

Abstract
Allopurinol (ALP) is a successful drug used in the treatment of gout. However, this drug has been implicated in hypersensitivity reactions that can cause severe to life‐threatening reactions such as Stevens–Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN). Individuals who carry the human leukocyte antigen (HLA)‐B*58:01 allotype are at higher risk of experiencing a hypersensitivity reaction (odds ratios ranging from 5.62 to 580.3 for mild to severe reactions, respectively). In addition to the parent drug, the metabolite oxypurinol (OXP) is implicated in triggering T cell‐mediated immunopathology via a labile interaction with HLA‐B*58:01. To date, there has been limited information regarding the T‐cell receptor (TCR) repertoire usage of reactive T cells in patients with ALP‐induced SJS or TEN and, in particular, there are no reports examining paired αβTCRs. Here, using in vitro drug‐treated PBMCs isolated from both resolved ALP‐induced SJS/TEN cases and drug‐naïve healthy donors, we show that OXP is the driver of CD8+ T cell‐mediated responses and that drug‐exposed memory T cells can exhibit a proinflammatory immunophenotype similar to T cells described during active disease. Furthermore, this response supported the pharmacological interaction with immune receptors (p‐i) concept by showcasing (i) the labile metabolite interaction with peptide/HLA complexes, (ii) immunogenic complex formation at the cell surface, and (iii) lack of requirement for antigen processing to elicit drug‐induced T cell responsiveness. Examination of paired OXP‐induced αβTCR repertoires highlighted an oligoclonal and private clonotypic profile in both resolved ALP‐induced SJS/TEN cases and drug‐naïve healthy donors.
Keywords: allopurinol, drug hypersensitivity reaction, oxypurinol, T‐cell receptor
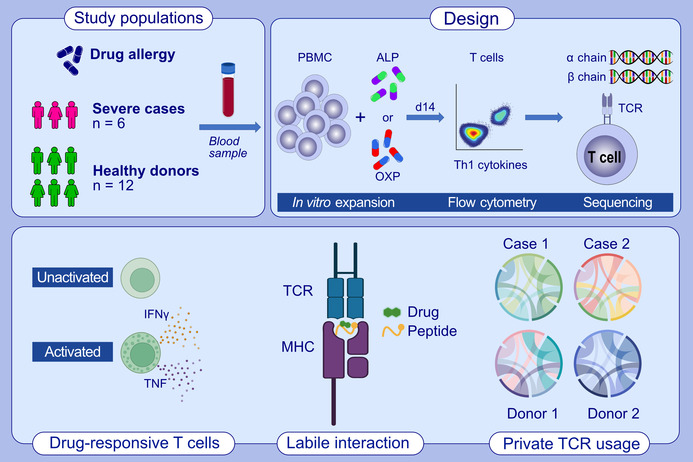
T cells from healthy drug‐naïve donors and patients with resolved allopurinol (ALP)‐induced hypersensitivity reactions were exposed to ALP and oxypurinol (OXP) to expand drug‐responsive T cells. Activated and cytokine‐producing drug‐responsive T cells were single‐cell sorted and sequenced to determine their T‐cell receptor repertoire clonotypes. We demonstrated that OXP is the driver of CD8+ T cell‐mediated responses and that drug‐exposed memory T cells can exhibit a proinflammatory immunophenotype similar to T cells described during active disease.Abbreviations: ALP, allopurinol; IFNγ, interferon gamma; MHC, major histocompatibility complex; OXP, oxypurinol; PBMC, peripheral blood mononuclear cells; TCR, T cell receptor; Th, T helper; TNF, tumour necrosis factor
Abbreviations
- ALP
allopurinol
- APC
antigen presenting cell
- cADRs
cutaneous adverse drug reactions
- DHR
drug hypersensitivity reaction
- DRESS
drug reaction with eosinophilia and systemic symptoms
- HLA
human leukocyte antigen
- IFNγ
interferon gamma
- MHC
major histocompatibility complex
- MPE
maculopapular exanthema
- OXP
oxypurinol
- PBMC
peripheral blood mononuclear cell
- SJS
Stevens–Johnson syndrome
- TCR
T‐cell receptor
- TEN
toxic epidermal necrolysis
- Th
T helper
- TNF
tumour necrosis factor
1. INTRODUCTION
Allopurinol (ALP) is an effective therapeutic agent for treatment of gout that acts by inhibiting the enzymatic action of xanthine oxidase, which is critical for production of uric acid. 1 ALP is rapidly metabolized into oxypurinol (OXP) in the liver and the latter is then excreted by the kidneys (mean half‐life of 23 h). 2 , 3 However, ALP has also been reported to cause a range of hypersensitivity responses leading to cutaneous adverse drug reactions (cADRs). These include a mild rash manifesting as maculopapular exanthema (MPE) 1 to severe pathology associated with Stevens–Johnson syndrome/toxic epidermal necrolysis (SJS/TEN) 4 and the rare, but life‐threatening, systemic allopurinol hypersensitivity syndrome (AHS) in 0.1% of patients. 5 , 6 , 7 ALP‐induced severe cADRs are associated with significant morbidity and have mortality rates of 10% for drug reaction with eosinophilia and systemic symptoms (DRESS), 8 23%–34% for SJS/TEN 9 and 9%–20% for AHS. 10
A strong risk association has been observed between human leukocyte antigen (HLA)‐B*58:01 and ALP‐induced hypersensitivities in diverse but predominantly Asian populations, 11 , 12 , 13 , 14 , 15 , 16 with odds ratios of 5.62 for MPE, 17 54.16 for DRESS 17 and 57.33–580.3 for SJS/TEN. 11 , 12 , 17 The immunopathology of ALP‐induced cADRs has been reported to be mediated by T‐cell receptor (TCR) recognition of HLA‐restricted presentation of the parent drug or metabolites. 18 Our current understanding of the mechanism underpinning T cell stimulation is that the parent drug or metabolite is presented by the HLA via the pharmacological interaction with immune receptors (p‐i) concept (reviewed in 19 ). The HLA‐B*58:01 positive predictive value for ALP‐induced hypersensitivities is between 9% and 11% in Han Chinese and <6% in Caucasians. 20 In contrast, 45% of ALP‐induced SJS/TEN patients with European ancestry do not carry the HLA‐B*58:01 allotype, 12 suggesting that there are other factors modifying risk.
The metabolite OXP is considered the primary molecule mediating the T cell hypersensitivity reactions, not because ALP is less immunogenic, but rather due to its rapid in vivo metabolism to OXP. 21 However, studies conducted in non‐physiological conditions, such as in vitro T cell cultures, show that ALP is not completely metabolized to OXP. 21 , 22 This enables direct assessment of both ALP‐specific and OXP‐specific T cells, which are highly dose‐dependent for induction and activation. 21 , 23 Additionally, OXP‐specific T cells derived from individuals carrying HLA‐B*58:01 have been shown to be highly restricted to this allele, whereas ALP‐specific T cells are less HLA‐B58 restricted for drug‐induced responses. 24 Moreover, exquisite specificity towards the inducing drug or metabolite has been observed, with in vitro expanded OXP‐specific T cells not activated by ALP and vice versa. 21 , 24 This differs from other examples of drug‐induced cADRs including carbamazepine, with T cells demonstrating cross‐reactivity towards a metabolite (i.e. carbamazepine‐10,11‐epoxide) or a structurally similar compound (i.e. oxcarbazepine). 25 , 26 , 27 Although, this narrow specificity in ALP responses has been challenged by an alternate study demonstrating reciprocal cross‐reactivity. 22 Interestingly, there also appears to be differential T cell subset involvement in ALP‐induced pathologies with CD4+ T cells detected in MPE, both CD4+ and CD8+ T cells implicated in DRESS, and exclusively CD8+ T cells involved in SJS/TEN. 28 , 29 , 30
There have been limited studies investigating the TCR repertoires of reactive T cells in ALP‐induced hypersensitivities. Collectively, these studies suggest that T cell responses to either the parent drug or metabolite are polyclonal. TCRβ chain repertoire investigations of blister cells from skin lesions and OXP‐specific T cell lines derived from ALP‐induced SJS/TEN patients have shown a polyclonal profile, with some variable regions of the TCRβ chain being expressed at high frequency (e.g. TRBV3‐1, 5‐1, 9, 29‐1). 23 An alternate study corroborated the absence of a public TCR clonotype for both ALP‐ and OXP‐specific T cells by TCR Vβ staining, with the drug‐specific TCR Vβ usage not being preserved across different sampling time points of individuals. 24 Here, we explored the TCR repertoire of patients that experienced either ALP‐induced SJS or TEN using paired α and β chain sequencing of individual T cell clones. First, we demonstrated that the OXP metabolite is the main driver of T cell‐mediated immune reactivity in these individuals. Second, we found no evidence of reciprocal cross‐reactivity between drug‐specific T cell lines expanded by either the parent drug, ALP or the metabolite, OXP. Third, we developed evidence that an OXP‐induced TCR is triggered in a manner consistent with the p‐i concept. Importantly, we report paired αβTCR repertoire profiling of drug‐exposed T cells in both patient and healthy donors revealing private oligoclonal TCR signatures.
2. MATERIALS AND METHODS
2.1. Study participants and HLA typing
Six HLA‐B*58:01‐positive individuals were recruited from Chulalongkorn University, Bangkok, Thailand (Table 1). Four of these patients experienced either SJS (CU002, CU003, CU006) or TEN (CU005) following administration of ALP, while two patients were not treated with ALP (CU001, CU004). In addition, both HLA‐B*58:01‐positive and HLA‐B*58:01‐negative ALP‐naïve healthy individuals were also recruited as controls (Table 1). All study participants provided written consent, with ethics approvals granted by Chulalongkorn University (Bangkok, Thailand; HREC 333/53 for Thai patients, denoted as CU), the Australian Bone Marrow Donor Registry (New South Wales, Australia; HREC 2013/04 for healthy individuals, denoted as AP) and Monash University (Victoria, Australia; HREC 7681 for Thai patients, denoted as CU and HREC 4717 for healthy individuals, denoted as AP). HLA typing of patients and healthy donors was performed by the HLA Laboratory, Immunology Division, Department of Microbiology, Faculty of Medicine, Chulalongkorn University (Bangkok, Thailand) and the Victorian Transplantation and Immunogenetics Service (Victoria, Australia), respectively.
TABLE 1.
Study cohort HLA and drug hypersensitivity information.
| Study ID | HLA‐B | ALP exposure | cADR | Collection date | Time post‐reaction |
|---|---|---|---|---|---|
| CU001 | 58:01 | No | No | 13/10/2010 | ‐ |
| CU002 | 58:01 | Yes | SJS | 29/09/2010 | 4 years |
| CU003 | 58:01 | Yes | SJS | 27/09/2010 | 6 years |
| CU004 | 58:01 | No | No | 13/10/2010 | ‐ |
| CU005 | 58:01 | Yes | TEN | 28/09/2010 | 5 years |
| CU006 | 58:01 | Yes | SJS | 10/04/2010 | 7 years |
| AP012 | 57:01, 58:01 | No | No | 23/07/2014 | ‐ |
| AP013 | 15:01, 58:01 | No | No | 24/07/2014 | ‐ |
| AP014 | 41:01, 58:01 | No | No | 30/07/2014 | ‐ |
| AP015 | 40:06, 58:01 | No | No | 31/07/2014 | ‐ |
| AP016 | 27:04, 58:01 | No | No | 13/08/2014 | ‐ |
| AP018 | 39:24, 58:01 | No | No | 27/08/2014 | ‐ |
| AP025 | 07:02, 58:01 | No | No | 22/09/2014 | ‐ |
| AP027 | 15:02, 58 | No | No | 30/09/2014 | ‐ |
| AP101 | 44, 58 | No | No | 12/03/2014 | ‐ |
| AP105 | 7, 8 | No | No | 20/03/2014 | ‐ |
| AP108 | 14:01, 51:01 | No | No | 31/03/2014 | ‐ |
| AP111 | 44, 57 | No | No | 18/06/2014 | ‐ |
Note: Patients are denoted as CU and healthy participants are denoted as AP. For CU patients, it is unknown whether HLA‐B*58:01 is homozygous or heterozygous.
Abbreviations: cADR, cutaneous adverse drug reaction; SJS, Stevens–Johnson syndrome; TEN, toxic epidermal necrolysis.
2.2. Generation of drug‐induced T cells
Peripheral blood mononuclear cells (PBMCs) were isolated from blood samples using Ficoll‐Paque (GE Healthcare) density gradient centrifugation and either used immediately or cryopreserved in foetal calf serum (FCS) containing 10% DMSO (Sigma‐Aldrich) at −196°C until required. For T cell stimulation assays, PBMCs were thawed in 37°C, washed twice in RPMI 1640 (Gibco, Life Technologies) and resuspended in complete medium (RPMI 1640 supplemented with 2 mM MEM nonessential amino acid solution (Gibco), 100 mM HEPES (Gibco), 2 mM L‐glutamine (Gibco), Penicillin/Streptomycin (Gibco), 50 μM 2‐mercaptoethanol (Sigma‐Aldrich) and 10% heat inactivated human blood group AB serum (Sigma‐Aldrich)). Drug‐induced T cell lines were generated from PBMCs following drug stimulation at a density of 5 million per 2 mL of complete medium in a 24‐well plate with 100 μg/mL of ALP or OXP. From Day 4, the T cell culture was supplemented with 50 U/mL of recombinant human IL‐2 (Peprotech) and cells were subcultured as required to ensure optimal expansion.
2.3. Antigen‐presenting cell lines
C1R.B*57:01, C1R.B*58:01 are B‐lymphoblastoid cell lines derived from the parental C1R cell line that express high levels of the introduced HLA allotype along with no detectable surface expression of endogenous HLA‐A, low levels of HLA‐B35 and normal levels of HLA‐Cw4. 31 All antigen‐presenting cell (APC) lines were cultured in RF10 (same as constituents as complete medium except 10% heat inactivated FCS (Sigma‐Aldrich)). Maintenance of transfected HLA expression during long‐term culture was facilitated by addition of 0.5 mg/mL Geneticin (Roche). HLA class I allele expression, as compared to parental C1R, was confirmed via flow cytometry after staining with a primary HLA class I pan‐specific monoclonal antibody W6/32 32 (produced in‐house from the W6/32 hybridoma) or HLA‐B57/B58‐specific monoclonal antibody 3E12 33 (kind gift from Prof James McCluskey, Peter Doherty Institute, University of Melbourne, Victoria, Australia) and secondary PE goat anti‐mouse IgG (Southern Biotech).
2.4. Characterization of drug‐induced T cells
Drug‐induced T cell phenotype and functionality were assessed using a combination of cell surface (subset biomarkers) and intracellular cytokine staining (proinflammatory Th1 cytokines; IFNγ and/or TNF). 34 Day 14 T cells (2 × 105) were stimulated with either Dynabeads® Human T‐Activator CD3/CD28 beads (positive control, Life Technologies), 100 μg/mL drug alone, APC in the continuous presence of drug (1 × 105; APC + 100 μg/mL drug) or drug‐pulsed APC incubated with 100 μg/mL of drug overnight then washed (1 × 105; APC/drug PW) and subsequently co‐cultured with T cells for 6 h. Brefeldin A (10 μg/mL; Sigma‐Aldrich) was added for the last 4 h of co‐culture. Cells were then surface labelled with LIVE/DEAD® fixable Aqua stain (Life Technologies), CD4 PE (clone RPA‐T4) and CD8 PerCP‐Cy5.5 (clone SK1), fixed in 1% paraformaldehyde (ProSciTech) and then permeabilized with 0.3% Saponin (Sigma‐Aldrich) containing IFNγ PE‐Cy7 (clone B27) and TNFα V450 (clone MAb11) before acquisition on a LSRII flow cytometer (Becton Dickinson (BD)). All monoclonal antibodies were purchased from BD and titrated for optimal staining efficiency. A maximum of 50,000 lymphocytes were acquired on a BD LSRII flow cytometer (FlowCore, Monash University) and analyzed using FlowJo software (version 10, BD). Representative gating strategy is shown in Figure S1.
2.5. Single‐cell TCR analysis of drug‐induced T cells
A single‐cell sort was performed to characterize the TCRαβ signature of drug‐induced T cells using the IFNγ Secretion Assay–Detection Kit (APC; Miltenyi Biotec), as described previously. 27 Briefly, cryopreserved Day 14 T cell lines (maximum of 5 × 106 cells) were incubated with either C1R.B*58:01 or drug‐pulsed (100 μg/mL) C1R.B*58:01 APCs at a 2:1 ratio in RH5 media (same constituents as complete medium, except 5% heat inactivated human blood group AB serum) for 4 h at 37°C, 5% CO2. Cells were washed in cold Wash Buffer (0.5% FCS, 2 mM EDTA pH 8.0 in PBS), centrifuged (285× g, 5 min, 4°C) and supernatant aspirated before addition of the IFNγ catch reagent antibody according to manufacturer's instructions. Cells were incubated on ice for 5 min and topped up to 10 mL with warm RH5 media, with a top up of drug (100 μg/mL) added to the drug‐pulsed APC tube. Cells were incubated for 45 min at 37°C with rotation. Cells were washed in cold Wash Buffer, centrifuged and supernatant aspirated prior to co‐staining with IFNγ allophycocyanin detection reagent and CD8 FITC (clone HIT8a, BD). Cells were incubated on ice for 20 min, washed in cold Wash Buffer, centrifuged and resuspended in 300 μL cold Wash Buffer.
Single‐cells were sorted on a BD Influx flow cytometer (FlowCore, Monash University) directly into 96‐well PCR plates (Bio‐Rad) based on CD8+IFNγ− for drug‐unresponsive T cells (negative control) and CD8+IFNγ+ for drug‐induced T cells and the plates stored at −80°C until required. TCRα and TCRβ gene segments were amplified using RT‐PCR followed by multiplex nested PCR and sequencing as described previously. 35 Both external and internal rounds of PCR included 40 TRAV and 27 TRBV forward primers, and a TRAC and TRBC reverse primer, as detailed elsewhere. 35 Sequences were analyzed according to the ImMunoGeneTics/V‐QUEry and STandardization (IMGT/VQUEST) web‐based tool. 36 All TCR nomenclature was according to Folch et al. 37 Complementarity determining region (CDR) 3 amino acid sequences described within the text start from CDR3‐position 3, which is equivalent to amino acid position 107 of the TRAV and TRBV segments and end at TRAJ‐position 10 or TRBJ‐position 6. All TCR analytical plots were generated based on concatenated pairings of CDR3A_TRAVJ and CDR3B_TRBVDJ using the ‘TCR_Explore’ program. 38
2.6. SKW3 reporter cells for TCR expression
Full‐length human TCRα and TCRβ cDNA were cloned into a self‐cleaving 2A peptide‐based pMIG vector facilitating their co‐expression as described previously. 39 A pMIG vector containing a specific TCR (4 μg) for the patient CU002/OXP was retrovirally transduced into SKW3.hCD8αβ GFP+ cells (kindly provided by Dr. Zhenjun Chen, Peter Doherty Institute for Infection and Immunity, University of Melbourne; hereafter referred to as SKW3). This reporter cell line is negative for endogenous TCRαβ but contains CD3 and its signalling components. Retroviral transductions was achieved with HEK293T packaging cells, pEQ‐pam3(−E) (4 μg) and pVSV‐G (2 μg) packaging vectors and Lipofectamine 3000 as previously described. 40 The original SKW3 parental cell line was kindly provided by Dr. Klaus Steube, Leibniz Institute DSMZ‐German Collection of Microorganisms and Cell Cultures (Braunschweig, Germany). The SKW3.CU002/OXP cell line was maintained in RF10 media.
2.7. Functional T cell assays
Activation of SKW3.CU002/OXP cells (1 x 105) were assessed using cell surface CD69 upregulation after 16–20 h incubation with either C1R parental, C1R.B*57:01 or C1R.B*58:01 targets (1:1 ratio) under different sets of conditions. SKW3.CU002/OXP cells were co‐stained with CD3 PE‐Cy7 (clone SK7), CD8 PerCP‐Cy5.5 (clone SK1), CD69 APC (clone L78) and LIVE/DEAD® fixable Aqua stain. For all experiments, stimulation with Dynabeads® Human T‐Activator CD3/CD28 beads served as a positive control, and SKW3.CU002/OXP cells alone as a negative control. Flow cytometry data were acquired and analyzed as described previously. 27 The CD69 expression profiles were measured as geometric mean fluorescence intensity (MFI) to provide more meaningful evaluation of changes in the relative amounts of expressed protein per cell. A maximum of 50,000 lymphocytes were acquired on a BD LSRII flow cytometer (FlowCore, Monash University) and analyzed using FlowJo software (version 10, BD). A representative gating strategy is shown in Figure S2.
2.8. Statistical analysis
All data were reported as mean ± standard error of the mean (SEM). Statistical analyses were performed using GraphPad Prism version 9.0 (GraphPad software). Statistical analyses were performed by using either a nonparametric one‐way ANOVA with post hoc Tukey multiple comparison test or an unpaired Student's t‐test (two‐tailed), with statistical significance defined as p‐value <.05.
3. RESULTS
3.1. T‐cell reactivity driven by the metabolite not the parent drug
Although studies have reported the in vitro expansion of both ALP‐ and OXP‐specific T cell lines either from patients with ALP‐induced cADR or healthy donors, 21 , 22 , 24 the immune response is mainly driven by the OXP metabolite. 23 Here, we sought to corroborate whether either ALP and/or OXP could induce T‐cell activation in both HLA‐B*58:01‐positive patients or drug‐naïve HLA‐B*58:01 healthy donors. Drug‐induced T cell lines were generated following PBMC stimulation with either 100 μg/mL ALP or OXP and in vitro expanded for 14 days (SJS or TEN patients) or re‐exposed to drug in up to three rounds of fortnightly restimulation (drug‐naïve healthy donors). Expanded T cells were then restimulated with either drug alone, C1R.B*58:01 alone (APC), APC pulsed with drug for 16 h and washed to remove drug prior to the assay (APC/drug PW) or APC with drug present throughout the assay (APC + drug). Media (untreated) and Dynabeads® Human T‐Activator CD3/CD28 (Life Technologies) were used as negative and positive controls, respectively.
For all four ALP‐exposed patients that experienced either SJS or TEN (Table 1; CU002, CU003, CU005, CU006) we did not observe activation of either CD8+ or CD4+ T cell lines directed towards ALP as measured by either IFNγ (Figure 1A,B) or TNF (Figure S3) production. However, stimulation with the OXP metabolite evoked significant CD8+ and/or CD4+ T cell immune reactivity in three of the four SJS patients (CU002, CU003, CU006) (Figure 1A,B and Figure S3). This activation was only observed when the drug was present continuously throughout the assay with washing of drug‐pulsed APCs abrogating Th1 cytokine production (Figure 1A,B). For CU005, high background levels (media alone) were observed following both ALP and OXP stimulation, therefore we were unable to assign ‘true’ positive responses from the background. As expected, no CD8+ or CD4+ T cell responses were observed for HLA‐B*58:01‐positive ALP naïve patients (CU001, CU004) (Figure 1A,B and Figure S3). Further dissection of the polyfunctionality of Th1 cytokine subsets produced by OXP‐induced CD8+ T cells showed that IFNγ was the main cytokine produced, representing up to 5.2% of total expanded CD8+ T cells in ALP‐exposed patients that experienced SJS or TEN (Figure 2).
FIGURE 1.
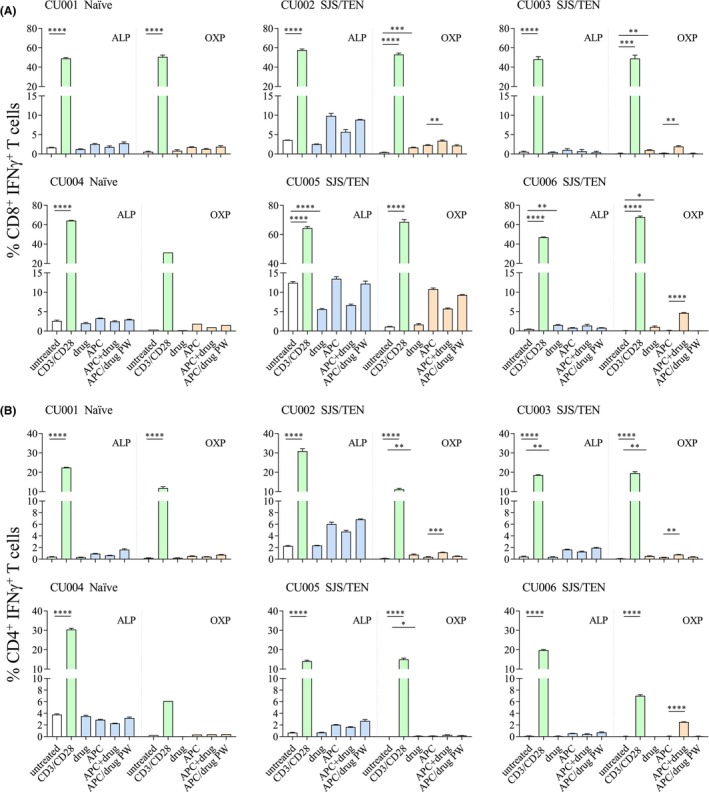
Functional T cell assessment of ALP‐exposed patients with parent drug and metabolite. Following in vitro exposure of PBMCs to drug, both CD4+ and CD8+ T cell functionality was assessed by intracellular staining for IFNγ production. ALP‐specific T cells (blue bars) and OXP‐specific T cells (orange bars) were represented as mean ± SEM obtained from technical triplicate measures. T cell data for CU004/OXP were considered a single measure due to low cell numbers. APC represented by C1R.B*58:01. Statistical analysis was performed with unpaired t‐test (two‐tailed) with significance denoted as *p < .05, **p < .01, ***p < .001, ****p < .0001.
FIGURE 2.
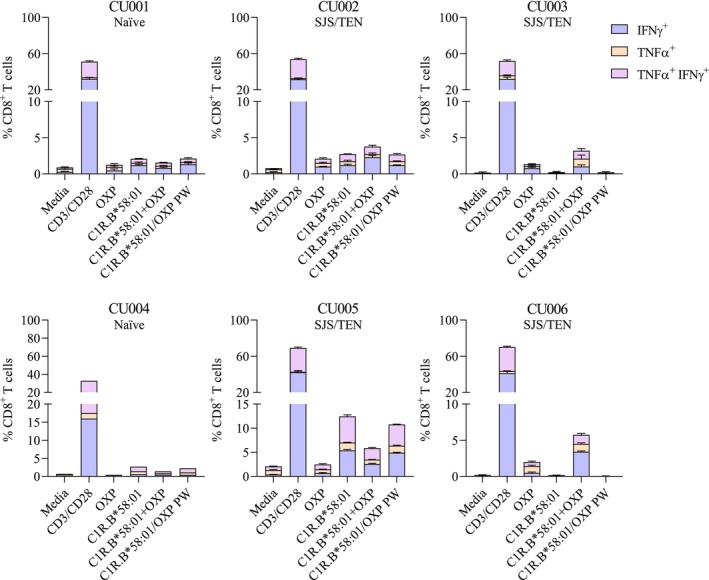
Dissecting Th1 cytokines produced by in vitro expanded OXP‐specific T cells. Profiling of d14 OXP‐specific CD8+ T cells derived from our patient cohort showed that most T cells preferentially produced IFNγ alone (purple) compared to either the alternate Th1 cytokine TNFα alone (orange) or dual expression of both cytokines (TNFα+IFNγ+; pink). Data are represented as mean ± SEM obtained from technical triplicate measures, except for CU004 dataset (n = 1).
We also examined whether HLA‐B*58:01‐positive and HLA‐B*58:01‐negative drug‐naïve healthy donors could be in vitro primed to generate drug‐induced T cell responses following repeated drug exposures. For ALP‐primed T cell cultures, we did not observe significant expansion of either CD4+ or CD8+ T cells following three rounds of stimulation in any of the drug‐naïve healthy individuals. Although, we did note high non‐specific T cell responses to APC alone for both AP111 (Round 1 [R1] of CD4+ T cells) and AP027 (R3 of CD8+ T cells). For OXP‐primed T cell cultures, we show statistically significant expansion of CD8+ T cells in R2, but not for CD4+ T cells. Interestingly, by R2 PBMC from both HLA‐B*58:01‐positive and HLA‐B*58:01‐negative healthy individuals could respond to either OXP alone or the C1R.B*58:01 APC in the presence of OXP. This response was lost for the HLA‐B*58:01‐negative healthy individuals by R3 (Figure 3). Collectively, both patient and drug‐naïve healthy individual data illustrated greater CD4+ and CD8+ T cell reactivity to the OXP metabolite compared to the parent drug.
FIGURE 3.
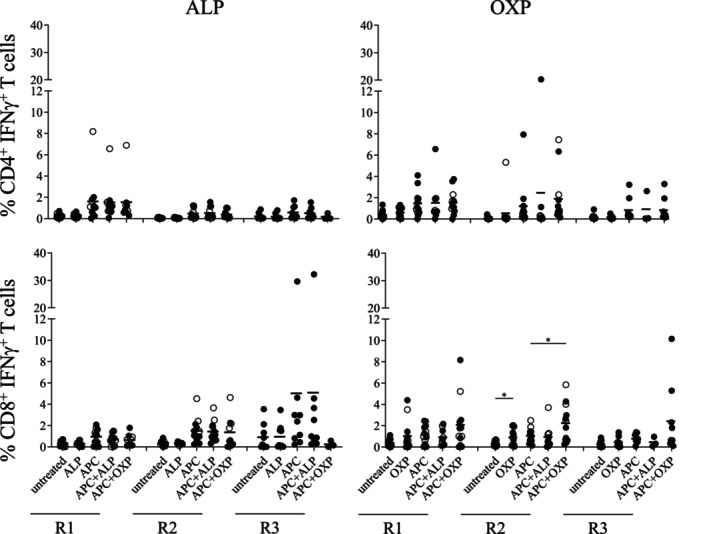
In vitro priming of drug‐specific T cells in drug‐naïve healthy individuals. Repeated drug exposure of PBMCs derived from drug‐naïve HLA‐B*58:01‐positive (black circles) or–B*58:01‐negative (white circles) individuals was performed to determine expansion and functionality of drug‐specific T cells. Individual data (n = 12) are shown as group data (bar = mean). Drug stimulation rounds were R1 (d12‐15 T cells), R2 (d22‐29 T cells) and R3 (d41‐43 T cells). APC represented by C1R.B*58:01. Statistical analysis was performed with a paired t‐test (two‐tailed) with significance denoted as *p < .05.
3.2. OXP‐induced T cells do not cross‐react
There are conflicting reports regarding the ability of either ALP‐ or OXP‐specific T cell lines to exhibit cross‐reactivity following stimulation by the alternate drug (i.e. ALP‐specific T cells stimulated by OXP and OXP‐specific T cells stimulated by ALP). 21 , 22 , 24 Here, we sought to determine whether OXP‐specific T cells expanded from our patient cohort could also cross‐react towards the parent drug. Briefly, Day 14 OXP‐induced T cells were restimulated by drug alone, APC or APC + drug. Examination of both CD4+ and CD8+ T cell responses in resolved ALP‐induced SJS/TEN cases failed to demonstrate cross‐reactivity between OXP and ALP as measured by Th1 cytokine production for both IFNγ (Figure 4) and TNF (data not shown). These data are consistent with studies also showing a lack of cross‐reactivity in OXP expanded T cell lines. 21 , 24
FIGURE 4.
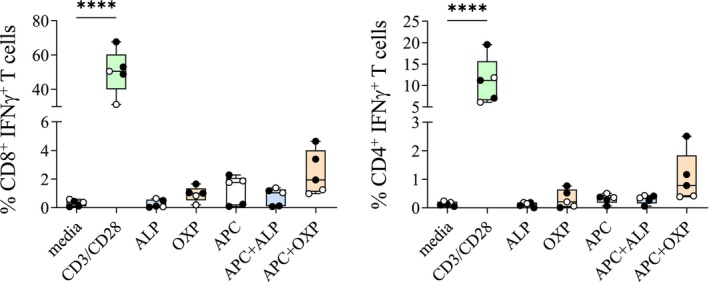
OXP‐specific T cells do not exhibit cross‐reactivity. The capacity of AHS patient‐derived OXP‐specific T cells to cross‐recognize the parent drug was evaluated using drug alone or drug in the presence of APC (C1R.B*58:01). HLA‐B*58:01‐positive ALP‐exposed (black circles) or ALP‐naive (white circles) are shown. Box and whiskers plot show minimum to maximum for all values. Statistical analysis was performed with nonparametric one‐way ANOVA with post hoc Tukey's multiple comparison test with significance denoted as ****p < .0001.
3.3. Oligoclonal TCR repertoires derived from drug‐induced T cells
To date there are only two publications examining the TCR repertoire usage of ALP‐ and/or OXP‐specific T cells in patients experiencing SJS/TEN. 23 , 41 Therefore, we sought to determine the level of TCR repertoire clonality in SJS patients (CU002, CU003, CU006) and drug‐naïve healthy donors (AP013, AP018). In addition, we examined whether drug‐induced TCR clonotypes were either public (common TCR observed across different individuals) or private (exclusive to one individual).
Here, in vitro expanded drug‐induced CD8+ T cells were restimulated for 4 h in the presence of either ALP or OXP and C1R.B*58:01, then single‐cell sorted based on their ability to produce IFNγ (two populations: non‐responsive to drug CD8+ IFNγ− T cells [hereafter referred to as CD8+] and responsive drug‐induced CD8+ IFNγ+ T cells [hereafter referred to as CD8+ IFNγ+]). A single‐cell RT‐PCR and multiplex nested PCR approach was then used to determine the paired αβTCR profiles of these sorted T cell populations with CDR3 resolution (Table S1). First, we examined the drug‐induced TCR repertoires of the two drug‐naïve healthy donors. For AP013/ALP, a single TCR represented 46.1% of the CD8+ IFNγ+ repertoire (TRAV8‐6TRAJ42_TRBV10‐3TRBJ2‐3). This TCR was also present in the non‐responsive CD8+ subset at low frequency demonstrating enrichment of this clonotype following drug treatment (Figure 5A). For AP018/ALP the highest representative clonotype for the responsive CD8+ IFNγ+ repertoire expressed the TCRβ chain (TRBV20‐1TRBJ1‐4_CDR3βCSARVYTEGYEKLFF), which was also observed in non‐responsive population but at a lower frequency (Figure 5A). For AP013/OXP, the CD8+ IFNγ+ TCR repertoire was mainly populated by two TRBV chains (BV7‐9 and BV20‐1). However, the BV20‐1 subset was shown to be heterogeneous, with different BVJ and CDR3β sequences. Interestingly, for the AP018/OXP CD8+ IFNγ+ TCR repertoire 40% of all sequences demonstrated a TCR bias towards the TRAV26‐2TRAJ49_TRBV7‐2/8TRBJ1‐1 clonotype. Closer examination of CDR3 sequences demonstrated that the α‐chain was composed of two different sequences, while the β‐chain was highly variable with up to seven different sequences (Table S1; column F for α‐chain and column L for β‐chain).
FIGURE 5.
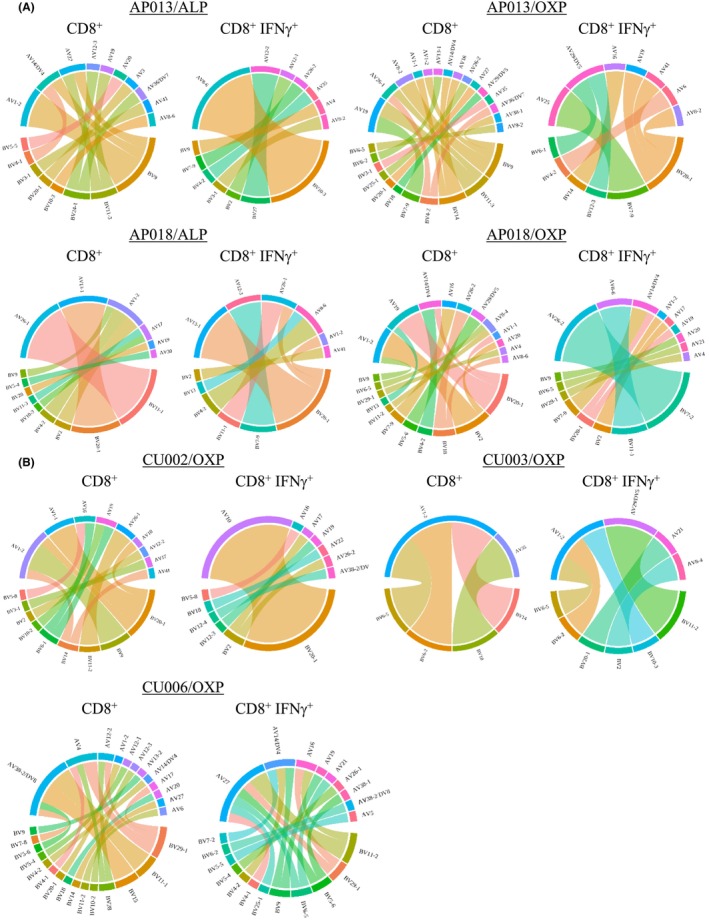
Profiling of drug‐specific αβTCR clonotypes shows oligoclonality. The αβTCR repertoire diversity of single cells derived from outgrown drug‐specific T cells were determined using multiplex RT‐PCR. αβTCR pairs were sequenced for both (A) ALP‐ and OXP‐specific CD8+ T cells from two drug‐naïve healthy individuals and (B) OXP‐specific CD8+ T cells from three patients depicted as chord diagrams. Each V gene segment represents a single CDR3 clonotype. Number of TCR pairs analyzed; AP013/ALP CD8+ (n = 14), CD8+IFNγ+ (n = 13), AP013/OXP CD8+ (n = 20), CD8+IFNγ+ (n = 9), AP018/ALP CD8+ (n = 23), CD8+IFNγ+ (n = 16), AP018/OXP CD8+ (n = 25), CD8+IFNγ+ (n = 21), CU002/OXP CD8+ (n = 25), CD8+IFNγ+ (n = 24), CU003/OXP CD8+ (n = 4), CD8+IFNγ+ (n = 7), CU006/OXP CD8+ (n = 23), CD8+IFNγ+ (n = 18).
Given that the OXP metabolite appears to drive T cell reactivity associated with cADRs, we evaluated the OXP‐specific TCR repertoires of ALP‐exposed patients that experienced SJS (CU002, CU003 and CU006). For CU002, the CD8+ IFNγ+ TCR repertoire comprised a dominant TCRβ chain (45.8% of total clonotypes) TRBV20‐1TRBJ1‐2_CDR3βCSARVGQGVSYGYTF that was predominately paired with TRAV10TRAJ33_CDR3αCVVRKSNYQLIW. This signature was observed in a single clonotype represented in the CD8+, demonstrating an eightfold increase in the drug responsive population compared to the non‐responsive population (Figure 5B). Strikingly, CU006/OXP contained a dominant TRAV27‐TRAJ12_CDR3αCAGRGMDSSYKLIF signature that associated with different TCRβ chains. This dominant TCRα chain was absent in CD8+ subset (Figure 5B). For CU003/OXP, few productive αβTCR sequences were identified, with only 5 and 7 for the CD8+ IFNγ+ and CD8+ populations, respectively. Therefore, the data are lacking to interpret the true clonality of the αβTCR signature (Figure 5B). Collectively, these data show that irrespective of drug specificity the TCR repertoires for both drug‐naïve and ALP‐exposed individuals were mostly oligoclonal.
3.4. Drug‐induced TCRs display private repertoires and decreasing diversity
Next, we examined the TCR clonotypes of both drug‐naïve healthy and ALP‐exposed patients to determine whether there were shared signatures across different individuals. The data showed a distinctive lack of overlap in TCR profiles, lending evidence that both ALP‐ and OXP‐induced TCR repertoires are private, which was true for both drug‐naïve healthy donors and patients (Figure 6). We also observed decreased diversity or alternate CDR3 α‐chain and β‐chain length distributions in the CD8+ IFNγ+ population for the majority of T cell clonotypes. However, this was not observed for CU003/OXP due to limited TCR sequencing information (Figure S4). Collectively, the data demonstrated a decrease in TCR repertoire diversity between non‐reactive and drug‐reactive subsets.
FIGURE 6.
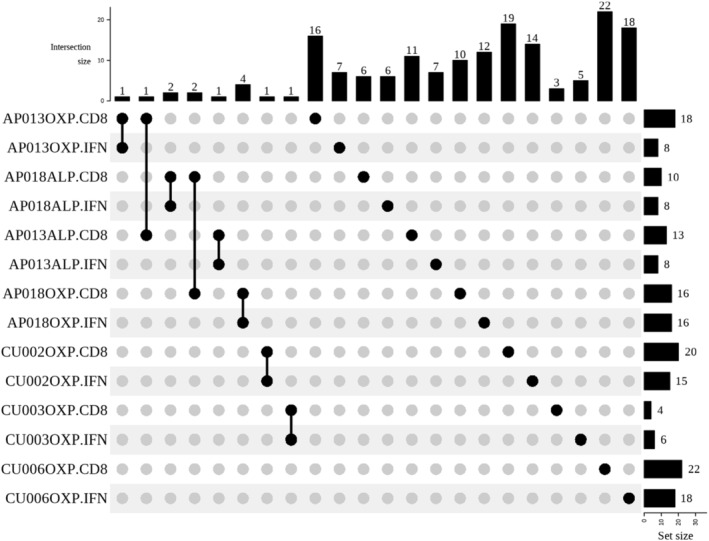
Private TCR repertoires are exhibited by both drug‐naïve and ALP‐exposed individuals. Repertoire analysis was performed using TCR_Explore program and demonstrated that the study cohort exhibited private drug‐specific TCR usage as visualized by the UpSet plot. There was minor overlap between CD8+ and CD8+ IFNγ+ populations within an individual, but this was not observed across the cohort.
3.5. OXP‐specific TCR activation is consistent with the p‐i concept
Studies have implicated the p‐i concept as a potential mechanism driving allopurinol‐induced T‐cell activation. 22 , 24 We explored whether the p‐i concept mediated activation of an OXP‐specific TCR derived from patient CU002. To do so, the CD8+ IFNγ+ TCR, TRAV10_TRAJ33_CDR3αCVVRKSNYQLIW_TRBV20‐1_TRBJ1‐2_CDR3βCSARVGQGVSYGYTF, was expressed in SKW3 reporter cells to generate SKW3.CU002/OXP. This reporter system provides the opportunity to examine TCR functionality in the absence of T‐T presentation (SKW3 lack HLA‐B*58:01), which can interfere with APC responses by augmenting immune reactivity, hindering differentiation of responses to different APCs. First, as anticipated the SKW3.CU002/OXP cells were only activated in the presence of drug and APC, with washing of drug‐pulsed APCs abrogating activation (Figure 7A), consistent with the T cell line data (Figure 1A). Second, unlike in T cell lines, no response was observed to drug alone, consistent with the lack of T‐T presentation by HLA‐B*58:01‐negative SKW3. Thus, without interference from T‐T presentation, we were able to determine that drug presentation was not mediated by APC expressing the alternate, yet closely related allotype, HLA‐B*57:01 (Figure 7A,B). The Arg97Val substitution between HLA‐B*58:01 and HLA‐B*57:01 has been proposed to reduce OXP binding. 23 , 24 Third, although fixation of C1R.B*58:01 APCs in the presence of drug showed a statistically significant reduction in TCR activation (p < .001), immune reactivity was still observed (Figure 7B). Collectively, the data support the non‐covalent and labile interaction between the drug, pMHC and the TCR complex.
FIGURE 7.
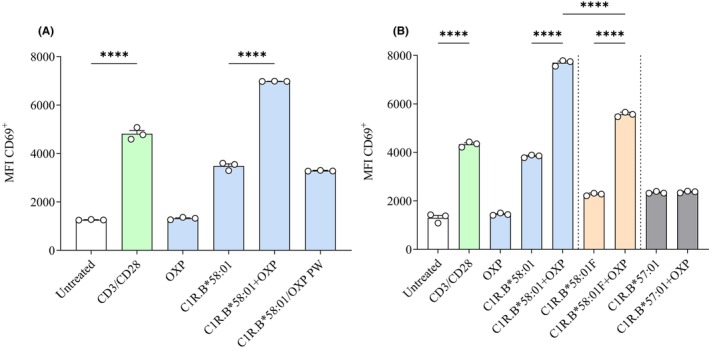
TCR activation supports the p‐i concept. SKW3.CU002/OXP cells were used to evaluate the mechanism of drug‐induced TCR activation. (A) SKW3.CU002/OXP cells were activated only in the presence of both drug and APC. (B) APC fixation did not affect drug‐induced TCR activation, although HLA specificity was required with C1R.B*57:01 APCs unable to generate immune reactivity. Data are represented as mean ± SEM obtained from triplicate measures. Statistical analysis was nonparametric one‐way ANOVA with post hoc Tukey's multiple comparison test with significance denoted as ****p < .0001.
4. DISCUSSION
In this study, we examined HLA‐B*58:01‐positive patients that had experienced either ALP‐induced SJS or TEN to determine (i) whether peripheral blood derived drug‐specific memory T cells (up to 7 years post‐reaction) could be reactivated by the parent drug, metabolite or both, (ii) do these memory T cells exhibit similar immunophenotypes and TCR repertoire signatures reported during the active phase of disease and (iii) compare drug induction responses in drug‐naïve individuals.
There is general consensus among previous studies that the main driver of T cell‐mediated AHS is the metabolite OXP in association with the HLA risk allotype, HLA‐B*58:01. 21 , 22 , 23 , 24 , 42 , 43 Our study corroborates these findings as we observed strong evidence that OXP was the primary mediator of T cell activation in both resolved AHS patients and drug‐naïve healthy donors. The principle mechanism of action for stimulation of ALP‐exposed T cells is considered to be the p‐i concept. 18 , 44 This mechanism features four defining parameters; (i) non‐covalent and labile interactions between the drug and TCR/peptide/HLA complex, 24 (ii) immunogenic complexes that are formed at cell surface, 24 (iii) drug removal from co‐cultures immediately abrogates T cell stimulation 24 and (iv) the classical HLA‐I antigen processing pathway is not required for de novo generation of neoepitopes. 22 , 24 Each of these parameters were fully supported by the data generated in this study.
In 2014, Yun et al. proposed a variation of the p‐i concept based on in silico modelling studies suggesting that ALP and OXP can be accommodated in the HLA‐B*58:01 peptide‐binding cleft. Modelling suggested that the parent drug and metabolite exhibit different HLA‐B*58:01 binding affinities, with OXP being stronger, based on key contacts with residue Arg97 and van der Waals interactions with residues neighbouring the F pocket in HLA‐B*58:01. 22 , 24 This is analogous to the occupation of the antigen binding cleft of HLA‐B*57:01 by abacavir that leads to presentation of an altered repertoire of peptides at the cell surface. 45 , 46 , 47 However, given that unlike for abacavir, T cell responses demonstrated lack requirement for active antigen processing pathways, Yun et al. 24 proposed that sporadic partial disengagement of the peptide and HLA‐B*58:01 complex may provide an opportunity for the drug to bind within the cleft at the cell surface to alter peptide conformation rather than the array of bound peptides. Our data do not resolve whether this is the mechanism of interaction of ALP and/or OXP with HLA‐B*58:01.
The ability of drug‐specific T cells, generated by either the parent drug or metabolite, to reciprocally respond to the alternate compound is a feature that has been associated with a number of commonly prescribed drugs that trigger drug hypersensitivity reactions. In this setting, T cell cross‐reactivity appears to be underpinned by recognition of either similar structural elements or spatial arrangements of atoms exhibited by both the parent drug and metabolite. For the anti‐seizure medication carbamazepine, T cells raised against either the parent drug or the reactive metabolite, carbamazepine‐10,11‐epoxide (ECBZ), show mutual cross‐reactivity based on structural similarities associated with a tricyclic aromatic core. 25 , 48
In direct contrast with these studies, evidence of T cell cross‐reactivity in ALP‐induced cADRs has been less convincing. In HLA‐B*58:01‐positive ALP‐naïve healthy donors, generation of ALP‐ or OXP‐induced T cells did not evoke cross‐reactivity when challenged with the reciprocal drug. 21 , 24 However, a clinical study examining HLA‐B*58:01‐positive patients (n = 14) with ALP‐induced cADRs generated drug‐specific T cell lines (8 ALP‐specific and 14 OXP‐specific) after multiple rounds of drug stimulation. 22 Conversely, this study demonstrated significant T cell cross‐reactivity between the parent drug and metabolite and attested these findings to the use of clinical patients compared to other studies that focused on healthy donors. In an attempt to address whether T cell cross‐reactivity could only be detected in patients with ALP‐induced cADRs or drug‐naïve individuals we examined both cohorts. Our data aligns with the studies by Yun et al., 21 , 24 with no cross‐reactivity observed in either direction irrespective of cohort examined.
To date only a handful of studies have been published examining the drug‐induced TCR repertoire of either ALP hypersensitive patients or primed healthy donors, furthermore these have been limited to unpaired TCRα and TCRβ chain analyses. The study by Yun et al. 24 examined in vitro primed ALP‐ and OXP‐specific T cells in drug‐naïve healthy donors and demonstrated that while major TRAV and TRBV clonotypes were determined in a single individual, a cross‐sectional analysis of 20 different Vβ repertoires failed to demonstrate a public nature of the clonotype. Interestingly, sampling of the same individual at different time points also showed that the drug‐specific TCR repertoire was not preserved. Therefore, private oligoclonal TCR signatures were generated following drug treatment. This differs from other drugs that cause cADRs such as abacavir and carbamazepine, which have been shown to induce either highly polyclonal TCRs 45 , 49 , 50 or more clonally restricted TCRs, 27 , 51 respectively.
The study by Chung et al. 23 demonstrated high frequency OXP‐specific TCRβ chains in ALP‐induced SJS/TEN patients (i.e. TRBV3‐1, 5‐1, 9, 29‐1) derived from blister cells. We also detected the presence of BV9 (AP018, CU006) and BV29‐1 (AP018, CU006) in OXP‐induced IFNγ+ TCRs derived from PBMC, thereby suggesting these Vβ sequences are not only found at the site of active immunopathology but also systemically. Moreover, the study by Pan et al. 41 examined both blister cells and PBMCs from ALP‐exposed SJS/TEN patients (n = 10) and showed the highest TCRβ chain frequencies (normalized mean values of corresponding gene of healthy donors' PBMC; n = 44) of TRBV3‐1, TRBV4‐2 and TRBV5‐1 in blister cells and TRBV3‐1, TRBV5.5, TRBV7‐3 and TRBV28 in PBMC. Examination of our OXP‐induced IFNγ+ TCR data also detected the presence of TRBV4‐2 (AP013, CU006) and TRBV5‐4 (CU006) in PBMC. While, it should be noted that there are two shared Vβ chains (TRBV3‐1 and TRBV5‐1) identified across the studies by Chung and Pan, 23 , 41 these were not the major Vβ clonotypes represented in our study. For instance, the dominant OXP‐induced IFNγ+ paired αβTCR clonotype identified for ALP‐SJS patient CU002 was TRAV10‐TRAJ33‐CDR3αCVVRKSNYQLIW TRBV20‐1‐TRBJ1‐2‐CDR3βCSARVGQGVSYGYTF, while ALP‐SJS patient CU006 displayed a dominant TRAV27‐TRAJ12‐CDR3αCAGRGMDSSYKLIF that was paired to multiple TCRβ chains. Overall, our data support that OXP‐induced TCRs are oligoclonal and private in nature. While we cannot exclude the possibility that there could be tissue‐specific TCR signatures at different locations (i.e. PBMC vs. blister fluid), there appears to be some overlap. This provides an encouraging opportunity to use PBMC‐derived TCR clonotypes to track the inflammatory status of AHS patients during active disease. Of note, a small number of unique TCR clones in the same individual were shared in both CD8+ and the CD8+ IFNγ+ populations for the same drug or within the CD8+ population across both drugs (i.e. AP013/ALP and OXP, AP018/ALP and OXP).
We also observed CD4+ T cells producing IFNγ following stimulation with OXP in both SJS patients and drug‐naïve healthy donors. This finding correlated with other studies reporting activated CD4+ T cells producing IFNγ, granulysin and CD107a in ALP‐exposed patients. 22 , 23 Overall, we demonstrated that drug‐responsive memory T cells from resolved patients could be reactivated in vitro several years following active disease and these cells showed a similar immunophenotype to studies that had examined active states of AHS. Furthermore, we confirmed in our cohort that the p‐i concept is the likely mechanism for drug‐exposed T cell stimulation. More importantly, this study for the first time reports paired αβTCR clonotypes in both drug‐naïve donors and resolved ALP‐SJS patients that provides evidence of oligoclonal and private repertoire usage.
AUTHOR CONTRIBUTIONS
NAM, PTI, JR, JV and AWP designed experiments and/or provided intellectual input. NAM, PTI, RH, JET and HF performed experiments. RR and AWP provided reagents and/or patient samples. NAM, PTI, RH and KAM analyzed data. NAM wrote the manuscript. All authors read and approved the manuscript.
FUNDING INFORMATION
PTI was supported by a National Health and Medical Research Council of Australia (NHMRC) Early Career Fellowship (1072159, 2014‐2017) and a Monash University Faculty of Medicine, Nursing and Health Sciences Senior Postdoctoral Fellowship (2020). JR and AWP are supported by an NHMRC Investigator awards. We acknowledge funding support from a NHMRC Project grant 1122099 (to AWP and JV).
CONFLICT OF INTEREST STATEMENT
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supporting information
Figure S1
Figure S2
Figure S3
Figure S4
Table S1
Data S1
ACKNOWLEDGEMENTS
The authors acknowledge the Monash University FlowCore Facility for flow cytometry instrumentation and technical support, the services and facilities of Micromon Genomics at Monash University, and the Monash Proteomics & Metabolomics Facility for the provision of mass spectrometry instrumentation, training and technical support. Micromon Computational resources were supported by the R@CMon/Monash Node of the NeCTAR Research Cloud, an initiative of the Australian Government's Super Science Scheme and the Education Investment Fund. Open access publishing facilitated by Monash University, as part of the Wiley ‐ Monash University agreement via the Council of Australian University Librarians.
Mifsud NA, Illing PT, Ho R, et al. The allopurinol metabolite, oxypurinol, drives oligoclonal expansions of drug‐reactive T cells in resolved hypersensitivity cases and drug‐naïve healthy donors. Allergy. 2023;78:2980‐2993. doi: 10.1111/all.15814
Contributor Information
Nicole A. Mifsud, Email: nicole.mifsud@monash.edu.
Anthony W. Purcell, Email: anthony.purcell@monash.edu.
DATA AVAILABILITY STATEMENT
The data that supports the findings of this study are available in the supplementary material of this article
REFERENCES
- 1. Stamp LK, Day RO, Yun J. Allopurinol hypersensitivity: investigating the cause and minimizing the risk. Nat Rev Rheumatol. 2016;12(4):235‐242. [DOI] [PubMed] [Google Scholar]
- 2. Day RO, Graham GG, Hicks M, McLachlan AJ, Stocker SL, Williams KM. Clinical pharmacokinetics and pharmacodynamics of allopurinol and oxypurinol. Clin Pharmacokinet. 2007;46(8):623‐644. [DOI] [PubMed] [Google Scholar]
- 3. Reiter S, Simmonds HA, Zollner N, Braun SL, Knedel M. Demonstration of a combined deficiency of xanthine oxidase and aldehyde oxidase in xanthinuric patients not forming oxipurinol. Clin Chim Acta. 1990;187(3):221‐234. [DOI] [PubMed] [Google Scholar]
- 4. Halevy S, Ghislain PD, Mockenhaupt M, et al. Allopurinol is the most common cause of Stevens‐Johnson syndrome and toxic epidermal necrolysis in Europe and Israel. J Am Acad Dermatol. 2008;58(1):25‐32. [DOI] [PubMed] [Google Scholar]
- 5. Chung WH, Chang WC, Stocker SL, et al. Insights into the poor prognosis of allopurinol‐induced severe cutaneous adverse reactions: the impact of renal insufficiency, high plasma levels of oxypurinol and granulysin. Ann Rheum Dis. 2015;74(12):2157‐2164. [DOI] [PubMed] [Google Scholar]
- 6. Excess of ampicillin rashes associated with allopurinol or hyperuricemia. A Report from the Boston Collaborative Drug Surveillance Program, Boston University Medical Center. N Engl J Med. 1972;286(10):505‐507. [DOI] [PubMed] [Google Scholar]
- 7. McInnes GT, Lawson DH, Jick H. Acute adverse reactions attributed to allopurinol in hospitalised patients. Ann Rheum Dis. 1981;40(3):245‐249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Husain Z, Reddy BY, Schwartz RA. DRESS syndrome: part II. Management and therapeutics. J Am Acad Dermatol. 2013;68(5):709.e1‐9 quiz 718‐720. [DOI] [PubMed] [Google Scholar]
- 9. Sekula P, Dunant A, Mockenhaupt M, et al. Comprehensive survival analysis of a cohort of patients with Stevens‐Johnson syndrome and toxic epidermal necrolysis. J Invest Dermatol. 2013;133(5):1197‐1204. [DOI] [PubMed] [Google Scholar]
- 10. Ramasamy SN, Korb‐Wells CS, Kannangara DR, et al. Allopurinol hypersensitivity: a systematic review of all published cases, 1950‐2012. Drug Saf. 2013;36(10):953‐980. [DOI] [PubMed] [Google Scholar]
- 11. Hung SI, Chung WH, Liou LB, et al. HLA‐B*5801 allele as a genetic marker for severe cutaneous adverse reactions caused by allopurinol. Proc Natl Acad Sci USA. 2005;102(11):4134‐4139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lonjou C, Borot N, Sekula P, et al. A European study of HLA‐B in Stevens‐Johnson syndrome and toxic epidermal necrolysis related to five high‐risk drugs. Pharmacogenet Genomics. 2008;18(2):99‐107. [DOI] [PubMed] [Google Scholar]
- 13. Kaniwa N, Saito Y, Aihara M, et al. HLA‐B locus in Japanese patients with anti‐epileptics and allopurinol‐related Stevens‐Johnson syndrome and toxic epidermal necrolysis. Pharmacogenomics. 2008;9(11):1617‐1622. [DOI] [PubMed] [Google Scholar]
- 14. Tassaneeyakul W, Jantararoungtong T, Chen P, et al. Strong association between HLA‐B*5801 and allopurinol‐induced Stevens‐Johnson syndrome and toxic epidermal necrolysis in a Thai population. Pharmacogenet Genomics. 2009;19(9):704‐709. [DOI] [PubMed] [Google Scholar]
- 15. Goncalo M, Coutinho I, Teixeira V, et al. HLA‐B*58:01 is a risk factor for allopurinol‐induced DRESS and Stevens‐Johnson syndrome/toxic epidermal necrolysis in a Portuguese population. Br J Dermatol. 2013;169(3):660‐665. [DOI] [PubMed] [Google Scholar]
- 16. Kang HR, Jee YK, Kim YS, et al. Positive and negative associations of HLA class I alleles with allopurinol‐induced SCARs in Koreans. Pharmacogenet Genomics. 2011;21(5):303‐307. [DOI] [PubMed] [Google Scholar]
- 17. Ng CY, Yeh YT, Wang CW, et al. Impact of the HLA‐B(*)58:01 allele and renal impairment on allopurinol‐induced cutaneous adverse reactions. J Invest Dermatol. 2016;136(7):1373‐1381. [DOI] [PubMed] [Google Scholar]
- 18. Zanni MP, von Greyerz S, Schnyder B, et al. HLA‐restricted, processing‐ and metabolism‐independent pathway of drug recognition by human alpha beta T lymphocytes. J Clin Invest. 1998;102(8):1591‐1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Illing PT, Mifsud NA, Purcell AW. Allotype specific interactions of drugs and HLA molecules in hypersensitivity reactions. Curr Opin Immunol. 2016;42:31‐40. [DOI] [PubMed] [Google Scholar]
- 20. Phillips EJ, Mallal SA. Pharmacogenetics of drug hypersensitivity. Pharmacogenomics. 2010;11(7):973‐987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yun J, Mattsson J, Schnyder K, et al. Allopurinol hypersensitivity is primarily mediated by dose‐dependent oxypurinol‐specific T cell response. Clin Exp Allergy. 2013;43(11):1246‐1255. [DOI] [PubMed] [Google Scholar]
- 22. Lin CH, Chen JK, Ko TM, et al. Immunologic basis for allopurinol‐induced severe cutaneous adverse reactions: HLA‐B*58:01‐restricted activation of drug‐specific T cells and molecular interaction. J Allergy Clin Immunol. 2015;135(4):1063‐1065 e5. [DOI] [PubMed] [Google Scholar]
- 23. Chung WH, Pan RY, Chu MT, et al. Oxypurinol‐specific T cells possess preferential TCR Clonotypes and express Granulysin in allopurinol‐induced severe cutaneous adverse reactions. J Invest Dermatol. 2015;135(9):2237‐2248. [DOI] [PubMed] [Google Scholar]
- 24. Yun J, Marcaida MJ, Eriksson KK, et al. Oxypurinol directly and immediately activates the drug‐specific T cells via the preferential use of HLA‐B*58:01. J Immunol. 2014;192(7):2984‐2993. [DOI] [PubMed] [Google Scholar]
- 25. Wei CY, Chung WH, Huang HW, Chen YT, Hung SI. Direct interaction between HLA‐B and carbamazepine activates T cells in patients with Stevens‐Johnson syndrome. J Allergy Clin Immunol. 2012;129(6):1562‐1569. [DOI] [PubMed] [Google Scholar]
- 26. Naisbitt DJ, Britschgi M, Wong G, et al. Hypersensitivity reactions to carbamazepine: characterization of the specificity, phenotype, and cytokine profile of drug‐specific T cell clones. Mol Pharmacol. 2003;63(3):732‐741. [DOI] [PubMed] [Google Scholar]
- 27. Mifsud NA, Illing PT, Lai JW, et al. Carbamazepine induces focused T cell responses in resolved Stevens‐Johnson syndrome and toxic epidermal necrolysis cases but does not perturb the Immunopeptidome for T cell recognition. Front Immunol. 2021;12:653710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yawalkar N, Egli F, Hari Y, Nievergelt H, Braathen LR, Pichler WJ. Infiltration of cytotoxic T cells in drug‐induced cutaneous eruptions. Clin Exp Allergy. 2000;30(6):847‐855. [DOI] [PubMed] [Google Scholar]
- 29. Chung WH, Hung SI, Yang JY, et al. Granulysin is a key mediator for disseminated keratinocyte death in Stevens‐Johnson syndrome and toxic epidermal necrolysis. Nat Med. 2008;14(12):1343‐1350. [DOI] [PubMed] [Google Scholar]
- 30. Nassif A, Bensussan A, Boumsell L, et al. Toxic epidermal necrolysis: effector cells are drug‐specific cytotoxic T cells. J Allergy Clin Immunol. 2004;114(5):1209‐1215. [DOI] [PubMed] [Google Scholar]
- 31. Zemmour J, Little AM, Schendel DJ, Parham P. The HLA‐A,B "negative" mutant cell line C1R expresses a novel HLA‐B35 allele, which also has a point mutation in the translation initiation codon. J Immunol. 1992;148(6):1941‐1948. [PubMed] [Google Scholar]
- 32. Barnstable CJ, Bodmer WF, Brown G, et al. Production of monoclonal antibodies to group a erythrocytes, HLA and other human cell surface antigens‐new tools for genetic analysis. Cell. 1978;14(1):9‐20. [DOI] [PubMed] [Google Scholar]
- 33. Kostenko L, Kjer‐Nielsen L, Nicholson I, et al. Rapid screening for the detection of HLA‐B57 and HLA‐B58 in prevention of drug hypersensitivity. Tissue Antigens. 2011;78(1):11‐20. [DOI] [PubMed] [Google Scholar]
- 34. Mifsud NA, Nguyen THO, Tait BD, Kotsimbos TC. Quantitative and functional diversity of cross‐reactive EBV‐specific CD8+ T cells in a longitudinal study cohort of lung transplant recipients. Transplantation. 2010;90(12):1439‐1449. [DOI] [PubMed] [Google Scholar]
- 35. Wang GC, Dash P, McCullers JA, Doherty PC, Thomas PG. T cell receptor alphabeta diversity inversely correlates with pathogen‐specific antibody levels in human cytomegalovirus infection. Sci Transl Med. 2012;4(128):128ra142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Brochet X, Lefranc MP, Giudicelli V. IMGT/V‐QUEST: the highly customized and integrated system for IG and TR standardized V‐J and V‐D‐J sequence analysis. Nucleic Acids Res. 2008;36:W503‐W508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Folch G, Scaviner D, Contet V, Lefranc MP. Protein displays of the human T cell receptor alpha, beta, gamma and delta variable and joining regions. Exp Clin Immunogenet. 2000;17(4):205‐215. [DOI] [PubMed] [Google Scholar]
- 38. Mullan KA, Zhang JB, Jones CM, et al. TCR_Explore: a novel webtool for T cell receptor repertoire analysis. Comput Struct Biotechnol J. 2023;21:1272‐1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Szymczak AL, Workman CJ, Wang Y, et al. Correction of multi‐gene deficiency in vivo using a single 'self‐cleaving' 2A peptide‐based retroviral vector. Nat Biotechnol. 2004;22(5):589‐594. [DOI] [PubMed] [Google Scholar]
- 40. Nguyen TH, Rowntree LC, Pellicci DG, et al. Recognition of distinct cross‐reactive virus‐specific CD8+ T cells reveals a unique TCR signature in a clinical setting. J Immunol. 2014;192(11):5039‐5049. [DOI] [PubMed] [Google Scholar]
- 41. Pan RY, Chu MT, Wang CW, et al. Identification of drug‐specific public TCR driving severe cutaneous adverse reactions. Nat Commun. 2019;10(1):3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Emmerson BT, Hazelton RA, Frazer IH. Some adverse reactions to allopurinol may be mediated by lymphocyte reactivity to oxypurinol. Arthritis Rheum. 1988;31(3):436‐440. [DOI] [PubMed] [Google Scholar]
- 43. Braden GL, Warzynski MJ, Golightly M, Ballow M. Cell‐mediated immunity in allopurinol‐induced hypersensitivity. Clin Immunol Immunopathol. 1994;70(2):145‐151. [DOI] [PubMed] [Google Scholar]
- 44. Pichler WJ. The p‐i concept: pharmacological interaction of drugs with immune receptors. World Allergy Organ J. 2008;1(6):96‐102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Illing PT, Vivian JP, Dudek NL, et al. Immune self‐reactivity triggered by drug‐modified HLA‐peptide repertoire. Nature. 2012;486(7404):554‐558. [DOI] [PubMed] [Google Scholar]
- 46. Norcross MA, Luo S, Lu L, et al. Abacavir induces loading of novel self‐peptides into HLA‐B*57: 01: an autoimmune model for HLA‐associated drug hypersensitivity. Aids. 2012;26(11):F21‐F29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ostrov DA, Grant BJ, Pompeu YA, et al. Drug hypersensitivity caused by alteration of the MHC‐presented self‐peptide repertoire. Proc Natl Acad Sci USA. 2012;109(25):9959‐9964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mifsud NA, Purcell AW, Chen W, Holdsworth R, Tait BD, McCluskey J. Immunodominance hierarchies and gender bias in direct T(CD8)‐cell alloreactivity. Am J Transplant. 2008;8(1):121‐132. [DOI] [PubMed] [Google Scholar]
- 49. Bell CC, Faulkner L, Martinsson K, et al. T‐cells from HLA‐B*57:01+ human subjects are activated with abacavir through two independent pathways and induce cell death by multiple mechanisms. Chem Res Toxicol. 2013;26(5):759‐766. [DOI] [PubMed] [Google Scholar]
- 50. Redwood AJ, Rwandamuriye F, Chopra A, et al. Single‐cell transcriptomics reveal polyclonal memory T‐cell responses in skin with positive abacavir patch test results. J Allergy Clin Immunol. 2019;144(5):1413‐1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ko TM, Chung WH, Wei CY, et al. Shared and restricted T‐cell receptor use is crucial for carbamazepine‐induced Stevens‐Johnson syndrome. J Allergy Clin Immunol. 2011;128(6):1266‐1276 e1211. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1
Figure S2
Figure S3
Figure S4
Table S1
Data S1
Data Availability Statement
The data that supports the findings of this study are available in the supplementary material of this article