

Abstract
The knotted configuration of lasso peptides confers thermal stability and proteolytic resistance, addressing two shortcomings of peptide-based drugs. However, low isolation yields hinder the discovery and development of lasso peptides. While testing Burkholderia sp. FERM BP-3421 as a bacterial host to produce the lasso peptide capistruin, an overproducer clone was previously identified. In this study, we show that an increase in plasmid copy number partially contributed to the overproducer phenotype. Further, we modulated plasmid copy number to recapitulate titers to an average of 160% relative to the overproducer, which is 1,000-fold higher than previously reported with E. coli, reaching up to 240 mg/L. To probe the applicability of the developed tools for lasso peptide discovery we targeted a new lasso peptide biosynthetic gene cluster from endosymbiont Mycetohabitans sp. B13, leading to the isolation of mycetolassin-15 and mycetolassin-18 in combined titers of 11 mg/L. These results validate Burkholderia sp. FERM BP-3421 as a production platform for lasso peptide discovery.
Keywords: lasso peptide, RiPP, natural product, secondary metabolite, bacteria, heterologous expression, Burkholderiaceae
Graphical Abstract
INTRODUCTION
Lasso peptides belong to the ribosomally synthesized and post-translationally modified peptide (RiPP) class of natural products. The distinctive structure of lasso peptides (lariat-like knot, Figure 1A) confers thermal stability and proteolytic resistance, welcome characteristics for peptide products.1 Lasso peptides are attracting increased attention due to their diverse potential applications and programmability via epitope grafting and genetically encoded libraries.2–4 However, the development of lasso peptides as drug leads is hampered by the lack of effective synthetic methods. In fact, only one successful chemical synthesis of a lasso peptide has been reported.5 However, the cryptand-imidazolium-supported, multi-linker method used to enable threading is not generalizable, and the overall yield was 2.5%.6 These challenges point to an enzymatic method to access lasso peptides. However, such procedures have also typically been low yielding.7–9
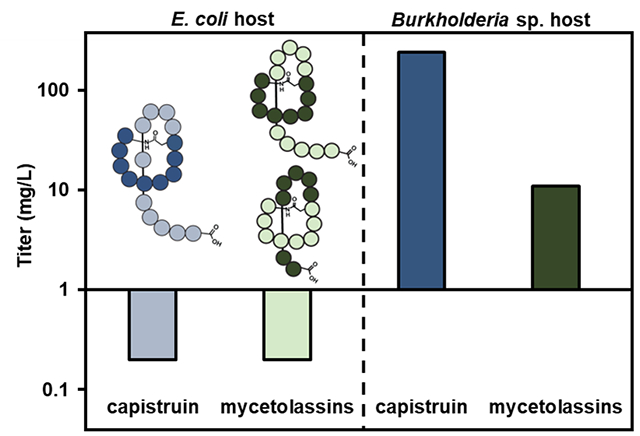
Figure 1. Lasso peptide biosynthesis and titers.
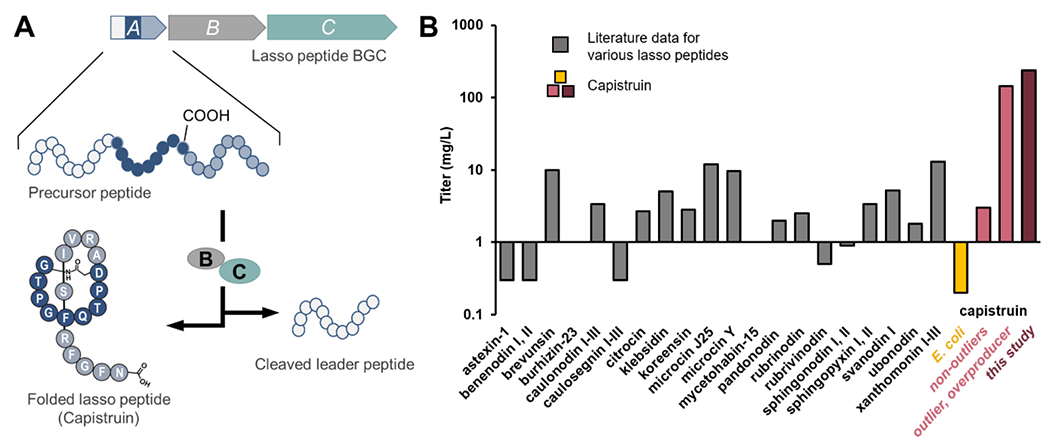
(A) Lasso peptide biosynthesis involves a minimum of three genes. Gene A encodes a precursor peptide; gene B, encodes a RiPP Recognition Element (RRE) domain and a leader peptidase that removes the leader sequence (white); and gene C, a lasso cyclase, catalyzes isopeptide bond formation with concomitant threading of the C-terminal tail. (B) Bar graph in log scale comparing reported titers of Pseudomonadota-derived lasso peptides (gray) after heterologous expression of the indicated BGCs (E. coli used in all cases, except for brevunsin, Sphingomonas subterranea; burhizin-23, Burkholderia gladioli; and koreensin, Sphingomonas subterranea – see Table S1 for details). Multiple titers are shown for capistruin expression. The titer previously reported with E. coli (0.2 mg/L, yellow) is shown for comparison with the titers we previously obtained with Burkholderia sp. FERM BP-3421(light pink). In the previous study, plasmid pSK020 containing the cap BGC was transferred into FERM BP-3421 and five clones were analyzed, four of which produced an average of 3 mg/L capistruin (non-outliers), and one of which was an outlier overproducer clone (>100 mg/L).7 This study’s recapitulation of high-titer capistruin production with FERM BP-3421 and further improvement is shown in dark pink (237 ± 6 mg/L).
Reported titers for heterologous expression of lasso peptides from Proteobacteria (now Pseudomonadota) vary from 0.2–13 mg/L, with E. coli being the most popular host (Figure 1B). The selection of host strain and the development of a host through strain engineering are key factors for successful and high-yielding production.10 Recently, β-Protobacteria, particularly members of the Burkholderia genus, have gained interest as both a source of and a means to access natural products.11,12 Burkholderia sp. FERM BP-3421 is a nonpathogenic soil isolate first used to produce autologous spliceostatins (nonribosomal peptide-polyketide) at industrial scale and with titers up to 6 g/L.13 In a previous study7 carried out to test FERM BP-3421 as an alternative host, an outlier, overproducer clone of the lasso peptide capistruin14 was identified after expression of the biosynthetic gene cluster (BGC) from Burkholderia thailandensis. The outlier clone consistently produced capistruin at an unprecedented average titer of >100 mg/L, at least one order of magnitude greater than previously reported titers of other lasso peptides from Pseudomonadota (Figure 1B).
In this report, we show that an increase in plasmid copy number (PCN) contributes to the overproducer phenotype. PCN modulation of expression plasmids in the Burkholderia host was then leveraged to further improve capistruin titer and to isolate and characterize two new lasso peptides.
RESULTS AND DISCUSSION
Host genome and plasmid sequencing reveals a 48-bp insertion in the low producer plasmid.
To obtain insight into the basis for high yield capistruin production by the outlier overproducer, we performed plasmid and draft genome sequencing to probe for mutations. Both the outlier overproducer and a non-outlier (hereafter termed “low producer”) clone were sequenced. Whole-plasmid sequencing has recently become available as an alternative to Sanger sequencing. Whole-plasmid sequencing of the pSK020 plasmid harboring the cap BGC7 revealed a 48-bp insertion between the ribosome binding site (RBS) and the −10 and −35 promoter regions in the low producer pSK020 plasmid (hereafter termed “pSK020+48”), whereas pSK020 from the overproducer was as designed without the insertion. The 48-bp insertion increases the distance between the PBAD promoter and the RBS and alters the sequence context for transcription and translation initiation, which is known to affect gene expression and subsequent production yield.15–17 Specifically, the 48-bp insertion disrupts the translation enhancer sequence located between the RBS and the promoter which is involved in ribosome interactions and mRNA stabilization (Figure S1A).16,17 The source of the insertion was the pUC57-Kan vector (Genscript) in which the cap BGC had been cloned after synthesis.7 The 48-bp insertion consists of inverted sequence repeats originating from the pUC57-Kan cloning vector (Figure S1B), which are not present in pSK020 from the overproducer.
To test for genomic mutations, whole genome draft sequencing was performed. Illumina sequence reads were mapped to the reference genome sequence for FERM BP-3421.18 Because we observed differences between the Illumina sequence and our reference genome for both the overproducer and low producer clones (Table S2), we hypothesized at least some of these differences were Illumina sequence errors rather than actual mutations. Illumina sequencing can result in an error rate as low as 10−5.19 Given that the size of our genome is ~7.7 Mbp, that presents the opportunity for ~100 potential sequence errors, which is on par with what we identified. An alternative hypothesis is that at least some of the sequence differences observed were actual mutations that contributed to the overproducer phenotype.
We next performed experiments to distinguish plasmid and genome influences on the overproducer phenotype. To test whether the 48-bp deletion is sufficient to explain the elevated titers, we transferred the correct pSK020 construct from the overproducer into the wild-type host. Although titers were higher than with the low producer, only 34% of the overproducer titers were reached (Figure S2).
If genomic mutations contribute to the overproducer phenotype, the overproducer cleared of the pSK020 plasmid could provide an improved host for the expression of lasso peptide BGCs. To test this hypothesis, we first cleared the overproducer clone of the pSK020 plasmid by growth on non-selective media. We then reintroduced pSK020+48 in the cleared host and observed the same titers as the low producers (Figure S3A). Furthermore, reintroduction of pSK020 in the cleared host resulted in titer improvement compared to the low producers but the titers reached only 21% of those of the overproducer (Figure S3B), comparable to the relative production observed when pSK020 was reintroduced in the wild type (Figure S2). The combined results established that genomic mutations were not responsible for overproduction and that the plasmid-cleared strain cannot be used as an improved chassis. We speculate that both the 48-bp pSK020 plasmid deletion and a reversible, host-based mechanism that is lost/reverts during plasmid clearing, together account for the overproducer phenotype. The nature of the reversible mechanism will be the subject of future studies.
An increase in plasmid copy number partially contributes to the overproducer phenotype.
An actionable observation during draft genome sequence comparison between low and overproducer clones was a normalized increase in coverage for the overproducer pSK020 plasmid which indicated a difference in PCN. We observed a 4-fold increase in normalized sequence reads of pSK020 plasmid from the overproducer when compared to the normalized sequence reads of pSK020+48 plasmid from the low producer (Figure 2A). Subsequent quantitative PCR analysis (Figure 2B) corroborated a ~4-fold increase in PCN, indicating that increased PCN correlates with the overproducer phenotype.
Figure 2. An increase in PCN contributes to the overproducer phenotype.
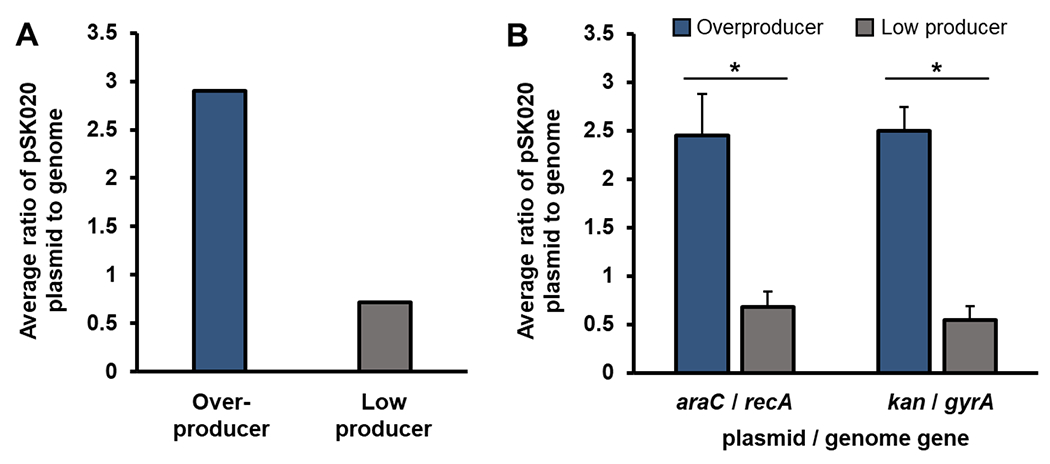
(A) Normalized PCN per genome equivalent determined by mapping Illumina sequence reads to the Burkholderia sp. reference genome.18 (B) Quantitative PCR. The average ratio of pSK020 plasmid to the genome was calculated based on two comparisons, the amplification of araC (cap plasmid pSK020) and recA (genome), and the amplification of the kanamycin resistance gene kan (cap plasmid pSK020) and gyrA (genome). The average of triplicate PCR ± standard deviation is shown. T-test was implemented for statistical analysis. *, P < 0.05.
Modulation of plasmid copy number to recapitulate and optimize capistruin titers.
To assess strain fitness, growth curves were generated with Burkholderia harboring either pSK020 (overproducer) or pSK020+48 (low producer). Results from growth assessment showed that the ~50-fold improvement in capistruin titers in the overproducer (Figure 3A) is accompanied by improved growth (Figure 3B). Although the mechanism remains unknown, both PCN and growth were improved in the overproducer. Thus, we next explored PCN modulation and the effect on growth to recapitulate capistruin titers.
Figure 3. Modulation of plasmid copy number to optimize capistruin titers.
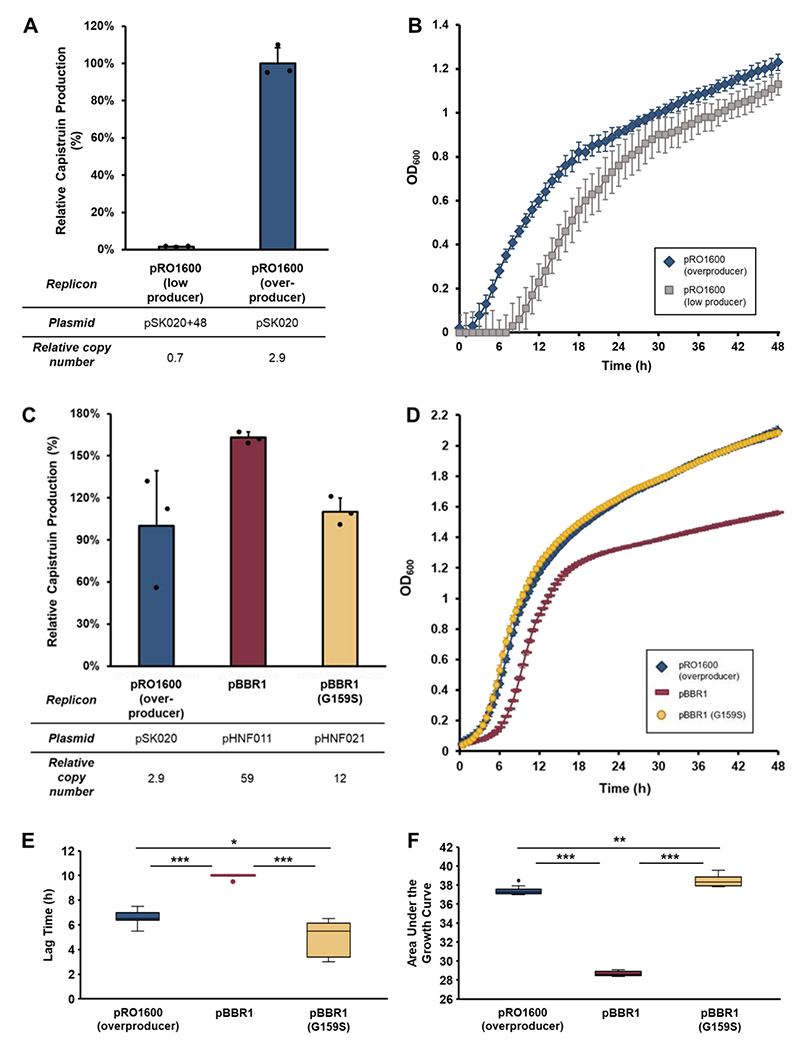
(A) Relative capistruin production between Burkholderia sp. FERM BP-4321 harboring the capistruin expression plasmid pSK020+48 and pSK020, both of which are expression vectors with a pRO1600 replicon. The average of N = 3 ± standard deviation is shown. (B) Growth comparison in production media between the Burkholderia host containing either the pSK020 or pSK020+48 capistruin expression plasmid. The average of N = 8 ± standard deviation is shown. (C) Comparison of capistruin production between the pSK020 (pRO1600 replicon) overproducer and either pHNF011 (containing a wild type pBBR1 replicon) or pHNF021 (containing a G159S mutated pBBR1 replicon). (D) Growth curves of Burkholderia containing either pSK020 (pRO1600 replicon) overproducer, pHNF011 (wild type pBBR1), or pHNF021 (mutated pBBR1) expression plasmids. The average of N = 10 ± standard deviation is shown. (E) Comparison of strain fitness when utilizing vectors with different plasmid replicons. The time that strains harboring alternate capistruin expression vectors take to reach the end of the lag phase and begin exponential growth is shown. (F) Comparison of the area under growth curve after 32 h for strains harboring alternate capistruin expression vectors. Analyses of lag time (E) and area under growth curve (F) are based on the growth curves in panel D. The wild-type strain of Burkholderia sp. FERM BP-3421 was used to produce capistruin in these studies. T-test was implemented for statistical analysis. *, P ≤ 0.05; **, P ≤ 0.001; ***, P ≤ 1.0E−5.
Normalized Illumina reads and quantitative PCR suggested that the pRO1600 replicon present in the pSK020 plasmids might be unstable in the Burkholderia sp. FERM BP-3421 host (except for the overproducer, average of <1 copy per genome equivalent, suggesting that some cells have no plasmid, Figure 2). While the pRO1600 replicon was estimated to be maintained at medium copy in Pseudomonas aeruginosa (10–25 copies per cell),20 PCN is host dependent.21,22
Only vectors with the pRO1600 replicon had been previously used in Burkholderia sp. FERM BP-3421.7,13 To test the copy number hypothesis further and attempt to recapitulate high titers, we obtained three additional vectors based on broad-host range replicons, i.e., RSF1010, RK2, and pBBR1.23–26 Illumina sequencing indicated a high PCN for all three vectors in Burkholderia sp. FERM BP-3421, measured as 190 (RSF1010), 430 (RK2), and 560 (pBBR1) copies per genome equivalent for the empty vectors (Figure S4). We thus decided to evaluate capistruin production using the highest PCN vector (pBBR1-based).
Although the pBBR1 replicon is reportedly low to medium copy number in other bacteria (~3 copies per cell for E. coli and ~30 for Pseudomonas),27 our data showed it to be the highest copy number vector in Burkholderia sp. FERM BP-3421. We cloned the cap BGC under control of the l-arabinose inducible PBAD promoter in the pBBR1-based vector pMo168 to generate pHNF011 (Figure S5). Illumina sequencing of total DNA showed that pHNF011 had 59 copies per genome equivalent. Increased plasmid size can result in decreased PCN,28,29 and transcription can also affect replication,30–32 likely explaining the order of magnitude reduction in PCN for the vector containing the cap BGC relative to the empty vector.
Satisfyingly, comparative metabolite analysis showed that pHNF011 increased the production of capistruin to overproducer levels by 160% (Figure 3C). However, cells harboring pHNF011 have a growth defect in production cultures compared to the overproducer. The growth defect is evident based on a prolonged lag phase and a reduction in the total area under the growth curve, indicating a lower overall cell density (Figure 3D–3F). To corroborate OD600 measurements, wet cell weights and colony forming units per milliliter (CFU/mL) counts were acquired from 48-h production cultures. In agreement with the growth curve measurements, cultures harboring pHNF011 had significantly reduced wet cell weight and CFU/mL as compared to the pSK020 overproducer and the pHNF021 cultures (Figure S6). The maintenance of high PCN vectors can negatively affect host fitness and lead to reduced production.29,33,34 Moreover, there was a correlation between host fitness and capistruin production observed with the pSK020 overproducer (Figure 3B).
A point mutation in the rep gene of the pBBR1 replicon has been shown to reduce copy number and eliminate phenotypic defects in Pseudomonas putida by reducing overall plasmid toxicity.34 To determine if PCN reduction would improve growth and increase capistruin production, we introduced the reported mutation (G to A at rep nucleotide position 468, encoding for Rep-G159S) to generate pHNF021 (Figure S7). Although the biochemical mechanism of PCN reduction is unknown, the fitness defect was indeed alleviated (Figures 3D–3F; Figure S6). However, capistruin titers were reduced compared to pHNF011 (Figure 3C). Still, pHNF021 recapitulated overproducer capistruin titers to an average of 110% (Figure 3C).
A mutant strain defective in autologous spliceostatin biosynthesis is an improved chassis.
Spliceostatins are autologous polyketide-nonribosomal peptides produced by Burkholderia sp. FERM BP-3421 at gram per liter scale under the growth conditions used in this study.13 We previously showed that expression of the cap BGC using pSK020+48 in a spliceostatin-defective mutant strain (fr9DEF−) led to 4-fold titer increase compared to the wild-type host.7 In the present study, we tested expression with the high-copy pBBR1-based plasmid pHNF011. Plasmid pHNF011 in the fr9DEF- background led to a significant 1.2-fold increase in capistruin titers compared to production in the wild-type host (Figure S8). While the relative increases appear modest in comparison with the low copy pSK020+48 plasmid, the absolute increase was superior (10 mg/L for pSK020+48, and 24 mg/L for pHNF011, on average). Thus, we used the spliceostatin-defective mutant in subsequent experiments because it outperformed the wild type with capistruin, and because the absence of spliceostatins facilitates heterologous product purification.
Lasso peptide discovery.
About 100 lasso peptides have been experimentally isolated and/or characterized. Bioinformatic tools, such as RODEO35 (Rapid ORF Description & Evaluation Online), in combination with an increasing number of publicly available genomes, enable the detection of underexplored biosynthetic landscapes. A previous study36 predicted 4,485 unique lasso peptide core sequences and 5,469 unique lasso peptide BGCs from publicly available genomes, showing that lasso peptides are significantly underexplored. We searched this set of predicted lasso peptides36 for BGCs encoded by Burkholderiaceae. Of the 38 unique lasso peptides predicted from Burkholderiaceae, seven have been experimentally characterized (Figure S9).9,14,37–39 We targeted the lasso peptide BGC predicted in the genome of fungal endosymbiont Mycetohabitans sp. (basonym Burkholderia sp.) B13 (Figure 4A).40 The Mycetohabitans sp. B13 BGC (mlsA1A2BCD) has similar organization and content compared to the cap BGC (capABCD), except it harbors two predicted precursor peptide (A) genes. The predicted core peptides of the mls BGC are closely related to the since isolated mycetohabins (Figure S9).39
Figure 4. Discovery of lasso peptides from Mycetohabitans sp. B13.
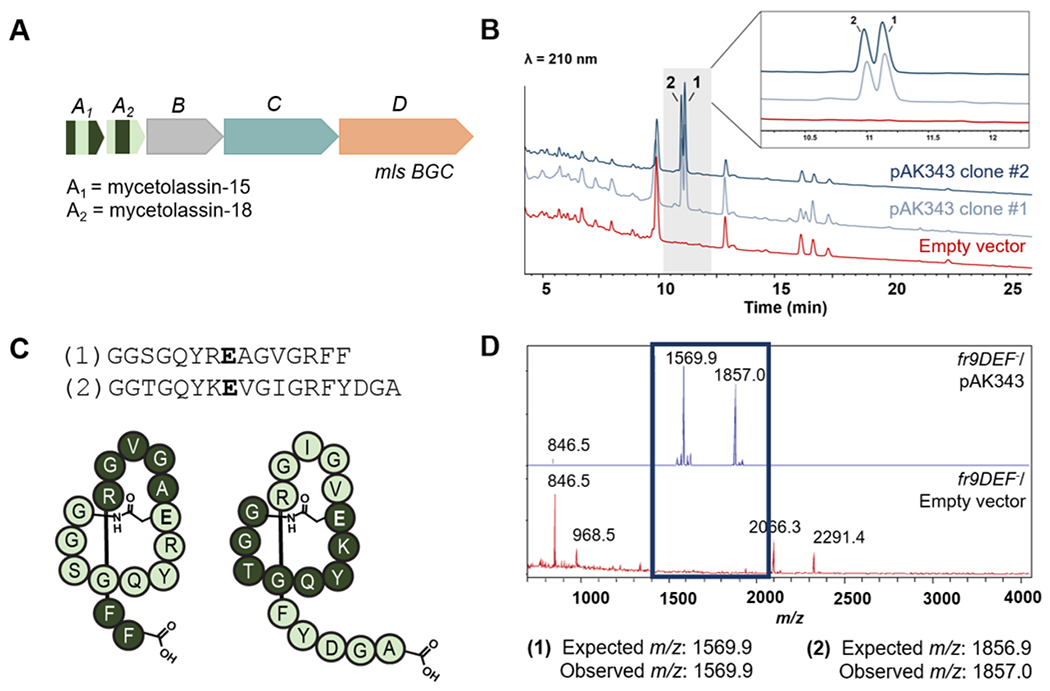
(A) Graphical representation of the mycetolassin BGC (NZ_FTPM01000001). Genes mlsA1 and mlsA2 encode for the precursor peptides of mycetolassin-15 and mycetolassin-18, respectively. (B) HPLC traces of culture extracts showing production of two lasso peptides. Detection at 210 nm. (C) Sequences and predicted structures of mycetolassin-15 (1) and mycetolassin-18 (2). (D) MALDI-TOF mass spectrometry showing detection of the two expected m/z features for [M+H]+.
We synthesized the mls BGC, as DNA fragments, altering any codons that were rare for either Burkholderia or E. coli to compare expression in both hosts. We first cloned genes mlsA1A2BC in the pRO1600-based vector pSK019 under control of the l-arabinose-inducible PBAD promoter to yield pAK343. As determined by whole plasmid sequencing, pSK019 and pAK343 are as designed and do not contain the extra 48-bp fragment found in the capistruin low producers. As the native RBS was the same for mlsA1 as for capA, we kept the sequence from the RBS to the start of mlsA1 gene consistent. We used the fr9DEF− strain as the host for these experiments because it outperformed the wild-type with capistruin. After extracting the cell pellet of the production cultures, two lasso peptides were detected by high-performance liquid chromatography (HPLC) (Figure 4B), consistent with the two precursor peptides encoded in the BGC (Figure 4C). The m/z features observed by matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry (MS) were consistent with the cyclized predicted core peptides of MlsA1 (1, herein mycetolassin-15) and of MlsA2 (2, herein mycetolassin-18), respectively (Figure 4D).
Mycetolassin-15 and mycetolassin-18 were HPLC purified from 1 L production cultures. Tandem MS experiments identified only y and b ions consistent with the removal of the C-terminal residues, which aligns with the expected N-terminal macrocyclization (Figures S10–11). For mycetolassin-18, additional low-intensity ions corresponding to fragmentation within the ring were present (Figure S11). Several assays were performed to evaluate if the lasso peptides were in a threaded or branched-cyclic conformation. First, mycetolassin-15 and mycetolassin-18 were treated with carboxypeptidase. Subsequent MS analysis showed that no truncations were observed for mycetolassin-15 and only the final two C-terminal residues of mycetolassin-18 were removed (Figure S12). After heating at 95 °C, mycetolassin-15 and mycetolassin-18 exhibited similar resistance to carboxypeptidase Y, indicative of thermostability. We also performed a hydrogen-deuterium exchange assay on purified mycetolassin-15 and mycetolassin-18 (solubility in water >20 mg/ml). While full deuteration is observed for unstructured peptides within 1 min,41 only a fraction of the exchangeable protons were deuterated on this timescale, and the sample was not fully deuterated within 24 h (Figure S13). The resistance to carboxypeptidase Y, H/D exchange, and thermal stability collectively support the isolated lasso peptides are in a globular, threaded conformation (Figure 4C).
Evaluation of plasmid copy number variation for mycetolassin production.
To test whether modulating PCN would lead to similar titer improvements as observed with capistruin, we subcloned the mls BGC into a pBBR1-based vector to yield pHNF007 (Figure S14A). PCN increased from 0.27 to 22 (~80-fold) whereas mycetolassin-15 and mycetolassin-18 combined titer increased by 2.4-fold compared to cultures harboring the pRO1600-based vector (Figure 5A). Growth was substantially improved with the pBBR1-based vector (Figure 5B and Figure S15).
Figure 5. Comparison of mycetolassin titers using different vector replicons.
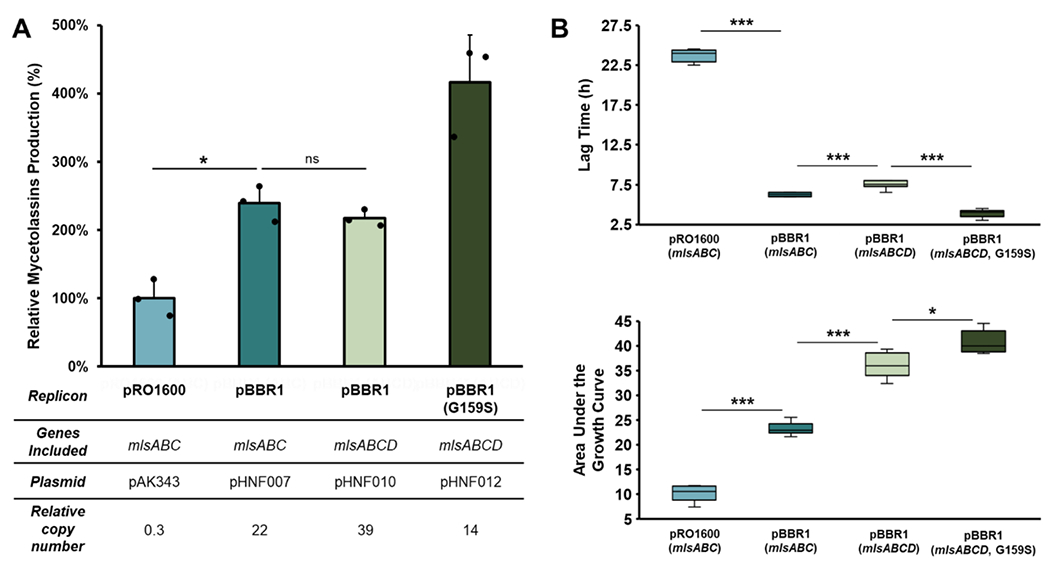
(A) Combined titers of mycetolassin-15 and mycetolassin-18 obtained using expression vectors with different replicons and with or without the transporter gene (mlsD) included in the construct. The average of N = 3 ± standard deviation is shown. (B) Comparison of strain fitness when utilizing mycetolassin expression vectors harboring different replicons. Growth analyses are based on a 48-h growth curve carried out in production media (Figure S15). Analysis of the time that the strain harboring the indicated mycetolassin expression vector takes to reach the end of lag phase and begin exponential growth phase (top). Comparison of the area under the growth curve after 32 h for strains harboring the indicated expression vector (bottom). Box and whisker plots of N = 10 are shown. The fr9DEF− mutant strain of Burkholderia sp. FERM BP-3421 was used for these studies. T-test was implemented for statistical analysis. *, P ≤ 0.05; **, P ≤ 0.001; ***, P ≤ 1.0E−5; ns, no significant difference.
To test if titer was impacted by the lack of the native transporter gene, the transporter gene (mlsD) was added to the construct to yield pHNF010 (Figure S14B). If mycetolassins have any antibiotic properties affecting the heterologous host, export from the cells may have increased titer. Including mlsD facilitated lasso peptide export as evidenced by an increase from 50% to 94% of the total titer of the mycetolassins isolated from the supernatant (Figure S16). However, inclusion of mlsD did not lead to a significant improvement in titers, suggesting that export was not yield limiting (Figure 5A). Growth with mlsD was delayed (longer lag phase) but the overall area under the growth curve was larger (Figure 5B). It is unclear why relative PCN with the transporter gene would be higher, but we speculate it may be related to improved overall growth.
Although the pBBR1 rep mutation did not increase capistruin titer, we evaluated if mycetolassin titer would be affected. Site-directed mutagenesis of pHNF010 generated pHNF012 and a significant fitness improvement was evident (Figure 5B). Further, production of mycetolassins was doubled compared to the wild-type rep vector (Figure 5A). In all, a 4-fold improvement was obtained compared to the initial pRO1600-based pAK343, corresponding to 11 mg/L isolated yield for the two lasso peptides combined (6 mg/L for mycetolassin-15 and 5 mg/L for mycetolassin-18).
While the fold changes observed with mycetolassins were less dramatic than with capistruin, it is important to note that different host backgrounds were used in each case (wild type for capistruin and fr9DEF− spliceostatin mutant for mycetolassins). Although speculative, the fact that the fr9DEF− mutant affords higher titers with a low PCN vector may explain the lower fold changes when modulating copy number in this host background. Additionally, differences in gene expression and the biological activities of the products may play a role. Nevertheless, the experiments with mycetolassins corroborated, as seen with capistruin, that a pBBR1-based expression system achieves greater titers than a pRO1600-based one in the Burkholderia host. The combined growth and copy number data highlighted that the pRO1600 replicon is not ideal for the Burkholderia host, as observed with capistruin (the exception being the outlier overproducer). Burkholderia harboring the pRO1600 expression vector displayed impaired plasmid maintenance with a normalized PCN of <1 and a significant growth defect relative to Burkholderia harboring pBBR1 vectors (Figure 5).
Curiously, however, the pBBR1 mutated rep vector was the most productive for mycetolassins while the wild type pBBR1 rep vector was the most productive for capistruin. Production improvement is multifactorial and context dependent. As previously mentioned, the lasso peptide produced, and the host used for expression may impact which vector is most effective.
The Burkholderia host outperforms E. coli with mycetolassins.
When the mls BGC was expressed in E. coli, only trace amounts of mycetolassin-15 and -18 were detected by MALDI-TOF MS, with an approximate titer of 200 μg/L mycetolassin-18 as determined by HPLC (Figure S17). This indicated a 25-fold increase in titer upon use of the Burkholderia host. Production of two new lasso peptides in higher titers validates our platform for lasso peptide discovery and production beyond capistruin.
Comparative analysis of the biosynthetic gene clusters for capistruin and mycetolassins.
Despite capistruin and the mycetolassins originating from bacteria of the same taxonomic family, there are significant BGC differences. The homologous proteins all share <30% sequence identity (29%, 26%, and 29% for the leader peptidases CapB and MlsB, lasso cyclases CapC and MlsC, and transporters CapD and MlsD, respectively). Varying expression efficiencies of the cap and mls BGCs likely contribute to the observed differences in lasso peptide titers.
Sequence similarity of mycetolassins to known lasso peptides.
While the core peptide sequence of capistruin is not similar to that of mycetolassins (Figure S9B), there are three lasso peptides reported in the literature that have similar sequences (Figure S9C): burhizin (7 out of 15 amino acids are identical for mycetolassin-15 and 10 out of 18 for mycetolassin-18 compared to burhizin); mycetohabin-15 (14 out of 15 for mycetolassin-15 and 10 out of 18 for mycetolassin-18); and mycetohabin-16 (12 out of 15 for mycetolassin-15 and 10 out of 18 for mycetolassin-18).9,39 Literature reports of the heterologous expression of burhizin and mycetohabin yielded 1 mg/L from Burkholderia gladioli and E. coli hosts, respectively. It is conceivable that with the successful expression of the mycetolassins at a higher combined titer of 11 mg/L, and capistruin at 240 mg/L, Burkholderia sp. FERM BP-3421 may provide a superior chassis to aid in further structural and biological characterization of additional lasso peptides.
Biological activity of mycetolassin-15 and mycetolassin-18.
We used purified mycetolassin-15, mycetolassin-18, and mycetolassin-18Δ2 (lacking two C-terminal residues) for bioactivity assessment. No antimicrobial activity was observed at up to 200 μM against an ESKAPE pathogen panel, consistent with previous reports on burhizin and mycetohabin.39 Because several lasso peptides from Pseudomonadota, including capistruin, have demonstrated inhibition of RNA polymerase, and other lasso peptides have required an import system to enter bacteria, we utilized a cell-free assay with cell lysate to test the inhibition of transcription and translation.36,42 This method effectively removes the barrier of cell entry for lasso peptides, and more directly assesses mode of action. Production of mCherry was assessed in the presence and absence of mycetolassins. While capistruin at 30 μM inhibited mCherry production, the mycetolassins had no effect, suggesting they are not transcription or translation inhibitors (Figure S18). Although the conservation of this family of lasso peptides in endosymbionts would suggest a specific role, the biological activity for mycetolassins remains unidentified, as it is true for the related lasso peptides burhizin and mycetohabins.9,39
Conclusions.
In this study, we have obtained insights into the basis of high titer capistruin production by an outlier Burkholderia sp. heterologous clone. An increase in PCN contributed to the overproducer phenotype as evidenced from genomic and quantitative PCR data. We demonstrated that the pRO1600 replicon initially used for capistruin heterologous expression7 is unstable and of low copy in Burkholderia, despite being of medium-copy in Pseudomonas.20,27 This study established high copy pBBR1-based vectors for Burkholderia sp. FERM BP-3421 as a chassis. Additional PCN modulation was implemented to optimize growth and recapitulate high titer capistruin production in a reproducible manner. We then tested the developed tools with an orphan lasso peptide BGC from the endosymbiont Mycetohabitans sp. B13, leading to the discovery of mycetolassin-15 and mycetolassin-18. Efforts to express lasso peptide BGCs from other bacterial taxa to probe the breadth of the host are currently ongoing.
METHODS
Strains, cultivation conditions, chemicals, and general procedures.
Burkholderia sp. FERM BP-3421 was cultivated in Luria-Bertani (LB) liquid media or on LB agar at 30 °C unless otherwise stated.13,43 Chemically competent E. coli DH5α (Invitrogen, cat. no. 18265017) and chemically competent E. coli NEB5α (NEB, cat. no. C2987H) were used for plasmid construction. E. coli cultures were grown at 30 °C in LB medium containing appropriate antibiotics. Bacterial strains were preserved in 20% sterile glycerol and stored at −80 °C.
Oligonucleotide primers used in PCR were designed using the bioinformatics software Geneious Prime and synthesized by Sigma-Aldrich or IDT. All primer sequences used for PCR amplifications are listed in Table S3. The restriction enzymes and T4 ligase used for cloning were from New England Biolabs. Q5 High-Fidelity DNA polymerase (NEB, cat. no. M0491) and Dream Taq polymerase (Thermo Fisher, cat. no. K1072) were used in PCR unless otherwise stated. PCR amplifications were performed using a Biorad T100 thermal cycler. The standard PCR thermal cycling conditions using Q5 polymerase were initial denaturation for 30 s at 98 °C, followed by 35 amplification cycles of 10 s denaturation at 98 °C, 30 s at the appropriate annealing temperature, and 2 min extension at 72 °C. The amplification cycles were followed by a final extension for 2 min at 72 °C. After thermal cycling, PCR were stored at 4 °C. Unless otherwise noted, 10 μL of the ligation mix was used to transform chemically competent E. coli DH5α or E. coli NEB5α by heat shock according to the manufacturer’s instructions. The selection of DH5α or NEB5α clones was performed on LB agar containing kanamycin (50 μg/mL). Transformed E. coli clones were verified by restriction digest and sequencing. Sanger sequencing was performed by the University of Illinois at Chicago (UIC) DNA sequencing core. Whole plasmid sequencing was performed by Primordium Labs.
Plasmid DNA isolation was performed with the Zymo Research plasmid miniprep kit (cat no. D4016). DNA cleanup and re-isolation during cloning was performed with the Zymo Research clean and concentrator kit (cat. no. D4014). Plasmid concentrations were verified by NanoDrop. Molecular biology procedures were executed according to manufacturer instructions.
Genomic DNA was isolated with the GenElute Bacterial Genomic DNA kit (Sigma Aldrich, cat. no. NA2120-1KT) according to manufacturer instructions for Gram-negative genomic DNA isolation. Isolation and DNA concentration were confirmed and determined by gel electrophoresis.
The following media were used for production cultures (concentrations in w/v): seed medium,13 containing peptone (Bacto Difco) 1%, yeast extract (Bacto Difco) 0.5%, and sodium chloride (Sigma-Aldrich) 0.5%; and 2S4G production medium, containing glycerol (Fisher Scientific) 4%, peptone (Bacto Difco) 2%, ammonium sulfate (Sigma-Aldrich) 0.2%, magnesium sulfate (Sigma-Aldrich) 0.006%, and calcium carbonate (Acros Organics) 0.2%.
Starter cultures for production contained 5 mL of seed medium and 500 μg/mL of kanamycin. Starter cultures were inoculated with 20 μL of a glycerol stock of strain FERM BP-3421 containing either the empty vector, pUCPneo (negative control) or the requisite expression plasmid. After incubation at 30 °C, 200 rpm for 1–2 d, starter cultures were used to inoculate 50 mL of 2S4G production medium contained in 250 mL Erlenmeyer flask to a starting OD600 of 0.01 (usually about 200–300 mL of starter culture was used to reach the desired OD). For plasmid maintenance and gene expression, production cultures were supplemented with kanamycin (500 μg/mL) and l-arabinose (100 mM, Sigma-Aldrich) at the time of inoculation. Production cultures were incubated at 30 °C, 200 rpm for 5 d. All production cultures were performed in triplicate. Culture titers were determined based on HPLC calibration curves. In brief, peptides purified to homogeneity by HPLC were weighed, and serial dilutions were generated and analyzed by HPLC. A calibration curve was obtained (area under the curve versus concentration) from which titers were calculated.
Host genome and sequence comparison between outlier overproducer and low producer clones.
The Burkholderia sp. FERM BP-3421 genome contains two chromosomes and two plasmids. The NCBI accession codes for chromosomes 1 and 2 are CP063681 and CP063680, respectively. The accession codes for plasmids 1 and 2 are CP063679 and CP063678, respectively. To assess the genomes of the pSK020 outlier overproducer clone and the pSK020+48 low producer clones for single nucleotide polymorphisms and indels, Illumina sequences of both genomes were analyzed with PATRIC.44 First, Illumina short reads of the overproducer and low producer were aligned to the Burkholderia sp. FERM BP-3421 reference genome with Bowtie2.45 Over 95% of reads were mapped to the reference genome for both. Next, the alignments were analyzed for variants with FreeBayes,46 results of which can be found in Table S2.
Plasmid construction.
The vector backbone used for construction of plasmids was the pBBR1-based pMo16824 (Addgene plasmid #27389; http://n2t.net/addgene:27389; RRID:Addgene_27389) Additionally, the RK2-based pSEVA227M plasmid from the SEVA collection25,26 and the RSF1010-based pAM4891 plasmid23 (Addgene plasmid #120080; http://n2t.net/addgene:120080; RRID: Addgene_120080) were used in the PCN experiments.
pKAE001.
The cap biosynthetic gene cluster (BGC) under control of the araC/PBAD promotor was subcloned from pSK0207—which at the time was not known to contain the +48 bp insertion but was later determined to contain the insertion—into the pMo168 vector backbone to construct pKAE001 (Figure S5). For amplification of the cap BGC and the araC/PBAD promotor, the PCR (25 μL) components consisted of: 10 ng of template plasmid DNA (pSK020),7 0.2 mM of dNTPs (Thermo Fischer), 12.5 μM forward primer P_CapBGC_BglII_f, 12.5 μM reverse primer P_CapBGC_NdeI_r, and Q5 polymerase (0.02 U/μL) in a final concentration of 1× Q5 reaction buffer and high GC enhancer that were supplied with the enzyme. An annealing temperature of 51 °C was used for amplification. The 5,884 bp cap BGC and araC/PBAD PCR product was digested with BglII and NdeI and ligated into the same sites of pMo168 to yield pKAE001. The constructed plasmid was verified by restriction digest and Sanger sequencing of the cap BGC. After the detection of the +48 bp insertion in the pSK020+48 plasmid from the low producer (and from E. coli where cloning was performed), pKAE001 was sequenced with whole plasmid sequencing, which revealed the presence of the same +48 bp insertion. While pKAE001 did contain the insertion, this high-copy plasmid containing the cap BGC proved useful in the construction of other plasmids used in this study.
pHNF011.
Due to the increased PCN afforded by the pBBR1-based pKAE001 vector, pKAE001 was isolated from Burkholderia sp. FERM BP-3421 cultures. To remove the 48 bp insert, restriction enzymes were utilized. The 48-bp insert is flanked on each side by KpnI restriction sites; therefore, pKAE001 plasmid was digested with KpnI and the sticky ends re-ligated to yield pHNF011. Transformed clones were verified by restriction digest and Primordium whole plasmid sequencing (Figure S5).
To design vector backbones that could be utilized in this study as well as in future cloning experiments, pHNF008 and pHNF009 were generated.
pHNF008.
The araC/PBAD promoter was added to the pMo168 vector backbone to generate pHNF008 (Figure S7A). The araC/PBAD promotor was amplified by PCR (25 μL) consisting of: 10 ng of template plasmid DNA (pSK019),7 1× final concentration of Dream Taq PCR master mix, 10 μM forward primer PBAD_NheI_f, and 10 μM reverse primer PBAD_BglII_r. The thermal cycling conditions were as follows: 3 min at 95 °C; 35 cycles of 95 °C for 30 s, 48 °C for 20 s, and 72 °C for 1 min; and a final hold at 72 °C for 5 min. The 1,300 bp araC/PBAD PCR product was digested with NheI and BglII and ligated in a T4 ligase reaction into the same sites of pMo168 to yield pHNF008. The resulting construct was confirmed by restriction digest and Sanger sequencing.
pHNF009.
The G159S amino acid substitution34 in the replicon of pHNF008 was introduced by site directed mutagenesis of the rep gene (GGC to AGC) utilizing the NEB Q5 site directed mutagenesis kit (E0554S) to generate pHNF009 (Figure S7B). The PCR (25 μL) consisted of 30 ng of pHNF008, 1× final concentration of Q5 Hot Start master mix, 12.5 μM of the forward primer P_pBBR1_G159S_f, and 12.5 μM of the reverse primer P_pBBR1_G159S_r. Primers were designed using NEBase Changer. The annealing temperature for amplification was 72 °C, and the extension time was extended from 2 to 5 min. The subsequent Kinase-Ligase-DpnI (KLD) reaction consisted of 1 μL of the PCR reaction, 1× final concentration of the KLD reaction buffer, and 1× KLD enzyme mix. 5 μL of the KLD mix was used to transform high-efficiency, chemically competent E. coli NEB5α by heat shock. The selection of NEB5α clones was performed on LB agar containing kanamycin (50 μg/mL), and pHNF009 clones were verified by Sanger sequencing and whole plasmid Primordium sequencing, which confirmed the intended GGC to AGC mutation in the rep gene (Figure S7C).
pHNF021.
To construct the pBBR1 Rep G159S capistruin expression plasmid, the cap BGC was digested from pKAE001 with NheI and SfiI and ligated into the same sites of digested pHNF009 to generate pHNF013 (Figure S7B). Similar to the construction of pHNF011, pHNF013 was further digested with KpnI and re-ligated to generate pHNF021 (Figure S7B). Constructs were confirmed by restriction digest and Primordium sequencing.
pAK343.
The mls BGC was purchased from GenScript as two genetic fragments, the first containing mlsA1A2B and the second containing mlsC. The intergenic regions between mlsA1 and mlsA2, and mlsA2 and mlsB, remained intact (see Figure S19 for sequences). The two gene fragments were amplified with primers that included the RBS site and overhangs for Gibson assembly: mlsA1A2B using P_mlsA1A2B_OLEwithpUC_f and P_mlsA1A2B_OLEwithmlsC_rm and mlsC using P_mlsC_OLEwithmlsAB_f and P_mlsC_OLEwithpUC_r. The annealing temperature used was 55 °C. pSK019,7 containing the araC/PBAD promoter and pRO1600 replicon, was linearized with SacI. The three fragments—pSK019 backbone, mlsA1A2BC, and mlsC—were assembled with NEBuilder HiFi DNA assembly master mix (NEB #E2621) to yield pAK343. An aliquot of 5 μL of the assembly was used to transform chemically competent E. coli DH5α by heat shock. The screened clones were verified by restriction digest and Sanger sequencing.
pHNF007.
The mls BGC under control of the araC/PBAD promoter was subcloned from pAK343 into pMo168 to construct pHNF007 (Figure S14A). For amplification of the mls BGC and the araC/PBAD promotor, the PCR (25 μL) consisted of 10 ng of pAK343, 0.2 mM of dNTPs (Thermo Fischer), 12.5 μM of the forward primer P_B13BGC_BglII_ f, 12.5 μM of the reverse primer P_B13BGC_HindIII_ r, and Q5 polymerase (0.02 U/μL) in a final concentration of 1× Q5 reaction buffer and high GC enhancer that were supplied with the enzyme. The annealing temperature used was 52 °C. The 4,238 bp PCR product was digested with BglII and HindIII and ligated into the same sites of pMo168 to yield pHNF007, which was confirmed by restriction digest.
pHNF010.
To generate pHNF010 the native transporter from the mls cluster was synthesized by Twist Bioscience and received cloned in pTwistAMPhigh (Figure S14B). The mlsD transporter gene (see Figure S19 for sequences) from pTwistAMPhigh was digested with HindIII and SphI and ligated into the same sites of pHNF007 to yield pHNF010, which was confirmed by restriction digest and Sanger sequencing.
pHNF012.
Site directed mutagenesis to create a point mutation in the rep gene was carried out on pHNF010 to generate pHNF012. The G159S point mutation in the rep gene (GGC to AGC) was introduced using the NEB Q5 site directed mutagenesis kit as described above for pHNF009 with the exception that 15 ng of pHNF010 was used in the PCR, annealing was performed at 70 °C, extension at 72 °C was increased from 5 to 6 min, and the subsequent KLD reaction used 2 μL of the PCR. pHNF012 clones were verified by Sanger sequencing as well as whole plasmid sequencing performed by Primordium labs. In addition to the desired GGC to AGC mutation, the following discrepancies were detected from whole plasmid sequencing in comparison to the reference sequence: a deletion of A at position 184; a deletion of CATACC from position 1941-1946; a point mutation of C to T at 2406; a point mutation of A to G at 6798. All the observed discrepancies were in the backbone of the plasmid except for the point mutation at 6798, which is located within the araC CDS. However, the point mutation does not change the AraC translated sequence.
Electroporation of plasmids into Burkholderia sp. FERM BP-3421.
LB (5 mL) cultures were inoculated with 20 μL of a FERM BP-3421 cryo stock that had been thawed on ice. After overnight incubation (30 °C, 200 rpm), fresh LB (50 mL) broth in a 250 mL Erlenmeyer flask was inoculated with 1 mL of starter culture. The 50 mL culture was incubated at 30 °C and 200 rpm until the OD600 reached 0.4–0.6 (about 4 h). The cells were harvested by centrifugation at 4 °C, 1800 × g for 5 min. After removal of the supernatant, the cell pellet was gently resuspended and washed twice with ice-cold, sterile 1 mM 4-(2-hydroxyethyl)-1-piperazineethanesulphonic acid (HEPES) buffer, pH 7.0 (Sigma-Aldrich). The first wash was performed with 50 mL of HEPES buffer, and the second with 25 mL. After the second wash, the cell pellet was resuspended in the remaining drops of buffer yielding approximately 150 μL of cell suspension. Electrocompetent FERM BP-3421 cells suspended in HEPES buffer (50 μL) were electroporated with 500–1000 ng of plasmid DNA. Electroporation was carried out in a 0.2 cm ice-cold electroporation cuvette using a Bio-Rad micropulser set to 2.5 kV. The resulting time constants ranged from 4.5–5.5 ms. Following electroporation, 950 μL of ice-cold LB medium was added to the cell and DNA mixture. The electroporated cells were transferred to a microcentrifuge tube and incubated horizontally at 30 °C at 200 rpm for 1 h. To select for the incoming plasmid, the cells were plated on LB agar containing kanamycin (500 μg/mL). Plates were incubated at 30 °C for 3–5 d. Transformed clones from the electroporation plates were confirmed by plasmid isolation and restriction digest.
Capistruin extraction.
Five-mL aliquots from production cultures were transferred to 15-mL conic tubes. The cell pellet and supernatant were separated by centrifugation (10 min, 1800 × g). The cell pellet was discarded, and the supernatant was mixed with 0.5% (5 g/L) XAD-16 resin (Alfa Aesar) and agitated on a tabletop shaker for 1 h at 500 rpm. After agitation, the resin and supernatant were separated by centrifugation (5 min, 1800 × g), and the resin was washed with one volume of water. After removing the water by centrifugation (5 min, 1800 × g), the resin was extracted with one volume of methanol (e.g., 5 mL of methanol for 5 mL cultures) by shaking for 30 min at 500 rpm. The methanol extract was gravity filtered through pre-wetted 11 μm filter paper (Whatman, cat. no. 1001-150) into vials to remove residual resin. All extracts were dried under reduced pressure.
Mycetolassins extraction.
Five-day production cultures were harvested by centrifugation (10 min, 1800 × g). The cell pellet was separated from the supernatant to be processed separately. The supernatant was processed in the same manner as the supernatant from the capistruin production cultures described above. The cell pellet was mixed with one volume of methanol, vortexed briefly to resuspend, and agitated for 1 h at 500 rpm. The cells were separated from the methanol by centrifugation (5 min, 1800 × g). The methanol extract was decanted into vials fitted with pre-wetted 11 μm filter paper to remove any residual cell pellet. All extracts were dried under reduced pressure. Titers shown in Figure 5 are the sum of product from the extraction of both the cell pellet and the supernatant. For scaled-up production cultures of pAK343 (1 L), only the cell pellet was extracted due to the lack of the transporter gene (mlsD) in the construct. For scaled-up production cultures of pHNF012 (0.5 L)—used to determine the optimized yield of mycetolassins—whole cultures (cells and supernatant) were extracted as mlsD is present in the construct. For relative expression studies, whole cultures were extracted.
HPLC analyses of crude extracts of capistruin and mycetolassins.
The crude extracts were dissolved in methanol for HPLC analyses. HPLC analyses were performed on an Agilent 1260 Infinity system equipped with a Kinetex C18 column (150 × 4.6 mm, 5 μm particle size, 100 Å pore size, Phenomenex). Elution of crude extracts was done in water containing 0.1% trifluoroacetic acid (TFA) (v/v) (solvent A) and acetonitrile (solvent B). The flow rate was set to 1 mL/min. The solvent gradient for analysis was as follows: initial hold at 20% B for 5 min, linear gradient from 20–55% B for 23 min, linear gradient from 55–100% for 5 min, and terminal hold at 100% B for 5 min. The detection wavelength range was 200–600 nm, and chromatograms were extracted at λ = 210 nm.
Isolation of mycetolassins.
Mycetolassin-15 and -18 were purified from 1 L and 0.5 L cultures using preparatory HPLC (Agilent 1260 Infinity II). The methanol extract was resuspended in 20% acetonitrile/water (50 mg/mL) and injected on a Thermo Betasil C18 semi-preparatory column in 1 mL injections. The compounds of interest were separated using mobile phase A (water, 0.1% TFA) and mobile phase B (acetonitrile, 0.1% TFA) over a gradient of 5 min at 15% B, 20 min 15–45% B, 5 min 45–90% B, and 4 mins 90%, at a flow rate of 5 mL min−1. All absorbances in the range of 190–450 nm were recorded. Fractions containing the compounds of interest based on UV and subsequent MALDI-TOF-MS analysis were combined and dried under reduced pressure. The retention time was 17–18 min for the mixture of mycetolassin-15 and mycetolassin-18.
Fractions containing mycetolassin-15 and mycetolassin-18 were next separated by analytical HPLC. The mixture of mycetolassin-15 and -18 was purified from the large-scale extraction was resuspended in 15% acetonitrile/water (1 mg/mL) and injected on a Macherey-Nagel Analytical Nucleodur C18 column in 0.5 mL injections. Mycetolassin-15 and -18 were separated using mobile phase A (water) and mobile phase B (acetonitrile) over a gradient of 5 min at 10% B, 5 min 10–20% B, 23 min 20–26% B, 2 min 26–90% B, and 4 min at 90%, at a flow rate of 1 mL min−1. Absorbance in the range of 190–450 nm was recorded. Fractions containing mycetolassin-15 and -18, based on UV and subsequent MALDI-TOF-MS analysis, were combined, respectively, and dried under reduced pressure. A C-terminal truncation of mycetolassin-18, described here as mycetolassin-18Δ2, was also isolated for bioactivity studies. The retention time was 16.5 min for mycetolassin-15 and 22 min for mycetolassin-18.
Characterization of mycetolassins.
Mass spectrometry.
High-resolution and tandem mass spectrometry (HRMS/MS) analyses of mycetolassins are presented in Figures S9 and S10. For these analyses, the purified peptides were resuspended into 60% acetonitrile. The eluents were directly infused into a ThermoFisher Scientific Orbitrap Fusion ESI-MS using an Advion TriVersa Nanomate 100. The MS was calibrated and tuned with Pierce LTQ Velos ESI Positive Ion Calibration Solution (ThermoFisher). The MS was operated using the following parameters: resolution, 100,000; isolation width (MS/MS), 1 m/z; normalized collision energy (MS/MS), 70; activation q value (MS/MS), 0.4; activation time (MS/MS), 30 ms. Fragmentation was performed using collision-induced dissociation (CID) at 35–70%. The resulting data were averaged and analyzed using the Qualbrowser application of Xcalibur software (Thermo-Fisher Scientific).
Carboxypeptidase Y digestion.
Purified mycetolassins were resuspended to 60% aq. acetonitrile. The sample was evaporated to dryness under vacuum and resuspended in 10 μL of 100 ng/μL carboxypeptidase Y in phosphate-buffer saline (PBS). Control reactions were resuspended in PBS. This mixture was briefly vortexed and allowed to react at room temperature for 18 h. Resulting mixtures were treated with 60% acetonitrile and analyzed by MALDI-TOF-MS with sinapinic acid (SA) as the matrix.
Heat stability.
To evaluate heat stability, mycetolassins in 60% acetonitrile were heat-treated at 95 °C for 3 h. Heating in the presence of organic solvent can amplify the unfolding pathway for less heat-tolerant lasso peptides.1 This product was then evaporated to dryness under vacuum and resuspended in 100 ng/μL carboxypeptidase Y in PBS or PBS only and the reaction occurred at room temperature for 18 h. The digested product was analyzed with MALDI-TOF-MS as above.
Hydrogen-deuterium exchange MS.
Hydrogen-deuterium exchange was performed using previously established methods.47 Briefly, purified peptide samples were evaporated to dryness under vacuum and resuspended in 20 μL 3:7 D2O:acetonitrile. At indicated time points, 1.5 μL aliquots were removed and quenched by spotting onto the MALDI target with 1.5 μL saturated sinapinic acid in 3:7 D2O:acetonitrile+ 0.1% formic acid (pH = 2). Samples were then analyzed by MALDI-TOF MS.
Antimicrobial assay.
Purified and lyophilized lasso peptide was re-dissolved in 10% methanol at a concentration of 500 μM. Paper disc diffusion assays were used to evaluate the antimicrobial activity of mycetolassin-15 and -18, as well as a C-terminal truncation of mycetolassin-18 (mycetolassin-16) against the panel of ESKAPE pathogens: Enterococcus faecium U503, Staphylococcus aureus NRS 384, Klebsiella pneumoniae ATCC 27736, Acinetobacter baumanii ATCC 19606, Pseudomonas aeruginosa PAO1, and Enterobacter cloacae ATCC 29893. A starter culture of indicator strain was grown in LB medium under aerobic conditions at 37 °C for 16 h. Agar plates were prepared by combining 40 mL of molten Mueller Hinton (MH) agar (cooled to 42 °C) with 4 mL of dense culture (starting OD600 ~0.1). The seeded agar was poured into a sterile Petri dish and allowed to solidify at room temperature after which 10 μL of the lasso peptide solution, or 10% methanol control, were spotted onto the agar plates. Plates were incubated at 37 °C for 16 h, and antimicrobial activity was qualitatively determined by the presence or absence of a zone of growth inhibition.
Inhibition of mCherry production.
Purified lasso peptides were resuspended in 10% methanol to 500 μM. A cell-free reaction was set up as previously described,36 with 100 ng plasmid encoding mCherry under endogenous σ70 control. Capistruin, mycetolassin-15, myceotolassin-18Δ2, mycetolassin-18 were added to a final concentration of 0 or 50 μM or 30 μM for capistruin. The reactions were conducted at 37 °C for 6 h with 800 rpm shaking for aeration. The samples were subjected to 15,000 × g for 1 min at room temperature to remove precipitate, diluted 1:4 in water, and the fluorescence emission at 600 nm was quantified (Tecan Infinite M2000) using an excitation of 550 nm (N = 4).
Production of mycetolassins in E. coli.
E. coli BL21 chemical competent cells were transformed with mls expression plasmid pAK343 as described above for DH5α. Overnight cultures were grown at 37 °C in LB media with kanamycin. 500 mL LB with kanamycin and 2% arabinose was inoculated overnight and grown for 2 d at 37 °C. The pellet and supernatant were extracted as described above. The expected masses for mycetolassin-15 and mycetolassin-18 were observed by MALDI-TOF MS in trace amounts (Figure S17A). Supernatant extracts (1% of extract, or ~5 mL eq culture) were injected on the HPLC using the same method described above with a Thermo betasil C18 analytical column (Figure S17B). No visible 280 nm peak at the retention time corresponding to either peptide was observed (25 and 35 min for mycetolassin-18 and -15 respectively). To quantify mycetolassin-18, a standard curve was constructed with a 500 μM stock solution (30, 10, 2, 0.5 μg injected) using the area of the 220 nm absorbance peak (Figure S17C). E. coli culture expressing pAK343 (1% of the extract, eq. ~5 mL culture) was injected onto the HPLC and the area of the 220 nm absorbance peak corresponding to retention time of myceotolassin-18 was compared to a standard curve to determine titer (N = 1). The peak area from the pellet extract corresponded to ~1 μg mycetolassin-18, for an extrapolated titer of ~200 μg/L.
Relative plasmid copy number determination.
Empty vectors containing the RSF1010 (pAM4891), RK2 (pSEVA227M), and pBBR1 (pMo168) replicons were transferred into Burkholderia sp. FERM BP-3421 by electroporation and clones were confirmed by plasmid isolation and restriction digest. Total genomic DNA (gDNA) was isolated from cells grown in 10 mL of LB liquid culture containing 500 μg/mL of kanamycin, and cultivated overnight at 30 °C, 200 rpm. Isolated gDNA was sequenced with Illumina technology by the SeqCenter (Pittsburgh, PA), using the 400 Mbp sequencing package. The resulting Illumina reads were mapped to a reference sequence containing the Burkholderia. sp. FERM BP-3421 genome and each of the vectors being evaluated using the Geneious software. The relative PCN per genome equivalent was calculated by dividing the average number of reads that mapped to the vector by the average number of reads that mapped to chromosomes 1 and 2. For example: the average number of reads mapped to the pSK020 plasmid for the overproducer was 1470; the number of reads mapped to chromosomes 1 and 2 for the overproducer are 487 and 545, respectively. The normalized PCN for chromosome 1 was calculated as 1470 / 487 = 3.02. The normalized PCN for chromosome 2 was calculated as 1470 / 545 = 2.70. The average of both values was calculated for the final normalized PCN value.
The relative PCN results obtained by mapping Illumina reads to the Burkholderia. sp. FERM BP-3421 pSK020 reference sequence were next validated by quantitative PCR (qPCR). qPCR experiments were performed with gDNA isolated from the Burkholderia. sp. FERM BP-3421 pSK020 low and overproducers. First, a stock solution of gDNA for each sample was prepared at a concentration of 10 ng/μL. To generate a calibration curve, stock solutions were diluted to 5 ng/μL, 2 ng/μL, 1 ng/μL, and 0.5 ng/μL. Cq (the number of cycles required for fluorescence to be detected above background fluorescence) was plotted against the −log of gDNA concentration. PCN was determined using qPCR by comparing the detected amplification of two regions of plasmid DNA (araC and kanR) to the amplification of two regions of chromosomal DNA (recA and gyrA). Primer pairs used to amplify araC and kanR are listed in Table S3.
The PCR was prepared with SYBR green mix (Applied Biosystems, cat. no. 4309155). In a 96-well plate (Applied Biosystems, cat. no. SG21111-5B), 5 μL of SYBR green mix was added to 5 μL of master mix, which contained 2 μL of water, 1 μL of 10 mM of forward primer, 1 μL of 10 mM of reverse primer, and 1 μL of gDNA template. The PCR conditions were: 1 min at 95 °C, 40 cycles of 20 s at 95 °C, 20 s at 60 °C, and a final 30 s at 72 °C. Analysis was done in triplicate. PCN relative to the chromosome was calculated with the following equation: relative plasmid number = 2−ΔCq, where ΔCq = Cqplasmid – Cqgenome. Copy number of the overproducer relative to the low producer was calculated using: 2−ΔΔCq, where ΔΔCq = Cqoverproducer – Cqlowproducer.
Because qPCR and Illumina sequencing results were equivalent for pSK020 (Figure 2), all subsequent relative copy number calculations were performed with Illumina sequencing only.
Growth comparison.
Cultures for growth curve acquisition were prepared in the same manner as production cultures, detailed under ‘Strains, cultivation conditions, chemicals, and general procedures.’ The pSK020 and pSK020+48 seed cultures were diluted to an OD600 of 0.1 with 2S4G production medium augmented with kanamycin 500 μg/mL and 100 mM of the araC/PBAD inducer l-arabinose. Diluted cultures were plated on a 96-well plate with 8 – 10 replicates for each culture. The same number of replicates of augmented 2S4G production medium were also plated as a blank. The growth curve was obtained using a BioTek Synergy HTX multimode microplate reader set to run for 48 h, continuously shaking at 282 rpm, 30 °C, reading OD600 every 30 min. The average of the OD600 of blanks for each time point was subtracted from the average of the corresponding time point for each production culture to obtain the normalized OD600 of the cultures.
To analyze growth, the time until start of exponential growth (i.e., duration of lag phase) and the total growth (i.e., area under the growth curve) were calculated. Previously published protocols for lag phase and area under the growth curve calculations were followed.33,48 The normalized OD600 values were converted to ln OD600 and the slope of consecutive windows of five time points (i.e., time points 1 – 5, 2 – 6, 3 – 7, etc.) were calculated for all time points of the 48-h run. From the five-point slopes, the maximum slope was determined, and the five-point slopes that were ≥85% of the maximum slope established the exponential phase of each growth curve. The first time point ≥85% of the maximum slope was considered the end of lag phase. Total growth was calculated by integrating the normalized OD600 values gathered from time point 30 min to 32 h.
Cell density comparison.
Production cultures were prepared in the same manner as detailed under ‘Strains, cultivation conditions, chemicals, and general procedures.’ The production cultures were removed from the incubator after 48 h and 10 mL aliquots were taken. An equal aliquot of PBS was added to the aliquot of production culture to facilitate cell harvesting from the glycerol-saturated production media. The cell pellet was harvested by centrifugation (10 min, 14000 × g). The majority of the supernatant was removed and discarded after initial centrifugation and a second centrifugation (5 min, 14000 × g) was preformed to separate any residual supernatant.
After wet cell weights were acquired, the harvested cells were gently and thoroughly resuspended in 10 mL of PBS. The cell resuspensions were serially diluted to a dilution factor of 10−7. From diluted cell suspensions, 100 – 200 μL was plated on LB agar augmented with kanamycin 500 μg/mL and incubated until colonies were visible (30 °C, 3 d).
Supplementary Material
ACKNOWLEDGMENTS
We thank the International Patent Organism Depository of the National Institute of Technology and Evaluation (Japan) for strain FERM BP-3421, E. Graziani and M. Noe (Pfizer, Groton, CT) for the fr9DEF− mutant of FERM BP-3421, and Dr. S. Zirah (Muséum National d’Histoire Naturelle, Paris, France) for the capistruin standard. Vector pAM4891 (pCV0003) was a kind gift from S. Golden (Addgene), pMo168 from M. Voskuil (Addgene), and pSEVA227M from V. de Lorenzo (SEVA collection). We thank S. Ramesh of the Mitchell laboratory for her assistance in acquiring the HRMS/MS data. Financial support for this work was provided by the National Institute of General Medical Sciences, National Institutes of Health (NIH) (GM129344 to A.S.E. and D.A.M. and F32GM145122 to A.M.K). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Footnotes
Supporting Information.
The Supporting Information is available free of charge on [insert link].
Previously reported heterologous production of lasso peptides from Pseudomonadota (Table S1), Identified differences between the reference genome and either pSK020 outlier overproducer or pSK020+48 low producer (Table S2), Primer sequence list for plasmid construction requiring PCR (Table S3), Schematic of pSK020+48 plasmid and sequence of the promoter region (Figure S1), Plasmid is not the only factor contributing to the overproducer phenotype (Figure S2), Assessment of the overproducer clone’s ability to recapitulate high yield capistruin production when used as a heterologous host (Figure S3), Normalized copy number of different replicons in Burkholderia sp. FERM BP-3421(Figure S4), Generation of pKAE001 and pHNF011 (Figure S5), Comparison of cell density between production cultures of Burkholderia sp. FERM BP-3421 harboring alternate capistruin expression vectors (Figure S6), Generation of pHNF021 (Figure S7), Production of capistruin in wild-type and spliceostatin-defective mutant of Burkholderia sp. FERM BP-3421 (Figure S8), Sequence similarity networks of predicted lasso core peptides (Figure S9), HR-ESI-MS/MS of purified mycetolassin-15 (Figure S10), HR-ESI-MS/MS of purified mycetolassin-18 (Figure S11), Proteolytic resistance of purified mycetolassins (Figure S12), Hydrogen-deuterium exchange of mycetolassin-15 and mycetolassin-18 (Figure S13), Generation of pHNF007 and pHNF010 (Figure S14), Growth curve comparison between production with alternate mls expression vectors (Figure S15), Export of mycetolassins with and without inclusion of transporter gene (Figure S16), Production of mycetolassins in E. coli (Figure S17), Inhibition of transcription and/or translation assessed by cell free production of mCherry (Figure S18), Sequences of mycetolassin genetic constructs mlsA1A2B, mlsC, and mlsD (Figure S19) (PDF).
Conflict of Interest Statement
D.A.M is a co-founder of Lassogen, Inc.
REFERENCES
- 1.Hegemann JD Factors governing the thermal stability of lasso peptides. ChemBioChem 2020, 21, 7–18. [DOI] [PubMed] [Google Scholar]
- 2.Knappe TA; Manzenrieder F; Mas-Moruno C; Linne U; Sasse F; Kessler H; Xie X; Marahiel MA Introducing lasso peptides as molecular scaffolds for drug design: Engineering of an integrin antagonist. Angew Chem Int Ed Engl 2011, 50, 8714–8717. [DOI] [PubMed] [Google Scholar]
- 3.Hegemann JD; De Simone M; Zimmermann M; Knappe TA; Xie X; Di Leva FS; Marinelli L; Novellino E; Zahler S; Kessler H; Marahiel MA Rational improvement of the affinity and selectivity of integrin binding of grafted lasso peptides. J Med Chem 2014, 57, 5829–5834. [DOI] [PubMed] [Google Scholar]
- 4.Pan SJ; Link AJ Sequence diversity in the lasso peptide framework: Discovery of functional microcin J25 variants with multiple amino acid substitutions. J Am Chem Soc 2011, 133, 5016–5023. [DOI] [PubMed] [Google Scholar]
- 5.Rowe SM; Spring DR The role of chemical synthesis in developing RiPP antibiotics. Chem Soc Rev 2021, 50, 4245–4245. [DOI] [PubMed] [Google Scholar]
- 6.Chen M; Wang S; Yu X Cryptand-imidazolium supported total synthesis of the lasso peptide BI-32169 and its D-enantiomer. Chem Commun 2019, 55, 3323–3326. [DOI] [PubMed] [Google Scholar]
- 7.Kunakom S; Eustáquio AS Heterologous production of lasso peptide capistruin in a Burkholderia host. ACS Synth Biol 2020, 9, 241–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maksimov MO; Pan SJ; James Link A Lasso peptides: Structure, function, biosynthesis, and engineering. Nat Prod Rep 2012, 29, 996–1006. [DOI] [PubMed] [Google Scholar]
- 9.Hegemann JD; Zimmermann M; Zhu S; Klug D; Marahiel MA Lasso peptides from proteobacteria: Genome mining employing heterologous expression and mass spectrometry. Biopolymers 2013, 100, 527–542. [DOI] [PubMed] [Google Scholar]
- 10.Wang G; Zhao Z; Ke J; Engel Y; Shi YM; Robinson D; Bingol K; Zhang Z; Bowen B; Louie K; et al. CRAGE enables rapid activation of biosynthetic gene clusters in undomesticated bacteria. Nat Microbiol 2019, 4, 2498–2510. [DOI] [PubMed] [Google Scholar]
- 11.Adaikpoh BI; Fernandez HN; Eustáquio AS Biotechnology approaches for natural product discovery, engineering, and production based on Burkholderia bacteria. Curr Opin Biotechnol 2022, 77, 102782–102782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kunakom S; Eustáquio AS Burkholderia as a source of natural products. J Nat Prod 2019, 82, 2018–2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eustáquio AS; Chang LP; Steele GL; O’Donnell CJ; Koehn FE Biosynthetic engineering and fermentation media development leads to gram-scale production of spliceostatin natural products in Burkholderia sp. Metab Eng 2016, 33, 67–75. [DOI] [PubMed] [Google Scholar]
- 14.Knappe TA; Linne U; Zirah S; Rebuffat S; Xie X; Marahiel MA Isolation and structural characterization of capistruin, a lasso peptide predicted from the genome sequence of Burkholderia thailandensis E264. J Am Chem Soc 2008, 130, 11446–11454. [DOI] [PubMed] [Google Scholar]
- 15.Stanssens P; Remaut E; Fiers W Alterations upstream from the Shine-Dalgarno region and their effect on bacterial gene expression. Gene 1985, 36, 211–223. [DOI] [PubMed] [Google Scholar]
- 16.Makrides SC Strategies for achieving high-level expression of genes in Escherichia coli. Microbiol Rev 1996, 60, 512–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kondo T; Yumura S Strategies for enhancing gene expression in Escherichia coli. Microbiol Rev 2020, 104, 3825–3834. [DOI] [PubMed] [Google Scholar]
- 18.Kunakom S; Adaikpoh BI; Tran TA; Eustáquio AS Complete genome sequence of soil bacterium Burkholderia sp. Strain FERM BP-3421, a producer of spliceostatins. Microbiol Resour Announc 2023, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ma X; Shao Y; Tian L; Flasch DA; Mulder HL; Edmonson MN; Liu Y; Chen X; Newman S; Nakitandwe J; et al. Analysis of error profiles in deep next-generation sequencing data. Genome Biol 2019, 20, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schweizer HP Escherichia-Pseudomonas shuttle vectors derived from pUC18/19. Gene 1991, 97, 109–112. [DOI] [PubMed] [Google Scholar]
- 21.Thomas CM Paradigms of plasmid organization. Mol Microbiol 2002, 37, 485–491. [DOI] [PubMed] [Google Scholar]
- 22.Del Solar G; Espinosa M Plasmid copy number control: An ever-growing story. Mol Microbiol 2002, 37, 492–500. [DOI] [PubMed] [Google Scholar]
- 23.Taton A; Unglaub F; Wright NE; Zeng WY; Paz-Yepes J; Brahamsha B; Palenik B; Peterson TC; Haerizadeh F; Golden SS; Golden JW Broad-host-range vector system for synthetic biology and biotechnology in cyanobacteria. Nucleic Acids Res 2014, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hamad MA; Zajdowicz SL; Holmes RK; Voskuil MI An allelic exchange system for compliant genetic manipulation of the select agents Burkholderia pseudomallei and Burkholderia mallei. Gene 2009, 430, 123–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Silva-Rocha R; Martínez-García E; Calles B; Chavarría M; Arce-Rodríguez A; De Las Heras A; Páez-Espino AD; Durante-Rodríguez G; Kim J; Nikel PI; et al. The Standard European Vector Architecture (SEVA): A coherent platform for the analysis and deployment of complex prokaryotic phenotypes. Nucleic Acids Res 2013, 41, D666–D675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Martínez-Garćía E; Aparicio T; Goñi-Moreno A; Fraile S; De Lorenzo V SEVA 2.0: An update of the Standard European Vector Architecture for de-/re-construction of bacterial functionalities. Nucleic Acids Res 2015, 43, D1183–D1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cook TB; Rand JM; Nurani W; Courtney DK; Liu SA; Pfleger BF Genetic tools for reliable gene expression and recombineering in Pseudomonas putida. J Ind Microbiol Biotechnol 2018, 45, 517–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Smith MA; Bidochka MJ Bacterial fitness and plasmid loss: The importance of culture conditions and plasmid size. Can J Microbiol 1998, 44, 351–355. [PubMed] [Google Scholar]
- 29.Friehs K Plasmid copy number and plasmid stability. Adv Biochem Engin/Biotechnol 2004, 86, 47–82. [DOI] [PubMed] [Google Scholar]
- 30.Stueber D; Bujard H Transcription from efficient promoters can interfere with plasmid replication and diminish expression of plasmid specified genes. EMBO J 1982, 1, 1399–1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Merrikh H; Zhang Y; Grossman AD; Wang JD Replication-transcription conflicts in bacteria. Nat Rev Microbiol 2012, 10, 449–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.St Germain C; Zhao H; Barlow JH Transcription-replication collisions—a series of unfortunate events. Biomolecules 2021, 11, 1249–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dorado-Morales P; Pilar Garcillán-Barcia M; Lasa I; Solano C Fitness cost evolution of natural plasmids of Staphylococcus aureus. mBio 2021, 12, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mi J; Sydow A; Schempp F; Becher D; Schewe H; Schrader J; Buchhaupt M Investigation of plasmid-induced growth defect in Pseudomonas putida. J Biotechnol 2016, 231, 167–173. [DOI] [PubMed] [Google Scholar]
- 35.Tietz JI; Schwalen CJ; Patel PS; Maxson T; Blair PM; Tai HC; Zakai UI; Mitchell DA A new genome-mining tool redefines the lasso peptide biosynthetic landscape. Nat Chem Biol 2017, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Si Y; Kretsch AM; Daigh LM; Burk MJ; Mitchell DA Cell-free biosynthesis to evaluate lasso peptide formation and enzyme-substrate tolerance. J Am Chem Soc 2021, 143, 5917–5927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xiu H; Wang M; Fage CD; He Y; Niu X; Han M; Li F; An X; Fan H; Song L; et al. Discovery and characterization of rubrinodin provide clues into the evolution of lasso peptides. Biochemistry 2022, 61, 595–607. [DOI] [PubMed] [Google Scholar]
- 38.Cheung-Lee WL; Parry ME; Zong C; Cartagena AJ; Darst SA; Connell ND; Russo R; Link AJ Discovery of ubonodin, an antimicrobial lasso peptide active against members of the Burkholderia cepacia complex. ChemBioChem 2020, 21, 1335–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bratovanov EV; Ishida K; Heinze B; Pidot SJ; Stinear TP; Hegemann JD; Marahiel MA; Hertweck C Genome mining and heterologous expression reveal two distinct families of lasso peptides highly conserved in endofungal bacteria. ACS Chem Biol 2020, 15, 1169–1176. [DOI] [PubMed] [Google Scholar]
- 40.Lastovetsky OA; Krasnovsky LD; Qin X; Gaspar ML; Gryganskyi AP; Huntemann M; Clum A; Pillay M; Palaniappan K; Varghese N; et al. Molecular dialogues between early divergent fungi and bacteria in an antagonism versus a mutualism. mBio 2020, 11, e02088–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dicaprio AJ; Firouzbakht A; Hudson GA; Mitchell DA Enzymatic reconstitution and biosynthetic investigation of the lasso peptide fusilassin. J Am Chem Soc 2019, 141, 290–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Raú R. l.; Salomón R. l. A.; Salomón S; Farís RN; Farís F. The peptide antibiotic microcin 25 is imported through the TonB pathway and the SbmA protein. J Bacteriol 1995, 177, 3323–3325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Eustáquio AS; Janso JE; Ratnayake AS; O’Donnell CJ; Koehn FE Spliceostatin hemiketal biosynthesis in Burkholderia spp. Is catalyzed by an iron/α-ketoglutarate-dependent dioxygenase. Proc Natl Acad Sci U S A 2014, 111, E3376–E3385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wattam AR; Abraham D; Dalay O; Disz TL; Driscoll T; Gabbard JL; Gillespie JJ; Gough R; Hix D; Kenyon R; et al. PATRIC, the bacterial bioinformatics database and analysis resource. Nucleic Acids Res 2014, 42, D581–D591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Langmead B; Salzberg SL Fast gapped-read alignment with Bowtie 2. Nat Methods 2012, 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Garrison E; Marth G Haplotype-based variant detection from short-read sequencing. arXiv preprint 2012, DOI: arXiv:1207.3907. [Google Scholar]
- 47.Pirrone GF; Wang H; Canfield N; Chin AS; Rhodes TA; Makarov AA Use of MALDI-MS combined with differential hydrogen-deuterium exchange for semiautomated protein global conformational screening. Anal Chem 2017, 89, 8351–8357. [DOI] [PubMed] [Google Scholar]
- 48.Hall BG; Acar H; Nandipati A; Barlow M Growth rates made easy. Mol Biol Evol 2013, 31, 232–238. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.