

Abstract
In recent years, the role of bioinformatics and computational biology together with omics techniques and transcriptomics has gained tremendous importance in biomedicine and healthcare, particularly for the identification of biomarkers for precision medicine and drug discovery. Differential gene expression (DGE) analysis is one of the most used techniques for RNA-sequencing (RNA-seq) data analysis. This tool, which is typically used in various RNA-seq data processing applications, allows the identification of differentially expressed genes across two or more sample sets. Functional enrichment analyses can then be performed to annotate and contextualize the resulting gene lists. These studies provide valuable information about disease-causing biological processes and can help in identifying molecular targets for novel therapies. This review focuses on differential gene expression (DGE) analysis pipelines and bioinformatic techniques commonly used to identify specific biomarkers and discuss the advantages and disadvantages of these techniques.
Keywords: Differential gene expression analysis, Pathway enrichment, Bioinformatic analyses, Biomarkers, Biomarkers discovery
Graphical Abstract

1. Introduction
With advances in precision medicine and therapies, there is a growing focus on the identification of disease-driver genes for more precise diagnosis and prognosis. Biomarkers are identifiers that could categorize a biological event or condition and monitor certain biological changes [1]. These can include genes, transcripts, proteins, and metabolites, all of which are termed biomarkers due to their ability to provide valuable insights into the diagnosis, prognosis, and disease therapy. According to the Biomarkers Definitions Working Group's specifications from 2001 (pp. 89–95) [2], biomarkers are pharmacological reactions to therapeutic intervention or objectively measurable indications of biological and pathological processes [3]. They are intended to replace a clinical endpoint and predict benefit or harm based on scientific evidence. Biomarkers have a variety of functions in healthcare and can have a substantial impact on patient care [4].
In recent years, bioinformatics has played a crucial role in omics techniques [5], [6], particularly in transcriptomics [7]. For example, RNA-sequencing (RNA-seq) techniques have provided vast amounts of data on gene expression levels across multiple conditions at high resolution [8]. This led to the need for the characterization of differentially expressed genes (DEGs) among various medical samples and biological contexts, including disease identification [9], tumorigenesis [10], microbial studies [11], and evaluation of therapeutic efficacy in patients.
In addition, machine learning has also begun to play an important role in the analysis of RNA sequencing data, particularly in the identification of significant genetic patterns that might not be evident with traditional methods. The use of advanced algorithms makes it possible to examine complex datasets and identify key genetic markers, which may have implications in the diagnosis and treatment of various diseases. Wenric et al. [12] showed that supervised learning methods can outperform traditional differential expression analysis in RNA-Seq for the identification of survival-related genes in various cancer datasets. In this study, the authors used the Random Forests [13] classification algorithm and an approach called EPS (extreme pseudo-samples), which employs Variational Autoencoders (VAE) [14] and regressors to classify genes [15]. The results indicated that out of 12 cancer datasets, these methods showed superior performance compared to differential expression analysis in 9 and 8 of the 12 datasets respectively. This demonstrates the potential of supervised learning-based gene selection methods in RNA-Seq studies. However, it is important to emphasize that these methods are still under development and do not replace, but rather complement, conventional genetic and transcriptomic analysis techniques, which remain the focus of this review [12].
Different software programs for R/Bioconductor perform statistical tests to identify which genes have a statistically significant difference between comparable samples (Table 1).
Table 1.
Differential gene expression analysis (DGE) tools.
| DGE Tool | Publish year | Distribution | Normalization | Description |
|---|---|---|---|---|
| DEGseq | 2009 | Binomial | None | Fisher's exact test and the likelihood ratio test are used in a random sampling model [16], [17] |
| edgeR | 2010 | Negative binomial | TMM [18] | Empirical Bayes estimate and either a Fisher's exact test tailored to over-dispersed data or a generalized linear model [8], [19], [20]. |
| baySeq | 2010 | Negative binomial | Internal | The posterior likelihood is empirically estimated using Bayesian statistics [21]. |
| DESeq | 2010 | Negative binomial | Deseq [22] | Shrinkage variance [22] |
| NOIseq | 2012 | None | RPKM [23] | Nonparametric test based on the signal-to-noise ratio [24] |
| PoissonSeq | 2012 | Poisson log-linear model | Internal | Score statistics [25] |
| SAMseq | 2013 | None | Internal | Mann–Whitney test with Poisson resampling [26] |
| EBSeq | 2013 | Negative binomial | Deseq [22] | Empirical Bayesian estimate of the posterior likelihood [27] |
| Deseq2 | 2014 | Negative binomial | Deseq [22] | Shrinkage variance with variance-based and Cook’s distance pre-filtering [28] |
| limma | 2015 | Log-normal | TMM [18] | Generalized linear model [29] |
| sleuth | 2017 | Linear model | TMM [18] | Estimates inferential variance through bootstrap pseudo-alignment techniques [30] |
Description of DGE tools (R/Bioconductor) packages, year of publication, type of distribution, type of normalization.
Among the various methods available, EdgeR and Deseq2 are some of the most used techniques for analyzing differential gene expression from RNA-seq data [31].
Once the differentially expressed genes have been identified, functional enrichment analysis can be performed to better understand the molecular mechanisms and pathways underlying the disease or condition analyzed [32]. These approaches require annotating genes with identifiers and cross-database descriptions, using a variety of instruments and software programs for pathway analysis and biomarker discovery [32]. However, several differential expression tools and pathway enrichment methods can be used depending on data's properties and study objectives [31].
This review will summarize the most common pipelines for differential gene expression analysis and bioinformatic studies for biomarker identification. We will also discuss the advantages and challenges associated with these techniques.
Recently, Costa-Silva et al. [33], provided a broad overview of advances in the field of differential gene expression analysis using RNA-Seq data. This review highlighted the various computational methodologies used to examine these data. In this review, we will further extend this analysis by specifically focusing on statistical insights into commonly used methods for DGE analysis, graphical representations essential for interpreting the results, tools, and databases for enriching biological pathways, and two examples of statistical analyses useful for complementing and improving the reliability of results.
2. Differential gene expression (DGE)
DGE analysis is a technique used in molecular biology to compare gene expression levels between two or more sample groups, such as healthy vs disease tissues or cells exposed to different treatments [34]. DGE analysis's primary objective is the identification of genes differentially expressed in settings being compared [35]. This tool can help in identifying genes involved in a particular biological process, disease, or response to treatment, thereby providing information on gene regulation and underlying biological mechanisms [36]. This multiple-steps analysis is frequently used in studies of disease, where it can help in the identification of biomarkers for diagnosis and prognosis or evaluate the effectiveness of specific treatments [37], [38].
The first step is the normalization and preprocessing of the data. Noise in the analysis is diminished by reducing variability related to technical issues, thereby assuring better comparability between results [31].
Once the data is clean and consistent, the next step is to select a model most suitable for the data [39]. Parametric methods, such as edgeR and DESeq2, are typically preferred for data that align well with specific statistical distributions like the negative binomial distribution, often used for RNA-Seq data [40]. On the other hand, non-parametric methods like NOIseq [24], [41] and SAMseq [26] are more suitable for datasets where these assumptions might not be valid or data distribution is more complex. These methods have more flexibility but may require larger sample sizes for obtaining reliable results. However, parametric methods, can be more efficient in analyses with small sample size, a common situation in RNA-Seq studies [40].
The third step goes deeper into the processed data to pinpoint genes with significant differences in expression. These differentially expressed genes can provide critical insights into disease mechanisms, potential drug targets, or constitute diagnostic or prognostic markers [39], [42].
The final step is the graphic visualization of the results. Once differentially expressed genes have been identified and their potential implications understood, it is then crucial to present these data in a clear and intuitive graphic format. Visualization allows researchers to easily observe trends, patterns, and anomalies in the data [43], [44], [45], [46].
2.1. Normalization
The accuracy and reliability of gene expression analysis largely hinges on the quality of the data analyzed [28], [47] as in fact normalisation is a pivotal step in data pre-processing, and it serves to modulate values so that they are directly comparable [31].
TMM (Trimmed Mean of M-values) and geometric mean methods are two widely recognized techniques among the myriad of normalization methods currently used [48].
Specifically, the TMM normalization is a simple and effective method for estimating relative RNA production levels from RNA-seq data, based on the assumption that most of the genes in the dataset are not differentially expressed between samples, i.e. their expression levels are relatively similar [18]. Then, this method estimates normalization factors that can adjust for differences in library size (total number of reads obtained from each sample) and composition (proportion of reads from different genes) between samples [49]. These differences can affect the accuracy of differential expression analysis, as in fact highly expressed genes or genes with different compositions might have a greater impact on the analysis results [49]. The TMM method can therefore help in eliminating the effect of sequencing depth on the analysis results by scaling the counts in order to have them comparable between samples [50]. This normalization process allows the accurate detection of differential expression in genes truly differentially expressed between samples, minimizing false-positive or false-negative results associated with differences in sequencing depth [47].
In their 2021 study, Liu S. et al. [31] applied the TMM method, using the 'calcNormFactors()' function of the edgeR package, to normalize RNA-seq data. This method corrects for discrepancies in library size and RNA composition, ensuring a more accurate comparison of gene expression levels between different samples.
In the context of RNA-seq data analysis, the geometric mean normalization method, often associated with the DESeq2 package, operates distinctly from the TMM method used in edgeR. This method involves calculating the geometric mean of expression values for each gene across all samples. The core principle of this approach is to normalize gene expression data by adjusting for variations in sequencing depth and distributional differences across samples, allowing therefore for more accurate comparisons of gene expression levels. The principles of this normalization method are addressed in the resources made available by the developers of DESeq2 package [28], [51]. Both methods are designed to tackle challenges in RNA-seq data analysis. However, they use different statistical approaches and are part of separate analytical packages.
2.2. edgeR and Deseq2 differential gene expression analysis techniques
EdgeR and Deseq2 are the most popular pipelines for analyzing differential gene expression from RNAseq data [31]. Table 2 lists the main features and advantages/disadvantages of these two pipelines.
Table 2.
Statistical model, advantages, disadvantages and applications of Deseq2 and edgeR pipelines.
| Features | DESeq2 [51] | edgeR [52] |
|---|---|---|
| Statistical Model | Negative binomial model | Negative binomial model |
| Advantages | Suitable for data sets with minor variability; user-friendly interface | Speed and robust approach for small data sets |
| Disadvantages | Lower speed; Less sensitivity for data sets with high biological variability | Reduced sensitivity for data sets with low biological variability |
| Typical Applications | Analyses under controlled experimental conditions with well-defined control groups [53]. | Analysis of complex datasets with high variability [54]. |
edgeR and Deseq2 are both based on the negative binomial distribution for modeling the count data in RNA sequencing experiments [22]. The parameters of the negative binomial distribution are determined solely by μ and φ. This approach assumes that the number of times a particular gene is read in an RNA sequencing experiment follows a certain pattern, known as the negative binomial distribution. This pattern is defined by two main factors: the average number of reads for a given gene (µij) and a factor called the dispersion parameter (φi), which accounts for variability in the data. These factors are both specific to each gene (i) and each sample (j) being studied. To identify DEGs, the accurate measurement of the dispersion parameter φ i for each gene is essential [19], [28]. Thus, it describes the probability of obtaining a certain number of counts in any given experimental condition, given the mean of the counts and a dispersion parameter, which takes into account the variance overlap. Variations in the estimate of φ i explain the primary discrepancies between edgeR and DESeq2 [55], [56].
The differences and similarities between these two important tools for analyzing differential gene expression in RNA-seq data have been extensively discussed and by Anders et al. [57].
2.2.1. The R package edgeR
EdgeR (Empirical Analysis of Digital Gene Expression data in R) [58] is a powerful and flexible tool for the analysis of RNA-seq, and is used in many applications as in fact it is effective in identifying differentially expressed genes with a low false discovery rate [19]. EdgeR is based on the conditional maximum likelihood (CML) method to calculate a common dispersion accounting for variability across genes [59]. Then, a modification of this approach is used to assist the estimate of gene-specific dispersion, and an empirical Bayes technique is used to reduce the dispersion closer to the common one [60]. The extent to which these specific dispersion estimates are adjusted towards a common value depends on how similar a gene in question is to other genes, in terms of its average expression level measured in log counts-per-million (log CPM). The counts per million (CPM) value is used instead of the read count, to remove the variation brought on by various sequencing depths [61].
EdgeR uses a combination of common and gene-specific dispersion, an approach which allows for efficient management of gene expression variability between different biological samples. This capability is particularly useful in situations with a small number of samples, where variability can be high [62]. A concrete example of this effectiveness is illustrated in the study by Chen et al. [63] In this study, the authors used edgeR to compare 5 normal samples, 5 with vascular calcification (VC) caused by uremia, and 4 samples with vitamin D3-induced vascular calcification. They identified a total of 650 DEGs in uremia-induced VC, including 405 up-regulated and 245 down-regulated genes, while they isolated in vitamin D3-induced VC 64 DEGs, including 42 up-regulated and 22 down-regulated genes. They then separately intersected the results of these two groups of up- and down-regulated DEGs to obtain a set of DEGs containing five down-regulated genes and nine up-regulated genes. This comparison that are allowed the identification of genes differentially expressed in VC, genes which might play a role in the development of this condition.
2.2.2. The R package DESeq2
DESeq2 (Differential Expression analysis for Sequence Count data) [28] uses a negative-binomial model similar to edgeR, but includes data-based shrinkage estimators for dispersion and l2FC. Specifically, it is based on a different approach to estimate the dispersion parameter based on a model which assumes a similar level of variability for genes with similar average expression levels. This approach is more appropriate when dealing with a large number of samples with relatively low biological variability [62]. Moreover, in DESeq2, gene-specific dispersion is narrowed down to a fitted smooth curve using an empirical Bayesian technique, based on the assuption that genes with comparable average expression levels have similar dispersion [28]. When the expression level is low, DESeq2 reduces l2FC estimates towards zero, overcoming the challenge in estimating l2FC for weakly expressed genes [64]. However, this narrowing process can produce conservative estimation statistics for the DGE test, underestimate dispersion, and reduce sensitivity while lowering the frequency of false positives [65]. Despite this, DESeq2 is still one of the most commonly used and well-validated methods for analyzing sequence count data [28].
DESeq2 is often preferred for analyzing datasets derived from a large number of samples with relatively low variability [62], as demonstrated in the study by Casarrubios et al. [66] In this study, the authors examined 16 pre-treatment and 36 post-treatment tissue samples derived from 41 patients with resectable stage IIIA NSCLC treated with neoadjuvant chemoimmunotherapy. Using DESeq2, they discovered differentially expressed genes between pathological complete response (CPR) and non-CPR samples and identified potential biomarkers and mechanisms associated with tumor response and recurrence.
2.3. Identification of differentially expressed genes
The identification of DEGs is a crucial process that requires careful cutoff selection [67]. The choice for measuring statistical significance, such as p-values [68], and p-values [39] adjusted with different techniques (padj) such as false discovery rate (FDR) [69], and magnitude of the difference, represented by the log2 fold change (l2FC) [42], is essential as in fact it directly affects reliability of results and interpretation of data. A p-value too high could lead to false negatives, while a value too low could result in false positives [70]. Similarly, the choice of an appropriate l2FC threshold is essential for the identification of gene expression changes which are biologically significant and not simply random variations [71], [72]. The choice of correction method for multiple tests, such as FDR or Bonferroni, has a significant impact on the identification of DEGs. While FDR offers a balance between discovery and reliability [73], Bonferroni's method is more conservative and reduces the risk of false positives [74]. To avoid a high number of false positives, it is necessary to correct for multiple testing and select the DEGs, setting a cutoff on the adjusted p-value [75]. Furthermore, the l2FC can be used to rank the genes more representative of differences between two experimental conditions [76]. For instance, Chen et al. [63] selected DEGs between calcified and normal vessels using a cutoff criterion of |l2FC| > 1 and an FDR threshold of 0.05. In the Casarrubios et al. [66] study, DEGs were identified using a cutoff of |l2FC| > 1.5 and a padj threshold of 0.05, using the Benjamini-Hochberg procedure, which is less conservative than Bonferroni's method and allows for the identification of significant differences between groups [77].
However, the choice of cutoffs for l2FC and p-value should be guided by the specificity of the data and the experimental conditions. This implies that there is no universal cutoff value, which has to be determined instead on a case-by-case basis [78], [79]. It is better to combine both cutoff criteria (l2FC and p-value/padj) for greater reliability in identifying DEGs [80]. To select appropriate cutoffs, it would be necessary to examine the distribution of data, identify appropriate thresholds reflecting biologically significant variations, and verify that the chosen cutoffs produce consistent and reproducible results [81].
2.4. Graphical representation
Once the differential expression analysis has been done on genes with an adjusted p-value minor of a certain cut-off selected, the graphic representation of the statistical analysis results is essential for the interpretation of the data [35], as in fact these graphical representations help in visualizing statistical significance [68], mean expression levels [82], and magnitude of comparison [50]. These representations include methods such as comparing fold-change to normalized mean counts (MA plots) [83]and p-value to fold-change (Volcano plots) [44].
2.4.1. MA and Volcano plot
The representation of l2FC vs. means expression between two treatments is frequently done using MA plots (Fig. 1) [83]. l2FC is plotted on the y-axis of a scatter plot, while normalized mean expression is plotted on the x-axis. Genes with different expression levels are shown as data points with extreme values along the y-axis. Lower mean expression values often exhibit greater l2FC variability than higher expression values. As a result, the data points fan out as the graph reads from right to left. Since there are established limits for l2FC, these are often represented on the MA plot by dotted lines. From these graphs, it is not possible to identify which genes are significant as the p-value is not evaluated [40], [84]. However, statistical significance does not always translate into biological or clinical relevance. A change in gene expression may be statistically significant but not biologically relevant [35]. l2FC, on the other hand, provides a measure of the magnitude of the change in gene expression between two conditions, and extreme l2FC values (both positive and negative) indicate marked differences in gene expression, which are likely more biologically relevant [35]. Thus, the M(A) graph is particularly useful because it combines information on the magnitude of change (on the y-axis) with the average abundance of gene expression (on the x-axis). This allows the visualization of both genes showing big changes in expression and those highly or poorly expressed in both conditions. This visualization avoids the confusion that might result from over-reliance on p-values and highlights biologically relevant differences [35].
Fig. 1.

MA plot. MA plot showing l2FC compared to mean expressions generated by Deseq2 R.
The comparison of padj and l2FC is commonly used to examine differences in gene expression between two conditions. This representation is often illustrated with a 'volcano plot' (Fig. 2), showing statistical significance versus magnitude of change [44]. On this graph, the y-axis represents the negative log in base 10 of the p-values, reflecting statistical significance, while the x-axis illustrates l2FC, providing an indication of the magnitude of the difference in expression for each gene. As smaller p-values indicate greater significance, high points on the y-axis represent genes with statistically significant differences. Similarly, extreme values of l2FC on the x-axis indicate marked differences in gene expression. Thus, genes located far from the origin in both directions are the ones showing differences in expression both significant and biologically relevant. It is crucial to note that, although volcano plots provide an indication of statistical significance (via the y-axis) and magnitude of the differences (via the x-axis), statistical significance does not necessarily mean biological importance. Therefore, it is essential to interpret the results in the context of the overall study and research objectives, to determine which changes in gene expression are truly biologically relevant [35].
Fig. 2.
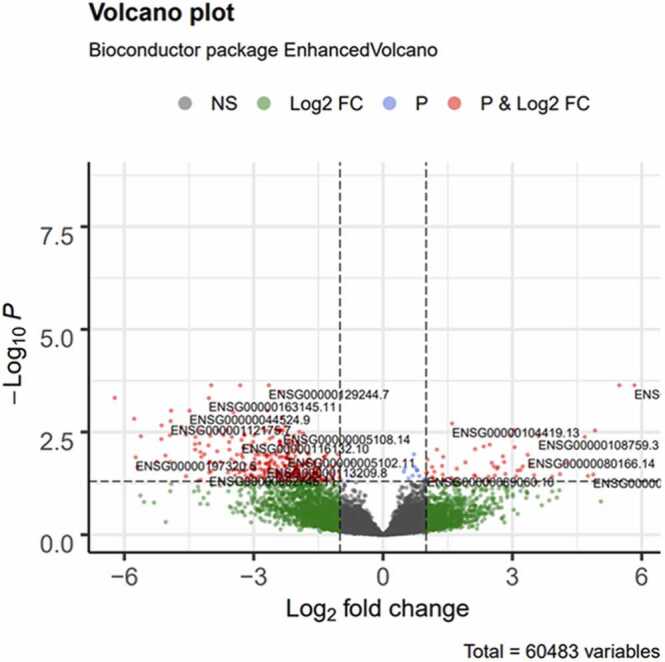
Volcano plot. Volcano plot generated by DESeq2 R.
When conducting hypothesis-driven studies, the identification of DEGs can be crucial for detecting genotypic differences between different sets of samples. Graphical visualisation of the resulting data can greatly facilitate the analysis and interpretation of these differences [85].
Lim, SH. et al. in 2021 [86]analyzed by DGE analysis changes in eyelid and buccal microbiomes between patients receiving long-term prostaglandin analogues for open-angle glaucoma (PG-OAG) and OAG-naive (Open-Angle Glaucoma patients without treatment) patients. The MA and volcano diagrams demonstrated that the PG-OAG group's relative abundances of a particular eyelid microbiome species were different from those of the naive-OAG group. When considering the eyelid microbiome of PG-OAG vs. OAG naïve, the MA graph specifically demonstrated the variance in measurements between the two samples by converting the data to M- and A-scale. The volcano plot of the PG-OAG patients revealed that Azomonas, Pseudomonas, and Granulicatella were abundant, while Delftia and Rothia were diminished when compared to OAG-naïve patients [86].
2.4.2. Heatmaps and Venn's diagrams
The aim of DGE analyses, is the identification of genes showing significant differences in expression levels between two or more groups. The number of DEGs between these groups provides a metric for assessing the extent of gene expression changes [31].
Heatmaps and Venn diagrams are visualization tools commonly used to display the results of DGE analysis [45], [46]. Heatmaps use color scales to map data values and create a grid with variables and observations along the two axes [31]. However, it is crucial to carefully use heatmaps. Indeed, if the heatmap is generated without proper statistical analysis and previous filtering, it could lead to incorrect or misleading interpretations instead of focusing on statistically relevant genes. Thus, researchers must be cautious when interpreting and presenting results using these graphical presentations, ensuring that they represent the most relevant and significant data [87].
On the other end, Venn diagrams represent the intersection and uniqueness of different data sets. Each circle in any Venn diagram represents a data set and the intersections between the circles show the similarities between these sets. This type of visualization is particularly useful to highlight similarities or differences between several groups or conditions concerning gene expression [46]. Yang Z. et al. in 2018 [88]analyzed exosomal miRNA profiles of SAT (Subcutaneous Adipose Tissue) and VAT (Visceral Adipose Tissues) from obese and lean patients. They discovered 10 exosome-derived DE-miRNAs (differentially expressed miRNAs) in SATs and 58 exosomal DE-miRNAs in VATs. The detected DE-miRNAs in SAT and VAT were different between patients with obesity and lean patients, according to heatmaps produced by the commonly used R package's 'pheatmap()' [89] function. One common exosomal DE-miRNA between SAT and VAT was specifically identified by Venn diagram analysis, but the remaining nine DE-mRNAs in SAT and 57 DE-miRNAs in VAT were tissue-specific. Utilizing the Venn diagram online tool, Lv X. et al. [90] compared two sets of differentially expressed genes and discovered 755 overlapped DEGs, comprising 590 up-regulated genes and 165 down-regulated genes. By using a Venn diagram to intersect the results of two separate groups of up- and down-regulated DEGs, Chen C. et al. (2022) [63]were able to produce a set of DEGs that contained five down-regulated genes with varying degrees of vascular calcification (VC) and nine up-regulated genes with varying degrees of VC.
3. Pathway enrichment analysis
Once differentially expressed genes are identified between two sets of samples, a tool called pathway enrichment analysis can be then used [91].
Pathway enrichment analysis is a computational tool to identify biological pathways or pathways significantly enriched in differentially expressed genes/proteins, or associated with a defined set of samples and/or diseases.
Pathway enrichment analysis [92] serves multiple purposes for biomarker identification and validation. First, it helps in pinpointing potential biomarkers by highlighting genes overrepresented in specific biological pathways [93]. It then helps in validating the biological significance of these candidate genes by analyzing their roles within enriched pathways and their interactions at protein level [94], [95]. In addition, this analysis links gene changes to relevant diseases, suggesting their potential as disease markers and insights into therapeutic targets [96], [97]. Finally, it supports the validation and refinement of biomarkers, ensuring their clinical relevance and accuracy [98], [99], [100]. For instance, Chen, G. et al 2021 performed pathway enrichment to investigate the biological function of DEGs in breast cancer. Up-regulated DEGs were significantly enriched in pathways such as p53 signaling, progesterone-mediated oocyte maturation, protein degradation, and uptake, and down-regulated DEGs were mostly enriched in AMPK, adipocytokine, and PPAR signaling pathways. They identified 12 genes correlated with prognosis of breast cancer patients using a protein-protein interaction (PPI) network analysis. Based on their results, the authors concluded that the identified pathways and genes play important roles in the development and prognosis of breast cancer. These findings provided valuable insights into the molecular mechanisms underlying breast cancer and contributed to the identification of potential prognostic biomarkers and therapeutic targets for breast cancer [98].
The construction of pathway enrichment networks is a key step in understanding the biology of complex systems and allows the identification of biological pathways involved in specific functions, such as regulation of apoptosis or cell signaling [101].
However, it is important to emphasize that the success of pathway enrichment analysis depends largely on the background distribution used in the analysis, i.e. the complete set of genes considered when determining pathway enrichment [101]. If the background is not representative or if it contains bias due to selection procedures, it could lead to incorrect conclusions. Otherwise, the analysis allows a complete and integrated view of biological functions and pathological mechanisms, providing insights for developing new therapies or diagnostic strategies [102].
Given its capability to consolidate large, high-dimensional datasets into key biological pathways, pathway enrichment analysis has become one of the main methodologies for analyzing and interpreting biological data [101].
In this respect, both databases and tools are key resources for facilitating and optimizing pathway enrichments.
3.1. Databases
WikiPathways [103] KEGG (Kyoto Encyclopaedia of Genes and Genomes) [104], GO (Gene Ontology) [105], Reactome [106], STRING (Search Tool for the Retrieval of Interacting Genes/Proteins) [107], Panther Pathways [108], [109], Biocarta [110], and HumanCyc [111] are some of the most widely used databases, which provide information on metabolic pathways, biological processes, and protein interactions useful to analyze gene expression data and identify pathways and biological functions associated with genes of interest [112] (Table 3).
Table 3.
Main bioinformatic databases for the analysis of biological pathways.
| Database | Description | URL |
|---|---|---|
| WikiPathways | An open science platform for the analysis of biological pathways developed, updated, and used by the research community. It allows users to collaboratively create, edit, and visualize biological pathways. WikiPathways comes with several features, including a zoomable pathway viewer and support for annotations of pathway ontologies. The content of the platform is available for free and has been adopted by external databases and tools. WikiPathways is also used as the base for centrally maintained databases such as Reactome [103]. | WikiPathways |
| KEGG | A manually curated database resource and one of the most established databases of biological pathways and genomic annotations. It provides a representation of metabolic and signaling pathways and gene-protein interactions and contains manually curated pathways based on scientific literature [104], [116], [117], [118]. It also provides an application programming interface (API) for data integration[119]. | KEGG |
| GO | Gene Ontology provides a unified annotation system to describe the roles of genes and proteins in any organism. It has three main categories: Biological Processes, Cellular Components and Molecular Functions. It is an important bioinformatic resource for the functional annotation of genes and proteins and is widely used to interpret gene expression and proteomic datasets. The GO is collaboratively maintained by several database projects such as UniProt and NCBI, providing a common language for describing gene and protein characteristics across different species. GO also contributes to powerful analysis tools for functional enrichment, allowing researchers to identify and better understand biological patterns [105]. | GO |
| Reactome | A manually curated biological pathway database, which provides information on more than 2000 human biological pathways. It also provides pathway enrichment analysis based on gene expression data [106], [120]. | Reactome |
| STRING | The STRING database contains all known and predicted protein-protein interactions, including both functional (proteins involved in the same biological processes or pathways) and physical interactions, annotated for different species. It also provides clustering and pathway enrichment analysis based on protein-protein interaction data and allow data visualization [107], [121]. It collects and scores evidence from a variety of sources, including automated text mining of scientific literature, databases of interaction experiments and annotated complexes/pathways, computational predictions of interactions based on co-expression and genomic context, and systematic transfers of evidence of interactions from one organism to another [115]. | STRING |
| Panther Pathways | A manually curated database of more than 1962 genes and 145 pathways. These pathways are divided into groups based on structural or functional annotations, which include metabolic, signaling, biosynthesis, and degradation processes. Literature curation and connection with curated pathways are used to quantify the relationships between proteins and pathways. Moreover, Panther Pathways offers visualizations of attribute and gene similarity [108], [109], [122], [123]. | Panther Pathways |
| Biocarta Pathways | 1396 genes, 254 pathways, and 4417 protein-pathway connections are present in this database. A valuable resource for experts in the field of biology and medical research. This site is constantly manually updated and provides in-depth details on pathways involved in important biological processes, including immune response, bone remodelling, gene regulation, apoptosis, and cell cycle regulation. The availability of detailed information on these biological pathways helps the understanding of the molecular mechanisms regulating these processes [110]. | Biocarta Pathways |
| HumanCyc | A database containing information about human metabolic processes, enzymes, and pathways. It provides a comprehensive collection of enzymatic reactions and metabolic pathways. It is a versatile reference resource for analyzing omics data. The database assigns enzymes to predicted metabolic pathways, placing genes in their biological context, thereby enabling measurement of metabolism. It contains 2709 human enzymes assigned to 896 bioreactions, with 622 enzymes assigned roles in 135 predicted metabolic pathways that closely match known human nutritional requirements. It allows analysis of gene expression, proteomics, and metabolomics data through an Omics Viewer. The database helps assess causal conclusions regarding the consequences of mutations, treatments, and modifications of gene regulation [111]. | HumanCyc |
Lv et al. [90] used DAVID [113] to perform enrichment analysis via GO [114] and KEGG [104] to highlight the biological functions of 755 DEGs. This analysis revealed significant enrichment of DEGs in several biological processes, including mitotic nuclear division, sister chromatid cohesion, and cell division. These findings provide valuable information on the specific cellular functions and processes that are influenced by DEGs. In addition, using the KEGG database analysis, the authors found that the most enriched pathways were PPAR and AMPK signaling, and oocyte meiosis pathways. Lastly, the authors used the STRING database [115] to map protein interactions between overlapping DEGs. The STRING database provides information on known or predicted interactions between proteins. The authors identified a total of 148 nodes (proteins) and 477 edges (protein interactions) using the STRING database. These nodes and edges represent the protein interactions corresponding to the overlapping DEGs in their study. In addition, the authors applied an enrichment test based on protein-protein interactions (PPI) using an enrichment p-value of less than 1.0e-16. This p-value indicates a high degree of statistical significance in the enrichment of protein interactions between overlapping DEGs. Overall, the use of the STRING database allowed the authors to visualize protein interactions between DEGs, providing information on protein networks and interconnections between proteins involved in the biological processes studied [115].
Overall, each database has differences and peculiarities and the choice of pathway database depends on the specific research question and the nature of the data [124] (Table 4).
Table 4.
Advantages, disadvantages, and context for the main bioinformatic databases for analyzing biological pathways.
| Database | Advantages | Disadvantages | Context of Use |
|---|---|---|---|
| WikiPathways [103] | Community-curated content; intuitive user interface; regular updates; open platform. | Limited coverage of less common species in research. | Useful for collaborative research and pathway analysis, particularly suitable for systems biology and omics studies. |
| KEGG [116], [117] | Extensive integrated data collection; detailed pathway information; analysis and mapping tools; broad species coverage. | Limited access to advanced features for non-registered users; interface not very intuitive. | Extensive use in bioinformatics for pathway analysis and genomic research, metabolomics and multi-omics data integration; used in a wide range of biological and medical studies. |
| GO [105] | Annotation of genes and proteins in many species; extensive integration with numerous other bioinformatic tools; tripartite structure covering biological processes, cellular components, and molecular functions. | Requires understanding of specific ontological categories for effective use; may not cover all possible gene functions, especially in less-studied species. | Used in a wide range of bioinformatic studies for functional annotation; particularly useful to identify underlying biological mechanisms; applied in comparative and functional studies across different species. |
| Reactome [120] | High-quality curated data; good data integration and visualization; biological event-oriented approach. | Mainly human-centred coverage; less detail for other species. | Preferred for studies on biological pathways and processes, especially in humans. |
| STRING [107] | Extensive database of protein interactions; integration of different data sources; user-friendly interface. | Possible false positives in predicted interactions; require experimental validation. | Used for the analysis of protein interaction networks and for proteomics studies. |
| Panther Pathways [122] | Integration of functional and evolutionary data; based on standardized ontologies; use of familiar gene models. | Limited coverage compared to other databases; more recent data may be missing. | Suitable for evolutionary and functional analyses of genes and proteins. |
| Biocarta [110] | Specificity in pathway detail; curated data. | Not frequently updated; limited coverage. | Used for detailed analysis of specific biological pathways, especially in biomedical studies. |
| HumanCyc [111] | Dedicated to human biochemistry; detailed in metabolic pathways; based on curated data. | Focused exclusively on humans; requires an understanding of biochemistry for effective use. | Used for the in-depth study of human metabolic pathways, particularly useful in biochemical and metabolomics research. |
3.2. Tools
Enrich-r [125], DAVID (Database for Annotation, Visualisation, and Integrated Discovery) [126], Metascape [127], GSEA (Gene Set Enrichment Analysis) [128], GSVA (Gene Set Variation Analysis) [129] and Cytoscape [130] are the most widely used bioinformatic analysis tools to identify biological pathways involved in any specific biological response or disease [131]. These tools (Table 5) offer a variety of functionalities ranging from pathway enrichment analysis, annotation and analysis of gene lists, visualization and analysis of biological networks, and examination of hierarchical relationships in terms of Gene Ontology.
Table 5.
Main bioinformatic tools for the analysis and visualisation of biological pathways and networks.
| Tool | Description | URL |
|---|---|---|
| Enrich-r | Web-based open-source bioinformatic analysis tool, which allows pathway enrichment analysis using over 100 publicly available databases. It also provides an intuitive user interface and offers many options for presenting results with precise statistical evidence [125]. It does not support the analysis of protein-protein interaction networks and is restricted to a single type of analysis and therefore not appropriate for more complex analysis [125]. | Enrich-r |
| DAVID | A bioinformatic analysis web-based tool integrating different sources of gene annotation and biological pathways, offering similar capabilities to Enrich-r, but with more user-friendly interfaces. It also provides clustering analysis based on gene and protein expression data [126]. | DAVID |
| Metascape | It offers a complete resource for gene list annotation and analysis. Metascape integrates functional enrichment, interactome analysis, gene annotation, and membership search to interrogate more than 40 separate knowledge bases in any single integrated gateway. Furthermore, despite the slow loading of results, it facilitates the comparison of data sets from many independent and orthogonal studies [127]. | Metascape |
| GSEA | Gene Set Enrichment Analysis (GSEA) is a tool used to determine whether a predefined set of genes shows statistically significant differences in gene expression between two biological conditions [138]. It is particularly useful for large-scale studies, such as analysis of high-throughput gene expression data [128], [139]. GSEA offers detailed analysis based on lists of genes sorted by p-value, fold change, or both, rather than on individual genes, allowing a comprehensive biological interpretation of the data [140]. GSEA software, available for free download, is accompanied by MSigDB, a collection of annotated gene sets. Although starting with the web version of the GSEA is more straightforward, downloading the software and MSigDB offers more flexibility for customized analyses [138] | GSEA |
| GSVA | A new gene enrichment analysis method for estimating variations in pathway activity in an unsupervised manner over a population of samples. GSVA is more robust and flexible than currently available sample enrichment methods for both microarray and RNA-seq data. It provides a more powerful analysis to detect even small variations in pathway activity and contributes to the need for enrichment analysis methods for RNA-seq data. It is a Bioconductor project component and an open-source R software package. A gene-expression matrix can be changed using the GSVA technique. It transforms a matrix in which each row denotes a gene and each column a sample into a matrix in which each row denotes a group of genes and each column a sample. This transformation allows for pathway-centric analysis, which focuses on analyzing groups of functionally related genes [129]. GSVA, like GSEA, has a user interface that requires expert users [128], [129], [139], [141]. | GSVA |
| Cytoscape | An open-source visualization and analysis software for biological networks with a wide range of functionality for creating biological networks, including tools for importing data, visualizing networks in different layout modes, and manipulating nodes and arcs [142]. In addition, it offers a wide range of plugins and analysis tools to perform various biological network analysis tasks, such as identifying network modules, analyzing pathways, and predicting protein-protein interactions [143], [144]. It is used in many applications, including biomarker identification, prediction of protein-protein interactions, studies on complex diseases, and drug design. | Cytoscape |
In Cytoscape, after constructing interaction networks of gene expression data, centrality metrics can be then used to identify hub genes within the network. Notably, Cytoscape's 'Correspondence' plugin offers advanced tools to examine these metrics. Centrality metrics include degree, betweenness, and proximity. The degree is the number of connections a gene has with other genes in the network, while betweenness indicates how often a gene is on the shortest path between two different genes in the network. Proximity, on the other hand, measures how close a gene is to other genes in the network. In this way, it is possible to identify genes that play a central role in gene regulation and biological processes [132], [133].
ClueGO is another example of Cytoscape plug-in [134] to visualize non-redundant biological keywords for large gene clusters in networks grouped by function. The plug-in uses kappa statistics to create a ClueGO network and reflects the relationships between keywords based on the similarity of associated genes. ClueGO can be updated with the latest files from Gene Ontology, KEGG, WikiPathways, and Reactome. In addition, the plug-in performs single cluster analysis and comparisons between several gene clusters. ClueGO has REST-enabled functions, which allows integration into analytical pipelines. New features include a new option for customized reference sets and ability to display lists of GO terms from other enrichment analyses. ClueGO facilitates the biological interpretation of large gene or protein lists by selecting representative terms from Gene Ontology [135]. However, it is essential to emphasize a significant limitation of traditional enrichment analyses using GO terms. Gene Ontology is inherently hierarchical, and different branches have different depths. Consequently, the conventional approach, which focuses on specific levels of the GO tree, may lead to analysis in which generic and specific GO terms are equally considered, introducing potential ambiguities or inaccuracies in the conclusions of the analysis. More appropriate approaches, which attempt to overcome this limitation, consider the context and proximity of terms in the GO tree, ensuring a more accurate and informed representation of functional enrichment [136]. The TopGO package [136] in R provides an example of this type of approach and offers many algorithms to perform enrichment analysis of Gene Ontology terms. One of these algorithms is the elim method, which considers hierarchical relationships between GO terms in its analysis. This means that, in assessing the significance of a particular GO term, the elim method considers neighboring terms in the GO graph [137]. The elim method provides more contextualized results by considering hierarchical relationships. It provides a better understanding of how different GO terms are related to each other and how they contribute to the overall enrichment analysis. This can be particularly useful for identifying biological processes or essential functions relevant to a given dataset [136].
Pro and Cons of each specific tools are described in Table 6.
Table 6.
Advantages, disadvantages and context of bioinformatic tools for the analysis and visualisation of biological pathways and networks.
| Tool | Advantages | Disadvantages | Context of Use |
|---|---|---|---|
| Enrich-r [125] | Extensive collection of gene set libraries; visual summaries; user-friendly interface, robust for different types of enrichment analysis including transcription, pathways, ontologies, diseases/drugs, cell types, and miscellaneous. | Overestimated results with large gene sets; lack of ID conversion tools. | Ideal for rapid, interactive analysis of gene/protein sets in transcriptomics and proteomics studies. Useful for identifying pathways and functions associated with diseases, drugs or cell types. |
| DAVID [126] | Broad taxonomic coverage; up-to-date annotations; gene ID conversion; free; intuitive interface; species parameter for list upload to minimize ambiguity. | General results; based on existing data; requires familiarity with biological databases. | In-depth analyses of gene sets with emphasis on detailed annotations and molecular interactions; comparative and functional studies on different species. |
| Metascape [127] | Automatic processing and recognition of various gene identifier; auto-clustering; supports multiple flexible file formats; user-friendly interface. | Generic results; may generate too many enriched pathways; limited support for species other than human and mouse. | Complex analyses requiring the integration of multiple omics data. Excellent for studies requiring enrichment analysis and automatic gene clustering. |
| GSEA [138] | Robust analysis method sensitive to the top and bottom of the gene list; handles large gene sets; support for gene lists from different model organisms. | It requires advanced skills in bioinformatics, systems biology and statistics; computationally intensive, requiring considerable processing resources. | Preferred for studies exploring subtle differences in gene expression between groups of samples, such as comparative studies between healthy and diseased conditions. Useful in oncology and genetics research. |
| GSVA [129] | pathway-centric analysis of molecular data; supports wide range standard analytical methods (i.e. functional enrichment, survival analysis, clustering); flexibility in input formats. | It requires expertise in R, bioinformatic and statistics; it does not consider correlations between genes, leading to an increased number of false-positive gene sets. | Suitable for pathway analysis in large-scale gene expression data, such as RNA-seq and microarray studies. Useful for studies requiring differentiated expression profile analysis. |
| Cytoscape [143], [144] | Open-source; runs on all operating systems that support Java; supports an ever-growing number of apps, continuously extending its capabilities and applications. | It requires memory and computational power for large networks; complex analyses may require additional tools (e.g. R/igraph). | Analysis of complex interactions in biological systems (e.g. signalling pathways, protein-protein interactions and gene relationships); integration of multi-omics data; translational research (e.g. understanding the molecular networks involved in various diseases, including cancer and genetic diseases). |
These bioinformatic tools are crucial for determining prospective biomarkers and therapeutic targets, as well as comprehending the functional implications of variations in gene expression [131]. Casarrubios et al. [66] performed a functional enrichment analysis using GSEA to characterize upregulated pathways in each pathological response group, revealing upregulation of pathways related to TCR co-expression, lymphocyte infiltrate (CCL21, CXCR4, GZMK, CD52, IL7R, LAMP3 and PTPRC), type II interferon signaling, and antigen processing in NSCLC tumors with CPR (pathological complete response) to neoadjuvant chemo-immunotherapy. In contrast, they discovered an increase of tumor markers, housekeeping genes, proliferation, and PD-1 signaling pathways in non-CPR (non-complete pathological response) cancers [66].
After identifying the most significant pathways associated with DEGs, it becomes crucial to explore different bioinformatic strategies to identify potential new biomarkers. These methods are designed to identify specific genes or molecular patterns that could serve as indicators or predictors of specific disease processes, paving the way for the development of targeted therapies and personalized medicine approaches.
4. Statistical analysis and result evaluation
Survival analysis and ROC are useful analyses for assessing the ability of a biomarker to predict outcomes, such as survival or mortality [145]. Survival analysis can be used to estimate the biomarker's link to survival or mortality, while ROC curves can be used to assess the biomarker's ability to distinguish between subjects with and without the disease [146]. In summary, survival analysis and ROC curves are useful statistical tools for assessing the relationship between variables and the outcome of interest in biomedical research [145].
4.1. ROC curve analysis
ROC curve is essential for comparing two different diagnostic tasks when performed on the same subjects, determining the best diagnostic threshold values (cutoff), and assessing the ability of the diagnostic test to distinguish the status of patients and determine, for example, whether a patient is sick or healthy [147]. ROC curves are graphically represented with true positive rate (sensitivity) on the y-axis and false positive rate (1-specificity) on the x-axis, providing a visual measure of the test's ability to discriminate between two conditions under exam [148]. AUC (area under the roc curve) is a useful measure of the test's overall diagnostic efficacy, with values between 0 and 1. Various methods are available to calculate the AUC [149], including the trapezoidal rule [150], non-parametric methods [151] and those based on statistical models [152]. The advantages of ROC curve analysis include its independence from disease prevalence in the population, which means that the AUC value remains the same regardless of disease incidence [153]. Another advantage is that the AUC does not depend on specific decision criteria or choices of diagnostic thresholds [148]. In conclusion, the AUC of the ROC curve provides a comprehensive and objective measure of the discriminatory ability of a diagnostic test. Being independent of disease prevalence and specific decision criteria, the AUC provides a reliable indication of test performance and facilitates comparison between different diagnostic tests [154]. However, it is important to emphasize that evaluating the effectiveness of diagnostic tests requires an appropriate study design, which should include a representative study population, with clearly defined inclusion and exclusion criteria to ensure that the patient’s pool reflects the diversity of the general population [155]. The inclusion in the study population of a wide range of cases and controls is crucial for accurate and reliable results and should be accompanied with appropriate randomization procedures to minimize bias and confounders [156]. Finally, an approach that includes comparison with a gold standard, appropriate statistical techniques for data analysis, and an adequate sample size can provide a comprehensive assessment of the effectiveness of a diagnostic test [147], [149], [153].
Chen et al. [63] assessed the ability of hub genes to distinguish calcified vessels from normal vessels by examining the expression profiles of hub genes in normal and uremia-induced VC samples. They determined that the area under the ROC curves of Sost, Ibsp, Fn1, Col1a1, and Spp1 were all near to one in the uremia-induced VC group in GSE146638 (Gene Expression Omnibus dataset), with the normal group serving as a control, indicating that these genes can discriminate between calcified and normal arteries. Thus, this analysis demonstrated that ROC curves can be used in biomedical research to assess the ability of a biomarker to distinguish between subjects with or without a certain disease. This can help in early diagnosis or monitoring its progression.
4.2. Survival statistical analysis
Statistical survival analysis are other important tools commonly used in clinical trials designed to identify specific biomarkers with predictive or diagnostic value for molecular-targeted therapies and personalized treatments [157], [158]. Accordingly, a statistical approach, like Cox regression analysis, is crucial for the evaluation of aetiological and prognostic hypotheses. It is based on the estimation of the HR related to a given risk factor or predictor for a given endpoint [159]. The Cox approach must take into account the number of patients who had the event in question when determining the number of variables to be tested (univariate, bivariate, or multivariate) [160].
For an effective application of Cox regression analysis, it is crucial to select a study design that involves longitudinal data collection [161], such as cohort studies [162] or nested case-control studies [163]. In these studies, enrolled patients are observed over extended periods, and events of interest (such as disease occurrence or mortality) are recorded over time. In this way, it is possible to analyze how risk factors or patient characteristics influence the probability of an event over time, a key element of Cox analysis [164], [165], [166]. Furthermore, it is essential that the enrolled pool includes an adequate number of events (such as deaths or relapses) to ensure sufficient statistical power [167], [168]. Variables should be selected on the basis of their clinical relevance and their plausible association with the event of interest and should be collected in a uniform and standardized manner to minimise the risk of bias [169].
The primary presumption of the traditional Cox hazard analysis is the proportionality of the risk [170]. Erroneous conclusions can be drawn by treating variables that become more pronounced as hazard factors during follow-up or even disappear altogether over time as their association with the hazard or risk under study diminishes or becomes negligible over time [171]. Thus, Cox analysis can be used to identify risk factors and biomarkers associated with a particular disease.
When combined with ROC curves, survival analysis can provide a deeper understanding of risk factors associated with specific diseases and improve the assessment of a biomarker's ability to anticipate the desired outcome [145].
Lv et al. [90], performed a survival analysis to assess whether the prognostic values of six mRNAs were independent of clinicopathological factors after selecting hub genes from the 755 differentially expressed mRNAs in triple-negative breast cancer and identifying the relative enrichment pathways of these genes. Specifically, the univariate Cox proportional hazard regression model showed that 16 out of 755 DEGs were substantially linked to survival time (P < 0.05), and the multivariate Cox proportional hazards regression model was used to create a predictive gene signature made up of six hub genes (6-mRNA) to predict overall survival. After that, using the median risk score as cut-off point, patients were separated into low- and high-risk categories of survival. According to the expression of the 6-mRNAs and the time-dependent ROC curve [172], TNBC patients with high-risk scores showed considerably shorter survival time than those with low-risk scores (P < 0.0001) [90]. Thus, through this analysis, it was confirmed that the identified genetic patterns were effective in predicting and diagnosing the disease. These results will form the basis not only for future prognosis but also for targeted therapy in TNBC [90]. Accordingly, Casarrubios et al. [66] performed ROC curve and survival analysis to assess the ability of tumor microenvironment gene expression profiles to predict response to neoadjuvant therapy and disease-free survival in NSCLC patients. The ROC curve analysis values with the highest likelihood ratio were utilized as thresholds to classify DEGs or immune cell subsets for each sample into high or low groups in the identification of individuals at high risk of recurrence following surgery. While progression-free survival (PFS) and overall survival (OS) were examined using the Kaplan-Meier curve, the log-rank test was used to compare groups. The results showed that gene expression profiles of the tumor microenvironment could significantly predict response to neoadjuvant therapy and disease-free survival in NSCLC patients [66].
5. Case studies
Several case studies used these pipelines to identify molecular signatures as potential biomarkers.
Han et al. [53] analyzed transcriptomic profiles and molecular signatures in platinum-sensitive and platinum-resistant ovarian carcinoma patients using DESeq2 software for normalization and differential gene expression analysis. Notably, differentially expressed genes were selected by meeting the criteria of an adjusted p-value (Benjamini-Hochberg) of less than 0.05 and an absolute log2 fold change greater than 1. They identified 263 genes differentially expressed between platinum-sensitive and resistant groups. Of these genes, 98 were upregulated and 165 downregulated and were represented by heatmap and volcano plot. In addition, the authors performed GO and KEGG enrichment pathway analyses to obtain information on the biological mechanism associated with platinum resistance and demonstrated that pathways significantly enriched were related to apoptosis, cell cycle, DNA damage repair, and epithelial-to-mesenchymal transition, which is an important mechanism regulating migration, invasion, and acquisition of chemoresistance. Because genes induced or suppressed in the platinum-resistant group were interlinked, the authors further characterized them via the STRING functional protein network and identified 3 key regulators (upregulated PACSIN3 and downregulated NTS and KIAA0319) related to the response to platinum-based chemotherapy.
In Suryawanshi et al. [173], the authors investigated the placental transcriptome and analyzed gene changes and differential gene expression between different sections of the placenta. They used Differential expression analysis with DESeq2 to compare gene expression levels between different trimesters of the placenta and between fetal and maternal side sections of the placenta at the end of pregnancy. The authors considered das differentially expressed genes the one with a P -Log10 value of 5 or more and a Log2 fold change of 1 or more. The analysis identified a total of 1120 differentially expressed genes in the placenta tissues of T1 and T3 (first and third trimester of pregnancy) samples. Specifically, 411 genes were upregulated in T1 placentas, while 709 genes were upregulated in T3 placentas. However, no significant differences in gene expression were observed between fetal and maternal side sections of the placenta in the third trimester, suggesting that gene expression patterns do not spatially change in the placenta. The authors used volcano plots to visualize differentially expressed genes between two groups of samples (genes expressed in the fetal versus the maternal side of T3 and genes expressed in T1 vs T3). The 30 best genes from each comparison were then chosen based on p-value, log2FC and BaseMean. These genes were then combined into a heatmap to visually identify common or differential gene expression patterns between sample groups. Finally, for pathway enrichment analysis, the authors removed genes with a BaseMean value below 20 to select genes with a higher expression level. Then, they analysed these genes for GO term enrichment using Enrichr, revealing that the biological processes enriched in T1 were related to cell division and proliferation, while in T3 were related to development and regulation of the vasculature. Thus, researchers can understand the molecular mechanisms underlying these disorders and potentially develop new diagnostic or therapeutic approaches by understanding the transcriptional changes that occur during different stages of pregnancy.
Chen et al. [174] used RNA-seq data from 169 glioblastoma multiforme (GBM) samples and RNA-seq data from 5 normal brain tissue samples downloaded from The Cancer Genome Atlas (TCGA) (portal.gdc.cancer.gov) database [175] to identify potential key nodes and molecular mechanisms associated with GBM progression [175]. They examined DEGs between GBM and normal samples using the edger package in R, with a significance cutoff of P < 0.01 and |log2 (fc)| > 4. A volcano graph was plotted to visualise the DEGs. The authors identified a total of 1483 DEGs (954 upregulated and 529 downregulated) and used the DAVID Database to analyze GO terms and enriched KEGG pathways in identified DEGs. A number of DEGs ≥ 2 and a significance cutoff of P < 0.05 were used to identify significant biological functions and signalling pathways. GO enrichment analysis revealed that upregulated DEGs were involved in biological processes such as anterior/posterior pattern specification, morphogenesis of the embryonic skeletal system, sister chromatid cohesion and cell division. Downregulated DEGs were significantly associated with chemical synaptic transmission, regulation of transmembrane ion transport and the γ-aminobutyric acid signaling pathway. KEGG pathway analysis indicated that upregulated DEGs were involved in pathways such as the cell cycle and the p53 signalling pathway, whereas downregulated genes were associated with retrograde endocannabinoid signalling, GABAergic synapse and glutamatergic synapse. Next, they used the STRING database to assess the DEGs interacting partners andCytoscape's CytoHubba tool to identify the top 10 significant genes based on their degrees of connectivity, including CDK1, CENPA, GNG3, BUB1, CCNB2, KIF2C, AURKB, BIRC5, CDCA8 and BUB1B. The genes identified in the PPI network may represent potential targets for the development of new treatments for GBM.
Clancy et al. [54] wanted to identify markers of severe response to SARS-CoV-2 through secondary transcriptomic analysis of biological material derived from human blood. They used MOdular Automated Reproducible Workflow for Preprocessing and Differential Analysis of RNA-seq Data (ARMOR) [52], [176] for the preprocessing and analysis of RNA-seq data. This workflow includes steps such as clipping of sequencing adapters and low-quality regions, calculation of quality control metrics, mapping and quantification of reads in the human GRCh38 transcriptome, and calculation of differential gene expression using edgeR. The authors used for differential gene expression analysis RNA-seq data from three studies based on blood samples from patients infected with SARS-CoV-2, stratified between severe and mild disease groups based on disease metadata. After calculating differential gene expression, they obtained 7941 DEGs genes which differed between severe and mild disease. Some highly significant DEGs were ASPH, MACIR/C5orf30, DGKH, and SLC26A6, genes involved in calcium homeostasis, immune response regulation, and metabolite transport. Gene ontology terms were determined using the DAVID database resource. DEGs were then subjected to signaling pathway analysis using the Signal Pathway Impact Analysis (SPIA) algorithm [177], [178], a tool for data analysis The signaling pathways used in the analysis were derived from publicly available versions of KEGG, Reactome, Pathway Interaction Database, BioCarta and Panther. This analysis identified nine significantly affected pathways, five of which were directly related to T-cell receptor (TCR) signaling, while a sixth described a Zap70 immunological synapse, the latter inhibited during severe COVID-19. Finally, to improve the reliability of their study, they constructed an ROC curve with all RNA sequencing reads and obtained an AUC value of over 96%. In this case, the AUC represents the percentage specificity and sensitivity of host transcriptomic data to predict disease severity, thereby indicating that the host transcriptional response strongly reflects disease severity.
6. Limitations
This review has highlighted the effectiveness of bioinformatics, computational biology, and statistical techniques in identifying biomarkers with prognostic and diagnostic values. However, significant challenges and limitations exist in these approaches.
Although RNA-Seq is a powerful technology for analyzing gene expression, it has limitations due to variability introduced during library preparation and sequencing, which can lead to differences in gene expression estimates between replicates [179]. This variability can be mitigated through appropriate normalization methods that correct for library size and reduce technical bias [180].
EdgeR and DESeq2, as highlighted in this review, are formidable tools for analyzing RNA-seq data butach comes with its own set of advantages and limitations, which affect theselection process based on the specific needs of the study andfactors like sample size, biological variability, and the underlying assumptions of the data distribution.
Pathway enrichment analysis is are a critical step following the identification of differentially expressed genes. This is a useful tool for identifying biological pathways involved in a particular biological function or disease. This review highlight various databases and tools that facilitate this analysis, highlighting their functionalities, the contexts in which they are most useful and limitations [181]. In particular, attention must be paid to the reliability of annotations [106], biological interpretation [182], and choice of analysis model [183]. Pathway databases and annotation resources are curated by experts, but some annotations might be outdated, while others might lack experimental validation. Using unreliable or incomplete annotations for pathway enrichment can lead to misleading results or omit key pathways relevant to the biological question posed [106]. While computational tools can identify statistically enriched pathways, the biological relevance of these pathways can sometimes be not clear. Using complementary data or experimental validation, careful interpretation can help in discerning the truly biologically relevant pathways from false positives [182]. Finally, different pathway enrichment methods and tools use various models and algorithms and some might be more suited for a particular data type or biological question than others. The choice of model can significantly influence the pathways identified, and no single model is universally optimal for all datasets or research questions [183].
We have also covered statistical analysis and evaluation of results, key to identifying potential biomarkers and to provide insights into their efficacy in distinguishing between different disease stages. Cox analysis and ROC curves are useful statistical techniques in biomedical research, but they must be used with caution and their limitations must be considered. Careful selection of patients [184], reduction of biological variability [185] and avoidance of overfitting [186] can help to ensure the reliability of the results of Cox analysis and ROC curves [187], [188]. The selection bias occurs when there is a systematic difference between those who were included in a study and those who were not [189]. Improper or biased patient selection can lead to potential confounding and inherent biological variability can introduce noise in the analysis. Thus, minimizing this variability, by focusing on homogeneous patient groups or using consistent measurement techniques, can improve the accuracy and reliability of the analysis. A further limitation may lie in the overfitting occurring when a statistical model captures not only the underlying patterns of data but also random noise. When applied to new data, an overfitted model is likely to perform poorly. In the context of Cox analysis, this may mean the inclusion of too many variables in the model. For ROC curves, overfitting can artificially inflate the area under the curve, leading to over-optimistic performance metrics. It is crucial to be aware of this risk and to take preventive measures. To avoid overfitting, regularisation techniques such as ridge regression [190] or LASSO (Least Absolute Shrinkage and Selection Operator) [191], [192] can be adopted during the model training phase. In Cox analysis regularization can help in reducing the weight of less relevant or redundant variables, limiting the complexity of the model and keeping only most influential variables [193]. For example, LASSO is effective in excluding insignificant variables by selecting an optimal subset of predictors [191], [192]. For ROC curves, regularisation can be used to prevent the model from over-fitting the training data, ensuring that the performance, as measured by the area under the curve (AUC), is realistic and reproducible on new datasets [194]. This can be achieved by implementing cross-validation techniques during the training process to evaluate the effectiveness of the model on different subsets of the dataset, ensuring that the model is well-trained and generalizable [195], [196]. Finally, continuous validation of the model on new data and critical review of the results are essential for maintaining the integrity and robustness of the analyses.
7. Conclusions
This review highlights the intricate and multifaceted nature of biomarker research, underscoring the importance of integrating bioinformatic tools and statistical analyses to comprehensively understand and utilize these tools in healthcare.
While there are challenges associated with these techniques the use of appropriate strategies to overcome methodological limitations, can produce accurate and reproducible analyses.
However, current developments in the field of computational biology and data analysis continue to enhance the accuracy and reliability of these analyses, bringing us ever closer to optimal solutions for biomarker research.
We expect future improvements in sequencing techniques and analysis algorithms to further accelerate the discovery and validation of novel biomarkers. This will have significant implications for diagnosis, prognosis, and therapy, bringing us ever closer to the idea of personalized medicine, in which treatments can be tailored to unique needs of each patient.
Ethics statement
This study was literature-based data and no ethical approval was needed.
Funding
The authors received the contribution from the University of Siena – Area ricerca, biblioteche, internazionalizzazione e terza missione, to support the open-access publication.
CRediT authorship contribution statement
Conception and design of study: Diletta Rosati, Maria Palmieri; acquisition of data: Diletta Rosati, Maria Palmieri; analysis and/or interpretation of data: Diletta Rosati, Maria Palmieri. Drafting the manuscript: Diletta Rosati, Maria Palmieri; revising the manuscript critically for important intellectual content: Elisa Frullanti, Andrea Morrione, Francesco Iannelli, Antonio Giordano. Approval of the version of the manuscript to be published (the names of all authors must be listed): Diletta Rosati, Maria Palmieri, Giulia Brunelli, Andrea Morrione, Francesco Iannelli, Elisa Frullanti, Antonio Giordano.
Author contributions
DR and MP searched databases, collected full-text papers, and wrote the paper. GB took care of the statistical part, EF, AM, FI and AG designed the review strategy and contributed to revising the article.
Declaration of Competing Interest
The authors have no conflict of interest.
Acknowledgements
The authors wish to thank the University of Siena for the contribution granted to support the open-access publication process of this study. The authors wish to thank the project “THE” - Tuscany Health ecosystem - Spoke 6, (CUP B63C22000680007) - MILESTONE n. 3: Implementation of artificial intelligence to foster precision medicine and personalized healthcare, funded by the Italian Ministry of University and Research within the project NextGenerationEU - PNRR Piano Nazionale di Ripresa e Resilienza Missione 4 Componente 2 – M4C2a.
Contributor Information
Diletta Rosati, Email: diletta.rosati2@unisi.it.
Maria Palmieri, Email: maria.palmieri@dbm.unisi.it.
Francesco Iannelli, Email: francesco.iannelli@unisi.it.
Elisa Frullanti, Email: elisa.frullanti@dbm.unisi.it.
Antonio Giordano, Email: antonio.giordano@temple.edu.
References
- 1.Dhillon A., Singh A., Bhalla V.K. A systematic review on biomarker identification for cancer diagnosis and prognosis in multi-omics: from computational needs to machine learning and deep learning. Arch Comput Methods Eng. 2023;30(2) [Google Scholar]
- 2.Biomarkers Definitions Working Group Biomarkers and surrogate endpoints: preferred definitions and conceptual framework. Clin Pharmacol Ther. 2001;69(3):89–95. doi: 10.1067/mcp.2001.113989. [DOI] [PubMed] [Google Scholar]
- 3.Ottenhoff T.H.M., Ellner J.J., Kaufmann S.H.E. Ten challenges for TB biomarkers. Tuberculosis. 2012;92(SUPPL.1) doi: 10.1016/S1472-9792(12)70007-0. [DOI] [PubMed] [Google Scholar]
- 4.Jain K.K., Jain K.K. Role of biomarkers in health care. Handb Biomark. 2010:115–188. [Google Scholar]
- 5.Goodwin S., McPherson J.D., McCombie W.R. Coming of age: ten years of next-generation sequencing technologies. Nat Rev Genet. 2016;17(6):333–351. doi: 10.1038/nrg.2016.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pettini F., Visibelli A., Cicaloni V., Iovinelli D., Spiga O. Multi-omics model applied to cancer genetics. Int J Mol Sci. 2021;22(11):5751. doi: 10.3390/ijms22115751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nagalakshmi U., Wang Z., Waern K., Shou C., Raha D., Gerstein M., et al. The transcriptional landscape of the yeast genome defined by RNA sequencing. Sci (N Y, N Y ) 2008;320(5881):1344–1349. doi: 10.1126/science.1158441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marioni J.C., Mason C.E., Mane S.M., Stephens M., Gilad Y. RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome Res. 2008;18(9):1509–1517. doi: 10.1101/gr.079558.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Melouane A., Ghanemi A., Aubé S., Yoshioka M., St-Amand J. Differential gene expression analysis in ageing muscle and drug discovery perspectives. Ageing Res Rev. 2018;41:53–63. doi: 10.1016/j.arr.2017.10.006. [DOI] [PubMed] [Google Scholar]
- 10.Wu M., Shang X., Sun Y., Wu J., Liu G. Integrated analysis of lymphocyte infiltration-associated lncRNA for ovarian cancer via TCGA, GTEx and GEO datasets. PeerJ. 2020;8 doi: 10.7717/peerj.8961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Andersson A.F., Lindberg M., Jakobsson H., Bäckhed F., Nyrén P., Engstrand L. Comparative analysis of human gut microbiota by barcoded pyrosequencing. PloS One. 2008;3(7) doi: 10.1371/journal.pone.0002836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wenric S., Shemirani R. Using supervised learning methods for gene selection in RNA-Seq case-control studies. Front Genet. 2018;9(AUG) doi: 10.3389/fgene.2018.00297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Breiman L. Random forests. Mach Learn. 2001;45:5–32. [Google Scholar]
- 14.Shemirani R., Wenric S., Kenny E., Ambite J.L. EPS: automated feature selection in case-control studies using extreme pseudo-sampling. Bioinforma (Oxf, Engl) 2021;37(19):3372–3373. doi: 10.1093/bioinformatics/btab214. [DOI] [PubMed] [Google Scholar]
- 15.Liu S., Lu M., Li H., Zuo Y. Prediction of gene expression patterns with generalized linear regression model. Front Genet. 2019;10:120. doi: 10.3389/fgene.2019.00120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mortazavi A., Williams B.A., McCue K., Schaeffer L., Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5(7):621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 17.Wang L., Feng Z., Wang X., Wang X., Zhang X. DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinforma (Oxf, Engl) 2009;26(1):136–138. doi: 10.1093/bioinformatics/btp612. [DOI] [PubMed] [Google Scholar]
- 18.Finotello F., Lavezzo E., Bianco L., Barzon L., Mazzon P., Fontana P., et al. Reducing bias in RNA sequencing data: a novel approach to compute counts. BMC Bioinform. 2014;15 Suppl 1(Suppl 1):S7. doi: 10.1186/1471-2105-15-S1-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Robinson M.D., McCarthy D.J., Smyth G.K. ‘EdgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26(1) doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li H., Lovci M.T., Kwon Y.S., Rosenfeld M.G., Fu X.D., Yeo G.W. Determination of tag density required for digital transcriptome analysis: application to an androgen-sensitive prostate cancer model. Proc Natl Acad Sci USA. 2008;105(51):20179–20184. doi: 10.1073/pnas.0807121105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hardcastle T.J., Kelly K.A. BaySeq: empirical bayesian methods for identifying differential expression in sequence count data. BMC Bioinforma. 2010;11 doi: 10.1186/1471-2105-11-422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Anders S., Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11(10) doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhao S., Ye Z., Stanton R. Misuse of RPKM or TPM normalization when comparing across samples and sequencing protocols. RNA. 2020;26(8) doi: 10.1261/rna.074922.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tarazona S., García F., Ferrer A., Dopazo J., Conesa A. NOIseq: a RNA-seq differential expression method robust for sequencing depth biases. EMBnet J. 2012;17:18–19. [Google Scholar]
- 25.Li J., Witten D.M., Johnstone I.M., Tibshirani R. Normalization, testing, and false discovery rate estimation for RNA-sequencing data. Biostat (Oxf, Engl) 2012;13(3):523–538. doi: 10.1093/biostatistics/kxr031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li J., Tibshirani R. Finding consistent patterns: a nonparametric approach for identifying differential expression in RNA-Seq data. Stat Methods Med Res. 2013;22(5):519–536. doi: 10.1177/0962280211428386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Leng N., Dawson J.A., Thomson J.A., Ruotti V., Rissman A.I., Smits B.M., et al. EBSeq: an empirical Bayes hierarchical model for inference in RNA-seq experiments. Bioinformatics. 2013;29(8):1035–1043. doi: 10.1093/bioinformatics/btt087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-Seq data with DESeq2. Genome Biol. 2014;15(12) doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ritchie M.E., Phipson B., Wu D., Hu Y., Law C.W., Shi W., et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7) doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pimentel H., Bray N.L., Puente S., Melsted P., Pachter L. Differential analysis of RNA-seq incorporating quantification uncertainty. Nat Methods. 2017;14(7):687–690. doi: 10.1038/nmeth.4324. [DOI] [PubMed] [Google Scholar]
- 31.Liu S., Wang Z., Zhu R., Wang F., Cheng Y., Liu Y. Three differential expression analysis methods for RNA sequencing: limma, EdgeR, DESeq2. J Vis Exp JoVE. 2021;(175) doi: 10.3791/62528. [DOI] [PubMed] [Google Scholar]
- 32.Udhaya Kumar S., Thirumal Kumar D., Bithia R., Sankar S., Magesh R., Sidenna M., et al. Analysis of differentially expressed genes and molecular pathways in familial hypercholesterolemia involved in atherosclerosis: a systematic and bioinformatics approach. Front Genet. 2020;11:734. doi: 10.3389/fgene.2020.00734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Costa-Silva J., Domingues D.S., Menotti D., Hungria M., Lopes F.M. Temporal progress of gene expression analysis with RNA-Seq data: a review on the relationship between computational methods. Comput Struct Biotechnol J. 2023;21:86–98. doi: 10.1016/j.csbj.2022.11.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kebschull M., Fittler M.J., Demmer R.T., Papapanou P.N. Differential expression and functional analysis of high-throughput -omics data using open source tools. Methods Mol Biol (Clifton, N J ) 2017;1537:327–345. doi: 10.1007/978-1-4939-6685-1_19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McDermaid A., Monier B., Zhao J., Liu B., Ma Q. Interpretation of differential gene expression results of RNA-seq data: review and integration. Brief Bioinforma. 2019;20(6):2044–2054. doi: 10.1093/bib/bby067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Singh K.P., Miaskowski C., Dhruva A.A., Flowers E., Kober K.M. Mechanisms and measurement of changes in gene expression. Biol Res Nurs. 2018;20(4):369–382. doi: 10.1177/1099800418772161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kakati T., Bhattacharyya D.K., Barah P., Kalita J.K. Comparison of methods for differential co-expression analysis for disease biomarker prediction. Comput Biol Med. 2019;113 doi: 10.1016/j.compbiomed.2019.103380. [DOI] [PubMed] [Google Scholar]
- 38.Wen H., Gallo R.A., Huang X., Cai J., Mei S., Farooqi A.A., et al. Incorporating differential gene expression analysis with predictive biomarkers to identify novel therapeutic drugs for fuchs endothelial corneal dystrophy. J Ophthalmol. 2021;2021:5580595. doi: 10.1155/2021/5580595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lindholm Carlström E., Niazi A., Etemadikhah M., Halvardson J., Enroth S., Stockmeier C.A., et al. Transcriptome analysis of post-mortem brain tissue reveals up-regulation of the complement cascade in a subgroup of schizophrenia patients. Genes. 2021;12(8):1242. doi: 10.3390/genes12081242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Soneson C., Delorenzi M. A comparison of methods for differential expression analysis of RNA-seq data. BMC Bioinform. 2013;14:91. doi: 10.1186/1471-2105-14-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tarazona S., Furió-Tarí P., Turrà D., Pietro A.D., Nueda M.J., Ferrer A., et al. Data quality aware analysis of differential expression in RNA-seq with NOISeq R/Bioc package. Nucleic Acids Res. 2015;43(21) doi: 10.1093/nar/gkv711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liang W., Sun F., Zhao Y., Shan L., Lou H. Identification of susceptibility modules and genes for cardiovascular disease in diabetic patients using WGCNA analysis. J Diabetes Res. 2020;2020:4178639. doi: 10.1155/2020/4178639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li Y., Liu H., Zhao Y., Yue D., Chen C., Li C., et al. Tumor-associated macrophages (TAMs)-derived osteopontin (OPN) upregulates PD-L1 expression and predicts poor prognosis in non-small cell lung cancer (NSCLC) Thorac Cancer. 2021;12(20):2698–2709. doi: 10.1111/1759-7714.14108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wodrich M.D., Sawatlon B., Busch M., Corminboeuf C. The genesis of molecular volcano plots. Acc Chem Res. 2021;54(5):1107–1117. doi: 10.1021/acs.accounts.0c00857. [DOI] [PubMed] [Google Scholar]
- 45.Yuan Y.H., Zhou J., Zhang Y., Xu M.D., Wu J., Li W., et al. Identification of key genes and pathways downstream of the β-catenin-TCF7L1 complex in pancreatic cancer cells using bioinformatics analysis. Oncol Lett. 2019;18(2):1117–1132. doi: 10.3892/ol.2019.10444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jia A., Xu L., Wang Y. Venn diagrams in bioinformatics. Brief Bioinforma. 2021;22(5):bbab108. doi: 10.1093/bib/bbab108. [DOI] [PubMed] [Google Scholar]
- 47.Rapaport F., Khanin R., Liang Y., Pirun M., Krek A., Zumbo P., et al. Comprehensive evaluation of differential gene expression analysis methods for RNA-seq data. Genome Biol. 2013;14(9):R95. doi: 10.1186/gb-2013-14-9-r95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Abbas-Aghababazadeh F., Li Q., Fridley B.L. Comparison of normalization approaches for gene expression studies completed with high-throughput sequencing. PloS One. 2018;13(10) doi: 10.1371/journal.pone.0206312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Robinson M.D., Oshlack A. A scaling normalization method for differential expression analysis of RNA-Seq data. Genome Biol. 2010;11(3) doi: 10.1186/gb-2010-11-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Evans C., Hardin J., Stoebel D.M. Selecting between-sample RNA-Seq normalization methods from the perspective of their assumptions. Brief Bioinforma. 2018;19(5) doi: 10.1093/bib/bbx008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Love, M, Anders, S, Huber, W. Analyzing RNA-seq data with DESeq2; 2023. 〈www.bioconductor.org/packages/release/bioc/vignettes/DESeq2/inst/doc/DESeq2.html〉. [DOI] [PMC free article] [PubMed]
- 52.Robinson, M., McCarthy, D. (2010) edgeR’s user guide. Bioconductor.Fhcrc.Org. 〈www.bioconductor.org/packages/devel/bioc/vignettes/edgeR/inst/doc/edgeRUsersGuide.pdf〉.
- 53.Han G.H., Shim J.E., Yun H., Kim J., Kim J.H., Cho H. RNA sequencing and bioinformatics analysis revealed PACSIN3 as a potential novel biomarker for platinum resistance in epithelial ovarian cancer. J gene Med. 2022;24(11) doi: 10.1002/jgm.3452. [DOI] [PubMed] [Google Scholar]
- 54.Clancy, J., Hoffmann, C.S., Pickett, B.E. (2023). Transcriptomics secondary analysis of severe human infection with SARS-CoV-2 identifies gene expression changes and predicts three transcriptional biomarkers in leukocytes. [DOI] [PMC free article] [PubMed]
- 55.Li D., Zand M.S., Dye T.D., Goniewicz M.L., Rahman I., Xie Z. An evaluation of RNA-seq differential analysis methods. PLoS One. 2022;17(9) doi: 10.1371/journal.pone.0264246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shahjaman M., Manir Hossain Mollah M., Rezanur Rahman M., Islam S.M.S., Nurul Haque Mollah M. Robust identification of differentially expressed genes from RNA-seq data. Genomics. 2020;112(2):2000–2010. doi: 10.1016/j.ygeno.2019.11.012. [DOI] [PubMed] [Google Scholar]
- 57.Anders S., McCarthy D.J., Chen Y., Okoniewski M., Smyth G.K., Huber W., et al. Count-based differential expression analysis of RNA sequencing data using R and Bioconductor. Nat Protoc. 2013;8(9):1765–1786. doi: 10.1038/nprot.2013.099. [DOI] [PubMed] [Google Scholar]
- 58.Robinson, M.D., et al. (2009) edgeR: Empirical analysis of digital gene expression data in R. Bioconductor. 1–6. bioconductor.org/packages/devel/bioc/manuals/edgeR/man/edgeR.pdf.
- 59.Robitzsch A. A comprehensive simulation study of estimation methods for the Rasch model. Stats. 2021;4(4) [Google Scholar]
- 60.Chen Y., Lun A.T.L., Smyth G.K. Differential expression analysis of complex RNA-Seq experiments using EdgeR. Stat Anal Gener Seq Data. 2014 [Google Scholar]
- 61.Lun A.T., Chen Y., Smyth G.K. It's DE-licious: a recipe for differential expression analyses of RNA-seq experiments using Quasi-Likelihood Methods in edgeR. Methods Mol Biol (Clifton, N J ) 2016;1418:391–416. doi: 10.1007/978-1-4939-3578-9_19. [DOI] [PubMed] [Google Scholar]
- 62.Schurch N.J., Schofield P., Gierliński M., Cole C., Sherstnev A., Singh V., et al. How many biological replicates are needed in an RNA-seq experiment and which differential expression tool should you use? RNA. 2016;22(6):839–851. doi: 10.1261/rna.053959.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chen C., Wu Y., Lu H.L., Liu K., Qin X. Identification of potential biomarkers of vascular calcification using bioinformatics analysis and validation in vivo. PeerJ. 2022;10 doi: 10.7717/peerj.13138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Stupnikov A., McInerney C.E., Savage K.I., McIntosh S.A., Emmert-Streib F., Kennedy R., et al. Robustness of differential gene expression analysis of RNA-seq. Comput Struct Biotechnol J. 2021;19:3470–3481. doi: 10.1016/j.csbj.2021.05.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mou T., Deng W., Gu F., Pawitan Y., Vu T.N. Reproducibility of methods to detect differentially expressed genes from single-cell RNA sequencing. Front Genet. 2020;10:1331. doi: 10.3389/fgene.2019.01331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Casarrubios M., Provencio M., Nadal E., Insa A., Del Rosario García-Campelo M., Lázaro-Quintela M., et al. Tumor microenvironment gene expression profiles associated to complete pathological response and disease progression in resectable NSCLC patients treated with neoadjuvant chemoimmunotherapy. J Immunother Cancer. 2022;10(9) doi: 10.1136/jitc-2022-005320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Baccarella A., Williams C.R., Parrish J.Z., Kim C.C. Empirical assessment of the impact of sample number and read depth on RNA-Seq analysis workflow performance. BMC Bioinforma. 2018;19(1):423. doi: 10.1186/s12859-018-2445-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Andrade C. The P value and statistical significance: misunderstandings, explanations, challenges, and alternatives. Indian J Psychol Med. 2019;41(3):210–215. doi: 10.4103/IJPSYM.IJPSYM_193_19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chumbley J.R., Friston K.J. False discovery rate revisited: FDR and topological inference using gaussian random fields. NeuroImage. 2009;44(1) doi: 10.1016/j.neuroimage.2008.05.021. [DOI] [PubMed] [Google Scholar]
- 70.Bonovas S., Piovani D. On p-values and statistical significance. J Clin Med. 2023;12(3):900. doi: 10.3390/jcm12030900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ji L., Chen S., Gu G., Zhou J., Wang W., Ren J., et al. Exploration of crucial mediators for carotid atherosclerosis pathogenesis through integration of microbiome, metabolome, and transcriptome. Front Physiol. 2021;12 doi: 10.3389/fphys.2021.645212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yin L., Xiao L., Gao Y., Wang G., Gao H., Peng Y., et al. Comparative bioinformatical analysis of pancreatic head cancer and pancreatic body/tail cancer. Med Oncol. 2020;37(5):46. doi: 10.1007/s12032-020-01370-0. [DOI] [PubMed] [Google Scholar]
- 73.Murray M.H., Blume J.D. FDRestimation: flexible false discovery rate computation in R. F1000Research. 2021;10:441. doi: 10.12688/f1000research.52999.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Menyhart O., Weltz B., Győrffy B. MultipleTesting.com: a tool for life science researchers for multiple hypothesis testing correction. PloS One. 2021;16(6) doi: 10.1371/journal.pone.0245824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Liu S., Abdellaoui A., Verweij K.J.H., van Wingen G.A. Gene expression has distinct associations with brain structure and function in major depressive disorder. Adv Sci. 2023;10(7) doi: 10.1002/advs.202205486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bian Z., Fan R., Xie L. A novel cuproptosis-related prognostic gene signature and validation of differential expression in clear cell renal cell carcinoma. Genes. 2022;13(5):851. doi: 10.3390/genes13050851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ghosh D. Incorporating the empirical null hypothesis into the Benjamini-Hochberg procedure. Stat Appl Genet Mol Biol. 2012;11(4) doi: 10.1515/1544-6115.1735. [DOI] [PubMed] [Google Scholar]
- 78.Tarazona S., García-Alcalde F., Dopazo J., Ferrer A., Conesa A. Differential expression in RNA-seq: a matter of depth. Genome Res. 2011;21(12):2213–2223. doi: 10.1101/gr.124321.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Souza C.S., Costa-Silva G.J., Roxo F.F., Foresti F., Oliveira C. Genetic and morphological analyses demonstrate that Schizolecis guntheri (Siluriformes: Loricariidae) is likely to be a species complex. Front Genet. 2018;9:69. doi: 10.3389/fgene.2018.00069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Dalman M.R., Deeter A., Nimishakavi G., Duan Z.H. Fold change and p-value cutoffs significantly alter microarray interpretations. BMC Bioinforma. 2012;13(Suppl 2(Suppl 2):S11. doi: 10.1186/1471-2105-13-S2-S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Costa-Silva J., Domingues D., Lopes F.M. RNA-Seq differential expression analysis: an extended review and a software tool. PloS One. 2017;12(12) doi: 10.1371/journal.pone.0190152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Farahbod M., Pavlidis P. Differential coexpression in human tissues and the confounding effect of mean expression levels. Bioinformtics. 2019;35(1):55–61. doi: 10.1093/bioinformatics/bty538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhao T., Wang Z. GraphBio: a Shiny Web App to easily perform popular visualization analysis for omics data. Front Genet. 2022;13 doi: 10.3389/fgene.2022.957317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gentleman R.C., Carey V.J., Bates D.M., Bolstad B., Dettling M., Dudoit S., et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5(10):R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Trapnell C., Williams B.A., Pertea G., Mortazavi A., Kwan G., van Baren M.J., et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28(5):511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lim S.H., Shin J.H., Lee J.W., Lee Y., Seo J.H. Differences in the eyelid and buccal microbiome of glaucoma patients receiving long-term administration of prostaglandin analog drops. Graefe'S Arch Clin Exp Ophthalmol = Albrecht Von Graefes Arch fur Klin und Exp Ophthalmol. 2021;259(10):3055–3065. doi: 10.1007/s00417-021-05218-9. [DOI] [PubMed] [Google Scholar]
- 87.Zhao Z., Yang H., Ji G., Su S., Fan Y., Wang M., et al. Identification of hub genes for early detection of bone metastasis in breast cancer. Front Endocrinol. 2022;13:1018639. doi: 10.3389/fendo.2022.1018639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yang Z., Wei Z., Wu X., Yang H. Screening of exosomal miRNAs derived from subcutaneous and visceral adipose tissues: determination of targets for the treatment of obesity and associated metabolic disorders. Mol Med Rep. 2018;18(3):3314–3324. doi: 10.3892/mmr.2018.9312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kolde, R. (2012) Package `pheatmap’. Bioconductor. Available from: 〈https://cran.r-project.org/package=pheatmap〉.
- 90.Lv X., He M., Zhao Y., Zhang L., Zhu W., Jiang L., et al. Identification of potential key genes and pathways predicting pathogenesis and prognosis for triple-negative breast cancer. Cancer Cell Int. 2019;19:172. doi: 10.1186/s12935-019-0884-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ma J., Shojaie A., Michailidis G. A comparative study of topology-based pathway enrichment analysis methods. BMC Bioinform. 2019;20(1) doi: 10.1186/s12859-019-3146-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Mujalli A., Banaganapalli B., Alrayes N.M., Shaik N.A., Elango R., Al-Aama J.Y. Myocardial infarction biomarker discovery with integrated gene expression, pathways and biological networks analysis. Genomics. 2020;112(6):5072–5085. doi: 10.1016/j.ygeno.2020.09.004. [DOI] [PubMed] [Google Scholar]
- 93.Siavoshi A., Taghizadeh M., Dookhe E., Piran M. Gene expression profiles and pathway enrichment analysis to identification of differentially expressed gene and signaling pathways in epithelial ovarian cancer based on high-throughput RNA-seq data. Genomics. 2022;114(1):161–170. doi: 10.1016/j.ygeno.2021.11.031. [DOI] [PubMed] [Google Scholar]
- 94.Ni M., Liu X., Wu J., Zhang D., Tian J., Wang T., et al. Identification of candidate biomarkers correlated with the pathogenesis and prognosis of non-small cell lung cancer via integrated bioinformatics analysis. Front Genet. 2018;9:469. doi: 10.3389/fgene.2018.00469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Li H., Zhou J., Zhou L., Zhang X., Shang J., Feng X., et al. Identification of the shared gene signatures and molecular pathways in systemic lupus erythematosus and diffuse large B-cell lymphoma. J Gene Med. 2023;25(12) doi: 10.1002/jgm.3558. [DOI] [PubMed] [Google Scholar]
- 96.Ouyang Y., Yin J., Wang W., Shi H., Shi Y., Xu B., et al. Downregulated gene expression spectrum and immune responses changed during the disease progression in patients With COVID-19. Clin Infect Dis Publ Infect Dis Soc Am. 2020;71(16):2052–2060. doi: 10.1093/cid/ciaa462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Rahman M.R., Islam T., Zaman T., Shahjaman M., Karim M.R., Huq F., et al. Identification of molecular signatures and pathways to identify novel therapeutic targets in Alzheimer's disease: insights from a systems biomedicine perspective. Genomics. 2020;112(2):1290–1299. doi: 10.1016/j.ygeno.2019.07.018. [DOI] [PubMed] [Google Scholar]
- 98.Chen G., Yu M., Cao J., Zhao H., Dai Y., Cong Y., et al. Identification of candidate biomarkers correlated with poor prognosis of breast cancer based on bioinformatics analysis. Bioengineered. 2021;12(1):5149–5161. doi: 10.1080/21655979.2021.1960775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Bansal R., Saxena U. Integrative analysis of potential biomarkers involved in the progression of papillary thyroid cancer. Appl Biochem Biotechnol. 2023;195(5):2917–2932. doi: 10.1007/s12010-022-04244-5. [DOI] [PubMed] [Google Scholar]
- 100.Fang Y., Zhan X. Identification of biomarkers associated with the prognoses of colorectal cancer patients. Digestion. 2023;104(2):148–162. doi: 10.1159/000528084. [DOI] [PubMed] [Google Scholar]
- 101.Reimand J., Isserlin R., Voisin V., Kucera M., Tannus-Lopes C., Rostamianfar A., et al. Pathway enrichment analysis and visualization of omics data using g:Profiler, GSEA, Cytoscape and EnrichmentMap. Nat Protoc. 2019;14(2):482–517. doi: 10.1038/s41596-018-0103-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Merico D., Isserlin R., Stueker O., Emili A., Bader G.D. Enrichment map: a network-based method for gene-set enrichment visualization and interpretation. PloS One. 2010;5(11) doi: 10.1371/journal.pone.0013984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Martens M., Ammar A., Riutta A., Waagmeester A., Slenter D.N., Hanspers K., et al. WikiPathways: connecting communities. Nucleic Acids Res. 2021;49(D1):D613–D621. doi: 10.1093/nar/gkaa1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kanehisa M., Furumichi M., Tanabe M., Sato Y., Morishima K. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017;45(D1):D353–D361. doi: 10.1093/nar/gkw1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Gene Ontology Consortium, Aleksander S.A., et al. The gene ontology knowledgebase in 2023. Genetics. 2023;224(1) doi: 10.1093/genetics/iyad031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Fabregat A., Jupe S., Matthews L., et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2018;46(D1):D649–D655. doi: 10.1093/nar/gkx1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Szklarczyk D., Gable A.L., et al. The STRING database in 2021: customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021;49(D1):D605–D612. doi: 10.1093/nar/gkaa1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Thomas P.D., Kejariwal A., Campbell M.J., Mi H., Diemer K., Guo N., et al. PANTHER: a browsable database of gene products organized by biological function, using curated protein family and subfamily classification. Nucleic Acids Res. 2003;31(1):334–341. doi: 10.1093/nar/gkg115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Mi H., Muruganujan A., Thomas P.D. PANTHER in 2013: modeling the evolution of gene function, and other gene attributes, in the context of phylogenetic trees. Nucleic Acids Res. 2013;41(Database issue):D377–D386. doi: 10.1093/nar/gks1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Adriaens M.E., Jaillard M., Waagmeester A., Coort S.L., Pico A.R., Evelo C.T. The public road to high-quality curated biological pathways. Drug Discov Today. 2008;13(19-20):856–862. doi: 10.1016/j.drudis.2008.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Trupp M., Altman T., Fulcher C.A., Caspi R., Krummenacker M., Paley S., et al. Beyond the genome (BTG) is a (PGDB) pathway genome database: HumanCyc. Genome Biol. 2010;11(Suppl 1):O12. [Google Scholar]
- 112.Stobbe M.D., Houten S.M., Jansen G.A., van Kampen A.H., Moerland P.D. Critical assessment of human metabolic pathway databases: a stepping stone for future integration. BMC Syst Biol. 2011;5:165. doi: 10.1186/1752-0509-5-165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Jiao X., Sherman B.T., et al. DAVID-WS: a stateful web service to facilitate gene/protein list analysis. Bioinformtics. 2012;28(13):1805–1806. doi: 10.1093/bioinformatics/bts251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Balakrishnan R., Harris M.A., Huntley R., Van Auken K., Cherry J.M. A guide to best practices for Gene Ontology (GO) manual annotation. Database J Biol Databases Curation. 2013;2013:bat054. doi: 10.1093/database/bat054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Mering C.V., Huynen M., Jaeggi D., Schmidt S., Bork P., Snel B. STRING: a database of predicted functional associations between proteins. Nucleic Acids Res. 2003;31(1):258–261. doi: 10.1093/nar/gkg034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kanehisa M., Furumichi M., Sato Y., Kawashima M., Ishiguro-Watanabe M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 2023;51(D1):D587–D592. doi: 10.1093/nar/gkac963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kanehisa M., Furumichi M., Sato Y., Ishiguro-Watanabe M., Tanabe M. KEGG: integrating viruses and cellular organisms. Nucleic Acids Res. 2021;49(D1):D545–D551. doi: 10.1093/nar/gkaa970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kanehisa M., Sato Y. KEGG Mapper for inferring cellular functions from protein sequences. Protein Sci a Publ Protein Soc. 2020;29(1):28–35. doi: 10.1002/pro.3711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Du J., Li M., Yuan Z., Guo M., Song J., Xie X., et al. A decision analysis model for KEGG pathway analysis. BMC Bioinforma. 2016;17 doi: 10.1186/s12859-016-1285-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Rothfels K., Milacic M., Matthews L., et al. Using the reactome database. Curr Protoc. 2023;3(4) doi: 10.1002/cpz1.722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Szklarczyk, Kirsch D., Koutrouli R., et al. The STRING database in 2023: protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 2023;51(D1):D638–D646. doi: 10.1093/nar/gkac1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Mi H., Ebert D., Muruganujan A., Mills C., Albou L.P., Mushayamaha T., et al. PANTHER version 16: a revised family classification, tree-based classification tool, enhancer regions and extensive API. Nucleic Acids Res. 2021;49(D1):D394–D403. doi: 10.1093/nar/gkaa1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Thomas P.D., Ebert D., Muruganujan A., Mushayahama T., Albou L.P., Mi H. PANTHER: Making genome-scale phylogenetics accessible to all. Protein Sci: a Publ Protein Soc. 2022;31(1):8–22. doi: 10.1002/pro.4218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Mubeen S., Hoyt C.T., Gemünd A., Hofmann-Apitius M., Fröhlich H., Domingo-Fernández D. The impact of pathway database choice on statistical enrichment analysis and predictive modeling. Front Genet. 2019;10:1203. doi: 10.3389/fgene.2019.01203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kuleshov M.V., Jones M.R., Rouillard A.D., Fernandez N.F., Duan Q., Wang Z., et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016;44(W1):W90–W97. doi: 10.1093/nar/gkw377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Sherman B.T., Hao M., Qiu J., Jiao X., Baseler M.W., Lane H.C., et al. DAVID: a web server for functional enrichment analysis and functional annotation of gene lists (2021 update) Nucleic Acids Res. 2022;50(W1):W216–W221. doi: 10.1093/nar/gkac194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Zhou Y., Zhou B., Pache L., Chang M., Khodabakhshi A.H., Tanaseichuk O., et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun. 2019;10(1):1523. doi: 10.1038/s41467-019-09234-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Shi J., Walker M. Gene Set Enrichment Analysis (GSEA) for interpreting gene expression profiles. Curr Bioinform. 2008;2(2) [Google Scholar]
- 129.Hänzelmann S., Castelo R., Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinforma. 2013;14:7. doi: 10.1186/1471-2105-14-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Singhal A., Cao S., Churas C., Pratt D., Fortunato S., Zheng F., et al. Multiscale community detection in Cytoscape. PLoS Comput Biol. 2020;16(10) doi: 10.1371/journal.pcbi.1008239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Huang daW., Sherman B.T., Lempicki R.A. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37(1):1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Liu Y., Gu H.Y., Zhu J., Niu Y.M., Zhang C., Guo G.L. Identification of hub genes and key pathways associated with bipolar disorder based on weighted gene co-expression network analysis. Front Physiol. 2019;10:1081. doi: 10.3389/fphys.2019.01081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Matin H., Taghian F., Chitsaz A. Artificial intelligence analysis to explore synchronize exercise, cobalamin, and magnesium as new actors to therapeutic of migraine symptoms: a randomized, placebo-controlled trial. Neurol Sci. 2022;43(7) doi: 10.1007/s10072-021-05843-6. [DOI] [PubMed] [Google Scholar]
- 134.Shannon P., Markiel A., Ozier O., Baliga N.S., Wang J.T., Ramage D., et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Mlecnik B., Galon J., Bindea G. Comprehensive functional analysis of large lists of genes and proteins. J Proteom. 2018;171:2–10. doi: 10.1016/j.jprot.2017.03.016. [DOI] [PubMed] [Google Scholar]
- 136.Rahnenfuhrer A.A.: (2023) Bioconductor – topGO. Available from: bioconductor.org/packages/release/bioc/html/topGO.html.
- 137.Alexa, A., Rahnenführer, J. (2023) Gene set enrichment analysis with topGO. Available from: bioconductor.org/packages/release/bioc/vignettes/topGO/inst/doc/topGO.pdf.
- 138.Canzler S., Hackermüller J. multiGSEA: a GSEA-based pathway enrichment analysis for multi-omics data. BMC Bioinform. 2020;21(1):561. doi: 10.1186/s12859-020-03910-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102(43):15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Reimand J., Isserlin R., Voisin V., et al. Pathway enrichment analysis and visualization of omics data using g:Profiler, GSEA, Cytoscape and EnrichmentMap. Nat Protoc. 2019;14(2):482–517. doi: 10.1038/s41596-018-0103-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Hänzelmann S., Castelo R., Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 2013;14:7. doi: 10.1186/1471-2105-14-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Saito R., Smoot M.E., Ono K., Ruscheinski J., Wang P.L., Lotia S., et al. A travel guide to Cytoscape plugins. Nat Methods. 2012;9(11):1069–1076. doi: 10.1038/nmeth.2212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Smoot M.E., Ono K., Ruscheinski J., Wang P.L., Ideker T. Cytoscape 2.8: new features for data integration and network visualization. Bioinforma (Oxf, Engl) 2011;27(3):431–432. doi: 10.1093/bioinformatics/btq675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Otasek D., Morris J.H., Bouças J., Pico A.R., Demchak B. Cytoscape automation: empowering workflow-based network analysis. Genome Biol. 2019;20(1):185. doi: 10.1186/s13059-019-1758-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.French B., Saha-Chaudhuri P., Ky B., Cappola T.P., Heagerty P.J. Development and evaluation of multi-marker risk scores for clinical prognosis. Stat Methods Med Res. 2016;25(1):255–271. doi: 10.1177/0962280212451881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Zheng Y., Cai T., Jin Y., Feng Z. Evaluating prognostic accuracy of biomarkers under competing risk. Biometrics. 2012;68(2):388–396. doi: 10.1111/j.1541-0420.2011.01671.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Polo T.C.F., Miot H.A. Use of ROC curves in clinical and experimental studies. J Vasc Bras. 2020;19 doi: 10.1590/1677-5449.200186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Verbakel J.Y., Steyerberg E.W., Uno H., De Cock B., Wynants L., Collins G.S., et al. ROC curves for clinical prediction models part 1. ROC plots showed no added value above the AUC when evaluating the performance of clinical prediction models. J Clin Epidemiol. 2020;126:207–216. doi: 10.1016/j.jclinepi.2020.01.028. [DOI] [PubMed] [Google Scholar]
- 149.Janssens A.C.J.W., Martens F.K. Reflection on modern methods: revisiting the area under the ROC Curve. Int J Epidemiol. 2020;49(4):1397–1403. doi: 10.1093/ije/dyz274. [DOI] [PubMed] [Google Scholar]
- 150.Mandrekar J.N. Receiver operating characteristic curve in diagnostic test assessment. J Thorac Oncol Publ Int Assoc Study Lung Cancer. 2010;5(9):1315–1316. doi: 10.1097/JTO.0b013e3181ec173d. [DOI] [PubMed] [Google Scholar]
- 151.Blangero Y., Rabilloud M., Laurent-Puig P., Le Malicot K., Lepage C., Ecochard R., et al. The area between ROC curves, a non-parametric method to evaluate a biomarker for patient treatment selection. Biom J Biom Z. 2020;62(6):1476–1493. doi: 10.1002/bimj.201900171. [DOI] [PubMed] [Google Scholar]
- 152.Huang, Y., Li, W., Macheret, F., Gabriel, R.A., Ohno-Machado, L.: A tutorial on calibration measurements and calibration models for clinical prediction models; (2021). [DOI] [PMC free article] [PubMed]
- 153.Nahm F.S. Receiver operating characteristic curve: overview and practical use for clinicians. Korean J Anesthesiol. 2022;75(1):25–36. doi: 10.4097/kja.21209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Yang Q., Wang S., Dai E., Zhou S., Liu D., Liu H., et al. Pathway enrichment analysis approach based on topological structure and updated annotation of pathway. Brief Bioinform. 2019;20(1):168–177. doi: 10.1093/bib/bbx091. [DOI] [PubMed] [Google Scholar]
- 155.Li B., Gatsonis C., Dahabreh I.J., Steingrimsson J.A. Estimating the area under the ROC curve when transporting a prediction model to a target population. Biometrics. 2023;79(3):2382–2393. doi: 10.1111/biom.13796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Huang Y., Pepe M.S. A parametric ROC model-based approach for evaluating the predictiveness of continuous markers in case-control studies. Biometrics. 2009;65(4):1133–1144. doi: 10.1111/j.1541-0420.2009.01201.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Zhao Q., Sun J. Cox survival analysis of microarray gene expression data using correlation principal component regression. Stat Appl Genet Mol Biol. 2007;6(1) doi: 10.2202/1544-6115.1153. [DOI] [PubMed] [Google Scholar]
- 158.Yu X., Wang T., Huang S., Zeng P. How can gene-expression information improve prognostic prediction in TCGA cancers: an empirical comparison study on regularization and mixed Cox models. Front Genet. 2020;11:920. doi: 10.3389/fgene.2020.00920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Kropko J., Harden J.J. Beyond the hazard ratio: generating expected durations from the Cox proportional hazards model. Br J Political Sci. 2020;50(1):303–320. [Google Scholar]
- 160.Abd ElHafeez S., D'Arrigo G., Leonardis D., Fusaro M., Tripepi G., Roumeliotis S. Methods to analyze time-to-event data: the Cox regression analysis. Oxid Med Cell Longev. 2021;2021:1302811. doi: 10.1155/2021/1302811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Cao J., Wang T., Li Z., Liu G., Liu Y., Zhu C., et al. Factors associated with death in bedridden patients in China: a longitudinal study. PloS One. 2020;15(1) doi: 10.1371/journal.pone.0228423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Fares A.F., Li Y., Jiang M., Brown M.C., et al. Association between duration of smoking abstinence before non-small-cell lung cancer diagnosis and survival: a retrospective, pooled analysis of cohort studies. Lancet Public Health. 2023;8(9):e691–e700. doi: 10.1016/S2468-2667(23)00131-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Nuño M.M., Gillen D.L. On estimation in the nested case-control design under nonproportional hazards. Scand J Stat. 2022;49 [Google Scholar]
- 164.Bengtsson V.W., Persson G.R., Berglund J.S., Renvert S. Periodontitis related to cardiovascular events and mortality: a long-time longitudinal study. Clin Oral Investig. 2021;25(6):4085–4095. doi: 10.1007/s00784-020-03739-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Zhang Y., Yang R., Dove A., Li X., Yang H., Li S., et al. Healthy lifestyle counteracts the risk effect of genetic factors on incident gout: a large population-based longitudinal study. BMC Med. 2022;20(1):138. doi: 10.1186/s12916-022-02341-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Luo T., Tu Y.F., Huang S., Ma Y.Y., Wang Q.H., Wang Y.J., et al. Time-dependent impact of type 2 diabetes mellitus on incident prodromal Alzheimer disease: a longitudinal study in 1395 participants. Eur J Neurol. 2023;30(9):2620–2628. doi: 10.1111/ene.15868. [DOI] [PubMed] [Google Scholar]
- 167.Abebe A., Kumela K., Belay M., Kebede B., Wobie Y. Mortality and predictors of acute kidney injury in adults: a hospital-based prospective observational study. Sci Rep. 2021;11(1):15672. doi: 10.1038/s41598-021-94946-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Riley R.D., Snell K.I.E., Martin G.P., Whittle R., Archer L., Sperrin M., et al. Penalization and shrinkage methods produced unreliable clinical prediction models especially when sample size was small. J Clin Epidemiol. 2021;132:88–96. doi: 10.1016/j.jclinepi.2020.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Chowdhury M.Z.I., Turin T.C. Variable selection strategies and its importance in clinical prediction modelling. Fam Med Community Health. 2020;8(1) doi: 10.1136/fmch-2019-000262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.De Neve J., Gerds T.A. On the interpretation of the hazard ratio in Cox regression. Biom J. 2020;62(3) doi: 10.1002/bimj.201800255. [DOI] [PubMed] [Google Scholar]
- 171.Babińska M., Chudek J., Chełmecka E., Janik M., Klimek K., Owczarek A. Limitations of Cox proportional hazards analysis in mortality prediction of patients with acute coronary syndrome. studies in logic. Gramm Rhetor. 2015;43(1):33–48. [Google Scholar]
- 172.Bansal A., Heagerty P.J. A comparison of landmark methods and time-dependent ROC methods to evaluate the time-varying performance of prognostic markers for survival outcomes. Diagn Progn Res. 2019;3(1) doi: 10.1186/s41512-019-0057-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Suryawanshi H., Max K., Bogardus K.A., Sopeyin A., Chang M.S., Morozov P., et al. Dynamic genome-wide gene expression and immune cell composition in the developing human placenta. J Reprod Immunol. 2022;151 doi: 10.1016/j.jri.2022.103624. [DOI] [PubMed] [Google Scholar]
- 174.Chen X., Pan Y., Yan M., Bao G., Sun X. Identification of potential crucial genes and molecular mechanisms in glioblastoma multiforme by bioinformatics analysis. Mol Med Rep. 2020;22(2):859–869. doi: 10.3892/mmr.2020.11160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.TCGA Research Network (2023) The Cancer Genome Atlas Program (TCGA). Available from: 〈https://www.cancer.gov/ccg/research/genome-sequencing/tcga〉.
- 176.Orjuela S., Huang R., Hembach K.M., Robinson M.D., Soneson C. ). ARMOR: an automated reproducible MOdular workflow for preprocessing and differential analysis of RNA-seq data. G3. 2019;9(7):2089–2096. doi: 10.1534/g3.119.400185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Bao, Z., Zhu, Y., Ge, Q., Gu, W., Dong, X., Bai, Y. (2020) Signaling pathway analysis combined with the strength variations of interactions between genes under different conditions.
- 178.Bao Z., Li X., Zan X., Shen L., Ma R., Liu W. Signalling pathway impact analysis based on the strength of interaction between genes. IET Syst Biol. 2016;10(4):147–152. doi: 10.1049/iet-syb.2015.0089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Bullard J.H., Purdom E., Hansen K.D., Dudoit S. Evaluation of statistical methods for normalization and differential expression in mRNA-Seq experiments. BMC Bioinforma. 2010;11:94. doi: 10.1186/1471-2105-11-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Tong L., Wu P.Y., Phan J.H., Hassazadeh H.R., SEQC Consortium, Tong W., et al. Impact of RNA-seq data analysis algorithms on gene expression estimation and downstream prediction. Sci Rep. 2020;10(1):17925. doi: 10.1038/s41598-020-74567-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Nguyen T.M., Shafi A., Nguyen T., Draghici S. Identifying significantly impacted pathways: a comprehensive review and assessment. Genome Biol. 2019;20(1):203. doi: 10.1186/s13059-019-1790-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Huang D.W., Sherman B.T., Lempicki R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1) doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 183.Tomczak A., Mortensen J.M., Winnenburg R., Liu C., Alessi D.T., Swamy V., et al. Interpretation of biological experiments changes with evolution of the Gene Ontology and its annotations. Sci Rep. 2018;8(1):5115. doi: 10.1038/s41598-018-23395-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Austin P.C., Ceyisakar I.E., Steyerberg E.W., Lingsma H.F., Marang-van de Mheen P.J. Ranking hospital performance based on individual indicators: can we increase reliability by creating composite indicators? BMC Med Res Methodol. 2019;19(1):131. doi: 10.1186/s12874-019-0769-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Lu Y., Johnston P.R., Dennis S.R., Monaghan M.T., John U., Spaak P., et al. Daphnia galeata responds to the exposure to an Ichthyosporean gut parasite by down-regulation of immunity and lipid metabolism. BMC Genom. 2018;19(1):932. doi: 10.1186/s12864-018-5312-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Ruppert D. The elements of statistical learning: data mining, inference, and prediction. J Am Stat Assoc. 2004;99(466) [Google Scholar]
- 187.Persson I., Khamis H. Bias of the Cox model hazard ratio. J Mod Appl Stat Methods. 2005;4(1) [Google Scholar]
- 188.Clark R.D., Webster-Clark D.J. Managing bias in ROC curves. J Comput-Aided Mol Des. 2008;22(3–4) doi: 10.1007/s10822-008-9181-z. [DOI] [PubMed] [Google Scholar]
- 189.Subramanian J., Simon R. Overfitting in prediction models - is it a problem only in high dimensions? Contemp Clin Trials. 2013;36(2) doi: 10.1016/j.cct.2013.06.011. [DOI] [PubMed] [Google Scholar]
- 190.van de Wiel M.A., van Nee M.M., Rauschenberger A. Fast cross-validation for multi-penalty high-dimensional ridge regression. J Comput Graph Stat. 2021;30 [Google Scholar]
- 191.Zhou D., Liu X., Wang X., Yan F., Wang P., Yan H., et al. A prognostic nomogram based on LASSO Cox regression in patients with alpha-fetoprotein-negative hepatocellular carcinoma following non-surgical therapy. BMC Cancer. 2021;21(1):246. doi: 10.1186/s12885-021-07916-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Zhang Z., Shen Z., Wang H., Ng S.K. A fast adaptive Lasso for the cox regression via safe screening rules. J Stat Comput Simul. 2021;91 [Google Scholar]
- 193.Wahid A., Khan D.M., Khan S.A., Hussain I., Khan Z. Robust regularization for high-dimensional Cox’s regression model using weighted likelihood criterion. Chemom Intell Lab Syst. 2021;213 [Google Scholar]
- 194.Fang R., Zhang R., et al. Prevent over-fitting and redundancy in physiological signal analyses for stress detection :In: Proceedings of the 2022 IEEE International Conference on Bioinformatics and Biomedicine (BIBM) 2022 [Google Scholar]
- 195.Hong F., Tian L., Devanarayan V. Improving the robustness of variable selection and predictive performance of regularized generalized linear models and Cox Proportional Hazard Models. Mathematics. 2023;11(3):557. doi: 10.3390/math11030557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Chang C.C., Chen C.H., Hsieh J.G., Jeng J.H. Iterated cross validation method for prediction of survival in diffuse large B-cell lymphoma for small size dataset. Sci Rep. 2023;13(1):1438. doi: 10.1038/s41598-023-28394-6. [DOI] [PMC free article] [PubMed] [Google Scholar]