

SUMMARY
Recent years have seen a remarkable growth in the field of protein-based medical treatments. Nevertheless, concerns have arisen regarding the cytotoxicity limitations, low affinity, potential immunogenicity, low stability, and challenges to modify these proteins. To overcome these obstacles, proximity-induced chemistry has emerged as a next-generation strategy for advancing protein therapeutics. This method allows site-specific modification of proteins with therapeutic agents, improving their effectiveness without extensive engineering. In addition, this innovative approach enables spatial control of the reaction based on proximity, facilitating the formation of irreversible covalent bonds between therapeutic proteins and their targets. This capability becomes particularly valuable in addressing challenges such as the low affinity frequently encountered between therapeutic proteins and their targets, as well as the limited availability of small molecules for specific protein targets. As a result, proximity-induced chemistry is reshaping the field of protein drug preparation and propelling the revolution in novel protein therapeutics.
Keywords: proximity-induced chemistry, protein drug, covalent drug, protein therapeutics, crosslinking
Graphical Abstract

eTOC Blurb
In this review, Cheng et al. provide an overview of the chemical mechanisms, functional groups, and introduction methods associated with proximity-induced chemistry. Additionally, they introduce innovative protein drugs developed via proximity-induced chemistry and discuss their therapeutic effectiveness across pre-clinical and clinical investigations.
Introduction
Protein therapeutics have emerged as a powerful approach in medicine, harnessing the diverse and dynamic roles of proteins in the body to address various diseases. Proteins play crucial roles in metabolic reactions, cell signaling, and immune responses, offering a tremendous opportunity for disease intervention1–4. The development of protein therapeutics dates back to 1982 with the first United States Food and Drug Administration (FDA)-approved recombinant protein humulin, and since then, the scientific community has recognized their immense potential1,5,6. With advancements in biotechnology, protein therapeutics have experienced significant growth, leading to the approval of over 200 therapeutic proteins by the FDA7,8. These therapeutics encompass a range of categories, including monoclonal antibodies, enzyme replacement therapy, growth factors, interferons, hormones, and fusion proteins. Protein therapeutics offer several advantages over traditional small molecule drugs, such as high target specificity, multifunctionality, reduced adverse effects, ability to target undruggable surfaces, and effectiveness in treating genetic mutations1,9,10. Despite revolutionizing the field of medicine, protein therapeutics still face limitations in terms of therapeutic efficacy. These limitations arise from their lack of inherent cytotoxic activity, relatively low affinity for their targets, relatively low stability, and potential immunogenicity. To address these limitations and improve the therapeutic potential of protein therapeutics, extensive research has been devoted to developing innovative chemical reactions that enable the modification of therapeutic proteins with diverse payloads.
The first-generation chemical modification of therapeutic proteins aims to enhance their potency by performing chemical reactions to create protein-small molecule conjugates. The most commonly used methods for chemical modification include N-hydroxysuccinimide (NHS) and maleimide-mediated chemistry, which attach functional molecules non-specifically to lysine or cysteine residues, respectively (Fig. 1A)11. Other classic modification methods utilizing reactive lysine and cysteine residues include isothiocyanate reaction, sulfonyl chloride reaction, vinyl sulfone reaction, amination reaction with aldehyde, and squaric acid reaction11. These methods are compatible with aqueous solutions, neutral pH, and room temperature. One of the major applications of these reactions is in the preparation of antibody-drug conjugates (ADCs) by conjugating toxins to antibodies. Several ADCs have been developed and approved by the FDA for various cancer treatments using the first-generation chemical modification approach12,13. However, these methods can only result in heterogeneous products with variable drug-to-antibody ratios (DARs) due to the random labeling nature of the modification, which cannot be further purified. As a result, these heterogeneous ADCs exhibit suboptimal therapeutic efficacy compared to their homogeneous counterparts14,15. This issue also applies to other therapeutic proteins, as the random labeling can lead to appendage blocking bio-reactive residues and causing a loss of protein activity, significantly limiting the utility of this non-specific strategy.
Figure 1.
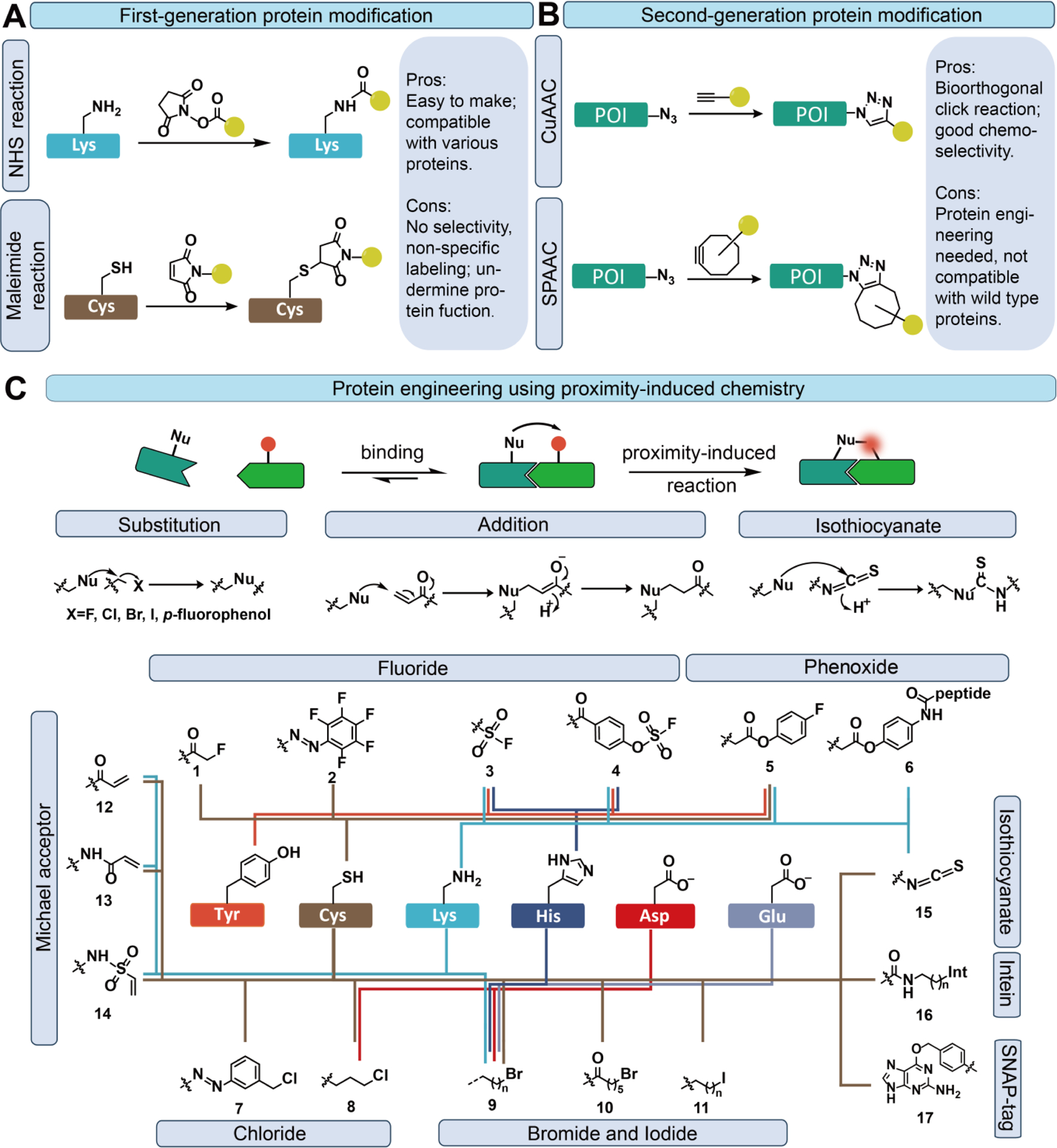
Protein modification approaches and representative reactions. (A) The first-generation protein modification approach employs N-hydroxysuccinimide (NHS) and maleimide-mediated reactions. (B) The second-generation protein modification approach incorporates copper-catalyzed azide-alkyne cycloaddition (CuAAC) and strain-promoted alkyne-azide cycloaddition (SPAAC) reactions. (C) Proximity-induced chemistry involves reactions between proximity-enabled reactive groups and specific natural amino acids in close proximity.
Due to the limitations associated with the first-generation protein modification, the second-generation approach has gained significant attention. This approach involves site-specific modification of target proteins using bioorthogonal click chemistry (Fig. 1B). Bioorthogonal chemistry comprises a set of chemical reactions that can occur within living systems without disrupting normal biochemical processes16,17. These reactions are bioorthogonal, meaning they are selective and do not react with other biomolecules in the system. Some commonly employed reactions in biological systems include oxime ligation18,19, Staudinger ligation20,21, copper-catalyzed azide-alkyne cycloaddition (CuAAC) reaction22,23, strain-promoted alkyne-azide cycloaddition (SPAAC) reaction24,25, and tetrazine-based inverse electron-demand Diels-Alder (IEDDA) reaction26,27. These methods exhibit selective reactivity towards target moieties, offering excellent regio-selectivity and compatibility with living organisms16,28. However, the second-generation protein modification approach requires the integration of two specific moieties onto the conjugation partners, often necessitating extensive engineering of the labeling proteins. Consequently, this poses additional limitations for the biomedical application of these techniques in vivo, particularly due to the difficulties associated with selectively labeling protein targets in live organisms.
The next-generation protein modification approach overcomes the requirement of engineering labeling proteins by enabling the site-specific covalent bond formation of a native amino acid residue through proximity-induced chemistry (Fig. 1C). Proximity, or the close physical closeness of molecules, plays a crucial role in driving two reactants to approach each other at the Ångström level. Bringing molecules into close proximity significantly enhances reaction rates, as the likelihood of an effective collision between molecules is a function that increases exponentially with proximity29,30. Proximity-induced chemistry facilitates chemical transformations between a proximity-enabled reactive group and a specific natural residue of proteins through the complex-induced proximity effect. To achieve high selectivity of the proximity-induced reaction, the proximity-enabled reactive group must remain stable and unreactive with residues on the protein surface, free amino acids, and other biomolecules under physiological conditions. This ensures that the proximity-enabled reactive moiety remains inactive until it reaches close proximity to the target site, where it selectively reacts with the desired natural residue in the binding pocket. Consequently, the use of proximity-induced chemistry for site-specific protein labeling eliminates the need for protein engineering to introduce unique chemical moieties. This advancement opens up exciting opportunities for preparing and developing novel protein therapeutics29. Furthermore, proximity-induced chemistry has been used to prepare covalent protein drugs. The concept of incorporating a proximity-enabled reactive group, typically a Michael acceptor, to selectively react with a cysteine residue in the binding site of the target protein has been previously employed to enhance the affinity of small molecule drugs. Initially, this approach was investigated to develop selective inhibitors for kinase mutants and has more recently been applied to optimize various inhibitors31. Several reviews have already covered the recent advancements in the development of these covalent inhibitors29,32–34, so we will not delve into those in this particular review. Instead, our focus will be on providing a comprehensive summary of the recent progress in utilizing proximity-induced chemistry to advance protein therapeutics.
1. THE CHEMISTRY BEHIND PROXIMITY-INDUCED PROTEIN THERAPEUTICS
The diversity of chemistry within natural proteins is limited due to the presence of only 20 canonical amino acids. Nucleophilic residues such as lysine, cysteine, and tyrosine are the most commonly used residues for carrying out chemical reactions (Fig. 1C and Table SI). The reactions with other weaker nucleophiles like histidine, aspartate, and glutamate have also been reported but to a lesser extent. These good nucleophilic residues are capable of participating in substitution reactions and nucleophilic addition reactions. When designing proximity-induced chemistry, the proximity-enabled reactive group is chosen to covalently react with one of these specific amino acid residues only in proximity. Among various proximity-induced chemistries, reactions with lysine residues are preferred because lysine is relatively abundant on protein surfaces and often found at the protein-protein interfaces of interest35. On the other hand, strong nucleophiles like cysteine are less common on protein surfaces and are frequently involved in disulfide bond cross-linking36. Apart from their intrinsic chemical properties, the nucleophilicities of amino acid residues can be profoundly influenced by their microenvironments.37,38 This means that amino acids nearby or adjacent to a nucleophilic residue can modulate its reactivity through various interactions, such as electrostatic forces, hydrogen bonding, metal ion coordination, and hydrophobic effects. For instance, while free cysteine has a pKa value of 8.3, its pKa can vary from as low as 3 to over 10 depending on its specific microenvironment in a protein. Consequently, the protein’s microenvironment can drastically alter the reactivity of nucleophilic amino acids, underscoring the importance of carefully selecting positions to introduce proximity-enabled reactive groups.
For an ideal proximity-enabled reactive moiety, it should exhibit high reactivity toward specific natural amino acid residues when they are in close proximity. However, it is crucial for this chemical moiety to remain inert under physiological conditions. Therefore, it is important to carefully modulate the reactivity of the chemical moieties used for proximity-induced chemistry. This involves finding a balance between reactivity and stability since stronger reactivity may make the moiety susceptible to broad chemical species in the natural system, leading to non-specific protein labeling.
1.1. Substitution reaction with fluoride as a leaving group
One major class of proximity-induced chemistry involves nucleophilic substitution reactions, where nucleophilic amino acid residues such as lysine, cysteine, and others attack weak electrophiles, leading to the displacement of a leaving group (Fig. 1C). In proximity-induced chemistry, the leaving groups predominantly consist of halogen atoms (F, Cl, and Br), as well as p-fluorophenyl. These leaving groups remain inert under aqueous and physiological conditions but become reactive when in proximity to nucleophilic residues, such as lysine or cysteine.
Fluoride, due to its small size and high charge density, is considered the least favorable leaving group among the reactive residues. In traditional SN2 reactions, F− rarely acts as a leaving group due to its low polarizability. However, when two residues come into close proximity, the reactivity of cysteine residues towards weak electrophiles containing fluoride as a leaving group is notably enhanced. To further improve the reactivity, α-carbonyl groups are introduced to facilitate the attack of cysteine residues with very high specificity (Fig. 1C and Table S1, 1)39. Despite the high specificity of these proximity-induced groups for cysteine, the yield is relatively low, with less than 50% efficiency due to the weak reactivity of fluoride40. Previous studies have demonstrated that perfluoroaromatic molecules react with cysteine thiolate via an SNAr mechanism41. Consequently, in a proximity-induced setting, the pentafluorophenyl-diazenyl (Fig. 1C and Table S1, 2) group has proven effective in achieving site-specific modification of cysteine residues, yielding complete conversion after 6 h at 25°C42,43.
The sulfur (VI) fluoride exchange (SuFEx) represents the latest set of click chemistry transformations44,45. SVI-F motifs, including arylfluorosulfates (ArOSO2F) and iminosulfuroxyfluoride (RN═S(O)F2), are two common forms that can be readily synthesized using sulfuryl fluoride (SO2F2) and thionyl tetrafluoride (O═SF4), respectively46. SuFEx reactions have recently been utilized for proximity-induced protein crosslinking, where SVI-F motifs react with lysine, histidine, or tyrosine residues on proteins via SuFEx chemistry when the two reactants are in close proximity. Building upon this principle, fluorosulfonyl (Fig. 1C and Table S1, 3) and arylfluorosulfonyl (Fig. 1C and Table S1, 4) were designed and successfully incorporated into proteins, demonstrating exceptional biocompatibility and proximity-induced activities against multiple residues, such as tyrosine, lysine, and histidine47,48. The crosslinking reactions with these residues exhibited outstanding specificities, resulting in high yields with an efficiency of over 75%47,48.
1.2. Substitution reaction with phenyl carbamate as a leaving group
Aryl carbamates, characterized as moderate electrophiles, are stable under physiological conditions but become susceptible to being attacked when in proximity to good nucleophiles. As a result, aryl carbamates have emerged as promising candidates for proximity-induced chemistry. Exploiting this property, phenyl carbamate and fluorophenyl carbamate (Fig. 1C and Table S1, 5) have been developed as proximity-induced crosslinkers for native protein residues49. The reactivities of these molecules were assessed in proximity-induced conditions, revealing that the phenyl carbamate group exhibits suboptimal reactivity, and the addition of a fluoro substituent to phenyl carbamate demonstrates remarkable reactivity towards cysteine, lysine, and tyrosine residues. The reaction is very impressive, resulting in more than 95% yields after 4–24 hours49. Another phenyl carbamate-based leaving group (Fig. 1C and Table S1, 6) is connected to an affinity peptide through an amide bond at the para position. The reactivity of proximity-enabled reactive moiety 6 is slightly lower than that of moiety 5, resulting in a labeling efficiency of 80% after 1 hour50. This reduced reactivity is likely attributed to the absence of an electron-withdrawing group on the phenyl carbamate moiety50.
1.3. Substitution reaction with chloride, bromide, and iodide as leaving groups
In addition to fluoride, other halogen atoms can be utilized as more reactive leaving groups in proximity-induced chemistry. When comparing reactive residues with different halogen atoms linked by the same alkyl chain, the covalent bond formation efficiency follows the order of I > Br > Cl > F, which aligns with the leaving ability of halides in SN2 reactions. Chlorine, bromine, and iodine substituents exhibit good reactivity for cysteine residue without the need for adjunct α-carbonyl groups. The alkyl halides (Fig. 1C and Table S1, 7, 8, 9, 10, 11) exhibit moderate cross-linking efficiency, ranging from 24% to 48% for the cysteine residue51,52. Among these chemical moieties, bromoethyl (Fig. 1C and Table S1, 9) has been extensively studied and recognized for its versatility, primarily attributed to its long aliphatic side chain that enhances reactivity51. Alkyl chloride (Fig. 1C and Table S1, 8) also served as a proximity-enabled reactive moiety in the Halo-tag system for site-specific protein labeling53. The Halo-tag protein, derived from bacterial haloalkane dehalogenase, was utilized in this process. Upon binding to the Halo-tag protein, the Halo-tag ligand with an alkyl chloride chain exhibited remarkable selectivity, reacting exclusively with the Asp residue in the binding pocket of the Halo-tag protein, resulting in over 90% labeling efficiency.54.
1.4. Addition reaction
Extensive research has been conducted on the irreversible, covalent inhibition of kinases and proteases through alkylation, acylation, phosphorylation, and other modifications of reactive amino acid residues55. Among the various strategies employed, the Michael addition of reactive residues with Michael acceptors is one of the widely used strategies56,57.
Michael acceptors, such as α, β-unsaturated ketones and vinyl sulfones, are frequently utilized for conjugate addition with nucleophiles like lysine and cysteine residues. Vinyl carbonyl, vinyl amidyl, and vinyl sulfonamidyl groups (Fig 1C and Table S1, 12–14) have been introduced into proteins58. These groups exhibit stability under physiological conditions and display relatively low non-specific reactivity towards other serum and cellular proteins. Notably, the protein with a vinyl sulfonamidyl group (Fig. 1C and Table S1, 14) demonstrates the highest reactivity, resulting in >95% covalent cross-linking of human epidermal growth factor receptor 2 (HER2) protein and its antibody58. Conversely, the protein with a vinyl amidyl group (Fig. 1C and Table S1, 13) exhibits moderate reactivity, while the protein with an N-alkylacrylamide group (Fig. 1C and Table S1, 12) shows relatively lower reactivity. These findings align with SVI-F probes, suggesting that the presence of an electron-withdrawing sulfone moiety with a sulfur (VI) center significantly enhances proximity-induced chemistry reactivity.
Phenyl isothiocyanates have been employed in Edman degradation peptide sequencing. Under mildly alkaline conditions, phenyl isothiocyanate reacts with an uncharged N-terminal amino group, forming a cyclic phenylthiocarbamide derivative59,60. The center carbon of the isothiocyanate group is electron-deficient, enabling attack by the amine group and the formation of a thiourea bridge. An isothiocyanate derivative (Fig. 1C and Table S1, 15) based on tyrosine demonstrates high efficiency (>80%) when proximal to a lysine residue61.
2. APPROACHES TO INTRODUCE PROXIMITY-ENABLED REACTIVE GROUPS INTO PROTEINS
The reactivity of proximity-induced chemistry relies on the distance and spatial geometry between the proximity-enabled reactive moieties within the binder and the specific natural residues of targeted proteins. Therefore, it is crucial to introduce these proximity-enabled reactive moieties into the binder in a site-specific manner. When the binder is a protein, the most commonly employed strategy involves using Genetic Code Expansion technology62–65. This technique allows for the incorporation of proximity-enabled reactive noncanonical amino acids (ncAAs) that can react with neighboring natural amino acid residues in close proximity39,49,51,58,66,67. Other strategies, such as split intein-mediated protein ligation, solid-phase peptide synthesis, and similar approaches, are utilized to bring the proximity-enabled reactive moieties closer to natural amino acids68–72. When the binder consists of small molecules, a structure-activity relationship analysis can be conducted to optimize the spatial geometry of the proximity-enabled reactive moieties in relation to the specific natural residue of the targeted protein. These efforts have resulted in the development of covalent-based protein tags like SNAP-tag, Halo-tag, and others53,73–75. Additionally, proximity-induced reactive peptides have been prepared using solid-phase peptide synthesis for antibody labeling purposes69,76–78. In this section, we provide a summary of these innovative approaches for the precise incorporation of proximity-induced reactive moieties into proteins.
2.1. Genetically encoded ncAAs with proximity-enabled reactive moieties into proteins
To achieve the site-specific introduction of unnatural reactive moieties into proteins in vivo, Genetic code expansion technology offers a highly effective approach62–65. Typically, a noncanonical amino acid (ncAA) is designed with a distinct chemical handle based on a tyrosine or pyrrolysine scaffold. This engineered ncAA can then be incorporated into proteins at specific sites in both prokaryotic and eukaryotic cells using a bioorthogonal aminoacyl-tRNA synthetase (aaRS)/tRNA pair that responds to the amber stop codon. Through the genetic incorporation of a ncAA equipped with a proximity-enabled reactive moiety, it becomes possible to establish precise and stable covalent linkages between proteins, enabling tailored crosslinking between ncAA and targeted amino acid residues in a site-specific manner (Fig. 2A). Furthermore, a diverse array of photo-activatable crosslinking ncAAs have found utility in protein engineering, propelling the development of innovative protein therapeutics. Several reviews have summarized the principles and utilities of these photo-activatable crosslinking ncAAs79,80. In this review, we will focus on proximity-enabled ncAAs that don’t require any post-treatment to elicit proximity-induced reactions.
Figure 2.
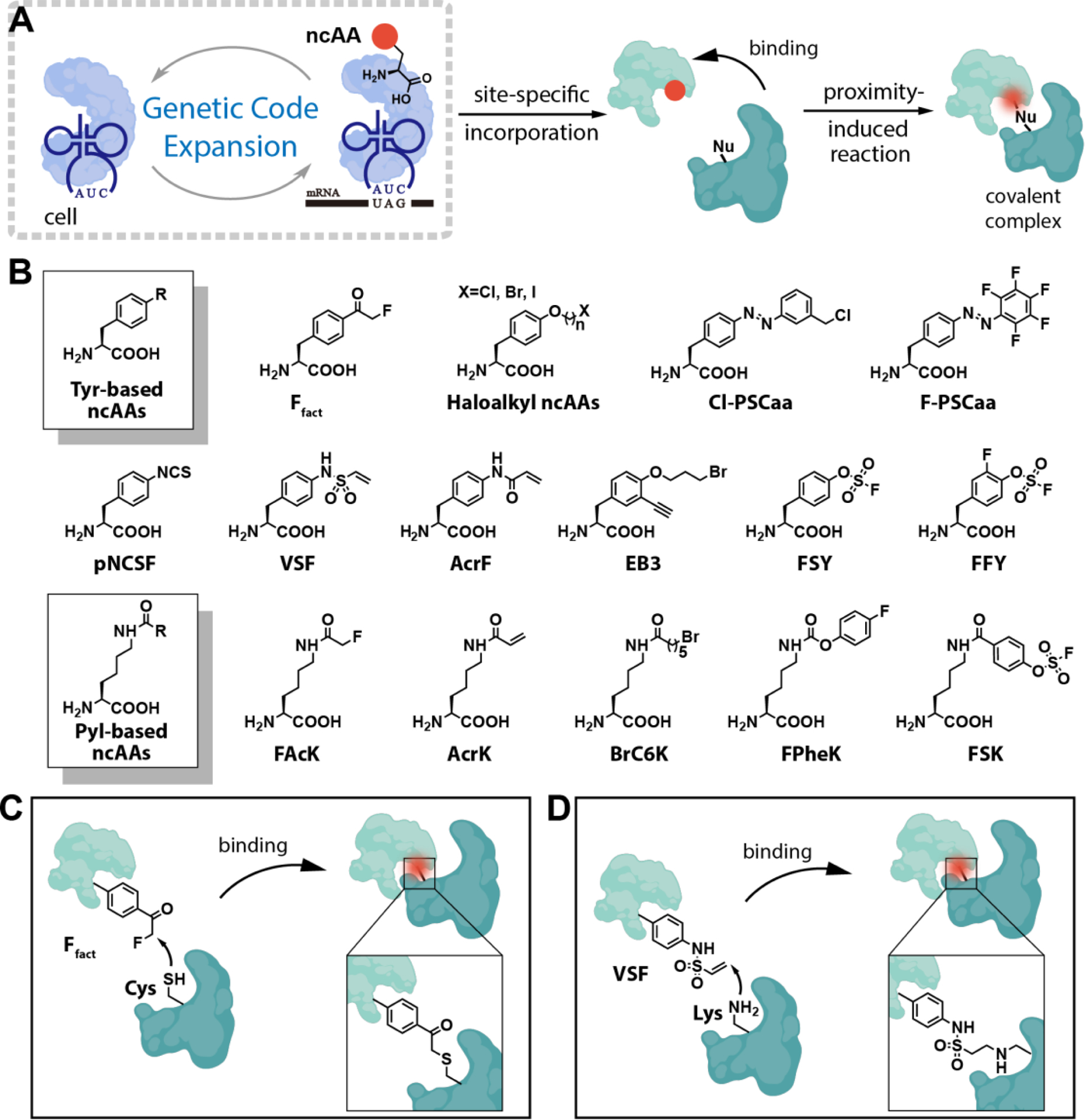
Genetic incorporation of ncAAs with proximity-enabled reactive moieties into proteins. (A) Genetic Code Expansion technology enables the incorporation of bioreactive ncAAs with proximity-enabled reactive moieties into proteins. These moieties can form covalent bonds with native residues on interacting proteins through proximity-induced reactions. (B) Structural of bioreactive ncAAs utilized in proximity-induced chemistry, based on either a tyrosine or pyrrolysine scaffold. (C) Ffact selectively reacts with the cysteine residue when two residues are in close proximity. (D) VSF selectively reacts with the lysine residue when two residues are in close proximity.
Based on the tyrosine scaffold, the first proximity-enabled reactive ncAA with a fluoroacetylphenyl moiety, fluoroacetylphenylalanine (Ffact) was designed and incorporated into proteins in Escherichia coli (Fig. 2B and 2C, Table 1)39. Ffact was designed to selectively react with nearby cysteine residues, allowing for precise crosslinking with the protein targets. In order to showcase its proximity-induced chemistry, Ffact was incorporated at position Asp36 within the ZSPA affibody (Afb) using the bioorthogonal Methanococcus jannaschii tyrosyl-tRNA synthetase (MjTyrRS)/tRNACUA system39. The wild-type Afb and the Z protein exhibited a binding affinity of 6 μM. Upon introducing Ffact, the resulting Afb mutant showed a crosslinking yield of over 60% with the Z protein containing an Asn6Cys mutation39,81. This study is particularly noteworthy as it marks the first successful example of Genetic Code Expansion technology to introduce a proximity-induced chemical moiety for protein crosslinking. Expanding on these discoveries, a series of haloalkyl ncAAs with varying side chain lengths and reactivities, including O-(3-chloropropyl)-tyrosine (CprY), O-(3-bromoethyl)-tyrosine (BetY), O-(3-bromopropyl)-tyrosine (BprY), and so on, were synthesized and integrated into proteins using a specific mutated Methanosarcina mazei pyrrolysyl-tRNA synthetase (MmPylRS)/tRNACUA variants (Fig. 2B and Table 1)51. Notably, the efficiency of crosslinking varied depending on the halogen atom present in the ncAAs, following the order of I>Br>Cl, which aligns with the halide leaving ability in SN2 reactions. Moreover, when comparing ncAAs with different side chain lengths, BetY, which possesses a two-carbon linker, demonstrated the highest crosslinking efficiency between the Afb mutant and Z protein51. Additionally, intramolecular proximity-induced crosslinking was employed to enhance protein stability. Intriguingly, the formation of a novel intramolecular covalent bond between BetY and a cysteine residue of Afb protein resulted in a remarkable increase in its Tm value from 46.7 ± 0.2 °C to 60.4 ± 0.7 °C51. This observation highlights the significant impact of proximity-induced crosslinking on bolstering protein stability.
Table 1.
NcAAs with proximity-enabled reactive moieties
| No | ncAA | Modified protein/Interacting protein | Target residues | Expression systems | Reference |
|---|---|---|---|---|---|
| 1 | Ffact | Afb/Z protein | Cys | Bacteria | 39 |
| 2 | VSF | Anti-HER2 Fab of Herceptin/Extracellular domain of HER2 | Lys | Bacteria | 58 |
| 3 | AcrF | Anti-HER2 Fab of Herceptin/Extracellular domain of HER2 | Lys | Bacteria | 58 |
| 4 | pNCSF | Z protein/Afb | Lys | Bacteria | 61 |
| 5 | Haloalkyl ncAAs | Afb/Z protein | Cys | Bacteria & Mammalian cells | 51 |
| 6 | Cl-PSCaa | The central helix of calmodulin (CaM)/CaM | Cys | Bacteria & Mammalian cells | 52 |
| 7 | F-PSCaa | CaM/CaM | Cys | Bacteria & Mammalian cells | 43 |
| 8 | EB3 | Z protein/Afb | Cys | Bacteria & Mammalian cells | 85 |
| 9 | FAcK | Afb/Z protein | Cys | Bacteria & Mammalian cells | 40 |
| 10 | BrC6K | Afb/Z protein or Afb/Afb | Cys, Lys, His, Glu, Asp | Bacteria & Mammalian cells | 66 |
| 11 | FSY | Z protein/Afb | Lys, His, Tyr | Bacteria & Mammalian cells | 47 |
| 12 | FFY | nanobody mNb6/Receptor binding domain (RBD) of the SARS-CoV-2 viral spike protein | Lys, His, Tyr | Bacteria & Mammalian cells | 86 |
| 13 | mPyTK | glutathione-S-transferase (GST) homodimer/GST homodimer | Glu, Asp | Bacteria & Mammalian cells | 87 |
| 14 | o-NBAK | GST homodimer/ GST homodimer | Lys | Bacteria & Mammalian cells | 88 |
| 15 | AcrK | Anti-HER2 Fab of Herceptin/Extracellular domain of HER2 | Lys | Bacteria & Mammalian cells | 58 |
| 16 | FPheK | Z protein/Afb | Cys, Lys, Tyr | Bacteria & Mammalian cells | 49 |
| 17 | FnbY | GST homodimer/ GST homodimer | Cys, Lys, His, Tyr, Trp, Met, Arg, Asn, Gln | Bacteria & Mammalian cells | 89 |
| 18 | FSK | Nanobody 7D12/Epidermal growth factor receptors (EGFR) | Lys, His, Tyr | Bacteria & Mammalian cells | 48 |
| 19 | FmnbY | Ubiquitin (Ub)/ Ubiquitin (Ub) | Cys, Lys, His, Tyr, Trp, Met, Arg, Asn, Gln | Bacteria & Mammalian cells | 90 |
To harness the benefits of proximity-induced Michael addition chemistry, various tyrosine and lysine analogs with Michael acceptors have been designed and integrated into proteins58. These ncAAs, such as p-acrylamido-phenylalanine (AcrF), p-vinylsulfonamido-phenylalanine (VSF), and N-acryloyl-lysine (AcrK), display selective reactivity towards nearby lysine residues (Fig. 2B, 2D, and Table 1). Among them, VSF has shown the most favorable proximity-induced reactivity, achieving up to 95% crosslinking efficiency. In order to demonstrate the utility of VSF, it was employed to crosslink an antibody with its antigen. Based on the co-crystalized complex structure, Tyr92 residue within the anti-human epidermal growth factor receptor 2 (HER2) fragment antigen-binding (Fab) region closely interacts with two lysine residues (Lys569 and Lys593) of HER2 at the binding interface. Accordingly, VSF was incorporated site-specifically at Tyr92 within the anti-HER2 Fab using a mutated MjTyrRS/tRNACUA system in E. coli (Table 1). The resulting mutant anti-HER2 Fab exhibited rapid covalent crosslinking with HER2 on the cell surface under physiological pH conditions58.
Sulfur (VI) -fluoride exchange (SuFEx) reactions have emerged as a novel class of click chemistry transformations involving the rapid replacement of an SVI-F bond with an S-O or S-N bond44,45. To achieve site-specific incorporation of SVI-F motifs into proteins, two designed compounds, namely fluorosulfate-tyrosine (FSY) and fluorosulfonyloxybenzoyl-lysine (FSK), enable proximity-induced sulfur-fluoride exchange (SuFEx) reactions with native Lys, His, and Tyr residues (Fig. 2B and Table 1)47,48. In a Zspa affibody (Afb) and Z protein binding model, the modification of Afb with FSY demonstrates cross-linking efficiencies of 59%, 53%, or 35% for Lys, His, or Tyr residues at position 7 of the Z protein, respectively. However, FSK exhibits a longer and more flexible aryl fluorosulfate-containing side chain compared to FSY. This unique property of FSK allows for the crosslinking of protein sites that are inaccessible with FSY48. Based on the Afb/Z protein pair, authors also studied the kinetic character of proximity-induced covalent bond formation, which consists of two steps, initial non-covalent protein binding and covalent bond formation (Fig. 2A).82 The Afb/Z protein complex demonstrated non-covalent binding with a dissociation constant (Kd) of approximately 6 uM. Upon the initiation of covalent bond formation, the equilibrium is disrupted, leading to a decrease in the Kd over time. This phenomenon is attributed to the relationship Kd = Koff/Kon, where Koff represents the dissociation rate constant and Kon represents the association rate constant. As the covalent bond forms, Koff decreases, contributing to the observed decline in Kd. The second-order rate constant for Afb/Z protein covalent protein complex formation was determined to be (1.32 ± 0.04) × 10−2 uM−1 h−1, indicating that the binding affinity is dramatically increased.
Building upon the versatility of Genetic Code Expansion and the utilization of proximity-induced chemistry towards natural residues, a wide range of proximity-enabled reactive ncAAs have been designed and genetically integrated into proteins (Fig. 2 and Table 1). In addition to the aforementioned proximity-enabled reactive ncAAs, other ncAAs containing phenyl isothiocyanate, fluorophenyl carbamate, and various other moieties have also been successfully introduced into proteins using Genetic Code Expansion for proximity-induced crosslinking of proteins (Fig 2B and Table 1)79,80,83,84. By expanding the repertoire of protein modification, this innovative approach opens up new avenues for precise and specific protein modification, facilitating the creation of advanced protein-based therapeutics with enhanced functionalities and therapeutic potential.
2.2. Innovative protein tags based on proximity-induced chemistry
Inspired by the reactions of natural self-labeling proteins or auto-processing proteins, several proximity-induced chemistry-based tags have been designed53,73,75,91. These proteins utilize their three-dimensional structures to bring proximity-induced chemical moieties to native residues in close proximity, thereby enabling covalent bond formation. These mechanisms have been utilized to create various protein tags (Fig. 3A).
Figure 3.
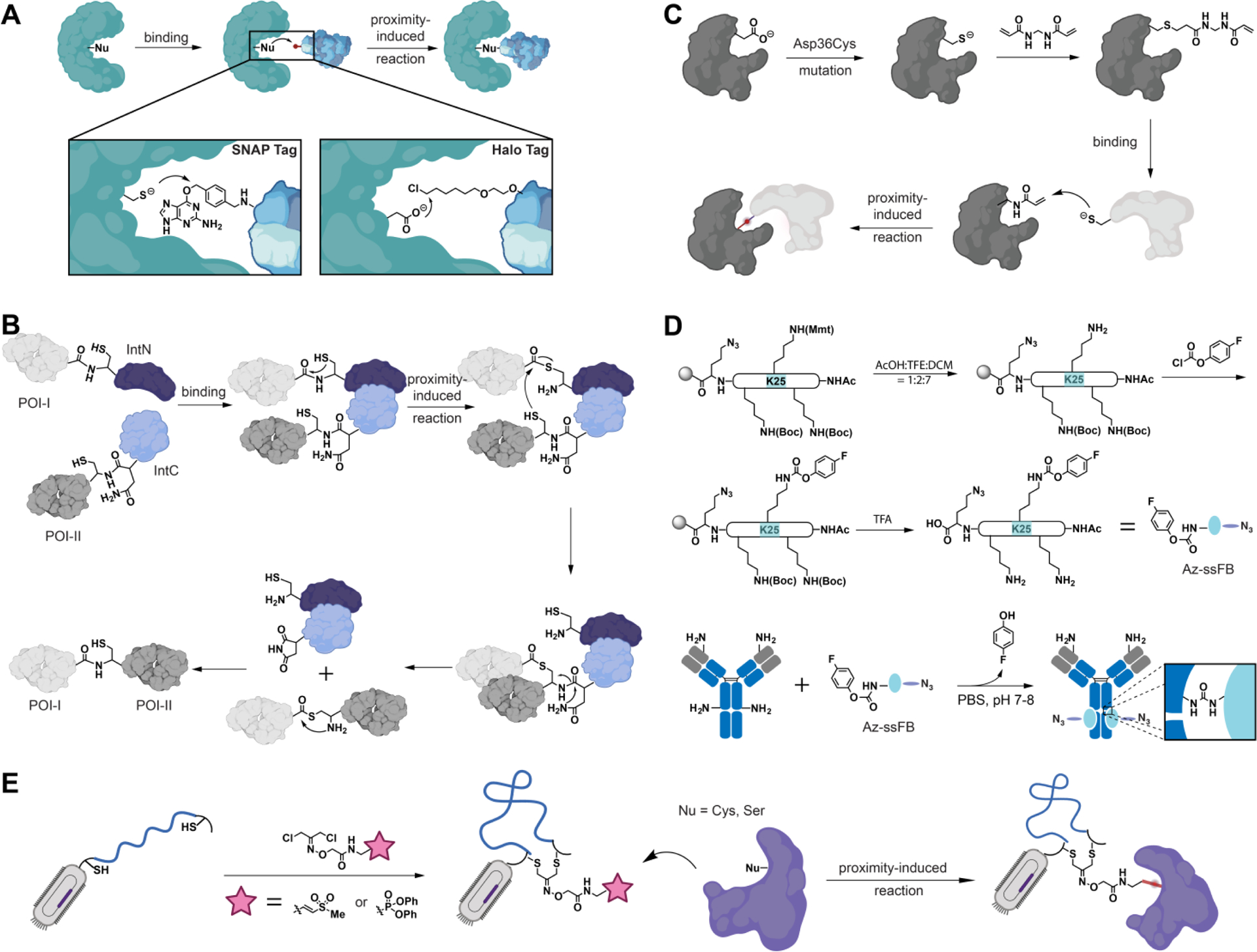
Strategies for Incorporating Proximity-Enabled Reactive Moieties into Proteins. (A) The proximity-induced chemistries between SNAP Tag or Halo Tag and their ligands. (B) Employing trans-splicing of split inteins to generate protein conjugates. (C) Post-modification of the affibody with acrylamide to enable proximity-induced chemistry with cysteine residues in close proximity. (D) Introduction of a proximity-enabled reactive group, fluorophenyl carbamate, into an antibody-binding peptide using solid-phase peptide synthesis. The resulting peptide enables proximity-induced chemistry with antibodies. (E) Two cysteine residues allow for the incorporation of proximity-responsive functional groups and this arrangement facilitates the detection of specific covalent binding agents through the utilization of display technologies.
One popular proximity-induced chemistry-based protein tag, called the SNAP-tag, was derived from human O6-alkylguanine-DNA alkyltransferase (hAGT)91. It facilitates the site-directed attachment of small molecules to proteins. When bound to O6-alkylguanine derivatives substituted at the 4 position of the benzyl ring, the SNAP-tag irreversibly transfers the benzyl moiety (Fig. 1C and Table S1, 17) of the substrate to its reactive cysteine residue, resulting in a covalent linkage between the SNAP-tag and the desired compounds (Fig. 3A). Expanding on the foundation of the SNAP-tag, the hAGT enzyme has also been evolved to enable covalent reactions with O2-benzylcytosine derivatives. This bioorthogonal tag, known as the CLIP-tag75, specifically reacts with O2-benzylcytosine substrates in close proximity, while the SNAP-tag selectively reacts with nearby O6-benzylguanines. Thus, these two innovative technologies enable simultaneous bioorthogonal labeling, thereby expanding the prospects for the development of innovative double-labeled protein drugs75. In addition to SNAP- and CLIP-tags, the Halo-tag is another extensively used self-labeling protein tag derived from bacterial haloalkane dehalogenase (Fig. 3A)53. When ligands with alkyl chloride (Fig. 1C and Table S1, 8) moiety bind to it, the Asp106 residue of the Xanthobacter dehalogenase mutant nucleophilically attacks the terminal chloride, resulting in the formation of a covalent alkyl-dehalogenase conjugate (Fig. 3A). Self-labeling enzyme tags have significantly transformed protein labeling techniques and the development of novel protein drugs. However, these tags do present certain challenges, such as limited labeling positions and the comparatively bulky size of the self-labeling protein.
Proximity-induced chemistries offer another category of protein reactions, exemplified by intein-mediated protein splicing70,92–94. Protein splicing is a naturally occurring process wherein an intein excises itself from a host protein through the cleavage and reformation of amide bonds in close proximity. This process is initiated by the attachment of an amide bond (Fig.1C and Table S1, 16) to a nearby nucleophilic residue (such as cysteine, serine, or threonine). The resulting thioester intermediate then undergoes trans-thioesterification and an S to N acyl shift, leading to the regeneration of the amide bond. Building upon this chemistry, various methods have been developed for therapeutic protein modifications, such as expressed protein ligation (EPL) and protein trans-splicing (PTS) (Fig. 3B)92–94. EPL is commonly employed to generate proteins with site-specific modifications, including phosphorylation, glycosylation, lipidation, ubiquitination, acetylation, and other desired alterations70,95. On the other hand, PTS has found applications in the synthesis of therapeutic cyclic proteins and protein conjugations. For instance, PTS has been utilized for the biosynthesis of cyclic peptide libraries, enabling the selection of α-synuclein inhibitors, as well as the production of site-specific antibody-drug conjugates and bispecific antibodies96.
2.3. Other strategies to introduce proximity-enabled reactive moieties into proteins
To create a protein that can form the covalent bond with specific residues of a target protein, a key step is to introduce proximity-enabled reactive groups in a site-specific manner. Besides Genetic Code Expansion, other strategies have been employed, such as post-chemical protein modification and solid-phase peptide synthesis, to synthesize polypeptides with well-defined proximity-enabled reactive moieties69,71,72,81,81,97.
Because of their low abundance in proteins, cysteine residues have been widely utilized for site-specific protein modification98–100. By employing the cysteine-based Michael addition reaction, acrylamide, a weak electrophile, was introduced specifically at the Asp36 residue of an affibody81. The post-chemical modification of acrylamide on the affibody facilitated the formation of covalent bonds with nearby nucleophilic residues (Cys, His, and Lys) on ZSPA (Fig. 3C). Importantly, this modified affibody displayed remarkable specificity towards the targeted protein, ZSPA, both in vitro and when expressed on mammalian cell surfaces. As a result of the enhanced specificity, the enzyme-linked immunosorbent assay (ELISA) demonstrated significantly improved detection sensitivity. Additionally, the modified affibody exhibited increased stability for cell surface imaging applications. These findings provide valuable insights into the development of post-chemical modification strategies to incorporate proximity-induced chemistry for protein engineering purposes.
Solid-phase peptide synthesis (SPPS) offers a versatile approach for incorporating proximity-enabled reactive groups into small polypeptides69,101. This can be achieved by either directly introducing a noncanonical amino acid as the proximity-enabled reactive group or performing post-chemical modification on a native lysine residue. In 2013, the small peptide BI-107D1 was discovered as a modest inhibitor for Siah, and a post-chemical modification using acrylamide was employed as the proximity-enabled reactive moiety71. The introduction of Lys-acrylamide onto the peptide facilitated the formation of a crosslink with a nearby cysteine residue situated in the binding pocket of the Siah mutant, resulting in a significant enhancement of its inhibitory efficiency. A similar strategy has been applied to prepare peptides with proximity-enabled reactive groups. Various electrophiles, such as acrylamide, chloroacetamide, and propiolamide, have been introduced into the Bim peptide to enhance its binding affinity against the oncogenic protein Bcl2A97. The introduced electrophiles in the modified peptide exhibited the ability to form stable covalent bonds with Cys55 of Bcl2A1, which is located in close proximity to the binding site. Consequently, the modified peptide effectively inhibited the activity of Bcl2A1 in U937 lymphoma and HeLa cells, surpassing the inhibitory efficacy of the native Bim peptide ligand. Furthermore, SPPS was utilized to introduce a proximity-enabled reactive group, sulfonyl fluoride, into SAHp53-8 peptides for inhibiting the p53-Mdm2/Mdm4 interaction72. The sulfonyl fluoride modification of SAHp53-8 enables the formation of covalent bonds with lysine and histidine residues of Mdm2 or Mdm4 upon binding. In addition to enhancing the affinity between therapeutic proteins and their targets, proximity-enabled reactive moieties have also been incorporated into antibody-binding peptides to enable the site-specific conjugation of antibodies. By employing SPPS, 4-fluorophenyl and N-hydroxysuccinimide ester were introduced into the FB and Fc-III peptides69,76,77. These modified antibody-affinity peptides can form covalent bonds with the lysine residues of antibodies upon binding, facilitating the site-specific modification of antibodies with drugs, fluorophores, and other proteins (Fig. 3D).
The cysteine bridging strategy has been employed to introduce novel chemical functionalities through the formation of covalent connections between two cysteine residues.102 This approach has also been harnessed for the integration of proximity-enabled reactive groups, thereby enabling the identification of covalent inhibitors using display technologies. To elaborate, a cyclic peptide library targeting the tobacco etch virus (TEV) protease was constructed using this strategy. The library was assembled by linking rebridging reagents, featuring a cysteine-reactive vinyl sulfone moiety, with two cysteine residues within the peptide library. Subsequent to this construction, the resulting cyclic peptide library underwent selection through a phage display process. The obtained peptide candidates adopted properly folded conformations that closely approached the TEV protease. This close proximity facilitated the reaction of the vinyl sulfone group with cysteine residues in the vicinity of the TEV protease, leading to the formation of covalent bonds. A similar methodology was employed, using a serine-reactive diphenylphosphonate moiety, to identify covalent inhibitors for fluorophosphonate-binding hydrolases F (Fig. 3E). This highlights the way in which incorporating proximity-enabled reactive groups into potential binders markedly streamlines the process of screening for specific covalent inhibitors using phage display technology.
3. HARNESSING PROXIMITY-INDUCED CHEMISTRY FOR PROTEIN THERAPEUTICS
Proximity-induced chemistry offers reaction selectivity by facilitating covalent bond formation between proximity-enabled reactive groups and specific natural residues within targeted proteins that are in close proximity. This approach eliminates the requirement for targeted protein engineering, UV irradiation, or additional chemical treatments, making it an optimal strategy for site-specific crosslinking of targeted proteins with desired molecules. It has found applications in preparing antibody modifications with payloads in a site-specific manner, as well as the development of covalent protein therapeutics with “infinitely high” affinity against specific protein targets. Taking advantage of binding between antibodies and peptides, a proximity-enabled reactive group has been introduced into antibody-binding peptides for the preparation of antibody-drug conjugates, bispecific antibodies with well-defined structures67,69,76–78. The therapeutic efficacy of these constructs has been successfully validated in vivo. Furthermore, proximity-induced chemistry allows for the formation of irreversible covalent bonds between therapeutic proteins and their targets, thereby overcoming the inherent low affinity between them. This strategy has been instrumental in the development of covalent protein inhibitors and nanobodies, leading to improved therapeutic efficacies in vitro and in vivo86,103,104. Most recently, this chemistry has been utilized to evolve epitope-specific antibodies capable of targeting specific antigenic epitopes, further expanding its application in antibody engineering105.
3.1. Preparation of well-defined antibody drugs using proximity-induced chemistry
Wild-type antibodies typically lack inherent cytotoxic activity. To enhance their therapeutic efficacy, antibodies have been conjugated to various potent payloads, including small molecular drugs, radio agents, immune cell-targeting antibodies, and others106–108. This strategy has led to the development of several FDA-approved ADCs. However, current approaches for preparing ADCs rely on N-hydroxy succinimidyl ester and maleimide conjugation strategies, resulting in heterogenous products with varying DARs106,107.
Recently, proximity-induced chemistry has emerged as a promising method for generating well-defined antibody conjugates. In an initial application of proximity-induced antibody conjugation (pClick) technology, a lysine-based proximity probe called 4-fluorophenyl carbamate lysine (FPheK) was employed to covalently link to a nearby lysine residue within an antibody (Fig. 2B and 4A)67,69. Specifically, the FPheK probe was introduced at the Glu25 position of an affinity peptide known as FB peptide, which possesses a defined binding site within the Fc region of the antibody. Upon binding to the antibody, the FPheK-modified FB peptide undergoes a spontaneous covalent bond formation reaction with a lysine residue on the antibody (Fig. 4A)76,77. This approach successfully enabled site-specific conjugation of fluorescein or toxins to Trastuzumab, an antibody used in the treatment of breast and gastric cancers67,69,109. Moreover, using this method, a prostate-specific membrane antigen (PSMA) inhibitor called DUPA was site-specifically conjugated to an anti-CD3 antibody, resulting in a bispecific antibody that targets both PMSA-positive prostate cancer cells and T cells69. The therapeutic efficacy of this bispecific antibody was demonstrated in a prostate xenograft mouse model. In addition, another proximity-enabled reactive group called phenyl azidoacetate was introduced into a different antibody-binding peptide known as Fc-III50. Upon binding to the antibody, the phenyl azidoacetate substitution at Glu8 of the Fc-III peptide can react with proximal Lys248 of the antibody. Although the reaction is rapid and proceeds to over 50% completion within 1 hour, it does not reach 100% yield, possibly due to the relatively low reactivity between phenyl azidoacetate and lysine residues.
Figure 4.
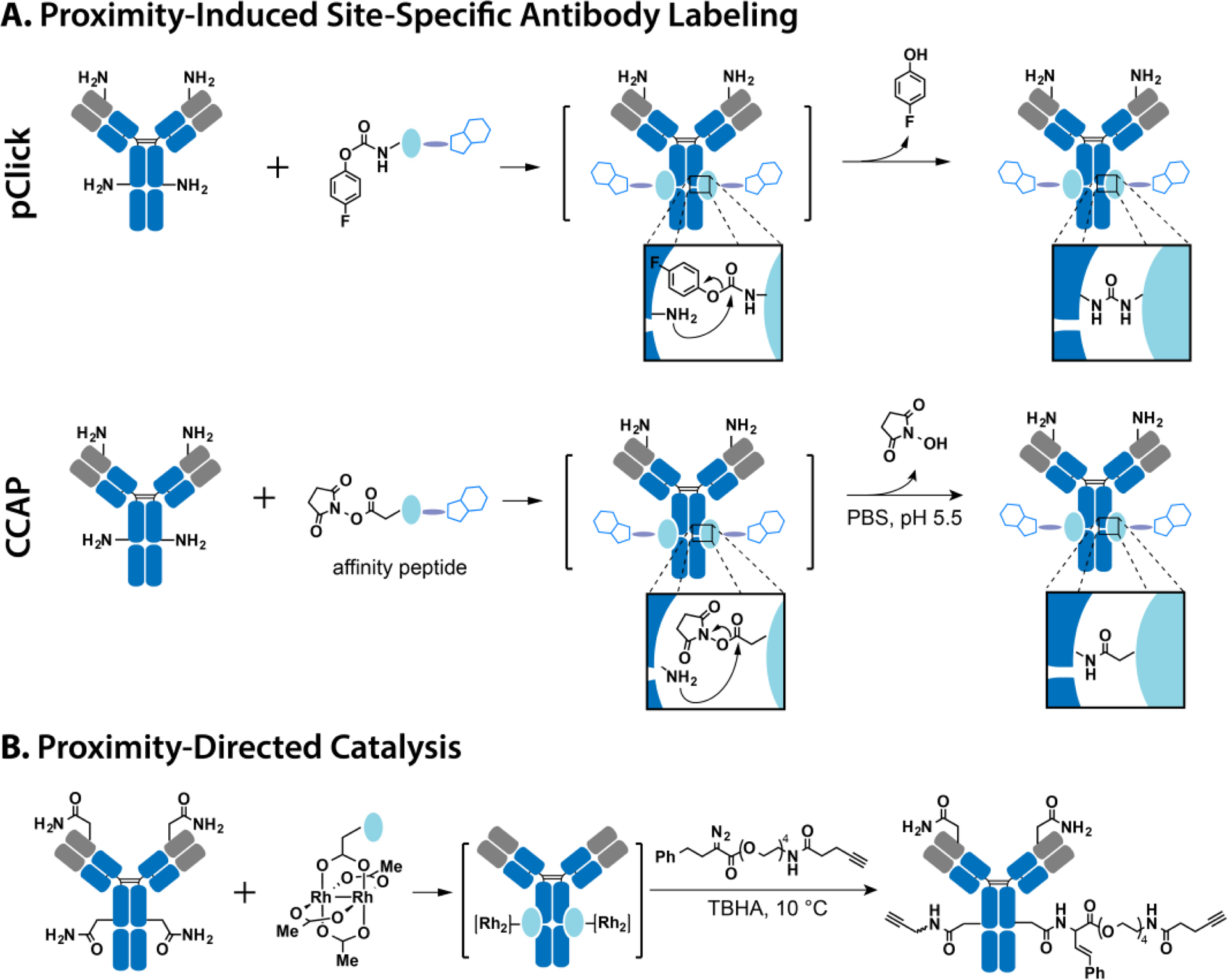
Preparation of site-specific antibody conjugates using proximity-induced chemistry. (A) The introduction of site-specific labeling and precise modifications in the antibody structure is achieved through proximity-induced chemistry by employing FPheK-modified FB protein and the affinity moiety Fc-III peptide. This crosslinking process occurs with nearby lysine residues on the antibody, utilizing pClick and CCAP technologies. (B) Utilizing a Fc-binding dirhodium metallopeptide catalyst, a cross-linking reaction is facilitated between asparagine residues on the antibody and an alkyne-bearing diazo reagent in close proximity. This strategy allows for the site-specific conjugation of functional moieties to the antibody.
The concept of proximity-induced chemistry involves the utilization of chemical moieties that remain inert under physiological conditions but exhibit high reactivity towards specific natural amino acid residues when in close proximity. This reactivity can be modulated by altering the pH or introducing catalysts in proximity. For instance, the NHS ester group demonstrates rapid reactivity with lysine residues at neutral pH, but its reactivity diminishes at lower pH levels110. Therefore, NHS ester has been incorporated into the Fc-III peptide. Under optimal pH conditions of 5.5, the Fc-III peptide modified with NHS ester exhibits selective reactivity against lysine residues while minimizing non-selective reactions (Fig. 4A)76,77. Upon binding to the antibody, it specifically reacts with the Lys248 residue, resulting in 100% labeling efficiency of 95%. This modification of trastuzumab antibody with emtansine demonstrates significant efficacy in treating HER2-positive tumors in vivo studies. Instead of relying on pH modulation, a metallocatalyst capable of facilitating the reaction between carboxamide and diazo groups was employed by bringing it into proximity with the desired asparagine residue of the antibody using an antibody-binding peptide78. An antibody-binding dirhodium metallocatalyst was created by introducing three glutamate residues into the Z domain of protein A via SPPS and subsequently metalating it with a heteroleptic dirhodium complex, Rh2(tfa)(OAc)3. When bound to the antibody, this metallocatalyst catalyzes the derivatization of the nearby Asn312 residue with an alkyne-bearing diazo reagent (Fig. 4B). This method proved to be compatible with human, pig, rabbit, and dog antibodies, demonstrating its broad applicability and compatibility.
3.2. Covalent Protein Therapeutics based on Proximity-induced chemistry
Previously, the concept of utilizing a proximity-enabled reactive group to selectively react with a natural residue within the target protein’s binding site has been successfully employed in the design of covalent small molecule drugs, offering advantages such as enhanced target selectivity, prolonged target engagement, the ability to overcome resistance mutations, and potentially increased potency111. More recently, this approach has been extended to the realm of protein-based therapeutics.
The first well-characterized covalent protein drug in both in vitro and in vivo is a human programmed cell death protein-1 (PD-1) fused with a proximity-enabled reactive ncAA called fluorosulfate-tyrosine (FSY, Fig. 2B and 5A)103. PD-1 is a transmembrane receptor that plays a crucial role in regulating T cell function112,113. Among the complex network of immune responses, programmed death-ligand 1 (PD-L1) emerges as a key ligand for PD-1, often found in an overexpressed state in various tumor types114–116. The affinity between PD-1 and PD-L1, with a Kd value of approximately 8.2 μM117,118. Incorporating FSY into PD-1 using the Genetic Code Expansion technology offers a fundamentally different mechanism compared to antibody blockage. PD-1 with FSY irreversibly forms a covalent bond with the histidine residue of PD-L1 on cancer cells upon binding. Once the bond is formed, therapeutic PD-1 is no longer able to dissociate from the PD-L1 target (Fig. 5A). Under normal circumstances, the interaction between T cell PD-1 and cancer cell PD-L1 inhibits T cell proliferation and activity. However, the irreversible binding of soluble PD-1 protein with FSY to PD-L1 effectively disrupts this interaction. As a result, PD-1 modified with FSY has been shown to significantly enhance the functional activities of human T cells and chimeric antigen receptor T cells in laboratory studies, demonstrating comparable efficacy to atezolizumab, an FDA-approved anti-PD-L1 antibody103. On the other hand, the noncovalent wild-type PD-1 did not exhibit similar effects. To further evaluate its therapeutic potential, PD-1 modified with FSY was administered in xenograft mouse models that were immune-humanized using either human peripheral blood mononuclear cells, human CAR-T cells, or a systematic bone marrow-liver-thymus humanization approach. In all three models, the PD-1 mutant with FSY showed potent inhibition of tumor growth, surpassing the therapeutic effect of PD-1 alone. These findings indicate the significant promise of covalent protein drugs and their potential to revolutionize cancer treatment.
Figure 5.
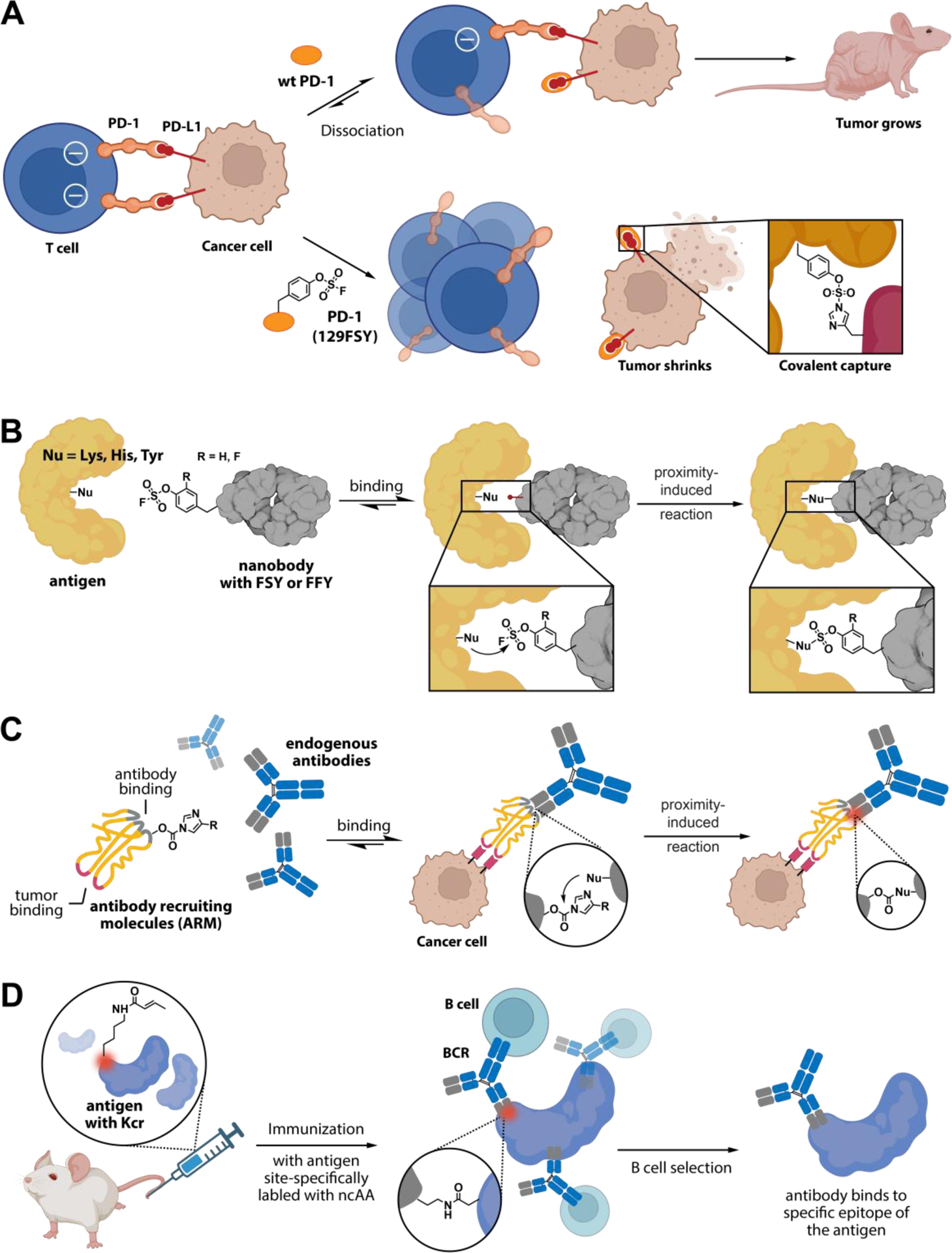
Preparation of covalent protein drugs using proximity-induced chemistry. (A) FSY-modified PD-1 covalently captures PD-L1 on tumor surfaces, leading to the restoration of T cell responses and the inhibition of tumor growth. (B) Antibodies with proximity-enabled reactive groups demonstrates stable crosslinking with the targeted antigen in close proximity, leading to superior efficacy in inhibiting the growth of tumor cells. (C) Proximity-enabled reactive moieties are integrated into antibody recruiting molecules, featuring target and antibody binding domains, to attract immune cells to tumor sites. (D) Immunization of mice with antigens with site-specific Kcr modification results in focused and enhanced antibody responses, specifically targeting the desired epitopes.
Creating antibodies with infinite affinity that possess excellent specificity against antigens without dissociation is an intriguing goal. This is particularly interesting because targeted therapeutic antibody drugs typically require high specificity and prolonged targeting to enhance efficacy. Previous approaches involved modifying antigens with proximity-enabled reactive groups to form covalent bonds with corresponding antibodies119. Expanding this concept in vivo, it is highly desirable to modify antibodies with proximity-enabled reactive groups instead of antigens due to the challenges associated with selectively labeling protein antigens in vivo. To address this, proximity-enabled reactive ncAAs have been incorporated into nanobodies using Genetic Code Expansion technology48,86. The resulting modified nanobodies can establish irreversible bonds with targeted protein antigens on cells, offering a potential strategy to enhance affinity, counteract mutational resistance, and improve pharmacodynamic properties. The global outbreak of COVID-19, caused by the novel coronavirus SARS-CoV-2, has emphasized the importance of developing innovative strategies to combat virus infection120. A crucial step in the viral cell entry process involves the binding of the viral spike protein to the human cellular angiotensin-converting enzyme 2 (ACE2) receptor121. To block the viral binding to the human ACE2 receptor, nanobodies incorporating a proximity-enabled reactive ncAA, fluorine-substituted fluorosulfate-tyrosine (FFY), have been developed86. Upon binding to the viral spike protein, these nanobodies with FFY initiate a proximity-induced covalent reaction between the fluorosulfate group of FFY and natural residues of ACE2. These covalent nanobodies targeting ACE2 demonstrate exceptional neutralization capabilities against various strains of SARS-CoV-2, surpassing the efficacy of their noncovalent wild-type counterparts. The covalent nanobodies exhibit potent neutralization activity against not only the wild-type SARS-CoV-2 strain (with a 41-fold increase in potency) but also its variants, including Alpha (23-fold), Delta (39-fold), Epsilon (38-fold), Lambda (24-fold), and Omicron (8- to 10-fold)86. Importantly, the superior potency of these covalent nanobodies effectively impedes viral infection by blocking the binding of SARS-CoV-2 to the ACE2 receptor. Utilizing a similar methodology, researchers have developed a covalent nanobody with a radionuclide that effectively targets HER2 for the treatment of HER2-positive breast cancer104. By incorporating a proximity-enabled reactive ncAA, FSY, into the nanobody, it acquires a high affinity for its target protein (Fig. 5B). Upon binding to HER2, the nanobody establishes a covalent linkage with the Lys150 residue of the HER2 receptor, resulting in irreversible cross-linking both in vitro and in vivo. Remarkably, the resulting covalent nanobody demonstrates significantly enhanced tumor accumulation in vivo, surpassing the accumulation of the wild-type nanobody. Building upon this success, researchers have also generated a radiolabeled covalent anti-HER2 nanobody. The 225Ac-labeled covalent nanobody exhibits superior efficacy in inhibiting the growth of HER2-expressing tumors in mice compared to the wild-type nanobody104. Importantly, this enhanced therapeutic agent demonstrates minimal toxicity in vital tissues. These findings underscore the immense potential of integrating proximity-enabled reactive ncAAs into nanobodies, opening up new avenues for targeted cancer therapy. Additionally, they provide valuable insights into the development of safer and more effective treatments.
Instead of modifying antibodies themselves, an approach involves introducing proximity-enabled reactive moieties into bifunctional molecules housing both a target binding domain (TBD) and an antibody binding domain (ABD, Fig. 5C)122–124. These hybrid constructs, termed antibody recruiting molecules (ARMs), possess the ability to form covalent connections with antibodies present in human serum, thereby enlisting them to target cancer cells. Within the tumor microenvironment, this complex interaction is expected to eliminate cancer cells through mechanisms such as antibody-dependent cellular cytotoxicity (ADCC) or antibody-dependent cellular phagocytosis (ADCP). Illustrating this concept with an initial example, an acyl imidazole was strategically positioned within an ARM featuring a dinitrophenyl moiety as the ABD122. This configuration enabled the recruitment of anti-antidinitrophenyl (DNP) antibodies, in addition to incorporating glutamate urea ligands known for binding to Prostate Specific Membrane Antigen (PSMA). The ARM, once linked to anti-DNP antibodies, underwent an interaction where the acyl imidazole was targeted by Lys59 of the anti-DNP antibody. This event facilitated the formation of a complex between the ARM and the anti-DNP antibody, offering a potential therapeutic avenue for treating PSMA-positive cancer cells. Despite achieving success in vitro experiments, further studies are needed to ascertain the efficacy of this approach in vivo.
The mouse hybridoma technology has established itself as a prominent and widely utilized method for generating monoclonal antibodies with high affinities125–127. However, selectively targeting a specific conformational epitope in a whole antigen in vivo remains a challenge. To overcome this challenge, the Genetic Code Expansion technology has been employed to genetically incorporate N-acryloyl-lysine (AcrK) or N-crotonyl-lysine (Kcr), both possessing cross-linking activities with natural protein residues, into specific epitopes of antigens (Fig. 2B)105. Upon binding to the B cell receptors (BCRs), the proximity-enabled reactive ncAAs, AcrK, and Kcr on the antigen exhibit an impressive capability to form covalent bonds with Lys or Cys residues situated in the binding pocket of BCRs (Fig. 5D). Consequently, the affinity between antigens and BCRs is significantly enhanced, resulting in a tighter and more robust binding. By immunizing mice with antigens containing AcrK or Kcr, researchers have successfully elicited and enriched antibody responses that selectively target the specific epitope with ncAA modification. By incorporating AcrK or Kcr into human interleukin-1β (hIL-1β) and Phaeodactylum tricornutum nucleoside triphosphate transporter 2 (PtNTT2) peptide, two antigen mutants have demonstrated the remarkable ability to induce robust epitope-directed antibody responses in mice105.
Conclusion
Protein therapeutics have revolutionized the medical field, offering promising treatment options for complex diseases. Over time, they have significantly advanced and contributed to the growing number of FDA-approved drugs1,5,6. However, despite their transformative impact, protein drugs encounter certain limitations compared to small-molecule drugs. These limitations encompass factors such as low stability, relatively low cytotoxicity, high manufacturing costs, potential immunogenicity, low cellular permeability, and challenges associated with targeting undruggable proteins. To overcome the challenges posed by weak affinity interactions between binding partners and undruggable protein targets, proximity-induced chemistry has emerged as a powerful tool in the development of covalent protein therapeutics with infinitely high affinity against their targets86,103,104. This innovative approach facilitates chemical transformations between a proximity-enabled reactive group and specific natural residues within targeted proteins that are in close proximity. By introducing proximity-induced chemistry, covalent protein therapeutics with reactive groups demonstrate enhanced binding affinity and therapeutic efficacy compared to their wild-type counterparts. Furthermore, proximity-induced chemistry has been utilized for site-specific protein labeling, enabling the introduction of diverse functional payloads to antibodies67,69,76–78,128. These modified antibody conjugates exhibit improved stability and cytotoxicity both in in vitro assays and in vivo mouse models. While these successes are notable, there is still much to explore regarding the mechanistic perspective of enhanced efficacy of covalent drugs and the additional benefits they offer, such as targeting undruggable surfaces, potentially increased selectivity, and reduced immunogenicity. Comprehensive studies on covalent protein therapeutics and their advantages in diverse organisms will unlock the full potential of proximity-induced chemistry in the development of innovative protein therapeutics.
The development of protein therapeutics using proximity-induced chemistry relies on the creation of novel proximity-enabled reactive groups and the implementation of effective strategies to incorporate these groups into proteins. Currently, more than 20 proximity-enabled reactive groups have been developed, enabling the targeting of a wide range of natural residues on proteins. When designing proximity-enabled reactive groups, striking a balance between reactivity and stability is crucial. Achieving a high reactivity is generally desirable for efficient proximity-induced crosslinking. However, higher reactivity can lead to reduced specificity, potentially resulting in random reactions with undesired residues. Additionally, the protein microenvironment strongly influences the reactivity between proximity-enabled reactive groups and natural protein residues. Consequently, the screening of proximity-enabled reactive groups with varying reactivity becomes a time-consuming and labor-intensive process in order to identify the most suitable group for a specific target. To expedite this process, computational approaches can be employed to enhance efficiency and streamline the selection of optimal proximity-enabled reactive groups for a specific target. To introduce proximity-enabled reactive groups into proteins in a site-specific manner, solid-phase peptide synthesis and Genetic Code Expansion technology have been widely employed for covalent peptide and protein preparation, respectively. Although Genetic Code Expansion technology enables the incorporation of proximity-enabled reactive groups, it may lead to lower protein yields compared to wild-type proteins. Therefore, further synthetic biology strategies are necessary to address these challenges and enhance protein yields in proximity-induced chemistry.
In conclusion, the advancement of protein therapeutics through proximity-induced chemistry relies on the synergistic contributions of chemistry, synthetic biology, and protein engineering. Initial studies on protein therapeutics based on proximity-induced chemistry have demonstrated promising therapeutic efficacy. Further evolution of protein therapy using proximity-induced chemistry holds great potential for the treatment of cancer and other diseases.
Supplementary Material
Highlights.
The molecular mechanism behind proximity-induced chemistry is thoroughly explored.
The proximity-enabled reactive groups are classified and summarized.
Approaches to introduce proximity-enabled groups into proteins are summarized.
The development of protein drugs by proximity-induced chemistry is discussed.
Significance.
Protein therapeutics have revolutionized the medical field, offering promising treatment options for complex diseases. Thriving protein therapeutics have largely contributed to a growing number of FDA-approved of over 200 therapeutic proteins. In this context, we made a comprehensive summary of mechanisms and achievements of proximity-induced chemistry on protein therapeutics to advance the field and guide future development. We first introduced the concept of proximity-induced chemistry, discussed the molecular mechanism behind proximity-induced chemistry, function groups that are able to carry out this reaction, and the methodology to incorporate reactive groups into proteins. Then we focused on the transition of this chemistry to boost the development of protein therapeutics and their therapeutic efficacy in pre-clinical and clinical studies. We summarized current achievements of proximity-induced chemistry as a novel strategy to develop covalent protein therapeutics with infinitely high affinity against their targets and site-specifically modified therapeutic protein with potent payload without requiring extensive target protein engineering. This review will be useful as a toolbox for the modification of native antibodies to generate more potent homogenous antibody conjugates and develop new antibody conjugation methods without requiring protein engineering of native antibodies. Moreover, it will open a new era of covalent protein drugs that drastically improve the therapeutic outcome of these drugs that suffer from low target affinity or stability and will boost the development of protein drugs that target undruggable targets.
Acknowledgments
This work was supported by the Cancer Prevention Research Institute of Texas (CPRIT RR170014 to H.X.), NIH (R01-CA277838, R35-GM133706, R21-CA255894, and R01-AI165079 to H.X.), the Robert A. Welch Foundation (C-1970 to H.X.), US Department of Defense (HT9425-23-1-0494 and W81XWH-21-1-0789 to H.X.), the John S. Dunn Foundation Collaborative Research Award (to H.X.), and the Hamill Innovation Award (to H.X.). H.X. is a Cancer Prevention & Research Institute of Texas (CPRIT) scholar in cancer research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare no competing interests.
Reference:
- 1.Leader B, Baca QJ, and Golan DE (2008). Protein therapeutics: a summary and pharmacological classification. Nat Rev Drug Discov 7, 21–39. 10.1038/nrd2399. [DOI] [PubMed] [Google Scholar]
- 2.Ganeshan K, and Chawla A (2014). Metabolic Regulation of Immune Responses. Annual Review of Immunology 32, 609–634. 10.1146/annurev-immunol-032713-120236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen B, Sun Y, Niu J, Jarugumilli GK, and Wu X (2018). Protein Lipidation in Cell Signaling and Diseases: Function, Regulation, and Therapeutic Opportunities. Cell Chemical Biology 25, 817–831. 10.1016/j.chembiol.2018.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hotamisligil GS, and Davis RJ (2016). Cell Signaling and Stress Responses. Cold Spring Harb Perspect Biol 8, a006072. 10.1101/cshperspect.a006072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ebrahimi SB, and Samanta D (2023). Engineering protein-based therapeutics through structural and chemical design. Nat Commun 14, 2411. 10.1038/s41467-023-38039-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dumontet C, Reichert JM, Senter PD, Lambert JM, and Beck A (2023). Antibody–drug conjugates come of age in oncology. Nat Rev Drug Discov, 1–21. 10.1038/s41573-023-00709-2. [DOI] [PubMed] [Google Scholar]
- 7.Wang L, Wang N, Zhang W, Cheng X, Yan Z, Shao G, Wang X, Wang R, and Fu C (2022). Therapeutic peptides: current applications and future directions. Sig Transduct Target Ther 7, 1–27. 10.1038/s41392-022-00904-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tong JTW, Harris PWR, Brimble MA, and Kavianinia I (2021). An Insight into FDA Approved Antibody-Drug Conjugates for Cancer Therapy. Molecules 26, 5847. 10.3390/molecules26195847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Verdine GL, and Walensky LD (2007). The Challenge of Drugging Undruggable Targets in Cancer: Lessons Learned from Targeting BCL-2 Family Members. Clinical Cancer Research 13, 7264–7270. 10.1158/1078-0432.CCR-07-2184. [DOI] [PubMed] [Google Scholar]
- 10.Kintzing JR, Filsinger Interrante MV, and Cochran JR (2016). Emerging Strategies for Developing Next-Generation Protein Therapeutics for Cancer Treatment. Trends in Pharmacological Sciences 37, 993–1008. 10.1016/j.tips.2016.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boutureira O, and Bernardes GJL (2015). Advances in Chemical Protein Modification. Chem. Rev 115, 2174–2195. 10.1021/cr500399p. [DOI] [PubMed] [Google Scholar]
- 12.Brown DG, and Wobst HJ (2021). A Decade of FDA-Approved Drugs (2010–2019): Trends and Future Directions. J. Med. Chem 64, 2312–2338. 10.1021/acs.jmedchem.0c01516. [DOI] [PubMed] [Google Scholar]
- 13.do Pazo C, Nawaz K, and Webster RM (2021). The oncology market for antibody–drug conjugates. Nature Reviews Drug Discovery 20, 583–584. 10.1038/d41573-021-00054-2. [DOI] [PubMed] [Google Scholar]
- 14.Junutula JR, Raab H, Clark S, Bhakta S, Leipold DD, Weir S, Chen Y, Simpson M, Tsai SP, Dennis MS, et al. (2008). Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nat Biotechnol 26, 925–932. 10.1038/nbt.1480. [DOI] [PubMed] [Google Scholar]
- 15.Hamblett KJ, Senter PD, Chace DF, Sun MMC, Lenox J, Cerveny CG, Kissler KM, Bernhardt SX, Kopcha AK, Zabinski RF, et al. (2004). Effects of Drug Loading on the Antitumor Activity of a Monoclonal Antibody Drug Conjugate. Clinical Cancer Research 10, 7063–7070. 10.1158/1078-0432.CCR-04-0789. [DOI] [PubMed] [Google Scholar]
- 16.Bertozzi CR (2011). A Decade of Bioorthogonal Chemistry. Acc. Chem. Res 44, 651–653. 10.1021/ar200193f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sletten EM, and Bertozzi CR (2009). Bioorthogonal Chemistry: Fishing for Selectivity in a Sea of Functionality. Angewandte Chemie International Edition 48, 6974–6998. 10.1002/anie.200900942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mahal LK, Yarema KJ, and Bertozzi CR (1997). Engineering Chemical Reactivity on Cell Surfaces Through Oligosaccharide Biosynthesis. Science 276, 1125–1128. 10.1126/science.276.5315.1125. [DOI] [PubMed] [Google Scholar]
- 19.Zhang Z, Smith BAC, Wang L, Brock A, Cho C, and Schultz PG (2003). A New Strategy for the Site-Specific Modification of Proteins in Vivo. Biochemistry 42, 6735–6746. 10.1021/bi0300231. [DOI] [PubMed] [Google Scholar]
- 20.Staudinger H, and Meyer J (1919). Über neue organische Phosphorverbindungen III. Phosphinmethylenderivate und Phosphinimine. Helvetica Chimica Acta 2, 635–646. 10.1002/hlca.19190020164. [DOI] [Google Scholar]
- 21.Saxon E, and Bertozzi CR (2000). Cell Surface Engineering by a Modified Staudinger Reaction. Science 287, 2007–2010. 10.1126/science.287.5460.2007. [DOI] [PubMed] [Google Scholar]
- 22.Tornøe CW, Christensen C, and Meldal M (2002). Peptidotriazoles on Solid Phase: [1,2,3]-Triazoles by Regiospecific Copper(I)-Catalyzed 1,3-Dipolar Cycloadditions of Terminal Alkynes to Azides. J. Org. Chem 67, 3057–3064. 10.1021/jo011148j. [DOI] [PubMed] [Google Scholar]
- 23.Rostovtsev VV, Green LG, Fokin VV, and Sharpless KB (2002). A Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective “Ligation” of Azides and Terminal Alkynes. Angewandte Chemie International Edition 41, 2596–2599. 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 24.Agard NJ, Baskin JM, Prescher JA, Lo A, and Bertozzi CR (2006). A Comparative Study of Bioorthogonal Reactions with Azides. ACS Chem. Biol 1, 644–648. 10.1021/cb6003228. [DOI] [PubMed] [Google Scholar]
- 25.Agard NJ, Prescher JA, and Bertozzi CR (2004). A Strain-Promoted [3 + 2] Azide−Alkyne Cycloaddition for Covalent Modification of Biomolecules in Living Systems. J. Am. Chem. Soc 126, 15046–15047. 10.1021/ja044996f. [DOI] [PubMed] [Google Scholar]
- 26.Blackman ML, Royzen M, and Fox JM (2008). Tetrazine Ligation: Fast Bioconjugation Based on Inverse-Electron-Demand Diels−Alder Reactivity. J. Am. Chem. Soc 130, 13518–13519. 10.1021/ja8053805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Devaraj NK, Weissleder R, and Hilderbrand SA (2008). Tetrazine-Based Cycloadditions: Application to Pretargeted Live Cell Imaging. Bioconjugate Chem. 19, 2297–2299. 10.1021/bc8004446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Patterson DM, Nazarova LA, and Prescher JA (2014). Finding the Right (Bioorthogonal) Chemistry. ACS Chem. Biol 9, 592–605. 10.1021/cb400828a. [DOI] [PubMed] [Google Scholar]
- 29.Stanton BZ, Chory EJ, and Crabtree GR (2018). Chemically induced proximity in biology and medicine. Science 359, eaao5902. 10.1126/science.aao5902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Karplus M (2000). Aspects of Protein Reaction Dynamics: Deviations from Simple Behavior. J. Phys. Chem. B 104, 11–27. 10.1021/jp993555t. [DOI] [Google Scholar]
- 31.Maly DJ, Allen JA, and Shokat KM (2004). A Mechanism-Based Cross-Linker for the Identification of Kinase−Substrate Pairs. J. Am. Chem. Soc 126, 9160–9161. 10.1021/ja048659i. [DOI] [PubMed] [Google Scholar]
- 32.Mullard A (2023). Proximity-inducing drugs get closer. Nature Reviews Drug Discovery 22, 254–257. 10.1038/d41573-023-00044-6. [DOI] [PubMed] [Google Scholar]
- 33.Kannt A, and Đikić I (2021). Expanding the arsenal of E3 ubiquitin ligases for proximity-induced protein degradation. Cell Chemical Biology 28, 1014–1031. 10.1016/j.chembiol.2021.04.007. [DOI] [PubMed] [Google Scholar]
- 34.Ng CSC, and Banik SM (2022). Recent advances in induced proximity modalities. Current Opinion in Chemical Biology 67, 102107. 10.1016/j.cbpa.2021.102107. [DOI] [PubMed] [Google Scholar]
- 35.Lins L, Thomas A, and Brasseur R (2003). Analysis of accessible surface of residues in proteins. Protein Science 12, 1406–1417. 10.1110/ps.0304803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pace NJ, and Weerapana E (2013). Diverse Functional Roles of Reactive Cysteines. ACS Chem. Biol 8, 283–296. 10.1021/cb3005269. [DOI] [PubMed] [Google Scholar]
- 37.Isom DG, Castañeda CA, Cannon BR, and García-Moreno E,B (2011). Large shifts in pKa values of lysine residues buried inside a protein. Proceedings of the National Academy of Sciences 108, 5260–5265. 10.1073/pnas.1010750108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Harms MJ, Castañeda CA, Schlessman JL, Sue GR, Isom DG, Cannon BR, and García-Moreno EB (2009). The pK(a) values of acidic and basic residues buried at the same internal location in a protein are governed by different factors. J Mol Biol 389, 34–47. 10.1016/j.jmb.2009.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xiang Z, Ren H, Hu YS, Coin I, Wei J, Cang H, and Wang L (2013). Adding an unnatural covalent bond to proteins through proximity-enhanced bioreactivity. Nat Methods 10, 885–888. 10.1038/nmeth.2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kobayashi T, Hoppmann C, Yang B, and Wang L (2016). Using Protein-Confined Proximity To Determine Chemical Reactivity. J. Am. Chem. Soc 138, 14832–14835. 10.1021/jacs.6b08656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Spokoyny AM, Zou Y, Ling JJ, Yu H, Lin Y-S, and Pentelute BL (2013). A Perfluoroaryl-Cysteine SNAr Chemistry Approach to Unprotected Peptide Stapling. J. Am. Chem. Soc 135, 5946–5949. 10.1021/ja400119t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hoppmann C, and Wang L (2019). Genetically encoding photoswitchable click amino acids for general optical control of conformation and function of proteins. Methods Enzymol 624, 249–264. 10.1016/bs.mie.2019.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hoppmann C, Maslennikov I, Choe S, and Wang L (2015). In Situ Formation of an Azo Bridge on Proteins Controllable by Visible Light. J. Am. Chem. Soc 137, 11218–11221. 10.1021/jacs.5b06234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chrominski M, Ziemkiewicz K, Kowalska J, and Jemielity J (2022). Introducing SuFNucs: Sulfamoyl-Fluoride-Functionalized Nucleosides That Undergo Sulfur Fluoride Exchange Reaction. Org. Lett 24, 4977–4981. 10.1021/acs.orglett.2c02034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dong J, Krasnova L, Finn MG, and Sharpless KB (2014). Sulfur(VI) Fluoride Exchange (SuFEx): Another Good Reaction for Click Chemistry. Angewandte Chemie International Edition 53, 9430–9448. 10.1002/anie.201309399. [DOI] [PubMed] [Google Scholar]
- 46.Hendrick CE, Jorgensen JR, Chaudhry C, Strambeanu II, Brazeau J-F, Schiffer J, Shi Z, Venable JD, and Wolkenberg SE (2022). Direct-to-Biology Accelerates PROTAC Synthesis and the Evaluation of Linker Effects on Permeability and Degradation. ACS Med. Chem. Lett 13, 1182–1190. 10.1021/acsmedchemlett.2c00124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang N, Yang B, Fu C, Zhu H, Zheng F, Kobayashi T, Liu J, Li S, Ma C, Wang PG, et al. (2018). Genetically Encoding Fluorosulfate-l-tyrosine To React with Lysine, Histidine, and Tyrosine via SuFEx in Proteins in Vivo. J. Am. Chem. Soc 140, 4995–4999. 10.1021/jacs.8b01087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu J, Cao L, Klauser PC, Cheng R, Berdan VY, Sun W, Wang N, Ghelichkhani F, Yu B, Rozovsky S, et al. (2021). A Genetically Encoded Fluorosulfonyloxybenzoyl-l-lysine for Expansive Covalent Bonding of Proteins via SuFEx Chemistry. J. Am. Chem. Soc 143, 10341–10351. 10.1021/jacs.1c04259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xuan W, Shao S, and Schultz PG (2017). Protein Crosslinking by Genetically Encoded Noncanonical Amino Acids with Reactive Aryl Carbamate Side Chains. Angewandte Chemie International Edition 56, 5096–5100. 10.1002/anie.201611841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yuan D, Zhang Y, Lim KH, Leung SKP, Yang X, Liang Y, Lau WCY, Chow KT, and Xia J (2022). Site-Selective Lysine Acetylation of Human Immunoglobulin G for Immunoliposomes and Bispecific Antibody Complexes. J. Am. Chem. Soc 144, 18494–18503. 10.1021/jacs.2c07594. [DOI] [PubMed] [Google Scholar]
- 51.Xiang Z, Lacey VK, Ren H, Xu J, Burban DJ, Jennings PA, and Wang L (2014). Proximity-Enabled Protein Crosslinking through Genetically Encoding Haloalkane Unnatural Amino Acids. Angewandte Chemie International Edition 53, 2190–2193. 10.1002/anie.201308794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hoppmann C, Lacey VK, Louie GV, Wei J, Noel JP, and Wang L (2014). Genetically Encoding Photoswitchable Click Amino Acids in Escherichia coli and Mammalian Cells. Angewandte Chemie International Edition 53, 3932–3936. 10.1002/anie.201400001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Los GV, Encell LP, McDougall MG, Hartzell DD, Karassina N, Zimprich C, Wood MG, Learish R, Ohana RF, Urh M, et al. (2008). HaloTag: A Novel Protein Labeling Technology for Cell Imaging and Protein Analysis. ACS Chem. Biol 3, 373–382. 10.1021/cb800025k. [DOI] [PubMed] [Google Scholar]
- 54.England CG, Luo H, and Cai W (2015). HaloTag Technology: A Versatile Platform for Biomedical Applications. Bioconjugate Chem. 26, 975–986. 10.1021/acs.bioconjchem.5b00191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Powers JC, Asgian JL, Ekici ÖD, and James KE (2002). Irreversible Inhibitors of Serine, Cysteine, and Threonine Proteases. Chem. Rev 102, 4639–4750. 10.1021/cr010182v. [DOI] [PubMed] [Google Scholar]
- 56.Santos MMM, and Moreira R Michael Acceptors as Cysteine Protease Inhibitors. Mini-Reviews in Medicinal Chemistry 7, 1040–1050. [DOI] [PubMed] [Google Scholar]
- 57.Reddick JJ, Cheng J, and Roush WR (2003). Relative Rates of Michael Reactions of 2’-(Phenethyl)thiol with Vinyl Sulfones, Vinyl Sulfonate Esters, and Vinyl Sulfonamides Relevant to Vinyl Sulfonyl Cysteine Protease Inhibitors. Org. Lett 5, 1967–1970. 10.1021/ol034555l. [DOI] [PubMed] [Google Scholar]
- 58.Furman JL, Kang M, Choi S, Cao Y, Wold ED, Sun SB, Smider VV, Schultz PG, and Kim CH (2014). A Genetically Encoded aza-Michael Acceptor for Covalent Cross-Linking of Protein–Receptor Complexes. J. Am. Chem. Soc 136, 8411–8417. 10.1021/ja502851h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Edman P, Högfeldt E, Sillén LG, and Kinell P-O (1950). Method for Determination of the Amino Acid Sequence in Peptides. Acta Chem. Scand 4, 283–293. 10.3891/acta.chem.scand.04-0283. [DOI] [Google Scholar]
- 60.Li JC, Nastertorabi F, Xuan W, Han GW, Stevens RC, and Schultz PG (2019). A Single Reactive Noncanonical Amino Acid Is Able to Dramatically Stabilize Protein Structure. ACS Chem. Biol 14, 1150–1153. 10.1021/acschembio.9b00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Genetic Incorporation of a Reactive Isothiocyanate Group into Proteins - Xuan - 2016 - Angewandte Chemie International Edition - Wiley Online Library. https://onlinelibrary.wiley.com/doi/10.1002/anie.201604891. [DOI] [PubMed]
- 62.Wang L, Xie J, and Schultz PG (2006). Expanding the Genetic Code. Annual Review of Biophysics and Biomolecular Structure 35, 225–249. 10.1146/annurev.biophys.35.101105.121507. [DOI] [PubMed] [Google Scholar]
- 63.Liu CC, and Schultz PG (2010). Adding New Chemistries to the Genetic Code. Annual Review of Biochemistry 79, 413–444. 10.1146/annurev.biochem.052308.105824. [DOI] [PubMed] [Google Scholar]
- 64.Chin JW (2017). Expanding and reprogramming the genetic code. Nature 550, 53–60. 10.1038/nature24031. [DOI] [PubMed] [Google Scholar]
- 65.Xiao H, and Schultz PG (2016). At the Interface of Chemical and Biological Synthesis: An Expanded Genetic Code. Cold Spring Harb Perspect Biol 8, a023945. 10.1101/cshperspect.a023945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chen X-H, Xiang Z, Hu YS, Lacey VK, Cang H, and Wang L (2014). Genetically Encoding an Electrophilic Amino Acid for Protein Stapling and Covalent Binding to Native Receptors. ACS Chem. Biol 9, 1956–1961. 10.1021/cb500453a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yu C, Tang J, Loredo A, Chen Y, Jung SY, Jain A, Gordon A, and Xiao H (2018). Proximity-Induced Site-Specific Antibody Conjugation. Bioconjugate Chem. 29, 3522–3526. 10.1021/acs.bioconjchem.8b00680. [DOI] [PubMed] [Google Scholar]
- 68.Han L, Chen J, Ding K, Zong H, Xie Y, Jiang H, Zhang B, Lu H, Yin W, Gilly J, et al. (2017). Efficient generation of bispecific IgG antibodies by split intein mediated protein trans-splicing system. Sci Rep 7, 8360. 10.1038/s41598-017-08641-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cao YJ, Yu C, Wu K-L, Wang X, Liu D, Tian Z, Zhao L, Qi X, Loredo A, Chung A, et al. (2021). Synthesis of precision antibody conjugates using proximity-induced chemistry. Theranostics 11, 9107–9117. 10.7150/thno.62444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Muir TW, Sondhi D, and Cole PA (1998). Expressed protein ligation: A general method for protein engineering. Proceedings of the National Academy of Sciences 95, 6705–6710. 10.1073/pnas.95.12.6705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Stebbins JL, Santelli E, Feng Y, De SK, Purves A, Motamedchaboki K, Wu B, Ronai ZA, Liddington RC, and Pellecchia M (2013). Structure-Based Design of Covalent Siah Inhibitors. Chemistry & Biology 20, 973–982. 10.1016/j.chembiol.2013.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hoppmann C, and Wang L (2016). Proximity-enabled bioreactivity to generate covalent peptide inhibitors of p53–Mdm4. Chem. Commun 52, 5140–5143. 10.1039/C6CC01226D. [DOI] [PubMed] [Google Scholar]
- 73.Juillerat A, Gronemeyer T, Keppler A, Gendreizig S, Pick H, Vogel H, and Johnsson K (2003). Directed Evolution of O6-Alkylguanine-DNA Alkyltransferase for Efficient Labeling of Fusion Proteins with Small Molecules In Vivo. Chemistry & Biology 10, 313–317. 10.1016/S1074-5521(03)00068-1. [DOI] [PubMed] [Google Scholar]
- 74.Mollwitz B, Brunk E, Schmitt S, Pojer F, Bannwarth M, Schiltz M, Rothlisberger U, and Johnsson K (2012). Directed Evolution of the Suicide Protein O6-Alkylguanine-DNA Alkyltransferase for Increased Reactivity Results in an Alkylated Protein with Exceptional Stability. Biochemistry 51, 986–994. 10.1021/bi2016537. [DOI] [PubMed] [Google Scholar]
- 75.Gautier A, Juillerat A, Heinis C, Corrêa IR, Kindermann M, Beaufils F, and Johnsson K (2008). An Engineered Protein Tag for Multiprotein Labeling in Living Cells. Chemistry & Biology 15, 128–136. 10.1016/j.chembiol.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 76.Kishimoto S, Nakashimada Y, Yokota R, Hatanaka T, Adachi M, and Ito Y (2019). Site-Specific Chemical Conjugation of Antibodies by Using Affinity Peptide for the Development of Therapeutic Antibody Format. Bioconjugate Chem. 30, 698–702. 10.1021/acs.bioconjchem.8b00865. [DOI] [PubMed] [Google Scholar]
- 77.Yamada K, Shikida N, Shimbo K, Ito Y, Khedri Z, Matsuda Y, and Mendelsohn BA (2019). AJICAP: Affinity Peptide Mediated Regiodivergent Functionalization of Native Antibodies. Angewandte Chemie International Edition 58, 5592–5597. 10.1002/anie.201814215. [DOI] [PubMed] [Google Scholar]
- 78.Ohata J, and Ball ZT (2017). A Hexa-rhodium Metallopeptide Catalyst for Site-Specific Functionalization of Natural Antibodies. J. Am. Chem. Soc 139, 12617–12622. 10.1021/jacs.7b06428. [DOI] [PubMed] [Google Scholar]
- 79.Wang N, and Wang L (2022). Genetically encoding latent bioreactive amino acids and the development of covalent protein drugs. Current Opinion in Chemical Biology 66, 102106. 10.1016/j.cbpa.2021.102106. [DOI] [PubMed] [Google Scholar]
- 80.Liu J, Yang B, and Wang L (2023). Residue selective crosslinking of proteins through photoactivatable or proximity-enabled reactivity. Current Opinion in Chemical Biology 74, 102285. 10.1016/j.cbpa.2023.102285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Holm L, Moody P, and Howarth M (2009). Electrophilic Affibodies Forming Covalent Bonds to Protein Targets*. Journal of Biological Chemistry 284, 32906–32913. 10.1074/jbc.M109.034322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yu B, Cao L, Li S, Klauser PC, and Wang L (2023). The proximity-enabled sulfur fluoride exchange reaction in the protein context. Chem. Sci 14, 7913–7921. 10.1039/D3SC01921G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wang L (2017). Genetically encoding new bioreactivity. New Biotechnology 38, 16–25. 10.1016/j.nbt.2016.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Cao L, and Wang L (2022). New covalent bonding ability for proteins. Protein Science 31, 312–322. 10.1002/pro.4228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yang B, Tang S, Ma C, Li S-T, Shao G-C, Dang B, DeGrado WF, Dong M-Q, Wang PG, Ding S, et al. (2017). Spontaneous and specific chemical cross-linking in live cells to capture and identify protein interactions. Nat Commun 8, 2240. 10.1038/s41467-017-02409-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yu B, Li S, Tabata T, Wang N, Cao L, Kumar GR, Sun W, Liu J, Ott M, and Wang L (2022). Accelerating PERx reaction enables covalent nanobodies for potent neutralization of SARS-CoV-2 and variants. Chem 8, 2766–2783. 10.1016/j.chempr.2022.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tian Y, Jacinto MP, Zeng Y, Yu Z, Qu J, Liu WR, and Lin Q (2017). Genetically Encoded 2-Aryl-5-carboxytetrazoles for Site-Selective Protein Photo-Cross-Linking. J. Am. Chem. Soc 139, 6078–6081. 10.1021/jacs.7b02615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hu W, Yuan Y, Wang C-H, Tian H-T, Guo A-D, Nie H-J, Hu H, Tan M, Tang Z, and Chen X-H (2019). Genetically Encoded Residue-Selective Photo-Crosslinker to Capture Protein-Protein Interactions in Living Cells. Chem 5, 2955–2968. 10.1016/j.chempr.2019.08.020. [DOI] [Google Scholar]
- 89.Liu J, Li S, Aslam NA, Zheng F, Yang B, Cheng R, Wang N, Rozovsky S, Wang PG, Wang Q, et al. (2019). Genetically Encoding Photocaged Quinone Methide to Multitarget Protein Residues Covalently in Vivo. J. Am. Chem. Soc 141, 9458–9462. 10.1021/jacs.9b01738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Liu J, Cheng R, Van Eps N, Wang N, Morizumi T, Ou W-L, Klauser PC, Rozovsky S, Ernst OP, and Wang L (2020). Genetically Encoded Quinone Methides Enabling Rapid, Site-Specific, and Photocontrolled Protein Modification with Amine Reagents. J. Am. Chem. Soc 142, 17057–17068. 10.1021/jacs.0c06820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Keppler A, Gendreizig S, Gronemeyer T, Pick H, Vogel H, and Johnsson K (2003). A general method for the covalent labeling of fusion proteins with small molecules in vivo. Nat Biotechnol 21, 86–89. 10.1038/nbt765. [DOI] [PubMed] [Google Scholar]
- 92.Muir TW (2003). Semisynthesis of Proteins by Expressed Protein Ligation. Annual Review of Biochemistry 72, 249–289. 10.1146/annurev.biochem.72.121801.161900. [DOI] [PubMed] [Google Scholar]
- 93.Muralidharan V, and Muir TW (2006). Protein ligation: an enabling technology for the biophysical analysis of proteins. Nat Methods 3, 429–438. 10.1038/nmeth886. [DOI] [PubMed] [Google Scholar]
- 94.Vila-Perelló M, and Muir TW (2010). Biological Applications of Protein Splicing. Cell 143, 191–200. 10.1016/j.cell.2010.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Evans TC, Benner J, and Xu MQ (1998). Semisynthesis of cytotoxic proteins using a modified protein splicing element. Protein Sci 7, 2256–2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Mootz HD (2009). Split Inteins as Versatile Tools for Protein Semisynthesis. ChemBioChem 10, 2579–2589. 10.1002/cbic.200900370. [DOI] [PubMed] [Google Scholar]
- 97.de Araujo AD, Lim J, Good AC, Skerlj RT, and Fairlie DP (2017). Electrophilic Helical Peptides That Bond Covalently, Irreversibly, and Selectively in a Protein–Protein Interaction Site. ACS Med. Chem. Lett 8, 22–26. 10.1021/acsmedchemlett.6b00395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Koniev O, and Wagner A (2015). Developments and recent advancements in the field of endogenous amino acid selective bond forming reactions for bioconjugation. Chem. Soc. Rev 44, 5495–5551. 10.1039/C5CS00048C. [DOI] [PubMed] [Google Scholar]
- 99.Francis MB, and Carrico IS (2010). New frontiers in protein bioconjugation. Current Opinion in Chemical Biology 14, 771–773. 10.1016/j.cbpa.2010.11.006. [DOI] [PubMed] [Google Scholar]
- 100.Ravasco JMJM, Faustino H, Trindade A, and Gois PMP (2019). Bioconjugation with Maleimides: A Useful Tool for Chemical Biology. Chemistry – A European Journal 25, 43–59. 10.1002/chem.201803174. [DOI] [PubMed] [Google Scholar]
- 101.Chen W, Dong J, Li S, Liu Y, Wang Y, Yoon L, Wu P, Sharpless KB, and Kelly JW (2016). Synthesis of Sulfotyrosine-Containing Peptides by Incorporating Fluorosulfated Tyrosine Using an Fmoc-Based Solid-Phase Strategy. Angewandte Chemie International Edition 55, 1835–1838. 10.1002/anie.201509016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Chen S, Lovell S, Lee S, Fellner M, Mace PD, and Bogyo M (2021). Identification of highly selective covalent inhibitors by phage display. Nat Biotechnol 39, 490–498. 10.1038/s41587-020-0733-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Li Q, Chen Q, Klauser PC, Li M, Zheng F, Wang N, Li X, Zhang Q, Fu X, Wang Q, et al. (2020). Developing Covalent Protein Drugs via Proximity-Enabled Reactive Therapeutics. Cell 182, 85–97.e16. 10.1016/j.cell.2020.05.028. [DOI] [PubMed] [Google Scholar]
- 104.Klauser PC, Chopra S, Cao L, Bobba KN, Yu B, Seo Y, Chan E, Flavell RR, Evans MJ, and Wang L (2023). Covalent Proteins as Targeted Radionuclide Therapies Enhance Antitumor Effects. ACS Cent. Sci 9, 1241–1251. 10.1021/acscentsci.3c00288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zhu C, Xu L, Chen L, Zhang Z, Zhang Y, Wu W, Li C, Liu S, Xiang S, Dai S, et al. (2023). Epitope-Directed Antibody Elicitation by Genetically Encoded Chemical Cross-Linking Reactivity in the Antigen. ACS Cent. Sci 10.1021/acscentsci.3c00265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Chari RVJ, Miller ML, and Widdison WC (2014). Antibody–Drug Conjugates: An Emerging Concept in Cancer Therapy. Angewandte Chemie International Edition 53, 3796–3827. 10.1002/anie.201307628. [DOI] [PubMed] [Google Scholar]
- 107.Fu Z, Li S, Han S, Shi C, and Zhang Y (2022). Antibody drug conjugate: the “biological missile” for targeted cancer therapy. Sig Transduct Target Ther 7, 1–25. 10.1038/s41392-022-00947-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Goldenberg DM, and Sharkey RM (2007). Novel radiolabeled antibody conjugates. Oncogene 26, 3734–3744. 10.1038/sj.onc.1210373. [DOI] [PubMed] [Google Scholar]
- 109.Tian Z, Wu L, Yu C, Chen Y, Xu Z, Bado I, Loredo A, Wang L, Wang H, Wu K-L, et al. (2021). Harnessing the power of antibodies to fight bone metastasis. Science Advances 7, eabf2051. 10.1126/sciadv.abf2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Trabjerg E, Keller A, and Leitner A (2022). pH Dependence of Succinimide-Ester-Based Protein Cross-Linking for Structural Mass Spectrometry Applications. ACS Meas. Sci. Au 2, 132–138. 10.1021/acsmeasuresciau.1c00032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ostrem JML, and Shokat KM (2022). Targeting KRAS G12C with Covalent Inhibitors. Annual Review of Cancer Biology 6, 49–64. 10.1146/annurev-cancerbio-041621-012549. [DOI] [Google Scholar]
- 112.Francisco LM, Sage PT, and Sharpe AH (2010). The PD-1 pathway in tolerance and autoimmunity. Immunological Reviews 236, 219–242. 10.1111/j.1600-065X.2010.00923.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Sharpe AH, and Pauken KE (2018). The diverse functions of the PD1 inhibitory pathway. Nat Rev Immunol 18, 153–167. 10.1038/nri.2017.108. [DOI] [PubMed] [Google Scholar]
- 114.Ishida Y, Agata Y, Shibahara K, and Honjo T (1992). Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. The EMBO Journal 11, 3887–3895. 10.1002/j.1460-2075.1992.tb05481.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Nishimura H, Nose M, Hiai H, Minato N, and Honjo T (1999). Development of Lupus-like Autoimmune Diseases by Disruption of the PD-1 Gene Encoding an ITIM Motif-Carrying Immunoreceptor. Immunity 11, 141–151. 10.1016/S1074-7613(00)80089-8. [DOI] [PubMed] [Google Scholar]
- 116.Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, Fitz LJ, Malenkovich N, Okazaki T, Byrne MC, et al. (2000). Engagement of the Pd-1 Immunoinhibitory Receptor by a Novel B7 Family Member Leads to Negative Regulation of Lymphocyte Activation. Journal of Experimental Medicine 192, 1027–1034. 10.1084/jem.192.7.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Zhang N, Tu J, Wang X, and Chu Q (2019). Programmed cell death-1/programmed cell death ligand-1 checkpoint inhibitors: differences in mechanism of action. Immunotherapy 11, 429–441. 10.2217/imt-2018-0110. [DOI] [PubMed] [Google Scholar]
- 118.Cowles SC, Sheen A, Santollani L, Lutz EA, Lax BM, Palmeri JR, Freeman GJ, and Wittrup KD (2022). An affinity threshold for maximum efficacy in anti-PD-1 immunotherapy. mAbs 14, 2088454. 10.1080/19420862.2022.2088454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Chmura AJ, Orton MS, and Meares CF (2001). Antibodies with infinite affinity. Proceedings of the National Academy of Sciences 98, 8480–8484. 10.1073/pnas.151260298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Xiu S, Dick A, Ju H, Mirzaie S, Abdi F, Cocklin S, Zhan P, and Liu X (2020). Inhibitors of SARS-CoV-2 Entry: Current and Future Opportunities. J. Med. Chem 63, 12256–12274. 10.1021/acs.jmedchem.0c00502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wu Y, Wang F, Shen C, Peng W, Li D, Zhao C, Li Z, Li S, Bi Y, Yang Y, et al. (2020). A noncompeting pair of human neutralizing antibodies block COVID-19 virus binding to its receptor ACE2. Science 368, 1274–1278. 10.1126/science.abc2241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Lake B, Serniuck N, Kapcan E, Wang A, and Rullo AF (2020). Covalent Immune Recruiters: Tools to Gain Chemical Control Over Immune Recognition. ACS Chem. Biol 15, 1089–1095. 10.1021/acschembio.0c00112. [DOI] [PubMed] [Google Scholar]
- 123.Kapcan E, Lake B, Yang Z, Zhang A, Miller MS, and Rullo AF (2021). Covalent Stabilization of Antibody Recruitment Enhances Immune Recognition of Cancer Targets. Biochemistry 60, 1447–1458. 10.1021/acs.biochem.1c00127. [DOI] [PubMed] [Google Scholar]
- 124.McCann HM, Lake BPM, Hoffman KS, Davola ME, Mossman KL, and Rullo AF (2022). Covalent Immune Proximity-Induction Strategy Using SuFEx-Engineered Bifunctional Viral Peptides. ACS Chem. Biol 17, 1269–1281. 10.1021/acschembio.2c00233. [DOI] [PubMed] [Google Scholar]
- 125.Köhler G, and Milstein C (1975). Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 256, 495–497. 10.1038/256495a0. [DOI] [PubMed] [Google Scholar]
- 126.Wade N (1982). Hybridomas: The Making of a Revolution. Science 215, 1073–1075. 10.1126/science.7038873. [DOI] [PubMed] [Google Scholar]
- 127.Parray HA, Shukla S, Samal S, Shrivastava T, Ahmed S, Sharma C, and Kumar R (2020). Hybridoma technology a versatile method for isolation of monoclonal antibodies, its applicability across species, limitations, advancement and future perspectives. International Immunopharmacology 85, 106639. 10.1016/j.intimp.2020.106639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Wu K-L, Yu C, Lee C, Zuo C, Ball ZT, and Xiao H (2021). Precision Modification of Native Antibodies. Bioconjugate Chem. 32, 1947–1959. 10.1021/acs.bioconjchem.1c00342. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.