

Summary
The RNA-binding protein PARP13 is a primary factor in the innate antiviral response, which suppresses translation and drives decay of bound viral and host RNA. PARP13 interacts with many proteins encoded by interferon-stimulated genes (ISG) to activate antiviral pathways including co-translational addition of ISG15, or ISGylation. We performed enhanced crosslinking immunoprecipitation (eCLIP) and RNA-seq in human cells to investigate PARP13’s role in transcriptome regulation for both basal and antiviral states. We find that the antiviral response shifts PARP13 target localization, but not its binding preferences, and that PARP13 supports the expression of ISGylation-related genes, including PARP13’s cofactor, TRIM25. PARP13 associates with TRIM25 via RNA-protein interactions, and we elucidate a transcriptome-wide periodicity of PARP13 binding around TRIM25. Taken together, our study implicates PARP13 in creating and maintaining a cellular environment poised for an antiviral response through limiting PARP13 translation, regulating access to distinct mRNA pools, and elevating ISGylation machinery expression.
Subject areas: Molecular biology, Immune response, Cell biology
Graphical abstract
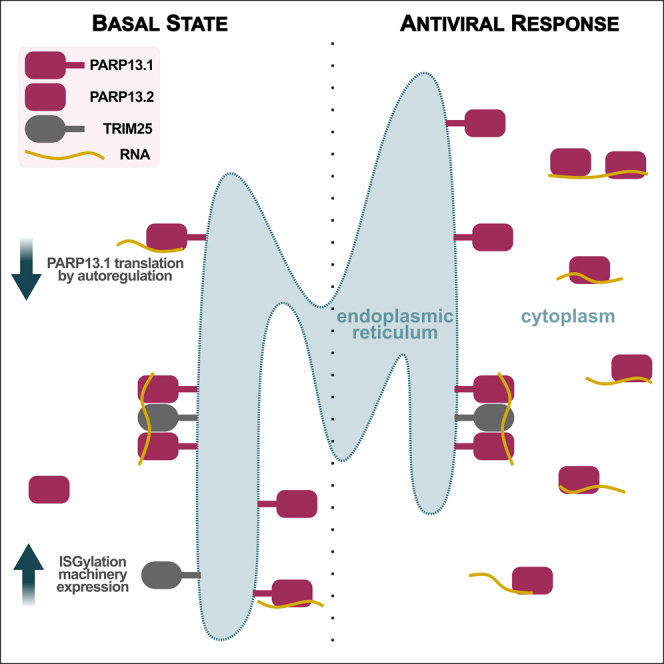
Highlights
-
•
PARP13 regulates PARP13.1 isoform, not virus-induced PARP13.2, via a unique 3′UTR
-
•
Antiviral response shifts PARP13 target localization, but not motif preference
-
•
PARP13 supports gene expression related to the ISGylation pathway, e.g., TRIM25
-
•
PARP13 binds periodically around antiviral co-factor TRIM25 transcriptome-wide
Molecular biology; Immune response; Cell biology
Introduction
The innate immune response is the first-line defense against viral pathogens. To confer a broad defense, components of the innate immune response target conserved aspects of pathogen structure, invasion, and proliferation. PARP13, also known as ZAP (zinc antiviral protein) and coded by the ZC3HAV1 gene, contributes to host defense against a plethora of RNA and DNA viruses, including members of Togaviridae (e.g., Sindbis virus), Coronaviridae (e.g., SARS-CoV-2), Herpesviridae (e.g., herpes simplex), and Retroviridae (e.g., human immunodeficiency virus) [reviewed1]. Upon viral infection, PARP13 activates innate immune pathways through interaction with the viral RNA-sensing protein RIG-I and enhancing its association with downstream effectors.2 PARP13 directly binds target viral RNA3,4 and can mediate both translation repression of bound RNAs by interfering with initiation5 and mRNA decay by recruiting the exosome and associated mRNA degradation machinery.6,7,8,9 PARP13-mediated decay requires translational repression, but translational repression can occur without degradation of target mRNAs.5
Humans have four PARP13 isoforms, with two being most abundant: the longer common isoform, PARP13.1, contains a catalytically inactive but antivirally important ADP-ribosyltransferase domain and membrane-localization CaaX motif at the C-terminus, both of which are absent in the shorter, cytoplasmic isoform, PARP13.2.10,11,12,13 PARP13.1 is constitutively expressed and localized to the endoplasmic reticulum (ER), and both isoforms are induced by viral infection or type I interferons (IFNs), with PARP13.2 more upregulated than PARP13.1.2,14 Both isoforms share an N-terminus that contains four zinc finger domains; these domains preferentially bind CG-rich viral RNA15,16 and are necessary for PARP13 antiviral activity.17,18 PARP13 also directly regulates the decay of human TRAILR4 mRNA through binding to its 3′UTR.19,20 Although PARP13 binds host RNAs, as shown by biochemical cross-linking experiments,19,20 the identities of genome-wide PARP13 host targets are not known.
PARP13 interacts with many proteins encoded by IFN-stimulated genes (ISGs) to perform its antiviral activities, such as activation of the pathway mediated by ISGylation—the co-translational conjugation of the ubiquitin-like protein, interferon-stimulating gene-15 (ISG15), onto proteins.21,22,23,24,25 In particular, TRIM25, an E3 ligase of ISG15, is the primary cofactor of PARP13 and is necessary for PARP13’s role in the immune response.24,26,27 TRIM25 binding to PARP13 requires the SPRY domain and N-terminus, respectively.24,26,28,29 Antiviral activity requires TRIM25 and PARP13 co-localization at membranes and the RNA-binding domains of both proteins.11,17,18,24,28,30,31 Ablation of TRIM25’s putative RNA-binding domain in its SPRY domain decreases PARP13 interaction,28 suggesting the interaction may be mediated via protein-RNA interaction and indicating the possibility of co-regulation of shared targets.
Here, we integrated RNA-seq and eCLIP-seq datasets to assess the role of PARP13 expression and RNA binding in transcriptome regulation in human cells. We explored transcriptomic signatures that correspond to basal PARP13 activity as well as shifts that occur exclusively in the presence of an antiviral response. PARP13 autoregulates its own long-isoform in basal states and contributes to the maintenance of the expression of transcripts essential for the ISGylation response. We also demonstrate that TRIM25 and PARP13 bind proximally on shared transcripts using spatial correlation analysis and that the TRIM25:PARP13 complex is stabilized by protein-RNA interactions.
Results
PARP13 regulates cellular homeostasis and the innate antiviral response
To study the role of PARP13 in basal and antiviral states, we performed RNA-seq on wild-type (WT) and PARP13 knock-out (KO) HEK293T cells treated with triphosphorylated single-stranded RNA (3p-RNA) as an RNA virus mimic or a single-stranded (ss)RNA of the same length that elicits no observable immune response2 (Figures 1 and S1A). WT cells demonstrated a strong, unidirectional transcriptomic shift upon 3p-RNA treatment versus ssRNA treatment, with many of the most differentially expressed genes (DEG) associated with the innate immune response (Figure 1A, y axis). In contrast, the transcriptomic response of PARP13 KO cells treated with 3p-RNA is dampened relative to WT, indicating the importance of PARP13 in the immune response (Figure 1A, x axis). To interrogate the role of PARP13 while controlling for immune perturbation state, we compared WT and PARP13 KO cell expression after both ssRNA and 3p-RNA treatment and observed many up- and downregulated DEGs (Figure 1B), suggesting that PARP13 plays an additional constitutive role outside of antiviral response. To validate our findings, we performed qPCR on a subset of genes with at least three biological replicates and confirmed that immune-associated gene expression upon 3p-RNA treatment is reduced in PARP13 KO cells relative to WT (Figures 1C and S1B). Consistent with the published result that TRAILR4 is upregulated in PARP13 KO cells,19 we also observed a ∼5-fold increase in TRAILR4 expression in PARP13 KO cells by qPCR (Figures 1C and S1B). Similarly, our RNA-seq data showed a 3.5-fold TRAILR4 increase in ssRNA-treated PARP13 KO cells and a 7-fold increase in 3p-RNA-treated cells (Figure 1B).
Figure 1.

PARP13 expression affects expression of many constitutive transcripts and is required for transcriptomic upregulation of innate immune response
(A) Log2 fold change expression data 24 h after treatment with +3p-RNA vs. +ssRNA in HEK293T WT and PARP13 KO cells (n = 2). A selection of innate immune response genes is highlighted in red.
(B) Log2 fold change expression data from HEK293T WT vs. PARP13 KO cell 24 h after treatment with +3p-RNA or +ssRNA (n = 2).
(C) Scaled average expression relative to WT + ssRNA measured by qPCR in WT HEK293T and PARP13 KO cells and normalized to GAPDH or TBP expression (n ≥ 3). Complete replicate information Figure S1B.
(D) IC pattern weights across RNA-seq samples. The color scale represents values of the linear mixing matrix centered at 0 where columns of S contain the independent components, A is a linear mixing matrix, and data matrix X is considered to be a linear combination of non-Gaussian components such that X = SA.
(E) Top GO terms that are enriched among genes that contribute to IC2, shown in (D).
(F) Top GO terms that are enriched among genes that contribute to IC3, shown in (D).
We observed that 138 DEGs of 3p-RNA-treated WT versus PARP13 KO cells (Figure 1B) are also differentially-expressed in 3p-RNA versus ssRNA treatment in WT cells (Figure 1A), and 388 DEGs of 3p-RNA-treated WT versus PARP13 KO cells are also DEGs in ssRNA-treated WT versus PARP13 KO (Figure 1B). The large number of overlapping DEGs reveals that there are multiple simultaneous biologically important signals in our data, which would make simple pairwise comparisons of DEG insufficient. We performed independent component analysis (ICA) across all RNA-seq samples to deconvolute simultaneous transcriptomic programs. Three independent components were adequate to separate samples into groups of anticipated biological relevance, suggesting that the biological signals in our experiment are robust and that replicates have comparatively minor technical variation (Figure 1D). We were able to identify genes that have consistent expression across all samples (IC1), genes that are differentially expressed between WT and PARP13 KO samples, regardless of treatment (IC2), and genes that are only differentially expressed in WT cells treated with 3p-RNA (IC3).
To investigate the functions of genes that drive IC2, we ordered genes by their weight in this individual component and performed gene ontology (GO) analysis. Transcripts persistently differentially expressed between WT and PARP13 KO samples are generally associated with the ER and are involved in stress responses (Figure 1E), which is consistent with the membrane localization and constitutive expression of PARP13.1 and recapitulates transcriptomic differences previously shown between untreated WT and PARP13 KO cells.11,12,19 GO analysis of ranked IC3 gene weights showed that genes only upregulated in WT cells treated with 3p-RNA are generally involved in the innate antiviral response (Figure 1F), which is consistent with the well-documented role of PARP13, where 3p-RNA-treated samples phenocopy an antiviral state [reviewed1].
PARP13 suppresses PARP13.1, but not PARP13.2, translation in the absence of an antiviral response
To study the role of PARP13 RNA-binding, we performed PARP13 eCLIP-seq32 on WT and PARP13 KO cells treated with 3p-RNA or ssRNA. To globally characterize PARP13’s RNA binding preferences across treatments, we identified binding sites in the PARP13 eCLIP-seq data for ssRNA- and 3p-RNA-treated samples. ssRNA-treated samples showed 1,405 peaks across 730 genes, and 3p-RNA-treated samples showed 1,950 peaks across 1,013 genes. The majority of PARP13 peaks mapped to coding regions and the 3′UTR of mRNAs, with little difference between treatments ( p = 0.03, Figure 2A).
Figure 2.

PARP13 selectively binds PARP13.1 and regulates its translation via the 3′ UTR
(A) Proportions of PARP13 binding sites that fall within the indicated gene regions of mRNAs.
(B) Log2 fold-change RNA expression of PARP13-bound genes in PARP13 KO versus WT +ssRNA-treated cells, separated by what region of the transcript PARP13 binds. Transcripts where PARP13 binds multiple types of gene regions were excluded. Colors correspond to the gene region legend on the left. T-test, ∗∗ = p < 0.01.
(C) RNA immunoprecipitation using GFP-trap beads against exogenous GFP-PARP13.1 in HEK293T cells. Error bars are +/− 1sd.
(D) Aligned RNA-seq (top) and eCLIP-seq (middle) reads within PARP13 gene region for +ssRNA and +3p-RNA treatments with gray boxes surrounding the PARP13.1-specific (left) and PARP13.2-specific (right) 3′UTRs. Red and blue alignments correspond to duplicate experiments. Two PARP13 binding peaks located within the PARP13.1 3′UTR identified by CLIPper (Methods) are indicated below the aligned reads. Bottom: a splicing map of the two PARP13 isoforms, which are transcribed right to left.
(E) Western blot of PARP13 expression in WT and PARP13 KO HEK293T cell lysates 24 h after +ssRNA or +3p-RNA treatment, with beta-actin as a loading control. Also Figure S2A.
(F) Luciferase construct map and design of luciferase expression experiments.
(G) Transcript levels of Renilla luciferase with either PARP13.1 or PARP13.2 3′UTR treated with ssRNA or 3p-RNA (n = 5). Within each experiment, Renilla expression was normalized to firefly expression and compared to normalized Renilla expression for a construct without a 3′UTR insert. ns = not significant.
(H) Luminescence of Renilla luciferase (n = 3) with the same normalization as (G). Paired t-test, ∗∗ = p < 0.01; ns = not significant.
PARP13 regulates mRNA stability via interactions with XRN1-dependent 5′-3′ decay and exosome-dependent 3′-5′ decay machinery and has been shown to target endogenous transcripts for decay via 3′UTR binding in the absence of an antiviral response.7,8,19 Proteins bound by PARP13 in ssRNA-treated samples demonstrated both increased and decreased expression in PARP13 KO cells compared to WT (Figure 2B). Expression of decay pathway proteins PARN, EXOSC5, EXOSC7, and XRN1 was consistent across cell lines. Notably, proteins bound by PARP13 within the 3′UTR showed a greater average difference in PARP13 KO versus WT gene expression when compared to proteins bound by PARP13 within the CDS (Figure 2B). Taken together, these data suggest that PARP13 does not facilitate decay of all targets, and binding location may influence how PARP13 affects target transcripts.
To verify eCLIP-seq identified targets, we performed qPCR on the immunoprecipitated (IP) RNA of GFP-PARP13.1 (Figure 2C). The negative control target transcript, HDAC3, showed no enrichment in the GFP-PARP13.1 IP over input, whereas the PARP13 target transcripts—PARP13, ATP6AP1, EIF1, SLC3A2, STT3A, TMED3, and XBP1—that were identified in both ssRNA and 3p-RNA treatments by eCLIP did. Since the GFP-PARP13.1 construct lacks a 3′UTR, we were able to design primers against 3′UTRs of select isoforms to assess endogenous PARP13 enrichment. Interestingly, PARP13.1 mRNA was enriched 40-fold more than PARP13.2 mRNA, indicating that the PARP13 protein does not bind both PARP13 isoform transcripts to the same extent.
We further characterized how PARP13 regulates its own transcripts. The RNA-seq data demonstrated that both isoforms were upregulated upon mock-viral infection, but PARP13.1 showed a 4.4-fold increase in expression based on aligned reads within its specific 3′UTR while PARP13.2 showed a 31.2-fold increase in reads based on its 3′UTR (Figure 2D, top), which is reflected in the relative protein expression levels of PARP13 isoforms (Figures 2E and S2A), consistent with prior publications.2 In contrast, eCLIP data show an enrichment for PARP13 binding within the PARP13.1 3′UTR but not within the PARP13.2 3′UTR over background in both treatments (Figure 2D, middle), which may account for the differential enrichment of PARP13 mRNAs seen in our RIP-qPCR results (Figure 2C).
Since the two PARP13 isoforms contain different 3′UTRs, we used a dual-reporter luciferase construct to assess the effect of PARP13 binding on these UTRs. We cloned the PARP13.1 or PARP13.2 3′UTR downstream of the Renilla luciferase gene, with the firefly luciferase under an independent promoter as a control (Figure 2F).33 By using WT and PARP13 KO cells, our dual luciferase system allowed us to assess the effect of PARP13 expression on the respective 3′UTRs at the mRNA and protein levels. We co-transfected the PARP13.1 or PARP13.2 3′UTR luciferase constructs into WT and PARP13 KO cells along with either ssRNA or 3p-RNA; 24 h later, we assessed the transcript expression by qPCR and luminescence of the Renilla luciferase and normalized with firefly luciferase expression. We observed that mRNA levels for luciferase constructs with either the PARP13.1 or PARP13.2 3′UTRs were the same in WT and PARP13 KO cells for both treatments (Figure 2G), suggesting that PARP13 binding to the PARP13.1 3′UTR does not drive its mRNA decay. Luminescence—and therefore protein levels—for the PARP13.2 3′UTR luciferase construct are the same in WT and PARP13 KO cells for both treatments (Figure 2H), suggesting the PARP13.2 isoform is not subject to PARP13 regulation. The lack of a PARP13-dependent shift in protein expression of the PARP13.2 3′UTR construct is consistent with our eCLIP data which showed that PARP13 does not bind the PARP13.2 3′UTR above background (Figure 2D). In contrast, protein levels for the PARP13.1 3′UTR construct are higher in PARP13 KO cells with ssRNA treatment but the same in WT and PARP13 KO cells with the 3p-RNA treatment (Figure 2H). The treatment-specific protein inhibition suggests that PARP13 binds the 3′UTR and inhibits PARP13.1 translation at basal state, but such inhibition is relieved upon antiviral response.
The antiviral response shifts PARP13 target localization, but not its binding preferences
Based on our eCLIP-seq data, 518 PARP13 targets were common across samples, but 212 PARP13 targets were unique to ssRNA-treated cells and 495 targets were unique to 3p-RNA-treated cells (Figure 3A). To determine whether the shift in PARP13 targets is attributable to differences in binding sequence preference, we calculated the enrichment of dinucleotides within PARP13 binding peaks, normalized to the dinucleotide frequency of all expressed transcripts. There was no shift in dinucleotide preference between treatments, with CG substantially enriched within PARP13-bound regions (Figure 3B). This CG binding preference is congruent with prior evidence from viral RNA studies.15,16 Identification of hexameric motifs within peaks also demonstrated the presence of an invariant CG dinucleotide and consistent binding preference across conditions (Figure 3C). The invariability of the binding motif across treatments can be explained by the fact that PARP13.1 and PARP13.2 share all RNA binding domains.
Figure 3.

Antiviral response shifts PARP13 target localization but not binding preferences
(A) Venn diagram of transcripts that have at least one significantly bound region across both treatments.
(B) Dinucleotide enrichment within PARP13-bound regions, normalized to dinucleotide expression of all transcribed RNAs.
(C) A hexameric motif calculated by HOMER34 that is enriched in samples with either +ssRNA (top) and +3p-RNA (bottom) treatments.
(D) Proportions of previously identified cellular localizations35 for PARP13-bound transcripts across both treatments.
PARP13.1 is membrane-associated and available to bind transcripts in all states, whereas PARP13.2 is cytoplasmic and constitutes a sizable proportion of the PARP13 population upon 3p-RNA treatment.2,11,12 Given that PARP13.1 is associated with ER-derived membranes and the RNA-seq data analyses implicated PARP13 in ER regulation (Figure 1G), we hypothesized there may be differences in target transcript localization of PARP13 isoforms. Since PARP13.2 is selectively upregulated upon 3p-RNA treatment (Figure 2D), we reasoned that PARP13 targets unique to the 3p-RNA-treated cells were predominantly occupied by PARP13.2, whereas targets identified in ssRNA treatment would be bound by PARP13.1. We explored the relationship with transcript localization to the ER using available data from APEX-RIP, which used engineered ascorbate peroxidase-catalyzed proximity biotinylation of endogenous proteins and RNA immunoprecipitation to isolate transcripts localized to a variety of subcellular compartments including the ER.35 Transcripts that are bound by PARP13 in ssRNA treatment are enriched in the ER (Fisher’s exact test, p = 1.67 × 10−111). In contrast, transcripts that are only PARP13-bound in 3p-RNA treatment are enriched in the cytoplasm (Fisher’s exact test, p = 6.71 × 10−120, Figure 3D). Of the 495 unique PARP13-bound targets identified in 3p-RNA-treated cells, 489 remain expressed in ssRNA-treated cells, so this shift in localization is not due to changes in target availability and may instead be explained by the upregulation of cytoplasmic PARP13.2 upon viral response.
PARP13 supports expression of transcripts related to the ISGylation and RIG-I pathway
Besides focusing on directly bound PARP13 targets, we are also interested in which genes are regulated in a PARP13-dependent manner and upregulated for antiviral responses. Based on our RNA-seq data, 12 genes were downregulated in PARP13 KO cells compared to WT cells at basal states and upregulated upon 3p-RNA treatment in WT cells (Table S1). Of these 12 genes, five are involved in ISGylation, which has broad antiviral effects and the expression of which is initiated by RIG-I-activated IRFs and NFkB [reviewed36]. Notably, ISG15, its E2 ligase UBE2L6, E3 ligase TRIM25, and its downstream activator RIG-I have all been shown to enhance the ability of PARP13 to mediate antiviral activities.23 GO analysis of IC3, a signal corresponding to genes that are only differentially expressed in WT cells treated with 3p-RNA, further demonstrated an enrichment for genes associated with ISG15 protein conjugation (Figures 1F and 1H). Given that PARP13 also binds the TRIM25, RIG-I, and unconjugated ISG15 proteins (Figure 4A),2,24,26,28,37,38 we aimed to directly assess the effect of PARP13 expression on the ISGylation pathway.
Figure 4.

Genes associated with ISGylation are dysregulated in PARP13 KO cells
(A) A schematic of the ISGylation pathway that includes ISG15, a ubiquitin-like protein; UBA7, the E1; UBE2L6, the E2; HERC5 and TRIM25, E3 ligases; and USP18 and USP41, which remove ISG15 from conjugated proteins. RIG-I, which is ubiquitinated by TRIM25, activates expression of ISG15. Genes that have been shown to promote PARP13 antiviral activity are highlighted in purple. Genes identified by the RNA-seq data to be down-regulated in PARP13 KO cells in control condition and upregulated upon mock viral infection in WT cells are highlighted in gold. Proteins shown to bind PARP13 are connected by dotted lines.
(B) qPCR of ISGylation genes in untreated WT and PARP13 KO HEK293T cells (n = 3). UBA7 expression was too low and beyond the limit of detection. Error bars are +/− 1sd.
(C) Western blot of PARP13 and TRIM25 expression in WT and PARP13 KO HEK293T cell lysates, with beta-actin as a loading control. Also Figure S2B.
(D) PARP13 KD time course in WT HEK293T cells assessing the expression of ISGylation genes shown in (B) to have reduced expression in PARP13 KO cells. Error bars are +/− 1sd.
Among genes involved in ISGylation, UBA7 and HERC5 had the same expression between WT and PARP13 KO cells in control treatment. qPCR using untreated WT and PARP13 KO samples recapitulated the RNA-seq findings, with UBE2L6, RIG-I, ISG15, USP18, USP41 and TRIM25, but not HERC5, downregulated in PARP13 KO cells (Figure 4B). eCLIP analyses identified that PARP13 binds to an intron of TRIM25 and the CDS of ISG15 upon 3p-RNA treatment. Corresponding to the reduced transcript expression observed, TRIM25 protein expression was reduced ∼50% in PARP13 KO cells (Figures 4C and S2B).
To exclude the possibility of clonal selection during the generation of PARP13 KO cells causing the observed ISGylation factor downregulation, we performed a PARP13 knockdown time course using siRNAs in WT cells. PARP13 expression decreased within 12 h post-siPARP13 treatment, followed by downregulation of ISG15, TRIM25, UBE2L6, USP18, and USP41 by 36 h post-treatment (Figure 4D). Though RIG-I did not demonstrate PARP13 expression-dependent downregulation within the 60-h time course, the remaining 17.5% PARP13.1 expression may be sufficient to maintain normal RIG-I expression. These data suggest that PARP13 expression, rather than change in RIG-1 expression, is responsible for the steady-state reduction of ISGylation gene expression observed in PARP13 KO cells.
Binding to common transcripts reinforces TRIM25:PARP13 interactions
The ISG15 E3 ligase TRIM25 is also a primary co-factor of PARP13 for immune regulation. Both proteins require their RNA-binding domains to perform their roles in the innate antiviral response.17,18,30,31 To interrogate whether the requirement of RNA binding for these proteins is related to their interaction with one another, we used CLIP-seq data to identify shared targets of TRIM25 and PARP13. Although no datasets of TRIM25 are published for 293T cells, comparison with CLIP-seq data from HeLa cells transiently expressing T7-tagged TRIM2528 revealed that 87% of all identified PARP13 targets were also bound by TRIM25, which is far greater than could be expected by chance (Figure 5A, Fisher’s exact test, p = 2.16 × 10−126). There is not a significant shift in the proportion of PARP13 targets across treatment conditions that are bound by TRIM25 (Figure 5B).
Figure 5.

TRIM25 and PARP13 interact via both RNA-dependent and RNA-independent contacts
(A) Venn diagram of previously identified TRIM25 targets from HeLa cells transiently expressing T7-tagged TRIM2528 and all PARP13 targets from HEK293T cells.
(B) Proportions of PARP13-bound transcripts across both treatments that are also targets of TRIM25 28.
(C) Spatial correlation of PARP13- and TRIM25-bound regions across the HEK293T transcriptome calculated by nearBynding.39 Black line indicates mean shuffled background signal with standard error (n = 10,000) and blue line represents mean spatial correlation signal with standard error (n = 3). Arrows are the same on the left and right plots and represent periodic regions of significant spatial correlation between TRIM25 and PARP13. Left: Correlation of TRIM25 binding peaks from HeLa cells and PARP13 binding peaks from ssRNA-treated HEK293T cells. Right: Correlation of TRIM25 binding peaks and PARP13 binding peaks from 3p-RNA-treated cells.
(D) Three possible models of TRIM25 and PARP13 binding proximity to explain the periodic signal observed in the spatial correlation in (C). 1. Multiple TRIM25 proteins bind near, upstream, and downstream a single PARP13 protein. 2. Multiple PARP13 proteins bind near, upstream, and downstream of a single TRIM25 protein. Or 3. a combination of 1 and 2, where multiple binding events of both proteins are proximal.
(E) Auto-correlation of TRIM25 binding. Spatial correlation of a binding site to itself equals 1 at position 0. The same auto-correlation with a magnified y-axis is shown in the top right.
(F and G) Auto-correlation of PARP13 binding with ssRNA treatment (F) and 3p-RNA treatment (G). Spatial correlation of a binding site to itself equals 1 at position 0, but peaks upstream and downstream show additional PARP13 binding sites nearby. The same auto-correlation with a magnified y-axis is shown in the top right.
(H) An example PARP13 and TRIM25 target, ATP6AP1. A splicing map of ATP6AP1 is highlighted by a red box corresponding to the region for which aligned PARP13 eCLIP-seq reads are shown for two replicates in ssRNA-treated cells. Significant binding peaks are indicated below for PARP13 (blue) and TRIM25 (red).
(I) Western blot for GFP and TRIM25 expression from co-immunoprecipitation using GFP-trap beads against exogenous GFP-PARP13 constructs in HEK293T cells. Also Figure S2C.
Although TRIM25 shares many targets with PARP13, it was unclear whether the proteins bind in proximity to each other. Using the transcriptome-wide cross-correlation tool nearBynding,39 we spatially compared the binding peaks of PARP13 and TRIM25 to determine the location of PARP13 binding relative to TRIM25. TRIM25-bound regions positively correlated with PARP13-bound regions identified for both ssRNA and 3p-RNA treatments at relative position 0, which shows that when PARP13 and TRIM25 bind the same transcript, they bind at approximately the same location (Figure 5C).
In addition, there were periodic intervals of positive spatial correlation both upstream and downstream of this primary site of colocalization, especially for correlation of TRIM25 with ssRNA PARP13 samples (Figure 5C, left). This phenomenon could be caused by 1) having multiple TRIM25 binding sites located upstream and/or downstream of a single PARP13 binding site at regular intervals, 2) having multiple PARP13 sites upstream and downstream of a single TRIM25 site, or 3) a mixture of these two scenarios (Figure 5D). To distinguish between these binding geometry possibilities, we auto-correlated the TRIM25 and PARP13 peak data. Auto-correlation of TRIM25-bound regions shows a single peak, suggesting that TRIM25 does not regularly bind a set distance away from itself upstream or downstream (Figure 5E). In contrast, auto-correlation of PARP13-bound regions for both ssRNA and 3p-RNA treatment shows small peaks upstream and downstream of the central signal, signifying that PARP13 tends to bind near itself at set intervals (Figures 5F and 5G). The proportion of binding peaks that contain the canonical “CG” motif (Figure 3C) and the proportion of binding peaks within different gene regions (Figure 2A) is the same for the subset of genes bound multiple times versus genes that were only bound once, indicating that the presence of proximal binding locations does not affect the binding sequence preference of PARP13. Previously published PARP13 CLIP data also show that PARP13 binds various viral transcripts at ∼200 nt intervals,16,20 which suggests that PARP13 often occupies multiple, regularly spaced sites. Therefore, the periodicity observed in the TRIM25 and PARP13 correlation is generally due to a single TRIM25 molecule binding near multiple PARP13 molecules (model 2 of Figures 5D and 5H).
To assess the degree to which PARP13 RNA binding contributes to its well-documented association with TRIM25,24,26,28,29,31 we performed co-immunoprecipitation experiments. We transfected GFP-tagged PARP13.1, PARP13.2, and PARP13.2(Q) mutant, which is RNA-binding-deficient due to quintuple mutations,19 into WT cells and immunoprecipitated using GFP antibodies. Consistent with prior literature, PARP13.1 bound TRIM25 better than PARP13.2 (Figures 5I and S2C).11,24 The RNA-binding-deficient PARP13.2(Q) construct bound TRIM25 less effectively than PARP13.2, suggesting that RNA binding does contribute to the interaction of PARP13 and TRIM25 (Figure 5I). However, PARP13.2(Q) pulled down still more TRIM25 than the vector control, demonstrating that RNA co-binding alone cannot explain the entirety of PARP13 and TRIM25’s interaction (Figure 5I). This is consistent with literature evidence demonstrating a reduction but not ablation of TRIM25:PARP13 interaction upon addition of RNase in whole cell lysate.29 Therefore, the stability of the TRIM25:PARP13 complex depends on both direct protein binding and protein-RNA-protein interactions.
Discussion
Integrative genomics provides a robust framework for analyzing cellular shifts, such as during antiviral response. Although recent studies have used diverse genomics methods to study the effect of PARP13, none have captured PARP13 RNA binding during both basal state and antiviral response, and the emphasis of the prior studies have been on the effect of PARP13 binding to viral RNA.15,16,20 Despite PARP13’s well-known role in RNA decay, there are few transcriptome-wide RNA-seq experiments comparing WT and PARP13 KD or KO cells19,20—most employ qPCR4,15—and none have deconvoluted transcriptomic signals to isolate the components of PARP13’s activity attributable to its role in the antiviral response versus cellular homeostasis. Here, we leverage eCLIP-seq and RNA-seq data to provide evidence that PARP13 may serve to create and maintain a cellular environment poised for an antiviral response through limiting PARP13 translation, controlling access to distinct mRNA pools, and regulating ISGylation machinery expression in the absence of infection.
We observe that PARP13 sequence-based binding specificity remains constant, regardless of antiviral response. However, the cellular localization of PARP13-bound transcripts shifts, likely due to a disproportionate upregulation of the cytoplasmic PARP13.2 isoform relative to the ER-derived membrane-associated PARP13.1 isoform. A context-specific shift in PARP13 localization would provide the opportunity for the cell to create a poised cytoplasmic PARP13-targeted RNA population, sequestered from regulation in healthy circumstances. Transcript localization may regulate which targets PARP13 has access to, so that certain transcripts are only bound and regulated by PARP13 in circumstances of antiviral response while other PARP13 targets are constitutively available for binding and regulation.
Autoregulation via binding to their own mRNA is common among RBPs, especially in response to stress. For example, some proteins bind their own pre-mRNA to affect splicing and drive transcript decay,40,41 some bind their own mRNA’s 3′UTR to improve stability,42 and others alter the structure of their mRNA’s 5′UTR to tune translation.43 Approximately one-third of PARP13 binding events are within 3′UTRs. The fact that PARP13.1 and PARP13.2 isoforms do not share 3′UTRs provides an opportunity for isoform-specific PARP13 regulation. Our observation that the 3′ UTR of PARP13.1 is only regulated by PARP13 in the absence of an antiviral response further supports a shift in PARP13 targeting and regulation between cells in a basal state and cells staging an antiviral response.
This study assesses, for the first time, the role of PARP13 on host gene pathways using an established innate immune response model.2,15,24 PARP13 not only directly binds multiple proteins of the ISGylation pathway but also affects the expression of their transcripts. Depletion of PARP13 globally downregulated expression of genes implicated in the innate antiviral response upon treatment with a viral mimic, in agreement with PARP13’s studied role as a stimulator of antiviral response signaling.2 But PARP13 depletion also depressed expression of many factors of the ISGylation pathway in basal state cells, suggesting that PARP13 plays an additional role in keeping the ISGylation response primed for stress responses. Since our data did not identify significant PARP13 binding sites within ISGylation machinery transcripts in ssRNA-treated cells, we infer that depletion of PARP13 affects ISGylation machinery expression indirectly. PARP13 may directly affect expression of a single protein such as a transcription factor or decay factor that regulates the expression of multiple proteins in the ISGylation pathway. The relationship between PARP13 and ISGylation is likely intricate, with cell-line and stimulus-dependent effects.19 For instance, PARP13 KO Huh7 cells exhibit prolonged ISG15 expression upon poly U/UC RNA treatment, but not with recombinant IFN-β.4 And PARP13 KD HeLa cells demonstrate significant upregulation of ISG15, HERC5, and RIG-I.19 Further studies should deconvolute the role of PARP13 in the primary activation of the ISGylation pathway in the basal state from its secondary effect supporting the ISGylation-dependent antiviral response.
A limitation of applying spatial correlation methods to assess TRIM25 and PARP13 co-localization is that it is impossible to discern whether all PARP13 sites are bound simultaneously, only that they are regularly spaced relative to TRIM25 binding sites and each other. A recent study suggests that PARP13 RNA binding may even compete with its binding to TRIM25.31 Our data cannot inform us whether TRIM25 binds different target transcripts post-viral infection, so it is possible that TRIM25 binding in 3p-RNA-treated cells may have a different relationship with PARP13 binding than what we are observing. The reason for PARP13’s binding periodicity is unclear and ought to be more deeply explored via low-throughput methods on its possible role in translation and decay. Since this periodicity is apparent across CLIP experiments from multiple labs, it is unlikely a technical artifact of any single CLIP-seq protocol or peak-calling algorithm.16,20 Both PARP13 and TRIM25 dimerize and bind with many proteins to form large complexes,3,44 but no modeling so far has predicted a nucleoprotein complex sufficiently large to explain the spatial correlation signal observed that spans hundreds of nucleotides. The effect of multiple binding sites along a single transcript has not been studied for many RBPs besides those that bind viral genomes for virion assembly,45,46 but kinetics studies of DAZL show its periodicity is cooperative and affects mRNA levels and ribosome association of bound proteins.47 Since PARP13 binding has also been implicated in mRNA decay and translational repression, multiple proximal binding sites may similarly modulate the effects of PARP13.
Limitations of the study
In this study, the RNA-seq and eCLIP-seq analysis were performed in biological duplicates. While we were able to demonstrate a strong correlation between samples and verify the expression changes of all tested genes in at least three replicates via qPCR, increasing the number of sequencing replicates would provide better confidence in the global transcriptome shifts observed across conditions, even though all conditions had comparable reproducibililty (Figure S1A). PARP13 KO cells provide information about steady-state effects of PARP13 deficiency; repeating our tests using the viral mimic 3p-RNA on cells treated with siPARP13 could provide additional information about the early direct events of PARP13 depletion and eliminate concerns about genomic artifacts of cell line drift between WT and mutant cell lines. Though we attempted to treat WT cells with siPARP13 and then 3p-RNA, the sequential transfections killed all cells, even when cells were given extended time to recover. This is likely due to the excessive, compounded stress of repeat lipofection, PARP13 depletion, and immune response. Lastly, we used the viral mimic to investigate the antiviral response, circumventing viral anti-host perturbations, but we acknowledge that this is an artificial system. Experiments with live viruses in future studies could provide further insights into how PARP13 regulates the ISGylation pathway and assess whether PARP13 KO cells exhibit increased susceptibility to viral infection than WT cells.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| TRIM25 polyclonal rabbit | Thermo Scientific | cat # 12573-1-AP; RRID: AB_2209732 |
| ISG15 monoclonal rabbit (7H29L24) | Thermo Scientific | cat # 703131; RRID: AB_2784562 |
| PARP13 (ZC3HAV1) polyclonal rabbit | ProteinTech | cat # 16820-1-AP; RRID: AB_2728733 |
| PARP13 (ZC3HAV1) polyclonal rabbit | Thermo Scientific | cat #PA5-31650; RRID: AB_2549123 |
| β-Actin monoclonal mouse (clone AC-15) | Sigma Aldrich | cat # A5441; RRID: AB_476744 |
| GFP mouse IgG1κ (clones 7.1 and 13.1) | Roche | cat # 11814460001; RRID: AB_390913 |
| OAS2 polyclonal rabbit | ProteinTech | cat # 19279-1-AP; RRID: AB_10642832 |
| Critical commercial assays | ||
| Dual-Luciferase Reporter Assay System | Promega | cat #E1910 |
| PowerUp SYBR Green master mix | Thermo Fisher | cat # A25742 |
| SuperScriptTM IV VILO Master Mix | Invitrogen | cat # 11754050 |
| TRIzol reagent | Invitrogen | cat # 15596026 |
| 100x Halt protease inhibitor cocktail | Thermo Fisher | cat # 78429 |
| SUPERase-In RNase inhibitor | Thermo Fisher | cat # AM2694 |
| 4x NuPAGE LDS sample buffer | Thermo Fisher | cat # NP0007 |
| 20x NuPAGE MOPS SDS running buffer | Thermo Fisher | cat # NP0001 |
| 20x NuPAGE transfer buffer | Thermo Fisher | cat # NP0006 |
| AmpliScribeTM T7 High Yield Transcription Kit | Lucigen | cat # AS3107 |
| Lipofectamine 2000 Transfection Reagent | Thermo Fisher | cat # 11668500 |
| 1X Dulbecco’s Modified Eagle Medium | Gibco | cat # 11995073 |
| fetal bovine serum | Sigma Aldrich | cat #F6765 |
| Opti-MEM(R) I Reduced-Serum Medium (1X) | Gibco | cat # 31985062 |
| Deposited data | ||
| TRIM25 CLIP-seq | Choudhury et al. 28 | GEO: GSE104949 |
| APEX-RIP | Kaewsapsak et al. 35 | GEO: GSE106493 |
| PARP13 eCLIP and RNA-seq data | This publication | GEO: GSE215251 |
| Experimental models: Cell lines | ||
| HEK 293T | ATCC | cat # CRL-3216; RRID: CVCL_0063 |
| PARP13 KO HEK 293T | Hayakawa et al. 2 | clone 32, c.1023_1047del25insGTT, c.1027_1046del20, c.1031_1032insATCCA and c.1033_1034insCAATC |
| Oligonucleotides | ||
| ssRNA | IDT | CACUUUCACUUCUCCCUUUUAGUUUCC |
| 3p-RNA | IDT | TAATACGACTCACTATAGGAAACTAAAAGGG AGAAGTGAAAGTG and CACTTTCACTTCTCC CTTTTAGTTTCCTATAGTGAGTCGTATTA |
| siPARP13 | Thermo Fisher | siRNA ID # HSS125638, cat # 1299001 |
| Recombinant DNA | ||
| EGFP-PARP13.1 | This paper | |
| EGFP-PARP13.2 | This paper | |
| EGFP-PARP13.2(Q) | This paper | |
| dual luciferase PARP13.1 3′UTR | This paper | |
| dual luciferase PARP13.2 3′UTR | This paper | |
| Software and algorithms | ||
| nearBynding | Busa et al. 39 | https://bioconductor.org/packages/nearBynding/; https://github.com/vbusa1/nearBynding |
| HOMER | Heinz et al. 34 | http://homer.ucsd.edu/homer/motif/ |
| GOrilla | Eden et al. 48 | http://cbl-gorilla.cs.technion.ac.il/ |
| Revigo | Supek et al. 49 | http://revigo.irb.hr/ |
| EMBOSS: compseq | Rice et al. 50 | http://emboss.sourceforge.net/apps/cvs/emboss/apps/compseq.html |
| CLIPper | Lovci et al. 51 | https://github.com/YeoLab/clipper/releases/tag/1.0 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Anthony Leung (anthony.leung@jhu.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
-
•
All sequencing data is deposited at GEO: GSE215251 and are publicly available as of the date of publication.
-
•
All original code used to generate figures is deposited on GitHub (https://github.com/vbusa1/PARP13_manuscript) and is available as of the date of publication.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental model
Cell culture
Human embryonic kidney (HEK) 293T cells (female, RRID: CVCL_0063) were maintained in 1X Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS) at 37°C in a 5% CO2 incubator. HEK293T PARP13 (GenBank: NM_024625.3) KO cells were gifted from Hayakawa et al. 2011. The PARP13 KO cells were treated for mycoplasma infection before experimental use.
Method details
ssRNA and 3p-RNA synthesis and transfection
A synthetic ssRNA (unphosphorylated RNA) oligo was used as a transfection control. A 3p-RNA (5′-triphosphorylated RNA viral mimic) oligo was produced by first annealing two synthetic oligos and transcribing the RNA fragment using T7 transcriptase with RNase inhibitor, followed by DNase I digestion. For transfection of HEK293T cells, 2.5 μL Lipofectamine 2000 and 125 μL Opti-MEM medium were combined with a mixture of 1 μg RNA (either ssRNA or 3p-RNA) and 125 μL Opti-MEM per well of a 6-well plate. After 5 min at room temperature, the solution was added drop-wise to cells grown to 70–90% confluence in 2 mL DMEM.
Dual luciferase assay
1000 HEK293T WT or PARP13 KO cells per well were seeded in a 96-well plate in 150 μL DMEM. 24 h later, 100 ng dual luciferase plasmid and 80 ng RNA treatment were combined with 120 μL Opti-MEM and 1 μL lipofectamine 2000 per well and incubated for 5 min at room temperature, and then were added to the cells (Figure 2C). 24 h later, media was removed from the wells and cells were washed with 1x PBS. Samples to be used for parallel RT-qPCR were stored in 100 μL TRIzol and stored at −20°C until RNA extraction. Dual luciferase reporter assay buffers were defrosted 2 h prior to experiment and diluted to 1× as relevant immediately before use. 20 μL per well of 1× lysis solution was added to cells and shaken at room temperature 15 min. To measure luminescence, 100 μL LAR II reagent was added to the lysed cells in each well and immediately measured to detect control firefly luciferase expression. 100 μL 1× Stop and Glo reagent was then added to each well and immediately measured to detect Renilla luciferase expression. Preliminary tests of untransfected cells demonstrated <1% luminescence compared to the lowest-luminescence transfected cells and so background is considered negligible in normalization calculations. To calculate luminescence, we normalized Renilla expression to firefly expression and compared it to normalized Renilla expression without a 3′UTR insert.
siRNA transfection
2.5 μL Lipofectamine 2000 and 125 μL Opti-MEM medium were combined with a mixture of 100 p.m. siPARP13 and 125 μL Opti-MEM per well of a 6-well plate. After 5 min at room temperature, the solution was added drop-wise to cells grown to ∼70% confluence in 2 mL DMEM.
GFP construct transfection and immunoprecipitation
12 μL Lipofectamine 2000 and 750 μL Opti-MEM Medium were combined with a mixture of 18 μg EGFP plasmid and 750 μL Opti-MEM per 10-cm plate. After 5 min at room temperature, the solution was added drop-wise to cells grown to 70–90% confluence. 24 h after transfection, cells were washed once with 1x PBS, scraped from the plate, and pelleted. Cells were lysed at 4°C for 15 min in 500 μL mRIPA buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 0.1% sodium deoxycholate, 1% NP-40, 1 mM EDTA pH 8.0, 1× Halt protease inhibitor cocktail, and 0.2 U/μL SUPERase-In RNase inhibitor). Samples were centrifuged at 20,000 × g for 10 min to separate cell debris and the supernatant was transferred to a new tube. 30 μL GFP-Trap bead slurry per sample was washed twice in 500 μL mRIPA buffer, resuspended in 500 μL mRIPA buffer, and added to cell supernatant. GFP-Trap beads were rotated with cell supernatant at 4°C for 2 h. Beads were spun down and washed twice. Beads were resuspended in 20 μL mRIPA buffer with either 6 μL NuPAGE LDS sample buffer for loading on a western blot or with 500 μL TRIzol for qRT-PCR.
Western blot
Samples were run on a denaturing polyacrylamide gel using NuPAGE MOPS SDS running buffer and transferred onto a nitrocellulose membrane at 20 V for 16 h using NuPAGE transfer buffer. Membrane was blocked with 6% milk in TBS for 45 min and then incubated with 1:1,000 primary antibody for 60 min in 6% milk in TBST. Membrane was washed three times with 6% milk in TBST and incubated with 1:10,000 secondary antibody for 90 min in 6% milk in TBST. Membrane was washed three times with TBS and imaged.
RT-PCR
Cells were harvested by suspension in TRIzol and RNA was isolated by phenol-chloroform extraction. Samples were reverse transcribed with SuperScript VILO cDNA synthesis kit and 10 μL RT-qPCR reactions were performed in triplicate with 1:200 dilution cDNA, 1 μM primer mix (IDT), and 1× PowerUp SYBR Green master mix on a 7500 Fast Real-Time PCR System (Applied Biosystems). Primer sequences for all target genes are available in Table S2.
RNA-seq analysis
RNA-seq was performed on WT and PARP13 KO HEK293T cells 24 h after transfection with either an ssRNA control or a 3p-RNA viral mimic in duplicate. 500,000 cells were seeded in 1 mL DMEM per well of a 12-well plate. The following day, 2 μL Lipofectamine 2000 and 25 μL Opti-MEM were combined with 500 ng RNA and 25 μL Opti-MEM per well and incubated for 5 min at room temperature, and then were added to the cells; two wells were pooled per sample. RNA was purified via phenol-chloroform extraction and RNA quality was measured via RT-qPCR. All data manipulation and visualization was performed in R. Adaptor-trimmed FASTQ RNA-seq reads were aligned using Bowtie2.52 Rsubread53 was used to identify genes corresponding to the peak loci, and DESeq254 was used to determine which transcripts were differentially expressed (FDR <0.05, fold-change >2). Gene set enrichment analysis was performed by GOrilla and simplified by Revigo.48,49
eCLIP-seq experimental procedures analysis
Enhanced CLIP-seq (eCLIP) was performed on WT and PARP13 KO HEK293T cells 24 h after transfection with either an ssRNA control or a 3p-RNA viral mimic in duplicate. 5 million cells were seeded in 10 mL DMEM per 10-cm dish. The following day, 20 μL Lipofectamine 2000 and 250 μL Opti-MEM were combined with 10 μg RNA and 250 μL Opti-MEM per dish and incubated for 5 min at room temperature, and then were added to the cells. Cells were washed with 1X PBS and UV cross-linked at 400 mJ/cm2. Cells were scraped from the plate, washed in 1X PBS, pelleted at 500 x g for 5 min at 4°C, and snap-frozen on liquid nitrogen. eCLIP was performed as previously published32 using anti-PARP13 antibody (Thermofisher Scientific, PA5-31650). Briefly, cells were lysed, and lysates sonicated and subjected to limited RNase digestion to fragment RNA. 2% of each lysate sample was stored at 4°C for preparation of a parallel size-matched input (SMInput) library. The remaining lysates were immunoprecipitated using PARP13 antibody and goat anti-rabbit IgG beads. RNA bound to washed immunoprecipitates (IPs) was dephosphorylated and 3ʹ-end ligated to an RNA adapter. Protein–RNA complexes from SMInputs and IPs were released from beads by heating to 70 deg C for 10 min in LDS sample buffer (Thermofisher Scientific), run on an SDS–polyacrylamide gel, and transferred to nitrocellulose membrane to remove non-covalently bound RNAs. Membrane regions comprising the molecular weight of PARP13.1 (∼78 kDa) to ∼150 kDa (corresponding to the protein molecular weight and ∼200 nt of RNA) were excised, and RNA was released from the membrane-bound complexes with proteinase K. SMInput samples were dephosphorylated and 3ʹ-end ligated to an RNA adaptor. All RNA samples (IPs and SMInputs) were reverse transcribed with AffinityScript (Agilent). cDNAs were 5ʹ-end ligated to a DNA adaptor containing a random-mer sequence, and yields quantified by qPCR. cDNA samples were PCR-amplified using standard Illumina adaptors and Q5 PCR mix (NEB) with 10–13 cycles (SMInputs) or 13–16 cycles (IPs). Resultant DNA libraries were size-fractionated on agarose gels to 175–350 bp. Library yields were 200–600 fmol each. Samples were sequenced to >20M read depth in single-end mode on an Illumina HiSeq instrument.
eCLIP-seq data analysis
eCLIP data analysis was performed as previously published.32 Briefly, after demultiplexing and adapter-trimming, reads were mapped to the human genome (hg19) using STAR (v2.4.0i). PCR duplicates were removed utilizing the random-mer sequence in the cDNA adaptor to yield usable reads. Binding cluster (peak) identification was performed on usable reads via CLIPper,51 (available at https://github.com/YeoLab/clipper/releases/tag/1.0) using PARP13 KO eCLIP reads as background signal. Reproducible peaks between replicates were determined by irreproducible discovery rate (IDR, https://www.encodeproject.org/software/idr/, https://github.com/YeoLab/merge_peaks). K-mer analysis was performed using tools from the Yeo Lab (https://github.com/YeoLab/clip_analysis_legacy/kmerdiff.py). Dinucleotide frequency of RNA-seq and CLIP-seq data was calculated by compseq from the EMBOSS suite.50 Hexameric motifs were calculated from CLIP-seq data using findMotifs.pl from HOMER.34
Quantification and statistical analysis
All statistical analyses were performed in R. The specific test used is listed directly before every reported p value in the text and/or figure legend. The n for each analysis is included in the respective figure legend.
Acknowledgments
We would like to thank Drs. Paul Chang and Akinori Takaoka for PARP13 plasmids and WT and KO cell lines, respectively. We also thank Dr. AJ Bhutkar for his suggestion of the use of independent component analyses in analyzing the RNA-seq data. This work was supported by Johns Hopkins President’s Frontier Award program and Johns Hopkins University Discovery Award to A.K.L.L., the Predoctoral Fellowship in Informatics from the Pharmaceutical Research and Manufacturers of America Foundation to V.F.B., National Institutes of Health T32GM07814 to V.F.B., R01GM104135 to A.K.L.L., and R01HG004659 and U41HG09889 to G.W.Y.
Author contributions
Conceptualization, V.F.B., Y.A., G.W.Y., and A.K.L.L.; Validation, V.F.B.; Analysis, V.F.B., Y.A., and B.A.Y.; Investigation, V.F.B., Y.A., and S.A.; Data curation, B.A.Y.; Writing—original draft, V.F.B. and A.K.L.L.; Writing—review and editing, V.F.B., Y.A., G.W.Y., and A.K.L.L.; Visualization, V.F.B.; Supervision, G.W.Y. and A.K.L.L.
Declaration of interests
G.W.Y. is co-founder, member of the Board of Directors, on the SAB, equity holder, and paid consultant for Locanabio and Eclipse BioInnovations. G.W.Y. is a visiting professor at the National University of Singapore. G.W.Y.’s interest(s) have been reviewed and approved by the University of California San Diego in accordance with its conflict-of-interest policies. The authors declare no other competing interests.
Published: February 16, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2024.109251.
Contributor Information
Gene W. Yeo, Email: ewyeo@health.ucsd.edu.
Anthony K.L. Leung, Email: anthony.leung@jhu.edu.
Supplemental information
References
- 1.Ficarelli M., Neil S.J.D., Swanson C.M. Targeted Restriction of Viral Gene Expression and Replication by the ZAP Antiviral System. Annu. Rev. Virol. 2021;8:265–283. doi: 10.1146/annurev-virology-091919-104213. [DOI] [PubMed] [Google Scholar]
- 2.Hayakawa S., Shiratori S., Yamato H., Kameyama T., Kitatsuji C., Kashigi F., Goto S., Kameoka S., Fujikura D., Yamada T., et al. ZAPS is a potent stimulator of signaling mediated by the RNA helicase RIG-I during antiviral responses. Nat. Immunol. 2011;12:37–44. doi: 10.1038/ni.1963. [DOI] [PubMed] [Google Scholar]
- 3.Chen S., Xu Y., Zhang K., Wang X., Sun J., Gao G., Liu Y. Structure of N-terminal domain of ZAP indicates how a zinc-finger protein recognizes complex RNA. Nat. Struct. Mol. Biol. 2012;19:430–435. doi: 10.1038/nsmb.2243. [DOI] [PubMed] [Google Scholar]
- 4.Schwerk J., Soveg F.W., Ryan A.P., Thomas K.R., Hatfield L.D., Ozarkar S., Forero A., Kell A.M., Roby J.A., So L., et al. RNA-binding protein isoforms ZAP-S and ZAP-L have distinct antiviral and immune resolution functions. Nat. Immunol. 2019;20:1610–1620. doi: 10.1038/s41590-019-0527-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhu Y., Wang X., Goff S.P., Gao G. Translational repression precedes and is required for ZAP-mediated mRNA decay. EMBO J. 2012;31:4236–4246. doi: 10.1038/emboj.2012.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen G., Guo X., Lv F., Xu Y., Gao G. p72 DEAD box RNA helicase is required for optimal function of the zinc-finger antiviral protein. Proc. Natl. Acad. Sci. USA. 2008;105:4352–4357. doi: 10.1073/pnas.0712276105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guo X., Ma J., Sun J., Gao G. The zinc-finger antiviral protein recruits the RNA processing exosome to degrade the target mRNA. Proc. Natl. Acad. Sci. USA. 2007;104:151–156. doi: 10.1073/pnas.0607063104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhu Y., Chen G., Lv F., Wang X., Ji X., Xu Y., Sun J., Wu L., Zheng Y.-T., Gao G. Zinc-finger antiviral protein inhibits HIV-1 infection by selectively targeting multiply spliced viral mRNAs for degradation. Proc. Natl. Acad. Sci. USA. 2011;108:15834–15839. doi: 10.1073/pnas.1101676108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ye P., Liu S., Zhu Y., Chen G., Gao G. DEXH-Box protein DHX30 is required for optimal function of the zinc-finger antiviral protein. Protein Cell. 2010;1:956–964. doi: 10.1007/s13238-010-0117-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gläsker S., Töller M., Kümmerer B.M. The alternate triad motif of the poly(ADP-ribose) polymerase-like domain of the human zinc finger antiviral protein is essential for its antiviral activity. J. Gen. Virol. 2014;95:816–822. doi: 10.1099/vir.0.060988-0. [DOI] [PubMed] [Google Scholar]
- 11.Kmiec D., Lista M.J., Ficarelli M., Swanson C.M., Neil S.J.D. S-farnesylation is essential for antiviral activity of the long ZAP isoform against RNA viruses with diverse replication strategies. PLoS Pathog. 2021;17 doi: 10.1371/journal.ppat.1009726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Charron G., Li M.M.H., MacDonald M.R., Hang H.C. Prenylome profiling reveals S-farnesylation is crucial for membrane targeting and antiviral activity of ZAP long-isoform. Proc. Natl. Acad. Sci. USA. 2013;110:11085–11090. doi: 10.1073/pnas.1302564110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li M.M.H., Aguilar E.G., Michailidis E., Pabon J., Park P., Wu X., de Jong Y.P., Schneider W.M., Molina H., Rice C.M., MacDonald M.R. Characterization of Novel Splice Variants of Zinc Finger Antiviral Protein (ZAP) J. Virol. 2019;93:e00715–e00719. doi: 10.1128/JVI.00715-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mao R., Nie H., Cai D., Zhang J., Liu H., Yan R., Cuconati A., Block T.M., Guo J.-T., Guo H. Inhibition of Hepatitis B Virus Replication by the Host Zinc Finger Antiviral Protein. PLoS Pathog. 2013;9 doi: 10.1371/journal.ppat.1003494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Takata M.A., Gonçalves-Carneiro D., Zang T.M., Soll S.J., York A., Blanco-Melo D., Bieniasz P.D. CG dinucleotide suppression enables antiviral defence targeting non-self RNA. Nature. 2017;550:124–127. doi: 10.1038/nature24039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Meagher J.L., Takata M., Gonçalves-Carneiro D., Keane S.C., Rebendenne A., Ong H., Orr V.K., MacDonald M.R., Stuckey J.A., Bieniasz P.D., et al. Structure of the zinc-finger antiviral protein in complex with RNA reveals a mechanism for selective targeting of CG-rich viral sequences. Proc. Natl. Acad. Sci. USA. 2019;116:24303–24309. doi: 10.1073/pnas.1913232116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gao G., Guo X., Goff S.P. Inhibition of Retroviral RNA Production by ZAP, a CCCH-Type Zinc Finger Protein. Science. 2002;297:1703–1706. doi: 10.1126/science.1074276. [DOI] [PubMed] [Google Scholar]
- 18.Guo X., Carroll J.-W.N., MacDonald M.R., Goff S.P., Gao G. The Zinc Finger Antiviral Protein Directly Binds to Specific Viral mRNAs through the CCCH Zinc Finger Motifs. J. Virol. 2004;78:12781–12787. doi: 10.1128/JVI.78.23.12781-12787.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Todorova T., Bock F.J., Chang P. PARP13 regulates cellular mRNA post-transcriptionally and functions as a pro-apoptotic factor by destabilizing TRAILR4 transcript. Nat. Commun. 2014;5:5362. doi: 10.1038/ncomms6362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gonzalez-Perez A.C., Stempel M., Wyler E., Urban C., Piras A., Hennig T., Ganskih S., Wei Y., Heim A., Landthaler M., et al. The Zinc Finger Antiviral Protein ZAP Restricts Human Cytomegalovirus and Selectively Binds and Destabilizes Viral UL4/UL5 Transcripts. mBio. 2021;12 doi: 10.1128/mBio.02683-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang Y., Burke C.W., Ryman K.D., Klimstra W.B. Identification and Characterization of Interferon-Induced Proteins That Inhibit Alphavirus Replication. J. Virol. 2007;81:11246–11255. doi: 10.1128/JVI.01282-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.MacDonald M.R., Machlin E.S., Albin O.R., Levy D.E. The Zinc Finger Antiviral Protein Acts Synergistically with an Interferon-Induced Factor for Maximal Activity against Alphaviruses. J. Virol. 2007;81:13509–13518. doi: 10.1128/JVI.00402-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Karki S., Li M.M.H., Schoggins J.W., Tian S., Rice C.M., MacDonald M.R. Multiple Interferon Stimulated Genes Synergize with the Zinc Finger Antiviral Protein to Mediate Anti-Alphavirus Activity. PLoS One. 2012;7 doi: 10.1371/journal.pone.0037398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li M.M.H., Lau Z., Cheung P., Aguilar E.G., Schneider W.M., Bozzacco L., Molina H., Buehler E., Takaoka A., Rice C.M., et al. TRIM25 Enhances the Antiviral Action of Zinc-Finger Antiviral Protein (ZAP) PLoS Pathog. 2017;13 doi: 10.1371/journal.ppat.1006145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Durfee L.A., Lyon N., Seo K., Huibregtse J.M. The ISG15 Conjugation System Broadly Targets Newly Synthesized Proteins: Implications for the Antiviral Function of ISG15. Mol. Cell. 2010;38:722–732. doi: 10.1016/j.molcel.2010.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zheng X., Wang X., Tu F., Wang Q., Fan Z., Gao G. TRIM25 Is Required for the Antiviral Activity of Zinc Finger Antiviral Protein. J. Virol. 2017;91 doi: 10.1128/JVI.00088-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zou W., Zhang D.-E. The Interferon-inducible Ubiquitin-protein Isopeptide Ligase (E3) EFP Also Functions as an ISG15 E3 Ligase. J. Biol. Chem. 2006;281:3989–3994. doi: 10.1074/jbc.M510787200. [DOI] [PubMed] [Google Scholar]
- 28.Choudhury N.R., Heikel G., Trubitsyna M., Kubik P., Nowak J.S., Webb S., Granneman S., Spanos C., Rappsilber J., Castello A., Michlewski G. RNA-binding activity of TRIM25 is mediated by its PRY/SPRY domain and is required for ubiquitination. BMC Biol. 2017;15 doi: 10.1186/s12915-017-0444-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gonçalves-Carneiro D., Takata M.A., Ong H., Shilton A., Bieniasz P.D. Origin and evolution of the zinc finger antiviral protein. PLoS Pathog. 2021;17 doi: 10.1371/journal.ppat.1009545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sanchez J.G., Sparrer K.M.J., Chiang C., Reis R.A., Chiang J.J., Zurenski M.A., Wan Y., Gack M.U., Pornillos O. TRIM25 binds RNA to modulate cellular anti-viral defense. J. Mol. Biol. 2018;430:5280–5293. doi: 10.1016/j.jmb.2018.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang E., Nguyen L.P., Wisherop C.A., Kan R.L., Li M.M.H. The Role of ZAP and TRIM25 RNA Binding in Restricting Viral Translation. Front. Cell. Infect. Microbiol. 2022;12 doi: 10.3389/fcimb.2022.886929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Van Nostrand E.L., Pratt G.A., Shishkin A.A., Gelboin-Burkhart C., Fang M.Y., Sundararaman B., Blue S.M., Nguyen T.B., Surka C., Elkins K., et al. Robust transcriptome-wide discovery of RNA binding protein binding sites with enhanced CLIP (eCLIP) Nat. Methods. 2016;13:508–514. doi: 10.1038/nmeth.3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fischer J.W., Busa V.F., Shao Y., Leung A.K.L. Structure-Mediated RNA Decay by UPF1 and G3BP1. Mol. Cell. 2020;78:70–84.e6. doi: 10.1016/j.molcel.2020.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Heinz S., Benner C., Spann N., Bertolino E., Lin Y.C., Laslo P., Cheng J.X., Murre C., Singh H., Glass C.K. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kaewsapsak P., Shechner D.M., Mallard W., Rinn J.L., Ting A.Y. Live-cell mapping of organelle-associated RNAs via proximity biotinylation combined with protein-RNA crosslinking. Elife. 2017;6 doi: 10.7554/eLife.29224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Villarroya-Beltri C., Guerra S., Sánchez-Madrid F. ISGylation – a key to lock the cell gates for preventing the spread of threats. J. Cell Sci. 2017;130:2961–2969. doi: 10.1242/jcs.205468. [DOI] [PubMed] [Google Scholar]
- 37.Zhao C., Denison C., Huibregtse J.M., Gygi S., Krug R.M. Human ISG15 conjugation targets both IFN-induced and constitutively expressed proteins functioning in diverse cellular pathways. Proc. Natl. Acad. Sci. USA. 2005;102:10200–10205. doi: 10.1073/pnas.0504754102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Youn J.-Y., Dunham W.H., Hong S.J., Knight J.D.R., Bashkurov M., Chen G.I., Bagci H., Rathod B., MacLeod G., Eng S.W.M., et al. High-Density Proximity Mapping Reveals the Subcellular Organization of mRNA-Associated Granules and Bodies. Mol. Cell. 2018;69:517–532.e11. doi: 10.1016/j.molcel.2017.12.020. [DOI] [PubMed] [Google Scholar]
- 39.Busa V.F., Favorov A.V., Fertig E.J., Leung A.K.L. Spatial correlation statistics enable transcriptome-wide characterization of RNA structure binding. Cell Rep. Methods. 2021;1 doi: 10.1016/j.crmeth.2021.100088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Konieczny P., Stepniak-Konieczna E., Taylor K., Sznajder Ł.J., Sobczak K. Autoregulation of MBNL1 function by exon 1 exclusion from MBNL1 transcript. Nucleic Acids Res. 2017;45:1760–1775. doi: 10.1093/nar/gkw1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pervouchine D., Popov Y., Berry A., Borsari B., Frankish A., Guigó R. Integrative transcriptomic analysis suggests new autoregulatory splicing events coupled with nonsense-mediated mRNA decay. Nucleic Acids Res. 2019;47:5293–5306. doi: 10.1093/nar/gkz193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wilbert M.L., Huelga S.C., Kapeli K., Stark T.J., Liang T.Y., Chen S.X., Yan B.Y., Nathanson J.L., Hutt K.R., Lovci M.T., et al. LIN28 binds messenger RNAs at GGAGA motifs and regulates splicing factor abundance. Mol. Cell. 2012;48:195–206. doi: 10.1016/j.molcel.2012.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang Y., Burkhardt D.H., Rouskin S., Li G.-W., Weissman J.S., Gross C.A. A stress response that monitors and regulates mRNA structure is central to cold-shock adaptation. Mol. Cell. 2018;70:274–286.e7. doi: 10.1016/j.molcel.2018.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sanchez J.G., Chiang J.J., Sparrer K.M.J., Alam S.L., Chi M., Roganowicz M.D., Sankaran B., Gack M.U., Pornillos O. Mechanism of TRIM25 Catalytic Activation in the Antiviral RIG-I Pathway. Cell Rep. 2016;16:1315–1325. doi: 10.1016/j.celrep.2016.06.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Askjaer P., Kjems J. Mapping of Multiple RNA Binding Sites of Human T-cell Lymphotropic Virus Type I Rex Protein within 5′- and 3′-Rex Response Elements. J. Biol. Chem. 1998;273:11463–11471. doi: 10.1074/jbc.273.19.11463. [DOI] [PubMed] [Google Scholar]
- 46.Brown R.S., Anastasakis D.G., Hafner M., Kielian M. Multiple capsid protein binding sites mediate selective packaging of the alphavirus genomic RNA. Nat. Commun. 2020;11:4693. doi: 10.1038/s41467-020-18447-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sharma D., Zagore L.L., Brister M.M., Ye X., Crespo-Hernández C.E., Licatalosi D.D., Jankowsky E. The kinetic landscape of an RNA-binding protein in cells. Nature. 2021;591:152–156. doi: 10.1038/s41586-021-03222-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Eden E., Navon R., Steinfeld I., Lipson D., Yakhini Z. GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinf. 2009;10:48. doi: 10.1186/1471-2105-10-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Supek F., Bošnjak M., Škunca N., Šmuc T. REVIGO Summarizes and Visualizes Long Lists of Gene Ontology Terms. PLoS One. 2011;6 doi: 10.1371/journal.pone.0021800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rice P., Longden I., Bleasby A. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. 2000;16:276–277. doi: 10.1016/S0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- 51.Lovci M.T., Ghanem D., Marr H., Arnold J., Gee S., Parra M., Liang T.Y., Stark T.J., Gehman L.T., Hoon S., et al. Rbfox proteins regulate alternative mRNA splicing through evolutionarily conserved RNA bridges. Nat. Struct. Mol. Biol. 2013;20:1434–1442. doi: 10.1038/nsmb.2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liao Y., Smyth G.K., Shi W. The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic Acids Res. 2019;47:e47. doi: 10.1093/nar/gkz114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15 doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All sequencing data is deposited at GEO: GSE215251 and are publicly available as of the date of publication.
-
•
All original code used to generate figures is deposited on GitHub (https://github.com/vbusa1/PARP13_manuscript) and is available as of the date of publication.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.