

Abstract
Latest advancements in the high-throughput single-cell genome (scDNA) and transcriptome (scRNA) sequencing technologies enabled cell-resolved investigation of tissue clones. However, it remains challenging to cluster and couple single cells for heterogeneous scRNA and scDNA data generated from the same specimen. In this study, we present a computational framework called CCNMF, which employs a novel Coupled-Clone Non-negative Matrix Factorization technique to jointly infer clonal structure for matched scDNA and scRNA data. CCNMF couples multi-omics single cells by linking copy number and gene expression profiles through their general concordance. It successfully resolved the underlying coexisting clones with high correlations between the clonal genome and transcriptome from the same specimen. We validated that CCNMF can achieve high accuracy and robustness using both simulated benchmarks and real-world applications, including an ovarian cancer cell lines mixture, a gastric cancer cell line, and a primary gastric cancer. In summary, CCNMF provides a powerful tool for integrating multi-omics single-cell data, enabling simultaneous resolution of genomic and transcriptomic clonal architecture. This computational framework facilitates the understanding of how cellular gene expression changes in conjunction with clonal genome alternations, shedding light on the cellular genomic difference of subclones that contributes to tumor evolution.
Introduction
Understanding how genomic content changes impact gene expression in individual cells is essential to further understand cell clone development in normal and diseased tissues. In particular, characterizing the clone-wise gene dosage effect, i.e. the sensitivity of cellular gene expression to the copy number profile shared by the group of cells, is critical to elucidate the functional consequence of diseases-associated genomic copy number variants (CNVs), a significant challenge in current structural variant research (1–3).
However, there is no technology available that can efficiently and accurately measure both DNA copy number and gene expression profiles of individual cells simultaneously. Although several technologies (4–6), like scTrio-seq have made an attempt to measure genomic and transcriptomic content of up to a few cells per batch, it remains a low-throughput technique. High-throughput single-cell sequencing technologies that are currently available can only measure either the transcriptome (7–10) or the genome (11–13) content of individual cells, but not both simultaneously.
For example, droplet-based single-cell RNA sequencing (scRNA) technology is routinely employed to measure cellular expression so as to assess the clonal development states of various tissues and cell systems (12,14). Recently, droplet-based single-cell DNA sequencing (scDNA) technology enabled cell-wise and genome-wide measurement of genomic alternations, such as copy number variants, in thousands of cells (12,15). High-quality single-cell copy number variants combined with single-cell gene expression profiles promise to further reveal the clonal heterogeneity in complex tissues and cell systems (16–20). Realizing the potential, however, will require high-fidelity co-clustering of heterogeneous single cells measured by scRNA and scDNA sequencing technologies.
Addressing this difficulty, we developed an efficient computational method – Coupled-Clone Non-negative Matrix Factorization, termed as CCNMF. It reasonably models the shared underlying clonal structure and the general concordance between cellular expression level and copy number states (12,21). CCNMF then employs machine learning algorithms to infer the most likely multi-modal integration solution. CCNMF takes two matrices as inputs: single-cell gene expression matrix obtained through scRNA-seq technology and single-cell copy number matrix obtained from scDNA-seq technology, both derived from the same biological specimen.
CCNMF was established on a model-based approach which couples single cells across scDNA and scRNA data by maximizing their global concordance between gene expression and copy number. CCNMF optimizes an objective function that simultaneously maximizes intra-clone compactness and inter-clone structure coherence, this coupled-clone nonnegative matrix factorization framework followed the co-clustering concept as introduced in (22). A coherent underlying clonal structure, i.e. the identity-linked cell clusters between scDNA and scRNA data, is thus inferred as the weight matrix optimally assigning all cells to their most likely cluster identity. Based on that, CCNMF accurately estimates the dosage effect per gene and cell cluster.
Before CCNMF, only a few methods were available for analyzing the combined scDNA and scRNA data. These methods mostly operate in a map-to-reference mechanism, i.e., data from one technology is mapped to the reference clonal structure derived from another technology (21,23–26). For examples, clonealign, an early attempt of integrative modeling gene dosage effect of DNA copy number and gene expression, statistically assigns scRNA gene expression states to a reference phylogenetic tree representing scDNA-derived clones, in a Bayesian way (21); Seurat, which mainly integrates multiple scRNA datasets, can also project other types of single cell data to the scRNA-derived clusters using the mutual nearest neighbor search (23); DENDRO infers single cell copy numbers from scRNA data and validates the result using the scDNA data (24).
However, these map-to-reference inference methods risk systematic bias because the choice of reference technology was largely arbitrary, and different choices significantly influence downstream analysis. Alternatively, the iNMF method (27) utilized a reference-free integrative nonnegative matrix factorization model to identify the shared cell types, as input scRNA-seq datasets of different samples or species, or of multi-modalities such as scATAC-seq and DNA methylation profiles.
CCNMF takes a data-driven approach, unbiased toward data and technology sources. CCNMF utilizes the coherence of the underlying clonal structure shared within the biological specimen to maximize the inference for true cell clonal identity and gene expression effects. Using both simulated and real cancer datasets, we validated that CCNMF can faithfully recover the underlying clonal structure, accurately identify clonal identity for all single cells, and statistically infer gene-wise dosage and expression changes that differentiate each clone. We applied CCNMF to characterize an ovarian cancer cell mixture, a gastric cancer cell line and a primary gastric cancer, and the results showed CCNMF is capable of identifying clonal structure and dosage effect in real cell systems. Thus, scDNA- and scRNA-seq combined with CCNMF analysis offers a new way to study the functional consequence of clonal gene dosage change and how it contributes to clonal development.
Materials and methods
Coupled factorization of scDNA and scRNA data
We utilized the coupled-clone nonnegative matrix factorization framework to identify the underlying clonal structure of scDNA and scRNA data from the same biological specimen. The input can be any matched scDNA and scRNA data generated by various technologies. Input matrix 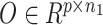 is the copy numbers of p genes and n1 cells from the scDNA; while the input matrix
is the copy numbers of p genes and n1 cells from the scDNA; while the input matrix 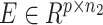 is the gene expression of p genes and n2 cells from the scRNA (Figure 1).
is the gene expression of p genes and n2 cells from the scRNA (Figure 1).
Figure 1.

The workflow of coupled-clone nonnegative matrix factorization (CCNMF). It took scDNA matrix 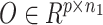 and scRNA matrix
and scRNA matrix 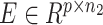 as input for inferring shared clonal structure between two modalities from the same bio-specimen. We applied a coupling matrix A ∈ Rp × p to link matrices
as input for inferring shared clonal structure between two modalities from the same bio-specimen. We applied a coupling matrix A ∈ Rp × p to link matrices 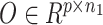 and
and 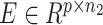 , which represented the global concordance between DNA copy number and gene expression.
, which represented the global concordance between DNA copy number and gene expression.
CCNMF was established on a powerful approach—nonnegative matrix factorization (NMF), which uncovers the latent low-dimensional representation for a given feature-by-sample matrix (28,29). Briefly, NMF factorizes the given feature by sample matrix into two non-negative matrices W and H, where W represents the latent structure of features (i.e. genes and CNVs), while H describes the weight of those features among samples (i.e. cells).
Besides NMF, the most important concept introduced in CCNMF is to couple the nonnegative factorization of matrices O and E. We additionally defined A ∈ Rp × p to represent the linked sensitivity of gene expression to copy number. The matrix A serves as a bridge to enforce the link between changes in copy number and gene expression level for correlated genes. A could be estimated by correlating tissue-specific RNA and DNA sequencing data (22) or by simply providing an identity matrix, as we used in CCNMF so far. The diagonal elements of the identity matrix represent the direct links between copy number and gene expression on the same genes. Hence, we simultaneously co-factorize the single-cell datasets O and E by minimizing the following objective function:
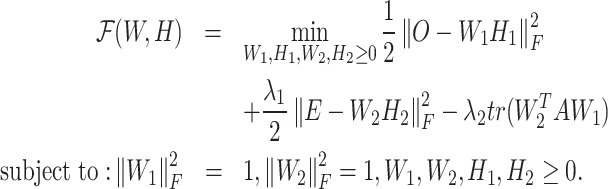 |
(1) |
where we denoted W1 ∈ Rp × k, W2 ∈ Rp × k and 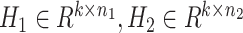 by shorthands W and H, tr() represents the trace of a matrix.
by shorthands W and H, tr() represents the trace of a matrix.
By minimizing the first two terms of the objective function in Equation (1), we ensured the respective NMF decompositions of O and E as O = W1H1 and E = W2H2. Notably, Wi (i = 1, 2) represents the mean matrix of clusters for the ni cells, while Hi is the weight matrices that softly assign ni single cells to the underlying identity linked cell clusters. Upon convergence, the weight matrix Hi provides the inferred cluster identities for all single cells by the maximum weight. Additionally, minimizing the cross term 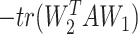 ensured the coherence of the inferred clone structure between the scRNA and scDNA data.
ensured the coherence of the inferred clone structure between the scRNA and scDNA data.
Optimization of the objective function
For optimization, we applied the alternating direction methods of multipliers (ADMM) (30,31) to the objective function Equation (1). Let μ1 and μ2 be the Lagrangian multipliers for W1 and W2, respectively, thus the transformed objective function was written as follows:
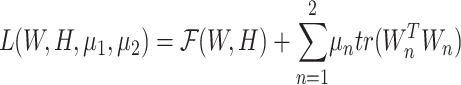 |
(2) |
To solve the transformed objective function, we first obtained required gradients by setting its first order derivatives to zeros. Then, we used the gradient descent algorithm to iteratively update and optimize the objective function until convergence by the following steps (for proof see Supplementary Methods):
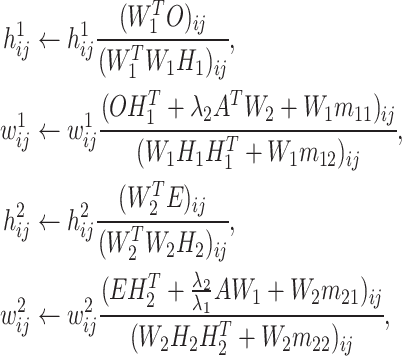 |
(3) |
where
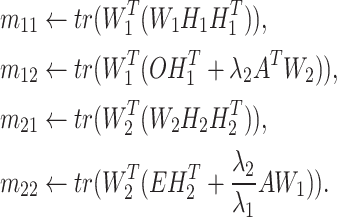 |
(4) |
Model inputs
The model had two hyper-parameters inputs as λ1 and λ2, which were used to initialize the iterative computation. Our experiences were that λ1 and λ2 are data-dependent. Nonetheless, they can be empirically determined by the input data. In practice, we used an automatic balancing strategy to determine the parameters, which ensured the initial values of the four terms of the objective function are within the same order.
The coupling matrix A is also expected as input, for which an identify matrix was supplied. The non-zero diagonal elements represent the strengths of linked copy number and gene expression on the same gene, while the zero non-diagonal elements mean cross-over between copy number and gene expression among genes were ignored. To incorporate a real informative prior for A, one may estimate it from known associations between copy number and gene expression using bulk sequencing data of the same tissue source.
Model selection
When the number of cluster k is unknown, CCNMF runs models with specified range of k. The optimal k was selected as the one minimizing the objective function.
Preprocessing matched scRNA & scDNA data
We preprocessed the scRNA data with several steps, including filtering out outlier genes and cells and normalization for sequencing depth using log transformation. We also performed a chromosome-level smoothing procedure for each cell in which the expression of genes along each chromosome was smoothed by average in a 101-gene window.
It is noted that the scDNA features were segmented into genome bins, while the scRNA features were already presented with genes. To ensure the consistency between scDNA and scRNA features, we associated multiple neighbor genome bins from scDNA with its corresponding gene using the following preprocessing steps. First, we aligned both scRNA and scDNA onto the same human genome assembly (GRCh37 or GRCh38). Then, we identified the one-to-multiple mapping between each gene and genome bins using the IRanges R package (32). Finally, bin-level copy number values of the scDNA matrix were merged into the gene level by taking the average copy number on the mapped bins.
Based on the gene-level single-cell CNV matrix, we developed a novel statistical approach to perform feature selection. The genes with the most highly variable CNVs across tumor cells after excluding replicating cells. In particular, we utilized an Expectation-Maximization (EM) algorithm to model the variances of CNVs for all genes as a mixture of normal distribution, then iteratively selected genes with higher variances as features. The selected genes were considered as potentially informative for ground-truth subclone construction and were used as common features basis for both modalities. Finally, we extracted the properly formed scRNA and scDNA matrices 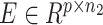 and
and 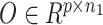 as input to CCNMF (as in Figure 1), where p genes were the selected features from the above process.
as input to CCNMF (as in Figure 1), where p genes were the selected features from the above process.
Simulation procedures
We generated the matched scRNA and scDNA data from the same clonal structure by presetting the ground truth genetic copy number (GCN) changes (as illustrated in Supplementary Figure S1). Notably, the ground truth GCN profile represented a specific clonal structure. First, we specifically set the first clone as normal cells with GCN vector V1 = [2, ⋅⋅⋅, 2] ∈ Rm, where m enumerates over all genome segmental bins. The second tumor clone’s associated GCN vector as V2 ∈ Rm, in which a fraction of V1 was replaced by randomly sampling from {0, 1, 3, 4} with varied probabilities. Similarly, V3 of the third clone was also simulated by the above procedure based on either V1 or V2. Finally, we obtained a ground truth GCN profile V = [V1, V2, V3] as the genomic change profile for the underlying clonal structure.
It was well known that DNA copy number is highly positively correlated with the gene expression levels for most (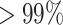 ) of expressed human genes (33). We calculated the ratio of gene-wise mean expression to mean copy number using the bulk RNA and DNA data generated by The Cancer Genome Atlas (TCGA) (https://www.cancer.gov/tcga). To simulate the scDNA data, we estimated their specific parameters and noise from the bulk datasets downloaded from cBioPortal (34,35). Specifically, (i) we estimated the probability transition matrix P(C|G = g) for observed integer copy number when given genetic changes using bulk CNV data, where C is the observed CNV, G is the genetic CNV, g (0, 1, 2, 3, ⋅⋅⋅) is the value of copy number (Supplementary Figure S2); (ii) we simulated copy number for per gene and per cell Dij ∼ multinomial(P(C|G)*P(V)) (Supplementary Figure S3) when given the clonal GCN matrix V; (iii) we then added outlier and dropout events (Supplementary Methods).
) of expressed human genes (33). We calculated the ratio of gene-wise mean expression to mean copy number using the bulk RNA and DNA data generated by The Cancer Genome Atlas (TCGA) (https://www.cancer.gov/tcga). To simulate the scDNA data, we estimated their specific parameters and noise from the bulk datasets downloaded from cBioPortal (34,35). Specifically, (i) we estimated the probability transition matrix P(C|G = g) for observed integer copy number when given genetic changes using bulk CNV data, where C is the observed CNV, G is the genetic CNV, g (0, 1, 2, 3, ⋅⋅⋅) is the value of copy number (Supplementary Figure S2); (ii) we simulated copy number for per gene and per cell Dij ∼ multinomial(P(C|G)*P(V)) (Supplementary Figure S3) when given the clonal GCN matrix V; (iii) we then added outlier and dropout events (Supplementary Methods).
We extended the associated genetic copy number (GCN) profile V to gene × cell matrix in which GCN determines the clone-wise genes expression and copies. We next used the Splatter pipeline (36) to simulate library effects, dropout, outlier events in scRNA-seq data based on the above extended clonal gene-wise matrix (Supplementary Methods). Splatter parameters were estimated from the same tumor tissue with RNA-seq and bulk CNV data downloaded from cBioPortal.
Simulated and real datasets
As the first benchmark, we simulated 46 scDNA- and scRNA-seq datasets. They were referred as the Sim data. The linear and bifurcated clone structure scenarios with 3 clones were simulated in this study. We varied common experimental parameters, such as the percentages of outliers, dropouts, and genome impacted by copy number changes. For each simulated dataset, we randomly generated cell-wise scDNA and scRNA data according to the specified scenario and parameters using the procedure as detailed previously (also see Supplementary Figure S1). Each of the obtained dataset in Sim, has 1000 cells and 2000 genes/CNV bins, and the three composing clones have 200, 400 and 400 cells each. The first clone was designed as normal cells, and the second and third clones represented by deletion and amplification events, respectively. We set the percentages of differentially acquired deletions and amplifications to affect 10–50% chromosome regions. We deposited the Sim data into GitHub.
For the real data benchmark, we obtained a mixture of high grade serous carcinoma (HGSC) cell lines for matched scDNA and scRNA, referred to as the OV data. It was sequenced by DLP scDNA-seq and the 10X Genomics scRNA-seq technologies and was downloaded from European Genome-Phenome archive with accession EGAD00001004553 (21). The mixture cells were made up from ascites (OV2295R) and solid tumors (TOV2295R). The scRNA subset included 1717 cells from ascites (OV2295R) and 4918 cells from solid tumors (TOV2295R), while the scDNA subset had 371 cells from OV2295R and 394 cells from TOV2295R.
As another real data application, we also downloaded the matched scRNA and scDNA data for NCI − N87 gastric cancer cell line from Gene Expression Omnibus (GSE142750) and National Institute of Health’s SRA (PRJNA498809) (12). We firstly processed the scDNA-seq data using Cellranger-DNA pipeline with reference genome GRCh38 according to described procedures in (12). The scDNA data includes 1005 single cells, each contains 154 423 copy number bins across the whole genome. The scRNA-seq data has 3246 cells with 135 13 genes per cell.
Finally, we also collected a primary gastric adenocarcinoma patient data, called P5931 (37), which was sequenced using 10× Genomics technologies for both scRNA and scDNA. The scDNA consists of 796 cells with 154 423 copy number bins across the whole genome. The scRNA of P5931 has in total 11 217 cells with 19 129 genes per cell. The data was downloaded from dbGAP repositories with accession number phs001711 (37).
Performance evaluations
To evaluate the performance of CCNMF given the ground truth cell cluster labels, we used the Adjusted Rand Index (ARI) (38,39). The ARI measures the similarity between the labels assigned by any two clustering schemes as follows:
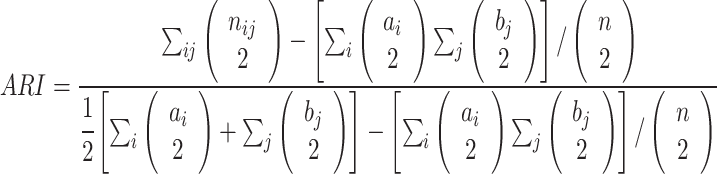 |
(5) |
where nij, ai, bj are values from the two-way contingency table describing the overlapping label counts between the two clustering schemes i and j. Here, nij is the number of overlapping label counts between the cluster i of the first scheme and the cluster j of the second scheme. Note ai = ∑jnij, and bj = ∑inij.
Results
The CCNMF toolkit
The CCNMF analytical framework has been implemented as an R package, with a workflow illustrated in Figure 1. CCNMF toolkit can accept scRNA and scDNA data in standard formats, including those generated by 10× Genomics scRNA-/scDNA-seq and DLP scDNA technologies. The toolkit executes the statistical framework and analytical steps as follows:
Aligns scRNA genes and scDNA genome bins to the same genome reference.
One-to-multiple mapping of scDNA bins to genes using location overlapping (>1 bp) to reduce the scDNA data to the same gene coordinates as the scRNA data.
Initializes the coupled term between scRNA (E) and scDNA data (O) using a coupling matrix (A), which is either an identity matrix or a user provided matrix with prior information.
Iteratively optimizes the objective function using a gradient descent algorithm until convergence.
Identifies the most coherent clonal structure by finding the maximum weights of the H matrices that represent the most likely cell clonality membership.
The outputs of CCNMF include: W matrices that represent the expression or copy number profile centroids of scRNA or scDNA clones; H matrices that represent the cell-wise membership weights toward clone for each of scRNA and scDNA cells. The toolkit is platform-independent and works with general R installation. It is made available as an open source software on Github with detailed readme, manual and examples.
CCNMF recovers the underlying clonal structures in simulation
We firstly evaluated the performance of CCNMF using simulated scDNA and scRNA datasets – Sim. The evaluation was based on Adjusted Rand Index (ARI), and the results were presented in Supplementary Tables S1-S3. Sim included two different scenarios, each with three parameters. In each scenario, ARI was assessed by varying the parameter of interest over its range while keeping the others as default. The default parameters for copy number fraction, outlier percentage and dropout percentage were 50%, 0 and 0, respectively.
Supplementary Table S1 showed the simulation results for the linear and bifurcate clonal structure scenarios, with various copy number fractions ranging from 10% to 50%, which was defined as the percentage of genome undergoing copy number changes. As shown in the table, CCNMF was able to recover the exact underlying clonal structure with the highest accuracy (ARI = 1) for all cases under both scenarios, except for one case with an ARI of 98%. With the decreasing of the copy number fraction, the clonal copy number difference becomes smaller, making it harder for CCNMF to resolve the clones accurately. The results demonstrated that such effect is only modest, as with only 10% of genome having copy number difference between the clones, CCNMF was still able to correctly uncover the underlying structure.
Supplementary Table S2 showed the simulation results with varying dropout percentages from 10% to 90%. Dropout percentage was defined as the percentage of cells with zero values for gene expression and copy number. The reason behind dropout could be the limited sensitivity of a technology or the gene was not present or expressed. Dropouts are very common in scRNA and scDNA data because of amplification bias and other random effects. In Sim, a dropout percentage at 10% means that 10% of all simulated gene expression or copy numbers were perturbed to be zeros. As shown in the table, CCNMF achieved high accuracy in recovering the underlying clonal structure for all cases under both scenarios. All resulted ARIs were 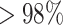 , except for one case with ARI of 81%.
, except for one case with ARI of 81%.
Supplementary Table S3 showed the simulation results for outlier percentage ranging from 10% to 90%. Outlier percentage was defined as the percent of cells with non-realistic copy numbers or expression values. In practice, these data points are typically deemed technical errors and are excluded from downstream analysis. In Sim, outlier percentage stands for the percent of all simulated scDNA and scRNA cells were perturbed to be outliers. It was obvious from the table that CCNMF was robust to the presence of outliers. When the outlier percentage was <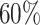 , CCNMF achieved high accuracy with all ARIs greater than
, CCNMF achieved high accuracy with all ARIs greater than 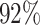 for both scenarios. In summary, our comprehensive simulation study demonstrated that CCNMF achieved good performance for resolving the coherent underlying clonal structures in scDNA and scRNA data with practical noise considerations.
for both scenarios. In summary, our comprehensive simulation study demonstrated that CCNMF achieved good performance for resolving the coherent underlying clonal structures in scDNA and scRNA data with practical noise considerations.
CCNMF detected cell origins for ovarian cancer mixture cell lines OV data
We conducted CCNMF analysis on the OV benchmark dataset consisting of a mixture of ovarian cancer cell lines. It was composed of two cell lines, OV-2295(R) and TOV-2295(R) from the same patient. OV-2295(R) was an ascites site cell line that is abnormal adjacent tissue but not cancerous, while TOV-2295(R) was a high-grade serous ovarian cancer cell line (40). The ground truth for individual cells were obtained from the original publication (40).
CCNMF successfully characterized two subclones in the OV mixture cells lines. The scDNA cells were classified into subclones C1 and C2, which comprised of 394 cells from TOV-2295(R) and 371 cells from OV-2295(R), respectively. Correspondingly, the scRNA cells were categorized into subclones R1 and R2, consisting of 4918 cells from TOV-2295(R) and 1717 cells from OV-2295(R), respectively. Notably, C1 and R1 were from the same cell line TOV-2295(R), while C2 and R2 were from cell line OV-2295(R), indicating corresponding relationship with scDNA subclones with scRNA subclones. The tSNE plots of Figure 2A and B demonstrated the clear separation of the two mixture cells lines by CCNMF. We compared the identified clones with the ground truth, resulting in an adjusted Rand index (ARI) of 1, indicating that CCNMF accurately recovered the underlying clonal structure of OV.
Figure 2.

The coherent clonal structure between scDNA and scRNA of the mixture ovarian cancer cell lines OV. (A) tSNE plot of scDNA clones. (B) tSNE plot of scRNA clones. (C) Heatmap shows CNV changes across scDNA clones. (D) Heatmap shows inferred CNV changes estimated by gene expression level across scRNA clones. (E) Heatmap of Pearson’s Correlation between single cells in coherent clones from scDNA and scRNA. Cells composing the same clone were coded in the same color. Each row represents a single cell and each column represents a genomic region for (C) and (D). The color in each dot of the heatmap represents the CNV status for (C) and (D). In panel (E), each row represents a single cell of scDNA, while each column represents a single cell of scRNA, the color in the heatmap represents the Pearson’s Correlation of cells between scDNA and scRNA.
In this high-grade ovarian cancer, the severe tumor heterogeneity led to a significant number of large CNVs across almost all chromosomes, except for chromosome 13 (Figure 2C). These features were indicators of general aneuploidy. Despite those widespread aneuploidy events, we were still able to identify geographical difference in some focal CNVs between two tumor biopsies/subclones, such as focal amplification events in C1 on chromosomes 1, 2, 3, 5 and 7 (Figure 2C). To explore further consistency between scDNA and scRNA clones, we applied the inferCNV package (41) (https://github.com/broadinstitute/inferCNV) to infer the large-scale CNVs in scRNA clones and visualized clonal pattens with cell identities from CCNMF (Figure 2D). It is important to note that the inferred CNVs were estimated based on the scRNA matrix and can only reflect relative gene expression levels between different clones. As shown in Figure 2D, the inferred CNVs in R1 were consistent with CNVs changes in C1, exhibiting inferred focal amplification events on chromosomes 1, 2, 3, 5 and 7. Despite the aneuploidy across the whole genome, we still observed slightly high correlations in the matched clones of the correlation heatmap (Figure 2E) calculated using Pearson’s correlation between pair-wise cells from scDNA and scRNA. Overall, CCNMF successfully captured the underlying cell line identities of the mixing cells.
CCNMF identified coherent clonal structure in gastric cancer cell line NCI − N87 data
To further investigate the performance of CCNMF, we analyzed the NCI − N87 gastric caner cell line to determine whether the clone structure can be detected by CCNMF. The large-scale scDNA data of NCI − N87 was composed of 1105 single cells. A substantial proportion of replicating cells existed in the scDNA which will cause analytical difficulty for clone reconstruction (12). It is because the most copy number changes in replicating cells were driven by ongoing DNA replication, which overwhelm the true clonal copy number variants.
To address this challenge, we filtered out replicating cells before joint clustering analysis. We identified the replicating cells by calculating the variance of copy number for each cell because the higher replication activity gives rise to a higher variance of observed intra-cell segments. We utilized the Expectation-Maximization algorithm to fit a two-component mixture normal distribution on the obtained copy number variance across all cells (Supplementary Figure S4). The cells were identified as replicating cells if they were assigned to a normal distribution with a larger mean, and were excluded from further analysis. Using this efficient filtering procedure, we successfully identified and removed the group of replicating cells with highly fluctuating copy numbers and retained 724 cells (Supplementary Figure S5).
To increase comparability between scRNA and scDNA, we also filtered out the replicating cells in scRNA by calculating cell cycle scores using the ‘CellCycleScoring’ function in Seurat. Ultimately, we selected 2168 scRNA cells in G0/G1 phase out of a total of 3246 cells and coupled them with 724 scDNA cells in G0/G1 phase.
We utilized CCNMF to analyze single-cell copy number and gene expression matrices with selected and shared genes/features. We successfully identified and characterized three subclones (C1, C2 and C3) in the NCI − N87 cell line for matched scDNA and scRNA, as shown in the tSNE plots (see Figures 3A and 3B). Out of the initial 1005 cells of scDNA, 281 cells were identified as replicating cells and were discarded from downstream clonal reconstruction. The remaining 724 cells were classified into three subclones, with 456, 91 and 177 cells in C1, C2 and C3, respectively. Notably, cells in C1 exhibited a consistent large CNV amplification on the chromosome 19, while cells in C3 shared a focal amplification event on the chromosome 3q arm. C3 involved a smaller proportion of cells without amplifications on chromosomes 3 and 19, and instead, shared lesser amplification events on the chromosome 11 (see Figure 3C). As an independent validation, our analysis successfully resolved the two major subclones and one minor subclones reported by an independent study (12), with C1 and C3 corresponding to the two major subclones and C2 corresponding the minor one.
Figure 3.

The coherent clonal structure between scDNA and scRNA of the gastric cancer cell line NCI-N87. (A) tSNE plot of scDNA clones. (B) tSNE plot of scRNA clones. (C) Heatmap shows CNV changes across scDNA clones. (D) Heatmap shows inferred CNV changes estimated by gene expression level across scRNA clones. (E) Heatmap of Pearson’s Correlation between single cells in coherent clones from scDNA and scRNA.
We identified three subclones (R1, R2 and R3) from 2168 scRNA cells, with 1337, 128 and 703 cells in each, respectively. To estimate the clonal large-scale CNVs, we applied the inferCNV package on gene expression with clone identities. We then visualized the inferred CNVs changes among the clones of scRNA by heatmap (Figure 3D). Notably, the three scRNA subclones (R1, R2 and R3) corresponded to the scDNA clones (C1, C2 and C3). The inferred CNVs in R1 were consistent with C1, which shared a amplification event on chromosome 20. The focal amplification event on chromosome 3q arm was shared by R3 and C3. The cells of R2 shared lesser amplification events on chromosome 11, similar to C2. It is worth noting that the amplification-like events on chromosome 1 and 4 observed in R3 represented the relatively higher gene expressions than R1 and R2, which were potentially diploid cells in C2. We calculated Pearson’s Correlation between pair-wise cells from scDNA and scRNA. It depicted a clear block pattern where cells in matched clones between two modalities had high correlations than cells in different clones, due to the clone-wise gene dosage (Figure 3E).
CCNMF characterized cohort clone structure for primary gastric cancer P5931
Besides analyzing tumor cell lines, we performed CCNMF on real-world application for primary gastric adenocarcinoma. The scRNA and scDNA datasets were derived from a primary gastric cancer patient P5931 (37). Primary tumors usually had higher degree of intracellular heterogeneity and more complex tumor microenvironment than cancer cell lines. Notably, after excluding 119 replicating cells and 376 normal cells (Supplementary Figure S6), a total of 301 cells in G0/G1 phase were retained of P5931 scDNA. Additionally, we identified a total of 2034 tumor epithelial cells in G0/G1 phase using the matched scRNA (37). Then we applied the CCNMF to find the underlying relative clone structure between 301 tumor G0/G1 cells in scDNA and 2034 tumor epithelial G0/G1 cells in scRNA for P5931.
Due to the heterogeneity in primary cancer specimen, there was no ground truth available of cell clone identities. Therefore, we interpreted the clonal structure based on the result of CCNMF. As shown in Figures 4A and B, two clones were overlaid with cell identities on tSNE plots for scDNA and scRNA. Subclones C1 and C2 contained 176 and 125 cells, respectively, were clearly separated in scDNA. The CNV clonal structure of P5931 was dominated by the significant copy number alternations affecting chromosomes 7 and 21 (Figure 4C). The somatic events shared by cells of C1 were chromosome 8 and 21 amplifications. Clearly, the chromosome 7 amplification and chromosome 21 deletion were the major defining somatic events that separated C2 from C1.
Figure 4.

The coherent clonal structure between scDNA and scRNA of the primary gastric cancer patient P5931. (A) tSNE plot of scDNA clones. (B) tSNE plot of scRNA clones. (C) Heatmap shows CNV changes across scDNA clones. (D) Heatmap shows inferred CNV changes estimated by gene expression level across scRNA clones. (E) Heatmap of Pearson’s Correlation between single cells in coherent clones from scDNA and scRNA.
Corresponding to the C1 and C2 subclones, CCNMF analysis detected two subclones R1 and R2 of the scRNA data. Subclone R1 was composed of 1050 cells, while subclone R2 consisted of 984 cells. We visualized the underlying large-scale CNV pattern of scRNA clones with cell identities from CCNMF (Figure 4D). Here, we applied the inferCNV package to infer the large-scale CNV on the clones of scRNA. The inferred CNV were estimated based on the relative gene expression levels among different clusters. In Figure 4D, the two clones R1 and R2 of scRNA correspond with C1 and C2 of scDNA, respectively. Here, the inferred CNVs in R1 were consistent with C1, which shared chromosome 8 and 20 amplifications. Also chromosome 7 amplification and chromosome 21 deletion were shared by R2 and C2. The Pearson’s Correlation was calculated between pair-wise cells between scDNA and scRNA. The correlation heatmap demonstrated that the cells in matched clones between the two modalities had high correlations due to the clone-wise gene dosage effect associated with CNV (Figure 4E).
Convergence and stability of CCNMF integration
We applied the ADMM to optimize the non-convex quadratic objective function of CCNMF as shown in Equation (1). The convergence of CCNMF was ensured by setting stringent criteria on the residual error, stopping the iterations once the residual error is smaller than 0.01. To demonstrate the convergence of CCNMF, we conducted numerical experiments, running CCNMF 10 times for each dataset. As shown in Supplementary Figures S7–S9, the objective function across 10 runs converges to a consistent minimum. CCNMF showed convergence in all three real datasets OV, NCI-N87 and P5931.
Furthermore, we assessed the stability of CCNMF’s results across the 10 runs for each dataset. The ARIs of scDNA clusters generated by CCNMF demonstrated perfect consistency with a value of 1 for the OV data, as well as the scRNA clusters. For NCI-N87 and P5931, the range of ARIs across 10 runs for both scDNA and scRNA was between 0.7 and 0.95 (Supplementary Figure S10). The high ARIs of different runs demonstrated the robustness and self-consistency of CCNMF’s outcomes.
Additionally, we evaluated the resolved subclones of scDNA using CCNMF by comparing them with the subclones from individual scDNA via NMF. We assumed that the subclones inferred from individual scDNA by NMF as the ground truth. CCNMF consistently reproduced the ground truth subclonal structures, validating its efficacy when integrated with scRNA (Supplementary Figure S11). Across the real datasets, the ARI was 1 for OV, 0.93 for NCI-N87 and 0.77 for P5931. The high ARIs suggest that CCNMF successfully resolved the underlying subclones of scDNA, further affirming its integration capability with scRNA.
CCNMF outperforms state-of-the-art methods in subclone integration
We benchmarked CCNMF with existing state-of-the-art single-cell integration methods, including iNMF (27), Seurat (23) and Clonealign (21), utilizing all three real datasets. iNMF and Seurat are widely used for single-cell multi-omics integration, such as scATAT-seq and scRNA-seq data. Meanwhile, Clonealign is an early method specifically designed to uncover the underlying clonal structure between scDNA-seq and scRNA-seq data. iNMF, operating within the NMF framework, is a reference-free integrative algorithm. Seurat and Clonealign are map-to-reference inference integration algorithms, where scRNA-seq cells are assigned to the constructed scDNA-seq subclones. In our comparison, we utilized NMF on individual scDNA data to construct subclones, which served as the reference input for Seurat and Clonealign. The default parameter settings were applied for across all three methods.
For the OV data, iNMF identified 7 scDNA clusters and 18 scRNA clusters, which over-clustered than ground truth of two cell lines. Both Seurat and Clonealign assigned the cells of scRNA to the referenced scDNA-seq subclones, which are consistent with the results obtained from CCNMF (Supplementary Figure S12). As the results of NCI-N87 data, while iNMF resolved the subclones of scDNA, it could not link them to the scRNA clusters due to over-clustering. Seurat was unable to separate C1 and C2 in scRNA, and Clonealign did not detect the minor subclone of scRNA (Supplementary Figure S13). With the P5931 dataset, iNMF recovered scDNA subclones of P5931, but it again could not link scRNA cells to the clonal structure. Seurat assigned all scRNA cells into only one cluster, which obviously failed to separate two subclones distinguished by CNVs on chromosome 7. Clonealign assigned half of the scRNA cells into two subclones only after filtering out approximately 1000 cells (Supplementary Figure S14). Overall, these findings strongly indicate that CCNMF generally outperforms other competitive methods in integrating matched scDNA and scRNA data for clonal analysis.
Discussion
Single-cell multi-omics technology enables the identification of cellular and genomic characteristics of cancer cells. The advancements in scDNA-seq technology allow for characterizing tumor subclone architectures by providing genomic DNA variation such as CNVs. Importantly, each subclone has distinct genomic alternations and cellular properties. Understanding the biological features and phenotypes of subclones is essential for precision cancer treatment. However, scDNA-seq data cannot be directly used to identify phenotypes. Single-cell RNA-seq provides gene expression informations that elucidates the biology of individual cells, but it is noisy to define specific cancer subclones. Inferring CNVs from scRNA-seq remains as a challenging area for obtaining biological insights of subclones (20,41,42). The inferred CNVs are based on changes in read depth or gene expression across the genome, and only provide limited reliability of copy number information of individual cells. Therefore, the matched scDNA-seq data is still needed as a ground truth of genome changes to study clonal biology such as clonal dosage effect.
It is important to note that currently, there is a lack of technologies that are capable of simultaneously measuring copy number variants and gene expression of the same cell with high throughput. Although G&T-seq (4), DR-seq (5) and scTrio-seq (6) can measure genome and transcriptome in up to a few cells per batch, they are in low throughput manner. High throughput scRNA-seq can only measure transcriptome, and scDNA-seq is only for genome content, but not for both modalities in a single cell. To overcome these limitations, the integration of matched scDNA-seq and scRNA-seq of the same specimen is a promising approach.
To facilitate the understanding of tumor clonal structure and the associated biological characteristics like clonal-wise gene dosage effect, we proposed a joint-clustering approach CCNMF for the integration of matched scRNA- and scDNA-seq data from the same specimen. CCNMF optimizes an objective function that simultaneously maximizes for intra-technology clonal compactness, inter-technology clonal coherence and expected dosage effect consistence. We demonstrated the utility of CCNMF by achieving high accuracy for resolving coherent clonal structure in simulation datasets with various noise levels. In real-world applications, CCNMF has been validated by identifying the underlying clonal structure in mixtures of ovarian cancer cell lines, a gastric caner cell line and a primary gastric cancer sample. The consistent CNV change patterns revealed with corresponding subclones between scDNA and scRNA were observed in results of CCNMF.
In summary, CCNMF uncoveres the coherent clonal architecture from the matched tumor single-cell genome and transcriptome data. The integration analysis showcased heterogeneous and clonal genetic nature of pathological tissues, which provides crucial dosage effect information for elucidating genetic cause and etiology of diseases. Furthermore, CCNMF can serve as a bioinformatics tool for performing single-cell level clonal dosage effect analysis for the community.
Data and code availability
The mixture of high grade serous carcinoma cell lines, OV data for DLP scDNA-seq and 10X Genomics scRNA-seq was downloaded from European Genome-Phenome archive with accession EGAD00001004553. The scRNA and scDNA data for NCI − N87 gastric cancer cell line were downloaded from Gene Expression Omnibus (GSE142750) and National Institute of Health’s SRA (PRJNA498809). The primary gastric adenocarcinoma patient P5931 data with 10X Genomics scRNA and scDNA was downloaded from dbGAP repositories with accession numbers phs001711. The R package of CCNMF is available at https://github.com/labxscut/CCNMF and https://doi.org/10.5281/zenodo.10570125.
Supplementary data
Supplementary Data are available at NARGAB Online.
Supplementary Material
Acknowledgements
Xiangqi Bai gratefully acknowledges the financial support from China Scholarship Council [201904910816].
Contributor Information
Xiangqi Bai, Division of Oncology, Department of Medicine, Stanford University School of Medicine, Stanford, CA 94305, USA.
Zhana Duren, Center for Human Genetics and Department of Genetics and Biochemistry, Clemson University, Greenwood, SC 29646, USA.
Lin Wan, NCMIS, LSC, Academy of Mathematics and Systems Science, Chinese Academy of Sciences, Beijing 100190, China; University of Chinese Academy of Sciences, Beijing 100049, China.
Li C Xia, Department of Statistics and Financial Mathematics, School of Mathematics, South China University of Technology, Guangzhou, 510006, China.
Funding
L.W. was supported by the National Key Research and Development Program of China [2022YFA1004801]; National Natural Science Foundation of China [12071466]; L.C.X. was supported by GuangDong Basic and Applied Basic Research Foundation [2022A1515-011426].
Conflict of interest statement. None declared.
References
- 1. Xia L.C., Bell J.M., Wood-Bouwens C., Chen J.J., Zhang N.R., Ji H.P. Identification of large rearrangements in cancer genomes with barcode linked reads. Nucleic Acids Res. 2018; 46:e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Xia L.C., Sakshuwong S., Hopmans E., Bell J., Grimes S., Siegmund D., Ji H., Zhang N. A genome-wide approach for detecting novel insertion-deletion variants of mid-range size. Nucleic Acids Res. 2016; 44:e126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Xia L.C., Ai D., Lee H., Andor N., Li C., Zhang N.R., Ji H.P. SVEngine: an efficient and versatile simulator of genome structural variations with features of cancer clonal evolution. GigaScience. 2018; 7:giy081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Macaulay I.C., Haerty W., Kumar P., Li Y.I., Hu T.X., Teng M.J., Goolam M., Saurat N., Coupland P., Shirley L.M. et al. G&T-seq: parallel sequencing of single-cell genomes and transcriptomes. Nat. Methods. 2015; 12:519–522. [DOI] [PubMed] [Google Scholar]
- 5. Dey S.S., Kester L., Spanjaard B., Bienko M., van Oudenaarden A. Integrated genome and transcriptome sequencing of the same cell. Nat. Biotechnol. 2015; 33:285–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hou Y., Guo H., Cao C., Li X., Hu B., Zhu P., Wu X., Wen L., Tang F., Huang Y. et al. Single-cell triple omics sequencing reveals genetic, epigenetic, and transcriptomic heterogeneity in hepatocellular carcinomas. Cell Res. 2016; 26:304–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tang F., Barbacioru C., Wang Y., Nordman E., Lee C., Xu N., Wang X., Bodeau J., Tuch B.B., Siddiqui A. mRNA-Seq whole-transcriptome analysis of a single cell. Nat. Methods. 2009; 6:377–382. [DOI] [PubMed] [Google Scholar]
- 8. Zheng G. X.Y., Terry J.M., Belgrader P., Ryvkin P., Bent Z.W., Wilson R., Ziraldo S.B., Wheeler T.D., McDermott G.P., Zhu J. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 2017; 8:14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Macosko E.Z., Basu A., Satija R., Nemesh J., Shekhar K., Goldman M., Tirosh I., Bialas A.R., Kamitaki N., Martersteck E.M. et al. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell. 2015; 161:1202–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Campbell J.N., Macosko E.Z., Fenselau H., Pers T.H., Lyubetskaya A., Tenen D., Goldman M., Verstegen A. M.J., Resch J.M., McCarroll S.A. et al. A molecular census of arcuate hypothalamus and median eminence cell types. Nat. Neurosci. 2017; 20:484–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zahn H., Steif A., Laks E., Eirew P., VanInsberghe M., Shah S.P., Aparicio S., Hansen C.L. Scalable whole-genome single-cell library preparation without preamplification. Nat. Methods. 2017; 14:167–173. [DOI] [PubMed] [Google Scholar]
- 12. Andor N., Lau B.T., Catalanotti C., Sathe A., Kubit M., Chen J., Blaj C., Cherry A., Bangs C.D., Grimes S.M. et al. Joint single cell DNA-seq and RNA-seq of gastric cancer cell lines reveals rules of in vitro evolution. NAR Genom. Bioinform. 2020; 2:lqaa016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Efremova M., Teichmann S.A. Computational methods for single-cell omics across modalities. Nat. Methods. 2020; 17:14–17. [DOI] [PubMed] [Google Scholar]
- 14. McCarthy D.J., Rostom R., Huang Y., Kunz D.J., Danecek P., Bonder M.J., Hagai T., Lyu R., Kilpinen H., Goncalves A. et al. Cardelino: computational integration of somatic clonal substructure and single-cell transcriptomes. Nat. Methods. 2020; 17:414–421. [DOI] [PubMed] [Google Scholar]
- 15. Velazquez-Villarreal E.I., Maheshwari S., Sorenson J., Fiddes I.T., Kumar V., Yin Y., Webb M.G., Catalanotti C., Grigorova M., Edwards P.A. et al. Single-cell sequencing of genomic DNA resolves sub-clonal heterogeneity in a melanoma cell line. Commun. Biol. 2020; 3:318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang Y., Waters J., Leung M.L., Unruh A., Roh W., Shi X., Chen K., Scheet P., Vattathil S., Liang H. et al. Clonal evolution in breast cancer revealed by single nucleus genome sequencing. Nature. 2014; 512:155–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Markowska M., Cakała T., Miasojedow B., Aybey B., Juraeva D., Mazur J., Ross E., Staub E., Szczurek E. CONET: copy number event tree model of evolutionary tumor history for single-cell data. Genome Biol. 2022; 23:128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zaccaria S., Raphael B.J. Characterizing allele- and haplotype-specific copy numbers in single cells with CHISEL. Nat. Biotechnol. 2021; 39:207–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wu C.Y., Lau B.T., Kim H.S., Sathe A., Grimes S.M., Ji H.P., Zhang N.R. Integrative single-cell analysis of allele-specific copy number alterations and chromatin accessibility in cancer. Nat. Biotechnol. 2021; 39:1259–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wu C.Y., Sathe A., Rong J., Hess P.R., Lau B.T., Grimes S.M., Ji H.P., Zhang N.R. Cancer subclone detection based on DNA copy number in single cell and spatial omic sequencing data. 2022; bioRxiv doi:08 July 2022, preprint: not peer reviewed 10.1101/2022.07.05.498882. [DOI]
- 21. Campbell K.R., Steif A., Laks E., Zahn H., Lai D., McPherson A., Farahani H., Kabeer F., O’Flanagan C., Biele J. et al. clonealign: statistical integration of independent single-cell RNA and DNA sequencing data from human cancers. Genome Biol. 2019; 20:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Duren Z., Chen X., Jiang R., Wang Y., Wong W.H. Modeling gene regulation from paired expression and chromatin accessibility data. Proc. Natl. Acad. Sci. U.S.A. 2017; 114:E4914–E4923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W.M., Hao Y., Stoeckius M., Smibert P., Satija R. Comprehensive integration of single-cell data. Cell. 2019; 177:1888–1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhou Z., Xu B., Minn A., Zhang N.R. DENDRO: genetic heterogeneity profiling and subclone detection by single-cell RNA sequencing. Genome Biol. 2020; 21:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Milite S., Bergamin R., Patruno L., Calonaci N., Caravagna G. A Bayesian method to cluster single-cell RNA sequencing data using copy number alterations. Bioinformatics. 2022; 38:2512–2518. [DOI] [PubMed] [Google Scholar]
- 26. Edrisi M., Huang X., Ogilvie H.A., Nakhleh L. Accurate integration of single-cell DNA and RNA for analyzing intratumor heterogeneity using MaCroDNA. Nat. Commun. 2023; 14:8262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Welch J.D., Kozareva V., Ferreira A., Vanderburg C., Martin C., Macosko E.Z. Single-cell multi-omic integration compares and contrasts features of brain cell identity. Cell. 2019; 177:1873–1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lee D.D., Seung H.S. Learning the parts of objects by non-negative matrix factorization. Nature. 1999; 401:788–791. [DOI] [PubMed] [Google Scholar]
- 29. Lee D., Seung H.S. Algorithms for non-negative matrix factorization. Advances in Neural Information Processing Systems. 2000; 13:MIT Press. [Google Scholar]
- 30. Boyd S., Parikh N., Chu E., Peleato B., Eckstein J. Distributed optimization and statistical learning via the alternating direction method of multipliers. Found. Trends Mach. Learn. 2010; 3:1–122. [Google Scholar]
- 31. Duren Z., Chen X., Zamanighomi M., Zeng W., Satpathy A.T., Chang H.Y., Wang Y., Wong W.H. Integrative analysis of single-cell genomics data by coupled nonnegative matrix factorizations. Proc. Natl. Acad. Sci. U.S.A. 2018; 115:7723–7728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lawrence M., Huber W., Pagès H., Aboyoun P., Carlson M., Gentleman R., Morgan M., Carey V. Software for Computing and Annotating Genomic Ranges. PLoS Computat. Biol. 2013; 9:e1003118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fehrmann R.S.N., Karjalainen J.M., Krajewska M., Westra H.-J., Maloney D., Simeonov A., Pers T.H., Hirschhorn J.N., Jansen R.C., Schultes E.A. et al. Gene expression analysis identifies global gene dosage sensitivity in cancer. Nat. Genet. 2015; 47:115. [DOI] [PubMed] [Google Scholar]
- 34. Cerami E., Gao J., Dogrusoz U., Gross B.E., Sumer S.O., Aksoy B.A., Jacobsen A., Byrne C.J., Heuer M.L. et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012; 2:401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gao J., Aksoy B.A., Dogrusoz U., Dresdner G., Gross B., Sumer S.O., Sun Y., Jacobsen A., Sinha R., Larsson E. et al. Integrative analysis of complex cancer genomics and clinical profiles using the cbioportal. Sci. Signal. 2013; 6:pl1–pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zappia L., Phipson B., Oshlack A. Splatter: simulation of single-cell RNA sequencing data. Genome Biol. 2017; 18:174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bai X., Lau B., Grimes S.M., Sathe A., Ji H.P. Single cell multi-omic mapping of subclonal architecture and pathway phenotype in primary gastric and metastatic colon cancers. 2022; bioRxiv doi:04 July 2022, preprint: not peer reviewed 10.1101/2022.07.03.498616. [DOI]
- 38. Rand W.M. Objective criteria for the evaluation of clustering methods. Publ. Am. Stat. Assoc. 1971; 66:846–850. [Google Scholar]
- 39. Kiselev V.Y., Kirschner K., Schaub M.T., Andrews T., Yiu A., Chandra T., Natarajan K.N., Reik W., Barahona M., Green A.R. SC3: consensus clustering of single-cell RNA-seq data. Nat. Methods. 2017; 14:483–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Letourneau I.J., Quinn M.C., Wang L.L., Portelance L., Caceres K.Y., Cyr L., Delvoye N., Meunier L., de Ladurantaye M., Shen Z., et al. Derivation and characterization of matched cell lines from primary and recurrent serous ovarian cancer. BMC Cancer. 2012; 12:379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tickle T., Tirosh I., Georgescu C., Brown M., Haas B. inferCNV of the trinity CTAT project. 2019; Cambridge, MA, USA: Klarman Cell Observatory, Broad Institute of MIT and Harvard; https://github.com/broadinstitute/inferCNV. [Google Scholar]
- 42. Gao R., Bai S., Henderson Y.C., Lin Y., Schalck A., Yan Y., Kumar T., Hu M., Sei E., Davis A. et al. Delineating copy number and clonal substructure in human tumors from single-cell transcriptomes. Nat. Biotechnol. 2021; 39:599–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mixture of high grade serous carcinoma cell lines, OV data for DLP scDNA-seq and 10X Genomics scRNA-seq was downloaded from European Genome-Phenome archive with accession EGAD00001004553. The scRNA and scDNA data for NCI − N87 gastric cancer cell line were downloaded from Gene Expression Omnibus (GSE142750) and National Institute of Health’s SRA (PRJNA498809). The primary gastric adenocarcinoma patient P5931 data with 10X Genomics scRNA and scDNA was downloaded from dbGAP repositories with accession numbers phs001711. The R package of CCNMF is available at https://github.com/labxscut/CCNMF and https://doi.org/10.5281/zenodo.10570125.