pixel classifier came from the manual annotation of slice A1. Then, the pathologist [47] used the threshold 0.5 to select the blood vessels (Figure 3A and Figure 3C).
After defining these histological structures, TransformerST was utilized to reveal the internal heterogeneity within visually homogeneous blood vessel and airway tissue regions. The cluster numbers of all comparison methods were set to 4. Figure 3B shows, for the first slice sample, SpaGCN, STAGATE and StLearn were able to distinguish the airways but failed to identify the tissue region of blood vessels. Surprisingly, BayesSpace failed to identify the significant tissue types such as blood vessels and airways (Figure 3B). Other comparison methods such as Mclust, Kmeans, CCST and Louvain had worse performance, which is contrary to the manual annotation (Figure 3A). Gitto could identify the major tissue types, but its result is very noisy. The most interesting finding is that TransformerST is able to identify the whole blood vessel regions and provide a more robust signal with detailed textural features (Figure 3B).
Moreover, we used the club cell tissues to evaluate the performance of TransformerST. As shown in Figure 3B, for the first slice sample, TransformerST, SpaGCN, Gitto, STAGATE and StLearn identified the club cell regions, an indicator of airways. We observed that the spatial expression patterns of club cells between the clusters were largely in line with the clinical annotations (Figure 3A). BayesSpace, CCST and non-spatial methods (Mclust, Kmeans and Louvain) failed to detect the spatial patterns of club cell structures. Comparing these results, it could be seen that spatial expression patterns acquired by TransformerST better reflect the club cell structures with detailed information on the boundaries.
The relative performance remains the same for the second slice sample (Figure 3C); TransformerST, StLearn, Gitto, STAGATE and SpaGCN were able to identify the heterogeneity within club cells structure (Figure 3d). As illustrated in Figure 3d, other methods, excluding TransformerST, displayed considerable noise and lacked clear spatial distinction between club cells. BayesSpace, Mclust, Louvain, CCST and Kmeans provided worse performance which violates the biological interpretation. The existing methods are not applicable to mouse lung tissue identification. TransformerST could identify the spatial patterns with histology images and provide finer details of manual annotations (Figure 3C).
ST super-resolution at enhanced resolution
Tissue identification and super-resolution in melanoma ST data at subspot resolution. We assessed the super-resolution performance at the subspot level using the publicly accessible melanoma ST dataset, as annotated and detailed in the study by Thrane et al. [14]. The manual annotation of melanoma, stroma and lymphoid regions (Figure 4A) were included to evaluate the performance of the TransformerST. Similar to manual annotations, we set the cluster number to 4. As the histology image is missing, both BayesSpace and TransformerST could enhance the resolution of ST expression to subspot resolution. We show the tissue identification results of the proposed method in both spot and subspot resolution in Figure 4A and Figure 4B. Comparison of the results of TransformerST with those of other methods (Mclust, Kmeans, Louvain, Gitto, SpaGCN, CCST, STAGATE and BayesSpace) confirms that TransformerST reveals similar patterns to the manual annotation.
Specifically, the melanoma tissue could be divided into two types, central tumor region and outer of the mixture of tumor and lymphoid tissue. Surprisingly, only TransformerST was able to identify the lymphoid regions at the original resolution (Figure 4A). The results of comparison methods could not identify lymphoid regions at the original resolution. The tissue identification results at enhanced resolution are in line with the finding that TransformerST identifies the lymphoid region in the tumor border with a higher resolution (Figure 4B). In accordance with a recent study, BayesSpace and STdeconvolve also identified the lymphoid regions of the tumor at the enhanced resolution (Figure 4B). The findings from this research suggest that while all the methods compared were able to discern the differences between the tumor’s edge and its center, they were unable to detect the lymphoid tissue at the initial resolution. TransformerST, STdeconvolve and BayesSpace provided enhanced resolution of tissue structures, which makes it possible to identify the lymphoid tissue. The observational results suggest TransformerST provides higher resolution and robust tissue identification results at both original and enhanced resolution.
Tissue identification and super-resolution in IDC Visium data at single-cell resolution. We assessed the performance of single-cell super-resolution using the IDC Visium data, which was stained with immunofluorescence for 4,6-diamidino-2-phenylindole (DAPI) and T cells staining CD3, as described in the study by Zhao et al. [28]. Pathologists, as referenced in the study by Zhao et al. [28], pinpointed regions predominantly characterized by invasive carcinoma (IC), carcinoma in situ and benign hyperplasia. These regions were included in the evaluation of clustering accuracy at spot resolution (Figure 5A and Figure 5B). Similar to the manual annotations, we clustered the IDC sample into five clusters at spot resolution. We used ARI to evaluate the clustering accuracy at spot resolution. The results of the clustering experiment at the original resolution indicate that TransformerST achieves the best clustering accuracy with an ARI of 0.493 (Figure 5A). The ARI is 0.42 for BayesSpace against 0.369 for SpaGCN, 0.357 for Mclust and 0.274 for Gitto. However, some comparison methods did not improve the clustering performance (ARI is only 0.257 for StLearn, 0.234 for STAGATE, 0.208 for CCST, 0.151 for Louvain and 0.101 for Kmeans).
We further improved the resolution of ST to highlight its biological significance using TransformerST, STdeconvolve and BayesSpace, as depicted in Figure 5C, Figure 5D and Figure 5E. In accordance with the BayesSpace paper [28], we set the cluster number 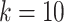 . As shown in Figure 5D and Figure 5E, TransformerST could identify four clusters (0,3,4,8) related to predominantly IC, one cluster (2) related to carcinoma regions and one cluster (7) identifies the benign hyperplasia regions. And clusters (1,5,6,9) are related to the unclassified regions. The result of ByesSpace was consistent with the previous report in [28]. It is hard to evaluate the cluster accuracy at enhanced resolution quantitatively. The results of the three methods show the spatial heterogeneity among tumors, which is inaccessible to histopathological analysis. However, we saw the visual difference between carcinoma and benign hyperplasia regions via TransformerST compared with BayesSpace and STdeconvolve. TransformerST exhibited a spatial organization more similar to manual annotations. BayesSpace could only increase the IDC data to subspot resolution; TransformerST could predict the heterogeneity within each tissue at single-cell resolution. STdeconvolve revealed the proportion of each cell type but failed to identify the location of cell patterns within each spot. The runtime of TransformerST at enhanced resolution is comparable with other methods for gene expression reconstruction, which uses 29 min (Table 2). TransformerST provides a more efficient approach to identifying the super-resolved tissue microenvironment than BayesSpace and STdeconvolve.
. As shown in Figure 5D and Figure 5E, TransformerST could identify four clusters (0,3,4,8) related to predominantly IC, one cluster (2) related to carcinoma regions and one cluster (7) identifies the benign hyperplasia regions. And clusters (1,5,6,9) are related to the unclassified regions. The result of ByesSpace was consistent with the previous report in [28]. It is hard to evaluate the cluster accuracy at enhanced resolution quantitatively. The results of the three methods show the spatial heterogeneity among tumors, which is inaccessible to histopathological analysis. However, we saw the visual difference between carcinoma and benign hyperplasia regions via TransformerST compared with BayesSpace and STdeconvolve. TransformerST exhibited a spatial organization more similar to manual annotations. BayesSpace could only increase the IDC data to subspot resolution; TransformerST could predict the heterogeneity within each tissue at single-cell resolution. STdeconvolve revealed the proportion of each cell type but failed to identify the location of cell patterns within each spot. The runtime of TransformerST at enhanced resolution is comparable with other methods for gene expression reconstruction, which uses 29 min (Table 2). TransformerST provides a more efficient approach to identifying the super-resolved tissue microenvironment than BayesSpace and STdeconvolve.
DISCUSSIONS
In our research, we introduce an innovative approach that leverages Transformer architectures to seamlessly integrate gene expression data, spatial coordinates and accompanying histological images (when provided). The proposed method, called TransformerST, stands out as the pioneering technique that elevates the resolution of ST to the single-cell level, all without the need for a scRNA-seq reference. Different from most of the existing ST analysis methods, TransformerST does not require linear PCA preprocessing and ensures the intricate understanding of the spatially dispersed tissue structures present in multimodal datasets, such as ST and 10x Visium. The innovative graph transformer model, equipped with multi-head attention, facilitates the integration of multimodal graph representations. This, in turn, uncovers the intricate relationships within the heterogeneity map, shedding light on the dynamics of tissue functionality. With the help of a cross-scale internal graph network, TransformerST enables the effective and efficient analysis of super-resolved tissue microenvironment at single-cell resolution. We assessed the efficacy of TransformerST using a variety of datasets, each produced using different ST techniques. When juxtaposed with leading-edge techniques, TransformerST demonstrates superior capability in discerning tissue clusters at both the spot level and single-cell resolution. TransformerST overcomes the limitation of the low resolution of current ST technology and provides an efficient way to explore the spatial neighboring relationship. The findings from our experiments underscore the significance of regional variability and the inherent relationship between structure and function within the dynamic tissue microenvironment. TransformerST could lower the computation complexity and memory usage than existing methods.
While the study of tissue type identification remains a pivotal aspect of contemporary ST analysis, our experimental findings highlight that a majority of the leading techniques fall short in accurately discerning the cellular diversity inherent to individual cell types. We expect TransformerST could help to provide a better resolution of ST data analysis. TransformerST could achieve super-resolved resolution of a single cell per subspot without the requirement of additional scRNA-seq reference. However, TransformerST could be easily adapted to incorporate additional single-cell references for deconvolution tasks. In the following assessments, including SVGs and meta-gene evaluations, TransformerST proved adept, revealing biological tissue structures that resonated well with manual annotations.
While TransformerST focuses on the ST and Visium platform, it could be easily applied to other platforms with slight modification. In summary, TransformerST presents a powerful and streamlined approach for a range of unsupervised ST analyses, including tissue identification, super-resolved gene expression reconstruction. For future work, we aim to enhance the accuracy of tissue type identification by estimating the contribution of cell-specific gene expression. Additionally, we plan to refine the graph transformer model to delve into the heterogeneity of tissue types within various micro-environments. Furthermore, we aspire to analyze meta-genes and SVGs utilizing TransformerST.
METHODS
Data description. TransformerST is evaluated using several publicly available datasets and one in-house dataset, most of which were obtained via the Visium platform. Specifically, the DLPFC dataset comprises 12 sections, with each section containing between 3498 to 4789 spots. The regions of the DLPFC layers and white matter were manually delineated by expert pathologists. To reconstruct gene expression at the enhanced resolution, we use the publicly available melanoma ST data which were annotated and described in Thrane et al. [14] We demonstrate the efficacy of our super-resolution approach at single-cell resolution by analyzing IDC Visium data, which have been subjected to immunofluorescence staining for 4,6-diamidino-2-phenylindole (DAPI) and T-cell marker CD3 in [28]. We conducted a simulation experiment using the Xenium in situ data from a human breast cancer block to demonstrate the performance of our method with respect to clustering and super-resolution capabilities.
In-house data preprocessing. For our in-house mouse lung data, the 10X Genomics Visium platform was used to perform the ST experiment. Following the extraction of mouse lungs, the left lobes were filled with a 1mL solution comprising an equal mix of sterile PBS and Tissue-Tek OCT compound (SAKURA FINETEK). Subsequently, they were frozen using an alcohol bath on dry ice. Until they were processed further, OCT blocks were kept at 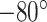 C Following the 10x Genomics Visium fresh frozen tissue processing protocol, OCT blocks were sliced to a thickness of 10
C Following the 10x Genomics Visium fresh frozen tissue processing protocol, OCT blocks were sliced to a thickness of 10 m and dimensions of 6.5 mm x 6.5 mm, mounted onto Visium slides, and subsequently stained with hematoxylin and eosin. An Olympus Fluoview 1000 fluorescence and tile scanning microscope was employed to capture H&E images. Following this, the tissue was removed from the slides, and library generation was carried out according to the protocol provided by 10x Genomics.
m and dimensions of 6.5 mm x 6.5 mm, mounted onto Visium slides, and subsequently stained with hematoxylin and eosin. An Olympus Fluoview 1000 fluorescence and tile scanning microscope was employed to capture H&E images. Following this, the tissue was removed from the slides, and library generation was carried out according to the protocol provided by 10x Genomics.
Every sequenced ST library was aligned to the mm10 mouse reference genome using the 10x Genomics’ Space Ranger software (version 1.2.2). UMI counts were then compiled for every spot. Tissue overlying spots were identified based on the images in order to distinguish them from the background. Upon generating the filtered UMI count matrices, only the barcodes linked to spots overlaying the tissue were retained. Furthermore, we manually removed spots identified by Space Ranger that were not covered by tissue. We then refined the UMI count matrices for each slice (A1: 32 285 genes  3689 spots; A2: 32 285 genes
3689 spots; A2: 32 285 genes 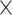 2840 spots; A3: 32 285 genes
2840 spots; A3: 32 285 genes 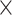 3950 spots; A4: 32 285 genes
3950 spots; A4: 32 285 genes 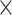 3765 spots).
3765 spots).
Public data preprocessing. All Visium samples were generated from 10x Genomics procured from BioIVT:ASTERAND. The remaining melanoma and breast cancer samples were obtained using the ST platform. We use the second replicate from biopsy 1 to detect the lymphoid sub-environment. For all datasets, raw gene expression counts expressed in fewer than three spots were filtered and eliminated. Seurat was introduced to find the top 3000 HVGs for each spot. The gene expression values are transformed into a natural log scale. We use both histology images (when available) and spatial gene expression to exploit tissue sub-environment at the super-resolved resolution.
Utilizing Vision and Graph Transformers for Single-Cell Resolution Enhancement. The methodology of our proposed approach is meticulously illustrated in Figure 1A, addressing a pivotal challenge in ST analysis: the discernment of spatial patterns in gene expression and the exploration of image-gene expression co-representation. To adeptly harness and utilize the spatial information encapsulated in ST, we enhance the resolution of ST data to a single-cell level, employing a structured, three-tiered process. This approach not only illuminates the intricate spatial patterns embedded within the gene expression data but also intricately explores the co-representation of image and gene expression, providing a nuanced, high-resolution insight into the cellular landscape of the tissue under investigation.
During the image processing stage, two distinct types of image patches are extracted: spot-centric and sliding-window patches. Spot-centric patches are extracted in alignment with each spot location, ensuring each spot is associated with a unique, non-overlapping patch. On the other hand, sliding-window patches are densely extracted within each spot region, producing overlapping image patches.
In the initial step, a Vision Transformer encoder is employed to learn the co-representation of image-gene expression, which is adept at predicting the gene expression of each spot from its corresponding spot-centric image patches, as illustrated in Figure 1A. Following this, the image patch embedding for each spot and its gene expression are concatenated to formulate a graph.
In the second step, we leverage the Graph Transformer, which adeptly links spatial information to spatial graphs. Simultaneously, the adaptive graph transformer is employed to aggregate gene expression based on the relationships of neighboring data points and the associated histology image, as illustrated in Figure 1B. An iterative unsupervised deep clustering model is introduced to detect heterogeneous tissue types at the original spot resolution, while the adaptive graph transformer facilitates the association of spatial patterns with gene expression at spot resolution.
In the pursuit of further enhancing the spatial gene expression resolution, the third step employs cross-scale internal graph networks, meticulously designed to fully leverage both gene expression and histology image data. These networks utilize the concatenated embedding and histology image patches as inputs, synthesizing gene expression at the single-cell resolution, as depicted in Figure 1C. This pivotal step is bifurcated into two sub-steps: graph reconstruction and patch aggregation. Both stages play a pivotal role in elevating spatial resolution and guaranteeing accuracy in gene expression prediction. This holistic method optimally harnesses gene expression and histological imagery, facilitating precise reconstruction and forecasting of spatial gene expression at a superior resolution.
Vision Transformer for Image-Gene Expression Co-Representation Learning in ST. In our research, we leverage the Vision Transformer (ViT) model to proficiently learn the encoding and decoding of image features extracted from histology images, which are crucial for comprehending the cellular structures and variations within tissue samples. Initially, the histology images are segmented into patches corresponding to the spot locations in the ST data, ensuring that the image features are localized and relevant to the respective gene expression profiles. Each patch, encapsulating localized morphological information, is then processed through the ViT model, which, with its transformer architecture, is adept at handling image data by dividing it into non-overlapping patches and linearly embedding them into the model. The ViT model is designed to forecast gene expression from associated image patches. A loss function is employed to reduce the discrepancy between the predicted and true gene expression, guaranteeing that the model establishes a reliable correlation between image attributes and gene expression. Subsequently, the learned image features are amalgamated with ST spot gene expression data, forming a comprehensive feature set that encapsulates both morphological and gene expression information. This enhanced combined feature set is then used to build a graph, where each node signifies a spatial spot and is defined by the integrated features. Edges in the graph denote spatial relationships and/or similarities in the feature space between the spots, thereby encapsulating the spatial dependencies and co-expression patterns prevalent in the tissue. This graph serves as a comprehensive visual summary of spatial transcriptomic data, enhanced with details from histological images. It forms the basis for advanced analyses, including clustering or classification of cellular structures and identification of spatially co-expressed gene sets. These steps enable a deeper exploration of the tissue's spatial molecular diversity.
The spatial gene expression data are represented by the matrix 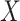 with dimensions
with dimensions 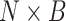 . Here,
. Here, 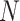 stands for the total number of spots, while
stands for the total number of spots, while 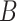 indicates the total genes present. When analyzing the histological image, we carefully segment patches that align with the dimensions and positioning of every spot. These segmented patches from a tissue section are then compiled and reshaped into an
indicates the total genes present. When analyzing the histological image, we carefully segment patches that align with the dimensions and positioning of every spot. These segmented patches from a tissue section are then compiled and reshaped into an 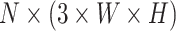 matrix, serving as the primary input for the Vision Transformer. In this context, the number 3 corresponds to the color channels, and
matrix, serving as the primary input for the Vision Transformer. In this context, the number 3 corresponds to the color channels, and 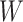 and
and 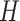 represent the patch’s width and height, respectively. We employ a modifiable layer, denoted as
represent the patch’s width and height, respectively. We employ a modifiable layer, denoted as 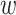 , to modify the histology image features from an
, to modify the histology image features from an 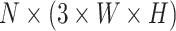 matrix to an
matrix to an 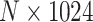 matrix labeled
matrix labeled 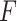 . Another essential input component is the
. Another essential input component is the 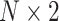 position matrix, which contains the
position matrix, which contains the 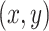 coordinates for each spot in the ST (ST) dataset. The x-coordinate data are converted into a one-hot encoded matrix, termed PP, with dimensions
coordinates for each spot in the ST (ST) dataset. The x-coordinate data are converted into a one-hot encoded matrix, termed PP, with dimensions 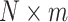 . Here,
. Here, 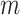 is the maximum count of x-coordinates spanning all tissue sections.
is the maximum count of x-coordinates spanning all tissue sections.
In the pursuit of establishing a robust model for image-gene co-expression representation learning, we introduce a two-step approach utilizing a Transformer model. Initially, a feature vector, 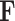 , is constructed, amalgamating histology image features and spatial coordinates, serving as the preliminary input for the subsequent Transformer model. The Transformer, designed to predict gene expression, outputs a representation, denoted as
, is constructed, amalgamating histology image features and spatial coordinates, serving as the preliminary input for the subsequent Transformer model. The Transformer, designed to predict gene expression, outputs a representation, denoted as 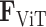 , which is subjected to a reconstruction loss,
, which is subjected to a reconstruction loss, 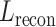 , when compared with the actual spot gene representation,
, when compared with the actual spot gene representation, 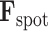 . Mathematically, the reconstruction loss is defined as
. Mathematically, the reconstruction loss is defined as
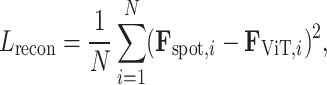 |
(1) |
where 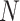 represents the number of spots, aiming to minimize the discrepancy between the predicted and actual gene expression representations. The optimization of the Transformer parameters,
represents the number of spots, aiming to minimize the discrepancy between the predicted and actual gene expression representations. The optimization of the Transformer parameters, 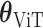 , is conducted by minimizing
, is conducted by minimizing 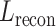 through iterative update rules in the training process, thereby enabling the model to accurately reconstruct the spot gene representation from the input features. The approach not only enables precise forecasting of spatial gene expression but also guarantees that the derived representations,
through iterative update rules in the training process, thereby enabling the model to accurately reconstruct the spot gene representation from the input features. The approach not only enables precise forecasting of spatial gene expression but also guarantees that the derived representations, 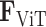 , align with the genuine gene expressions,
, align with the genuine gene expressions, 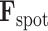 . By combining these,
. By combining these, 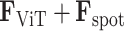 , for each node feature, it offers a thorough and precise framework for examining the tissue’s spatial molecular diversity.
, for each node feature, it offers a thorough and precise framework for examining the tissue’s spatial molecular diversity.
To elucidate the transformer mechanism, the Multihead Attention mechanism in Transformer models is designed to enhance the model’s capability to focus on different positions, or words, in the input sequence simultaneously, thereby capturing various types of information and dependencies from the input. For every attention head, denoted by 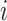 , the mathematical representation of the mechanism is given by
, the mathematical representation of the mechanism is given by
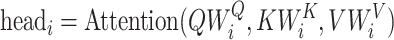 |
(2) |
In this representation, 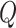 ,
, 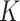 and
and 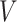 stand for the query, key and value matrices, respectively. The weight matrices for the
stand for the query, key and value matrices, respectively. The weight matrices for the 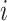 -th head for query, key, and value projections are given by
-th head for query, key, and value projections are given by 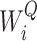 ,
, 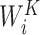 and
and 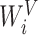 . The function
. The function 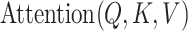 signifies the scaled dot-product attention mechanism.
signifies the scaled dot-product attention mechanism.
The results from each of the heads are merged together and then undergo a linear transformation to yield the ultimate output.
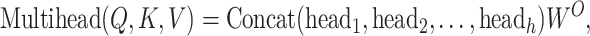 |
(3) |
where 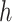 is the number of heads and
is the number of heads and 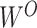 is the final linear transformation weight matrix.
is the final linear transformation weight matrix.
Graph reconstruction for spatial gene expression. TransformerST reconstructs the cell–cell relationship using an undirected graph 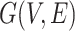 . Each vertex
. Each vertex 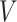 symbolizes the spot, characterized by the output of the Vision Transformer given by
symbolizes the spot, characterized by the output of the Vision Transformer given by 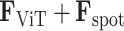 . And the edge
. And the edge 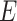 measures the weighted relationships between two vertices. We map each spot back to the histology image and define the corresponding pixel using similar smooth and rescale steps in SpaGCN [30]. The adjacency matrix
measures the weighted relationships between two vertices. We map each spot back to the histology image and define the corresponding pixel using similar smooth and rescale steps in SpaGCN [30]. The adjacency matrix 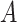 is constructed by calculating the Euclidean distance between vertices using image coordinates. For each spot, the top 20 neighbors are selected to form this matrix.
is constructed by calculating the Euclidean distance between vertices using image coordinates. For each spot, the top 20 neighbors are selected to form this matrix.
Adaptive graph-transformer for spatial embedding. The proposed method utilizes the adaptive graph transformer model to embed the spatial relationship of neighboring spots. The proposed method concatenates the gene expression embedding 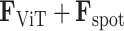 and edge weights to cluster each spot. In the subsequent analysis, the Graph Transformer layer is employed in conjunction with the multi-head attention model to aggregate the features of all nodes. The multi-head attention mechanism takes in three components: the query, key and value. For each edge and for each layer
and edge weights to cluster each spot. In the subsequent analysis, the Graph Transformer layer is employed in conjunction with the multi-head attention model to aggregate the features of all nodes. The multi-head attention mechanism takes in three components: the query, key and value. For each edge and for each layer 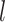 , the multi-head attention is defined as follows:
, the multi-head attention is defined as follows:
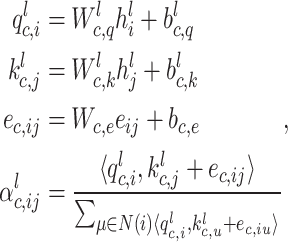 |
(4) |
where 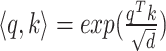 denotes the scaled exponential dot-product. Here,
denotes the scaled exponential dot-product. Here, 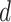 signifies the dimensionality of each head’s hidden state. We use the learnable parameters
signifies the dimensionality of each head’s hidden state. We use the learnable parameters 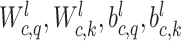 to transform each source feature
to transform each source feature 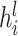 and distant feature
and distant feature 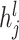 into query vector
into query vector 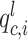 and key vector
and key vector 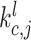 . The additional edge feature
. The additional edge feature 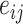 is also added into the key vector
is also added into the key vector 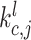 .
.
The message aggregation from 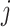 to
to 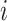 is defined as follows:
is defined as follows:
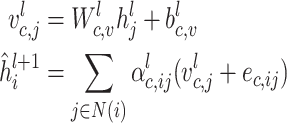 |
(5) |
A gated residual connection between layers is adopted to prevent over-smoothing.
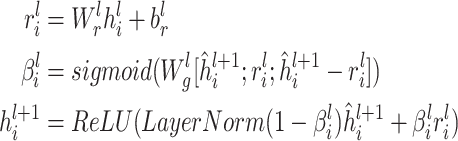 |
(6) |
The output from the final layer is derived by taking the average of the outputs from all the attention heads.
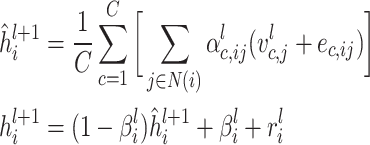 |
(7) |
Adaptive Graph transformer representation learning The previous ST clustering method only considers the spatial information to construct the graph representation. We present an adaptive Graph Transformer model designed to capture both the spatial and feature representations of the entire graph. The model is formulated as follows:
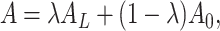 |
(8) |
where 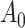 is the initial adjacency matrix, while
is the initial adjacency matrix, while 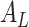 denotes the adjacency matrix that is iteratively learned at each step. The initial adjacency matrix is constructed using the
denotes the adjacency matrix that is iteratively learned at each step. The initial adjacency matrix is constructed using the 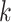 nearest neighborhood using the histology image. The adaptive updating mechanism helps to learn the global and local representation of ST data. The hyperparameter
nearest neighborhood using the histology image. The adaptive updating mechanism helps to learn the global and local representation of ST data. The hyperparameter 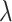 serves to strike a balance between the spatial and feature-based graph structures, ensuring that neither dominates the learning process.
serves to strike a balance between the spatial and feature-based graph structures, ensuring that neither dominates the learning process.
Identifying tissue types with iterative clustering. Based on the outputs of the Graph Transformer encoder, the proposed method iteratively identifies the tissue type in an unsupervised manner. The beginning of our proposed approach draws inspiration from Louvain’s technique. The clustering process is segmented into two distinct stages. In the preliminary stage, we designate a soft cluster category, denoted as 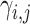 , to every spot embedding represented by
, to every spot embedding represented by 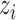 in the manner described below:
in the manner described below:
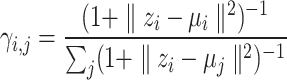 |
(9) |
Subsequently, we fine-tune the clusters using an auxiliary target distribution, denoted as 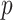 , which is derived from
, which is derived from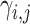
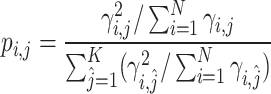 |
(10) |
Similar to the previous iterative clustering algorithm in scRNA-seq analysis, the loss function is formulated using the Kullback–Leibler (KL) divergence.
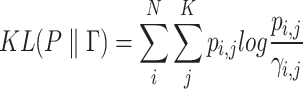 |
(11) |
Reconstructing the super-resolved gene expression at the sub-spot resolution. In order to explore the tissue sub-environment at the enhanced resolution, we partition each spot to a single-cell resolution, leveraging the associated histological image for guidance. If the histology is missing in real-time applications, we adopt the setting of BayesSpace [28], each ST spot is divided into nine smaller subspots, while each Visium data spot is split into six subspots. Given that the diameter of ST spots is 100 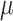 m and that of Visium spots is 55
m and that of Visium spots is 55 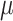 m, TransformerST is capable of attaining gene expression at a single-cell resolution, as opposed to the traditional approach that amalgamates data from dozens of cells. The proposed super-resolved reconstruction components are divided into two steps, histology image super-resolution, and spatial gene expression reconstruction.
m, TransformerST is capable of attaining gene expression at a single-cell resolution, as opposed to the traditional approach that amalgamates data from dozens of cells. The proposed super-resolved reconstruction components are divided into two steps, histology image super-resolution, and spatial gene expression reconstruction.
In the formulation of the internal cross-scale super-resolution model, we commence with a preprocessing phase on the histology image, extracting image patches—termed ‘spot-centric patches’—based on each spot location, ensuring a unique, non-overlapping patch is associated with each spot. Subsequently, for every spot region, we extract patches with increased density, producing overlapping image patches, dubbed ‘sliding-window patches’. The model aims to predict gene expression at a single-cell resolution. We model the internal cross-scale relationship between each sliding-window image patch at the original spot resolution and its corresponding spot-centric patch neighbors, forming a graph. In this graph, each sliding-window image patch becomes a vertex, and the edge signifies the weighted connection between the spot-centric patch and the sliding-window image patch. The proposed method unfolds in two segments: graph construction and patch aggregation. Employing the mapping function, we can identify the 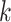 nearest neighboring spot-centric image patches. Consequently, the reconstructed graph yields
nearest neighboring spot-centric image patches. Consequently, the reconstructed graph yields 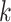 spot mapping pairs of spot-centric and sliding-window patches. Following this, we utilize the patch aggregation model to amalgamate
spot mapping pairs of spot-centric and sliding-window patches. Following this, we utilize the patch aggregation model to amalgamate 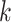 spot-centric patches, conditioned on the similarity distance. With the patch aggregation model, we introduce learnable weights for the
spot-centric patches, conditioned on the similarity distance. With the patch aggregation model, we introduce learnable weights for the 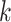 nearest neighbors, enabling us to use the weights and
nearest neighbors, enabling us to use the weights and 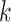 spot-centric image patches to estimate the gene expression at the center of each sliding-window segment. Given the limitations of current ST technology, obtaining ground truth data at the enhanced resolution is challenging. We hypothesize that the spatial gene expression at the spot resolution represents the averaged mixture of its corresponding single-cell segments. Instead of directly calculating the reconstruction loss at the enhanced resolution, we average the single-cell components into a spot to steer the training process.
spot-centric image patches to estimate the gene expression at the center of each sliding-window segment. Given the limitations of current ST technology, obtaining ground truth data at the enhanced resolution is challenging. We hypothesize that the spatial gene expression at the spot resolution represents the averaged mixture of its corresponding single-cell segments. Instead of directly calculating the reconstruction loss at the enhanced resolution, we average the single-cell components into a spot to steer the training process.
Graph reconstruction. In our approach, we initiate by extracting two specific types of image patches: spot−centric patches 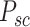 and sliding−window patches
and sliding−window patches 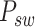 , denoted mathematically as
, denoted mathematically as
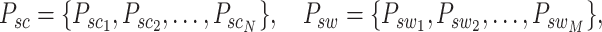 |
(12) |
where each patch is a 3D matrix of dimensions 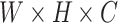 , representing the width, height and the number of channels (typically 3 for RGB images), respectively. After the extraction process, we utilize a Vision Transformer as described in Equation 1 to derive the embedded features of the patches. These features, representing both spot-centric and sliding-window patches, are captured in dimensions
, representing the width, height and the number of channels (typically 3 for RGB images), respectively. After the extraction process, we utilize a Vision Transformer as described in Equation 1 to derive the embedded features of the patches. These features, representing both spot-centric and sliding-window patches, are captured in dimensions 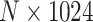 , where
, where 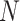 signifies the total number of patches:
signifies the total number of patches:
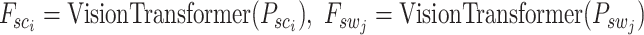 |
(13) |
Subsequently, we explore the internal cross-scale relationship between sliding-window patches 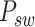 and their corresponding spot-centric patches
and their corresponding spot-centric patches 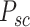 by constructing a graph. Each vertex in this graph represents a sliding-window patch, and edges represent the weighted connections to its
by constructing a graph. Each vertex in this graph represents a sliding-window patch, and edges represent the weighted connections to its 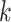 neighboring spot-centric patches. The Euclidean distance, defined as
neighboring spot-centric patches. The Euclidean distance, defined as
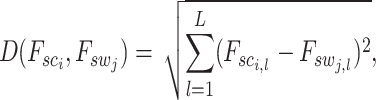 |
(14) |
is utilized to determine these neighbors, where 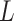 is the length of the embedded feature vectors, and
is the length of the embedded feature vectors, and 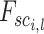 and
and 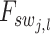 are the
are the 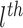 elements of the embedded features
elements of the embedded features 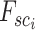 and
and 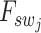 , respectively. The
, respectively. The 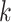 neighboring spot-centric patches for a given sliding-window patch
neighboring spot-centric patches for a given sliding-window patch 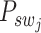 are identified by selecting the
are identified by selecting the 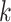 patches
patches 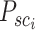 that minimize the Euclidean distance
that minimize the Euclidean distance 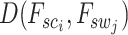 . This methodology facilitates the exploration and modeling of the spatial relationships between different resolution scales in the histological image, providing a foundation for predicting gene expression at a single-cell resolution.
. This methodology facilitates the exploration and modeling of the spatial relationships between different resolution scales in the histological image, providing a foundation for predicting gene expression at a single-cell resolution.
Patch aggregation. We weight the 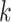 neighboring patches on the similarity distance and aggregate the enhanced gene expression as
neighboring patches on the similarity distance and aggregate the enhanced gene expression as
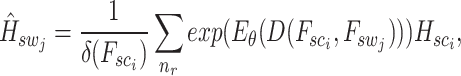 |
(15) |
where 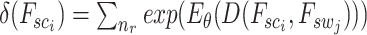 denotes the normalization factor.
denotes the normalization factor. 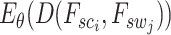 is used to estimate the aggregation weight for each neighboring patch. The output feature for each spot situated at location
is used to estimate the aggregation weight for each neighboring patch. The output feature for each spot situated at location 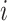 is denoted by
is denoted by 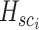 in Equation 7. Additionally,
in Equation 7. Additionally, 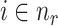 signifies the
signifies the 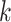 nearest neighbor patches of patch
nearest neighbor patches of patch 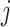 , with
, with 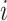 being the central spot of that patch.
being the central spot of that patch. 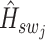 denotes the central feature embedding of the sliding window patch
denotes the central feature embedding of the sliding window patch 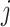 . The term
. The term 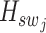 represents the intermediate output from our super-resolution model, which can be utilized for clustering at a single-cell resolution. Furthermore,
represents the intermediate output from our super-resolution model, which can be utilized for clustering at a single-cell resolution. Furthermore, 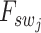 can be viewed as the super-resolved gene expression for each individual cell.
can be viewed as the super-resolved gene expression for each individual cell.
The loss functions for the reconstruction of the Vision Transformer pertaining to spot gene expression, denoted as 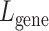 , and for the image patches, denoted as
, and for the image patches, denoted as 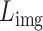 , are formulated as follows:
, are formulated as follows:
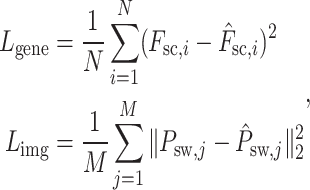 |
(16) |
where 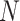 is the number of spots,
is the number of spots, 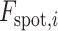 is the actual gene expression of spot
is the actual gene expression of spot 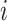 ,
, 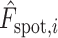 is the predicted gene expression of spot
is the predicted gene expression of spot 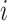 ,
, 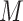 is the number of sliding window image patches,
is the number of sliding window image patches, 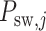 is the original sliding window image patch
is the original sliding window image patch 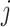 and
and 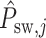 is the reconstructed sliding window image patch
is the reconstructed sliding window image patch 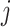 . The total loss,
. The total loss, 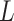 , used to train the model is a combination of these two losses, typically weighted to balance their contributions during training:
, used to train the model is a combination of these two losses, typically weighted to balance their contributions during training:
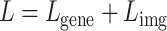 |
(17) |
Key Points
Advanced Model Integration: The TransformerST model employs both graph and vision transformer architectures to synergize histological imagery with spatial gene expression data, facilitating a novel image-gene co-representation not achieved by conventional methods.
Super-Resolution with TransformerST: The TransformerST model’s cross-scale super-resolution feature facilitates the achievement of single-cell resolution in ST data without requiring single-cell references. This enhances the clarity of data from lower resolution methods such as
 Visium.
Visium.TransformerST’s High-Dimensional Gene Expression Reconstruction: TransformerST enhances the computational efficiency of reconstructing original, high-dimensional gene expression patterns, offering both speed and precision that refine the single-cell resolution data analysis beyond the capabilities of traditional PCA-based methods.
Versatile and High-Quality Performance: The TransformerST model has been validated for its robust performance and exceptional accuracy, showcasing adaptability across diverse ST platforms, including STomics and
 Visium.
Visium.
Supplementary Material
ACKNOWLEDGMENTS
This work was partially supported by National Science Foundation IIS 1845666, 1852606, 1838627, 1837956, 1956002, and National Institutes of Health P01AI106684, R01HL137709, P30CA008748, U19AG055373. It was also supported in part by UPMC Children’s Hospital of Pittsburgh, and the University of Pittsburgh Center for Research Computing through the resources provided.
Author Biographies
Chongyue Zhao is a postdoctoral researcher in the Department of Pediatrics at the University of Pittsburgh. His research interests include machine learning, computer vision, biomedical data science, and spatial transcriptomics.
Zhongli Xu is an MD-PhD candidate at Tsinghua University and a Visiting Research Scholar at the University of Pittsburgh. His research focuses on Biostatistics and Immunology.
Xinjun Wang is an Assistant Attending Biostatistician at Memorial Sloan Kettering Cancer Center. His research focuses on single-cell multi-omics data and subgroup analysis with differential treatment effects in cancer research.
Shiyue Tao is a graduate student in the Department of Biostatistics at the University of Pittsburgh. Her research interests include single-cell multi-omics integrative analysis and statistical genetics.
William A. MacDonald is the Assistant Director of the Health Sciences Sequencing Core at UPMC Children's Hospital of Pittsburgh.
Kun He is a postdoctoral fellow at the University of Pittsburgh. He recieved his PhD in Biomedical Engineer from Shanghai Jiao Tong University. His research interest is Immunology.
Amanda C. Poholek is an Assistant Professor at the University of Pittsburgh School of Medicine, affiliated with both the Department of Pediatrics, and the Department of Immunology. Her lab is interested in exploring how tissue-specific environmental factors alter transcriptomes and epigenomes of T cells that control their differentiation in the context of diseases such as Th2 differentiation in allergic asthma and T cell exhaustion in the tumor.
Kong Chen is an Assistant Professor at the Department of Medicine, University of Pittsburgh. His lab focuses on studying memory Th17 responses using mouse models of Klebsiella pneumoniae, P. aeruginosa, and Rhesus macaque models of SIV and Streptococcus pneumoniae.
Heng Huang is the Brendan Iribe Endowed Professor in Computer Science at the University of Maryland College Park. His lab focuses on Machine Learning and Data Science, Bioinformatics and Computational Biology, Computer Vision and Machine Perception, Natural Language Processing, and AI and Robotics.
Wei Chen is a Professor at the University of Pittsburgh with affiliations in Pediatrics, Biostatistics, and Human Genetics. His lab focuses on creating statistical tools for high-throughput genomic data to study complex diseases such as age-related macular degeneration (AMD), childhood asthma, and chronic obstructive pulmonary disease (COPD).
Contributor Information
Chongyue Zhao, Department of Pediatrics, University of Pittsburgh, Pittsburgh, 15224, Pennsylvania, USA.
Zhongli Xu, Department of Pediatrics, University of Pittsburgh, Pittsburgh, 15224, Pennsylvania, USA; School of Medicine, Tsinghua University, Beijing, 100084, Beijing, China.
Xinjun Wang, Department of Epidemiology and Biostatistics, Memorial Sloan Kettering Cancer Center, New York, 10065, New York, USA.
Shiyue Tao, Department of Pediatrics, University of Pittsburgh, Pittsburgh, 15224, Pennsylvania, USA; Department of Biostatistics, University of Pittsburgh, Pittsburgh, 15261, Pennsylvania, USA.
William A MacDonald, Health Sciences Sequencing Core at UPMC Children’s Hospital of Pittsburgh, Department of Pediatrics , University of Pittsburgh, Pittsburgh, 15224, Pennsylvania, USA.
Kun He, Division of Pediatric Rheumatology, Department of Pediatrics , University of Pittsburgh, Pittsburgh, 15224, Pennsylvania, USA.
Amanda C Poholek, Division of Pediatric Rheumatology, Department of Pediatrics , University of Pittsburgh, Pittsburgh, 15224, Pennsylvania, USA; Department of Immunology , University of Pittsburgh, Pittsburgh, 15224, Pennsylvania, USA; Health Sciences Sequencing Core at UPMC Children’s Hospital of Pittsburgh, Department of Pediatrics , University of Pittsburgh, Pittsburgh, 15224, Pennsylvania, USA.
Kong Chen, Department of Medicine, University of Pittsburgh, Pittsburgh, 15213, Pennsylvania, USA.
Heng Huang, Department of Computer Science, University of Maryland, College Park, 20742, Maryland, USA.
Wei Chen, Department of Pediatrics, University of Pittsburgh, Pittsburgh, 15224, Pennsylvania, USA; Department of Biostatistics, University of Pittsburgh, Pittsburgh, 15261, Pennsylvania, USA.
CODE AVAILABILITY
TransformerST is implemented in Python. The source code can be downloaded from the website: https://github.com/Zhaocy-Research/TransformerST.
DATA AVAILABILITY
We use several publicly available data which could be acquired using the following websites or accession numbers: (1) LIBD human dorsolateral prefrontal cortex data (DLPFC) (http://research.libd.org/spatialLIBD/); (2) Melanoma ST data (https://www.spatialresearch.org/wp-content/uploads/2019/03/ST-Melanoma-Datasets_1.zip); (3) Human epidermal growth factor receptor(HER) 2 amplified (HER+) invasive ductal carcinoma (IDC) sample [28]; (4) Our in-house mouse lung data are deposited in Gene Expression Omnibus (GEO) (GSE190225)
References
- 1. Moncada R, Barkley D, Wagner F, et al. Integrating microarray-based spatial transcriptomics and single-cell rna-seq reveals tissue architecture in pancreatic ductal adenocarcinomas. Nat Biotechnol 2020;38(3):333–42. [DOI] [PubMed] [Google Scholar]
- 2. Chen W-T, Lu A, Craessaerts K, et al. Spatial transcriptomics and in situ sequencing to study alzheimer’s disease. Cell 2020;182(4):976–991.e19. [DOI] [PubMed] [Google Scholar]
- 3. Lubeck E, Coskun AF, Zhiyentayev T, et al. Single-cell in situ rna profiling by sequential hybridization. Nat Methods 2014;11(4):360–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shah S, Lubeck E, Zhou W, Cai L. in situ transcription profiling of single cells reveals spatial organization of cells in the mouse hippocampus. Neuron 2016;92(2):342–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Eng C-HL, Lawson M, Zhu Q, et al. Transcriptome-scale super-resolved imaging in tissues by rna seqfish+. Nature 2019;568(7751):235–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Moffitt JR, Bambah-Mukku D, Eichhorn SW, et al. Molecular, spatial, and functional single-cell profiling of the hypothalamic preoptic region. Science 2018;362(6416):eaau5324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chen KH, Boettiger AN, Moffitt JR, et al. Spatially resolved, highly multiplexed rna profiling in single cells. Science 2015;348(6233):aaa6090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wang X, Allen WE, Wright MA, et al. Three-dimensional intact-tissue sequencing of single-cell transcriptional states. Science 2018;361(6400):eaat5691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lee JH, Daugharthy ER, Scheiman J, et al. Highly multiplexed subcellular rna sequencing in situ. Science 2014;343(6177):1360–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ståhl PL, Salmén F, Vickovic S, et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science 2016;353(6294):78–82. [DOI] [PubMed] [Google Scholar]
- 11. Rodriques SG, Stickels RR, Goeva A, et al. Slide-seq: a scalable technology for measuring genome-wide expression at high spatial resolution. Science 2019;363(6434):1463–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Stickels RR, Murray E, Kumar P, et al. Highly sensitive spatial transcriptomics at near-cellular resolution with slide-seqv2. Nat Biotechnol 2021;39(3):313–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Vickovic S, Eraslan G, Salmén F, et al. High-definition spatial transcriptomics for in situ tissue profiling. Nat Methods 2019;16(10):987–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Thrane K, Eriksson H, Maaskola J, et al. Spatially resolved transcriptomics enables dissection of genetic heterogeneity in stage iii cutaneous malignant melanoma. Cancer Res 2018;78(20):5970–9. [DOI] [PubMed] [Google Scholar]
- 15. Berglund E, Maaskola J, Schultz N, et al. Spatial maps of prostate cancer transcriptomes reveal an unexplored landscape of heterogeneity. Nat Commun 2018;9(1):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hao Y, Hao S, Andersen-Nissen E, et al. Integrated analysis of multimodal single-cell data. Cell 2021;184:3573–3587.e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cable DM, Murray E, Zou LS, et al. Robust decomposition of cell type mixtures in spatial transcriptomics. Nat Biotechnol 2021;40(4):517–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Andersson A, Bergenstråhle J, Asp M, et al. Single-cell and spatial transcriptomics enables probabilistic inference of cell type topography. Commun Biol 2020;3(1):565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Elosua-Bayes M, Nieto P, Mereu E, et al. SPOTlight: seeded NMF regression to deconvolute spatial transcriptomics spots with single-cell transcriptomes. Nucleic Acids Res 2021;49(9):e50–0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dong R, Yuan G-C. SpatialDWLS: accurate deconvolution of spatial transcriptomic data. Genome Biol 2021;22(1):145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kleshchevnikov V, Shmatko A, Dann E, et al. Comprehensive mapping of tissue cell architecture via integrated single cell and spatial transcriptomics bioRxiv. 2020:2020–11.
- 22. Kiemen A, Braxton AM, Grahn MP, et al. in situ characterization of the 3d microanatomy of the pancreas and pancreatic cancer at single cell resolution bioRxiv. 2020:2020–12.
- 23. Nguyen QH, Pervolarakis N, Nee K, Kessenbrock K. Experimental considerations for single-cell rna sequencing approaches. Front Cell Dev Biol 2018;6:108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rozenblatt-Rosen O, Stubbington MJ, Regev A, Teichmann SA. The human cell atlas: from vision to reality. Nature 2017;550(7677):451–3. [DOI] [PubMed] [Google Scholar]
- 25. Yao Z, Liu H, Xie F, et al. A transcriptomic and epigenomic cell atlas of the mouse primary motor cortex. Nature 2021;598(7879):103–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Consortium, H . The human body at cellular resolution: the nih human biomolecular atlas program. Nature 2019;574(7777):187–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Haque A, Engel J, Teichmann SA, Lönnberg T. A practical guide to single-cell rna-sequencing for biomedical research and clinical applications. Genome Med 2017;9(1):1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhao E, Stone MR, Ren X, et al. Spatial transcriptomics at subspot resolution with bayesspace. Nat Biotechnol 2021;39(11):1375–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Li J, Chen S, Pan X, et al. Cell clustering for spatial transcriptomics data with graph neural networks. Nat Comput Sci 2022;2(6):399–408. [DOI] [PubMed] [Google Scholar]
- 30. Hu J, Li X, Coleman K, et al. Spagcn: integrating gene expression, spatial location and histology to identify spatial domains and spatially variable genes by graph convolutional network. Nat Methods 2021;18(11):1342–51. [DOI] [PubMed] [Google Scholar]
- 31. Dong K, Zhang S. Deciphering spatial domains from spatially resolved transcriptomics with an adaptive graph attention auto-encoder. Nat Commun 2022;13(1):1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zong Y, Yu T, Wang X, et al. Const: an interpretable multi-modal contrastive learning framework for spatial transcriptomics. bioRxiv 2022–01. 2022. 10.1101/2022.01.14.476408. [DOI]
- 33. Xu C, Jin X, Wei S, et al. Deepst: identifying spatial domains in spatial transcriptomics by deep learning. Nucleic Acids Res 2022;50(22):e131–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pham D, Tan X, Xu J, et al. Robust mapping of spatiotemporal trajectories and cell–cell interactions in healthy and diseased tissues. Nat Commun 2023;14(1):7739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Miller BF, Huang F, Atta L, et al. Reference-free cell type deconvolution of multi-cellular pixel-resolution spatially resolved transcriptomics data. Nat Commun 2022;13(1):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Xie R, Pang K, Bader GD, Wang B. Spatially resolved gene expression prediction from h&e histology images via bi-modal contrastive learning. Advances in Neural Information Processing Systems. 2024;36. [Google Scholar]
- 37. Xiao X, Kong Y, Li R, et al. Transformer with convolution and graph-node co-embedding: an accurate and interpretable vision backbone for predicting gene expressions from local histopathological image. Med Image Anal 2024;91:103040. [DOI] [PubMed] [Google Scholar]
- 38. Shang L, Zhou X. Spatially aware dimension reduction for spatial transcriptomics. Nat Commun 2022;13(1):7203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Martin PC, Kim H, Lövkvist C, et al. Vesalius: high-resolution in silico anatomization of spatial transcriptomic data using image analysis. Mol Syst Biol 2022;18(9):e11080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Dehghan A, Razzaghi P, Abbasi K, Gharaghani S. Tripletmultidti: multimodal representation learning in drug-target interaction prediction with triplet loss function. Expert Syst Appl 2023;232:120754. [Google Scholar]
- 41. Rafiei F, Zeraati H, Abbasi K, et al. Deeptrasynergy: drug combinations using multimodal deep learning with transformers. Bioinformatics 2023;39(8):btad438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Palhamkhani F, Alipour M, Dehnad A, et al. Deepcompoundnet: enhancing compound–protein interaction prediction with multimodal convolutional neural networks. J Biomol Struct Dyn 2023;1–10. 10.1080/07391102.2023.2291829. [DOI] [PubMed] [Google Scholar]
- 43. Andersson A, Bergenstråhle J, Asp M, et al. Single-cell and spatial transcriptomics enables probabilistic inference of cell type topography. Commun Biol 2020;3(1):1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. He B, Bergenstråhle L, Stenbeck L, et al. Integrating spatial gene expression and breast tumour morphology via deep learning. Nat Biomed Eng 2020;4(8):827–34. [DOI] [PubMed] [Google Scholar]
- 45. Chen A, Liao S, Cheng M, et al. Spatiotemporal transcriptomic atlas of mouse organogenesis using dna nanoball-patterned arrays. Cell 2022;185(10):1777–1792.e21. [DOI] [PubMed] [Google Scholar]
- 46. Maynard KR, Collado-Torres L, Weber LM, et al. Transcriptome-scale spatial gene expression in the human dorsolateral prefrontal cortex. Nat Neurosci 2021;24(3):425–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Xu Z, Wang X, Fan L, et al. Integrative analysis of spatial transcriptome with single-cell transcriptome and single-cell epigenome in mouse lungs after immunization. iScience 2022;25(9):104900. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
We use several publicly available data which could be acquired using the following websites or accession numbers: (1) LIBD human dorsolateral prefrontal cortex data (DLPFC) (http://research.libd.org/spatialLIBD/); (2) Melanoma ST data (https://www.spatialresearch.org/wp-content/uploads/2019/03/ST-Melanoma-Datasets_1.zip); (3) Human epidermal growth factor receptor(HER) 2 amplified (HER+) invasive ductal carcinoma (IDC) sample [28]; (4) Our in-house mouse lung data are deposited in Gene Expression Omnibus (GEO) (GSE190225)