

ABSTRACT
Mycobacteria, including the infamous pathogen Mycobacterium tuberculosis, are high-GC Gram-positive bacteria with a distinctive cell envelope. Although there is a typical inner membrane, the mycobacterial cell envelope is unusual in having its peptidoglycan layer connected to a polymer of arabinogalactan, which in turn is covalently attached to long-chain mycolic acids that help form a highly impermeable mycobacterial outer membrane. This complex double-membrane, or diderm, cell envelope imparts mycobacteria with unique requirements for protein export into and across the cell envelope for secretion into the extracellular environment. In this article, we review the four protein export pathways known to exist in mycobacteria: two conserved systems that exist in all types of bacteria (the Sec and Tat pathways) and two specialized systems that exist in mycobacteria, corynebacteria, and a subset of low-GC Gram-positive bacteria (the SecA2 and type VII secretion pathways). We describe the progress made over the past 15 years in understanding each of these mycobacterial export pathways, and we highlight the need for research to understand the specific steps of protein export across the mycobacterial outer membrane.
INTRODUCTION
Mycobacteria are Actinobacteria, which is a phylum of high-GC Gram-positive bacteria. Among the wide range of Mycobacterium spp. are several important pathogens. Most notable of these is Mycobacterium tuberculosis, the causative agent of tuberculosis (TB). Although the number of TB cases and deaths show a declining trend in the past decade, the World Health Organization recently warned that “current actions and investments in research are falling far short” (http://www.who.int/mediacentre/news/releases/2016/tuberculosis-investments-short/en/). Over 1.3 million deaths and up to 10.0 million new infections were attributed to M. tuberculosis in 2017, which makes M. tuberculosis the most deadly infectious agent in the world, surpassing HIV and the malaria parasite (1). One issue facing efforts to control TB is that the live attenuated bacillus Calmette-Guérin (BCG) vaccine strain is not able to provide lifelong protection against M. tuberculosis (2). Another problem is the increased incidence of infections caused by multidrug-resistant M. tuberculosis strains. Consequently, major research efforts focus on understanding M. tuberculosis pathogenesis and physiology to advance the development of new anti-TB vaccines and antibiotics.
M. tuberculosis is a member of the order Corynebacteriales, which is an order that is characterized by its resistance to many classic antibiotics and its ability to withstand highly stressful environments, such as oxidative stress, desiccants, and common disinfectants (3, 4). These features can be explained by the presence of a unique cell envelope, which differs from that of both typical Gram-positive (low-GC), bacteria also called Firmicutes, and Gram-negative bacteria. While members of the Corynebacteriales have a traditional inner membrane, their peptidoglycan layer is connected to an additional polymer of arabinose and galactose (i.e., an arabinogalactan layer), which in turn is linked to mycolic acids, unusually long fatty acids that can contain up to 90 carbon atoms (Fig. 1) (5–8). It is now widely accepted that these mycolic acids are the main constituents of a second (outer) membrane, which also contains noncovalently linked (free) lipids such as phthiocerol dimycocerosates and phenolic glycolipids. Finally, mycobacteria have a more loosely attached capsular layer, which mainly consists of polysaccharides and proteins (9). Although the presence of an outer membrane in mycobacteria was initially proposed in 1982 (10), its existence was not confirmed until 2008 by electron microscopy (EM) imaging (7, 11). Thus, although the order Corynebacteriales belongs to the group of high-GC Gram-positive bacteria, their double-membrane, or diderm, cell envelope is physically more similar to the inner and outer membrane-containing cell envelope of Gram-negative bacteria, because EM images of the two reveal a resemblance. However, despite the resemblance, the mycolic acid outer membrane of Corynebacteriales differs dramatically from the Gram-negative outer membrane, the latter being composed of phospholipids and lipopolysaccharides.
FIGURE 1.
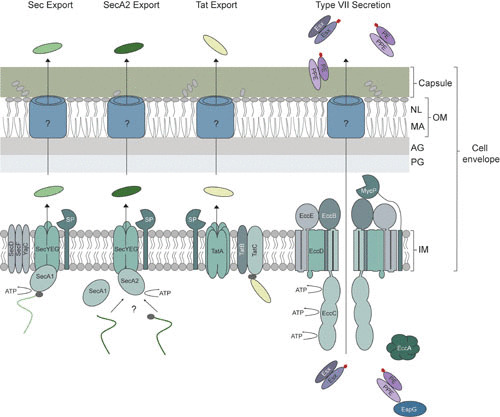
Model of the mycobacterial cell envelope and export systems. Although the mycobacterial cell envelope contains a traditional cytosolic (inner) membrane, the peptidoglycan layer is covalently linked to an arabinogalactan layer consisting of arabinose and galactose, which in turn is covalently linked to mycolic acids. These unusually long fatty acids (up to 90 carbon atoms) are one of the main components of the outer membrane, which also contains noncovalently bound (free) lipids. The final layer of the cell envelope is the capsule, mainly consisting of polysaccharides and proteins. The cell envelope is highly impermeable and unique to other Gram-positive bacteria. To export proteins into and across the cell envelope, mycobacteria have four systems, Sec, SecA2, Tat, and type VII secretion (T7S). The Sec pathway exports unfolded proteins across the inner membrane, and the substrates bind to an ATPase, SecA1, which targets the substrates to the translocation channel consisting of SecYEG and provides energy for translocation. The additional membrane components SecD, SecF, and YajC increase the efficiency of export. Upon translocation across the inner membrane, the N-terminal signal peptide (depicted in gray) is removed by a signal peptidase (SP). Less is known about the SecA2 pathway. Substrates are dependent on the ATPase SecA2; however, studies show that they also utilize the SecYEG channel and possibly SecA1, as well. The list of SecA2-dependent exported proteins includes examples with and without a signal peptide. The third export system, the Tat pathway, exports folded proteins. The Tat substrates, containing an N-terminal signal peptide, with a pair of arginine residues, is targeted to the membrane components TatBC, which then recruit homo-oligomers of TatA for subsequent transportation across the membrane. Similar to Sec, the signal peptide is removed by a signal peptidase. The T7S system consists of five conserved membrane components, of which EccBCDE form the secretion complex. EccC is the ATPase, providing the required energy for the secretion process. Although the mycosin protease (MycP) is not an integral component of the secretion complex, it associates with the complex and is essential for successful secretion. The substrates (Esx and PE-PPE) are secreted as heterodimers and are targeted to the secretion complex by a conserved secretion signal that includes a YxxxD/E motif (depicted in red), whereas the cytosolic chaperone EspG is required for directing the PE-PPE pairs to their specific T7S. The role of the second conserved cytosolic component, EccA, remains uncertain. It is currently unknown how Sec, SecA2, Tat, and T7S substrates are secreted across the outer membrane into the capsular layer or culture supernatant. IM, inner membrane; PG, peptidoglycan layer; AG, arabinogalactan layer; MA, mycolic acids; NL, noncovalently bound lipids; OM, outer membrane.
The firm bond of the long mycolic acids to the underlying arabinogalactan and peptidoglycan structures results in very low fluidity of the outer membrane, which contributes to the high impermeability of the Corynebacteriales cell envelope (5). Although this characteristic provides excellent protection from extreme environmental conditions, it also requires Corynebacteriales species to have pathways to import and export molecules. In line with the notion that the mycolic acid-containing membrane is functionally comparable to the outer membrane of Gram-negative bacteria, both mycobacteria and corynebacteria possess outer membrane porins for importing hydrophilic nutrients through the outer membrane (12–14). At the same time, the unique cell envelope of Corynebacteriales necessitates the existence of special pathways for secreting proteins into the environment.
In this article, we will review the different pathways mycobacteria use to export proteins from the cytoplasm into the cell envelope or beyond into the extracellular environment (i.e., to secrete them across the outer membrane) (Fig. 1). Four types of protein export pathways are currently known in mycobacteria: two highly conserved systems that exist across Gram-positive and Gram-negative bacteria (the Sec and Tat pathways) and two specialized systems that exist in mycobacteria, corynebacteria, and a subset of low-GC Gram-positive bacteria (the SecA2 and type VII secretion [T7S] pathways). The Sec, Tat, and SecA2 pathways serve to transport proteins across the inner membrane, and whether an exported protein remains in the putative periplasmic space between the two membranes or reaches the capsule or extracellular environment is determined by currently unknown secondary mechanisms. T7S pathways differ in that, so far, periplasmic localization of substrates has not been observed, suggesting that this pathway may secrete proteins beyond the outer membrane. However, here too, the mechanism for crossing the outer membrane remains a mystery.
THE MYCOBACTERIAL SEC PATHWAY
Bacteria export a large proportion of their proteome to the cell envelope or extracellular environment (∼20 to 35%) (15, 16). In all bacteria, the majority of this protein export is carried out by the general Sec pathway (16). Mycobacteria have ∼1,000 predicted Sec-exported proteins (this number includes proteins exported across as well as into the inner membrane), many of which have been experimentally demonstrated to be exported (17, 18). Among the many proteins exported by the Sec pathway are proteins with essential functions in the cell envelope (i.e., nutrient acquisition, cell wall biogenesis); consequently, in all bacteria the Sec pathway is essential for viability (19). Further, there are Sec-exported proteins with critical roles in host-pathogen interactions, making the Sec pathway critical for bacterial pathogenesis as well (17, 18, 20). The Sec pathway is highly conserved in bacteria, and most of our understanding of this pathway comes from studies of the Escherichia coli system (for recent reviews of the Sec system see references 21 and 22). Mycobacteria possess conserved orthologues of all the critical Sec pathway components, and as expected, these Sec orthologues are either proven or predicted by saturating mutagenesis transposon-site hybridization or transposon insertion sequencing studies to be essential (23). While only a few mycobacterial Sec proteins have been directly studied, the results so far indicate that the Sec pathway of mycobacteria functions similarly to the Sec pathways of other bacteria.
The Sec pathway transports proteins across the inner membrane. Sec-exported proteins can remain in the cell envelope or through subsequent mechanisms transit across the additional cell envelope layers and be fully secreted. Sec-exported proteins travel across the inner membrane through a channel composed of the integral membrane proteins SecY, SecE, and SecG (Fig. 1), in which SecY forms the central core (24). Sec-exported proteins are in an unfolded state during their translocation through the channel (25). SecY and SecE are sufficient to achieve translocation, while SecG improves the efficiency of export (26–28). There are also additional membrane components that improve Sec export efficiency: SecD, SecF, and YajC (29). The SecA ATPase provides energy for protein translocation (30, 31). SecA binds to Sec-exported proteins in the cytoplasm, targets them to SecYEG in the membrane, and harnesses energy from repeated rounds of ATP binding and hydrolysis to drive stepwise export of unfolded proteins through the SecYEG channel in the inner membrane (32). For mycobacteria, the second step a Sec-exported protein may take to cross the outer membrane remains completely unknown.
While the majority of bacteria have a single essential SecA, mycobacteria and a subset of other Gram-positive bacteria have two SecA proteins (named SecA1 and SecA2) (33–35). As demonstrated in mycobacteria, the two SecAs have unique functions. SecA1 functions in the general Sec pathway, as described above. SecA2 is a specialized SecA and is discussed later in the article. Consistent with SecA1 being the SecA of the essential general Sec pathway, SecA1 of Mycobacterium smegmatis and M. tuberculosis is essential for viability (33, 36; L. Rank and M. Braunstein, unpublished). SecA1 exports proteins with Sec signal peptides (introduced below) (36) and has ATPase activity, as expected for it being the canonical SecA (37).
Sec export is a posttranslational process. Following protein synthesis, Sec-exported proteins exist in the cytoplasm as unfolded preproteins with N-terminal signal peptides that are ∼25 to 30 amino acids long (38). The Sec signal peptide is composed of a positively charged N-terminus, a hydrophobic central domain, and an uncharged polar C-terminus containing a signal peptidase cleavage site (39). For lipoproteins exported by the Sec pathway, there is also a lipobox motif at the C-terminus of the signal peptide with an invariant cysteine that serves as the site of lipid attachment and anchoring of the protein to the cell envelope postexport (40). In association with export, the signal peptide is removed from the preprotein by one of two signal peptidases (LepB for the majority of Sec preproteins and LspA for lipoprotein preproteins specifically), which serves to produce the cleaved, mature protein species on the extracytoplasmic side of the membrane (39). As is the case in all other bacteria studied to date, LepB of M. tuberculosis is essential (41). On the other hand, LspA of M. tuberculosis is not essential, although it is important for pathogenesis (42, 43). The dispensability of LspA is also the case for Gram-positive Firmicutes, while in Gram-negative bacteria, LspA is essential (44).
The general principles of signal peptides established for other bacteria also apply to mycobacterial proteins exported by the Sec pathway, as shown by in silico prediction programs for Sec signal peptides being good predictors of M. tuberculosis-exported proteins (18) and by comparing in silico predictions to experimentally determined N-terminal signal peptides of M. tuberculosis (45).
In addition to possessing a signal peptide, another required feature of Sec-exported proteins is that they must be unfolded to be exported (25, 46, 47). Cytosolic chaperones can help preproteins maintain an unfolded and translocation-competent state, and the SecA-SecYEG translocase may also play a role (48, 49). Gram-negative bacteria have a SecB chaperone that maintains a subset of preproteins in an unfolded state compatible with transport through SecYEG (49, 50). SecB also has a role in delivering preproteins to SecA (51). However, SecB is not present in Gram-positive bacteria. In M. tuberculosis, there is a SecB-like protein (Rv1957); however, Rv1957 is not conserved across mycobacteria, and the available data indicate that Rv1957 is a chaperone for the HigBA toxin-antitoxin system and not a protein export chaperone (52).
In addition to the above described posttranslational export of proteins across the inner membrane, the Sec pathway also participates in cotranslational export of transmembrane domain (TMD)-containing integral membrane proteins into the membrane (53). With membrane proteins, once the TMD emerges from the ribosome, the nascent polypeptide is recognized by the signal recognition particle (SRP), (comprising the Ffh protein and the 4.5S RNA), which then delivers the ribosome-mRNA-nascent protein complex to the SRP receptor FtsY (54). The SRP receptor in turn delivers the nascent protein to the SecYEG channel for translocation. For integral membrane proteins, there is a lateral gate in SecY that allows TMDs to transfer out of the channel and integrate into the inner membrane through a process that is facilitated by the integral membrane YidC protein (55). Specifically, for membrane proteins with large periplasmic domains, SecA may assist in exporting these domains across the membrane (56). Both Ffh and FtsY are GTPases, and GTP hydrolysis is required for membrane protein delivery (57). Using purified proteins, M. tuberculosis Ffh and FtsY are shown to possess functions of their E. coli counterparts; they possess GTPase activity and interact with one another and with the 4.5S RNA of M. tuberculosis (58, 59). Working with a conditional yidC depletion strain of M. tuberculosis, YidC was demonstrated to be required for growth and membrane protein localization, as expected for a YidC ortholog (60). However, in the fast-growing M. smegmatis, yidC depletion has no effect on growth (60).
THE MYCOBACTERIAL Tat PATHWAY
The twin-arginine translocase (Tat) pathway is another conserved export system of bacteria. Again, our understanding of the Tat pathway comes largely from studies of the E. coli system (for a recent review see reference 61). Similar to the Sec pathway, after transport from the cytoplasm, Tat-exported proteins can either remain in the cell envelope or be fully secreted via a subsequent mechanism. Here too, the pathway for crossing the outer membrane remains a mystery. Proteins exported by the Tat pathway are synthesized as preproteins with cleavable N-terminal signal peptides. However, as the name of the pathway reflects, Tat signal peptides are distinguished from Sec signal peptides by a Tat motif containing a pair of twin arginine residues (see below) (62). Compared to the Sec pathway, fewer total proteins are exported by the Tat pathway. Some Streptomyces strains are estimated to have ∼150 proteins exported by the Tat pathway, and M. tuberculosis may have as many as 95 proteins (63), whereas other bacteria have far fewer or lack a Tat pathway completely (64, 65). Most notably, the Tat pathway is fundamentally different from the Sec pathway in that folded, rather than unfolded, proteins are exported (66). The studies conducted so far of the mechanism of Tat export in mycobacteria indicate that the Tat system of mycobacteria functions similarly to the Tat pathways of other bacteria.
Like in E. coli, in high-GC Gram-positive actinobacteria, including mycobacteria, the Tat pathway is made up of three integral membrane proteins: TatA, TatB, and TatC (Fig. 1) (67–69). TatA and TatB are small homologous membrane proteins, each containing a single TMD. TatC is a larger integral membrane protein (70). In Firmicutes, however, the Tat pathway is a streamlined system with only TatA and TatC (65).
Tat preproteins are recognized by a complex of TatB and TatC, with TatC recognizing the twin arginine signal peptide (71, 72). Once a preprotein is bound to TatBC, homo-oligomers of TatA are recruited to the complex (71) and the Tat preprotein is transported across the membrane. Tat export is a posttranslational process, and the energy required to drive Tat transport comes from the proton motive force (73, 74). The mechanism of transit across the membrane remains a significant unknown. Some models propose that TatA oligomers form a substrate-fitted protein-conducting channel (75) that accommodates folded proteins of different sizes and shapes. An alternative model proposes that TatA multimers lead to destabilization of the membrane in the vicinity of the TatBC-substrate complex and thereby allow TatC-driven translocation of the substrate without the need of a channel (76). Following export, Tat preproteins undergo signal peptide cleavage by the same signal peptidases that work with the Sec pathway: LepB or LspA (77, 78).
Tat signal peptides resemble Sec signal peptides in having a positively charged N-terminus, a hydrophobic central domain, and an uncharged polar C-terminus containing a cleavage site. However, Tat signal peptides are distinguished from Sec signal peptides by the presence of a Tat motif with twin arginines (R-R-X-Φ-Φ [Φ = hydrophobic]) (62). Substitution of the twin arginines with other residues prevents export of most Tat-dependent proteins, but there are a few examples of Tat-exported proteins with only one arginine (79–81). In mycobacteria, Tat signal peptides appear to follow the same rules established in other bacteria. So far, 18 M. tuberculosis proteins are confirmed as having functional Tat signal peptides (for a list see reference 23). Working with the endogenous β-lactamase of M. tuberculosis (BlaC), which is a Tat-exported protein, the requirement of the twin RR pair for mycobacterial Tat export was confirmed (63, 67). Since β-lactamases must be exported to protect bacteria against β-lactam antibiotics, the requirement of the Tat pathway for BlaC export contributes to the natural resistance of M. tuberculosis to β-lactams.
In addition to a Tat signal peptide, the second required feature of Tat-dependent proteins is that they are folded prior to export. Unfolded or misfolded proteins are generally unacceptable for export, suggesting that the Tat export system has a “proofreading” ability that restricts export to properly folded proteins (66, 82).
In addition to fully exporting proteins across the membrane, the Tat pathway plays a role in the delivery of a small subset of integral membrane proteins into the inner membrane. These cases either involve a protein with a Tat signal peptide and a C-terminal TMD, with the latter becoming integrated into the membrane during translocation, or a protein with a noncleavable N-terminal Tat signal peptide that remains anchored in the membrane (65, 83, 84). Most recently, examples of integral membrane proteins that require both the Sec and Tat pathways to deliver and transport across the membrane multiple periplasmic domains were identified. The role of the Tat pathway in this interesting subset of proteins is to translocate periplasmic domains that, similar to many soluble Tat substrates, must bind a cofactor in the cytoplasm and, therefore, fold prior to export. An internal Tat motif is present in such Tat-dependent periplasmic domains. The first example of a dual Sec- and Tat-dependent membrane protein was the Streptomyces Rieske iron-sulfur binding protein, which is a subunit of the cytochrome bc1 complex (85).
In general, the Tat pathway is not essential for bacterial viability, and tat mutants are viable, at least in standard laboratory media (65). This is not the case, however, for M. tuberculosis, which provides one of the few examples of a bacterium with an essential Tat pathway (69). In fact, even in the fast-growing and nonpathogenic mycobacterium M. smegmatis, the Tat system is not essential, although tatA, tatB, and tatC deletion mutants have in vitro growth defects (67, 68). A possible explanation for the essentiality of the M. tuberculosis Tat pathway is that, like the case in Streptomyces, the mycobacterial Tat pathway may be required for export of the iron-sulfur-bound periplasmic domain of the Rieske membrane protein (i.e., QcrA) subunit of cytochrome bc1 since in M. tuberculosis the cytochrome bc1 complex is essential (86). In support of this possibility, the iron-sulfur binding domain of M. tuberculosis QcrA has an internal Tat motif (RRKLI).
THE MYCOBACTERIAL SecA2 PATHWAY
Mycobacteria are unusual in having two SecA homologues, but they are not the only bacteria with two SecAs. Along with other Actinobacteria (e.g., corynebacteria) (87), there is a subset of Firmicutes, including Listeria (88), Clostridioides (89), Staphylococcus (90), some Streptococci (91–93), and some Bacilli (94) with two SecAs. In all these cases, SecA1 is the name given to the canonical and essential SecA ATPase of the general Sec pathway, and SecA2 refers to a specialized SecA that promotes export of a relatively small set of proteins. It is important to note that not all SecA2 systems are similar or evolutionarily conserved (35, 95). SecA2 systems can be divided into at least two groups: mycobacteria, Listeria monocytogenes, and Clostridioides difficile provide examples of SecA2 systems in which SecA2 appears to work with the canonical SecYEG channel of the general Sec pathway to export a small but diverse set of proteins (89, 96, 97). These systems are called multisubstrate SecA2 systems or SecA2-only systems, the latter name referring to their lack of a second SecY (SecY2) in the system. Staphylococcus and Streptococci are examples of bacteria with SecA2 systems that include an accessory SecY (SecY2) and additional accessory Sec proteins (98). These latter systems export a single large glycosylated substrate, and they are called accessory Sec (aSec) or SecA2-SecY2 systems and will not be discussed further here.
Composition of the Mycobacterial SecA2 Pathway
The SecA2 pathway exists in all mycobacteria. In contrast to SecA1 of the general Sec pathway, the role of SecA2 in mycobacterial protein export is limited to a smaller set of proteins, and SecA2 is not essential for mycobacterial growth in vitro (99–102). In pathogenic mycobacteria, SecA2 is required for pathogenesis (99–101). Early studies of the SecAs of mycobacteria demonstrated that SecA2 and SecA1 are functionally distinct proteins. Even if SecA2 is overexpressed, it cannot replace the requirement for SecA1, and if SecA1 is overexpressed, it cannot carry out the function of SecA2 (33). The functional differences between SecA1 and SecA2 remain a big unknown. Both SecAs have ATPase activity (37), and the crystal structures of M. tuberculosis SecA1 and SecA2 reveal a surprisingly high level of structural similarity, with both SecAs possessing the functional domains of canonical SecAs (103).
As shown with SecA2 variants harboring amino acid substitutions in the ATP binding site of M. smegmatis SecA2 (K129R) or M. tuberculosis SecA2 (K115R), ATP binding is required for SecA2 to carry out its function in protein export and pathogenesis (37, 104). Interestingly, secA2 K129R in M. smegmatis is dominant negative and associated with more severe phenotypes than a secA2 null mutant. Moreover, SecA2 K129R is associated with reduced levels of the sole SecY of mycobacteria, and suppressors of the severe secA2 K129R phenotypes map to secY (96). These results support a model whereby the severe phenotypes of SecA2 K129R result from a detrimental interaction with the canonical SecYEG channel of the general Sec pathway. By extension, these effects of SecA2 K129R argue for SecA2 exporting proteins through the same SecYEG channel that is used by SecA1 (Fig. 1). Data from L. monocytogenes and C. difficile are consistent with SecA2 working with the canonical SecYEG channel in these other multisubstrate SecA2-only systems as well (89, 97).
Along with SecY, SecA1 may also be important for mycobacterial SecA2-dependent export. SecA proteins are known to dimerize, and a recent study demonstrated heterodimer (as well as homodimer) formation in vitro with M. tuberculosis SecA1 and SecA2 proteins purified from E. coli (105). While it remains to be demonstrated if SecA1-SecA2 heterodimers are biologically relevant, another finding in support of a role of SecA1 in the SecA2 pathway is that export of SecA2-dependent substrates in M. smegmatis is compromised when SecA1 is depleted. However, it is also possible that this latter result reflects a role for SecA1 in delivering components of the SecYEG channel to their membrane location (104).
The convergence of SecA1 and SecA2 on the same translocation channel raises the question of what distinguishes the SecA2 pathway from the general Sec pathway. We expect there are distinctive features of SecA2 substrates and additional components of the SecA2 pathway that distinguish it from the general Sec pathway.
The Multisubstrates of the Mycobacterial SecA2 Pathway
Through a combination of discovery proteomics and direct testing of candidates, proteins that are exported by the SecA2 pathway have been identified in M. tuberculosis, Mycobacterium marinum, and M. smegmatis (99, 101, 102, 106). Across these species, there are currently 15 validated examples of proteins that are exported in a SecA2-dependent manner (Table 1). Some of these proteins localize to the cell envelope, and others are fully secreted. From proteomics experiments, there are additional candidates for SecA2-dependent proteins that remain to be validated. The current list of SecA2-dependent proteins includes examples of proteins with or without predicted N-terminal signal peptides or TMDs. In the SecA2-only system of L. monocytogenes, the list of proteins identified as SecA2 dependent also includes examples with or without signal peptides (107). The list of proteins exported by the mycobacterial SecA2 pathway reveals functional themes.
TABLE 1.
Validated examples of SecA2-dependent exported proteins of mycobacteria
| Protein | Signal peptide/transmembranea | Species | Function | Reference |
|---|---|---|---|---|
| Ms1704 | SP | M. smegmatis | SBP | 106 |
| Ms1712 | SP | M. smegmatis | SBP | 106 |
| PhoS1 (Rv0928) | SP | M. tuberculosis | SBP | 102 |
| DppA (Mmar_5154) | SP | M. marinum | SBP | 101 |
| Mce1A (Rv0169) | TM | M. tuberculosis | Mce transporter component | 102 |
| Mce1C (Rv0171) | SP | M. tuberculosis | Mce transporter component | 102 |
| Mce1E (Rv0173) | SP | M. tuberculosis | Mce transporter component | 102 |
| Mce1F (Rv0174) | TM | M. tuberculosis | Mce transporter component | 102 |
| SapM (Rv3310) | SP | M. tuberculosis | Phosphatase effector | 111 |
| PknG (Rv0410c) | None | M. tuberculosis M. marinum | Kinase effector | 101, 102 |
| LipO (Rv1426c) | SP | M. tuberculosis | Predicted esterase (possible effector) | 102 |
| SodA (Rv3846) | None | M. tuberculosis | Superoxide dismutase | 99, 293, 294 |
| KatG (Rv1908c) | None | M. tuberculosis | Catalase-peroxidase | 99 |
| LipA (Mmar_2284) | SP | M. marinum | Cell wall hydrolase | 99 |
| MMAR_3060 | SP | M. marinum | Conserved hypothetical | 101 |
SP, Signal peptide; TM, transmembrane.
Solute binding proteins (SBPs), a category of transporter proteins, are exported by the SecA2 pathways of M. tuberculosis, M. marinum, and M. smegmatis (99, 101, 102, 106). SBPs are exported into the cell envelope to carry out functions in delivering solutes to membrane-localized ABC (ATP-binding cassette) transporters for import into the cell. The first SBPs identified as exported by the SecA2 pathway are the M. smegmatis Msmeg1704 and Msmeg1712 proteins (106), which are predicted to function in sugar import. SBPs are also identified as being exported by the SecA2 pathways of M. tuberculosis and M. marinum (101, 102). Of 15 SBPs identified by quantitative proteomics of M. tuberculosis, 13 are detected as being exported in a SecA2-dependent manner (102). SBPs are also on the list of SecA2-exported proteins for the L. monocytogenes SecA2-only system (107). It is worth noting that SBPs exist across bacteria (i.e., they are not always exported by SecA2 systems). The SecA2-dependent SBPs of mycobacteria possess recognizable N-terminal signal peptides containing a lipobox motif, with the majority having predicted Sec signal peptides. However, 4 of the 13 SecA2-dependent SBPs of M. tuberculosis are predicted to have Tat signal peptides (102).
Mce proteins, another category of transporter proteins, are also exported in a SecA2-dependent manner in mycobacteria (101, 102). Comparative proteomics of cell envelope fractions of wild-type versus secA2 mutant M. tuberculosis and M. marinum strains identified numerous components of cell envelope-localized Mce transporters as SecA2-dependent; Mce components are also SecA2-dependent in M. smegmatis (L. Rank and M. Braunstein, unpublished). These exported Mce components possess either a signal peptide or transmembrane domain. Mce transporters are thought to function similarly to ABC transporters in that they recognize an extracytoplasmic solute (in this case, a lipid) and deliver it to a membrane complex that imports it across the membrane using ATP hydrolysis (108). In an interesting similarity to SecA2-dependent SBPs, each Mce transporter (M. tuberculosis has four Mce transporters) includes six exported Mce proteins that are thought to function like SBPs. It remains to be resolved if SecA2 is required for export of numerous individual components of Mce transporter complexes or if the role of SecA2 is to export one or a small number of Mce proteins that are required to stabilize the entire Mce complex in the cell envelope. In the latter scenario, a defect in the export of a single Mce protein may account for the observed reduction in abundance of numerous Mce components in a secA2 mutant.
Effectors of phagosome maturation arrest are another category of protein exported by the SecA2 pathway. A hallmark of pathogenic mycobacteria that survive in macrophages, such as M. tuberculosis and M. marinum, is their ability to arrest the phagosomes in which they reside from maturing and fusing with degradative lysosomes (109). As a result, pathogenic mycobacteria are in a phagosomal environment that is permissive for replication and sequestered from antigen-presenting compartments that drive immune responses. Phagosome maturation arrest is a complex process involving numerous M. tuberculosis proteins (i.e., effectors) that must be exported to reach their site of action at the host-pathogen interface. In M. tuberculosis and M. marinum, the SecA2 pathway is required for phagosome maturation arrest (101, 110), and SapM and PknG are two SecA2-dependent effectors of this process (101, 111). SapM is a secreted phosphatase that dephosphorylates phosphatidyl inositol-3-phosphate and thereby limits phagosomal recruitment of host factors that drive downstream phagosome fusion and maturation events (112). PknG is a eukaryotic-like serine threonine kinase that is found in both the cytoplasm and cell envelope of pathogenic mycobacteria (113). From its exported location, PknG has a poorly defined role in phagosome maturation arrest (114), and from its cytoplasmic location it has better-characterized roles in metabolism and redox homeostasis (113, 115, 116). Through experiments in which wild-type levels of exported SapM and/or PknG were restored to the secA2 mutant of M. tuberculosis or M. marinum, the role of the SecA2 pathway in exporting these proteins was proven to be required for phagosome maturation arrest (101, 111). However, since the combined effect of restored SapM and PknG export is not sufficient to fully restore phagosome maturation arrest to the secA2 mutant, additional SecA2-dependent effectors must exist. A candidate for an additional factor is the M. tuberculosis lipoprotein LipO, which was identified in a screen for phagosome maturation arrest effectors of M. tuberculosis (117). LipO, which was initially detected as being SecA2 dependent by comparative proteomics (102), was recently validated as being exported in a SecA2-dependent manner (K. Zulauf and M. Braunstein, unpublished). Although there is a clear role for the SecA2 pathway in exporting effectors of phagosome maturation arrest, it is important to note that not all effectors of this process are exported by the SecA2 pathway.
Proteins lacking signal peptides are another category of SecA2-dependent protein. PknG, SodA (superoxide dismutase), and KatG (catalase-peroxidase) make up this subset of SecA2-dependent proteins (99, 101, 102). Although there are multiple reports of all three of these proteins being exported by mycobacteria (i.e., localized to the cell envelope or secreted fractions), they lack any obvious signal peptide or TMD, which makes them unconventional exported proteins (113, 114, 118–120). At the same time, these proteins are also known to reside in the mycobacterial cytoplasm, where they have physiological functions (113, 115, 116, 118, 121). Interestingly, the SecA2-only pathway of L. monocytogenes is also reported to export a SodA protein lacking a signal peptide (122). However, nothing is known about the recognition of proteins lacking signal peptides by the SecA2 pathway, and it remains possible that the effect of SecA2 on their export is indirect. For example, there may be signal peptide-containing proteins exported by SecA2 that are themselves responsible for export of these unconventional proteins.
The Defining Features of SecA2 Substrates
While there are exceptions, the majority of SecA2-exported proteins possess N-terminal signal peptides. The role of the signal peptide in export of SecA2 substrates was evaluated with Msmeg1704, Msmeg1712 (123), and SapM (K. Zulauf and M. Braunstein, unpublished). As is the case for proteins exported by the general Sec pathway, the signal peptide on these SecA2 substrates is necessary for protein export to occur, and the signal peptide is cleaved in association with export to release the mature protein (106). Experiments in which the signal peptide of a SecA2-dependent and SecA1-dependent substrate are swapped demonstrate that the signal peptide does not determine whether a protein is exported by the SecA2 pathway (123). Rather, the mature domains of SecA2-exported proteins impart the requirement for SecA2 in their export.
The defining feature of the mature domain that dictates the need for SecA2 in export may be a propensity to fold in the cytoplasm prior to export. In support of this idea, when the Sec signal peptide of Msmeg1704 is exchanged for a signal peptide that directs preproteins for export through the Tat pathway, Msmeg1704 is exported by the Tat pathway (123). Since the Tat pathway requires preproteins to be folded prior to export, this suggests that the mature domain of a SecA2 substrate can fold in the cytoplasm prior to export.
Since there is no SecB protein export chaperone in mycobacteria, one possibility is that the SecA2 pathway is adapted to facilitate export through the SecYEG channel of problematic substrates that can fold in the cytoplasm. Along these lines, a mycobacterial protein export chaperone named SatS was recently identified as playing a role in the SecA2 pathway (124). SatS stabilizes and prevents aggregation of a subset of SecA2 substrates. Thus, SatS may serve to keep SecA2 substrates in an unfolded state compatible with transport through SecYEG and thereby help the SecA2 pathway export proteins that have the potential for folding in the cytoplasm.
MYCOBACTERIAL T7S PATHWAYS
Substrates of the T7S pathway are among the most abundantly secreted proteins, indicating their ability to efficiently cross the mycobacterial outer membrane (125). Although conserved throughout the phylum Actinobacteria (see below), this secretion pathway was first described in mycobacteria, and in earlier literature T7S was referred to by multiple names, such as ESAT-6 secretion system (Ess), WXG100 secretion system (Wss), and Snm (secretion in mycobacteria) system (126, 127). However, with the visualization of the diderm cell envelope and the observation that T7S substrates are efficiently secreted into the extracellular environment and therefore need to pass two membranes, the term T7S, in line with the specialized secretion systems found in Gram-negative bacteria, became the accepted standard name (128, 129). Mycobacteria can contain up to five T7S systems, named ESX-1 through ESX-5 (125, 130). Of these five systems, the ESX-1 system was the first one to be discovered, being responsible for the secretion of the EsxA and EsxB proteins, also known as ESAT-6 and CFP-10, respectively, in M. tuberculosis (131, 132). The importance of the ESX-1 system for the virulence of M. tuberculosis is clearly demonstrated by the observation that deletion of a large portion of the esx-1 gene cluster, resulting in a nonfunctional system, is the decisive determinant for attenuation of the vaccine strain Mycobacterium bovis BCG (133–135). Further studies, in both M. tuberculosis and the closely related fish pathogen M. marinum, showed that the ESX-1 system is crucial for intracellular survival in macrophages by mediating lysis of the phagosomal membrane (136). This membrane permeabilization has been linked to phagosomal escape (137, 138). Additionally, phagosome permeabilization by the ESX-1 system is likely required for the delivery of secreted effectors from the phagosomal lumen to the host cell cytosol, including effectors secreted from mycobacteria by other ESX systems, SecA, SecA2, or Tat. Although this permeabilization has been mainly attributed to the ESX-1-secreted protein EsxA, different studies provide conflicting results (138–142). Further research is therefore required to pinpoint the ESX-1 substrate that is responsible for the phagosomal membrane rupture (138–142). Besides its pivotal role in the virulence of pathogenic mycobacteria, ESX-1 is also present and functional in nonpathogenic mycobacteria such as the fast-growing M. smegmatis. Intriguingly, the ESX-1 system of M. smegmatis seems to have a very different role, of promoting DNA conjugation, which will be discussed later (143). The diverse roles of ESX-1 are probably caused by a difference in the ESX-1 substrates that are secreted by different species. Indeed, M. smegmatis lacks several ESX-1 substrates that are present in M. tuberculosis and M. marinum and have been linked to virulence (136).
The role of the ESX-1 system in virulence and pathogenicity has been a major focus of TB-related research for more than a decade. The function and importance of several of the other ESX systems have only been described in the past few years. Strikingly, while the ESX-1 system is dispensable for growth in culture, both the ESX-3 and ESX-5 systems are necessary for bacterial viability in M. tuberculosis. The essentiality of the ESX-3 system is due to its role in iron and zinc uptake (144, 145). Possibly linked to its role in iron acquisition, ESX-3 is one of the most conserved ESX systems, being present in all mycobacterial species, of which genome sequences are available. In addition to this, the ESX-3 substrates EsxH, PE15, and PPE20 also play a role in immune modulation, e.g., EsxH is required for inhibition of phagosome maturation (146, 147). The ESX-5 system, on the other hand, is only present in the subgroup of so-called slow-growing mycobacteria, which contains most pathogenic species, such as M. tuberculosis, Mycobacterium leprae, and M. marinum (148). Although it is still unknown what the exact role of the ESX-5 system is, its essentiality for growth has been linked to outer membrane permeability and nutrient uptake in M. marinum (149). Furthermore, the ESX-5 system has been shown to be involved in immune modulation of the host as well (150–154). The characterization of the exact roles of the ESX-5 system is complicated by the large number (>100) of substrates that it secretes.
The roles of the ESX-2 and ESX-4 systems and the substrates they secrete remain unknown. Several studies have shown that these systems are not essential for virulence or in vitro growth of M. tuberculosis (155, 156), although a transposon insertion sequencing study found that mutations in the ESX-2 and ESX-4 systems resulted in a reduced fitness in dendritic cells (157). In contrast to this, a recent study by Laencina et al. showed that the ESX-4 system of the opportunistic pathogen Mycobacterium abscessus is not only functional but even plays a critical role in virulence (158), because ESX-4 transposon and deletion mutants showed reduced survival both in amoeba and murine cells. This phenotype was suggested to be the result of impaired phagosomal rupture, a feature generally attributed to the ESX-1 system in M. tuberculosis and M. marinum. Because M. abscessus lacks the ESX-1 system, ESX-4 may carry out its role in this species. Finally, in M. smegmatis, ESX-4 seems to be, together with ESX-1, involved in DNA conjugation as well (see below) (159, 160).
In addition to these five ESX systems that are genomically encoded, many other esx loci have been identified that are located on plasmids (161–163). In addition, homologous esx gene clusters are present throughout the phylum Actinobacteria, not only in species with a diderm cell envelope similar to that in mycobacteria, such as in corynebacteria and nocardia, but also in species that lack a mycolic acid-containing outer membrane (130, 164, 165). Lastly, more distantly related systems are present and functional in a subset of Firmicutes, such as Staphylococcus aureus, Streptococcus intermedius, and Bacillus subtilis. Because most T7S components present in Actinobacteria are not conserved in the Firmicutes systems, Firmicutes systems are referred to as type VIIb secretion systems, opposed to the type VIIa systems of Actinobacteria (130, 166, 167).
Composition and Mechanism of Mycobacterial ESX Systems
The ESX systems of mycobacteria contain a set of conserved components. During the early years of T7S-related research, genome annotation numbers were often used to refer to these components. To avoid confusion, researchers in the T7S field proposed a systematic nomenclature based on the genetic composition of the systems, which is now the accepted standard (128). Based on this nomenclature, genes that are conserved in at least four of the mycobacterial ESX systems are defined as ecc, for esx conserved component. The system-specific genes, in most cases encoding secreted substrates, are designated esp, for esx-specific protein. There are two exceptions to this rule, mycP and espG. Although the mycosin proteases (MycP) are conserved in all ESX-systems, the term “mycosin” was already an established name and was therefore not altered. For the second exception, EspG was originally not identified as a conserved component due to low homology between the different espG genes. However, the EspG proteins, present in four of the ESX systems, have now been shown to be functional homologues (168, 169). For the major substrate groups, the previously defined terms were kept. The first of these groups are called the Esx proteins, because “EsxA” (ESAT-6) and “EsxB” (CFP-10) were already frequently used terms for two members of this group. The second, larger group contains the so-called PE and PPE proteins, referring to the presence of a proline-glutamic acid (PE) or proline-proline-glutamic acid (PPE) N-terminal amino acid motif (128, 130, 154, 170).
ESX substrates
All mycobacterial ESX systems are encoded by genes that are clustered in distinctive genomic loci. In the middle of these gene clusters is always a pair of esx genes. These esx genes, which are cotranscribed, encode small proteins of the WXG100 family, named after the conservation of a tryptophan and glycine residue separated by a random amino acid and their length of roughly 100 amino acids (166, 171). The esx genes are not limited to the ESX loci; additional esx gene pairs can be found throughout the genome sequence (130, 148). The two Esx proteins that are encoded from the bicistronic transcript form heterodimers in the cytosol and are codependent on each other for secretion (Fig. 1) (172). For example, the ESX-1 system contains the well-described protein pair EsxA-EsxB, which are believed to be secreted as a dimer (173, 174). However, whereas EsxA has been shown to play a major role in virulence, this has not been shown for EsxB (138, 175). From this observation, it is postulated that EsxB is the chaperone for EsxA and that their interaction is required for successful secretion. Because their interaction is required for successful secretion, an esxB mutant shows a similar phenotype as an esxA mutant (139, 176). The heterodimer EsxA-EsxB is the first Esx pair for which a high-resolution structure has been solved (174). The structure shows the presence of a helix-turn-helix motif in both proteins, where the turn contains the WXG motif and the double helices interact in an antiparallel orientation with the double helices of the partner. Furthermore, it shows that the N- and C-terminal tails are flexible. Predicted structural similarities suggest that the EsxA-EsxB structure can serve as a model for the other Esx proteins, which was confirmed by the crystal structures of the ESX-3-associated heterodimeric substrates EsxG-EsxH and EsxR-EsxS (177, 178).
Genes encoding the second substrate group, the PE and PPE proteins, are also present in four of the esx loci; only the ESX-4 gene cluster does not contain any pe or ppe genes. However, most pe and ppe genes are not associated with these loci (130, 148, 154, 179). In fact, pathogenic mycobacteria contain up to 99 pe and 69 ppe genes and, e.g., in M. tuberculosis approximately 10% of the encoding genome belongs to pe and ppe genes. While the ESX-1 and ESX-3 systems are responsible for the secretion of some PE and PPE proteins (147, 154, 180), the majority of the PE and PPE proteins are secreted through the ESX-5 system (150). Similar to the Esx proteins, several pe and ppe genes are clustered in a bicistronic operon, consisting of one pe and one ppe gene, and at least some of the corresponding proteins form heterodimers (169, 181). These heterodimers are often dependent on each other for stability and solubility (Fig. 1) (169, 181, 182).
Substrates belonging to the PE/PPE protein group are often bound to the cell surface, which is in contrast to the Esx proteins that are mainly secreted into the culture medium (183, 184). These substrates consist of a distinctive N- and C-terminal domain: whereas the N-terminal PE and PPE domains, with a length of ∼110 and 180 amino acids, respectively, are highly conserved and are involved in the secretion process, the C-terminal domains are less conserved and are thought to be involved in protein-specific functions. The resolved crystal structures of PE25-PPE41 and PE18-PPE15 reveal structural similarities between Esx and PE-PPE heterodimers (182, 185). While the PE domain only consists of two alpha helices, the PPE domain contains five alpha-helices, of which helices α2 and α3 mediate dimerization with the PE partner, forming a four-helix bundle similar to Esx pairs. Unlike Esx dimers, only the PPE protein contains a WXG motif in the turn between these two helices. The fourth and fifth alpha helices of the PPE domain form an extending hydrophobic tip that is recognized by the EspG component, which will be discussed below (169, 180, 181, 185). The size of the more variable C-terminal domains of PE and PPE proteins can be large; for example, the subgroup of PE_PGRS (polymorphic GC-rich repetitive sequence) proteins can have C-terminal domains of over 1,400 amino acids (148). Only a minority of these C-termini contain predicted functional domains, e.g., executing predicted lipase activity. Consequently, a function has been described for only a few PE-PPE proteins (186, 187).
The final group of substrates, the Esp proteins, are specific for the ESX-1 system (188–191). Although less is currently known about the secretion of these substrates, EspA and EspC are cotranscribed and have been shown to be codependent on each other for secretion and interact to form a heterodimer (188, 192), similar to what is likely the case for EspE and EspF (184, 188, 193). In contrast to this, another ESX-1 substrate, EspB, has a PE-PPE dimer-like fold, forming a double helix-turn-helix by itself, suggesting that it is secreted as a monomer (194, 195). Whether the final Esp substrates, EspK and EspJ, are secreted as monomers or heterodimers remains unknown (190, 191).
An intriguing feature of ESX-1 substrates is that there are some cases of unpaired substrates being codependent on each other for secretion; i.e., the secretion of EspA-EspC is affected by mutations in esxA-esxB, and vice versa (176, 188, 196). This codependence of ESX-1 substrates complicates the understanding of the mechanism of secretion by the ESX-1 system, because a mutation in, e.g., specific chaperones, will also affect the secretion of ESX-1 substrates that do not interact with this chaperone. Furthermore, it makes it difficult to pinpoint specific functions for individual ESX-1 substrates (132, 176, 197). Some level of codependence of substrates for secretion has also been reported for ESX-3 and ESX-5 substrates. For ESX-3, deletion of esxH has been shown to abolish secretion of ESX-3-dependent PE-PPE proteins, although this does not happen the other way around (147). The ESX-5 substrate, PPE38, is required for secretion of the two largest ESX-5 substrate groups, PPE_MPTR and PE_PGRS (198). Although the mechanism for this dependency on PPE38 is currently unknown, PPE38 appears to be of great importance because the emerging M. tuberculosis Beijing isolates share a polymorphism in the ppe38 locus (198). In addition, the ESX-5 substrate PPE10 affects the integrity of the capsular layer of mycobacteria (see below) (199), indirectly affecting the proper localization of other substrates, not only of ESX-5 but also of the ESX-1 system (199).
ESX secretion signal and the cytosolic chaperone EspG
Unlike Sec or Tat substrates, the ESX substrates do not contain an N-terminal signal peptide; instead, the Esx, PE, and Esp substrates have a C-terminal secretion signal directly downstream of a helix-turn-helix. In the case of the Esx and PE-PPE heterodimers, only one of the proteins has this secretion signal: EsxB of the EsxA-EsxB pair (172) and the PE protein in the case of the PE-PPE pairs (195). Two elements of the secretion signal have been identified. One is a conserved motif, consisting of a tyrosine residue and a residue with a negative charge separated by three random amino acids (YxxxD/E). This motif is required for secretion of T7S substrates (195). The other element is downstream of the YxxxD/E motif at the extreme C-terminus of the protein. These approximately seven amino acids, which include additional hydrophobic residues, are also required for secretion (172). In addition, these C-terminal seven amino acids interact with the C-terminal domain of the ESX-1 component EccCb1 of M. tuberculosis, as demonstrated by yeast two-hybrid analysis and immunoprecipitation (172). Structural studies demonstrate a similar interaction between a peptide containing the last 23 residues of EsxB and EccC of the T7S system of Thermomonospora curvata (200). It is noteworthy that only the last seven amino acids of this peptide were visible in the structure. The YxxxD/E motif was disordered in the structure, suggesting that the motif is not involved in the binding of this peptide to EccC (200). However, it remains unclear how the entire signal interacts with EccC in the context of the full substrate and the complete secretion system. Sequence alignments across the wider array of WXG100 subfamilies also identify a C-terminal consensus sequence, aligned to the side of alpha helix 2, consisting of a conserved HxxxD/E motif (like that of YxxxD/E) and hydrophobic residues downstream of this motif (201). Swapping the C-terminal 15 amino acids, including the YxxxD/E secretion motif of two PE substrates, secreted by ESX-1 or ESX-5 did not affect their secretion or by which system they were transported (195). This shows that, although the C-terminal secretion signal is essential for secretion, it is not involved in system specificity. Instead, a recent study by Phan et al. showed that the fourth and fifth helix of the PPE domain, recognized by the EspG chaperone, contains the signal for system-specific recognition (180).
Although EspG was at first considered an ESX-1-specific protein, it has now been established to be a conserved component; in fact, only the ESX-4 system does not contain an espG gene. Although the EspG proteins share a low amino acid conservation, the solved tertiary structures of EspG1, EspG3, and EspG5 are very similar (169, 181, 202). Previous studies illustrated the importance of EspG5 in the general secretion process, because it is required for the secretion of several ESX-5 substrates in M. marinum (151). More information about its role in the secretion process was obtained by the observation that the deletion of espG1 decreases the stability of PPE68 in M. tuberculosis (203), which indicated that the EspG proteins might function as a chaperone. Importantly, the chaperone of the ESX-5 system, EspG5, only interacts with PE-PPE pairs secreted by the ESX-5 system in M. marinum (169). Similar to this, EspG1 was shown to only bind to an ESX-1 dependent PE-PPE heterodimer and not to PE-PPE pairs secreted by the ESX-5 system (168). This is in line with the observation that EspG1 and EspG5 increase the solubility of their cognate PE-PPE substrates in vitro (181). Because EspG is strictly cytoplasmic, it was hypothesized that EspG is required for the recruitment of the PE-PPE pairs to the membrane-embedded secretion complex, after which it dissociates from these substrates (Fig. 1) (168).
The regions involved in the interaction of EspG and PE-PPE dimers were shown by the crystal structure of EspG5 in complex with the PE25-PPE41 dimer (169, 181). The structure showed that EspG5 only binds to the hydrophobic tip of helices α4 and α5 of the PPE domain of PPE41 and does not interact with the PE protein, including the general secretion signal. This corresponds with an earlier study showing that the YxxxD/E motif of the PE protein is not required for binding of the PE-PPE pair to EspG (195). The observation that binding of EspG5 does not introduce conformational changes to the PE-PPE dimer, but increases the solubility of the protein pair (181), suggests that EspG prevents self-aggregation via the hydrophobic tip of the PPE protein (169, 181).
Because EspG proteins specifically interact with PE-PPE pairs of their respective system, it was already postulated that EspG might direct system specificity of these substrates (169). To confirm this, Phan et al. investigated the role of the region involved in EspG1 binding, hereafter called the EspG1 binding domain, of the ESX-1 substrate PE35-PPE68_1 in M. marinum (180). Indeed, PE35-PPE68_1 could be rerouted to the ESX-5 system by replacing the EspG1 binding domain of PPE68_1 with the equivalent EspG5 binding domain of the ESX-5 substrate PPE18. This domain replacement makes the PE35-PPE68_1 protein pair independent of both EspG1 and the ESX-1 membrane complex but, instead, dependent on EspG5 and the ESX-5 complex for secretion. These findings show not only that EspG is required for the solubility of PE-PPE complexes, but also that it specifically directs these protein pairs to their respective ESX system. The additional observation in several studies that EspG1 is also required for the secretion of Esx and Esp proteins in M. marinum is probably an indirect effect of the substrate codependence, because Esx pairs lack an EspG binding domain (180, 204). Finally, recent findings show that the ESX-1 substrate EspE interacts with EspH, a small ESX-1-associated protein that is not secreted, in the cytosol of M. marinum, which is required for the stable expression and secretion of EspE-EspF (204). Interestingly, several ESX-1-associated proteins show homology and conserved predicted structural features to EspH (i.e., EspD and EspL), indicating that these Esp proteins might serve as specific chaperones for specific Esp substrates.
EccA
The second ESX conserved cytosolic component is EccA, belonging to the family of AAA+ (ATPases associated with diverse cellular activities) proteins. AAA+ ATPases have been linked to a wide range of mechanisms, such as protein homeostasis, signaling pathways, and the assembly and disassembly of protein complexes (205, 206). A distinctive feature of AAA+ proteins is that they usually form hexameric rings with an opening in the center. Although not a common feature of prokaryotic secretion systems, the type VI secretion system also contains an AAA+ ATPase, which is involved in the disassembly of the secretion apparatus (205). EccA is absent in the ESX-4 system, similar to the cytosolic chaperone EspG. All EccA homologs have an identical composition, consisting of distinctive N- and C-terminal domains separated by a linker region. The C-terminal domain contains the typical AAA+ ATPase characteristics, such as the walker and oligomerization motifs, whereas the N-terminal domain contains 12 antiparallel α helices arranged into 6 tandem tetratricopeptide repeat motifs, known for their involvement in protein-protein interactions (207–209). Full-length EccA1 indeed has been shown to form hexamers, although it has (relatively low) ATPase activity in vitro (207). Furthermore, it was confirmed that oligomerization and ATPase activity are mediated by the C-terminal domain. Because expression of only the C-terminal domain results in higher ATPase activity than full-length EccA1, it is thought that the N-terminal domain plays a regulatory role in this activity (207).
The role of EccA in secretion remains unclear. Early studies, showing that EccA1 is essential for secretion of EsxA and EsxB in M. tuberculosis and M. marinum, suggested that EccA proteins might play a role as a chaperone in the ESX secretion process (140, 210). Similarly, EccA5 was also found to be required for the secretion of ESX-5 substrates in M. marinum (153, 211). However, other studies of M. tuberculosis, M. marinum, and M. smegmatis showed that EccA1 and EccA5 are dispensable for ESX-1- and ESX-5-mediated secretion, respectively, suggesting that the involvement of this component varies between different strains and/or growth conditions (127, 212). The latter notion is supported by the recent finding that the importance of EccA1 for secretion of EsxA in M. marinum is dependent on the growth condition (204). Although yeast two- hybrid experiments indicated that a peptide corresponding to the C-terminus of EsxB interacts with EccA1 (213), this interaction could not be observed with full-length EsxB, nor does EccA1 affect the conformation or solubility of EsxB (207). On the other hand, both yeast two-hybrid assays and in vivo protein pull-downs in M. marinum showed that EccA1 is able to interact with EspC (188). This interaction can be blocked by the removal of the C-terminus of EspC, which also blocks the secretion of EspC. However, EccA1 could not be shown to interact with EsxA or EsxB in this same study. Although this could mean that EccA1 exclusively interacts with the Esp proteins and targets them to the membrane-embedded secretion machinery, the role of the EccA component in the other ESX systems that do not secrete Esp substrates remains unknown.
ESX membrane components, complex formation, and structure
The ESX systems of mycobacteria contain five conserved membrane components, EccB, EccC, EccD, EccE, and MycP, which are expected to be involved in the transport of ESX substrates across the cell envelope. All these components have been shown to be essential for secretion for ESX-1 (132, 214, 215), ESX-3 (216), and ESX-5 (149, 153, 212, 217). EccD is the most hydrophobic of these five components, predicted to have up to 11 TMDs, whereas the other components have only one or two TMDs and contain relatively large N- or C-terminal hydrophilic domains. EccD is postulated to be the core membrane pore of the secretion machinery due to its hydrophobicity. The structure of the ∼110-amino acid N-terminal soluble domain of EccD1 has been solved and shows a ubiquitin-like fold with a negatively charged groove, suggesting a role in the recruitment of other components of the secretion complex or possibly as a gating element of the secretion channel (218). The structure of the C-terminal soluble domain of EccB1 shows an elongated shape with a hydrophobic core, suggesting a more structural role of this protein that likely forms a hexamer (218, 219). In addition to this, the C-terminal domain of EccB1 was shown to have ATPase activity in vitro, although it is uncertain whether this has functional relevance because this domain is predicted to be located in the periplasm, where ATP is absent (219). There is no crystal structure available for EccE; however, it is predicted to have two TMDs and a C-terminal soluble domain of unclear localization.
EccC is the most conserved of all T7S components, because it is the only component that is also found in the type VIIb secretion systems in Firmicutes (see below) (220). EccC contains three predicted functional domains, which all show homology to the family of FtsK-SpoIIIE ATPases (221), named after the two best-studied members, FtsK and SpoIIIE (222), both mediating DNA transport. Another notable protein of this family is the type IV coupling protein, involved in the recognition of substrates and protein transport in type IV secretion systems (223). Just like the AAA+ ATPases, a major part of the FtsK-SpoIIIE ATPases also forms ring-like hexamers. However, because EccC contains three nucleotide binding domains (NBDs), opposed to one, it might have different structural features.
Although EccC has been proposed to be the motor of the secretion complex, energizing the translocation of substrates across the cell envelope, its function remains unclear. Of the different EccC proteins, EccC1 has been most extensively investigated. In contrast to other EccC proteins, which are encoded by a single gene, EccC1 has the unusual feature that it is encoded by two genes, eccCa1 and eccCb1. However, both yeast two-hybrid experiments and protein pull-downs showed that it most likely functions as a single unit (132, 212, 224). EccCb1 has been shown by the Cox lab to interact with the C-terminus of EsxB that contains the general secretion motif (188). This suggests that it could play a similar role as the type IV coupling protein in mediating substrate recognition and subsequent transport. The same group subsequently solved the crystal structure of the cytoplasmic domain of EccC from the actinobacterium T. curvata (200). The structure revealed that NBD2 and NBD3 were both bound to ATP, while NBD1 was in a nucleotide-free state, indicating a low ATPase activity of NBD2 and NBD3. They additionally produced a cocrystal of a truncated version of EccC with a peptide representing the last 23 amino acids of the C-terminus of EsxB bound to NBD3. The ATPase activity of EccC, shown to be held in an auto-inhibited state of NBD1 by interactions with NBD2, could not be induced by binding of the C-terminus of EsxB, suggesting that other substrates or components are required for this (200). Interestingly, they did find that EsxB binding resulted in the multimerization of EccC in both dimers and higher-order oligomers. From these findings the authors proposed a working model in which binding of multiple substrates induces EccC multimerization and subsequent activation of NBD1. The suggestion that NBD1 is the main domain responsible for energizing protein translocation is supported by another study that found that ATP binding to NBD1 of EccC5 of M. marinum is essential for secretion, while mutations in the ATP pocket of NBD2 and NBD3 only reduced secretion efficiency (149).
Evidence that the conserved membrane components form a large membrane-embedded secretion complex was first provided for the ESX-5 system of both M. marinum and M. bovis BCG (212). First, the five components with predicted TMDs were indeed shown to be localized to the cell envelope, of which four, EccB5, EccC5, EccD5, and EccE5, were shown to be part of a 1.5-MDa membrane-embedded complex, postulated to form the ESX-5 membrane channel (Fig. 1). Subsequent antibody pull-downs on EccB5 confirmed that these four proteins are the main constituents of this complex, with a notable absence of the fifth membrane component, MycP5. Finally, limited proteolytic digests indicated that the N-terminal part of EccE5 and the middle and C-terminal part of EccC5 are the most exposed domains of the complex. Additional studies using protein pull-downs of Strep-tagged EccCb1 and EccC5 of M. marinum showed that the composition of the ESX-1 complex is comparable to that of the ESX-5 complex, also forming a complex of approximately 1.5 MDa (224). Mass spectrometry analysis confirmed the presence of the four Ecc components in both ESX complexes. Although MycP1 and MycP5 were present in the ESX-1 and ESX-5 complexes, respectively, they were both below the set threshold levels, indicating that the MycP proteins are not integral components of these assemblies.
More recently, the first image of a mycobacterial T7S system was published, determined by negative-stain single-particle analysis (225). For this, the ESX-5 system of the slightly thermophilic and slow-growing Mycobacterium xenopi was overproduced in and purified from M. smegmatis, which does not naturally contain an ESX-5 system. The overproduction in M. smegmatis resulted in a relatively stable and functional system with a similar composition and size as the complexes that were previously isolated for M. bovis BCG and M. marinum (212, 224). Furthermore, the reconstituted system in M. smegmatis allowed the investigation of the minimal requirements for complex formation. This showed that the ESX-5 complex assembles independently of the presence of ESX-5 substrates, opposed to the previously proposed mechanism of EsxB-dependent multimerization of EccC (200).
The EM images of negatively stained purified complexes and subsequent three-dimensional reconstruction resulted in a 13-Å structure, revealing a 6-fold symmetry and a size of 28 (width) × 16 (height) nm, with a 5-nm-wide central pore. Interestingly, based on previously solved crystal structures of Esx and PE-PPE heterodimers, the central pore is wide enough to allow the secretion of folded, dimeric ESX-substrates (201). The combination of the observed symmetry with mass spectrometry-based stoichiometry determination suggests that the four individual components are each present in 6 copies, resulting in a 24-subunit complex with a mass of 1.8 MDa, opposed to the previously estimated 1.5 MDa (212, 224).
The raw EM images of single particles showed “beads-on-a-string-like” densities extending from the main body of the complex. These extrusions have a similar shape as that previously shown for the cytosolic portion of EccC of T. curvata (200), and specific gold labeling confirmed them to be the EccC subunits. The seemingly random orientation of these EccC extrusions suggests that they are flexible, which was also observed by small-angle X-ray analysis, resulting in their absence in averaged complexes. Segmentation analysis in combination with the available crystal structure of the soluble domain of EccB1 allowed for the fitting of EccB5 at the periplasmic side of the complex, while gold-labeling showed that the C-terminal domain of EccE5 is also periplasmic and located at the periphery of the complex (218, 225).
A distinct and interesting feature is that the dimensions of the ESX-5 complex dictate that it can only span the inner membrane of mycobacteria, because it is not tall enough to span both the inner and mycobacterial outer membrane (184). This is in contrast to the complexes of type I, II, III, IV, and VI secretion systems in Gram-negative bacteria, which span both the inner and outer membrane. It therefore remains unknown how T7S substrates can cross the outer membrane of mycobacteria. However, several possibilities have been proposed, which will be discussed below.
The fifth membrane component, the mycosin protease, or MycP, is not an integral part of the core ESX secretion complex, as shown by protein pull-down analysis (212, 224, 225). However, it is highly conserved in T7S systems throughout the phylum Actinobacteria (4), indicating that it does play an important role within T7S. Mycosins are subtilisin-like serine proteases, which had been identified and indicated to be cell envelope associated for M. tuberculosis before T7S was discovered (226, 227). Because mycosins have a predicted N-terminal signal sequence and a C-terminal TMD, the protease domain is most likely located in the periplasm.
Crystal structures of MycP1 from Mycobacterium thermoresistible and M. smegmatis and a structure of MycP3 from M. smegmatis all show a similar subtilisin-like alpha/beta structure, consisting of a seven-stranded beta sheet surrounded by eight alpha helices (228, 229). The active site, consisting of an aspartic acid, histidine, and serine residue, is in the active conformation in all mycosin structures (228). One interesting feature is the presence of an atypical N-terminal extension. Subtilisins usually contain an N-terminal propeptide that blocks the active site of the protease and is cleaved and degraded upon protein maturation (230). In contrast, the “propeptide” of mycosins does not block the active site of the protease, but it is instead tightly wrapped around the protease domain. In vivo data furthermore showed that the N-terminal domain remains attached to the protease without blocking protease activity (228), while in vitro analysis showed that the N-terminal extension is most likely involved in protein stability (231, 232). Additional unique features of mycosins are the presence of three extended loops in the protease domain, surrounding the active site and extending the active site pocket (228, 229). Finally, the substrate binding grooves of MycP1 and MycP3 show significant differences to each other, in both the surface hydrophobicity and the depth of interacting sites, which is also influenced by differences in the three extended loops of the protease domain (228). These differences are probably the basis for substrate specificity of the mycosins.
Although the catalytic site is conserved in mycosins, as is their predicted active conformation, only one mycosin substrate has so far been identified: MycP1 has been shown to process the ESX-1 substrate EspB near its C-terminus, both in vitro and in vivo, in M. marinum and M. tuberculosis (214, 224, 228). Interestingly, the proteolytic inactive mutant showed an increased secretion of ESX-1 substrates, indicating that although MycP1 is required for secretion, its proteolytic activity is not (214, 224). In line with this observation, while MycP1 is crucial for intracellular growth and virulence, a proteolytic inactive mutant of MycP1 resulted in only a slight attenuated phenotype in a mouse infection model (214). This disparity suggests that mycosins have an additional role in ESX systems, besides being involved in the processing of substrates. This second role has been identified to be in the stabilization of the core membrane-embedded ESX-secretion complex, because in the absence of MycP, its respective ESX complex is highly unstable (224). Furthermore, it was shown that although mycosins are not a stable component of the ESX complex, they do loosely associate with it (Fig. 1). It is currently unknown whether this decreased stability is the only way that mycosins directly affect secretion; hopefully, additional interaction studies between the ESX complex and MycP can shed more light on this. Although proteolytic activity is not required for secretion, the decreased virulence of an active site mutant of MycP1 could potentially be of interest for the development of novel drugs that block protease activity (233).
Outer membrane translocation of substrates
Because the EM structure of the isolated ESX-5 membrane complex indicates that this core secretion channel only spans the inner membrane (225), the outer membrane channel in T7S remains unknown. While these findings suggest that T7S is a two-step process in mycobacteria, i.e., mediated by two separate membrane channels, one in the cytoplasmic membrane and one in the mycobacterial outer membrane, a one-step mechanism for T7S, i.e., a single membrane channel that mediates protein export across the complete cell envelope, is still possible, because outer membrane components could be lost during the complex purifications. However, the five conserved membrane components EccB, EccC, EccD, EccE, and MycP are most likely not involved in outer membrane translocation, because all these components can also be widely found in T7S systems of Actinobacteria that lack mycolic acids, the major constituent of the mycobacterial outer membrane. Therefore, in both scenarios, a one-step and a two-step mechanism, the proteins mediating outer membrane translocation remain unknown. Notably, there is no conserved component within the different esx clusters that could be involved in this second translocation step, with the exception of the PE and PPE proteins, which are present in most ESX systems and have only been found in Corynebacteriales.
The identification of outer membrane proteins involved in T7S secretion is complicated by the fact that very little is known about mycobacterial outer membrane proteins in general (234). In fact, for mycobacteria, only the outer membrane porin MspA of M. smegmatis, which is not present in slow-growing species, has been properly characterized and its crystal structure solved (12, 235). The 96-Å-tall MspA porin is an octamer, showing an 8-fold rotational symmetry with a central pore (12). The individual subunits contain a 134-amino acid domain forming the bulk of the porin. This globular domain consists of two 4-stranded antiparallel beta sheets connected by a 50-residue loop at the base of the structure, forming two 16-stranded beta-barrels. While four highly homologous Msp porins, MspA, B, C, and D, are critical for the sufficient uptake of nutrients such as glucose and phosphate in M. smegmatis, no such porins are present in M. tuberculosis. In M. tuberculosis, CpnT and SpmT are the best candidates for outer membrane proteins, because they have been shown to be involved in nutrient and metabolite uptake (236–240) and possibly form a channel or porin (241, 242). However, thus far, the structures of these channels remain unknown. Most attention has been given to the identification of mycobacterial outer membrane proteins that form a beta-barrel, analogous to the Gram-negative bacteria. However, research on PorB, one of the porins present in Corynebacterium glutamicum (243–245), shows a very different structure for an outer membrane porin (14). The core of this protein consists of 70 residues forming 4 alpha helices connected by disulfide bridges, showing that mycolic acid-embedded proteins do not necessarily need to form beta-barrel-like structures.
None of the mycobacterial outer membrane proteins identified so far have been linked to protein export; however, there are indications that specific ESX substrates may be involved in the outer membrane translocation step. For ESX-1, there are currently two substrates suggested to be involved in this second secretion step: EspB and EspC. Both EspB and EspC have been shown to be required for secretion of EsxA/EsxB in pathogenic mycobacteria (246, 247), whereas EspB is still secreted in the absence of EspA-EspC or when EsxA-EsxB is not secreted (188, 246).
As already mentioned, EspB has the specific feature that it forms a PE-PPE-like fold by itself and is therefore probably secreted as a monomer (194, 248). Interestingly, this substrate has been shown to multimerize upon secretion and to form heptameric tubular-shaped particles after overexpression in E. coli, as visualized by negative-stain EM (248). Modeling the solved crystal structure of monomeric EspB without the C-terminal portion that is cleaved off by MycP1 results in an arrangement where the hydrophobic helical tips of EspB are tightly packed, possibly allowing membrane-embedding activity. This has led to the hypothesis that EspB might be within the (outer) membrane and involved in the secretion of ESX substrates. The cleaved off C-terminal fragment of EspB was shown to be dispensable for the formation of these ring-shaped oligomers. By direct comparison of structures with and without this region, the C-terminus was visualized as an appendage protruding from the top of the ring (248). A model was subsequently proposed in which the C-terminal cleavage of EspB by MycP1 has a regulatory function for passage through this potential channel, which could help to explain the increased secretion of ESX-1 substrates in an active site mutant of mycP1 (214, 224).
EspC is the second ESX-1 substrate proposed to be involved in the outer membrane translocation. EspC, which is secreted as a heterodimer together with EspA, has been shown to localize to the cell surface of M. tuberculosis and forms filamentous structures when overproduced in E. coli (192). Based on these findings, EspC was postulated to form filaments that have a role analogous to the type III injectisome needle proteins, mediating contact-dependent phagosomal lysis (142, 249). However, while EspC is required for the secretion of EsxA and EsxB in M. tuberculosis (188), EspC is absent in M. smegmatis, while this species does secrete EsxA-EsxB. Possibly another unknown protein could replace the function of EspC in M. smegmatis.
It still needs to be seen whether EspB and/or EspC are involved in the outer membrane translocation step within T7S. In any case, this would only provide a model for the secretion of ESX-1 proteins, because EspB is not required for secretion of ESX-5 substrates (250). This would suggest that each ESX system has its own specific outer membrane components. The only current hypothesis for the outer membrane channels of ESX-3 and ESX-5 is that this role is performed by members of the PE/PPE families. This hypothesis is supported by their localization to the cell envelope and the notion that they are not widely found outside mycolic acid-containing species.
Role of Mycobacterial ESX Systems in Conjugation and Cell Physiology
While the mycobacterial ESX systems are known to play critical roles in virulence, e.g., in immune modulation (ESX-3 and ESX-5) and phagosomal escape (ESX-1), more recently, several crucial roles in cell physiology and nutrient uptake have been additionally found and described. Here, we will discuss several of those roles and the involved ESX systems (Fig. 2).
FIGURE 2.
Roles of ESX systems in DNA transfer, and cell physiology and nutrient uptake. (A) The pRAW plasmid-encoded ESX-P1 system, present in certain strains of M. marinum, is involved in DNA conjugation, allowing the transfer of the plasmid to other slow-growing mycobacteria (161). Similar plasmids are found in other mycobacteria, and they have been linked to the evolution and distribution of esx gene clusters within mycobacteria (162, 256). In addition, the ESX-1 and ESX-4 systems of M. smegmatis are involved in distributive conjugal transfer, allowing the horizontal gene transfer of random large genomic fragments (143, 159). (B) A second major role of the ESX-systems is in cell physiology. While the ESX-1 system has been linked to the integrity of the cell envelope and the biosynthesis of mycolic acids (196, 210, 264), the ESX-3 and ESX-5 systems are involved in the uptake of metabolites and nutrients, respectively. The ESX-3 system and two of its substrate pairs (EsxG-EsxH and PE5-PPE4) have been specifically linked to the uptake of iron and zinc (144, 145, 147), while the ESX-5 system has been linked to the utilization of fatty acids and possibly other nutrients (149) Finally, the ESX-5 system secretes a substrate (PPE10) that is required for the integrity of the capsular layer (199). IM, inner membrane; OM, outer membrane.
One of the first nonvirulent roles attributed to an ESX-system was the role of the ESX-1 system in horizontal gene transfer in M. smegmatis (143). The DNA transfer is unidirectional, chromosomally encoded, and allows for the transfer of large genomic fragments. This form of conjugation is called distributive conjugal transfer and has so far not been described for other mycobacteria, including M. tuberculosis (159). An interesting observation is that, while recipient strains are dependent on the ESX-1 system for DNA uptake (143), esx-1 mutants in the donor strains are hyper-conjugative (251). In addition to the ESX-1 system, the ESX-4 system is also required for receiving DNA by the recipient, because a deletion of eccC4 or eccD4 blocked conjugation (159). The presence of ESX-4 was only required for the recipient strain, because the same genes could be deleted in the donor strain without affecting DNA transfer. A more recent study showed that SigM, the regulator of ESX-4, is highly upregulated in the recipient when in contact with a donor cell, suggesting that ESX-4 is specifically induced upon cell-cell contact (160). The exact role of ESX-1 and ESX-4 in conjugation by M. smegmatis remains unclear. The fact that conjugation takes place in a contact-dependent manner (159) might indicate that putative surface-localized ESX-1 and ESX-4 substrates might be required for initial donor-acceptor engagement and/or communication.
More recently, a conjugative plasmid has been described for M. marinum, the pRAW plasmid (161). This plasmid contains not only an esx gene cluster, designated esx-P1, but also genes predicted to encode type IV secretion-like proteins, known to be involved in DNA conjugation in Gram-negative and Gram-positive bacteria (161, 252, 253). The pRAW plasmid could be transferred very efficiently, but only between different slow-growing species, whereas no transfer was observed to fast-growing mycobacteria, such as M. smegmatis (161). Conjugation of the pRAW plasmid was shown to depend on the presence of an intact esx-P1 cluster and on the type IV-like genes (161). Interestingly, since then, similar esx gene cluster-containing plasmids have been found in various nontuberculous mycobacteria, both pathogenic and nonpathogenic (161, 254, 255). The wide distribution of these esx-containing plasmids has been linked to a possible role of plasmids in the evolution and distribution of esx gene clusters within mycobacteria (162, 256). Because ESX-P1 is most closely related to the ESX-5 system, pRAW-like plasmids might have been involved in acquiring the ESX-5 system in slow-growing mycobacteria.
DNA exchange and/or cell-to-cell contact is not the only function assigned to ESX systems. Another critical role of these systems is in the uptake of nutrients. In fact, both the ESX-3 and ESX-5 systems are involved in this, explaining why these systems are required for bacterial viability in slow-growing species (149, 155, 217, 257). The ESX-3 system is transcriptionally controlled by the iron-dependent transcriptional repressor (IdeR), and in M. tuberculosis, additionally by the zinc uptake repressor (Zur), indicating that the ESX-3 system is linked to the uptake of metal ions (258, 259). Indeed, the ESX-3 system is upregulated at low iron levels (145, 257, 260), and more recent work confirmed that the ESX-3 system is actively involved in the uptake of iron and, in the case of M. tuberculosis, also of zinc (144). The ESX-3 system was additionally shown to be essential for growth of M. tuberculosis, not only in low-iron or -zinc conditions, but also for optimal growth in high-iron and -zinc concentrations (144). For M. tuberculosis, six substrates of the ESX-3 system have been identified: EsxG-EsxH, PE5-PPE4, and PE15-PPE20. Interestingly, only EsxG-EsxH and PE5-PPE4 appear to be involved in iron uptake, while PE15-PPE20 is involved in immune modulation (147). In contrast to M. tuberculosis, the deletion of the ESX-3 system of M. smegmatis only affected bacterial growth when fxbA was also deleted. FxbA is an enzyme required for the biosynthesis of the high-affinity iron chelator or siderophore exochelin, which is not produced by M. tuberculosis (144, 145, 216). Importantly, the production of a second siderophore, mycobactin, only affected growth in this fxbA mutant, linking the function of ESX-3 to iron uptake through mycobactin (147). Later, it was shown that the addition of hemin rescues the growth of an esx-3 mutant in M. tuberculosis (144), supporting the hypothesis that ESX-3 is involved in iron uptake (145).
Both directed knockout experiments and transposon insertion sequencing experiments have shown that the central components of the ESX-5 system are required for growth of M. marinum and M. tuberculosis (149, 217). Using a modified screen, transposon mutants could be identified that allowed the deletion of ESX-5 (149). These mutants contained a transposon in either the mas or ppsD gene, both involved in the synthesis of the phthiocerol dimycocerosates and phenolic glycolipids. The absence of these outer membrane lipids results in a more permeable cell envelope (149). To test whether a more permeable outer membrane was responsible for the observed difference in esx-5 essentiality, an independent mechanism was devised. Indeed, heterologous expression of the outer membrane porin MspA from M. smegmatis in M. marinum also allowed the generation of esx-5 mutants (149). Because the ESX-5 system is only present in slow-growing mycobacteria, while MspA-like outer membrane porins are only present in fast-growing species, it was proposed that the ESX-5 system is able to substitute the function of these porins in nutrient uptake (149). Growth assays utilizing different carbon sources showed a difference in the growth rate of wild-type M. marinum compared to an esx-5 mutant for Tween-80, suggesting that the ESX-5 system is involved in the utilization of fatty acids (149). ESX-5 mutants were subsequently shown to be unable to internalize fluorescently labeled fatty acids (149). In addition to its role in uptake, the ESX-5 system could additionally be involved in the processing of Tween-80, releasing the oleic acid, as one of its substrates, LipY, degrades long-chain triacylglycerols (261). The role of the ESX-5 system in nutrient uptake is further substantiated by its upregulation under phosphate-limiting conditions by the Pst-SenX3-RegX3 system (262). This would also mean that specific ESX-3 and ESX-5 substrates might form the outer membrane channels that mediate the uptake of specific nutrients.
The ESX-5 system has a second physiological role, because it is required for the integrity of the capsule of M. marinum via its substrate PPE10 (199). Mutations in the esx-5 system components or in ppe10 resulted in a reduced capsular layer and, subsequently, reduced amounts of glycolipids and surface-localized proteins, such as EspE and PE_PGRS proteins (199). As a result of these morphological differences, a ppe10 mutant showed a reduced hemolytic activity and reduced ubiquitin binding during infection of host cells, indicative of phagosomal rupture and cytosolic translocation, respectively, as read-outs for ESX-1 functionality (199). A study also found a link between the ESX-1 system and cell envelope integrity, because esx-1 mutants in M. tuberculosis have been shown to have an increased susceptibility to detergents (196), although another study showed that ESX-1 mutations did not impact cell envelope integrity (263). Finally, studies of M. marinum have suggested a link between the ESX-1 system and mycolic acid biosynthesis (210, 264). In addition to these physiological roles, substrates of ESX-5, mainly the PE_PGRS proteins, have been implicated in host-pathogen interactions by various groups, although some of these results are still ambiguous (265).
T7S in Other Species
As already mentioned, T7S is also found outside of the mycobacterial family, not only throughout the phylum of Actinobacteria, including corynebacteria, nocardia, and Streptomyces species, but also in more distantly related systems that can be found in the Firmicutes. The T7S systems found in Actinobacteria are highly similar to the ESX systems of mycobacteria, all containing an Esx protein pair and four of the five conserved membrane components, EccB, EccC, EccD, and MycP; the membrane component EccE is only present in some T7S systems. In many cases pe/ppe gene pairs can also be identified (4). Interestingly, although EccE has been shown to be an integral component of both the ESX-1 and ESX-5 membrane complexes, the structure of the ESX-5 complex of M. xenopi showed that EccE5 is present at the periphery of this complex, and in the absence of EccE5, complexes could still successfully be purified (225). This makes it reasonable that secretion complexes can be functional without the EccE component in some T7S systems of Actinobacteria. The strong conservation of MycP is also rather unexpected, because MycP is not an integral component of the core secretion complex in mycobacterial ESX systems of (212, 224, 225). However, because MycP is required for a stable ESX complex and successful secretion in mycobacteria (214, 224), it is possible that this is a conserved feature in all Actinobacteria. In addition to this, the association of MycP to the core complex might be stronger in the T7S systems of other Actinobacteria. This hypothesis is supported by the T7S gene cluster found in Bifidobacterium dentium, where there are two mycP copies, one of which is part of a chimeric gene together with a complete homologue of eccB. This gene potentially produces a chimeric protein, in which the periplasmic domain of EccB is fused to the protease domain of MycP (4).
Within the Actinobacteria, T7S is not limited to species containing mycolic acids, i.e., to species with a diderm cell envelope, because it is also present, for instance, in Streptomyces species such as S. coelicolor. These mycolic acid-lacking bacteria can also contain the five conserved membrane components (164), supporting the model of the ESX membrane complex mediating secretion across the inner membrane only (225). Consequently, components, or substrates, that mediate secretion across the mycobacterial outer membrane should not be present in these monoderm species.
The T7S systems found in the Firmicutes show a lower similarity to the ESX systems of mycobacteria. In fact, only the FtsK-SpoIIIE ATPase and (potential) substrates belonging to the WXG100 protein family are conserved (128, 129, 166). In addition to these conserved features, the Firmicutes systems contain a set of unique genes, encoding either substrates or system-specific (functional) components. The T7S systems within different Firmicutes species also show relatively high variation among each other, both in substrates and in (predicted) structural components.
While functional systems have been confirmed for L. monocytogenes, Streptococcus agalactiae, S. intermedius, Bacillus anthracis, and B. subtilis (166, 220, 266–269), most information on T7S in Firmicutes has been obtained for the T7S system of S. aureus, also called Ess (ESAT-6 secretion system) (220). The Ess system contains five structural components, conserved throughout different S. aureus strains (270), i.e., EssA, EssB, EssC, EsaA (Esa, ESAT-6 secretion accessory), and EsaB. Whereas EsaB is cytoplasmic, the other four components are membrane proteins (270), with EssC being the conserved FtsK-SpoIIIE ATPase. Interestingly, the C-terminal ATPase domain of EssC shows four variants within different S. aureus strains that cluster with genes encoding different potential substrates. It was recently shown that this domain is indeed involved in specific substrate recognition (270, 271). The role of EsaB is uncertain, because it is not required for Ess secretion in all S. aureus strains. An interesting feature of EsaB is the ubiquitin-like fold, resembling the soluble domain of EccD of mycobacteria (218, 272, 273). However, it is unknown how and if these proteins share a comparable function.
Recently, several papers were published describing the first structural data on the Ess secretion complex. Whereas one study only found homo-oligomers of EsaA, EssB, and EssC (274), other studies showed a complex consisting of EssC, EsaA, EssB, and EssA (275, 276). It is possible that both the heteromeric and homo-oligomeric complexes are present in S. aureus, because Ess secretion in S. aureus is highly variable both in different strains and at different growth phases (272). In addition, a flotillin-like protein has been found to catalyze the heteromeric oligomerization of the Ess complex (275). This opens the possibility that scaffold proteins are involved in complex assembly upon their specific upregulation during, e.g., different growth phases or during infection (272, 277). Based on these three studies, the Ess complex seems less stable than the ESX-complexes found in mycobacteria, as for instance, shown by the strong effect of different mild detergents on the oligomerization state (274). This considerably complicates the isolation and characterization of the Ess secretion complex.
The Ess system of S. aureus contains two proteins of the WXG100 family, EsxA and EsxB, which are, unlike the homologous proteins in mycobacteria, not produced from bicistronic transcripts and not cosecreted as a heterodimer (278). Interestingly, EsxA is reported to either self-interact or interact with the substrate EsxC, and EsxB interacts with EsxD (279, 280). All four of these secreted proteins have been linked to the pathogenicity of S. aureus (272, 273, 279). Inactivation of the Ess system results in a less persistent infection and reduced bacterial survival (220, 272, 273); more specifically, Ess-mediated secretion of EsxA and EsxB is required for the formation of staphylococcal abscesses and the blocking of apoptosis, indicating a role in intracellular survival (220, 273, 281, 282). Unlike the ESX-3 and ESX-5 systems of mycobacteria, which are required for bacterial viability, the Ess system of S. aureus is dispensable for in vitro growth (220, 273).
The B. subtilis T7S system (also known in the literature as the YukE-Yue system) resembles the Ess system of S. aureus. The notable difference is that it only seems to secrete a single substrate, the WXG100 protein YukE, which forms a homodimer (167, 283, 284). Similar to the Ess system, the YukE-Yue system also contains five functional components, the membrane proteins YukC, YukBA, YueB, and YueC and the cytoplasmic YukD, the latter resembling EsaB of S. aureus (167, 283, 285). YukBA is the FtsK/SpoIIIE ATPase of the system. A yueB mutant shows reduced conjugative DNA transfer, similar to the ESX-1 system M. smegmatis (286). WXG100 genes have also been found in the pathogenic strains Bacillus cereus, Bacillus thuringiensis, and B. anthracis, and at least several of their protein products are actively secreted (167, 267, 283). For example, B. anthracis contains six WXG100 genes, of which the gene products EsxB and EsxW are secreted during in vitro growth, while the other genes might only be expressed during infection (267).
One distinguishing feature of the T7S systems of Firmicutes compared to the ESX systems of mycobacteria is their involvement in bacterial competition by mediating the secretion of toxins. This intriguingly shows that Firmicutes use T7S systems in bacterial warfare similarly to how Gram-negative bacteria use type VI secretion systems. For example, S. aureus secretes a fifth substrate, EsaD, which functions as an antibacterial DNase (287). The toxic effects of EsaD are blocked by EsaG, an antitoxin produced also by S. aureus strains lacking EsaD (287). A T7S system in S. intermedius secretes so-called LXG proteins, with N-termini homologous to WXG100 proteins, that function as antibacterial toxins in a contact-dependent manner (266). LXG toxins with RNase activity have also been found in B. subtilis (288) and shown to have antibacterial properties when produced in E. coli. Although they contain a typical N-terminal WXG100 motif, it is not known whether these proteins are dependent on the YukE-Yue system for secretion (288). A genomic study found that predicted T7S substrate genes with putative toxin activities are present in a range of different species, indicating that T7S systems might be widely used by Firmicutes in bacterial competition (289).
OUTLOOK
In the past 15 years, considerable effort has been made to understand the different export pathways of mycobacteria. A major focus of research in this area has been the identification of substrates of the different systems and their roles in mycobacterial physiology and virulence. In parallel, we are steadily obtaining insight into the components of these systems and their mechanisms of substrate recognition and transport. However, a major topic that needs to be addressed is the issue of how proteins transit the specific mycolic acid-containing second membrane for release into the extracellular environment. While T7S substrates are among the most abundant proteins found in culture supernatants, there are also signal peptide-containing proteins that are efficiently secreted into the extracellular environment (17, 18, 63, 120, 290–292), emphasizing the importance of an outer membrane translocation step. Protein transport across this highly impermeable outer membrane should be tightly regulated to maintain the permeability barrier, because this is crucial for pathogenic mycobacteria to withstand the harsh conditions of their intracellular niche. As discussed in this article, for T7S, some specific substrates might be involved in the second membrane transport event; however, the mechanism of outer membrane transport for the Sec, Tat, and SecA2 substrates is a complete mystery. Research focused on identifying and characterizing the proteins that are localized to this special mycobacterial outer membrane is urgently needed to gain insight into these processes.
ACKNOWLEDGMENTS
Our work is supported by a VIDI grant (864.12.006) from the Netherlands Organization for Scientific Research (NWO) (to E.N.G.H.) and by grant AI054540 from the National Institute of Allergy and Infectious Disease (NIAID) (to M.B.).
We thank Brittany Miller and Wilbert Bitter for critical reading of the manuscript.
Contributor Information
Vincent J. C. van Winden, Department of Medical Microbiology and Infection Control, Amsterdam UMC, Vrije Universiteit, Amsterdam, The Netherlands
Edith N. G. Houben, Section of Molecular Microbiology, Amsterdam Institute for Molecules, Medicines, and Systems, Vrije Universiteit, Amsterdam, The Netherlands
Miriam Braunstein, Department of Microbiology and Immunology, University of North Carolina—Chapel Hill, Chapel Hill, NC 27599.
Vincent A. Fischetti, The Rockefeller University, New York, NY
Richard P. Novick, Skirball Institute for Molecular Medicine, NYU Medical Center, New York, NY
Joseph J. Ferretti, Department of Microbiology & Immunology, University of Oklahoma Health Science Center, Oklahoma City, OK
Daniel A. Portnoy, Department of Molecular and Cellular Microbiology, University of California, Berkeley, Berkeley, CA
Miriam Braunstein, Department of Microbiology and Immunology, University of North Carolina-Chapel Hill, Chapel Hill, NC.
Julian I. Rood, Infection and Immunity Program, Monash Biomedicine Discovery Institute, Monash University, Melbourne, Australia
REFERENCES
- 1.WHO. 2018. Global tuberculosis report. World Health Organization, Geneva, Switzerland. [PubMed] [Google Scholar]
- 2.Roy A, Eisenhut M, Harris RJ, Rodrigues LC, Sridhar S, Habermann S, Snell L, Mangtani P, Adetifa I, Lalvani A, Abubakar I. 2014. Effect of BCG vaccination against Mycobacterium tuberculosis infection in children: systematic review and meta-analysis. BMJ 349:g4643 10.1136/bmj.g4643. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brennan PJ, Nikaido H. 1995. The envelope of mycobacteria. Annu Rev Biochem 64:29–63 10.1146/annurev.bi.64.070195.000333. [DOI] [PubMed] [Google Scholar]
- 4.Houben ENG, Korotkov KV, Bitter W. 2014. Take five: type VII secretion systems of mycobacteria. Biochim Biophys Acta 1843:1707–1716 10.1016/j.bbamcr.2013.11.003. [PubMed] [DOI] [PubMed] [Google Scholar]
- 5.Barry CE III, Lee RE, Mdluli K, Sampson AE, Schroeder BG, Slayden RA, Yuan Y. 1998. Mycolic acids: structure, biosynthesis and physiological functions. Prog Lipid Res 37:143–179 10.1016/S0163-7827(98)00008-3. [DOI] [PubMed] [Google Scholar]
- 6.Brennan PJ. 2003. Structure, function, and biogenesis of the cell wall of Mycobacterium tuberculosis. Tuberculosis (Edinb) 83:91–97 10.1016/S1472-9792(02)00089-6. [DOI] [PubMed] [Google Scholar]
- 7.Hoffmann C, Leis A, Niederweis M, Plitzko JM, Engelhardt H. 2008. Disclosure of the mycobacterial outer membrane: cryo-electron tomography and vitreous sections reveal the lipid bilayer structure. Proc Natl Acad Sci USA 105:3963–3967 10.1073/pnas.0709530105. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ortalo-Magné A, Lemassu A, Lanéelle MA, Bardou F, Silve G, Gounon P, Marchal G, Daffé M. 1996. Identification of the surface-exposed lipids on the cell envelopes of Mycobacterium tuberculosis and other mycobacterial species. J Bacteriol 178:456–461 10.1128/jb.178.2.456-461.1996. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jackson M. 2014. The mycobacterial cell envelope-lipids. Cold Spring Harb Perspect Med 4:a021105 10.1101/cshperspect.a021105. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Minnikin DE. 1982. Lipids: complex lipids, their chemistry, biosynthesis and roles, p 95–184. In Ratledge C, Stanford J (ed), The Biology of Mycobacteria. Academic Press, London, UK. [Google Scholar]
- 11.Zuber B, Chami M, Houssin C, Dubochet J, Griffiths G, Daffé M. 2008. Direct visualization of the outer membrane of mycobacteria and corynebacteria in their native state. J Bacteriol 190:5672–5680 10.1128/JB.01919-07. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Faller M, Niederweis M, Schulz GE. 2004. The structure of a mycobacterial outer-membrane channel. Science 303:1189–1192 10.1126/science.1094114. [PubMed] [DOI] [PubMed] [Google Scholar]
- 13.Schiffler B, Barth E, Daffé M, Benz R. 2007. Corynebacterium diphtheriae: identification and characterization of a channel-forming protein in the cell wall. J Bacteriol 189:7709–7719 10.1128/JB.00864-07. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ziegler K, Benz R, Schulz GE. 2008. A putative alpha-helical porin from Corynebacterium glutamicum. J Mol Biol 379:482–491 10.1016/j.jmb.2008.04.017. [PubMed] [DOI] [PubMed] [Google Scholar]
- 15.Saleh MT, Fillon M, Brennan PJ, Belisle JT. 2001. Identification of putative exported/secreted proteins in prokaryotic proteomes. Gene 269:195–204 10.1016/S0378-1119(01)00436-X. [DOI] [PubMed] [Google Scholar]
- 16.Orfanoudaki G, Economou A. 2014. Proteome-wide subcellular topologies of E. coli polypeptides database (STEPdb). Mol Cell Proteomics 13:3674–3687 10.1074/mcp.O114.041137. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miller BK, Zulauf KE, Braunstein M. 2017. The Sec pathways and exportomes of Mycobacterium tuberculosis. Microbiol Spectr 5:TBTB2-0013–2016. 10.1128/microbiolspec.TBTB2-0013-2016. [PubMed] [DOI] [PubMed] [Google Scholar]
- 18.Perkowski EF, Zulauf KE, Weerakoon D, Hayden JD, Ioerger TR, Oreper D, Gomez SM, Sacchettini JC, Braunstein M. 2017. The exit strategy: an approach for identifying bacterial proteins exported during host infection. MBio 8:e00333-17 10.1128/mBio.00333-17. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vrontou E, Economou A. 2004. Structure and function of SecA, the preprotein translocase nanomotor. Biochim Biophys Acta Mol Cell Res 1694:67–80 10.1016/j.bbamcr.2004.06.003. [PubMed] [DOI] [PubMed] [Google Scholar]
- 20.Finlay BB, Falkow S. 1997. Common themes in microbial pathogenicity revisited. Microbiol Mol Biol Rev 61:136–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Crane JM, Randall LL. 2017. The Sec system: protein export in Escherichia coli. Ecosal Plus 7. 10.1128/ecosalplus.ESP-0002-2017. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tsirigotaki A, De Geyter J, Šoštaric N, Economou A, Karamanou S. 2017. Protein export through the bacterial Sec pathway. Nat Rev Microbiol 15:21–36 10.1038/nrmicro.2016.161. [PubMed] [DOI] [PubMed] [Google Scholar]
- 23.Ligon LS, Hayden JD, Braunstein M. 2012. The ins and outs of Mycobacterium tuberculosis protein export. Tuberculosis (Edinb) 92:121–132 10.1016/j.tube.2011.11.005. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zimmer J, Nam Y, Rapoport TA. 2008. Structure of a complex of the ATPase SecA and the protein-translocation channel. Nature 455:936–943 10.1038/nature07335. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wickner W, Driessen AJM, Hartl FU. 1991. The enzymology of protein translocation across the Escherichia coli plasma membrane. Annu Rev Biochem 60:101–124 10.1146/annurev.bi.60.070191.000533. [PubMed] [DOI] [PubMed] [Google Scholar]
- 26.Brundage L, Hendrick JP, Schiebel E, Driessen AJM, Wickner W. 1990. The purified E. coli integral membrane protein SecY/E is sufficient for reconstitution of SecA-dependent precursor protein translocation. Cell 62:649–657 10.1016/0092-8674(90)90111-Q. [DOI] [PubMed] [Google Scholar]
- 27.Akimaru J, Matsuyama S, Tokuda H, Mizushima S. 1991. Reconstitution of a protein translocation system containing purified SecY, SecE, and SecA from Escherichia coli. Proc Natl Acad Sci USA 88:6545–6549 10.1073/pnas.88.15.6545. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nishiyama K, Hanada M, Tokuda H. 1994. Disruption of the gene encoding p12 (SecG) reveals the direct involvement and important function of SecG in the protein translocation of Escherichia coli at low temperature. EMBO J 13:3272–3277 10.1002/j.1460-2075.1994.tb06628.x. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Duong F, Wickner W. 1997. The SecDFyajC domain of preprotein translocase controls preprotein movement by regulating SecA membrane cycling. EMBO J 16:4871–4879 10.1093/emboj/16.16.4871. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lill R, Cunningham K, Brundage LA, Ito K, Oliver D, Wickner W. 1989. SecA protein hydrolyzes ATP and is an essential component of the protein translocation ATPase of Escherichia coli. EMBO J 8:961–966 10.1002/j.1460-2075.1989.tb03458.x. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chatzi KE, Sardis MF, Economou A, Karamanou S. 2014. SecA-mediated targeting and translocation of secretory proteins. Biochim Biophys Acta 1843:1466–1474 10.1016/j.bbamcr.2014.02.014. [PubMed] [DOI] [PubMed] [Google Scholar]
- 32.Economou A, Wickner W. 1994. SecA promotes preprotein translocation by undergoing ATP-driven cycles of membrane insertion and deinsertion. Cell 78:835–843 10.1016/S0092-8674(94)90582-7. [DOI] [PubMed] [Google Scholar]
- 33.Braunstein M, Brown AM, Kurtz S, Jacobs WRJ Jr. 2001. Two nonredundant SecA homologues function in mycobacteria. J Bacteriol 183:6979–6990 10.1128/JB.183.24.6979-6990.2001. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Feltcher ME, Braunstein M. 2012. Emerging themes in SecA2-mediated protein export. Nat Rev Microbiol 10:779–789 10.1038/nrmicro2874. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bensing BA, Seepersaud R, Yen YT, Sullam PM. 2014. Selective transport by SecA2: an expanding family of customized motor proteins. Biochim Biophys Acta 1843:1674–1686 10.1016/j.bbamcr.2013.10.019. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Guo XV, Monteleone M, Klotzsche M, Kamionka A, Hillen W, Braunstein M, Ehrt S, Schnappinger D. 2007. Silencing essential protein secretion in Mycobacterium smegmatis by using tetracycline repressors. J Bacteriol 189:4614–4623 10.1128/JB.00216-07. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hou JM, D’Lima NG, Rigel NW, Gibbons HS, McCann JR, Braunstein M, Teschke CM. 2008. ATPase activity of Mycobacterium tuberculosis SecA1 and SecA2 proteins and its importance for SecA2 function in macrophages. J Bacteriol 190:4880–4887 10.1128/JB.00412-08. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.von Heijne G. 1988. Transcending the impenetrable: how proteins come to terms with membranes. Biochim Biophys Acta 947:307–333 10.1016/0304-4157(88)90013-5. [DOI] [PubMed] [Google Scholar]
- 39.Paetzel M, Karla A, Strynadka NCJ, Dalbey RE. 2002. Signal peptidases. Chem Rev 102:4549–4580 10.1021/cr010166y. [PubMed] [DOI] [PubMed] [Google Scholar]
- 40.Nakayama H, Kurokawa K, Lee BL. 2012. Lipoproteins in bacteria: structures and biosynthetic pathways. FEBS J 279:4247–4268 10.1111/febs.12041. [PubMed] [DOI] [PubMed] [Google Scholar]
- 41.Ollinger J, O’Malley T, Ahn J, Odingo J, Parish T. 2012. Inhibition of the sole type I signal peptidase of Mycobacterium tuberculosis is bactericidal under replicating and nonreplicating conditions. J Bacteriol 194:2614–2619 10.1128/JB.00224-12. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sander P, Rezwan M, Walker B, Rampini SK, Kroppenstedt RM, Ehlers S, Keller C, Keeble JR, Hagemeier M, Colston MJ, Springer B, Böttger EC. 2004. Lipoprotein processing is required for virulence of Mycobacterium tuberculosis. Mol Microbiol 52:1543–1552 10.1111/j.1365-2958.2004.04041.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 43.Banaiee N, Kincaid EZ, Buchwald U, Jacobs WR Jr, Ernst JD. 2006. Potent inhibition of macrophage responses to IFN-gamma by live virulent Mycobacterium tuberculosis is independent of mature mycobacterial lipoproteins but dependent on TLR2. J Immunol 176:3019–3027 10.4049/jimmunol.176.5.3019. [PubMed] [DOI] [PubMed] [Google Scholar]
- 44.Hutchings MI, Palmer T, Harrington DJ, Sutcliffe IC. 2009. Lipoprotein biogenesis in Gram-positive bacteria: knowing when to hold ’em, knowing when to fold ’em. Trends Microbiol 17:13–21 10.1016/j.tim.2008.10.001. [PubMed] [DOI] [PubMed] [Google Scholar]
- 45.Leversen NA, de Souza GA, Målen H, Prasad S, Jonassen I, Wiker HG. 2009. Evaluation of signal peptide prediction algorithms for identification of mycobacterial signal peptides using sequence data from proteomic methods. Microbiology 155:2375–2383 10.1099/mic.0.025270-0. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bassford PJ Jr, Silhavy TJ, Beckwith JR. 1979. Use of gene fusion to study secretion of maltose-binding protein into Escherichia coli periplasm. J Bacteriol 139:19–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Randall LL, Hardy SJS. 1986. Correlation of competence for export with lack of tertiary structure of the mature species: a study in vivo of maltose-binding protein in E. coli. Cell 46:921–928 10.1016/0092-8674(86)90074-7. [DOI] [PubMed] [Google Scholar]
- 48.Nouwen N, Berrelkamp G, Driessen AJM. 2007. Bacterial Sec-translocase unfolds and translocates a class of folded protein domains. J Mol Biol 372:422–433 10.1016/j.jmb.2007.07.003. [PubMed] [DOI] [PubMed] [Google Scholar]
- 49.Bechtluft P, Nouwen N, Tans SJ, Driessen AJM. 2010. SecB: a chaperone dedicated to protein translocation. Mol Biosyst 6:620–627 10.1039/B915435C. [PubMed] [DOI] [PubMed] [Google Scholar]
- 50.Hartl FU, Lecker S, Schiebel E, Hendrick JP, Wickner W. 1990. The binding cascade of SecB to SecA to SecY/E mediates preprotein targeting to the E. coli plasma membrane. Cell 63:269–279 10.1016/0092-8674(90)90160-G. [DOI] [PubMed] [Google Scholar]
- 51.Fekkes P, van der Does C, Driessen AJM. 1997. The molecular chaperone SecB is released from the carboxy-terminus of SecA during initiation of precursor protein translocation. EMBO J 16:6105–6113 10.1093/emboj/16.20.6105. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bordes P, Cirinesi A-M, Ummels R, Sala A, Sakr S, Bitter W, Genevaux P. 2011. SecB-like chaperone controls a toxin-antitoxin stress-responsive system in Mycobacterium tuberculosis. Proc Natl Acad Sci USA 108:8438–8443 10.1073/pnas.1101189108. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Luirink J, von Heijne G, Houben E, de Gier J-W. 2005. Biogenesis of inner membrane proteins in Escherichia coli. Annu Rev Microbiol 59:329–355 10.1146/annurev.micro.59.030804.121246. [PubMed] [DOI] [PubMed] [Google Scholar]
- 54.Valent QA, de Gier J-WL, von Heijne G, Kendall DA, ten Hagen-Jongman CM, Oudega B, Luirink J. 1997. Nascent membrane and presecretory proteins synthesized in Escherichia coli associate with signal recognition particle and trigger factor. Mol Microbiol 25:53–64 10.1046/j.1365-2958.1997.4431808.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 55.Egea PF, Stroud RM. 2010. Lateral opening of a translocon upon entry of protein suggests the mechanism of insertion into membranes. Proc Natl Acad Sci USA 107:17182–17187 10.1073/pnas.1012556107. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dalbey RE, Chen M. 2004. Sec-translocase mediated membrane protein biogenesis. Biochim Biophys Acta 1694:37–53 10.1016/j.bbamcr.2004.03.009. [PubMed] [DOI] [PubMed] [Google Scholar]
- 57.Grudnik P, Bange G, Sinning I. 2009. Protein targeting by the signal recognition particle. Biol Chem 390:775–782 10.1515/BC.2009.102. [PubMed] [DOI] [PubMed] [Google Scholar]
- 58.Palaniyandi K, Veerasamy M, Narayanan S. 2012. Characterization of Ffh of Mycobacterium tuberculosis and its interaction with 4.5S RNA. Microbiol Res 167:520–525 10.1016/j.micres.2012.03.002. [PubMed] [DOI] [PubMed] [Google Scholar]
- 59.Venkatesan A, Palaniyandi K, Sharma D, Bisht D, Narayanan S. 2018. Characterization of FtsY, its interaction with Ffh, and proteomic identification of their potential substrates in Mycobacterium tuberculosis. Can J Microbiol 64:243–251 10.1139/cjm-2017-0385. [PubMed] [DOI] [PubMed] [Google Scholar]
- 60.Thakur P, Gantasala NP, Choudhary E, Singh N, Abdin MZ, Agarwal N. 2016. The preprotein translocase YidC controls respiratory metabolism in Mycobacterium tuberculosis. Sci Rep 6:24998 10.1038/srep24998. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Berks BC. 2015. The twin-arginine protein translocation pathway. Annu Rev Biochem 84:843–864 10.1146/annurev-biochem-060614-034251. [PubMed] [DOI] [PubMed] [Google Scholar]
- 62.Berks BC. 1996. A common export pathway for proteins binding complex redox cofactors? Mol Microbiol 22:393–404 10.1046/j.1365-2958.1996.00114.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 63.McDonough JA, McCann JR, Tekippe EM, Silverman JS, Rigel NW, Braunstein M. 2008. Identification of functional Tat signal sequences in Mycobacterium tuberculosis proteins. J Bacteriol 190:6428–6438 10.1128/JB.00749-08. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chater KF, Biró S, Lee KJ, Palmer T, Schrempf H. 2010. The complex extracellular biology of Streptomyces. FEMS Microbiol Rev 34:171–198 10.1111/j.1574-6976.2009.00206.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 65.Palmer T, Berks BC. 2012. The twin-arginine translocation (Tat) protein export pathway. Nat Rev Microbiol 10:483–496 10.1038/nrmicro2814. [PubMed] [DOI] [PubMed] [Google Scholar]
- 66.DeLisa MP, Tullman D, Georgiou G. 2003. Folding quality control in the export of proteins by the bacterial twin-arginine translocation pathway. Proc Natl Acad Sci USA 100:6115–6120 10.1073/pnas.0937838100. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.McDonough JA, Hacker KE, Flores AR, Pavelka MS Jr, Braunstein M. 2005. The twin-arginine translocation pathway of Mycobacterium smegmatis is functional and required for the export of mycobacterial beta-lactamases. J Bacteriol 187:7667–7679 10.1128/JB.187.22.7667-7679.2005. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Posey JE, Shinnick TM, Quinn FD. 2006. Characterization of the twin-arginine translocase secretion system of Mycobacterium smegmatis. J Bacteriol 188:1332–1340 10.1128/JB.188.4.1332-1340.2006. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Saint-Joanis B, Demangel C, Jackson M, Brodin P, Marsollier L, Boshoff H, Cole ST. 2006. Inactivation of Rv2525c, a substrate of the twin arginine translocation (Tat) system of Mycobacterium tuberculosis, increases β-lactam susceptibility and virulence. J Bacteriol 188:6669–6679 10.1128/JB.00631-06. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Behrendt J, Standar K, Lindenstrauss U, Brüser T. 2004. Topological studies on the twin-arginine translocase component TatC. FEMS Microbiol Lett 234:303–308 10.1111/j.1574-6968.2004.tb09548.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 71.Alami M, Lüke I, Deitermann S, Eisner G, Koch HG, Brunner J, Müller M. 2003. Differential interactions between a twin-arginine signal peptide and its translocase in Escherichia coli. Mol Cell 12:937–946 10.1016/S1097-2765(03)00398-8. [DOI] [PubMed] [Google Scholar]
- 72.Rollauer SE, Tarry MJ, Graham JE, Jääskeläinen M, Jäger F, Johnson S, Krehenbrink M, Liu SM, Lukey MJ, Marcoux J, McDowell MA, Rodriguez F, Roversi P, Stansfeld PJ, Robinson CV, Sansom MSP, Palmer T, Högbom M, Berks BC, Lea SM. 2012. Structure of the TatC core of the twin-arginine protein transport system. Nature 492:210–214 10.1038/nature11683. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yahr TL, Wickner WT. 2001. Functional reconstitution of bacterial Tat translocation in vitro. EMBO J 20:2472–2479 10.1093/emboj/20.10.2472. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bageshwar UK, Musser SM. 2007. Two electrical potential-dependent steps are required for transport by the Escherichia coli Tat machinery. J Cell Biol 179:87–99 10.1083/jcb.200702082. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gohlke U, Pullan L, McDevitt CA, Porcelli I, de Leeuw E, Palmer T, Saibil HR, Berks BC. 2005. The TatA component of the twin-arginine protein transport system forms channel complexes of variable diameter. Proc Natl Acad Sci USA 102:10482–10486 10.1073/pnas.0503558102. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Brüser T, Sanders C. 2003. An alternative model of the twin arginine translocation system. Microbiol Res 158:7–17 10.1078/0944-5013-00176. [PubMed] [DOI] [PubMed] [Google Scholar]
- 77.Giménez MI, Dilks K, Pohlschröder M. 2007. Haloferax volcanii twin-arginine translocation substates include secreted soluble, C-terminally anchored and lipoproteins. Mol Microbiol 66:1597–1606 10.1111/j.1365-2958.2007.06034.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 78.Lüke I, Handford JI, Palmer T, Sargent F. 2009. Proteolytic processing of Escherichia coli twin-arginine signal peptides by LepB. Arch Microbiol 191:919–925 10.1007/s00203-009-0516-5. [PubMed] [DOI] [PubMed] [Google Scholar]
- 79.Stanley NR, Palmer T, Berks BC. 2000. The twin arginine consensus motif of Tat signal peptides is involved in Sec-independent protein targeting in Escherichia coli. J Biol Chem 275:11591–11596 10.1074/jbc.275.16.11591. [PubMed] [DOI] [PubMed] [Google Scholar]
- 80.Hinsley AP, Stanley NR, Palmer T, Berks BC. 2001. A naturally occurring bacterial Tat signal peptide lacking one of the ‘invariant’ arginine residues of the consensus targeting motif. FEBS Lett 497:45–49 10.1016/S0014-5793(01)02428-0. [DOI] [PubMed] [Google Scholar]
- 81.Ignatova Z, Hörnle C, Nurk A, Kasche V. 2002. Unusual signal peptide directs penicillin amidase from Escherichia coli to the Tat translocation machinery. Biochem Biophys Res Commun 291:146–149 10.1006/bbrc.2002.6420. [PubMed] [DOI] [PubMed] [Google Scholar]
- 82.Robinson C, Matos CFRO, Beck D, Ren C, Lawrence J, Vasisht N, Mendel S. 2011. Transport and proofreading of proteins by the twin-arginine translocation (Tat) system in bacteria. Biochim Biophys Acta 1808:876–884 10.1016/j.bbamem.2010.11.023. [PubMed] [DOI] [PubMed] [Google Scholar]
- 83.Hatzixanthis K, Palmer T, Sargent F. 2003. A subset of bacterial inner membrane proteins integrated by the twin-arginine translocase. Mol Microbiol 49:1377–1390 10.1046/j.1365-2958.2003.03642.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 84.De Buck E, Vranckx L, Meyen E, Maes L, Vandersmissen L, Anné J, Lammertyn E. 2007. The twin-arginine translocation pathway is necessary for correct membrane insertion of the Rieske Fe/S protein in Legionella pneumophila. FEBS Lett 581:259–264 10.1016/j.febslet.2006.12.022. [PubMed] [DOI] [PubMed] [Google Scholar]
- 85.Keller R, de Keyzer J, Driessen AJM, Palmer T. 2012. Co-operation between different targeting pathways during integration of a membrane protein. J Cell Biol 199:303–315 10.1083/jcb.201204149. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Matsoso LG, Kana BD, Crellin PK, Lea-Smith DJ, Pelosi A, Powell D, Dawes SS, Rubin H, Coppel RL, Mizrahi V. 2005. Function of the cytochrome bc1-aa3 branch of the respiratory network in mycobacteria and network adaptation occurring in response to its disruption. J Bacteriol 187:6300–6308 10.1128/JB.187.18.6300-6308.2005. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Caspers M, Freudl R. 2008. Corynebacterium glutamicum possesses two secA homologous genes that are essential for viability. Arch Microbiol 189:605–610 10.1007/s00203-008-0351-0. [PubMed] [DOI] [PubMed] [Google Scholar]
- 88.Lenz LL, Portnoy DA. 2002. Identification of a second Listeria secA gene associated with protein secretion and the rough phenotype. Mol Microbiol 45:1043–1056 10.1046/j.1365-2958.2002.03072.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 89.Fagan RP, Fairweather NF. 2011. Clostridium difficile has two parallel and essential Sec secretion systems. J Biol Chem 286:27483–27493 10.1074/jbc.M111.263889. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Siboo IR, Chaffin DO, Rubens CE, Sullam PM. 2008. Characterization of the accessory Sec system of Staphylococcus aureus. J Bacteriol 190:6188–6196 10.1128/JB.00300-08. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bensing BA, Sullam PM. 2002. An accessory sec locus of Streptococcus gordonii is required for export of the surface protein GspB and for normal levels of binding to human platelets. Mol Microbiol 44:1081–1094 10.1046/j.1365-2958.2002.02949.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 92.Chen Q, Wu H, Fives-Taylor PM. 2004. Investigating the role of secA2 in secretion and glycosylation of a fimbrial adhesin in Streptococcus parasanguis FW213. Mol Microbiol 53:843–856 10.1111/j.1365-2958.2004.04116.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 93.Bandara M, Skehel JM, Kadioglu A, Collinson I, Nobbs AH, Blocker AJ, Jenkinson HF. 2017. The accessory Sec system (SecY2A2) in Streptococcus pneumoniae is involved in export of pneumolysin toxin, adhesion and biofilm formation. Microbes Infect 19:402–412 10.1016/j.micinf.2017.04.003. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Nguyen-Mau SM, Oh SY, Kern VJ, Missiakas DM, Schneewind O. 2012. Secretion genes as determinants of Bacillus anthracis chain length. J Bacteriol 194:3841–3850 10.1128/JB.00384-12. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Rigel NW, Braunstein M. 2008. A new twist on an old pathway: accessory Sec [corrected] systems. Mol Microbiol 69:291–302 10.1111/j.1365-2958.2008.06294.x. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ligon LS, Rigel NW, Romanchuk A, Jones CD, Braunstein M. 2013. Suppressor analysis reveals a role for SecY in the SecA2-dependent protein export pathway of mycobacteria. J Bacteriol 195:4456–4465 10.1128/JB.00630-13. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Durack J, Burke TP, Portnoy DA. 2015. A prl mutation in SecY suppresses secretion and virulence defects of Listeria monocytogenes secA2 mutants. J Bacteriol 197:932–942 10.1128/JB.02284-14. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Takamatsu D, Bensing BA, Sullam PM. 2004. Genes in the accessory sec locus of Streptococcus gordonii have three functionally distinct effects on the expression of the platelet-binding protein GspB. Mol Microbiol 52:189–203 10.1111/j.1365-2958.2004.03978.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 99.Braunstein M, Espinosa BJ, Chan J, Belisle JT, Jacobs WR Jr. 2003. SecA2 functions in the secretion of superoxide dismutase A and in the virulence of Mycobacterium tuberculosis. Mol Microbiol 48:453–464 10.1046/j.1365-2958.2003.03438.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 100.Watkins BY, Joshi SA, Ball DA, Leggett H, Park S, Kim J, Austin CD, Paler-Martinez A, Xu M, Downing KH, Brown EJ. 2012. Mycobacterium marinum SecA2 promotes stable granulomas and induces tumor necrosis factor alpha in vivo. Infect Immun 80:3512–3520 10.1128/IAI.00686-12. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.van der Woude AD, Stoop EJM, Stiess M, Wang S, Ummels R, van Stempvoort G, Piersma SR, Cascioferro A, Jiménez CR, Houben ENG, Luirink J, Pieters J, van der Sar AM, Bitter W. 2014. Analysis of SecA2-dependent substrates in Mycobacterium marinum identifies protein kinase G (PknG) as a virulence effector. Cell Microbiol 16:280–295 10.1111/cmi.12221. [PubMed] [DOI] [PubMed] [Google Scholar]
- 102.Feltcher ME, Gunawardena HP, Zulauf KE, Malik S, Griffin JE, Sassetti CM, Chen X, Braunstein M. 2015. Label-free quantitative proteomics reveals a role for the Mycobacterium tuberculosis SecA2 pathway in exporting solute binding proteins and Mce transporters to the cell wall. Mol Cell Proteomics 14:1501–1516 10.1074/mcp.M114.044685. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Swanson S, Ioerger TR, Rigel NW, Miller BK, Braunstein M, Sacchettini JC. 2015. Structural similarities and differences between two functionally distinct SecA proteins, Mycobacterium tuberculosis SecA1 and SecA2. J Bacteriol 198:720–730 10.1128/JB.00696-15. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Rigel NW, Gibbons HS, McCann JR, McDonough JA, Kurtz S, Braunstein M. 2009. The accessory SecA2 system of mycobacteria requires ATP binding and the canonical SecA1. J Biol Chem 284:9927–9936 10.1074/jbc.M900325200. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Prabudiansyah I, Kusters I, Driessen AJM. 2015. In vitro interaction of the housekeeping SecA1 with the accessory SecA2 protein of Mycobacterium tuberculosis. PLoS One 10:e0128788 10.1371/journal.pone.0128788. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Gibbons HS, Wolschendorf F, Abshire M, Niederweis M, Braunstein M. 2007. Identification of two Mycobacterium smegmatis lipoproteins exported by a SecA2-dependent pathway. J Bacteriol 189:5090–5100 10.1128/JB.00163-07. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lenz LL, Mohammadi S, Geissler A, Portnoy DA. 2003. SecA2-dependent secretion of autolytic enzymes promotes Listeria monocytogenes pathogenesis. Proc Natl Acad Sci USA 100:12432–12437 10.1073/pnas.2133653100. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Casali N, Riley LW. 2007. A phylogenomic analysis of the Actinomycetales mce operons. BMC Genomics 8:60 10.1186/1471-2164-8-60. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Upadhyay S, Mittal E, Philips JA. 2018. Tuberculosis and the art of macrophage manipulation. Pathog Dis 76:fty037 10.1093/femspd/fty037. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Sullivan JT, Young EF, McCann JR, Braunstein M. 2012. The Mycobacterium tuberculosis SecA2 system subverts phagosome maturation to promote growth in macrophages. Infect Immun 80:996–1006 10.1128/IAI.05987-11. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Zulauf KE, Sullivan JT, Braunstein M. 2018. The SecA2 pathway of Mycobacterium tuberculosis exports effectors that work in concert to arrest phagosome and autophagosome maturation. PLoS Pathog 14:e1007011 10.1371/journal.ppat.1007011. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Vergne I, Chua J, Lee H-H, Lucas M, Belisle J, Deretic V. 2005. Mechanism of phagolysosome biogenesis block by viable Mycobacterium tuberculosis. Proc Natl Acad Sci USA 102:4033–4038 10.1073/pnas.0409716102. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Cowley S, Ko M, Pick N, Chow R, Downing KJ, Gordhan BG, Betts JC, Mizrahi V, Smith DA, Stokes RW, Av-Gay Y. 2004. The Mycobacterium tuberculosis protein serine/threonine kinase PknG is linked to cellular glutamate/glutamine levels and is important for growth in vivo. Mol Microbiol 52:1691–1702 10.1111/j.1365-2958.2004.04085.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 114.Walburger A, Koul A, Ferrari G, Nguyen L, Prescianotto-Baschong C, Huygen K, Klebl B, Thompson C, Bacher G, Pieters J. 2004. Protein kinase G from pathogenic mycobacteria promotes survival within macrophages. Science 304:1800–1804 10.1126/science.1099384. [PubMed] [DOI] [PubMed] [Google Scholar]
- 115.Wolff KA, de la Peña AH, Nguyen HT, Pham TH, Amzel LM, Gabelli SB, Nguyen L. 2015. A redox regulatory system critical for mycobacterial survival in macrophages and biofilm development. PLoS Pathog 11:e1004839 10.1371/journal.ppat.1004839. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Rieck B, Degiacomi G, Zimmermann M, Cascioferro A, Boldrin F, Lazar-Adler NR, Bottrill AR, le Chevalier F, Frigui W, Bellinzoni M, Lisa MN, Alzari PM, Nguyen L, Brosch R, Sauer U, Manganelli R, O’Hare HM. 2017. PknG senses amino acid availability to control metabolism and virulence of Mycobacterium tuberculosis. PLoS Pathog 13:e1006399 10.1371/journal.ppat.1006399. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Pethe K, Swenson DL, Alonso S, Anderson J, Wang C, Russell DG. 2004. Isolation of Mycobacterium tuberculosis mutants defective in the arrest of phagosome maturation. Proc Natl Acad Sci USA 101:13642–13647 10.1073/pnas.0401657101. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Zhang Y, Lathigra R, Garbe T, Catty D, Young D. 1991. Genetic analysis of superoxide dismutase, the 23 kilodalton antigen of Mycobacterium tuberculosis. Mol Microbiol 5:381–391 10.1111/j.1365-2958.1991.tb02120.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 119.Harth G, Lee BY, Horwitz MA. 1997. High-level heterologous expression and secretion in rapidly growing nonpathogenic mycobacteria of four major Mycobacterium tuberculosis extracellular proteins considered to be leading vaccine candidates and drug targets. Infect Immun 65:2321–2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Sonnenberg MG, Belisle JT. 1997. Definition of Mycobacterium tuberculosis culture filtrate proteins by two-dimensional polyacrylamide gel electrophoresis, N-terminal amino acid sequencing, and electrospray mass spectrometry. Infect Immun 65:4515–4524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Rouse DA, DeVito JA, Li Z, Byer H, Morris SL. 1996. Site-directed mutagenesis of the katG gene of Mycobacterium tuberculosis: effects on catalase-peroxidase activities and isoniazid resistance. Mol Microbiol 22:583–592 10.1046/j.1365-2958.1996.00133.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 122.Archambaud C, Nahori MA, Pizarro-Cerda J, Cossart P, Dussurget O. 2006. Control of Listeria superoxide dismutase by phosphorylation. J Biol Chem 281:31812–31822 10.1074/jbc.M606249200. [PubMed] [DOI] [PubMed] [Google Scholar]
- 123.Feltcher ME, Gibbons HS, Ligon LS, Braunstein M. 2013. Protein export by the mycobacterial SecA2 system is determined by the preprotein mature domain. J Bacteriol 195:672–681 10.1128/JB.02032-12. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Miller BK, Hughes R, Ligon LS, Rigel NW, Malik S, Anjuwon-Foster BR, Sacchettini JC, Braunstein M. 2019. Mycobacterium tuberculosis SatS is a chaperone for the SecA2 protein export pathway. eLife 8:e40063 10.7554/eLife.40063. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Abdallah AM, Gey van Pittius NC, Champion PA, Cox J, Luirink J, Vandenbroucke-Grauls CM, Appelmelk BJ, Bitter W. 2007. Type VII secretion: mycobacteria show the way. Nat Rev Microbiol 5:883–891 10.1038/nrmicro1773. [PubMed] [DOI] [PubMed] [Google Scholar]
- 126.Brodin P, Rosenkrands I, Andersen P, Cole ST, Brosch R. 2004. ESAT-6 proteins: protective antigens and virulence factors? Trends Microbiol 12:500–508 10.1016/j.tim.2004.09.007. [PubMed] [DOI] [PubMed] [Google Scholar]
- 127.Converse SE, Cox JS. 2005. A protein secretion pathway critical for Mycobacterium tuberculosis virulence is conserved and functional in Mycobacterium smegmatis. J Bacteriol 187:1238–1245 10.1128/JB.187.4.1238-1245.2005. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Bitter W, Houben ENG, Bottai D, Brodin P, Brown EJ, Cox JS, Derbyshire K, Fortune SM, Gao LY, Liu J, Gey van Pittius NC, Pym AS, Rubin EJ, Sherman DR, Cole ST, Brosch R. 2009. Systematic genetic nomenclature for type VII secretion systems. PLoS Pathog 5:e1000507 10.1371/journal.ppat.1000507. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Bitter W, Houben ENG, Luirink J, Appelmelk BJ. 2009. Type VII secretion in mycobacteria: classification in line with cell envelope structure. Trends Microbiol 17:337–338 10.1016/j.tim.2009.05.007. [PubMed] [DOI] [PubMed] [Google Scholar]
- 130.Gey Van Pittius NC, Gamieldien J, Hide W, Brown GD, Siezen RJ, Beyers AD. 2001. The ESAT-6 gene cluster of Mycobacterium tuberculosis and other high G+C Gram-positive bacteria. Genome Biol 2:RESEARCH0044. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Sørensen AL, Nagai S, Houen G, Andersen P, Andersen AB. 1995. Purification and characterization of a low-molecular-mass T-cell antigen secreted by Mycobacterium tuberculosis. Infect Immun 63:1710–1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Stanley SA, Raghavan S, Hwang WW, Cox JS. 2003. Acute infection and macrophage subversion by Mycobacterium tuberculosis require a specialized secretion system. Proc Natl Acad Sci USA 100:13001–13006 10.1073/pnas.2235593100. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Lewis KN, Liao R, Guinn KM, Hickey MJ, Smith S, Behr MA, Sherman DR. 2003. Deletion of RD1 from Mycobacterium tuberculosis mimics bacille Calmette-Guérin attenuation. J Infect Dis 187:117–123 10.1086/345862. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Philipp WJ, Nair S, Guglielmi G, Lagranderie M, Gicquel B, Cole ST. 1996. Physical mapping of Mycobacterium bovis BCG Pasteur reveals differences from the genome map of Mycobacterium tuberculosis H37Rv and from M. bovis. Microbiology 142:3135–3145 10.1099/13500872-142-11-3135. [PubMed] [DOI] [PubMed] [Google Scholar]
- 135.Pym AS, Brodin P, Brosch R, Huerre M, Cole ST. 2002. Loss of RD1 contributed to the attenuation of the live tuberculosis vaccines Mycobacterium bovis BCG and Mycobacterium microti. Mol Microbiol 46:709–717 10.1046/j.1365-2958.2002.03237.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 136.Simeone R, Bobard A, Lippmann J, Bitter W, Majlessi L, Brosch R, Enninga J. 2012. Phagosomal rupture by Mycobacterium tuberculosis results in toxicity and host cell death. PLoS Pathog 8:e1002507 10.1371/journal.ppat.1002507. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.van der Wel N, Hava D, Houben D, Fluitsma D, van Zon M, Pierson J, Brenner M, Peters PJ. 2007. M. tuberculosis and M. leprae translocate from the phagolysosome to the cytosol in myeloid cells. Cell 129:1287–1298 10.1016/j.cell.2007.05.059. [PubMed] [DOI] [PubMed] [Google Scholar]
- 138.Smith J, Manoranjan J, Pan M, Bohsali A, Xu J, Liu J, McDonald KL, Szyk A, LaRonde-LeBlanc N, Gao LY, Pan M, Bohsali A, Xu J, Liu J, Mcdonald KL, Szyk A, Laronde-leblanc N, Gao L. 2008. Evidence for pore formation in host cell membranes by ESX-1-secreted ESAT-6 and its role in Mycobacterium marinum escape from the vacuole. Infect Immun 76:5478–5487 10.1128/IAI.00614-08. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Hsu T, Hingley-Wilson SM, Chen B, Chen M, Dai AZ, Morin PM, Marks CB, Padiyar J, Goulding C, Gingery M, Eisenberg D, Russell RG, Derrick SC, Collins FM, Morris SL, King CH, Jacobs WR Jr. 2003. The primary mechanism of attenuation of bacillus Calmette-Guerin is a loss of secreted lytic function required for invasion of lung interstitial tissue. Proc Natl Acad Sci USA 100:12420–12425 10.1073/pnas.1635213100. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Gao LY, Guo S, McLaughlin B, Morisaki H, Engel JN, Brown EJ. 2004. A mycobacterial virulence gene cluster extending RD1 is required for cytolysis, bacterial spreading and ESAT-6 secretion. Mol Microbiol 53:1677–1693 10.1111/j.1365-2958.2004.04261.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 141.Houben D, Demangel C, van Ingen J, Perez J, Baldeón L, Abdallah AM, Caleechurn L, Bottai D, van Zon M, de Punder K, van der Laan T, Kant A, Bossers-de Vries R, Willemsen P, Bitter W, van Soolingen D, Brosch R, van der Wel N, Peters PJ. 2012. ESX-1-mediated translocation to the cytosol controls virulence of mycobacteria. Cell Microbiol 14:1287–1298 10.1111/j.1462-5822.2012.01799.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 142.Conrad WH, Osman MM, Shanahan JK, Chu F, Takaki KK, Cameron J, Hopkinson-Woolley D, Brosch R, Ramakrishnan L. 2017. Mycobacterial ESX-1 secretion system mediates host cell lysis through bacterium contact-dependent gross membrane disruptions. Proc Natl Acad Sci USA 114:1371–1376 10.1073/pnas.1620133114. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Coros A, Callahan B, Battaglioli E, Derbyshire KM. 2008. The specialized secretory apparatus ESX-1 is essential for DNA transfer in Mycobacterium smegmatis. Mol Microbiol 69:794–808. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Serafini A, Pisu D, Palù G, Rodriguez GM, Manganelli R. 2013. The ESX-3 secretion system is necessary for iron and zinc homeostasis in Mycobacterium tuberculosis. PLoS One 8:e78351 10.1371/journal.pone.0078351. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Siegrist MS, Unnikrishnan M, McConnell MJ, Borowsky M, Cheng T-Y, Siddiqi N, Fortune SM, Moody DB, Rubin EJ. 2009. Mycobacterial Esx-3 is required for mycobactin-mediated iron acquisition. Proc Natl Acad Sci USA 106:18792–18797 10.1073/pnas.0900589106. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Portal-Celhay C, Tufariello JM, Srivastava S, Zahra A, Klevorn T, Grace PS, Mehra A, Park HS, Ernst JD, Jacobs WR Jr, Philips JA. 2016. Mycobacterium tuberculosis EsxH inhibits ESCRT-dependent CD4+ T-cell activation. Nat Microbiol 2:16232 10.1038/nmicrobiol.2016.232. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Tufariello JM, Chapman JR, Kerantzas CA, Wong K-W, Vilchèze C, Jones CM, Cole LE, Tinaztepe E, Thompson V, Fenyö D, Niederweis M, Ueberheide B, Philips JA, Jacobs WR Jr. 2016. Separable roles for Mycobacterium tuberculosis ESX-3 effectors in iron acquisition and virulence. Proc Natl Acad Sci USA 113:E348–E357 10.1073/pnas.1523321113. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Gey van Pittius NC, Sampson SL, Lee H, Kim Y, van Helden PD, Warren RM. 2006. Evolution and expansion of the Mycobacterium tuberculosis PE and PPE multigene families and their association with the duplication of the ESAT-6 (esx) gene cluster regions. BMC Evol Biol 6:95 10.1186/1471-2148-6-95. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Ates LS, Ummels R, Commandeur S, van de Weerd R, Sparrius M, Weerdenburg E, Alber M, Kalscheuer R, Piersma SR, Abdallah AM, Abd El Ghany M, Abdel-Haleem AM, Pain A, Jiménez CR, Bitter W, Houben ENG. 2015. Essential role of the ESX-5 secretion system in outer membrane permeability of pathogenic mycobacteria. PLoS Genet 11:e1005190 10.1371/journal.pgen.1005190. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Abdallah AM, Savage NDL, van Zon M, Wilson L, Vandenbroucke-Grauls CMJE, van der Wel NN, Ottenhoff THM, Bitter W. 2008. The ESX-5 secretion system of Mycobacterium marinum modulates the macrophage response. J Immunol 181:7166–7175 10.4049/jimmunol.181.10.7166. [PubMed] [DOI] [PubMed] [Google Scholar]
- 151.Abdallah AM, Verboom T, Weerdenburg EM, Gey van Pittius NC, Mahasha PW, Jiménez C, Parra M, Cadieux N, Brennan MJ, Appelmelk BJ, Bitter W. 2009. PPE and PE_PGRS proteins of Mycobacterium marinum are transported via the type VII secretion system ESX-5. Mol Microbiol 73:329–340 10.1111/j.1365-2958.2009.06783.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 152.Abdallah AM, Bestebroer J, Savage NDL, de Punder K, van Zon M, Wilson L, Korbee CJ, van der Sar AM, Ottenhoff THM, van der Wel NN, Bitter W, Peters PJ. 2011. Mycobacterial secretion systems ESX-1 and ESX-5 play distinct roles in host cell death and inflammasome activation. J Immunol 187:4744–4753 10.4049/jimmunol.1101457. [PubMed] [DOI] [PubMed] [Google Scholar]
- 153.Bottai D, Di Luca M, Majlessi L, Frigui W, Simeone R, Sayes F, Bitter W, Brennan MJ, Leclerc C, Batoni G, Campa M, Brosch R, Esin S. 2012. Disruption of the ESX-5 system of Mycobacterium tuberculosis causes loss of PPE protein secretion, reduction of cell wall integrity and strong attenuation. Mol Microbiol 83:1195–1209 10.1111/j.1365-2958.2012.08001.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 154.Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, Gordon SV, Eiglmeier K, Gas S, Barry CE III, Tekaia F, Badcock K, Basham D, Brown D, Chillingworth T, Connor R, Davies R, Devlin K, Feltwell T, Gentles S, Hamlin N, Holroyd S, Hornsby T, Jagels K, Krogh A, McLean J, Moule S, Murphy L, Oliver K, Osborne J, Quail MA, Rajandream MA, Rogers J, Rutter S, Seeger K, Skelton J, Squares R, Squares S, Sulston JE, Taylor K, Whitehead S, Barrell BG. 1998. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393:537–544 10.1038/31159. [PubMed] [DOI] [PubMed] [Google Scholar]
- 155.Sassetti CM, Boyd DH, Rubin EJ. 2003. Genes required for mycobacterial growth defined by high density mutagenesis. Mol Microbiol 48:77–84 10.1046/j.1365-2958.2003.03425.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 156.Sassetti CM, Rubin EJ. 2003. Genetic requirements for mycobacterial survival during infection. Proc Natl Acad Sci USA 100:12989–12994 10.1073/pnas.2134250100. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Mendum TA, Wu H, Kierzek AM, Stewart GR. 2015. Lipid metabolism and type VII secretion systems dominate the genome scale virulence profile of Mycobacterium tuberculosis in human dendritic cells. BMC Genomics 16:372 10.1186/s12864-015-1569-2. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Laencina L, Dubois V, Le Moigne V, Viljoen A, Majlessi L, Pritchard J, Bernut A, Piel L, Roux A-L, Gaillard J-L, Lombard B, Loew D, Rubin EJ, Brosch R, Kremer L, Herrmann J-L, Girard-Misguich F. 2018. Identification of genes required for Mycobacterium abscessus growth in vivo with a prominent role of the ESX-4 locus. Proc Natl Acad Sci USA 115:E1002–E1011 10.1073/pnas.1713195115. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Gray TA, Clark RR, Boucher N, Lapierre P, Smith C, Derbyshire KM. 2016. Intercellular communication and conjugation are mediated by ESX secretion systems in mycobacteria. Science 354:347–350 10.1126/science.aag0828. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Clark RR, Judd J, Lasek-Nesselquist E, Montgomery SA, Hoffmann JG, Derbyshire KM, Gray TA. 2018. Direct cell-cell contact activates SigM to express the ESX-4 secretion system in Mycobacterium smegmatis. Proc Natl Acad Sci USA 115:E6595–E6603 10.1073/pnas.1804227115. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Ummels R, Abdallah AM, Kuiper V, Aâjoud A, Sparrius M, Naeem R, Spaink HP, van Soolingen D, Pain A, Bitter W. 2014. Identification of a novel conjugative plasmid in mycobacteria that requires both type IV and type VII secretion. MBio 5:e01744-14 10.1128/mBio.01744-14. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Newton-Foot M, Warren RM, Sampson SL, van Helden PD, Gey van Pittius NC. 2016. The plasmid-mediated evolution of the mycobacterial ESX (type VII) secretion systems. BMC Evol Biol 16:62 10.1186/s12862-016-0631-2. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Boritsch EC, Khanna V, Pawlik A, Honoré N, Navas VH, Ma L, Bouchier C, Seemann T, Supply P, Stinear TP, Brosch R. 2016. Key experimental evidence of chromosomal DNA transfer among selected tuberculosis-causing mycobacteria. Proc Natl Acad Sci USA 113:9876–9881 10.1073/pnas.1604921113. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Akpe San Roman S, Facey PD, Fernandez-Martinez L, Rodriguez C, Vallin C, Del Sol R, Dyson P. 2010. A heterodimer of EsxA and EsxB is involved in sporulation and is secreted by a type VII secretion system in Streptomyces coelicolor. Microbiology 156:1719–1729 10.1099/mic.0.037069-0. [PubMed] [DOI] [PubMed] [Google Scholar]
- 165.Gago G, Diacovich L, Arabolaza A, Tsai S-C, Gramajo H. 2011. Fatty acid biosynthesis in actinomycetes. FEMS Microbiol Rev 35:475–497 10.1111/j.1574-6976.2010.00259.x. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Pallen MJ. 2002. The ESAT-6/WXG100 superfamily: and a new Gram-positive secretion system? Trends Microbiol 10:209–212 10.1016/S0966-842X(02)02345-4. [DOI] [PubMed] [Google Scholar]
- 167.Huppert LA, Ramsdell TL, Chase MR, Sarracino DA, Fortune SM, Burton BM. 2014. The ESX system in Bacillus subtilis mediates protein secretion. PLoS One 9:e96267 10.1371/journal.pone.0096267. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Daleke MH, van der Woude AD, Parret AHA, Ummels R, de Groot AM, Watson D, Piersma SR, Jiménez CR, Luirink J, Bitter W, Houben ENG. 2012. Specific chaperones for the type VII protein secretion pathway. J Biol Chem 287:31939–31947 10.1074/jbc.M112.397596. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Ekiert DC, Cox JS. 2014. Structure of a PE-PPE-EspG complex from Mycobacterium tuberculosis reveals molecular specificity of ESX protein secretion. Proc Natl Acad Sci USA 111:14758–14763 10.1073/pnas.1409345111. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Tekaia F, Gordon SV, Garnier T, Brosch R, Barrell BG, Cole ST. 1999. Analysis of the proteome of Mycobacterium tuberculosis in silico. Tuber Lung Dis 79:329–342 10.1054/tuld.1999.0220. [PubMed] [DOI] [PubMed] [Google Scholar]
- 171.Unnikrishnan M, Constantinidou C, Palmer T, Pallen MJ. 2017. The Enigmatic Esx proteins: looking beyond mycobacteria. Trends Microbiol 25:192–204 10.1016/j.tim.2016.11.004. [PubMed] [DOI] [PubMed] [Google Scholar]
- 172.Champion PAD, Stanley SA, Champion MM, Brown EJ, Cox JS. 2006. C-terminal signal sequence promotes virulence factor secretion in Mycobacterium tuberculosis. Science 313:1632–1636 10.1126/science.1131167. [PubMed] [DOI] [PubMed] [Google Scholar]
- 173.Renshaw PS, Lightbody KL, Veverka V, Muskett FW, Kelly G, Frenkiel TA, Gordon SV, Hewinson RG, Burke B, Norman J, Williamson RA, Carr MD. 2005. Structure and function of the complex formed by the tuberculosis virulence factors CFP-10 and ESAT-6. EMBO J 24:2491–2498 10.1038/sj.emboj.7600732. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Renshaw PS, Panagiotidou P, Whelan A, Gordon SV, Hewinson RG, Williamson RA, Carr MD. 2002. Conclusive evidence that the major T-cell antigens of the Mycobacterium tuberculosis complex ESAT-6 and CFP-10 form a tight, 1:1 complex and characterization of the structural properties of ESAT-6, CFP-10, and the ESAT-6*CFP-10 complex. Implications for pathogenesis and virulence. J Biol Chem 277:21598–21603 10.1074/jbc.M201625200. [PubMed] [DOI] [PubMed] [Google Scholar]
- 175.de Jonge MI, Pehau-Arnaudet G, Fretz MM, Romain F, Bottai D, Brodin P, Honoré N, Marchal G, Jiskoot W, England P, Cole ST, Brosch R. 2007. ESAT-6 from Mycobacterium tuberculosis dissociates from its putative chaperone CFP-10 under acidic conditions and exhibits membrane-lysing activity. J Bacteriol 189:6028–6034 10.1128/JB.00469-07. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Fortune SM, Jaeger A, Sarracino DA, Chase MR, Sassetti CM, Sherman DR, Bloom BR, Rubin EJ. 2005. Mutually dependent secretion of proteins required for mycobacterial virulence. Proc Natl Acad Sci USA 102:10676–10681 10.1073/pnas.0504922102. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Ilghari D, Lightbody KL, Veverka V, Waters LC, Muskett FW, Renshaw PS, Carr MD. 2011. Solution structure of the Mycobacterium tuberculosis EsxG·EsxH complex: functional implications and comparisons with other M. tuberculosis Esx family complexes. J Biol Chem 286:29993–30002 10.1074/jbc.M111.248732. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Arbing MA, Kaufmann M, Phan T, Chan S, Cascio D, Eisenberg D. 2010. The crystal structure of the Mycobacterium tuberculosis Rv3019c-Rv3020c ESX complex reveals a domain-swapped heterotetramer. Protein Sci 19:1692–1703 10.1002/pro.451. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Akhter Y, Ehebauer MT, Mukhopadhyay S, Hasnain SE. 2012. The PE/PPE multigene family codes for virulence factors and is a possible source of mycobacterial antigenic variation: perhaps more? Biochimie 94:110–116 10.1016/j.biochi.2011.09.026. [PubMed] [DOI] [PubMed] [Google Scholar]
- 180.Phan TH, Ummels R, Bitter W, Houben ENG. 2017. Identification of a substrate domain that determines system specificity in mycobacterial type VII secretion systems. Sci Rep 7:42704 10.1038/srep42704. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Korotkova N, Freire D, Phan TH, Ummels R, Creekmore CC, Evans TJ, Wilmanns M, Bitter W, Parret AHA, Houben ENG, Korotkov KV. 2014. Structure of the Mycobacterium tuberculosis type VII secretion system chaperone EspG5 in complex with PE25-PPE41 dimer. Mol Microbiol 94:367–382 10.1111/mmi.12770. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Strong M, Sawaya MR, Wang S, Phillips M, Cascio D, Eisenberg D. 2006. Toward the structural genomics of complexes: crystal structure of a PE/PPE protein complex from Mycobacterium tuberculosis. Proc Natl Acad Sci USA 103:8060–8065 10.1073/pnas.0602606103. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.van der Woude AD, Mahendran KR, Ummels R, Piersma SR, Pham TV, Jiménez CR, de Punder K, van der Wel NN, Winterhalter M, Luirink J, Bitter W, Houben ENG. 2013. Differential detergent extraction of Mycobacterium marinum cell envelope proteins identifies an extensively modified threonine-rich outer membrane protein with channel activity. J Bacteriol 195:2050–2059 10.1128/JB.02236-12. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Sani M, Houben ENG, Geurtsen J, Pierson J, de Punder K, van Zon M, Wever B, Piersma SR, Jiménez CR, Daffé M, Appelmelk BJ, Bitter W, van der Wel N, Peters PJ. 2010. Direct visualization by cryo-EM of the mycobacterial capsular layer: a labile structure containing ESX-1-secreted proteins. PLoS Pathog 6:e1000794 10.1371/journal.ppat.1000794. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Chen X, Cheng HF, Zhou J, Chan CY, Lau KF, Tsui SKW, Au SW. 2017. Structural basis of the PE-PPE protein interaction in Mycobacterium tuberculosis. J Biol Chem 292:16880–16890 10.1074/jbc.M117.802645. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Deb C, Daniel J, Sirakova TD, Abomoelak B, Dubey VS, Kolattukudy PE. 2006. A novel lipase belonging to the hormone-sensitive lipase family induced under starvation to utilize stored triacylglycerol in Mycobacterium tuberculosis. J Biol Chem 281:3866–3875 10.1074/jbc.M505556200. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Sultana R, Vemula MH, Banerjee S, Guruprasad L. 2013. The PE16 (Rv1430) of Mycobacterium tuberculosis is an esterase belonging to serine hydrolase superfamily of proteins. PLoS One 8:e55320 10.1371/journal.pone.0055320. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Champion PAD, Champion MM, Manzanillo P, Cox JS. 2009. ESX-1 secreted virulence factors are recognized by multiple cytosolic AAA ATPases in pathogenic mycobacteria. Mol Microbiol 73:950–962 10.1111/j.1365-2958.2009.06821.x. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.McLaughlin B, Chon JS, MacGurn JA, Carlsson F, Cheng TL, Cox JS, Brown EJ. 2007. A mycobacterium ESX-1-secreted virulence factor with unique requirements for export. PLoS Pathog 3:e105 10.1371/journal.ppat.0030105. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Champion MM, Williams EA, Pinapati RS, Champion PAD. 2014. Correlation of phenotypic profiles using targeted proteomics identifies mycobacterial esx-1 substrates. J Proteome Res 13:5151–5164 10.1021/pr500484w. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Bosserman RE, Nicholson KR, Champion MM, Champion PA. 2019. A new ESX-1 substrate in M. marinum that is required for hemolysis but not host cell lysis. J Bacteriol 201:e00760-18. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Lou Y, Rybniker J, Sala C, Cole ST. 2017. EspC forms a filamentous structure in the cell envelope of Mycobacterium tuberculosis and impacts ESX-1 secretion. Mol Microbiol 103:26–38 10.1111/mmi.13575. [PubMed] [DOI] [PubMed] [Google Scholar]
- 193.Carlsson F, Joshi SA, Rangell L, Brown EJ. 2009. Polar localization of virulence-related Esx-1 secretion in mycobacteria. PLoS Pathog 5:e1000285 10.1371/journal.ppat.1000285. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Korotkova N, Piton J, Wagner JM, Boy-Röttger S, Japaridze A, Evans TJ, Cole ST, Pojer F, Korotkov KV. 2015. Structure of EspB, a secreted substrate of the ESX-1 secretion system of Mycobacterium tuberculosis. J Struct Biol 191:236–244 10.1016/j.jsb.2015.06.003. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Daleke MHM, Ummels R, Bawono P, Heringa J, Vandenbroucke-Grauls CMJE, Luirink J, Bitter W. 2012. General secretion signal for the mycobacterial type VII secretion pathway. Proc Natl Acad Sci USA 109:11342–11347 10.1073/pnas.1119453109. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Garces A, Atmakuri K, Chase MR, Woodworth JS, Krastins B, Rothchild AC, Ramsdell TL, Lopez MF, Behar SM, Sarracino DA, Fortune SM. 2010. EspA acts as a critical mediator of ESX1-dependent virulence in Mycobacterium tuberculosis by affecting bacterial cell wall integrity. PLoS Pathog 6:e1000957 10.1371/journal.ppat.1000957. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Chen JM, Boy-Röttger S, Dhar N, Sweeney N, Buxton RS, Pojer F, Rosenkrands I, Cole ST. 2012. EspD is critical for the virulence-mediating ESX-1 secretion system in Mycobacterium tuberculosis. J Bacteriol 194:884–893 10.1128/JB.06417-11. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Ates LS, Dippenaar A, Ummels R, Piersma SR, van der Woude AD, van der Kuij K, Le Chevalier F, Mata-Espinosa D, Barrios-Payán J, Marquina-Castillo B, Guapillo C, Jiménez CR, Pain A, Houben ENG, Warren RM, Brosch R, Hernández-Pando R, Bitter W. 2018. Mutations in ppe38 block PE_PGRS secretion and increase virulence of Mycobacterium tuberculosis. Nat Microbiol 3:181–188 10.1038/s41564-017-0090-6. [PubMed] [DOI] [PubMed] [Google Scholar]
- 199.Ates LS, van der Woude AD, Bestebroer J, van Stempvoort G, Musters RJP, Garcia-Vallejo JJ, Picavet DI, Weerd R, Maletta M, Kuijl CP, van der Wel NN, Bitter W. 2016. The ESX-5 system of pathogenic mycobacteria is involved in capsule integrity and virulence through its substrate PPE10. PLoS Pathog 12:e1005696 10.1371/journal.ppat.1005696. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Rosenberg OS, Dovala D, Li X, Connolly L, Bendebury A, Finer-Moore J, Holton J, Cheng Y, Stroud RM, Cox JS. 2015. Substrates control multimerization and activation of the multi-domain ATPase motor of type VII secretion. Cell 161:501–512 10.1016/j.cell.2015.03.040. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Poulsen C, Panjikar S, Holton SJ, Wilmanns M, Song YH. 2014. WXG100 protein superfamily consists of three subfamilies and exhibits an α-helical C-terminal conserved residue pattern. PLoS One 9:e89313 10.1371/journal.pone.0089313. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Tuukkanen AT, Freire D, Chan S, Arbing MA, Reed RW, Evans TJ, Zenkeviciutė G, Kim J, Kahng S, Sawaya MR, Chaton CT, Wilmanns M, Eisenberg D, Parret AHA, Korotkov KV. 2019. Structural variability of EspG chaperones from mycobacterial ESX-1, ESX-3 and ESX-5 type VII secretion systems. J Mol Biol 431:289–307 10.1016/j.jmb.2018.11.003. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Bottai D, Majlessi L, Simeone R, Frigui W, Laurent C, Lenormand P, Chen J, Rosenkrands I, Huerre M, Leclerc C, Cole ST, Brosch R. 2011. ESAT-6 secretion-independent impact of ESX-1 genes espF and espG1 on virulence of Mycobacterium tuberculosis. J Infect Dis 203:1155–1164 10.1093/infdis/jiq089. [PubMed] [DOI] [PubMed] [Google Scholar]
- 204.Phan TH, van Leeuwen LM, Kuijl C, Ummels R, van Stempvoort G, Rubio-Canalejas A, Piersma SR, Jiménez CR, van der Sar AM, Houben ENG, Bitter W. 2018. EspH is a hypervirulence factor for Mycobacterium marinum and essential for the secretion of the ESX-1 substrates EspE and EspF. PLoS Pathog 14:e1007247 10.1371/journal.ppat.1007247. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Bönemann G, Pietrosiuk A, Diemand A, Zentgraf H, Mogk A. 2009. Remodelling of VipA/VipB tubules by ClpV-mediated threading is crucial for type VI protein secretion. EMBO J 28:315–325 10.1038/emboj.2008.269. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Basler M, Pilhofer M, Henderson GP, Jensen GJ, Mekalanos JJ. 2012. Type VI secretion requires a dynamic contractile phage tail-like structure. Nature 483:182–186 10.1038/nature10846. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Luthra A, Mahmood A, Arora A, Ramachandran R. 2008. Characterization of Rv3868, an essential hypothetical protein of the ESX-1 secretion system in Mycobacterium tuberculosis. J Biol Chem 283:36532–36541 10.1074/jbc.M807144200. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Gaur A, Sharma VK, Shree S, Rai N, Ramachandran R. 2017. Characterization of EccA3, a CbbX family ATPase from the ESX-3 secretion pathway of M. tuberculosis. Biochim Biophys Acta Proteins Proteomics 1865:715–724 10.1016/j.bbapap.2017.04.001. [PubMed] [DOI] [PubMed] [Google Scholar]
- 209.Wagner JM, Evans TJ, Korotkov KV. 2014. Crystal structure of the N-terminal domain of EccA1 ATPase from the ESX-1 secretion system of Mycobacterium tuberculosis. Proteins 82:159–163 10.1002/prot.24351. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Joshi SA, Ball DA, Sun MG, Carlsson F, Watkins BY, Aggarwal N, McCracken JM, Huynh KK, Brown EJ. 2012. EccA1, a component of the Mycobacterium marinum ESX-1 protein virulence factor secretion pathway, regulates mycolic acid lipid synthesis. Chem Biol 19:372–380 10.1016/j.chembiol.2012.01.008. [PubMed] [DOI] [PubMed] [Google Scholar]
- 211.Abdallah AM, Verboom T, Hannes F, Safi M, Strong M, Eisenberg D, Musters RJP, Vandenbroucke-Grauls CMJE, Appelmelk BJ, Luirink J, Bitter W. 2006. A specific secretion system mediates PPE41 transport in pathogenic mycobacteria. Mol Microbiol 62:667–679 10.1111/j.1365-2958.2006.05409.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 212.Houben ENG, Bestebroer J, Ummels R, Wilson L, Piersma SR, Jiménez CR, Ottenhoff THM, Luirink J, Bitter W. 2012. Composition of the type VII secretion system membrane complex. Mol Microbiol 86:472–484 10.1111/j.1365-2958.2012.08206.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 213.Teutschbein J, Schumann G, Möllmann U, Grabley S, Cole ST, Munder T. 2009. A protein linkage map of the ESAT-6 secretion system 1 (ESX-1) of Mycobacterium tuberculosis. Microbiol Res 164:253–259 10.1016/j.micres.2006.11.016. [PubMed] [DOI] [PubMed] [Google Scholar]
- 214.Ohol YM, Goetz DH, Chan K, Shiloh MU, Craik CS, Cox JS. 2010. Mycobacterium tuberculosis MycP1 protease plays a dual role in regulation of ESX-1 secretion and virulence. Cell Host Microbe 7:210–220 10.1016/j.chom.2010.02.006. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Brodin P, Majlessi L, Marsollier L, de Jonge MI, Bottai D, Demangel C, Hinds J, Neyrolles O, Butcher PD, Leclerc C, Cole ST, Brosch R. 2006. Dissection of ESAT-6 system 1 of Mycobacterium tuberculosis and impact on immunogenicity and virulence. Infect Immun 74:88–98 10.1128/IAI.74.1.88-98.2006. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Siegrist MS, Steigedal M, Ahmad R, Mehra A, Dragset MS, Schuster BM, Philips JA, Carr SA, Rubin EJ. 2014. Mycobacterial Esx-3 requires multiple components for iron acquisition. MBio 5:e01073-14 10.1128/mBio.01073-14. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Di Luca M, Bottai D, Batoni G, Orgeur M, Aulicino A, Counoupas C, Campa M, Brosch R, Esin S. 2012. The ESX-5 associated ecc5-ecc5 locus is essential for Mycobacterium tuberculosis viability. PLoS One 7:e52059 10.1371/journal.pone.0052059. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Wagner JM, Chan S, Evans TJ, Kahng S, Kim J, Arbing MA, Eisenberg D, Korotkov KV. 2016. Structures of EccB1 and EccD1 from the core complex of the mycobacterial ESX-1 type VII secretion system. BMC Struct Biol 16:5 10.1186/s12900-016-0056-6. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Zhang XL, Li DF, Fleming J, Wang LW, Zhou Y, Wang DC, Zhang XE, Bi LJ. 2015. Core component EccB1 of the Mycobacterium tuberculosis type VII secretion system is a periplasmic ATPase. FASEB J 29:4804–4814 10.1096/fj.15-270843. [PubMed] [DOI] [PubMed] [Google Scholar]
- 220.Burts ML, Williams WA, DeBord K, Missiakas DM. 2005. EsxA and EsxB are secreted by an ESAT-6-like system that is required for the pathogenesis of Staphylococcus aureus infections. Proc Natl Acad Sci USA 102:1169–1174 10.1073/pnas.0405620102. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Barre FX. 2007. FtsK and SpoIIIE: the tale of the conserved tails. Mol Microbiol 66:1051–1055 10.1111/j.1365-2958.2007.05981.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 222.Burton B, Dubnau D. 2010. Membrane-associated DNA transport machines. Cold Spring Harb Perspect Biol 2:a000406 10.1101/cshperspect.a000406. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Atmakuri K, Cascales E, Christie PJ. 2004. Energetic components VirD4, VirB11 and VirB4 mediate early DNA transfer reactions required for bacterial type IV secretion. Mol Microbiol 54:1199–1211 10.1111/j.1365-2958.2004.04345.x. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.van Winden VJC, Ummels R, Piersma SR, Jiménez CR, Korotkov KV, Bitter W, Houben ENG. 2016. Mycosins are required for the stabilization of the ESX-1 and ESX-5 type VII secretion membrane complexes. MBio 7:e0147-16 10.1128/mBio.01471-16. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Beckham KSH, Ciccarelli L, Bunduc CM, Mertens HDT, Ummels R, Lugmayr W, Mayr J, Rettel M, Savitski MM, Svergun DI, Bitter W, Wilmanns M, Marlovits TC, Parret AHA, Houben ENG. 2017. Structure of the mycobacterial ESX-5 type VII secretion system membrane complex by single-particle analysis. Nat Microbiol 2:17047 10.1038/nmicrobiol.2017.47. [PubMed] [DOI] [PubMed] [Google Scholar]
- 226.Brown GD, Dave JA, Gey van Pittius NC, Stevens L, Ehlers MRW, Beyers AD. 2000. The mycosins of Mycobacterium tuberculosis H37Rv: a family of subtilisin-like serine proteases. Gene 254:147–155 10.1016/S0378-1119(00)00277-8. [DOI] [PubMed] [Google Scholar]
- 227.Dave JA, Gey van Pittius NC, Beyers AD, Ehlers MRW, Brown GD. 2002. Mycosin-1, a subtilisin-like serine protease of Mycobacterium tuberculosis, is cell wall-associated and expressed during infection of macrophages. BMC Microbiol 2:30 10.1186/1471-2180-2-30. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Wagner JM, Evans TJ, Chen J, Zhu H, Houben ENG, Bitter W, Korotkov KV. 2013. Understanding specificity of the mycosin proteases in ESX/type VII secretion by structural and functional analysis. J Struct Biol 184:115–128 10.1016/j.jsb.2013.09.022. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Solomonson M, Huesgen PF, Wasney GA, Watanabe N, Gruninger RJ, Prehna G, Overall CM, Strynadka NCJ. 2013. Structure of the mycosin-1 protease from the mycobacterial ESX-1 protein type VII secretion system. J Biol Chem 288:17782–17790 10.1074/jbc.M113.462036. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Shinde U, Inouye M. 1995. Folding mediated by an intramolecular chaperone: autoprocessing pathway of the precursor resolved via a substrate assisted catalysis mechanism. J Mol Biol 247:390–395 10.1006/jmbi.1994.0147. [PubMed] [DOI] [PubMed] [Google Scholar]
- 231.Sun D, Liu Q, He Y, Wang C, Wu F, Tian C, Zang J. 2013. The putative propeptide of MycP1 in mycobacterial type VII secretion system does not inhibit protease activity but improves protein stability. Protein Cell 4:921–931 10.1007/s13238-013-3089-7. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.van Winden VJC, Damen MPM, Ummels R, Bitter W, Houben ENG. 2019. Protease domain and transmembrane domain of the type VII secretion mycosin protease determine system-specific functioning in mycobacteria. J Biol Chem 294:4806–4814 10.1074/jbc.RA118.007090. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Frasinyuk MS, Kwiatkowski S, Wagner JM, Evans TJ, Reed RW, Korotkov KV, Watt DS. 2014. Pentapeptide boronic acid inhibitors of Mycobacterium tuberculosis MycP1 protease. Bioorg Med Chem Lett 24:3546–3548 10.1016/j.bmcl.2014.05.056. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Niederweis M, Danilchanka O, Huff J, Hoffmann C, Engelhardt H. 2010. Mycobacterial outer membranes: in search of proteins. Trends Microbiol 18:109–116 10.1016/j.tim.2009.12.005. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Mahfoud M, Sukumaran S, Hülsmann P, Grieger K, Niederweis M. 2006. Topology of the porin MspA in the outer membrane of Mycobacterium smegmatis. J Biol Chem 281:5908–5915 10.1074/jbc.M511642200. [PubMed] [DOI] [PubMed] [Google Scholar]
- 236.Niederweis M. 2003. Mycobacterial porins: new channel proteins in unique outer membranes. Mol Microbiol 49:1167–1177 10.1046/j.1365-2958.2003.03662.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 237.Trias J, Jarlier V, Benz R. 1992. Porins in the cell wall of mycobacteria. Science 258:1479–1481 10.1126/science.1279810. [PubMed] [DOI] [PubMed] [Google Scholar]
- 238.Wolschendorf F, Mahfoud M, Niederweis M. 2007. Porins are required for uptake of phosphates by Mycobacterium smegmatis. J Bacteriol 189:2435–2442 10.1128/JB.01600-06. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Stephan J, Bender J, Wolschendorf F, Hoffmann C, Roth E, Mailänder C, Engelhardt H, Niederweis M. 2005. The growth rate of Mycobacterium smegmatis depends on sufficient porin-mediated influx of nutrients. Mol Microbiol 58:714–730 10.1111/j.1365-2958.2005.04878.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 240.Wolschendorf F, Ackart D, Shrestha TB, Hascall-Dove L, Nolan S, Lamichhane G, Wang Y, Bossmann SH, Basaraba RJ, Niederweis M. 2011. Copper resistance is essential for virulence of Mycobacterium tuberculosis. Proc Natl Acad Sci USA 108:1621–1626 10.1073/pnas.1009261108. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Danilchanka O, Sun J, Pavlenok M, Maueröder C, Speer A, Siroy A, Marrero J, Trujillo C, Mayhew DL, Doornbos KS, Muñoz LE, Herrmann M, Ehrt S, Berens C, Niederweis M. 2014. An outer membrane channel protein of Mycobacterium tuberculosis with exotoxin activity. Proc Natl Acad Sci USA 111:6750–6755 10.1073/pnas.1400136111. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Speer A, Sun J, Danilchanka O, Meikle V, Rowland JL, Walter K, Buck BR, Pavlenok M, Hölscher C, Ehrt S, Niederweis M. 2015. Surface hydrolysis of sphingomyelin by the outer membrane protein Rv0888 supports replication of Mycobacterium tuberculosis in macrophages. Mol Microbiol 97:881–897 10.1111/mmi.13073. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Costa-Riu N, Maier E, Burkovski A, Krämer R, Lottspeich F, Benz R. 2003. Identification of an anion-specific channel in the cell wall of the Gram-positive bacterium Corynebacterium glutamicum. Mol Microbiol 50:1295–1308 10.1046/j.1365-2958.2003.03754.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 244.Lichtinger T, Burkovski A, Niederweis M, Krämer R, Benz R. 1998. Biochemical and biophysical characterization of the cell wall porin of Corynebacterium glutamicum: the channel is formed by a low molecular mass polypeptide. Biochemistry 37:15024–15032 10.1021/bi980961e. [PubMed] [DOI] [PubMed] [Google Scholar]
- 245.Hünten P, Costa-Riu N, Palm D, Lottspeich F, Benz R. 2005. Identification and characterization of PorH, a new cell wall channel of Corynebacterium glutamicum. Biochim Biophys Acta 1715:25–36 10.1016/j.bbamem.2005.07.011. [PubMed] [DOI] [PubMed] [Google Scholar]
- 246.Chen JM, Zhang M, Rybniker J, Boy-Röttger S, Dhar N, Pojer F, Cole ST. 2013. Mycobacterium tuberculosis EspB binds phospholipids and mediates EsxA-independent virulence. Mol Microbiol 89:1154–1166 10.1111/mmi.12336. [PubMed] [DOI] [PubMed] [Google Scholar]
- 247.Pang X, Samten B, Cao G, Wang X, Tvinnereim AR, Chen XL, Howard ST. 2013. MprAB regulates the espA operon in Mycobacterium tuberculosis and modulates ESX-1 function and host cytokine response. J Bacteriol 195:66–75 10.1128/JB.01067-12. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Solomonson M, Setiaputra D, Makepeace KAT, Lameignere E, Petrotchenko EV, Conrady DG, Bergeron JR, Vuckovic M, DiMaio F, Borchers CH, Yip CK, Strynadka NCJ. 2015. Structure of EspB from the ESX-1 type VII secretion system and insights into its export mechanism. Structure 23:571–583 10.1016/j.str.2015.01.002. [PubMed] [DOI] [PubMed] [Google Scholar]
- 249.Loquet A, Sgourakis NG, Gupta R, Giller K, Riedel D, Goosmann C, Griesinger C, Kolbe M, Baker D, Becker S, Lange A. 2012. Atomic model of the type III secretion system needle. Nature 486:276–279 10.1038/nature11079. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Mattow J, Schaible UE, Schmidt F, Hagens K, Siejak F, Brestrich G, Haeselbarth G, Müller E-C, Jungblut PR, Kaufmann SHE. 2003. Comparative proteome analysis of culture supernatant proteins from virulent Mycobacterium tuberculosis H37Rv and attenuated M. bovis BCG Copenhagen. Electrophoresis 24:3405–3420 10.1002/elps.200305601. [PubMed] [DOI] [PubMed] [Google Scholar]
- 251.Flint JL, Kowalski JC, Karnati PK, Derbyshire KM. 2004. The RD1 virulence locus of Mycobacterium tuberculosis regulates DNA transfer in Mycobacterium smegmatis. Proc Natl Acad Sci USA 101:12598–12603 10.1073/pnas.0404892101. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Goessweiner-Mohr N, Arends K, Keller W, Grohmann E. 2013. Conjugative type IV secretion systems in Gram-positive bacteria. Plasmid 70:289–302 10.1016/j.plasmid.2013.09.005. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Bhatty M, Laverde Gomez JA, Christie PJ. 2013. The expanding bacterial type IV secretion lexicon. Res Microbiol 164:620–639 10.1016/j.resmic.2013.03.012. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Wang J, McIntosh F, Radomski N, Dewar K, Simeone R, Enninga J, Brosch R, Rocha EP, Veyrier FJ, Behr MA. 2015. Insights on the emergence of Mycobacterium tuberculosis from the analysis of Mycobacterium kansasii. Genome Biol Evol 7:856–870 10.1093/gbe/evv035. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Kim BJ, Kim BR, Lee SY, Seok SH, Kook YH, Kim BJ. 2013. Whole-genome sequence of a novel species, Mycobacterium yongonense DSM 45126T. Genome Announc 1:e00604-13 10.1128/genomeA.00604-13. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Dumas E, Christina Boritsch E, Vandenbogaert M, Rodríguez de la Vega RC, Thiberge JM, Caro V, Gaillard JL, Heym B, Girard-Misguich F, Brosch R, Sapriel G. 2016. Mycobacterial pan-genome analysis suggests important role of plasmids in the radiation of type VII secretion systems. Genome Biol Evol 8:387–402 10.1093/gbe/evw001. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Serafini A, Boldrin F, Palù G, Manganelli R. 2009. Characterization of a Mycobacterium tuberculosis ESX-3 conditional mutant: essentiality and rescue by iron and zinc. J Bacteriol 191:6340–6344 10.1128/JB.00756-09. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Maciag A, Dainese E, Rodriguez GM, Milano A, Provvedi R, Pasca MR, Smith I, Palù G, Riccardi G, Manganelli R. 2007. Global analysis of the Mycobacterium tuberculosis Zur (FurB) regulon. J Bacteriol 189:730–740 10.1128/JB.01190-06. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Rodriguez GM, Voskuil MI, Gold B, Schoolnik GK, Smith I. 2002. ideR, An essential gene in Mycobacterium tuberculosis: role of IdeR in iron-dependent gene expression, iron metabolism, and oxidative stress response. Infect Immun 70:3371–3381 10.1128/IAI.70.7.3371-3381.2002. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Tinaztepe E, Wei JR, Raynowska J, Portal-Celhay C, Thompson V, Philips JA. 2016. Role of metal-dependent regulation of ESX-3 secretion in intracellular survival of Mycobacterium tuberculosis. Infect Immun 84:2255–2263 10.1128/IAI.00197-16. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Daniel J, Maamar H, Deb C, Sirakova TD, Kolattukudy PE. 2011. Mycobacterium tuberculosis uses host triacylglycerol to accumulate lipid droplets and acquires a dormancy-like phenotype in lipid-loaded macrophages. PLoS Pathog 7:e1002093 10.1371/journal.ppat.1002093. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Elliott SR, Tischler AD. 2016. Phosphate starvation: a novel signal that triggers ESX-5 secretion in Mycobacterium tuberculosis. Mol Microbiol 100:510–526 10.1111/mmi.13332. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Chen JM, Zhang M, Rybniker J, Basterra L, Dhar N, Tischler AD, Pojer F, Cole ST. 2013. Phenotypic profiling of Mycobacterium tuberculosis EspA point mutants reveals that blockage of ESAT-6 and CFP-10 secretion in vitro does not always correlate with attenuation of virulence. J Bacteriol 195:5421–5430 10.1128/JB.00967-13. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Loots DT, Swanepoel CC, Newton-Foot M, Gey van Pittius NC. 2016. A metabolomics investigation of the function of the ESX-1 gene cluster in mycobacteria. Microb Pathog 100:268–275 10.1016/j.micpath.2016.10.008. [PubMed] [DOI] [PubMed] [Google Scholar]
- 265.Sampson SL. 2011. Mycobacterial PE/PPE proteins at the host-pathogen interface. Clin Dev Immunol 2011:497203 10.1155/2011/497203. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Whitney JC, Peterson SB, Kim J, Pazos M, Verster AJ, Radey MC, Kulasekara HD, Ching MQ, Bullen NP, Bryant D, Goo YA, Surette MG, Borenstein E, Vollmer W, Mougous JD. 2017. A broadly distributed toxin family mediates contact-dependent antagonism between Gram-positive bacteria. eLife 6:1–24 10.7554/eLife.26938. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Garufi G, Butler E, Missiakas D. 2008. ESAT-6-like protein secretion in Bacillus anthracis. J Bacteriol 190:7004–7011 10.1128/JB.00458-08. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Way SS, Wilson CB. 2005. The Mycobacterium tuberculosis ESAT-6 homologue in Listeria monocytogenes is dispensable for growth in vitro and in vivo. Infect Immun 73:6151–6153 10.1128/IAI.73.9.6151-6153.2005. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Shukla A, Pallen M, Anthony M, White SA. 2010. The homodimeric GBS1074 from Streptococcus agalactiae. Acta Crystallogr Sect F Struct Biol Cryst Commun 66:1421–1425 10.1107/S1744309110036286. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Warne B, Harkins CP, Harris SR, Vatsiou A, Stanley-Wall N, Parkhill J, Peacock SJ, Palmer T, Holden MTG. 2016. The Ess/type VII secretion system of Staphylococcus aureus shows unexpected genetic diversity. BMC Genomics 17:222 10.1186/s12864-016-2426-7. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Jäger F, Kneuper H, Palmer T. 2018. EssC is a specificity determinant for Staphylococcus aureus type VII secretion. Microbiology 164:816–820 10.1099/mic.0.000650. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Kneuper H, Cao ZP, Twomey KB, Zoltner M, Jäger F, Cargill JS, Chalmers J, van der Kooi-Pol MM, van Dijl JM, Ryan RP, Hunter WN, Palmer T. 2014. Heterogeneity in ess transcriptional organization and variable contribution of the Ess/type VII protein secretion system to virulence across closely related Staphylocccus aureus strains. Mol Microbiol 93:928–943 10.1111/mmi.12707. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Burts ML, DeDent AC, Missiakas DM. 2008. EsaC substrate for the ESAT-6 secretion pathway and its role in persistent infections of Staphylococcus aureus. Mol Microbiol 69:736–746 10.1111/j.1365-2958.2008.06324.x. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Jäger F, Zoltner M, Kneuper H, Hunter WN, Palmer T. 2016. Membrane interactions and self-association of components of the Ess/type VII secretion system of Staphylococcus aureus. FEBS Lett 590:349–357 10.1002/1873-3468.12065. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Mielich-Süss B, Wagner RM, Mietrach N, Hertlein T, Marincola G, Ohlsen K, Geibel S, Lopez D. 2017. Flotillin scaffold activity contributes to type VII secretion system assembly in Staphylococcus aureus. PLoS Pathog 13:e1006728 10.1371/journal.ppat.1006728. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Aly KA, Anderson M, Ohr RJ, Missiakas D. 2017. Isolation of a membrane protein complex for type VII secretion in Staphylococcus aureus. J Bacteriol 199:e00482-17. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Lopez MS, Tan IS, Yan D, Kang J, McCreary M, Modrusan Z, Austin CD, Xu M, Brown EJ. 2017. Host-derived fatty acids activate type VII secretion in Staphylococcus aureus. Proc Natl Acad Sci USA 114:11223–11228 10.1073/pnas.1700627114. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Schulthess B, Bloes DA, Berger-Bächi B. 2012. Opposing roles of σB and σB-controlled SpoVG in the global regulation of esxA in Staphylococcus aureus. BMC Microbiol 12:17 10.1186/1471-2180-12-17. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Anderson M, Aly KA, Chen YH, Missiakas D. 2013. Secretion of atypical protein substrates by the ESAT-6 secretion system of Staphylococcus aureus. Mol Microbiol 90:734–743 10.1111/mmi.12395. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Sundaramoorthy R, Fyfe PK, Hunter WN. 2008. Structure of Staphylococcus aureus EsxA suggests a contribution to virulence by action as a transport chaperone and/or adaptor protein. J Mol Biol 383:603–614 10.1016/j.jmb.2008.08.047. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Anderson M, Chen YH, Butler EK, Missiakas DM. 2011. EsaD, a secretion factor for the Ess pathway in Staphylococcus aureus. J Bacteriol 193:1583–1589 10.1128/JB.01096-10. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Korea CG, Balsamo G, Pezzicoli A, Merakou C, Tavarini S, Bagnoli F, Serruto D, Unnikrishnan M. 2014. Staphylococcal Esx proteins modulate apoptosis and release of intracellular Staphylococcus aureus during infection in epithelial cells. Infect Immun 82:4144–4153 10.1128/IAI.01576-14. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Baptista C, Barreto HC, São-José C. 2013. High levels of DegU-P activate an Esat-6-like secretion system in Bacillus subtilis. PLoS One 8:e67840 10.1371/journal.pone.0067840. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Sysoeva TA, Zepeda-Rivera MA, Huppert LA, Burton BM. 2014. Dimer recognition and secretion by the ESX secretion system in Bacillus subtilis. Proc Natl Acad Sci USA 111:7653–7658 10.1073/pnas.1322200111. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.van den Ent F, Löwe J. 2005. Crystal structure of the ubiquitin-like protein YukD from Bacillus subtilis. FEBS Lett 579:3837–3841 10.1016/j.febslet.2005.06.002. [PubMed] [DOI] [PubMed] [Google Scholar]
- 286.Rösch TC, Golman W, Hucklesby L, Gonzalez-Pastor JE, Graumann PL. 2014. The presence of conjugative plasmid pLS20 affects global transcription of its Bacillus subtilis host and confers beneficial stress resistance to cells. Appl Environ Microbiol 80:1349–1358 10.1128/AEM.03154-13. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Cao Z, Casabona MG, Kneuper H, Chalmers JD, Palmer T. 2016. The type VII secretion system of Staphylococcus aureus secretes a nuclease toxin that targets competitor bacteria. Nat Microbiol 2:16183 10.1038/nmicrobiol.2016.183. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Holberger LE, Garza-Sánchez F, Lamoureux J, Low DA, Hayes CS. 2012. A novel family of toxin/antitoxin proteins in Bacillus species. FEBS Lett 586:132–136 10.1016/j.febslet.2011.12.020. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Zhang D, de Souza RF, Anantharaman V, Iyer LM, Aravind L. 2012. Polymorphic toxin systems: comprehensive characterization of trafficking modes, processing, mechanisms of action, immunity and ecology using comparative genomics. Biol Direct 7:18 10.1186/1745-6150-7-18. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Horwitz MA, Lee BW, Dillon BJ, Harth G. 1995. Protective immunity against tuberculosis induced by vaccination with major extracellular proteins of Mycobacterium tuberculosis. Proc Natl Acad Sci USA 92:1530–1534 10.1073/pnas.92.5.1530. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Andersen P, Askgaard D, Ljungqvist L, Bennedsen J, Heron I. 1991. Proteins released from Mycobacterium tuberculosis during growth. Infect Immun 59:1905–1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Målen H, Berven FS, Fladmark KE, Wiker HG. 2007. Comprehensive analysis of exported proteins from Mycobacterium tuberculosis H37Rv. Proteomics 7:1702–1718 10.1002/pmic.200600853. [PubMed] [DOI] [PubMed] [Google Scholar]
- 293.Hinchey J, Lee S, Jeon BY, Basaraba RJ, Venkataswamy MM, Chen B, Chan J, Braunstein M, Orme IM, Derrick SC, Morris SL, Jacobs WR Jr, Porcelli SA. 2007. Enhanced priming of adaptive immunity by a proapoptotic mutant of Mycobacterium tuberculosis. J Clin Invest 117:2279–2288 10.1172/JCI31947. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Gouzy A, Larrouy-Maumus G, Bottai D, Levillain F, Dumas A, Wallach JB, Caire-Brandli I, de Chastellier C, Wu TD, Poincloux R, Brosch R, Guerquin-Kern JL, Schnappinger D, Sório de Carvalho LP, Poquet Y, Neyrolles O. 2014. Mycobacterium tuberculosis exploits asparagine to assimilate nitrogen and resist acid stress during infection. PLoS Pathog 10:e1003928 10.1371/journal.ppat.1003928. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]