

Abstract
Ferroptosis is an iron‐dependent cell death program that is characterized by excessive lipid peroxidation. Triggering ferroptosis has been proposed as a promising strategy to fight cancer and overcome drug resistance in antitumor therapy. Understanding the molecular interactions and structural features of ferroptosis‐inducing compounds might therefore open the door to efficient pharmacological strategies against aggressive, metastatic, and therapy‐resistant cancer. We here summarize the molecular mechanisms and structural requirements of ferroptosis‐inducing small molecules that target central players in ferroptosis. Focus is placed on (i) glutathione peroxidase (GPX) 4, the only GPX isoenzyme that detoxifies complex membrane‐bound lipid hydroperoxides, (ii) the cystine/glutamate antiporter system Xc − that is central for glutathione regeneration, (iii) the redox‐protective transcription factor nuclear factor erythroid 2‐related factor (NRF2), and (iv) GPX4 repression in combination with induced heme degradation via heme oxygenase‐1. We deduce common features for efficient ferroptotic activity and highlight challenges in drug development. Moreover, we critically discuss the potential of natural products as ferroptosis‐inducing lead structures and provide a comprehensive overview of structurally diverse biogenic and bioinspired small molecules that trigger ferroptosis via iron oxidation, inhibition of the thioredoxin/thioredoxin reductase system or less defined modes of action.
Keywords: drug‐resistant cancer, ferroptosis, natural product, small molecule, structural requirement
1. INTRODUCTION
Ferroptosis, an alternative cell death program to apoptosis, promises access to anticancer strategies that selectively kill malignant cells and are effective against aggressive, drug‐resistant tumors. 1 , 2 Challenges in targeting ferroptosis arise from interference with redox‐dependent signaling cascades whose regulatory mechanisms, unlike kinase signaling cascades, are less investigated and not completely understood. Cutting‐edge reports on chemical probes that inhibit central proteins in ferroptosis, that is, glutathione peroxidase (GPX) 4 and system Xc −, provided essential insights into ferroptotic pathways and disclosed additional targets. 2 , 3 , 4 Recent review articles add to the growing literature that links ferroptosis to human disease and emphasize the critical involvement of redox‐dependent mechanisms. 1 , 5 , 6 , 7 , 8 , 9 , 10 While the induction of ferroptosis is considered a promising strategy to fight aggressive, metastatic, and therapy‐resistant cancer, current drug development is hampered by the rapidly increasing but still insufficiently structured knowledge about molecular mechanisms and structural requirements of ferroptosis‐inducing small molecules.
Ferroptosis is a strictly regulated necrotic form of programmed cell death (PD) that differs on a cellular and molecular level from other cell death programs like apoptosis, anoikis, autophagy, and regulated necrosis (necroptosis, pyroptosis). 11 Morphological signs of ferroptosis are visible in but not limited to mitochondria, which decrease in size, gain membrane density, and lose clearly structured mitochondrial cristae. In addition, large membrane blebs are frequently detected. 12 Ferroptotic cell death critically depends on the iron‐dependent peroxidation of membrane lipids. 13 , 14 , 15 , 16 While moderate membrane peroxidation also occurs in other cell death pathways, excessive phospholipid peroxidation is a hallmark of ferroptosis.
Phospholipids with arachidonic acid (20:4) and adrenic acid (22:4) are preferentially converted into hydroperoxides, with phosphatidylethanolamines seemingly more prone to oxidative modification than other phospholipid classes. 11 , 13 Lipid hydroperoxides subsequently degrade to highly reactive metabolites like malondialdehyde (MDA), 4‐hydroxynonenal, and other Michael acceptors 11 , 17 that covalently bind to cysteine‐containing stress sensors and likely contribute to membrane dysfunction in ferroptosis (Figure 1). The integrity of cellular membranes is reduced during ferroptosis by microperforations and microruptures, potentially as a direct result of lipid peroxidation, though the exact mechanisms remain obscure. 18 , 19 Ferroptotic pathways are engaged in manifold pathologies like degenerative diseases of the central nervous system, organ injury as well as cancer, and emerging evidence suggests that their targeting might help to overcome drug resistance in anticancer therapy. 1 , 5 , 11 , 20 , 21 The number of proteins known to participate in ferroptosis is continuously increasing, as is the information about small molecules that interfere with ferroptotic pathways.
Figure 1.
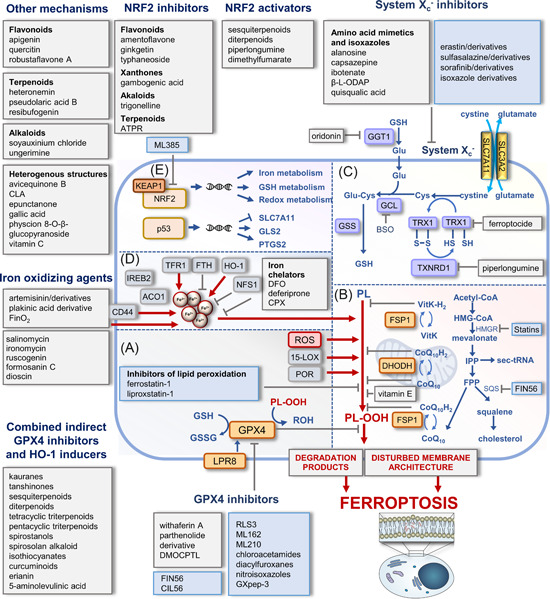
Major metabolic and regulatory pathways in ferroptosis targeted by small molecules. (A) Membrane peroxidation in ferroptosis depends on enzymatic and nonenzymatic mechanisms. LOX isoenzymes and oxidoreductases (POR) with Fe2+/Fe3+ in their active center introduce oxygen into polyunsaturated fatty acids (PUFAs), and free metal ions, in particular Fe2+, convert hydrogen peroxide into hydroxyl radicals via the Fenton reaction. GPX4 reduces lipid hydroperoxides, counteracts ferroptosis, and relies on the biosynthesis and regeneration of its substrate glutathione. (B) Additional protection against membrane peroxidation offers endogenous lipophilic radical traps such as ubiquinol and vitamin K (VitK), which are regenerated by FSP1 and DHODH in the cytosol and mitochondria, respectively. The mevalonate pathway is central for the biosynthesis of (i) cholesterol, (ii) the lipophilic radical trap CoQ10, and (iii) selenocysteine transfer RNA (sec‐tRNA), which inserts selenocysteine into GPX4. (C) System Xc − regulates cellular GSH levels by importing cystine in exchange for glutamate (Glu). Intracellular cystine is reduced to cysteine, which subsequently enters GSH biosynthesis and serves as a cofactor for GPX4. (D) Labile iron levels are kept within narrow thresholds by co‐ordinated regulation of iron uptake via the transferrin receptor and iron storage within ferritin. Other factors in the control of labile iron levels sequester iron into iron–sulfur clusters (NSF1) or liberate iron from heme oxygenase‐1 (HO‐1). The transmembrane glycoprotein CD44 mediates the endocytosis of iron‐bound hyaluronate. The scheme illustrates points of attack of selected small molecules that induce ferroptosis, with a focus on drug candidates, tool compounds, and natural products. The color of the boxes distinguishes between biogenic/bioinspired (gray) and synthesized small molecules (blue). (E) Central ferroptotic genes are under the control of the transcription factors NRF2 and p53. Mitochondrium in (B) was adapted from “Resting Metabolic Activity vs. Stimulated Metabolic Activity,” by BioRender.com (2020). Retrieved from https://app.biorender.com/biorender-templates. 2,2′‐BP, 2,2′‐bipyridine; 5‐ALA, 5‐aminolevulinic acid; ACO1, aconitase 1; ATPR, 4‐amino‐2‐trifluoromethyl‐phenyl retinate; BHT, butylhydroxy toluol; BSO, l ‐buthionine‐S,R‐sulfoximine; CLA conjugated linolenic acids; CPX, ciclopirox; DFO, DHODH, dihydroorotate dehydrogenase; deferoxamine; DMF, dimethyl fumarate; FPP, farnesyl pyrophosphate; FSP1, ferroptosis suppressor protein 1; FTH, ferritin heavy chain; GCL, glutamate cysteine ligase; GGT1, γ‐glutamyl transpeptidase 1; GLS2, glutaminase 2; GPX4, glutathione peroxidase 4; GSH, reduced glutathione; GSS, glutathione synthetase; GSSG, glutathione disulfide; HMG‐CoA, 3‐hydroxy‐3‐methylglutaryl‐coenzyme A; HMGR, 3‐hydroxy‐3‐methylglutaryl‐CoA reductase; IPP, isopentenyl pyrophosphate; IREB2, iron responsive element binding protein 2; KEAP1, kelch‐like ECH‐associated protein 1; LOX, lipoxygenase; NFS1, mitochondrial cysteine desulfurase; NRF2, nuclear factor erythroid 2‐related factor 2; PL, phospholipid; PL‐OOH, phospholipid hydroperoxide; PL‐OH, phospholipid alcohol; POR, cytochrome P450 oxidoreductase; ROS, reactive oxygen species; SQS squalene synthase; TFR, transferrin receptor; TRX, thioredoxin; TXNRD1, thioredoxin reductase 1; β‐ l ‐ODAP, β‐N‐oxalyl‐l‐α‐β‐diaminopropionic acid. [Color figure can be viewed at wileyonlinelibrary.com]
Research on designed ligands that trigger ferroptosis is still in its infancy, and many questions remain unanswered: How to selectively modify and fine‐tune redox‐dependent signaling cascades with small molecules? How to design target‐selective ferroptosis inducers? Are multitarget compounds of advantage? Which ferroptotic pathways are predestined targets against therapy‐resistant cancer? How does ferroptosis achieve selective lethality of cancer cells? What underlies the synergism between ferroptosis inducers and conventional chemotherapeutics? The rapidly increasing number of small molecules that trigger ferroptosis will help to answer these questions when channeled into targeted drug discovery and design. Previous review articles addressed the molecular mechanisms and signaling pathways in ferroptotic cell death and, in this context, referred to ferroptosis‐inducing screening hits and synthetic derivatives, phytochemicals, or tool compounds, without in‐depth discussing biomedical and medicinal chemical aspects. 5 , 10 , 16 , 21 , 22 , 23 , 24 , 25 , 26 , 27 , 28 , 29 , 30 , 31 , 32 , 33 Here, we address ferroptosis from the small molecule inducers’ point of view and provide a comprehensive overview of synthesized, biogenic and bioinspired compounds from diverse sources that induce ferroptosis through mechanisms related to redox signaling, that is, by targeting GPX4, system Xc −, nuclear factor erythroid 2‐related factor 2 (NRF2), and selected NRF2 target genes. We critically discuss ferroptosis‐inducing chemical probes and drug candidates with regard to their structural requirements, molecular mechanisms, and ferroptotic profiles. Moreover, we highlight challenges in proferroptotic drug design and development, propose overarching modes of action, and categorize less‐defined ferroptosis inducers based on structural and functional considerations. On this basis, we propose likely modes of action for less‐defined ferroptosis inducers and highlight challenges in proferroptotic drug design and development.
2. POTENTIAL TARGETS IN FERROPTOSIS RELATED TO REDOX SIGNALING
2.1. Membrane peroxidation and iron metabolism
Membrane peroxidation in ferroptosis is mediated by enzymatic and nonenzymatic mechanisms. 34 Lipoxygenases (LOXs), which have Fe2+/Fe3+ in their active center, introduce oxygen into polyunsaturated fatty acids (PUFAs) in a controlled enzymatic reaction with clear regio‐ and stereospecificity that differs between isoenzymes. With the exception of 15‐LOX that also accepts membrane phospholipids, human LOX isoenzymes (5‐LOX, 12‐LOX) are limited to nonesterified PUFAs as substrates. 11 In addition to 15‐LOX, 35 cytochrome P450 oxidoreductase and NADH‐cytochrome b5 reductase 1 participate in enzymatic membrane peroxidation during ferroptosis. 18 , 36 Nonenzymatic peroxidation of membrane‐bound PUFAs, on the other hand, is associated with the availability of free redox‐active metal ions, that is, ferrous iron, that converts hydrogen and alkyl peroxides into hydroxyl and alkoxy radicals via the Fenton reaction, respectively. 5 The import, export, and storage of iron, as well as the turnover and control of the labile iron pool, have, therefore, a strong impact on ferroptosis sensitivity, and iron chelators that decrease the availability of free iron efficiently inhibit ferroptotic cell death. 37
Iron metabolism is orchestrated by multiple transcription factors, with many of them being linked to ferroptosis, including transcription factor NRF2, yes‐associated protein 1 (YAP1), heat shock transcription factor 1, and hypoxia‐induced factor (HIF) 1/2. 37 To maintain labile iron levels despite elevated intracellular iron concentrations, cells engage two iron regulatory proteins, IRP1 and IRP2, that tightly coordinate the expression of transferrin receptor (responsible for iron uptake) and ferritin (an iron storage protein). 38 Sequestering iron into iron–sulfur cluster (ISC) is another efficient cellular strategy to prevent iron‐mediated Fenton reaction and subsequent lipid peroxidation. 39 , 40 The biosynthesis of ISCs relies on sulfur supply by mitochondrial cysteine desulfurase (NFS1), which has recently been discovered to protect from ferroptosis. 39 Suppression of NFS1 activates the canonical iron starvation response and accordingly increases labile iron levels by inducing transferrin receptor expression and decreasing ferritin protein levels. Two other ISC‐modulating proteins, frataxin and CDGSH iron–sulfur domain‐containing protein 2 are upregulated in diverse tumors, most likely to protect cancer cells from ferroptosis by decreasing free iron levels. 41 Other important factors controlling the cellular abundance of iron are (i) the iron export protein ferroportin, (ii) prominin 2 that mediates the formation of ferritin‐containing multivesicular bodies, (iii) the kinase ataxia telangiectasia mutated, that regulates ferritin availability as well as (iv) the ferrous ion membrane transport protein solute carrier family 11 member 2. 2
2.2. NRF2, p53, and other transcription factors
NRF2 protects from oxidative and electrophilic stress and is linked to enhanced cell survival 42 and ferroptosis resistance. 43 Protein levels of NRF2 are tightly controlled by kelch‐like ECH‐associated protein 1 (KEAP1), which forms a complex with NRF2 that is ubiquitinylated by the E3 ubiquitin ligase and subsequently degraded. 44 Oxidative or electrophilic modifications of cysteine side chains in KEAP1 inhibit NRF2 ubiquitination and allow newly translated NRF2 to translocate into the nucleus and induce NRF2 target gene expression. 45 , 46 NRF2 orchestrates the expression of genes involved in iron and redox metabolism, 47 with many of them (e.g., GPX4, SLC7A11, heme oxygenase [HO]‐1, glutamate‐cysteine ligase catalytic subunit [GCLC], as well as the ferroptosis suppressor protein [FSP] 1 and GTP cyclohydrolase 1 [GCH1]) being closely linked to ferroptosis (Figure 1). 48 , 49 , 50 , 51
A large set of small molecules modulates NRF2 activation 48 , 52 or interferes with other major transcription factors at the heart of ferroptosis, such as p53, 53 HIF1, 54 YAP1, 55 , 56 activating transcription factor (ATF) 3, activating transcription factor 4 (ATF 4), and specificity protein (SP) 1. 57 The section “NRF2 inhibitors” discusses those NRF2 inhibitors that have been demonstrated to trigger ferroptosis. There are also few reports on ferroptosis‐inducing compounds that upregulate p53, 58 a transcription factor that is most commonly mutated in cancer and critically involved in tumorigenesis, metastasis, and drug resistance. 59 p53 is a tumor suppressor protein that induces redox stress and sensitizes cells to ferroptosis 60 by coordinating the expression of a large set of target genes including the ferroptosis‐related genes SLC7A11, mitochondrial glutaminase (GLS2), and prostaglandin‐endoperoxide synthase 2 (cyclo‐oxygenase‐2 [COX‐2]). 61 It should be noted that the effects of p53 are highly dependent on the cell type and context and that p53 activation can both sensitize to and inhibit ferroptosis. 61 , 62 Whether small molecules that interfere with HIF1, 63 , 64 YAP1, 65 , 66 , 67 ATF3/4, or specificity protein 1 (SP1) 68 induce cell death through a ferroptotic mechanism is poorly understood.
2.3. GPX4 and glutathione regeneration
The selenoprotein GPX4 reduces and thereby detoxifies phospholipid hydroperoxides to the corresponding alcohols (Figure 1). 5 Deletion or selective inhibition of GPX4, for example, by the tetrahydro‐β‐carboline RSL3 (23), elevates lipid hydroperoxide levels and induces ferroptotic cell death. 69 GPX4 activity essentially depends on the supply of the cosubstrate GSH and therefore relies on GSH biosynthesis and regeneration as well as the cellular import of cystine via the cystine/glutamate antiporter system Xc − (Figure 1). 70 The antiporter (i) consists of two subunits, SLC7A11 (xCT) and SLC3A2 (CD98hc or 4F2hc) that are linked via a disulfide bridge, (ii) regulates the cellular redox status, and (iii) counteracts ferroptosis. 70 SLC7A11, which is unlike SLC3A2 not shared by other transporters of the heteromeric amino acid transporter family, mediates substrate specificity. After cellular uptake, cystine is reduced to cysteine and subsequently transferred to GSH biosynthesis (Figure 1).
Suppression of GPX4 levels rather than direct inhibition of GPX4 has been suggested as an efficient strategy to promote ferroptosis. 2 The availability of GPX4 is controlled at different stages. On the on hand, the expression of GPX4 and other key regulators in ferroptosis is orchestrated by transcription factors 2 , 57 , 71 : AP‐2γ (TFAP2C), ATF4, SP1, nuclear factor‐κB (NF‐κB), androgen receptor (AR), and CCAAT/enhancer binding protein induce and early growth response protein 1 (EGR1), and sterol regulatory element‐binding protein‐1 repress GPX4 expression. Effects are controversial for NRF2, with deletion and knockdown of the transcription factor either enhancing or diminishing GPX4 transcription. 50 , 72 , 73
The insertion of the catalytically active selenocysteine into GPX4 is regulated by the availability of selenocysteine‐transfer RNA during protein biosynthesis. 74 The lipoprotein receptor LRP8 (also known as ApoER2) serves as receptor for the uptake of selenoprotein P, a selenium‐rich protein that delivers selenium to extrahepatic tissues. 75 Selenium, as well as GPX4 levels, accordingly decrease upon knockout of LRP8, the latter being specific for cancer cells. 76 Conclusively, a cancer‐specific vulnerability toward ferroptotic cell death can be obtained by knock‐out or pharmacological inhibition of LRP8.
Another strategy to adjust cellular GPX4 levels relies on controlled protein degradation following E3 ligase‐dependent ubiquitination. 77 Ubiquitin‐independent degradation takes place in lysosomes and comprises chaperone‐mediated autophagy, a process that drives the degradation of selective (e.g., oxidized) proteins. 78 Important for the lysosomal enrichment of GPX4 are the 124NVKFD128 or 187QVIEK191 recognition motifs that enable interaction with heat‐shock protein (HSP) 70. 78
GPX4 enzyme activity is also regulated by posttranslational modification, such as the succination of GPX4 at cysteine 93 (mono‐ and disuccination), which reduces enzymatic activity. 79 Other posttranslational modifications like SUMOylation and phosphorylation affect GPX4 activity, 77 but the exact amino acid residues modified and biological consequences remain yet to be elucidated. 77
2.4. Endogenous inhibitors of lipid peroxidation
Several cellular antiferroptotic redox cycles work independent from GPX4 to keep ferroptosis at bay, 11 , 80 including (i) FSP1/coenzyme (Co) Q10 (also known as ubiquinone‐10), (ii) dihydroorotate dehydrogenase (DHODH)/CoQ10, and (iii) GCH1/tetrahydrobiopterin (BH4) (Figure 1B). Using NAD(P)H as a redox cofactor, FSP1 reduces CoQ10 at intracellular membranes to ubiquinol (Figure 1B), 5 , 81 , 82 , 83 which scavenges lipophilic radicals 84 and regenerates endogenous antioxidants such as tocopherols. 82 In addition to CoQ10, FSP1 restores the reduced form of vitamin K and derivatives, which are structurally closely related to CoQ10 and represent a group of naphthoquinones with radical‐trapping and antioxidative properties. CoQ10, vitamin K as well as plant‐derived phylloquinone and menaquinone accordingly form (upon reduction) a robust endogenous ferroptosis‐suppressing system. 85 While FSP1 is located at diverse (intra)cellular membranes, DHODH reduces mitochondrial CoQ10 at the outer side of the inner mitochondrial membrane. 86 The biosynthesis of CoQ10 can be inhibited by statins that target 3‐hydroxy‐3‐methylglutaryl‐CoA reductase (HMGR), the rate‐limiting enzyme of the mevalonate pathway (Figure 1B). 87 Similar to endogenous hydroquinones, the widely used ferroptosis inhibitors ferrostatin‐1 and liproxstatin‐1 act as lipophilic radical traps that protect membranes from peroxidation. 88 , 89 , 90 Regeneration to the active reduced form occurs with the consumption of CoQ10. 91 Other endogenous antiferroptotic factors include the GCH1‐tetrahydrobiopterin (BH4) system that works analogous to FSP1/CoQ10, 92 inducible nitric oxide (NO) synthase 2 that triggers NO production under proinflammatory conditions and the endosomal sorting complex required for transport that participates in the formation of multilaminar vesicles. 62
2.5. Shaping the PUFA composition of membranes
Ferroptosis sensitivity is associated with PUFA biosynthesis and the incorporation of PUFAs into membrane phospholipids 5 and decreases with their release from phospholipids by Ca2+‐independent phospholipase A2. 93 Long‐chain PUFAs are preferentially found in the sn‐2 position of phospholipids, where they are either incorporated during de novo fatty acid biosynthesis by lysophosphatidic acid acyltransferases or through the concerted action of lysophospholipid acyltransferase and phospholipase A2 isoenzymes within the remodeling pathway (Land's cycle). 94 According to their preference for PUFAs as substrates, acetyl‐CoA synthetase long‐chain family member (ACSL) 4, and lysophosphatidylcholine acyltransferase 3 (LPCAT3) increase the PUFA ratio of membranes and render them more susceptible to lipid peroxidation in ferroptosis. 13 , 95 , 96 , 97 ACSL4 esterifies PUFAs with CoA and the resulting thioesters then serve as the substrate of LPCAT3 that incorporates them into phosphatidylcholines and phosphatidylethanolamines. 98 , 99 Pharmacological or genetic inhibition of Acsl4 and Lpcat3 accordingly inhibits ferroptosis in mouse embryonic fibroblasts (MEFs) with tamoxifen‐inducible Gpx4 disruption (Pfa1 cells). 13 , 95 , 100 Another isoenzyme, ACSL1, has recently been described to mediate ferroptosis by conjugated linoleic acids. 101 Monounsaturated fatty acids (MUFAs) lack the bis‐allylic double bond of PUFAs and protect from membrane peroxidation. The biosynthesis of MUFA‐containing phospholipids via stearoyl‐CoA desaturase 1 96 , 102 and ACSL3 2 accordingly exerts antiferroptotic effects and contributes to stress adaption, partially through the lipokine PI(18:1/18:1). 103 , 104 , 105
2.6. Other pathways
Ferroptosis is closely linked to (i) cell–cell contact‐mediated signal transduction, for example, via the E‐cadherin–NF2–Hippo–YAP/TAZ axis, 55 , 106 (ii) cell metabolism, 5 , 11 among others via the energy sensor AMP‐activated kinase (AMPK) 13 , 96 and glutamate/glutamine metabolism, 107 and (iii) the production of reactive oxygen species (ROS). Major sources for (lipid) ROS are nicotinamide adenine dinucleotide phosphate (NADPH) oxidases (NOXs), LOX isoenzymes, cytochrome P450 oxidoreductases, and electron leakage from the mitochondrial electron transport chain. 36 , 88 , 108 , 109 Ferroptosis is also modulated by epigenetic mechanisms. For example, the histone‐lysine N‐methyltransferase 2B (MLL4) promotes ferroptosis by rewiring the expression of ferroptosis‐related gene sets, 110 whereas the ubiquitin carboxyl‐terminal hydrolase BRCA1‐associated protein 1 supports ferroptosis by repressing SLC7A11 besides participating in multiple cellular processes related to DNA damage repair, cell cycle control, and the immune response. 111 These pleiotropic enzymes, pathways, and regulatory factors are not the focus of this article, either because few is known about how their interaction with small molecules influences ferroptosis or because they are covered by recent review articles. 1 , 5 , 9 , 11 , 112
There are also many links between ferroptosis and immunoregulation existing. Recent reports suggest that ferroptosis is involved in the physiological processes of the immune system to suppress tumor growth. 2 , 113 Activated CD8+ cells, for example, induce tumor cell ferroptosis by secreting interferon γ (IFNγ), which subsequently downregulates SLC7A11 and upregulates ACSL4 in cancer cells. 41 Moreover, the oxidized phospholipid, 1‐steaoryl‐2‐15‐HpETE‐sn‐glycero‐3‐phosphatidylethanolamine was identified as a key “eat me” signal for the phagocytotic clearance of cancer cells in vivo. 114
3. FERROPTOSIS, CANCER, AND CHEMORESISTANCE
3.1. Ferroptosis and cancer
Mounting evidence suggests the induction of ferroptosis as a promising strategy for the treatment of various types of cancer, including therapy‐resistant forms. 1 , 115 Cancer cells are more susceptible to ferroptosis than nonmalignant cells for several reasons: they (i) have persistently high ROS levels as a consequence of genetic alterations and aberrant proliferation, 116 (ii) have elevated intracellular iron levels, 88 , 117 , 118 and (iii) rely on oncogenic signal transduction that is crosslinked to ferroptotic pathways. 1 Given the strong differences in ferroptosis sensitivity between cell types, it is obvious that the efficacy of antiferroptotic therapy varies between tumors from different origins and genotypes. The applicability of ferroptosis‐inducing drugs to treat individual tumors, therefore, requires careful evaluation in terms of personalized medicine.
Cancer cells upregulate endogenous antioxidative systems, for example, GPXs, superoxide dismutases (SODs), GSH, catalase, peroxiredoxins (PRDXs), and thioredoxins, to maintain redox balance and ensure cell survival despite elevated ROS production. 70 By keeping oxidative stress below a certain threshold, they avoid irreparable cell damage leading to the induction of PD as a consequence of lipid, protein, or DNA (per)oxidation. 119 Note that cell death by oxidative stress is neither unique to ferroptosis nor cancer cells. ROS induction or the inhibition of antioxidative enzymes evokes various forms of cell death like apoptosis, necrosis, and autophagy and is deleterious to cancer and normal cells. However, the increased ROS levels of cancer cells likely confer to their higher vulnerability toward pro‐oxidants as compared to nontransformed cells. 120 , 121 , 122 In addition, the elevated iron levels in tumors as compared to healthy tissues 123 , 124 , 125 may contribute to cancer‐specific ROS formation and lipid peroxidation via the Fenton reaction. 126 It is tempting to speculate that cells with high basal ROS levels are more sensitive to ferroptosis and that the targeted induction of ferroptotic pathways (that channel ROS damage to membranes) adds to this selectivity. 127 , 128 , 129 Interestingly, endogenous ROS concentrations are even further elevated in cancer cells resistant to chemotherapy or radiotherapy, which might render them more sensitive to ROS‐driven ferroptosis. 127 , 128
3.2. Ferroptosis and tumor resistance
Drug resistance mechanisms of cancer cells impact ferroptosis sensitivity. 1 Most relevant in this context are: (i) the adaption to the microenvironment and activated defense systems, 130 (ii) mutated drug target genes or oncogenes that confer protection, 131 (iii) as well as mechanisms that regulate the cellular differentiation status, including stemness or epithelial‐to‐mesenchymal transition (EMT). 58 Another major strategy of cancer cells to acquire drug resistance is the expression of transport proteins, for example, multidrug resistance proteins like P‐glycoprotein, that pump chemotherapeutics out of the cells. In some cases, therapy‐resistant cancer cells are even more sensitive to ferroptosis than less aggressive malignant cells. 87 The exact mechanisms that render therapy‐resistant cancer cells sensitive to ferroptosis are incompletely understood. Many ferroptosis‐related factors (e.g., NRF2, SLC7A11) are upregulated by tumor resistance mechanisms, 70 among which EMT seems to play a prominent role. 87 In consequence, aggressive cancer cells that are resistant to other forms of PD (e.g., apoptosis) often remain vulnerable to ferroptosis. While it is out of the scope of this review to outline all the complex interconnections between ferroptosis, cancer and drug resistance, we want to point out three promising drug targets at this interface, namely NRF2, SLC7A11, and GPX4, and refer to comprehensive review articles in this field. 21 , 42 , 48 , 70 , 132 For all three proteins, a growing number of ligands and indirect modulators has been reported during recent years, which are described in Sections 5, 6, and 8.
4. INDUCING FERROPTOSIS BY SMALL MOLECULES
Since the term ferroptosis has been coined in 2012, the field prospered and important discoveries on the complex ferroptosis machinery have been made. With some delay and following a better understanding of the relevance of ferroptosis in cancer and drug resistance, research on ferroptosis‐inducing small molecules has been intensified, resulting in an exponentially growing number of published articles on this field. Ferroptosis‐inducing small molecules promise novel therapeutic options for the treatment of (therapy‐resistant) cancer. 133 They interact with transport proteins (e.g., system Xc −), enzymes (e.g., GPX4), or transcription factors (e.g., NRF2) or nonenzymatically initiate ROS formation via the Fenton reaction. Rational drug development against these targets is hampered by nonclassical binding modes and insufficient structural information about putative binding pockets. For example, there are several electrophilic inhibitors of GPX4 described that covalently react with the active site selenocysteine, 69 but these compounds lack selectivity and have poor pharmacodynamic properties. 4 While these inhibitors establish stable covalent bonds with the enzyme, the initial positioning of the ligand is kinetically disfavored without pronounced noncovalent protein–ligand interactions. The majority of approved drugs target well‐defined binding sites, either competitively at substrate or ligand binding pockets or substrate independently at allosteric sites. This classical mode of action of noncovalent inhibitors is extended by the warhead concept that aims at covalently coupling the ligand to cysteine residues (or other reactive groups) close to the binding site. 134 Combining specific noncovalent interactions with an irreversible modification promises superior potency as well as selectivity. Ferroptosis‐inducing small molecules with such features are of high pharmacological interest and represent potential chemical tools for investigating redox‐regulated processes in cell signaling. 135 Along these lines, oxidative protein modifications have recently been suggested as another level in the regulation of signaling cascades in addition to phosphorylation. 136 In support of this hypothesis, the reactivity of cysteine residues within the cysteinome varies by seven magnitudes of order. 136 Little is known so far how redox‐dependent cysteine modifications achieve target selectively under physiological conditions, which renders the design of selective inhibitors or activators challenging.
With the erastin analogue PRLX 93936 (21), a selective inhibitor of system Xc − entered clinical trials already in 2007 (www.clinicaltrials.gov, NCT00528047), with another study following in 2012 (NCT01695590). More recent clinical studies (based on single or combinatory treatments) focused on dimethyl fumarate (three clinical trials for cancer treatment, phase I–III, 2015–2016, NCT02784834, NCT02546440, and NCT02337426) and the natural products artemisinin/artesunate (eight clinical trials for cancer, phase I–II, 2008‐2021, NCT02633098, NCT03093129, NCT00764036, NCT02353026, NCT02354534, NCT03792516, NCT03100045, NCT04098744, NCT02786589, NCT05478239, NCT02304289), phenylethyl isothiocyanate (PEITC, 71) (seven clinical trials, phase I–III, 2004‐2021, NCT00968461, NCT00005883, NCT00691132, NCT01790204, NCT03700983, NCT03034603, and NCT02468882), and β‐elemene (56) (two clinical trials, phase II–III, 2012‐2021, NCT02629757, NCT03123484), and not further specified elemenes (six clinical trials, phase I–IV, NCT04674527, NCT04401059, NCT03166553, NCT03167775, NCT04397432, NCT01679847). Major breakthroughs were, however, hampered by the poor druggability of key players in ferroptosis, that is, GPX4 and NRF2, both of which lack a classical binding pocket for “drug‐like” small molecules that dominate high‐throughput screening. Secondary metabolites from plants and other organisms cover a broader chemical space and include complex structures with multiple stereogenic centers, diversified polycyclic ring systems, and substitution patterns that might overcome these limitations. 3 , 137 We consider natural products as a rich source of novel lead structures for targets in ferroptosis, which are otherwise difficult to address, like GPX4 and NRF2, or where the target is either unknown or not associated with ferroptosis. The number of small molecules that induce ferroptosis, trigger membrane peroxidation, or target NRF2 signaling is rapidly increasing, as is the preclinical evidence for their efficacy in suppressing drug‐resistant cancer. 47 , 48 , 138 There is a high demand for small molecules that induce ferroptosis, whether for use as chemical probes to unravel ferroptosis‐associated pathways or as drug candidates to fight aggressive cancer. 135
5. INHIBITORS OF SYSTEM XC −
Before being recognized as a major target in ferroptosis, system Xc − was already known to support tumor growth, survival, and drug resistance and to contribute to neurological dysfunction. 139 , 140 , 141 , 142 , 143 Efforts have been undertaken to identify small molecule system Xc − inhibitors for therapeutic use. 54 High‐throughput screening led to the identification of the potent system Xc − inhibitor erastin (18) and disclosed additional modes of action for the approved multikinase inhibitor and anticancer drug sorafenib (22), which shows moderate system Xc −‐inhibitory activity. 54
5.1. Quisqualic acid (1) and isoxazole derivatives
Compound 1 from Quisqualis indica and ibotenate (2) from Amanita muscaria and Amanita pantherine are analogues of α‐amino‐3‐hydroxy‐5‐methyl‐4‐isoxazole‐4‐propionic acid (AMPA, 4) (Figure 2), an endogenous activator of ionotropic glutamate receptors (AMPA receptors). 144 They are substrates of system Xc − and inhibit the antiporter (K i = 5 µM). 144 System Xc − transports ibotenate with comparable efficiency to the endogenous substrate cystine but is less efficient for bromohomoibotenate (3) and 1. Nuclear magnetic resonance analysis suggests that the endogenous substrate cystine (5) adopts a ring‐like conformation that is stabilized by an intracellular hydrogen bond between sulfur and the amino group, an orientation that aligns with the pentacyclic system of 1 and 2 (Figure 2). 144
Figure 2.
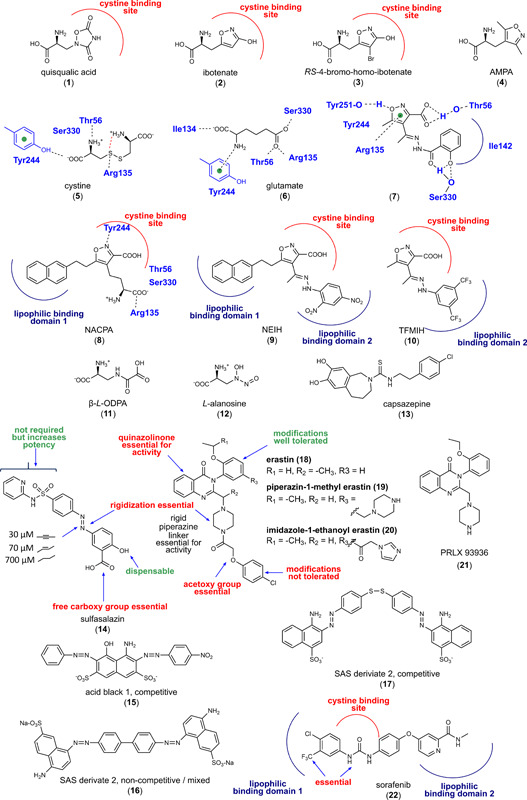
Inhibitors of system Xc −. [Color figure can be viewed at wileyonlinelibrary.com]
According to docking studies on an Xc − homology model, the α‐amino acid head group of 5 and glutamate (6) interact with Tyr244 of SLC7A11, whereas the distal carboxy groups bind to Thr56, Arg135, and Ser330, with Arg135 being located near to the Xc − glutamate binding site (Figure 2). 145 Within a series of isoxazole derivatives synthesized by the authors, compound 7 was most active and inhibited glutamate uptake by 50% at 500 µM. 145 Computational calculations indicate four principal interactions that govern the binding of compound 7 to SLC7A11: (i) a hydrogen bond between isoxazole‐3‐carboxylate and Thr56, (ii) π–π interaction of the isoxazole ring with Arg135, (iii) lipophilic interactions with Ile142, Tyr244, and Il134, and (iv) a hydrogen bond between the phenolic hydroxy group and Ser133.
Structural optimization of the conformationally restrained isoxazole scaffold yielded derivatives with bulky lipophilic residues in 3‐ and/or 4‐position (NACPA (8), NEIH (9), and TFMIH (10); K i = 3–100 µM) (Figure 2), 146 which efficiently inhibit system Xc − but are no longer substrates. Interestingly, the substitution pattern determines whether system Xc − inhibition is competitive (one lipophilic substituent) or noncompetitive (two lipophilic substituents). The authors propose that the isoxazoles target two lipophilic pockets adjacent to the substrate binding site. Given the structural similarities to glutamate, it is not surprising that 1 and derivatives show many cross‐reactivities, in particular with glutamate receptors of the central nervous system. 147
5.2. β‐N‐oxalyl‐l‐α‐β‐diaminopropionic acid (11)
The excitotoxin 11 (Figure 2) from Lathyrus species (e.g., L. sativus, L. clymenum, and L. cicera) activates non‐N‐methyl‐d‐aspartate receptors (K D = 1.3 µM). 148 , 149 Later, compound 9 was identified as a competitive inhibitor and alternative substrate of system Xc − in rat LRM55 glia cells and human SNB‐19 glioma cells (80%‐85% inhibition of glutamate transport at 500 µM), and it has been speculated that an intracellular accumulation of 11 enlarges the target profile to thus far unknown proteins. 149
5.3. l‐Alanosine (12)
The amino acid antimetabolite 12 (Figure 2) from Streptomyces alanosinicus carries a 3‐hydroxynitrosamine group in the side chain and has pronounced antitumoral activity in vitro and in vivo. 150 Correlations between SLC7A11 gene expression and the potency of 1400 anticancer agents over 60 human cancer cell lines revealed 12 as inhibitor and substrate of system Xc − with half maximal inhibitory concentration (IC50) values ranging from 0.86 to 53 µM in different carcinoma cell lines.
5.4. Capsazepine (13)
The thiourea derivative 13 (Figure 2) derives from capsaicin, the active ingredient of chili pepper, and competitively inhibits vanilloid receptor‐1 (TRPV‐1), though with moderate potency (IC50 = 0.42 µM). 151 Compound 13 reversibly binds to system Xc − (EC50 = 3 µM), elevates ROS levels in human MDA‐MB‐231 breast cancer cells (at 25 µM), and triggers cell death at high concentrations (10% at 25 µM 13; 90% at 100 µM 13). 152
5.5. Sulfasalazin (14)
The Food and Drug Administration (FDA)‐approved anti‐inflammatory drug 14 (brand name Azulfidine, Salazopyrin, Sulazine, etc.) was identified in 1985 as a moderate inhibitor of system Xc − (IC50 = 30 µM) (Figure 2). 153 Several clinical studies with 14 for the (supportive) treatment of breast cancer and brain tumors are ongoing (www.clinicaltrials.gov, NCT03847311, NCT01577966, NCT04205357). While the diazo group of 14 has to be cleaved to yield the anti‐inflammatory active compound mesalazine, this cleavage is detrimental to system Xc − inhibition. 154 The poor metabolic stability of the diazo group thus limits the application of 14 as anticancer drug. 153 structure–activity relationship (SAR) studies revealed that the replacement of the diazo group by a linear alkyne (Figure 2) is tolerated without loss of activity (IC50 = 30 µM) and improves metabolic stability. Replacement by ethenediyl (Figure 2) is accepted with slightly decreased inhibitory potency (IC50 = 70 µM), whereas the saturated ethane bridge (Figure 2) abolishes system Xc − inhibition (IC50 = 700 µM), which indicates that rigidization is essential for activity. The free carboxylic acid of 14 is also important for target interaction; methyl esters are not active. Removal of the phenolic alcohol group is tolerated and potentially enhances metabolic stability by eliminating phase II metabolism. Together, the structural optimization of 14 yielded derivatives with improved metabolic stability, but their affinity to system Xc − remained low. Accordingly, compound 14 less potently inhibits cystine uptake in human HT‐1080 fibrosarcoma and human Calu‐1 non‐small‐cell lung cancer (NSCLC) cells (EC50 = 32 µM) than erastin (EC50 = 0.3 µM). 54 We conclude that 14 induces ferroptosis in various cancer cell lines but has major limitations as a drug (template) for ferroptosis‐based anticancer therapy.
Supposedly inspired by the isoxazole derivatives 8, 9, and 10, analogues of 14 with lipophilic moieties (15–17) were designed that might address the same lipophilic binding pockets as proposed for the substituted isoxazoles (Figure 2). 155 With increasing length and lipophilicity of the substituents, the binding mode switches from competitive to noncompetitive, similar to what was observed for aryl‐substituted isoxazoles, which further strengthens the hypothesis that allosteric sites are located close to the substrate pocket. The thus optimized derivatives of 14 gained inhibitory potency (2.5‐fold) but are of limited therapeutic value for the treatment of glioblastoma because they cannot cross the blood‐brain barrier. 155
5.6. Erastin (18) and analogues 19 and 20
By screening small molecules that activate iron‐dependent cell death in oncogenic‐RAS‐harboring cancer cells, 18 (Figure 2) was identified as system Xc − inhibitor with the novel scaffold. 88 , 118 Compound 18 (10 µM) induces ferroptosis in N‐RAS‐mutant HT‐1080 fibrosarcoma cells by inhibiting system Xc − and lowering the cellular levels of GSH, 156 , 157 which is needed by GPX4 as cofactor to reduce lipid hydroperoxides. With an IC50 value of 1.4 µM, compound 18 inhibits cystine uptake in Slc7a11‐overexpressing MEFs more potent than other systems Xc − inhibitors (IC50 = 26.1 µM for 14, 4.4 µM for 2‐(S)‐(4‐carboxyphenyl)glycine [CPG] and 15.4 µM for 1). 158 The potency of ferroptosis inducers, including 18, strongly differs between cell lines and experimental settings. For example, human BJ‐TERT/LT/ST/RASG12V tumorigenic fibroblasts are considerably more sensitive to 18 (EC50 = 1.25 µM) than primary BJ fibroblasts (EC50 > 5 µM, selectivity > 8). 159 Modifications of 18 are only tolerated at the ethoxy‐phenyl moiety, with a biphenyl substituent increasing the potency 10‐fold (Figure 2). Modifications at other sites, in particular at the rigid piperazine linker, reduced or abrogated system Xc − inhibition (40% inhibition of glutamate release at 10 µM in human HT‐1080 fibrosarcoma cells). 54 , 160 Note that a rigid linear structural element is also present in derivatives of 14 and in the isoxazoles 8, 9, and 10 (where isoxazole was suggested to mimic cystine) and seemingly represents a common feature of potent system Xc − inhibitors. It is tempting to speculate that these rigidized inhibitors occupy the same binding site.
Compound 18 irreversibly inhibits system Xc −, 158 which is unique among system Xc − inhibitors and not shared by 14, CPG, and 1. 158 Thus, 18 is a serendipitously discovered targeted covalent inhibitor (TCI). 134 These inhibitors have a bond‐forming group with relatively low reactivity and additional residues that noncovalently bind the target. 134 The additional residues are required for the exact positioning of the inhibitor, which, after binding, reacts rapidly with the noncatalytic residue, in most cases a cysteine. The acetoxy group is the most likely point of attack for reactive cysteines in 18, which might explain why this moiety is indispensable for inhibitory activity. Of note, the acetoxy linker resembles the chloroacetamide group of GPX4 inhibitors (Figure 3), which covalently bind to selenocysteine and cysteine. Where 18 exactly binds system Xc − is not known. Mutation of six endogenous cysteine residues (Cys86, Cys158, Cys197, Cys271, C327, and Cys435) in SLC7A11 did not result in a loss of inhibitory activity, which suggests that 18 either reacts with alternative sulfhydryl groups or binds to amino acid residues that were not investigated. 158 Further studies are required to elucidate the binding mode of 18.
Figure 3.
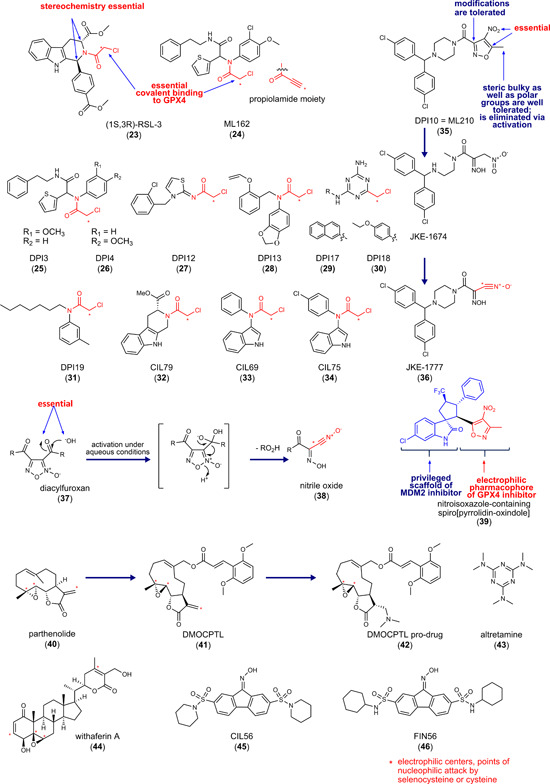
Inhibitors of glutathione peroxidase 4. MDM2, mouse double minute 2. [Color figure can be viewed at wileyonlinelibrary.com]
The poor water solubility and metabolic stability of 18 limit its application in vivo. Introduction of piperazin‐1‐methyl (19) or imidazole‐1‐ethanoyl (imidazole ketone erastin 20) at 5‐position of the ethoxy‐phenyl ring yields derivatives of 18 with 16‐fold (19) and threefold (20) improved water solubility (Figure 2). 54 In addition, system Xc − inhibitory activity was strongly enhanced for 20 as compared to 18 in human BJeLR fibroblasts (20: IC50 = 3 nM; 18: IC50 = 625 nM). 10 When given intraperitoneally (ip) at 23 or 40 mg/kg, 20 reduced tumor size in a human B cell lymphoma SUDHL6 cell xenograft model in mice. 161 Referring to the pharmacokinetic profile, the authors stated that 20 might not be suited to counter rapid tumor growth in vivo. The analogue of 18, PRLX 93936 (21), was subjected to two clinical trials investigating safety, efficiency, and pharmacokinetics in patients with advanced solid tumors and multiple myeloma in 2007 and 2012 (www.clinicaltrials.gov, NCT00528047 and NCT01695590). The outcome has not been published. Notably, compound 21 lacks the 4‐chloro‐phenoxyethanoyl moiety, which is considered to be essential for system Xc − inhibition by 18.
5.7. Sorafenib (22) and analogues
Compound 22 is a multikinase inhibitor with anticancer properties that inhibits system Xc − as subordinate target (Figure 2). 54 SARs studies with 87 analogues of 22 indicate that similar features are required for system Xc − inhibition than for binding of 22 to kinases like B‐RAF. Accordingly, the ferroptosis‐inducing activity was diminished by removing the lipophilic meta‐CF3 group that points toward the hydrophobic pocket of the kinase or by modifying the urea structure that forms two hydrogen bonds between the urea substructure and Glu501 of B‐RAF. 162 These findings allow for two interpretations. Either 22 binds to a pocket of system Xc −, which resembles the interaction site at the kinase, or inhibits a yet unknown kinase that lowers Xc − antiporter activity. 54 Of note, the urea structure is also an integral part of the 1‐oxa‐2,4‐diazolidine‐3,5‐dione ring of 1 that was replaced by isoxazole in 8, 9, and 10. These scaffolds mimic the preferred conformation of cystine and were proposed to target the system Xc − substrate binding site and additionally extend to lipophilic allosteric regions. 147 We speculate that 22, which consists of a urea core with two lipophilic substituents, shares this binding (Figure 2). Recently, analogues of 22 were described that trigger multiple forms of cell death, including ferroptosis, apoptosis, and autophagy, depending on compound concentration and incubation time. 163
Novel synthetic small molecule inhibitors of system Xc − with cytotoxic effects on various cancer cell lines (lung, cervix, muscle, colon, breast, and prostate; EC50 = 5–10 µM) were identified in 2019. Strongest effects were observed for cells with the mesenchymal phenotype (e.g., human HT1080 fibrosarcoma cells and human Rh30 and Rh41 rhabdomyosarcoma cells), whereas epithelial cells (MCF7, HCT116) were only slightly affected. 164
5.8. Other inhibitors of system Xc −
Cystine uptake via system Xc − can be competitively blocked by compounds that mimic substrates of the antiporter, such as l ‐glutamate, α‐aminoadipate, l ‐homocysteate, l ‐serine‐O‐sulfate, and l ‐cystathionine 165 as well as CPG, 2‐(S)‐(4‐sulfophenyl)glycine, 2‐(S)‐[4‐(4‐carboxyphenyl)phenyl]glycine, and 2‐(S)‐(5‐sulfothien‐2‐yl)glycine. 144 , 158 Moreover, mercurial reagents, such as p‐chloromercuribenzoic acid and p‐chloromercuribenzenesulfonic acid potently inhibit system Xc − activity by interacting with Cys327 of SLC7A11. 158
6. GPX4 INHIBITORS
Eight glutathione peroxidase (GPX) isoenzymes exist in humans; five of them are selenoproteins with selenocysteine in the active center. 166 Selenocysteines are markedly more nucleophilic than cysteines, 167 which adds to their enzymatic reactivity and renders them prone to electrophilic attacks. GPX4 is unique among GPX isoenzymes for its ability to accept complex phospholipid hydroperoxides as substrates, 168 which can be explained by the monomeric rather than tetrameric structure and the lack of a surface‐exposed loop lining the active site that is present in other GPX isoenzymes. 169 The flat binding cleft of GPX4 consists of long, apolar residues that surround the catalytic tetrad (Sec46, Gln81, Trp136, and Asn137) and extends into a positively charged surface consisting of basic amino acids (K48, K135, and R152). 169 , 170 With regard to the substrates of GPX4, the apolar region (Met156, Pro155, Gly154, I1e29, Leu130, Ala133, I1e34, Phe78, and Gly79) likely supports the binding of lipid hydroperoxides, whereas the positively charged area might interact with the negatively charged phosphate in phospholipids. Interestingly, GPX4 has a structural motif that consists of Cys148, Gln123, and Trp119 and resembles the catalytic tetrad but is located on the opposite side of the enzyme. 169 Site‐directed mutagenesis studies suggest that this motif does not contribute to catalysis, though it might be involved in binding ligands that allosterically modulate GPX4 activity. In fact, two synthetic activators of GPX4 have recently been found to bind to this putative allosteric site. 171 , 172
6.1. Direct inhibitors of GPX4
6.1.1. Chloroacetamide derivatives
RSL3 (23) (Figure 3) was identified in tumorous fibroblast‐derived cell lines as a small molecule with oncogenic RAS‐selective lethality (RSL) in 2008, and GPX4 was identified as a target in 2014. 69 , 118 In contrast to 18‐like compounds, which lower GPX4 activity by depleting the cosubstrate GSH, (1S,3R)‐RSL3 (23) directly inhibits GPX4 by covalently binding to Sec46 in the active center via the chloroacetamide motif. 69 (1S,3R)‐RSL3 (50 nM) suppresses GPX4 activity in human COH‐BR1 breast cancer cells that overexpress GPX4 (L7G4 variant). The two stereogenic centers of 23 are important for inhibitory activity. While the (1S,3R)‐stereoisomer potently inhibits GPX4, the (1R,3S)‐enantiomer, and the (1S,3S)‐ and (1R,3R)‐diastereomers are more than 100‐fold less active. Moreover, they do not show a preference for killing human BJ‐derived fibroblasts with an oncogenic RAS mutation (H‐RASG12V) over BJ‐derived fibroblasts with wild‐type H‐RAS.
In a large‐scale screen with over 1,000,000 compounds, 14 ferroptosis‐inducing compounds were identified with similar chemical structure, 7 of them have an electrophilic chloroacetamide moiety and inhibit GPX4 (Figure 3; 24–31). 69 The compounds selectively suppress cell growth in human H‐RASG12V‐expressing BJ fibroblasts with GI50 values ranging from 0.02 to 0.3 µM (compare with 23: GI50 = 0.15 µM).
The hit compound ML162 (24) was subjected to SARs studies, which focused on the substitution pattern at the thiophene, chloromethoxy, or phenethylamine moiety but did not lead to more potent or selective analogs. ML162 surpassed 23 in inducing cytotoxicity (0.025 vs. 0.1 µM, respectively), although both compounds were comparably efficient in inhibiting the GPX4‐dependent degradation of phosphatidylcholine hydroperoxides in the cellular context. 69 , 173
Another screen for cytotoxic agents with 3169 compounds identified 10 compounds that induce ferroptosis. Three of them (32–34) possess again an electrophilic chloroacetamide moiety and closely resemble the structure of 23, with compound 32 only lacking the 4‐(methoxycarbonyl)phenyl moiety and consequently one of the stereogenic centers. 174
Nonfavorable pharmacodynamic properties as well as high reactivity to off‐target proteins are major disadvantages of strong electrophiles like chloroacetamides. 4 Weak electrophiles like acrylamides or fumarates might improve target selectivity but are not compatible with covalent GPX4 inhibition, despite the high nucleophilicity of selenocysteine, 4 likely due to slow reaction kinetics. Increasing the enzyme resident time by exploiting noncovalent interactions might circumvent these limitations.
The 4‐nitro‐1,2‐oxazol derivative DPI10 (also named ML210, 35), identified in the above‐mentioned large‐scale screen, 4 inhibits GPX4 in a cellular system despite lacking an electrophilic chloroacetamide moiety but fails to reduce GPX4 activity under cell‐free conditions. Subsequent SARs studies guided the structural optimization toward compound 35 which selectively induces ferroptosis in H‐RASG12V BJ cells. 173 Compound 35 is a masked electrophile with a nitroisoxazole system that is converted to the metabolite JKE‐1674 and subsequently to the reactive nitrile oxide (36) (Figure 3), which forms a covalent adduct with GPX4. 4 Masking the reactive electrophile as prodrug improves the selectivity of 35 as compared to 23 and 24, which both show high off‐target activity in a spectrometry‐based proteomics screen. 4 Notably, the proteins interacting with 35 differ from those 23 and 24 binds to. Compound 34 mainly interacts with abundant proteins, for example, tubulin, whereas 23 and 24 both have a more defined off‐target profile. Masking the reactive group in 35 also makes the inhibitor more stable, however at the expense of inhibitory activity. 4 Replacement of chloroacetamide in 23, 24, 28, and 31, by nitroisoxazole resulted in inactive compounds except for 31. 175 This finding indicates that concrete structural elements apparently underlie the intracellular enzymatic activation of the heterocyclic nitro group as nitril oxide. Eaton et al. 176 described further nitrile oxide prodrugs with a diacylfuroxan scaffold (37) (Figure 3), but the proteome‐wide reactivity of these compounds rather excludes the masking of nitrile oxide as diacylfluroxan is an effective mean to enhance the selectivity for GPX4. The prodrugs seem inherently unstable and are subjected to multiple decomposition pathways. Together, masked electrophiles are valuable probes to study GPX4 function but have not been developed into promising drug candidates thus far. 176
Compound 24, the most potent GPX4 inhibitor identified to date, was used as a template in a systematic study to search for possible replacements for the chloroacetamide warhead. 175 Among the 25 electrophilic groups tested, only propiolamides retained the ability to induce ferroptotic cell death. In derivatives of 35, chloroacetamide, propiolamide, and trimethylsilyl propiolamide warheads are tolerated as substitutes for the original 5‐methyl‐4‐nitro‐3‐isoxazolyl moiety. Propiolamides, which are highly reactive and effectively bind thiol nucleophiles, were expected to face similar challenges in selectivity, pharmacokinetics, and stability to chloroacetamides. It was therefore surprising that cotreatment with the ferroptosis inhibitor ferrostatin‐1 raised the IC50 value of the propiolamide derivative to 24 stronger than for the lead compound, which might hint toward a superior GPX4 selectivity. 175
Because of the challenging bacterial expression of selenocysteine‐containing proteins, most crystal structures available for GPX4 used the GPX4U46C mutant that lacks the catalytic selenocysteine. Moosmayer et al. 177 recently disclosed the crystal structure of wild‐type human GPX4 in complex with the covalent inhibitor 24 (Figure 4). Compound 24 does interestingly not only covalently bind to Sec46 but also to Cys66, which underlines the limited selectivity of chloroacetamide warheads. This finding implies that 24 likely reacts with a broad spectrum of proteins within the cysteinome. Further studies were conducted on the GPX4C66S mutant, in which the reactive cysteine Cys66 was replaced by serine to prevent heterogeneous covalent modifications. 177 Compound 24 is located at the shallow surface pit near the catalytic tetrad, where it interacts with Gln81, Trp136, and Asn137 of the catalytic tetrad. Note that GPX4 inhibitory peptide GPX4 binding peptides (GXpep)‐3 described later in this section occupies a different surface area of GPX4 (Figure 4). 177 The cocrystal structure of GPX4 with 24 confirmed the lack of a well‐defined deeper binding pocket but revealed a subpocket adjacent to Sec46, which is located between Lys48 and Trp136 and might be occupied by inhibitors to enhance selectivity. 177
Figure 4.
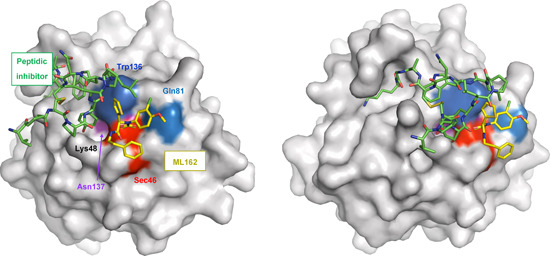
Human GPX4 (C66S) in complex with ML162 (S enantiomer). The surface of GPX4 with bound (S)‐ML162 (depicted with yellow carbons) (PDB entry 6HKQ) was superimposed to a structure that shows the peptide inhibitor GXpep3 (depicted with green carbons) bound to GPX4 (U46C) (PDB entry 5h5s). The surface of the catalytic tetrad is colored red (Sec46), blue (Gln81), dark blue (Trp136), and purple (Asn137). Two different orientations are shown. Reproduced with permission of the International Union of Crystallography. 177 GPX4, glutathione peroxidase 4; PDB, Protein Data Bank. [Color figure can be viewed at wileyonlinelibrary.com]
An elegant cell‐based study on the binding mode of diverse direct and indirect GPX4 inhibitors recently addressed the impact of the most reactive cysteine residues in GPX4 (Cys2, Cys10, Cys37, Sec46, Cys66, Cys75, Cys75, Cys107, and Cys148) on enzyme activity. 178 Interestingly, it was found that GPX4 inhibition does not exclusively rely on the covalent coupling to active site Sec46 but also involves covalent modification of Cys66 (which is close to the active site) and Cys10. 178 For example, the direct GPX4 inhibitors RSL3 and ML162 selectively target Sec46 and Cys66 and, in case of ML162, additionally bind to the less reactive GPX4Sec46Cys mutant, for which the active center selenocysteine was replaced by a less reactive cysteine. Covalent binding to Cys66 induces conformational changes and subjects GPX4 to degradation. In addition, Cys10, as well as Cys66, are important for the regulation of GPX4 activity under limited GSH supply, when cells were treated with the system Xc − inhibitor 20. It was suggested that GPX4 forms a locked inactive dimeric form of GPX4 under limited GSH conditions via Cys10 (GPX4–Cys10–Sec46–GPX4) that is regenerated into active GPX4 by reductive cleavage involving Cys66 from a third GPX4 protein. Other indirect GPX4 inhibitors, that is, FIN56 and FINO2, do not bind to Cys66. In summary, a complex regulatory system exists in cells that fine‐tunes the activity of GPX4 under distinct cellular conditions.
6.1.2. Nitroisoxazole derivatives
Novel nitroisoxazole‐containing spiro[pyrrolidine‐oxindoles] (39) were designed as dual inhibitors of GPX4 and mouse double minute 2 (MDM2) (Figure 3) to induce both ferroptosis and apoptosis in cancer cells. Cellular thermal shift assays indicate that these compounds covalently bind to GPX4. The dual inhibitors exhibit inhibitory effects on MDM2 and efficiently reduced the tumor volume in a murine MCF‐7 xenograft model at 25 mg/kg (ip). 179
6.1.3. GPX4 inhibitory peptide GXpep3 (Val–Pro–Cys–Pro–Tyr–Leu–Pro–Leu–Trp–Asn–Cys–Ala–Gly–Lys)
The crystal structure of GPX4 in complex with the GXpep 1–3 has been resolved for a mutant enzyme (Cys37Ala, Cys93Arg, Cys134Glu, Cys175Val, Cys64Ser, Cys102Ser, and Sec73Cys, and an N‐terminal SUMO‐tag) and revealed noncovalent interaction. 180 GXpep1 and GXpep2 bind to a site far from the active center of GPX4, do not induce conformational changes, and are inactive. In contrast, GXpep3 occupies a cavity close to the catalytic site and inhibits GPX4 activity with an IC50 of 10 µM (Figure 4). The interaction of GXpep‐3 and GPX4 involves (i) hydrogen bonds between the C‐terminal residue (Lys14) of GXpep3 and the main‐chain of Ile156 and side‐chain of Lys162 and Arg179 in GPX4, (ii) hydrophobic interactions between the Trp9 side chain of GXpep‐3 and Trp163 and Pro182 of GPX4 as well as (iii) a water‐mediated hydrogen bond between the Tyr5 side chain of the inhibitor and the main‐chain of Asn164 in GPX4 close to the active center. Peptides with substituted Tyr5 side chain that covalently target selenocysteine Sec73 have been proposed to possess superior GPX4 inhibitory activity, 180 which is in line with the TCI concept described in Section 5. 134 Thus, the noncovalent interactions of the peptide with GPX4 might align the inhibitor in the binding cleft, thereby favoring a subsequent reaction between the electrophilic group and the protein.
6.1.4. Parthenolide derivative DMOCPTL (41)
The naturally occurring anticancer germacranolide‐type sesquiterpene lactone parthenolide (40) was used as a template to design 41, which has, like 40, an exocyclic α,β‐unsaturated carbonyl function as well as an epoxide as reactive groups (Figure 3). 181 Compound 41 reduces the viability of the human triple‐negative breast cancer cell lines MDA‐MB‐231 and SUM159 (EC50 = 0.2–0.5 µM). The ferroptosis inhibitors deferoxamine (DFO) and ferrostatin‐1 as well as the apoptosis inhibitor Z‐VAD‐FMK attenuated the cytotoxic effect of 41, whereas inhibition of autophagy (3‐methyladenine [3‐MA]) and necroptosis (necrostatin‐1) was without effect. While the induction of apoptosis by 41 was ascribed to an upregulation of EGR1, the drop in GPX4 protein levels seems to be responsible for ferroptosis induction. 181 Of note, the transcription factor EGR1 is known to be a negative regulator of GPX4 protein expression. 71 Compound 41 directly binds to GPX4 and promotes ubiquitination and subsequent degradation of the enzyme, as confirmed by pull‐down assays and GPX4/compound 41 colocalization studies using a fluorescence‐labeled derivative of 41. Docking studies suggest that 41 is tightly bound to the active site of GPX4 with the styryl ring of 41 extending into a hydrophobic pocket. Proposed key interactions between 41 and GPX4 are (i) strong hydrophobic interactions of the styryl ring with Trp163 and Pro182 of GPX4 and (ii) hydrogen bonds between the oxygen of the methoxy group and the Lys162 and Ile156 side chains of GPX4. Whether 41 has additional targets besides GPX4 that contribute to ferroptosis induction is unknown. Speculations about further targets are supported by the parental compound 40 that activates NRF2 (Figure 3) 132 , 182 , 183 and inhibits thioredoxin reductase (IC50 = 2.5–5 µM). 184 Inhibition of the latter by 40 has been linked to apoptosis in human Hela cervix carcinoma cells. 184 Considering the important role of the thioredoxin/thioredoxin reductase system in GSH regeneration and protection from ferroptosis, 3 , 185 we speculate that the ferroptosis‐inducing activity of 40 and potentially 41 at least partially relies on the inhibition of these pathways. Due to the poor pharmacokinetics of 41, a prodrug (42, Figure 3) was subjected to preclinical studies. In mice bearing tumors derived from mouse 4T1 breast cancer cells, the prodrug 42 (1 mg/kg, iv, 6× every other day) decreased Gpx4 and increased Egr1 protein levels and inhibited tumor growth. Oral application (50 mg/kg) of 42 prolonged survival without any obvious toxic effects (drop in body weight). Together, compound 42 is a promising candidate for the development of drugs directed against breast cancer, which might include aggressive, metastatic forms like triple‐negative tumors.
6.1.5. Altretamine (43)
The FDA‐approved anticancer drug 43 was recently identified as a GPX4 inhibitor by network perturbation analysis, which combines hybrid computational and experimental studies (DeMAND) and aims at elucidating genome‐wide molecular mechanisms of small molecules. 186 Compound 43 inhibits the GPX4‐dependent reduction of phosphatidylcholine hydroperoxide in a cell‐free assay, although at a relatively high concentration (500 µM). 186 Considering that 43 is an alkylating agent, the GPX4‐inhibitory activity might derive from the covalent modification of selenocysteine in the active center.
6.1.6. Withaferin A (44)
Withania somnifera and other members of the Solanaceae family (e.g., Acnistus arborescens) possess anti‐inflammatory, antioxidative, and stress‐adaptive properties and are traditionally used in ayurvedic medicine to treat tumors and ulcers. 187 The bufadienolide 44 (Figure 3), isolated from the roots of these plants, covalently binds to GPX4 at micromolar concentrations (10 µM). 188 Mutagenesis studies indicate that 44 interacts preferentially with cysteines but not with the selenocysteine of GPX4. 188 Compound 44 exhibits three reactive sites, which are attacked by nucleophilic cysteines, two α,β‐unsaturated carbonyl groups, and a highly reactive epoxide. Moreover, compound 44 affects various other pathways (e.g., NRF2 and NF‐κB signaling, cytoskeleton dynamics, cell cycle regulation, and kinase cascades), whose contribution to ferroptotic cell death versus other forms of cell death remains to be investigated. 189
6.1.7. Comparative discussion
Taken together, potent GPX4 inhibitors have been identified that covalently bind to selenocysteine in the active center. Since these inhibitors require reactive electrophilic groups with rapid reaction kinetics and hardly engage noncovalent interactions, they lack selectivity and metabolic stability. To date, none of the GPX4 inhibitors is applicable for in vivo use due to low solubility and poor pharmacokinetics. 31 To circumvent these drawbacks, pro‐drugs have been developed that are intracellularly converted into electrophiles, however at the expense of potency. 4 GPX4 has a flat binding pocket, which allows the enzyme to accept bulky phospholipids (embedded into bilayers) as substrates. Identification of a conventional small‐molecule inhibitor that forms noncovalent interactions with GPX4 might therefore be challenging. A large database screen with over 1,000,000 compounds actually identified only nine electrophilic inhibitors that covalently bind to the catalytic selenocysteine of GPX4. Additional noncovalent interactions were low or absent. Note that inhibitors that simply rely on an electrophilic moiety for GPX4 inhibition are poorly compatible with selectivity, as confirmed by cysteinome analysis. 4 On the other hand, it should be taken into account that databases used for screening rather include conventional small molecule inhibitors, whereas noncovalent inhibitors of GPX4 might demand structural features that are more complex and not covered. GPX4 inhibition by the peptide GXpep3 indicates that the development of noncovalent inhibitors of GPX4 is feasible and also the parthenolide derivative seems to form noncovalent interactions with GPX4 according to modeling studies. 180 , 181 Noncovalent binding to GPX might be accomplished by mimicking either the cosubstrate GSH, which is a small peptide containing three amino acids, or the natural substrates of GPX4, that is, oxidized PUFAs. The high chemical diversity of natural products might make a difference and pave the way to success in this challenging field. Promising for targeting GPX4 appear peptide‐like structures and oxylipins, in particular when combined with electrophilic warheads, such as α,β‐unsaturated ketones.
6.2. Indirect inhibitors of GPX4
Interference with GPX4 expression is an indirect strategy to lower GPX4 activity. Inhibitors of NRF2, which we discuss in detail in Section 8 employ this mechanism. The small molecules CIL56 (45) and FIN56 (46) (Figure 3) also lead to a drop in GPX4 protein levels but engage a different mechanism. The two compounds potently induce ferroptotic cell death in human HT1080 fibrosarcoma cells (EC50 = 0.1–0.4 µM), with 46 being more selective toward oncogenic H‐RAS‐mutant cells (H‐RASG12V). 174 Both compounds stimulate GPX4 degradation by a nonproteasomal mechanism, which might involve the activation of acetyl‐CoA carboxylase (ACC) 1, as suggested from compensation studies using the acetyl‐CoA carboxylase ACC1 inhibitor 5‐tetradecyloxy‐2‐furonic acid. 100 , 174 The exact mechanisms remain elusive. Compound 46 also activates squalene synthase (Figure 1), thereby depleting coenzyme Q10. Further studies are required to evaluate the potential of 45 and 46 for in vivo application. 31 Note that the natural product derivative FINO2 (97) described in Section 9.1 also indirectly inhibits GPX4 activity.
6.3. Combined indirect GPX4 inhibitors and HO‐1 inducers
The role of NRF2 activation on ferroptosis is bivalent. NRF2 activators have been described to counteract lipid peroxidation and ferroptosis 190 but also to have antitumoral activity associated to ferroptotic cell death. 48 The latter is counterintuitive since ferroptosis relies on the oxidative damage of cellular lipids against which classical NRF2 target genes provide protection. 138 The enigmatic mechanisms behind the proferroptotic activity might be related to the point of attack of NRF2 activators within the NRF2 signaling pathway, the responsiveness of the selected model system, cell type‐specific NRF2 target gene profiles, or off‐target effects. Among the critical determinants that define the cellular fate upon NRF2 activation is HO‐1 as well as a simultaneous suppression of GPX4 levels. The NRF2‐target gene HO‐1 degrades heme to biliverdin/bilirubin, carbon monoxide, and ferrous iron, which then regulate cell death or mediate cytoprotection. 191 The ferroptosis‐inducing activity of HO‐1 predominantly depends on the release of ferrous iron, whereas biliverdin, bilirubin, and carbon monoxide have overall antioxidant, antiapoptotic, anti‐inflammatory, and stress‐protective activity. 192 HO‐1 seems to be a double‐edged sword, with both tumor‐inhibiting and ‐promoting activity depending on the metabolic status of the cancer cells and the tumor microenvironment. Studies on the pharmacological or genetic induction of HO‐1 support this hypothesis. On the one hand, HO‐1 expression enhanced ferroptotic cancer cell death upon cystine deprivation (compound 18). 193 On the other hand, HO‐1‐stimulating small molecules rather had the opposite effect and prevented ferroptosis in noncancer cells. 194 , 195
6.3.1. Curcumin (47) and derivatives 48 and 49
The yellow dye from curcuma, 47 (Figure 5), is a Michael acceptor and pan–assay interference compound, for which a broad spectrum of activities has been described in vitro and in vivo, including antioxidative, anticancer, and antimetastatic properties. 196 Compound 47 induces cell death in a variety of cancer cell lines, mainly by inducing apoptosis. Ferroptosis has recently been suggested to contribute to cell death induction by 47 in human breast cancer cell lines (EC50 = 50–100 µM), based on the protective effect of ferrostatin‐1 and DFO. 197 Thus, compound 47 induced the accumulation of intracellular iron, enhanced lipid peroxidation, decreased glutathione levels, and regulated ferroptosis target genes on a transcriptome level. Accordingly, the piperidone‐based C5‐curcuminoid EF24 (48) (Figure 5) (EC50 = 1–2 µM) and the cyclobutanmethyl‐substituted C7‐curcuminoid ALZ003 (49) (Figure 5) (at 2–10 µM) decreased the viability of human U2OS and Saos‐2 osteosarcoma cells 198 and human U87MG glioma cells, 199 respectively, but did not show cytotoxic effects on primary human astrocytes. 199 While 49 also induced apoptosis at micromolar concentrations (2–5 µM) in addition to ferroptosis, 199 the cytotoxic effects of 48 were exclusively diminished by ferrostatin‐1 but neither by apoptosis (Z‐VAD‐FMK), pyroptosis (necrosulfonamide), nor autophagy inhibitor (MRT68921). 198 Cell death induction by 49 was ascribed to ubiquitination‐dependent degradation of AR, which represses GPX4 expression and subsequently raises lipid hydroperoxide levels. 199 Knockdown of AR induced lipid hydroperoxide formation, whereas overexpression upregulated GPX4 and had the opposite effect. Importantly, AR expression is elevated in human glioblastoma compared to normal brain tissue and further increased in temozolomide‐resistant cells. Accordingly, both temozolomide‐sensitive and ‐resistant glioblastomas were significantly inhibited by 49, while cytotoxic effects were not observed in normal astrocytes. 199
Figure 5.
Combined indirect glutathione peroxidase 4 inhibitors and heme oxygenase‐1 inducers. PEITC, phenylethyl isothiocyanate; TCBQ, tetrachloro‐1,4‐benzoquinone. [Color figure can be viewed at wileyonlinelibrary.com]
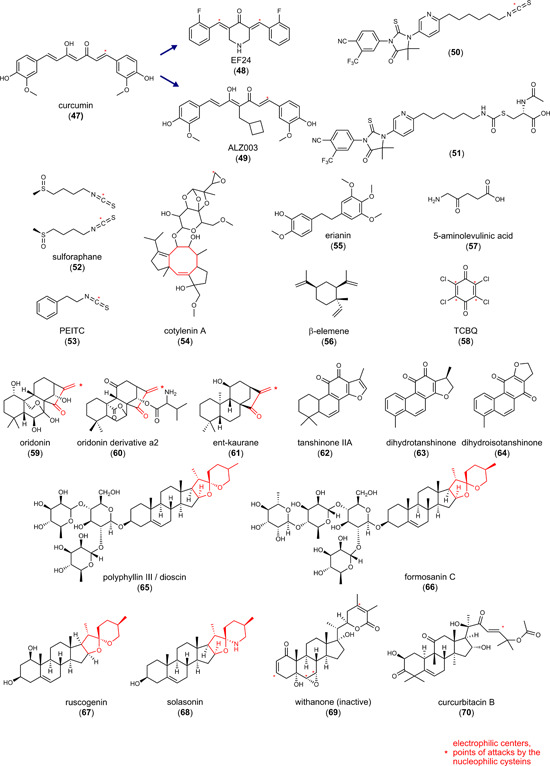
6.3.2. Hybrid AR antagonists (50 and 51)
The AR is known to (i) contribute to redox homeostasis, 200 (ii) be upregulated in tumor tissues, and (iii) promote drug resistance, 201 but its role in ferroptosis is insufficiently understood. The combination of structural elements for AR inhibition (enzalutamide) and AR degradation (PEITC, 53) yielded isothiocyanate 50 (Figure 5), which was further optimized to the N‐acetylcysteine derivative 51 (Figure 5). 202 Within 72 h, the hybrid compounds reduced the viability of human VCaP prostate cancer cells that abundantly express full‐length wild‐type AR as well as a splice variant (50: EC50 = 3.9 µM; 51: EC50 = 6.1 µM). Compound 51 decreased the expression of the AR, elevated free iron levels, induced lipid peroxidation, and triggered the expression of the NRF2 target gene HO‐1. Cotreatment with the GSH biosynthesis inhibitor buthionine sulfoximine (BSO) (to compensate for higher GSH biosynthesis and regeneration upon NRF2 activation) enhanced these effects and showed pronounced cytotoxic activity already within 24 h, which was prevented either by ferrostatin‐1 or the iron chelator DFO. As described for 79, the upregulation of HO‐1 seems to be central for the ferroptosis‐inducing activity of 51, as the HO‐1 inhibitor zinc protoporphyrin (ZnPP)‐9 diminished the response. Qin et al. 202 speculated that the combined treatment of 51 and BSO unfolds its anticancer properties by downregulating the AR and inducing ferroptosis by NRF2‐dependent expression of HO‐1.
Most recently, overexpression and knockdown studies revealed AR as a major regulator of GPX4 expression in the luminal AR (LAR) subtype of triple‐negative breast cancer cells, which strongly express AR and GPX4. 203 AR has been proposed to directly bind to the GPX4 promotor. Based on these findings, we speculate that impaired GPX4 expression contributes to the ferroptosis‐inducing activity of compounds 50 and 51.
These findings strengthen the mechanistic link between AR signaling, GPX4, and ferroptosis and we anticipate that AR inhibition and degradation also contribute to the ferroptosis‐inducing activity of the hybrid isothiocyanate‐ and N‐acetylcysteine‐containing AR antagonists 50 and 51 described above. Compound 48 seems to engage a similar mechanism, given the decrease of GPX4 protein expression. 198 On the other hand, compounds 50 and 51 induced ferroptosis by upregulating HO‐1, 202 , 204 and also 47 and 48 enhanced HO‐1 expression, 198 which closes another gap. Based on these studies, we hypothesize that hybrid isothiocyanate‐containing AR antagonists (50, 51) and curcuminoids (47–49) share a common ferroptotic mechanism: GPX4 is downregulated through suppression of AR (and potentially further mechanisms), while HO‐1 expression is induced, likely as a result of impaired AR signaling and/or NRF2 activation. Note that 47 is a KEAP1 inhibitor that protects NRF2 from proteasomal degradation. 205 In a murine xenograft model using intracranially transplanted wild‐type and temozolomide‐resistant human U87MG glioblastoma cells, compound 49 (20–80 mg/kg, iv) significantly reduced tumor size and prolonged survival.
6.3.3. Sulforaphane (52)
Mustard oil glycosides are abundant in cruciferous vegetables and taken up by food. 134 , 206 When getting in contact with the enzyme myrosinase upon cell damage, mustard oil glycosides are converted into isothiocyanates. These electrophiles have manifold targets and, amongst others, reversibly bind to KEAP1 at Cys151. 134 , 206 The isothiocyante 52 (Figure 5) is a major component responsible for the proposed anticarcinogenic effects of broccoli (Brassica oleracea, Brassicaceae) 207 and represents one of the most potent natural activators of the NRF2 pathway. Preclinical in vivo studies suggest (neuro)protective effects of 52 in Parkinson's disease (5 mg/kg, ip), 208 Huntington's disease (0.5 mg/kg, daily injection), 209 Alzheimer's disease (30 mg/kg, ip, daily) 210 and multiple sclerosis, as well as traumatic injuries (5–50 mg/kg, ip). 211 In addition to these protective effects on nontransformed cells, 52 triggers the death of cancer cells from different origins. 212 In small‐cell lung cancer, compound 52 (20 µM) induced lipid peroxidation and ferroptosis, which was diminished by ferrostatin‐1 and DFO. 212 Cytotoxic effects of 52 on normal 16 HBE bronchial epithelial cells were instead very low. Along these lines, compound 52 decreased the viability of cancerous but not normal prostate epithelial cells. 213 The exact mechanisms responsible for the selective lethality of the electrophilic compound 52 against cancer cells remain elusive. We suggest that 52 differentially affect redox‐dependent signaling pathways depending on the cellular iron and redox status. In this context, compound 52 increased the cellular iron pool, depleted GSH, and decreased the expression of the NRF2 target gene SLC7A11. 212 Given that 52 is a potent NRF2 activator 214 that strongly induces HO‐1 expression, 213 it is tempting to speculate that 52 unfolds its ferroptosis‐inducing activity in cancer cells, at least partially, by upregulating HO‐1.
6.3.4. Phenylethyl isothiocyanate (53)
The phenylethy isothiocyanate 53 (Figure 5) is prevalent in cruciferous vegetables and commonly found in Brassica species. 215 Antioxidative, anti‐inflammatory, and cytoprotective activities are well documented for compound 53; the latter in diverse in vitro (20 µM) and in vivo (1 mg/kg, −12.5 mg/kg, ip) organ injury models. 216 , 217 On the other hand, compound 53 induces NRF2 signaling, 218 as known for other isothiocyanates, and triggers multiple forms of cell death, including ferroptosis in murine K7M2 osteosarcoma cells. 219 Inhibitors of ferroptosis (ferrostatin‐1, liproxstatin‐1, DFO), apoptosis (Z‐VAD‐FMK) and necroptosis (necrostatin‐1) diminished the cytotoxic effects of 53 (EC50 = 30 µM) to a similar extent, whereas inhibition of autophagy (bafilomycin A1, 3‐MA) was less effective. Treatment with 53 resulted in ROS formation and lipid peroxidation, increased the labile iron pool, and lowered cellular GSH levels. Mechanistic studies revealed an enhanced ferritin heavy chain (FTH) 1 and SLC7A11 messenger RNA (mRNA) and decreased GPX4 protein expression but did not address HO‐1. 219 It was found in another study that 53 (10 µM) induces HO‐1 expression in human HCT116 human colon adenocarcinoma cells, which was blocked by coincubation with the iron chelator DFO. At high dosage, compound 53 (60 mg/kg, ig, once daily) reduced tumor weight in a K7M2 xenograft mouse model. 219 Increasing the dose to 90 mg/kg surprisingly lowered the antitumoral efficacy. Clinical trials (phase I–III, 2004‐2011) indicate that 53 is well‐tolerated (www.clinicaltrials.gov, NCT00968461, NCT00005883, NCT00691132, NCT01790204, NCT03700983, NCT03034603, and NCT02468882).
The fusicoccane diterpene glycoside cotylenin A (54) (Figure 5) isolated from the fungus Alternaria brassicicola 215 sensitizes cancer cells to 53. Treatment of human MIAPaCa‐2 and PANC‐1 pancreatic cancer cells with either 53 (4–6 µM) or 54 (24–32 µM) moderately triggered ROS production (twofold to fivefold), but only 53 decreased cell viability (by 40%). The combination of 53 and 54 showed strong synergistic effects, both inducing ROS formation (20‐fold) and inducing cell death (80%). Cytotoxic effects were prevented by N‐acetyl cysteine or the water‐soluble α‐tocopherol derivative trolox, and cell death induction was significantly attenuated by the membrane radical trapping agents ferrostatin‐1 and liproxstatin‐1 or the iron chelator DFO. Inhibition of other forms of cell death like apoptosis (Z‐VAD‐FMK, Q‐VD‐OPH), necroptosis (necrostatin‐1), or autophagy (3‐metyladenine and chloroquine) was not effective, which indicates that the cytotoxic effects of a combinatory treatment with 53 and 54 selectively unfolds its activity by inducing ferroptotic cell death.
6.3.5. Erianin (55)
The substituted dibenzylic compound 55 (Figure 5), isolated from Dendrobium chrysotoxum, substantially reduced the viability and slightly the proliferation of human H460 and H1299 lung cancer cells at concentrations of 50–100 nM. 220 However, compound 55 also has cytoprotective activities, as shown for normal rat NRK‐52E kidney epithelial cells under high glucose‐induced oxidative injury. 221 In cancer cells, low concentrations of compound 55 (12.5–100 nM) augmented intracellular ROS and MDA levels, depleted intracellular GSH, and decreased the expression of GPX4, SLC7A11, and SLC40A1. Very high concentrations of compound 55 (320 µM) additionally lowered the expression of NRF2 as well as of the NRF2 target proteins FTH1 and HO‐1. 222
Cotreatment with cysteine derivatives (N‐acetyl cysteine or GSH) and ferroptosis inhibitors (liproxstatin‐1 or ferrostatin‐1) diminished the drop in cell viability and proliferation by 55, 220 , 222 whereas inhibition of apoptosis (Z‐VAD‐FMK), necroptosis (necrostatin‐1), or autophagy (chloroquine) failed to suppress the cytotoxic effect. 222 Interestingly, the calmodulin (CaM) antagonist calmidazolium attenuated the inhibitory effect of 55 on the proliferation of lung cancer cells, which indicates that changes in Ca2+/CaM signaling regulate 55‐induced ferroptosis. 220 Compound 55 is effective in vivo and significantly suppressed tumor growth at high doses (100 mg/kg, ip, once daily) in murine xenograft models, either using H460 220 or human KU‐19–19 bladder carcinoma cells. 222
Since 55 has been reported to enhance the expression of NRF2 and its target genes HO‐1, SOD1, and SOD2 in vivo (20 mg/kg, ip, once daily), 223 we consider it likely that the NRF2/HO‐1 axis contributes to the ferroptotic mechanism of the compound. Whether the NRF2/HO‐1 pathway is activated in vivo (as shown in vitro for low and potentially pharmacologically relevant 55 concentrations) or downregulated (as observed for high compound concentrations) remains unclear. In this context, the expression of two other (ferroptosis‐related) NRF2 target genes, GPX4 and SLC7A11, is decreased by 55 in cell‐based studies (at low concentrations) 220 and in tumors from murine xenografts. 222 Such an opposing regulation of SLC7A11 and GPX4 relative to major NRF2 target genes (GCLC, GCL modifier subunit [GCLM], NAD(P)H quinone dehydrogenase 1 [NQO1], HO‐1) has, however, previously been observed, for example, for phorbol 12‐myristate 13‐acetate (PMA)‐treated human THP‐1 monocytic leukemia cells, 224 and is not necessarily an indicator for NRF2 inhibition.
6.3.6. β‐Elemene (56)
The natural sesquiterpene 56 (Figure 5) is prevalent in diverse medicinal plants, such as the Chinese herb Curcuma wenyujin. 225 At high concentrations (612 µM), 56 induced cell death in K‐RAS‐mutant HCT116 colorectal cancer cells (CRC) when combined with cetuximab (25 µg/ml), a monoclonal antibody against epidermal growth factor receptor (EGFR). Cytotoxic effects were strongly reduced by the ferroptosis inhibitors ferrostatin‐1, liproxstatin‐1, or DFO but neither by the apoptosis inhibitor Z‐VAD‐FMK, the necroptosis inhibitor necrostatin‐1 nor the autophagy inhibitor 3‐MA. The combinatory treatment with 56 and cetuximab depleted intracellular GSH, downregulated GPX4, SLC7A11, and SLC40A1 expression, and increased ROS and MDA levels. Expression of HO‐1, another NRF2 target gene, was instead upregulated, which is in line with our assumption that the NRF2/HO‐1 axis contributes to ferroptosis induction. Compound 56 also affects multiple other signaling pathways with important roles in tumorigenesis, among others Wnt‐β‐catenin (at 200 µM), Notch (at 250 µM), mitogen‐activated protein kinase (MAPK; at 100–400 µM), and autophagy cascades (at 50–250 µM), which likely contribute to the overall antitumoral activity. In an orthotopic colon cancer xenograft model in mice, compound 56 together with cetuximab (50 mg/kg, iv, every 6 days, each) triggered cell death of drug‐resistant cancer cells (HCT116‐luc cells) and improved survival. 225 Mechanistically, the combination of 56 and cetuximab lowered the expression of GPX4, SLC7A11, and SLC40A1 in grafted K‐RAS‐mutant HCT116 CRC cells. 225 Compound 56 is currently under investigation in phase 2 and 3 clinical trials for the treatment of cancer, including glioblastoma and lung cancer, either as monotherapy or in combination with tyrosine kinase inhibitors to treat tumors with acquired drug resistance (www.clinicaltrials.gov, NCT02629757, NCT03123484). Conclusively, 56 has pronounced anticancer properties but it is unknown to which extent ferroptosis contributes to cell death induction in vitro and in vivo. 226
6.3.7. 5‐Aminolevulinic acid (57)
The naturally occurring, nonproteinogenic amino acid 57 (Figure 5) has anticancer properties and is in clinical use for photodynamic therapy of glioblastoma, skin, and bladder cancer. 227 , 228 , 229 Cancer cells preferentially metabolize 57 into the photoactive protoporphyrin IX, which produces ROS upon light irradiation, whereas nontransformed cells lack the machinery for the effective conversion of 57 into the toxic metabolite. 230 Note that 57 decreased cancer cell survival at high concentrations (EC50 = 200–500 µM) also without being photoactivated. Direct cytotoxic effects of 57 on human KYSE‐30 esophageal squamous carcinoma cells (EC50 = 400 µM) were attenuated by ferrostatin‐1 but not by the apoptosis inhibitor Z‐VAD‐FMK. Despite the high effective concentrations in vitro, oral gavage of 57 (30 mg/kg, daily) efficiently reduced the growth of KYSE‐30‐grafted tumors in mice, which is likely related to the favorable bioavailability and high turnover of amino acids by cancer cells. Mechanistically, 57 decreased GPX4 and increased HO‐1 protein expression both in vitro (at 50–100 µM) and in vivo (at 30 mg/kg, po, daily).
6.3.8. Tetrachloro‐1,4‐benzoquinone (58)
The chlorinated benzoquinone 58 (Figure 5) increased MDA levels, elevated the labile iron pool, and induced cell death in rat PC12 adrenal gland cells (40% cell viability at 20 µM), which was diminished by ferrostatin‐1. 231 Compound 58 has previously been reported to activate the NRF2 pathway 232 and to increase SLC7A11 and FTH1 and decrease GPX4 expression. 231
6.3.9. Oridonin (59)
The ent‐kaurene‐type diterpenoid 59 (Figure 5), a major active component of extracts from Isodon rubescens, 233 shares the saturated phenanthrene scaffold and the α,β‐unsaturated ketone bridge with 61. Compound 59 (27 µM) decreased the viability of human TE1 esophageal cancer cells (by 50%), which was diminished by cotreatment with DFO, 233 and inhibited the growth of esophageal squamous cell carcinoma in vivo in patient‐derived xenografts (40 mg/kg, po). 234 Labile iron, MDA, and ROS levels were significantly increased by 59 (15–30 µM), while GPX4 activity and GSH/glutathione disulfide (GSSG) ratios were decreased. 233 Cytotoxic effects of 59 seem to depend on the inhibition of the γ‐glutamyl cycle, which is important for GSH regeneration. 235 Thus, 59 formed covalent adducts with free cysteine in a cell‐free assay and inhibited the activity of γ‐glutamyl transpeptidase 1 (GGT1), which converts extracellular GSH to cysteinylglycine—a crucial step for the reuse of GSH‐derived amino acids for intracellular GSH biosynthesis. 235 Whether cysteine adducts serve as inhibitory product mimetics of GGT1 or whether 59 directly binds to GGT1 or other proteins with exposed nucleophilic (seleno)cysteines has not been addressed. In support of the latter hypothesis, compound 59 was identified as a weak inhibitor of thioredoxin reductase (IC50 = 50–100 µM). 236 Note that multiple other mechanisms independent from ferroptosis have been described for 59, which might add to its antitumoral properties. For example, compound 59 interferes with the cell cycle, apoptotic pathways, and autophagy at micromolar concentrations (15–54 µM) in various cancer cell lines. 234 , 237
6.3.10. Oridonin derivative a2 (60)
The valinyl ester 60 (Figure 5), which differs from 59 by the degree and position of ring oxygenation, reduced cell proliferation in gastric cancer cell lines (GC50 = 1–15 µM) in an iron‐dependent manner. 238 Similar to the parental compound 59 the derivative 60 (at 2.5–10 µM) increased labile iron levels and lipid ROS production and decreased GPX4 expression. Other NRF2 target genes like SLC7A11, HO‐1, GCLC, and FTH1 were instead upregulated. Compound 60 (5–20 mg/kg, iv) reduced the tumor size in patient‐derived tumor xenograft mouse models on GC (by 30%–80%), being more efficient than 5‐fluoro uracil.
6.3.11. ent‐Kauranes (61)
The ent‐kaurane family (Figure 5) comprises complex tetracyclic diterpenoids that are found in liverwoods (Jungermannia species) as well as higher plants (Isodon species). 239 Compound 61 and analogues induced cell death in various cell lines (EC50 = 0.9–5.1 µM), including human A549 lung carcinoma cells, HepG2 hepatocarcinoma, HBE bronchial epithelial, 768‐O renal adenocarcinoma, and A2780 ovarian cancer cells. 239 Ferroptosis and apoptosis contribute to cell death to a similar extent, as suggested by inhibitor studies using ferrostatin‐1, DFO, and Z‐VAD‐FMK. The α,β‐unsaturated ketone is essential for the cytotoxic activity of 61 and directly reacts with GSH. Compound 61 accordingly lowered cellular GSH (EC50 = 2.5 µM) and raised lipid hydroperoxide levels. In addition, 61 interacts with PRDX1/2, as shown for a biotinylated derivative by immunoprecipitation. Compensation studies in PRDX1/2‐overexpressing A549 cells suggested PRDX1/2 as a functional target of 61. When combined with cisplatin (32.5 µM), compound 61 (1.25 µM) synergistically induced membrane peroxidation and cell death in vitro. Accordingly, coadministration of 61 (10 mg/kg, ip) and cisplatin (4 mg/kg, ip) every 3 days showed superior antitumoral efficacy in a cisplatin‐resistant A549 cell xenograft model in mice.
In summary, the ent‐kauranes 59–61 share an exocyclic α,β‐unsaturated ketone, which predestines them to interact with (seleno)cysteine redox switches in proteins. The physiological relevance of such modifications is supported by recent studies showing that compound 61 forms covalent adducts with PRDX1/2 and GSH, respectively. Less understood is whether electrophilic ent‐kauanes share a common ferroptotic mechanism. Recent reports suggested that (i) compound 61 targets PRDX1/2, (ii) compound 59 inhibits the activity of GGT1, and (iii) its derivative 60 downregulates GPX4 expression. In addition, the major ferroptosis regulator NRF2 is an established target of kaurane diterpenoids (5–25 µM) 240 , 241 but rather considered to mediate their organ‐protective functions against injury. 242 Given the bivalent function of NRF2 in ferroptosis, we propose that excessive activation of the NRF2/HO‐1 axis along with GPX4 repression by ent‐kauranes instead initiates cell death. Experimental proof for this hypothesis is lacking so far. In view of the close structural similarity of 59–61 together with the fact that the studies on 61, 239 59, 233 and 60 238 did not exclude other putative targets, we further speculate that ent‐kauranes unfold their ferroptosis‐inducing activities through a common mechanism, namely by covalently targeting redox‐regulated proteins, including PRDX1/2, GGT1, NRF2, and TXNRD.
6.3.12. Tanshinone IIA (62)
The partially saturated phenanthrofuran 62 (Figure 5) from the Chinese herb Salvia miltiorrhiza (Danshen) induced cell death in human GC cells, that is, BGC‐823 (EC50 = 2.8 µM) and NCI‐H87 cells (EC50 = 3.1 µM) 243 —effects that were diminished by ferrostatin‐1. Compound 62 increased lipid hydroperoxide levels, lowered SLC7A11 protein expression, and reduced cellular GSH concentrations, which was ascribed to an upregulation of p53. Silencing of p53 strongly diminished these proferroptotic adaptions and prevented compound 62‐induced ferroptotic cell death. These studies point to a major role of p53 in mediating the cytotoxic effect of 62. In a BGC‐823 xenograft mouse model, compound 62 (50 mg/kg, ip every other day) significantly reduced the tumor weight (2.5‐fold), upregulated p53 expression and lipid peroxidation, and decreased GSH levels. Effects of 62 on tumor growth and ROS production were prevented by ferrostatin‐1 in vivo, which strongly suggests that the mechanism of 62 relies on the induction of ferroptotic cell death. On the other hand, compound 62 also stimulated apoptosis in human MGC803 and SGC7901 GC cells at comparable concentrations (EC50 = 1.5 µM) 244 than needed to trigger ferroptosis. 243 These data suggest that compound 62 induces a mixed cell death program of apoptosis and ferroptosis, with a preference for one or the other mechanism dependent on the cell line. Causative for the decrease in cancer cell stemness is the induction of ferroptosis, at least in SGC‐7901 and BGC‐823 GC cells at high concentrations of 62 (EC50 = 250–550 µM). 245 Cancer stem cells promote tumor reconstitution and propagation and are linked to chemoresistance, cancer recurrence, and metastasis. 246 Three clinical trials currently investigate the efficacy of 62 in the treatment of polycystic ovary syndrome (NCT01452477), pulmonary hypertension (NCT01637675), and acute myocardial infarct (NCT02524964) (www.clinicaltrail.gov). A fourth study investigates an extract enriched in 62, tetraarsenic tetrasulfide, and indirubin in promyelocytic leukemia (NCT02200978). Besides being an inductor of ferroptosis or apoptosis, compound 62 (at 50 nM) also potently inhibited 18‐ and 23‐induced ferroptotic cell death, namely in normal human coronary artery endothelial cells by activating NRF2 signaling. 247 This finding underscores the beneficial bivalent properties of NRF2 activators in normal versus cancer cells.
6.3.13. 15,16‐Dihydrotanshinone I (63)
Another bioactive compound from the tanshinone series, 63 (Figure 5), was isolated from tanshen (Salvia miltiorrhiza) 248 and differs from 62 by the degree of unsaturation and a number of methyl substituents. Compound 63 inhibited cell proliferation in human U251 and U87 glioma (GC50 = 10 µM), 248 breast cancer (GC50 = 1 µM), 249 colon cancer (GC50 = 12.5 µM), 250 and GC cells (GC50 = 12.5 µM), 251 but seems to be less potent than 62, at least in GC cells, where a direct comparison is possible. Studies on glioma cells showed that protein expression of GPX4, as well as cellular GSH/GSSG ratios, are significantly decreased by 63, while ACSL4 expression and MDA levels were elevated, though both effects only manifested at high concentrations of 63 (100 µM). 248 The 63‐induced drop of GPX4 and GSH and increase in MDA levels was reduced by ferrostatin‐1, which suggests that 63 combines low micromolar cytostatic activity with a moderate potential to induce ferroptosis in glioma cells. On the other hand, 63 rather induced apoptosis in breast and colon cancer cells 249 , 250 and suppressed cell proliferation in GC cells via a c‐Jun N‐terminal kinase/p38 MAPK‐dependent pathway. 251 Because of the high concentrations of 63 (100 µM) that were applied to study ferroptosis as compared to the low EC50 values (1–12 µM) required to stimulate apoptosis, we consider the latter as the predominate form of cell death induced by 63.
6.3.14. Dihydroisotanshinone I (64)
While 62 and 63 have a phenanthro[1,2‐b]furan core, compound 64 (Figure 5) isolated from Salvia miltiorrhiza is a phenanthro[3,2‐b]furan. 252 Compound 64 suppressed the proliferation of human breast cancer cell lines (MCF‐7 by 80%; MDA‐MB‐231 by 70%) at low micromolar concentrations (5 µM) within 24 h. Higher concentrations of 64 or prolonged incubation times were required to inhibit cell viability measured as mitochondrial dehydrogenase activity. 253 Compound 64 (10 µM) significantly reduced the protein expression and activity of GPX4, decreased the cellular GSH/GSSG ratio, and substantially increased MDA levels, 252 which suggests that ferroptosis contributes to cell death induction. However, compound 64 (5 µM) also rapidly activated caspase‐3, a major executive caspase in apoptotic cell death. 253 Administration of 64 to mice (30 mg/kg, ip, every 2 days) effectively suppressed the growth of MCF‐7‐ and HCT‐116‐grafted tumors. 254
Together, tanshinone derivatives have in common that they induce ferroptosis as well as apoptosis but there are also substantial differences. While 62 and 64 trigger both cell death programs to a similar extent at single‐digit micromolar concentrations, compound 63 shows signs of ferroptosis only at high concentrations (100 µM) and rather induces apoptosis. Of note, tanshinones have antioxidative and anti‐inflammatory properties and are cytoprotective by activating the NRF2 axis. 247 , 255
6.3.15. Polyphyllin III/dioscin (65) and formosanin C (66)
The diosgenin saponins 65 and 66 (Figure 5) from Paris formosana and Dioscorea species have immunoregulatory, anti‐inflammatory, antibacterial, and anticancer properties. 256 , 257 , 258 For compound 65, it has been shown that cell death induction relies on multiple mechanisms, involving apoptosis, ferroptosis, autophagy, and pyroptosis. 258 , 259 The extent by which the different pathways contribute to cell death varies between cell lines. In human hepatocarcinoma cells (HepG2 and Hep3B, 66, EC50 = 2.5–10 µM), 260 melanoma cells (65, EC50 = 5 µM), 261 and MDA‐MB‐231 breast cancer cells (65, 72 h: EC50 = 2.5 µM; 24 h: EC50 = 8 µM), ferroptosis was identified as major cell death pathway activated by 65 and 66. 257 The decrease in cell viability by 65 was accompanied by an accumulation of lipid hydroperoxides. Ferrostatin‐1 diminished the cytotoxic effect of 65, whereas cotreatment with apoptosis (Z‐VAD‐FMK), necroptosis (necrosulfonamide), and autophagy inhibitors (3‐MA) was not effective. 257 Why only the iron chelator ciclopirox but not DFO impaired breast cancer cell death by compound 65 is not fully understood. 257 Cells treated with 65 develop typical morphological and molecular hallmarks of ferroptosis: the mitochondrial membrane density and lipid peroxidation increase, the number of mitochondrial cristae decrease, and GSH levels drop. 257 Synergistic cytotoxic effects on melanoma cells were observed for the combination of 65 with various chemotherapeutic agents (rapamycin, cisplatin, dacarbazine, and vemurafenib). On the other hand, compound 65 only slightly affected the viability of normal human immortal keratinocytes (HaCaT cells). 261
In melanoma cells, lipid peroxidation and iron accumulation by 65 seem to depend on the downregulation of ferroportin (an NRF2 target gene) and upregulation of transferrin; two major proteins in iron metabolism, which regulate the iron transport from and into cells. 261 Ferroportin represents the sole cellular efflux transporter for nonheme iron, 37 and transferrin is an iron transport protein that is critically involved in cellular iron supply. 262 Interestingly, compound 65 hardly affected the cellular availability of two other iron regulatory NRF2 target proteins, the transferrin receptor, and ferritin, 261 whereas another study observed more pronounced cytotoxic effects of 66 on HepG2 versus Hep3B cells and ascribed this difference to a lower expression of FTH1. 260 Ferritin consists of two subunits, ferritin light chain (FTL) and FTH1, and is an iron storage protein that nucleates oxidized iron, thereby limiting iron for the Fenton reaction. While FTH1 has an iron‐binding site as well as ferroxidase activity, FTL lacks enzymatic properties. 260 , 263 Since FTH1 expression is associated with autophagy, the authors speculated that the cytotoxic effects of 66 are mediated by the induction of autophagy and ferritinophagy. Given that 66 differs from 65 only by an additional rhamnopyranoside in the glycon, the different responses on cellular ferritin levels are surprising, and it cannot be excluded that the apparent failure of 65 to decrease ferritin expression might be related to experimental issues, for example, the antibody specificity for either FTL or FTH. In fact, Lin et al. 260 applied an antibody directed against total ferritin to investigate the effect of 65 on ferritin expression, whereas Xie et al. 261 used an anti‐FTH1 antibody for studies on compound 66.
The cytotoxic activity of 65 in human MDA‐MB‐231 breast cancer cells partially depends on the upregulation of ACSL4, as suggested by knockdown experiments. 257 Moreover, 65 induced the expression of the system Xc − component SLC7A11 via the transcription factor Krüppel‐like factor (KFL) 4, which counteracts the induction of ferroptotic cell death. Silencing of either KLF4 or system Xc − or inhibition of SLC7A11 by 14 sensitized MDA‐MB‐231 cells to 65‐induced ferroptosis. Administration of 65 (10 mg/kg, ip) to mice with grafted MDA‐MB‐231 cells accordingly reduced the tumor burden, and cotreatment with 14 (200 mg/kg) prevented the upregulation of KFL4 and SLC7A11 and exhibited superior antitumoral activity. Of note, KLF4 has recently been shown to activate the NRF2 axis, 264 which raises the question of whether KLF4‐dependent NRF2 activation represents a common mechanism that favors induction rather than protection from ferroptosis by installing a specific NRF2‐target gene profile (low GPX4, high SLC7A11, and HO‐1). In support of this hypothesis, compound 65 decreased GPX4 protein levels. We speculate that 65 acts similar to the HO‐1‐inducers described above, even though the consequences of HO‐1 remain elusive.
As observed for other ferroptosis‐inducing NRF2 activators, compound 65 inhibited stress‐induced cell death in normal cells via the Nrf2/HO‐1‐axis, for example, in primary rat type II alveolar epithelial cells (115–230 nM), 265 rat NRK‐52E kidney epithelial cells (115–230 nM), 266 rat H9C2 cardiomyoblasts (60–230 nM), 267 and mouse AML‐12 liver cells (60–230 nM). 268 Animal models on organ injuries confirmed that 65 is, among others, protective against lung ischemia/reperfusion injury, 265 doxorubicin‐induced nephrotoxicity, 266 doxorubicin‐induced cardiotoxicity, 267 and doxorubicin‐triggered hepatotoxicity. 268
6.3.16. Ruscogenin (67)
The pentacyclic triterpene 67 with spiroketal scaffold (Figure 5) from the root of Ophiopogon japonicus increased the labile iron pool, elevated ROS levels and induced ferroptosis in human pancreatic cancer cell lines (BxPC‑3, SW1990, PANC‑1, and AsPC‑1; EC50 = 10 µM). 262 Accumulation of free iron in cancer cells was ascribed to an upregulation of transferrin and downregulation of ferroportin. The iron chelator DFO accordingly diminished the cytotoxic effect of 67. In mice with grafted BxPC‐3 cells, compound 67 (5 and 10 mg/kg, iv, twice a week) efficiently reduced the tumor weight (by 25%–50%).
6.3.17. Solasonine aglycon (68)
The aglycon of the major steroidal alkaloid solasonine (Figure 5) from Solanum melongena induced ferroptotic cell death in human HepG2 hepatocarcinoma cells (at 15 ng/ml = 0.036 µM), which was suppressed by cotreatment with ferrostatin‐1 or the iron chelator DFO. 269 Compound 47 efficiently downregulated GPX4 protein expression, rose cellular ROS levels and inhibited at 15 mg/kg (single dose; application route is not given) tumor growth (4.5‐fold) in a murine xenograft model of HepG2 cells.
Note that the HO‐1‐inducing terpenoids 65, 66, and 67 shares the spiroketal scaffold with 100 and 101 (see Section 9.3). The latter two compounds were reported to induce ferroptosis by iron chelation and ferritin degradation in lysosomes. Given the structural similarity, we assume that common ferroptosis‐inducing mechanisms exist for these spiroketals, which might include NRF2/HO‐1 activation and interference with iron metabolism, though the experimental proof is still lacking.
6.3.18. Withaferin A (44)
In addition to its ability to directly bind to GPX4 (at 10 µM), compound 44 induced ferroptosis in human IMR‐32 neuroblastoma cells already at low concentrations (1 µM) by enhancing HO‐1 protein expression via NRF2. 188 Inhibition of HO‐1 by ZnPP protected from compound 44‐induced cytotoxicity. Compound 44 decreased the expression of the NRF2 target gene GPX4, 188 and increased lipid peroxidation, which was diminished by ferrostatin‐1 and DFO. 270 Increasing concentrations of 44 (10 µM) interestingly attenuated the upregulation of HO‐1. 188 Compound 44 (1 µM) covalently binds KEAP1 at Cys151 and four other cysteines in the C‐terminal domain, thereby activating NRF2. 270 The positioning of the epoxide at C5/C6 of ring B seems to be essential for NRF2 activation since the structurally related withanolide 69 (epoxide at C6/C7, (Figure 5) is inactive. Note that 44 affects a multitude of other signaling pathways, which makes it difficult to define functional mechanisms that contribute to ferroptosis induction.
6.3.19. Cucurbitacin B (70)
The tetracyclic triterpenoid 70 (Figure 5) present in Cucurbitaceae and Scrophulariaceae families induces cell death in the human breast, ovarian, liver, lung, and CRC lines, being most active on human CNE1 nasopharyngeal carcinoma cells (EC50 = 16 nM). 271 The ferroptosis inhibitors DFO, ferrostatin‐1, or ciclopirox neutralize the cytotoxic effects. Compound 70 evokes morphological changes that are characteristic of ferroptosis, which include shrunken mitochondria, increased mitochondrial membrane density, and a reduced number of mitochondrial cristae. Labile iron and lipid hydroperoxide levels were substantially elevated upon treatment with 70 (50 nM), whereas GSH levels as well as GPX4 and YAP protein expression (at 0.1 µM) were decreased. 271 The latter is a proto‐oncogenic transcriptional coactivator that upregulates multiple ferroptosis regulators and promotes ferroptosis. 55 Moreover, compound 70 caused a G2/M cell cycle arrest, most likely by disrupting microtubule dynamics, which was proposed to contribute to ferroptosis induction. 271 Further studies are needed to elucidate the role of microtubule (70) as drug targets in the context of ferroptosis. Besides its ferroptotic activity, compound 70 (at 0.5 µM) also has cytoprotective functions by activating the NRF2/HO‐1 axis, as shown for cocultures of primary mouse neurons and astroglia. 272 In a CNE1 mouse xenograft model, compound 70 (1 mg/kg, ip, 3× weekly) reduced tumor growth (sixfold) more efficiently than the first‐line drug gemcitabine (25 mg/kg, 3× weekly). The high antitumoral efficacy in vivo together with potentially low toxicity render 70 a promising ferroptosis‐inducing drug candidate and lead structure for further structural optimization.
6.3.20. Glycyrrhetinic acid (71)
The oleanene‐type pentacyclic triterpene saponine 71 (Figure 5), a major bioactive compound isolated from Glycyrrhiza glabra, possesses broad antitumoral activities in different types of cancer. 273 Remarkably, compound 71 induced ferroptotic cell death in human MDA‐MB‐231 breast cancer cells (EC50 = 25 µM) without substantially stimulating apoptosis or necroptosis. On the one hand, the cytotoxic effects of 71 were ascribed to an upregulation of the NOX subunit p47phox and a subsequent increase in NOX activity. On the other hand, 71 impaired the expression of SLC7A11, and decreased cellular GSH levels and GPX activity (without affecting GPX4 protein expression).
Inhibitory effects on ferroptosis are also evident for 71. In TNF‑α/D‐galactosamine‐challenged normal human L02 liver cells, compound 71 restored NRF2, GPX4, and HO‐1 levels, reduced lipid peroxidation, and diminished the decrease of cell viability, however only at very high concentrations (1–2 mM). 274 To which extent ferroptosis contributes to cell death induction by TNF‑α/D‐galactosamine has not been addressed in this study. Despite the highly effective concentrations in vitro, compound 71 (15–60 mg/kg, po) significantly reduced hepatic lipid peroxidation, lowered the ferrous iron content, and partially restored GSH levels as well as GPX4, NRF2, and HO‐1 expression in a mouse model of LPS/D‐galactosamine‑induced acute liver injury. 274
6.3.21. Oleanolic acid (72)
The pentacyclic triterpenoid 72 (Figure 5) from the olive tree exerts organ‐protective functions by targeting NRF2, 275 but can also trigger ferroptosis. 276 Thus, compound 72 reduced the viability of human HeLa cervix carcinoma cells (EC50 = 10–20 µM) and inhibited tumor growth, when these cells are grafted in mice (40 and 80 mg/kg, ip). 276 Labile iron, ROS as well as MDA levels were increased and GSH levels and GPX4 expression decreased. Inhibitory effects of 72 on cell viability and proliferation were ascribed to an upregulation of ACSL4, an acyl‐CoA synthetase isoenzyme that activates PUFAs for incorporation into cellular lipids. Knockdown of ACSL4 lowered compound 72‐induced cytotoxic effects and ROS formation and, interestingly, partially restored GPX4 protein levels.
6.3.22. Corosolic acid (73)
The pentacyclic triterpenoid 73 (Figure 5) isolated from Lagerstroemia speciose has high structural similarity to 72 and is cytotoxic for various cancer cells. 277 Compound 73 (IC50 = 5‐10 µM) induced ROS as well as lipid hydroperoxide formation and triggered cell death in human Caki renal carcinoma cells. Prominent cytotoxic effects were also observed in other human renal carcinoma, breast cancer and hepatocellular carcinoma cell lines but not in primary human mesangial cells, which suggests that cancer cells are more sensitive to 73 than nontransformed cells. Despite the accumulation of lipid hydroperoxides, cell death seems not to be driven by ferroptosis because DFO and ferrostatin‐1 failed to block the cytotoxic effects of 73, which is not readily understood but might be related to the intracellular site where membrane peroxidation takes place. 278 In support of this hypothesis, α‐tocopherol, with potentially different subcellular distribution, prevented 73‐induced ROS formation, lipid peroxidation as well as cell death. Alternative cell death programs, such as apoptosis and necroptosis, seem not to contribute to the cytotoxic activity of 73.
6.3.23. Aridanin (74), ardisiacrispin B (75), albiziabioside A (76, AlbA), and its derivatives 77 and 78
Three further oleanane‐type natural products (74 from Tetrapleura tetraptera, 75 from Ardisia kivuensis, and 76 from Albizia inundata) (Figure 5) have recently been reported to raise intracellular ROS levels and induce ferroptosis. 279 , 280 , 281 Compounds 74 and 75 are cytotoxic against a broad range of human breast cancer, colorectal carcinoma, glioma, lymphoblastoma, and mouse melanoma cell lines as well as several drug‐resistant phenotypes, either with P‐glycoprotein and BCRP (ABC transporters) overexpressed, p53 deleted, or EGFR or B‐RAF mutation‐activated. EC50 values for cell death induction were in the low micromolar range. The cytotoxic effects of the two natural products were partially decreased by the iron chelator DFO and the lipid peroxidation inhibitor ferrostatin‐1, which suggests that ferroptosis contributes to cell death induction. Compounds 75 and 74 additionally induced apoptosis and, as shown for 74, necroptosis. 279 , 280
The natural oleanane triterpenoid saponin 76 exhibits selective lethality on diverse cancer cell lines as compared to nontransformed cells 282 but the potency is moderate (16–22 µM). To obtain synergistic cytotoxic effects on cancer cells, 76 was coupled to the phosphoinositide‐dependent kinase inhibitor dichloroacetic acid (DCA). 281 Alb‐DCA (77) (EC50 = 0.43–3.9 µM) was more active than the parental 76 and selective for cancer cells, revealing only weak cytotoxic effects on human brain microvascular endothelial cells (76: EC50 = 37.1 µM, 77: EC50 = 38.2 µM) and normal human hepatic cells (LO2, 76: EC50 = 33.2 µM, 77: EC50 = 53.1 µM). 281 Another approach to optimize the structure of 76 focused on esters of the carboxylic acid at C17 and yielded compound AlbA‐D13 (78). 282 This derivative showed superior cytotoxicity to 76 in human colorectal, hepatocarcinoma, breast cancer, lung cancer, and gastric adenocarinoma cell lines (EC50 = 5–11 µM) and was sixfold more selective for cancer than nontransformed cells, that is, brain microvascular endothelial, colon epithelial and liver cells as well as keratinocytes. Compound 77‐ and 78‐induced cytotoxicity was reduced by DFO or ferrostatin‐1, which points toward a ferroptotic component of cell death. Moreover, compounds 78 (5 µM), 76 (10 µM), and more effectively the conjugate 77 (2 µM) elevated MDA levels and decreased GPX4 expression in human HCT116 CRCs 282 or in human MCF‐7 breast cancer cells. 281 Compound 77 (2 mg/kg, iv, every second day), 76 (10 mg/kg, iv, every second day), and 78 (20 mg/kg, iv, daily) significantly inhibited tumor growth in an MCF‐7 (76 and 77) or HCT116 (78), murine xenograft model, with 77 being more efficient than 5‐fluoro uracil. Notably, compound 78 significantly upregulated p53, which is required to exert cytotoxic activity. In 2016, Wei et al. 283 described further derivatives of 76, which selectively killed HCT116 cells (EC50 = 7.6 µM) and were neither cytotoxic to cancer cells from other origin nor normal cells investigated (up to 50 µM), though ferroptosis has not been addressed.
The molecular targets that underlie ferroptosis induction by the pentacyclic triterpenoids 74, and 75 as well as 76–78 are poorly understood. Given the well‐documented NRF2‐inducing activity of 71–73 and the characteristic decrease of GPX4 expression by 76–78 together with the p53‐stimulating activity of 78, we speculate that all members of this small molecule class share with the combined activation of the NRF2/HO‐1 and p53 a common (though not necessarily exclusive) ferroptotic mechanism. Further studies are needed to clarify whether the postulated NRF2 activation substantially contributes to ferroptosis induction by this compound class.
6.3.24. Comparative discussion
In summary, structurally diverse small molecules trigger ferroptosis by inducing HO‐1 expression, most likely as result of NRF2 activation. At a first glance, it seems paradoxical to induce ferroptosis by activating NRF2, which upregulates adaptive pathways against oxidative and electrophilic stress, such as GSH biosynthesis. Many of the compounds in this section accordingly have cytoprotective functions in nonmalignant cells in addition to their ferroptosis‐inducing abilities, and two of them (62, 71) have actually been reported to prevent ferroptosis in normal cells. 247 , 274 , 284 The cytotoxic activity of HO‐1‐inducing small molecules on cancer cells seems to rely on the concomitant drop in GPX4 levels, which is shared by the compounds here presented (Figure 6).
Figure 6.
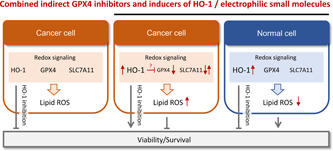
Molecular characteristics related to cancer cell‐selective lethality shared by small molecules indirectly inhibiting GPX4 and inducing HO‐1. Genetic or pharmacological inhibition of HO‐1 decreases the viability of cancer cells under basal conditions. When cancer cells are, however, exposed to electrophilic small molecules that activate NRF2 and evoke a strong expression of HO‐1, the accumulation of lipid hydroperoxides and a drop in cell survival are diminished by selective HO‐1 inhibition. Whether the often associated increase of HO‐1 and decrease of GPX4 are independent events or functionally linked is poorly understood. In nontransformed, normal cells, the NRF2/HO‐1 axis instead rather protects from lipid peroxidation and cell death. This antiferroptotic activity seems at least partially to be mediated by HO‐1. GPX4, glutathione peroxidase 4; HO‐1, heme oxygenase‐1; NRF2, nuclear factor erythroid 2‐related factor 2; ROS, reactive oxygen species. [Color figure can be viewed at wileyonlinelibrary.com]
Different mechanisms seem to contribute to the drop of GPX4 levels. First, electrophilic compounds that activate the NRF2/HO‐1 axis also covalently bind to GPX4, thereby promoting proteasomal degradation, as shown for compound 41. 181 However, other mechanisms seem to contribute as well, since compound 44 decreased GPX4 protein levels at concentrations below those required for targeting GPX4. 188 Second, curcuminoids (47–49) as well as the hybride isothiocyanates 50 and 51 downregulate GPX4 by antagonizing AR. 199 , 202 The important role of AR in regulating GPX4 expression has recently been confirmed by AR overexpression in HEK‐293 cells and AR knockdown in ferroptosis‐resistant triple‐negative LAR breast cancer cells, and this counter‐regulation correlates with ferroptosis sensitivity. 203 Third, GPX4 levels are suppressed likely following p53 activation, as suggested for tanshinones (62–64) and compounds 78 and 92. 243 , 282 , 285 Compound 92 is described in Section 8 because the evidence is missing that the HO‐1/NRF2 axis is targeted.
Small molecules that lower GPX4 and induce HO‐1 expression gain antitumoral activity when combined with inhibitors of GSH biosynthesis or regeneration. In support of HO‐1 promoting ferroptosis, its inhibition by ZnPP protected human HT‐1080 fibrosarcoma cells from the system Xc − inhibitor 18. 193 Silencing of HO‐1 instead enhanced 18‐induced cytotoxicity in human HepG2 cancer cells, which rather points toward antiferroptotic properties of HO‐1. 43 Increasing evidence suggests that HO‐1 expression offers selective cytotoxicity against cancer cells over nontransformed cells. The latter even seem to benefit from abundant HO‐1 expression in terms of survival. On the other hand, moderate HO‐1 expression also sustains cancer cell survival by counteracting apoptosis and stimulating angiogenesis, 286 and HO‐1 inhibition has been suggested as a promising strategy in anticancer therapy. 287 Taken together, activators of the NRF2/HO‐1 axis that additionally reduce GPX4 expression have promising cancer cell selectivity and, when given as combination therapy with conventional drugs, might even protect normal cells. However, their cytotoxic effects seem to depend on the genetic background and metabolic state of the cells as well as the extent and potential duration of HO‐1 upregulation. 288 The bivalent role of HO‐1 on ferroptosis is not fully understood but seems to be tightly associated with the iron state of cancer cells. 289 We speculate that the higher iron status of cancer cells compared to normal cells as well as their dependency on GPX4‐protective mechanisms against lipid peroxidation contribute to the selective toxicity of combined GPX4‐suppressing and HO‐1‐inducing compounds toward malignant cells. Future studies will be required to explore the potential and safety of drug candidates that simultaneously target GPX4 and the NRF2/HO‐1 axis in cancer patients.
A drop in GPX4 levels is expected to be more challenging for cancer cells than for normal cells because of their high metabolic turnover that is associated with oxidative stress. Nonetheless, many covalent GPX4 inhibitors are toxic to both cancer and normal cells, potentially because they are confronted by poor selectivity. Preferentially decreasing GPX4 protein levels might therefore be a more proficient and safer strategy to induce ferroptosis than direct inhibition of GPX4 through covalent binding. 2 The exact mechanisms that deplete cells from GPX4 are only partially understood. Distinct ferroptosis inducers (45 and 46) indirectly inhibit GPX4 by reducing the enzyme's cellular availability, for example, by stimulating nonproteasomal degradation. On the other hand, the curcuminoids 47–49 and the hybrid AR antagonists 50 and 51 likely decrease GPX4 levels by interfering with AR signaling.
Recent evidence suggests that GPX4 inhibition may also affect cells of the immune system and negatively impact antitumor immunity. 290 Two immune‐suppressive strategies have been proposed to limit or even outweigh the anticancer activity of the direct GPX4 inhibitor 23 in vivo. On the one hand, compound 23 kills tumor‐infiltrating CD8+ T cells (besides cancer cells) that are important for the antitumor immune response. On the other hand, ferroptotic cell death fosters the release of immune‐suppressive factors (including prostaglandin E2) by pathophysiologically activated neutrophils, which further dampens the immune response. Future studies are needed to clarify whether these negative effects are (i) related to direct inhibition of GPX4, (ii) might be circumvented by compounds with superior cancer cell selectivity, and (iii) are limited to distinct cancer entities. These new insights into cancer–immune interactions also offer access to new therapeutic strategies. Silver‐coated NRF2‐targeting ZVI‐nano particles (ZVI@Ag), for example, induce ferroptosis in human A549 lung cancer cells while simultaneously increasing antitumor immunity in immune‐competent mice. Mechanistically, ZVI@Ag (i) reduces the number of regulatory T cells, a subpopulation of CD4+ cells with immunosuppressive properties, (ii) enhances the cytolytic activity of CD8+ cells by downregulating the negative immune checkpoints PD‐1 and cytotoxic T‐lymphocyte‐associated protein 4, and (iii) reprograms protumor M2 to antitumor M1 macrophages. 291 The biogenic ferroptosis inducer 102 also appears to enhance anticancer immunity by increasing the populations of CD8+ T cells, natural killer cells, and natural killer T cells. 292 Altogether, these findings suggest that the detrimental immune‐suppressive effects of 23 are related to direct GPX4 inhibition rather than being a general feature of ferroptosis‐inducing small molecules. Critical issues that need to be tackled are the in vivo efficacy of ferroptosis‐inducing compounds in immunocompetent model systems along with the ferroptosis susceptibility of different types of immune cells.
7. ACTIVATORS OF THE NRF2/HO‐1 AXIS
This chapter summarizes small molecule NRF2 activators, which do not fall into the categories described in Section 6.3, either because they do not additionally repress GPX4 expression or because the experimental proof is (still) lacking.
7.1. Bay 11‐7085 (79)
The 2‐sulfonyl‐acrylnitril derivative 79 (Figure 5) inhibits IκBα and has antiproliferative and proapoptotic activity in various cancer cell lines. 204 The cytotoxic effects of 79 on human MDA‐MB‐231 breast cancer cells are independent of NF‐κB signaling and related to ferroptosis induction (EC50 = 5 µM). As suggested from HO‐1 inhibition by ZnPP, compound 79 activates NRF2 and triggers ferroptosis through the markedly upregulated NRF2 target gene HO‐1. Compound 79 further increased SLC7A11 protein expression, as expected from NRF2 activation, but did not elevate the expression of GPX4 (within 8 h). Inhibition of system Xc − by 18 enhanced the cytotoxic effect of 79, likely by compensating for the upregulation of SLC7A11.
7.2. Piperlongumine (80)
The alkaloid 80 (Figure 7; 5–10 µM) isolated from Piper longum induced cell death in pancreatic cancer and breast cancer cells but hardly affected normal human MCF‐10A breast epithelial cells. 293 , 294 Compound 80 has two electrophilic α,β‐unsaturated carbonyl functions, directly bind to KEAP1 and activate NRF2. The selective cytotoxicity against cancer cells has been ascribed to an upregulation of HO‐1 protein expression via the NRF2 axis. 295 Compound 80 upregulated HO‐1 both in human MCF‐7 breast cancer cells as well as nontransformed MCF‐10A cells, but with different consequences on cell death. Thus, pharmacological inhibition or knockdown of HO‐1 decreased the cytotoxic effect of 80 in MCF‐7 cells but enhanced cell death induction in MCF‐10A cells. These findings suggest that HO‐1 expression is protective in normal cells but death promoting in cancer cells, which might provide an explanation for the cancer cell‐selective cytotoxicity of 80 and other NRF2‐activating compounds. 295 Lipophilic radical traps (ferrostatin‐1, liproxstatin‐1) or iron chelators (DFO, ciclopirox) but not apoptosis (Z‐VAD‐FMK) or necroptosis inhibitors (necrostatin‐1) impaired the cytotoxic activity of 80 on human PANC‑1 pancreatic cancer cells. 293 While this profile points toward a ferroptotic mechanism, it should be noted that also the water‐soluble antioxidant N‐acetyl cysteine blocked cell death induction by 80. The increase of intracellular ROS levels upon treatment with 80 was ascribed to the direct inhibition of glutathione S‐transferase and the decrease of intracellular GSH levels. Another study reported that 80 binds to (K d = 22 µM) and inhibits thioredoxin reductase 1 (IC50 = 5–10 µM) and induces ROS‐mediated apoptosis in human GC cells (EC50 = 10–15 µM). 296 The NRF2 target gene thioredoxin reductase 1 is a flavoprotein that uses NADPH to regenerate thioredoxin and is implicated in the protection from ferroptosis. 185 , 297 Docking studies suggest a covalent coupling of 80 to Cys498 in the redox‐active center of the enzyme. As observed for other ROS‐inducing natural products, 80 also induces other forms of cell death besides ferroptosis, including apoptosis, autophagy, and necrosis, seemingly dependent on the cell type/line and experimental condition. 298 , 299
Figure 7.
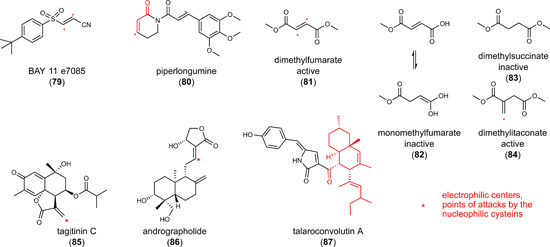
Inducers of the nuclear factor erythroid 2‐related factor 2/heme oxygenase‐1 axis [Color figure can be viewed at wileyonlinelibrary.com]
7.3. Dimethyl fumarate (81, brand name Tecifidera)
The dimethyl ester of fumarate 81 (Figure 5), an intermediate of the citric acid cycle, was initially approved for the treatment of psoriasis and in this course randomly discovered to ameliorate symptoms of multiple sclerosis. 137 Compound 81 is in clinical use in multiple sclerosis and under clinical investigation (single and combination therapy) for the treatment of various types of cancer, for example, refractory chronic lymphocytic leukemia, small lymphocytic lymphoma, cutaneous T cell lymphoma and glioblastoma (www.clinicaltrial.gov, NCT02784834, NCT02337426, NCT02337426, NCT02546440).
The impact of 81 on ferroptosis is ambiguous. 300 On the one hand, compound 81 (20 µM) inhibited ferroptosis in nontumorous damaged or stressed cells, along with increasing GSH levels and decreasing lipid peroxidation. 284 Compound 81 also elevated GSH levels in mouse MLE‐12 lung epithelial cells undergoing seawater‐induced ferroptosis, which was ascribed to an activation of the Nrf2 pathway. 284 On the other hand, the germinal center B‐cell‐like (GCB) subtype of the diffuse large B‐cell lymphoma (DLBCL) cell line has low GSH and GPX4 levels and strongly expresses 5‐LOX, which renders these subtype susceptible to ferroptotic cell death by 81 (20 µM). 301 The activated B‐cell‐like (ABC) subtype is also vulnerable to 81 (20 µM), but cell death does not involve ferroptosis and depends on the inhibition of NF‐κB and Janus kinase (JAK)/signal transducer and activator of transcription 3 signaling.
The Michael acceptor 81 influences cell signaling through its reactivity toward nucleophiles. The less reactive monomethyl fumarate (82) and the nonelectrophilic dimethyl succinate (83) accordingly failed to impair DLBCL cell proliferation, whereas the structurally related Michael acceptor dimethyl itaconate (84) was comparably active (Figure 5). 301 Chemical proteomic mapping identified 40 cysteine residues among 2400 cysteines of the cysteinome that are modified by 81. 302 Succination of IκB kinase at Cys179 and JAK at the highly conserved Cys257 explains the suppression of NF‐κB and JAK survival signaling by 81, and depletion of GSH by covalent coupling with 81 accounts for the induction of ferroptosis in the GCB subtype of lymphoma cells. Interestingly, compound 81 (25 µM) enhanced the mRNA expression of the NRF2 target gene HO‐1 in human primary peripheral blood mononuclear cells, 303 and we hypothesize that this mechanism contributes to the ferroptosis‐inducing activity and cancer cell selectivity of 81. However, other mechanisms contribute to the cytotoxicity of 81 as well, and they are not necessarily limited to ferroptosis. For example, compound 81 induced cell death in human colorectal HCT116 and SW480 cancer cells (at 25–100 µM) through a nonferroptotic pathway dependent on HIF‐2α, 304 which underlines the importance of the cellular origin, (epi)genetic background, and/or metabolic state in defining the antitumoral mechanism and effectiveness of (electrophilic) small molecules. When administered to mice with xenografted HCT116 and SW480 cells, compound 81 (10 mg/kg, ip) sensitized the tumor to a hypoxic mimetic but failed to reduce tumor weight on its own. 304
7.4. Tagitinin C (85)
The heliangolide‐type sesquiterpene lactone 85 (Figure 7) is commonly found in the Asteraceae family of plants and known for its antitumor, antiparasitic, antiviral, antifibrotic, and cardioprotective properties. 305 Compound 85 triggered cell death in human CRCs (EC50 = 15 µM) and showed synergistic cytotoxicity when combined with the Xc − inhibitor 18. 306 Cell death was attenuated by DFO and ferrostatin‐1 within 12 h but neither by inhibitors of apoptosis (Z‐VAD‐FMK), autophagy (3‐MA), nor necroptosis (necrostatin‐1). Compound 85 rapidly induced lipid peroxidation and reduced GSH levels, which was suggested to be mediated by protein kinase R‐like ER kinase (PERK)‐dependent NRF2 activation. Consequently, the NRF2 target genes HO‐1, SLC7A11, NQO1, FTH1, GCLC, and GCLM were upregulated, while GPX4 mRNA levels did not substantially change. Of note, DFO and ferrostatin‐1 failed to prevent compound 85‐induced cell death at 24 or 48 h of incubation, which indicates that other forms of cell death dominate the cytotoxic activity at later time points.
7.5. Andrographis extract standardized to 20% andrographolide (86)
Protective effects of the diterpenoid andrographolide (Figure 7), which is a major bioactive component of andrographis extracts, have been described in various models of organ injuries in vitro (5–25 µM) 307 and in vivo (20 mg/kg sc, 10 mg/kg ip, 30–120 mg/kg po). 308 On the other hand, Andrographis paniculata extracts were cytotoxic on human HCT116 and SW480 CRCs (EC50 = 10 and 20 µg/ml, respectively) and sensitized cell lines with acquired resistance to 5‐fluoro uracil. 309 Cell death triggered by the andrographis extract was ascribed to a caspase 9‐dependent induction of apoptosis and the initiation of ferroptosis via the NRF2/HO‐1 axis. The relative contribution of these cell death programs to the overall loss in cell viability is still elusive. The andrographis extract (125 mg/kg, ip, every second day) effectively reduced tumor weight in a xenograft mouse model with 5‐fluoro uracil‐resistant HCT116 cells (fourfold). Coadministration of the extract and 5‐fluoro uracil (30 mg/kg, ip) was only marginally more efficient. Another study showed that compound 86 (25–100 mg/kg, ig administration) protects from 5‐fluoro uracil‐induced intestinal mucositis in tumor‐bearing mice with grafted H22 hepatocellular carcinoma cells. 310 These findings are in line with the dual role of NRF2/HO‐1‐inducing small molecules in anticancer treatment. 48 , 191 On the one hand, NRF2/HO‐1 activators seem to synergize with conventional chemotherapeutics to reduce cancer cell survival. On the other hand, they beneficially reduce the cytotoxicity of anticancer drugs in (distinct) nontransformed cells.
7.6. Talaroconvolutin A (87)
The azaphilone 87 (Figure 7) was isolated from the endophytic fungus Talaromyces purpureogenus inhabiting Panax notoginseng. 311 Compound 87 efficiently triggered cell death in various CRC cell lines (human HCT116, SW480, and SW620; EC50 = 8–12 µM) without stimulating apoptosis. ROS levels increased and morphological changes characteristic for ferroptosis were induced, which includes membrane perforations and ruptures, shrunken mitochondria and a decline in the number of mitochondrial cristae. 311 Cytotoxic effects of 87 were attenuated by the ferroptosis inhibitor ferrostatin‐1 and the iron chelator deferiprone. Compound 87 enhanced lipid peroxidation and changed the expression of proteins that are tightly associated with ferroptosis (e.g., FTL, SLC7A11, HO‐1, epidermis‐type lipoxygenase 3, GSS, ACSL5). Effects on NRF2 were not investigated but the expression profile strongly suggests that 87 interferes with the NRF2 axis. The authors ascribed the ferroptotic activity of 87 to the downregulation of SLC7A11 (a component of system Xc −) and upregulation of epidermis‐type lipoxygenase 3 (a lipoxygenase isoenzyme i.e. highly expressed in epidermis), as confirmed by knockdown and overexpression studies. Moreover, compound 87 (6 mg/kg, ip, every other day) decreased tumor weight by 30% in a murine xenograft model using human HCT116 cells.
8. NRF2 INHIBITORS
Manifold signaling pathways regulate NRF2 activation, 42 as exemplarily outlined in Figure 8. Of particular importance are the E3 ubiquitin ligase complexes CUL3‐RBX1‐KEAP1, SCF/β‐TrCP, and HRD1, which essentially control the availability of NRF2 by marking the transcription factor for ubiquitin‐dependent degradation. 312 Binding of specific factors to either KEAP1 or NRF2 interrupts the interaction of the two proteins, which prevents NRF2 from being degraded. 42 The CDK inhibitor p21 for example, competes with KEAP1 for the binding to NRF2, whereas the phosphorylated autophagy substrate p62 binds to KEAP1 and disrupts the interaction with NRF2. 42 NRF2 transcription is in addition enhanced by diverse oncogenic and stress‐activated pathways (e.g., via K‐RAS, B‐RAF, phosphatidylinositol 3‐kinase/protein kinase B, extracellular signal‐regulated kinase, p38 MAPK), the GRP78‐PERK branch of the unfolded protein response as well as the energy sensor AMPK. 132 Not only the expression but also the activity of the transcription factor NRF2 is also under tight control. NRF2 is regulated by (i) the import into and export from the nucleus, (ii) phosphorylation, (iii) transactivation by dimerization with different transcription factors or nuclear receptors, and (iv) the affinity to antioxidant response elements (ARE), the DNA binding sites for NRF2. 42
Figure 8.
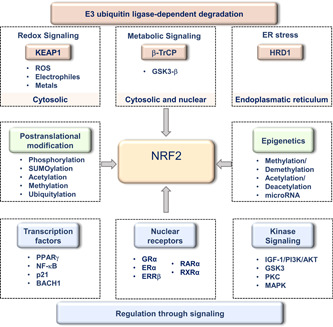
NRF2‐regulatory pathways. The transcription factor NRF2 consists of multiple domains that are under tight regulatory control. In complexes with KEAP1, NRF2 is ubiquitinated and degraded by the proteasome. Electrophilic and oxidative modifications of cysteine residues in KEAP1 lower the affinity to NRF2, and factors like p21 or NF‐κB hamper the binding of KEAP1 to NRF2. Ubiquitin‐dependent NRF2 degradation is further determined through the availability of the substrate recognition component of the SKP1‐cullin 1‐F‐box protein E3 ligase complex β‐TrCP, which responds to metabolic changes and is regulated by GSK3‐β. Another NRF2‐regulatory factor is the E3 ubiquitin‐protein ligase HRD1 which participates in ER‐associated degradation during ER stress. A variety of signaling cascades regulate the expression, nuclear availability, DNA‐binding affinity, and transactivation activity of NRF2, and also posttranslational and epigenetic modifications essentially contribute to the regulation of NRF2 activity. These pathways and factors represent potential targets for NRF2‐inhibiting small molecules. AKT, protein kinase B; BACH1, BTB domain and CNC homolog 1; ERRβ, estrogen‐related receptor β; ERα, estrogen receptor α; GRα, glucocorticoid receptor α; GSK3‐β, glycogen synthase kinase‐3β; HRD1, HMG‐CoA reductase degradation protein 1; IGF‐1, insulin‐like growth factor 1; MAPK, mitogen‐activated protein kinase; NF‐κB, nuclear factor κ‐light‐chain‐enhancer of activated B cells; NRF2, nuclear factor erythroid 2‐related factor 2; PI3K, phosphatidylinositol 3‐kinase; PKC, protein kinase C, PPARγ, peroxisome proliferator‐activated receptor γ; RARα, retinoic acid receptor α; RXRα, retinoid X receptor α. [Color figure can be viewed at wileyonlinelibrary.com]
The heterogeneity in the mechanisms that regulate NRF2 signaling is reflected by the diversity of small molecules that interfere with NRF2 activation. 48 , 132 For a comprehensive overview of natural products that target NRF2, we want to refer to recent review articles. 46 , 48 , 132 , 312 , 313 , 314 , 315 , 316 , 317 , 318 , 319 , 320 Here, we place focus on those NRF2 inhibitors, which explicitly trigger ferroptotic cell death, and on ferroptosis‐stimulating compounds, which we anticipate to inhibit NRF2 based on their effect on NRF2 target gene expression. Ferroptosis‐inducing natural products, for which inhibition of NRF2 is less obvious, are instead listed in Section 9.
8.1. ML385 (88)
In a large‐scale screen with 400,000 compounds, 88 (Figure 9) was recently discovered as an inhibitor of NRF2. 321 Compound 88 enhanced seawater exposure‐induced ferroptosis (at 20 µM), increased MDA and lipid hydroperoxide formation and decreased GSH levels as well as Gpx4 mRNA expression in MLE‐12 mouse lung epithelial cells. 284 Mechanistically, compound 88 directly binds to the Neh1 DNA binding domain of NRF2 and disrupts the binding of the NRF2–V‐Maf avian musculoaponeurotic fibrosarcoma oncogene homologue complex to the ARE sequence within the NRF2 target gene promoters. 321 Note that 88 is the only known direct NRF2 inhibitor. Other compounds indirectly suppress NRF2 signal transduction by targeting upstream pathways or inducing proteasomal degradation (Figure 8).
Figure 9.
Nuclear factor erythroid 2‐related factor 2 inhibitors
The strong impact of KEAP1 mutations on the cellular susceptibility to 88 supports that NRF2 inhibition is causative for ferroptosis induction. Thus, compound 88 more effectively suppressed colony formation in human H460 lung carcinoma cells with inactivating KEAP1D262H mutation and high NRF2 activity (GC50 = 1.5 µM) than in the wild‐type counterpart (H460 cells with knock‐in of wild‐type KEAP1; GC50 = 8–16 µM). Moreover, 88 inhibited the expression of the NRF2‐target gene NQO1 in lung cancer cells (by 55% at 5 µM) 321 and showed pronounced cytotoxic activity (at 10 µM) in a cell line with inactivating KEAP1G333C mutation. Growth‐inhibitory effects on nontumorigenic lung epithelial cells (BEAS2B) were instead not evident up to a concentration of 25 µM.
In therapy‐resistant NSCLC with KEAP1 deficiency, 88 (5 µM) substantially enhanced the cytotoxicity of doxorubicin and taxol. 321 When combined with the antitumoral drugs paclitaxel, doxorubicin, or carboplatin, 88 synergistically attenuated colony formation of human A549 and H460 lung cancer cells and induced caspase activation. This finding suggests that 88, which does not induce apoptosis per se, triggers supportive mechanisms, potentially at the crosslink between ferroptosis and apoptosis, where also NRF2 is located.
Compound 88 (30 mg/kg, ip daily) suppressed tumor growth in xenografts of A549 and H460 cells in mice comparably to carboplatin (5 mg/kg, ip), and the combination of the two cytotoxic agents showed synergistic effects. The latter was ascribed to an accumulation of carboplatin in the tumor, most likely as a result of the impaired NRF2‐dependent upregulation of ABCG2 (BCRP) and potentially other efflux transporters. 321 , 322
8.2. All‐trans retinoic acid (89) and the derivative 4‐amino‐2‐trifluoromethyl‐phenyl retinate (90)
Both compounds 89 and 90 (10 µM, Figure 9) downregulated NRF2 protein levels (89: by 30% at 10 µM, 90: by 50% at 10 µM) and induced cell death (EC50 = 1–10 µM) in human NB4 acute myeloid leukemia (AML) cells. 323 The cytotoxic effect of 90 was associated with an increase in lipid hydroperoxide and MDA levels (0.1–10 µM), a depletion of the GSH pool, and a reduced expression of NRF2 target genes, such as GPX4, ferritin, and SOD1, which was reversed by ferrostatin‐1. 323 Effects of 89 on ferroptosis markers were not investigated. In a xenograft mouse model using NB4 cells, compound 90 (10 mg/kg, ip) efficiently decreased GPX4 and elevated COX‐2 expression in the tumor. In addition, NRF2 forms a complex with the retinoic acid receptor in the presence of 89 (Figure 9), which disables the binding to the ARE sequence and thereby disrupts NRF2 signaling, 42 a mechanism that likely also applies to 90. Together, compounds 89 and 90 inhibit NRF2‐dependent gene expression and, as shown for 90, induce ferroptosis. 323
8.3. Trigonelline (91)
The coffee alkaloid 91 (Figure 9) is a metabolite of niacin and present in many plants including Coffea species (Rubiaceae) and Trigonella foenum‐graecum (Fabaceae). 324 Compound 91 decreased nuclear NRF2 protein levels in the human pancreatic carcinoma cell lines Panc1, Colo357, H6c7, and MiaPaca2 as well as NRF2 activity in a reporter gene assay at submicromolar concentrations (0.1–1 µM), which was ascribed to an impaired import of NRF2 into the nucleus. 324 Neither nuclear NRF2 export nor total cellular NRF2 protein levels were influenced by 91. By inhibiting NRF2, 91 sensitized pancreatic cancer cell lines as well as grafted tumors in mice to apoptosis upon cotreatment with the topoisomerase II inhibitor etoposide. The potential of 91 (150–300 µM) to overcome ferroptosis‐resistant mechanisms in cancer was confirmed in human head and neck cancer cell lines. 325 Compound 91 markedly enhanced their sensitivity to the GPX4 inhibitor 23, further increased lipid peroxidation by interfering with NRF2 signaling, and decreased tumor growth in a mouse xenograft model. 325 Together, combination therapy of conventional anticancer drugs and 91 promises superior efficacy in the treatment of resistant tumors.
8.4. Gambogenic acid (92)
The bioactive natural product 92 (Figure 9) isolated from the dry resin of Garcinia hanburyi (Gamboge) showed marked toxicity in human A375 (EC50 = 1.5 µM) and A2058 (EC50 = 0.9 µM) melanoma cells that undergo transforming growth factor(TGF)‐β‐induced EMT. Nonstimulated cells with lower metastatic capacity (without TGF‐β treatment) were instead less sensitive (A375: EC50 = 4 µM; A2058: EC50 = 1.5 µM). 285 EMT is a dedifferentiation process of epithelial cells, in which they lose their polarity and obtain features of mesenchymal stem cells, such as the capacity to differentiate into multiple tissue lineages. Cells undergoing EMT are characterized by decreased cell–cell contacts, an elevated migratory and invasive potential, and acquired resistance against cell death by apoptosis. 326 On the other hand, cancer cells with mesenchymal features are more susceptible to ferroptosis, 87 , 327 , 328 most likely due to an upregulation of enzymes involved in the activation and membrane incorporation of long‐chain PUFAs. 87 , 329
Cell death induced by compound 92 is inhibited by the ferroptosis inhibitor ferrostatin‐1, the iron‐chelating agent DFO as well as an autophagy inhibitor but not by an inhibitor of apoptosis. 285 The morphology and microstructure of mitochondria accordingly show hallmarks of ferroptosis (i.e., smaller mitochondria and an increase in membrane density) as well as autophagy (i.e., presence of autophagy bodies). Compound 92 increased membrane peroxidation (levels of membrane hydroperoxides and the lipid peroxidation end product MDA) and decreased GPX4 and SLC7A11 protein expression, which was ascribed to an upregulation of p53. 285 Moreover, GSH levels were decreased, as was the activity of SOD. These findings strongly suggest an efficient suppression of NRF2 signaling by 92 and hint toward a putative role of p53 in mediating this effect.
8.5. Typhaneoside (93)
The bioactive component 93 (Figure 9; 40 µM), isolated from pollen of Typha angustata, triggered cell death in AML cells (Kas‐1, HL60, and NB4). 330 The loss of cell viability was accompanied by elevated lipid hydroperoxidation, depleted GSH levels, and reduced GPX4 mRNA expression. Cotreatment with ferrostatin‐1 or DFO diminished the cytotoxic effect. While this profile is in line with NRF2 inhibition, experimental proof is lacking, and the cytotoxic mechanism of 93 remains speculative. Despite a relatively low potency in vitro, Compound 93 showed favorable in vivo activity in an HL60 cell BALB/c xenograft mouse model (10–40 mg/kg, ip, once daily). However, the role of ferroptosis in the overall cytotoxicity of 93 is still diffuse, and other forms of cell death, i.e., autophagy and apoptosis, participate as well.
8.6. Ginkgetin (94)
The biflavonoid 94 (Figure 9) from Ginkgo biloba synergizes with the anticancer drug cisplatin in the treatment of therapy‐resistant NSCLC. Compound 94 induced autophagic, apoptotic and to a limited extent ferroptotic cell death, and enhanced cytotoxicity by cisplatin through a ferroptotic mechanism. 331 Cancer cells seem to be more sensitive to compound 94‐induced cell death than nontransformed cells. Thus, compound 94 suppressed more potently the viability of human A549 epithelial lung cancer (EC50 = 5 µM) than of human nontumorigenic BEAS‐2B epithelial lung cells (EC50 = 150 µM), whereas cisplatin was equally effective on both cell lines (EC50 = 7.5 µM). The cytotoxicity of 94 was reduced by GPX4 overexpression and marginally by the ferroptosis inhibitors UAMC‐3203 (a derivative of ferrostatin‐1) and DFO. Effects were more pronounced for cotreatment with 94 and cisplatin. Lou et al. proposed that the cell death programs are hierarchically linked under these conditions, with ferroptosis promoting apoptosis, as indicated by a loss in mitochondrial membrane potential, serine externalization, and the activation of initiator and effector caspases. 331 Compound 94 (5 µM) elevated the labile iron pool, ROS formation, and lipid peroxidation, while decreasing GPX4 and HO‐1 expression as well as the cellular GSH to GSSG ratio. The role of NRF2 in this process has been challenged because NRF2 activators, either sulforaphane (52) or dimethyl fumarate (81), did not diminish 94‐induced cytotoxicity. On the other hand, cisplatin (5 µM) increased ARE‐mediated NRF2 transcriptional activity (2.6‐fold), and cotreatment with 94 abolished this effect. In an NSCLC xenograft mouse model using human A549 lung cancer cells, a single treatment with either 94 (30 mg/kg, ip, once daily) or cisplatin (3 mg/kg, ip, 2–3 times per week) reduced tumor weight by 50%. Cotreatment of cisplatin and 94 was considerably more effective (80% reduction of tumor weight), which underlines the potential of combining conventional anticancer drugs with proferroptotic agents in the treatment of NSCLC.
8.7. Amentoflavone (95)
The bioactive biflavonoid 95 (Figure 9) from Selaginalla species stimulated cell death in human U251 and U373 glioma cells (at 10–20 µM) and, when given ip (40–80 mg/kg), effectively reduced the size of U251 xenograft tumors in mice (by 50%–75%), while not affecting the viability of primary human astrocytes in vitro. 332 DFO or ferrostatin‐1 is protected from cytotoxic effects by amentoflavone, which points toward a ferroptotic mechanism. The authors suggest that the induction of ferroptosis is related to a decreased expression of FTH, which evokes changes in iron homeostasis that are associated with an mammalian target of rapamycin (mTOR)/AMPK/p70SK‐dependent induction of autophagy. Compound 95 (20 µM) further increased iron, MDA and lipid hydroperoxides and decreased cellular GSH levels. Whether 95 acts via the NRF2 axis was not investigated. However, we consider such a mechanism likely due to structural similarities of 95 and 94 (Figure 9). The two compounds share the same dimeric scaffold and only differ in the methylation of polyphenolic hydroxyl groups.
8.8. Comparative discussion
In summary, an emerging number of studies demonstrate a beneficial role of NRF2 inhibitors in drug‐resistant cancer therapy, both for cultured cell lines in vitro and grafted cancer cells in vivo. Except for 88, all inhibitors derive from natural sources or are natural product‐inspired compounds. Another synthetic NRF2 inhibitor, AEM1, was recently identified in a large‐scale screen, 333 but neither was the target/binding site of AEM1 identified nor the effect on ferroptosis investigated. The exact mechanisms of how natural products interfere with the NRF2 axis often remain obscure, and heterogeneous indirect mechanisms are likely involved. Several natural products like brusatol—a well‐known natural NRF2 inhibitor—and 91 have nonselective anticancer activity and inhibit NRF2 signaling both in normal and tumor‐derived cells. 333 Other natural products like 94 instead exhibit selectivity toward cancer cells as compared to normal cells, thereby being superior to the synthetic drug candidate 88. However, it should be taken into account that comparisons in the selectivity for cancer cells over nontransformed cells can only be crude estimates when based on different experimental systems with varying susceptibility to ferroptosis. Our overall impression is that compounds more specifically interfering with NRF2 signaling may have advantages in inducing selective cancer cell lethality. The mechanisms are not fully understood but likely involve a reduced capacity to adapt to redox stress and detoxify xenobiotics (e.g., conventional anticancer drugs), which is important for tumor resistance. 48 In fact, a broad spectrum of cancer cells from different origin rely on an activated NRF2 axis. 138 Considering that many of the here described small molecules have multiple targets, detailed mechanistic studies are urgently needed to estimate the relevance of NRF2 inhibition for their ferroptosis‐inducing and/or antitumoral activity.
9. FERROPTOSIS‐INDUCING NATURAL PRODUCTS WITH DIVERSE MECHANISMS
Phospholipid and neutral lipid hydroperoxides accumulate during ferroptosis and are degraded to metabolites with truncated fatty acid chains and reactive aldehydes that may disrupt the membrane architecture. 101 Polyunsaturated acyl chains are oxidized by nonenzymatic mechanisms, LOX, or dependent on cytochrome P450 oxidoreductases to lipid hydroperoxides. 36 , 96 , 334 Defense systems against membrane peroxidation involve GPX4 that reduces peroxidized fatty acids to nonreactive alcohols using GSH as cofactor 335 and radical‐trapping antioxidants, such as CoQ10. 82 , 336 Reported small molecules that induce ferroptosis either inhibit GPX4, deplete GSH, oxidize iron, 32 , 336 , 337 or incorporate PUFAs into phospholipids or neutral lipids. 101 In Sections 5 and 6, we discuss system Xc − and GPX4 inhibitors that directly interfere with lipid hydroperoxide detoxification via GPX4 or indirectly by depleting intracellular GSH pools. Section 6.3 highlights potential NRF2 activators that exert their ferroptosis‐inducing activity by inducing HO‐1 expression and lowering GPX4 levels along with engaging multiple other mechanisms, such as an interference with GSH biosynthesis or iron metabolism. Additional small molecules that activate or inhibit NRF2 but do not fit into these categories (either because they do not target system Xc −/GPX4 or experimental proof is lacking) are summarized in Sections 7 and 8. The following section concentrates on natural products and derivatives, which induce ferroptosis, either by oxidizing ferrous iron, interfering with the antioxidative defense system, or through other known or unknown mechanisms. The latter is a heterogeneous group that requires more detailed investigation and may include, among others, GPX4, system Xc − and NRF2 inhibitors as well as iron‐oxidizing compounds or modulators of the membrane fatty acid composition. Sensitizing to ferroptosis was not a sufficient criterion for being included in this chapter if the natural product does not possess marked cytotoxic activity itself at micromolar concentrations. Included are only natural products that have explicitly been shown to trigger ferroptosis (e.g., in compensation experiments with ferroptosis inhibitors), even though many other natural products exist that induce cell death and/or interfere with pathways that are critical for ferroptosis. We also excluded natural products that employ multiple cell death programs, where in sum ferroptosis is only marginally involved.
Besides natural products, an increasing number of established anticancer drugs have been identified to induce ferroptotic cell death. 338 However, the contribution of ferroptosis to their overall cytotoxic activity is often low (e.g., for cisplatin), with only a few exceptions. Ferroptosis is substantially induced by siramesine (sigma receptor agonist) in combination with lapatinib (an inhibitor of the tyrosine kinases EGFR and HER2), 339 neratinib (a potent irreversible pan‐tyrosine kinase inhibitor), 340 and apatinib (a selective inhibitor of the tyrosine kinase vascular endothelial growth factor receptor‐2). 341 Moreover, multiple FDA‐approved drugs with recorded cytotoxic/anticancer properties like the hypoglycemic drug metformin 342 or the antifungal drug itraconazole 343 have been reported to induce ferroptosis in addition to their primary mode of action. Recent review articles give a comprehensive overview of the current knowledge in these fields. 344 , 345
9.1. Compounds that trigger iron oxidation
ROS formation (which is triggered by pleiotropic natural products) is not sufficient to induce ferroptosis, 88 , 346 and an increase of ROS levels or the inhibition of antioxidative enzymes might also be deleterious for noncancerous cells resulting in severe side‐effects during anticancer therapy. ROS inducers acting on ferrous iron and enhancing the Fenton reaction effectively trigger lipid peroxidation, preferentially induce ferroptosis as compared to other cell death programs, and might show selectivity for cancer cells due to higher iron levels in tumors as compared to healthy tissue. 124 , 125 Even general ROS inducers might have some preference for cancer cells, given their increased ROS tonus as compared to normal cells. 6 Nevertheless, ROS‐inducing drug candidates have to be carefully evaluated for their cytotoxicity on normal cells, tissues, and organs.
9.1.1. Plakinic acid (96) and its synthetic derivative FINO2 (97)
The natural product 96 (Figure 10) found in sponges (Plactortis species) served as a lead scaffold for the synthesis of endoperoxides, yielding 97. 337 The dioxolane 97 induced lipid hydroperoxide generation and iron‐dependent cell death in various cancer cell lines, surpassing artemisinin, a naturally occurring endoperoxide described below. 347 Ferroptosis inhibitors (ferrostatin‐1, liproxstatin‐1, DFO, baicalain, trolox) fully prevented cell death by 97 (2–10 µM) in human BJ‐ELR fibroblast cancer cells and human HT108 fibrosarcoma cells. 337 , 347 Neither apoptosis, necroptosis nor autophagy were triggered by the endoperoxide. Compound 97 oxidized ferrous iron, induced lipid peroxidation and lowered GPX4 activity through an unknown mechanism, without reducing GSH levels. Direct inhibition of GPX4 has been excluded, and also GPX4 protein expression was only slightly decreased. 337 , 347 Compound 97 effectively induced cell death in oncogenic, transformed human BJ‐ELR fibroblasts (EC50 = 2 µM), 337 and cell lines with inactive or lost p53 gene, such as human NCI‐H522 and HOP‐92 NSCLC cells and HL‐60 leukemia cells (EC50 = 1.2–2.2 µM). 347 Cancer cells seem to be more susceptible to 97 than normal cells, as suggested from the high effective concentrations required to induce cell death in immortalized (noncancerous) human BJ‐hTERT fibroblasts (noncytotoxic at 20 µM). 347 Compound 97 requires both the endoperoxide and the proximate polar head group (alcohol) to induce cytotoxicity, and neither replacing the alcohol with nonpolar substituents nor increasing the distance between the alcohol and endoperoxide is tolerated (Figure 9). 337 Modifications of the tert‐butyl moiety were instead accepted. These structural requirements suggest that 97 has to acquire a defined orientation at membrane interphases that place the endoperoxide in close proximity to the bis‐allylic double bonds of esterified PUFAs. It is tempting to speculate that 97 is anchored in cellular membranes and that the polar head group is orientated toward the aqueous phase while the lipophilic core extends toward the inner membrane, where the endoperoxide initiates the generation of hydroxyl radicals via the Fenton reaction.
Figure 10.
Inducers of (lipid) reactive oxygen species formation [Color figure can be viewed at wileyonlinelibrary.com]
9.1.2. Artemisinin (98), its derivatives artesunate (99), arteether (100), artemether (101), and the metabolite, dihydroartemisinin (102)
The sesquiterpene lactone 98 (Figure 9) from Artemisia annua has been used in traditional Chinese medicine for two millennia and is FDA‐approved as antimalarial drug, as is artesunate, a derivative with improved water solubility. 10 , 348 The pharmacological activity of 98 and its derivatives have been intensively investigated during the last 2 decades, and several phase I/II trials have been conducted (www.clinicaltrial.gov, NCT02633098, NCT03093129, NCT00764036, NCT02353026, NCT02354534, NCT03792516, NCT03100045, NCT04098744, NCT02786589, NCT05478239, NCT02304289). 349 These studies indicate prominent antitumoral efficacy combined with generally low toxicity and adverse effects and highlight the endoperoxide (1,2,4‐trioxane) as an essential pharmacophore. 348 Interested readers are encouraged to consult a recent survey article that describes the potential of 98 in anticancer therapy. 348 , 350 , 351
Here, we focus on mechanisms likely contributing to the proferroptotic activity of 98. Compound 98 and multiple derivatives exhibit potent cytotoxic activity in cancer cell lines and tumor models, which was partially ascribed to the induction of ferroptotic cell death. 348 , 352 , 353 , 354 , 355 , 356 The antimalarial potential of 98 is linked to its ability to react with free ferrous ions. 348 Most likely, the endoperoxide is cleaved by ferrous iron, leading to the formation of reactive free radicals via the Fenton reaction. The combination of 98 with ferrous iron accordingly increased the cytotoxic activity of cancer cells, as expected from an iron‐dependent mechanism. 352 Further proof that labile iron plays an important role in the mechanism of 98 comes from the cotreatment with the iron chelator DFO that diminished the anticancer activity of 102. 357 Of note, compound 102 and its derivatives induced a plethora of other mechanisms involved in ferroptosis but also other forms of cell death, including DNA damage, modulation of gene expression, and interference with mTOR, NF‐κB, MAPK, and Wnt/β‐catenin signaling. 349 Since many of this survival, oncogenic and stress‐adaptive pathways contribute to tumor resistance, polypharmacological inhibitors like 97 might be advantageous for the treatment of therapy‐resistant cancer. 348 Along these lines, artesunate efficiently killed apoptosis‐resistant pancreatic ductal adenocarcinoma cells by triggering ferroptosis. 352
The metabolite of 98, compound 102, was recently reported to upregulate GPX4 expression as part of an antiferroptotic feedback loop. 358 Compound 102 activated PERK that led to an upregulation of ATF4 and subsequent expression of the heat‐shock protein HSPA5. 358 Inhibition of PERK, ATF4, and HSPA5 (either using a pharmacological inhibitor or siRNA) substantially increased the sensitivity of cultured and grafted glioma cells to 102, which indicates that targeting the PERK branch of the unfolded protein response might be an effective means to increase the vulnerability of tumors against ferroptosis induction by 98 and derivatives.
9.2. Modulators of cysteinyl–cystinyl homeostasis
A large variety of natural products suppresses redox‐dependent processes. Compounds that inactivate thioredoxin or inhibit thioredoxin reductases are described in detail in recent survey articles. 297 , 359 , 360 Other natural products target GSH, PRDX1/2, GGT1, and further players in redox homeostasis. 361 Thioredoxin reductases are, like GPX4, selenoproteins with a nucleophilic, active site selenocysteine, which forms covalent adducts with electrophilic compounds. 359 Among the natural products that potently inhibit thioredoxin reductases are phenylpropanoids and polyphenols (e.g., 47 and curcuminoids), 362 quinoids (e.g., plumbagin and 80), 296 terpenoids (e.g., 40, 59 and mitomycin), 184 , 236 and diverse other structural classes (e.g., gambogic acid). 297 Note that distinct thioredoxin reductase inhibitors (47, the curcumin derivative 48, 80) also activate NRF2 as described in Section 6.3. Thioredoxin reductase inhibition most likely involves covalent binding of the inhibitor to selenocysteine and/or cysteines of the active site, as originally proposed for 80 (see Section 7). 296 Such electrophilic groups are also present in NRF2 activators that covalently bind to the NRF2 inhibitor KEAP1. 48 It is, therefore, not surprising that compounds with electrophilic groups target both proteins, KEAP1 and thioredoxin reductase. Other factors involved in redox homeostasis, such as PRDXs and GGT1, also contain redox‐active cysteines, either in the active center or as redox sensors, which offer prominent sites for electrophilic attack, as exploited by 61 and 59 (see Section 6.3). The large majority of small molecules that target cysteinyl‐cystinyl homeostasis (47, 48, 59, 61, 80) also induce HO‐1 expression and are therefore listed in Sections 6.3 and 7. In this section, we discuss the thioredoxin‐targeting ferroptocide (99), which seems not to share this dual mechanism.
9.2.1. Ferroptocide (104)
Applying the complexity‐to‐diversity (CtD) strategy to the diterpene natural product pleuromutilin (103) yielded compound 104 (Figure 11), a ferroptosis inducer with varying cytotoxic potency between cell lines (human ovarian ES‐2 cells: EC50 = 1.6 µM; MDA‐MB‐231: EC50 = 25 µM). 3 The CtD strategy aims at producing complex and diverse compound libraries from natural products by ring distortion reactions that create novel scaffolds. 3 SAR studies indicate that the electrophilic chloroacetoxy group is essential for the cytotoxic activity of 104, whereas the secondary alcohol as well as N4 and N9 of the partially saturated benzo[a]triazole ring system are promising sites for structural optimization. Compound 104 selectively inactivated thioredoxin and increased membrane peroxidation in human ES‐2 ovarian clear cell carcinoma cells similar to thioredoxin silencing. 3 Thioredoxins are important redox‐cofactors of the antioxidative defense system, which (i) lower the redox tonus, (ii) decrease oxidative protein modifications, and (iii) reduce cystine to cysteine for GSH biosynthesis. 147 The five cysteines in thioredoxins represent possible sites for covalent binding of the reactive chloroacetoxy moiety of 104, two of them form the redox‐active cysteine pair in the active center (Cys32 and Cys35). 3 Site‐directed mutagenesis identified Cys32 and Cys35 as well as the adjacent cysteine Cys73 as sites of modification. Although the chloroacetoxy moiety of 104 resembles the chloroacetamide in GPX4 inhibitors (Section “GPX4 inhibitors”), compound 104 does not inhibit GPX4. 3
Figure 11.
Ferroptosis‐inducing ferroptocide covalently targets thioredoxin [Color figure can be viewed at wileyonlinelibrary.com]
Thioredoxin 1 and thioredoxin reductase 1 are upregulated in various forms of cancer 363 and associated with excessive proliferation, metastasis, and protection from apoptosis. 360 Moreover, there are several lines of evidence that the thioredoxin system is involved in drug resistance. 364 Targeting the thioredoxin system thus seems to be another promising strategy to trigger ferroptosis and combat (therapy‐resistant) cancer.
9.3. Natural products that trigger ferroptosis through other mechanisms
9.3.1. Salinomycin (105) and the derivative ironomycin (106)
The tricyclic spiroketal 105 (Figure 12) was originally isolated from Streptomyces albus and is a monovalent ionophor and antibiotic. 365 Compound 105 (EC50 = 1 µM) and the more potent derivative 106 (EC50 = 0.1 µM) induced ferroptotic cell death in transformed human HMLER mammary epithelial cells (with high CD44 expression) but were hardly active on human primary breast cells. The cytotoxic effect was attenuated by ferrostatin‐1 but not by apoptosis (Z‐VAD‐FMK) or necroptosis inhibitors (necrostatin‐1). Compounds 105 and 106 were enriched in lysosomes and efficiently bound ferrous iron, which depleted iron in the cytosol. Enhanced lysosomal degradation of ferritin further increased the lysosomal iron load, and the free ferrous/ferric ions seemingly elevated ROS formation preferentially in lysosomes as well as cellular lipid peroxidation. Of note, both ionophores have a rather low affinity to ferrous iron and form iron complexes that are still Fenton‐active in contrast to the tight Fe2+ complexes of DFO and DFO that are redox‐inactive. Both, compounds 105 (3 mg/kg, ip, daily) and 106 (1 mg/kg, ip, daily) efficiently inhibited tumor growth in a Werner syndrome patient‐derived SV40 fibroblast cell xenograft model in mice.
Figure 12.
Increasing the lysosomal iron load or inducing spontaneous lipid peroxidation [Color figure can be viewed at wileyonlinelibrary.com]
9.3.2. PUFAs and conjugated linolenic acids
PUFAs like dihomo‐γ‐linolenic acid (≥500 µM) and fragmented oxidized phosphatidylcholines (1–10 µM), for example, 1‐palmitoyl‐2‐(5'‐oxo‐valeroyl)‐sn‐glycero‐3‐phosphocholine, have been shown to induce ferroptosis in human fibrosarcoma cells 366 and rat primary cardiomyocytes, 367 respectively. Conjugated linolenic acid (CLA) with three conjugated double bonds, such as α‐eleostearic acid (107) (Figure 12), have antioxidative, antiatherogenic, antiobese, anti‐inflammatory, and antitumoral activities 368 and are more effective in inducing ferroptosis than PUFAs with isolated double bonds. They are synthesized in high amounts in bitter gourd (Momordica charantia) 369 and promegranate seeds 370 and are also produced by distinct (intestinal) bacteria. 371 Compound 107 is incorporated into triglycerides by ACSL1, increases lipid peroxidation, and induces cell death in many human cancer cell lines (>100 investigated), including triple‐negative breast cancer cells (EC50 = 10 µM). 101 The mechanism by which triglyceride‐bound 107 causes cell death is not fully understood. It has been hypothesized that 107 undergoes spontaneous or enzymatic oxidation when its concentration exceeds a distinct threshold, which then propagates autooxidation by chain reaction that is facilitated by the conjugated system and potentially leads to polymerization. The anticancer activity of CLA was studied in an aggressive triple‐negative breast cancer orthotopic xenograft model, where compound 102‐rich tung oil (100 µl, po, 5× per week) decreased tumor mass as well as lung metastatic invasion.
9.4. Natural products that trigger ferroptosis through unknown mechanisms
9.4.1. Resibufogenin (108)
The bufadienolide 108 (20–40 µM) (Figure 13) isolated from Asiatic toad (Bufo gargarizans) increased MDA, iron, and ROS levels, decreased GPX4 expression, reduced the availability of GSH and triggered ferroptosis in human HT29 and SW480 CRC cells. 372 Administration of 108 (5 and 10 mg/kg, ip) to mice suppressed growth and metastasis of human SW480 CRC cells through RIP3‐mediated necroptosis in a xenograft model. 373 Another study using this CRC cancer model reported that oxidative stress is induced and tumorigenicity is reduced at high dosage of 108 (80 mg/kg, injected in the tumor each day). 372 It seems that 108 engages different cell death programs, and ferroptosis might not be the predominant form in vivo. Note that 108 has the same bufadienolide scaffold as 44, a natural GPX4 inhibitor described in Section 6. Moreover, similar to 44, it contains a highly reactive endoperoxide albeit at a different position.
Figure 13.
Inducers of (lipid) reactive oxygen species formation that trigger ferroptosis through less characterized mechanisms [Color figure can be viewed at wileyonlinelibrary.com]
9.4.2. Heteronemin (109)
The pentacyclic triterpenoid 109 (Figure 13) found in the sponge Hippospongia species showed profound cytotoxicity in various cancer cell lines (EC50 = 0.1–2.4 µM). 374 , 375 Compound 109 induced caspase‐dependent apoptosis as well as ferroptosis. 375 The latter was diminished by ferrostatin‐1 as well as liproxstatin‐1 and associated with reduced GPX4 protein levels. 375 The development of 109 as a drug candidate was hampered by severe toxic effects in mice already at 1 mg/kg (application route not given) and a lethal dose of 5 mg/kg (application route not given).
9.4.3. Pseudolaric acid A (110)
The diterpene acid 110 (2 µM, (Figure 13) from the root and trunk bark of Pseudolarix amabilis 376 induced lipid peroxidation and cell death (EC50 = 1 µM) in rat C6 and human SHG‐44 glioma cells. MDA generation was prevented by ferrostatin‐1 or the iron chelator DFO. Compound 110 (10 mg/kg, daily for 8 days, ip) was also active on grafted C6 glioma cells in mice and markedly reduced the tumor volume (eightfold). 376 Compound 110 decreased SLC7A11, cysteine, and GSH levels enhanced the expression of NOX4, which reduces oxygen to superoxide radical anions, rose the labile iron pool, and surprisingly upregulated GPX4 in diverse rat and human glioma cell lines (C6, SHG‐44, U87, U251) and/or grafted tumors in mice. Note that an increase in GPX4 expression was also observed for the artemisinin metabolite 102. 358 In addition, compound 110 enhanced p53 expression and phosphorylation but showed similar cytotoxicity and ferroptotic signatures (SLC7A11, GPX4 and transferrin expression) in p53 wild‐type (U87) and p53 mutant (U251) cells, 376 which questions a major role of p53 in compound 110‐induced ferroptosis.
9.4.4. Epunctanone (111), ungerimine (112), and soyauxinium chloride (113)
The benzophenone 111 from Garcinia epunctata and the two alkaloids 112 from Crinum zeylanicum and 113 from Araliopsis soyauxii (Figure 13) have recently been identified to induce ferroptosis. 279 , 280 , 377 , 378 , 379 These compounds have cytotoxic activity (EC50 = 3.6–111 µM) against diverse cancer cell lines from the breast, colon, lymphatic tissue, brain, and skin, which includes drug‐resistant phenotypes that either overexpress ABC transporters (P‐glycoprotein and BCRP), have activating mutations in EGFR or B‐RAF, or have p53 deleted. The iron chelator DFO and the lipid peroxidation inhibitor ferrostatin‐1 decreased the cytotoxic effects of the natural products, which suggests a major role of ferroptosis in cell death induction. Whether the observed increase in intracellular ROS levels is associated with enhanced membrane peroxidation was not investigated. Other forms of cell death were triggered likewise, albeit with variable profiles for the different natural products. Compound 111 induced apoptosis (necroptosis and autophagy were not investigated), 280 , 377 112 activated apoptosis and autophagy, 378 and 113 initiated apoptosis as well as necroptosis. 279 , 379 Note that 111 shares a prenylated bicyclic ring system with 87 (see Section 7) and that we proposed above that 87 modulates NRF2 activation based on changes in NRF2 target gene expression. Respective mechanistic hints are lacking for 113.
9.4.5. Vitamin C (114)
Preclinical studies suggest that the antioxidant 114 (Figure 13) also acts as pro‐oxidant, disturbs the cellular redox balance, and has anticancer activity at supraphysiological concentrations in the millimolar range. 380 Compound 114 (1 mM) exhibits cytotoxic, antiproliferative activity in human ATC 8505 C thyroid carcinoma cells, 381 which was diminished by the iron chelator DFO but neither by apoptosis (Z‐VAD‐FMK) nor necroptosis inhibitors (necrostatin‐1). 381 Mechanistically, GPX4 levels were decreased and MDA levels as well as the labile iron pool increased, which was ascribed to ferritinophagy and subsequent FTH degradation.
9.4.6. Flavonoids—quercetin (115) and apigenin (116)
The flavonoids 115 and 116 (Figure 13) are cytotoxic for diverse cancer cell lines at micromolar concentrations, including human HepG2 and HepB3 hepatocarcinoma, HTC116 CRC and MDA‐MB‐231 breast cancer cells (EC50 < 100 µM). 382 , 383 Compound 115 (50 µM) enhanced lipid peroxidation and elevated cellular iron levels, which was prevented by the ferroptosis inhibitors DFO, ferrostatin‐1, and liproxstatin‐1 as well as the autophagy inhibitor bafilomycin A1. 382 Cellular iron accumulation was ascribed to an activation of the transcription factor EB that leads to lysosomal degradation of ferritin (ferritinophagy). Moreover, compound 115 decreased GPX4 protein expression. The structurally related flavonoid 116 induced ferroptotic cell death in multiple myeloma cell lines (EC50 = 10–30 µM) along with apoptosis and autophagy. 383
9.4.7. Biflavonoids—robustaflavone A (117)
Several biflavonoids from Selaginella trichoclada (used in traditional Chinese medicine to treat jaundice and burns) induced cell death in human MCF‐7 breast cancer cells, with 117 (Figure 13) being most potent (EC50 = 12 µM). 384 Cytotoxic effects of 117 were associated with enhanced lipid hydroperoxide formation and slightly diminished by ferrostatin‐1 or DFO, which hints toward a limited involvement of ferroptosis in cell death induction. Compound 117 downregulated Nedd4, an E3‐ubiquitin ligase that subjects voltage‐dependent anion channel 2 (VDAC2) to proteasomal degradation, and elevated VDAC2 protein levels, seemingly both via impaired protein degradation and enhanced mRNA expression. VDAC2 is an outer membrane pore in mitochondria and has recently been shown to increase the sensitivity of cancer cells to system Xc − inhibition. 385
Due to the structural similarity to 94, we proposed in Section 8 that the biflavonoid 95 inhibits NRF2 signaling, and compound 117 only differs from 95 by the site of oxidative coupling. While the apigenin moieties of 95 are linked between C‐3 of the hydroxyphenyl ring and C‐8 of chromene, the bond between the two apigenins is formed between C‐3 and C‐6 in 117. This apparent small difference has strong impact on the 3D‐structure of the molecules, resulting either in a compact (95) or curved, rod‐like structure (117). Since the knowledge about the ferroptotic mechanisms of 117 are limited, we decided not to group this compound together with other biflavonoids in Section 7, although we cannot exclude a common mode of action.
9.4.8. Physcion 8‐O‐β‐glucopyranoside (118)
The anthraquinone glycoside 118 (Figure 13) isolated from Rumex japonicus possesses antiapoptotic, antiproliferative and antimetastatic properties. 386 Compound 118 elevated cellular iron and MDA levels and induces death in human MGC‐803 and MKN‐45 gastric carcinoma cells (EC50 = 40 µM), 387 which was prevented by ferrostatin‐1 but not by the apoptosis inhibitor Z‐VAD‐FMK or the necroptosis inhibitor necrostatin‐1. 387 Knockdown studies indicate that ferroptosis induction by 118 depends on miR‐103a‐3p upregulation and subsequent expression of GLS2. The mitochondrial GLS2 converts glutamine to glutamate, is involved in cancer metabolic reprogramming and has been shown to promote lipid peroxidation and ferroptosis. 388
9.4.9. Napthoquinone—avicequinone B (119)
The naphthoquinone 119 (Figure 13) from the mangrove tree Avicennia alba reduced the viability of human breast, colorectal, and lung adenocarcinoma cells (EC50 = 3–12 µM). 389 Transcriptome analysis revealed that 119 regulates ferroptosis‐related genes (e.g., SLC7A11 and GCL), however, in the opposite direction as expected for ferroptosis induction. The authors speculate about unknown targets and feedback mechanisms. 389
9.4.10. Gallic acid (120)
3,4,5‐Trihydroxybenzoic acid (120; Figure 13) is prevalent in Quercus species and has recently been proposed to induce ferroptosis besides apoptosis and necroptosis at high concentrations (300 µM) in human HeLa epithelial adenocarcinoma cells, H446 lung carcinoma epithelial cells, SH‐SY5Y neuroblastoma cells, MDA‐MB‐231 breast cancer cells and A375 melanoma cancer cells. 390 , 391 The contribution of ferroptosis to cell death induction remains enigmatic. On the one hand, gallic acid decreased the mRNA expression of distinct ferroptosis‐related genes (e.g., GPX4 and p53), 390 and it was speculated based on docking studies that 120 directly binds to several key factors in ferroptosis, including GPX4, p53 and NRF2. 390 The iron chelator DFO diminished the cytotoxic effects of 120. 392 On the other hand, 120 did not evoke substantial lipid peroxidation, and the lipophilic radical trap ferrostatin‐1 failed to prevent cell death. 392 Together, 120 induces cell death through multiple mechanisms and further studies are necessary to define the degree, to which ferroptosis is involved.
9.4.11. Comparative discussion
An increasing number of compounds have been identified during recent years that induce membrane peroxidation and promote ferroptotic cell death independent from GPX4 and system Xc − inhibition. Many of them are natural products or bioinspired small molecules that induce besides ferroptosis also other forms of PD like apoptosis, necroptosis or autophagy. Such polypharmacological molecules might have advantages regarding clinical efficacy in anticancer therapy by hindering the tumors to develop escape routes and acquire resistance mechanisms. 7 , 393 On the other hand, increasing evidence suggests that compounds preferentially inducing ferroptosis might have advantages in discriminating between cancer and nontransformed cells, which expands to diverse therapy‐resistant phenotypes and is important for safety.
Major ferroptosis‐inducing mechanisms exploited by the natural products addressed in this chapter involve effects on (i) iron oxidation and the Fenton reaction (97, artemisinins), (ii) the thioredoxin system (104), (iii) spontaneous lipid autoxidation (CLA‐like 107), (iv) iron storage and transport (this section: 105, 106 and 65, 66 in Section 6.3), and (v) glutaminolysis (119). It should be noted that many of the here described natural products have promiscuous activities, which might essentially add to their partly encouraging activity against (therapy‐resistant) cancer but hamper conclusions about the individual targets that contribute to cell death induction. For iron‐oxidizing endoperoxides (97, artemisinins), it is well documented that they drive lipid hydroperoxide formation through the Fenton reaction, 337 though also here the exact interactions and mechanisms of subcellular membrane peroxidation are not always fully understood. For ferroptosis inducers that indirectly stimulate the accumulation of lipid hydroperoxides, it is even more difficult to assign which of the proposed mechanisms (i) have pharmacological relevance, (ii) interfere with the sensitive redox balance of membranes and (iii) contribute to ferroptosis induction under physiological settings. Functional studies based on compensation experiments that link proposed targets with cell death induction were successfully conducted only for few studies and are inconclusive (104) 3 or lacking for others (e.g., 110). 376 For many further small molecules, the molecular targets are still enigmatic or, if known at all, not obviously linked to ferroptosis.
Among the diverse compounds in this section, two major groups can be classified based on their structure: (1) the artemisinins 98–102 and the compounds 103, 110 (this section), and 54 (see Section 6.3) share an eight‐membered ring system, and 2) compounds 105 and 106 (this section) as well as 65, 66, 67, and 68 (see Section 6.3) have a spiroketal or an aza‐analogous spiroketal scaffold. The mechanisms by which the former induce ferroptosis are diverse and range from triggering the Fenton reaction to inactivating thioredoxin, whereas the spiroketals 65–68, 105, and 106 have been reported to interfere with iron metabolism. Further studies might help to better understand the bioactivity and clinical potential of these anticancer drug candidates and potentially provide novel insights into the regulation of ferroptosis‐protective pathways.
10. CONCLUSION AND FUTURE PERSPECTIVES
Long before system Xc − has been identified as a key player in ferroptosis, inhibitors have been developed and the first anticancer drug candidate entered clinical trials. The interest in small molecules that target system Xc − rapidly increased after iron‐dependent lipid peroxidation has been recognized as a central part of a strictly regulated cell death program, and the term ferroptosis has been coined. Inhibitors of ferroptosis promise protection against neurodegenerative diseases, 20 , 21 , 394 whereas ferroptosis inducers are directed against hyperproliferative disorders like cancer. 10 , 58 , 329 Research in the latter field experienced a boost after aggressive, metastatic, and therapy‐resistant cancer cells were found to be highly sensitive to ferroptosis. 1 GPX4, the only GPX isoenzyme that detoxifies lipid hydroperoxides, was early considered a promising target to trigger ferroptosis in tumors. However, major advances in drug development were hampered for several reasons, one of them being the lack of a clearly defined inhibitor binding pocket at the enzyme. Major breakthroughs in GPX4 inhibitor development may be expected from ligands combining specific noncovalent inhibition with an electrophilic warhead that covalently binds to the active site selenocysteine.
Direct GPX4 and system Xc − inhibitors are nowadays dominated by screening hits from synthetic compound libraries and structurally optimized derivatives, but early system Xc − inhibitors (1 and 2) were actually plant secondary metabolites 144 and most NRF2 inhibitor/activators still originate from natural sources or are bioinspired. The high number of ferroptosis‐inducing natural products is not surprising when considering that ferroptosis is an ancient, evolutionally conserved cell death program that is shared by plants and animals. 395
Triggering ferroptosis is generally considered an effective means to induce cell death in therapy‐resistant cancer cells. 1 Among the ferroptosis inducers that efficiently discriminate between cancer and nontransformed cells are small molecules that induce lipid hydroperoxide formation either by (i) inhibiting GPX4, system Xc − or the thioredoxin/thioredoxin reductase system, (ii) inactivating NRF2, (iii) repressing GXP4 in combination with inducing HO‐1 expression, or (iv) triggering iron oxidation and the Fenton reaction. For example, the NRF2 modulators 94 and 56 act synergistically with conventional anticancer drugs and show high cytotoxic activity against drug‐resistant cancer cells, in particular toward those carrying mutations within the NRF2 axis. On the other hand, activation of the NRF2/HO‐1 axis seems to be an effective strategy, when combined with diminished GPX4 expression. Further studies are necessary to explore the functional links.
Overall, ferroptotic cell death is heterogenic and highly context‐dependent, which demands for personalized ferroptosis‐related anticancer strategies. Factors that shape ferroptosis sensitivity are the fatty acid composition of (sub)cellular membranes (in particular the balance between esterified PUFAs and MUFAs) and the capacity of antioxidant systems, including GPX4/GSH, FSP1/DHODH‐CoQ10, and GCH1/BH4. The number of known regulatory proteins in ferroptosis is dynamically increasing, and links to potential drug targets are emerging. For example, KEAP1 deletion (and thereby activation of NRF2) dramatically reduced GPX4 levels in human H1299 lung cancer cells but, in contrast to GPX4 deficiency, neither raised lipid peroxidation nor enhanced cell death. The failure of KEAP1 disruption to induce ferroptosis was ascribed to a concomitant induction of FSP1 that compensates for the loss of GPX4. 50 On the other hand, cancer‐related processes, like EMT or metabolic reprogramming (e.g., by upregulating AR), are located at the crossroad to ferroptosis and regulate hallmarks in ferroptosis, such as the membrane PUFA proportion or the availability of GPX4. 51 , 203 These cancer‐specific discrepancies render distinct (therapy‐resistant) tumors more vulnerable toward ferroptosis‐inducing strategies than others, though not necessarily always toward the same targets or target combinations. 199 , 202 , 203 The recent progress in the understanding of the complex defense network against ferroptosis now provides access to pleiotropic putative targets for ferroptosis‐triggering small molecules. Whether these points of attack are already exploited by some of the here described ferroptosis inducers and whether their pharmacological manipulation is promising in light of antitumoral efficacy and safety, requires further investigation.
We hypothesize that ferroptosis inducers limit the capacity of cancer cells to hijack their targets and upregulate them under oxidative stress. In addition, many of these small molecules have multiple activities that likely add to the overall anticancer properties and might help to overcome tumor resistance. The exact mechanisms behind are largely obscure, as are the consequences on the tumor microenvironment, immune surveillance, stemness, vasculation, metastasis, energy metabolism, membrane composition as well as the redox lipidome; the latter requiring sensitive, high‐end mass spectrometric redox lipidomics. 396
Classical strategies employed in drug design require the knowledge of defined targets as well as structural information about the binding sites and can therefore not easily be applied to ferroptosis‐inducing small molecules. GPX4 lacks a classical binding pocket, NRF2 is mainly regulated via redox‐dependent processes and specific target sites are absent or not defined, and the molecular targets are unknown for many other reported ferroptosis inducers. Future studies are needed to expand our current understanding of redox sensors and redox‐controlled mechanisms in ferroptosis. The increasing awareness that HO‐1 plays a central role in NRF2‐dependent ferroptosis is an important step in this direction. Such insights will be the catalyst to further accelerate the discovery, design, and rational optimization of ferroptosis‐modulating small molecules that specifically interfere with single or multiple key targets in ferroptosis and potentially other cell death pathways.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Supporting information
Supporting information.
ACKNOWLEDGMENTS
Research activities of the authors related to the subject of this article were funded in part by the Austrian Science Fund (FWF) (P 36299, I 4968), the German Research Council (Grant Numbers KO4589/7‐1, GRK 1715, FOR 2558 [KI 1590/3‐2]), the Phospholipid Research Center Heidelberg (Grant Number AKO‐2019‐070/2‐1), and the University of Jena (Grant Number AZ2.113‐A1_2018‐02). For the purpose of open acces, the authors have applied a CC BY public copyright licence to any Author Accepted Manuscript version arising from this submission.
Biographies
Solveigh C. Koeberle is currently the principal investigator at the Michael Popp Institute, University of Innsbruck, Austria. In addition, she is holding an interim professorship in medicinal chemistry at the University of Regensburg, Germany. After her PhD degree in medicinal chemistry (University of Tuebingen), she worked as a postdoc at the University of Tokyo, the Leibniz Institute on Aging Research, and the University of Jena. Her current research interests lie in the area of (redox‐dependent) signal transduction and the development of selective chemical probes in the context of inflammation and cancer. Currently, she aims to identify natural products that induce ferroptosis and investigate the underlying mechanism.
Anna P. Kipp is a professor of molecular nutritional physiology at the Friedrich Schiller University Jena. Her research focuses on the interactions of essential trace elements including selenium, copper, and zinc, and the direct and indirect effects of trace elements on redox signaling. This connection is going to be studied with regard to the healthy intestine, during inflammation and cancer development focusing on selenoproteins, especially glutathione peroxidases.
Hermann Stuppner studied pharmaceutical sciences at the University of Innsbruck. After receiving his PhD in pharmaceutical biology from the University of Munich, he was a postdoctoral fellow at the Department of Developmental and Cell Biology of the University of California, Irvine. Since 2001, he has been a full professor of pharmacognosy at the Institute of Pharmacy of the University of Innsbruck. His main research interests are isolation and structural elucidation of secondary metabolites from higher plants with anti‐inflammatory and antitumor activity, analysis and quality assessment of (medicinal) plants and phytopharmaceuticals, and discovery of pharmacologically active natural products by means of computer‐aided models.
Andreas Koeberle is a full professor for new phyto entities at the University of Innsbruck and a member of the Center for Molecular Biosciences Innsbruck. As a biochemist and medicinal chemist, he has more than 12 years of experience in studying the link between lipid diversity, inflammation, and cancer. His research strives for the development of putative anti‐inflammatory and anticancer (bioinspired) drug candidates and is acknowledged by multiple national and international awards. He applies mass spectrometric functional lipidomics for target identification and lead discovery and aims at finding new strategies to overcome nonresolved inflammation and tumor resistance. His current research focuses on the molecular mechanisms and preclinical development of natural products that target lipid signaling in ferroptosis.
Koeberle SC, Kipp AP, Stuppner H, Koeberle A. Ferroptosis‐modulating small molecules for targeting drug‐resistant cancer: challenges and opportunities in manipulating redox signaling. Med Res Rev. 2023;43:614‐682. 10.1002/med.21933
Contributor Information
Solveigh C. Koeberle, Email: solveigh.koeberle@uibk.ac.at.
Andreas Koeberle, Email: andreas.koeberle@uibk.ac.at.
DATA AVAILABILITY STATEMENT
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
REFERENCES
- 1. Friedmann Angeli JP, Krysko DV, Conrad M. Ferroptosis at the crossroads of cancer‐acquired drug resistance and immune evasion. Nat Rev Cancer. 2019;19:405‐414. 10.1038/s41568-019-0149-1 [DOI] [PubMed] [Google Scholar]
- 2. Stockwell BR. Ferroptosis turns 10: emerging mechanisms, physiological functions, and therapeutic applications. Cell. 2022;185:2401‐2421. 10.1016/j.cell.2022.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Llabani E, Hicklin RW, Lee HY, et al. Diverse compounds from pleuromutilin lead to a thioredoxin inhibitor and inducer of ferroptosis. Nat Chem. 2019;11:521‐532. 10.1038/s41557-019-0261-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Eaton JK, Furst L, Ruberto RA, et al. Selective covalent targeting of GPX4 using masked nitrile‐oxide electrophiles. Nat Chem Biol. 2020;16:497‐506. 10.1038/s41589-020-0501-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jiang X, Stockwell BR, Conrad M. Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol. 2021;22:266‐282. 10.1038/s41580-020-00324-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhang J, Duan D, Song ZL, Liu T, Hou Y, Fang J. Small molecules regulating reactive oxygen species homeostasis for cancer therapy. Med Res Rev. 2021;41:342‐394. 10.1002/med.21734 [DOI] [PubMed] [Google Scholar]
- 7. Ke B, Tian M, Li J, Liu B, He G. Targeting programmed cell death using small‐molecule compounds to improve potential cancer therapy. Med Res Rev. 2016;36:983‐1035. 10.1002/med.21398 [DOI] [PubMed] [Google Scholar]
- 8. Tuo Q, Zhang S, Lei P. Mechanisms of neuronal cell death in ischemic stroke and their therapeutic implications. Med Res Rev. 2021;42:259‐305. 10.1002/med.21817 [DOI] [PubMed] [Google Scholar]
- 9. Stockwell BR, Jiang X, Gu W. Emerging mechanisms and disease relevance of ferroptosis. Trends Cell Biol. 2020;30:478‐490. 10.1016/j.tcb.2020.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Liang C, Zhang X, Yang M, Dong X. Recent progress in ferroptosis inducers for cancer therapy. Adv Mater. 2019;31:1904197. 10.1002/adma.201904197 [DOI] [PubMed] [Google Scholar]
- 11. Stockwell BR, Friedmann Angeli JP, Bayir H, et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell. 2017;171:273‐285. 10.1016/j.cell.2017.09.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kajiwara K, Beharier O, Chng CP, et al. Ferroptosis induces membrane blebbing in placental trophoblasts. J Cell Sci. 2022;135:jcs255737. 10.1242/jcs.255737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kagan VE, Mao G, Qu F, et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol. 2017;13:81‐90. 10.1038/nchembio.2238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Doll S, Conrad M. Iron and ferroptosis: a still ill‐defined liaison. IUBMB Life. 2017;69:423‐434. 10.1002/iub.1616 [DOI] [PubMed] [Google Scholar]
- 15. Guo JD, Lifan He, Xueying L, et al. A combined model of human iPSC‐derived liver organoids and hepatocytes reveals ferroptosis in DGUOK mutant mtDNA depletion syndrome. Adv Sci. 2021;8:2004680. 10.1002/advs.202004680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yan H, Zou T, Tuo Q, et al. Ferroptosis: mechanisms and links with diseases. Signal Transduct Target Ther. 2021;6:49. 10.1038/s41392-020-00428-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gaschler MM, Stockwell BR. Lipid peroxidation in cell death. Biochem Biophys Res Commun. 2017;482:419‐425. 10.1016/j.bbrc.2016.10.086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yan B, Ai Y, Sun Q, et al. Membrane damage during ferroptosis is caused by oxidation of phospholipids catalyzed by the oxidoreductases POR and CYB5R1. Mol Cell. 2021;81:355‐369. 10.1016/j.molcel.2020.11.024 [DOI] [PubMed] [Google Scholar]
- 19. Pedrera L, Espiritu RA, Ros U, et al. Ferroptotic pores induce Ca2+ fluxes and ESCRT‐III activation to modulate cell death kinetics. Cell Death Differ. 2020;28:1644‐1657. 10.1038/s41418-020-00691-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Angeli JPF, Shah R, Pratt DA, Conrad M. Ferroptosis inhibition: mechanisms and opportunities. Trends Pharmacol Sci. 2017;38:489‐498. 10.1016/j.tips.2017.02.005 [DOI] [PubMed] [Google Scholar]
- 21. Seibt TM, Proneth B, Conrad M. Role of GPX4 in ferroptosis and its pharmacological implication. Free Radic Biol Med. 2019;133:144‐152. 10.1016/j.freeradbiomed.2018.09.014 [DOI] [PubMed] [Google Scholar]
- 22. Zheng K, Dong Y, Yang R, Liang Y, Wu H, He Z. Regulation of ferroptosis by bioactive phytochemicals: implications for medical nutritional therapy. Pharmacol Res. 2021;168:105580. 10.1016/j.phrs.2021.105580 [DOI] [PubMed] [Google Scholar]
- 23. Conrad M, Angeli JPF, Vandenabeele P, Stockwell BR. Regulated necrosis: disease relevance and therapeutic opportunities. Nat Rev Drug Discov. 2016;15:348‐366. 10.1038/nrd.2015.6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chen X, Kang R, Kroemer G, Tang D. Targeting ferroptosis in pancreatic cancer: a double‐edged sword. Trends in Cancer. 2021;7:891‐901. 10.1016/j.trecan.2021.04.005 [DOI] [PubMed] [Google Scholar]
- 25. Florean C, Song S, Dicato M, Diederich M. Redox biology of regulated cell death in cancer: a focus on necroptosis and ferroptosis. Free Radic Biol Med. 2019;134:177‐189. 10.1016/j.freeradbiomed.2019.01.008 [DOI] [PubMed] [Google Scholar]
- 26. Hassannia B, Vandenabeele P, Vanden Berghe T. Targeting ferroptosis to iron out cancer. Cancer Cell. 2019;35:830‐849. 10.1016/j.ccell.2019.04.002 [DOI] [PubMed] [Google Scholar]
- 27. Li N, Jiang W, Wang W, Xiong R, Wu X, Geng Q. Ferroptosis and its emerging roles in cardiovascular diseases. Pharmacol Res. 2021;166:105466. 10.1016/j.phrs.2021.105466 [DOI] [PubMed] [Google Scholar]
- 28. Mou Y, Wang J, Wu J, et al. Ferroptosis, a new form of cell death: opportunities and challenges in cancer. J Hematol Oncol. 2019;12:34. 10.1186/s13045-019-0720-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wang H, Cheng Y, Mao C, et al. Emerging mechanisms and targeted therapy of ferroptosis in cancer. Mol Ther. 2021;29:2185‐2208. 10.1016/j.ymthe.2021.03.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wang H, Lin D, Yu Q, et al. A promising future of ferroptosis in tumor therapy. Front Cell Dev Biol. 2021;9:629150. 10.3389/fcell.2021.629150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Feng H, Stockwell BR. Unsolved mysteries: how does lipid peroxidation cause ferroptosis. PLoS Biol. 2018;16:e2006203. 10.1371/journal.pbio.2006203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Conrad M, Pratt DA. The chemical basis of ferroptosis. Nat Chem Biol. 2019;15:1137‐1147. 10.1038/s41589-019-0408-1 [DOI] [PubMed] [Google Scholar]
- 33. Han C, Liu Y, Dai R, Ismail N, Su W, Li B. Ferroptosis and its potential role in human diseases. Front Pharmacol. 2020;11:239. 10.3389/fphar.2020.00239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dixon SJ, Stockwell BR. The role of iron and reactive oxygen species in cell death. Nat Chem Biol. 2014;10:9‐17. 10.1038/nchembio.1416 [DOI] [PubMed] [Google Scholar]
- 35. Ma XH, Liu JHZ, Liu CY, et al. ALOX15‐launched PUFA‐phospholipids peroxidation increases the susceptibility of ferroptosis in ischemia‐induced myocardial damage. Signal Transduct Target Ther. 2022;7:288. 10.1038/s41392-022-01090-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zou Y, Li H, Graham ET, et al. Cytochrome P450 oxidoreductase contributes to phospholipid peroxidation in ferroptosis. Nat Chem Biol. 2020;16:302‐309. 10.1038/s41589-020-0472-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chen X, Yu C, Kang R, Tang D. Iron metabolism in ferroptosis. Front Cell Dev Biol. 2020;8:590226. 10.3389/fcell.2020.590226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pantopoulos K. Iron metabolism and the IRE/IRP regulatory system: an update. Ann NY Acad Sci. 2004;1012:1‐13. 10.1196/annals.1306.001 [DOI] [PubMed] [Google Scholar]
- 39. Alvarez SW, Sviderskiy VO, Terzi EM, et al. NFS1 undergoes positive selection in lung tumours and protects cells from ferroptosis. Nature. 2017;551:639‐643. 10.1038/nature24637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Alvarez SW, Possemato R. Leveraging the iron‐starvation response to promote ferroptosis. Oncotarget. 2018;9:10830‐10831. 10.18632/oncotarget.24395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lei G, Zhuang L, Gan B. Targeting ferroptosis as a vulnerability in cancer. Nat Rev Cancer. 2022;22:381‐396. 10.1038/s41568-022-00459-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Menegon S, Columbano A, Giordano S. The dual roles of NRF2 in cancer. Trends Mol Med. 2016;22:578‐593. 10.1016/j.molmed.2016.05.002 [DOI] [PubMed] [Google Scholar]
- 43. Sun X, Ou Z, Chen R, et al. Activation of the p62‐Keap1‐NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology. 2016;63:173‐184. 10.1002/hep.28251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ren D, Villeneuve NF, Jiang T, et al. Brusatol enhances the efficacy of chemotherapy by inhibiting the Nrf2‐mediated defense mechanism. Proc Natl Acad Sci USA. 2011;108:1433‐1438. 10.1073/pnas.1014275108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tebay LE, Robertson H, Durant ST, et al. Mechanisms of activation of the transcription factor Nrf2 by redox stressors, nutrient cues, and energy status and the pathways through which it attenuates degenerative disease. Free Radic Biol Med. 2015;88:108‐146. 10.1016/j.freeradbiomed.2015.06.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lu MC, Ji JA, Jiang ZY, You QD. The Keap1‐Nrf2‐ARE pathway as a potential preventive and therapeutic target: an update. Med Res Rev. 2016;36:924‐963. 10.1002/med.21396 [DOI] [PubMed] [Google Scholar]
- 47. Anandhan A, Dodson M, Schmidlin CJ, Liu P, Zhang DD. Breakdown of an ironclad defense system: the critical role of NRF2 in mediating ferroptosis. Cell Chem Biol. 2020;27:436‐447. 10.1016/j.chembiol.2020.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Panieri E, Buha A, Telkoparan‐Akillilar P, et al. Potential applications of NRF2 modulators in cancer therapy. Antioxidants. 2020;9:193. 10.3390/antiox9030193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kang YP, Mockabee‐Macias A, Jiang C, et al. Non‐canonical glutamate‐cysteine ligase activity protects against ferroptosis. Cell Metab. 2021;33:174‐189. 10.1016/j.cmet.2020.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Koppula P, Lei G, Zhang Y, et al. A targetable CoQ‐FSP1 axis drives ferroptosis‐ and radiation‐resistance in KEAP1 inactive lung cancers. Nat Commun. 2022;13:2206. 10.1038/s41467-022-29905-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Nishizawa H, Yamanaka M, Igarashi K. Ferroptosis: regulation by competition between NRF2 and BACH1 and propagation of the death signal. FEBS J. Published online February 2, 2022. 10.1111/febs.16382 [DOI] [PubMed] [Google Scholar]
- 52. Uddin MJ, Kim EH, Hannan MA, Ha H. Pharmacotherapy against oxidative stress in chronic kidney disease: promising small molecule natural products targeting Nrf2‐HO‐1 signaling. Antioxidants. 2021;10:258. 10.3390/antiox10020258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Qin JJ, Li X, Hunt C, Wang W, Wang H, Zhang R. Natural products targeting the p53‐MDM2 pathway and mutant p53: recent advances and implications in cancer medicine. Genes Dis. 2018;5:204‐219. 10.1016/j.gendis.2018.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Dixon SJ, Patel DN, Welsch M, et al. Pharmacological inhibition of cystine‐glutamate exchange induces endoplasmic reticulum stress and ferroptosis. eLife. 2014;3:e02523. 10.7554/eLife.02523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wu J, Minikes AM, Gao M, et al. Intercellular interaction dictates cancer cell ferroptosis via NF2‐YAP signalling. Nature. 2019;572:402‐406. 10.1038/s41586-019-1426-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Johnson R, Halder G. The two faces of Hippo: targeting the Hippo pathway for regenerative medicine and cancer treatment. Nat Rev Drug Discov. 2014;13:63‐79. 10.1038/nrd4161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Dai C, Chen X, Li J, Comish P, Kang R, Tang D. Transcription factors in ferroptotic cell death. Cancer Gene Ther. 2020;27:645‐656. 10.1038/s41417-020-0170-2 [DOI] [PubMed] [Google Scholar]
- 58. Bebber CM, Müller F, Prieto Clemente L, Weber J, von Karstedt S. Ferroptosis in cancer cell biology. Cancers. 2020;12:164. 10.3390/cancers12010164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Muller PAJ, Vousden KH. Mutant p53 in cancer: new functions and therapeutic opportunities. Cancer Cell. 2014;25:304‐317. 10.1016/j.ccr.2014.01.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Jiang L, Kon N, Li T, et al. Ferroptosis as a p53‐mediated activity during tumour suppression. Nature. 2015;520:57‐62. 10.1038/nature14344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ji H, Wang W, Li X, et al. p53: a double‐edged sword in tumor ferroptosis. Pharmacol Res. 2022;177:106013. 10.1016/j.phrs.2021.106013 [DOI] [PubMed] [Google Scholar]
- 62. Liu Y, Gu W. p53 in ferroptosis regulation: the new weapon for the old guardian. Cell Death Differ. 2022;29:895‐910. 10.1038/s41418-022-00943-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Semenza GL. Targeting HIF‐1 for cancer therapy. Nat Rev Cancer. 2003;3:721‐732. 10.1038/nrc1187 [DOI] [PubMed] [Google Scholar]
- 64. Bhattarai D, Xu X, Lee K. Hypoxia‐inducible factor‐1 (HIF‐1) inhibitors from the last decade (2007 to 2016): a “structure‐activity relationship” perspective. Med Res Rev. 2018;38:1404‐1442. 10.1002/med.21477. [DOI] [PubMed] [Google Scholar]
- 65. Ma L, Jiang H, Xu X, et al. Tanshinone IIA mediates SMAD7‐YAP interaction to inhibit liver cancer growth by inactivating the transforming growth factor beta signaling pathway. Aging. 2019;11:9719‐9737. 10.18632/aging.102420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Trott A, West JD, Klaić L, et al. Activation of heat shock and antioxidant responses by the natural product celastrol: transcriptional signatures of a thiol‐targeted molecule. Mol Biol Cell. 2008;19:1104‐1112. 10.1091/mbc.E07-10-1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Chen C, Zhu D, Zhang H, et al. YAP‐dependent ubiquitination and degradation of β‐catenin mediates inhibition of Wnt signalling induced by physalin F in colorectal cancer. Cell Death Dis. 2018;9:591. 10.1038/s41419-018-0645-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Safe S, Kasiappan R. Natural products as mechanism‐based anticancer agents: Sp transcription factors as targets. Phytother Res. 2016;30:1723‐1732. 10.1002/ptr.5669 [DOI] [PubMed] [Google Scholar]
- 69. Yang WS, SriRamaratnam R, Welsch ME, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156:317‐331. 10.1016/j.cell.2013.12.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Lin W, Wang C, Liu G, et al. SLC7A11/xCT in cancer: biological functions and therapeutic implications. Am J Cancer Res. 2020;10:3106‐3126. [PMC free article] [PubMed] [Google Scholar]
- 71. Stoytcheva ZR, Berry MJ. Transcriptional regulation of mammalian selenoprotein expression. Biochim Biophys Acta. 2009;1790:1429‐1440. 10.1016/j.bbagen.2009.05.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Osburn WO, Wakabayashi N, Misra V, et al. Nrf2 regulates an adaptive response protecting against oxidative damage following diquat‐mediated formation of superoxide anion. Arch Biochem Biophys. 2006;454:7‐15. 10.1016/j.abb.2006.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Takahashi N, Cho P, Selfors LM, et al. 3D culture models with CRISPR screens reveal hyperactive NRF2 as a prerequisite for spheroid formation via regulation of proliferation and ferroptosis. Mol Cell. 2020;80:828‐844. 10.1016/j.molcel.2020.10.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Labunskyy VM, Hatfield DL, Gladyshev VN. Selenoproteins: molecular pathways and physiological roles. Physiol Rev. 2014;94:739‐777. 10.1152/physrev.00039.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Burk RF, Hill KE. Regulation of selenium metabolism and transport. Annu Rev Nutr. 2015;35:109‐134. 10.1146/annurev-nutr-071714-034250 [DOI] [PubMed] [Google Scholar]
- 76. Li Z, Ferguson L, Deol KK, et al. Ribosome stalling during selenoprotein translation exposes a ferroptosis vulnerability. Nat Chem Biol. 2022;18:751‐761. 10.1038/s41589-022-01033-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Cui C, Yang F, Li Q. Post‐translational modification of GPX4 is a promising target for treating ferroptosis‐related diseases. Front Mol Biosci. 2022;9:901565. 10.3389/fmolb.2022.901565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Wu Z, Geng Y, Lu X, et al. Chaperone‐mediated autophagy is involved in the execution of ferroptosis. Proc Natl Acad Sci USA. 2019;116:2996‐3005. 10.1073/pnas.1819728116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Kerins MJ, Milligan J, Wohlschlegel JA, Ooi A. Fumarate hydratase inactivation in hereditary leiomyomatosis and renal cell cancer is synthetic lethal with ferroptosis induction. Cancer Sci. 2018;109:2757‐2766. 10.1111/cas.13701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Hadian K, Stockwell BR. SnapShot: ferroptosis. Cell. 2020;181:1188. 10.1016/j.cell.2020.04.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Battaglia AM, Chirillo R, Aversa I, Sacco A, Costanzo F, Biamonte F. Ferroptosis and cancer: mitochondria meet the “Iron Maiden” cell death. Cells. 2020;9:1505. 10.3390/cells9061505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Doll S, Freitas FP, Shah R, et al. FSP1 is a glutathione‐independent ferroptosis suppressor. Nature. 2019;575:693‐698. 10.1038/s41586-019-1707-0 [DOI] [PubMed] [Google Scholar]
- 83. Stockwell BR. A powerful cell‐protection system prevents cell death by ferroptosis. Nature. 2019;575:597‐598. 10.1038/d41586-019-03145-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Frei B, Kim MC, Ames BN. Ubiquinol‐10 is an effective lipid‐soluble antioxidant at physiological concentrations. Proc Natl Acad Sci USA. 1990;87:4879‐4883. 10.1073/pnas.87.12.4879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Mishima E, Ito J, Wu Z, et al. A non‐canonical vitamin K cycle is a potent ferroptosis suppressor. Nature. 2022;608:778‐783. 10.1038/s41586-022-05022-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Mao C, Liu X, Zhang Y, et al. DHODH‐mediated ferroptosis defence is a targetable vulnerability in cancer. Nature. 2021;593:586‐590. 10.1038/s41586-021-03539-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Viswanathan VS, Ryan MJ, Dhruv HD, et al. Dependency of a therapy‐resistant state of cancer cells on a lipid peroxidase pathway. Nature. 2017;547:453‐457. 10.1038/nature23007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Dixon SJ, Lemberg KM, Lamprecht MR, et al. Ferroptosis: an iron‐dependent form of nonapoptotic cell death. Cell. 2012;149:1060‐1072. 10.1016/j.cell.2012.03.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Friedmann Angeli JP, Schneider M, Proneth B, et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol. 2014;16:1180‐1191. 10.1038/ncb3064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Zilka O, Shah R, Li B, et al. On the mechanism of cytoprotection by ferrostatin‐1 and liproxstatin‐1 and the role of lipid peroxidation in ferroptotic cell death. ACS Cent Sci. 2017;3:232‐243. 10.1021/acscentsci.7b00028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Sheng X, Shan C, Liu J, Yang J, Sun B, Chen D. Theoretical insights into the mechanism of ferroptosis suppression via inactivation of a lipid peroxide radical by liproxstatin‐1. Phys Chem Chem Phys. 2017;19:13153‐13159. 10.1039/c7cp00804j [DOI] [PubMed] [Google Scholar]
- 92. Kraft VAN, Bezjian CT, Pfeiffer S, et al. GTP cyclohydrolase 1/tetrahydrobiopterin counteract ferroptosis through lipid remodeling. ACS Cent Sci. 2020;6:41‐53. 10.1021/acscentsci.9b01063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Sun WY, Tyurin VA, Mikulska‐Ruminska K, et al. Phospholipase iPLA2β averts ferroptosis by eliminating a redox lipid death signal. Nat Chem Biol. 2021;17:465‐476. 10.1038/s41589-020-00734-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Kita Y, Shindou H, Shimizu T. Cytosolic phospholipase A2 and lysophospholipid acyltransferases. Biochim Biophys. 2019;1864:838‐845. 10.1016/j.bbalip.2018.08.006. [DOI] [PubMed] [Google Scholar]
- 95. Doll S, Proneth B, Tyurina YY, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol. 2017;13:91‐98. 10.1038/nchembio.2239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci USA. 2016;113:E4966‐E4975. 10.1073/pnas.1603244113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Tang D, Kroemer G. Peroxisome: the new player in ferroptosis. Signal Transduct Target Ther. 2020;5:273. 10.1038/s41392-020-00404-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Shindou H, Shimizu T. Acyl‐CoA:lysophospholipid acyltransferases. J Biol Chem. 2009;284:1‐5. 10.1074/jbc.R800046200 [DOI] [PubMed] [Google Scholar]
- 99. Li D, Li Y. The interaction between ferroptosis and lipid metabolism in cancer. Signal Transduct Target Ther. 2020;5:108. 10.1038/s41392-020-00216-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Dixon SJ, Winter GE, Musavi LS, et al. Human haploid cell genetics reveals roles for lipid metabolism genes in nonapoptotic cell death. ACS Chem Biol. 2015;10:1604‐1609. 10.1021/acschembio.5b00245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Beatty A, Singh T, Tyurina YY, et al. Ferroptotic cell death triggered by conjugated linolenic acids is mediated by ACSL1. Nat Commun. 2021;12:2244. 10.1038/s41467-021-22471-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Tesfay L, Paul BT, Konstorum A, et al. Stearoyl‐CoA desaturase 1 protects ovarian cancer cells from ferroptotic cell death. Cancer Res. 2019;79:5355‐5366. 10.1158/0008-5472.CAN-19-0369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Thürmer M, Gollowitzer A, Pein H, et al. PI(18:1/18:1) is a SCD1‐derived lipokine that limits stress signaling. Nat Commun. 2022;13:2982. 10.1038/s41467-022-30374-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Koeberle A, Pergola C, Shindou H, et al. Role of p38 mitogen‐activated protein kinase in linking stearoyl‐CoA desaturase‐1 activity with endoplasmic reticulum homeostasis. FASEB J. 2015;29:2439‐2449. 10.1096/fj.14-268474 [DOI] [PubMed] [Google Scholar]
- 105. Koeberle A, Shindou H, Harayama T, Yuki K, Shimizu T. Polyunsaturated fatty acids are incorporated into maturating male mouse germ cells by lysophosphatidic acid acyltransferase 3. FASEB J. 2012;26:169‐180. 10.1096/fj.11-184879 [DOI] [PubMed] [Google Scholar]
- 106. Yang WH, Ding CKC, Sun T, et al. The Hippo pathway effector TAZ regulates ferroptosis in renal cell carcinoma. Cell Rep. 2019;28:2501‐2508.e4. 10.1016/j.celrep.2019.07.107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Gao M, Monian P, Quadri N, Ramasamy R, Jiang X. Glutaminolysis and transferrin regulate ferroptosis. Mol Cell. 2015;59:298‐308. 10.1016/j.molcel.2015.06.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Xie Y, Zhu S, Song X, et al. The tumor suppressor p53 limits ferroptosis by blocking DPP4 activity. Cell Rep. 2017;20:1692‐1704. 10.1016/j.celrep.2017.07.055 [DOI] [PubMed] [Google Scholar]
- 109. Yang WH, Huang Z, Wu J, Ding CKC, Murphy SK, Chi JT. A TAZ‐ANGPTL4‐NOX2 axis regulates ferroptotic cell death and chemoresistance in epithelial ovarian cancer. Mol Cancer Res. 2020;18:79‐90. 10.1158/1541-7786.MCR-19-0691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Egolf S, Zou J, Anderson A, et al. MLL4 mediates differentiation and tumor suppression through ferroptosis. Sci Adv. 2021;7:eabj9141. 10.1126/sciadv.abj9141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Zhang Y, Shi J, Liu X, et al. BAP1 links metabolic regulation of ferroptosis to tumour suppression. Nature Cell Biol. 2018;20:1181‐1192. 10.1038/s41556-018-0178-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Li J, Cao F, Yin H, et al. Ferroptosis: past, present and future. Cell Death Dis. 2020;11:88. 10.1038/s41419-020-2298-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Friedmann Angeli JP, Xavier da Silva TN, Schilling B. CD8+ T cells PUF(A)ing the flames of cancer ferroptotic cell death. Cancer Cell. 2022;40:346‐348. 10.1016/j.ccell.2022.03.003 [DOI] [PubMed] [Google Scholar]
- 114. Luo X, Gong HB, Gao HY, et al. Oxygenated phosphatidylethanolamine navigates phagocytosis of ferroptotic cells by interacting with TLR2. Cell Death Differ. 2021;28:1971‐1989. 10.1038/s41418-020-00719-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Liu J, Xia X, Huang P. xCT: A critical molecule that links cancer metabolism to redox signaling. Mol Ther. 2020;28:2358‐2366. 10.1016/j.ymthe.2020.08.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Xia X, Fan X, Zhao M, Zhu P. The relationship between ferroptosis and tumors: a novel landscape for therapeutic approach. Curr Gene Ther. 2019;19:117‐124. 10.2174/1566523219666190628152137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Hou W, Xie Y, Song X, et al. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy. 2016;12:1425‐1428. 10.1080/15548627.2016.1187366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Yang WS, Stockwell BR. Synthetic lethal screening identifies compounds activating iron‐dependent, nonapoptotic cell death in oncogenic‐RAS‐harboring cancer cells. Chem Biol. 2008;15:234‐245. 10.1016/j.chembiol.2008.02.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Moloney JN, Cotter TG. ROS signalling in the biology of cancer. Semin Cell Dev Biol. 2018;80:50‐64. 10.1016/j.semcdb.2017.05.023 [DOI] [PubMed] [Google Scholar]
- 120. Singh A, Misra V, Thimmulappa RK, et al. Dysfunctional KEAP1‐NRF2 interaction in non‐small‐cell lung cancer. PLoS Med. 2006;3:e420. 10.1371/journal.pmed.0030420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Wang J, Luo B, Li X, et al. Inhibition of cancer growth in vitro and in vivo by a novel ROS‐modulating agent with ability to eliminate stem‐like cancer cells. Cell Death Dis. 2017;8:e2887. 10.1038/cddis.2017.272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Lee HY, Parkinson EI, Granchi C, et al. Reactive oxygen species synergize to potently and selectively induce cancer cell death. ACS Chem Biol. 2017;12:1416‐1424. 10.1021/acschembio.7b00015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Zhao F, WANG H, KUNDA P, CHEN X, LIU QL, LIU T. Artesunate exerts specific cytotoxicity in retinoblastoma cells via CD71. Oncol Rep. 2013;30:1473‐1482. 10.3892/or.2013.2574 [DOI] [PubMed] [Google Scholar]
- 124. Raggi C, Gammella E, Correnti M, et al. Dysregulation of iron metabolism in cholangiocarcinoma stem‐like cells. Sci Rep. 2017;7:17667. 10.1038/s41598-017-17804-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Manz DH, Blanchette NL, Paul BT, Torti FM, Torti SV. Iron and cancer: recent insights. Ann NY Acad Sci. 2016;1368:149‐161. 10.1111/nyas.13008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Xie C, Cen D, Ren Z, et al. FeS@BSA nanoclusters to enable H2S‐amplified ROS‐based therapy with MRI guidance. Adv Sci. 2020;7:1903512. 10.1002/advs.201903512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Gorrini C, Harris IS, Mak TW. Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov. 2013;12:931‐947. 10.1038/nrd4002 [DOI] [PubMed] [Google Scholar]
- 128. Zou Z, Chang H, Li H, Wang S. Induction of reactive oxygen species: an emerging approach for cancer therapy. Apoptosis. 2017;22:1321‐1335. 10.1007/s10495-017-1424-9 [DOI] [PubMed] [Google Scholar]
- 129. Chen Q, Liu L, Lu Y, et al. Tumor microenvironment‐triggered aggregated magnetic nanoparticles for reinforced image‐guided immunogenic chemotherapy. Adv Sci. 2019;6:1802134. 10.1002/advs.201802134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Ye J, Zhang R, Wu F, et al. Non‐apoptotic cell death in malignant tumor cells and natural compounds. Cancer Lett. 2018;420:210‐227. 10.1016/j.canlet.2018.01.061 [DOI] [PubMed] [Google Scholar]
- 131. Tiribelli M, Latagliata R, Luciano L, et al. Impact of BCR‐ABL mutations on response to dasatinib after imatinib failure in elderly patients with chronic‐phase chronic myeloid leukemia. Ann Hematol. 2013;92:179‐183. 10.1007/s00277-012-1591-2 [DOI] [PubMed] [Google Scholar]
- 132. Panieri E, Saso L. Potential applications of NRF2 inhibitors in cancer therapy. Oxid Med Cell Longev. 2019;2019:1‐34. 10.1155/2019/8592348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Xie Y, Hou W, Song X, et al. Ferroptosis: process and function. Cell Death Differ. 2016;23:369‐379. 10.1038/cdd.2015.158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Gehringer M, Laufer SA. Emerging and Re‐Emerging warheads for targeted covalent inhibitors: applications in medicinal chemistry and chemical biology. J Med Chem. 2019;62:5673‐5724. 10.1021/acs.jmedchem.8b01153 [DOI] [PubMed] [Google Scholar]
- 135. Stockwell BR, Jiang X. The chemistry and biology of ferroptosis. Cell Chem Biol. 2020;27:365‐375. 10.1016/j.chembiol.2020.03.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Jones DP, Go YM. Mapping the cysteine proteome: analysis of redox‐sensing thiols. Curr Opin Chem Biol. 2011;15:103‐112. 10.1016/j.cbpa.2010.12.014 [DOI] [PubMed] [Google Scholar]
- 137. Atanasov AG, Zotchev SB, Dirsch VM, Supuran CT. Natural products in drug discovery: advances and opportunities. Nat Rev Drug Discov. 2021;20:200‐216. 10.1038/s41573-020-00114-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Rojo de la Vega M, Chapman E, Zhang DD. NRF2 and the hallmarks of cancer. Cancer Cell. 2018;34:21‐43. 10.1016/j.ccell.2018.03.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Okuno S, Sato H, Kuriyama‐Matsumura K, et al. Role of cystine transport in intracellular glutathione level and cisplatin resistance in human ovarian cancer cell lines. Br J Cancer. 2003;88:951‐956. 10.1038/sj.bjc.6600786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Buckingham SC, Campbell SL, Haas BR, et al. Glutamate release by primary brain tumors induces epileptic activity. Nat Med. 2011;17:1269‐1274. 10.1038/nm.2453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Ishimoto T, Nagano O, Yae T, et al. CD44 variant regulates redox status in cancer cells by stabilizing the xCT subunit of system Xc − and thereby promotes tumor growth. Cancer Cell. 2011;19:387‐400. 10.1016/j.ccr.2011.01.038 [DOI] [PubMed] [Google Scholar]
- 142. Yae T, Tsuchihashi K, Ishimoto T, et al. Alternative splicing of CD44 mRNA by ESRP1 enhances lung colonization of metastatic cancer cell. Nat Commun. 2012;3:883. 10.1038/ncomms1892 [DOI] [PubMed] [Google Scholar]
- 143. Timmerman LA, Holton T, Yuneva M, et al. Glutamine sensitivity analysis identifies the xCT antiporter as a common triple‐negative breast tumor therapeutic target. Cancer Cell. 2013;24:450‐465. 10.1016/j.ccr.2013.08.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Bridges RJ, Natale NR, Patel SA. System Xc − cystine/glutamate antiporter: an update on molecular pharmacology and roles within the CNS. Br J Pharmacol. 2012;165:20‐34. 10.1111/j.1476-5381.2011.01480.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Matti AA, Mirzaei J, Rudolph J, et al. Microwave accelerated synthesis of isoxazole hydrazide inhibitors of the system Xc − transporter: initial homology model. Bioorg Med Chem Lett. 2013;23:5931‐5935. 10.1016/j.bmcl.2013.08.080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Newell JL, Keyari CM, McDaniel SW, et al. Novel di‐aryl‐substituted isoxazoles act as noncompetitive inhibitors of the system Xc − cystine/glutamate exchanger. Neurochem Int. 2014;73:132‐138. 10.1016/j.neuint.2013.11.012 [DOI] [PubMed] [Google Scholar]
- 147. Lewerenz J, Hewett SJ, Huang Y, et al. The cystine/glutamate antiporter system Xc − in health and disease: from molecular mechanisms to novel therapeutic opportunities. Antioxid Redox Signal. 2013;18:522‐555. 10.1089/ars.2011.4391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Bridges R, Stevens D, Kahle J, Nunn P, Kadri M, Cotman C. Structure‐function studies on N‐oxalyl‐diamino‐dicarboxylic acids and excitatory amino acid receptors: evidence that beta‐l‐ODAP is a selective non‐NMDA agonist. J Neurosci. 1989;9:2073‐2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Warren BA, Patel SA, Nunn PB, Bridges RJ. The lathyrus excitotoxin β‐N‐oxalyl‐l‐α,β‐diaminopropionic acid is a substrate of the l‐cystine/l‐glutamate exchanger system Xc − . Toxicol Appl Pharmacol. 2004;200:83‐92. 10.1016/j.taap.2004.04.001 [DOI] [PubMed] [Google Scholar]
- 150. Huang Y, Dai Z, Barbacioru C, Sadée W. Cystine‐glutamate transporter SLC7A11 in cancer chemosensitivity and chemoresistance. Cancer Res. 2005;65:7446‐7454. 10.1158/0008-5472.CAN-04-4267 [DOI] [PubMed] [Google Scholar]
- 151. Walpole CSJ, Bevan S, Bovermann G, et al. The discovery of capsazepine, the first competitive antagonist of the sensory neuron excitants capsaicin and resiniferatoxin. J Med Chem. 1994;37:1942‐1954. 10.1021/jm00039a006 [DOI] [PubMed] [Google Scholar]
- 152. Fazzari J, Balenko M, Zacal N, Singh G. Identification of capsazepine as a novel inhibitor of system xc(‐) and cancer‐induced bone pain. J Pain Res. 2017;10:915‐925. 10.2147/JPR.S125045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Gout P, Buckley A, Simms C, Bruchovsky N. Sulfasalazine, a potent suppressor of lymphoma growth by inhibition of the x(c)‐ cystine transporter: a new action for an old drug. Leukemia. 2001;15:1633‐1640. 10.1038/sj.leu.2402238 [DOI] [PubMed] [Google Scholar]
- 154. Shukla K, Thomas AG, Ferraris DV, et al. Inhibition of transporter‐mediated cystine uptake by sulfasalazine analogs. Bioorg Med Chem Lett. 2011;21:6184‐6187. 10.1016/j.bmcl.2011.07.081 [DOI] [PubMed] [Google Scholar]
- 155. Nehser M, Dark J, Schweitzer D, et al. System Xc − antiporter inhibitors: azo‐linked amino‐naphthyl‐sulfonate analogues of sulfasalazine. Neurochem Res. 2020;45:1375‐1386. 10.1007/s11064-019-02901-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Zuo S, Yu J, Pan H, Lu L. Novel insights on targeting ferroptosis in cancer therapy. Biomark Res. 2020;8:50. 10.1186/s40364-020-00229-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Li Y, Yan H, Xu X, Liu H, Wu C, Zhao L. Erastin/sorafenib induces cisplatin‐resistant non‐small cell lung cancer cell ferroptosis through inhibition of the Nrf2/xCT pathway. Oncol Lett. 2019;19:323‐333. 10.3892/ol.2019.11066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Sato M, Kusumi R, Hamashima S, et al. The ferroptosis inducer erastin irreversibly inhibits system Xc − and synergizes with cisplatin to increase cisplatin's cytotoxicity in cancer cells. Sci Rep. 2018;8:968. 10.1038/s41598-018-19213-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Dolma S, Lessnick SL, Hahn WC, Stockwell BR. Identification of genotype‐selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell. 2003;3:285‐296. 10.1016/s1535-6108(03)00050-3 [DOI] [PubMed] [Google Scholar]
- 160. Wilhelm SM, Carter C, Tang L, et al. BAY 43‐9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004;64:7099‐7109. 10.1158/0008-5472.CAN-04-1443 [DOI] [PubMed] [Google Scholar]
- 161. Zhang Y, Zhao S, Fu Y, et al. Computational repositioning of dimethyl fumarate for treating alcoholic liver disease. Cell Death Dis. 2020;11:641. 10.1038/s41419-020-02890-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Buchstaller HP, Burgdorf L, Finsinger D, et al. Design and synthesis of isoquinolines and benzimidazoles as RAF kinase inhibitors. Bioorg Med Chem Lett. 2011;21:2264‐2269. 10.1016/j.bmcl.2011.02.108 [DOI] [PubMed] [Google Scholar]
- 163. Chen JN, Li T, Cheng L, et al. Synthesis and in vitro anti‐bladder cancer activity evaluation of quinazolinyl‐arylurea derivatives. Eur J Med Chem. 2020;205:112661. 10.1016/j.ejmech.2020.112661 [DOI] [PubMed] [Google Scholar]
- 164. Taylor WR, Fedorka SR, Gad I, et al. Small‐molecule ferroptotic agents with potential to selectively target cancer stem cells. Sci Rep. 2019;9:5926. 10.1038/s41598-019-42251-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Kobayashi S, Sato M, Kasakoshi T, et al. Cystathionine is a novel substrate of cystine/glutamate transporter. J Biol Chem. 2015;290:8778‐8788. 10.1074/jbc.M114.625053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Brigelius‐Flohé R, Maiorino M. Glutathione peroxidases. Biochim Biophys Acta. 2013;1830:3289‐3303. 10.1016/j.bbagen.2012.11.020. [DOI] [PubMed] [Google Scholar]
- 167. Mannes AM, Seiler A, Bosello V, Maiorino M, Conrad M. Cysteine mutant of mammalian GPx4 rescues cell death induced by disruption of the wild‐type selenoenzyme. FASEB J. 2011;25:2135‐2144. 10.1096/fj.10-177147 [DOI] [PubMed] [Google Scholar]
- 168. Brigelius‐Flohé R. Tissue‐specific functions of individual glutathione peroxidases. Free Radic Biol Med. 1999;27:951‐965. 10.1016/s0891-5849(99)00173-2 [DOI] [PubMed] [Google Scholar]
- 169. Scheerer P, Borchert A, Krauss N, et al. Structural basis for catalytic activity and enzyme polymerization of phospholipid hydroperoxide glutathione peroxidase‐4 (GPx4). Biochemistry. 2007;46:9041‐9049. 10.1021/bi700840d [DOI] [PubMed] [Google Scholar]
- 170. Flohé L, Toppo S, Cozza G, Ursini F. A comparison of thiol peroxidase mechanisms. Antioxid Redox Signal. 2011;15:763‐780. 10.1089/ars.2010.3397 [DOI] [PubMed] [Google Scholar]
- 171. Li C, Deng X, Xie X, Liu Y, Friedmann Angeli JP, Lai L. Activation of glutathione peroxidase 4 as a novel anti‐inflammatory strategy. Front Pharmacol. 2018;9:1120. 10.3389/fphar.2018.01120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Li C, Deng X, Zhang W, et al. Novel allosteric activators for ferroptosis regulator glutathione peroxidase 4. J Med Chem. 2019;62:266‐275. 10.1021/acs.jmedchem.8b00315 [DOI] [PubMed] [Google Scholar]
- 173. Weïwer M, Bittker JA, Lewis TA, et al. Development of small‐molecule probes that selectively kill cells induced to express mutant RAS. Bioorg Med Chem Lett. 2012;22:1822‐1826. 10.1016/j.bmcl.2011.09.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Shimada K, Skouta R, Kaplan A, et al. Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat Chem Biol. 2016;12:497‐503. 10.1038/nchembio.2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Eaton JK, Furst L, Cai LL, Viswanathan VS, Schreiber SL. Structure‐activity relationships of GPX4 inhibitor warheads. Bioorg Med Chem Lett. 2020;30:127538. 10.1016/j.bmcl.2020.127538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Eaton JK, Ruberto RA, Kramm A, Viswanathan VS, Schreiber SL. Diacylfuroxans are masked nitrile oxides that inhibit GPX4 covalently. J Am Chem Soc. 2019;141:20407‐20415. 10.1021/jacs.9b10769 [DOI] [PubMed] [Google Scholar]
- 177. Moosmayer D, Hilpmann A, Hoffmann J, et al. Crystal structures of the selenoprotein glutathione peroxidase 4 in its apo form and in complex with the covalently bound inhibitor ML162. Acta Crystallogr D Struct Biol. 2021;77:237‐248. 10.1107/S2059798320016125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Liu H, Forouhar F, Lin AJ, et al. Small‐molecule allosteric inhibitors of GPX4. Cell Chem Biol. 2022;29:1680‐1693. 10.1016/j.chembiol.2022.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Liu SJ, Zhao Q, Peng C, et al. Design, synthesis, and biological evaluation of nitroisoxazole‐containing spiro[pyrrolidin‐oxindole] derivatives as novel glutathione peroxidase 4/mouse double minute 2 dual inhibitors that inhibit breast adenocarcinoma cell proliferation. Eur J Med Chem. 2021;217:113359. 10.1016/j.ejmech.2021.113359 [DOI] [PubMed] [Google Scholar]
- 180. Sakamoto K, Sogabe S, Kamada Y, et al. Discovery of GPX4 inhibitory peptides from random peptide T7 phage display and subsequent structural analysis. Biochem Biophys Res Commun. 2017;482:195‐201. 10.1016/j.bbrc.2016.11.035 [DOI] [PubMed] [Google Scholar]
- 181. Ding Y, Chen X, Liu C, et al. Identification of a small molecule as inducer of ferroptosis and apoptosis through ubiquitination of GPX4 in triple negative breast cancer cells. J Hematol Oncol. 2021;14:19. 10.1186/s13045-020-01016-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Kim CY, Kang B, Suh HJ, Choi HS. Parthenolide, a feverfew‐derived phytochemical, ameliorates obesity and obesity‐induced inflammatory responses via the Nrf2/Keap1 pathway. Pharmacol Res. 2019;145:104259. 10.1016/j.phrs.2019.104259 [DOI] [PubMed] [Google Scholar]
- 183. Wu RP, Hayashi T, Cottam HB, et al. Nrf2 responses and the therapeutic selectivity of electrophilic compounds in chronic lymphocytic leukemia. Proc Natl Acad Sci USA. 2010;107:7479‐7484. 10.1073/pnas.1002890107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Duan D, Zhang J, Yao J, Liu Y, Fang J. Targeting thioredoxin reductase by parthenolide contributes to inducing apoptosis of HeLa cells. J Biol Chem. 2016;291:10021‐10031. 10.1074/jbc.M115.700591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Mandal PK, Seiler A, Perisic T, et al. System x(c)‐ and thioredoxin reductase 1 cooperatively rescue glutathione deficiency. J Biol Chem. 2010;285:22244‐22253. 10.1074/jbc.M110.121327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Woo JH, Shimoni Y, Yang WS, et al. Elucidating compound mechanism of action by network perturbation analysis. Cell. 2015;162:441‐451. 10.1016/j.cell.2015.05.056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Hassannia B, Logie E, Vandenabeele P, Vanden Berghe T, Vanden Berghe W. Withaferin A: from ayurvedic folk Medicine to preclinical anti‐cancer drug. Biochem Pharmacol. 2020;173:113602. 10.1016/j.bcp.2019.08.004 [DOI] [PubMed] [Google Scholar]
- 188. Hassannia B, Wiernicki B, Ingold I, et al. Nano‐targeted induction of dual ferroptotic mechanisms eradicates high‐risk neuroblastoma. J Clin Invest. 2018;128:3341‐3355. 10.1172/JCI99032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Dom M, Vanden Berghe W, Van Ostade X. Broad‐spectrum antitumor properties of withaferin A: a proteomic perspective. RSC Med Chem. 2020;11:30‐50. 10.1039/c9md00296k [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Dodson M, Castro‐Portuguez R, Zhang DD. NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol. 2019;23:101107. 10.1016/j.redox.2019.101107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Chiang SK, Chen SE, Chang LC. A dual role of heme oxygenase‐1 in cancer cells. Int J Mol Sci. 2018;20:39. 10.3390/ijms20010039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Luu Hoang KN, Anstee JE, Arnold JN. The diverse roles of heme oxygenase‐1 in tumor progression. Front Immunol. 2021;12:658315. 10.3389/fimmu.2021.658315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Kwon MY, Park E, Lee SJ, Chung SW. Heme oxygenase‐1 accelerates erastin‐induced ferroptotic cell death. Oncotarget. 2015;6:24393‐24403. 10.18632/oncotarget.5162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Liu XJ, Lv YF, Cui WZ, et al. Icariin inhibits hypoxia/reoxygenation‐induced ferroptosis of cardiomyocytes via regulation of the Nrf2/HO‐1 signaling pathway. FEBS Open Bio. 2021;11:2966‐2976. 10.1002/2211-5463.13276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Wu A, Feng B, Yu J, et al. Fibroblast growth factor 21 attenuates iron overload‐induced liver injury and fibrosis by inhibiting ferroptosis. Redox Biol. 2021;46:102131. 10.1016/j.redox.2021.102131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Koeberle A, Werz O. Multi‐target approach for natural products in inflammation. Drug Discov Today. 2014;19:1871‐1882. 10.1016/j.drudis.2014.08.006 [DOI] [PubMed] [Google Scholar]
- 197. Li R, Zhang J, Zhou Y, et al. Transcriptome investigation and in vitro verification of curcumin‐induced HO‐1 as a feature of ferroptosis in breast cancer cells. Oxid Med Cell Longev. 2020;2020:1‐18. 10.1155/2020/3469840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Lin H, Chen X, Zhang C, et al. EF24 induces ferroptosis in osteosarcoma cells through HMOX1. Biomed Pharmacother. 2021;136:111202. 10.1016/j.biopha.2020.111202 [DOI] [PubMed] [Google Scholar]
- 199. Chen TC, Chuang JY, Ko CY, et al. AR ubiquitination induced by the curcumin analog suppresses growth of temozolomide‐resistant glioblastoma through disrupting GPX4‐mediated redox homeostasis. Redox Biol. 2020;30:101413. 10.1016/j.redox.2019.101413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Golovine KV, Makhov PB, Teper E, et al. Piperlongumine induces rapid depletion of the androgen receptor in human. Prostate. 2013;73:23‐30. 10.1002/pros.22535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Kohli M, Ho Y, Hillman DW, et al. Androgen receptor variant AR‐V9 is coexpressed with AR‐V7 in prostate cancer metastases and predicts abiraterone resistance. Clin Cancer Res. 2017;23:4704‐4715. 10.1158/1078-0432.CCR-17-0017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Qin Z, Ou S, Xu L, et al. Design and synthesis of isothiocyanate‐containing hybrid androgen receptor (AR) antagonist to downregulate AR and induce ferroptosis in GSH‐deficient prostate cancer cells. Chem Biol Drug Des. 2021;97:1059‐1078. 10.1111/cbdd.13826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Yang F, Xiao Y, Ding JH, et al. Ferroptosis heterogeneity in triple‐negative breast cancer reveals an innovative immunotherapy combination strategy. Cell Metab. 2023;35:84‐100. 10.1016/j.cmet.2022.09.021 [DOI] [PubMed] [Google Scholar]
- 204. Chang LC, Chiang SK, Chen SE, Yu YL, Chou RH, Chang WC. Heme oxygenase‐1 mediates BAY 11‐7085 induced ferroptosis. Cancer Lett. 2018;416:124‐137. 10.1016/j.canlet.2017.12.025 [DOI] [PubMed] [Google Scholar]
- 205. Shin JW, Chun KS, Kim DH, et al. Curcumin induces stabilization of Nrf2 protein through Keap1 cysteine modification. Biochem Pharmacol. 2020;173:113820. 10.1016/j.bcp.2020.113820 [DOI] [PubMed] [Google Scholar]
- 206. Dinkova‐Kostova AT, Fahey JW, Kostov RV, Kensler TW. KEAP1 and done? Targeting the NRF2 pathway with sulforaphane. Trends Food Sci Technol. 2017;69:257‐269. 10.1016/j.tifs.2017.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Zhang Y, Talalay P, Cho CG, Posner GH. A major inducer of anticarcinogenic protective enzymes from broccoli: isolation and elucidation of structure. Proc Natl Acad Sci USA. 1992;89:2399‐2403. 10.1073/pnas.89.6.2399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Morroni F, Tarozzi A, Sita G, et al. Neuroprotective effect of sulforaphane in 6‐hydroxydopamine‐lesioned mouse model of Parkinson's disease. Neurotoxicology. 2013;36:63‐71. 10.1016/j.neuro.2013.03.004 [DOI] [PubMed] [Google Scholar]
- 209. Liu Y, Hettinger CL, Zhang D, Rezvani K, Wang X, Wang H. Sulforaphane enhances proteasomal and autophagic activities in mice and is a potential therapeutic reagent for Huntington's disease. J Neurochem. 2014;129:539‐547. 10.1111/jnc.12647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Kim HV, Kim HY, Ehrlich HY, Choi SY, Kim DJ, Kim Y. Amelioration of Alzheimer's disease by neuroprotective effect of sulforaphane in animal model. Amyloid. 2013;20:7‐12. 10.3109/13506129.2012.751367 [DOI] [PubMed] [Google Scholar]
- 211. Alfieri A, Srivastava S, Siow RCM, et al. Sulforaphane preconditioning of the Nrf2/HO‐1 defense pathway protects the cerebral vasculature against blood‐brain barrier disruption and neurological deficits in stroke. Free Radic Biol Med. 2013;65:1012‐1022. 10.1016/j.freeradbiomed.2013.08.190 [DOI] [PubMed] [Google Scholar]
- 212. Iida Y, Okamoto‐Κatsuyama M, Maruoka S, et al. Effective ferroptotic small‐cell lung cancer cell death from SLC7A11 inhibition by sulforaphane. Oncol Lett. 2020;21:71. 10.3892/ol.2020.12332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Clarke JD, Hsu A, Yu Z, Dashwood RH, Ho E. Differential effects of sulforaphane on histone deacetylases, cell cycle arrest and apoptosis in normal prostate cells versus hyperplastic and cancerous prostate cells. Mol Nutr Food Res. 2011;55:999‐1009. 10.1002/mnfr.201000547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Jabbarzadeh Kaboli P, Afzalipour Khoshkbejari M, Mohammadi M, et al. Targets and mechanisms of sulforaphane derivatives obtained from cruciferous plants with special focus on breast cancer—contradictory effects and future perspectives. Biomed Pharmacother. 2020;121:109635. 10.1016/j.biopha.2019.109635 [DOI] [PubMed] [Google Scholar]
- 215. Kasukabe T, Honma Y, Okabe‐Kado J, Higuchi Y, Kato N, Kumakura S. Combined treatment with cotylenin A and phenethyl isothiocyanate induces strong antitumor activity mainly through the induction of ferroptotic cell death in human pancreatic cancer cells. Oncol Rep. 2016;36:968‐976. 10.3892/or.2016.4867 [DOI] [PubMed] [Google Scholar]
- 216. Sant'Ana M, Souza HR, Possebon L, et al. Effect of piperlongumine during exposure to cigarette smoke reduces inflammation and lung injury. Pulm Pharmacol Ther. 2020;61:101896. 10.1016/j.pupt.2020.101896 [DOI] [PubMed] [Google Scholar]
- 217. Wang H, Liu J, Gao G, Wu X, Wang X, Yang H. Protection effect of piperine and piperlonguminine from Piper longum L. alkaloids against rotenone‐induced neuronal injury. Brain Res. 2016;1639:214‐227. 10.1016/j.brainres.2015.07.029 [DOI] [PubMed] [Google Scholar]
- 218. Soundararajan P, Kim J. Anti‐carcinogenic glucosinolates in cruciferous vegetables and their antagonistic effects on prevention of cancers. Molecules. 2018;23:2983. 10.3390/molecules23112983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Lv H, Zhen C, Liu J, Shang P. PEITC triggers multiple forms of cell death by GSH‐iron‐ROS regulation in K7M2 murine osteosarcoma cells. Acta Pharmacol Sin. 2020;41:1119‐1132. 10.1038/s41401-020-0376-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Chen P, Wu Q, Feng J, et al. Erianin, a novel dibenzyl compound in dendrobium extract, inhibits lung cancer cell growth and migration via calcium/calmodulin‐dependent ferroptosis. Signal Transduct Target Ther. 2020;5:51. 10.1038/s41392-020-0149-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Chen MF, Liou SS, Kao ST, Liu IM. Erianin protects against high glucose‐induced oxidative injury in renal tubular epithelial cells. Food Chem Toxicol. 2019;126:97‐105. 10.1016/j.fct.2019.02.021 [DOI] [PubMed] [Google Scholar]
- 222. Xiang Y, Chen X, Wang W, et al. Natural product erianin inhibits bladder cancer cell growth by inducing ferroptosis via NRF2 inactivation. Front Pharmacol. 2021;12:775506. 10.3389/fphar.2021.775506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Yang A, Sun Z, Liu R, et al. Transferrin‐conjugated erianin‐loaded liposomes suppress the growth of liver cancer by modulating oxidative stress. Front Oncol. 2021;11:727605. 10.3389/fonc.2021.727605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Wolfram T, Weidenbach LM, Adolf J, et al. The trace element selenium is important for redox signaling in phorbol ester‐differentiated THP‐1 macrophages. Int J Mol Sci. 2021;22:11060. 10.3390/ijms222011060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Chen P, Li X, Zhang R, et al. Combinative treatment of β‐elemene and cetuximab is sensitive to KRAS mutant colorectal cancer cells by inducing ferroptosis and inhibiting epithelial‐mesenchymal transformation. Theranostics. 2020;10:5107‐5119. 10.7150/thno.44705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Zhai B, Zeng Y, Zeng Z, et al. Drug delivery systems for elemene, its main active ingredient beta‐elemene, and its derivatives in cancer therapy. Int J Nanomedicine. 2018;13:6279‐6296. 10.2147/IJN.S174527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. Nguyen K, Khachemoune A. An update on topical photodynamic therapy for clinical dermatologists. J Dermatolog Treat. 2019;30:732‐744. 10.1080/09546634.2019.1569752 [DOI] [PubMed] [Google Scholar]
- 228. Nordmann NJ, Michael AP. 5‐Aminolevulinic acid radiodynamic therapy for treatment of high‐grade gliomas: a systematic review. Clin Neurol Neurosurg. 2021;201:106430. 10.1016/j.clineuro.2020.106430 [DOI] [PubMed] [Google Scholar]
- 229. Inoue K. 5‐Aminolevulinic acid‐mediated photodynamic therapy for bladder cancer. Int J Urol. 2017;24:97‐101. 10.1111/iju.13291 [DOI] [PubMed] [Google Scholar]
- 230. Shishido Y, Amisaki M, Matsumi Y, et al. Antitumor effect of 5‐Aminolevulinic acid through ferroptosis in esophageal squamous cell carcinoma. Ann Surg Oncol. 2020;28:3996‐4006. 10.1245/s10434-020-09334-4 [DOI] [PubMed] [Google Scholar]
- 231. Liu Z, Lv X, Yang B, Qin Q, Song E, Song Y. Tetrachlorobenzoquinone exposure triggers ferroptosis contributing to its neurotoxicity. Chemosphere. 2021;264:128413. 10.1016/j.chemosphere.2020.128413 [DOI] [PubMed] [Google Scholar]
- 232. Su C, Zhang P, Song X, et al. Tetrachlorobenzoquinone activates Nrf2 signaling by Keap1 cross‐linking and ubiquitin translocation but not Keap1‐Cullin3 complex dissociation. Chem Res Toxicol. 2015;28:765‐774. 10.1021/tx500513v [DOI] [PubMed] [Google Scholar]
- 233. Zhang J, Wang N, Zhou Y, et al. Oridonin induces ferroptosis by inhibiting gamma‐glutamyl cycle in TE1 cells. Phytother Res. 2021;35:494‐503. 10.1002/ptr.6829 [DOI] [PubMed] [Google Scholar]
- 234. Song M, Liu X, Liu K, et al. Targeting AKT with oridonin inhibits growth of esophageal squamous cell carcinoma in vitro and patient‐derived xenografts in vivo. Mol Cancer Ther. 2018;17:1540‐1553. 10.1158/1535-7163.MCT-17-0823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Kageyama S, Ii H, Taniguchi K, et al. Mechanisms of tumor growth inhibition by depletion of γ‐glutamylcyclotransferase (GGCT): a novel molecular target for anticancer therapy. Int J Mol Sci. 2018;19:2054. 10.3390/ijms19072054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236. Gao FH, LIU F, WEI W, et al. Oridonin induces apoptosis and senescence by increasing hydrogen peroxide and glutathione depletion in colorectal cancer cells. Int J Mol Med. 2012;29:649‐655. 10.3892/ijmm.2012.895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237. Liu QQ, Chen K, Ye Q, Jiang XH, Sun YW. Oridonin inhibits pancreatic cancer cell migration and epithelial‐mesenchymal transition by suppressing Wnt/β‐catenin signaling pathway. Cancer Cell Int. 2016;16:57. 10.1186/s12935-016-0336-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238. Liu Y, Song Z, Liu Y, et al. Identification of ferroptosis as a novel mechanism for antitumor activity of natural product derivative a2 in gastric cancer. Acta Pharm Sin B. 2021;11:1513‐1525. 10.1016/j.apsb.2021.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239. Sun Y, Qiao Y, Liu Y, et al. ent‐kaurane diterpenoids induce apoptosis and ferroptosis through targeting redox resetting to overcome cisplatin resistance. Redox Biol. 2021;43:101977. 10.1016/j.redox.2021.101977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240. Yang H, Lv H, Li H, Ci X, Peng L. Oridonin protects LPS‐induced acute lung injury by modulating Nrf2‐mediated oxidative stress and Nrf2‐independent NLRP3 and NF‐κB pathways. Cell Commun Signal. 2019;17:62. 10.1186/s12964-019-0366-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241. González‐Burgos E, Carretero ME, Gómez‐Serranillos MP. Kaurane diterpenes from Sideritis spp. exert a cytoprotective effect against oxidative injury that is associated with modulation of the Nrf2 system. Phytochemistry. 2013;93:116‐123. 10.1016/j.phytochem.2013.03.017 [DOI] [PubMed] [Google Scholar]
- 242. Casas‐Grajales S, Reyes‐Gordillo K, Cerda‐García‐Rojas CM, Tsutsumi V, Lakshman MR, Muriel P. Rebaudioside A administration prevents experimental liver fibrosis: an in vivo and in vitro study of the mechanisms of action involved. JAT. 2019;39:1118‐1131. 10.1002/jat.3797 [DOI] [PubMed] [Google Scholar]
- 243. Guan Z, Chen J, Li X, Dong N. Tanshinone IIA induces ferroptosis in gastric cancer cells through p53‐mediated SLC7A11 down‐regulation. Biosci Rep. 2020;40:BSR20201807. 10.1042/BSR20201807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244. Dong X, Dong J, Peng G, Hou X, Wu G. Growth‐inhibiting and apoptosis‐inducing effects of tanshinone II A on human gastric carcinoma cells. J Huazhong Univ Sci Technol Med Sci. 2007;27:706‐709. 10.1007/s11596-007-0623-y [DOI] [PubMed] [Google Scholar]
- 245. Ni H, Ruan G, Sun C, et al. Tanshinone IIA inhibits gastric cancer cell stemness through inducing ferroptosis. Environ Toxicol. 2021;37:192‐200. 10.1002/tox.23388 [DOI] [PubMed] [Google Scholar]
- 246. Ahmed N, Escalona R, Leung D, Chan E, Kannourakis G. Tumour microenvironment and metabolic plasticity in cancer and cancer stem cells: perspectives on metabolic and immune regulatory signatures in chemoresistant ovarian cancer stem cells. Sem Cancer Biol. 2018;53:265‐281. 10.1016/j.semcancer.2018.10.002 [DOI] [PubMed] [Google Scholar]
- 247. He L, Liu YY, Wang K, et al. Tanshinone IIA protects human coronary artery endothelial cells from ferroptosis by activating the NRF2 pathway. Biochem Biophys Res Commun. 2021;575:1‐7. 10.1016/j.bbrc.2021.08.067 [DOI] [PubMed] [Google Scholar]
- 248. Tan S, Hou X, Mei L. Dihydrotanshinone I inhibits human glioma cell proliferation via the activation of ferroptosis. Oncol Lett. 2020;20:122. 10.3892/ol.2020.11980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249. Tsai SL, Suk FM, Wang CI, et al. Anti‐tumor potential of 15,16‐dihydrotanshinone I against breast adenocarcinoma through inducing G1 arrest and apoptosis. Biochem Pharmacol. 2007;74:1575‐1586. 10.1016/j.bcp.2007.08.009 [DOI] [PubMed] [Google Scholar]
- 250. Wang L, Hu T, Shen J, et al. Dihydrotanshinone I induced apoptosis and autophagy through caspase dependent pathway in colon cancer. Phytomedicine. 2015;22:1079‐1087. 10.1016/j.phymed.2015.08.009 [DOI] [PubMed] [Google Scholar]
- 251. Cheng R, Chen J, Wang Y, Ge Y, Huang Z, Zhang G. Dihydrotanshinone induces apoptosis of SGC7901 and MGC803 cells via activation of JNK and p38 signalling pathways. Pharm Biol. 2016;54:3019‐3025. 10.1080/13880209.2016.1199045 [DOI] [PubMed] [Google Scholar]
- 252. Lin YS, Shen YC, Wu CY, et al. Danshen improves survival of patients with breast cancer and dihydroisotanshinone I induces ferroptosis and apoptosis of breast cancer cells. Front Pharmacol. 2019;10:1226. 10.3389/fphar.2019.01226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253. Nizamutdinova IT, Lee GW, Son KH, et al. Tanshinone I effectively induces apoptosis in estrogen receptor‐positive (MCF‐7) and estrogen receptor‐negative (MDA‐MB‐231) breast cancer cells. Int J Oncol. 2008;33:485‐491. [PubMed] [Google Scholar]
- 254. Lin YY, Lee IY, Huang WS, et al. Danshen improves survival of patients with colon cancer and dihydroisotanshinone I inhibit the proliferation of colon cancer cells via apoptosis and skp2 signaling pathway. J Ethnopharmacol. 2017;209:305‐316. 10.1016/j.jep.2017.08.011 [DOI] [PubMed] [Google Scholar]
- 255. Tao S, Justiniano R, Zhang DD, Wondrak GT. The Nrf2‐inducers tanshinone I and dihydrotanshinone protect human skin cells and reconstructed human skin against solar simulated UV. Redox Biol. 2013;1:532‐541. 10.1016/j.redox.2013.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256. Wu RT, Chiang HC, Fu WC, Chien KY, Chung YM, Horng LY. Formosanin‐C, an immunomodulator with antitumor activity. Int J Immunopharmacol. 1990;12:777‐786. 10.1016/0192-0561(90)90042-l [DOI] [PubMed] [Google Scholar]
- 257. Zhou Y, Yang J, Chen C, et al. Polyphyllin ‐induced ferroptosis in MDA‐MB‐231 triple‐negative breast cancer cells can be protected against by KLF4‐mediated upregulation of xCT. Front Pharmacol. 2021;12:670224. 10.3389/fphar.2021.670224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258. Wang C, Huo X, Wang L, et al. Dioscin strengthens the efficiency of adriamycin in MCF‐7 and MCF‐7/ADR cells through autophagy induction: more than just down‐regulation of MDR1. Sci Rep. 2016;6:28403. 10.1038/srep28403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259. Kim EA, Jang JH, Lee YH, et al. Dioscin induces caspase‐independent apoptosis through activation of apoptosis‐inducing factor in breast cancer cells. Apoptosis. 2014;19:1165‐1175. 10.1007/s10495-014-0994-z [DOI] [PubMed] [Google Scholar]
- 260. Lin PL, Tang HH, Wu SY, Shaw NS, Su CL. Saponin formosanin C‐induced ferritinophagy and ferroptosis in human hepatocellular carcinoma cells. Antioxidants. 2020;9:682. 10.3390/antiox9080682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261. Xie Y, Chen G. Dioscin induces ferroptosis and synergistic cytotoxicity with chemotherapeutics in melanoma cells. Biochem Biophys Res Commun. 2021;557:213‐220. 10.1016/j.bbrc.2021.04.024 [DOI] [PubMed] [Google Scholar]
- 262. Song Z, Xiang X, Li J, et al. Ruscogenin induces ferroptosis in pancreatic cancer cells. Oncol Rep. 2019;43:516‐524. 10.3892/or.2019.7425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263. Terman A, Kurz T. Lysosomal iron, iron chelation, and cell death. Antioxid Redox Signal. 2013;18:888‐898. 10.1089/ars.2012.4885 [DOI] [PubMed] [Google Scholar]
- 264. Liu M, Hu C, Xu Q, et al. Methylseleninic acid activates Keap1/Nrf2 pathway via up‐regulating miR‐200a in human oesophageal squamous cell carcinoma cells. Biosci Rep. 2015;35:e00256. 10.1042/BSR20150092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265. Dong L, Yin L, Li R, et al. Dioscin alleviates lung ischemia/reperfusion injury by regulating FXR‐mediated oxidative stress, apoptosis, and inflammation. Eur J Pharmacol. 2021;908:174321. 10.1016/j.ejphar.2021.174321 [DOI] [PubMed] [Google Scholar]
- 266. Zhang Y, Xu Y, Qi Y, et al. Protective effects of dioscin against doxorubicin‐induced nephrotoxicity via adjusting FXR‐mediated oxidative stress and inflammation. Toxicology. 2017;378:53‐64. 10.1016/j.tox.2017.01.007 [DOI] [PubMed] [Google Scholar]
- 267. Zhao L, Tao X, Qi Y, Xu L, Yin L, Peng J. Protective effect of dioscin against doxorubicin‐induced cardiotoxicity via adjusting microRNA‐140‐5p‐mediated myocardial oxidative stress. Redox Biol. 2018;16:189‐198. 10.1016/j.redox.2018.02.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268. Song S, Chu L, Liang H, et al. Protective effects of dioscin against doxorubicin‐induced hepatotoxicity via regulation of Sirt1/FOXO1/NF‐κb signal. Front Pharmacol. 2019;10:1030. 10.3389/fphar.2019.01030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269. Jin M, Shi C, Li T, Wu Y, Hu C, Huang G. Solasonine promotes ferroptosis of hepatoma carcinoma cells via glutathione peroxidase 4‐induced destruction of the glutathione redox system. Biomed Pharmacother. 2020;129:110282. 10.1016/j.biopha.2020.110282 [DOI] [PubMed] [Google Scholar]
- 270. Heyninck K, Sabbe L, Chirumamilla CS, et al. Withaferin A induces heme oxygenase (HO‐1) expression in endothelial cells via activation of the Keap1/Nrf2 pathway. Biochem Pharmacol. 2016;109:48‐61. 10.1016/j.bcp.2016.03.026 [DOI] [PubMed] [Google Scholar]
- 271. Huang S, Cao B, Zhang J, et al. Induction of ferroptosis in human nasopharyngeal cancer cells by cucurbitacin B: molecular mechanism and therapeutic potential. Cell Death Dis. 2021;12:237. 10.1038/s41419-021-03516-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272. Park SY, Kim YH, Park G. Cucurbitacins attenuate microglial activation and protect from neuroinflammatory injury through Nrf2/ARE activation and STAT/NF‐κB inhibition. Neurosci Lett. 2015;609:129‐136. 10.1016/j.neulet.2015.10.022 [DOI] [PubMed] [Google Scholar]
- 273. Wen Y, Chen H, Zhang L, et al. Glycyrrhetinic acid induces oxidative/nitrative stress and drives ferroptosis through activating NADPH oxidases and iNOS, and depriving glutathione in triple‐negative breast cancer cells. Free Radic Biol Med. 2021;173:41‐51. 10.1016/j.freeradbiomed.2021.07.019 [DOI] [PubMed] [Google Scholar]
- 274. Wang Y, Chen Q, Shi C, Jiao F, Gong Z. Mechanism of glycyrrhizin on ferroptosis during acute liver failure by inhibiting oxidative stress. Mol Med Rep. 2019;20:4081‐4090. 10.3892/mmr.2019.10660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275. Peng XP, Li XH, Li Y, Huang XT, Luo ZQ. The protective effect of oleanolic acid on NMDA‐induced MLE‐12 cells apoptosis and lung injury in mice by activating SIRT1 and reducing NF‐κB acetylation. Int Immunopharmacol. 2019;70:520‐529. 10.1016/j.intimp.2019.03.018 [DOI] [PubMed] [Google Scholar]
- 276. Xiaofei J, Mingqing S, Miao S, et al. Oleanolic acid inhibits cervical cancer Hela cell proliferation through modulation of the ACSL4 ferroptosis signaling pathway. Biochem Biophys Res Commun. 2021;545:81‐88. 10.1016/j.bbrc.2021.01.028 [DOI] [PubMed] [Google Scholar]
- 277. Woo S, Seo S, Min K, et al. Corosolic acid induces non‐apoptotic cell death through generation of lipid reactive oxygen species production in human renal carcinoma caki cells. Int J Mol Sci. 2018;19:1309. 10.3390/ijms19051309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278. Gaschler MM, Hu F, Feng H, Linkermann A, Min W, Stockwell BR. Determination of the subcellular localization and mechanism of action of ferrostatins in suppressing ferroptosis. ACS Chem Biol. 2018;13:1013‐1020. 10.1021/acschembio.8b00199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279. Mbaveng AT, Chi GF, Bonsou IN, et al. N‐acetylglycoside of oleanolic acid (aridanin) displays promising cytotoxicity towards human and animal cancer cells, inducing apoptotic, ferroptotic and necroptotic cell death. Phytomedicine. 2020;76:153261. 10.1016/j.phymed.2020.153261 [DOI] [PubMed] [Google Scholar]
- 280. Mbaveng AT, Ndontsa BL, Kuete V, et al. A naturally occuring triterpene saponin ardisiacrispin B displayed cytotoxic effects in multi‐factorial drug resistant cancer cells via ferroptotic and apoptotic cell death. Phytomedicine. 2018;43:78‐85. 10.1016/j.phymed.2018.03.035 [DOI] [PubMed] [Google Scholar]
- 281. Wei G, Sun J, Luan W, et al. Natural product albiziabioside A conjugated with pyruvate dehydrogenase kinase inhibitor dichloroacetate to induce apoptosis‐ferroptosis‐M2‐TAMs polarization for combined cancer therapy. J Med Chem. 2019;62:8760‐8772. 10.1021/acs.jmedchem.9b00644 [DOI] [PubMed] [Google Scholar]
- 282. Wei G, Sun J, Hou Z, et al. Novel antitumor compound optimized from natural saponin albiziabioside A induced caspase‐dependent apoptosis and ferroptosis as a p53 activator through the mitochondrial pathway. Eur J Med Chem. 2018;157:759‐772. 10.1016/j.ejmech.2018.08.036 [DOI] [PubMed] [Google Scholar]
- 283. Wei G, Cui S, Luan W, et al. Natural product‐based design, synthesis and biological evaluation of albiziabioside A derivatives that selectively induce HCT116 cell death. Eur J Med Chem. 2016;113:92‐101. 10.1016/j.ejmech.2015.12.034 [DOI] [PubMed] [Google Scholar]
- 284. Qiu Y, Wan B, Liu G, et al. Nrf2 protects against seawater drowning‐induced acute lung injury via inhibiting ferroptosis. Respir Res. 2020;21:232. 10.1186/s12931-020-01500-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285. Wang M, Li S, Wang Y, Cheng H, Su J, Li Q. Gambogenic acid induces ferroptosis in melanoma cells undergoing epithelial‐to‐mesenchymal transition. Toxicol Appl Pharmacol. 2020;401:115110. 10.1016/j.taap.2020.115110 [DOI] [PubMed] [Google Scholar]
- 286. Hjortso M, Andersen M. The expression, function and targeting of haem oxygenase‐1 in cancer. Curr Cancer Drug Targets. 2014;14:337‐347. 10.2174/1568009614666140320111306 [DOI] [PubMed] [Google Scholar]
- 287. Chau LY. Heme oxygenase‐1: emerging target of cancer therapy. J Biomed Sci. 2015;22:22. 10.1186/s12929-015-0128-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288. Bolloskis MP, Carvalho FP, Loo G. Iron depletion in HCT116 cells diminishes the upregulatory effect of phenethyl isothiocyanate on heme oxygenase‐1. Toxicol Appl Pharmacol. 2016;297:22‐31. 10.1016/j.taap.2016.02.024 [DOI] [PubMed] [Google Scholar]
- 289. Suttner DM, Dennery PA. Reversal of HO‐1 related cytoprotection with increased expression is due to reactive iron. FASEB J. 1999;13:1800‐1809. 10.1096/fasebj.13.13.1800 [DOI] [PubMed] [Google Scholar]
- 290. Kim R, Hashimoto A, Markosyan N, et al. Ferroptosis of tumour neutrophils causes immune suppression in cancer. Nature. 2022;612:338‐346. 10.1038/s41586-022-05443-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291. Hsieh CH, Hsieh HC, Shih FH, et al. An innovative NRF2 nano‐modulator induces lung cancer ferroptosis and elicits an immunostimulatory tumor microenvironment. Theranostics. 2021;11:7072‐7091. 10.7150/thno.57803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292. Zhang H, Zhuo Y, Li D, et al. Dihydroartemisinin inhibits the growth of pancreatic cells by inducing ferroptosis and activating antitumor immunity. Eur J Pharmacol. 2022;926:175028. 10.1016/j.ejphar.2022.175028 [DOI] [PubMed] [Google Scholar]
- 293. Yamaguchi Y, Kasukabe T, Kumakura S. Piperlongumine rapidly induces the death of human pancreatic cancer cells mainly through the induction of ferroptosis. Int J Oncol. 2018;52:1011‐1022. 10.3892/ijo.2018.4259 [DOI] [PubMed] [Google Scholar]
- 294. Dhillon H, Mamidi S, McClean P, Reindl KM. Transcriptome analysis of piperlongumine‐treated human pancreatic cancer cells reveals involvement of oxidative stress and endoplasmic reticulum stress pathways. J Med Food. 2016;19:578‐585. 10.1089/jmf.2015.0152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295. Lee HN, Jin HO, Park JA, et al. Heme oxygenase‐1 determines the differential response of breast cancer and normal cells to piperlongumine. Mol Cells. 2015;38:327‐335. 10.14348/molcells.2015.2235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296. Zou P, Xia Y, Ji J, et al. Piperlongumine as a direct TrxR1 inhibitor with suppressive activity against gastric cancer. Cancer Lett. 2016;375:114‐126. 10.1016/j.canlet.2016.02.058 [DOI] [PubMed] [Google Scholar]
- 297. Zhang J, Zhang B, Li X, Han X, Liu R, Fang J. Small molecule inhibitors of mammalian thioredoxin reductase as potential anticancer agents: an update. Med Res Rev. 2019;39:5‐39. 10.1002/med.21507 [DOI] [PubMed] [Google Scholar]
- 298. Wang Y, Wang JW, Xiao X, et al. Piperlongumine induces autophagy by targeting p38 signaling. Cell Death Dis. 2013;4:e824. 10.1038/cddis.2013.358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299. Chen Y, Liu JM, Xiong XX, et al. Piperlongumine selectively kills hepatocellular carcinoma cells and preferentially inhibits their invasion via ROS‐ER‐MAPKs‐CHOP. Oncotarget. 2015;6:6406‐6421. 10.18632/oncotarget.3444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300. Saidu NEB, Kavian N, Leroy K, et al. Dimethyl fumarate, a two‐edged drug: current status and future directions. Med Res Rev. 2019;39:1923‐1952. 10.1002/med.21567 [DOI] [PubMed] [Google Scholar]
- 301. Schmitt A, Xu W, Bucher P, et al. Dimethyl fumarate induces ferroptosis and impairs NF‐κB/STAT3 signaling in DLBCL. Blood. 2021;138:871‐884. 10.1182/blood.2020009404 [DOI] [PubMed] [Google Scholar]
- 302. Blewett MM, Xie J, Zaro BW, et al. Chemical proteomic map of dimethyl fumarate‐sensitive cysteines in primary human T cells. Sci Signal. 2016;9:rs10. 10.1126/scisignal.aaf7694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303. Lehmann JCU, Listopad JJ, Rentzsch CU, et al. Dimethylfumarate induces immunosuppression via glutathione depletion and subsequent induction of heme oxygenase 1. J Invest Dermatol. 2007;127:835‐845. 10.1038/sj.jid.5700686 [DOI] [PubMed] [Google Scholar]
- 304. Singhal R, Mitta SR, Das NK, et al. HIF‐2α activation potentiates oxidative cell death in colorectal cancers by increasing cellular iron. J Clin Invest. 2021;131:e143691. 10.1172/JCI143691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305. Liao MH, Tsai YN, Yang CY, et al. Anti‐human hepatoma Hep‐G2 proliferative, apoptotic, and antimutagenic activity of tagitinin C from Tithonia diversifolia leaves. J Nat Med. 2013;67:98‐106. 10.1007/s11418-012-0652-0 [DOI] [PubMed] [Google Scholar]
- 306. Wei R, Zhao Y, Wang J, et al. Tagitinin C induces ferroptosis through PERK‐Nrf2‐HO‐1 signaling pathway in colorectal cancer cells. Int J Biol Sci. 2021;17:2703‐2717. 10.7150/ijbs.59404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307. Xu Y, Tang D, Wang J, Wei H, Gao J. Neuroprotection of andrographolide against microglia‐mediated inflammatory injury and oxidative damage in PC12 neurons. Neurochem Res. 2019;44:2619‐2630. 10.1007/s11064-019-02883-5 [DOI] [PubMed] [Google Scholar]
- 308. Li Y, Xiang LL, Miao JX, Miao MS, Wang C. Protective effects of andrographolide against cerebral ischemiareperfusion injury in mice. Int J Mol Med. 2021;48:186. 10.3892/ijmm.2021.5019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309. Sharma P, Shimura T, Banwait JK, Goel A. Andrographis‐mediated chemosensitization through activation of ferroptosis and suppression of β‐catenin/Wnt‐signaling pathways in colorectal cancer. Carcinogenesis. 2020;41:1385‐1394. 10.1093/carcin/bgaa090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310. Xiang DC, Yang JY, Xu YJ, et al. Protective effect of andrographolide on 5‐Fu induced intestinal mucositis by regulating p38 MAPK signaling pathway. Life Sci. 2020;252:117612. 10.1016/j.lfs.2020.117612 [DOI] [PubMed] [Google Scholar]
- 311. Xia Y, Liu S, Li C, et al. Discovery of a novel ferroptosis inducer‐talaroconvolutin A—killing colorectal cancer cells in vitro and in vivo. Cell Death Dis. 2020;11:988. 10.1038/s41419-020-03194-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312. Dodson M, de la Vega MR, Cholanians AB, Schmidlin CJ, Chapman E, Zhang DD. Modulating NRF2 in disease: timing is everything. Annu Rev Pharmacol Toxicol. 2019;59:555‐575. 10.1146/annurev-pharmtox-010818-021856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313. Zhao F, Ci X, Man X, Li J, Wei Z, Zhang S. Food‐derived pharmacological modulators of the Nrf2/ARE pathway: their role in the treatment of diseases. Molecules. 2021;26:1016. 10.3390/molecules26041016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314. Dayalan Naidu S, Dinkova‐Kostova AT. KEAP1, a cysteine‐based sensor and a drug target for the prevention and treatment of chronic disease. Open Biol. 2020;10:200105. 10.1098/rsob.200105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315. Ma Q, He X. Molecular basis of electrophilic and oxidative defense: promises and perils of Nrf2. Pharmacol Rev. 2012;64:1055‐1081. 10.1124/pr.110.004333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316. Cuadrado A, Rojo AI, Wells G, et al. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat Rev Drug Discov. 2019;18:295‐317. 10.1038/s41573-018-0008-x [DOI] [PubMed] [Google Scholar]
- 317. Cuadrado A, Manda G, Hassan A, et al. Transcription factor NRF2 as a therapeutic target for chronic diseases: a systems medicine approach. Pharmacol Rev. 2018;70:348‐383. 10.1124/pr.117.014753 [DOI] [PubMed] [Google Scholar]
- 318. Kumar H, Kim IS, More SV, Kim BW, Choi DK. Natural product‐derived pharmacological modulators of Nrf2/ARE pathway for chronic diseases. Nat Prod Rep. 2014;31:109‐139. 10.1039/c3np70065h [DOI] [PubMed] [Google Scholar]
- 319. Magesh S, Chen Y, Hu L. Small molecule modulators of Keap1‐Nrf2‐ARE pathway as potential preventive and therapeutic agents. Med Res Rev. 2012;32:687‐726. 10.1002/med.21257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320. Zhang DD, Chapman E. The role of natural products in revealing NRF2 function. Nat Prod Rep. 2020;37:797‐826. 10.1039/c9np00061e [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321. Singh A, Venkannagari S, Oh KH, et al. Small molecule inhibitor of NRF2 selectively intervenes therapeutic resistance in KEAP1‐deficient NSCLC tumors. ACS Chem Biol. 2016;11:3214‐3225. 10.1021/acschembio.6b00651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322. Singh A, Wu H, Zhang P, Happel C, Ma J, Biswal S. Expression of ABCG2 (BCRP) is regulated by Nrf2 in cancer cells that confers side population and chemoresistance phenotype. Mol Cancer Ther. 2010;9:2365‐2376. 10.1158/1535-7163.MCT-10-0108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323. Du Y, Bao J, Zhang M, et al. Targeting ferroptosis contributes to ATPR‐induced AML differentiation via ROS‐autophagy‐lysosomal pathway. Gene. 2020;755:144889. 10.1016/j.gene.2020.144889 [DOI] [PubMed] [Google Scholar]
- 324. Arlt A, Sebens S, Krebs S, et al. Inhibition of the Nrf2 transcription factor by the alkaloid trigonelline renders pancreatic cancer cells more susceptible to apoptosis through decreased proteasomal gene expression and proteasome activity. Oncogene. 2013;32:4825‐4835. 10.1038/onc.2012.493 [DOI] [PubMed] [Google Scholar]
- 325. Shin D, Kim EH, Lee J, Roh JL. Nrf2 inhibition reverses resistance to GPX4 inhibitor‐induced ferroptosis in head and neck cancer. Free Radic Biol Med. 2018;129:454‐462. 10.1016/j.freeradbiomed.2018.10.426 [DOI] [PubMed] [Google Scholar]
- 326. Sequist LV, Waltman BA, Dias‐Santagata D, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med. 2011;3:75ra26. 10.1126/scitranslmed.3002003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327. Hangauer MJ, Viswanathan VS, Ryan MJ, et al. Drug‐tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature. 2017;551:247‐250. 10.1038/nature24297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328. Tsoi J, Robert L, Paraiso K, et al. Multi‐stage differentiation defines melanoma subtypes with differential vulnerability to drug‐induced iron‐dependent oxidative stress. Cancer Cell. 2018;33:890‐904. 10.1016/j.ccell.2018.03.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329. Xu T, Ding W, Ji X, et al. Molecular mechanisms of ferroptosis and its role in cancer therapy. J Cell Mol Med. 2019;23:4900‐4912. 10.1111/jcmm.14511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330. Zhu HY, Huang ZX, Chen GQ, Sheng F, Zheng YS. Typhaneoside prevents acute myeloid leukemia (AML) through suppressing proliferation and inducing ferroptosis associated with autophagy. Biochem Biophys Res Commun. 2019;516:1265‐1271. 10.1016/j.bbrc.2019.06.070 [DOI] [PubMed] [Google Scholar]
- 331. Lou JS, Zhao LP, Huang ZH, et al. Ginkgetin derived from Ginkgo biloba leaves enhances the therapeutic effect of cisplatin via ferroptosis‐mediated disruption of the Nrf2/HO‐1 axis in EGFR wild‐type non‐small‐cell lung cancer. Phytomedicine. 2021;80:153370. 10.1016/j.phymed.2020.153370 [DOI] [PubMed] [Google Scholar]
- 332. Chen Y, Li N, Wang H, et al. Amentoflavone suppresses cell proliferation and induces cell death through triggering autophagy‐dependent ferroptosis in human glioma. Life Sci. 2020;247:117425. 10.1016/j.lfs.2020.117425 [DOI] [PubMed] [Google Scholar]
- 333. Bollong MJ, Yun H, Sherwood L, Woods AK, Lairson LL, Schultz PG. A small molecule inhibits deregulated NRF2 transcriptional activity in cancer. ACS Chem Biol. 2015;10:2193‐2198. 10.1021/acschembio.5b00448 [DOI] [PubMed] [Google Scholar]
- 334. Wenzel SE, Tyurina YY, Zhao J, et al. PEBP1 wardens ferroptosis by enabling lipoxygenase generation of lipid death signals. Cell. 2017;171:628‐641. 10.1016/j.cell.2017.09.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335. Conrad M, Friedmann Angeli JP. Glutathione peroxidase 4 (Gpx4) and ferroptosis: what's so special about it. Mol Cell Oncol. 2015;2:e995047. 10.4161/23723556.2014.995047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336. Bersuker K, Hendricks JM, Li Z, et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature. 2019;575:688‐692. 10.1038/s41586-019-1705-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337. Gaschler MM, Andia AA, Liu H, et al. FINO2 initiates ferroptosis through GPX4 inactivation and iron oxidation. Nat Chem Biol. 2018;14:507‐515. 10.1038/s41589-018-0031-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338. Guo J, Xu B, Han Q, et al. Ferroptosis: a novel anti‐tumor action for cisplatin. Cancer Res Treat. 2018;50:445‐460. 10.4143/crt.2016.572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339. Ma S, Henson ES, Chen Y, Gibson SB. Ferroptosis is induced following siramesine and lapatinib treatment of breast cancer cells. Cell Death Dis. 2016;7:e2307. 10.1038/cddis.2016.208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340. Nagpal A, Redvers RP, Ling X, et al. Neoadjuvant neratinib promotes ferroptosis and inhibits brain metastasis in a novel syngeneic model of spontaneous HER2(+ve) breast cancer metastasis. Breast Cancer Res. 2019;21:94. 10.1186/s13058-019-1177-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341. Zhao L, Peng Y, He S, et al. Apatinib induced ferroptosis by lipid peroxidation in gastric cancer. Gastric Cancer. 2021;24:642‐654. 10.1007/s10120-021-01159-8 [DOI] [PubMed] [Google Scholar]
- 342. Hou Y, Cai S, Yu S, Lin H. Metformin induces ferroptosis by targeting miR‐324‐3p/GPX4 axis in breast cancer. Acta Biochim Biophys Sin. 2021;53:333‐341. 10.1093/abbs/gmaa180 [DOI] [PubMed] [Google Scholar]
- 343. Xu Y, Wang Q, Li X, Chen Y, Xu G. Itraconazole attenuates the stemness of nasopharyngeal carcinoma cells via triggering ferroptosis. Environ Toxicol. 2021;36:257‐266. 10.1002/tox.23031 [DOI] [PubMed] [Google Scholar]
- 344. Su Y, Zhao B, Zhou L, et al. Ferroptosis, a novel pharmacological mechanism of anti‐cancer drugs. Cancer Lett. 2020;483:127‐136. 10.1016/j.canlet.2020.02.015 [DOI] [PubMed] [Google Scholar]
- 345. Elgendy SM, Alyammahi SK, Alhamad DW, Abdin SM, Omar HA. Ferroptosis: an emerging approach for targeting cancer stem cells and drug resistance. Crit Rev Oncol Hematol. 2020;155:103095. 10.1016/j.critrevonc.2020.103095 [DOI] [PubMed] [Google Scholar]
- 346. Skouta R, Dixon SJ, Wang J, et al. Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. J Am Chem Soc. 2014;136:4551‐4556. 10.1021/ja411006a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347. Abrams RP, Carroll WL, Woerpel KA. Five‐membered ring peroxide selectively initiates ferroptosis in cancer cells. ACS Chem Biol. 2016;11:1305‐1312. 10.1021/acschembio.5b00900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348. Efferth T. From ancient herb to modern drug: Artemisia annua and artemisinin for cancer therapy. Sem Cancer Biol. 2017;46:65‐83. 10.1016/j.semcancer.2017.02.009 [DOI] [PubMed] [Google Scholar]
- 349. Xu C, Zhang H, Mu L, Yang X. Artemisinins as anticancer drugs: novel therapeutic approaches, molecular mechanisms, and clinical trials. Front Pharmacol. 2020;11:529881. 10.3389/fphar.2020.529881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350. Wong YK, Xu C, Kalesh KA, et al. Artemisinin as an anticancer drug: recent advances in target profiling and mechanisms of action. Med Res Rev. 2017;37:1492‐1517. 10.1002/med.21446 [DOI] [PubMed] [Google Scholar]
- 351. Meng Y, Ma N, Lyu H, et al. Recent pharmacological advances in the repurposing of artemisinin drugs. Med Res Rev. 2021;41:3156‐3181. 10.1002/med.21837 [DOI] [PubMed] [Google Scholar]
- 352. Eling N, Reuter L, Hazin J, Hamacher‐Brady A, Brady NR. Identification of artesunate as a specific activator of ferroptosis in pancreatic cancer cells. Oncoscience. 2015;2:517‐532. 10.18632/oncoscience.160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353. Chen GQ, Benthani FA, Wu J, Liang D, Bian ZX, Jiang X. Artemisinin compounds sensitize cancer cells to ferroptosis by regulating iron homeostasis. Cell Death Differ. 2020;27:242‐254. 10.1038/s41418-019-0352-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354. Lin R, Zhang Z, Chen L, et al. Dihydroartemisinin (DHA) induces ferroptosis and causes cell cycle arrest in head and neck carcinoma cells. Cancer Lett. 2016;381:165‐175. 10.1016/j.canlet.2016.07.033 [DOI] [PubMed] [Google Scholar]
- 355. Ooko E, Saeed MEM, Kadioglu O, et al. Artemisinin derivatives induce iron‐dependent cell death (ferroptosis) in tumor cells. Phytomedicine. 2015;22:1045‐1054. 10.1016/j.phymed.2015.08.002 [DOI] [PubMed] [Google Scholar]
- 356. Roh JL, Kim EH, Jang H, Shin D. Nrf2 inhibition reverses the resistance of cisplatin‐resistant head and neck cancer cells to artesunate‐induced ferroptosis. Redox Biol. 2017;11:254‐262. 10.1016/j.redox.2016.12.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 357. Chan HW, Singh NP, Lai HC. Cytotoxicity of dihydroartemisinin toward Molt‐4 cells attenuated by N‐tert‐butyl‐alpha‐phenylnitrone and deferoxamine. Anticancer Res. 2013;33:4389‐4393. [PubMed] [Google Scholar]
- 358. Chen Y, Mi Y, Zhang X, et al. Dihydroartemisinin‐induced unfolded protein response feedback attenuates ferroptosis via PERK/ATF4/HSPA5 pathway in glioma cells. J Exp Clin Cancer Res. 2019;38:402. 10.1186/s13046-019-1413-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 359. Zhang B, Liu Y, Li X, Xu J, Fang J. Small molecules to target the selenoprotein thioredoxin reductase. Chem Asian J. 2018;13:3593‐3600. 10.1002/asia.201801136 [DOI] [PubMed] [Google Scholar]
- 360. Jia JJ, Geng WS, Wang ZQ, Chen L, Zeng XS. The role of thioredoxin system in cancer: strategy for cancer therapy. Cancer Chemother Pharmacol. 2019;84:453‐470. 10.1007/s00280-019-03869-4 [DOI] [PubMed] [Google Scholar]
- 361. Fang J, Seki T, Maeda H. Therapeutic strategies by modulating oxygen stress in cancer and inflammation. Adv Drug Deliv Rev. 2009;61:290‐302. 10.1016/j.addr.2009.02.005 [DOI] [PubMed] [Google Scholar]
- 362. Zou P, Xia Y, Chen W, et al. EF24 induces ROS‐mediated apoptosis via targeting thioredoxin reductase 1 in gastric cancer cells. Oncotarget. 2016;7:18050‐18064. 10.18632/oncotarget.7633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363. Jaganjac M, Milkovic L, Sunjic SB, Zarkovic N. The NRF2, thioredoxin, and glutathione system in tumorigenesis and anticancer therapies. Antioxidants. 2020;9:1151. 10.3390/antiox9111151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364. Biaglow JE, Miller RA. The thioredoxin reductase/thioredoxin system: novel redox targets for cancer therapy. Cancer Biol Ther. 2005;4:13‐20. 10.4161/cbt.4.1.1434 [DOI] [PubMed] [Google Scholar]
- 365. Mai TT, Hamaï A, Hienzsch A, et al. Salinomycin kills cancer stem cells by sequestering iron in lysosomes. Nat Chem. 2017;9:1025‐1033. 10.1038/nchem.2778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 366. Perez MA, Magtanong L, Dixon SJ, Watts JL. Dietary lipids induce ferroptosis in caenorhabditiselegans and human cancer cells. Dev Cell. 2020;54:447‐454. 10.1016/j.devcel.2020.06.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 367. Stamenkovic A, O'Hara KA, Nelson DC, et al. Oxidized phosphatidylcholines trigger ferroptosis in cardiomyocytes during ischemia‐reperfusion injury. Am J Physiol Heart Circ Physiol. 2021;320:H1170‐H1184. 10.1152/ajpheart.00237.2020 [DOI] [PubMed] [Google Scholar]
- 368. Yuan GF, Chen XE, Li D. Conjugated linolenic acids and their bioactivities: a review. Food Funct. 2014;5:1360‐1368. 10.1039/c4fo00037d [DOI] [PubMed] [Google Scholar]
- 369. Kohno H, Yasui Y, Suzuki R, Hosokawa M, Miyashita K, Tanaka T. Dietary seed oil rich in conjugated linolenic acid from bitter melon inhibits azoxymethane‐induced rat colon carcinogenesis through elevation of colonic PPAR? expression and alteration of lipid composition. Int J Cancer. 2004;110:896‐901. 10.1002/ijc.20179 [DOI] [PubMed] [Google Scholar]
- 370. Shabbir MA, Khan MR, Saeed M, Pasha I, Khalil AA, Siraj N. Punicic acid: A striking health substance to combat metabolic syndromes in humans. Lipids Health Dis. 2017;16:99. 10.1186/s12944-017-0489-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371. Salsinha AS, Pimentel LL, Fontes AL, Gomes AM, Rodríguez‐Alcalá LM. Microbial production of conjugated linoleic acid and conjugated linolenic acid relies on a multienzymatic system. Microbiol Mol Biol Rev. 2018;82:e00019. 10.1128/MMBR.00019-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 372. Shen LD, Qi WH, Bai JJ, et al. RETRACTED: resibufogenin inhibited colorectal cancer cell growth and tumorigenesis through triggering ferroptosis and ROS production mediated by GPX4 inactivation. Anat Rec. 2021;304:313‐322. 10.1002/ar.24378 [DOI] [PubMed] [Google Scholar]
- 373. Han Q, Ma Y, Wang H, et al. Resibufogenin suppresses colorectal cancer growth and metastasis through RIP3‐mediated necroptosis. J Transl Med. 2018;16:201. 10.1186/s12967-018-1580-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 374. Saikia M, Retnakumari AP, Anwar S, et al. Heteronemin, a marine natural product, sensitizes acute myeloid leukemia cells towards cytarabine chemotherapy by regulating farnesylation of Ras. Oncotarget. 2018;9:18115‐18127. 10.18632/oncotarget.24771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 375. Chang WT, Bow YD, Fu PJ, et al. A marine terpenoid, heteronemin, induces both the apoptosis and ferroptosis of hepatocellular carcinoma cells and involves the ROS and MAPK pathways. Oxid Med Cell Longev. 2021;2021:1‐12. 10.1155/2021/7689045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 376. Wang Z, Ding Y, Wang X, et al. Pseudolaric acid B triggers ferroptosis in glioma cells via activation of Nox4 and inhibition of xCT. Cancer Lett. 2018;428:21‐33. 10.1016/j.canlet.2018.04.021 [DOI] [PubMed] [Google Scholar]
- 377. Mbaveng AT, Fotso GW, Ngnintedo D, et al. Cytotoxicity of epunctanone and four other phytochemicals isolated from the medicinal plants Garcinia epunctata and Ptycholobium contortum towards multi‐factorial drug resistant cancer cells. Phytomedicine. 2018;48:112‐119. 10.1016/j.phymed.2017.12.016 [DOI] [PubMed] [Google Scholar]
- 378. Mbaveng AT, Bitchagno GTM, Kuete V, Tane P, Efferth T. Cytotoxicity of ungeremine towards multi‐factorial drug resistant cancer cells and induction of apoptosis, ferroptosis, necroptosis and autophagy. Phytomedicine. 2019;60:152832. 10.1016/j.phymed.2019.152832 [DOI] [PubMed] [Google Scholar]
- 379. Mbaveng AT, Noulala CGT, Samba ARM, et al. The alkaloid, soyauxinium chloride, displays remarkable cytotoxic effects towards a panel of cancer cells, inducing apoptosis, ferroptosis and necroptosis. Chem Biol Interact. 2021;333:109334. 10.1016/j.cbi.2020.109334 [DOI] [PubMed] [Google Scholar]
- 380. Ngo B, Van Riper JM, Cantley LC, Yun J. Targeting cancer vulnerabilities with high‐dose vitamin C. Nat Rev Cancer. 2019;19:271‐282. 10.1038/s41568-019-0135-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 381. Wang X, Xu S, Zhang L, et al. Vitamin C induces ferroptosis in anaplastic thyroid cancer cells by ferritinophagy activation. Biochem Biophys Res Commun. 2021;551:46‐53. 10.1016/j.bbrc.2021.02.126 [DOI] [PubMed] [Google Scholar]
- 382. Wang ZX, Ma J, Li XY, et al. Quercetin induces p53‐independent cancer cell death through lysosome activation by the transcription factor EB and reactive oxygen species‐dependent ferroptosis. Br J Pharmacol. 2021;178:1133‐1148. 10.1111/bph.15350 [DOI] [PubMed] [Google Scholar]
- 383. Adham AN, Abdelfatah S, Naqishbandi AM, Mahmoud N, Efferth T. Cytotoxicity of apigenin toward multiple myeloma cell lines and suppression of iNOS and COX‐2 expression in STAT1‐transfected HEK293 cells. Phytomedicine. 2021;80:153371. 10.1016/j.phymed.2020.153371 [DOI] [PubMed] [Google Scholar]
- 384. Xie Y, Zhou X, Li J, et al. Identification of a new natural biflavonoids against breast cancer cells induced ferroptosis via the mitochondrial pathway. Bioorg Chem. 2021;109:104744. 10.1016/j.bioorg.2021.104744 [DOI] [PubMed] [Google Scholar]
- 385. Yang Y, Luo M, Zhang K, et al. Nedd4 ubiquitylates VDAC2/3 to suppress erastin‐induced ferroptosis in melanoma. Nat Commun. 2020;11:433. 10.1038/s41467-020-14324-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 386. XunLi, Liu Y, Chu S, et al. Physcion and physcion 8‐O‐β‐glucopyranoside: a review of their pharmacology, toxicities and pharmacokinetics. Chem Biol Interact. 2019;310:108722. 10.1016/j.cbi.2019.06.035 [DOI] [PubMed] [Google Scholar]
- 387. Niu Y, Zhang J, Tong Y, Li J, Liu B. RETRACTED: physcion 8‐O‐β‐glucopyranoside induced ferroptosis via regulating miR‐103a‐3p/GLS2 axis in gastric cancer. Life Sci. 2019;237:116893. 10.1016/j.lfs.2019.116893 [DOI] [PubMed] [Google Scholar]
- 388. Luo M, Wu L, Zhang K, et al. miR‐137 regulates ferroptosis by targeting glutamine transporter SLC1A5 in melanoma. Cell Death Differ. 2018;25:1457‐1472. 10.1038/s41418-017-0053-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 389. Ocampo Y, Caro D, Rivera D, Piermattey J, Gaitán R, Franco LA. Transcriptome changes in colorectal cancer cells upon treatment with avicequinone B. Adv Pharm Bull. 2020;10:638‐647. 10.34172/apb.2020.077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 390. Hong Z, Tang P, Liu B, et al. Ferroptosis‐related genes for overall survival prediction in patients with colorectal cancer can be inhibited by gallic acid. Int J Biol Sci. 2021;17:942‐956. 10.7150/ijbs.57164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 391. Khorsandi K, Kianmehr Z, Hosseinmardi Z, Hosseinzadeh R. Anti‐cancer effect of gallic acid in presence of low level laser irradiation: ROS production and induction of apoptosis and ferroptosis. Cancer Cell Int. 2020;20:18. 10.1186/s12935-020-1100-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 392. Tang HM, Cheung PCK. Gallic acid triggers iron‐dependent cell death with apoptotic, ferroptotic, and necroptotic features. Toxins. 2019;11:492. 10.3390/toxins11090492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 393. Cheng YT, Yang CC, Shyur LF. Phytomedicine‐modulating oxidative stress and the tumor microenvironment for cancer therapy. Pharmacol Res. 2016;114:128‐143. 10.1016/j.phrs.2016.10.022 [DOI] [PubMed] [Google Scholar]
- 394. Proneth B, Conrad M. Ferroptosis and necroinflammation, a yet poorly explored link. Cell Death Differ. 2019;26:14‐24. 10.1038/s41418-018-0173-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 395. Conrad M, Kagan VE, Bayir H, et al. Regulation of lipid peroxidation and ferroptosis in diverse species. Genes Dev. 2018;32:602‐619. 10.1101/gad.314674.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 396. Aldrovandi M, Fedorova M, Conrad M. Juggling with lipids, a game of Russian roulette. Trends Endocrinol Metab. 2021;32:463‐473. 10.1016/j.tem.2021.04.012 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.