

Abstract
Although μ-opioid peptide (MOP) receptor agonists are effective analgesics available in clinical settings, their serious adverse effects put limits on their use. The marked increase in abuse and misuse of prescription opioids for pain relief and opioid overdose mortality in the past decade has seriously impacted society. Therefore, safe analgesics that produce potent analgesic effects without causing MOP receptor-related adverse effects are needed. This review highlights the potential therapeutic targets for the treatment of opioid abuse and pain based on available evidence generated through preclinical studies and clinical trials. To ameliorate the abuse-related effects of opioids, orexin-1 receptor antagonists and mixed nociceptin/MOP partial agonists have shown promising results in translational aspects of animal models. There are several promising non-opioid targets for selectively inhibiting pain-related responses, including nerve growth factor inhibitors, voltage-gated sodium channel inhibitors, and cannabinoid- and nociceptin-related ligands. We have also discussed several emerging and novel targets. The current medications for opioid abuse are opioid receptor-based ligands. Although neurobiological studies in rodents have discovered several non-opioid targets, there is a translational gap between rodents and primates. Given that the neuroanatomical aspects underlying opioid abuse and pain are different between rodents and primates, it is pivotal to investigate the functional profiles of these non-opioid compounds compared to those of clinically used drugs in non-human primate models before initiating clinical trials. More pharmacological studies of the functional efficacy, selectivity, and tolerability of these newly discovered compounds in non-human primates will accelerate the development of effective medications for opioid abuse and pain.
1. Introduction
Although nociceptive somatosensory systems, such as pain, are indispensable to detect abnormalities or disorders in organs and protect tissue damage, intractable pain associated with several chronic diseases should be effectively relieved. Pain is considered a burden that largely reduces quality of life and impairs productivity globally (Cohen, Vase, & Hooten, 2021; Collaborators et al., 2018). In the US, over 100 million adults experience chronic pain annually, and the annual total cost of pain, including direct costs, decreased wages, and lost productivity, largely eclipsed that of any other diseases such as heart diseases, cancer, and diabetes (Holmes, 2016). Thus, there is an unmet need to establish effective treatment strategies for intractable pain. People recognized different types of pain, such as burning and stabbing, and the opium poppy (Papaver somniferum L.) was used to relieve pain for thousands of years (Brook, Bennett, & Desai, 2017). In 1805, morphine was identified as the active pain-killing ingredient of the opium poppy. The discovery of morphine has had a huge impact that contributed to not only scientific knowledge but also human lives (Pain, 2016). Till date, this ingredient remains one of the most potent analgesics, and several of its derivatives or related compounds that act on μ-opioid peptide (MOP) receptors are widely used for the management of severe pain associated with serious diseases, such as cancer and myocardial infarction (Degenhardt et al., 2019; Kreek, Reed, & Butelman, 2019).
Opioid receptors were identified and cloned in the 1990s after several years of biochemical, physiological, and pharmacological research (Chen, Mestek, Liu, Hurley, & Yu, 1993; Evans, Keith, Morrison, Magendzo, & Edwards, 1992; Yasuda et al., 1993). Classical opioid receptors consist of three subtypes—MOP, δ-opioid peptide (DOP), and κ-opioid peptide (KOP) receptors. These receptors are widely distributed in the peripheral and central nervous system (CNS) and are particularly located in pain-processing regions, such as the dorsal root ganglia (DRG), spinal dorsal horn (SDH), and brain in mammals (Corder, Castro, Bruchas, & Scherrer, 2018; Fields, 2004). Under physiological conditions, endogenous opioid peptides activate cognate opioid receptors to reduce pain and influence reward processing and mood through Gi-protein coupled receptor (GPCR) signaling. This in turn inhibits adenylate cyclase and voltage-gated calcium channels and activates inward potassium channels, resulting in a reduction in synaptic transmission (Darcq & Kieffer, 2018). Among the opioid receptors, the MOP receptor is known to be the sole receptor responsible for the analgesic effects of morphine. This observation is based on rodent studies that demonstrated that the deletion of the MOP receptor eliminates the analgesic effects of morphine (Matthes et al., 1996). The MOP receptor is also a key therapeutic target for clinically used opioids (such as oxycodone, fentanyl, methadone, buprenorphine), indicating that activation of the MOP receptor has substantial utility to relieve severe pain. However, MOP receptors also produce serious dose-limiting adverse effects such as abuse liability, respiratory depression, constipation, itching sensation, and physical dependence (Gunther et al., 2018). Although MOP receptor ligands are the strongest analgesics currently known to humans, these adverse effects often compromise therapeutic effects and cause serious problems, such as opioid use disorder (OUD).
Recently, an epidemic of opioid abuse and overdoses emerged in North America and a part of Europe. The marked increase in abuse and misuse of prescription opioids for pain relief and opioid overdose mortality in the past decade has had considerable impact on society (Dart et al., 2015; Degenhardt et al., 2014). Given that MOP receptor agonists are the only drugs that can relieve unbearable pain associated with cancer or some other medical conditions, it is pivotal to develop novel safe opioid analgesics that produce potent analgesic effects without causing MOP receptor-related adverse effects (Volkow & Collins, 2017). In this review, we have compiled evidence from preclinical and clinical studies to uncover the regulatory mechanisms of opioid abuse and analgesia and propose newly discovered targets as potential therapeutics for the treatment of opioid abuse and pain.
2. Opioid abuse
2.1. Background of opioid abuse
Opioid abuse is a brain disorder associated with behavioral, psychological, and neurochemical manifestations (Kreek et al., 2019). Although a break-through finding in the 1970s revealed the importance of dopamine systems, for several decades, key components of drug addiction have been considered responsible for dysregulation of dopaminergic neurotransmitter systems (Nutt, Lingford-Hughes, Erritzoe, & Stokes, 2015). Dopamine systems normally contribute to various brain functions. Further, dopamine neurons play an important role in attention and working memory and are necessary for regulating motivation, reward, and similar motor functions (Bromberg-Martin, Matsumoto, & Hikosaka, 2010). Moreover, dopamine is associated with the pathogenesis of a proportion of psychosis and involved in positive mood in humans (Cousins, Butts, & Young, 2009). Notably, the mesolimbic dopaminergic system, known as the reward system, originates from the ventral tegmental area (VTA) and projects into the corticolimbic regions, such as the nucleus accumbens (NAC) and the prefrontal cortex (PFC) (Kourrich, Calu, & Bonci, 2015). The majority of NAC neurons are medium spiny neurons that receive excitatory glutamatergic inputs from the PFC, thalamus, amygdala, and VTA; therefore, they are considered key reward-related neural circuits (Kourrich et al., 2015). Based on these data, drug addiction is hypothesized to be due to drugs that induce dopamine release and they are considered to have a risk of abuse. There is a significant causal link between dopamine levels and actions in the brain and addiction to psychostimulants (such as amphetamine and cocaine) (Bello et al., 2011; Sulzer, 2011). However, studies in rodents have demonstrated that blockade of dopamine receptors do not attenuate the rewarding effects of opioids (Fields & Margolis, 2015; Hnasko, Sotak, & Palmiter, 2005), and clinical trials could not support that this blockade was effective for the treatment of drug addiction in humans (Lingford-Hughes, Welch, Peters, Nutt, & British Association for Psychopharmacology, 2012; Rothman, 1994).
Preclinical animal studies have suggested that the opioid system largely contributes to reward and aversion and that dysregulation of opioid neurotransmission is crucial for drug abuse (Darcq & Kieffer, 2018). MOP receptors are widely distributed throughout the mesocorticolimbic circuitry in the brain (Corder et al., 2018). The prevailing disinhibition hypothesis proposes that activation of MOP receptors expressed by GABAergic interneurons in the VTA reduces local inhibition, subsequently resulting in the activation of dopamine neurons (Fields & Margolis, 2015; Morales & Margolis, 2017). However, as discussed above, such a hypothesis has not been confirmed as several brain regions that receive dopaminergic input, such as NAC, have abundant MOP receptors (Cui et al., 2014). Furthermore, dopamine-independent opioid reward based on MOP receptor-expressing neurons driving opioid reward outside the VTA has been demonstrated (Hnasko et al., 2005). MOP and KOP receptors oppositely regulate hedonic homeostasis, and MOP and KOP receptor agonists lead to euphoria and dysphoria, respectively. Unlike MOP receptors, KOP receptors in dopamine neurons of the VTA counteract MOP receptor-mediated activity in the NAC and PFC (Margolis et al., 2006) and disrupt behaviors. KOP receptors in the medium spiny neurons of the NAC regulate the input to the amygdala to negatively regulate motivational processes (Tejeda et al., 2017). Accumulating evidence suggests that nociceptin/orphanin FQ (N/OFQ) peptide (NOP) and its cognate receptor system, known as the fourth opioid receptor subtype, play unique roles in negatively regulating craving for abused drugs (Kiguchi, Ding, & Ko, 2020; Parker et al., 2019). Although evidence on the contribution of DOP receptors in opioid abuse is limited, four opioid receptor subtypes have been implicated in drug-dependent motivation underlying reward and abuse potential. Given that addiction is a complex mixture of behaviors and other neurological factors, there is no rationale to support that a single neurotransmitter (dopamine) could be associated with all aspects of addiction, suggesting that addiction can be considered a multiple-neurotransmitter disorder (Kreek et al., 2019; Nutt et al., 2015; Rasmussen, White, & Acri, 2019).
2.2. Treatment of opioid abuse
Currently there are three medications available for the treatment of OUD. These medications are methadone (a full MOP receptor agonist), buprenorphine (a low-efficacy partial MOP receptor agonist), and naltrexone (a MOP receptor antagonist). Each of these medications has its own unique pharmacological properties that require considerable knowledge and skills for appropriate prescribing (Kreek et al., 2019; Saxon, 2013; Schuckit, 2016). The appropriate use of these medications has also been limited by stigma, insufficient medical education, and inadequate resources (Kreek et al., 2019). Scientists have developed different strategies to address the opioid crisis. While reviewing the literature, we selected five approaches to update readers with recent findings. It should be noted that given the complex nature of opioid addiction, it is not realistic to expect a magic bullet to treat OUD. Successful treatment outcomes for patients with OUD require integration of both pharmacotherapy and psychosocial interventions (Rasmussen et al., 2019; Volkow & Blanco, 2020). Here, we have focused on opioid-related compounds with improved side-effect profiles and those that could suppress the abuse liability of opioids.
2.3. G protein signaling-biased MOP receptor agonists
MOP receptor activation initiates two different signaling pathways—G protein signaling and β-arrestin recruitment (Whistler, Chuang, Chu, Jan, & von Zastrow, 1999; Zhang et al., 1998). Earlier studies using β-arrestin2 knockout mice have shown that the antinociceptive effect of morphine could be improved, that is, potentiation of morphine can be enhanced, tolerance to it can be slowly developed, and side effects such as respiratory depression and constipation due to its usage can be reduced (Bohn et al., 1999; Bohn, Gainetdinov, Lin, Lefkowitz, & Caron, 2000; Raehal, Walker, & Bohn, 2005). Therefore, a G protein signaling-biased ligand devoid of β-arrestin recruitment may exert enhanced analgesia with improved safety and tolerability (Luttrell, Maudsley, & Bohn, 2015; Schmid & Bohn, 2009; Violin, Crombie, Soergel, & Lark, 2014). Oliceridine (TRV130) was the first reported G protein-biased MOP receptor agonist (Chen et al., 2013). In rodents, oliceridine, a potent analgesic across several pain models, causes less respiratory depression and gastrointestinal dysfunction (DeWire et al., 2013). Although oliceridine has progressed to clinical trials and has been approved by the US Food and Drug Administration (FDA), its therapeutic window is not as wide as originally expected. Namely, an analgesic effect is seen and accompanied by respiratory depression (Soergel et al., 2014; Viscusi et al., 2016).
Recently, scientists have discovered a series of G protein signaling-biased MOP agonists as new chemical entities with different bias factors, i.e., the degree of separation measured in the G protein signaling and β-arrestin2 recruitment assays. Through computational docking of over 3 million molecules against the MOP receptor structure and further optimization, PZM21 was discovered as a G protein-biased MOP receptor agonist. This compound exerted antinociceptive effects without rewarding effects and respiratory depression in mice (Manglik et al., 2016). Studies have also shown that PZM21 produces morphine-like respiratory depression in mice (Hill et al., 2018) and oxycodone-like reinforcing effects in monkeys (Ding et al., 2020), indicating that there is no difference in the two major side effects of opioids (respiratory depression and abuse liability) between PZM21 and the other clinically used opioid analgesics.
SR-17018 was identified as a G protein-biased MOP agonist with the highest bias factor and minimal respiratory depression even at higher doses (Schmid et al., 2017). However, in another study, SR-17018, PZM21, and oliceridine displayed low intrinsic efficacy similar to that of buprenorphine in MOP receptor regulation, regardless of the downstream signaling pathways (Gillis et al., 2020). Given the correlation between the therapeutic windows and their intrinsic efficacies at the MOP receptor, it is reasonable to attribute the improved side-effect profile of G protein-biased MOP agonists to their low intrinsic efficacy (Azevedo Neto et al., 2020; Gillis et al., 2020). In particular, these G protein-biased MOP agonists with low intrinsic efficacy can function like buprenorphine, which explains their effectiveness in attenuating the abuse-related effects of opioids in animal models. Although substitution with SR-17018 in morphine-tolerant mice restored the antinociceptive effect of morphine in the hot plate test (Grim et al., 2020), additional studies are required to investigate the degree to which SR-17018 is different from buprenorphine in terms of its analgesic and adverse effects in different animal models. With regard to the retained abuse liability of G protein-biased MOP agonists (Ding et al., 2020; Zamarripa et al., 2018), it is difficult to ascertain how biased G protein signaling can substantially refine opioid therapeutics in response to the opioid crisis.
2.4. Orexin-1 receptor antagonists
Accumulated evidence on the orexin receptors has opened an exciting avenue for the treatment of drug abuse (Baimel et al., 2015; James, Mahler, Moorman, & Aston-Jones, 2017). Orexin neurons in the lateral hypothalamus project to several brain regions involved in reward and drug-seeking behavior (Harris, Wimmer, & Aston-Jones, 2005; Hopf, 2020). For example, the VTA has been demonstrated to be an important site of orexin signaling in drug abuse ( James et al., 2017). Blockade of the orexin-1 receptor (OX1R) in the VTA attenuates the rewarding property of MOP receptor agonists (Zarepour, Fatahi, Sarihi, & Haghparast, 2014). Importantly, SB-334867, an OX1R antagonist, reduced heroin self-administration in rats (Smith & Aston-Jones, 2012). A recent study further demonstrated that SB-334867 decreased oxycodone self-administration, but the orexin-2 receptor (OX2R) antagonist TCSOX229 does not affect oxycodone self-administration in rats (Matzeu & Martin-Fardon, 2020). In addition, OX1R is critical for the expression of morphine withdrawal, and increased numbers of orexin-producing cells may play a role in maintaining opioid addiction (Georgescu et al., 2003; Sharf, Sarhan, & Dileone, 2008; Thannickal et al., 2018).
Using behavioral economics procedures, preclinical studies have also demonstrated that SB-334867 reduces motivation to consume highly abused opioid fentanyl and attenuates cue-induced reinstatement of fentanyl seeking (Fragale, Pantazis, James, & Aston-Jones, 2019). Considerable evidence supports that orexin-based therapies may represent an effective treatment strategy for addiction across a various drugs of abuse, including opioids, stimulants, alcohol, and nicotine (Fragale et al., 2021; Hopf, 2020). According to the available literature, OX1R antagonists are effective in inhibiting the motivation for drug rewards. In contrast, OX2R antagonists and dual orexin antagonists (DORAs) are useful for facilitating sleep (Fragale et al., 2021; Perrey & Zhang, 2020). Although ample evidence supports the therapeutic potential of OX1R antagonists in the treatment of opioid abuse in animal models, there is currently no FDA-approved OX1R antagonist. Nevertheless, a DORA, suvorexant, has been approved by the FDA for the treatment of insomnia. Several clinical trials have investigated the functional efficacy of suvorexant in patients with OUD and other substance use disorders (Fragale et al., 2021; James et al., 2020). Given that sleep impairment is often associated with OUD, it is worthwhile to wait for results of ongoing clinical trials of suvorexant. Further, preclinical studies rigorously comparing the functional efficacy and therapeutic window of OX1R antagonists and DORAs in the context of OUD-related endpoints will facilitate the future development of orexin-based ligands as a new treatment approach for OUD.
2.5. NOP receptor agonists
Recent research on the NOP receptor has opened another avenue as a potential medication for the dynamic epidemic of opioid abuse (Lin & Ko, 2013; Rasmussen et al., 2019; Toll, Bruchas, Calo, Cox, & Zaveri, 2016). Briefly, activation of the NOP receptor inhibits sensory processing, dopamine release, and neural transmission, which are considered viable targets for both pain and addiction (Kiguchi et al., 2020; Schroder, Lambert, Ko, & Koch, 2014; Toll et al., 2016). NOP agonists alone do not produce rewarding or aversive effects (Ko et al., 2009; Le Pen, Wichmann, Moreau, & Jenck, 2002). In an earlier study, the effects of opioids, measured by conditioned place preference, was blocked by selective NOP receptor agonists (Murphy, Lee, & Maidment, 1999). However, selective NOP receptor agonists did not display functional selectivity, that is, their sedative doses attenuated the reinforcing effects mediated by both opioids and food, measured by the self-administration assay (Podlesnik et al., 2011; Sukhtankar, Lagorio, & Ko, 2014). Given that opioid-induced rewarding and reinforcing effects are not fully mediated by dopamine signaling (Fields & Margolis, 2015; Nutt et al., 2015), selective NOP agonists may have mild-to-moderate efficacy in suppressing the abuse liability of opioids.
Clinical trials have documented that buprenorphine is an effective medication for OUD as it can reduce opioid craving and relapse risk (Kakko et al., 2019; Reimer, Vogelmann, Trümper, & Scherbaum, 2020). However, buprenorphine can produce moderate euphoric effects and physical dependency. It is also associated with an additional risk of diversion and misuse or abuse of medication (Johanson, Arfken, di Menza, & Schuster, 2012; Lavonas et al., 2014). Nonetheless, added naloxone in the commonly available formulations (e.g., suboxone) is expected to decrease abuse potential ( Jones et al., 2017). Given that NOP receptor agonists synergistically enhance the antinociceptive effect of buprenorphine and partially inhibit MOP receptor-mediated reinforcing effects (Cremeans, Gruley, Kyle, & Ko, 2012; Ding et al., 2021), a ligand with mixed NOP/MOP receptor agonist activities may have a wider therapeutic window with fewer side effects (Kiguchi et al., 2020; Lin & Ko, 2013). This hypothesis is supported by the functional profile of BU08028, a buprenorphine analog, with additional NOP receptor binding affinity and efficacy, which shows improved analgesic potency and a lack of reinforcing effects and physical dependency (Ding et al., 2016). The promising therapeutic profile of mixed NOP/MOP partial agonists is not compound specific as this class of compounds in different chemical structures include safe analgesics without abuse potential and physical dependency in non-human primates (NHPs) (Ding et al., 2018; Kiguchi et al., 2019). More importantly, ligands with dual actions (such as partial MOP agonists and NOP agonists) can inhibit the reinforcing effects of oxycodone without affecting the reinforcing effects of food pellets in NHP self-administration models, possessing a functional selectivity for inhibiting opioid abuse-related effects (Ding et al., 2018). Although cebranopadol, a mixed NOP/MOP full agonist, has been developed as a safe analgesic (Calo & Lambert, 2018; Tzschentke, Linz, Koch, & Christoph, 2019), its moderate reinforcing strength may limit its use in a broader therapeutic context (Ding et al., 2021). Nonetheless, given the clinical utility of buprenorphine, its analogs with additional NOP receptor agonist activity and other “centrally penetrant” combined NOP/MOP partial agonists possess great potential for treating OUD. In particular, more preclinical studies should be undertaken to further investigate the functional efficacy of such ligands with different brain-penetrant degrees in the context of OUD-related endpoints in NHP models (Ding & Ko, 2021).
2.6. Emerging targets
After the discovery of trace amine-associated receptor 1 (TAAR1) and subsequent development of selective small-molecule TAAR1 agonists (Borowsky et al., 2001; Sotnikova, Caron, & Gainetdinov, 2009), there has been a growing interest in the therapeutic potential of this receptor (Tonelli & Cichero, 2020). TAAR1 agonists are known to reduce the firing rate of dopaminergic neurons (Revel et al., 2011) and have great potential as therapeutic targets for neuropsychiatric diseases and drug addiction (Liu & Li, 2018; Schwartz et al., 2018). Although TAAR1 agonists have been extensively demonstrated as a promising pharmacotherapy for psychostimulant addiction (Liu, Wu, & Li, 2020), little is known about whether these agonists can modulate the abuse-related effects of opioids. Importantly, a recent study demonstrated that RO5263397, a TAAR1 partial agonist, attenuates morphine intake and motivation to self-administer morphine and decreases cue- and drug-induced reinstatement of morphine-seeking behavior in rats (Liu et al., 2021). Selective inhibition of the reinforcing effects of morphine by RO5263397 without changing the antinociceptive effect of morphine warrants additional animal and human studies to develop an optimal treatment strategy to curb opioid abuse.
Ibogaine, an alkaloid extracted from plants from the family Apocynaceae such as Tabernanthe iboga, is classified as a hallucinogen owing to its psychedelic property (Iyer, Favela, Zhang, & Olson, 2021). Although human studies on the safety and efficacy of ibogaine are lacking, anecdotal reports and animal studies support that it can reduce opioid cravings and prevent relapse (Belgers et al., 2016). Ibogaine has been reported to cause acute motor impairment and cardiotoxicity in animals. However, additional studies are required to determine its risks and potential benefits. As ibogaine is a psychoplastogen that can promote functional and structural neural plasticity in addiction-related circuitry, chemists have concentrated on the function-oriented synthesis of analogs of iboga alkaloids (Iyer et al., 2021). Importantly, tabernanthalog was discovered through a psychoplastogenic pharmacophore of ibogaine; it does not possess the toxicity and hallucinogenic effects of ibogaine (Cameron et al., 2021). Systemic administration of tabernanthalog reduced both heroin and sucrose self-administration, indicating that its inhibitory effect could be due to non-selective disruption of the operant response. Nonetheless, pretreatment with tabernanthalog prevented only cue-induced reinstatement of heroin and not sucrose-seeking behavior in rats (Cameron et al., 2021). These new findings encourage additional behavioral and neurochemical studies on tabernanthalog to determine its functional efficacy and selectivity across different animal models of OUD.
3. Pain
3.1. Background of pain
Primary sensory neurons transmit electrical impulses that encode specific sensory information to the CNS (the SDH and brainstem). Among heterogeneous sensory neurons, C- or Aδ-fibers are nociceptors that respond to noxious (such as thermal, mechanical, and chemical) stimuli that are essential for the recognition of pain, while Aβ-fibers play a role in sensing tactile information (Moehring, Halder, Seal, & Stucky, 2018; Peirs & Seal, 2016). For the detection and transmission of nociceptive information, specialized cation channels, such as transient receptor potential vanilloid 1 (TRPV1) and voltage-gated sodium channels (Navs), are necessary to amplify these signals for generation of action potential (Waxman & Zamponi, 2014). Subsequently, propagation of action potentials to the central terminals of sensory neurons leads to the release of neurotransmitters (such as glutamate and neuropeptides), which activate secondary CNS neurons by binding to postsynaptic cognate receptors. Primary sensory neurons input corresponding laminae in the SDH, which is the region for integration of peripheral input and descending supraspinal regulation. Most C- and Aδ-fibers form synapses in the superficial laminae (I and II), and Aβ-fibers project to deeper laminae (III–V) (Braz, Solorzano, Wang, & Basbaum, 2014; Moehring et al., 2018). The majority of SDH neurons consist of excitatory or inhibitory interneurons that locally modulate pain processing, while the other minor population is neurokinin-1 (NK1) receptor-expressing projection neurons located in laminae I (Peirs & Seal, 2016). Projection neurons convey pain information to several supraspinal areas such as the thalamus, parabrachial nucleus, amygdala, and cortex (Todd, 2010). Recent studies have uncovered complicated pain-processing mechanisms at the peripheral and SDH levels, suggesting that these areas could be reasonable therapeutic targets for pain (Woolf, 2020).
Pain is classified into three types—nociceptive, neuropathic, and nociplastic according to its etiology, pathophysiology, anatomical presentation, intensity, and duration (Raja et al., 2020). Nociceptive pain is caused by the activation of nociceptors and is important for defensive behaviors under physiological conditions, while continued noxious stimuli lead to pathological nociceptive pain. For example, osteoarthritis (OA) causes intense joint pain based on hyperactivation of nociceptors (Basbaum, Bautista, Scherrer, & Julius, 2009). To deal with such pain, a pharmacotherapy directly targeting molecules expressed on nociceptors, for instance, TRPV1 and other cation channels, might be effective (Moran, McAlexander, Biro, & Szallasi, 2011). In the event of tissue injury and inflammation, alterations in nociceptor function cause hypersensitivity and spontaneous pain. Nevertheless, if the inflammation is acute and transient, non-steroidal anti-inflammatory drugs can effectively suppress inflammatory pain (Woolf, 2020). Neuropathic pain is elicited by lesions or disorders in the somatosensory nervous system and present allodynia (pain caused by innoxious stimuli) and hyperalgesia, which are exaggerated responses to noxious stimuli (Attal, Bouhassira, & Baron, 2018). Compared to inflammatory pain, neuropathic pain is normally intractable and long lasting because of structural and functional alterations occurring in the nervous system, at least in part, through neuro-immune mechanisms ( Ji, Chamessian, & Zhang, 2016). Nociplastic pain is defined as pain occurring in the absence of noxious stimuli, inflammation, or damage to the nervous system (Fitzcharles et al., 2021). Thus, it is very challenging to appropriately control neuropathic and nociplastic pain because of their complicated mechanisms. To appropriately deal with different types of pain, the extent to which the component of pain is mechanistically linked needs to be determined.
3.2. Treatment of pain
Currently, there are a limited number of options that produce clinically meaningful analgesia, and efforts to develop novel effective analgesics are ongoing (Yekkirala, Roberson, Bean, & Woolf, 2017). Traditionally used analgesics, such as opioids, non-steroidal anti-inflammatory drugs, acetaminophen, and antiepileptics, were discovered first for their analgesic effects, after which the target molecules were identified. In contrast, serotonin-norepinephrine re-uptake inhibitors and gabapentinoids have been identified based on preclinical studies of pain-regulatory mechanisms (Woolf, 2020). Although it is necessary to use these drugs as front-line treatment, they do not sufficiently relieve some types of pain because of their dose-limiting adverse effects or diversity of pathophysiology (Attal & Bouhassira, 2015; Cohen et al., 2021). Given the complicated mechanisms of pain processing, it might be reasonable to discover novel analgesic targets based on preclinical studies. TRPV1, NK1 receptor, Navs, and nerve growth factor (NGF) have been nominated as candidates for analgesics that block excessive input of somatosensory nociceptive information into the SDH. Despite numerous studies demonstrating that such molecules (i.e., TRPV1 and NK1 receptor) significantly contribute to several types of pain in rodents (Nilius & Szallasi, 2014; Steinhoff, von Mentzer, Geppetti, Pothoulakis, & Bunnett, 2014; Yekkirala et al., 2017), inhibitors for TRPV1 or NK1 receptor failed clinical human studies because of their lower efficacy than originally anticipated or unexpected adverse effects. These facts suggest that there are difficulties in developing novel analgesics that are safe and clinically effective. Hence, reliance on this approach alone may not be appropriate to identify clinically effective therapeutic targets. Human diseases and biological responses are not well simulated in rodents because of anatomical and functional gaps between rodents and humans (Balsters, Zerbi, Sallet, Wenderoth, & Mars, 2020; Seok et al., 2013; Warren et al., 2020). It is very important to conduct preclinical studies that replicate human pain conditions using multiple outcome measures and different species (Fig. 1).
Fig. 1.
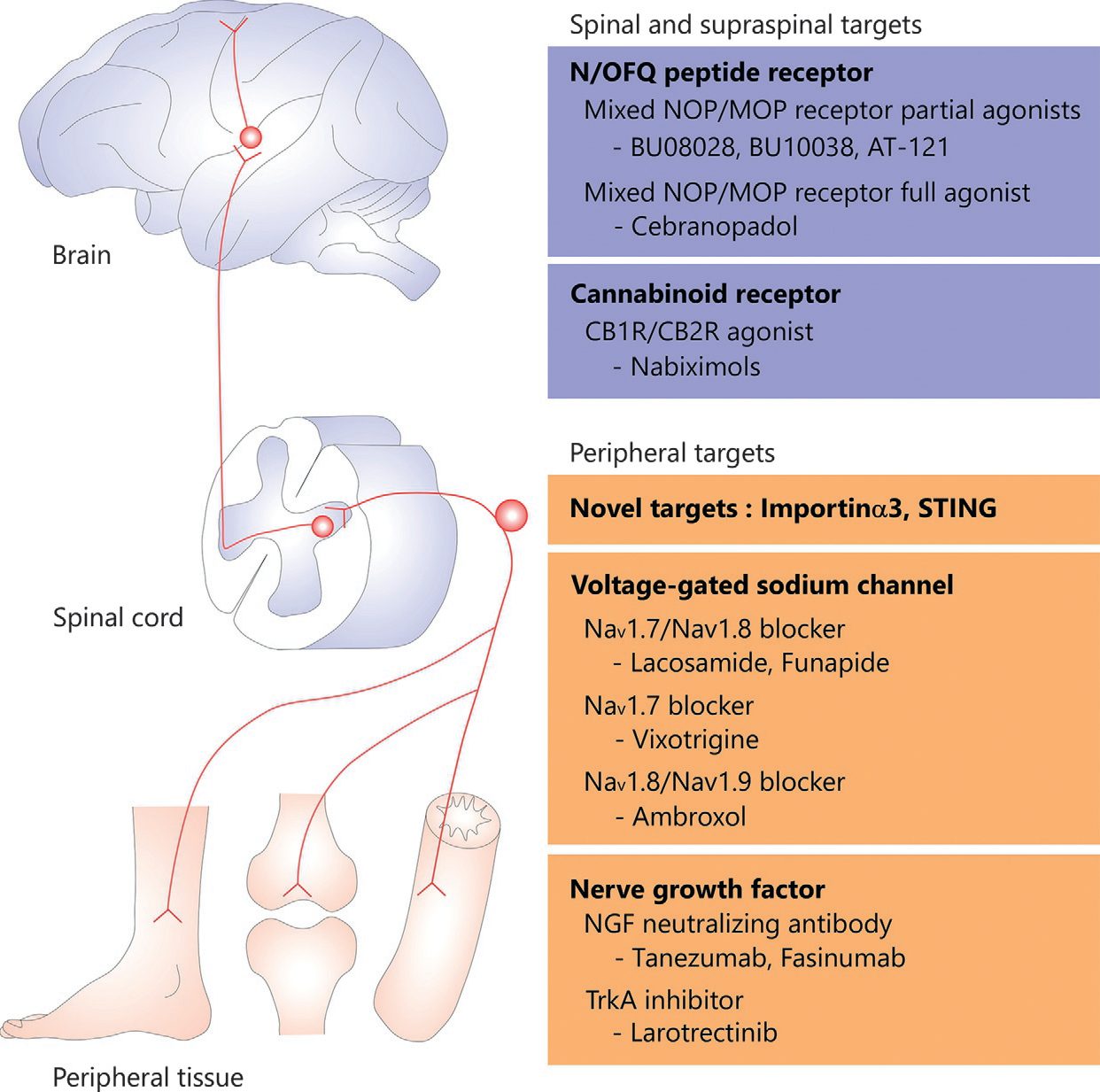
Potential therapeutic targets for the treatment of pain. Primary sensory neurons transmit electrical impulses that encode specific sensory information to the spinal cord, subsequently projection neurons in the dorsal horn convey pain information to the brain. Given the complicated mechanisms of pain processing, it might be reasonable to discover novel analgesic targets based on preclinical studies. At the same time, it is also very important to conduct preclinical studies that replicate human pain conditions. CB1R; cannabinoid 1 receptor, CB2R; cannabinoid 2 receptor, MOP; μ- opioid peptide, Nav; voltage-gated sodium channel, NGF; nerve growth factor, N/OFQ; nociceptin/orphanin FQ, NOP; nociceptin/orphanin FQ peptide, STING; stimulator of interferon genes, TrkA; tyrosine kinase A.
3.3. Treatment of pain by peripheral actions
3.3.1. NGF inhibitors
Accumulating evidence suggests that neurotrophins, such as NGF and brain-derived neurotrophic factor, largely contribute to several types of pain through peripheral and central mechanisms ( Ji, Xu, & Gao, 2014). NGF is the most characterized neurotrophin that activates nociceptive sensory neurons to convey pain signals from the periphery to the CNS, and its effects on pain processing have been well investigated (Denk, Bennett, & McMahon, 2017). Although NGF underlies the development of sensory neurons, it is also required to maintain the homeostasis of neurons and other tissues. NGF forms a ligand-receptor complex with the high-affinity receptor tyrosine kinase A (TrkA), and the complex is translocated to the nuclei of DRG neurons. Subsequently, phosphorylation of the NGF-TrkA complex enhances gene transcription (Denk et al., 2017; Ehlers, Kaplan, Price, & Koliatsos, 1995). In response to nociceptive (such as thermal, mechanical, and chemical) stimuli, substance P and calcitonin gene-related peptide are upregulated by NGF-TrkA signaling. Although these neuropeptides are transported to the central terminals of sensory neurons, they can also be released from peripheral terminals to act as proinflammatory molecules in tissue (Wise, Seidel, & Lane, 2021). Moreover, NGF increases the excitability of DRG neurons when nociceptors, such as TRPV1, are activated, resulting in peripheral sensitization ( Ji, Samad, Jin, Schmoll, & Woolf, 2002). Given that NGF is a multifunctional molecule that affects not only the C- and Aδ-fibers of DRG neurons but also the bone and other tissues, musculoskeletal diseases are closely related to NGF signaling among the different types of chronic pain (Lane & Corr, 2017). Notably, NGF plays an important role in bone metabolism and regeneration as documented in animal studies. Peripherally released NGF facilitates bone formation by increasing the number of osteoblasts in mice with bone fractures (Wise et al., 2021).
NGF inhibitors attenuate OA-related pain in mice, suggesting that inhibition of NGF signaling might have favorable effects on musculoskeletal pain in humans (Lane & Corr, 2017; Wise et al., 2021). NGF is expressed on accumulating lymphocytes and monocytes in the synovial fluid in patients with rheumatoid arthritis or OA (Barthel et al., 2009; Stoppiello et al., 2014), which is supported by findings that substance P and pro-inflammatory cytokines upregulate NGF in cultured synovial cells. Furthermore, NGF is expressed in bone-associated cells (such as subchondral mononuclear cells, osteoclasts, and chondrocytes) in tissues of patients with OA. The expression levels of NGF are associated with age and synovitis scores, suggesting a correlation between NGF and chronic pain caused by OA (Aso et al., 2019). Based on these findings, a number of small compounds and antibodies that inhibit NGF and TrkA functions have been tested in both preclinical and clinical studies. Larotrectinib, which inhibits TrkA, TrkB, and TrkC, has been approved for the treatment of solid tumors (Drilon et al., 2018). As the therapeutic potential of NGF antibodies has also been demonstrated in animal models of bone cancer pain, it is pivotal to further undertake clinical studies to assess the effectiveness of NGF inhibitors (Lucchesi et al., 2017). However, other small molecules, such as ASP7962, a TrkA antagonist, did not show the desired efficacy in patients with knee OA (Watt et al., 2019). Although, several monoclonal antibodies have been investigated, a majority of these molecules have not entered clinical trials because of lower efficacy or unexpected adverse effects, such as osteonecrosis. Tanezumab and fasinumab are currently being evaluated in clinical trials for OA and chronic low back pain with promising results for further consideration (Wise et al., 2021). Therefore, preclinical and clinical studies have supported the therapeutic potential of NGF-TrkA signaling for chronic pain associated with musculoskeletal diseases.
3.3.2. Nav inhibitors
Among the different types of Navs that are expressed in humans, Nav1.7, Nav1.8, and Nav1.9 are localized on DRG neurons and are crucial for peripheral signaling in nociceptive pain (Dib-Hajj, Yang, Black, & Waxman, 2013; Goodwin & McMahon, 2021). Navs consist of an ion-conducting pore-forming α subunit and one to two β subunits; their trafficking and gating properties are due to the α subunit. These Navs are divided into the tetrodotoxin (TTX)-sensitive subtype (Nav1.7) and the TTX-resistant subtype (Nav1.8 and Nav1.9). TTX-sensitive subtypes are activated more rapidly and show faster kinetics than TTX-resistant subtypes (Zeisel et al., 2018). Previous studies have indicated that mutations in the genes encoding these Navs are largely associated with sensitivity to pain in humans. This is because mutation leads to gain of function in Nav1.7, which in turn activates Nav1.8, which enhances pain (Bennett, Clark, Huang, Waxman, & Dib-Hajj, 2019). Furthermore, loss of function in Nav1.7 results in loss of pain sensation due to different nociceptive stimuli without affecting other neurological functions (Cox et al., 2006; Goldberg et al., 2007). Although the mechanisms linking the loss of Nav1.7 function and insensitivity to pain are not fully understood, it may be correlated to the fact that the majority of inactivating mutations of Nav1.7 lead to a complete loss of channel function (Cox et al., 2006; McDermott et al., 2019). Nav1.8 mutations have been identified in individuals with painful peripheral neuropathy (Faber et al., 2012), and a mutation causing a moderate loss of function in Nav1.8 has been associated with reduced pain sensitivity (Duan et al., 2016). Based on preclinical studies in rodent models, Nav1.8 contributes to neuropathic pain (Lai et al., 2002). Given that many preclinical and human studies have emphasized the potential role of Nav1.8 in regulating pain at the peripheral level, it is essential to validate Nav1.8 as a potential therapeutic target for pain (Alsaloum, Higerd, Effraim, & Waxman, 2020).
Moreover, locally administered non-selective Nav blockers, such as lidocaine, suppress pain originating from different nociceptive stimuli. Funapide, an inhibitor of both Nav1.7 and Nav1.8, has been investigated for the treatment of inherited erythromelalgia, neuropathic pain, and postherpetic neuralgia (Goldberg et al., 2012; Price et al., 2017). Although funapide showed promising results in clinical studies, multiple trials have failed to meet the endpoint (Alsaloum et al., 2020). Lacosamide, which is prescribed as an anticonvulsant inhibiting Nav1.7 and Nav1.8, has been tested in clinical studies for Nav1.7-related small fiber neuropathy (de Greef et al., 2019), and currently available data suggest that this compound is effective in the treatment of painful neuropathy (Alsaloum et al., 2020). Moreover, various selective blockers of Nav1.7 have also been tested in patients with chronic pain. For example, vixotrigine was tested for the treatment of lumbosacral radiculopathy and trigeminal neuralgia. The results of phase II trials suggest that vixotrigine has the potential to be an effective and safe treatment for small fiber neuropathy (Alsaloum et al., 2020). Ambroxol, a widely used mucoactive agent for bronchopulmonary diseases (Malerba & Ragnoli, 2008), inhibits TTX-resistant Nav1.8 and Nav1.9, rather than TTX-sensitive Nav1.7, in DRG neurons (Weiser & Wilson, 2002). Ambroxol not only attenuates neuropathic pain, as seen in preclinical studies using various rodent models (Belkouch et al., 2014; Gaida, Klinder, Arndt, & Weiser, 2005), but also exerts analgesic effects at the peripheral level, as seen in clinical studies (Chenot, Weber, & Friede, 2014). In a recent phase III randomized controlled trial, ambroxol attenuated oropharyngeal pain (Sousa, Lakha, Brette, & Hitier, 2019). Nevertheless, the use of ambroxol for the treatment of neuropathic pain has only been supported by case reports, necessitating controlled clinical trials for neuropathic pain. Furthermore, other Nav1.8 blockers, such as PF-0431082 and VX-150, manufactured by different pharmaceutical companies have also been tested in clinical trials. These new compounds, at least in part, demonstrate reasonable effects on several types of pain, and it is important to keep an eye on the results of the ongoing clinical studies.
3.3.3. Novel targets
More recently, newly discovered molecules regulating sensory processing have been identified in animal models of chronic pain. Importin α3, which is a key regulator of nucleocytoplasmic transport, induces nuclear import of c-Fos in DRG neurons (Marvaldi et al., 2020; Panayotis, Karpova, Kreutz, & Fainzilber, 2015). Importin α3 knockout or sensory neuron-specific knockdown attenuated responses to noxious stimuli and the maintenance phase of neuropathic pain in rodents (Marvaldi et al., 2020). Although c-Fos-related mechanisms underlie both peripheral and central levels of pain processing (Coggeshall, 2005), peripheral sensory neurons are targets of importin α3-dependent regulation of pain. Given that the expression patterns of both importin α3 and c-Fos are conserved between mice and humans (Ray et al., 2018), importin α3 might be a potential target for pain. Stimulator of interferon genes (STING) is an endoplasmic reticulum-bound DNA sensor that induces type I interferons and other cytokines that regulate immune responses (Hopfner & Hornung, 2020; Zhang et al., 1998). As STING is highly expressed in peripheral nociceptors, it was hypothesized that it may regulate nociception through interferon signaling. Although the loss of STING functions exhibited hypersensitivity to nociceptive stimuli and heightened nociceptor excitability, the intrathecal activation of STING exerted analgesic effects in mice with chemotherapy-induced peripheral neuropathy, nerve injury-induced neuropathic pain, and bone cancer pain (Donnelly et al., 2021). Importantly, the STING pathway exerts long-lasting analgesic effects in NHPs, and interferons directly suppress the excitability of nociceptors in mice and that of NHPs in humans through modulation of sodium and calcium channel function (Donnelly et al., 2021), indicating that STING can also be a novel peripheral target for chronic pain. Collectively, these results suggest that novel pain-regulatory molecules exhibiting different peripheral mechanisms are ideal candidates for the development of potent and non-opioid-based analgesics.
4. Treatment of pain by spinal and supraspinal actions
4.1. Cannabinoid receptor agonists
It has been traditionally known that cannabinoids (CBs), such as Δ9-tetrahydrocannabinol (THC) and cannabidiol (CBD), purified from Cannabis sativa have therapeutic effects against several neurological disorders (Friedman, French, & Maccarrone, 2019). Based on binding studies using radiolabeled THC, CB1 and CB2 receptors (CB1R and CB2R, respectively) have been identified as target receptors of naturally isolated CBs (Devane, Dysarz 3rd, Johnson, Melvin, & Howlett, 1988; Matsuda, Lolait, Brownstein, Young, & Bonner, 1990). Subsequently, the biochemical and physiological characteristics of endogenous ligands for CB1R and CB2R, which are anandamide and 2-arachidonoyl-glycerol, have been worked out (Devane et al., 1992; Sugiura et al., 1995). Similar to the opioid receptors, both CB1R and CB2R are GPCRs. Although CB1Rs are abundantly expressed in the brain, CB2Rs are highly expressed in the immune system and involved in several physiological functions (Cristino, Bisogno, & Di Marzo, 2020). As CB1Rs are located on presynaptic terminals of excitatory and inhibitory neurons, activation of CB1R signaling can inhibit voltage-gated calcium channels and vesicular release of neurotransmitters (such as GABA and glutamate) underlying regulation of mood, cognition, and perception in animals and humans (Bara, Ferland, Rompala, Szutorisz, & Hurd, 2021; Cristino et al., 2020). In particular, accumulating evidence from studies using animal models suggests that CB1R agonists produce antinociception through the activation of CB1Rs at spinal or supraspinal levels (Donvito et al., 2018), indicating that CB1R agonists may be potential targets for centrally acting analgesics. Moreover, CB1Rs are also expressed on astrocytes that are not only involved in the regulation of synaptic plasticity in different brain areas and but also further located on progenitor stem cells and differentiate into neurons or astrocytes (Prenderville, Kelly, & Downer, 2015). Given that the patterns of CB1R activation and distribution may be altered in several CNS regions under pathological conditions (Cristino et al., 2020), CB1R may be considered a potential target for the treatment of neurological diseases. In contrast, CB2Rs are mainly expressed in CNS microglia in patients with multiple sclerosis and amyotrophic lateral sclerosis (Aymerich et al., 2018), and the activation of CB2Rs attenuates the expression of inflammatory cytokines associated with neurodegenerative disorders (Cassano et al., 2017). Thus, CB2Rs may have different functional properties and diverse therapeutic mechanisms than CB1Rs.
In view of the substantial anatomical and neurochemical overlap between opioid and CB systems (Le Foll, 2021; Rios, Gomes, & Devi, 2006), it was hypothesized that there is a therapeutic benefit of combining opioids and CBs for the treatment of pain. In this regard, a number of studies have demonstrated that non-analgesic doses of THC enhance opioid analgesia in rodents and NHPs (Babalonis & Walsh, 2020). The synergistic effects of opioid and CB combinations appeared to be rather selective for pain processing in animals, and the other unfavorable effects of CBs (such as hypothermia and hypoactivity) and opioids (such as respiratory depression) were not potentiated by such a combination (Welch, 2009). However, these findings have not been clearly confirmed in human studies. In fact, a combination of CBs and opioids demonstrated diverse results depending on the experimental setting in humans (Babalonis & Walsh, 2020). In addition, no substantial data are available to suggest that CBs are highly effective analgesics for different pain conditions, including chronic pain. However, considerable evidence suggests that the expression levels of CB1R and CB2R are altered not only in animal models but also in patients with multiple sclerosis and that the activation of CB1Rs and CB2Rs has a beneficial therapeutic effect (Baker et al., 2000; Cristino et al., 2020; Maresz et al., 2007). Nabiximols, a cannabis extract that includes THC and CBD, has shown promising results in clinical studies and has been approved in several countries for the treatment of neuropathic pain and spasticity in multiple sclerosis (Giacoppo, Bramanti, & Mazzon, 2017). Moreover, as the effects of nabiximols are based on neuroimmune regulation, treatment with nabiximols reduces circulating levels of inflammatory cytokines that usually enhance pathological pain (Orefice et al., 2016). Nabiximols are currently being investigated for the treatment of painful conditions associated with CNS disorders, such as stroke and glioblastoma (Marinelli et al., 2017). Since nabiximols have shown promising therapeutic effects in clinical studies for these diseases (Friedman et al., 2019), it is likely that CBs can also be useful for several types of neurological disorders.
4.2. NOP receptor agonists
As NOP receptors are located on the pain-processing pathways (the DRG, spinal cord, and brain), several studies have been conducted to understand the functional profiles of NOP receptors in regulating pain (Corder et al., 2018; Schroder et al., 2014). Similar to MOP receptors, presynaptic and postsynaptic NOP receptors can inhibit excitatory glutamatergic transmission via GPCR signaling in the SDH and brain (Winters, Christie, & Vaughan, 2019), indicating that NOP receptor activation may produce antinociception. In rodents, NOP receptor activation produces both dose- and pain modality-dependent bi-directional (pronociceptive or antinociceptive) effects at the spinal or supraspinal levels. (Schroder et al., 2014). Nevertheless, as peptidic and non-peptidic NOP receptor agonists suppress neuropathic pain in rodents (Kiguchi, Ding, Kishioka, & Ko, 2020; Toll et al., 2016), NOP receptor-related ligands may be effective for the treatment of neuropathic pain. In contrast, intrathecal, intracisternal, or systemic administration of NOP receptor agonists only produces antinociception in NHPs (Ding et al., 2015; Kiguchi & Ko, 2019; Ko et al., 2009). Notably, N/OFQ was found to be the most potent endogenous peptide among all opioid peptides, such as β-endorphin and enkephalins, for spinal analgesia in inflammatory pain conditions (Lee & Ko, 2015). Importantly, intrathecal administration of NOP receptor agonists produces potent antinociceptive effects without eliciting morphine-like adverse effects, such as itching, in NHPs (Kiguchi et al., 2020). Moreover, the combination of SCH221510, a NOP receptor agonist, and buprenorphine significantly enhanced the analgesic effect and did not cause adverse effects in NHPs (Cremeans et al., 2012). These results indicate the probability of synergistic analgesia by the co-activation of both NOP and MOP receptors in humans.
Given the preferable functional properties of the NOP receptor agonists, it has been hypothesized that mixed NOP/MOP receptor agonists may be safer opioid analgesics, without the undesirable adverse effects. Based on the understanding of the structure-activity relationship of NOP receptors, several candidates for novel ligands, acting on both NOP and MOP receptors with different affinities and partial agonist efficacies, have been investigated in preclinical studies (Kiguchi, Ding, Kishioka, & Ko, 2020; Zaveri & Meyer, 2019). In rodents, intrathecal administration of BU08028 (Khroyan et al., 2011) and SR16435 exerted antinociceptive and antiallodynic effects through the activation of NOP and MOP receptors (Sukhtankar, Zaveri, Husbands, & Ko, 2013). Recently, BU10038 was developed as a naltrexone-derived bifunctional NOP/MOP receptor agonist. Intrathecal BU10038 produced potent and long-lasting antinociceptive and antiallodynic effects compared to morphine in NHPs, and intrathecal BU10038 did not cause adverse effects, such as itching sensation, and tolerance. Unlike spinal analgesia, systemically administered NOP receptor agonists demonstrate integrated effects of peripheral, spinal, and supraspinal actions (Kiguchi et al., 2019). Systemic administration of BU08028 exerted morphine-comparable antinociceptive effects, but showed rewarding effects in rodents (Khroyan et al., 2011). However, it did not elicit reinforcing effects in NHPs (Ding et al., 2016), indicating that a translational gap exists with respect to the efficacy of mixed NOP/MOP receptor agonists between rodents and NHPs. A non-morphinan NOP/MOP receptor agonist, AT-121, produced potent antinociceptive and antiallodynic effects, which were almost 100-fold more potent than those of morphine, and was antagonized by either NOP or MOP receptor antagonist in NHPs (Ding et al., 2018). Moreover, the systemic administration of AT-121 lacks dose-limiting adverse effects such as abuse liability, itching sensation, respiratory depression, cardiovascular dysfunction, physical dependence, and opioid-induced hyperalgesia (Ding et al., 2018). Although these NOP/MOP receptor agonists have different binding profiles to NOP and MOP receptors (Kiguchi, Ding, Kishioka, & Ko, 2020), mixed NOP/MOP receptor partial agonists seem to have ideal functional profiles and, thus, may be developed as novel non-addictive and safe analgesics.
In rodents, cebranopadol has a half-life of 4.5h and oral bioavailability of 13–23% and exerts dose-dependent antinociceptive effects via NOP and MOP receptor activation after systemic administration (Calo & Lambert, 2018; Linz et al., 2014). Notably, cebranopadol was >100-fold more potent and long-lasting than morphine-induced analgesia, producing potent anti-allodynic effects in different models of neuropathic pain; unlike morphine, it does not affect respiratory and motor functions at analgesic doses (Calo & Lambert, 2018; Linz et al., 2014). The beneficial effects of cebranopadol can be translated to NHPs as it produces potent antinociceptive and antihypersensitive effects compared to fentanyl after systemic or intrathecal administration with reduced side effects, such as a lack of respiratory depression and itching (Ding et al., 2021). Although cebranopadol caused reinforcing effects in the fixed-ratio schedule of self-administration, its reinforcing strength was lower than that of fentanyl (Ding et al., 2021). Given that MOP receptor activation mainly contributes to cebranopadol-induced antinociception in NHPs, it is necessary to consider the detectable reinforcing effects of cebranopadol for clinical use. In phase I and phase II clinical trials for analgesic indications, the pharmacokinetic properties of cebranopadol have been demonstrated in patients (Calo & Lambert, 2018; Kleideiter, Piana, Wang, Nemeth, & Gautrois, 2018). Although oral administration of cebranopadol produced drug-liking effects, they were less or shorter than those of the usual MOP receptor full agonist hydromorphone (Gohler et al., 2019). This observation was consistent with that of preclinical study on NHPs (Ding et al., 2021). In clinical trials, cebranopadol produced significant analgesic efficacy in patients with low back pain, cancer pain, or diabetic neuropathy, with fewer undesirable side effects such as miosis, respiratory depression, constipation, dizziness, and nausea (Christoph, Eerdekens, Kok, Volkers, & Freynhagen, 2017; Eerdekens et al., 2019; Kiguchi et al., 2020). Collectively, these findings support the concept of coactivation of NOP and MOP receptors for enhanced pain relief with reduced side effects.
5. Conclusions
This review highlights the recent progress in well-known targets and discusses exciting findings of emerging and novel targets for the treatment of opioid abuse and pain. Given that opioids are the most commonly prescribed analgesics for the treatment of moderate-to-severe pain, it is challenging to develop non-opioid analgesics that can replace opioids in clinical settings (Azzam, McDonald, & Lambert, 2019; Corbett, Henderson, McKnight, & Paterson, 2006). Nevertheless, biomedical science has evolved rapidly to discover innovative medications to treat opioid abuse and/or as safe, non-addictive interventions to manage pain (Rasmussen et al., 2019; Volkow & Collins, 2017; Woolf, 2020). There is a translational gap in the development of effective medications for opioid abuse and pain as behavioral and physiological responses to opioid-related ligands in rodents cannot be translated to primates (Ding & Ko, 2021; Kiguchi, Ding, & Ko, 2016). Rhesus macaques have genetic similarities to the human genome (Rhesus Macaque Genome et al., 2007) and this species plays a critical role in modeling human diseases and physiological responses that are not well simulated in rodents (Balsters et al., 2020; Seok et al., 2013; Warren et al., 2020). Although rodent studies have documented promising novel targets, few non-opioid targets have been validated in primates. To date, NHP studies have demonstrated that MOP receptor- or mixed opioid receptor subtype-based ligands are effective in alleviating opioid-induced adverse effects (Ding & Ko, 2021). Given the mounting evidence that neuroanatomical aspects of opioid and non-opioid receptors differ between rodents and primates (Bianchi et al., 2012; Hawkinson et al., 2007; Shiers et al., 2021; Yu et al., 2019), it is important to investigate the functional profiles of novel compounds extensively in NHP models before initiating expensive clinical trials. More pharmacological studies of the functional efficacy, selectivity, and tolerability of the novel compounds in NHP models will accelerate the development of effective medications for opioid abuse and pain to advance human medicine.
Acknowledgments
Funding was provided by the US National Institutes of Health, National Institute on Drug Abuse (DA053343, DA044450, and DA049580). The contents of this study are solely the responsibility of the authors and do not necessarily represent the official views of US federal agencies. We thank Editage for editing the manuscript.
Abbreviations
- CB
cannabinoid
- CB1R
cannabinoid 1 receptor
- CB2R
cannabinoid 2 receptor
- CBD
cannabidiol
- CNS
central nervous system
- DOP
δ-opioid peptide
- DORA
dual orexin antagonist
- DRG
dorsal root ganglia
- FDA
Food and Drug Administration
- GPCR
G-protein coupled receptor
- KOP
κ-opioid peptide
- MOP
μ-opioid peptide
- N/OFQ
nociceptin/orphanin FQ
- NAC
nucleus accumbens
- Nav
voltage-gated sodium channel
- NGF
nerve growth factor
- NHP
non-human primate
- NK1
neurokinin-1
- NOP
nociceptin/orphanin FQ peptide
- OA
osteoarthritis
- OUD
opioid use disorder
- OX1R
orexin-1 receptor
- OX2R
orexin-2 receptor
- PFC
prefrontal cortex
- SDH
spinal dorsal horn
- STING
stimulator of interferon genes
- TAAR1
trace amine-associated receptor 1
- THC
Δ9-tetrahydrocannabinol
- TrkA
tyrosine kinase A
- TRPV1
transient receptor potential vanilloid 1
- TTX
tetrodotoxin
- VTA
ventral tegmental area
Footnotes
Conflict of interest
N.K. and M.C.K. declare that there is no conflict of interest.
References
- Alsaloum M, Higerd GP, Effraim PR, & Waxman SG (2020). Status of peripheral sodium channel blockers for non-addictive pain treatment. Nature Reviews. Neurology, 16(12), 689–705. 10.1038/s41582-020-00415-2. [DOI] [PubMed] [Google Scholar]
- Aso K, Shahtaheri SM, Hill R, Wilson D, McWilliams DF, & Walsh DA (2019). Associations of symptomatic knee osteoarthritis with histopathologic features in subchondral bone. Arthritis & Rhematology, 71(6), 916–924. 10.1002/art.40820. [DOI] [PubMed] [Google Scholar]
- Attal N, & Bouhassira D (2015). Pharmacotherapy of neuropathic pain: which drugs, which treatment algorithms? Pain, 156(Suppl 1), S104–S114. 10.1097/01.j.pain.0000460358.01998.15. [DOI] [PubMed] [Google Scholar]
- Attal N, Bouhassira D, & Baron R (2018). Diagnosis and assessment of neuropathic pain through questionnaires. Lancet Neurology, 17(5), 456–466. 10.1016/S1474-4422(18)30071-1. [DOI] [PubMed] [Google Scholar]
- Aymerich MS, Aso E, Abellanas MA, Tolon RM, Ramos JA, Ferrer I, et al. (2018). Cannabinoid pharmacology/therapeutics in chronic degenerative disorders affecting the central nervous system. Biochemical Pharmacology, 157, 67–84. 10.1016/j.bcp.2018.08.016. [DOI] [PubMed] [Google Scholar]
- Azevedo Neto J, Costanzini A, De Giorgio R, Lambert DG, Ruzza C, & Calò G (2020). Biased versus partial agonism in the search for safer opioid analgesics. Molecules, 25(17), 3780. 10.3390/molecules25173870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azzam AAH, McDonald J, & Lambert DG (2019). Hot topics in opioid pharmacology: Mixed and biased opioids. British Journal of Anaesthesia, 122(6), e136–e145. 10.1016/j.bja.2019.03.006. [DOI] [PubMed] [Google Scholar]
- Babalonis S, & Walsh SL (2020). Therapeutic potential of opioid/cannabinoid combinations in humans: Review of the evidence. European Neuropsychopharmacology, 36, 206–216. 10.1016/j.euroneuro.2020.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baimel C, Bartlett SE, Chiou LC, Lawrence AJ, Muschamp JW, Patkar O, et al. (2015). Orexin/hypocretin role in reward: Implications for opioid and other addictions. British Journal of Pharmacology, 172(2), 334–348. 10.1111/bph.12639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker D, Pryce G, Croxford JL, Brown P, Pertwee RG, Huffman JW, et al. (2000). Cannabinoids control spasticity and tremor in a multiple sclerosis model. Nature, 404(6773), 84–87. 10.1038/35003583. [DOI] [PubMed] [Google Scholar]
- Balsters JH, Zerbi V, Sallet J, Wenderoth N, & Mars RB (2020). Primate homologs of mouse cortico-striatal circuits. eLife, 9. 10.7554/eLife.53680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bara A, Ferland JN, Rompala G, Szutorisz H, & Hurd YL (2021). Cannabis and synaptic reprogramming of the developing brain. Nature Reviews. Neuroscience, 22(7), 423–438. 10.1038/s41583-021-00465-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barthel C, Yeremenko N, Jacobs R, Schmidt RE, Bernateck M, Zeidler H, et al. (2009). Nerve growth factor and receptor expression in rheumatoid arthritis and spondyloarthritis. Arthritis Research & Therapy, 11(3), R82. 10.1186/ar2716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basbaum AI, Bautista DM, Scherrer G, & Julius D (2009). Cellular and molecular mechanisms of pain. Cell, 139(2), 267–284. 10.1016/j.cell.2009.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belgers M, Leenaars M, Homberg JR, Ritskes-Hoitinga M, Schellekens AF, & Hooijmans CR (2016). Ibogaine and addiction in the animal model, a systematic review and meta-analysis. Translational Psychiatry, 6(5), e826. 10.1038/tp.2016.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belkouch M, Dansereau MA, Tetreault P, Biet M, Beaudet N, Dumaine R, et al. (2014). Functional up-regulation of Nav1.8 sodium channel in Abeta afferent fibers subjected to chronic peripheral inflammation. Journal of Neuroinflammation, 11, 45. 10.1186/1742-2094-11-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bello EP, Mateo Y, Gelman DM, Noain D, Shin JH, Low MJ, et al. (2011). Cocaine supersensitivity and enhanced motivation for reward in mice lacking dopamine D2 autoreceptors. Nature Neuroscience, 14(8), 1033–1038. 10.1038/nn.2862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett DL, Clark AJ, Huang J, Waxman SG, & Dib-Hajj SD (2019). The role of voltage-gated sodium channels in pain signaling. Physiological Reviews, 99(2), 1079–1151. 10.1152/physrev.00052.2017. [DOI] [PubMed] [Google Scholar]
- Bianchi BR, Zhang XF, Reilly RM, Kym PR, Yao BB, & Chen J (2012). Species comparison and pharmacological characterization of human, monkey, rat, and mouse TRPA1 channels. The Journal of Pharmacology and Experimental Therapeutics, 341(2), 360–368. 10.1124/jpet.111.189902. [DOI] [PubMed] [Google Scholar]
- Bohn LM, Gainetdinov RR, Lin FT, Lefkowitz RJ, & Caron MG (2000). Mu-opioid receptor desensitization by beta-arrestin-2 determines morphine tolerance but not dependence. Nature, 408(6813), 720–723. 10.1038/35047086. [DOI] [PubMed] [Google Scholar]
- Bohn LM, Lefkowitz RJ, Gainetdinov RR, Peppel K, Caron MG, & Lin FT (1999). Enhanced morphine analgesia in mice lacking beta-arrestin 2. Science, 286(5449), 2495–2498. [DOI] [PubMed] [Google Scholar]
- Borowsky B, Adham N, Jones KA, Raddatz R, Artymyshyn R, Ogozalek KL, et al. (2001). Trace amines: identification of a family of mammalian G protein-coupled receptors. Proceedings of the National Academy of Sciences of the United States of America, 98(16), 8966–8971. 10.1073/pnas.151105198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braz J, Solorzano C, Wang X, & Basbaum AI (2014). Transmitting pain and itch messages: a contemporary view of the spinal cord circuits that generate gate control. Neuron, 82(3), 522–536. 10.1016/j.neuron.2014.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bromberg-Martin ES, Matsumoto M, & Hikosaka O (2010). Dopamine in motivational control: rewarding, aversive, and alerting. Neuron, 68(5), 815–834. 10.1016/j.neuron.2010.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brook K, Bennett J, & Desai SP (2017). The chemical history of morphine: An 8000-year journey, from resin to de-novo synthesis. Journal of Anesthesia History, 3(2), 50–55. 10.1016/j.janh.2017.02.001. [DOI] [PubMed] [Google Scholar]
- Calo G, & Lambert DG (2018). Nociceptin/orphanin FQ receptor ligands and translational challenges: focus on cebranopadol as an innovative analgesic. British Journal of Anaesthesia, 121(5), 1105–1114. 10.1016/j.bja.2018.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron LP, Tombari RJ, Lu J, Pell AJ, Hurley ZQ, Ehinger Y, et al. (2021). A non-hallucinogenic psychedelic analogue with therapeutic potential. Nature, 589(7842), 474–479. 10.1038/s41586-020-3008-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassano T, Calcagnini S, Pace L, De Marco F, Romano A, & Gaetani S (2017). Cannabinoid receptor 2 signaling in neurodegenerative disorders: From pathogenesis to a promising therapeutic target. Frontiers in Neuroscience, 11, 30. 10.3389/fnins.2017.00030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Mestek A, Liu J, Hurley JA, & Yu L (1993). Molecular cloning and functional expression of a mu-opioid receptor from rat brain. Molecular Pharmacology, 44(1), 8–12. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/8393525. [PubMed] [Google Scholar]
- Chen XT, Pitis P, Liu G, Yuan C, Gotchev D, Cowan CL, et al. (2013). Structure-activity relationships and discovery of a G protein biased mu opioid receptor ligand, [(3-methoxythiophen-2-yl)methyl]({2-[(9R)-9-(pyridin-2-yl)-6-oxaspiro-[4.5] decan- 9-yl]ethyl})amine (TRV130), for the treatment of acute severe pain. Journal of Medicinal Chemistry, 56(20), 8019–8031. 10.1021/jm4010829. [DOI] [PubMed] [Google Scholar]
- Chenot JF, Weber P, & Friede T (2014). Efficacy of Ambroxol lozenges for pharyngitis: A meta-analysis. BMC Family Practice, 15, 45. 10.1186/1471-2296-15-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christoph A, Eerdekens MH, Kok M, Volkers G, & Freynhagen R (2017). Cebranopadol, a novel first-in-class analgesic drug candidate: First experience in patients with chronic low back pain in a randomized clinical trial. Pain, 158(9), 1813–1824. 10.1097/j.pain.0000000000000986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coggeshall RE (2005). Fos, nociception and the dorsal horn. Progress in Neurobiology, 77(5), 299–352. 10.1016/j.pneurobio.2005.11.002. [DOI] [PubMed] [Google Scholar]
- Cohen SP, Vase L, & Hooten WM (2021). Chronic pain: An update on burden, best practices, and new advances. Lancet, 397(10289), 2082–2097. 10.1016/S0140-6736(21)00393-7. [DOI] [PubMed] [Google Scholar]
- Collaborators U. S. B. o. D, Mokdad AH, Ballestros K, Echko M, Glenn S, Olsen HE, et al. (2018). The State of US Health, 1990–2016: Burden of diseases, injuries, and risk factors among US states. JAMA, 319(14), 1444–1472. 10.1001/jama.2018.0158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbett AD, Henderson G, McKnight AT, & Paterson SJ (2006). 75 years of opioid research: The exciting but vain quest for the Holy Grail. British Journal of Pharmacology, 147(Suppl 1), S153–S162. 10.1038/sj.bjp.0706435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corder G, Castro DC, Bruchas MR, & Scherrer G (2018). Endogenous and exogenous opioids in pain. Annual Review of Neuroscience, 41, 453–473. 10.1146/annurev-neuro-080317-061522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cousins DA, Butts K, & Young AH (2009). The role of dopamine in bipolar disorder. Bipolar Disorders, 11(8), 787–806. 10.1111/j.1399-5618.2009.00760.x. [DOI] [PubMed] [Google Scholar]
- Cox JJ, Reimann F, Nicholas AK, Thornton G, Roberts E, Springell K, et al. (2006). An SCN9A channelopathy causes congenital inability to experience pain. Nature, 444(7121), 894–898. 10.1038/nature05413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cremeans CM, Gruley E, Kyle DJ, & Ko MC (2012). Roles of mu-opioid receptors and nociceptin/orphanin FQ peptide receptors in buprenorphine-induced physiological responses in primates. The Journal of Pharmacology and Experimental Therapeutics, 343(1), 72–81. 10.1124/jpet.112.194308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cristino L, Bisogno T, & Di Marzo V (2020). Cannabinoids and the expanded endocannabinoid system in neurological disorders. Nature Reviews. Neurology, 16(1), 9–29. 10.1038/s41582-019-0284-z. [DOI] [PubMed] [Google Scholar]
- Cui Y, Ostlund SB, James AS, Park CS, Ge W, Roberts KW, et al. (2014). Targeted expression of mu-opioid receptors in a subset of striatal direct-pathway neurons restores opiate reward. Nature Neuroscience, 17(2), 254–261. 10.1038/nn.3622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darcq E, & Kieffer BL (2018). Opioid receptors: drivers to addiction? Nature Reviews. Neuroscience, 19(8), 499–514. 10.1038/s41583-018-0028-x. [DOI] [PubMed] [Google Scholar]
- Dart RC, Surratt HL, Cicero TJ, Parrino MW, Severtson SG, Bucher-Bartelson B, et al. (2015). Trends in opioid analgesic abuse and mortality in the United States. The New England Journal of Medicine, 372(3), 241–248. 10.1056/NEJMsa1406143. [DOI] [PubMed] [Google Scholar]
- de Greef BTA, Hoeijmakers JGJ, Geerts M, Oakes M, Church TJE, Waxman SG, et al. (2019). Lacosamide in patients with Nav1.7 mutations-related small fibre neuropathy: A randomized controlled trial. Brain, 142(2), 263–275. 10.1093/brain/awy329. [DOI] [PubMed] [Google Scholar]
- Degenhardt L, Charlson F, Mathers B, Hall WD, Flaxman AD, Johns N, et al. (2014). The global epidemiology and burden of opioid dependence: results from the global burden of disease 2010 study. Addiction, 109(8), 1320–1333. 10.1111/add.12551. [DOI] [PubMed] [Google Scholar]
- Degenhardt L, Grebely J, Stone J, Hickman M, Vickerman P, Marshall BDL, et al. (2019). Global patterns of opioid use and dependence: harms to populations, interventions, and future action. Lancet, 394(10208), 1560–1579. 10.1016/S0140-6736(19)32229-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denk F, Bennett DL, & McMahon SB (2017). Nerve growth factor and pain mechanisms. Annual Review of Neuroscience, 40, 307–325. 10.1146/annurev-neuro-072116-031121. [DOI] [PubMed] [Google Scholar]
- Devane WA, Dysarz FA 3rd, Johnson MR, Melvin LS, & Howlett AC (1988). Determination and characterization of a cannabinoid receptor in rat brain. Molecular Pharmacology, 34(5), 605–613. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/2848184. [PubMed] [Google Scholar]
- Devane WA, Hanus L, Breuer A, Pertwee RG, Stevenson LA, Griffin G, et al. (1992). Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science, 258(5090), 1946–1949. 10.1126/science.1470919. [DOI] [PubMed] [Google Scholar]
- DeWire SM, Yamashita DS, Rominger DH, Liu G, Cowan CL, Graczyk TM, et al. (2013). A G protein-biased ligand at the mu-opioid receptor is potently analgesic with reduced gastrointestinal and respiratory dysfunction compared with morphine. The Journal of Pharmacology and Experimental Therapeutics, 344(3), 708–717. 10.1124/jpet.112.201616. [DOI] [PubMed] [Google Scholar]
- Dib-Hajj SD, Yang Y, Black JA, & Waxman SG (2013). The Na(V)1.7 sodium channel: from molecule to man. Nature Reviews. Neuroscience, 14(1), 49–62. 10.1038/nrn3404. [DOI] [PubMed] [Google Scholar]
- Ding H, Czoty PW, Kiguchi N, Cami-Kobeci G, Sukhtankar DD, Nader MA, et al. (2016). A novel orvinol analog, BU08028, as a safe opioid analgesic without abuse liability in primates. Proceedings of the National Academy of Sciences of the United States of America, 113(37), E5511–E5518. 10.1073/pnas.1605295113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding H, Hayashida K, Suto T, Sukhtankar DD, Kimura M, Mendenhall V, et al. (2015). Supraspinal actions of nociceptin/orphanin FQ, morphine and substance P in regulating pain and itch in non-human primates. British Journal of Pharmacology, 172(13), 3302–3312. 10.1111/bph.13124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding H, Kiguchi N, Perrey DA, Nguyen T, Czoty PW, Hsu FC, et al. (2020). Antinociceptive, reinforcing, and pruritic effects of the G-protein signalling-biased mu opioid receptor agonist PZM21 in non-human primates. British Journal of Anaesthesia, 125(4), 596–604. 10.1016/j.bja.2020.06.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding H, Kiguchi N, Yasuda D, Daga PR, Polgar WE, Lu JJ, et al. (2018). A bifunctional nociceptin and mu opioid receptor agonist is analgesic without opioid side effects in nonhuman primates. Science Translational Medicine, 10(456), eaar3483. 10.1126/scitranslmed.aar3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding H, & Ko MC (2021). Translational value of non-human primates in opioid research. Experimental Neurology, 338, 113602. 10.1016/j.expneurol.2021.113602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding H, Trapella C, Kiguchi N, Hsu FC, Calo G, & Ko MC (2021). Functional profile of systemic and intrathecal cebranopadol in nonhuman primates. Anesthesiology, 135(3), 482–493. 10.1097/ALN.0000000000003848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnelly CR, Jiang C, Andriessen AS, Wang K, Wang Z, Ding H, et al. (2021). STING controls nociception via type I interferon signalling in sensory neurons. Nature, 591(7849), 275–280. 10.1038/s41586-020-03151-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donvito G, Nass SR, Wilkerson JL, Curry ZA, Schurman LD, Kinsey SG, et al. (2018). The endogenous cannabinoid system: A budding source of targets for treating inflammatory and neuropathic pain. Neuropsychopharmacology, 43(1), 52–79. 10.1038/npp.2017.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drilon A, Laetsch TW, Kummar S, DuBois SG, Lassen UN, Demetri GD, et al. (2018). Efficacy of Larotrectinib in TRK fusion-positive cancers in adults and children. The New England Journal of Medicine, 378(8), 731–739. 10.1056/NEJMoa1714448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan G, Han C, Wang Q, Guo S, Zhang Y, Ying Y, et al. (2016). A SCN10A SNP biases human pain sensitivity. Molecular Pain, 12. 10.1177/1744806916666083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eerdekens MH, Kapanadze S, Koch ED, Kralidis G, Volkers G, Ahmedzai SH, et al. (2019). Cancer-related chronic pain: Investigation of the novel analgesic drug candidate cebranopadol in a randomized, double-blind, noninferiority trial. European Journal of Pain, 23(3), 577–588. 10.1002/ejp.1331. [DOI] [PubMed] [Google Scholar]
- Ehlers MD, Kaplan DR, Price DL, & Koliatsos VE (1995). NGF-stimulated retrograde transport of trkA in the mammalian nervous system. The Journal of Cell Biology, 130(1), 149–156. 10.1083/jcb.130.1.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans CJ, Keith DE Jr., Morrison H, Magendzo K, & Edwards RH (1992). Cloning of a delta opioid receptor by functional expression. Science, 258(5090), 1952–1955. 10.1126/science.1335167. [DOI] [PubMed] [Google Scholar]
- Faber CG, Lauria G, Merkies IS, Cheng X, Han C, Ahn HS, et al. (2012). Gain-of-function Nav1.8 mutations in painful neuropathy. Proceedings of the National Academy of Sciences of the United States of America, 109(47), 19444–19449. 10.1073/pnas.1216080109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields H (2004). State-dependent opioid control of pain. Nature Reviews. Neuroscience, 5(7), 565–575. 10.1038/nrn1431. [DOI] [PubMed] [Google Scholar]
- Fields HL, & Margolis EB (2015). Understanding opioid reward. Trends in Neurosciences, 38(4), 217–225. 10.1016/j.tins.2015.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzcharles MA, Cohen SP, Clauw DJ, Littlejohn G, Usui C, & Hauser W (2021). Nociplastic pain: Towards an understanding of prevalent pain conditions. Lancet, 397(10289), 2098–2110. 10.1016/S0140-6736(21)00392-5. [DOI] [PubMed] [Google Scholar]
- Fragale JE, James MH, Avila JA, Spaeth AM, Aurora RN, Langleben D, et al. (2021). The insomnia-addiction positive feedback loop: Role of the orexin system. Frontiers of Neurology and Neuroscience, 45, 117–127. 10.1159/000514965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fragale JE, Pantazis CB, James MH, & Aston-Jones G (2019). The role of orexin-1 receptor signaling in demand for the opioid fentanyl. Neuropsychopharmacology, 44(10), 1690–1697. 10.1038/s41386-019-0420-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman D, French JA, & Maccarrone M (2019). Safety, efficacy, and mechanisms of action of cannabinoids in neurological disorders. Lancet Neurology, 18(5), 504–512. 10.1016/S1474-4422(19)30032-8. [DOI] [PubMed] [Google Scholar]
- Gaida W, Klinder K, Arndt K, & Weiser T (2005). Ambroxol, a Nav1.8-preferring Na(+) channel blocker, effectively suppresses pain symptoms in animal models of chronic, neuropathic and inflammatory pain. Neuropharmacology, 49(8), 1220–1227. 10.1016/j.neuropharm.2005.08.004. [DOI] [PubMed] [Google Scholar]
- Georgescu D, Zachariou V, Barrot M, Mieda M, Willie JT, Eisch AJ, et al. (2003). Involvement of the lateral hypothalamic peptide orexin in morphine dependence and withdrawal. The Journal of Neuroscience, 23(8), 3106–3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giacoppo S, Bramanti P, & Mazzon E (2017). Sativex in the management of multiple sclerosis-related spasticity: An overview of the last decade of clinical evaluation. Multiple Sclerosis and Related Disorders, 17, 22–31. 10.1016/j.msard.2017.06.015. [DOI] [PubMed] [Google Scholar]
- Gillis A, Gondin AB, Kliewer A, Sanchez J, Lim HD, Alamein C, et al. (2020). Low intrinsic efficacy for G protein activation can explain the improved side effect profiles of new opioid agonists. Science Signaling, 13(625), eaaz3140. 10.1126/scisignal.aaz3140. [DOI] [PubMed] [Google Scholar]
- Gillis A, Kliewer A, Kelly E, Henderson G, Christie MJ, Schulz S, et al. (2020). Critical assessment of G protein-biased agonism at the μ-opioid receptor. Trends in Pharmacological Sciences, 41(12), 947–959. 10.1016/j.tips.2020.09.009. [DOI] [PubMed] [Google Scholar]
- Gohler K, Sokolowska M, Schoedel KA, Nemeth R, Kleideiter E, Szeto I, et al. (2019). Assessment of the abuse potential of cebranopadol in nondependent recreational opioid users: A phase 1 randomized controlled study. Journal of Clinical Psychopharmacology, 39(1), 46–56. 10.1097/JCP.0000000000000995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg YP, MacFarlane J, MacDonald ML, Thompson J, Dube MP, Mattice M, et al. (2007). Loss-of-function mutations in the Nav1.7 gene underlie congenital indifference to pain in multiple human populations. Clinical Genetics, 71(4), 311–319. 10.1111/j.1399-0004.2007.00790.x. [DOI] [PubMed] [Google Scholar]
- Goldberg YP, Price N, Namdari R, Cohen CJ, Lamers MH, Winters C, et al. (2012). Treatment of Na(v)1.7-mediated pain in inherited erythromelalgia using a novel sodium channel blocker. Pain, 153(1), 80–85. 10.1016/j.pain.2011.09.008. [DOI] [PubMed] [Google Scholar]
- Goodwin G, & McMahon SB (2021). The physiological function of different voltage-gated sodium channels in pain. Nature Reviews. Neuroscience, 22(5), 263–274. 10.1038/s41583-021-00444-w. [DOI] [PubMed] [Google Scholar]
- Grim TW, Schmid CL, Stahl EL, Pantouli F, Ho JH, Acevedo-Canabal A, et al. (2020). A G protein signaling-biased agonist at the μ-opioid receptor reverses morphine tolerance while preventing morphine withdrawal. Neuropsychopharmacology, 45(2), 416–425. 10.1038/s41386-019-0491-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunther T, Dasgupta P, Mann A, Miess E, Kliewer A, Fritzwanker S, et al. (2018). Targeting multiple opioid receptors - improved analgesics with reduced side effects? British Journal of Pharmacology, 175(14), 2857–2868. 10.1111/bph.13809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris GC, Wimmer M, & Aston-Jones G (2005). A role for lateral hypothalamic orexin neurons in reward seeking. Nature, 437(7058), 556–559. 10.1038/nature04071. [DOI] [PubMed] [Google Scholar]
- Hawkinson JE, Szoke BG, Garofalo AW, Hom DS, Zhang H, Dreyer M, et al. (2007). Pharmacological, pharmacokinetic, and primate analgesic efficacy profile of the novel bradykinin B1 Receptor antagonist ELN441958. The Journal of Pharmacology and Experimental Therapeutics, 322(2), 619–630. 10.1124/jpet.107.120352. [DOI] [PubMed] [Google Scholar]
- Hill R, Disney A, Conibear A, Sutcliffe K, Dewey W, Husbands S, et al. (2018). The novel mu-opioid receptor agonist PZM21 depresses respiration and induces tolerance to antinociception. British Journal of Pharmacology, 175(13), 2653–2661. 10.1111/bph.14224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnasko TS, Sotak BN, & Palmiter RD (2005). Morphine reward in dopamine-deficient mice. Nature, 438(7069), 854–857. 10.1038/nature04172. [DOI] [PubMed] [Google Scholar]
- Holmes D (2016). The pain drain. Nature, 535(7611), S2–S3. 10.1038/535S2a. [DOI] [PubMed] [Google Scholar]
- Hopf FW (2020). Recent perspectives on orexin/hypocretin promotion of addiction-related behaviors. Neuropharmacology, 168, 108013. 10.1016/j.neuropharm.2020.108013. [DOI] [PubMed] [Google Scholar]
- Hopfner KP, & Hornung V (2020). Molecular mechanisms and cellular functions of cGAS-STING signalling. Nature Reviews. Molecular Cell Biology, 21(9), 501–521. 10.1038/s41580-020-0244-x. [DOI] [PubMed] [Google Scholar]
- Iyer RN, Favela D, Zhang G, & Olson DE (2021). The iboga enigma: the chemistry and neuropharmacology of iboga alkaloids and related analogs. Natural Product Reports, 38(2), 307–329. 10.1039/d0np00033g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James MH, Fragale JE, Aurora RN, Cooperman NA, Langleben DD, & Aston-Jones G (2020). Repurposing the dual orexin receptor antagonist suvorexant for the treatment of opioid use disorder: why sleep on this any longer? Neuropsychopharmacology, 45(5), 717–719. 10.1038/s41386-020-0619-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James MH, Mahler SV, Moorman DE, & Aston-Jones G (2017). A decade of orexin/hypocretin and addiction: Where are we now? Current Topics in Behavioral Neurosciences, 33, 247–281. 10.1007/7854_2016_57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji RR, Chamessian A, & Zhang YQ (2016). Pain regulation by non-neuronal cells and inflammation. Science, 354(6312), 572–577. 10.1126/science.aaf8924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji RR, Samad TA, Jin SX, Schmoll R, & Woolf CJ (2002). p38 MAPK activation by NGF in primary sensory neurons after inflammation increases TRPV1 levels and maintains heat hyperalgesia. Neuron, 36(1), 57–68. 10.1016/s0896-6273(02)00908-x. [DOI] [PubMed] [Google Scholar]
- Ji RR, Xu ZZ, & Gao YJ (2014). Emerging targets in neuroinflammation-driven chronic pain. Nature Reviews. Drug Discovery, 13(7), 533–548. 10.1038/nrd4334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johanson CE, Arfken CL, di Menza S, & Schuster CR (2012). Diversion and abuse of buprenorphine: Findings from national surveys of treatment patients and physicians. Drug and Alcohol Dependence, 120(1–3), 190–195. 10.1016/j.drugalcdep.2011.07.019. [DOI] [PubMed] [Google Scholar]
- Jones JD, Manubay JM, Mogali S, Metz VE, Madera G, Martinez S, et al. (2017). Abuse liability of intravenous buprenorphine vs. buprenorphine/naloxone: Importance of absolute naloxone amount. Drug and Alcohol Dependence, 179, 362–369. 10.1016/j.drugalcdep.2017.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakko J, Alho H, Baldacchino A, Molina R, Nava FA, & Shaya G (2019). Craving in opioid use disorder: From neurobiology to clinical practice. Frontiers in Psychiatry, 10, 592. 10.3389/fpsyt.2019.00592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khroyan TV, Polgar WE, Cami-Kobeci G, Husbands SM, Zaveri NT, & Toll L (2011). The first universal opioid ligand, (2S)-2-[(5R,6R,7R,14S)-N-cyclopropylmethyl-4,5-epoxy-6,14-ethano-3-hydroxy-6-meth oxymorphinan-7-yl]-3,3-dimethylpentan-2-ol (BU08028): characterization of the in vitro profile and in vivo behavioral effects in mouse models of acute pain and cocaine-induced reward. The Journal of Pharmacology and Experimental Therapeutics, 336(3), 952–961. 10.1124/jpet.110.175620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiguchi N, Ding H, Cami-Kobeci G, Sukhtankar DD, Czoty PW, DeLoid HB, et al. (2019). BU10038 as a safe opioid analgesic with fewer side-effects after systemic and intrathecal administration in primates. British Journal of Anaesthesia, 122(6), e146–e156. 10.1016/j.bja.2018.10.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiguchi N, Ding H, Kishioka S, & Ko MC (2020). Nociceptin/Orphanin FQ Peptide Receptor-Related Ligands as Novel Analgesics. Current Topics in Medicinal Chemistry, 20(31), 2878–2888. 10.2174/1568026620666200508082615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiguchi N, Ding H, & Ko MC (2016). Central N/OFQ-NOP receptor system in pain modulation. Advances in Pharmacology, 75, 217–243. 10.1016/bs.apha.2015.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiguchi N, Ding H, & Ko MC (2020). Therapeutic potentials of NOP and MOP receptor coactivation for the treatment of pain and opioid abuse. Journal of Neuroscience Research. 10.1002/jnr.24624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiguchi N, & Ko MC (2019). Effects of NOP-related ligands in nonhuman primates. Handbook of Experimental Pharmacology, 254, 323–343. 10.1007/164_2019_211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleideiter E, Piana C, Wang S, Nemeth R, & Gautrois M (2018). Clinical pharmacokinetic characteristics of cebranopadol, a novel first-in-class analgesic. Clinical Pharmacokinetics, 57(1), 31–50. 10.1007/s40262-017-0545-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko MC, Woods JH, Fantegrossi WE, Galuska CM, Wichmann J, & Prinssen EP (2009). Behavioral effects of a synthetic agonist selective for nociceptin/orphanin FQ peptide receptors in monkeys. Neuropsychopharmacology, 34(9), 2088–2096. 10.1038/npp.2009.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kourrich S, Calu DJ, & Bonci A (2015). Intrinsic plasticity: An emerging player in addiction. Nature Reviews. Neuroscience, 16(3), 173–184. 10.1038/nrn3877. [DOI] [PubMed] [Google Scholar]
- Kreek MJ, Reed B, & Butelman ER (2019). Current status of opioid addiction treatment and related preclinical research. Science Advances, 5(10), eaax9140. 10.1126/sciadv.aax9140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai J, Gold MS, Kim CS, Bian D, Ossipov MH, Hunter JC, et al. (2002). Inhibition of neuropathic pain by decreased expression of the tetrodotoxin-resistant sodium channel, NaV1.8. Pain, 95(1–2), 143–152. 10.1016/s0304-3959(01)00391-8. [DOI] [PubMed] [Google Scholar]
- Lane NE, & Corr M (2017). Osteoarthritis in 2016: Anti-NGF treatments for pain—Two steps forward, one step back? Nature Reviews Rheumatology, 13(2), 76–78. 10.1038/nrrheum.2016.224. [DOI] [PubMed] [Google Scholar]
- Lavonas EJ, Severtson SG, Martinez EM, Bucher-Bartelson B, Le Lait MC, Green JL, et al. (2014). Abuse and diversion of buprenorphine sublingual tablets and film. Journal of Substance Abuse Treatment, 47(1), 27–34. 10.1016/j.jsat.2014.02.003. [DOI] [PubMed] [Google Scholar]
- Le Foll B (2021). Opioid-sparing effects of cannabinoids: Myth or reality? Progress in Neuro-Psychopharmacology & Biological Psychiatry, 106, 110065. 10.1016/j.pnpbp.2020.110065. [DOI] [PubMed] [Google Scholar]
- Le Pen G, Wichmann J, Moreau JL, & Jenck F (2002). The orphanin receptor agonist RO 64–6198 does not induce place conditioning in rats. Neuroreport, 13(4), 451–454. 10.1097/00001756-200203250-00018. [DOI] [PubMed] [Google Scholar]
- Lee H, & Ko MC (2015). Distinct functions of opioid-related peptides and gastrin-releasing peptide in regulating itch and pain in the spinal cord of primates. Scientific Reports, 5, 11676. 10.1038/srep11676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin AP, & Ko MC (2013). The therapeutic potential of nociceptin/orphanin FQ receptor agonists as analgesics without abuse liability. ACS Chemical Neuroscience, 4(2), 214–224. 10.1021/cn300124f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingford-Hughes AR, Welch S, Peters L, Nutt DJ, & British Association for Psychopharmacology, E. R. G. (2012). BAP updated guidelines: evidence-based guidelines for the pharmacological management of substance abuse, harmful use, addiction and comorbidity: recommendations from BAP. Journal of Psychopharmacology, 26(7), 899–952. 10.1177/0269881112444324. [DOI] [PubMed] [Google Scholar]
- Linz K, Christoph T, Tzschentke TM, Koch T, Schiene K, Gautrois M, et al. (2014). Cebranopadol: a novel potent analgesic nociceptin/orphanin FQ peptide and opioid receptor agonist. The Journal of Pharmacology and Experimental Therapeutics, 349(3), 535–548. 10.1124/jpet.114.213694. [DOI] [PubMed] [Google Scholar]
- Liu JF, & Li JX (2018). TAAR1 in addiction: Looking beyond the tip of the iceberg. Frontiers in Pharmacology, 9, 279. 10.3389/fphar.2018.00279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Seaman R Jr., Johnson B, Wu R, Vu J, Tian J, et al. (2021). Activation of trace amine-associated receptor 1 selectively attenuates the reinforcing effects of morphine. British Journal of Pharmacology, 178(4), 933–945. 10.1111/bph.15335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Wu R, & Li JX (2020). TAAR1 and psychostimulant addiction. Cellular and Molecular Neurobiology, 40(2), 229–238. 10.1007/s10571-020-00792-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucchesi M, Lanzetta G, Antonuzzo A, Rozzi A, Sardi I, Favre C, et al. (2017). Developing drugs in cancer-related bone pain. Critical Reviews in Oncology/Hematology, 119, 66–74. 10.1016/j.critrevonc.2017.08.005. [DOI] [PubMed] [Google Scholar]
- Luttrell LM, Maudsley S, & Bohn LM (2015). Fulfilling the promise of “biased” G protein-coupled receptor agonism. Molecular Pharmacology, 88(3), 579–588. 10.1124/mol.115.099630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malerba M, & Ragnoli B (2008). Ambroxol in the 21st century: Pharmacological and clinical update. Expert Opinion on Drug Metabolism & Toxicology, 4(8), 1119–1129. 10.1517/17425255.4.8.1119. [DOI] [PubMed] [Google Scholar]
- Manglik A, Lin H, Aryal DK, McCorvy JD, Dengler D, Corder G, et al. (2016). Structure-based discovery of opioid analgesics with reduced side effects. Nature, 537(7619), 185–190. 10.1038/nature19112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maresz K, Pryce G, Ponomarev ED, Marsicano G, Croxford JL, Shriver LP, et al. (2007). Direct suppression of CNS autoimmune inflammation via the cannabinoid receptor CB1 on neurons and CB2 on autoreactive T cells. Nature Medicine, 13(4), 492–497. 10.1038/nm1561. [DOI] [PubMed] [Google Scholar]
- Margolis EB, Lock H, Chefer VI, Shippenberg TS, Hjelmstad GO, & Fields HL (2006). Kappa opioids selectively control dopaminergic neurons projecting to the prefrontal cortex. Proceedings of the National Academy of Sciences of the United States of America, 103(8), 2938–2942. 10.1073/pnas.0511159103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinelli L, Balestrino M, Mori L, Puce L, Rosa GM, Giorello L, et al. (2017). A randomised controlled cross-over double-blind pilot study protocol on THC:CBD oromucosal spray efficacy as an add-on therapy for post-stroke spasticity. BMJ Open, 7(9), e016843. 10.1136/bmjopen-2017-016843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marvaldi L, Panayotis N, Alber S, Dagan SY, Okladnikov N, Koppel I, et al. (2020). Importin alpha3 regulates chronic pain pathways in peripheral sensory neurons. Science, 369(6505), 842–846. 10.1126/science.aaz5875. [DOI] [PubMed] [Google Scholar]
- Matsuda LA, Lolait SJ, Brownstein MJ, Young AC, & Bonner TI (1990). Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature, 346(6284), 561–564. 10.1038/346561a0. [DOI] [PubMed] [Google Scholar]
- Matthes HW, Maldonado R, Simonin F, Valverde O, Slowe S, Kitchen I, et al. (1996). Loss of morphine-induced analgesia, reward effect and withdrawal symptoms in mice lacking the mu-opioid-receptor gene. Nature, 383(6603), 819–823. 10.1038/383819a0. [DOI] [PubMed] [Google Scholar]
- Matzeu A, & Martin-Fardon R (2020). Targeting the orexin system for prescription opioid use disorder: Orexin-1 receptor blockade prevents oxycodone taking and seeking in rats. Neuropharmacology, 164, 107906. 10.1016/j.neuropharm.2019.107906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDermott LA, Weir GA, Themistocleous AC, Segerdahl AR, Blesneac I, Baskozos G, et al. (2019). Defining the functional role of NaV1.7 in human nociception. Neuron, 101(5), 905–919. e908 10.1016/j.neuron.2019.01.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moehring F, Halder P, Seal RP, & Stucky CL (2018). Uncovering the cells and circuits of touch in normal and pathological settings. Neuron, 100(2), 349–360. 10.1016/j.neuron.2018.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales M, & Margolis EB (2017). Ventral tegmental area: cellular heterogeneity, connectivity and behaviour. Nature Reviews. Neuroscience, 18(2), 73–85. 10.1038/nrn.2016.165. [DOI] [PubMed] [Google Scholar]
- Moran MM, McAlexander MA, Biro T, & Szallasi A (2011). Transient receptor potential channels as therapeutic targets. Nature Reviews. Drug Discovery, 10(8), 601–620. 10.1038/nrd3456. [DOI] [PubMed] [Google Scholar]
- Murphy NP, Lee Y, & Maidment NT (1999). Orphanin FQ/nociceptin blocks acquisition of morphine place preference. Brain Research, 832(1–2), 168–170. [DOI] [PubMed] [Google Scholar]
- Nilius B, & Szallasi A (2014). Transient receptor potential channels as drug targets: From the science of basic research to the art of medicine. Pharmacological Reviews, 66(3), 676–814. 10.1124/pr.113.008268. [DOI] [PubMed] [Google Scholar]
- Nutt DJ, Lingford-Hughes A, Erritzoe D, & Stokes PR (2015). The dopamine theory of addiction: 40 years of highs and lows. Nature Reviews. Neuroscience, 16(5), 305–312. 10.1038/nrn3939. [DOI] [PubMed] [Google Scholar]
- Orefice NS, Alhouayek M, Carotenuto A, Montella S, Barbato F, Comelli A, et al. (2016). Oral palmitoylethanolamide treatment is associated with reduced cutaneous adverse effects of interferon-beta1a and circulating proinflammatory cytokines in relapsing-remitting multiple sclerosis. Neurotherapeutics, 13(2), 428–438. 10.1007/s13311-016-0420-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pain S (2016). Painful progress. Nature, 535(7611), S18–S19. 10.1038/535S18a. [DOI] [PubMed] [Google Scholar]
- Panayotis N, Karpova A, Kreutz MR, & Fainzilber M (2015). Macromolecular transport in synapse to nucleus communication. Trends in Neurosciences, 38(2), 108–116. 10.1016/j.tins.2014.12.001. [DOI] [PubMed] [Google Scholar]
- Parker KE, Pedersen CE, Gomez AM, Spangler SM, Walicki MC, Feng SY, et al. (2019). A paranigral VTA nociceptin circuit that constrains motivation for reward. Cell, 178(3), 653–671. e619. 10.1016/j.cell.2019.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peirs C, & Seal RP (2016). Neural circuits for pain: Recent advances and current views. Science, 354(6312), 578–584. 10.1126/science.aaf8933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrey DA, & Zhang Y (2020). Therapeutics development for addiction: Orexin-1 receptor antagonists. Brain Research, 1731, 145922. 10.1016/j.brainres.2018.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podlesnik CA, Ko MC, Winger G, Wichmann J, Prinssen EP, & Woods JH (2011). The effects of nociceptin/orphanin FQ receptor agonist Ro 64–6198 and diazepam on antinociception and remifentanil self-administration in rhesus monkeys. Psychopharmacology, 213(1), 53–60. 10.1007/s00213-010-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prenderville JA, Kelly AM, & Downer EJ (2015). The role of cannabinoids in adult neurogenesis. British Journal of Pharmacology, 172(16), 3950–3963. 10.1111/bph.13186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price N, Namdari R, Neville J, Proctor KJ, Kaber S, Vest J, et al. (2017). Safety and efficacy of a topical sodium channel inhibitor (TV-45070) in patients with postherpetic neuralgia (PHN): A randomized, controlled, proof-of-concept, crossover study, with a subgroup analysis of the Nav1.7 R1150W genotype. The Clinical Journal of Pain, 33(4), 310–318. 10.1097/AJP.0000000000000408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raehal KM, Walker JK, & Bohn LM (2005). Morphine side effects in beta-arrestin 2 knockout mice. The Journal of Pharmacology and Experimental Therapeutics, 314(3), 1195–1201. 10.1124/jpet.105.087254. [DOI] [PubMed] [Google Scholar]
- Raja SN, Carr DB, Cohen M, Finnerup NB, Flor H, Gibson S, et al. (2020). The revised International Association for the Study of Pain definition of pain: concepts, challenges, and compromises. Pain, 161(9), 1976–1982. 10.1097/j.pain.0000000000001939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen K, White DA, & Acri JB (2019). NIDA’s medication development priorities in response to the opioid crisis: ten most wanted. Neuropsychopharmacology, 44(4), 657–659. 10.1038/s41386-018-0292-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray P, Torck A, Quigley L, Wangzhou A, Neiman M, Rao C, et al. (2018). Comparative transcriptome profiling of the human and mouse dorsal root ganglia: an RNA-seq-based resource for pain and sensory neuroscience research. Pain, 159(7), 1325–1345. 10.1097/j.pain.0000000000001217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimer J, Vogelmann T, Trümper D, & Scherbaum N (2020). Impact of buprenorphine dosage on the occurrence of relapses in patients with opioid dependence. European Addiction Research, 26(2), 77–84. 10.1159/000505294. [DOI] [PubMed] [Google Scholar]
- Revel FG, Moreau JL, Gainetdinov RR, Bradaia A, Sotnikova TD, Mory R, et al. (2011). TAAR1 activation modulates monoaminergic neurotransmission, preventing hyperdopaminergic and hypoglutamatergic activity. Proceedings of the National Academy of Sciences of the United States of America, 108(20), 8485–8490. 10.1073/pnas.1103029108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhesus Macaque Genome S, Analysis C, Gibbs RA, Rogers J, Katze MG, Bumgarner R, et al. (2007). Evolutionary and biomedical insights from the rhesus macaque genome. Science, 316(5822), 222–234. 10.1126/science.1139247. [DOI] [PubMed] [Google Scholar]
- Rios C, Gomes I, & Devi LA (2006). Mu opioid and CB1 cannabinoid receptor interactions: reciprocal inhibition of receptor signaling and neuritogenesis. British Journal of Pharmacology, 148(4), 387–395. 10.1038/sj.bjp.0706757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman RB (1994). A review of the effects of dopaminergic agents in humans: Implications for medication development. NIDA Research Monograph, 145, 67–87. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/8742808. [PubMed] [Google Scholar]
- Saxon AJ (2013). Treatment of opioid dependence. In Research and Development of Opioid-Related Ligands (pp. 61–102). American Chemical Society Publications. [Google Scholar]
- Schmid CL, & Bohn LM (2009). Physiological and pharmacological implications of beta-arrestin regulation. Pharmacology & Therapeutics, 121(3), 285–293. 10.1016/j.pharmthera.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid CL, Kennedy NM, Ross NC, Lovell KM, Yue Z, Morgenweck J, et al. (2017). Bias factor and therapeutic window correlate to predict safer opioid analgesics. Cell, 171(5), 1165–1175. e1113 10.1016/j.cell.2017.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder W, Lambert DG, Ko MC, & Koch T (2014). Functional plasticity of the N/OFQ-NOP receptor system determines analgesic properties of NOP receptor agonists. British Journal of Pharmacology, 171(16), 3777–3800. 10.1111/bph.12744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuckit MA (2016). Treatment of opioid-use disorders. The New England Journal of Medicine, 375(4), 357–368. 10.1056/NEJMra1604339. [DOI] [PubMed] [Google Scholar]
- Schwartz MD, Canales JJ, Zucchi R, Espinoza S, Sukhanov I, & Gainetdinov RR (2018). Trace amine-associated receptor 1: A multimodal therapeutic target for neuropsychiatric diseases. Expert Opinion on Therapeutic Targets, 22(6), 513–526. 10.1080/14728222.2018.1480723. [DOI] [PubMed] [Google Scholar]
- Seok J, Warren HS, Cuenca AG, Mindrinos MN, Baker HV, Xu W, et al. (2013). Genomic responses in mouse models poorly mimic human inflammatory diseases. Proceedings of the National Academy of Sciences of the United States of America, 110(9), 3507–3512. 10.1073/pnas.1222878110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharf R, Sarhan M, & Dileone RJ (2008). Orexin mediates the expression of precipitated morphine withdrawal and concurrent activation of the nucleus accumbens shell. Biological Psychiatry, 64(3), 175–183. 10.1016/j.biopsych.2008.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiers SI, Sankaranarayanan I, Jeevakumar V, Cervantes A, Reese JC, & Price TJ (2021). Convergence of peptidergic and non-peptidergic protein markers in the human dorsal root ganglion and spinal dorsal horn. Journal of Comparative Neurology, 529(10), 2771–2788. 10.1002/cne.25122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith RJ, & Aston-Jones G (2012). Orexin/hypocretin 1 receptor antagonist reduces heroin self-administration and cue-induced heroin seeking. The European Journal of Neuroscience, 35(5), 798–804. 10.1111/j.1460-9568.2012.08013.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soergel DG, Subach RA, Burnham N, Lark MW, James IE, Sadler BM, et al. (2014). Biased agonism of the mu-opioid receptor by TRV130 increases analgesia and reduces on-target adverse effects versus morphine: A randomized, double-blind, placebo-controlled, crossover study in healthy volunteers. Pain, 155(9), 1829–1835. 10.1016/j.pain.2014.06.011. [DOI] [PubMed] [Google Scholar]
- Sotnikova TD, Caron MG, & Gainetdinov RR (2009). Trace amine-associated receptors as emerging therapeutic targets. Molecular Pharmacology, 76(2), 229–235. 10.1124/mol.109.055970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa R, Lakha DR, Brette S, & Hitier S (2019). A randomized, double-blind, placebo-controlled study to assess the efficacy and safety of ambroxol hard-boiled lozenges in patients with acute pharyngitis. Pulm Ther, 5(2), 201–211. 10.1007/s41030-019-00100-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinhoff MS, von Mentzer B, Geppetti P, Pothoulakis C, & Bunnett NW (2014). Tachykinins and their receptors: Contributions to physiological control and the mechanisms of disease. Physiological Reviews, 94(1), 265–301. 10.1152/physrev.00031.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoppiello LA, Mapp PI, Wilson D, Hill R, Scammell BE, & Walsh DA (2014). Structural associations of symptomatic knee osteoarthritis. Arthritis & Rhematology, 66(11), 3018–3027. 10.1002/art.38778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiura T, Kondo S, Sukagawa A, Nakane S, Shinoda A, Itoh K, et al. (1995). 2-Arachidonoylglycerol: a possible endogenous cannabinoid receptor ligand in brain. Biochemical and Biophysical Research Communications, 215(1), 89–97. 10.1006/bbrc.1995.2437. [DOI] [PubMed] [Google Scholar]
- Sukhtankar DD, Lagorio CH, & Ko MC (2014). Effects of the NOP agonist SCH221510 on producing and attenuating reinforcing effects as measured by drug self-administration in rats. European Journal of Pharmacology, 745, 182–189. 10.1016/j.ejphar.2014.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukhtankar DD, Zaveri NT, Husbands SM, & Ko MC (2013). Effects of spinally administered bifunctional nociceptin/orphanin FQ peptide receptor/mu-opioid receptor ligands in mouse models of neuropathic and inflammatory pain. The Journal of Pharmacology and Experimental Therapeutics, 346(1), 11–22. 10.1124/jpet.113.203984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulzer D (2011). How addictive drugs disrupt presynaptic dopamine neurotransmission. Neuron, 69(4), 628–649. 10.1016/j.neuron.2011.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tejeda HA, Wu J, Kornspun AR, Pignatelli M, Kashtelyan V, Krashes MJ, et al. (2017). Pathway- and cell-specific kappa-opioid receptor modulation of excitation-inhibition balance differentially gates D1 and D2 accumbens neuron activity. Neuron, 93(1), 147–163. 10.1016/j.neuron.2016.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thannickal TC, John J, Shan L, Swaab DF, Wu MF, Ramanathan L, et al. (2018). Opiates increase the number of hypocretin-producing cells in human and mouse brain and reverse cataplexy in a mouse model of narcolepsy. Science Translational Medicine, 10(447). 10.1126/scitranslmed.aao4953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd AJ (2010). Neuronal circuitry for pain processing in the dorsal horn. Nature Reviews. Neuroscience, 11(12), 823–836. 10.1038/nrn2947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toll L, Bruchas MR, Calo G, Cox BM, & Zaveri NT (2016). Nociceptin/orphanin FQ receptor structure, signaling, ligands, functions, and interactions with opioid systems. Pharmacological Reviews, 68(2), 419–457. 10.1124/pr.114.009209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonelli M, & Cichero E (2020). Trace amine associated receptor 1 (TAAR1) modulators: a patent review (2010-present). Expert Opinion on Therapeutic Patents, 30(2), 137–145. 10.1080/13543776.2020.1708900. [DOI] [PubMed] [Google Scholar]
- Tzschentke TM, Linz K, Koch T, & Christoph T (2019). Cebranopadol: A novel first-in-class potent analgesic acting via NOP and opioid receptors. Handbook of Experimental Pharmacology, 254, 367–398. 10.1007/164_2019_206. [DOI] [PubMed] [Google Scholar]
- Violin JD, Crombie AL, Soergel DG, & Lark MW (2014). Biased ligands at G-protein-coupled receptors: promise and progress. Trends in Pharmacological Sciences, 35(7), 308–316. 10.1016/j.tips.2014.04.007. [DOI] [PubMed] [Google Scholar]
- Viscusi ER, Webster L, Kuss M, Daniels S, Bolognese JA, Zuckerman S, et al. (2016). A randomized, phase 2 study investigating TRV130, a biased ligand of the mu-opioid receptor, for the intravenous treatment of acute pain. Pain, 157(1), 264–272. 10.1097/j.pain.0000000000000363. [DOI] [PubMed] [Google Scholar]
- Volkow ND, & Blanco C (2020). The changing opioid crisis: Development, challenges and opportunities. Molecular Psychiatry, 26(1), 218–233. 10.1038/s41380-41020-40661-41384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkow ND, & Collins FS (2017). The role of science in addressing the opioid crisis. The New England Journal of Medicine, 377(4), 391–394. 10.1056/NEJMsr1706626. [DOI] [PubMed] [Google Scholar]
- Warren WC, Harris RA, Haukness M, Fiddes IT, Murali SC, Fernandes J, et al. (2020). Sequence diversity analyses of an improved rhesus macaque genome enhance its biomedical utility. Science, 370(6523). 10.1126/science.abc6617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watt FE, Blauwet MB, Fakhoury A, Jacobs H, Smulders R, & Lane NE (2019). Tropomyosin-related kinase A (TrkA) inhibition for the treatment of painful knee osteoarthritis: results from a randomized controlled phase 2a trial. Osteoarthritis and Cartilage, 27(11), 1590–1598. 10.1016/j.joca.2019.05.029. [DOI] [PubMed] [Google Scholar]
- Waxman SG, & Zamponi GW (2014). Regulating excitability of peripheral afferents: Emerging ion channel targets. Nature Neuroscience, 17(2), 153–163. 10.1038/nn.3602. [DOI] [PubMed] [Google Scholar]
- Weiser T, & Wilson N (2002). Inhibition of tetrodotoxin (TTX)-resistant and TTX-sensitive neuronal Na(+) channels by the secretolytic ambroxol. Molecular Pharmacology, 62(3), 433–438. 10.1124/mol.62.3.433. [DOI] [PubMed] [Google Scholar]
- Welch SP (2009). Interaction of the cannabinoid and opioid systems in the modulation of nociception. International Review of Psychiatry, 21(2), 143–151. 10.1080/09540260902782794. [DOI] [PubMed] [Google Scholar]
- Whistler JL, Chuang HH, Chu P, Jan LY, & von Zastrow M (1999). Functional dissociation of mu opioid receptor signaling and endocytosis: implications for the biology of opiate tolerance and addiction. Neuron, 23(4), 737–746. [DOI] [PubMed] [Google Scholar]
- Winters BL, Christie MJ, & Vaughan CW (2019). Electrophysiological Actions of N/OFQ. Handbook of Experimental Pharmacology, 254, 91–130. 10.1007/164_2019_205. [DOI] [PubMed] [Google Scholar]
- Wise BL, Seidel MF, & Lane NE (2021). The evolution of nerve growth factor inhibition in clinical medicine. Nature Reviews Rheumatology, 17(1), 34–46. 10.1038/s41584-020-00528-4. [DOI] [PubMed] [Google Scholar]
- Woolf CJ (2020). Capturing novel non-opioid pain targets. Biological Psychiatry, 87(1), 74–81. 10.1016/j.biopsych.2019.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda K, Raynor K, Kong H, Breder CD, Takeda J, Reisine T, et al. (1993). Cloning and functional comparison of kappa and delta opioid receptors from mouse brain. Proceedings of the National Academy of Sciences of the United States of America, 90(14), 6736–6740. 10.1073/pnas.90.14.6736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yekkirala AS, Roberson DP, Bean BP, & Woolf CJ (2017). Breaking barriers to novel analgesic drug development. Nature Reviews. Drug Discovery, 16(8), 545–564. 10.1038/nrd.2017.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H, Zhao T, Liu S, Wu Q, Johnson O, Wu Z, et al. (2019). MRGPRX4 is a bile acid receptor for human cholestatic itch. eLife, 8, e48431. 10.7554/eLife.48431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamarripa C, Edwards SR, Qureshi HN, Yi JN, Blough BE, & Freeman KB (2018). The G-protein biased mu-opioid agonist, TRV130, produces reinforcing and antinociceptive effects that are comparable to oxycodone in rats. Drug and Alcohol Dependence, 192, 158–162. 10.1016/j.drugalcdep.2018.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarepour L, Fatahi Z, Sarihi A, & Haghparast A (2014). Blockade of orexin-1 receptors in the ventral tegmental area could attenuate the lateral hypothalamic stimulation-induced potentiation of rewarding properties of morphine. Neuropeptides, 48(3), 179–185. 10.1016/j.npep.2014.04.003. [DOI] [PubMed] [Google Scholar]
- Zaveri NT, & Meyer ME (2019). NOP-targeted nonpeptide ligands. Handbook of Experimental Pharmacology, 254, 37–67. 10.1007/164_2019_213. [DOI] [PubMed] [Google Scholar]
- Zeisel A, Hochgerner H, Lonnerberg P, Johnsson A, Memic F, van der Zwan J, et al. (2018). Molecular architecture of the mouse nervous system. Cell, 174(4), 999–1014. e1022 10.1016/j.cell.2018.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Ferguson SS, Barak LS, Bodduluri SR, Laporte SA, Law PY, et al. (1998). Role for G protein-coupled receptor kinase in agonist-specific regulation of mu-opioid receptor responsiveness. Proceedings of the National Academy of Sciences of the United States of America, 95(12), 7157–7162. [DOI] [PMC free article] [PubMed] [Google Scholar]