

Abstract
Solute-linked carrier 26 (SLC26) isoforms constitute a conserved family of anion transporters with 10 distinct members. Except for SLC26A5 (prestin), all can operate as multifunctional anion exchangers, with three members (SLC26A7, SLC26A9, and SLC26A11) also capable of functioning as chloride channels. Several SLC26 isoforms can specifically mediate Cl−/HCO3− exchange. These include SLC26A3, A4, A6, A7, A9, and A11, all of which are expressed in the kidney except for SLC26A3 (DRA), which is predominantly expressed in the intestine. SLC26 Cl−/HCO3− exchanger isoforms display unique nephron segment distribution patterns with distinct subcellular localization in the kidney tubules. Together with studies in pathophysiologic states and the examination of genetically engineered mouse models, the evolving picture points to important roles for the SLC26 family in health and disease states. This review summarizes recent advances in the characterization of the SLC26 Cl−/HCO3− exchangers in the kidney with emphasis on their essential role in diverse physiological processes, including chloride homeostasis, oxalate excretion and kidney stone formation, vascular volume and blood pressure regulation, and acid-base balance.
Keywords: Hypertension, volume depletion, salt excretion, oxalate stone, renal tubular acidosis
Introduction
Chloride absorption is an essential function of epithelial cells in the kidney and gastrointestinal tract and plays an important role in vascular volume homeostasis. This process is primarily mediated via apical chloride/base exchangers and is coupled to the secretion of HCO3− or mono or divalent anions. In the kidney, apical chloride/base exchangers have been identified in the proximal tubule and the cortical collecting duct (CCD) (1–4). The apical chloride/base exchangers work in tandem with the sodium-absorbing transporters- the Na+/H+ exchanger NHE3 or the epithelial sodium channel ENaC- to mediate the absorption of salt and the secretion of acid or base equivalents in kidney tubules (1–9).
Cloning and functional expression studies have identified a new class of anion exchangers that belong to a gene superfamily known as solute-linked carrier 26 (SLC26) (10–17). This family is genetically distinct from the SLC4 anion exchangers (AE1, AE2, AE3, and AE4) and comprises 10 distinct members (SLC26A1–11) (18–20). Several SLC26 isoforms show highly restricted and distinct tissue distribution patterns, whereas other isoforms are widely distributed (10–20). Subcellular localization studies have demonstrated apical, basolateral, or endosomal localization of SLC26 isoforms in the kidney and other organs (18–20). It is now accepted that the apical chloride/base exchange in epithelial tissues is predominantly mediated via SLC26 isoforms.
All SLC26 isoforms—except for SLC26A5 (prestin)—are versatile anion exchangers and show remarkable ability to transport various anions. Modes of transport mediated by SLC26 members include the exchange of chloride for bicarbonate, hydroxyl, sulfate, formate, iodide, or oxalate with variable specificity (21–32). Several SLC26 family members can specifically function as Cl−/HCO3− exchangers. These include SLC26A3 (DRA), SLC26A4 (pendrin), SLC26A6 (PAT1 or CFEX), SLC26A7, SLC26A9, and SLC26A11 (21–32). In addition to mediating chloride/base exchange, several SLC26 isoforms can also function as chloride channels, including SLC26A7, SLC26A9, and SLC26A11 (30–35). Figure 1 is a dendrogram of the SLC26 family of anion exchangers and shows the relative degree of homology at the amino acid level among various isoforms. Among the SLC26 isoforms that can function as Cl−/HCO3− exchangers, all except SLC26A3 (DRA) are expressed in the kidney. Table 1 shows the distribution of SLC26 Cl−/HCO3− exchangers in the kidney and gastrointestinal tract. Figure 2 is a schematic diagram based on published literature and depicts the nephron segment distribution of SLC26A4 (pendrin), SLC26A6 (PAT-1; CFEX), SLC26A7, SLC26A9, and SLC26A11 (KBAT).
Figure 1. A dendrogram of the SLC26 isoforms.
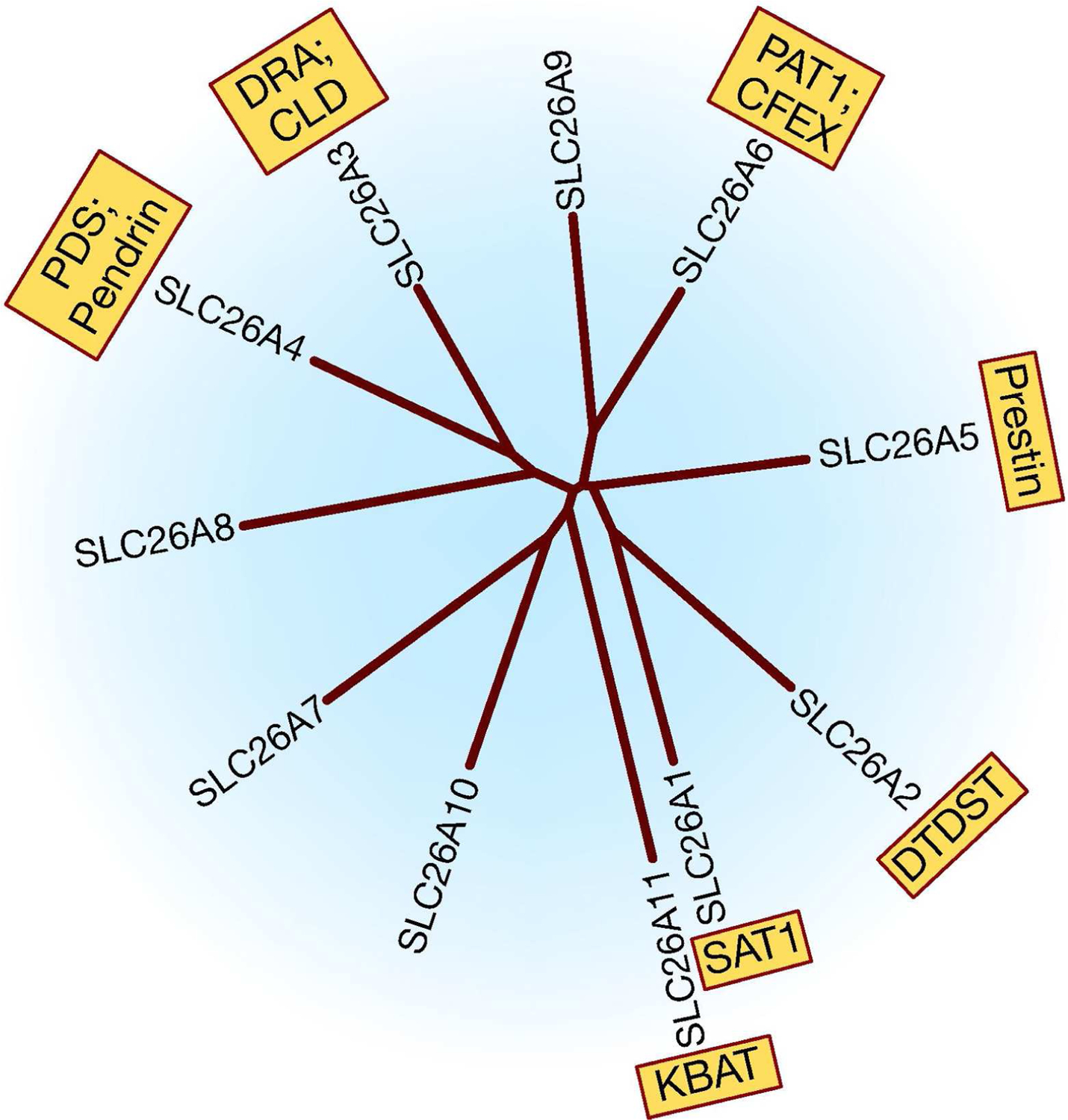
SAT1: Sulfate Anion Transporter 1; DTDST: diastrophic dysplasia sulfate transporter; DRA: Down Regulated in Adenoma; CLD; Chloride Losing Diarrhea; PDS: Pendred Syndrome; PAT1: Putative Anion Transporter 1; CFEX: Chloride-Formate Exchange; KBAT: Kidney Brain Anion Transporter. SLC26A10 is a pseudogene and therefore not discussed.
Table 1. Distribution of SLC26 Cl−/HCO3− exchangers in the kidney and gastrointestinal (G.I) tract.
SLC26A3 (DRA) shows abundant expression in the gastrointestinal tract and no or very low expression in the kidney, whereas SLC26A4 (pendrin) is abundantly detected in the kidney with no or little expression in the gastrointestinal tract.
| Kidney | G.I. Tract | |
|---|---|---|
| SLC26A3 | − | ++++ |
| SLC26A4 | +++ | − |
| SLC26A6 | ++++ | ++++ |
| SLC26A7 | +++ | +++ |
| SLC26A9 | + | ++++ |
| SLC26A11 | ++++ | + |
The expression intensity in gastrointestinal tract is assessed by mRNA expression levels in the intestine, stomach or both.
Figure 2. A schematic diagram depicting the nephron segment distribution of SLC26A4 (pendrin), SLC26A6 (PAT-1; CFEX), SLC26A7, SLC26A9, and SLC26A11 (KBAT).
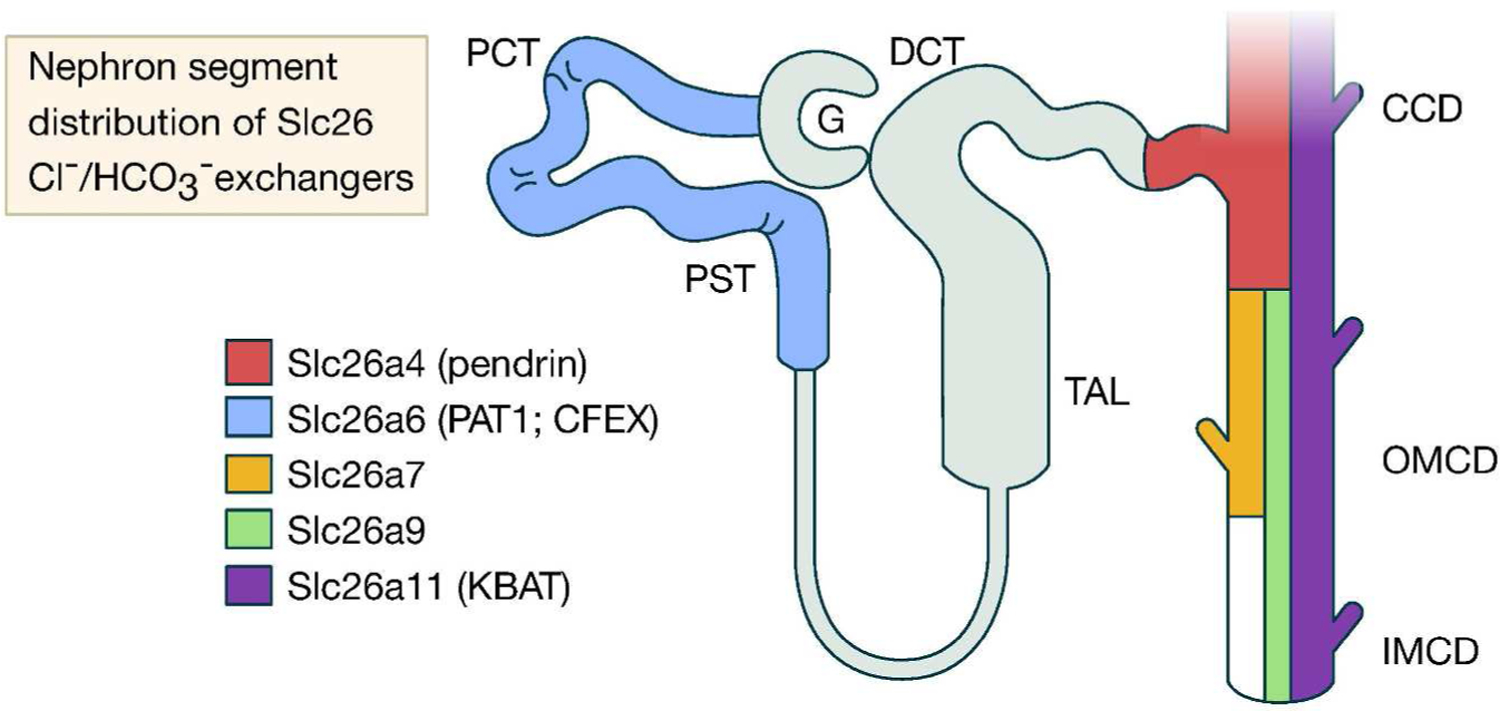
SLC26A6 is primarily detected in the proximal tubule whereas the other SLC26 Cl−/HCO3− exchangers are expressed in the collecting duct.
The diversity in functional modes of SLC26 isoforms and their distinct distribution pattern in epithelial cells support important roles for SLC26 paralogs in the physiological function of the kidney and other tissues. SLC26 isoforms express a unique sequence on their C terminus, known as the sulfate transporter and anti-sigma factor antagonist (STAS) domain, which plays an important role in SLC26 function and regulation (36). Given the ability of SLC26 isoforms to function in multiple chloride transport modes, the question as to which of these functional modes is dominant in the kidney, intestine or other organs is a fascinating and at the same time complicated one. It is very plausible that the indigenous milieu (presence of oxalate, bicarbonate or other anions) as well as the presence of specific functional partners and tissue specific signaling mechanisms all play important roles in determining the functional mode of an SLC26 isoform in kidney tubules.
Mutations in several SLC26 isoforms are linked to autosomal recessive genetic disorders, with SLC26A2 associated with chondrodysplasias (11), SLC26A3 linked to chloride-losing diarrhea (12), and SLC26A4 connected to Pendred syndrome (13), confirming the important roles of SLC26 isoforms in normal physiology and human pathophysiology.
SLC26 Isoforms and the Kidney
In the following sections, the expression, localization, and regulation of SLC26 Cl−/HCO3− exchangers in the kidney will be reviewed, and their role in kidney physiology, systemic acid-base balance, vascular volume homeostasis, and blood pressure control will be discussed.
SLC26A4 (Pendrin, PDS).
Cloning, functional expression, and localization.
The SLC26A4 gene was first discovered by positional cloning studies in patients with Pendred syndrome (13), an autosomal recessive hereditary disorder characterized by goiter and deafness (37). The protein was named pendrin to indicate its association with Pendred syndrome (13). SLC26A4 (pendrin) is located on chromosome 7 next to SLC26A3 (DRA) gene, another member of the family (13). Pendrin encodes an mRNA of ~5 kb and a protein of ~95 to 100 kDa in humans, and is abundantly expressed in the thyroid follicular cells (13, 37).
Pendrin can function in Cl−/HCO3− exchange, Cl−/OH− exchange, and Cl−/formate exchange modes (22). While pendrin mRNA is detected in the CCD and the proximal tubule (22), its protein is only detected on the apical membrane of B-IC and non-A, non-B-IC cells in the connecting tubule and CCDs (38–40). Figure 3 is a schematic diagram depicting the localization of pendrin in B-IC cells in the CCD.
Figure 3. A schematic diagram depicting the localization of pendrin in cortical collecting duct cells.
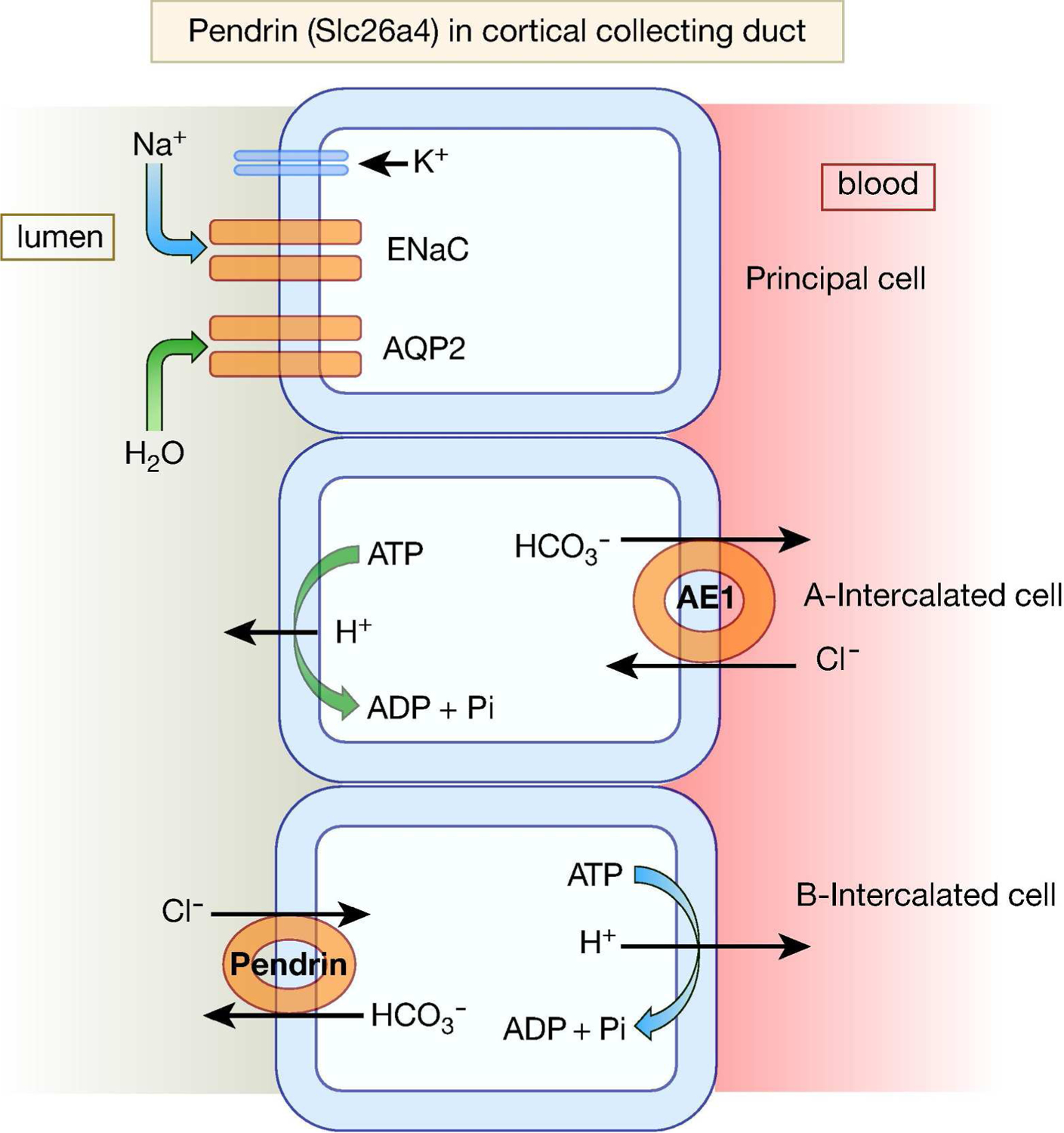
Pendrin protein is primarily detected on the apical membrane of B-and non-A, non-B intercalated cells in the cortical collecting duct.
Pendrin-deficient mice: Phenotypic presentation at baseline and in pathophysiologic states.
Pendrin knockout (KO) mice show decreased ability to excrete bicarbonate in response to bicarbonate loading, consistent with playing an important role in bicarbonate secretion in the kidney (38). Intracellular pH measurements in microperfused CCDs demonstrated that pendrin ablation inhibited apical Cl−/HCO3− exchange activity in B-IC cells and bicarbonate secretion in the CCD (41). Taken together, these results demonstrate that pendrin is the main apical Cl−/HCO3− exchanger in the kidney collecting duct.
Genetic deletion of pendrin does not cause any excessive diuresis under basal conditions (38, 41, 42). Indeed, deafness is the only major phenotypic presentation in pendrin-deficient mice and in humans with Pendred syndrome (43). Kidney functions, including salt excretion, are normal in pendrin null mice (41, 42, 44). Studies in pendrin null mice, however, indicate that this exchanger plays an important role in chloride reabsorption in a salt-depleted state or in the presence of excess aldosterone (42, 44). In carefully performed studies, Verlander et al demonstrated that the aldosterone analog deoxycorticosterone acetate (DOCA), which increases systemic blood pressure in the presence of enhanced salt intake, causes translocation of pendrin to the apical membrane in mouse kidney CCD (42). The same treatment with DOCA and salt failed to increase systemic blood pressure in pendrin KO mice (42). These results convincingly show that pendrin, working in tandem with ENaC, enhances salt absorption in the collecting duct in the presence of increased aldosterone and raises systemic blood pressure (8, 42).
In separate studies, Wall et al showed that pendrin KO mice display signs of volume depletion and develop metabolic alkalosis and hypotension when subjected to salt restriction (44). A recent report indicated the development of metabolic alkalosis in a patient with Pendred syndrome during volume depletion (45).
Regulation of pendrin in acid base and electrolyte disorders.
Pendrin is adaptively downregulated in acidosis and upreguated in bicarbonate loading (46, 47). These results are consistent with a role for pendrin in conserving bicarbonate in acidosis and secreting bicarbonate in alkalosis. Alternatively, it was demonstrated that the downregulation of pendrin in acidosis could be secondary to increased chloride intake (in the form of NH4Cl) and its upregulation in alkalosis could be due to reduced chloride intake (in the form of chloride free, Na-HCO3−) (48, 49). Whether the systemic pH (acidosis or alkalosis) has a direct regulatory role on pendrin expression needs carefully controlled experiments. Pendrin is downregulated in potassium depletion (46), suggesting that it may contribute to decreased bicarbonate excretion in hypokalemia.
Pendrin and cystic fibrosis.
Recent studies have demonstrated that CFTR physically interacts with and activates pendrin in parotid duct, tracheal epithelial and thyroid follicular cells (50, 51). This phenomenon might have ramifications for patients with cystic fibrosis, who on occasion can present with metabolic alkalosis, specifically at a young age (52, 53). It is plausible that pendrin, which plays an important role in chloride absorption and bicarbonate secretion in volume-depleted states, remains inactive in patients with cystic fibrosis, resulting in further loss of chloride and retention of HCO3− by the kidney.
Pendrin deficient mice and hypercalciuria.
A recent study demonstrated that the expression levels of TRPV5, Calbindin 28 and Na/Ca exchanger, which are important to calcium absorption in the distal nephron (54, 55), were all decreased in kidneys of pendrin null mice, presumably due to the luminal acidic pH, which has been shown to inactivate TRPV5 (41, 56, 57). These changes were associated with a significant hypercalciuria (57), and were all reversed in response to bicarbonate loading (57). Humans with unexplained acidic urine and hypercalciuria could have reduced pendrin activity (57).
Pendrin-dependent, sodium absorption pathways in the collecting duct: Electrogenic vs. electroneutral pathway.
Published studies indicate that pendrin-mediated Cl−/HCO3− exchange is coupled to sodium absorption via ENaC (8, 42). However, that view has been revisited. A recent publication indicated that pendrin can work in tandem with the sodium-dependent Cl−/HCO3− exchanger (NDCBE) in B intercalated cells to absorb sodium (58, 59). Such a coordinated mechanism would be electroneutral and could provide an ENaC-independent salt reabsorption pathway in the collecting duct (58, 59). It is plausible that both ENaC-dependent and independent, pendrin-mediated salt absorption coexist in the collecting duct and are differentially regulated in pathophysiologic states.
Essential role of pendrin in salt absorption in the setting of Na-Cl cotransporter inactivation.
Based on published literature, investigators have concluded that pendrin is predominantly active during salt (or chloride) depletion and/or in response to increased aldosterone levels (8, 42, 44). A recent study tested the hypothesis that pendrin and NCC compensate for loss of function of the other, therefore masking the role that each plays in salt absorption under baseline conditions. Toward this end, the investigators generated pendrin/NCC double KO mice (60), which displayed severe salt wasting under basal conditions (60). As a result, animals developed profound volume depletion, renal failure, and metabolic alkalosis, which were all corrected with salt replacement (60). These results indicate that pendrin plays an important role in salt absorption, specifically in the setting of NCC inactivation (60). They further indicate that pendrin can be a new target for novel diuretics that, in conjunction with inhibitors of NCC can be an effective regimen for patients with fluid overload (60, 61). The schematic diagram in Figure 4 depicts the synergistic effects of pendrin and NCC inhibition in salt and water excretion. A recent study showed that hydrochlorothiazide treatment caused severe volume depletion, along with metabolic alkalosis and decreased kidney function in a patient with Pendred syndrome (62), supporting the conclusion that a functional pendrin blunts the diuretic effect of NCC inhibition subsequent to treatment with thiazides.
Figure 4. A schematic diagram highlighting the synergistic effects of pendrin and Na-Cl cotransport (NCC) inhibition on salt and water excretion.
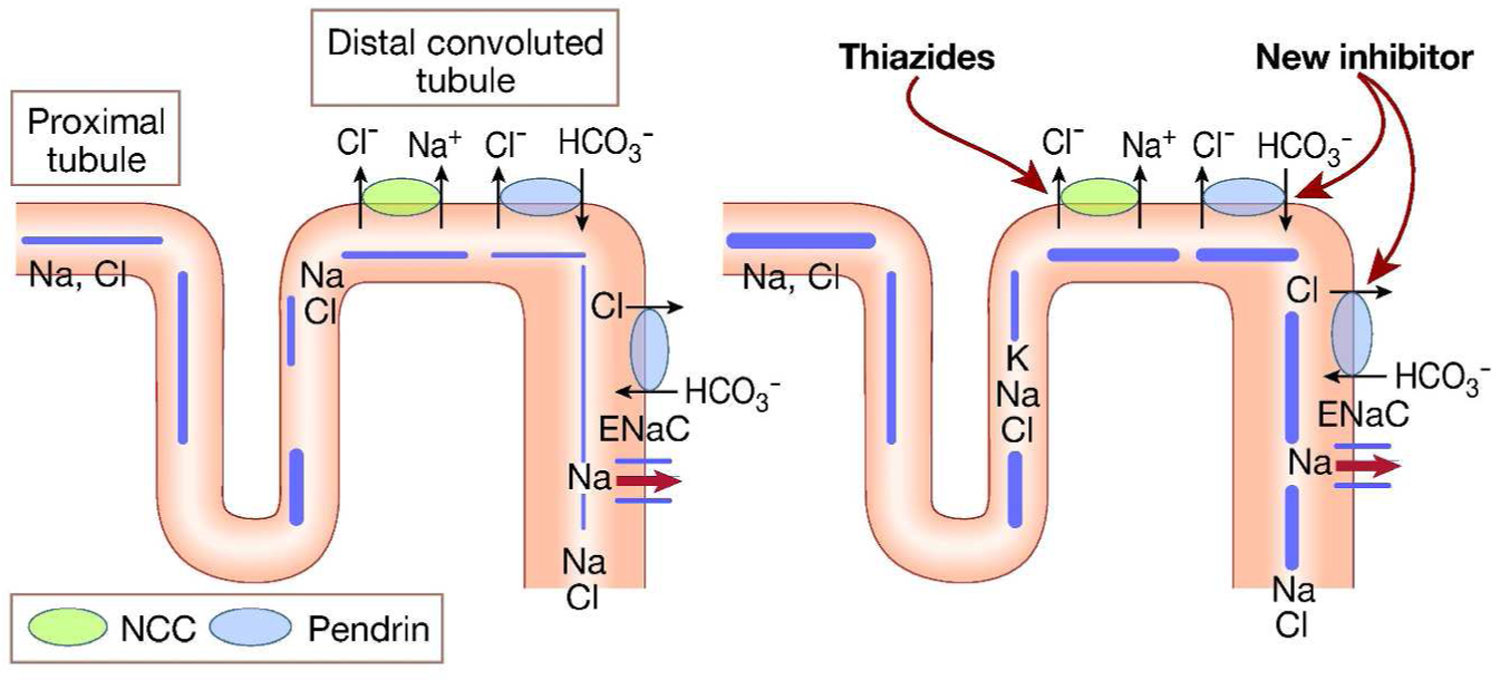
Combined inactivation or inhibition of pendrin and NCC should result in severe salt wasting.
SLC26A6 (PAT1, CFEX).
Cloning, functional expression, and localization.
SLC26A6 was cloned on the basis of homology to the genes encoding DRA (SLC26A3) and pendrin (SLC26A4) (15). SLC26A6 maps to chromosome 3, encodes a 738-amino-acid protein and is detected in kidney, pancreas, and several other tissues (15). Slc26a6 localizes to the apical membrane of kidney proximal tubule and small intestinal villi (26, 63, 64). SLC26A6 can transport Cl−/HCO3− exchange, Cl−/OH− exchange, chloride/oxalate exchange, and chloride/formate exchange (23–26), resembling apical anion exchange activities in the proximal tubule and the small intestine (1, 63, 64). Figure 5 presents immunofluorescence labeling of Slc26a6 (PAT1) depicting apical membrane localization in the proximal tubule.
Figure 5. An immunofluorescence labeling of Slc26a6 (PAT1) in the kidney proximal tubule.
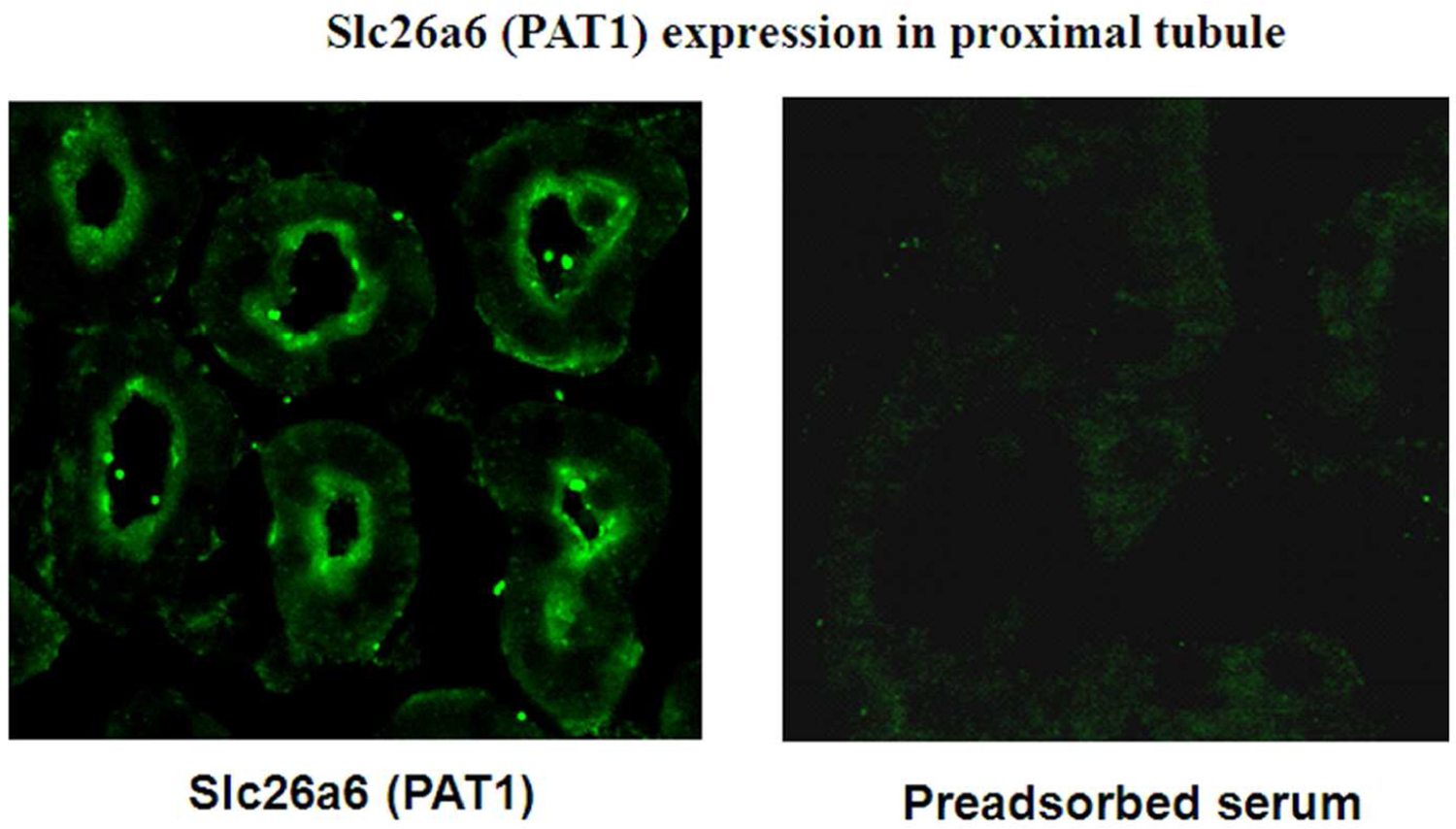
SLC26A6 is exclusively detected on the apical membrane of kidney proximal tubules.
Slc26a6 (PAT1)-deficient mice: Phenotypic presentation at baseline and in pathophysiologic states.
To determine the role of Slc26a6 in trans-tubular NaCl transport in the proximal tubule, Wang et al generated mice with a genetic deletion of Slc26a6 (PAT1) (65). Slc26a6 null mice have normal kidney function under basal conditions (65). Microperfusion studies demonstrated that Slc26a6 is the major apical Cl−/HCO3− exchanger in the proximal tubule straight (S3) segment (65). In vivo microperfusion studies in proximal convoluted tubules indicated that while the baseline rate of Jv (net volume absorption) was comparable in wild-type and null mice, the oxalate-stimulated volume absorption was completely abolished in PAT1 null mice (65). This finding strongly suggests that the Cl−/oxalate exchanger that was proposed to mediate NaCl absorption in the proximal tubule (1) is mediated in its entirety by SLC26A6 (65). The increment in Jv induced by formate was not significantly different between Slc26a6 null mice and WT littermates, suggesting that Slc26a6 is not the chloride/formate exchanger that was functionally identified in the proximal tubule (1).
Slc26a6 (PAT1)-deficient mice and kidney stones.
In addition to studies examining the role of Slc26a6 in NaCl absorption in the proximal tubule, studies on two independently generated Slc26a6-null mice demonstrated the important role of this exchanger in oxalate homeostasis. These studies indicated that Slc26a6-null mice have a 4-fold increase in urine oxalate excretion (66, 67). The hyperoxaluria was not due to increased secretion of oxalate in the kidney, but rather resulted from enhanced absorption of oxalate from the intestine (66, 67). These results support the view that Slc26a6 can predominantly function in Cl−/oxalate exchange mode in mouse ileum by secreting oxalate into the lumen in exchange for chloride absorption (66, 67).
An intriguing observation in one of the Slc26a6-null mouse models was the high frequency of kidney stones (66). The stones were detectable macroscopically and were present in a majority of male and a minority of female null mice. Histological examination of the kidney showed that stones were predominantly composed of calcium oxalate and were primarily detected in the lumen of cortical tubules and in the urinary space of the Slc26a6-null mice (66). Taken together, these studies demonstrate that Slc26a6 plays essential roles both in proximal tubule NaCl transport and in the prevention of hyperoxaluria and calcium oxalate nephrolithiasis.
Slc26a6 (PAT1) and fructose-induced hypertension: Relevance to metabolic syndrome.
The worldwide increase in the incidence of metabolic syndrome correlates with a marked increase in total fructose intake in the form of high-fructose corn syrup, beverages, and table sugar. Increased dietary fructose intake in rodents has been shown to recapitulate many aspects of metabolic syndrome by causing hypertension, insulin resistance, and hyperlipidemia. Unexpectedly, DNA microarray experiments revealed a significant increase in the expression of Glut5, a fructose transporter (69), in the intestine of Slc26a6 null mice (68). PAT1 and Glut5 co-localized on the apical membrane of villi in the jejunum (70), the main site for the absorption of salt and fructose.
To test the possibility that fructose may stimulate salt absorption by activating PAT1,, the proximal jejunum was perfused in vivo with isotonic perfusate and net fluid absorption was examined before and after the addition of fructose (70). The results demonstrated that luminal fructose increased the salt absorption in intestine of wild-type mice, a response that was significantly blunted in PAT1 null mice (70). Complementary studies in intact animals showed that increased dietary fructose intake for ~12 weeks also enhances the absorption of salt in the kidney and causes hypertension, an observation that was abrogated in PAT1 null mice (70). Animals with a genetic deletion of Glut5 failed to demonstrate enhanced salt absorption in the presence of fructose, indicating that both Glut5 and PAT1 are essential for the generation of fructose-induced hypertension (71). Taken together, these studies indicate that dietary fructose stimulates salt absorption in both the jejunum and the kidney proximal tubule, resulting in a state of salt overload and thus causing hypertension (68, 70–72).
SLC26A7
Cloning, functional expression, and localization.
Slc26a7 was cloned on the basis of homology to other Slc26 isoforms (16). In humans, SLC26A7 maps to chromosome 8, encodes a 656-amino-acid protein and is abundantly detected in the kidney (16). Functional expression studies demonstrated that both human and mouse SLC26A7 can transport Cl−/HCO3− exchange (28). SLC26A7 also showed affinity for oxalate and sulfate transport (16). Expression and immunolocalization studies localized Slc26a7 on the basolateral membrane of A intercalated cells in the outer medullary collecting duct (OMCD) (73). No Slc26a7 labeling was detected in plasma membrane of the CCD or other nephron segments (73). The localization of Slc26a7 in the kidney was intriguing since the anion exchanger AE1 (Slc4a1), which can function as a Cl−/HCO3− exchanger, is also localized to the same membrane domain in the OMCD (73). Patch-clamp studies in cultured cells expressing SLC26A7 show that SLC26A7 can also function as a chloride channel (34). Based on studies in Slc26a7 null mice, it seems that Slc26a7 can function predominantly in Cl−/HCO3− exchange mode in the kidney (see below).
Regulation of Slc26a7 in pathophysiologic states.
Hypertonicity and potassium depletion.
Using epitope-tagged SLC26A7 cDNA and transient expression approach in MDCK cells (74), Xu et al reported that SLC26A7 is detected in the cytoplasmic compartment in cultured cells in isotonic medium (74). This contrasted with membrane localization of this transporter in the kidney in vivo (73). Since the physiologic milieu in the kidney outer medulla is hypertonic, the experiments were repeated in a high-osmolality media. Contrary to its cytoplasmic distribution in an isotonic environment, in medium that was made hypertonic for 16 h, SLC26A7 was detected predominantly in the plasma membrane (74). SLC26A7 was localized to the transferrin receptor-positive endosomes (74) and its trafficking to the plasma membrane in hypertonic media was blocked by mitogen-activated kinase inhibitors (74). SLC26A7 also showed increased targeting to the plasma membrane in potassium-depleted medium vs. cytoplasmic distribution in normal potassium isotonic medium (4 mEq/L) (74). These results suggest that SLC26A7 is present in endosomes, and its targeting to the basolateral membrane is increased in hypertonicity or potassium depletion.
Regulation of Slc26a7 by the antidiuretic hormone (ADH).
To examine the role of ADH in the expression of Slc26a7, Petrovic et al injected Brattleboro rats, which have central diabetes insipidus, with dDAVP, an ADH analog that normalizes their concentrating defect (75). In the absence of dDAVP, Slc26a7 expression was almost absent in kidneys of Brattleboro rats (75). However, treatment with dDAVP heavily induced the expression of Slc26a7 (75). Interestingly, the labeling of the Cl−/HCO3− exchanger AE1 remained unchanged in dDAVP-treated Brattleboro rats (75). The induction of Slc26a7 by vasopressin is likely an attempt by OMCD cells to regulate their cell volume and maintain HCO3− absorption in a state associated with increased interstitial medullary tonicity.
Differential regulation of Slc26a7 and AE1 in pathophysiologic states: Effect of increasing medullary interstitium osmolality.
To determine whether Slc26a7 and AE1 are differentially regulated, Barone et al examined the expression and regulation of AE1 and Slc26a7 in water deprivation, a condition known to increase the osmolality of the medulla, and in potassium depletion (76, 77). In rats that were subjected to 3 days of water deprivation, Slc26a7 abundance in OMCD cells increased by ~3-fold, whereas AE1 abundance decreased by ~50% vs. control animals (76). These results suggested that AE1 and Slc26a7 are differentially regulated in OMCD in water deprivation. It was proposed that SLC26A7 may play an important role in bicarbonate reabsorption and/or cell volume regulation in OMCD under hypertonic conditions. Both AE1 and Slc26a7 showed enhanced expression in the OMCD of rats subjected to potassium depletion (77).
Slc26a7-deficient mice: Phenotypic presentation at baseline and in pathophysiologic states.
Slc26a7 is expressed on the basolateral membrane of acid-secreting cells in the renal OMCD and in gastric parietal cells. To ascertain its role in kidney and stomach physiology, Xu et al generated mice with a genetic deletion of Slc26a7. Interestingly, Slc26a7-null mice develop distal renal tubular acidosis, as manifested by metabolic acidosis and alkaline urine pH (78). In the kidney, basolateral Cl−/HCO3− exchange activity in acid-secreting intercalated cells in the OMCD was significantly decreased in hypertonic medium, but was reduced only mildly in isotonic medium (78). Changing from a hypertonic to isotonic medium (relative hypotonicity) decreased the membrane abundance of Slc26a7 in kidney cells in vivo and in vitro (78). In the stomach, stimulated acid secretion was significantly impaired in isolated gastric mucosa and in the intact organ (78). It was concluded that Slc26a7 plays an important role in bicarbonate absorption in the kidney OMCD and in acid secretion in the stomach (78). It is very plausible that AE1 is mostly active during isotonic or hypotonic conditions, whereas Slc26a7 is the dominant bicarbonate transporter under hypertonic states.
SLC26A9
Cloning, functional expression, and localization.
Slc26a9 was cloned on the basis of homology to other Slc26 isoforms (16). In humans, SLC26A9 maps to chromosome 1, and encodes a 791-amino-acid protein (16). SLC26A9 (human)/Slc26a9 (mouse) is abundantly expressed in the stomach and lung, with lower levels in the kidney (29, 79). Slc26a9 can function in three distinct modes, including electrogenic Cl−/HCO3− exchange, chloride channel, and sodium chloride cotransport (29, 30, 32, 33, 35, 79). In the stomach, Slc26a9 is located on the apical membrane of surface epithelial cells and in the tubulovesicles of gastric parietal cells and regulates gastric acid secretion (29, 30). Little is known about the role of Slc26a9 in the kidney. A recent publication reported that Slc26a9 is predominantly detected in the medulla and cultured medullary collecting duct cells (80) and located on the apical membrane of a subset of cells in the OMCD and the initial portion of the IMCD (80). Double immunofluorescence experiments with Slc26a9 and AQP2 antibodies indicated that Slc26a9 is localized on the apical membrane of principal cells (80).
Slc26a9-deficient mice: Phenotypic presentation at baseline and in pathophysiologic states.
A recent study utilizing Slc26a9 knockout mice showed a significant reduction in renal chloride excretion vs. wild-type animals when fed a diet high in salt or subjected to water deprivation (80). Systemic arterial pressure measurements indicated that Slc26a9 KO (Slc26a9−/−) mice are hypertensive under baseline conditions and increase their blood pressure further within 48 hours of switching to a high salt diet (80). These results suggest that Slc26a9 plays an important role in renal chloride/fluid excretion and arterial pressure regulation. It was proposed that impaired SLC26A9 activity in humans may interfere with the excretion of excess salt and result in hypertension (80).
An important finding of these studies was the identification of Slc26a9 as a regulator of renal salt (chloride and sodium) excretion and blood pressure. These conclusions are based on the following findings: (1) localization of Slc26a9 to the apical membrane of principal cells in the medullary collecting duct, (2) decreased chloride and sodium excretion in Slc26a9 KO mice in response to water deprivation, (3) impaired chloride and sodium excretion in Slc26a9 KO mice upon switching to a high salt diet, and (4) elevated systemic arterial pressure in Slc26a9 null mice (80). Taken together, these results suggest that Slc26a9 mediates chloride excretion and regulates systemic arterial pressure. We propose that Slc26a9 predominantly functions as a chloride channel in medullary collecting duct cells. Figure 6 is a schematic diagram depicting Slc26a7 and Slc26a9 in the OMCD (and IMCD). Other than systemic hypertension, the most dramatic impact of Slc26a9 deletion is on gastric acid production. Slc26a9 deletion resulted in the complete loss of gastric acid secretion, indicating an essential role of this channel/transporter in gastric acid secretion.
Figure 6. A schematic diagram depicting the localization of Slc26a7 and Slc26a9 in the kidney OMCD (and IMCD).
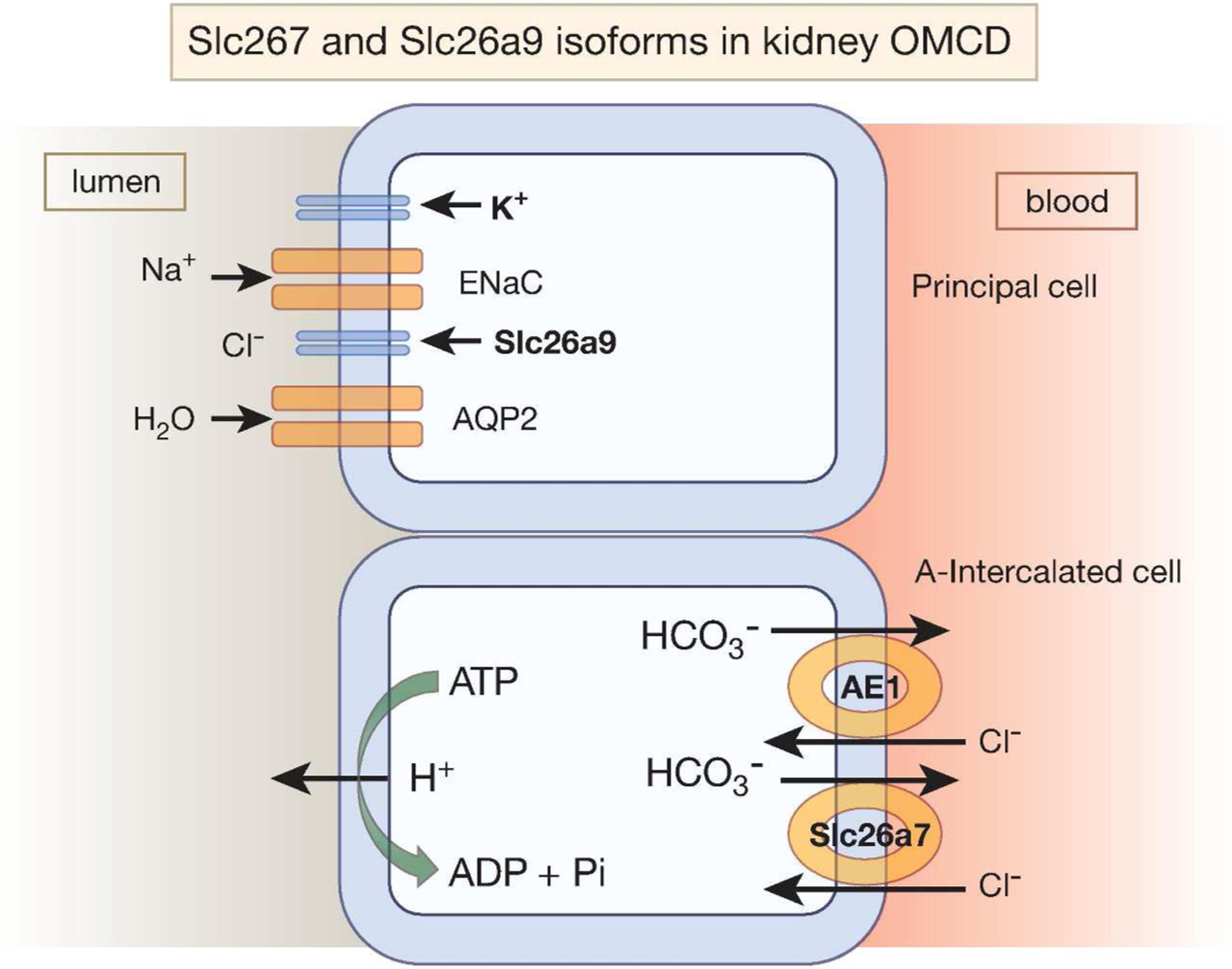
SLC26A7 is primarily detected on the basolateral membrane of A-intercalated cells in the OMCD whereas SLC26A9 is located on the apical membrane of principal cells in OMCD and the initial portion of IMCD. Slc26a9 is likely functioning as a chloride channel and not a Cl−/HCO3− exchange, given the electrochemical gradients for chloride and bicarbonate across the apical membrane. Slc26a7 is likely a Cl−/HCO3− exchanger based on the presence of distal renal tubular acidosis and functional studies in microperfused OMCD in Slc26a7 null mice.
SLC26A11 (KBAT)
Cloning, functional expression, and localization.
SLC26A11 was cloned on the basis of sequence homology to the high-affinity sulfate transporter SUL2 in yeast (GenBank Acc. NP013193) (17). SLC26A11 is expressed in several tissues, with the highest levels in placenta, kidney, and brain (17). Human SLC26A11 is located on chromosome 17, encodes a 2901-bp cDNA and a 606-amino-acid protein (17) and is detected in the endoplasmic reticulum, Golgi apparatus, and the plasma membrane (17).
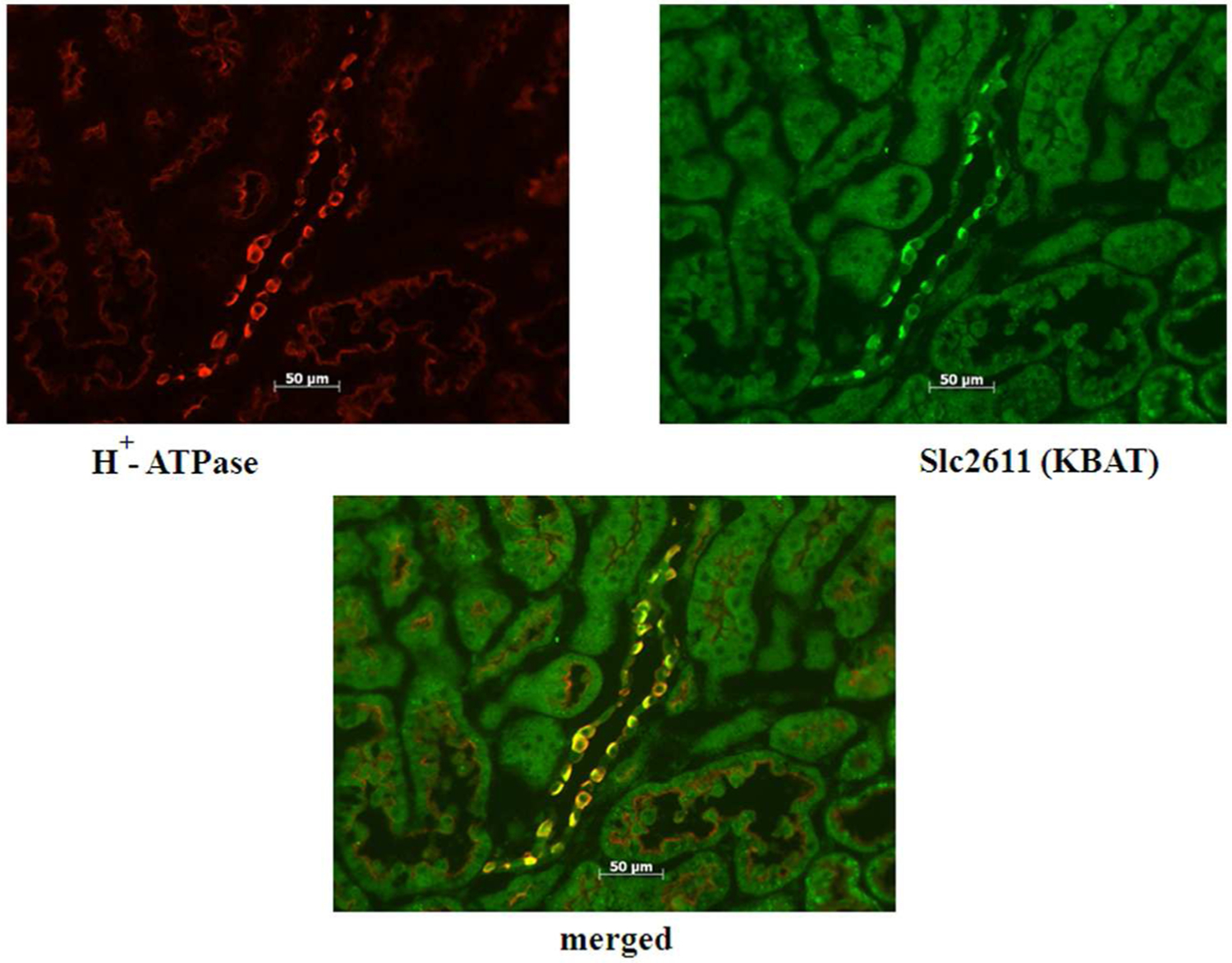
Slc26a11 is exclusively detected in the collecting duct and shows remarkable co-localization with H+-ATPase (31). Figure 7 (top panel) is an immunofluorescence labeling and shows the localization of AQP2 and Slc26a11 to distinct cell types in mouse CCD. Figure 7 (bottom panel) is an immunofluorescence labeling in the CCD, and shows remarkable co-localization of Slc26a11 with H+-ATPase on the apical membrane of A-intercalated cells and the basolateral membrane of B intercalated cells.
Figure 7. Localization of Slc26a11 in the kidney.
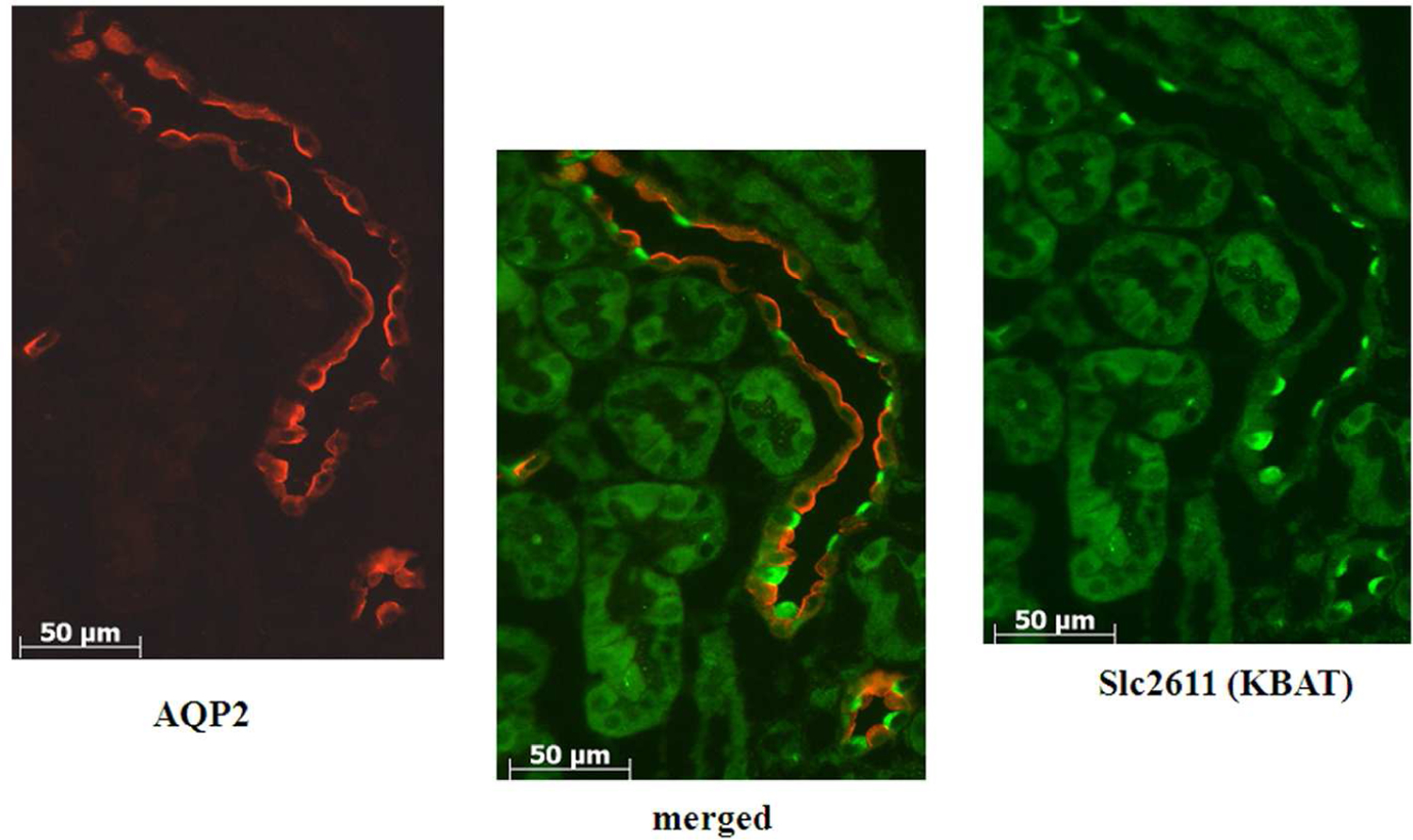
Top panel: Double immunofluorescence labeling of Slc26a11 and AQP2 in the kidney CCD. Slc26a11 and AQP2 localize to two distinct cell types in the collecting duct.
Bottom panel: Double immunofluorescence labeling of Slc26a11 and H+-ATPase in the kidney CCD. Slc26a11 co-localizes with H+-ATPase in the collecting duct.
Functional identity of Slc26a11.
KBAT was able to function as an electrogenic chloride-extruding pathway, as measured by 36Cl flux studies, as well as in Cl−/HCO3− exchange mode, as determined by the pH-sensitive dye BCECF (31). A recent patch-clamp study demonstrated that KBAT is indeed a chloride channel (81). KBAT-mediated chloride channel was active under baseline states and not further stimulated by cAMP, intracellular calcium, or co-expression with CFTR (81).
Vacuolar H+-ATPase, expressed in intercalated cells (82, 83) is an electrogenic pump and can create an electrical potential difference across the membrane by secreting H+ (acid). Studies in cultured kidney cells indicated that KBAT enhanced the H+-ATPase-mediated acid extrusion by a chloride-dependent mechanism (31). These studies are in agreement with published reports indicating that chloride modulates H+-ATPase activity (84) and that chloride channel(s) control bicarbonate absorption in the collecting duct (85). Slc26a11 (KBAT) can facilitate acid secretion in the collecting duct, either by dissipating the membrane potential resulting from H+ secretion with electrogenic chloride transport or directly through mechanism(s) that warrant further examination (31). Figure 8 is a schematic diagram depicting Slc2611 localization in cortical collecting duct cells.
Fig. 8. A schematic diagram depicting Slc26a11 as an apical chloride channel in intercalated cells in the CCD.
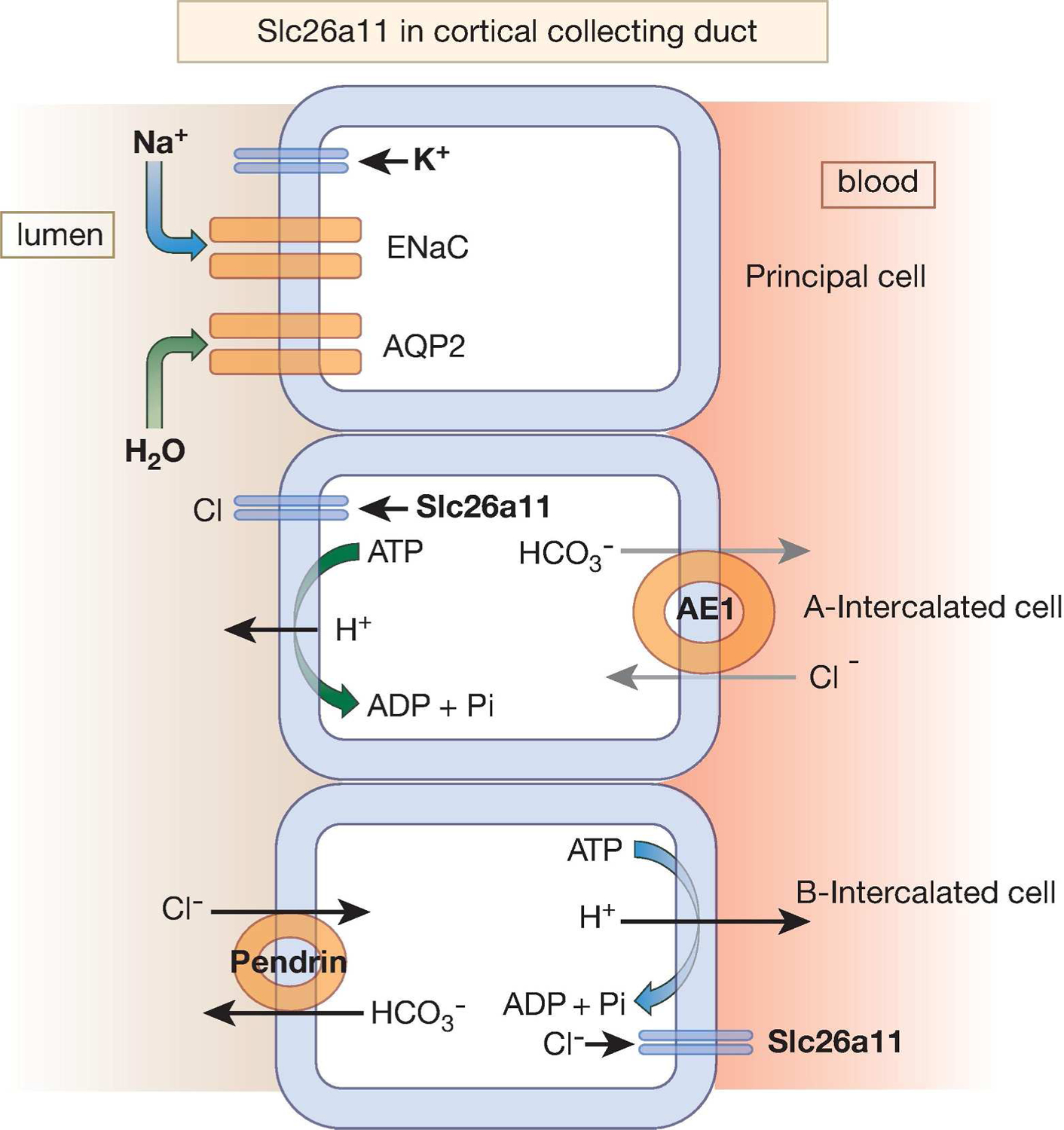
Slc26a11 is expressed on the apical membrane of A-intercalated cells and the basolateral membrane of B-intercalated cells in the CCD. Whether the basolateral Slc26a11 can also function as a chloride channel remains speculative.
SLC26 isoforms exhibit greater sequence diversity between mouse and human orthologs than is found among the SLC4 bicarbonate transporters. While these intrinsic differences must be considered when extrapolating results from mouse to human pathophysiology, examination of genetically engineered mouse models deficient in several SLC26 isoforms (Slc26a2, Slc26a3, and Slc26a4) has revealed the recapitulation of disease phenotypes in humans with mutations in these genes, including deafness and predisposition to dehydration in Slc26a4 (pendrin) KO mice (37, 43, 44, 45), congenital chloride diarrhea in Slc26a3 KO mice (27), and chondrodysplasia in mice with a partial loss of function in Slc26a2 (86). In conclusion, SLC26 isoforms are a new, rapidly evolving family of anion (chloride, bicarbonate, oxalate) transporters and/or channels and play important roles in kidney salt absorption, acid base balance, vascular volume homeostasis, and blood pressure regulation.
Acknowledgement
Studies in the author’s laboratory were supported by Merit Review awards (1I01BX001000-01A1 and 5I01BX001000-03) from the Department of Veterans Affairs, NIH grants RO1 DK62809 and R56DK62809, Center for Genetics of Transport and Epithelial Biology from University of Cincinnati and by grants from DCI Inc. and US Renal Care (to M.S).
Footnotes
Disclosure:
The author declares no conflict of interest.
References:
- 1.Aronson PS, Giebisch G. Mechanisms of chloride transport in the proximal tubule. Am J Physiol. 1997. Aug;273(2 Pt 2):F179–92. Review. [DOI] [PubMed] [Google Scholar]
- 2.Sindić A, Chang MH, Mount DB, Romero MF. Renal physiology of SLC26 anion exchangers. Curr Opin Nephrol Hypertens. 16(5):484–90, 2007. Review. [DOI] [PubMed] [Google Scholar]
- 3.Soleimani M. Expression, regulation and the role of SLC26 Cl-/HCO3- exchangers in kidney and gastrointestinal tract. Novartis Found Symp. 273:91–102, 2006. Review. [PubMed] [Google Scholar]
- 4.Baum M. Developmental changes in proximal tubule NaCl transport. Pediatr Nephrol. 23(2):185–94, 2008. Review. [DOI] [PubMed] [Google Scholar]
- 5.Alper SL, Chernova MN, Stewart AK. Regulation of Na+-independent Cl-/HCO3- exchangers by pH. JOP. 2(4 Suppl):171–5, 2001. Review. [PubMed] [Google Scholar]
- 6.Wagner CA, Mohebbi N, Capasso G, Geibel JP. The anion exchanger pendrin (SLC26A4) and renal acid-base homeostasis. Cell Physiol Biochem. 28(3):497–504, 2011. Review. [DOI] [PubMed] [Google Scholar]
- 7.Wang X, Armando I, Upadhyay K, Pascua A, Jose PA. The regulation of proximal tubular salt transport in hypertension: an update. Curr Opin Nephrol Hypertens. 18(5):412–20, 2009. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wall SM, Pech V. Pendrin and sodium channels: relevance to hypertension. J Nephrol. 23 Suppl 16:S118–23, 2010. Review. [PubMed] [Google Scholar]
- 9.Pech V, Pham TD, Hong S, Weinstein AM, Spencer KB, Duke BJ, Walp E, Kim YH, Sutliff RL, Bao HF, Eaton DC, Wall SM. Pendrin modulates ENaC function by changing luminal HCO3-. J Am Soc Nephrol. 21(11):1928–41, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bissig M, Hagenbuch B, Stieger B, Koller T, Meier PJ. Functional expression cloning of the canalicular sulfate transport system of rat hepatocytes. J Biol Chem. 269:3017–3021, 1994. [PubMed] [Google Scholar]
- 11.Hastbacka J, de la Chapelle A, Mahtani MM, Clines G, Reeve-Daly MP, Daly M, Hamilton BA, Kusumi K, Trivedi B, Weaver A, Coloma A, Lovett M, Buckler A, Kaitila I, Lander ES. The diastrophic dysplasia gene encodes a novel sulfate transporter: positional cloning by fine-structure linkage disequilibrium mapping. Cell. 78:1073–1087, 1994. [DOI] [PubMed] [Google Scholar]
- 12.Hoglund P, Haila S, Socha J, Tomaszewski L, Saarialho-Kere U, Karjalainen-Lindsberg M-L, Airola K, Holmberg C, de la Chapelle A, Kere J. Mutations of the Down-regulated in adenoma (DRA) gene cause congenital chloride diarrhea Nature Genet. 14:316–319, 1996. [DOI] [PubMed] [Google Scholar]
- 13.Everett LA, Glaser B, Beck JC, Idol JR, Buchs A, Heyman M, Adawi F, Hazani E, Nassir E, Baxevanis AD, Sheffield VC, Green ED. Pendred syndrome is caused by mutations in a putative sulphate transporter gene (PDS). Nature Gen. 17:411–422, 1997. [DOI] [PubMed] [Google Scholar]
- 14.Zheng J, Shen W, He DZ, Long KB, Madison LD, Dallos P. Prestin is the motor protein of cochlear outer hair cells. Nature. 405:149–155, 2000. [DOI] [PubMed] [Google Scholar]
- 15.Lohi H, Kujala M, Kerkela E, Saarialho-Kere U, Kestila M, Kere J. Mapping of five new putative anion transporter genes in human and characterization of SLC26A6, a candidate gene for pancreatic anion exchanger. Genomics. 70:102–112, 2000. [DOI] [PubMed] [Google Scholar]
- 16.Lohi H, Kujala M, Makela S, Lehtonen E, Kestila M, Saarialho-Kere U, Markovich D, and Kere J Functional characterization of three novel tissue-specific anion exchangers SLC26A7, -A8, and -A9. J Biol Chem. 277, 14246–14254, 2002. [DOI] [PubMed] [Google Scholar]
- 17.Vincourt JB, Jullien D, Amalric F, and Girard JP Molecular and functional characterization of SLC26A11, a sodium-independent sulfate transporter from high endothelial venules. FASEB J 17, 890–892, 2003. [DOI] [PubMed] [Google Scholar]
- 18.Romero MF, Chang MH, Plata C, Zandi-Nejad K, Mercado A, Broumand V, Sussman CR, and Mount DB Physiology of electrogenic SLC26 paralogues. Novartis Found Symp 273, 126–138, 2006. [PubMed] [Google Scholar]
- 19.Soleimani M, Xu J. SLC26 chloride/base exchangers in the kidney in health and disease. Semin Nephrol. 26(5):375–85, 2006. [DOI] [PubMed] [Google Scholar]
- 20.Alper SL. Molecular physiology and genetics of Na+-independent SLC4 anion exchangers. J Exp Biol. 212(Pt 11):1672–83, 2009. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Melvin JE, Park K, Richardson L, Schultheis PJ, Shull GE. Mouse Down-Regulated in Adenoma (DRA) is an intestinal Cl(−)/HCO(3)(−) exchanger and is upregulated in colon of mice lacking the NHE-3 Na(+)/H(+) exchanger. J Bio Chem. 274:22855–22861, 1999. [DOI] [PubMed] [Google Scholar]
- 22.Soleimani M, Greeley T, Petrovic S, Wang Z, Amlal H, Kopp P, and Burnham CE Pendrin: an apical Cl-/OH-/HCO3- exchanger in the kidney cortex. Am J Physiol Renal Physiol 280, F356–F364, 2001. [DOI] [PubMed] [Google Scholar]
- 23.Jiang Z, Grichtchenko II, Boron WF, Aronson PS. Specificity of anion exchange mediated by mouse Slc26a6. J Biol Chem. 277(37):33963–7, 2002. [DOI] [PubMed] [Google Scholar]
- 24.Xie Q, Welch R, Mercado A, Romero MF, Mount DB. Molecular characterization of the murine Slc26a6 anion exchanger: functional comparison with Slc26a1. Am J Physiol Renal Physiol. 283:F826–F838, 2002. [DOI] [PubMed] [Google Scholar]
- 25.Chernova MN, Jiang L, Friedman DJ, Darman RB, Lohi H, Kere J, Vandorpe DH, Alper SL. Functional comparison of mouse slc26a6 anion exchanger with human SLC26A6 polypeptide variants: differences in anion selectivity, regulation, and electrogenicity. J Biol Chem. 280(9):8564–80, 2005. [DOI] [PubMed] [Google Scholar]
- 26.Wang Z, Petrovic S, Mann E, Soleimani M. Identification of an apical Cl(−)/HCO3(−) exchanger in the small intestine. Am J Physiol Gastrointest Liver Physiol. 282(3):G573–9, 2002. [DOI] [PubMed] [Google Scholar]
- 27.Schweinfest CW, Spyropoulos DD, Henderson KW, Kim JH, Chapman JM, Barone S, Worrell RT, Wang Z, and Soleimani M Slc26a3 (dra)-deficient mice display chloride-losing diarrhea, enhanced colonic proliferation, and distinct up-regulation of ion transporters in the colon. J Biol Chem 281, 37962–37971, 2006. [DOI] [PubMed] [Google Scholar]
- 28.Petrovic S, Ju X, Barone S, Seidler U, Alper SL, Lohi H, Kere J, Soleimani M. Identification of a basolateral Cl-/HCO3- exchanger specific to gastric parietal cells. Am J Physiol Gastrointest Liver Physiol. 284(6):G1093–103, 2003. [DOI] [PubMed] [Google Scholar]
- 29.Xu J, Henriksnäs J, Barone S, Witte D, Shull GE, Forte JG, Holm L, Soleimani M. SLC26A9 is expressed in gastric surface epithelial cells, mediates Cl-/HCO3- exchange, and is inhibited by NH4+. Am J Physiol Cell Physiol. 289(2):C493–505, 2005 [DOI] [PubMed] [Google Scholar]
- 30.Xu J, Song P, Miller ML, Borgese F, Barone S, Riederer B, Wang Z, Alper SL, Forte JG, Shull GE, Ehrenfeld J, Seideler U, Soleimani M. Deletion of the chloride transporter Slc26a9 causes loss of tubulovesicles in parietal cells and impairs acid secretion in the stomach. Proc Natl Acad Sci U S A 105(46), 17955–17960, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xu J, Barone S, Li H, Holiday S, Zahedi K, Soleimani M. Slc26a11, a chloride transporter, localizes with the vacuolar H(+)-ATPase of A-intercalated cells of the kidney. Kidney Int. 80(9):926–37, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen AP, Chang MH, Romero MF. Functional analysis of nonsynonymous single nucleotide polymorphisms in human SLC26A9. Hum Mutat 33(8):1275–84, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bertrand CA, Zhang R, Pilewski JM, Frizzell RA. SLC26A9 is a constitutively active, CFTR-regulated anion conductance in human bronchial epithelia. J Gen Physiol. 133(4):421–38, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim KH, Shcheynikov N, Wang Y, and Muallem S SLC26A7 is a Cl− channel regulated by intracellular pH. J Biol Chem 280, 6463–6470, 2005. [DOI] [PubMed] [Google Scholar]
- 35.Dorwart MR, Shcheynikov N, Wang Y, Stippec S, and Muallem S SLC26A9 is a Cl− channel regulated by the WNK kinases. J Physiol 584, 333–345, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sharma AK, Rigby AC, Alper SL. STAS domain structure and function. Cell Physiol Biochem. 28(3):407–22, 2011. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bizhanova A, Kopp P. Genetics and phenomics of Pendred syndrome. Mol Cell Endocrinol. 322(1–2):83–90, 2010. Review. [DOI] [PubMed] [Google Scholar]
- 38.Royaux IE, Wall SM, Karniski LP, Everett LA, Suzuki K, Knepper MA, Green ED Pendrin, encoded by the Pendred syndrome gene, resides in the apical region of renal intercalated cells and mediates bicarbonate secretion. Proc Natl Acad Sci U S A 98, 4221–4226, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim YH, Kwon TH, Frische S, Kim J, Tisher CC, Madsen KM, Nielsen S. Immunocytochemical localization of pendrin in intercalated cell subtypes in rat and mouse kidney. Am J Physiol Renal Physiol. 283(4):F744–54, 2002. [DOI] [PubMed] [Google Scholar]
- 40.Wall SM, Hassell KA, Royaux IE, Green ED, Chang JY, Shipley GL, Verlander JW. Localization of pendrin in mouse kidney. Am J Physiol Renal Physiol. 284(1):F229–41, 2003. [DOI] [PubMed] [Google Scholar]
- 41.Amlal H, Petrovic S, Xu J, Wang Z, Sun X, Barone S, Soleimani M. Deletion of the anion exchanger Slc26a4 (pendrin) decreases apical Cl(−)/HCO3(−) exchanger activity and impairs bicarbonate secretion in kidney collecting duct. Am J Physiol Cell Physiol. 299(1):C33–41, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Verlander JW, Hassell KA, Royaux IE, Glapion DM, Wang ME, Everett LA, Green ED, and Wall SM Deoxycorticosterone upregulates PDS (Slc26a4) in mouse kidney: role of pendrin in mineralocorticoid-induced hypertension. Hypertension 179, 356–362, 2003. [DOI] [PubMed] [Google Scholar]
- 43.Wangemann P. The role of pendrin in the development of the murine inner ear. Cell Physiol Biochem. 28:527–34, 2011. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wall SM, Kim YH, Stanley L, Glapion DM, Everett LA, Green ED, Verlander JW. NaCl restriction upregulates renal Slc26a4 through subcellular redistribution: role in Cl- conservation. Hypertension. 2004 Dec;44(6):982–7, 2004. [DOI] [PubMed] [Google Scholar]
- 45.Kandasamy N, Fugazzola L, Evans M, Chatterjee K, Karet F. Life-threatening metabolic alkalosis in Pendred syndrome. Eur J Endocrinol. 165(1):167–70, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wagner CA, Finberg KE, Stehberger PA, Lifton RP, Giebisch GH, Aronson PS, Geibel JP. Regulation of the expression of the Cl-/anion exchanger pendrin in mouse kidney by acid-base status. Kidney Int. 62(6):2109–17, 2002. [DOI] [PubMed] [Google Scholar]
- 47.Petrovic S, Wang Z, Ma L, Soleimani M. Regulation of the apical Cl-/HCO3- exchanger pendrin in rat cortical collecting duct in metabolic acidosis. Am J Physiol Renal Physiol. 284(1):F103–12, 2003. [DOI] [PubMed] [Google Scholar]
- 48.Quentin F, Chambrey R, Trinh-Trang-Tan MM, Fysekidis M, Cambillau M, Paillard M, Aronson PS, Eladari D. The Cl-/HCO3- exchanger pendrin in the rat kidney is regulated in response to chronic alterations in chloride balance. Am J Physiol Renal Physiol. 287(6):F1179–88, 2004. [DOI] [PubMed] [Google Scholar]
- 49.Vallet M, Picard N, Loffing-Cueni D, Fysekidis M, Bloch-Faure M, Deschênes G, Breton S, Meneton P, Loffing J, Aronson PS, Chambrey R, Eladari D. Pendrin regulation in mouse kidney primarily is chloride-dependent. J Am Soc Nephrol. 17(8):2153–63, 2006. [DOI] [PubMed] [Google Scholar]
- 50.Shcheynikov N, Yang D, Wang Y, Zeng W, Karniski LP, So I, Wall SM, Muallem S. The Slc26a4 transporter functions as an electroneutral Cl-/I-/HCO3- exchanger: role of Slc26a4 and Slc26a6 in I- and HCO3- secretion and in regulation of CFTR in the parotid duct. J Physiol. 586(16):3813–24, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Garnett JP, Hickman E, Burrows R, Hegyi P, Tiszlavicz L, Cuthbert AW, Fong P, Gray M. Novel role for pendrin in orchestrating bicarbonate secretion in cystic fibrosis transmembrane conductance regulator (CFTR)-expressing airway serous cells. J Biol Chem. 286(47):41069–82, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bates CM, Baum M, Quigley R. Cystic fibrosis presenting with hypokalemia and metabolic alkalosis in a previously healthy adolescent. J Am Soc Nephrol. 8(2):352–5, 1997. [DOI] [PubMed] [Google Scholar]
- 53.Baird JS, Walker P, Urban A, Berdella M. Metabolic alkalosis and cystic fibrosis. Chest. 122(2):755–6, 2002. [DOI] [PubMed] [Google Scholar]
- 54.Hoenderop JG, Hartog A, Stuiver M, Doucet A, Willems PH, Bindels RJ. Localization of the epithelial Ca(2+) channel in rabbit kidney and intestine. J Am Soc Nephrol. 11(7):1171–8, 2000. [DOI] [PubMed] [Google Scholar]
- 55.Hoenderop JG, Nilius B, Bindels RJ. Molecular mechanism of active Ca2+ reabsorption in the distal nephron. Annu Rev Physiol 64:529–549, 2002. [DOI] [PubMed] [Google Scholar]
- 56.Bonny O, Rubin A, Huang CL, Frawley WH, Pak CY, Moe OW. Mechanism of urinary calcium regulation by urinary magnesium and pH. J Am Soc Nephrol. 19(8):1530–7, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Barone S, Amlal H, Xu J, Soleimani M. Deletion of the Cl-/HCO3- exchanger pendrin downregulates calcium-absorbing proteins in the kidney and causes calcium wasting. Nephrol Dial Transplant. 27(4):1368–79, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Leviel F, Hübner CA, Houillier P, Morla L, El Moghrabi S, Brideau G, Hassan H, Parker MD, Kurth I, Kougioumtzes A, Sinning A, Pech V, Riemondy KA, Miller RL, Hummler E, Shull GE, Aronson PS, Doucet A, Wall SM, Chambrey R, Eladari D. The Na+-dependent chloride-bicarbonate exchanger SLC4A8 mediates an electroneutral Na+ reabsorption process in the renal cortical collecting ducts of mice. J Clin Invest. 120(5):1627–35, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Eladari D, Hübner CA. Novel mechanisms for NaCl reabsorption in the collecting duct. Curr Opin Nephrol Hypertens. 20(5):506–11, 2011. Review. [DOI] [PubMed] [Google Scholar]
- 60.Soleimani M, Barone S, Xu J, Shull GE, Siddiqui F, Zahedi K, Amlal H. Double knockout of pendrin and Na-Cl cotransporter (NCC) causes severe salt wasting, volume depletion, and renal failure. Proc Natl Acad Sci U S A. 109(33):13368–73, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Amlal H, Soleimani M. Pendrin as a novel target for diuretic therapy. Cell Physiol Biochem. 28(3):521–6, 2011. Review. [DOI] [PubMed] [Google Scholar]
- 62.Pela I, Bigozzi M, Bianchi B. Profound hypokalemia and hypochloremic metabolic alkalosis during thiazide therapy in a child with Pendred syndrome. Clin Nephrol. 69(6):450–3, 2008. [DOI] [PubMed] [Google Scholar]
- 63.Knauf F, Yang CL, Thomson RB, Mentone SA, Giebisch G, Aronson PS. Identification of a chloride-formate exchanger expressed on the brush border membrane of renal proximal tubule cells. Proc Natl Acad Sci U S A. 98(16):9425–30, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Petrovic S, Ma L, Wang Z, Soleimani M. Identification of an apical Cl-/HCO3- exchanger in rat kidney proximal tubule. Am J Physiol Cell Physiol. 285(3):C608–17, 2003. [DOI] [PubMed] [Google Scholar]
- 65.Wang Z, Wang T, Petrovic S, Tuo B, Riederer B, Barone S, Lorenz JN, Seidler U, Aronson PS, Soleimani M. Renal and intestinal transport defects in Slc26a6-null mice. Am J Physiol Cell Physiol. 288(4):C957–65, 2005. [DOI] [PubMed] [Google Scholar]
- 66.Jiang Z, Asplin JR, Evan AP, Rajendran VM, Velazquez H, Nottoli TP, Binder HJ, Aronson PS. Calcium oxalate urolithiasis in mice lacking anion transporter Slc26a6. Nat Genet. 38(4):474–8, 2006. [DOI] [PubMed] [Google Scholar]
- 67.Freel RW, Hatch M, Green M, Soleimani M. Ileal oxalate absorption and urinary oxalate excretion are enhanced in Slc26a6 null mice. Am J Physiol Gastrointest Liver Physiol. 290(4):G719–28, 2006. [DOI] [PubMed] [Google Scholar]
- 68.Soleimani M. Dietary ▪ fructose, salt absorption and ▪ hypertension ▪ in ▪ metabolic syndrome : Toward a new paradigm. Acta Physiol. 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Douard V, Ferraris RP. Regulation of the fructose transporter GLUT5 in health and disease. Am J Physiol Endocrinol Metab. 295(2):E227–37, 2008. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Singh AK, Amlal H, Haas PJ, Dringenberg U, Fussell S, Barone SL, Engelhardt R, Zuo J, Seidler U, and Soleimani M. Fructose- induced hypertension: essential role of chloride and ▪ fructose absorbing transporters PAT1 and Glut5. Kidney Int 74: 438–447, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Barone S, Fussell SL, Singh AK, Lucas F, Xu J, Kim C, Wu X, Yu Y, Amlal H, Seidler U, Zuo J, Soleimani M. Slc2a5 (Glut5) is essential for the absorption of fructose in the intestine and generation of fructose-induced hypertension. J Biol Chem. 84(8):5056–66, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Soleimani M, Alborzi P. The role of salt in the pathogenesis of fructose-induced hypertension. Int J Nephrol. 2011;2011:392708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Petrovic S, Barone S, Xu J, Conforti L, Ma L, Kujala M, Kere J, Soleimani M. SLC26A7: a basolateral Cl-/HCO3- exchanger specific to intercalated cells of the outer medullary collecting duct. Am J Physiol Renal Physiol. 286(1):F161–9, 2004. [DOI] [PubMed] [Google Scholar]
- 74.Xu J, Worrell RT, Li HC, Barone SL, Petrovic S, Amlal H, Soleimani M. Chloride/bicarbonate exchanger SLC26A7 is localized in endosomes in medullary collecting duct cells and is targeted to the basolateral membrane in hypertonicity and potassium depletion. J Am Soc Nephrol. 17(4):956–67, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Petrovic S, Amlal H, Sun X, Karet F, Barone S, and Soleimani M. Vasopressin induces expression of the Cl/HCO3- exchanger ▪ SLC26A7 in kidney medullary ▪ collecting ducts of Brattleboro rats. Am J Physiol Renal Physiol 290: F1194–F1201, 2006. [DOI] [PubMed] [Google Scholar]
- 76.Barone S, Amlal H, Xu J, Kujala M, Kere J, Petrovic S, Soleimani M. Differential regulation of basolateral Cl-/HCO3- exchangers SLC26A7 and AE1 in kidney outer medullary collecting duct. J Am Soc Nephrol. 15(8):2002–11, 2004. [DOI] [PubMed] [Google Scholar]
- 77.Barone S, Amlal H, Kujala M, Xu J, Karet F, Blanchard A, Kere J, Soleimani M. Regulation of the basolateral chloride/base exchangers AE1 and SLC26A7 in the kidney collecting duct in potassium depletion. Nephrol Dial Transplant. 22(12):3462–70, 2007. [DOI] [PubMed] [Google Scholar]
- 78.Xu J, Song P, Nakamura S, Miller M, Barone S, Alper SL, Riederer B, Bonhagen J, Arend LJ, Amlal H, Seidler U, and Soleimani M Deletion of the chloride transporter Slc26a7 causes distal renal tubular acidosis and impairs gastric acid secretion. J Biol Chem 284(43), 29470–29479, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chang MH, Plata C, Zandi-Nejad K, Sindić A, Sussman CR, Mercado A, Broumand V, Raghuram V, Mount DB, and Romero MF Slc26a9--anion exchanger, channel and Na+ transporter. J Membr Biol 228(3), 125–140, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Amlal H, Xu J, Barone S, Zahedi K, and Soleimani M The Chloride Channel/Transporter Slc26a9 Regulates the Systemic Arterial Pressure and Renal Chloride Excretion. J. Molecular Medicine (accepted for publication). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Rahmati N, Kunzelmann K, Kongsuphol P, Xu J, Barone S, De Zeeuw CI, Soleimani M Slc26a11 is a chloride channel in the brain: expression in purkinje cells and stimulation of V H+-ATPase. Under review. [DOI] [PMC free article] [PubMed]
- 82.Schwartz GJ, Barasch J, Al-Awqati Q. Plasticity of functional epithelial polarity. Nature 318: 368–371, 1985. [DOI] [PubMed] [Google Scholar]
- 83.Brown D, Paunescu TG, Breton S, Marshansky V. Regulation of the V-ATPase in kidney epithelial cells: dual role in acid-base homeostasis and vesicle trafficking. J Exp Biol 212: 1762–1772, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wagner CA, Giebisch G, Lang F, Geibel JP. Angiotensin II stimulates vesicular H+-ATPase in rat proximal tubular cells. Proc Natl Acad Sci U S A. 95(16):9665–8, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Carraro-Lacroix LR, Lessa LM, Fernandez R, Malnic G. Physiological implications of the regulation of vacuolar H+-ATPase by chloride ions. Braz J Med Biol Res. 42(2):155–63, 2009. [DOI] [PubMed] [Google Scholar]
- 86.Forlino A, Piazza R, Tiveron C, Della Torre S, Tatangelo L, Bonafè L, Gualeni B, Romano A, Pecora F, Superti-Furga A, Cetta G, Rossi A. A diastrophic dysplasia sulfate transporter (SLC26A2) mutant mouse: morphological and biochemical characterization of the resulting chondrodysplasia phenotype. Hum Mol Genet. 14(6):859–71, 2005. [DOI] [PubMed] [Google Scholar]