

Abstract

We describe the gram-scale synthesis of (−)-(1R,2S,3R,4R,5S,6S)-1,3-di(diamino)-1,3-diazido-2,5,6-tri-O-benzylstreptamine from streptomycin by (i) hydrolysis of the two streptomycin guanidine residues, (ii) reprotection of the amines as azides, (iii) protection of all alcohols as benzyl ethers, and (iv) glycosidic bond cleavage with HCl in methanol. Protocols for regioselective monodebenzylation and regioselective reduction of a single azide in the product are also described, providing four optically pure building blocks for exploitation in novel aminoglycoside synthesis.
The continued spread of multidrug resistant infectious diseases demands the equally continuous development of novel anti-infective agents with which to combat them.1−4 The aminoglycoside antibiotics (AGAs), with their well-understood mechanisms of action and resistance,5−11 excellent activity against Gram-negative pathogens,7,12 and wide commercial availability, are excellent starting materials for further development and as such have undergone something of a renaissance in recent years.13,14 These advantages are offset by nephrotoxic and ototoxic side effects15,16 such that next-generation AGA development programs target circumvention of resistance mechanisms concomitant with the reduction of these undesirable side effects.17−20
Although the first AGA to be isolated and developed, streptomycin 1, is based on the streptamine core 2, most common AGAs are constructed around 2-deoxystreptamine 3 and hence belong to the 2-deoxystreptamine class, which is subdivided into the 4,5-disubstituted and 4,6-disubstituted 2-deoxystreptamine AGAs, exemplified by neomycin B 4 and gentamicin C1a5, respectively. The hybrimycins, isolated from a mutant of Streptomyces fradiae,21−26 are 2-hydroxy analogs of the 4,5-AGAs that display parent-like levels of antibacterial activity and, at the target level, of inhibition of ribosomal protein synthesis,21,23,24 with hybrimycin A16 serving as an example of the class. In the 4,6-AGAs, the enzymatically derived 2-hydroxygentamicins 7 are reported to have comparable activity toward wild-type strains as the gentamicins 5 themselves21,24 and even greater activity than the gentamicins toward resistant strains.27 More recently, the 4,6-AGA, 2-hydroxyarbekacin 8, has been reported to have excellent antibacterial activity against MRSA and Pseudomonas aeruginosa. Most importantly though in the context of the need for next generation AGAs with reduced toxicity it was reported that the 2-hydroxygentamicin 7 shows reduced toxicity in mice compared to the parent gentamicin,27 and that 2-hydroxyarbekacin 8 is less nephrotoxic than arbekacin 9 in vitro and in a rat model.28 Similarly, it is known streptomycin 1 itself displays low nephrotoxicity compared to other AGAs (Figure 1).29,30
Figure 1.

Streptamine and 2-deoxystreptamine and derived aminoglycoside antibiotics.
The retention of antibacterial activity by the 2-hydroxy variants of the AGAs coupled with the potential for reduced nephrotoxicity28−30 suggests a broader exploration of the 2-hydroxy AGAs, which in turn gives rise to the need for effective synthetic methods and building blocks. Streptamine 2 is commercial, or can be obtained by several straightforward literature methods from commercial inositol,31−35 but its use as starting material would require desymmetrization of this meso-compound in order to provide any enantiomerically pure AGA targets, and while progress has been made in desymmetrizing glycosylations of meso-diols, including inositol derivatives,36−40 these methods are not sufficient to enable the practical synthesis of meaningful quantities of compound.
Desymmetrizing glycosylation reactions of 2-deoxystreptamine derivatives themselves have been described by Nagorny and co-workers with catalysis by readily available chiral phosphoric acid catalysts but selectivities are not ideal, leading to the need for impractical chromatographic separations.41 Potentially, desymmetrization of the diazidostreptamine isomer 10, available in four straightforward steps from myo-inositol,31−34 by ketal formation with d-camphor dimethyl acetal as described for myo-inositol42 followed by glycosylation could be of use, but ultimately we considered that the most practical way forward is exploitation of the desymmetrization of streptamine by nature in the form of streptomycin 1. This approach is clearly related to the use of 2-hydroxygentamicin C1a 7 by Takahashi and co-workers as a substrate for the synthesis of 2-hydroxyarbekacin 8, but as streptomycin 1 is widely commercially available on a large scale compared to the minor gentamicin component 8 clearly has broader potential. Accordingly, we describe here a straightforward gram-scale synthesis of a suitably regioselectively protected derivative of streptamine suitable for immediate use in glycosylation at the 4-position, as well as protocols for its regioselective deprotection giving ready access to several useful building blocks for use in medicinal chemistry campaigns.
Commercially available streptomycin sulfate 1 was reduced to dihydrostreptomycin with aqueous sodium borohydride as previously described.43 This was followed without purification by deguanylation in refluxing saturated aqueous barium hydroxide over 36 h44 and, again without purification by copper sulfate-catalyzed treatment with Stick’s reagent (imidazole-1-sulfonyl azide)45−48 in aqueous methanol for 16 h to give, after chromatographic purification, the anticipated 1,3-di(deamino)-1,3-diazidodihydrostreptomycin 11 and N′-methyl-1,3-di(deamino)-1,3-diazidodihydrostreptomycin derivative 12 in 52% and 14% yield, respectively, over the three steps. The isolation and characterization of 12 confirms the previously reported49 presence of N′-methylstreptomycin as an impurity in the commercial drug, a fact that was further supported by detection of an M+14 peak with formula C22H41N7O12 and 15% abundance relative to 1 in the ESIHRMS spectrum of the commercial material employed in this study. Compound 11 was then treated with benzyl bromide and sodium hydride in DMF at room temperature for 14 h to afford perbenzylated derivative 13 in 74% yield. Following earlier reports on the selective cleavage of the ribofuranosyl bond in streptomycin itself with either methanol or ethyl mercaptan in the presence of hydrogen chloride into streptidine and streptobiosaminide derivatives,50,51 compound 13 was heated to reflux with 3 N HCl in methanol for 16 h. After neutralization of the reaction mixture and removal of the volatiles the crude reaction was acetylated to facilitate chromatographic purification, which ultimately yielded the streptamine derivative 14 and the methyl streptobiosaminide 15 in 83% and 81% yields, respectively, the latter as essentially a single anomer whose configuration was established on the basis of NOE correlations and a 3JH1,H2 coupling constant of 4.1 Hz (Figure 2).52 Finally, the target desymmetrized streptamine mono-ol 16 was obtained in 92% yield on a gram scale by treatment of 14 with sodium methoxide (Scheme 1).
Figure 2.

Diagnostic scalar coupling constant and NOE interactions for the assignment of the configuration of 15.
Scheme 1. Synthesis of Streptamine Derivative 16 from Streptomycin 1.
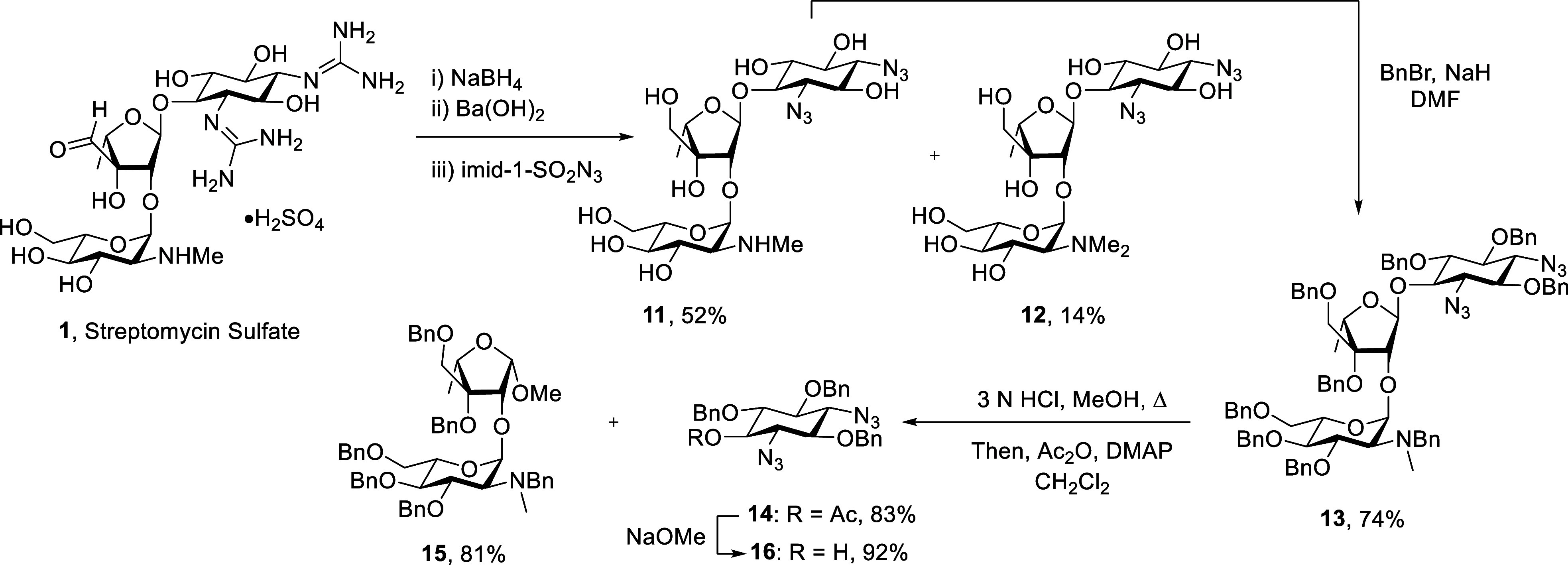
With 16 available on gram scale, we turned to selective partial deprotection so as to provide a range of building blocks for use in the synthesis of novel AGAs and other systems. White light photolysis of 16 in acetonitrile in the presence of iodobenzene diacetate and iodine cleanly provided a diol 17 in 71% yield that was converted to the diacetate 18 in 90% yield to facilitate spectral interpretation (Scheme 2). The formation of 17 is the result of initial alkoxy radical generation followed by δ-hydrogen atom abstraction from the adjacent benzylic methylene group and eventually trapping by iodine and then hydrolysis;53 no evidence was found for the formation of a benzylidene acetal, presumably because it would span a trans-1,2-diol as opposed to the more usual cis-diol.54 Further selective functionalization of 17 is expected to follow the pattern established for related 1,2-diols.20,55−58 Reaction of 16 with boron trichloride in dichloromethane at −20 °C59 cleanly gave diols 19 and meso-20 in 65 and 14% yield, respectively, with the regioselectivity an apparent function of proximity to the strongly electron-withdrawing azido groups. As with 17, diols 19 and 20 were converted to their respective diacetates 21 and 22 for ease of structural elucidation (Scheme 2). Finally, treatment of 16 with stannous chloride and lithium iodide60 in ethyl acetate at 70 °C resulted in apparent proximity-induced regioselective reduction of a single of the two azido groups, whose location was identified following treatment of the crude reaction mixture with an excess of Boc2O and DMAP when the N-Boc oxazolidinone 23 was isolated in 67% yield (Scheme 2).
Scheme 2. Regioselective Partial Deprotections of 16.

Overall, we have provided a convenient scalable route to four optically pure, regioselectively protected streptamine building blocks suitable for use in next-generation AGA syntheses from readily available streptomycin in a minimum of steps.
Experimental Section
General Experimental Methods
All reagents were purchased from commercial sources and used without further purification. Reaction mixtures were heated with the aid of an appropriately sized thermostatically controlled aluminum heating block. Thin-layer chromatography was carried out with 250 μm glass-backed silica plates, and the spots were revealed by UV absorption (254 nm) and by charring with a 20:80 v/v solution of sulfuric acid in ethanol or with a ceric ammonium molybdate solution. All organic solutions were concentrated under a vacuum at 30–45 °C on a rotary evaporator. Purification of crude residues was performed manually as well as by flash column chromatography. Specific rotations were recorded on an automatic polarimeter in CHCl3, MeOH at 589 nm and 23 ± 1 °C with a path length of 10 cm. Nuclear magnetic resonance (NMR) spectra of all compounds were obtained in CDCl3, CD3OD, or C6D6 at 500, 600, or 900 MHz, with chemical shifts (δ) calculated with respect to the residual solvent peak and given in ppm. Multiplicities are abbreviated as follows: s (singlet), d (doublet), t (triplet), q (quartet), dd (doublet of doublet), bs (broad singlet), and m (multiplet). Peak assignments were based on two-dimensional NMR (COSY, HSQC, and HMBC) experiments, and the configurational assignments were determined with the aid of selective 1D NOESY NMR experiments. High-resolution electrospray ionization (ESI) mass spectrometry spectra were recorded using a Thermo Scientific Orbitrap mass analyzer.
1,3-Di(deamino)-1,3-diazidodihydrostreptomycin (11) and N′-Methyl-1,3-di(deamino)-1,3-diazidodihydrostreptomycin (12)
Streptomycin sulfate (10.0 g, 14.7 mmol, purchased from Sigma-Aldrich, CAS No. 3810-74-0) was dissolved in deionized water (70 mL) with stirring, and the pH was adjusted to 8.0 using triethylamine (∼0.6 mL). Sodium borohydride (0.34 g, 8.8 mmol) dissolved in deionized water (10 mL) was added dropwise to the stirred reaction mixture over a period of 5 min at room temperature (a slight elevation in reaction temperature was observed), which was then stirred at room temperature for 0.5 h before it was acidified to pH 1.5 by slow addition of 6 N H2SO4 (∼0.6 mL). After standing at room temperature for 10 min, the reaction mixture was added with stirring to methanol (350 mL) resulting in a white precipitate that was collected by vacuum filtration, washed with methanol (100 mL × 2), and dried under vacuum for 3 h to give crude dihydrostreptomycin sulfate (10.7 g) as confirmed by ESI mass spectrometry. This crude product (10.7 g, 14.7 mmol) was dissolved in deionized water (40 mL), treated with saturated aqueous Ba(OH)2 (250 mL), and stirred for 10 min at room temperature. The white precipitate of barium sulfate was filtered off, and the filtrate was heated to 125 °C for 36 h, after which it was cooled to room temperature, and the excess barium hydroxide was neutralized by addition of carbon dioxide (dry ice). The precipitates were filtered off and washed with water (100 mL), and the combined filtrate and washings were concentrated under reduced pressure at 60 °C to obtain the crude di(deguanidinyl)dihydrostreptomycin carbonate as a thick syrup (6.98 g) as confirmed by ESI mass spectrometry. This crude product (6.98 g, 14.0 mmol) was taken up in deionized water (80 mL), treated with NaHCO3 (11.7 g, 139.7 mmol) and CuSO4·5H2O (0.35 g, 1.40 mmol), and cooled to 0 °C in an ice bath before imidazole-1-sulfonyl azide hydrochloride (7.32 g, 34.9 mmol) was added portionwise with stirring over 10 min. The reaction mixture was held at 0 °C for 0.5 h, then allowed to come to room temperature, and stirred for 16 h. After completion, the reaction mixture was recooled to 0 °C in an ice bath, butylamine (0.5 mL) was added dropwise, and stirring was continued for 0.5 h before the solvents were evaporated under reduced pressure (below 40 °C) to give a crude mixture that was purified by silica gel column chromatography eluting with ammonical methanol in dichloromethane (gradients 25%, 30%, 35%, and 40%) to obtain compound 11 (4.54 g, 52% overall) and 12 (1.26 g, 14% overall).
Compound 11:
Rf = 0.3 in 40% ammonical MeOH in CH2Cl2; [α]21D = −77.0 (c = 2.5, MeOH); 1H NMR (900 MHz, CD3OD) δ 5.56 (d, J = 3.5 Hz, 1H, H1″), 5.40 (d, J = 1.8 Hz, 1H, H1′), 4.37 (d, J = 1.8 Hz, 1H, H2′), 4.25 (q, J = 6.4 Hz, 1H, H4′), 3.90–3.85 (m, 2H, H3″ and H6a″), 3.81 (dd, J = 12.0, 4.7 Hz, 1H, H6b″), 3.76 (ddd, J = 10.4, 4.7, 2.3 Hz, 1H, H4″), 3.64–3.56 (m, 2H, H6′), 3.48 (t, J = 9.4 Hz, 1H, H5″), 3.43 (t, J = 10.0 Hz, 1H, H4), 3.39 (t, J = 9.3 Hz, 1H, H2), 3.32–3.29 (m, 2H, H5 and H6), 3.25 (t, J = 9.9 Hz, 1H, H3), 3.22–3.18 (t, J = 9.9 Hz, 1H, H1), 3.18–3.16 (m, 1H, H2″), 2.88 (s, 3H, H5′), 1.22 (d, J = 6.4 Hz, 3H, H5′); 13C{1H} NMR (226 MHz, CD3OD) δ 107.4 (C1′), 95.4 (C1″), 86.0 (C2′), 82.1(C3′), 79.2 (C6), 79.0 (C4′), 74.9 (C4″), 74.7 (C2), 74.0 (C5), 73.8 (C1), 71.3 (C3″), 71.1 (C5″), 69.5 (C3), 68.7 (C4), 65.5 (C6′), 63.4 (C2″), 62.0 (C6′), 33.0 (NCH3), 13.9 (C5′); HRMS (ESI-TOF) m/z: [M + H]+ calculated for C19H34O12N7 552.2260; found 552.2240.
Compound 12:
Rf = 0.4 in 40% ammonical MeOH in CH2Cl2; [α]21D = −88.4 (c = 1.0, MeOH); 1H NMR (900 MHz, CD3OD) δ 5.59 (d, J = 3.3 Hz, 1H, H1″), 5.37 (d, J = 1.9 Hz, 1H, H1′), 4.36 (d, J = 1.9 Hz, 1H, H2′), 4.26 (q, J = 6.3 Hz, 1H, H4′), 4.05 (ddd, J = 10.6, 8.5, 1H, H3″), 3.87 (dd, J = 11.7, 1.9 Hz, 1H, H6a″), 3.79 (dd, J = 11.9, 4.8 Hz, 1H, H6b″), 3.76 (ddd, J = 10.0, 4.4, 2.0 Hz, 1H, H5″), 3.63–3.57 (m, 2H, H6′), 3.49 (t, J = 9.2 Hz, 1H, H4″), 3.42 (t, J = 9.9 Hz, 1H, H2), 3.38 (t, J = 9.2 Hz, 1H, H5), 3.35–3.32 (m, 2H, H2″ and H6), 3.28 (t, J = 9.7 Hz, 1H, H4), 3.24 (t, J = 9.9 Hz, 1H, H3), 3.19 (t, J = 9.6 Hz, 1H, H1), 3.06 (s, 6H, N(CH3)2), 2.62 (d, J = 1.4 Hz, 1H), 1.20 (d, J = 6.3 Hz, 3H, H5′); 13C{1H} NMR (226 MHz, CD3OD) δ 107.4 (C1′), 95.9 (C1″), 84.9 (C2″), 81.9 (C3′), 79.3 (C4′), 79.1 (C4), 74.8 (C5″), 74.4 (C5), 74.0 (C6), 73.9 (C1), 72.1 (C4″), 69.5 (C3), 69.1 (C3″), 68.7 (C2), 67.6 (C2′), 65.5 (C6′), 62.1 (C6″), 42.1 (N(CH3)2, 14.0 (C5′); HRMS (ESI-TOF) m/z: [M + H]+: calculated for C20H36O12N7 566.2416; found 566.2416.
1,3-Di(deamino)-1,3-diazido-2,5,6-tri-O-benzyl-4-O-(3′,6′-di-O-benzyl-2′-O-(2″-N-benzyl-3″,4″,6″-tri-O-benzyldihydrostreptomycin (13)
Sodium hydride (4.96 g, 124.0 mmol, 60% in mineral oil) was added in two portions to an ice-cold stirred solution of 11 (4.56 g, 8.27 mmol) in DMF (120 mL) under an Ar atmosphere, followed after 10 min by dropwise addition of benzyl bromide (11.9 mL, 99.2 mmol) over 10 min. The reaction mixture was allowed to come to room temperature and was stirred for 16 h, after which it was quenched by dropwise addition of methanol (5 mL), diluted with ethyl acetate (200 mL), and washed with ice-cold water (200 mL × 2) and brine (200 mL), and the organic layer was dried over Na2SO4 and concentrated under reduced pressure. The crude reaction mixture was purified by silica gel column chromatography eluting with ethyl acetate in hexane (gradients 5%, 10%, and 15%) to obtain 13 (8.32 g, 74%): Rf = 0.35 in 15% EtOAc in hexane; [α]21D = −31.5 (c = 1.2, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.47–7.17 (m, 45H, Ar), 5.76 (d, J = 3.5 Hz, 1H, H1′), 5.14 (d, J = 3.3 Hz, 1H, H1″), 4.97–4.71 (m, 12H, 6 × CH2Ph), 4.59–4.34 (m, 4H, 2 × CH2Ph), 4.54 (d, J = 3.5 Hz, 1H, H2′), 4.31 (q, J = 6.4 Hz, 1H, H4′), 4.09 (dd, J = 10.9, 8.5 Hz, 1H, H3″), 3.89 (dt, J = 10.1, 2.6 Hz, 1H, H5″), 3.82 (s, 2H, NCH2Ph), 3.76 (dd, J = 10.1, 8.5 Hz, 1H, H4″), 3.70–3.61 (m, 3H, H6″ and H4), 3.54 (d, J = 9.9 Hz, 1H, H6a′), 3.43–3.35 (m, 4H, H6b′ and H2, H5, H6), 3.27 (t, J = 9.7 Hz, 1H, H3), 3.1 5 (t, J = 9.9 H z, 1H, H1), 2.94 (dd, J = 10.9, 3.4 Hz, 1H, H2″), 2.39 (s, 3H, NCH3), 1.13 (d, J = 6.5 Hz, 3H, H5′); 13C{1H} NMR (126 MHz, CDCl3) δ 140.6, 139.4, 139.1, 138.6, 138.5, 138.0, 138.0, 137.7, 137.2, 128.8, 128.6, 128.6, 128.5, 128.5, 128.4, 128.4, 128.3, 128.3, 128.2, 128.2, 128.1, 128.0, 127.9, 127.8, 127.7, 127.6, 127.5, 127.5, 127.5, 127.3, 127.1, 126.8, 126.7 (aromatic), 105.9 (C1′), 101.5 (C1″), 84.6 (C3′), 84.0 (C2′), 82.4 (C2), 81.0 (C3), 80.4 (C1), 79.8(C4″), 79.0 (C3″), 78.2 (C4′), 76.0 (C4), 75.9, 75.5, 75.4, 74.5, 73.7, 73.7, 73.4 (8 × CH2Ph), 71.3 (C5″), 69.2 (C6′), 68.6 (C6″), 67.3 (C5), 67.3 (C6), 67.1 (CH2Ph), 65.3 (C2″), 62.0 (NCH2Ph), 38.6 (NCH3), 13.3 (C5′); HRMS (ESI-TOF) m/z: [M + H]+ calculated for C82H88O12N7 1362.6391; found 1362.6390.
4-O-Acetyl-1,3-di(deamino)-1,3-diazido-2,5,6-tri-O-benzylstreptamine (14) and Methyl 3′,6′-Di-O-benzyl-2-O-(2″-N-benzyl-3″,4″,6″-tri-O-benzyl)-β-streptobiosaminide (15)
Compound 13 (4.0 g, 2.45 mmol) was suspended in 3 N HCl in methanol (40 mL), and dichloromethane (4.0 mL) was added until a clear solution was obtained. The reaction mixture was then heated to reflux with stirring for 16 h before it was cooled in an ice bath, and triethylamine (5 mL) was added dropwise. The solvents were evaporated under reduced pressure below 45 °C, and the resulting thick syrup was taken up in ethyl acetate (80 mL), washed with saturated aqueous NaHCO3 (80 mL × 2) and brine (80 mL), dried over Na2SO4, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography eluting with ethyl acetate in hexane (gradients 5%, 10%, 15%, and 20%) to obtain an inseparable mixture of 15 and 16 (4.35 g), which was dissolved in CH2Cl2 (20 mL), treated with acetic anhydride (0.6 mL, 5.90 mmol) and DMAP (0.25 g, 2.1 mmol), and stirred at room temperature for 0.5 h. After completion, the solvents were removed under reduced pressure and the residue was purified by silica gel column chromatography eluting with ethyl acetate in hexane (gradient 5%, 10%, 15% and 20%) to give 14 (1.33 g, 83%) and 15 (2.12 g, 81%).
Compound 14:
Rf = 0.55 in 20% EtOAc in hexane; [α]21D = −4.1 (c = 1.0, CHCl3); 1H NMR (900 MHz, CDCl3) δ 7.43–7.23 (m, 15H, Ar), 4.92 (t, J = 10.2 Hz, 1H, H4), 4.86–4.79 (m, 5H, CH2Ph), 4.63 (d, J = 11.4 Hz, 1H, CH2Ph), 3.52 (t, J = 9.6 Hz, 1H, H5), 3.49 (t, J = 10.3 Hz, 1H, H1), 3.46 (t, J = 10.3 Hz, 1H, H3), 3.37 (t, J = 9.7 Hz, 1H, H2), 3.21 (t, J = 9.9 Hz, 1H, H6), 1.99 (s, 3H, CH3); 13C{1H} NMR (226 MHz, CDCl3) δ 169.9 (C=O), 137.9, 137.5, 137.1, 128.8, 128.7, 128.7, 128.7, 128.5, 128.5, 128.3, 128.1, 127.8 (aromatic), 81.3 (C5), 80.9 (C2), 79.4 (C6), 76.3 (CH2Ph), 76.2 (CH2Ph), 75.8 (CH2Ph), 71.8 (C4), 67.4 (C1), 65.3 (C3), 20.9 (CH3); HRMS (ESI-TOF) m/z: [M + Na]+ calculated for C29H30O5N6Na 565.2169; found 565.2156.
Compound 15:
Rf = 0.45 in 20% EtOAc in hexane; [α]21D = −41.8 (c = 1.0, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.40–7.11 (m, 30H, aromatic), 5.11 (d, J = 4. One Hz, 1H, H1′), 5.06 (d, J = 3.3 Hz, 1H, H1″), 4.96 (ABq, J = 11.2 Hz, 2H, CH2Ph), 4.85–4.63 (m, 4H, 2 × CH2Ph), 4.58–4.44 (m, 4H, 2 × CH2Ph), 4.42 (d, J = 4.1 Hz, 1H, H2′), 4.38 (q, J = 6.4 Hz, 1H, H4′), 4.05 (dd, J = 10.9, 8.3 Hz, 1H, H3″), 3.90–3.75 (m, 5H, NCH2Ph, H4″, H5″ and H6a″), 3.65 (dd, J = 10.5, 1.9 Hz, 1H, H6b″), 3.58 (d, J = 9.9 Hz, 1H, H6a′), 3.48 (d, J = 9.9 Hz, 1H, H6b′), 3.40 (s, 3H, OCH3), 2.99 (dd, J = 10.9, 3.3 Hz, 1H, H2″), 2.40 (s, 3H, NCH3), 1.31 (d, J = 6.5 Hz, 3H, H5′); 13C{1H} NMR (126 MHz, CDCl3) δ 140.6, 139.4, 139.0, 138.4, 138.0, 137.9, 128.5, 128.5, 128.4, 128.4, 128.3, 128.2, 128.1, 127.9, 127.8, 127.8, 127.8, 127.7, 127.6, 127.2, 126.9, 126.8 (aromatic), 107.2 (C1′), 102.2 (C1″), 85.8 (C3′), 83.9 (C2′), 80.3 (C4″), 78.9 (C3″), 77.6 (C4′), 74.8 (C5″), 73.7, 73.7, 73.6, 71.3 (4 × CH2Ph), 69.1 (C6′), 68.5 (C6″), 67.6 (CH2Ph), 65.8 (C2″), 61.1 (CH2Ph), 56.0 (OCH3), 38.9 (NCH3), 13.4 (C5′); HRMS (ESI-TOF) m/z: [M + H]+ calculated for C56H64O9N 894.4575; found 894.4568.
1,3-Di(deamino)-1,3-diazido-2,5,6-tri-O-benzylstreptamine (16)
Sodium methoxide (0.26 g, 4.90 mmol) was added to a stirred solution of 14 (1.33 g, 2.45 mmol) in anhydrous CH2Cl2:MeOH (1:1 v/v, 16 mL) at room temperature and stirring continued for 6 h before the reaction mixture was neutralized with Amberlite IRC120 H+ resin and filtered through a cotton wool plug and the filtrate concentrated under reduced pressure. The residue was subjected to silica gel column chromatography eluting with ethyl acetate in hexane (gradients 10%, 20%) to give 16 (1.12 g, 92%): Rf = 0.45 in 20% EtOAc in hexane; [α]21D = −4.4 (c = 1.0, CHCl3); 1H NMR (900 MHz, CDCl3) δ 7.39–7.24 (m, 15H, Ar), 4.85 (d, J = 11.2 Hz, 1H, CH2Ph), 4.81–4.74 (m, 4H, 2 × CH2Ph), 4.68 (d, J = 11.2 Hz, 1H, CH2Ph), 3.42 (t, J = 10.0 Hz, 1H, H1), 3.33 (t, J = 9.9. Hz, 1H, H5) 3.35–3.28 (m, 2H, H3, H4), 3.23 (t, J = 9.6 Hz, 1H, H2), 3.08 (t, J = 9.5 Hz, 1H, H6); 13C{1H} NMR (226 MHz, CDCl3) δ 138.0, 137.5, 137.1, 128.7, 128.6, 128.6, 128.6, 128.6, 128.3, 128.3, 128.3, 128.3, 128.2, 128.1, 128.0, 127.9 (aromatic), 82.9 (C5), 80.8 (C2), 79.5 (C6), 75.9 (CH2Ph), 75.9 (CH2Ph), 75.7 (CH2Ph), 72.7 (C4), 67.6 (C1), 66.8 (C3); HRMS (ESI-TOF) m/z: [M + Na]+ calculated for C27H28O4N6Na 523.2064; found 523.2062.
1,3-Di(deamino)-1,3-diazido-2,6-di-O-benzylstreptamine (17)
A solution of 16 (24 mg, 0.05 mmol) and (diacetoxyiodo)benzene (23 mg, 0.07 mmol) in anhydrous acetonitrile (0.8 mL) was stirred with shielding from ambient light for 0.5 h before iodine (7 mg, 0.03 mmol) was added, and the reaction mixture was irradiated with white light (300 W, tungsten lamp) for 2 h. The reaction mixture was cooled to room temperature, diluted with EtOAc, and washed with saturated aqueous Na2S2O3. The aqueous layer was extracted with EtOAc, and the combined organic layer was washed with brine (20 mL), dried over Na2SO4, and concentrated under reduced pressure. The crude mixture was purified by silica gel column chromatography eluting with ethyl acetate in hexane (gradient 20%, 30%, and 40%) to afford diol 17 (13.7 mg, 71%) as a colorless thick syrup: Rf = 0.35 in 40% EtOAc in hexane; [α]21D = −49.6 (c = 1.0, CHCl3); 1H NMR (500 MHz, benzene-d6) δ 7.44 (d, J = 7.6 Hz, 2H, Ar), 7.37 (d, J = 7.5 Hz, 2H, Ar), 7.23–7.18 (m, 4H, Ar), 7.14–7.10 (m, 2H, Ar), 4.77 (d, J = 11.3 Hz, 1H, CH2Ph), 4.69 (d, J = 12.4 Hz, 3H, CH2Ph), 3.06 (t, J = 9.3 Hz, 1H, H5), 3.01 (t, J = 9.9 Hz, 1H, H1), 2.86 (t, J = 9.8 Hz, 1H, H3), 2.79 (td, J = 9.6, 5.1 Hz, 2H, H4 and H6), 2.71 (t, J = 9.7 Hz, 1H, H2), 2.29 (s, 1H, OH), 2.23 (s, 1H, OH); 13C{1H} NMR (126 MHz, benzene-d6) δ 138.7, 138.1, 128.8, 128.7, 128.7, 128.4, 128.4, 128.2, 128.0 (aromatic), 80.1 (C2), 79.5 (C6), 75.6 (C5), 75.3 (CH2Ph), 75.2 (CH2Ph), 72.8 (C4), 67.4 (C1), 67.1 (C3); HRMS (ESI-TOF) m/z: [M + Na]+ calculated for C20H22N6O4Na 433.1600; found 433.1586.
4,5-Di-O-acetyl-1,3-di(deamino)-1,3-diazido-2,6-di-O-benzylstreptamine (18)
Diol 17 (13.7 mg) was treated with Ac2O (20 μL, 0.201 mmol) and DMAP (5 mg, 0.040 mmol) in CH2Cl2 (1.0 mL) for 0.5 h at room temperature before it was quenched with MeOH (50 μL). The solvents were removed under reduced pressure, and the residue was purified by silica gel column chromatography to obtain diester 18 (15 mg, 90%) as a colorless oil: Rf = 0.40 in 20% EtOAc in hexane; [α]21D = −52.4 (c = 1.0, CHCl3); 1H NMR (500 MHz, benzene-d6) δ 7.40 (d, J = 7.5 Hz, 2H, Ar), 7.30 (d, J = 7.5 Hz, 2H, Ar), 7.25–7.17 (m, 4H, Ar), 7.13–7.09 (m, 2H, Ar), 5.08 (t, J = 9.7 Hz, 1H, H5), 4.86 (t, J = 10.3 Hz, 1H, H4), 4.68–4.58 (m, 3H, CH2Ph), 4.45 (d, J = 11.4 Hz, 1H, CH2Ph), 2.95–2.88 (m, 2H, H1 and H3), 2.85 (t, J = 9.9 Hz, 1H, H6), 2.65 (t, J = 9.8 Hz, 1H, H2), 1.74 (s, 3H, CH3), 1.62 (s, 3H, CH3); 13C{1H} NMR (126 MHz, benzene-d6) δ 169.3 (C=O), 169.2 (C=O), 138.2, 137.9, 128.7, 128.7, 128.6, 128.4, 128.3, 128.2, 128.0, 128.0 (aromatic), 78.9 (C6), 78.7 (C2), 75.7 (CH2Ph), 75.5 (CH2Ph), 73.1 (C5), 70.8 (C4), 67.0 (C1), 64.8 (C3), 20.2 (CH3), 20.2 (CH3); HRMS (ESI-TOF) m/z: [M + Na]+ calculated for C24H26N6O6Na 517.1806; found 517.1801.
1,3-Di(deamino)-1,3-diazido-5,6-di-O-benzylstreptamine (19) and 1,3-Di(deamino)-1,3-diazido-2,5-di-O-benzylstreptamine (20)
BCl3 (220 μL, 1 M in CH2Cl2, 0.22 mmol) was added to a stirred solution of 16 (50 mg, 0.10 mmol) in anhydrous CH2Cl2 (2.0 mL) cooled to −20 °C. After stirring for 2 h, the reaction was quenched by addition of MeOH (100 μL), the reaction mixture was diluted with CH2Cl2 and washed with saturated aqueous NaHCO3, and the aqueous layer was extracted with CH2Cl2. The combined organic layer was washed with brine, dried over Na2SO4, and concentrated under reduced pressure to give a residue that was purified by silica gel column chromatography eluting with ethyl acetate in hexane (gradients 10%, 20%, and 30%) to obtain 19 (27 mg, 65%) and 20 (6 mg, 14%) both as colorless thick syrups.
19:
Rf = 0.30 in 30% EtOAc in hexane; [α]21D = −58.8 (c = 1.0, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.42–7.28 (m, 10H, Ar), 4.93 (d, J = 11.3 Hz, 1H, PhCH2), 4.86 (s, 2H, PhCH2), 4.76 (d, J = 11.3 Hz, 1H, PhCH2), 3.48–3.39 (m, 3H, H5, H4 and H1), 3.34 (td, J = 9.2, 3.6 Hz, 2H, H2 and H6), 3.25 (t, J = 9.8, Hz, 1H, H3), 2.69 (s, 1H, OH), 2.53 (s, 1H, OH); 13C{1H} NMR (126 MHz, CDCl3) δ 138.0, 137.5, 128.9, 128.7, 128.4, 128.3, 128.3, 128.1 (aromatic), 83.4 (C6), 80.9 (C5), 76.0 (CH2Ph), 75.9 (CH2Ph), 73.1 (C4), 72.0 (C2), 67.5 (C3), 66.6 (C1); HRMS (ESI-TOF) m/z: [M + Na]+ calculated for C20H22N6O4Na 433.1600; found 433.1586.
20:
Rf = 0.35 in 30% EtOAc in hexane; 1H NMR (600 MHz, C6D6) δ 7.45–7.41 (m, 2H, Ar), 7.29–7.16 (m, 6H, Ar), 7.15–7.09 (m, 2H, Ar), 4.69 (s, 2H, CH2Ph), 4.65 (s, 2H, CH2Ph), 2.96–2.90 (m, 2H, H4 and H6), 2.88–2.79 (m, 3H, H1, H3, H5), 2.69 (t, J = 9.7 Hz, 1H, H2), 1.90 (s, 2H, 2 × OH); 13C{1H} NMR (151 MHz, C6D6) δ 138.9, 138.1, 128.8, 128.7, 128.6, 128.6, 128.3, 128.1, 128.1, 128.0 (aromatic), 82.0 (C5), 79.2 (C2), 75.5 (CH2Ph), 74.9 (CH2Ph), 73.1 (C4), 73.1 (C6), 67.5 (C1), 67.5(C3); HRMS (ESI-TOF) m/z: [M + Na]+ calculated for C20H22N6O4Na 433.1600; found 433.1588.
2,4-Di-O-acetyl-1,3-di(deamino)-1,3-diazido-5,6-di-O-benzylstreptamine (21)
The diol 19 (26 mg, 0.06 mmol) was treated with Ac2O (20 μL, 0.20 mmol) and DMAP (5 mg, 0.04 mmol) in anhydrous CH2Cl2 (1.0 mL) and stirred for 0.5 h at room temperature before concentration under reduced pressure to give a residue that was subjected to silica gel column chromatography eluting with ethyl acetate in hexane (gradients 10% and 20%) to obtain 21 (24.8 mg, 92%) as a colorless oil: Rf = 0.40 in 20% EtOAc in hexane; [α]21D = −50.6 (c = 1.0, CHCl3); 1H NMR (500 MHz, benzene-d6) δ 7.35–7.30 (m, 2H, Ar), 7.24 (d, J = 7.5 Hz, 2H, Ar), 7.20–7.16 (m, 4H, Ar), 7.13–7.06 (m, 2H, Ar), 5.06 (t, J = 10.2 Hz, 1H, H4), 4.88 (t, J = 10.4 Hz, 1H, H2), 4.67 (d, J = 11.6 Hz, 1H, CH2Ph), 4.62–4.53 (m, 2H, CH2Ph), 4.50 (d, J = 11.6 Hz, 1H, CH2Ph), 3.13 (t, J = 9.6 Hz, 1H, H5), 2.95 (t, J = 10.2 Hz, 1H, H1), 2.85 (t, J = 10.5 Hz, 1H, H3), 2.78 (t, J = 9.7 Hz, 1H, H6), 1.76 (s, 3H, CH3), 1.65 (s, 3H, CH3); 13C{1H} NMR (126 MHz, benzene-d6) δ 168.8 (C=O), 168.7 (C=O), 138.5, 138.3, 128.7, 128.7, 128.4, 128.4, 128.2, 128.0, 127.9, 127.7 (aromatic), 81.2 (C5), 80.3 (C6), 76.0 (CH2Ph), 75.3 (CH2Ph), 71.4 (C4), 70.4 (C2), 65.2 (C1), 63.3 (C3), 20.3 (CH3), 20.2 (CH3); HRMS (ESI-TOF) m/z: [M + Na]+ calculated for C24H26N6O6Na 517.1806; found 517.1809.
4,6-Di-O-acetyl-1,3-di(deamino)-1,3-diazido-2,5-di-O-benzylstreptamine (22)
The diol 20 (6.5 mg, 0.015 mmol) was treated with Ac2O (20 μL, 0.20 mmol) and DMAP (5 mg, 0.04 mmol) in anhydrous CH2Cl2 (1.0 mL) and stirred for 0.5 h at room temperature before concentration under reduced pressure to give a residue that was subjected to silica gel column chromatography eluting with ethyl acetate in hexane (gradients 10% and 20%) to obtain 22 (7 mg, 88%) as a colorless oil: Rf = 0.35 in 20% EtOAc in hexane; 1H NMR (500 MHz, benzene-d6) δ 7.39 (d, J = 7.6 Hz, 2H, Ar), 7.25–7.17 (m, 6H, Ar), 7.15–7.03 (m, 2H, Ar), 5.02 (t, J = 10.2 Hz, 2H, H2 and H4), 4.55 (s, 2H, CH2Ph), 4.45 (s, 2H, CH2Ph), 3.15 (t, J = 9.8 Hz, 1H, H5), 2.89 (t, J = 10.2 Hz, 2H, H1 and H3), 2.54 (t, J = 9.9 Hz, 1H, H6), 1.68 (s, 6H, 2 × CH3); 13C{1H} NMR (126 MHz, benzene-d6) δ 168.7 (2 × C=O), 138.3, 137.9, 128.7, 128.7, 128.7, 128.4, 128.2, 128.0 (aromatic), 79.4 (C5), 78.9 (C2), 76.0 (CH2Ph), 74.4 (CH2Ph), 71.4 (C4 and C6), 65.4 (C1 and C3), 20.3 (2 × CH3); HRMS (ESI-TOF) m/z: [M + Na]+ calculated for C24H26N6O6Na 517.1806; found 517.1809.
1,3-Di(deamino)-1-azido-3-N-tert-butylcarboxy-2,5,6-tri-O-benzyl-3,4-oxazolidinostreptamine (23)
A mixture of CrCl2 (41 mg, 0.34 mmol) and LiI (112 mg, 0.84 mmol) in moist ethyl acetate (2.5 mL) was heated to 70 °C for 0.5 h, then the mono-ol 16 (42 mg, 0.083 mmol) dissolved in ethyl acetate (1.0 mL) was added dropwise, and stirring was continued for 4 h at same temperature. The reaction mixture was cooled to room temperature, diluted with EtOAc (30 mL), and washed with saturated aqueous Na2S2O3 (30 mL), and the aqueous layer was extracted with EtOAc (20 mL × 2). The organic layers were combined, washed with brine (30 mL), dried over Na2SO4, and concentrated under reduced pressure to obtain a crude product (46 mg) which was taken up in EtOAc (1.5 mL), treated with Boc2O (15 mg, 0.17 mmol) and DMAP (5 mg, 0.04 mmol), and stirred for 1 h. The reaction was quenched by addition of MeOH (100 μL), the solvents were removed under vacuum, and the residue was subjected to silica gel column chromatography eluting with ethyl acetate in hexane (gradient 5%, 10%, and 20%) to afford compound 23 (34 mg, 67%) as a colorless thick syrup: Rf = 0.30 in 20% EtOAc in hexane; [α]21D = −86.8 (c = 1.0, CHCl3); 1H NMR (500 MHz, benzene-d6) δ 7.50 (d, J = 7.6 Hz, 2H, Ar), 7.37–7.17 (m, 10H, Ar), 7.15–7.07 (m, 3H, Ar), 4.80–4.73 (m, 2H, CH2Ph), 4.68–4.59 (m, 2H, CH2Ph), 4.51–4.45 (m, 2H, CH2Ph), 3.49 (t, J = 9.7 Hz, 1H, H3), 3.29 (t, J = 9.6 Hz, 1H, H5), 3.12 (t, J = 9.1 Hz, 1H, H1), 3.10–2.99 (m, 2H, H4 and H6), 2.93 (t, J = 9.3 Hz, 1H, H2), 1.43 (s, 9H, 3 × CH3); 13C{1H} NMR (126 MHz, benzene-d6) δ 153.5 (C=O), 151.5 (C=O), 138.1, 137.9, 137.7, 137.6, 129.3 128.4, 128.3, 128.3, 128.0, 127.8, 127.7 (aromatic), 83.8 (quat), 81.7 (C6), 79.5 (C5), 79.4 (C2), 76.0 (C4), 75.4 (CH2Ph), 73.8 (CH2Ph), 73.3 (CH2Ph), 68.7 (C1), 59.0 (C3), 27.5 (3 × CH3); HRMS (ESI-TOF) m/z: [M + Na]+ calculated for C33H36N4O7Na 623.2476; found 623.2464.
Acknowledgments
We thank the NIH (AI172807) for support of this project.
Data Availability Statement
The data underlying this study are available in the published article and its Supporting Information.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.joc.3c02922.
Copies of NMR spectra of all compounds (PDF)
The authors declare the following competing financial interest(s): DC is a cofounder of and equity holder in Juvabis, a biotech startup developing aminoglycoside antibiotics.
Supplementary Material
References
- Wright P. M.; Seiple I. B.; Myers A. G. The Evolving Role of Chemical Synthesis in Antibacterial Drug Discovery. Angew. Chem., Int. Ed. 2014, 53, 8840–8863. 10.1002/anie.201310843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan-Krohn T.; Manetsch R.; O’Doherty G. A.; Kirby J. E. New Strategies and Structural Considerations in Development of Therapeutics for Carbapenem-Resistant Enterobacteriaceae. Translational Res. 2020, 220, 14–32. 10.1016/j.trsl.2020.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Privalsky T. M.; Soohoo A. M.; Wang J.; Walsh C. T.; Wright G. D.; Gordon E. M.; Gray N. S.; Khosla C. Prospects for Antibacterial Discovery and Development. J. Am. Chem. Soc. 2021, 143, 21127–21142. 10.1021/jacs.1c10200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright G. D. Solving the Antibiotic Crisis. ACS Infect. Dis. 2015, 1, 80–84. 10.1021/id500052s. [DOI] [PubMed] [Google Scholar]
- Haddad J.; Kotra L. P.; Mobashery S.. Aminoglycoside Antibiotics: Structures and Mechanisms of Action. In Glycochemsitry: Principles, Synthesis, and Applications; Wang P. G., Bertozzi C. R., Eds.; Dekker, 2001; pp 307–351. [Google Scholar]
- Lin J.; Zhou D.; Steitz T. A.; Polikanov Y. S.; Gagnon M. G. Ribosome-Targeting Antibiotics: Modes of Action, Mechanisms of Resistance, and Implications for Drug Design. Annu. Rev. Biochem. 2018, 87, 451–478. 10.1146/annurev-biochem-062917-011942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong E. S.; Kostrub C. F.; Cass R. T.; Moser H. E.; Serio A. W.; Miller G. H.. Aminoglycosides. In Antibiotic Discovery and Development; Dougherty T. J., Pucci M. J., Eds.; Springer Science+Business Media, 2012; pp 229–269. [Google Scholar]
- Garneau-Tsodikova S.; Labby K. J. Mechanisms of Resistance to Aminoglycoside Antibiotics: Overview and Perspectives. Med. Chem. Commun. 2016, 7, 11–27. 10.1039/C5MD00344J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnet S.; Blanchard J. S. Molecular Insights into Aminoglycoside Action and Resistance. Chem. Rev. 2005, 105, 477–498. 10.1021/cr0301088. [DOI] [PubMed] [Google Scholar]
- Zárate S. G.; De la Cruz Claure M. L.; Benito-Arenas R.; Revuelta R.; Santana A. G.; Bastida A. Overcoming Aminoglycoside Enzymatic Resistance: Design of Novel Antibiotics and Inhibitors. Molecules 2018, 23, 284–301. 10.3390/molecules23020284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeremia L.; Deprez B. E.; Dey D.; Conn G. L.; Wuest W. M. Ribosome-Targetting Antibiotics and Resistance via Ribosomal RNA Methylation. RSC Med. Chem. 2023, 14, 624–643. 10.1039/D2MD00459C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert T.Aminoglycosides and Gram-Negative Bacteria. In Antibiogram; Courvalin P., Leclercq R., Rice L. B., Eds.; ASM Press, 2010; pp 225–242. [Google Scholar]
- Takahashi Y.; Igarashi M. Destination of Aminoglycoside Antibiotics in the ‘Post-Antibiotic Era’. J. Antibiot. 2018, 71, 4–14. 10.1038/ja.2017.117. [DOI] [PubMed] [Google Scholar]
- Böttger E. C.; Crich D. Aminoglycosides: Time for Resurrection of a Neglected Class of Antibacterials?. ACS Infect. Dis. 2020, 6, 168–172. 10.1021/acsinfecdis.9b00441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jospe-Kaufman M.; Siomin L.; Fridman M. The Relationship Between the Structure and Toxicity of Aminoglycoside Antibiotics. Bioorg. Med. Chem. Lett. 2020, 30, 127218 10.1016/j.bmcl.2020.127218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J.; Hemachandran S.; Cheng A. G.; Ricci A. J. Identifying Targets to Prevent Aminoglycoside Ototoxicity. Mol. Cell. Neurosci. 2022, 120, 103722 10.1016/j.mcn.2022.103722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsushita T.; Sati G. C.; Kondasinghe N.; Pirrone M. G.; Kato T.; Waduge P.; Kumar H. S.; Cortes Sanchon A.; Dobosz-Bartoszek M.; Shcherbakov D.; et al. Design, Multigram Synthesis, and in Vitro and in Vivo Evaluation of Propylamycin: A Semisynthetic 4,5-Deoxystreptamine Class Aminoglycoside for the Treatment of Drug-Resistant Enterobacteriaceae and Other Gram-Negative Pathogens. J. Am. Chem. Soc. 2019, 141, 5051–5061. 10.1021/jacs.9b01693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonousi A.; Quirke J. C. K.; Waduge P.; Janusic T.; Gysin M.; Haldimann K.; Xu S.; Hobbie S. N.; Sha S.-H.; Schacht J.; et al. An Advanced Apralog with Increased in-vitro and in-vivo Activity toward Gram-negative Pathogens and Reduced ex-vivo Cochleotoxicity. Chem. Med. Chem. 2021, 16, 335–339. 10.1002/cmdc.202000726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otsuka Y.; Umemura E.; Takamiya Y.; Ishibashi T.; Hayashi C.; Yamada K.; Igarashi M.; Shibasaki M.; Takahashi Y. Aprosamine Derivatives Active against Multidrug-Resistant Gram Negative Bacteria. ACS Infect. Dis. 2023, 9, 886–898. 10.1021/acsinfecdis.2c00557. [DOI] [PubMed] [Google Scholar]
- Zada S. L.; Baruch B. B.; Simhaev L.; Engel H.; Fridman M. Chemical Modifications Reduce Auditory Cell Damage Induced by Aminoglycoside Antibiotics. J. Am. Chem. Soc. 2020, 142, 3077–3087. 10.1021/jacs.9b12420. [DOI] [PubMed] [Google Scholar]
- Shier W. T.; Rinehart K. L.; Gottlieb D. Preparation of Four New Antibiotics from a Mutant of Streptomyces Fradiae. Proc. Natl. Acad. Sci., USA 1969, 63, 198–204. 10.1073/pnas.63.1.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shier W. T.; Ogawa S.; Hichens M.; Rinehart K. L. Chemistry and Biochemistry of the Neomycins. XVII Bioconversion of Aminocyclitols to Aminocyclitol Antibiotics. J. Antibiot. 1973, 26, 551–561. 10.7164/antibiotics.26.551. [DOI] [PubMed] [Google Scholar]
- Shier W. T.; Schaefer P. C.; Gottlieb D.; Rinehart K. L. Use of Mutants in the Study of Aminocyclitol Antibiotic Biosynthesis and the Preparation of the Hybrimycin C Complex. Biochemistry 1974, 13, 5073–5078. 10.1021/bi00722a002. [DOI] [PubMed] [Google Scholar]
- Davies J. Structure-Activity Relations among the Aminoglycoside Antibiotics: Comparison of the Neomycins and Hybrimycins. Biochim. Biophys. Acta 1970, 222, 674–676. 10.1016/0304-4165(70)90197-2. [DOI] [PubMed] [Google Scholar]
- Kojima M.; Satoh A. Microbial Semi-Synthesis of Aminoglycoside Antibiotics by Mutants of S. ribosidificus and S. kanamyceticus. J. Antibiot. 1973, 26, 784–786. 10.7164/antibiotics.26.784. [DOI] [PubMed] [Google Scholar]
- Borders D. B.; Hargreaves R. T.; Van Lear G. E.; Kirby J. P. Detection of Naturally Occurring Hybrimycin-Type Antibiotics by Mass Spectroscopy. J. Antibiot. 1982, 35, 1107–1110. 10.7164/antibiotics.35.1107. [DOI] [PubMed] [Google Scholar]
- Rosi D.; Goss W. A.; Daum S. J. Mutational Biosynthesis by Idiotrophs of Micromonospora purpurea. 1. Conversion of Aminocyclitols to New Aminoglycoside Antibiotics. J. Antiobiotics 1977, 30, 88–97. 10.7164/antibiotics.30.88. [DOI] [PubMed] [Google Scholar]
- Takahashi Y.; Umemura E.; Kobayashi Y.; Murakami S.; Nawa T.; Morinaka A.; Miyake T.; Sibasaki M. Discovery of 2-Hydroxyarbekacin, A New Aminoglycoside Antibiotic with Reduced Nephrotoxicity. J. Antibiot. 2018, 71, 345–347. 10.1038/ja.2017.60. [DOI] [PubMed] [Google Scholar]
- Sato R.; Tanigawa Y.; Kaku M.; Aikawa N.; Shimizu K. Pharmacokinetic-Pharmacodynamic Relationships of Arbekacin for Treatment of Patients Infected with Methicillin-Resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 2006, 50, 3763–3769. 10.1128/AAC.00480-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. H.; Lee C.-S. Clinical Usefulness of Arbekacin. Infect. Chemother. 2016, 48, 1–11. 10.3947/ic.2016.48.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtenthaler F. W.; Leinert H.; Suami T. Synthesis of Streptamine and Actinamine. Chem. Ber. 1967, 100, 2383–2388. 10.1002/cber.19671000736. [DOI] [PubMed] [Google Scholar]
- Ogawa S.; Abe T.; Sano H.; Kotera K.; Suami T. Aminocyclitols XIV. Synthesis of Streptamine and Actinamine. Bull. Chem. Soc. Jpn. 1967, 40, 2405–2409. 10.1246/bcsj.40.2405. [DOI] [Google Scholar]
- Suami T.; Ogawa S. Aminocyclitols IX. The Facile Synthesis of Streptamine. Bull. Chem. Soc. Jpn. 1965, 38, 2026–2026. 10.1246/bcsj.38.2026. [DOI] [PubMed] [Google Scholar]
- Suami T.; Sano H. Aminocyclitols XIX. The Facile Synthesis of Actinamine. Tetrahedron Lett. 1968, 9, 2655–2657. 10.1016/S0040-4039(00)89666-5. [DOI] [Google Scholar]
- Posternak T.The Cyclitols; Holden-Day, 1962. [Google Scholar]
- Bohé L.; Crich D. Double Diastereoselection, Regioselectivity, and the Importance of Donor-Acceptor Complementarity in the Stereoselectivity of Glycosylation Reactions. Trends. Glycosci. Glycotech. 2010, 22, 1–15. 10.4052/tigg.22.1. [DOI] [Google Scholar]
- Patil P. S.; Hung S.-C. Total Synthesis of Phosphatidylinositol Dimannoside: A Cell-Envelope Component of Mycobacterium tubercolosis. Chem.—Eur. J. 2009, 15, 1091–1094. 10.1002/chem.200802189. [DOI] [PubMed] [Google Scholar]
- Uhlmann P.; Vasella A. Regioselective Glycosylation of Diols and Triols: Intra- and Intermolecular Hydrogen Bonds. Helv. Chim. Acta 1992, 75, 1979–1994. 10.1002/hlca.19920750623. [DOI] [Google Scholar]
- Uriel C.; Gómez A. M.; López J. C.; Fraser-Reid B. Reciprocal Donor-Acceptor Selectivity: the Influence of the Donor O-2 Substituent in the Regioselective Mannosylation of myo-Inositol Orthopentanoate. Eur. J. Org. Chem. 2009, 2009, 403–411. 10.1002/ejoc.200800991. [DOI] [Google Scholar]
- Tatai J.; Fügedi P. A New, Powerful Glycosylation Method: Activation of Thioglycosides with Dimethyl Disulfide-Triflic Anhydride. Org. Lett. 2007, 9, 4647–4650. 10.1021/ol702139u. [DOI] [PubMed] [Google Scholar]
- Lee J.; Borovika A.; Khomutnyka Y.; Nagorny P. Chiral Phosphoric Acid-Catalyzed Desymmetrizative Glycosylation of 2-Deoxystreptamine and its Application to Aminoglycoside Synthesis. Chem. Commun. 2017, 53, 8976–8979. 10.1039/C7CC05052F. [DOI] [PubMed] [Google Scholar]
- Bruzik K. S.; Tsai M.-D. Efficient and Systematic Syntheses of Enantiomerically Pure and Regiospecifically Protected myo-Inositols. J. Am. Chem. Soc. 1992, 114, 6361–6374. 10.1021/ja00042a011. [DOI] [Google Scholar]
- Kaplan M. A.; Fardig O. B.; Hooper I. R. The Reduction of Streptomycin with Sodium Borohydride. J. Am. Chem. Soc. 1954, 76, 5161–5162. 10.1021/ja01649a057. [DOI] [Google Scholar]
- Polglase W. J. Alkaline Degradation of Dihydrostreptomycin. J. Org. Chem. 1962, 27, 1923–1923. 10.1021/jo01052a532. [DOI] [Google Scholar]
- Goddard-Borger E. D.; Stick R. V. An Efficient, Inexpensive, and Shelf-Stable Diazotransfer Reagent: Imidazole-1-sulfonyl Azide Hydrochloride. Org. Lett. 2007, 9, 3797–3800. 10.1021/ol701581g. [DOI] [PubMed] [Google Scholar]
- Fischer N.; Goddard-Borger E. D.; Greiner R.; Klapotke T. M.; Skelton B. W.; Stierstorfer J. Senstivities of Some Imidazole-1-sulfonyl Azide Salts. J. Org. Chem. 2012, 77, 1760–1764. 10.1021/jo202264r. [DOI] [PubMed] [Google Scholar]
- Potter G. T.; Jayson G. C.; Miller G. J.; Gardiner J. M. An Updated Synthesis of the Diazo-Transfer Reagent Imidazole-1-sulfonyl Azide Hydrogen Sulfate. J. Org. Chem. 2016, 81, 3443–3446. 10.1021/acs.joc.6b00177. [DOI] [PubMed] [Google Scholar]
- Ye H.; Liu R.; Li D.; Liu Y.; Yuan H.; Guo W.; Zhou L.; Cao X.; Tian H.; Shen J.; et al. A Safe and Facile Route to Imidazole-1-sulfonyl Azide as a Diazotransfer Reagent. Org. Lett. 2013, 15, 18–21. 10.1021/ol3028708. [DOI] [PubMed] [Google Scholar]
- Kawano S.-i. Analysis of Impurities in Streptomycin and Dihydrostreptomycin by Hydrophilic Interaction Chromatography/Electrospray Ionization Quadrupole Ion Trap/Time-of-Flight Mass Spectrometry. Rapid Commun. Mass Spec. 2009, 23, 907–914. 10.1002/rcm.3936. [DOI] [PubMed] [Google Scholar]
- Brink N. G.; Kuehl F. A.; Folkers K. A. Degradation of Streptomycin to Streptobiosamine Derivatives. Science 1945, 102, 506–507. 10.1126/science.102.2655.506. [DOI] [PubMed] [Google Scholar]
- Kuehl F. A.; Flynn E. H.; Brink N. G.; Folkers K. The Degradation of Streptomycin and Dihydrostreptomycin with Ethyl Mercaptan. J. Am. Chem. Soc. 1946, 68, 2096–2101. 10.1021/ja01214a065. [DOI] [PubMed] [Google Scholar]
- Taha H. A.; Richards M. R.; Lowary T. L. Conformational Analysis of Furanoside-Containing Mono- and Oligosaccharides. Chem. Rev. 2013, 113, 1851–1876. 10.1021/cr300249c. [DOI] [PubMed] [Google Scholar]
- Teduka T.; Togo H. I2-Mediated Photochemical Preparation of 2-Substituted 1,3-Dioxolanes and Tetrahydrofurans from Alcohols with Polymer-Supported Hypervalent Iodine Reagent, PSDIB. Synlett 2005, 2005, 923–926. 10.1055/s-2005-864802. [DOI] [Google Scholar]
- Schuler M.; Tatibouët A.. Strategies Toward Protection of 1,2- and 1,3-Diols in Carbohydrate Chemistry. In Protecting Groups: Strategies and Application in Carbohydrate Chemistry; Vidal S., Ed.; Wiley, 2019; pp 307–335. [Google Scholar]
- Haddad J.; Liu M.-Z.; Mobashery S.. Methodologies in Syntheses of Aminoglycoside Antibiotics. In Glycochemistry: Principles, Synthesis, and Applications; Wang P. G., Bertozzi C. R., Eds.; Dekker, 2001; pp 353–424. [Google Scholar]
- Wang J.; Chang C.-W. T.. Design, Chemical Synthesis, and Antibacterial Activity of Kanamycin and Neomycin Class Aminoglycoside Antibiotics. In Aminoglycoside Antibiotics; Arya D. P., Ed.; Wiley, 2007; pp 141–180. [Google Scholar]
- Berkov-Zrihen Y.; Fridman M.. Synthesis of Aminoglycosides. In Modern Synthetic Methods in Carbohydrate Chemistry; From Monosaccharides to Complex Glycoconjugates; Werz D. B., Vidal S., Eds.; Wiley, 2014; pp 161–190. [Google Scholar]
- Quirke J. C. K.; Rajasekaran P.; Sarpe V. A.; Sonousi A.; Osinnii I.; Gysin M.; Haldimann K.; Fang Q.-J.; Shcherbakov D.; Hobbie S. N.; et al. Apralogs: Apramycin 5-O-Glycosides and Ethers with Improved Antibacterial Activity and Ribosomal Selectivity and Reduced Susceptibility to the Aminoacyltranserferase (3)-IV Resistance Determinant. J. Am. Chem. Soc. 2020, 142, 530–544. 10.1021/jacs.9b11601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie J.; Ménand M.; Valéry J. M. Regioselective Debenzylation of C-Glycosyl Compounds by Boron Trichloride. Carbohydr. Res. 2005, 340, 481–487. 10.1016/j.carres.2004.12.001. [DOI] [PubMed] [Google Scholar]
- Falck J.R.; Barma D.K.; Venkataraman S. K.; Baati R.; Mioskowski C. Regioselective De-O-benzylation of Monosaccharides. Tetrahedron Lett. 2002, 43, 963–966. 10.1016/S0040-4039(01)02306-1. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this study are available in the published article and its Supporting Information.