

Abstract
Exposure to hepatotoxic chemicals is involved in liver disease-related morbidity and mortality worldwide. The liver responds to damage by triggering compensatory hepatic regeneration. Physical agent or chemical-induced liver damage disrupts hepatocyte proteostasis, including endoplasmic reticulum (ER) homeostasis. Post liver injury ER experiences a homeostatic imbalance, followed by active ER stress response signaling. Activated ER stress response causes selective upregulation of stress response genes and downregulation of many hepatocyte genes. Acetaminophen overdose, CCl4, acute and chronic alcohol exposure, and physical injury activates the ER stress response, but details about the cellular consequences of the ER stress response on liver regeneration remain unclear. The current data indicate that inhibiting the ER stress response after partial hepatectomy-induced liver damage promotes liver regeneration, whereas inhibiting the ER stress response after chemical-induced hepatotoxicity impairs liver regeneration. This review summarizes key findings and emphasizes the knowledge gaps in role of ER stress in injury and regeneration.
Keywords: Liver regeneration, ER stress response, unfolded protein response, liver injury, chemical induced liver injury, partial hepatectomy
Graphical Abstract
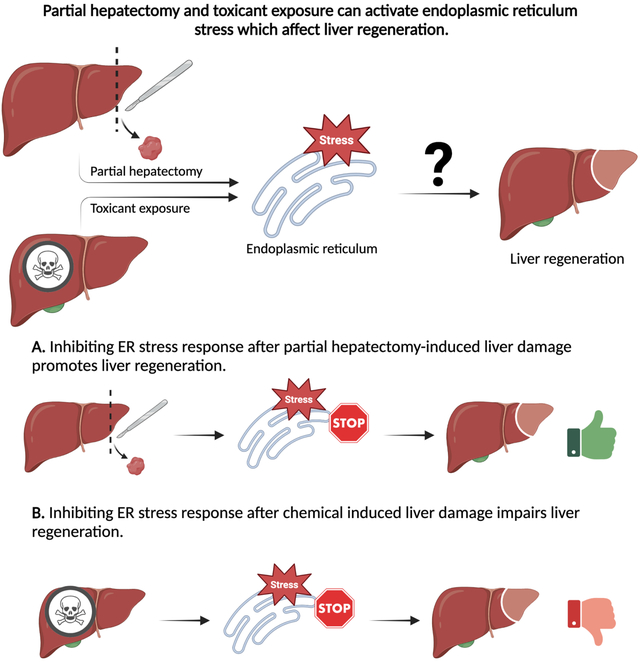
Lay summary
Liver injury induced by chemicals is a global and common problem. The liver is vulnerable to damage because of its central role in chemical detoxification. Following injury, the liver tries to repair itself by regenerating the damaged portion. The endoplasmic reticulum (ER) is part of cells that is necessary for normal functioning if cells including those in the liver. During liver injury, ER activates a stress response that affects the expression of genes, affecting how well the liver can regenerate. Various drugs like acetaminophen, alcohol, and even physical injuries can activate ER stress. A limitation in our understanding is how ER stress plays a role during liver regeneration. Studies suggest blocking the ER stress response can help the liver heal after a physical injury. In the case of chemical injuries blocking the stress response can worsen the outcome. This review highlights the role of ER stress response in liver regeneration and highlights potential lines of future investigation.
A. Introduction
The liver is one of the largest organs in human body and performs several important functions including macronutrient metabolism, detoxification of xenobiotics, bile acid synthesis, and lipid and cholesterol metabolism.1 The liver is anatomically divided into four lobes and functionally into eight by the portal vein and hepatic artery.2 It is made up of different cell types including approximately 60% hepatocytes and approximately 40% nonparenchymal cells, which include cholangiocytes, hepatic stellate cells (HSCs), Kupffer cells, and liver sinusoidal endothelial cells.1,3 Hepatocytes are the workhorse of the liver and perform all the major liver functions described above.4,5 The anatomical and physiological arrangement of the liver exposes it to significant concentrations of toxicants and infectious agents which can result in hepatocyte damage and liver injury. However, the liver has a remarkable capacity to undergo regeneration to refurbish the damaged liver and regain normal function.6
Maintenance of cellular protein homeostasis (proteostasis) is a key to cellular health. To maintain cellular proteostasis, cells employ various mechanisms to control protein synthesis, folding, intracellular trafficking, and compartmentalization along with regulated protein degradation.7 The endoplasmic reticulum (ER) plays a critical role in regulating proteostasis, protein synthesis, and protein trafficking.8 An imbalance between newly synthesized polypeptide chains entering the ER and folded proteins exiting the ER can result in the accumulation of unfolded or misfolded proteins in the ER.9 This accumulation of unfolded proteins can trigger an ER stress response - unfolded protein response (UPR).10 The severity and duration of stress experienced by the cells guide whether the cells will adapt to the stress or undergo cell death.11
Liver injury caused by hepatotoxicant exposure or by physical damage as in partial hepatectomy (PH) is associated with increased histological damage and necrosis. This damage activates proliferation in the remnant uninjured hepatocytes to compensate for and restore the lost liver tissue. Hepatocytes are under an active state of proliferation during liver regeneration which is associated with an increase in cellular protein synthesis.12 It is likely that the rise in protein synthesis during liver regeneration can result in the accumulation of unfolded proteins in the ER lumen which can activate hepatocyte UPR. In hepatocytes, many genes are repressed due to activated ER stress response signaling, whereas expression of stress response genes, which support cell survival during stress conditions are selectively upregulated.13,14 The cellular and hepatological effects of ER stress on liver regeneration is an underexplored area of research. In this review, we attempt to summarizing the key findings and highlighting the knowledge gaps in our understanding of the role ER stress plays in liver regeneration. Although a large part of our discussion focuses on the role of ER stress response in liver parenchymal cells, readers are encouraged to visit Maiers and Malhi, 201915 for a thorough discussion on the role of ER stress in hepatic stellate cells, and Zhou et al. 202216, for a discussion on the role of ER stress response in Kupffer cells. Finally, since chronic liver injury and constitutively active ER stress is associated with cellular apoptosis, readers are directed to visit the recent review by Zhang et al. 202217, on ER stress mediated cell death in conditions of liver injury.
B. Mechanistic insights into ER stress response
The ER provides conditions that support protein folding, which is monitored by the members of ER protein folding machinery which ensures proteins are properly folded and packaged in ER exit vesicles. When unfolded proteins accumulate in the ER lumen the ER protein folding machinery activates the UPR. The active UPR also augments the targeted degradation of unfolded proteins through ER-associated degradation (ERAD).18, 19 UPR functions by relaying the information of unfolded proteins to the cell by three transmembrane protein sensors: protein kinase R-like endoplasmic reticulum kinase (PERK) encoded by eukaryotic translation initiation factor 2 alpha kinase 3 (EIF2AK3); inositol-requiring enzyme 1 (IRE1α) encoded by endoplasmic reticulum to nucleus signaling 1 (ERN1) and activating transcription factor 6 (ATF6).10 Each of these protein sensors has a unique mechanism of propagating the ER stress response (Fig. 1).
Fig. 1:
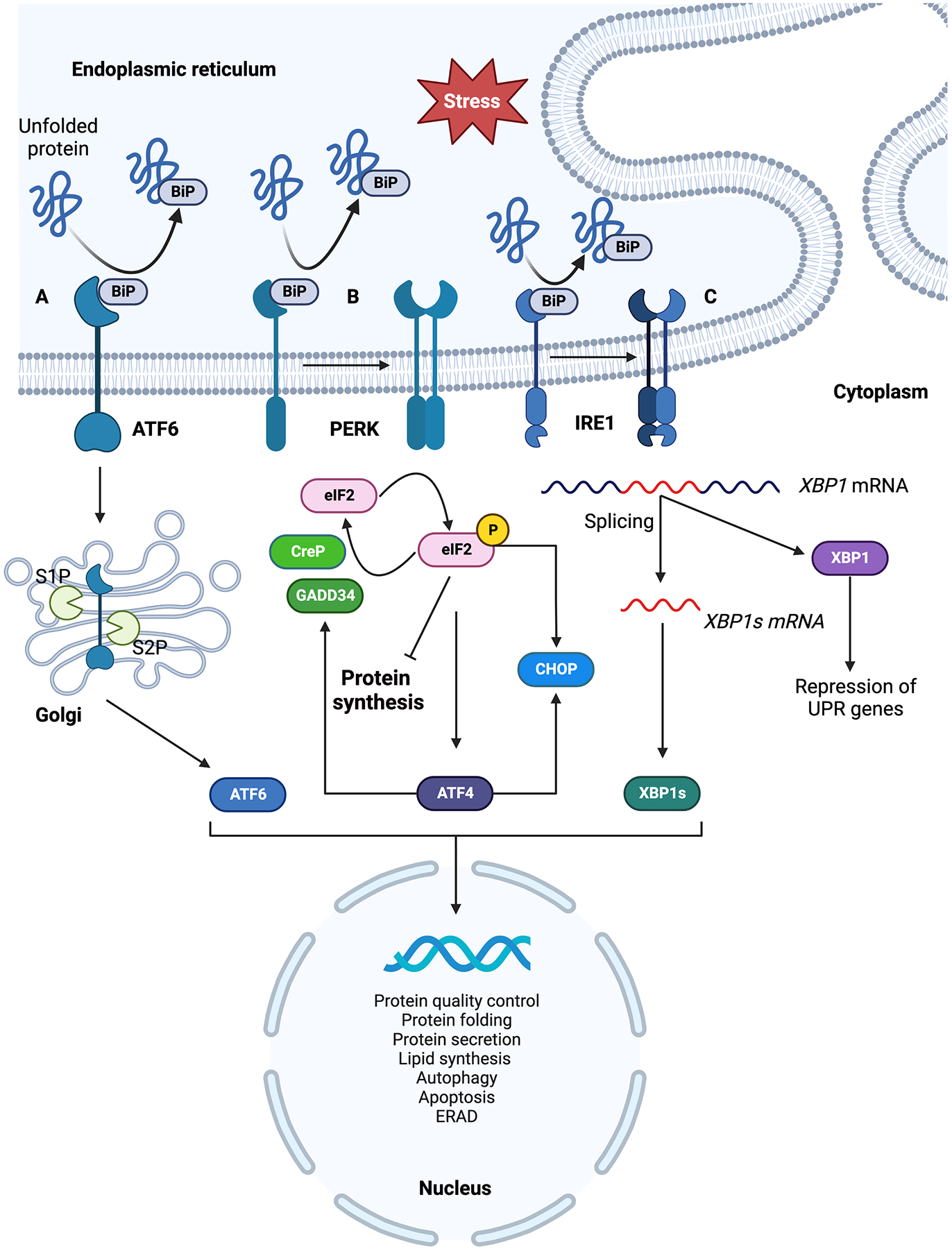
Summary of three branches of ER stress response signaling pathways. On accumulation of unfolded proteins in the ER lumen, BiP dissociates from the ER luminal domain of three ER membrane localized sensors; ATF6, IRE1 and PERK resulting in their activation. A) BiP dissociated ATF6 translocate to the Golgi where it is activated by proteolytic cleavage and the activated ATF6 travels to the nucleus and induces expression of downstream ER stress response genes. B) PERK is activated by oligomerization and trans autophosphorylation upon BiP dissociation. Active PERK phosphorylates eIF2α resulting in inhibition of protein synthesis and selective translation of ATF4 which induces ER stress response genes. Phosphorylated eIF2α and ATF4 selectively translate CHOP. Phosphorylated eIF2α is dephosphorylated by CReP and GADD34 phosphatases. C) IRE1 is activated by oligomerization and trans autophosphorylation which selectively affect mRNA splicing. IRE1 mediated splicing activates XBP1 which induces the expression of ER stress response genes. Both spliced and precursor mRNAs are expressed in cells, spliced XBP1 acts as an activator of UPR target genes while its unspliced precursor represses UPR gene expression. Figure generated using BioRender.com.
Under normal physiological conditions, these UPR sensors are inactive and bound to binding immunoglobin protein (BiP), a member of the heat shock protein family, on their ER luminal domains. Accumulation of unfolded proteins in the ER lumen beyond a threshold dissociates BiP from the luminal side of the ER stress sensors activating them to trigger ER stress response.20,21 An alternative mechanism proposed for BiP dissociation involves activation of UPR sensors by direct binding of unfolded proteins to their luminal domains and dissociating BiP.22,23 UPR activation has significant cellular consequences including transcriptional reprogramming, translation inhibition, selective translation of stress response genes and depending upon the severity and duration of stress, cell death.10,11,24
1. ATF6
ATF6 is an ER membrane-localized transcription factor bound to BiP under inactive conditions.10 ATF6α functions as a transcription activator while ATF6β has not been shown to have an effect on the ATF6α-mediated gene expression.25 Accumulation of unfolded proteins activates ATF6 by dissociation of BiP from its ER luminal domain translocating ATF6 from ER membrane to Golgi.26 Two Golgi proteases S1P and S2P (site-1 and site-2 proteases) cleave the translocated ATF6 at two sites liberating its N-terminal domain to migrate into the nucleus and activating UPR response genes (Fig. 1A).27,28 Golgi proteases used in the processing of ATF6 are also used by the liver in lipid metabolism and processing of sterol response element binding proteins (SREBPs).28–30 Although important in proteotoxic stress response, ATF6 is also activated by sphingolipids which trigger ER lipid biosynthetic genes through mechanisms distinct from the proteotoxic stress response.31 In the liver, ATF6 activation has been seen to play a role in hepatocarcinogenesis and liver regeneration.32,33
2. IRE1
IRE1 is a bifunctional transmembrane protein with kinase and endoribonuclease activities.34,35 Under inactive conditions, its monomeric form is bound to BiP on its ER luminal domain.21 Unfolded protein accumulation in the ER lumen dissociates BiP from IRE1’s ER luminal domain allowing the oligomerization of IRE1 followed by trans-autophosphorylation which activates the protein.36 The now active IRE1 acts as an endoribonuclease, splicing the UPR specific transcription- factor X-box binding protein 1 (XBP1) mRNA, which is followed by ligation of 5’ and 3’ ends of spliced mRNA (Fig. 1B).37,38 In metazoans, both precursor and spliced XBP1 mRNAs upon translation have different functional properties. Precursor mRNAs encode a protein that represses the expression of UPR target genes whereas the protein produced from spliced mRNA acts as a potent activator of UPR target genes.39 This newly spliced XBP1 mRNA on translation acts as a transcription activator for UPR response genes associated with ER chaperone and ER secretory genes along with genes for ERAD. In addition to activating UPR target genes, IRE1 plays an important role in mediating the death of ER-stressed cells by recruiting tumor necrosis factor receptor (TRAF) and activating Jun N-terminal kinase (JNK) while also interacting with other components of cell death machinery like caspase-12.40 It has been seen that activated IRE1 can degrade nonessential mRNAs in a process called regulated IRE1α dependent decay (RIDD).41–43 RIDD has been noted to be associated with cell survival and cell death in nonalcoholic fatty liver disease (NAFLD) and nonalcoholic steatohepatitis (NASH).44 Transport and Golgi organization (TANGO1) is an ER membrane exit site resident protein required for the secretion of collagens and expansion of transport vesicles to accommodate large cargo from the ER.45 TANGO1 is one of the downstream targets of the spliced XBP1. Under ER stress conditions, spliced XBP1 upregulates TANGO1 expression in the liver resulting in liver fibrosis. Experiments with the abrogation of TANGO1 or chronic unresolved ER stress have resulted in apoptosis46 These observations suggest that ER stress plays an active role in liver fibrosis.
3. PERK
PERK is a transmembrane protein kinase which inhibits protein synthesis under stress conditions. It is an ER resident transmembrane protein kinase with ER luminal domain similar to IRE1 and a cytoplasmic kinase domain.47 Under normal physiological conditions the ER luminal domain of PERK is bound to BiP. With the accumulation of unfolded proteins in the ER lumen, PERK is activated by oligomerization and trans-autophosphorylation and acts as a kinase phosphorylating Ser51 residue on the alfa subunit of eukaryotic translation initiation factor 2 (eIF2) (Fig. 1C). eIF2 is an important translation initiation factor required for the delivery of initiating tRNA (Met-tRNAi) to the translation initiation complex. Eukaryotic translation initiation factor 2B (eIF2B) is a guanine exchange factor for eIF2. Phosphorylated eIF2α acts as an inhibitor by binding to eIF2B irreversibly and not undergoing guanine exchange causing a drop in the available active pools of eIF2 ultimately resulting in inhibition of translation.48 Such conditions of translation inhibition due to phosphorylated eIF2α selectively favor the translation of stress response genes like activating transcription factor 4 (ATF4) (Fig. 1C) which activates yet another set of stress response genes including inducible eIF2α phosphatase, growth arrest and DNA-damage- inducible protein-34 (GADD34) and a transcription factor, C/EBP homologous protein (CHOP).48 Experiments with overexpression of CHOP have been shown to arrest the cell cycle and increase cellular apoptosis while CHOP deletion has been shown to reduce apoptosis under conditions of ER stress. Furthermore, an inverse apoptotic relationship is observed between CHOP and antiapoptotic factor Bcl-2 expression. taken together, these and many other observations highlight the prominent role of CHOP in apoptosis.49 To cope with the translation inhibition conditions, a constitutively expressed eIF2α phosphatase, a constitutive repressor of eIF2α phosphorylation (CReP) continues to dephosphorylate eIF2α to restore protein synthesis independent of stress.50–53 The retention of phosphorylated eIF2α and inhibition of translation protect the cell by reducing the damaging effects of ER stress. Pharmacological modulators like GSK2606414, and GSK2656157 inhibit PERK, eIF2B inhibitor ISRIB, and eIF2α phosphatase inhibitors: Salubrinal, Guanabenz, and Sephin1, protect cells from the adverse effects of ER stress.54 Mouse models of hepatic steatosis- and obesity-induced NAFLD have shown that consumption of a high-fat diet can trigger ER stress response through PERK-mediated phosphorylation of eIF2α.55 Administration of Salubrinal showed attenuation of obesity and hepatic steatosis by reducing the severity of ER stress through increased levels of phosphorylated eIF2α. Rise in ATF4 levels due to phosphorylated eIF2α promoted autophagy.55 On similar lines, the PERK- eIF2α-ATF4 branch of ER stress response has also been shown to play a hepatoprotective role in alcohol-induced liver damage.56 This suggests that the PERK- eIF2α-ATF4 branch of ER stress response is important in protecting the liver from damage caused by ER stress.
Overall, in a cell under ER stress, the three branches of UPR (ATF6, IRE1, and PERK) work in a complex interconnected manner and together contribute to cell survival against ER stress.57–60 More research is needed to elucidate the complex interrelationship between the three UPR branches and their role in cell survival under stress conditions.
C. The role of ER stress during liver pathogenesis and regeneration after chemical induced liver injury
Toxicant-mediated tissue injury and tissue’s response to the inflicted injury are key aspects in determining the progression or regression of the toxicant-induced liver damage. The physiological effect of pharmacological agents and toxicants is determined mainly by their dose. Once toxicant exposure occurs, the toxic effects depend on the absorption, distribution, metabolism, and excretion of the chemical. It is probable that the majority of toxins at some level can disrupt the cellular ER homeostasis and activate UPR. More investigation into the involvement of the ER stress response due to chemical exposure is therefore necessary. Since reviewing the involvement of ER stress in liver injury induced by every chemical is beyond the scope of this review, we have chosen to review ER stress modulation by chemicals that have either clinical (acetaminophen and alcohol) or experimental (thapsigargin, tunicamycin, and carbon tetrachloride) significance (Fig. 2).
Fig. 2:
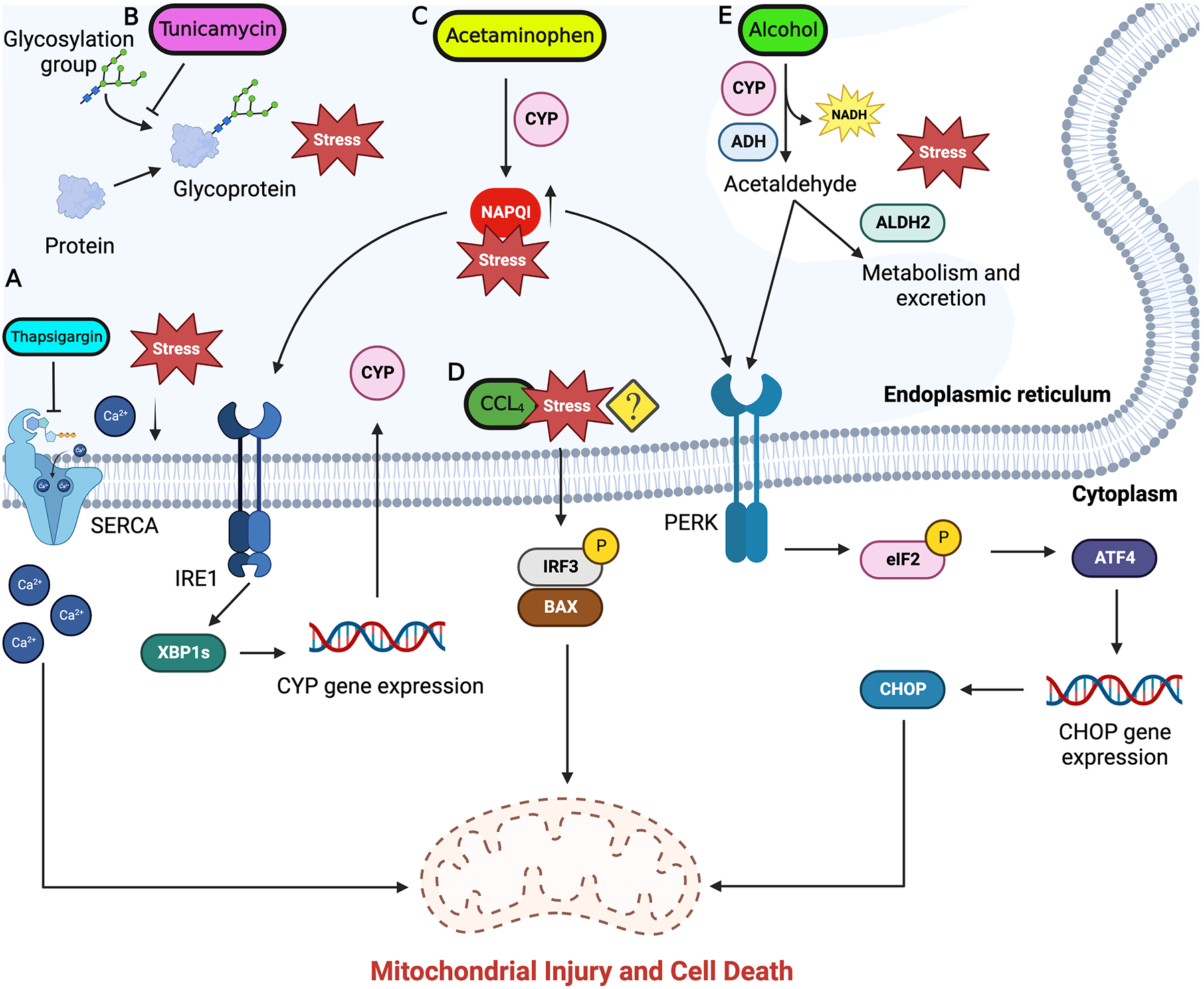
ER stress response signal flow in chemical induced liver injury. Subthreshold level concentrations of hepatotoxicants like acetaminophen, alcohol, CCl4 are metabolized by the ER resident enzymes. Buildup of these toxins beyond the threshold levels is associated with induction of ER stress. A) Thapsigargin inhibit SERCA pump on ER membrane and unbalance the ER calcium homeostasis and induce ER stress. B) Tunicamycin inhibits protein glycosylation and protein assembly causing accumulation of unfolded proteins resulting in activation of ER stress. C) Toxic levels of acetaminophen result in accumulation of reactive intermediate NAPQI triggering ER stress through IRE1 and PERK branches. D) The mechanism of ER stress induction by CCl4 is still not completely understood but is seen to activate apoptosis through IRF3 and BAX. E) Alcohol imbalances ER redox balance and trigger ER stress through PERK which induces ISR and subsequent CHOP expression. Downstream signaling from UPR activate mitochondria mediated apoptosis in hepatocytes which can progress to liver fibrosis and cirrhosis. Figure generated using BioRender.com.
1. Thapsigargin and Tunicamycin induced ER stress
The Product of the Mediterranean plant Thapsia garganica, thapsigargin is a potent inducer of ER stress. Thapsigargin induces ER stress by inhibiting sarcoplasmic/endoplasmic reticulum calcium ATPase (SERCA) which causes depletion in ER calcium pools affecting ER homeostasis and triggering ER stress response while also initiating downstream ER-mediated apoptotic signaling (Fig. 2A). Inhibition of SERCA increases the cytosolic calcium deposition, which in turn triggers apoptotic signaling.61 Detailed insights into the role of thapsigargin in inducing ER stress in liver hepatocytes are limited and will require further investigation. Product of Streptomyces lysosuperificus, tunicamycin is a potent antibiotic against many gram-positive bacteria, fungi, yeast, and viruses. Tunicamycin interferes with the first ER step in the synthesis of N-glycoproteins causing impaired protein glycosylation in the ER and resulting in misfolded proteins, triggering unfolded protein response in the ER (Fig. 2B).62–64 Due to their physiological toxicity, thapsigargin and tunicamycin are not of pathophysiological relevance, however, they have been widely used experimentally as a model compounds to induce ER stress in liver and in cultured hepatocytes to study the role of ER stress response signaling.65,66
Due to their ability to induce ER stress, thapsigargin and tunicamycin have been explored as potential candidates against cancer.67–69 However, given their physiological toxicity, there still needs to be a deeper understanding of their effects on human physiology and their concentration-dependent effects on the biological system. For example, it has been seen that sub-toxic concentrations of thapsigargin have a cytoprotective role against the influenza virus.70 It is therefore necessary to develop temporal dose-response studies to address irreversible slow changes in cellular functioning in response to low-concentration doses of thapsigargin and tunicamycin. Such studies also need to be developed for other toxins discussed later in this section.
2. Acetaminophen induced ER stress
Acetaminophen (APAP) is the most widely used analgesic and antipyretic agent in the world. Overdose of APAP is the most common cause of acute liver failure in the Western world leading to thousands of hospitalizations and hundreds of deaths.71 APAP is metabolized by the drug-metabolizing enzyme Cytochrome P450 2E1 (CYP2E1) into a reactive metabolite N-acetyl-p-benzoquinone imine (NAPQI) (Fig. 2C) which under normal conditions is detoxified by cellular processes through conjugation with glutathione (GSH).71 Under overdose conditions NAPQI accumulates at toxic levels due to rapid GSH depletion and alters cellular redox balance. The distorted redox balance results in imbalanced mitochondrial membrane potential, increasing reactive oxygen species (ROS) and peroxynitrite species concentration while disrupting Ca2+ homeostasis, and cellular proteostasis, causing DNA damage, impairing mitochondrial function, and finally culminating into hepatocyte necrosis.71–73 Studies have indicated that ER stress response signaling is active in APAP-induced hepatotoxicity.74 Spliced XBP1 activates the expression of CYP1A2 and CYP2E1 which convert APAP to its reactive intermediate NAPQI (Fig. 2C).75 XBP1-deficient mouse models have shown constitutively active IRE1α signaling which had hepatoprotective effects by suppressing the expression of CYP1A2 and CYP2E1 and cleavage of existing CYP1A2 and CYP2E1 mRNA through RIDD.75 Acetaminophen overdose resulted in increased expression of CHOP. Whole-body CHOP knockout mice showed decreased APAP toxicity and had a better regenerative response.76
A common observation in these studies is the latent activation of ER stress response against acetaminophen toxicity. Multiple factors can underly this latent activation. One possible explanation can be that the ER stress response is a result of the cellular damage happening immediately after acetaminophen exposure. Another aspect of latent ER stress activation can be the unspecific binding of accumulated NAPQI to ER proteins interfering with ER homeostasis.77–79 Mechanistic insights into ER stress response activation and downstream consequences of ER stress signaling on hepatocyte recovery or death or recovery from acetaminophen and other toxin exposure will be an important and interesting area for further investigation.
3. Carbon tetrachloride induced ER stress
Carbon tetrachloride (CCl4) is an organic solvent heavily used in industry for degreasing and is a well-studied hepatotoxicant. Treatment of CCl4 is also used as an experimental model to study acute and chronic liver injury. CCl4 toxicity is associated with centrilobular hepatocyte necrosis, which results in subsequent dose-dependent induction of liver regeneration.80 However, repeat dosing of CCl4 can induce liver fibrosis and scar formation which can develop into liver cirrhosis. The progression of scar tissue results in cessation of liver regeneration, whereas cessation of CCl4 administration is associated with reactivation of liver parenchyma regeneration.81 CCl4 exposure leads to activation of ER stress signaling but mechanisms are not completely known (Fig. 2D). Following CCl4 exposure, cytoplasmic interferon regulatory factor 3 (IRF3) and BAX (a proapoptotic factor) complex formation is detected which is seen to migrate to the mitochondria and activate caspase-mediated hepatocyte apoptosis and subsequent liver fibrosis.82
4. Alcohol induced ER stress
a. Metabolism of alcohol in the liver
Uncontrolled consumption of alcohol is recognized as a global issue of public health. Hepatocyte resident alcohol dehydrogenase (ADH) and CYP2E1 are the main enzymes involved in metabolism of alcohol. ADH catalyzes alcohol oxidation through NAD+ reduction converting alcohol to acetaldehyde. This reaction results in the production of acetaldehyde and NADH which are heavily reactive and toxic (Fig. 2E). Acetaldehyde is further oxidized to relatively safer acetate by mitochondria resident aldehyde dehydrogenase 2 (ALDH2) through the reduction of NAD+.83 Excess consumption of alcohol can result in increased activity of ADH and ALDH2 which is associated with increased concentration of NADH resulting in imbalanced cellular redox potential (NAD+/NADH ratio). This is followed by a metabolic shift from oxidative to reductive and increased fatty acid synthesis which contributes to fatty liver disease.83 CYP2E1 follows a similar oxidative reaction converting alcohol to acetaldehyde.84 Besides ADH and CYP2E1, cellular catalase has also been noted to oxidize alcohol to acetaldehyde.85
b. Acute alcohol consumption and its effect on the liver
Acute alcohol consumption is a major cause of alcoholic liver damage, however, the damage is reversible. Acute alcohol consumption can impact liver function by overwhelming liver’s capacity to process alcohol efficiently. This can result in oxidative stress, inflammation, and damage to liver cells.86 Acute alcohol consumption can disrupt the balance of lipid metabolism in the liver, leading to the accumulation of fat droplets and the development of alcoholic fatty liver disease (AFLD).87 Moreover, the breakdown of alcohol by the liver enzyme alcohol dehydrogenase generates toxic byproducts, such as acetaldehyde, which can cause DNA and protein damage and impair liver function.83,88,89 Another consequence of acute alcohol consumption is the activation of immune cells in the liver, triggering an inflammatory response. This chronic inflammation can lead to the development of alcoholic hepatitis, characterized by liver cell injury and inflammation.86,90 If left untreated, alcoholic hepatitis can progress to more severe conditions such as liver fibrosis and cirrhosis. Interestingly, variations in genes involved in alcohol metabolism and antioxidant defense systems can influence an individual’s risk of developing liver diseases associated with alcohol consumption.91 Understanding these genetic and other host-associated factors can help identify individuals who may be more vulnerable to the harmful effects of acute alcohol consumption. An interesting gut-liver axis is seen to function in alcohol-induced liver damage where consumed alcohol coupled with lipopolysaccharide (LPS) from the gut microbiome translocates to the liver and activates Kupffer cells inducing liver inflammation. Furthermore, the LPS-alcohol combination can activate ROS formation which results in the worsening of hepatocellular damage.92
c. Chronic alcohol consumption and its effect on the liver
Chronic alcohol consumption has profound and detrimental effects on the liver and has been associated with the development of alcoholic liver disease (ALD). ALD encompasses a spectrum of conditions, ranging from alcoholic fatty liver to alcoholic hepatitis and ultimately, liver cirrhosis.93 Chronic alcohol consumption disrupts lipid metabolism, resulting in the accumulation of fat within liver cells, a characteristic feature of alcoholic fatty liver disease.94 Oxidative stress induction has been seen as one of the consequences of chronic alcohol consumption, leading to the production of ROS and lipid peroxidation, contributing to liver cell injury and inflammation.95 Additionally, chronic alcohol exposure activates hepatic stellate cells, promoting collagen deposition and fibrosis, which are key mechanisms underlying the development of liver cirrhosis.93,95,96 Furthermore, chronic alcohol consumption and subsequent ALD are also associated with impaired liver regenerative potential through disruption of the balance between liver cell proliferation and cell death.95 This impaired regenerative process contributes to the development of liver fibrosis, cirrhosis, and ultimately, liver failure.93,95 Excessive alcohol consumption is initially followed by liver steatosis which can progress to steatohepatitis. Continued consumption of alcohol can lead to liver fibrosis which can progress to liver cirrhosis and hepatocellular carcinoma (HCC).97
d. Role of the ER stress response
Acute and chronic alcohol consumption triggers ER stress response in the liver and can contribute to ALD. Alcohol disrupts ER homeostasis, leading to the accumulation of unfolded or misfolded proteins within the ER lumen, activating ER stress.98 It has been suggested that ER stress can contribute to the development of liver steatosis or fatty liver disease, an early manifestation of ALD.93,98 ER, stress-induced activation of the transcription factor SREBP-1c promotes lipid synthesis and accumulation, contributing to the development of hepatic steatosis.83 Additionally, ER stress-mediated dysregulation of lipid metabolism pathways, such as lipolysis and fatty acid oxidation, further exacerbates liver lipid accumulation.11,99 Prolonged and excessive ER stress can overwhelm the UPR, resulting in sustained activation of inflammatory pathways and cell death. It has been seen that alcohol-induced oxidative stress and lipid accumulation can trigger ER stress by disrupting calcium homeostasis and impairing protein folding machinery resulting in the development of hepatic steatosis and ALD.100
Multiple ER stress response genes have been identified to be up-regulated during alcohol-induced liver toxicity indicating ER stress response signaling as an active contributor to alcohol-mediated liver injury.101 Alcohol toxicity causes hepatocyte apoptosis102 which is driven by the PERK-eIF2-ATF4 branch of ER stress signaling leading to CHOP activation followed by hepatocyte apoptosis.103 Furthermore, studies in primary cultured human hepatocytes exposed to alcohol showed activation of the integrated stress response (ISR).104 ISR is a cellular stress response system in which various extracellular and intracellular stresses are identified by four kinases; PERK, PKR, HRI, and GCN2, which give a common output of eIF2 phosphorylation and inhibition of cellular protein synthesis.105 It will be interesting to see how active ISR can have detrimental effects on liver regeneration.
5. Role of ER stress in liver regeneration after chemical induced liver injury
The liver responds to chemical-induced injury by compensatory proliferative activation of hepatocytes and nonparenchymal cells through complex signaling networks.106,107 These newly formed cells make up for the lost liver tissue and restore normal liver function.71,108,109 Multiple studies discussed in this section highlight the role of ER stress response signaling in liver regeneration after chemical-induced liver injury.
Studies conducted using chemical-induced hepatotoxicity models suggest that abrogating ER stress response results in impaired liver regeneration. CCl4-induced liver toxicity models on the background of IRE1α deletion showed poor regenerative response and diminished STAT3 phosphorylation. Whereas, when IRE1α expression was restored, sustained STAT3 phosphorylation levels were detected.110 STAT3 is an important signal transducer in hepatocyte proliferative signaling111 and therefore, these results suggest that ER stress signals interact with other cellular signaling pathways and plays an important role in activating cell proliferation following chemical-induced liver injury. Mechanistic insights into why IRE1α signaling is important come from studies on downstream promotion of genes due to IRE1α spliced XBP1 signaling. Interleukin-24 (IL-24) is a known negative regulator of cell proliferation and has a hepatoprotective effect.112 Spliced XBP1 is seen to promote the expression of IL-24 which accumulates on the ER membrane and inhibits PERK-eIF2-ATF4 branch of ER stress.113 This PERK-eIF2-ATF4 inhibition has two consequences; first, it allows for continued protein synthesis necessary for hepatocyte adaptation to the incurred chemical injury and liver regeneration; second, it downregulates the CHOP expression and subsequent apoptosis-protecting remnant hepatocytes and allowing liver regeneration.113
D. The role of ER stress in regeneration after partial hepatectomy
1. Liver injury and regeneration dynamics after partial hepatectomy
Due to the lobular structure of the liver, surgical resection of one of its lobes provides a clean model to study liver regeneration. Seminal experiments demonstrated the potential of the liver to regenerate to normal physiological size after 2/3rd PH making it the most widely used model to study liver regeneration.114 Although 2/3rd PH is widely used, studies exceeding this limit have shown to have lethality due to inefficiency and lack of functionality of the remnant liver to sustain normal physiological functioning and regenerative potential.115 PH is followed by recalibration of hepatic blood flow with the blood now circulating through the remnant liver. This can increase the blood pressure in the remnant liver causing portal hyperperfusion and focal hemorrhage which can progress in the adjacent parenchyma. Progression of poor hepatic circulation and associated damage can cause functional dearterialization, overall, drastically damaging the liver and impairing liver regeneration.116
The liver undertakes multiple regenerative processes to modulate its size corresponding to the host’s physiological conditions. For example, liver size increases during pregnancy while decreases in cases of cachexia. Furthermore, liver size is seen to be modulated by the host physiology through yet unexplored factors.117–119 This phenomenon of regulated liver size during regeneration in correspondence to the host physiology suggests a “hepatostat” like regulatory function.120 The restored liver after PH does not involve the regrowth of its lost liver lobe, but instead is associated with the expansion of existing liver lobes to the critical mass before hepatectomy.121 Multiple signaling pathways have been identified initiating and regulating liver regeneration after PH.106,107,122
In a healthy liver, majority of cells are in a quiescent state with a minimal population in an active state of proliferation, dividing with long intervals.123 However, upon injury compensatory regeneration is stimulated and a significant number of cells, both hepatocytes and nonparenchymal cells, enter the cell cycle and undergo cell proliferation to replace the dead cells. Liver regeneration is a complex process relying on internal and external signals that control the nature and extent of regeneration.109,124 The purpose of liver regeneration is to retain the lost stability of liver functioning which is critical for maintaining physiological homeostasis. Interestingly, under conditions of excessive liver parenchymal cell damage, nonparenchymal cells are seen to undergo active regeneration and transdifferentiate into hepatocytes through genome-wide alterations (epigenetic and transcriptomic) and signaling pathway rewiring.125–129
2. Role of ER stress in liver regeneration after PH
PH is associated with an increased risk of hepatic steatosis and inflammatory liver failure due to the generation of ROS, and excessive apoptosis and disturbed hepatic circulation. Chemicals that modulate ER stress such as 4-phenyl butyric acid (PBA) and tauroursodeoxycholic acid (TUDCA) have noted hepatoprotective effects against PH and ischemia-reperfusion injury.130–132 Experiments investigating the roles of PBA and TUDCA on ER stress response and liver regeneration post-PH suggested that all three branches of ER stress response and their associated downstream signaling are activated in livers post-PH and ischemia reperfusion.133,134 Steatotic livers exhibit a reduced response to ER stress signaling than non-steatotics livers. PBA and TUDCA resulted in suppression of IRE1 and PERK branches of ER stress response subsequently inhibiting apoptosis and inflammation and improving liver regeneration.133 Although these experiments highlight a correlation between ER stress inhibition and improved liver regeneration, it is important to note that the mechanism of action of PBA and TUDCA is not completely elucidated and a deeper investigation into their mechanism will help in uncovering how they modulate ER stress in hepatocytes and affect the liver regeneration post PH. Phenyl butyric acid (PBA) has emerged as a promising therapeutic agent with diverse applications in neurodegenerative diseases, cancer, and metabolic disorders. As a chemical chaperone and modulator of cellular stress response pathways, PBA exhibits multiple beneficial effects. It has the ability to reduce protein aggregation in neurological conditions, alleviate endoplasmic reticulum (ER) stress, induce cellular differentiation and apoptosis, and enhance metabolic function. These findings highlight the potential of PBA as a targeted intervention for improved patient outcomes.135 Tauroursodeoxycholic acid (TUDCA) has been extensively studied for its hepatoprotective effects in liver diseases such as non-alcoholic fatty liver disease (NAFLD), cholestasis, and liver fibrosis. Moreover, TUDCA has demonstrated promising neuroprotective properties, making it a potential therapeutic intervention for neurodegenerative disorders like Parkinson’s and Alzheimer’s disease. Additionally, TUDCA’s anti-inflammatory and anti-apoptotic characteristics offer potential therapeutic avenues for various inflammatory and apoptotic-related conditions. The diverse therapeutic applications of TUDCA underscore its significance as a candidate for further research and clinical exploration in different disease contexts.136
Mouse models with 90% hepatectomy followed by co-administration of prostaglandin and somatostatin combination showed their combinatorial hepatoprotective effect to be more pronounced than their individual administration. This protective effect was seen to be through inhibition of ER stress response which inhibited cellular apoptosis and promoted liver regeneration.137 It is important to note that prostaglandins and somatostatins carry out a vast spectrum of functions in the body and their hepatoprotective effects seen in these experiments through suppression of ER stress response can be a part of a multifaceted cell-wide action, the effect of which is reduced hepatic damage after PH. We lack detailed mechanistic insights into the action of prostaglandins and somatostatins on the hepatocyte ER and liver and more investigations are warranted.
Overall, these observations suggest that activation of ER stress response following PH is associated with increased hepatic damage and poor liver regeneration while drugs suppressing the ER stress-induced hepatocyte damage are hepatoprotective. The potential clinical applications of such drug interventions would require a deeper understanding of their mechanisms of action, their clinical efficiency and safety, and fundamentally, how the ER stress response affects liver regeneration after PH.
3. UFMylation, cell death and regeneration
A recently identified cyclin-dependent kinase 5 activator, CDK5RAP3, is especially expressed in the liver along with other organs of the body, contributes to a multitude of cellular processes and interestingly also is a component of the UFMylation system. UFMylation, also known as ubiquitin-fold modifier 1 (UFM1) conjugation, is a post-translational modification pathway involving the attachment of UFM1 protein to the target proteins.138 UFMylation has been shown to play a role in regulating ER homeostasis and the cellular response to ER stress. Studies have demonstrated that UFM1 and its conjugating enzymes are involved in the maintenance of ER protein folding capacity and the unfolded protein response (UPR), a cellular mechanism activated during ER stress.138,139 Since UFMylation is a part of post-translational modification in the ER, CDK5RAP3 is seen to play an important role in ER homeostasis.140 CDK5RAP3 during liver injury due to PH has been shown to have a hepatoprotective role by preventing the activation of ER stress response and maintaining ER homeostasis along with maintaining normal lipid metabolism.141 CDK5RAP3 deletion has been shown to disrupt ER homeostasis through an impaired post-translation modification altogether contributing to the activation of UPR.140,141 However, it must be noted that the complete understanding of the cell wide functions of CDK5RAP3 and the ramifications of its deletion in relation to ER stress induction and liver regeneration need to be further investigated.
Overall, such experiments suggest that hepatocytes have upstream regulatory factors, the loss of which triggers ER stress-mediated hepatocyte apoptosis to perhaps avoid errors that might result in unregulated hepatocyte proliferation and hepatocellular carcinoma.
Actively proliferating hepatocytes in response to PH-induced liver damage require continued synthesis of proliferation dependent proteins. Studies in fibroblast cell lines showed that activation of the PERK-eIF2-ATF4 branch of ER stress response results in a drop in cellular protein synthesis and cause cell cycle arrest at the G2/M phase.142,143 ER stress response is also associated with proliferation-promoting and inhibitory signaling. Liver regeneration is associated with active cell proliferation through oncogenic Ras signaling. It has been seen that IRE1a-mediated ER stress response signaling is activated by Ras-mediated proliferative signaling. This observation comes from experiments that showed that abrogating ER stress through the reduction of IRE1a-mediated Xbp1 splicing resulted in growth arrest and premature senescence.144 Corresponding to this, it has been seen that ER stress is associated with ubiquitin proteasomal degradation of p53 and further supporting cell proliferation.145
Canopy homolog 2 (CNYP2) is a recently identified mediator of PERK signaling. Under stress conditions CNYP2 bind to the ER luminal domain of PERK by dissociating BiP and activates the PERK-eIF2-ATF4 branch of ER stress response. CNYP2 expression is activated by CHOP which is activated by ATF4 signaling. The expressed CNYP2 then gets localized in the ER lumen.146 Recently, mechanistic insights into CNPY2-mediated cell cycle enhancement were revealed which suggest the activation of PERK by CNPY2, activates multiple signaling pathways that inhibit p53, alleviating its proliferation inhibitory effect and promoting rapid cell proliferation in the liver.147
It is important to note that impaired liver regeneration in response to toxicant-induced injury or PH or error-prone cell proliferation is associated with the development of hepatocellular carcinoma. These studies highlight the importance of maintaining a fine balance of proliferative and proliferation inhibitory signaling in liver regeneration and the important role played by ER stress response signaling in this process.
E. Future lines of investigation on the role of ER stress in liver regeneration
ER plays a central role in cellular homeostasis and different environmental conditions like toxicant exposure and physical injury have been associated with distorting cellular homeostasis. Recent studies have begun to elucidate the role of ER stress response in modulating cellular processes in response to stress conditions. Many observations in the past decade have suggested that ER plays an important role in regulating liver regeneration. Based on the limited literature on the topic, we highlight the following as important areas of investigation.
Previously discussed studies have largely applied single dose or repeat dosing to induce liver injury following which the process of liver regeneration is monitored. Future studies need to focus on dose-dependent ER stress response prior to associated damage. Furthermore, continued monitoring of ER stress response through liver regeneration can help elucidate the liver regeneration regulatory role of ER stress response.
As previously discussed, a complete understanding of how different toxins affect hepatocytes and directly or indirectly result in activating ER stress response followed by its implications on liver regeneration is necessary. Furthermore, how toxicants targeting cellular locations other than ER can trigger a cascade of response upstream of ER eventually activating ER stress needs to be further resolved.
Multiple studies have highlighted context dependent PERK-eIF2-ATF4 signaling associated cellular changes in response to oncogene and tumor suppressor gene signaling.144,145,148,149 Since oncogene and tumor suppressor gene signaling is active during liver regeneration and considering their links with the PERK-eIF2-ATF4 branch of ER stress response, this signaling crosstalk needs to be further explored.
Our discussion highlights that inhibition of ER stress response in PH support liver regeneration while, inhibition in chemical-induced liver injury results in impaired regeneration. The three arms of UPR act in a sequential fashion with PERK-eIF2-ATF4 acting last.150,151,38,60 It would be interesting to explore how sequential activation of the three branches of ER stress response and associated downstream effects in PH and chemical-induced liver injury contribute to the contextual nature of ER stress in liver regeneration. This apparently contextual nature demands further elucidation of differential mechanisms involved in PH and toxicant damage hepatocyte response. Furthermore, we need to understand how the remnant injured or uninjured cells perceive the damage and in turn modulate their ER stress response and initiate liver regeneration.
Mitochondria mediated metabolism and energy production is important in regulating liver regeneration. Important links in ER-mitochondria crosstalk have been observed at different levels from physical contact, mitochondria associated membranes (MAMs) between the two organelles to signaling crosstalk.152–154 ER-mitochondria calcium crosstalk is important in maintaining cellular calcium homeostasis. ER release of calcium signals to mitochondria is crucial to regulate mitochondrial functions like metabolism, energy production and apoptosis.155 ER stress response signalling proteins like PERK are seen to interact with mitochondria under stress. this is seen to regulate mitochondrial protein homeostasis, ER mitochondria calcium signaling, and apoptosis.10,156–158 Recently it has been seen that mitochondria to ER crosstalk can be mediated by NADPH production and redox regulation of GSH. Mechanistic insights suggest active redox cycling of GSH is associated with the inhibition of ER stress.159 This highlights the potential importance of investigating the processes involved in ER-mitochondria crosstalk and their broader implications on regulating liver regeneration.
Finally, we believe that it is important to elucidate the mechanistic role of ER stress response in liver regeneration under different liver insults.
F. Conclusion
Over time, we have gained good insights into the role of ER stress response in PH and drug-induced liver toxicity.109 We believe that in the future, an even clearer picture of ER stress in various other liver diseases will emerge.160 ER plays a central role in cellular homeostasis and different environmental conditions like toxicant exposure and physical injury have been associated with distorting cellular homeostasis. Recent studies have begun to elucidate the role of ER stress response in modulating cellular processes in response to stress conditions. Multiple observations in the past decade have suggested that ER plays an important role in regulating liver regeneration. Based on the limited literature on this topic, multiple studies suggest that inhibiting the ER stress response after PH-induced liver damage promotes liver regeneration, whereas chemical-induced hepatotoxicity demonstrated that inhibiting the ER stress response impairs liver regeneration. Therefore, we see an apparent contextual nature to the role of ER stress response signaling in liver regeneration. Finally, we would like to highlight that while extensive mechanistic data are available in rodent models, more research on role of UPR is needed human liver diseases.
Acknowledgments
The authors would like to thank Dr. Thomas Rutkowski for helpful guidance on the writing and figure design of this manuscript. All the intext and graphical abstract images were generated using BioRender.com.
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
References
- 1.Trefts E, Gannon M, Wasserman DH. The liver. Curr Biol. 2017;27(21):R1147–R1151. doi: 10.1016/j.cub.2017.09.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bismuth H Surgical anatomy and anatomical surgery of the liver. World J Surg. 1982;6(1):3–9. doi: 10.1007/BF01656368 [DOI] [PubMed] [Google Scholar]
- 3.Blouin A, Bolender RP, Weibel ER. Distribution of organelles and membranes between hepatocytes and nonhepatocytes in the rat liver parenchyma. A stereological study. J Cell Biol. 1977;72(2):441–455. doi: 10.1083/jcb.72.2.441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schulze RJ, Schott MB, Casey CA, Tuma PL, McNiven MA. The cell biology of the hepatocyte: A membrane trafficking machine. J Cell Biol. 2019;218(7):2096–2112. doi: 10.1083/jcb.201903090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gunawan BK, Kaplowitz N. Mechanisms of Drug-Induced Liver Disease. Clin Liver Dis. 2007;11(3):459–475. doi: 10.1016/j.cld.2007.06.001 [DOI] [PubMed] [Google Scholar]
- 6.Forbes SJ, Newsome PN. Liver regeneration-mechanisms and models to clinical application. Nat Rev Gastroenterol Hepatol. 2016;13(8):473–485. doi: 10.1038/nrgastro.2016.97 [DOI] [PubMed] [Google Scholar]
- 7.Sala AJ, Bott LC, Morimoto RI. Shaping proteostasis at the cellular, tissue, and organismal level. J Cell Biol. 2017;216(5):1231–1241. doi: 10.1083/jcb.201612111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang M, Kaufman RJ. Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature. 2016;529(7586):326–335. doi: 10.1038/nature17041 [DOI] [PubMed] [Google Scholar]
- 9.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8(7):519–529. doi: 10.1038/nrm2199 [DOI] [PubMed] [Google Scholar]
- 10.Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334(6059):1081–1086. doi: 10.1126/science.1209038 [DOI] [PubMed] [Google Scholar]
- 11.Rutkowski DT, Kaufman RJ. That which does not kill me makes me stronger: adapting to chronic ER stress. Trends Biochem Sci. 2007;32(10):469–476. doi: 10.1016/j.tibs.2007.09.003 [DOI] [PubMed] [Google Scholar]
- 12.Skullman S, Wirén M, Garlick PJ, McNurlan MA, Larsson J. Protein synthesis in regenerating rat liver during malnutrition. J Hepatol. 1994;21(2):174–181. doi: 10.1016/S0168-8278(05)80391-5 [DOI] [PubMed] [Google Scholar]
- 13.Dubois V, Gheeraert C, Vankrunkelsven W, et al. Endoplasmic reticulum stress actively suppresses hepatic molecular identity in damaged liver. Mol Syst Biol. 2020;16(5):1–27. doi: 10.15252/msb.20199156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Argemí J, Kress TR, Chang HCY, et al. X-box Binding Protein 1 Regulates Unfolded Protein, Acute-Phase, and DNA Damage Responses During Regeneration of Mouse Liver. Gastroenterology. 2017;152(5):1203–1216.e15. doi: 10.1053/j.gastro.2016.12.040 [DOI] [PubMed] [Google Scholar]
- 15.Maiers JL, Malhi H. Endoplasmic Reticulum Stress in Metabolic Liver Diseases and Hepatic Fibrosis. Semin Liver Dis. 2019;39(2):235–248. doi: 10.1055/s-0039-1681032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhou L, Shen H, Li X, Wang H. Endoplasmic reticulum stress in innate immune cells - a significant contribution to non-alcoholic fatty liver disease. Front Immunol. 2022;13(July):1–14. doi: 10.3389/fimmu.2022.951406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang J, Guo J, Yang N, Huang Y, Hu T, Rao C. Endoplasmic reticulum stress-mediated cell death in liver injury. Cell Death Dis. 2022;13(12). doi: 10.1038/s41419-022-05444-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vembar SS, Brodsky JL. One step at a time: Endoplasmic reticulum-associated degradation. Nat Rev Mol Cell Biol. 2008;9(12):944–957. doi: 10.1038/nrm2546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ellgaard L, Helenius A. Quality control in the endoplasmic reticulum. Nat Rev Mol Cell Biol. 2003;4(3):181–191. doi: 10.1038/nrm1052 [DOI] [PubMed] [Google Scholar]
- 20.Carrara M, Prischi F, Nowak PR, Kopp MC, Ali MMU. Noncanonical binding of BiP ATPase domain to Ire1 and Perk is dissociated by unfolded protein CH1 to initiate ER stress signaling. Elife. 2015;2015(4):1–16. doi: 10.7554/eLife.03522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat Cell Biol. 2000;2(6):326–332. doi: 10.1038/35014014 [DOI] [PubMed] [Google Scholar]
- 22.Credle JJ, Finer-Moore JS, Papa FR, Stroud RM, Walter P. On the mechanism of sensing unfolded protein in the endoplasmic reticulum. Proc Natl Acad Sci U S A. 2005;102(52):18773–18784. doi: 10.1073/pnas.0509487102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Korennykh A, Walter P. Structural basis of the unfolded protein response. Annu Rev Cell Dev Biol. 2012;28:251–277. doi: 10.1146/annurev-cellbio-101011-155826 [DOI] [PubMed] [Google Scholar]
- 24.Tabas I, Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol. 2011;13(3):184–190. doi: 10.1038/ncb0311-184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yamamoto K, Sato T, Matsui T, et al. Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6alpha and XBP1. Dev Cell. 2007;13(3):365–376. doi: 10.1016/j.devcel.2007.07.018 [DOI] [PubMed] [Google Scholar]
- 26.Schindler AJ, Schekman R. In vitro reconstitution of ER-stress induced ATF6 transport in COPII vesicles. Proc Natl Acad Sci U S A. 2009;106(42):17775–17780. doi: 10.1073/pnas.0910342106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Haze K, Yoshida H, Yanagi H, Yura T, Mori K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol Biol Cell. 1999;10(11):3787–3799. doi: 10.1091/mbc.10.11.3787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ye J, Rawson RB, Komuro R, et al. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol Cell. 2000;6(6):1355–1364. doi: 10.1016/S1097-2765(00)00133-7 [DOI] [PubMed] [Google Scholar]
- 29.Brown MS, Goldstein JL. A proteolytic pathway that controls the cholesterol content of membranes, cells, and blood. Proc Natl Acad Sci U S A. 1999;96(20):11041–11048. doi: 10.1073/pnas.96.20.11041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.You M, Crabb DW. Molecular mechanisms of alcoholic fatty liver: Role of sterol regulatory element-binding proteins. Alcohol. 2004;34(1):39–43. doi: 10.1016/j.alcohol.2004.07.004 [DOI] [PubMed] [Google Scholar]
- 31.Tam AB, Roberts LS, Chandra V, et al. The UPR Activator ATF6 Responds to Proteotoxic and Lipotoxic Stress by Distinct Mechanisms. Dev Cell. 2018;46(3):327–343.e7. doi: 10.1016/j.devcel.2018.04.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ramdas Nair A, Lakhiani P, Zhang C, Macchi F, Sadler KC. A permissive epigenetic landscape facilitates distinct transcriptional signatures of activating transcription factor 6 in the liver. Genomics. 2022;114(1):107–124. doi: 10.1016/j.ygeno.2021.11.034 [DOI] [PubMed] [Google Scholar]
- 33.Shuda M, Kondoh N, Imazeki N, et al. Activation of the ATF6, XBP1 and grp78 genes in human hepatocellular carcinoma: A possible involvement of the ER stress pathway in hepatocarcinogenesis. J Hepatol. 2003;38(5):605–614. doi: 10.1016/S0168-8278(03)00029-1 [DOI] [PubMed] [Google Scholar]
- 34.Cox JS, Shamu CE, Walter P. Transcriptional induction of genes encoding endoplasmic reticulum resident proteins requires a transmembrane protein kinase. Cell. 1993;73(6):1197–1206. doi: 10.1016/0092-8674(93)90648-A [DOI] [PubMed] [Google Scholar]
- 35.Tirasophon W, Welihinda AA, Kaufman RJ. A stress response pathway from the endoplasmic reticulum to the nucleus requires a novel bifunctional protein kinase/endoribonuclease (Ire1p) in mammalian cells. Genes Dev. 1998;12(12):1812–1824. doi: 10.1101/gad.12.12.1812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shamu CE, Walter P. Oligomerization phosphorylation of the Ire1p kinase during intracellular signaling from the endoplasmic reticulum to the nucleus. EMBO J. 1996;15(12):3028–3039. doi: 10.1002/j.1460-2075.1996.tb00666.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Calfon M, Zeng H, Urano F, et al. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA [published correction appears in Nature 2002 Nov 14;420(6912):202]. Nature. 2002;415(6867):92–96. doi: 10.1038/415092a [DOI] [PubMed] [Google Scholar]
- 38.Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107(7):881–891. doi: 10.1016/s0092-8674(01)00611-0 [DOI] [PubMed] [Google Scholar]
- 39.Yoshida H, Oku M, Suzuki M, Mori K. pXBP1(U) encoded in XBP1 pre-mRNA negatively regulates unfolded protein response activator pXBP1(S) in mammalian ER stress response. J Cell Biol. 2006;172(4):565–575. doi: 10.1083/jcb.200508145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yoneda T, Imaizumi K, Oono K, et al. Activation of Caspase-12, an Endoplastic Reticulum (ER) Resident Caspase, through Tumor Necrosis Factor Receptor-associated Factor 2-dependent Mechanism in Response to the ER Stress. J Biol Chem. 2001;276(17):13935–13940. doi: 10.1074/jbc.M010677200 [DOI] [PubMed] [Google Scholar]
- 41.Han D, Lerner AG, Vande Walle L, et al. IRE1α Kinase Activation Modes Control Alternate Endoribonuclease Outputs to Determine Divergent Cell Fates. Cell. 2009;138(3):562–575. doi: 10.1016/j.cell.2009.07.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hollien J, Weissman JS. Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science (80-). 2006;313(5783):104–107. doi: 10.1126/science.1129631 [DOI] [PubMed] [Google Scholar]
- 43.Hollien J, Lin JH, Li H, Stevens N, Walter P, Weissman JS. Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. J Cell Biol. 2009;186(3):323–331. doi: 10.1083/jcb.200903014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dasgupta D, Nakao Y, Mauer AS, et al. IRE1A Stimulates Hepatocyte-Derived Extracellular Vesicles That Promote Inflammation in Mice With Steatohepatitis. Gastroenterology. 2020;159(4):1487–1503.e17. doi: 10.1053/j.gastro.2020.06.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Aridor M A tango for coats and membranes: New insights into ER-to-Golgi traffic. Cell Rep. 2022;38(3):1–13. doi: 10.1016/j.celrep.2021.110258 [DOI] [PubMed] [Google Scholar]
- 46.Maiers JL, Kostallari E, Mushref M, et al. The unfolded protein response mediates fibrogenesis and collagen I secretion through regulating TANGO1 in mice. Hepatology. 2017;65(3):983–998. doi: 10.1002/hep.28921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic- reticulum-resident kinase. Nature. 1999;397(6716):271–274. doi: 10.1038/16729 [DOI] [PubMed] [Google Scholar]
- 48.Costa-Mattioli M, Walter P. The integrated stress response: From mechanism to disease. Science. 2020;368(6489). doi: 10.1126/science.aat5314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Oyadomari S, Mori M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004;11(4):381–389. doi: 10.1038/sj.cdd.4401373 [DOI] [PubMed] [Google Scholar]
- 50.Harding HP, Novoa I, Zhang Y, et al. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell. 2000;6(5):1099–1108. doi: 10.1016/S1097-2765(00)00108-8 [DOI] [PubMed] [Google Scholar]
- 51.Novoa I, Zeng H, Harding HP, Ron D. Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2α. J Cell Biol. 2001;153(5):1011–1021. doi: 10.1083/jcb.153.5.1011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jousse C, Oyadomari S, Novoa I, et al. Inhibition of a constitutive translation initiation factor 2alpha phosphatase, CReP, promotes survival of stressed cells. J Cell Biol. 2003;163(4):767–775. doi: 10.1083/jcb.200308075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Palam LR, Baird TD, Wek RC. Phosphorylation of eIF2 facilitates ribosomal bypass of an inhibitory upstream ORF to enhance CHOP translation. J Biol Chem. 2011;286(13):10939–10949. doi: 10.1074/jbc.M110.216093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Marciniak SJ, Chambers JE, Ron D. Pharmacological targeting of endoplasmic reticulum stress in disease. Nat Rev Drug Discov. 2022;21(2):115–140. doi: 10.1038/s41573-021-00320-3 [DOI] [PubMed] [Google Scholar]
- 55.Li J, Li X, Liu D, et al. Phosphorylation of eIF2α signaling pathway attenuates obesity-induced non-alcoholic fatty liver disease in an ER stress and autophagy-dependent manner. Cell Death Dis. 2020;11(12). doi: 10.1038/s41419-020-03264-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Song Q, Chen Y, Wang J, et al. ER stress-induced upregulation of NNMT contributes to alcohol-related fatty liver development. J Hepatol. 2020;73(4):783–793. doi: 10.1016/j.jhep.2020.04.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Teske BF, Wek SA, Bunpo P, et al. The eIF2 kinase PERK and the integrated stress response facilitate activation of ATF6 during endoplasmic reticulum stress. Mol Biol Cell. 2011;22(22):4390–4405. doi: 10.1091/mbc.E11-06-0510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gonen N, Sabath N, Burge CB, Shalgi R. Widespread PERK-dependent repression of ER targets in response to ER stress. Sci Rep. 2019;9(1):1–12. doi: 10.1038/s41598-019-38705-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Walter F, Schmid J, Dussmann H, Concannon CG, Prehn JHM. Imaging of single cell responses to ER stress indicates that the relative dynamics of IRE1/XBP1 and PERK/ATF4 signalling rather than a switch between signalling branches determine cell survival. Cell Death Differ. 2015;22(9):1502–1516. doi: 10.1038/cdd.2014.241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Walter F, O’Brien A, Concannon CG, Düssmann H, Prehn JHM. ER stress signaling has an activating transcription factor 6 (ATF6)-dependent “off-switch.” J Biol Chem. 2018;293(47):18270–18284. doi: 10.1074/jbc.RA118.002121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Treiman M, Caspersen C, Christensen SB. A tool coming of age: Thapsigargin as an inhibitor of sarco-endoplasmic reticulum Ca2+-ATPases. Trends Pharmacol Sci. 1998;19(4):131–135. doi: 10.1016/S0165-6147(98)01184-5 [DOI] [PubMed] [Google Scholar]
- 62.Ericson MC, Gafford JT, Elbein AD. Tunicamycin inhibits GlcNAc-lipid formation in plants. J Biol Chem. 1977;252(21):7431–7433. doi: 10.1016/s0021-9258(17)40981-1 [DOI] [PubMed] [Google Scholar]
- 63.Heifetz A, Keenan RW, Elbein AD. Mechanism of Action of Tunicamycin on the UDP-GlcNAc:Dolichyl-Phosphate GlcNAc-1 -Phosphate Transferase. Biochemistry. 1979;18(11):2186–2192. doi: 10.1021/bi00578a008 [DOI] [PubMed] [Google Scholar]
- 64.Bieberich E. Synthesis, Processing, and Function of N-glycans in N-glycoproteins. :47–70. doi: 10.1007/978-1-4939-1154-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Oslowski CM, Urano F. Measuring ER stress and the unfolded protein response using mammalian tissue culture system. Methods Enzymol. 2011;490(C):71–92. doi: 10.1016/B978-0-12-385114-7.00004-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Foufelle F, Fromenty B. Role of endoplasmic reticulum stress in drug-induced toxicity. Pharmacol Res Perspect. 2016;4(1):e00211. Published 2016 Feb 4. doi: 10.1002/prp2.211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jaskulska A, Janecka AE, Gach-Janczak K. Thapsigargin—from traditional medicine to anticancer drug. Int J Mol Sci. 2021;22(1):1–12. doi: 10.3390/ijms22010004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wang X, Xiong W, Tang Y. Tunicamycin suppresses breast cancer cell growth and metastasis via regulation of the protein kinase B/nuclear factor-κB signaling pathway. Oncol Lett. 2018;15(4):4137–4142. doi: 10.3892/ol.2018.7874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.You S, Li W, Guan Y. Tunicamycin inhibits colon carcinoma growth and aggressiveness via modulation of the ERK-JNK-mediated AKT/mTOR signaling pathway. Mol Med Rep. 2018;17(3):4203–4212. doi: 10.3892/mmr.2018.8444 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 70.Goulding LV, Yang J, Jiang Z, et al. Thapsigargin at non-cytotoxic levels induces a potent host antiviral response that blocks influenza a virus replication. Viruses. 2020;12(10). doi: 10.3390/v12101093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bhushan B, Apte U. Liver Regeneration after Acetaminophen Hepatotoxicity: Mechanisms and Therapeutic Opportunities. Am J Pathol. 2019;189(4):719–729. doi: 10.1016/j.ajpath.2018.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jaeschke H, Bajt ML. Intracellular signaling mechanisms of acetaminophen-induced liver cell death. Toxicol Sci. 2006;89(1):31–41. doi: 10.1093/toxsci/kfi336 [DOI] [PubMed] [Google Scholar]
- 73.Bajt ML, Cover C, Lemasters JJ, Jaeschke H. Nuclear translocation of endonuclease G and apoptosis-inducing factor during acetaminophen-induced liver cell injury. Toxicol Sci. 2006;94(1):217–225. doi: 10.1093/toxsci/kfl077 [DOI] [PubMed] [Google Scholar]
- 74.Chen S, Melchior WB Jr, Guo L. Endoplasmic reticulum stress in drug- and environmental toxicant-induced liver toxicity. J Environ Sci Health C Environ Carcinog Ecotoxicol Rev. 2014;32(1):83–104. doi: 10.1080/10590501.2014.881648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hur KY, So JS, Ruda V, et al. IRE1 α activation protects mice against acetaminophen-induced hepatotoxicity. J Exp Med. 2012;209(2):307–318. doi: 10.1084/jem.20111298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Uzi D, Barda L, Scaiewicz V, et al. CHOP is a critical regulator of acetaminophen-induced hepatotoxicity. J Hepatol. 2013;59(3):495–503. doi: 10.1016/j.jhep.2013.04.024 [DOI] [PubMed] [Google Scholar]
- 77.Pumford NR, Roberts DW, Benson RW, Hinson JA. Immunochemical quantitation of 3-(cystein-S-yl)acetaminophen protein adducts in subcellular liver fractions following a hepatotoxic dose of acetaminophen. Biochem Pharmacol. 1990;40(3):573–579. doi: 10.1016/0006-2952(90)90558-3 [DOI] [PubMed] [Google Scholar]
- 78.Weis M, Morgenstern R, Cotgreave IA, Nelson SD, Moldéus P. N-acetyl-p-benzoquinone imine-induced protein thiol modification in isolated rat hepatocytes. Biochem Pharmacol. 1992;43(7):1493–1505. doi: 10.1016/0006-2952(92)90207-y [DOI] [PubMed] [Google Scholar]
- 79.Zhou L, McKenzie BA, Eccleston ED Jr, et al. The covalent binding of [14C]acetaminophen to mouse hepatic microsomal proteins: the specific binding to calreticulin and the two forms of the thiol:protein disulfide oxidoreductases. Chem Res Toxicol. 1996;9(7):1176–1182. doi: 10.1021/tx960069d [DOI] [PubMed] [Google Scholar]
- 80.Rao PS, Mangipudy RS, Mehendale HM. Tissue injury and repair as parallel and opposing responses to CCl4 hepatotoxicity: A novel dose-response. Toxicology. 1997;118(2–3):181–193. doi: 10.1016/S0300-483X(97)03617-2 [DOI] [PubMed] [Google Scholar]
- 81.Iredale JP. Models of liver fibrosis: Exploring the dynamic nature of inflammation and repair in a solid organ. J Clin Invest. 2007;117(3):539–548. doi: 10.1172/JCI30542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Iracheta-Vellve A, Petrasek J, Gyongyosi B, et al. Endoplasmic reticulum stress-induced hepatocellular death pathways mediate liver injury and fibrosis via stimulator of interferon genes. J Biol Chem. 2016;291(52):26794–26805. doi: 10.1074/jbc.M116.736991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Donohue TM. Alcohol-induced steatosis in liver cells. World J Gastroenterol. 2007;13(37):4974–4978. doi: 10.3748/wjg.v13.i37.4974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Dilger K, Metzler J, Bode JC, Klotz U. CYP2E1 activity in patients with alcoholic liver disease. J Hepatol. 1997;27(6):1009–1014. doi: 10.1016/S0168-8278(97)80144-4 [DOI] [PubMed] [Google Scholar]
- 85.Aragon CMG, Rogan F, Amit Z. Ethanol metabolism in rat brain homogenates by a catalase-H2O2 system. Biochem Pharmacol. 1992;44(1):93–98. doi: 10.1016/0006-2952(92)90042-H [DOI] [PubMed] [Google Scholar]
- 86.Massey VL, Arteel GE. Acute alcohol-induced liver injury. Front Physiol. 2012;3 JUN(June):1–8. doi: 10.3389/fphys.2012.00193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Crabb DW, Liangpunsakul S. Alcohol and lipid metabolism. J Gastroenterol Hepatol. 2006;21 Suppl 3:S56–60. [DOI] [PubMed] [Google Scholar]
- 88.Seitz HK, Stickel F. Molecular mechanisms of alcohol-mediated carcinogenesis. Nat Rev Cancer. 2007;7(8):599–612. [DOI] [PubMed] [Google Scholar]
- 89.Tuma DJ, Hoffman T, Sorrell MF. The chemistry of acetaldehyde-protein adducts. Alcohol Alcohol Suppl. 1991;1:271–276. [PubMed] [Google Scholar]
- 90.Choudhury M, Friedman SL. Alcoholic liver disease and hepatitis C: a frequently underestimated combination. World J Gastroenterol. 2009;15(28):3462–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Yuan Y, Tian L, Guo Q, et al. Role of genetic factors in alcoholic liver disease. J Transl Int Med. 2019;7(2):51–57. [Google Scholar]
- 92.Szabo G Gut-liver axis in alcoholic liver disease. Gastroenterology. 2015;148(1):30–36. doi: 10.1053/j.gastro.2014.10.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.O’Shea RS, Dasarathy S, McCullough AJ. Alcoholic liver disease. Hepatology. 2010;51(1):307–328. [DOI] [PubMed] [Google Scholar]
- 94.Jou J, Choi SS, Diehl AM. Mechanisms of disease progression in alcoholic liver disease. Semin Liver Dis. 2008;28(4):370–379. [DOI] [PubMed] [Google Scholar]
- 95.Tsukamoto H, Lu SC. Current concepts in the pathogenesis of alcoholic liver injury. FASEB J. 2001;15(8):1335–1349. [DOI] [PubMed] [Google Scholar]
- 96.Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. 2005;115(2):209–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Osna NA, Donohue TM Jr, Kharbanda KK. Alcoholic Liver Disease: Pathogenesis and Current Management. Alcohol Res. 2017;38(2):147–161. [PMC free article] [PubMed] [Google Scholar]
- 98.Malhi H, Kaufman RJ. Endoplasmic reticulum stress in liver disease. J Hepatol. 2011;54(4):795–809. doi: 10.1016/j.jhep.2010.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Rutkowski DT. Liver function and dysfunction – a unique window into the physiological reach of ER stress and the unfolded protein response. FEBS J. 2019;286(2):356–378. doi: 10.1111/febs.14389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Tsedensodnom O, Vacaru AM, Howarth DL, Yin C, Sadler KC. Ethanol metabolism and oxidative stress are required for unfolded protein response activation and steatosis in zebrafish with alcoholic liver disease. DMM Dis Model Mech. 2013;6(5):1213–1226. doi: 10.1242/dmm.012195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ji C Mechanisms of alcohol-induced endoplasmic reticulum stress and organ injuries. Biochem Res Int. 2012;2012. doi: 10.1155/2012/216450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Deaciuc IV, Fortunato F, D’Souza NB, Hill DB, McClain CJ. Chronic alcohol exposure of rats exacerbates apoptosis in hepatocytes and sinusoidal endothelial cells. Hepatol Res. 2001;19(3):306–324. doi: 10.1016/S1386-6346(00)00112-1 [DOI] [PubMed] [Google Scholar]
- 103.Ji C, Mehrian-Shai R, Chan C, Hsu YH, Kaplowitz N. Role of CHOP in hepatic apoptosis in the murine model of intragastric ethanol feeding. Alcohol Clin Exp Res. 2005;29(8):1496–1503. doi: 10.1097/01.alc.0000174691.03751.11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Magne L, Blanc E, Legrand B, et al. ATF4 and the integrated stress response are induced by ethanol and cytochrome P450 2E1 in human hepatocytes. J Hepatol. 2011;54(4):729–737. doi: 10.1016/j.jhep.2010.07.023 [DOI] [PubMed] [Google Scholar]
- 105.Pavitt GD. Regulation of translation initiation factor eIF2B at the hub of the integrated stress response. Wiley Interdiscip Rev RNA. 2018;9(6):1–22. doi: 10.1002/wrna.1491 [DOI] [PubMed] [Google Scholar]
- 106.Michalopoulos GK. Liver regeneration. Liver Biol Pathobiol. 2020;12:566–584. [Google Scholar]
- 107.Michalopoulos GK, Bhushan B. Liver regeneration: biological and pathological mechanisms and implications. Nat Rev Gastroenterol Hepatol. 2021;18(1):40–55. doi: 10.1038/s41575-020-0342-4 [DOI] [PubMed] [Google Scholar]
- 108.Clemens MM, McGill MR, Apte U. Mechanisms and Biomarkers of Liver Regeneration after Drug-Induced Liver Injury. Vol 85. 1st ed. Elsevier Inc.; 2019. doi: 10.1016/bs.apha.2019.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Apte U, Bhushan B, Dadhania V. Hepatic Defenses Against Toxicity: Liver Regeneration and Tissue Repair. Vol 9. Third Edit. Elsevier; 2018. doi: 10.1016/b978-0-12-801238-3.64918-8 [DOI] [Google Scholar]
- 110.Liu Y, Shao M, Wu Y, et al. Role for the endoplasmic reticulum stress sensor IRE1α in liver regenerative responses. J Hepatol. 2015;62(3):590–598. doi: 10.1016/j.jhep.2014.10.022 [DOI] [PubMed] [Google Scholar]
- 111.Cressman DE, Diamond RH, Taub R. Rapid activation of the Stat3 transcription complex in liver regeneration. Hepatology. 1995;21(5):1443–1449. doi: 10.1016/0270-9139(95)90068-3 [DOI] [PubMed] [Google Scholar]
- 112.Wang HH, Huang JH, Sue MH, et al. Interleukin-24 protects against liver injury in mouse models. EBioMedicine. 2021;64:1–12. doi: 10.1016/j.ebiom.2021.103213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Wang J, Hu B, Zhao Z, et al. Intracellular XBP1-IL-24 axis dismantles cytotoxic unfolded protein response in the liver. Cell Death Dis. 2020;11(1). doi: 10.1038/s41419-019-2209-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Higgins GM, Anderson RM. Experimental pathology of the liver: Restoration of the liver of the white rat following partial surgical removal. Arch pathol. 1931;12:186–202. [Google Scholar]
- 115.Demetriou AA, Reisner A, Sanchez J, Levenson SM, Moscioni AD, Chowdhury JR. Transplantation of microcarrier-attached hepatocytes into 90% partially hepatectomized rats. Hepatology. 1988;8(5):1006–1009. doi: 10.1002/hep.1840080505 [DOI] [PubMed] [Google Scholar]
- 116.Demetris AJ, Kelly DM, Eghtesad B, et al. Pathophysiologic observations and histopathologic recognition of the portal hyperperfusion or small-for-size syndrome. Am J Surg Pathol. 2006;30(8):986–993. doi: 10.1097/00000478-200608000-00009 [DOI] [PubMed] [Google Scholar]
- 117.Van Thiel DH, Gavaler JS, Kam I, et al. Rapid growth of an intact human liver transplanted into a recipient larger than the donor. Gastroenterology. 1987;93(6):1414–1419. doi: 10.1016/0016-5085(87)90274-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kawasaki S, Makuuchi M, Ishizone S, Matsunami H, Terada M, Kawarazaki H. Liver regeneration in recipients and donors after transplantation. Lancet. 1992;339(8793):580–581. doi: 10.1016/0140-6736(92)90867-3 [DOI] [PubMed] [Google Scholar]
- 119.Kam I, Lynch S, Svanas G, et al. Evidence that host size determines liver size: studies in dogs receiving orthotopic liver transplants. Hepatology. 1987;7(2):362–366. doi: 10.1002/hep.1840070225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Michalopoulos GK. Liver regeneration after partial hepatectomy: Critical analysis of mechanistic dilemmas. Am J Pathol. 2010;176(1):2–13. doi: 10.2353/ajpath.2010.090675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Michalopoulos GK, DeFrances M. Liver regeneration. Adv Biochem Eng Biotechnol. 2005;93(April):101–134. doi: 10.1007/b99968 [DOI] [PubMed] [Google Scholar]
- 122.MICHALOPOULOS GK. Liver Regeneration. J Cell Physiol. 2007;211(3)(May):736–747. doi: 10.1002/JCP17299801 [DOI] [Google Scholar]
- 123.Chen F, Jimenez RJ, Sharma K, et al. Broad Distribution of Hepatocyte Proliferation in Liver Homeostasis and Regeneration. Cell Stem Cell. 2020;26(1):27–33.e4. doi: 10.1016/j.stem.2019.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Michalopoulos GK. Principles of liver regeneration and growth homeostasis. Compr Physiol. 2013;3(1):485–513. doi: 10.1002/cphy.c120014 [DOI] [PubMed] [Google Scholar]
- 125.Raven A, Lu WY, Man TY, et al. Cholangiocytes act as facultative liver stem cells during impaired hepatocyte regeneration. Nature. 2017;547(7663):350–354. doi: 10.1038/nature23015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Lu WY, Bird TG, Boulter L, et al. Hepatic progenitor cells of biliary origin with liver repopulation capacity. Nat Cell Biol. 2015;17(8):971–983. doi: 10.1038/ncb3203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Choi TY, Ninov N, Stainier DY, Shin D. Extensive conversion of hepatic biliary epithelial cells to hepatocytes after near total loss of hepatocytes in zebrafish. Gastroenterology. 2014;146(3):776–788. doi: 10.1053/j.gastro.2013.10.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Deng X, Zhang X, Li W, et al. Chronic Liver Injury Induces Conversion of Biliary Epithelial Cells into Hepatocytes. Cell Stem Cell. 2018;23(1):114–122.e3. doi: 10.1016/j.stem.2018.05.022 [DOI] [PubMed] [Google Scholar]
- 129.Kamimoto K, Kaneko K, Kok CY, Okada H, Miyajima A, Itoh T. Heterogeneity and stochastic growth regulation of biliary epithelial cells dictate dynamic epithelial tissue remodeling. Elife. 2016;5:e15034. Published 2016 Jul 19. doi: 10.7554/eLife.15034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Xie Q, Khaoustov VI, Chung CC, et al. Effect of tauroursodeoxycholic acid on endoplasmic reticulum stress-induced caspase-12 activation. Hepatology. 2002;36(3):592–601. doi: 10.1053/jhep.2002.35441 [DOI] [PubMed] [Google Scholar]
- 131.Falasca L, Tisone G, Palmieri G, et al. Protective role of tauroursodeoxycholate during harvesting and cold storage of human liver. Transplantation. 2001;71(9):1268–1276. doi: 10.1097/00007890-200105150-00015 [DOI] [PubMed] [Google Scholar]
- 132.Vilatoba M, Eckstein C, Bilbao G, et al. Sodium 4-phenylbutyrate protects against liver ischemia reperfusion injury by inhibition of endoplasmic reticulum-stress mediated apoptosis. Surgery. 2005;138(2):342–351. doi: 10.1016/j.surg.2005.04.019 [DOI] [PubMed] [Google Scholar]
- 133.Ben Mosbah I, Alfany-Fernández I, Martel C, et al. Endoplasmic reticulum stress inhibition protects steatotic and non-steatotic livers in partial hepatectomy under ischemia-reperfusion. Cell Death Dis. 2010;1(7):1–12. doi: 10.1038/cddis.2010.29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Ben Mosbah I, Duval H, Mbatchi SF, et al. Intermittent selective clamping improves rat liver regeneration by attenuating oxidative and endoplasmic reticulum stress. Cell Death Dis. 2014;5(3):1–11. doi: 10.1038/cddis.2014.65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Kolb PS, Ayaub EA, Zhou W, Yum V, Dickhout JG, Ask K. The therapeutic effects of 4-phenylbutyric acid in maintaining proteostasis. Int J Biochem Cell Biol. 2015;61:45–52. doi: 10.1016/j.biocel.2015.01.015 [DOI] [PubMed] [Google Scholar]
- 136.Vang S, Longley K, Steer CJ, Low WC. The Unexpected Uses of Urso- and Tauroursodeoxycholic Acid in the Treatment of Non-liver Diseases. Glob Adv Heal Med. 2014;3(3):58–69. doi: 10.7453/gahmj.2014.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Jia C, Dai C, Bu X, et al. Co-administration of prostaglandin E1 with somatostatin attenuates acute liver damage after massive hepatectomy in rats via inhibition of inflammatory responses, apoptosis and endoplasmic reticulum stress. Int J Mol Med. 2013;31(2):416–422. doi: 10.3892/ijmm.2012.1213 [DOI] [PubMed] [Google Scholar]
- 138.Gerakis Y, Quintero M, Li H, Hetz C. The UFMylation System in Proteostasis and Beyond. Trends Cell Biol. 2019;29(12):974–986. doi: 10.1016/j.tcb.2019.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Wei Y, Xu X. UFMylation: A Unique & Fashionable Modification for Life. Genomics Proteomics Bioinformatics. 2016;14(3):140–146. doi: 10.1016/j.gpb.2016.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Sheng L, Li J, Rao S, Yang Z, Huang Y. Cyclin-Dependent Kinase 5 Regulatory Subunit Associated Protein 3: Potential Functions and Implications for Development and Disease. Front Oncol. 2021;11(October):1–13. doi: 10.3389/fonc.2021.760429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Yang S, Yang R, Wang H, Huang Y, Jia Y. CDK5RAP3 Deficiency Restrains Liver Regeneration after Partial Hepatectomy Triggering Endoplasmic Reticulum Stress. Am J Pathol. 2020;190(12):2403–2416. doi: 10.1016/j.ajpath.2020.08.011 [DOI] [PubMed] [Google Scholar]
- 142.Lee D, Hokinson D, Park S, et al. ER stress induces cell cycle arrest at the g2/m phase through eif2α phosphorylation and GADD45α. Int J Mol Sci. 2019;20(24):1–13. doi: 10.3390/ijms20246309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Hamanaka RB, Bennett BS, Cullinan SB, Diehl JA. PERK and GCN2 contribute to eIF2alpha phosphorylation and cell cycle arrest after activation of the unfolded protein response pathway. Mol Biol Cell. 2005;16(12):5493–5501. doi: 10.1091/mbc.e05-03-0268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Blazanin N, Son J, Craig-Lucas AB, et al. ER stress and distinct outputs of the IRE1α RNase control proliferation and senescence in response to oncogenic Ras. Proc Natl Acad Sci U S A. 2017;114(37):9900–9905. doi: 10.1073/pnas.1701757114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Pluquet O, Qu LK, Baltzis D, Koromilas AE. Endoplasmic reticulum stress accelerates p53 degradation by the cooperative actions of Hdm2 and glycogen synthase kinase 3beta. Mol Cell Biol. 2005;25(21):9392–9405. doi: 10.1128/MCB.25.21.9392-9405.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Hong F, Liu B, Wu BX, et al. CNPY2 is a key initiator of the PERK-CHOP pathway of the unfolded protein response. Nat Struct Mol Biol. 2017;24(10):834–839. doi: 10.1038/nsmb.3458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Hong F, Lin CY, Yan J, et al. Canopy Homolog 2 contributes to liver oncogenesis by promoting unfolded protein response–dependent destabilization of tumor protein P53. Hepatology. 2022;(October 2020):1587–1601. doi: 10.1002/hep.32318 [DOI] [PubMed] [Google Scholar]
- 148.Hart LS, Cunningham JT, Datta T, et al. ER stress-mediated autophagy promotes Myc-dependent transformation and tumor growth. J Clin Invest. 2012;122(12):4621–4634. doi: 10.1172/JCI62973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Denoyelle C, Abou-Rjaily G, Bezrookove V, et al. Anti-oncogenic role of the endoplasmic reticulum differentially activated by mutations in the MAPK pathway. Nat Cell Biol. 2006;8(10):1053–1063. doi: 10.1038/ncb1471 [DOI] [PubMed] [Google Scholar]
- 150.Teske BF, Wek SA, Bunpo P, et al. The eIF2 kinase PERK and the integrated stress response facilitate activation of ATF6 during endoplasmic reticulum stress. Mol Biol Cell. 2011;22(22):4390–4405. doi: 10.1091/mbc.E11-06-0510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Gonen N, Sabath N, Burge CB, Shalgi R. Widespread PERK-dependent repression of ER targets in response to ER stress. Sci Rep. 2019;9(1):1–12. doi: 10.1038/s41598-019-38705-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Raffaello A, Mammucari C, Gherardi G, Rizzuto R. Calcium at the Center of Cell Signaling: Interplay between Endoplasmic Reticulum, Mitochondria, and Lysosomes. Trends Biochem Sci. 2016;41(12):1035–1049. doi: 10.1016/j.tibs.2016.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Vance JE. MAM (mitochondria-associated membranes) in mammalian cells: lipids and beyond. Biochim Biophys Acta. 2014;1841(4):595–609. doi: 10.1016/j.bbalip.2013.11.014 [DOI] [PubMed] [Google Scholar]
- 154.Yoboue ED, Sitia R, Simmen T. Redox crosstalk at endoplasmic reticulum (ER) membrane contact sites (MCS) uses toxic waste to deliver messages. Cell Death Dis. 2018;9(3):331. Published 2018 Feb 28. doi: 10.1038/s41419-017-0033-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Marchi S, Patergnani S, Missiroli S, et al. Mitochondrial and endoplasmic reticulum calcium homeostasis and cell death. Cell Calcium. 2018;69:62–72. doi: 10.1016/j.ceca.2017.05.003 [DOI] [PubMed] [Google Scholar]
- 156.Fan Y, Simmen T. Mechanistic Connections between Endoplasmic Reticulum (ER) Redox Control and Mitochondrial Metabolism. Cells. 2019;8(9):1071. Published 2019 Sep 12. doi: 10.3390/cells8091071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Rainbolt TK, Saunders JM, Wiseman RL. Stress-responsive regulation of mitochondria through the ER unfolded protein response. Trends Endocrinol Metab. 2014;25(10):528–537. doi: 10.1016/j.tem.2014.06.007 [DOI] [PubMed] [Google Scholar]
- 158.Gutiérrez T, Simmen T. Endoplasmic reticulum chaperones tweak the mitochondrial calcium rheostat to control metabolism and cell death. Cell Calcium. 2018;70:64–75. doi: 10.1016/j.ceca.2017.05.015 [DOI] [PubMed] [Google Scholar]
- 159.Gansemer ER, McCommis KS, Martino M, et al. NADPH and Glutathione Redox Link TCA Cycle Activity to Endoplasmic Reticulum Homeostasis [published correction appears in iScience. 2022 Dec 05;25(12):105716]. iScience. 2020;23(5):101116. doi: 10.1016/j.isci.2020.101116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Ajoolabady A, Kaplowitz N, Lebeaupin C, et al. Endoplasmic reticulum stress in liver diseases. Hepatology. 2022;(March):1–21. doi: 10.1002/hep.32562 [DOI] [PMC free article] [PubMed] [Google Scholar]