

ABSTRACT
The MinION sequencer was launched by the Oxford Nanopore Technologies start-up as a disruptive technology for genome sequencing based on single-molecule synthesis. Its characteristics as a portable device, low cost, and simple library preparation have made it a good candidate for field researchers. MinION has been used to sequence a number of microorganisms, such as bacteria, viruses, and fungi. Based on the experience that characterized the Ebola virus genetic diversity in Guinea during the 2014-2015 outbreak, the ZiBRA (Zika in Brazil Real-time Analysis) project aimed to sequence a large number of Zika virus genomes during a mobile laboratory trip in northeast Brazil to provide important epidemiological information about the spread of this disease in this country. In response to the positive and rapid results obtained by the ZiBRA project, the Brazilian Ministry of Health and many leading institutions, such as the Pan American Health Organization and WHO, have shown interest in expanding the strategy used in this project to other countries dealing with arbovirus infection.
INTRODUCTION
Rapid identification of causative agents is important for rapid responses during outbreak investigations. Whole-genome sequence determination combined with bioinformatic analysis is being used as a genomic surveillance approach to answer epidemiological questions, to trace the adaptation of pathogens to their environment, and to identify the main sources of its transmission (1).
Next-generation benchtop devices like Illumina and ION Torrent have been used to generate large amounts of data in past outbreaks (2–4); however, in areas with limited access to infrastructure, a lack of devices for field research represents the main challenge. In this scenario, as part of this Microbiology Spectrum Curated Collection: Advances in Molecular Epidemiology of Infectious Diseases, this review discusses the launch of the MinION, a portable device that can be used in the field and is proving very promising.
The MinION sequencer has been released by the start-up Oxford Nanopore Technologies (ONT) as a disruptive technology for a single molecule synthesis-based genome sequencing (Fig. 1). Its characteristics as a portable device, low cost, simple library preparation make it a great candidate for field researchers (5). In the 4 years since its launch, MinION has been used to sequence a range of microorganisms, such as bacteria, viruses, and fungi from human and other animal clinical samples and also for some interesting specimens, like snake venom (5).
FIGURE 1.

MinION device, Oxford Nanopore Technologies, available at https://nanoporetech.com/products/minion. Image courtesy of Oxford Nanopore Technologies.
Considering the Zika virus (ZIKV) epidemic that has affected Latin America in the last few years and the evidence of association between the infection caused by this arbovirus and microcephaly in newborns and Guillain-Barré syndrome (GBS) in adults, the attention of authorities has turned to the importance of genomic surveillance of circulating arboviruses in this continent.
Arboviruses are a group of zoonotic viruses that circulate in the wild through biological transmission between a susceptible vertebrate host and hematophagous arthropods, mainly mosquitoes. These viruses are predominantly in tropical countries and present tropism for small vessels and the central nervous system. They can cause polymorphic diseases classically characterized by encephalitis and hemorrhage, although more often, nonspecific fever is observed (6). The incubation period of infection is usually 7 to 10 days, after which the presentation is sudden, with high fever, headache, pain, and general malaise lasting for 2 or 3 days (7). Besides classical fever, related syndromes can occur and are characterized by macular rash and polyadenopathy. Hemorrhagic cases are complicated by significant bleeding; in those cases, mortality rates range from 10 to 20% (Southeast Asia and the Caribbean) and encephalitis can be present varying from mild uncomplicated meningitis to severe cases.
Infections caused by dengue virus (DENV), chikungunya virus (CHIKV), and ZIKV are considered important challenges for public health. They have as main vectors mosquitoes from the genus Aedes, such as Aedes aegypti and A. albopictus. These present similar morphological characteristics, are of great importance for the urban environment (8, 9), and are also associated with large epidemics with consequent increase of financial costs related to diagnosis and treatment.
A set of conditions contributes to the adaptation of Aedes mosquitoes to an environment. In Brazil, for example, the main factor we can point to is the tropical climatic condition, which is hot and humid in most of the country, but with alternating periods of rain and drought. This increases the speed of mosquito proliferation. Another feature that contributes to this is the conditions of poverty for major portions of the population that inhabit peripheries of large urban centers. The lack of access to basic sanitation activities such as distribution of piped water and regular collection of garbage also predisposes to the maintenance of mosquito circulation in those areas. In addition to the scenario caused by DENV, endemic in most of the country and responsible for epidemics for decades, the recent introductions of CHIKV and mainly of ZIKV in Brazil have concerned authorities (10).
In this context, generation of complete genome sequences of arboviruses makes possible advances in molecular and clinical epidemiology studies of infectious diseases they cause. Therefore, it allows monitoring of viral diversity, which is fundamental to understanding the emergence and spread of epidemics/endemics as well as identification of strains associated with greater epidemic potential and also evaluation of genotypes that have greater coverage for potential vaccines.
ZIKA VIRUS
ZIKV is morphologically classified as a flavivirus and was first isolated from blood of a Rhesus sentinel monkey in 1947 in Zika Forest in Uganda, Africa (11). In 1948 the virus was also isolated from Aedes africanus mosquitoes captured in the same area (12). Between 1951 and 1952, serological studies demonstrated the presence of antibodies against ZIKV in human populations in Uganda and Nigeria, as well as communities in India, suggesting that the virus was already circulating outside Africa (12, 13). Between 1953 and 1983, evidence of ZIKV-associated infections was reported for human populations in India (14), Southeast Asia (15, 16), and African countries such as the Central African Republic, Egypt, Gabon, Sierra Leone, and Tanzania (12, 17–19).
Initially, there was no clear evidence that ZIKV caused human diseases (18), although studies with newborn mice had shown neurotrophism in those animals (12). Human infections were then confirmed in 1953 in Nigeria (18), and in the same year ZIKV was definitively established as a human pathogen after experimental infections and natural symptoms of fever and exanthema (20, 21).
In the 1950s, A. aegypti was identified as the probable vector involved in ZIKV transmission, after a successful transmission from an infected human volunteer to mosquitoes (21). Following that, ZIKV could be isolated from several Aedes species, including A. albopictus (22–25).
For decades following the discovery of ZIKV, serum epidemiological surveys continued to evidence the presence of this virus in human populations in Africa, India, and Southeast Asia (13, 16, 26–30). The evidence of active circulation of the virus was also confirmed by viral isolation from mosquitoes and human and nonhuman primate samples (13, 20, 31–40). Few clinical cases associated with ZIKV were reported before 2007, and up to that time it had been considered a nonconcerning virus for public health (13, 20, 29, 37). However, in 2007, the first clinically recognized ZIKV epidemic occurred on Yap Island within the Federated States of Micronesia (41). Several patients were initially positive for DENV, but uncommon clinical presentations led physicians involved to refer patient samples to the Centers for Disease Control and Prevention (CDC), where results were positive for ZIKV infection.
During the epidemic, approximately 73% of the population was infected (41, 42). After the explosion of this epidemic, ZIKV was sporadically isolated from Southeast Asian residents’ and travelers’ blood samples (43–45). Other epidemiologically significant ZIKV outbreaks were recorded in the region by the end of 2011.
Between October 2013 and April 2014, French Polynesia experienced a large epidemic wave when an estimated 66% of the population was infected. In that epidemic, between November 2013 and February 2014, 42 cases of GBS were reported. The increase in GBS cases raised concerns of an association between the syndrome and ZIKV (46, 47). Those were the first findings of neurological sequelae associated with ZIKV infection. Although there was no categorical evidence, retrospective analysis suggested an association of ZIKV infections and cases of microcephaly (48).
Following the epidemic in French Polynesia, in 2014, ZIKV spread throughout the South Pacific, including outbreaks on the New Caledonia islands, Easter Island, and the Cook Islands (49). In the Americas, the first confirmed cases of ZIKV infection occurred at the end of 2014 in northeastern Brazil (50, 51) and simultaneously in Haiti (52). Over the next few months, the virus spread rapidly throughout Brazil (53), with a significant increase in GBS and microcephaly cases in regions affected by the epidemic (80).
In 2015, 91,387 cases of Zika were reported in Brazil, and until epidemiological week 6 (13 February 2016), 22 of the 26 federate states reported autochthonous cases of ZIKV infections, confirming a ZIKV epidemic occurring in the country, and a health emergency was declared by the WHO (7, 53–55).
GENOMIC SURVEILLANCE
Despite the large number of cases reported, until the beginning of 2016, only 55 complete ZIKV genomes were available from GenBank (http://www.ncbi.nlm.nih.gov/), of which 7 were Brazilian strains related to microcephaly, febrile disease, and blood donors. These data contributed to the identification of the Asian genotype as the cause of the epidemic in the country, as well as to determining the probable date of ZIKV introduction in Brazil, estimated to May 2014, which matches the timing of the FIFA Confederations Cup sporting event (56).
In the last few years, several studies have reported simultaneous epidemics of DENV, CHIKV, and ZIKV in the same geographical area (49, 57), especially in northeastern Brazil. Concomitant infections have potential deleterious effects on the fetus in pregnant women, and the rapid species and genotype identification of replicating virus is crucial.
Before 2016, the genotyping methods most appropriate for precise classification of genetic regions of those arboviruses remained poorly defined. Before that year, there was a lack of a nationwide specific ZIKV surveillance (7). Thus, the generation and sequencing of complete genomes of those circulating arboviruses in infected population were shown to be extremely important.
Genomic data generated by real-time sequencing can provide important information about how and when viruses were introduced in a particular site, their pattern and determinants of dissemination in neighboring locations, and the extent of genetic diversity, making it possible to establish an effective surveillance framework for tracking the spread of infections to other geographical regions.
The shortage of complete genomic sequences was a limiting point for the study of virus genetic divergence as well as of population dynamics (genotypes and subgenotypes), pathogenesis, and vectors associated with virus transmission among human populations. In that context, the launch of the ZiBRA (Zika in Brazil Real-time Analysis) project as a result of a multicenter collaboration between the University of Oxford, University of Birmingham, Evandro Chagas Institute, University of São Paulo, and Oswaldo Cruz Foundation was shown to be a promising approach to generate a substantial number of ZIKV complete genome sequences through MinION in a mobile laboratory.
Based on a previous example of mobile genomic surveillance during the Ebola outbreak in Guinea in 2014-2015 (58), the ZiBRA project aimed to generate a large number of ZIKV complete genome sequences from the northeast of Brazil covering a broad geographical region, including historical samples, and from patients with a range of clinical presentations. The method consisted of genome-tiling PCR to enrich ZIKV material in clinical samples followed by library preparation prior to MinION loading (59, 60).
The ZiBRA team, working together with Central Laboratory (LACEN) personnel, tested 1,330 clinical samples for ZIKV RNA across the states Rio Grande do Norte, Paraíba, Recife, Maceió, and Bahia and captured 850 mosquitoes from urban and periurban fields in each place along the way. The project also involved capacity building, as multiple team members were trained to perform the whole protocol on later trips (10, 59).
After the original trip that took place in June 2016, the ZiBRA project was extended; to date, it has performed five genomic surveillance trips to track not only ZIKV but also CHIKV, yellow fever virus (YFV) in recent outbreaks, and Oropouche virus (OROV) infection. As the trips occur, more people are being trained to continue performing genomic surveillance throughout all the country and in some places in Africa, like Angola and Cabo Verde (unpublished data). In addition, the productivity of trips is increasing each time, with generation of around 60 complete genome sequences in only 5 days. In addition, they are developing faster protocols and more than 12 barcodes per run; 48- and 96-barcode protocols suggest that this number will increase soon.
SEQUENCING METHODOLOGIES AND MinION INNOVATION
To understand the innovation brought about by MinION, let us take a quick look at the trajectory of sequencing methodologies since their beginning (Fig. 2).
FIGURE 2.

Overview of the second- and third-generation sequencing technologies. Sanger’s method was the first-generation sequencing method, followed by 454 and Solexa techniques, which marked the second generation. After Helicos and Pacific Biosciences releases, Illumina and ION Torrent were used to generate a large amount of data. More recently, nanopore sequencing techniques have interested researchers working on field research and real-time analysis.
In 1953, Watson and Crick solved the three-dimensional structure of DNA (61); in 1965, Robert Holley and colleagues determined the complete nucleotide sequence of an alanine tRNA, comprising the first nucleic acid structure known (62). Meanwhile, in parallel, Frederick Sanger and colleagues developed a two-dimensional fractionation procedure for radioactive nucleotide detection (63). Following the work of Holley and Sanger, many researchers made progress in endeavoring to sequence portions of different genomes, for example, Min-Jou and Fiers with MS2 bacteriophage (64, 65), Wu and Kaiser with cohesive ends of bacteriophage lambda (66), and finally, in 1977, Maxam and Gilbert with the development of chemical cleavage technique that established the first sequencing methodology (67).
Although the achievement of Maxam and Gilbert was important, it was Sanger with the dideoxy technique that changed the course of sequence methodologies (68). The remarkable work of Sanger introduced the chain termination technique, which used dideoxynucleotides. These are otherwise complete triphosphate nucleotides that lack a 3′ hydroxyl group. This prevents chain elongation during polymerase replications and produces short segments ending at each nucleotide position, allowing the recognition of each base in the sequence (69).
Together, improvements to this technique and the release of sequencing machines based on Sanger’s method were known as first-generation sequencing. Sanger’s method was considered the standard and most influential sequencing method for two decades since it was first presented in 1977 (70).
Twenty years later, another sequencing method was proposed using luminescent measurement of pyrophosphate synthesis and commercially available as 454 Life Science/Roche machines (71). By that time, sequencing technologies were categorized as second-generation sequencing and involved a technique in which a single run generates a large volume of data by massively parallel sequencing but still using the pyrosequencing approach (72).
454 machines use emulsion PCR in DNA-coated beads that amplify a clonal population of the same DNA molecule in each one. Following the success of parallel sequencing technique released by 454, we highlight the Solexa method (later Illumina), which also uses pyrosequencing but instead of bead-based emulsion-PCR, it binds DNA molecules to a flow cell through adapters (73). Later, Genome Analyzer (GA) made improvements on sequencing technologies, including paired-end data and recording both sequences’ ends of DNA cluster. GA, in turn, was followed by HiSeq, which produces good depth and length, and MySeq, which, although it produces low throughput, is low cost (74).
The third-generation DNA sequencing is comprised of techniques that differ from previous ones, as it is capable of single-molecule sequencing (SMS). The first described SMS equipment was from Helicos BioSciences; it operates in the same manner as Illumina but does not require amplification.
Following sequencer evolution, Pacific Biosciences released single-molecule real-time sequencing that uses nanostructures called zero-mode waveguides, which are basically holes in a metallic film that uses light properties to determine the sequence (75).
Currently, third-generation nanopore sequencing techniques have interested researchers working on field research and real-time analysis. The most promising representative for this approach is the MinION, since its use allows analysis of a wide variety of biological samples in addition to allowing monitoring at the same time as the sequences are generated. The portability of the MinION device is its main feature. Ease of transport plus simple handling makes this device an excellent candidate for field research. MinION is smaller than current smartphones and can be carried in a pocket, which is a great ally for this methodology to be introduced not only in the field but also in fixed laboratories.
MinION SEQUENCING APPROACH: HOW DOES IT WORK?
MinION allows analysis of multiple biological molecules such as DNA, RNA, and proteins. The major feature of the MinION sequencing approach is the use of nanopores. Nanopores are nanoscale holes that can be created by proteins and are present at biological membranes or in solid materials known as solid-state nanopores. In ONT systems, nanopores are inserted into an electric resistant membrane created from synthetic polymers and a potential is applied across membrane, resulting in current flowing only through the aperture of nanopores. When single molecules enter a nanopore, disruptions characteristic of each nucleotide or amino acid are produced in the current, and by measuring those disruptions, the sequence can be determined (ONT).
The MinION device operates with coupling of a flow cell that includes a sensing chip, which contains an array of wells consisting of individually addressed electronic channels. A DNA library loaded into MinION, i.e., a library that contains DNA molecules to be sequenced, is previously mixed with copies of a processive enzyme. As the DNA-enzyme complex approaches the nanopore, the single-strand DNA fits into the nanopore aperture, and as each base passes through the nanopore it creates a disruption in the current that can be used to determine all the bases of the strand. When a nanopore has processed a complete read, the enzyme detaches and the nanopore starts to process a new read. Nanopores can process reads of hundreds of kilobases. The signal is measured by the application-specific integrated circuit (ASIC) in the flow cell and processed by Minknow software, which also controls the MinION. It allows changing parameters and following up the experiment in real time, as it provides feedback of read length and number of events. The computer with Minknow software is connected and sends data to Metrichor, a cloud bioinformatics platform which processes the output file’s format fast5 and then sends it back to the computer as the sequencing is in progress (1). In the field, where the Internet connection is poor or not available, offline basecaller can be used. As the run is completed, data can be analyzed using bioinformatics tools.
ZIKV AND MinION
ZIKV sequencing with MinION comprises several steps starting from sample collection to analysis (Fig. 3). As a result of a protocol developed by Joshua Quick and colleagues (60) and tested for several groups and also during a ZiBRA trip in June 2016 (60), we were able to propose a ZIKV-specific surveillance scheme which is now extended to other viruses such CHIKV, YFV, OROV, and Mayaro virus.
FIGURE 3.
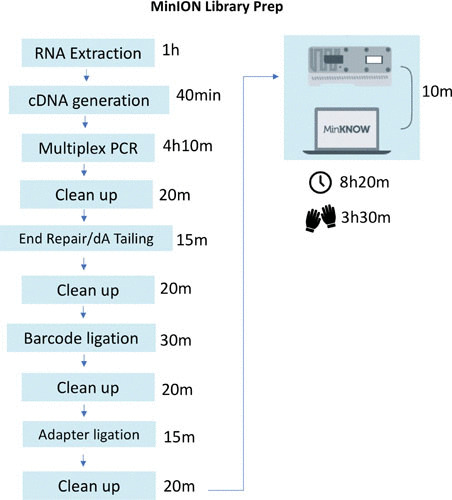
ZIKV MinION sampling and library prep workflow. Shown are average times for ZIKV sampling and library preparation using the Primal Scheme-derived multiplex PCR and Oxford Nanopore Technologies SKL-108 sequencing kit. From RNA extraction to library load on a flow cell, the time spent is around 8 h, with at least 3 h of hands-on time.
The development of this protocol comprised the following steps: multiplex primer pool design, multiplex PCR, sequencing on MinION, bioinformatic analysis, and quality control (QC) (60).
Multiplex Primer Pool Design
A set of 35 pairs of primers that generate overlapping products ∼400 bp in length to cover the ZIKV complete genome is used for its amplification. Those primers were generated on the online tool Primal Scheme, developed by Quick and colleagues (60) and available at http://primal.zibraproject.org. Primal Scheme provides a complete pipeline for the development of efficient multiplex primer schemes and can be used to generate a set of primers to amplify any genome of interest. Parameters such as amplicon length, length of overlap, and melting temperature can be set by users (Fig. 4).
FIGURE 4.
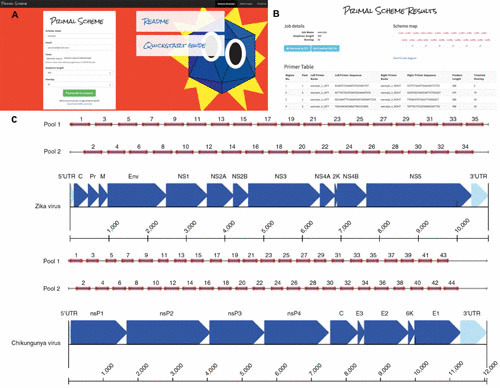
Overview of multiplex primer design using the Primal Scheme online primer design tool. (A) Submission box for online primer design tool. (B) Primer table of results. (C) Schematic showing expected amplicon products for each pool in genomic context for the Zika Asian scheme. Adapted from reference (60), with permission.
Multiplex PCR
Instead of using a single-plex approach, this protocol was designed to perform two multiplex PCRs with lower primer concentrations and a longer annealing/extension time. In addition, alternate target genome regions are amplified in each primer pool; thus, neighboring amplicons do not overlap.
Sequencing on MinION
A native barcoding kit (ONT) is used to sequence up to 12 samples per flow cell, and a two-dimensional barcoded library is prepared and loaded onto R9 or R9.4 flow cells. MinION sequencing run time can be determined by the user, as data generation can be monitored in real time. For 12 samples, 4 h is usually enough.
Bioinformatic Analysis and QC
MinION bioinformatic pipelines were developed by the same group (https://github.com/zibraproject/zika-pipeline). The pipeline consists of primer trimming, alignment, variant calling, and consensus generation steps. To perform QC, inclusion of positive samples on MinION runs is recommended to check if the protocol is generating the expected results. In addition, negative sequencing controls are also recommended to check for contamination.
One disadvantage of Oxford Nanopore sequencing is the high error rate (>15%) compared with that of Illumina (77, 78). The error rate can be calculated by counting the number of mismatches positions in a gapped alignment of a read to a reference sequence. In this way it measures substitution, insertion, and deletion errors (79). Previous studies have shown that when used in conjunction with Illumina MiSeq data, the contiguity of MinION de novo assembly can be enhanced (79). Other approaches to decrease error rates are read overlapping, correction of genomic alignments, and the use of a reasonable reference genome. On the ZiBRA project we use overlapping primers in order to have low error rates, and bioinformatic analysis is also performed to maximize the contiguity of de novo assembly.
DEPLOYMENT OF PROTOCOL IN BRAZIL: A BRIEF REVIEW OF OUR EXPERIENCE
ZIKV detection in Brazil is a challenge, as the cocirculation of other arboviruses with similar clinical symptoms, such as DENV and CHIKV, may mask its presence. Also, the frequency of mild disease caused by this infection can lead it to be underreported (59).
The probable cause of the dearth of ZIKV genome sequence is the low viral load in serum after the onset of symptoms, since peak viremia occurs within a week before (76). In order to overcome this difficulty, clinical samples are enriched by PCR (60).
In Brazil, samples from clinical ZIKV diagnostics are sent to local Central Laboratories of Public Health (LACEN) in each state, which perform molecular diagnostics mainly by real-time PCR. The focus of our approach for ZIKV genomic surveillance is those laboratories, which can be accessed by previous authorization from Brazilian Ministry of Health (MS). Currently, with the expansion of genomic surveillance, other institutions participate in the process and their teams are also trained.
Samples available were retested or tested for the first time during the ZiBRA trip. Now, with collaborative effort from laboratories, positive samples are preselected based on cycle threshold value, host species, and location, so we can go ahead and perform ZIKV multiplex PCR.
After PCR enrichment, samples are forwarded to library preparation according to the ONT Native Barcoding Protocol, available at https://community.nanoporetech.com. Ready libraries are then loaded to the MinION sequencer, and generated data are analyzed by bioinformaticians within, at most, a week.
As soon as the sequences are generated and analyzed, technical reports containing epidemiological data of samples, as well as the results of the phylogenetic and phylogeographic analyses, are sent to the MS and Pan American Health Organization (PAHO), confirming the commitment made and reaffirming the proposal to provide rapid responses for outbreak management. Under this prerogative, ZIKV (and other arboviruses) genomic surveillance is being deployed in several laboratory units throughout the country. So far, besides laboratories visited along the ZiBRA trip route, personnel from the Ezequiel Dias Foundation (FUNED) and the Federal University of Minas Gerais (both in Minas Gerais), Leonidas e Maria Deane Institute in Amazonas (FIOCRUZ-AM), Adolfo Lutz Institute (IAL), University of São Paulo and State University of São Paulo (UNESP) (both in São Paulo), Oswaldo Cruz Institute (IOC-FIOCRUZ) in Rio de Janeiro, and LACEN-BA in Bahia have been trained to perform genomic surveillance.
CONCLUSION
It is known that greater knowledge regarding the genetic characteristics of ZIKV is imperative to control its dissemination. The occurrence of a recent epidemic, associated with cases of microcephaly and congenital infections in Brazil, pointed to the need for a systematic and continuous monitoring of this arbovirus. Considering genomic surveillance as an approach to do this, complete viral genome sequencing allows monitoring of viral diversity, which is fundamental to understand the origin and the events of viral introduction and the dispersion, as well as for the identification of strains or genotypes with greater epidemic potential and with greater coverage by potential vaccines.
During the ZiBRA trip, we were able to generate 54 partial and complete ZIKV genomes. One of those sequences represents the earliest confirmed ZIKV infection in the country (10). Our analysis shows that northeast Brazil had great importance in the establishment of ZIKV in the Americas, as it was present by February 2014 and is likely to have disseminated from there. We also demonstrated that duration of predetection is cryptic for viral transmission (10).
In this study, molecular analyses performed in several LACENs (northeast region) showed that the precise diagnosis of ZIKV still poses a challenge to public health, since a large majority of samples with positive results obtained during the trip have had negative ZIKV detection in previous tests. In response to the positive and rapid-return results obtained by the ZiBRA project, in addition to the Brazilian Ministry of Health, many world reference institutions, such as the PAHO and WHO, have shown interest in expanding the strategy used in this project to other countries dealing with arbovirus infection as well as others, like Paraguay.
On the basis of our previous experience, we believe that the use of real-time sequencing is the most appropriate strategy for this expansion. The development of new protocols allowing simultaneous running of more samples will allow greater coverage of the information in less time, also allowing improvement of studies in the effort to contain the dispersion of these viruses and damage to public health.
Also, the establishment of a network of laboratories working together for the rapid response to the outbreak of epidemics is essential for their management. Within the network, in the event of an increase in cases or outbreaks of ZIKV and/or other arboviruses, the recruitment of trained personnel during the ZiBRA project will allow the rapid generation of sequences, besides allowing the training of the team of the laboratories visited for the management of subsequent outbreaks. This also reinforces one of the aims of the ZIBRA project, which is capacity building to continue the genomic surveillance activities.
Studies involving more in-depth molecular analysis of the circulating sequences across the country and the tracking of viral dispersion may help the Ministry of Health to appropriately adopt measures to control the epidemic and to monitor the dynamics of the evolution of circulating viral strains. These data will allow the country to understand the impact of mutation patterns in the control of viral replication, directing subsidies to the appropriate choice of preventive actions and therapeutic interventions and bringing a direct benefit to the patient and community.
Footnotes
This article is part of a curated collection.
Contributor Information
Jaqueline Goes de Jesus, Laboratory of Experimental Pathology, Gonçalo Moniz Institute, Oswaldo Cruz Foundation, Salvador, Bahia 40296-710, Brazil.
Marta Giovanetti, Laboratory of Flaviviruses, Oswaldo Cruz Institute, Oswaldo Cruz Foundation, Rio de Janeiro, Rio de Janeiro 21040-900, Brazil; Laboratory of Cellular and Molecular Genetics, ICB, Federal University of Minas Gerais, Belo Horizonte, Minas Gerais 31270-901, Brazil.
Nuno Rodrigues Faria, Department of Zoology, University of Oxford, Oxford OX1 3SZ, United Kingdom.
Luiz Carlos Junior Alcantara, Laboratory of Flaviviruses, Oswaldo Cruz Institute, Oswaldo Cruz Foundation, Rio de Janeiro, Rio de Janeiro 21040-900, Brazil; Laboratory of Cellular and Molecular Genetics, ICB, Federal University of Minas Gerais, Belo Horizonte, Minas Gerais 31270-901, Brazil.
Lee W. Riley, Divisions of Infectious Diseases and Vaccinology, School of Public Health, University of California, Berkeley, Berkeley, CA
Ronald E. Blanton, Center for Global Health & Diseases, Case Western Reserve University, Cleveland, OH
CURATED COLLECTION
REFERENCES
- 1.Walter MC, Zwirglmaier K, Vette P, Holowachuk SA, Stoecker K, Genzel GH, Antwerpen MH. 2017. MinION as part of a biomedical rapidly deployable laboratory. J Biotechnol 250:16–22. 10.1016/j.jbiotec.2016.12.006. [PubMed] [DOI] [PubMed] [Google Scholar]
- 2.Antwerpen MH, Prior K, Mellmann A, Höppner S, Splettstoesser WD, Harmsen D. 2015. Rapid high resolution genotyping of Francisella tularensis by whole genome sequence comparison of annotated genes (“MLST+”). PLoS One 10:e0123298. 10.1371/journal.pone.0123298. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mellmann A, Harmsen D, Cummings CA, Zentz EB, Leopold SR, Rico A, Prior K, Szczepanowski R, Ji Y, Zhang W, McLaughlin SF, Henkhaus JK, Leopold B, Bielaszewska M, Prager R, Brzoska PM, Moore RL, Guenther S, Rothberg JM, Karch H. 2011. Prospective genomic characterization of the German enterohemorrhagic Escherichia coli O104:H4 outbreak by rapid next generation sequencing technology. PLoS One 6:e22751. 10.1371/journal.pone.0022751. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Meinel DM, Kuehl R, Zbinden R, Boskova V, Garzoni C, Fadini D, Dolina M, Blümel B, Weibel T, Tschudin-Sutter S, Widmer AF, Bielicki JA, Dierig A, Heininger U, Konrad R, Berger A, Hinic V, Goldenberger D, Blaich A, Stadler T, Battegay M, Sing A, Egli A. 2016. Outbreak investigation for toxigenic Corynebacterium diphtheriae wound infections in refugees from northeast Africa and Syria in Switzerland and Germany by whole genome sequencing. Clin Microbiol Infect 22:1003.e1–1003.e8. 10.1016/j.cmi.2016.08.010. [PubMed] [DOI] [PubMed] [Google Scholar]
- 5.Mikheyev AS, Tin MM. 2014. A first look at the Oxford Nanopore MinION sequencer. Mol Ecol Resour 14:1097–1102. 10.1111/1755-0998.12324. [PubMed] [DOI] [PubMed] [Google Scholar]
- 6.World Health Organization. 2016. WHO Director-General summarizes the outcome of the Emergency Committee regarding clusters of microcephaly and Guillain-Barré syndrome. World Health Organization, Geneva, Switzerland. https://www.who.int/news-room/detail/01-02-2016-who-director-general-summarizes-the-outcome-of-the-emergency-committee-regarding-clusters-of-microcephaly-and-guillain-barr%C3%A9-syndrome. [Google Scholar]
- 7.Lessler JTOC, Ott CT, Carcelen AC, Konikoff JM, Williamson J, Bi Q, Kucirka LM, Cummings DAT, Reich NG, Chaisson LH. 2016. Times to key events in the course of Zika infection and their implications: a systematic review and pooled analysis. Bull World Health Organ 94:841–849. 10.2471/BLT.16.1740540. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Webb JL, Jr. 2016. Aedes aegypti suppression in the Americas: historical perspectives. Lancet 388:556–557. 10.1016/S0140-6736(16)31225-9. [DOI] [PubMed] [Google Scholar]
- 9.Kraemer MU, Sinka ME, Duda KA, Mylne AQ, Shearer FM, Barker CM, Moore CG, Carvalho RG, Coelho GE, Van Bortel W, Hendrickx G, Schaffner F, Elyazar IR, Teng HJ, Brady OJ, Messina JP, Pigott DM, Scott TW, Smith DL, Wint GR, Golding N, Hay SI. 2015. The global distribution of the arbovirus vectors Aedes aegypti and Ae. albopictus. eLife 4:e08347. 10.7554/eLife.08347. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Faria NR, et al. 2017. Establishment and cryptic transmission of Zika virus in Brazil and the Americas. Nature 546:406–410. 10.1038/nature22401. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dick GW. 1952. Zika virus. II. Pathogenicity and physical properties. Trans R Soc Trop Med Hyg 46:521–534. 10.1016/0035-9203(52)90043-6. [DOI] [PubMed] [Google Scholar]
- 12.Dick GW. 1953. Epidemiological notes on some viruses isolated in Uganda; yellow fever, Rift Valley fever, Bwamba fever, West Nile, Mengo, Semliki Forest, Bunyamwera, Ntaya, Uganda S and Zika viruses. Trans R Soc Trop Med Hyg 47:13–48. 10.1016/0035-9203(53)90021-2. [DOI] [PubMed] [Google Scholar]
- 13.Fagbami AH. 1979. Zika virus infections in Nigeria: virological and seroepidemiological investigations in Oyo State. J Hyg (Lond) 83:213–219. 10.1017/S0022172400025997. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Smithburn KC. 1952. Neutralizing antibodies against certain recently isolated viruses in the sera of human beings residing in East Africa. J Immunol 69:223–234. [PubMed] [Google Scholar]
- 15.Smithburn KC. 1954. Antigenic relationships among certain arthropod-borne viruses as revealed by neutralization tests. J Immunol 72:376–388. [PubMed] [Google Scholar]
- 16.Pond WL. 1963. Arthropod-borne virus antibodies in sera from residents of South-East Asia. Trans R Soc Trop Med Hyg 57:364–371. 10.1016/0035-9203(63)90100-7. [DOI] [PubMed] [Google Scholar]
- 17.Kokernot RH, Szlamp EL, Levitt J, McIntosh BM. 1965. Survey for antibodies against arthropod-borne viruses in the sera of indigenous residents of the Caprivi Strip and Bechuanaland Protectorate. Trans R Soc Trop Med Hyg 59:553–562. 10.1016/0035-9203(65)90158-6. [DOI] [PubMed] [Google Scholar]
- 18.MacNamara FN. 1954. Zika virus: a report on three cases of human infection during an epidemic of jaundice in Nigeria. Trans R Soc Trop Med Hyg 48:139–145. 10.1016/0035-9203(54)90006-1. [DOI] [PubMed] [Google Scholar]
- 19.Smithburn KC. 1952. Studies on certain viruses isolated in the tropics of Africa and South America; immunological reactions as determined by cross-neutralization tests. J Immunol 68:441–460. [PubMed] [Google Scholar]
- 20.Simpson DI. 1964. Zika virus infection in man. Trans R Soc Trop Med Hyg 58:335–338. 10.1016/0035-9203(64)90201-9. [DOI] [PubMed] [Google Scholar]
- 21.Bearcroft WG. 1956. Zika virus infection experimentally induced in a human volunteer. Trans R Soc Trop Med Hyg 50:442–448. 10.1016/0035-9203(56)90090-6. [DOI] [PubMed] [Google Scholar]
- 22.Faye O, Freire CC, Iamarino A, Faye O, de Oliveira JV, Diallo M, Zanotto PM, Sall AA. 2014. Molecular evolution of Zika virus during its emergence in the 20th century. PLoS Negl Trop Dis 8:e2636. 10.1371/journal.pntd.0002636. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grard G, Caron M, Mombo IM, Nkoghe D, Mboui Ondo S, Jiolle D, Fontenille D, Paupy C, Leroy EM. 2014. Zika virus in Gabon (Central Africa)—2007: a new threat from Aedes albopictus? PLoS Negl Trop Dis 8:e2681. 10.1371/journal.pntd.0002681. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vogel G. 2016. Infectious disease. Mosquito hunters search for Zika vectors. Science 352:1152–1153. 10.1126/science.352.6290.1152. [PubMed] [DOI] [PubMed] [Google Scholar]
- 25.Wong PS, Li MZ, Chong CS, Ng LC, Tan CH. 2013. Aedes (Stegomyia) albopictus (Skuse): a potential vector of Zika virus in Singapore. PLoS Negl Trop Dis 7:e2348. 10.1371/journal.pntd.0002348. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Darwish MA, Hoogstraal H, Roberts TJ, Ahmed IP, Omar F. 1983. A sero-epidemiological survey for certain arboviruses (Togaviridae) in Pakistan. Trans R Soc Trop Med Hyg 77:442–445. 10.1016/0035-9203(83)90106-2. [DOI] [PubMed] [Google Scholar]
- 27.Bowen ET, Simpson DI, Platt GS, Way H, Bright WF, Day J, Achapa S, Roberts JM. 1973. Large scale irrigation and arbovirus epidemiology, Kano Plain, Kenya. II. Preliminary serological survey. Trans R Soc Trop Med Hyg 67:702–709. 10.1016/0035-9203(73)90041-2. [DOI] [PubMed] [Google Scholar]
- 28.Rodhain F, Gonzalez JP, Mercier E, Helynck B, Larouze B, Hannoun C. 1989. Arbovirus infections and viral haemorrhagic fevers in Uganda: a serological survey in Karamoja district, 1984. Trans R Soc Trop Med Hyg 83:851–854. 10.1016/0035-9203(89)90352-0. [DOI] [PubMed] [Google Scholar]
- 29.Olson JG, Ksiazek TG, Suhandiman, Triwibowo. 1981. Zika virus, a cause of fever in Central Java, Indonesia. Trans R Soc Trop Med Hyg 75:389–393. 10.1016/0035-9203(81)90100-0. [DOI] [PubMed] [Google Scholar]
- 30.Olson JG, Ksiazek TG, Gubler DJ, Lubis SI, Simanjuntak G, Lee VH, Nalim S, Juslis K, See R. 1983. A survey for arboviral antibodies in sera of humans and animals in Lombok, Republic of Indonesia. Ann Trop Med Parasitol 77:131–137. 10.1080/00034983.1983.11811687. [PubMed] [DOI] [PubMed] [Google Scholar]
- 31.Haddow AJ, Williams MC, Woodall JP, Simpson DI, Goma LK. 1964. Twelve isolations of Zika virus from Aedes (Stegomyia) africanus (Theobald) taken in and above a Uganda forest. Bull World Health Organ 31:57–69. [PMC free article] [PubMed] [Google Scholar]
- 32.Lee VH, Moore DL. 1972. Vectors of the 1969 yellow fever epidemic on the Jos Plateau, Nigeria. Bull World Health Organ 46:669–673. [PMC free article] [PubMed] [Google Scholar]
- 33.Marchette NJ, Garcia R, Rudnick A. 1969. Isolation of Zika virus from Aedes aegypti mosquitoes in Malaysia. Am J Trop Med Hyg 18:411–415. 10.4269/ajtmh.1969.18.411. [PubMed] [DOI] [PubMed] [Google Scholar]
- 34.McCrae AW, Kirya BG. 1982. Yellow fever and Zika virus epizootics and enzootics in Uganda. Trans R Soc Trop Med Hyg 76:552–562. 10.1016/0035-9203(82)90161-4. [DOI] [PubMed] [Google Scholar]
- 35.McIntosh BM, Worth CB, Kokernot RH. 1961. Isolation of Semliki Forest virus from Aedes (Aedimorphus) argenteopunctatus (Theobald) collected in Portuguese East Africa. Trans R Soc Trop Med Hyg 55:192–198. 10.1016/0035-9203(61)90025-6. [DOI] [PubMed] [Google Scholar]
- 36.Monlun E, et al. 1993. Surveillance of the circulation of arbovirus of medical interest in the region of eastern Senegal. Bull Soc Pathol Exot 86:21–28. (In French.) [PubMed] [Google Scholar]
- 37.Moore DL, Causey OR, Carey DE, Reddy S, Cooke AR, Akinkugbe FM, David-West TS, Kemp GE. 1975. Arthropod-borne viral infections of man in Nigeria, 1964–1970. Ann Trop Med Parasitol 69:49–64. 10.1080/00034983.1975.11686983. [PubMed] [DOI] [PubMed] [Google Scholar]
- 38.Fagbami A. 1977. Epidemiological investigations on arbovirus infections at Igbo-Ora, Nigeria. Trop Geogr Med 29:187–191. [PubMed] [Google Scholar]
- 39.Weinbren MP, Williams MC. 1958. Zika virus: further isolations in the Zika area, and some studies on the strains isolated. Trans R Soc Trop Med Hyg 52:263–268. 10.1016/0035-9203(58)90085-3. [DOI] [PubMed] [Google Scholar]
- 40.Althouse BM, Hanley KA, Diallo M, Sall AA, Ba Y, Faye O, Diallo D, Watts DM, Weaver SC, Cummings DA. 2015. Impact of climate and mosquito vector abundance on sylvatic arbovirus circulation dynamics in Senegal. Am J Trop Med Hyg 92:88–97. 10.4269/ajtmh.13-0617. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Duffy MR, Chen TH, Hancock WT, Powers AM, Kool JL, Lanciotti RS, Pretrick M, Marfel M, Holzbauer S, Dubray C, Guillaumot L, Griggs A, Bel M, Lambert AJ, Laven J, Kosoy O, Panella A, Biggerstaff BJ, Fischer M, Hayes EB. 2009. Zika virus outbreak on Yap Island, Federated States of Micronesia. N Engl J Med 360:2536–2543. 10.1056/NEJMoa0805715. [PubMed] [DOI] [PubMed] [Google Scholar]
- 42.Lanciotti RS, Kosoy OL, Laven JJ, Velez JO, Lambert AJ, Johnson AJ, Stanfield SM, Duffy MR. 2008. Genetic and serologic properties of Zika virus associated with an epidemic, Yap State, Micronesia, 2007. Emerg Infect Dis 14:1232–1239. 10.3201/eid1408.080287. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Buathong R, Hermann L, Thaisomboonsuk B, Rutvisuttinunt W, Klungthong C, Chinnawirotpisan P, Manasatienkij W, Nisalak A, Fernandez S, Yoon IK, Akrasewi P, Plipat T. 2015. Detection of Zika virus infection in Thailand, 2012–2014. Am J Trop Med Hyg 93:380–383. 10.4269/ajtmh.15-0022. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fonseca K, Meatherall B, Zarra D, Drebot M, MacDonald J, Pabbaraju K, Wong S, Webster P, Lindsay R, Tellier R. 2014. First case of Zika virus infection in a returning Canadian traveler. Am J Trop Med Hyg 91:1035–1038. 10.4269/ajtmh.14-0151. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kwong JC, Druce JD, Leder K. 2013. Zika virus infection acquired during brief travel to Indonesia. Am J Trop Med Hyg 89:516–517. 10.4269/ajtmh.13-0029. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cao-Lormeau VM, Roche C, Teissier A, Robin E, Berry AL, Mallet HP, Sall AA, Musso D. 2014. Zika virus, French Polynesia, South Pacific, 2013. Emerg Infect Dis 20:1085–1086. 10.3201/eid2011.141380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cao-Lormeau VM, Blake A, Mons S, Lastere S, Roche C, Vanhomwegen J, Dub T, Baudouin L, Teissier A, Larre P, Vial AL, Decam C, Choumet V, Halstead SK, Willison HJ, Musset L, Manuguerra JC, Despres P, Fournier E, Mallet HP, Musso D, Fontanet A, Neil J, Ghawché F. 2016. Guillain-Barré syndrome outbreak associated with Zika virus infection in French Polynesia: a case-control study. Lancet 387:1531–1539. 10.1016/S0140-6736(16)00562-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cauchemez S, Besnard M, Bompard P, Dub T, Guillemette-Artur P, Eyrolle-Guignot D, Salje H, Van Kerkhove MD, Abadie V, Garel C, Fontanet A, Mallet HP. 2016. Association between Zika virus and microcephaly in French Polynesia, 2013–15: a retrospective study. Lancet 387:2125–2132. 10.1016/S0140-6736(16)00651-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Roth A, Mercier A, Lepers C, Hoy D, Duituturaga S, Benyon E, Guillaumot L, Souares Y. 2014. Concurrent outbreaks of dengue, chikungunya and Zika virus infections—an unprecedented epidemic wave of mosquito-borne viruses in the Pacific 2012–2014. Euro Surveill 19:20929. 10.2807/1560-7917.ES2014.19.41.20929. [PubMed] [DOI] [PubMed] [Google Scholar]
- 50.Zanluca C, Melo VC, Mosimann AL, Santos GI, Santos CN, Luz K. 2015. First report of autochthonous transmission of Zika virus in Brazil. Mem Inst Oswaldo Cruz 110:569–572. 10.1590/0074-02760150192. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Campos GS, Bandeira AC, Sardi SI. 2015. Zika virus outbreak, Bahia, Brazil. Emerg Infect Dis 21:1885–1886. 10.3201/eid2110.150847. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lednicky J, Beau De Rochars VM, El Badry M, Loeb J, Telisma T, Chavannes S, Anilis G, Cella E, Ciccozzi M, Rashid M, Okech B, Salemi M, Morris JG, Jr. 2016. Zika virus outbreak in Haiti in 2014: molecular and clinical data. PLoS Negl Trop Dis 10:e0004687. 10.1371/journal.pntd.0004687. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.World Health Organization. 2016. Zika virus microcephaly and Guillain-Barré syndrome situation report 26 February 2016. World Health Organization, Geneva, Switzerland. http://www.who.int/emergencies/zika-virus/situation-report-26-02-2016.pdf. [Google Scholar]
- 54.BRASIL Ministério da Saúde, Secretaria de Vigilância em Saúde. 2016. Boletim Epidemiológico: Monitoramento dos casos de dengue, febre de chikungunya e febre pelo vírus Zika até a Semana Epidemiológica 21, 2016. 47:27. [Google Scholar]
- 55.BRASIL Ministério da Saúde, Secretaria de Vigilância em Saúde. 2016. Boletim Epidemiológico: Monitoramento dos casos de dengue, febre de chikungunya e febre pelo vírus Zika até a Semana Epidemiológica 6, 2016. 47:7. [Google Scholar]
- 56.Faria NR, et al. 2016. Zika virus in the Americas: early epidemiological and genetic findings. Science 352:345–349. 10.1126/science.aaf5036. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cardoso CW, Paploski IA, Kikuti M, Rodrigues MS, Silva MM, Campos GS, Sardi SI, Kitron U, Reis MG, Ribeiro GS. 2015. Outbreak of exanthematous illness associated with Zika, Chikungunya, and dengue viruses, Salvador, Brazil. Emerg Infect Dis 21:2274–2276. 10.3201/eid2112.151167. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Quick J, et al. 2016. Real-time, portable genome sequencing for Ebola surveillance. Nature 530:228–232. 10.1038/nature16996. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Faria N, Sabino E, Nunes M, Carlos Junior Alcantara L, J Loman N, G Pybus O. 2016. Mobile real-time surveillance of Zika virus in Brazil. Genome Med 8:97. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Quick J, Grubaugh ND, Pullan ST, Claro IM, Smith AD, Gangavarapu K, Oliveira G, Robles-Sikisaka R, Rogers TF, Beutler NA, Burton DR, Lewis-Ximenez LL, de Jesus JG, Giovanetti M, Hill SC, Black A, Bedford T, Carroll MW, Nunes M, Alcantara LC, Jr, Sabino EC, Baylis SA, Faria NR, Loose M, Simpson JT, Pybus OG, Andersen KG, Loman NJ. 2017. Multiplex PCR method for MinION and Illumina sequencing of Zika and other virus genomes directly from clinical samples. Nat Protoc 12:1261–1276. 10.1038/nprot.2017.066. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Watson JD, Crick FHC. 1953. Molecular structure of nucleic acids; a structure for deoxyribose nucleic acid. Nature 171:737–738. 10.1038/171737a0. [PubMed] [DOI] [PubMed] [Google Scholar]
- 62.Holley RW, Apgar J, Everett GA, Madison JT, Marquisee M, Merrill SH, Penswick JR, Zamir A. 1965. Structure of a ribonucleic acid. Science 147:1462–1465. 10.1126/science.147.3664.1462. [PubMed] [DOI] [PubMed] [Google Scholar]
- 63.Sanger F, Brownlee GG, Barrell BG. 1965. A two-dimensional fractionation procedure for radioactive nucleotides. J Mol Biol 13:373–398. 10.1016/S0022-2836(65)80104-8. [DOI] [PubMed] [Google Scholar]
- 64.Min Jou W, Haegeman G, Ysebaert M, Fiers W. 1972. Nucleotide sequence of the gene coding for the bacteriophage MS2 coat protein. Nature 237:82–88. 10.1038/237082a0. [PubMed] [DOI] [PubMed] [Google Scholar]
- 65.Fiers W, Contreras R, Duerinck F, Haegeman G, Iserentant D, Merregaert J, Min Jou W, Molemans F, Raeymaekers A, Van den Berghe A, Volckaert G, Ysebaert M. 1976. Complete nucleotide sequence of bacteriophage MS2 RNA: primary and secondary structure of the replicase gene. Nature 260:500–507. 10.1038/260500a0. [PubMed] [DOI] [PubMed] [Google Scholar]
- 66.Wu R, Kaiser AD. 1968. Structure and base sequence in the cohesive ends of bacteriophage lambda DNA. J Mol Biol 35:523–537. 10.1016/S0022-2836(68)80012-9. [DOI] [PubMed] [Google Scholar]
- 67.Maxam AM, Gilbert W. 1977. A new method for sequencing DNA. Proc Natl Acad Sci U S A 74:560–564. 10.1073/pnas.74.2.560. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sanger F, Air GM, Barrell BG, Brown NL, Coulson AR, Fiddes CA, Hutchison CA, Slocombe PM, Smith M. 1977. Nucleotide sequence of bacteriophage ɸ X174 DNA. Nature 265:687–695. 10.1038/265687a0. [PubMed] [DOI] [PubMed] [Google Scholar]
- 69.Heather JM, Chain B. 2016. The sequence of sequencers: the history of sequencing DNA. Genomics 107:1–8. 10.1016/j.ygeno.2015.11.003. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pettersson E, Lundeberg J, Ahmadian A. 2009. Generations of sequencing technologies. Genomics 93:105–111. 10.1016/j.ygeno.2008.10.003. [PubMed] [DOI] [PubMed] [Google Scholar]
- 71.Ambardar S, Gupta R, Trakroo D, Lal R, Vakhlu J. 2016. High throughput sequencing: an overview of sequencing chemistry. Indian J Microbiol 56:394–404. 10.1007/s12088-016-0606-4. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nilakanta H, Drews KL, Firrell S, Foulkes MA, Jablonski KA. 2014. A review of software for analyzing molecular sequences. BMC Res Notes 7:830. 10.1186/1756-0500-7-830. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Buermans HP, den Dunnen JT. 2014. Next generation sequencing technology: advances and applications. Biochim Biophys Acta 1842:1932–1941. 10.1016/j.bbadis.2014.06.015. [PubMed] [DOI] [PubMed] [Google Scholar]
- 74.Balasubramanian S. 2011. Sequencing nucleic acids: from chemistry to medicine. Chem Commun (Camb) 47:7281–7286. 10.1039/c1cc11078k. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Levene MJ, Korlach J, Turner SW, Foquet M, Craighead HG, Webb WW. 2003. Zero-mode waveguides for single-molecule analysis at high concentrations. Science 299:682–686. 10.1126/science.1079700. [PubMed] [DOI] [PubMed] [Google Scholar]
- 76.Petersen LR, Jamieson DJ, Powers AM, Honein MA. 2016. Zika virus. N Engl J Med 374:1552–1563. 10.1056/NEJMra1602113. [PubMed] [DOI] [PubMed] [Google Scholar]
- 77.Laver T, Harrison J, O’Neill PA, Moore K, Farbos A, Paszkiewicz K, Studholme DJ. 2015. Assessing the performance of the Oxford Nanopore Technologies MinION. Biomol Detect Quantif 3:1–8. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lu H, Giordano F, Ning Z. 2016. Oxford Nanopore MinION Sequencing and Genome Assembly. Genomics, Proteomics Bioinformatics 14:5. 10.1016/j.gpb.2016.05.004. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Plesivkova D, Richards R, Harbison S. 2019. A review of the potential of the MinION™ single-molecule sequencing system for forensic applications. WIREs Forensic Sci 1:e1323. 10.1002/wfs2.1323. [DOI] [Google Scholar]
- 80.Musso D, Gubler DJ. 2016. Zika virus. Clin Microbiol Rev 29:487–524. 10.1128/CMR.00072-15. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]