

Abstract
Protein kinase C epsilon (PKCε) regulates behavioural responses to ethanol and plays a role in anxiety‐like behaviour, but knowledge is limited on downstream substrates of PKCε that contribute to these behaviours. We recently identified brain‐specific serine/threonine‐protein kinase 1 (BRSK1) as a substrate of PKCε. Here, we test the hypothesis that BRSK1 mediates responses to ethanol and anxiety‐like behaviours that are also PKCε dependent. We used in vitro kinase assays to further validate BRSK1 as a substrate of PKCε and used Brsk1 −/− mice to assess the role of BRSK1 in ethanol‐ and anxiety‐related behaviours and in physiological responses to ethanol. We found that BRSK1 is phosphorylated by PKCε at a residue identified in a chemical genetic screen of PKCε substrates in mouse brain. Like Prkce −/− mice, male and female Brsk1 −/− mice were more sensitive than wild‐type to the acute sedative‐hypnotic effect of alcohol. Unlike Prkce −/− mice, Brsk1 −/− mice responded like wild‐type to ataxic doses of ethanol. Although in Prkce −/− mice ethanol consumption and reward are reduced in both sexes, they were reduced only in female Brsk1 −/− mice. Ex vivo slice electrophysiology revealed that ethanol‐induced facilitation of GABA release in the central amygdala was absent in male Brsk1 −/− mice similar to findings in male Prkce −/− mice. Collectively, these results indicate that BRSK1 is a target of PKCε that mediates some PKCε‐dependent responses to ethanol in a sex‐specific manner and plays a role distinct from PKCε in anxiety‐like behaviour.
Keywords: alcohol, anxiety, protein kinase c epsilon
Protein kinase C epsilon (PKCε) signalling is linked to behavioural responses to ethanol andanxiety‐like behaviour. Here, we used knockout mice to evaluate the role of a PKCε substrate,brain‐specific serine/threonine‐protein kinase 1 (BRSK1), in these behaviours. We found thatBRSK1 influences these behaviours in a sex‐specific manner; notably, ethanol consumption andreward were reduced in female BRSK1 knockout mice, while anxiety‐like behaviour wasincreased in male BRSK1 knockout mice.
1. INTRODUCTION
Current evidence indicates that protein kinase C epsilon (PKCε) regulates ethanol consumption, preference, intoxication and reward, and anxiety like behaviour. Prkce −/− mice drink substantially less ethanol than wild‐type mice and show reduced conditioned place preference for ethanol. 1 , 2 , 3 , 4 Prkce −/− mice also show reduced anxiety‐like behaviour compared with wild‐type mice. 5 The ethanol‐related behaviours do not result from developmental changes since inducible transgenic expression of PKCε in the amygdala and striatum restores normal responses to intoxication and increases drinking in Prkce −/− mice to levels observed in wild‐type mice. 6 Similarly, knockdown of PKCε by RNA interference in the amygdala reduces anxiety‐like behaviour of wild‐type mice. 7 In wild‐type mice, knockdown of PKCε by RNA interference in the amygdala also reduces ethanol consumption, and microinjection of a peptide inhibitor of PKCε in the amygdala or nucleus accumbens reduces binge‐like ethanol drinking. 8 , 9 In addition, a small‐molecule inhibitor of PKCε reduces intermittent ethanol consumption in wild‐type mice. 10 A role for PKCε in alcohol use disorder (AUD) is suggested by a study of lymphoblastoid cell lines (LCLs) from 21 AUD subjects and 21 controls which showed that PRKCE mRNA transcripts were increased 1.4‐fold in AUD cases compared with controls. 11 Taken together, these results suggest that PKCε is a novel target for medications development for AUD and possibly anxiety‐related disorders.
Studies in vitro using purified proteins have provided evidence of an ethanol binding site on PKCε that inhibits the activity of PKCε, but other in vitro studies have shown that ethanol has no effect on PKC activity. 12 , 13 In wild‐type and Prkce −/− mice, the ED50 of ethanol for inducing ataxia is the same consistent with the lack of an acute effect of ethanol on PKCε in vivo. In the same study, increased phosphorylation of PKCε at S729, which is required for full kinase activity was evident in the cerebellum only 60 min after an ethanol injection. 4 , 14 These results suggest that ethanol indirectly activates PKCε through actions on upstream signalling pathways.
In the brain, PKCε is enriched in presynaptic terminals where it plays a role in release of synaptic vesicles. 15 There are several limbic regions associated with AUD and anxiety where PKCε is implicated in the release of neurotransmitters. In hippocampal granule cells, PKCε is important for the exocytosis of glutamate, and this action is dependent on an actin binding domain in PKCε. 16 In the nucleus accumbens of Prkce −/− mice, dopamine release is impaired. 2 In the central amygdala, ethanol‐stimulated gamma‐aminobutyric acid (GABA) release is absent in Prkce −/− mice and blocked by PKCε inhibitors in wild‐type mice. 10 , 17
How PKCε signalling regulates neurotransmitter release is not known. Recently, we conducted a chemical genetic screen for PKCε substrates in mouse brain. 18 Our analysis revealed several substrates whose function is associated with synapses or synaptic vesicles. One of these is brain serine/threonine kinase 1 (BRSK1) which is a synaptic vesicle associated, serine–threonine, protein kinase. BRSK1 is an ortholog of the C. elegans protein SAD‐1, which was first identified for its role in synaptic vesicle clustering and axonal development. 19 , 20 , 21 In mammals, BRSK1 plays important roles in neuronal polarization and nerve terminal maturation. 22 , 23 , 24 , 25 , 26 , 27 , 28 BRSK1 also regulates synaptic vesicle release in hippocampal granule cells and may function by regulating the readily releasable pool of vesicles. 29 , 30 , 31 This association with vesicle release makes BRSK1 a promising candidate for further investigation into its role in PKCε signalling.
Here we used Brsk1 −/− global knockout mice to examine the role of BRSK1 in responses to ethanol and in anxiety‐like behaviour. We report that BRSK1 has a sex specific effect on ethanol and anxiety related behaviours and plays a role in ethanol‐induced facilitation of GABA release in the central nucleus of the amygdala. Our findings indicate that BRSK1 is involved in several but not all effects of PKCε on behavioural and physiological responses to ethanol, and that the role BRSK1 plays in anxiety is likely unrelated to PKCε signalling.
2. MATERIALS AND METHODS
2.1. BRSK1 expression vectors and protein purification
The BRSK1‐GST‐tagged peptide constructs (12 amino acids in length) corresponding to the PKCε phosphorylation site (S559) and the corresponding alanine mutant (S559A) were cloned into pGEX‐6p‐2 vectors (Sigma‐Aldrich; Cat No. GE28‐9546‐50) in frame with the GST purification tag sequence and transformed into BL21(DE3) Escherichia coli cells for expression. Following expression, GST‐tagged peptide constructs were purified using a Pierce GST Spin Purification Kit (Thermo Fisher Scientific; Cat No. 16106).
BRSK1‐FLAG‐tagged full length protein constructs were cloned into the vector pCMV6‐Entry (OriGene; Cat. No. PS100001) in frame with the FLAG purification tag sequence and were transformed into DH5α cells (Thermo Fisher Scientific; Cat. No. 18265017) for plasmid purification. Purified plasmid (10 μg) was transfected into 60–90% confluent COS‐7 cells (ATCC; Cat. No. CRL‐1651) on a 10 cm plate using Lipofectamine 3000 transfection reagent (Thermo Fisher Scientific; Cat. No. L3000008). Following transfection, the cells were incubated at 37°C in an atmosphere of 95% air: 5% CO2 for 24–48 h before purification using a FLAG® Immunoprecipitation Kit (Sigma‐Aldrich; Cat No. FLAGIPT1).
2.2. In‐vitro thiophosphorylation
PKCε (100 ng) was added to a 40 μl kinase reaction containing PKC kinase buffer (20mM HEPES, 10mM MgCl2, 0.1mM EGTA, 0.3% Triton‐X‐100) with 0.5 μg/μl phosphatidylserine, 1mM PMA, 1 ng/μl BSA, and 1 mM of ATPγS. Phosphatidylserine was prepared by evaporating away chloroform from 250 μl of 10 mg/ml stock solution with a stream of argon and resuspending in 500 μl of PKC kinase buffer. This mixture was incubated in a 27°C water bath for 10–15 min. After this incubation, 5 μg of GST‐tagged BRSK1 peptide was added to the reaction, which was allowed to proceed for 30 min at 37°C in a Thermo‐Mixer shaking at 1000 RPM. Then 500 mM EDTA was added to a final concentration of 100mM to quench the reaction, followed by addition of 2.5 μl of 50mM p‐nitrobenzyl mesylate (PNBM) in DMSO (final concentration: 2.5mM PNBM, 5% DMSO). After 1 h of incubation at room temperature with constant nutation, 12.5 μl of 5X Laemmli sample buffer were added, and samples were analysed by western blotting.
2.3. In‐vitro phosphorylation
PKCε (100 ng) was added to a 40 μl kinase reaction containing PKC kinase buffer with 0.5 μg/μl phosphatidylserine, 1mM PMA, 1 ng/μl BSA, and 1mM of ATP. This mixture was incubated in a 37 °C water bath for 10–15 min. Then 2 μg of FLAG‐tagged BRSK1 wild‐type or S559A mutant protein were added to the reaction. The kinase reaction proceeded for 30 min at 37°C in a Thermo‐Mixer shaking at 1000 RPM. After 100 mM EDTA was added to quench the reaction, samples were analysed by SDS‐PAGE and western blotting.
2.4. Western blotting
GST‐tagged BRSK1 peptide or FLAG‐tagged BRSK1 full‐length proteins (0.1 μg each) were separated by SDS‐PAGE and transferred to LF‐PVDF membranes. Blots from the in‐vitro thiophosphorylation kinase reaction were blocked in 5% nonfat dry milk, TBST (20 mM Tris, 137 mM NaCl, 0.1% Tween 20) for 30 min at room temperature and then incubated overnight at 4°C with anti‐GST antibody from mouse (Invitrogen; #MA4‐004; 1:1000) and anti‐thiophosphate ester antibody from rabbit (Abcam; ab133473; 1:5000) in TBST with 5% nonfat dry milk. After five washes with TBST, 5 min each, blots were incubated with goat anti‐mouse IgG StarBright B520 (BioRad; #12005867; 1:5000) and goat anti‐rabbit IgG StarBright B700 (BioRad; #12004162; 1:5000) in TBS with 5% nonfat dry milk and 0.02% SDS for 1 h at room temperature. Blots from the in‐vitro phosphorylation kinase reaction were blocked in TBST with 5% BSA for 30 min at room temperature and then incubated overnight at 4°C with anti‐FLAG antibody from mouse (Sigma‐Aldrich; #F1804; 1:5000), and anti‐BRSK1 S(P)559 antibody from rabbit (custom antibody from Pacific Immunology, 1:1000) in TBST with 5% BSA. Blots were then washed five times in TBST and incubated in goat anti‐mouse IgG StarBright B520 (1:5000) and goat anti‐rabbit IgG StarBright B700 (1:5000) in TBS with 3% BSA and 0.02% SDS for 1 h at room temperature. All blots were washed six times with TBST, 5 min per wash, before imaging. Blots were imaged on a BioRad ChemiDoc MP station and analysed with Image Lab software (version 6.1, BioRad). Thiophosphorylation and phosphorylation signals were normalized to GST or FLAG signals respectively, and signals from mutants were quantified relative to signals from wild‐type peptide or protein.
2.5. Animals
Studies were conducted in male and female 8‐ to 16‐week‐old C57BL/6J wild‐type and C57BL/6J Brsk1 −/− mice obtained from Jackson Labs (Bar Harbor, ME). Mice were grouped‐housed in temperature‐ and humidity‐controlled rooms with free access to food (LabDiet, #5053) and water under a 12‐h light/dark reverse cycle, lights on at 11 PM. Experimental rooms were maintained at an ambient temperature of 21 ± 1°C with 40–60% humidity. Experiments were approved by the Institutional Animal Care and Use Committees at The University of Texas at Austin and at Scripps Research, and complied with the ARRIVE guidelines and the National Institutes of Health Guide for the Care and Use of Laboratory Animals. 32
2.6. Loss of righting reflex
Brsk1 −/− mice and their wild‐type littermates (10–12 mice per genotype per sex) were administered 3.6 g/kg ethanol (20% v/v in saline) by i.p. injection. Once mice were sedated, they were placed in a supine position on a flat bed of nestlets and timing began. When the mice were able to right themselves three times within 30 s, the time at the third righting was recorded as the latency to recover the righting reflex.
2.7. Ethanol‐induced rotarod ataxia
Brsk1 −/− mice and their wild‐type littermates (10–12 mice per genotype per sex) were moved to the procedure room to acclimate to the environment and sound of the rotarod 1 h before the experiment. Mice were trained on the rotarod at a fixed speed of 10 RPM until they could stay on the rotarod for 60 s. Each mouse was given a maximum of six attempts to remain on the rotarod for 60 s; if they failed this training, they were removed from the experiment. Mice that remained on the rotarod for 60 s were administered 2 g/kg ethanol (20% v/v in saline) by i.p. injection and placed on the rotarod every 15 min. Their latency to fall was measured, and they were considered recovered when they remained on the rotarod for 60 s.
2.8. Two‐bottle choice intermittent access ethanol drinking
Mice were individually housed and allowed to acclimate to two water bottles 1 week before the start of the experiment. A modified two‐bottle choice, intermittent access (2BC‐IA) drinking procedure was used to determine ethanol consumption and preference. 33 Mice were given 24‐hour access to two bottles, one containing water and the other ethanol 3 h into the dark cycle on Monday, Wednesday, and Friday (MWF). The first week mice were given increasing concentrations of ethanol (3%, 6%, 10% [v/v]) each day on MWF. The following weeks, mice were given a 15% (v/v) ethanol solution on MWF and given access to only water on other days. On days that ethanol was available, bottle positions were alternated to control side preferences. Ethanol and water bottles were weighed after drinking days, and animals were weighed once per week to calculate consumption. The study concluded after 31 days (14 ethanol drinking sessions).
2.9. Saccharin and quinine consumption
A cohort of Brsk1 −/− mice and their wild‐type littermates (6–12 mice per genotype per sex) were acclimated to two bottles with water only for one week. Thereafter, mice were given access to water and 0.75mM saccharin bottles for two days. After two days, the saccharin concentration was increased to 1.5mM and mice were given access for another two days. The position of the water and saccharin bottles was switched each day to control for side preferences. Three days later, mice were given access to water and 15 μM quinine bottles for two days. After two days, the quinine concentration was increased to 30 μM and mice were given access for another two days. The position of the water and quinine bottles was also switched each day to control for side preferences. The bottles were weighed after days two and four. Saccharin and quinine consumption was measured and averaged over 24 h for both concentrations.
2.10. Ethanol conditioned place preference
Brsk1 −/− mice and their wild‐type littermates (17–20 mice per genotype per sex) were conditioned in an apparatus consisting of a rectangular chamber equipped with two different floor textures (rods and holes) separated by a plexiglass barrier with a central opening. Mice were acclimated to the conditioning room for 30 min before the start of the experiment each day. On the day before conditioning, the mice were pre‐tested in the apparatus by injecting saline (i.p.) and allowing 30 min of free access to both chambers. Mice that showed a strong unconditioned preference (>70%) for either side were removed from the experiment. Each treatment group was counterbalanced by sex and genotype for order of conditioning treatment (saline or ethanol on day one), conditioning context paring, and orientation of the apparatus. Mice were conditioned once each day, 3 h into their dark cycle, for eight days. They received alternating injections (i.p.) of saline or ethanol (1 g/kg) immediately prior to being confined to one of the chamber sides for 5 min with a plexiglass barrier blocking entry to the other side. The saline and ethanol paired sides were different and did not change. On the day following the final conditioning (day 9), mice were injected with saline (i.p.) and tested for conditioned place preference by allowing free access to both compartments for 30 min as on the initial pre‐test day. The time spent in each compartment was determined electronically by monitoring the disruption of infrared photo beams. Results were analysed in two ways. First, the time spent in the ethanol paired side during the pretest session was subtracted from time spent in that same side during the test session to calculate a CPP score, which was compared within and between genotypes. Next, we compared the amount of time spent on the rod floor when it was paired with ethanol (rod+) versus when it was paired with saline (rod–) during the test session for each genotype. Sexes were analysed separately. The identification of outliers in the two analyses was carried out using the ROUT method. 34
2.11. Elevated plus maze
The elevated plus maze (EPM) consisted of two open arms (50 × 10 cm) and two enclosed arms (50 × 10 × 40 cm) connected by a central area measuring 10 × 10 cm, 50 cm above the floor. Mice (14–16 per sex per genotype) were allowed to acclimate to the procedure room in their home cage, 1 h before the start of the experiment. At the beginning of each trial, mice were placed in the centre of the EPM facing one open arm. The trials lasted 5 min and were performed under red lighting. Trials were recorded on camera. Three‐point animal tracking was used to measure distance moved and time spent in the open and closed arms of the EPM with EthoVision XT17 (Noldus, US) software. Sexes were analysed separately.
2.12. Blood ethanol clearance
Mice (five mice per genotype per sex) were administered 4 g/kg ethanol (20% v/v in saline) intraperitoneally. Retro‐orbital blood samples were taken at 30, 60, 120, 180, and 240 min after injection with a heparinized capillary tube. The capillary tubes were sealed and spun at 3 100 g for 6 min in a tabletop centrifuge. Plasma samples were stored at −20°C until blood ethanol concentrations (BECs) were determined in 5 μl aliquots with an AM1 Ethanol Analyser (Analox Instruments). The machine was calibrated using a 200 mg/dl commercial standard every 15 samples and BECs were determined using commercially available reagents according to the manufacturer's instructions. Samples were averaged from duplicate runs and expressed as mg/dl.
2.13. Electrophysiology
Preparation of acute brain slices and electrophysiological recordings were performed as described previously 17 , 35 , 36 from six male Brsk1 −/− mice and five male wild‐type mice. Coronal slices (300 μm) that included the central amygdala (CeA) were prepared from anaesthetised mice in an ice‐cold, high sucrose cutting solution (sucrose 206 mM; KCl 2.5 mM; CaCl2 0.5 mM; MgCl2 7 mM; NaH2PO4 1.2 mM; NaHCO3 26 mM; glucose 5 mM and HEPES 5 mM). Slices were superfused (flow rate 2–4 ml/min) with artificial cerebrospinal fluid (aCSF; NaCl 130 mM; KCl 3.5 mM; NaH2PO4 1.25 mM; MgSO4·7H2O 1.5 mM; CaCl2 2.0 mM; NaHCO3 24 mM and glucose 10 mM) equilibrated with 95%O2/5% CO2. Whole‐cell patch‐clamp recordings of GABAergic miniature inhibitory post‐synaptic currents (mIPSCs) were recorded in neurons from the medial subdivision of the CeA clamped at −60 mV. Patch pipettes (3–6 MΩ) were filled with an internal solution composed of the following (in mM): 145 KCl, 0.5 EGTA, 2 MgCl2, 10 HEPES, 2 Mg‐ATP and 0.2 Na‐GTP. Action potential independent GABAergic activity was pharmacologically isolated with 6,7‐dinitroquinoxaline‐2,3‐dione (DNQX, 20 μM), DL‐2‐amino‐5‐phosphonovalerate (DL‐AP5, 30 μM), CGP 55845A (1 μM), and tetrodotoxin (TTX, 1 μM). In all experiments, cells with a series resistance >25 MΩ were excluded from analysis, and series resistance was continuously monitored during gap‐free recording with a 10‐mV pulse. Cells in which series resistance changed >25% during the experiment were excluded. Data were analysed using Easy Electrophysiology v2.5.0 with three‐minute bins of gap‐free recording. We applied all drugs by bath superfusion.
2.14. Drugs
CGP 55845A, DL‐AP5 and DNQX were purchased from Tocris (Ellisville, MO). Ethanol (95%) was purchased from Remet (La Mirada, CA). Drugs were added to the aCSF from stock solutions to obtain known concentrations in the superfusate.
2.15. Statistical analysis
All data are shown as mean ± SEM values and were analysed with GraphPad Prism version 10.1.2 (GraphPad Software, San Diego, CA) and Statistica version 13.5 (TIBCO Software, Palo Alto, CA). For the phosphorylation reactions, each experimental condition was repeated at least three times. For western blotting, data are shown as the average signal across three to six replicates relative to the wild‐type conditions. For ethanol related behaviours and electrophysiology, data were analysed by two‐tailed t‐test, three‐way repeated measures ANOVA, or two‐way repeated measures ANOVA with post‐hoc Sidak's multiple comparisons test as appropriate. Effect sizes were calculated as partial η2, which is appropriate for ANOVAs. 37
3. RESULTS
3.1. PKCε phosphorylates BRSK1 in vitro
In a chemical genetic screen of mouse brain lysates, we found that BRSK1 was a putative direct PKCε substrate, with phosphorylation sites at serines 555 and 559. 18 To confirm that BRSK1 is a PKCε substrate, we examined thiophosphorylation of mutant and wild‐type GST‐tagged BRSK1 peptides and phosphorylation of FLAG‐tagged full‐length BRSK1 proteins. PKCε thiophosphorylated GST and both GST‐tagged peptides. (Figure 1A). We observed two thiophosphorylated bands in the tagged peptides and only one for GST alone. The lower band in the tagged peptides corresponded to GST, which can be phosphorylated by PKCε. 38 The upper band in the S559A peptide was reduced compared with the WT peptide, and the residual signal was presumed due to thiophosphorylation at S555, since this residue was present in the tagged peptides (Figure 1B). Additionally, PKCε phosphorylated full‐length BRSK1 at S559 as measured with an anti‐phospho S(P) S559‐BRSK1 antibody (Figure 1C). The signal from this antibody was detected when both PKCε and the wild‐type BRSK1 protein were present in the reaction but was reduced in the S559A mutant or when PKCε was not present (Figure 1D).
FIGURE 1.
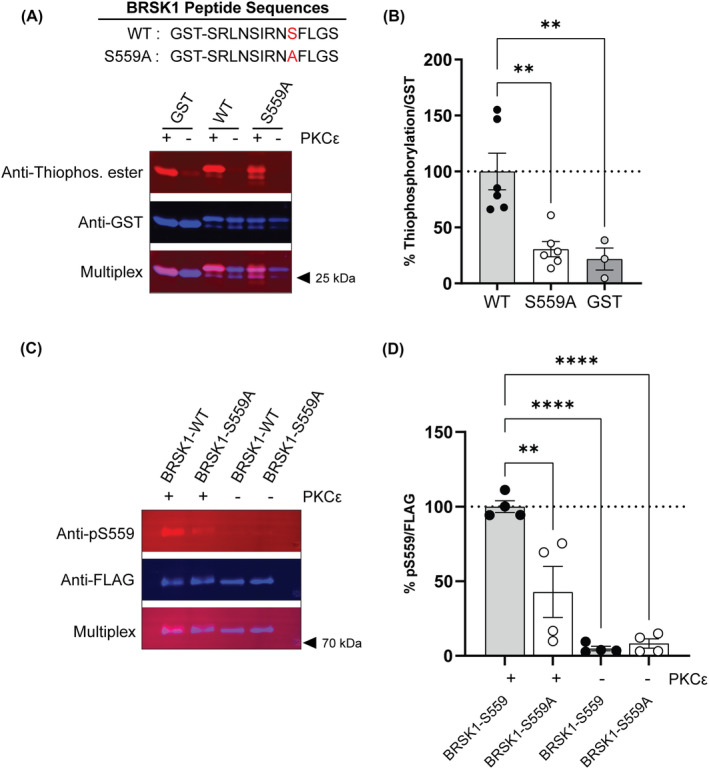
PKCε phosphorylates BRSK1 in vitro at S559. A) The average ratio of thiophosphorylation/GST signal expressed relative to the wild‐type (WT) BRSK1 peptide (n = 3–6). Thiophosphorylation of the S559A BRSK1 peptide was reduced compared with the WT BRSK1 peptide [F(2, 12) = 11.33, p = 0.0017, one‐way ANOVA]. B) Representative fluorescent western blot of a thiophosphorylation reaction with WT and S559A GST‐tagged BRSK1 peptides, and GST alone. C) The average ratio of pS559/FLAG signal expressed relative to the BRSK1‐WT full length FLAG‐tagged protein (n = 4). Immunoreactivity to pS559 was reduced in BRSK1‐S559A compared with BRSK1‐WT protein [F(3, 12) = 24.24, p < 0.0001, one‐way ANOVA]. D) Representative fluorescent western blot of pS559 immunoreactivity in BRSK1‐WT and BRSK1‐S559A full‐length FLAG‐tagged proteins. In B and D, the + and – indicate the presence or absence of PKCε in the reaction. ** p < 0.01 and **** p < 0.0001 by post‐hoc Sidak's multiple comparisons test in A) and C).
3.2. Increased duration of ethanol‐induced loss of the righting in Brsk1 −/− mice
Recovery from ethanol‐induced loss of righting is a measure of the sedative‐hypnotic effect of ethanol and is prolonged in Prkce −/− mice. 1 If BRSK1 mediates this effect of PKCε, we reasoned that Brsk1 −/− mice would show the same phenotype as Prkce −/− mice. We found that male and female Brsk1 −/− mice recovered more slowly from ethanol‐induced loss of righting after receiving a sedative dose (3.8 g/kg) of ethanol [Fgenotype (1,40) = 13.89, p = 0.0006; Fgenotype × sex (1,40) = 0.2251, p = 0.6377; large effect size, partial η2 = 0.258] (Figure 2A).
FIGURE 2.
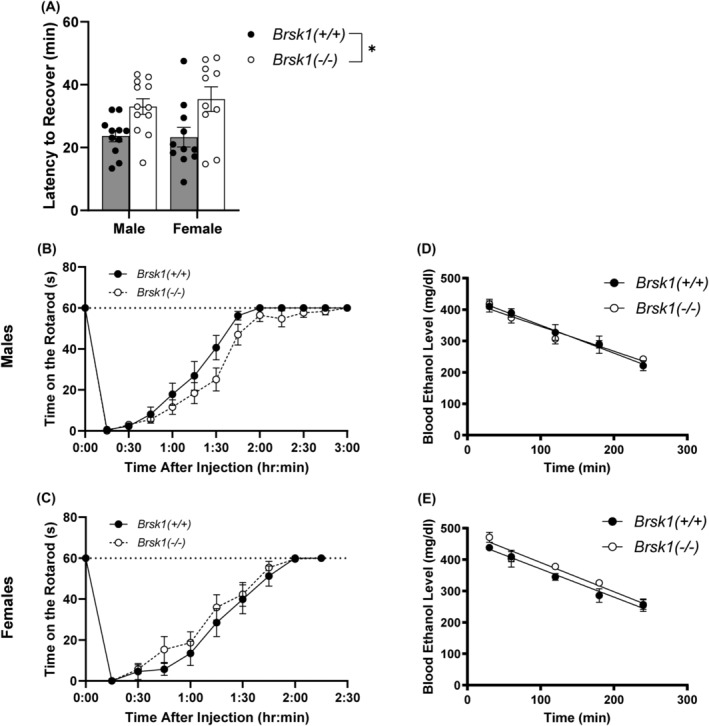
Brsk1 −/− mice show prolonged recovery from ethanol induced loss of righting but no change in ethanol‐induced ataxia or clearance. A) Ethanol (3.6 g/kg i.p.) increased LORR duration in both male (n = 12) and female (n = 10) Brsk1 −/− mice compared with male (n = 11) and female (n = 11) wild‐type (Brsk1 +/+ ) mice after an acute dose of ethanol (3.6 kg/g i.p.), * p < 0.001. (B, C) Male (B) and female (C) wild‐type and Brsk1 −/− mice showed similar recovery from ethanol‐induced ataxia (2 g/kg i.p.). (D, E) Blood ethanol clearance (4 g/kg i.p.) was similar between male (D) and female (E) wild‐type and Brsk1 −/− mice.
3.3. Ethanol‐induced ataxia is unaltered in Brsk1 −/− mice
Lower doses of ethanol produce ataxia and Prkce −/− mice show prolonged recovery from ethanol‐induced ataxia. 1 To determine if Brsk1 −/− mice share this phenotype, we trained wild‐type and Brsk1 −/− mice on a fixed‐speed rotarod and measured time for them to recover from an ataxic dose (2 g/kg) of ethanol. There was no difference in ethanol‐induced ataxia between genotypes [Fgenotype (1, 36) = 0.2963, p = 0.5896; Fgenotype × time (12, 432) = 0.5799, p = 0.8587; Fgenotype × sex (1, 36) = 3.334, p = 0.0762] (Figure 2B and C).
3.4. Ethanol clearance is not altered in Brsk1 −/− mice
To determine if the observed difference in ethanol‐induced loss of righting was due to differences in ethanol metabolism between wild‐type and Brsk1 −/− mice, we measured blood ethanol clearance. There was no difference between genotypes in ethanol clearance in [Fgenotype (1,16) = 0.5720, p = 0.4605; Fgenotype × time (4, 64) = 0.9762, p = 0.4269] (Figure 2D and E). Female mice of both genotypes had higher blood ethanol levels compared with male mice of both genotypes [Fsex (1, 16) = 6.109, p = 0.0251].
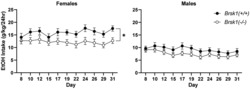
3.5. Ethanol consumption and preference is reduced in female Brsk1 −/− mice
Prkce −/− mice show reduced ethanol consumption and preference for ethanol. 1 , 2 If BRSK1 functions downstream of PKCε, we predicted that Brsk1 −/− mice would also show reduced ethanol consumption and preference. Brsk1 −/− mice and wild‐type littermates were subjected to a two‐bottle choice, intermittent access (2BC‐IA) procedure (Figure 3). 33 No differences were observed during the initial week as ethanol concentrations were increased, and these results are not included in the analysis. Beginning with the second week, Brsk1 −/− mice of both sexes consumed less (15%) ethanol [Fgenotype (1, 81) = 7.271, p = 0.0085; medium effect size, partial η2 = 0.082; Fgenotype × day (10, 810) = 1.342, p = 0.2036; Fgenotype × sex (1, 81) = 0.8520, p = 0.3587] and more water [Fgenotype (1,81) = 9.675, p = 0.0026; medium effect size, partial η2 = 0.107; Fgenotype × day (10, 810) = 1.048, p = 0.4007, Fgenotype × sex (1, 81) = 0.5254, p = 0.4706] than wild‐type littermates of both sexes. Brsk1 −/− mice of both sexes also showed reduced ethanol preference [Fgenotype (1, 81) = 9.345, p = 0.0030; medium effect size, partial η2 = 0.103; Fgenotype × day (10, 810) = 1.539, p = 0.1206; Fgenotype × sex (1, 81) = 0.1848, p = 0.6684] without a change in total fluid consumption.
FIGURE 3.
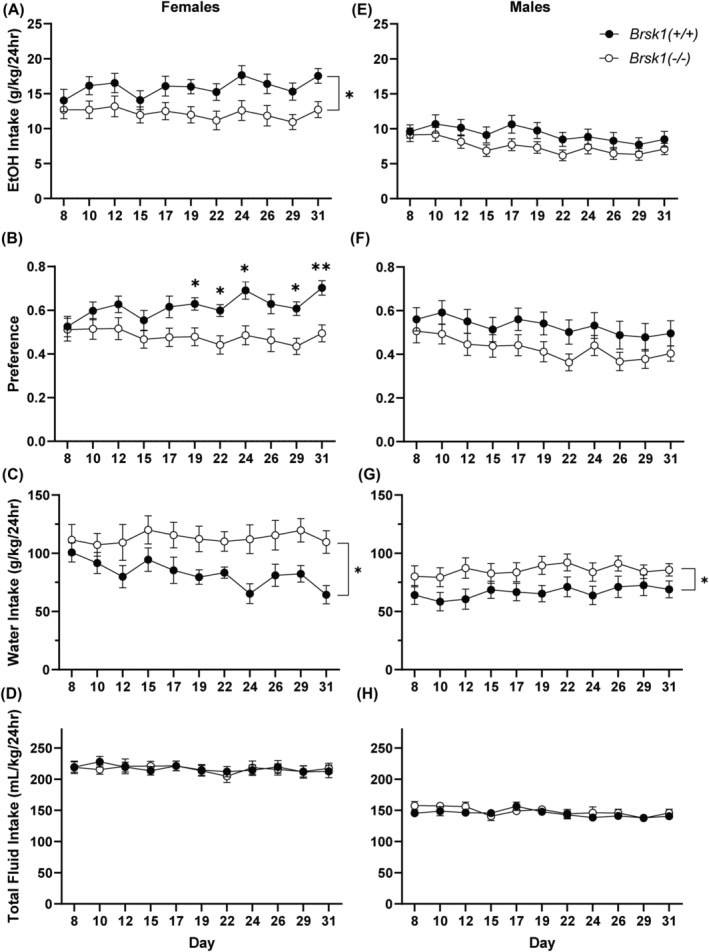
Female Brsk1 −/− mice show reduced ethanol consumption and preference. Ethanol consumption (A) and preference (B) was reduced in female Brsk1 −/− mice (n = 25) compared with their wild‐type littermates (n = 17). Water consumption was increased (C) and total fluid consumption was unchanged (D) in female Brsk1 −/− versus wild‐type mice. Ethanol consumption (E) and preference (F) in male Brsk1 −/− mice (n = 24) and their wild‐type littermates (n = 19) were similar. Water consumption was slightly increased (G) but total fluid consumption was similar (H) in male Brsk1 −/− versus wild‐type mice. * p < 0.05 in (A), (C), and (G); * p < 0.05 and ** p < 0.01 compared with wild‐type littermates on the same day by post‐hoc Sidak's multiple comparisons test in B).
When stratified by sex, female Brsk1 −/− mice consumed less ethanol [Fgenotype (1,40) = 4.962, p = 0.0316; Fgenotype × day (1,40) = 1.395, p = 0.18] (Figure 3A) and more water [Fgenotype (1,40) = 5.348, p = 0.0260; Fgenotype × day (1,40) = 1.452, p = 0.155] (Figure 3C) than wild‐type littermates. Female Brsk1 −/− mice also showed reduced ethanol preference [Fgenotype × day (1,40) = 2.344, p = 0.0107] (Figure 3B) without a change in total fluid consumption (Figure 3D). In contrast to females, male Brsk1 −/− mice did not show a significant change in ethanol consumption [Fgenotype (1,41) = 2.245, p = 0.1417; Fgenotype × day (1,41) = 0.7323, p = 0.694] (Figure 3E), although water consumption was slightly increased [Fgenotype (1,41) = 4.385, p = 0.0425; Fgenotype × day (1,41) = 0.3979, p = 0.95] (Figure 3G) compared with wild‐type male littermates. Ethanol preference was also not significantly reduced in male Brsk1 −/− mice [Fgenotype (1,41) = 3.131, p = 0.0843; Fgenotype × day (1,41) = 0.2854, p = 0.9843] (Figure 3F), and total fluid consumption was almost identical between male mice of both genotypes (Figure 3H).
3.6. Saccharin and quinine consumption and preference are not altered in Brsk1 −/− mice
To determine if the reduction in ethanol consumption and preference in female Brsk1 −/− mice was due to differences in taste perception or neophobia, we measured saccharin and quinine consumption at two concentrations (saccharin: 0.75mM and 1.5mM; quinine: 15 μM and 30 μM) in a separate cohort of ethanol‐naïve, female Brsk1 −/− and wild‐type mice (Figure S1). There were no differences in saccharin consumption [Fgenotype (1,15) = 0.282, p = 0.6398; Fgenotype × concentration (1,15) = 0.1852, p = 0.673] (Figure S1A) or saccharin preference [Fgenotype (1,15) = 0.6852, p = 0.4208; Fgenotype × concentration (1,15) = 0.4666, p = 0.505] (Figure S1B) between genotypes. There were also no differences in quinine consumption [Fgenotype (1,15) = 0.5423, p = 0.4728; Fgenotype × concentration (1,15) = 0.9015, p = 0.3574] (Figure S1C) or quinine preference [Fgenotype (1,15) = 0.2315, p = 0.6374; Fgenotype × concentration (1,15) = 0.3817, p = 0.5460] (Figure S1D) between genotypes. Similarly there were no differences in saccharin consumption [Fgenotype (1,16) = 2.182, p = 0.1591; Fgenotype × concentration (1,16) = 0.0219, p = 0.884] (Figure S1E), saccharin preference [Fgenotype (1,16) = 1.60, p = 0.224; Fgenotype × concentration (1,16) = 0.3477, p = 0.5637] (Figure S1F), quinine consumption [Fgenotype (1, 16) = 0.1045, p = 0.7507; Fgenotype × concentration (1,16) = 0.1298, p = 0.7233] (Figure S1G), or quinine preference [Fgenotype (1,16) = 0.2241, p = 0.6423; Fgenotype × concentration (1,16) = 0.1648, p = 0.6901] (Figure S1H) between male mice of either genotype.
3.7. Ethanol conditioned place preference (CPP) in Brsk1 −/− mice varies in a genotype and sex dependent manner
Unlike wild‐type mice, Prkce −/− mice do not show CPP for a low dose of ethanol (1 g/kg), but do show it for a higher dose (2 g/kg). 3 If BRSK1 plays a role downstream of PKCε in ethanol conditioned place preference, then Brsk1 −/− mice should also show impaired CPP for 1 g/kg ethanol. During the analysis, one subject in the female wild‐type group was identified as an outlier by the large amount of time spent on the rod side when paired with saline (ROD‐) compared with other subjects in this group. This mouse was removed from analysis of the CPP data. Female wild‐type mice developed ethanol CPP ([t(15) = 2.255, p = 0.0395; one sample t‐test against hypothetical mean of 0 = no CPP] but Brsk1 −/− female mice did not [t(19) = 0.4661, p = 0.6465]. When genotypes were compared, CPP scores trended toward a difference between wild‐type and Brsk1 −/− female mice [t(34) = 1.781, p = 0.0838] (Figure 4A). In contrast, we found that male wild‐type mice did not develop CPP to 1 g/kg ethanol ([t(19) = 1.024, p = 0.3187; one sample t‐test against hypothetical mean of 0 = no CPP]. Male Brsk1 −/− mice did develop CPP ([t(17) = 3.309, p = 0.0042] but CPP scores were not different between wild‐type and Brsk1 −/− male mice [t(36) = 1.019, p = 0.3148] (Figure 4C). When we compared both sexes and genotypes for pairing with ethanol or saline on the rod floor, we found that only wild‐type females spent more time on the rod floor when it was paired with ethanol than when it was paired with saline [Fgenotype × rod pairing × sex (1,66) = 8.022, p = 0.0061; medium effect size, partial η2 = 0.108]. (Figure 4B and D).
FIGURE 4.
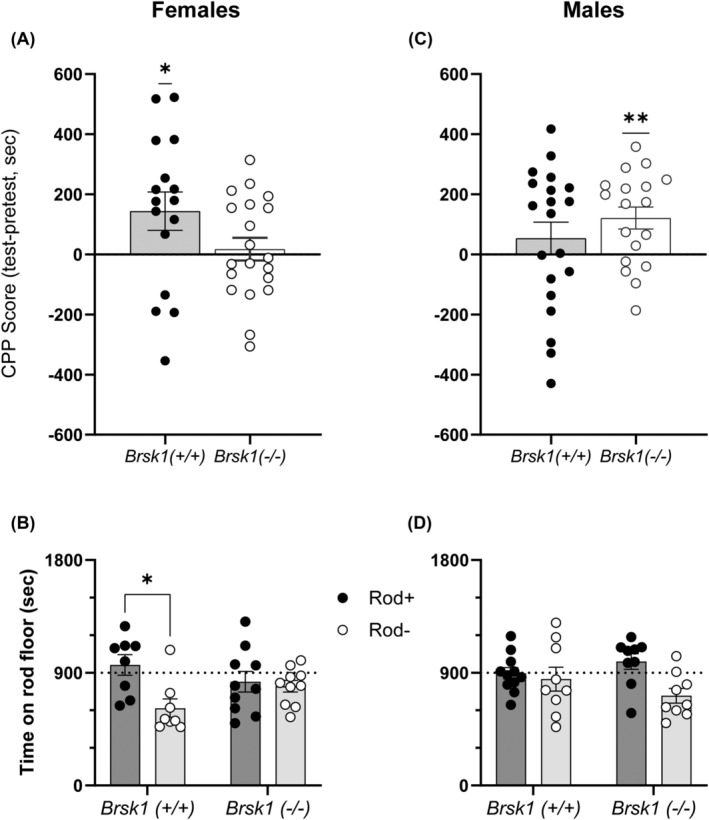
CPP in Brsk1 −/− mice varies in a genotype and sex‐dependent manner. A) Ethanol CPP score in female Brsk1 −/− mice (n = 20) and wild‐type littermates (n = 17). B) Time spent on rod floor when paired with ethanol (Rod+) or saline (Rod−) in female Brsk1 −/− mice (n = 10 Rod+, 10 Rod−) and wild‐type littermates (n = 11 Rod+, 9 Rod−). C) Ethanol CPP score in male Brsk1 −/− mice (n = 18) and wild‐type (n = 20) littermates. D) Time spent on rod floor when paired with ethanol (Rod+) or saline (Rod−) in male Brsk1 −/− mice (n = 9 Rod+, 9 Rod−) and wild‐type (n = 8 Rod+, 9 Rod−) littermates. * p < 0.05 and ** p < 0.01 by one sample t‐test in A) and C); * p < 0.05 by post‐hoc Sidak's multiple comparisons test in B).
3.8. Male Brsk1 −/− mice display increased anxiety‐like behaviour in the elevated plus maze (EPM)
Prkce −/− mice display reduced anxiety‐like behaviour compared with wild‐type mice and genetic knockdown of PKCε in the amygdala reduces anxiety in wild‐type mice. 5 , 7 Brsk1 −/− mice should also display reduced anxiety‐like behaviour if BRSK1 functions downstream of PKCε signalling in regulating this behaviour. Brsk1 −/− and wild‐type mice were observed on an EPM to measure anxiety‐like behaviour. Unexpectedly, we found that male Brsk1 −/− mice spent less time in the open arms [Fgenotype (1, 56) = 6.492, p = 0.0136; Fsex (1, 56) = 5.827, p = 0.0191; medium effect sizes, partial η2 genotype = 0.104, partial η2 sex = 0.094] and made less entries into the open arms [Fgenotype × sex (1, 56) = 6.677, p = 0.0124; medium effect size, partial η2 = 0.107] than wild‐type male mice (Figure 5A, B). In contrast, there were no differences in the time spent or entries made into the open arms between female Brsk1 −/− and wild‐type mice. There were also no locomotion differences observed between Brsk1 −/− and wild‐type mice of either sex as measured by the number of entries into the closed arms and distance moved on the EPM (Figure 5C, D).
FIGURE 5.
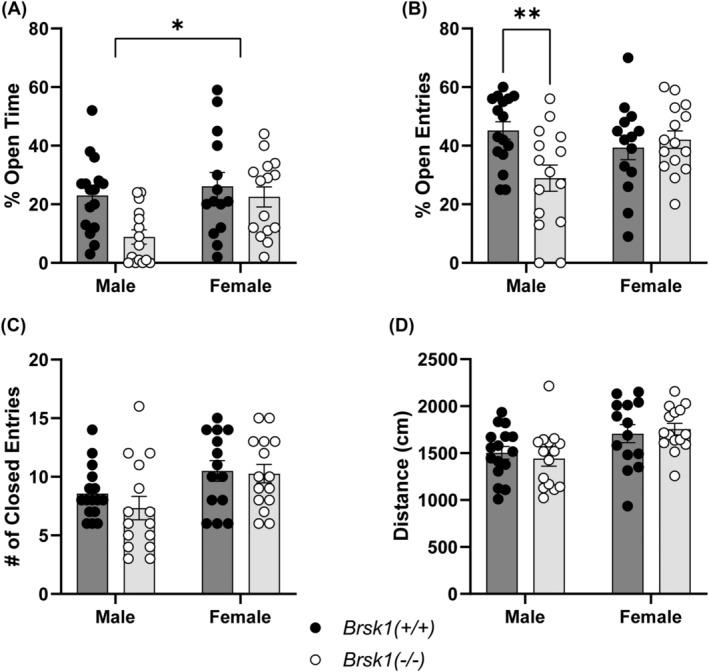
Male Brsk1 −/− mice display increased anxiety‐like behaviour in the elevated plus maze. A) Percent of time spent in the open arms of the EPM by Brsk1 −/− (n = 15 male, 15 female) mice and wild‐type (n = 16 male, 14 female) littermates, * p < 0.05. B) Percent of entries made into the open arms of the EPM by Brsk1 −/− mice and wild‐type littermates. C) Number of entries made into the closed arms of the EPM by Brsk1 −/− mice and wild‐type littermates. D) Total distance moved on the EPM by Brsk1 −/− mice and wild‐type littermates. ** p < 0.01 by post hoc Sidak's multiple comparisons test in B).
3.9. Ethanol‐facilitation of GABA release in the CeA is absent in male Brsk1 −/− mice
Ethanol‐facilitation of GABA release in the CeA is a PKCε‐dependent process since it is absent in Prkce −/− male mice and is blocked by pharmacological inhibition of PKCε in wild‐type male mice. 10 , 17 Since BRSK1 plays a role in regulating the probability of neurotransmitter release, 29 , 31 we examined if BRSK1 functions in regulating the ethanol‐induced facilitation of GABA release in the CeA of male Brsk1 −/− and wild‐type littermates. We recorded GABAA receptor mediated mIPSCs in CeA brain slices in the presence or absence of 44 mM ethanol (Figure 6A). At baseline, before the application of ethanol, there were no differences in the frequency (Figure 6B), amplitude (Figure 6C), or rise and decay kinetics (Figure 6D, E) of mIPSCs between genotypes. In a subset of cells, we applied 44mM ethanol for 9–15 min and found that it evoked a significant increase in the frequency of mIPSCs in wild‐type mice but not in Brsk1 −/− mice (Figure 6F). There were no differences between the genotypes in any of the other parameters during ethanol application (amplitude, rise, and decay time) (Figure 6F).
FIGURE 6.
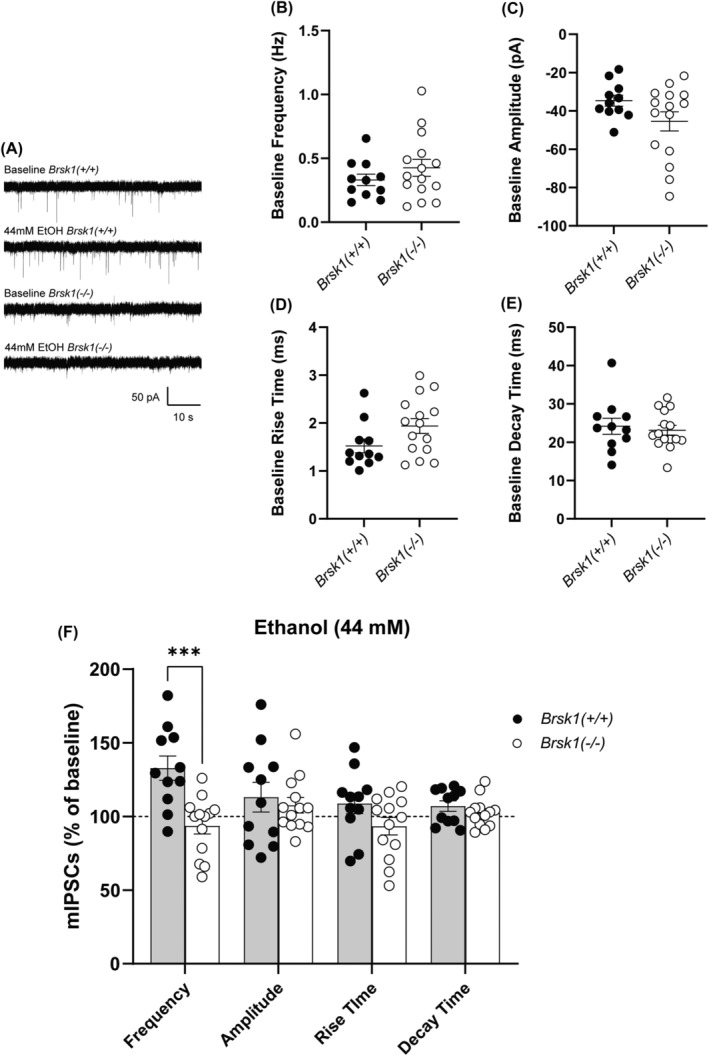
Ethanol‐facilitated GABA release is absent in the male Brsk1 −/− CeA. A) Representative traces of mIPSCs from a CeA neuron of wild‐type and a CeA neuron of Brsk1 −/− mice at baseline and in the presence of 44 mM ethanol. Baseline frequency [t(24) = 1.099, p = 0.2827]. (B) Amplitude [t(24) = 1.708, p = 0.1006], (C) rise time [t(24) = 1.897, p = 0.0700], (D) and (E) decay time [t(24) = 0.4562, p = 0.6524] of mIPSCs in the CeA of Brsk1 −/− (n = 15 cells) and wild‐type (n = 11 cells) mice. F) Effect of acute 44 mM ethanol application (displayed as a percentage of baseline) on frequency [t(22) = 4.049, p = 0.0005], amplitude [t(22) = 0.5107, p = 0.6147], and kinetics (rise [t(22) = 1.715, p = 0.1004] and decay [t(22) = 1.041, p = 0.3091] time) of mIPSCs in the CeA of wild‐type (n = 11 cells) and Brsk1 −/− (n = 13 cells) mice, *** p < 0.001 compared with wild‐type littermates by two‐tailed, unpaired t‐test.
4. DISCUSSION
PKCε regulates responses to ethanol and has become a potential drug target for the treatment of alcohol use disorder. 1 , 2 , 3 , 4 , 5 , 6 , 8 , 9 , 10 , 38 , 39 , 40 , 41 We sought to identify downstream substrates of PKCε that mediate its behavioural effects with the goal of revealing additional therapeutic targets for treatment. Here we evaluated one such substrate, BRSK1, using in vitro kinase assays and Brsk1 −/− mice. We found several behavioural and physiological responses to in Brsk1 −/− mice that are similar, but not identical, to those previously observed in Prkce −/− mice.
Using a chemical genetic screen with mouse brain lysates, we identified serine 559 (S559) of BRSK1 as a putative site of PKCε phosphorylation. 18 Here, we used a combination of in vitro kinase assays and a phospho‐specific antibody to further confirm that PKCε phosphorylates BRSK1 at S559. These experiments provide additional evidence that BRSK1 is a true direct PKCε substrate. Substrates of PKCε have been identified previously using similar methods. 38 , 42 , 43 , 44 , 45 However, only one has been linked to ethanol‐related responses. That substrate is the γ2 subunit of GABA type A (GABAA) receptors, which is phosphorylated by PKCε at serine 327; phosphorylation of this residue reduces the sensitivity of α1β2γ2 containing GABAA receptors to ethanol. 38
PKCε regulates the duration of ethanol intoxication; male Prkce −/− mice take longer than wild‐type mice to recover from sedative‐hypnotic or ataxic doses of ethanol. 1 , 4 Here we found that both male and female Brsk1 −/− mice also show prolonged recovery from a sedative‐hypnotic dose of ethanol, but not from an ataxic dose. This finding suggests that the sedative‐hypnotic effect of ethanol involves neural circuits regulated by BRSK1 whereas ethanol's effect on coordination involves other circuits and PKCε substrates. Our previous work found that the γ2 subunit of GABAA receptors is a cerebellar PKCε substrate that modulates ethanol‐induced ataxia, which could explain the difference in responses to ataxic doses of ethanol between Prkce −/− and Brsk1 −/− mice. 38
Several studies have demonstrated a clear role for PKCε signalling in ethanol consumption and preference in male 1 , 2 , 6 , 8 , 9 , 10 and in female mice. 10 We found that Brsk1 −/− mice of both sexes showed reduced ethanol consumption and preference. However, when the data were stratified by sex, the reduction in ethanol consumption and preference was more significant in female Brsk1 −/− mice. Thus, unlike PKCε, BRSK1 appears to play a more sex‐specific role in regulating ethanol intake in females more so than in males. This apparent sex difference is particularly compelling since female C57BL/6J mice consume much more ethanol than males. 33 , 46 , 47 , 48 The reasons for this sex difference is an active area of study, and there is an extensive literature on sex differences in the neurobiology of alcohol use disorder (reviewed in 49 ). Notably, women with alcohol use disorder are at higher risk for negative health outcomes compared with men who consume comparable levels of alcohol. 50 , 51 , 52 , 53 , 54 , 55 , 56 , 57 , 58 Why female Brsk1 −/− mice drink less is unclear but there is a growing literature on BRSK1's involvement in ovarian function, age of menopause onset, cervical cancer, and breast cancer. 59 , 60 , 61 , 62 , 63 Future work is needed to reveal the functional mechanisms involving BRSK1 that regulate ethanol drinking in females.
The reduction in ethanol intake we observed in female Brsk1 −/− mice may be related to reduced rewarding effects of ethanol since female Brsk1 −/− mice failed to develop CPP for 1 g/kg ethanol. In contrast, male Brsk1 −/− mice showed a lower reduction in ethanol intake which may be related to their development of CPP for 1 g/kg ethanol. However, results for CPP in males are not easy to interpret since male wild‐type mice did not develop clear CPP for ethanol. This result in wild type males is consistent with previous reports showing low levels of CPP for ethanol in C57BL/6J mice compared with other strains. 64 , 65 , 66 , 67 Further studies in mouse strains more sensitive to the acute rewarding effects of ethanol, such as the DBA/2J strain, could help in assessing ethanol reward in Brsk1 −/− males. Additionally, previous behavioural studies were done with Prkce −/− mice on a 129/S4 background or a 129/S4 × C57BL/6J hybrid background. The work in this study was done with mice on an inbred C57BL/6J background. This difference in genetic background is a limitation in our study when we compare our present results in Brsk1 −/− mice with previous findings in Prkce −/− mice.
Unexpectedly, we observed increased anxiety like behaviour specifically in male Brsk1 −/− mice, unlike the reduced anxiety observed in both sexes of Prkce −/− mice. 5 BRSK1 appears to play a sex specific role in regulating anxiety in a manner opposite to PKCε. This finding suggests that the regulation of anxiety by BRSK1 is distinct from PKCε signalling. Increased anxiety‐like phenotypes have previously been associated with increased ethanol consumption in several rodent models. 68 , 69 , 70 , 71 , 72 , 73 , 74 Thus, the disparity between ethanol consumption in male and female Brsk1 −/− mice could be related to increased anxiety‐like behaviour in male Brsk1 −/− mice leading them to consume ethanol at higher levels.
Ethanol‐facilitation of vesicular GABA release in the CeA was blocked in brain slices from male Brsk1 −/− mice, similar to what has been observed in CeA slices from male Prkce −/− mice. 17 This finding is consistent with both kinases playing roles in modulating the release of neurotransmitters. 2 , 10 , 16 , 17 , 29 , 30 , 31 We previously reported a role for PKCε in both action potential‐dependent and action potential‐independent GABA transmission using both Prkce −/− mice and pharmacological approaches. 10 , 17 We demonstrated the dual role of PKCε in regulating baseline GABA transmission as well as ethanol‐induced facilitation of GABA release. In the current study, we limited our investigation to the role of BRSK1 in action potential‐independent GABA release and found that BRSK1 regulates ethanol‐induced vesicular GABA release but unlike PKCε does not regulate baseline vesicular GABA release. A limitation of the current study is that we only used male Brsk1 −/− mice to measure GABA release in the CeA. However, it is important to note that CeA neurons from female rats and mice are largely insensitive to the acute effects of ethanol on GABA release. 75 , 76 , 77 Since this phenotype was observed in male Brsk1 −/− brain slices, but male Brsk1 −/− mice did not show altered ethanol intake, the PKCε signalling pathways that regulate these two phenotypes can be dissociated, with GABA release being BRSK1‐dependent and ethanol consumption being BRSK1‐independent in male mice. Future studies are needed to shed light on the mechanisms underlying these differences.
In summary, our results demonstrate a role for BRSK1 in behavioural and physiological responses to ethanol and further establish BRSK1 as a substrate of PKCε. PKCε signalling has a strong connection to alcohol and anxiety related behaviours, and our findings indicate a role for BRSKI in a subset of these behaviours, some in a sex specific manner. Identifying additional downstream substrates of PKCε that are involved should generate additional targets for the development of new, potentially sex‐specific treatments for AUD and anxiety.
AUTHOR CONTRIBUTIONS
Michael P. Dugan: Protein/peptide purification, kinase assays, behavioural experiments, data analyses for all experiments, writing—original draft. Rajani Maiya: Peptide purification, kinase assays, review, and editing. Caleb Fleisher: Assisted with drinking and conditioned place preference experiments. Michal Bajo: Electrophysiology. Angela Snyder: Electrophysiology. Ashwin Koduri: Assisted with cell culture work and drinking experiments. Sathvik Srinivasan: Assisted with cell culture work, loss of righting, rotarod, and drinking experiments. Marisa Roberto: Electrophysiology recordings and analysis, data interpretation, review, and editing. Robert O. Messing: Conceptualization, supervision, funding acquisition, writing—review and editing.
CONFLICT OF INTEREST STATEMENT
The authors declare that there are no competing interests associated with the manuscript.
Supporting information
Figure S1. Supporting Information.
ACKNOWLEDGEMENTS
We would like to acknowledge and thank Michela Marinelli PhD for assistance in performing the statistical analysis.
Dugan MP, Maiya R, Fleischer C, et al. Brain‐specific serine/threonine‐protein kinase 1 is a substrate of protein kinase C epsilon involved in sex‐specific ethanol and anxiety phenotypes. Addiction Biology. 2024;29(3):e13388. doi: 10.1111/adb.13388
Funding information This work was supported by National Institutes of Health Grant AA013588 and funds provided by the M. June and J. Virgil Waggoner Chair in Molecular Biology to R.O.M., and a University of Texas at Austin Bruce‐Jones Fellowship and an National Institutes of Health F31 AA030218 award to M.P. D., and AA013498, and AA029841 to M.R. and T32 AA007456 to A.E.S.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Hodge CW, Mehmert KK, Kelley SP, et al. Supersensitivity to allosteric GABA a receptor modulators and alcohol in mice lacking PKCε. Nat Neurosci. 1999;2(11):997‐1002. doi: 10.1038/14795 [DOI] [PubMed] [Google Scholar]
- 2. Olive MF, Mehmert KK, Messing RO, Hodge CW. Reduced operant ethanol self‐administration and in vivo mesolimbic dopamine responses to ethanol inPKCε‐deficient mice. Eur J Neurosci. 2000;12(11):4131‐4140. doi: 10.1046/j.1460-9568.2000.00297.x [DOI] [PubMed] [Google Scholar]
- 3. Newton PM, Messing RO. Increased sensitivity to the aversive effects of ethanol in PKCε null mice revealed by place conditioning. Behav Neurosci. 2007;121(2):439‐442. doi: 10.1037/0735-7044.121.2.439 [DOI] [PubMed] [Google Scholar]
- 4. Wallace MJ, Newton PM, Oyasu M, et al. Acute functional tolerance to ethanol mediated by protein kinase C ɛ. Neuropsychopharmacology. 2007;32(1):127‐136. doi: 10.1038/sj.npp.1301059 [DOI] [PubMed] [Google Scholar]
- 5. Hodge CW, Raber J, McMahon T, et al. Decreased anxiety‐like behavior, reduced stress hormones, and neurosteroid supersensitivity in mice lacking protein kinase Cε. J Clin Invest. 2002;110(7):1003‐1010. doi: 10.1172/JCI15903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Choi D‐S, Wang D, Dadgar J, Chang WS, Messing RO. Conditional Rescue of Protein Kinase C ε regulates ethanol preference and hypnotic sensitivity in adult mice. J Neurosci Society for Neuroscience. 2002;22(22):9905‐9911. doi: 10.1523/JNEUROSCI.22-22-09905.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lesscher HMB, McMahon T, Lasek AW, et al. Amygdala protein kinase C epsilon regulates corticotropin‐releasing factor and anxiety‐like behavior. Genes Brain Behav. 2008;7(3):323‐333. doi: 10.1111/j.1601-183X.2007.00356.x [DOI] [PubMed] [Google Scholar]
- 8. Lesscher HMB, Wallace MJ, Zeng L, et al. Amygdala protein kinase C epsilon controls alcohol consumption. Genes Brain Behav. 2009;8(5):493‐499. doi: 10.1111/j.1601-183X.2009.00485.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cozzoli DK, Courson J, Rostock C, et al. Protein kinase C epsilon activity in the nucleus Accumbens and central nucleus of the amygdala mediates binge alcohol consumption. Biol Psychiatry. 2016;79(6):443‐451. doi: 10.1016/j.biopsych.2015.01.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Blasio A, Wang J, Wang D, et al. Novel small‐molecule inhibitors of protein kinase C epsilon reduce ethanol consumption in mice. Biol Psychiatry. 2018;84(3):193‐201. doi: 10.1016/j.biopsych.2017.10.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. McClintick JN, Brooks AI, Deng L, et al. Ethanol treatment of lymphoblastoid cell lines from alcoholics and non‐alcoholics causes many subtle changes in gene expression. Alcohol. 2014;48(6):603‐610. doi: 10.1016/j.alcohol.2014.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Das J, Pany S, Rahman GM, Slater SJ. PKCϵ has an alcohol‐binding site in its second cysteine‐rich regulatory domain. Biochem J. 2009;421(3):405‐413. doi: 10.1042/BJ20082271 [DOI] [PubMed] [Google Scholar]
- 13. Machu TK, Olsen RW, Browning MD. Ethanol has no effect on CAMP‐dependent protein kinase‐, protein kinase C‐, or Ca2+‐calmodulin‐dependent protein kinase II‐stimulated phosphorylation of highly purified substrates in vitro. Alcohol Clin Exp Res. 1991;15(6):1040‐1044. doi: 10.1111/j.1530-0277.1991.tb05208.x [DOI] [PubMed] [Google Scholar]
- 14. Parekh D, Ziegler W, Yonezawa K, Hara K, Parker PJ. Mammalian TOR controls one of two kinase pathways acting upon nPKCdelta and nPKCepsilon. J Biol Chem. 1999;274(49):34758‐34764. doi: 10.1074/jbc.274.49.34758 [DOI] [PubMed] [Google Scholar]
- 15. Minami H, Owada Y, Suzuki R, Handa Y, Kondo H. Localization of mRNAs for novel, atypical as well as conventional protein kinase C (PKC) isoforms in the brain of developing and mature rats. J Mol Neurosci. 2000;15(2):121‐135. doi: 10.1385/JMN:15:2:121 [DOI] [PubMed] [Google Scholar]
- 16. Prekeris R, Mayhew MW, Cooper JB, Terrian DM. Identification and localization of an actin‐binding motif that is unique to the epsilon isoform of protein kinase C and participates in the regulation of synaptic function. J Cell Biol. 1996;132(1‐2):77‐90. doi: 10.1083/jcb.132.1.77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bajo M, Cruz MT, Siggins GR, Messing R, Roberto M. Protein kinase C epsilon mediation of CRF‐ and ethanol‐induced GABA release in central amygdala. Proc Natl Acad Sci. 2008;105(24):8410‐8415. doi: 10.1073/pnas.0802302105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dugan MP, Ferguson LB, Hertz NT, et al. Chemical genetic identification of PKC epsilon substrates in mouse brain. Mol Cell Proteomics MCP. 2023;22(4):100522. doi: 10.1016/j.mcpro.2023.100522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Crump JG, Zhen M, Jin Y, Bargmann CI. The SAD‐1 kinase regulates presynaptic vesicle clustering and axon termination. Neuron. 2001;29(1):115‐129. doi: 10.1016/S0896-6273(01)00184-2 [DOI] [PubMed] [Google Scholar]
- 20. Hung W, Hwang C, Po MD, Zhen M. Neuronal polarity is regulated by a direct interaction between a scaffolding protein, Neurabin, and a presynaptic SAD‐1 kinase in Caenorhabditis elegans. Development. 2007;134(2):237‐249. doi: 10.1242/dev.02725 [DOI] [PubMed] [Google Scholar]
- 21. Kim JSM, Hung W, Narbonne P, Roy R, Zhen M. C. Elegans STRADα and SAD cooperatively regulate neuronal polarity and synaptic organization. Development. 2010;137(1):93‐102. doi: 10.1242/dev.041459 [DOI] [PubMed] [Google Scholar]
- 22. Kishi M. Mammalian SAD kinases are required for neuronal polarization. Science. 2005;307(5711):929‐932. doi: 10.1126/science.1107403 [DOI] [PubMed] [Google Scholar]
- 23. Barnes, A. P. , Lilley, B. N. , Pan, Y. A. , Plummer, L. J. , Powell, A. W. , Raines, A. N. , Sanes J. R., Polleux F. (2007) LKB1 and SAD kinases define a pathway required for the polarization of cortical neurons. Cell 129, 549–563. doi: 10.1016/j.cell.2007.03.025, 3. [DOI] [PubMed] [Google Scholar]
- 24. Müller M, Lutter D, Püschel AW. Persistence of the cell‐cycle checkpoint kinase Wee1 in SadA‐ and SadB‐deficient neurons disrupts neuronal polarity. J Cell Sci. 2010;123(2):286‐294. doi: 10.1242/jcs.058230 [DOI] [PubMed] [Google Scholar]
- 25. Lilley BN, Krishnaswamy A, Wang Z, Kishi M, Frank E, Sanes JR. SAD kinases control the maturation of nerve terminals in the mammalian peripheral and central nervous systems. SAD Kinases Control Matur Nerve Termin Mamm Peripher Cent Nerv Syst. 2014;111(3):1138‐1143. doi: 10.1073/pnas.1321990111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lilley BN, Krishnaswamy A, Wang Z, Kishi M, Frank E, Sanes JR. SAD kinases control the maturation of nerve terminals in the mammalian peripheral and central nervous systems. Proc Natl Acad Sci. 2014;111(3):1138‐1143. doi: 10.1073/pnas.1321990111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sample V, Ramamurthy S, Gorshkov K, Ronnett GV, Zhang J. Polarized activities of AMPK and BRSK in primary hippocampal neurons. Mol Biol Cell. 2015;26(10):1935‐1946. doi: 10.1091/mbc.E14-02-0764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dhumale P, Menon S, Chiang J, Püschel AW. The loss of the kinases SadA and SadB results in early neuronal apoptosis and a reduced number of progenitors. PLOS ONE (Sato, M, Ed). 2018;13(4):e0196698. doi: 10.1371/journal.pone.0196698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Inoue E, Mochida S, Takagi H, et al. SAD: a presynaptic kinase associated with synaptic vesicles and the active zone cytomatrix that regulates neurotransmitter release. Neuron. 2006;50(2):261‐275. doi: 10.1016/j.neuron.2006.03.018 [DOI] [PubMed] [Google Scholar]
- 30. Mochida S, Hida Y, Tanifuji S, et al. SAD‐B phosphorylation of CAST controls active zone vesicle recycling for synaptic depression. Cell Rep. 2016;16(11):2901‐2913. doi: 10.1016/j.celrep.2016.08.020 [DOI] [PubMed] [Google Scholar]
- 31. Watabe AM, Nagase M, Hagiwara A, et al. SAD‐B kinase regulates pre‐synaptic vesicular dynamics at hippocampal Schaffer collateral synapses and affects contextual fear memory. J Neurochem. 2016;136(1):36‐47. doi: 10.1111/jnc.13379 [DOI] [PubMed] [Google Scholar]
- 32. du Sert NP, Hurst V, Ahluwalia A, et al. The ARRIVE guidelines 2.0: updated guidelines for reporting animal research. PLoS Biol. 2020;18(7):e3000410. doi: 10.1371/journal.pbio.3000410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hwa LS, Chu A, Levinson SA, Kayyali TM, DeBold JF, Miczek KA. Persistent escalation of alcohol drinking in C57BL/6J mice with intermittent access to 20% ethanol. Alcohol Clin Exp Res. 2011;35(11):1938‐1947. doi: 10.1111/j.1530-0277.2011.01545.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Motulsky HJ, Brown RE. Detecting outliers when fitting data with nonlinear regression – a new method based on robust nonlinear regression and the false discovery rate. BMC Bioinformatics. 2006;7(1):123. doi: 10.1186/1471-2105-7-123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Roberto M, Cruz MT, Gilpin NW, et al. Corticotropin releasing factor‐induced amygdala gamma‐aminobutyric acid release plays a key role in alcohol dependence. Biol Psychiatry. 2010;67(9):831‐839. doi: 10.1016/j.biopsych.2009.11.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Steinman MQ, Kirson D, Wolfe SA, et al. Importance of sex and trauma context on circulating cytokines and amygdalar GABAergic signaling in a comorbid model of posttraumatic stress and alcohol use disorders. Mol Psychiatry. 2021;26(7):3093‐3107. doi: 10.1038/s41380-020-00920-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ialongo C. Understanding the effect size and its measures. Biochem Med. 2016;26(2):150‐163. doi: 10.11613/BM.2016.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Qi Z‐H, Song M, Wallace MJ, et al. Protein kinase Cϵ regulates γ‐Aminobutyrate type a receptor sensitivity to ethanol and benzodiazepines through phosphorylation of γ2 subunits. J Biol Chem. 2007;282(45):33052‐33063. doi: 10.1074/jbc.M707233200 [DOI] [PubMed] [Google Scholar]
- 39. Maiya R, McMahon T, Wang D, et al. Selective chemical genetic inhibition of protein kinase C epsilon reduces ethanol consumption in mice. Neuropharmacology. 2016;107:40‐48. doi: 10.1016/j.neuropharm.2016.02.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Olive MF, Mehmert KK, Nannini MA, Camarini R, Messing RO, Hodge CW. Reduced ethanol withdrawal severity and altered withdrawal‐induced c‐fos expression in various brain regions of mice lacking protein kinase C‐epsilon. Neuroscience. 2001;103(1):171‐179. doi: 10.1016/S0306-4522(00)00566-2 [DOI] [PubMed] [Google Scholar]
- 41. Olive MF, Mcgeehan AJ, Kinder JR, et al. The mGluR5 antagonist 6‐Methyl‐2‐(phenylethynyl)pyridine decreases ethanol consumption via a protein kinase Cϵ‐dependent mechanism. Mol Pharmacol. 2005;67(2):349‐355. doi: 10.1124/mol.104.003319 [DOI] [PubMed] [Google Scholar]
- 42. Newton PM, Messing RO. The substrates and binding partners of protein kinase Cε. Biochem J. 2010;427(2):189‐196. doi: 10.1042/BJ20091302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wu D‐F, Chandra D, McMahon T, et al. PKCε phosphorylation of the sodium channel NaV1.8 increases channel function and produces mechanical hyperalgesia in mice. J Clin Invest. 2012;122(4):1306‐1315. doi: 10.1172/JCI61934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Oehrlein SA, Parker PJ, Herget T. Phosphorylation of GAP‐43 (growth‐associated protein of 43 kDa) by conventional, novel and atypical isotypes of the protein kinase C gene family: differences between oligopeptide and polypeptide phosphorylation. Biochem J. 1996;317(1):219‐224. doi: 10.1042/bj3170219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Überall F, Giselbrecht S, Hellbert K, et al. Conventional PKC‐α, novel PKC‐ε and PKC‐θ, but not atypical PKC‐λ are MARCKS kinases in intact NIH 3T3 fibroblasts*. J Biol Chem. 1997;272(7):4072‐4078. doi: 10.1074/jbc.272.7.4072 [DOI] [PubMed] [Google Scholar]
- 46. Yoneyama N, Crabbe JC, Ford MM, Murillo A, Finn DA. Voluntary ethanol consumption in 22 inbred mouse strains. Alcohol. 2008;42(3):149‐160. doi: 10.1016/j.alcohol.2007.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Middaugh LD, Frackelton WF, Boggan WO, Onofrio A, Shepherd CL. Gender differences in the effects of ethanol on C57BL/6 mice. Alcohol. 1992;9(3):257‐260. doi: 10.1016/0741-8329(92)90062-F [DOI] [PubMed] [Google Scholar]
- 48. Middaugh LD, Kelley BM, Bandy A‐LE, McGroarty KK. Ethanol consumption by C57BL/6 mice: influence of gender and procedural variables. Alcohol. 1999;17(3):175‐183. doi: 10.1016/S0741-8329(98)00055-X [DOI] [PubMed] [Google Scholar]
- 49. Flores‐Bonilla A, Richardson HN. Sex differences in the neurobiology of alcohol use disorder. Alcohol Res. 2020;40(2):04. doi: 10.35946/arcr.v40.2.04 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. White AJ, DeRoo LA, Weinberg CR, Sandler DP. Lifetime alcohol intake, binge drinking behaviors, and breast cancer risk. Am J Epidemiol. 2017;186(5):541‐549. doi: 10.1093/aje/kwx118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Peltier MR, Verplaetse TL, Mineur YS, et al. Sex differences in stress‐related alcohol use. Neurobiol Stress. 2019;10:100149. doi: 10.1016/j.ynstr.2019.100149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Mann K, Ackermann K, Croissant B, Mundle G, Nakovics H, Diehl A. Neuroimaging of gender differences in alcohol dependence: are women more vulnerable? Alcohol Clin Exp Res. 2005;29(5):896‐901. doi: 10.1097/01.ALC.0000164376.69978.6B [DOI] [PubMed] [Google Scholar]
- 53. Åberg F, Helenius‐Hietala J, Puukka P, Jula A. Binge drinking and the risk of liver events: a population‐based cohort study. Liver Int. 2017;37(9):1373‐1381. doi: 10.1111/liv.13408 [DOI] [PubMed] [Google Scholar]
- 54. Schwarzinger M, Thiébaut SP, Baillot S, Mallet V, Rehm J. Alcohol use disorders and associated chronic disease – a national retrospective cohort study from France. BMC Public Health. 2017;18(1):43. doi: 10.1186/s12889-017-4587-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hydes TJ, Burton R, Inskip H, Bellis MA, Sheron N. A comparison of gender‐linked population cancer risks between alcohol and tobacco: how many cigarettes are there in a bottle of wine? BMC Public Health. 2019;19(1):316. doi: 10.1186/s12889-019-6576-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wilsnack RW, Wilsnack SC, Gmel G, Kantor LW. Gender Differences in Binge Drinking. Alcohol Res Curr Rev. 2018;39:57‐76. [PMC free article] [PubMed] [Google Scholar]
- 57. Kerr‐Corrêa F, Igami TZ, Hiroce V, Tucci AM. Patterns of alcohol use between genders: a cross‐cultural evaluation. J Affect Disord. 2007;102(1‐3):265‐275. doi: 10.1016/j.jad.2006.09.031 [DOI] [PubMed] [Google Scholar]
- 58. Erol A, Karpyak VM. Sex and gender‐related differences in alcohol use and its consequences: contemporary knowledge and future research considerations. Drug Alcohol Depend. 2015;156:1‐13. doi: 10.1016/j.drugalcdep.2015.08.023 [DOI] [PubMed] [Google Scholar]
- 59. Stolk L, Zhai G, van Meurs JBJ, et al. Loci at chromosomes 13, 19 and 20 influence age at natural menopause. Nat Genet. 2009;41(6):645‐647. doi: 10.1038/ng.387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Qin Y, Sun M, You L, et al. ESR1, HK3 and BRSK1 gene variants are associated with both age at natural menopause and premature ovarian failure. Orphanet J Rare Dis. 2012;7(1):5. doi: 10.1186/1750-1172-7-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Wang H, Liu X‐B, Chen J‐H, et al. Decreased expression and prognostic role of cytoplasmic BRSK1 in human breast carcinoma: correlation with Jab1 stability and PI3K/Akt pathway. Exp Mol Pathol. 2014;97(2):191‐201. doi: 10.1016/j.yexmp.2014.07.012 [DOI] [PubMed] [Google Scholar]
- 62. van der Kooi A‐LLF, van Dijk M, Broer L, et al. Possible modification of BRSK1 on the risk of alkylating chemotherapy‐related reduced ovarian function. Hum Reprod Oxf Engl. 2021;36(4):1120‐1133. doi: 10.1093/humrep/deaa342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Liu G, Li L, Shang D, Zhou C, Zhang C. BRSK1 confers cisplatin resistance in cervical cancer cells via regulation of mitochondrial respiration. J Cancer Res Clin Oncol. 2023;149(11):8803‐8815. doi: 10.1007/s00432-023-04821-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Lim JP, Zou ME, Janak PH, Messing RO. Responses to ethanol in C57BL/6 versus C57BL/6 × 129 hybrid mice. Brain Behav. 2012;2(1):22‐31. doi: 10.1002/brb3.29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Cunningham CL. Genetic relationship between ethanol‐induced conditioned place preference and other ethanol phenotypes in 15 inbred mouse strains. Behav Neurosci. 2014;128(4):430‐445. doi: 10.1037/a0036459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Cunningham CL, Shields CN. Effects of sex on ethanol conditioned place preference, activity and variability in C57BL/6J and DBA/2J mice. Pharmacol Biochem Behav. 2018;173:84‐89. doi: 10.1016/j.pbb.2018.07.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Cunningham CL, Niehus DR, Malott DH, Prather LK. Genetic differences in the rewarding and activating effects of morphine and ethanol. Psychopharmacology (Berl). 1992;107(2‐3):385‐393. doi: 10.1007/BF02245166 [DOI] [PubMed] [Google Scholar]
- 68. Spanagel R, Montkowski A, Allingham K, Shoaib M, Holsboer F, Landgraf R. Anxiety: a potential predictor of vulnerability to the initiation of ethanol self‐administration in rats. Psychopharmacology (Berl). 1995;122(4):369‐373. doi: 10.1007/BF02246268 [DOI] [PubMed] [Google Scholar]
- 69. Izídio GS, Ramos A. Positive association between ethanol consumption and anxiety‐related behaviors in two selected rat lines. Alcohol. 2007;41(7):517‐524. doi: 10.1016/j.alcohol.2007.07.008 [DOI] [PubMed] [Google Scholar]
- 70. Hayton SJ, Mahoney MK, Olmstead MC. Behavioral traits predicting alcohol drinking in outbred rats: an investigation of anxiety, novelty seeking, and cognitive flexibility. Alcohol Clin Exp Res. 2012;36(4):594‐603. doi: 10.1111/j.1530-0277.2011.01668.x [DOI] [PubMed] [Google Scholar]
- 71. Radwanska K, Kaczmarek L. Characterization of an alcohol addiction‐prone phenotype in mice. Addict Biol. 2012;17(3):601‐612. doi: 10.1111/j.1369-1600.2011.00394.x [DOI] [PubMed] [Google Scholar]
- 72. Bahi A. Increased anxiety, voluntary alcohol consumption and ethanol‐induced place preference in mice following chronic psychosocial stress. Stress. 2013;16(4):441‐451. doi: 10.3109/10253890.2012.754419 [DOI] [PubMed] [Google Scholar]
- 73. Pelloux Y, Costentin J, Duterte‐Boucher D. Differential involvement of anxiety and novelty preference levels on oral ethanol consumption in rats. Psychopharmacology (Berl). 2015;232(15):2711‐2721. doi: 10.1007/s00213-015-3910-5 [DOI] [PubMed] [Google Scholar]
- 74. Jadhav KS, Magistretti PJ, Halfon O, Augsburger M, Boutrel B. A preclinical model for identifying rats at risk of alcohol use disorder. Sci Rep. 2017;7(1):9454. doi: 10.1038/s41598-017-09801-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Kirson D, Khom S, Rodriguez L, et al. Sex differences in acute alcohol sensitivity of Naïve and alcohol dependent central amygdala GABA synapses. Alcohol Alcohol Oxf Oxfs. 2021;56(5):581‐588. doi: 10.1093/alcalc/agab034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Rodriguez L, Kirson D, Wolfe SA, et al. Alcohol dependence induces CRF sensitivity in female central amygdala GABA synapses. Int J Mol Sci. 2022;23(14):7842. doi: 10.3390/ijms23147842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Agoglia AE, Tella J, Herman MA. Sex differences in corticotropin releasing factor peptide regulation of inhibitory control and excitability in central amygdala corticotropin releasing factor receptor 1‐neurons. Neuropharmacology. 2020;180:108296. doi: 10.1016/j.neuropharm.2020.108296 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Supporting Information.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.