

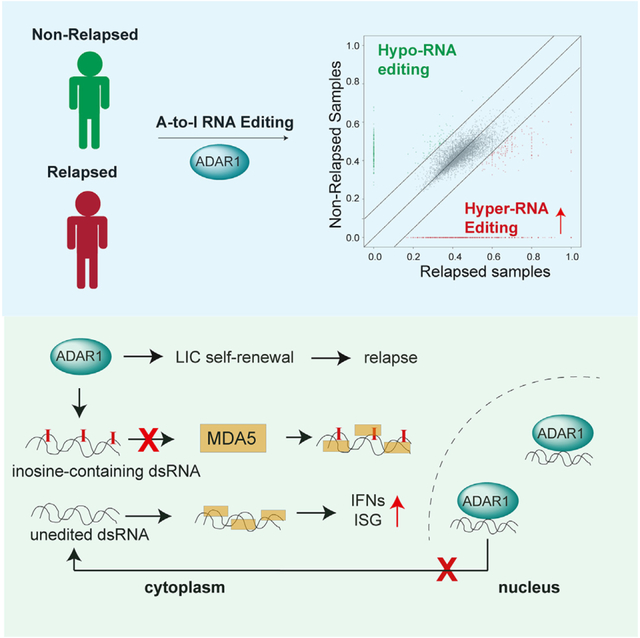
SUMMARY
Leukemia-initiating cells (LICs) are regarded as the origin of leukemia relapse and therapeutic resistance. Identifying direct stemness determinants that fuel LIC self-renewal is critical for developing targeted approaches. Here, we show that the RNA-editing enzyme ADAR1 is a crucial stemness factor that promotes LIC self-renewal by attenuating aberrant double-stranded RNA (dsRNA) sensing. Elevated adenosine-to-inosine editing is a common attribute of relapsed T cell acute lymphoblastic leukemia (T-ALL) regardless of molecular subtype. Consequently, knockdown of ADAR1 severely inhibits LIC self-renewal capacity and prolongs survival in T-ALL patient-derived xenograft models. Mechanistically, ADAR1 directs hyper-editing of immunogenic dsRNA to avoid detection by the innate immune sensor melanoma differentiation-associated protein 5 (MDA5). Moreover, we uncover that the cell-intrinsic level of MDA5 dictates the dependency on the ADAR1-MDA5 axis in T-ALL. Collectively, our results show that ADAR1 functions as a self-renewal factor that limits the sensing of endogenous dsRNA. Thus, targeting ADAR1 presents an effective therapeutic strategy for eliminating T-ALL LICs.
In brief
Rivera et al. showed that widespread adenosine-to-inosine RNA editing by the RNA-editing enzyme ADAR1 is associated with leukemia relapse in patients with T-ALL. ADAR1 promotes LIC stemness via regulation of stem cell gene expression and suppression of MDA5-directed dsRNA sensing by RNA hyper-editing and RNA-editing-independent mechanisms.
Graphical Abstract
INTRODUCTION
T cell acute lymphoblastic leukemia (T-ALL) is an aggressive hematological malignancy that frequently occurs in children, adolescents, and young adults. Approximately 10%–20% of patients with T-ALL will experience relapse months or years following remission and will often become refractory to further treatments.1,2 The survival of relapsed/refractory patients is very poor, with an overall survival rate of less than 25%.3 Relapsed patients often have enriched pools of leukemia-initiating cells (LICs) with enhanced pro-survival and self-renewal capacities, suggesting a potential vulnerable population for effective targeted therapies with less toxicity.4–6
An emerging research topic in LIC biology is the identification of RNA-modifying enzymes that may cooperate with genetic lesions to provide advantages in important LIC functions.7 ADAR enzymes catalyze the transition of adenosine (A) to inosine (I) in precursor double-stranded RNA (dsRNA) that are extensively detected in the mammalian transcriptome.8–10 Epitranscriptomic A-to-I RNA-editing events are widespread in the cancer transcriptome and are critical for the transition from pre-leukemic cells to fully functional LICs.7,11–13
A-to-I RNA editing has a wide range of effects on RNA biology including gene expression, splicing, RNA degradation and translation, and microRNA (miRNA) biogenesis and/or 3′ UTR targeting.11,14–17 The best documented functional roles of ADAR1 are suppression of the interferon (IFN) response18,19 and RNA editing of self-dsRNA to prevent abnormal activation of cytosolic self-dsRNA sensing.17,20 The presence of cellular dsRNA can be the result of direct viral infection or from endogenous sources including mitochondrial dsRNA and transposable elements such as short interspersed nuclear elements.21,22 Both viral dsRNA and endogenous dsRNA can trigger anti-viral innate immune responses via detection by cytosolic dsRNA sensors such as melanoma differentiation-associated protein 5 (MDA5) and protein kinase R (PKR).23–26 ADAR1 introduces A-to-I RNA modifications, which destabilize the dsRNA structure and then avoid recognition by MDA5 and PKR as well as prevent the activation of IFN signaling.27,28 Indeed, concurrent deletion of MDA5 and PKR can completely rescue embryo death and reverse the IFN signatures due to loss of ADAR1.29–31 ADAR1 has two isoforms, the inflammation-induced p150 that is expressed in the cytoplasm and the constitutively expressed nuclear p110, which have diverse cellular functions.15,32–34 The p150 isoform is thought to be the main regulator of the MDA5 pathway and is the major contributor to LIC generation in myeloid leukemia.15,32,33,35 In addition, recent reports suggest that the Zα-RNA-binding region specific to the ADAR1 p150 isoform is responsible for the induction of IFN-stimulated genes (ISGs) in hematopoietic cells.31,36,37
Compared to myeloid leukemia, the role of ADAR1 in lymphoid malignancies is not well understood. In this study, we applied both a three-dimensional human thymic organoid system38 and a T-ALL patient-derived xenograft (PDX) model to examine the function of ADAR1 in the context of T-ALL LIC maintenance. We found that the inflammation-induced ADAR1 p150 isoform is highly expressed within the LIC compartment. A thorough comparison of the A-to-I RNA-editing landscape between non-relapsed and relapsed patient cohorts revealed hyper-editing within ISGs. Moreover, depletion of ADAR1 inhibits LIC self-renewal and survival through both MDA5-dependent and -independent pathways based on the intrinsic expression of MDA5. Our findings indicate that deregulated RNA editing is a critical process in the generation of LICs that has important implications for T-ALL chemoresistance and therapeutic outcomes.
RESULTS
ADAR1-controlled RNA epitranscriptome in relapsed T-ALL
Here, we investigate if RNA modifications by ADAR1 contribute to T-ALL relapse. The three isoforms within the ADAR family of RNA deaminases (ADAR1, ADAR2, and ADAR3) play different roles depending on the particular cancer type.39 We analyzed the expression of ADAR family genes in the NCI TARGET T-ALL dataset and discovered that the most abundant RNA editase is ADAR1 (Figure 1A). In contrast, ADAR2 is expressed at very low levels, and ADAR3 expression is below detection; therefore, both are unlikely to play any significant roles in T-ALL. By comparing the isoform expression between normal hematopoietic stem and progenitor cells (HSPCs) and T-ALL samples, the ADAR1 p150 isoform is overexpressed in T-ALL, while the p110 expression remains constant (Figure 1B). These data were confirmed in fluorescence-activated cell sorting (FACS)-isolated T-ALL LICs by RT-qPCR detection of ADAR1 isoforms (Figure S1A) and by intracellular flow cytometry to detect ADAR1 protein expression (Figure 1C). Interestingly, ADAR1 is expressed predominantly in the immature CD34+Lin− population, which is enriched for T-ALL LICs, instead of the more differentiated CD34−Lin+ fractions. Together, these findings raise the possibility that ADAR1 may play an important role in LIC maintenance.
Figure 1. Relapsed T-ALL acquires a distinct RNA editome in contrast to non-relapsed T-ALL.
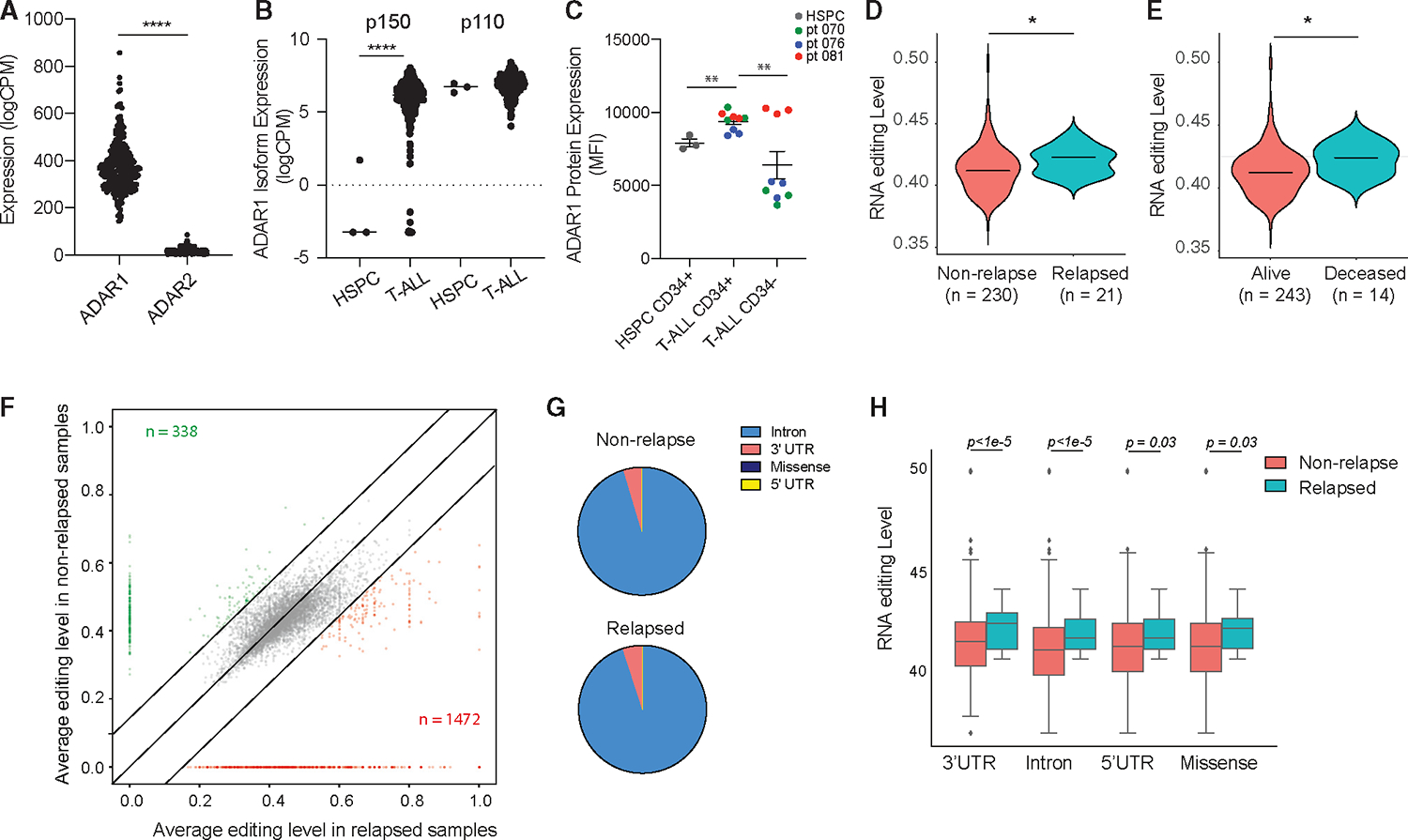
(A) Expression of ADAR1 and ADAR2 in T-ALL patient by RNA sequencing (n = 256).
(B) Isoform expression of ADAR1 p150 and p110 between HSPCs (n = 3) and T-ALL (n = 256).
(C) Quantification of ADAR1 expression in cord blood HSPCs (CD34+Lin−), T-ALL LICs (CD34+Lin−), and non-LICs (CD34−Lin+). n = 3 cord blood HSPC and 3 T-ALL samples.
(D and E) Overall RNA editing between relapsed and non-relapsed patients (D) or between mortality status (E) in violin plots.
(F) Comparison of RNA-editing level between relapsed and non-relapsed cohort display under-edited sites (green) and overedited sites (red) with editinglevels >0.2 and detected in >10% of patients in each group.
(G) Pie chart showing RNA-editing locations in non-relapsed and relapsed T-ALL.
(H) Elevated RNA-editing levels across all categories of editing locations between non-relapsed (n = 230) and relapsed groups (n = 21).
Statistical analysis was calculated by unpaired Student’s t test.
Next, we applied the A-to-I RNA-editing bioinformatic pipeline to the TARGET T-ALL dataset by calculating the percentage of guanosine (G) reads at A at known RNA-editing sites (Is are read as Gs).12 The editing events were restricted to those detected at a minimum of 10% of samples and >10 reads per site to avoid false positives. The median variant allele frequency (VAF) of non-relapsed and relapsed patients was calculated to represent the overall RNA-editing level in each cohort. To understand if RNA editing contributes to T-ALL relapse, ADAR1 expression and the overall RNA-editing levels were compared between relapsed and non-relapsed patient groups (Figures 1D–1E and S1B). Surprisingly, we did not observe any significant differences in ADAR1 expression between the two cohorts (Figure S1B). Instead, a significantly increased incidence of A-to-I RNA modifications (VAF) is associated with both increased risk of relapse and leukemia-associated mortality (Figures 1D and 1E). A total of 338 under-edited and 1,472 over-edited sites were found in relapsed patients compared to non-relapsed samples (Figure 1F; Table S4). As previously reported,7,40,41 A-to-I RNA editing occurs predominately in intronic regions, followed by 3′ UTRs, 5′ UTRs, and lastly missense or coding regions (Figure 1G). Of note, the percentage of intronic editing is more striking than previously published in human editome datasets,17,28,36 which may suggest a cell-type-specific intronic editing pattern in T-ALL. Moreover, the increased VAF editing levels were observed in all locations, indicating no preference for location-specific hyper-editing by ADAR1 during relapse (Figure 1H).
Certain molecular subtypes and early T cell precursor (ETP) status have been associated with more aggressive disease and a higher chance of developing relapse.42,43 We also examined if RNA editing is distinctive among different T-ALL subtypes based on the specific genomic lesions44–46 (Figure S1C). However, there was very little difference in ADAR1 expression, overall A-to-I RNA-editing levels, total number of editing sites, and editing location among the various molecular subtypes. Similarly, no difference in RNA-editing level is associated with ETP status or sex (Figures S1D–S1E). Together, these data indicate that ADAR1 expression and elevated A-to-I RNA editing are common attributes of relapsed T-ALL regardless of the genetic mutation status, sex, and ETP status.
Reduction of ADAR1 impairs T-ALL LIC survival and self-renewal capacity
The significant upregulation of ADAR1 together with the elevated A-to-I RNA-editing levels in relapsed T-ALL cohorts suggest a potential role of ADAR1 in disease relapse and maintenance of LIC properties. LICs exhibit characteristics comparable to those of normal stem cells, such as self-renewal capacity, which enables them to resist chemotherapy and induce relapse.47–49 To examine ADAR1’s function in T-ALL LICs, we adapted the artificial thymic organoid (ATO)38 system for leukemic cell expansion and established in vivo PDX models with high human leukemic engraftment (Figures 2 and S2A). Similar to previous reports of co-culture of T-ALL cells with MS5-DLL,50 the MS5-DLL4 ATO system permits successful T-ALL LIC expansion in vitro (Figure S2A). Primary T-ALL cells cultured in ATOs display a 2-fold expansion by week 6 and more than 20-fold by week 10 (Figures 2B and 2C). To rule out the potential expansion of normal HSPCs from T-ALL samples, we cultured cord-blood-derived HSPCs in ATOs to compare the propagation rate to T-ALL LICs. As previously reported,38 HSPCs expand until 5 weeks, and then cell number decreases between weeks 5 and 10, suggesting that the expanded cells are indeed T-ALL leukemic clones (Figure 2B).
Figure 2. Loss of ADAR1 impairs T-ALL LIC functions.
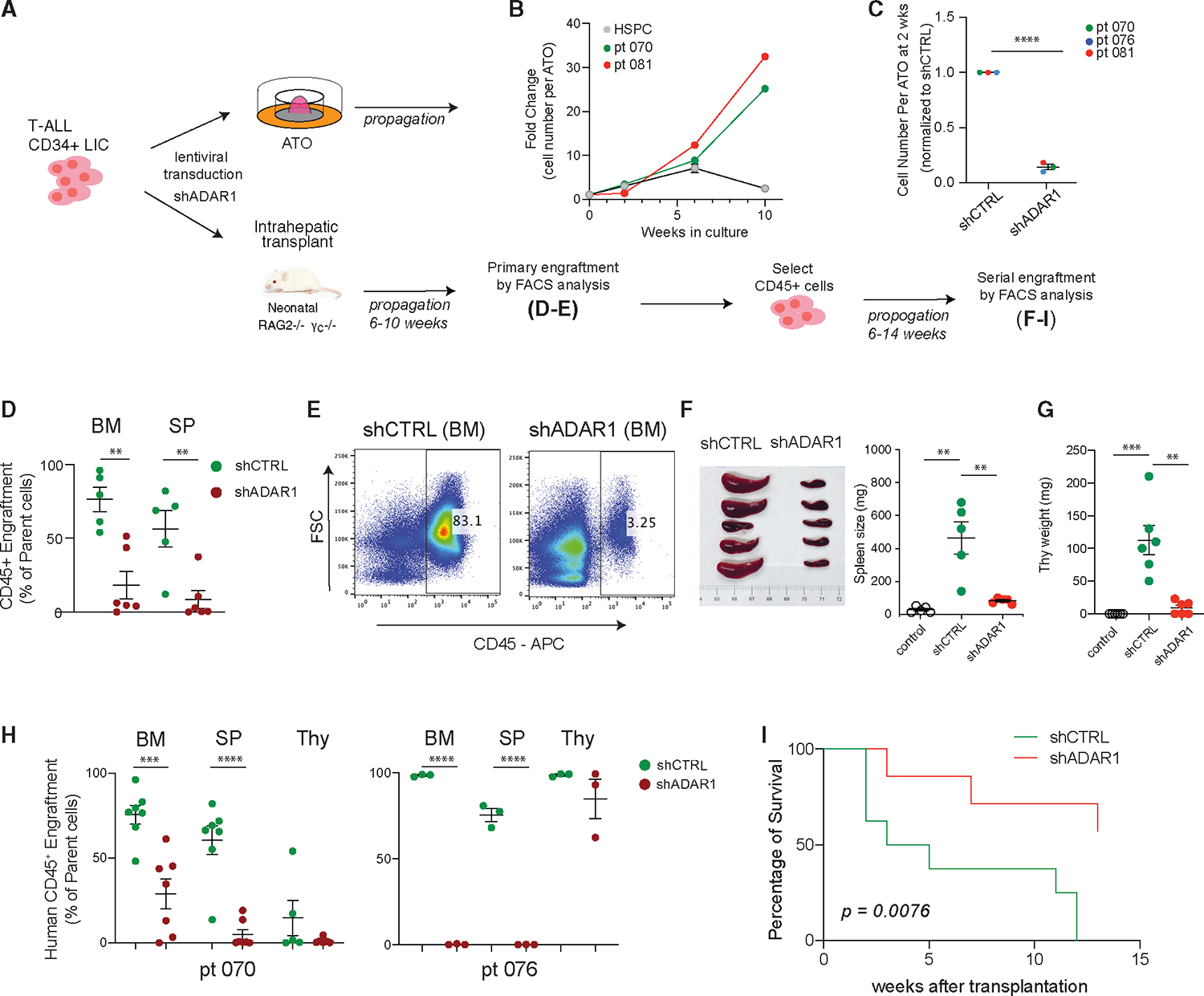
(A) Experimental setup for assessing ADAR1’s effects on T-ALL propagation in ATO and PDX models.
(B) Growth curve of T-ALL samples (n = 2, patient 081 and patient 076) in ATO co-culture system. Cord blood CD34+ HSPCs (n = 2) are also shown. Error bars represent mean with SEM.
(C) Human CD45+ cells were quantified by flow cytometry in non-targeting lentiviral control (shCTRL [shRNA control]) or shADAR1 conditions (n = 3 T-ALL samples).
(D and E) Effects of ADAR1 knockdown on primary transplant of LICs (n = 5 mice for shCTRL and n = 6 mice for shADAR1). An example of flow cytometry showing human CD45+ engraftment in bone marrow is shown in (E).
(F–I) Serial transplant assay of shCTRL and shADAR1.
(F–G) The weights of spleen (F) (n = 5 mice per group) and thymus (G) (n = 6 mice per group) were measured after serial transplant.
(H) Human CD45+ frequencies in the bone marrow, spleen, and thymus of two T-ALL PDX models (n = 3–7 mice per group).
(I) Loss of ADAR1 is associated with improved mouse survival (patient 076, n = 7–8 mice per group). p value was determined by the Mantel-Cox log-rank test.
(C–H) Error bars represent mean with SEM. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001, unpaired Student’s t test.
In PDX models, abundant human CD45+ leukemic engraftment was observed in bone marrow, spleen, and thymic hematological niches within 6–10 weeks after intrahepatic transplant into neonatal Rag2−/−γc−/− mice (Figures 2D–2H). Of note, thymic engraftment by T-ALL leukemic clones can be biased toward the growth of human T-ALL leukemia cells and can present a very high engraftment rate since Rag2−/−γc−/− mice do not develop a thymus naturally. To detect if any HSPCs are present in these engrafted cells, we performed an in vitro colony-formation assay, which permits myeloid lineage differentiation and quantification of self-renewal capacity of HSPCs by colony-replating assay51 (Figure S2B). CD34+Lin− T-ALL LICs and the differentiated CD34−Lin+ T-ALL subpopulations were FACS sorted from bone marrow and placed in colony formation media. Compared to normal CD34+Lin− HSPCs, CD34+Lin− T-ALL LICs produce a significantly reduced number of primary colonies, and this is further reduced in CD34−Lin+ T-ALL cells. More strikingly, we could not detect any secondary colony formation by either CD34+Lin− or CD34−Lin+ T-ALL cells. Collectively, these data demonstrate that the in vitro ATO and in vivo PDX systems are suitable for studying T-ALL leukemic clones without interference from healthy HSPCs.
To study the effects of ADAR1 on self-renewal capacity in LICs, ADAR1 was knocked down by short hairpin RNA (shRNA; shADAR1) in patient-derived enriched LICs (CD34+Lin−) followed by culture in ATOs or transplantation into PDX models (Figures 2A and 2B). Consistent with an anti-apoptotic role of ADAR1,19,52–54 we observed a significant increase in apoptosis activity in ADAR1-knockdown cells compared to non-targeting lentiviral (shCTRL [shRNA control]) control cells (Figure S2C). This is accompanied by decreased leukemia cell propagation (>70% reduction) in both ATO and primary engrafted PDX mice (Figures 2C–2E and S2D). However, the most striking effects were seen in serial transplant recipients. Equal numbers of live bone marrow cells derived from shCTRL or shADAR1 mice were transplanted into secondary mice recipients to assess the self-renewal capacity of LICs. Spleen and thymus weights of mice injected with shADAR1 cells returned to the same level of non-transplanted litter controls (Figures 2F and 2G). In addition, ADAR1 knockdown strongly impedes serial leukemic engraftments in bone marrow and spleen niches (Figure 2H). Because of the marked differences in leukemia burden between shADAR1 and control conditions, we evaluated the survival potential between these two groups. We observed significantly improved survival in shADAR1 mice (p < 0.0076; Figure 2I). Together, these data suggest that ADAR1 contributes to self-renewal and survival of T-ALL LICs.
Loss of ADAR1 reduces hyper-editing events
Since the TARGET dataset is based on bulk cell sequencing, LIC-specific events could be masked. To gain better insights into LIC-specific molecular targets and pathways regulated by ADAR1, we performed RNA sequencing studies on enriched T-ALL LICs (CD34+Lin−) with ADAR1 knockdown (Figures 3 and S3). Since loss of ADAR1 leads to reduced cell survival (Figure 2), the lentivirus-to-cell ratio was carefully titrated to obtain approximately 50% reduction of ADAR1 (Figure S3A). This allows for sufficient cell recovery after transduction for sequencing analysis. A total of 661 genes are differentially expressed upon ADAR1 knockdown, including 56 downregulated and 605 upregulated genes (Figure 3A; Table S3). A close examination of the “lymphoblastic leukemia” and “acute undifferentiated leukemia” pathways revealed that several critical self-renewal genes (e.g., CD34, CD44, LMO2, JAK3, and TAL1) were downregulated in ADAR1-deficient LICs (Figures 3B, 3C, and S3B).55–57 We further confirmed decreased expression of a set of these genes (CD34, CD44, and LMO2) by RT-qPCR after 48 h of ADAR1 knockdown in T-ALL LICs (Figure 3D). Interestingly, A-to-I RNA editing is rarely detected in these transcripts regardless of the direction of differential expression, except for three editing sites within the LMO2 intronic region in scramble control cells (Table S4). Similarly, the most extensively edited genes are often not differentially expressed (e.g., IL17RA and EIF2AK2), suggesting indirect regulation of ADAR1 on gene expression (Figures 3E and 3SC). These data indicate that ADAR1 likely promotes self-renewal gene expression independent from A-to-I RNA editing of the specific self-renewal gene.
Figure 3. ADAR1 downregulation reduces LIC stemness gene expression.
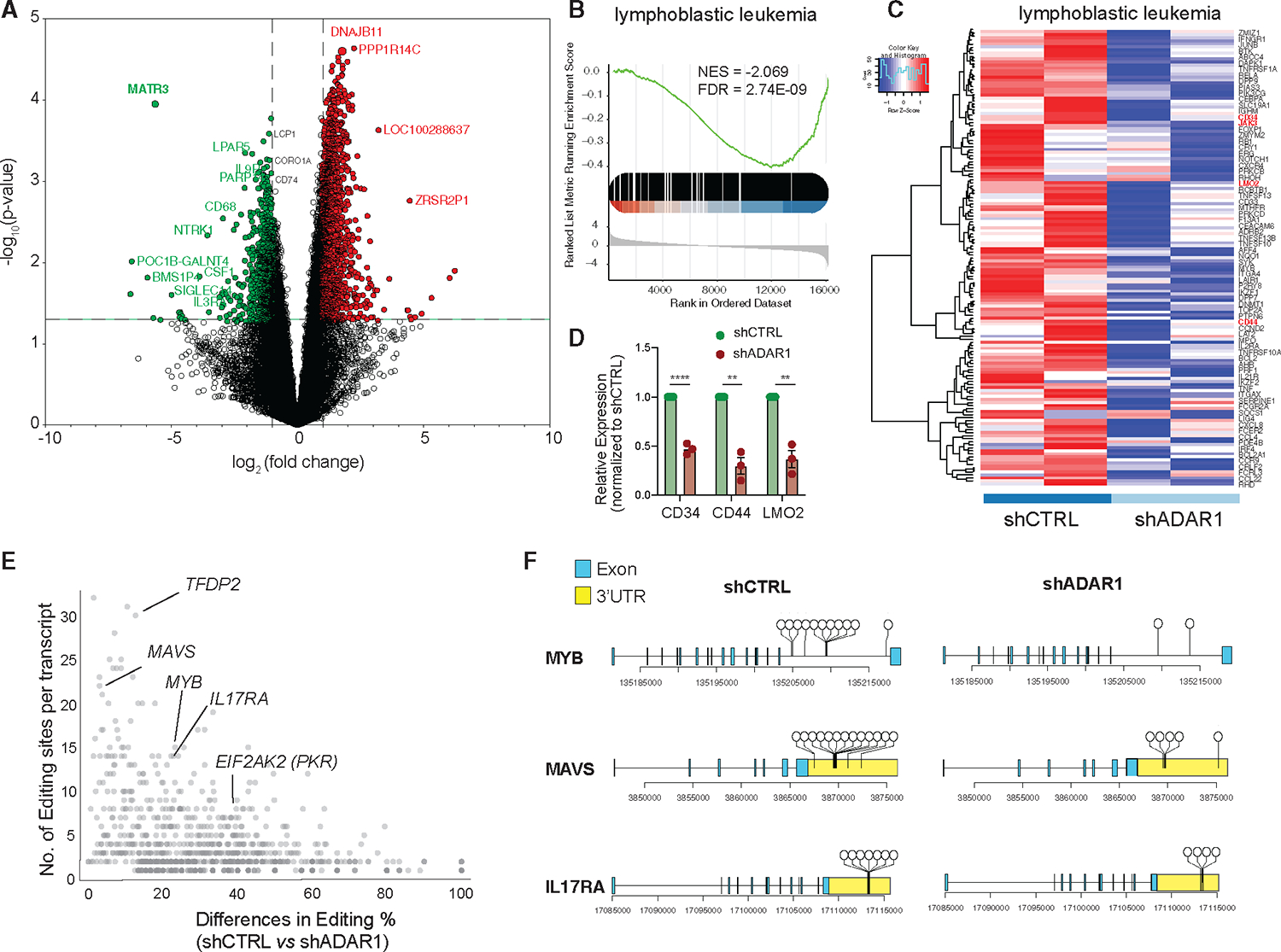
(A) Volcano plot depicting significantly differentially expressed genes in T-ALL LICs with ADAR1 knockdown (n = 2 samples).
(B and C) Gene enrichment scores (B) and heatmap (C) of “lymphoblastic leukemia” pathway.
(D) Expression of CD34, CD44, and LMO2 was quantified by RT-qPCR (n = 3, patients 070, 076, and 081) after 48 h lentiviral transduction of shADAR1.
(E) Analysis of differential level of RNA editing and number of A-to-I editing sites per transcript between scramble shCTRL and ADAR1 knockdown.
(F) Dandelion plot showing RNA-editing location in MYB, MAVS, and IL17RA. Each dot represents a unique RNA-editing event.
Error bars represent mean with SEM. **p < 0.01 and ****p < 0.0001, unpaired Student’s t test.
We also profiled the RNA editome landscape in FACS-sorted LICs of two patients with T-ALL prior to and after ADAR1 knockdown (Figures S3D–G). Reduced ADAR1 led to a small but significant decrease in overall editing levels (Figure S3D). However, the most profound effect was the reduction in the number of editing events (~50%) (Figure S3E). The total number of edits decreased from 1,698 in the control to 901 in the shADAR1 condition with a predominant drop in Alu-enriched intronic editing sites (Figure S3F). However, the A-to-I editing level within intronic regions is not altered (Figure S3G). Since ADAR1 has a tendency to edit in clusters, a phenotype termed hyper-editing,15,58,59 we calculated the number of edits and changes in editing level between control and shADAR1 per each transcript. Hyper-editing is widespread in intronic regions (e.g., MYB) and 3′ UTRs of mRNA transcripts (e.g., MAVS and IL17RA) (Figures 3E and 3F). Therefore, ADAR1 knockdown in T-ALL LICs reduces hyper-editing events rather than the editing level at a particular site.
T-ALL LICs pose different dependencies on dsRNA sensing by MDA5 pathway
ADAR1-directed RNA editing negatively regulates IFN production and ISG activation by preventing accumulation of endogenous dsRNAs, which are detected by the MDA5-MAVS dsRNA-sensing pathway.17,27,60,61 These roles of ADAR1 are the foundation for many of its important functions, such as preventing embryonic lethality, suppressing apoptosis during oncogenesis, and overcoming resistance to immunotherapy.20,29,62 However, dsRNA sensing of immunostimulatory transcripts as a mechanism in ADAR1-regulated LIC self-renewal has never been fully characterized.
To investigate this functionally, we performed concurrent knockdown of MDA5 (mCherry-labeled shRNA) and ADAR1 (EGFP-labeled shRNA) in PDX T-ALL LICs (Figure 4A). We detected comparable levels of reduction in ADAR1 and MDA5 (>75% and >90%, respectively) in the successfully dual-transduced cells (mCherry+EFGP+) in the three patient models (Figure S4A). The mCherry+EFGP+ cells were transplanted into immunocompromised Rag2−/−γc−/− mice and then serially transplanted to quantify the self-renewal capacity of LICs. Surprisingly, co-knockdown of ADAR1 and MDA5 exhibits diverse rescue effects on self-renewal in the three PDX models tested. A partial rescue of serial leukemia engraftment was detected in co-knockdown compared to the ADAR1-deficient-alone condition in PDX-070 (Figure 4B). In PDX-081, a complete rescue was observed in bone marrow and spleen niches, which is associated with the reversal of improved survival advantages by loss of ADAR1 alone (Figures 4C and S4B). In contrast, no differences in serial leukemia engraftment or spleen weight were observed in PDX-076 (Figures 4D and 4E). These data indicate that ADAR1-directed RNA editing controls LIC self-renewal through dsRNA sensing in at least a portion of patients with T-ALL.
Figure 4. Concurrent knockdown of MDA5 and ADAR1 rescues self-renewal to various degrees in different T-ALL models.
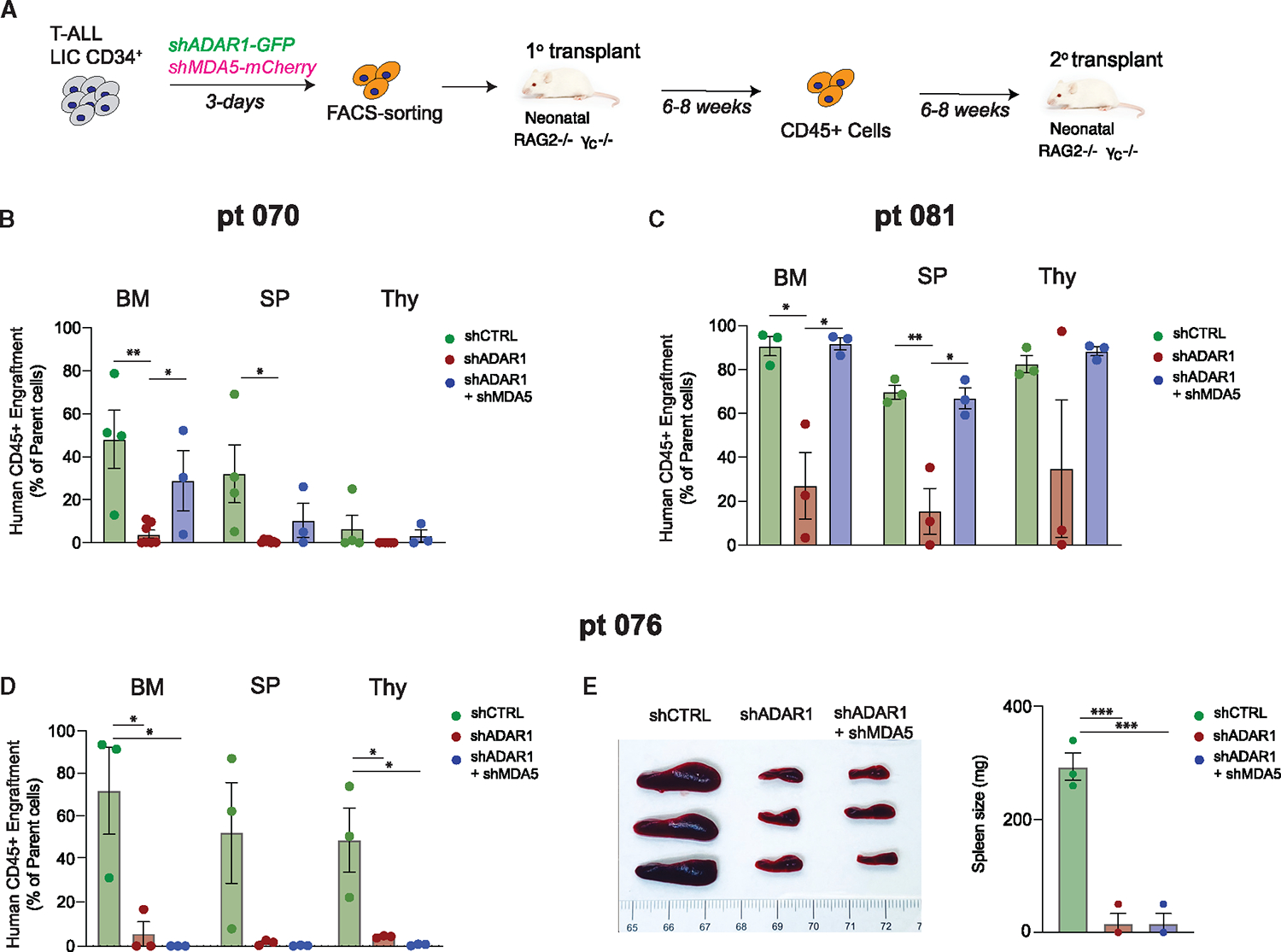
(A) Experimental setup. T-ALL CD34+ cells were transduced with shCTRL, shADAR1, or shADAR1 and shMDA5 lentivirus in combination. Transduced cells were sorted based on GFP+mCherry+ (GFP for shADAR1 and mCherry for shMDA5), and serial transplant potential was measured in recipient Rag2−/−γc−/− mice.
(B–D) Serial leukemia engraftment and representative bone marrow FACS plots of patient 070 (B), patient 081 (C), and patient 076 (D) were determined for shCTRL, shADAR1, and shADAR1 in combination with shMDA5 (3–8 mice per condition).
(E) Images of spleen (left) and spleen weights (right) in serial transplanted mice were determined after an 8 week engraftment interval (n = 3 mice per group).
*p < 0.05, **p < 0.01, and ***p < 0.001, unpaired Student’s t test.
Next, we explored potential mechanisms guiding the difference in response to co-knockdown of MDA5 and ADAR1. Curiously, differential gene expression in the retinoic-acid-inducible gene I (Rig-I)-like signaling and cytosolic sensing pathways was detected between samples 070 and 076 (Figure S4C). Further gene expression analysis of mediators in IFN signaling pathways and ISGs shows variable levels of cell-intrinsic IFN signaling, with sample 081 having higher levels of ISG15, IFIT1, IRF7, and IRF9 than samples 070 and 076, which could suggest differences in intrinsic IFN signaling properties and dependency on ADAR1 to suppress aberrant dsRNA sensing among patients (Figure S4D). The inflammation-inducible ADAR1 p150 isoform is thought of as the main regulator of RNA editing in the cytoplasm and therefore is responsible for regulating dsRNA sensing by MDA5,25,29,35 while nuclear-localized ADAR1 p110 is largely dispensable for MDA5 signaling.63 Thus, the intrinsic expression of p150 and MDA5 dsRNA sensing in patients with T-ALL might dictate the level of dependency on the MDA5 pathway. To test this hypothesis, we measured the expression of ADAR1 isoforms and MDA5 in FACS-enriched LICs in the three T-ALL samples (Figure 5). Patient 070 did not yield enough LICs, and therefore we could not complete the analysis. Patient 081 had significantly elevated expression of ADAR1 p150, and p110, as well as MDA5, compared to patient 076 (Figures 5A–5C). In contrast, the level of another dsRNA sensor, PKR, showed no difference between patients. Coupled with the differential rescue effects of MDA5 and ADAR1 co-knockdown, our data support that patient 081 relies on the ADAR1 p150-MDA5 axis for promoting self-renewal, while patient 076 likely depends on both the p150-MDA5 axis and p110-dependent mechanisms.
Figure 5. Basal expression of MDA5 controls dependency on the ADAR1-MDA5 axis.
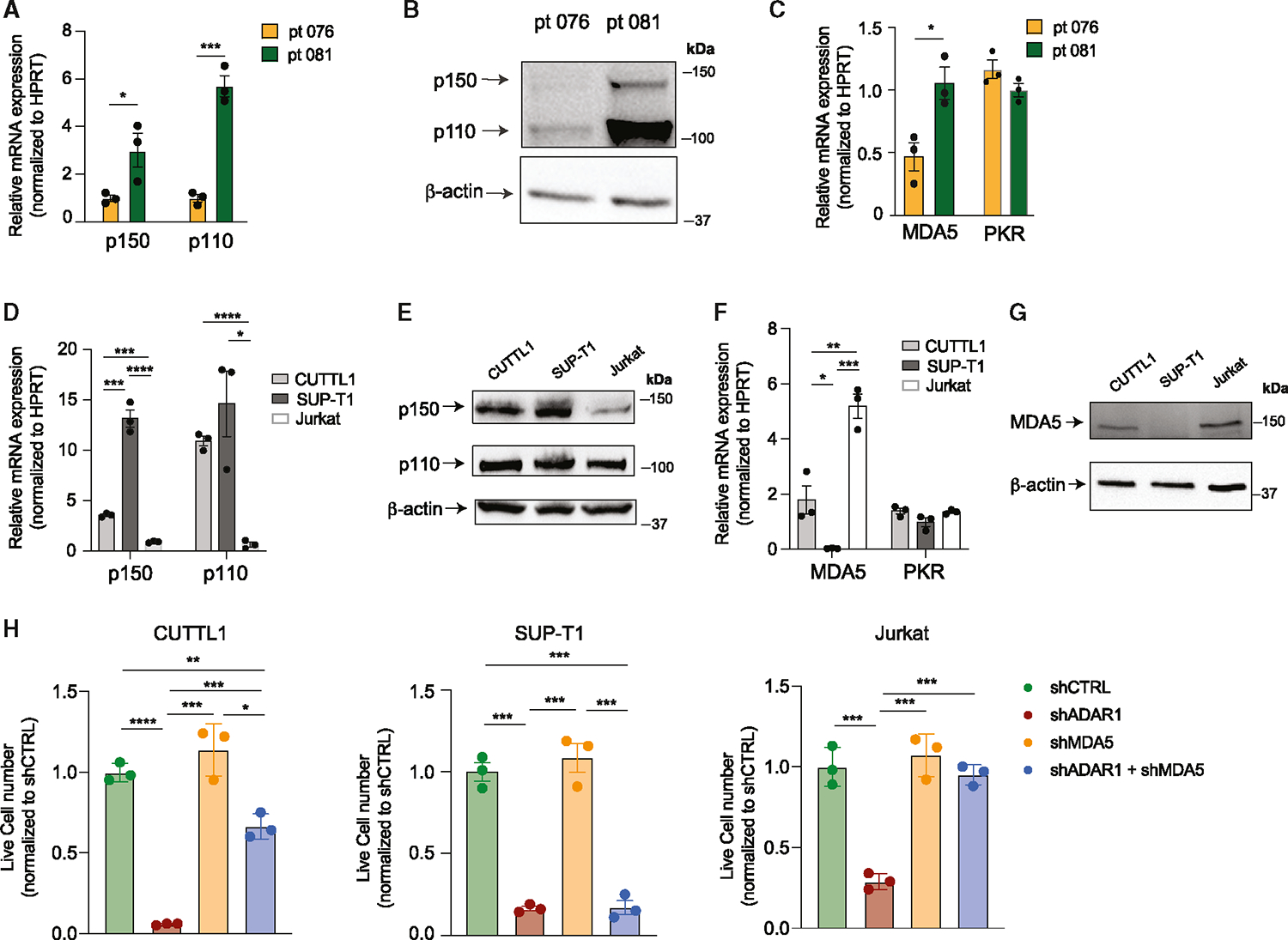
(A and B) Expression of ADAR1 isoforms in patient 076 and patient 081 was measured by RT-qPCR and western blot in Lin−CD34+ LIC-enriched population (n = 3 independent experiments).
(C) Expression of MDA5 was determined in Lin−CD34+ LIC-enriched cells of patient 076 and patient 081 (n = 3 independent experiments).
(D and E) Expression of ADAR1 isoforms in three T-ALL cell lines, CUTTL1, SUP-T1, and Jurkat (n = 3 independent experiments).
(F and G) MDA5 and PKR mRNA expression (F) and protein level (G) were determined in T-ALL cell lines (n = 3 independent experiments).
(H) Cell counts of shRNA control, shADAR1, shMDA5, and co-knockdown of shADAR1 and shMDA5 were assessed 3 days post-lentiviral transduction. Data from three independent experiments are shown.
Error bars represent mean with SEM. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001, unpaired Student’s t test.
We next sought to validate whether this isoform-specific dependency of ADAR1 is also presented in T-ALL cell lines. We first evaluated the endogenous expression of the p150 and p110 isoforms, and dsRNA sensors MDA5 and PKR, in CUTTL1, SUP-T1, and Jurkat T-ALL cell lines. SUP-T1 has the highest expression of the p150 isoform, followed by CUTTL1 and then Jurkat cells (Figures 5D and 5E). The protein expression of p110 is comparable among the three cell lines as shown by western blot analysis. MDA5 is expressed at the highest level in Jurkat cells and lowly expressed in CUTTL1 and SUP-T1, while no significant difference in PKR expression is seen among the cell lines (Figures 5F and 5G). Next, we performed ADAR1 knockdown alone, MDA5 knockdown alone, or MDA5 and ADAR1 co-knockdown to examine if partial or complete rescue effects were presented. Successfully transduced cells were FACS-sorted based on shMDA5 (mCherry+) and shADAR1 (EGFP+) signals, and cell propagation and apoptosis rate were evaluated (Figures 5H and S5A–S5D). While knockdown of MDA5 alone does not significantly alter cell proliferation, silencing of ADAR1 reduced cell proliferation and induced apoptosis in all cell lines (Figures 5H, S5C, and S5D). Interestingly, similar to T-ALL PDX models, co-knockdown of ADAR1 and MDA5 completely rescued the effects of ADAR1 knockdown in Jurkat, while only partial rescue was observed in CUTTL1. Consistent with the low MDA5 expression, no significant difference was observed in ADAR1 knockdown alone compared to the co-knockdown with MDA5 in SUP-T1 cells. Thus, the phenotypic differences in p150-MDA5 dependency predominately reflect the level of MDA5 expression, as opposed to p150 level, in T-ALL models.
ADAR1 RNA-editing-independent activity promotes nuclear localization of dsRNA
ADAR1 can operate as a dsRNA-binding protein with functions independent of editing activity to suppress aberrant IFN signaling and to promote cancer progression.15,41,64,65 Since Jurkat cells show the highest level of cell-intrinsic MDA5, we utilized it to investigate the RNA-editing-dependent and -independent mechanisms of ADAR1 via MDA5-directed sensitivity to IFN treatment. Since shRNA-directed ADAR1 knockdown may still permit ADAR1 activation by IFN treatment, we generated ADAR1-knockout (KO) cells (Figure 6A). A wild-type “addback” cell line (ADAR1WT) and a catalytically inactive mutant of the ADAR1 p150 isoform (ADAR1E912A)15,18,60 were also generated by re-expressing WT or mutant ADAR1 p150 isoforms in KO cells. As expected, the overexpression constructs produce both p150 and p110 isoforms due to leaky ribosome scanning.34 To activate the IFN response, these cells were treated with various doses of IFNα, −β, and −γ for 48 h to examine changes in ADAR1 expression (Figures 6B, S6A, and S6B). IFN treatment upregulates ADAR1 in WT cells, while KO and ADAR1WT addback cells had little response. As expected, exposure to IFNβ predominately upregulates the p150 isoform rather than p110 (Figure 6C). Moreover, loss of ADAR1 induces apoptosis upon IFNβ treatment as demonstrated by elevated levels of cleaved PARP1 and a reduced percentage of viable cells (Figures 6D and S6C). The loss of cell viability is completely reversed by re-expression of ADAR1WT and partly rescued by ADAR1E912A expression in IFNα, −β, or −γ treatments, suggesting that both RNA-editing-dependent and -independent activity of ADAR1 is necessary to suppress IFN-induced apoptosis (Figure 6E).
Figure 6. RNA-editing-dependent and -independent mechanisms of ADAR1 suppress aberrant dsRNA sensing.
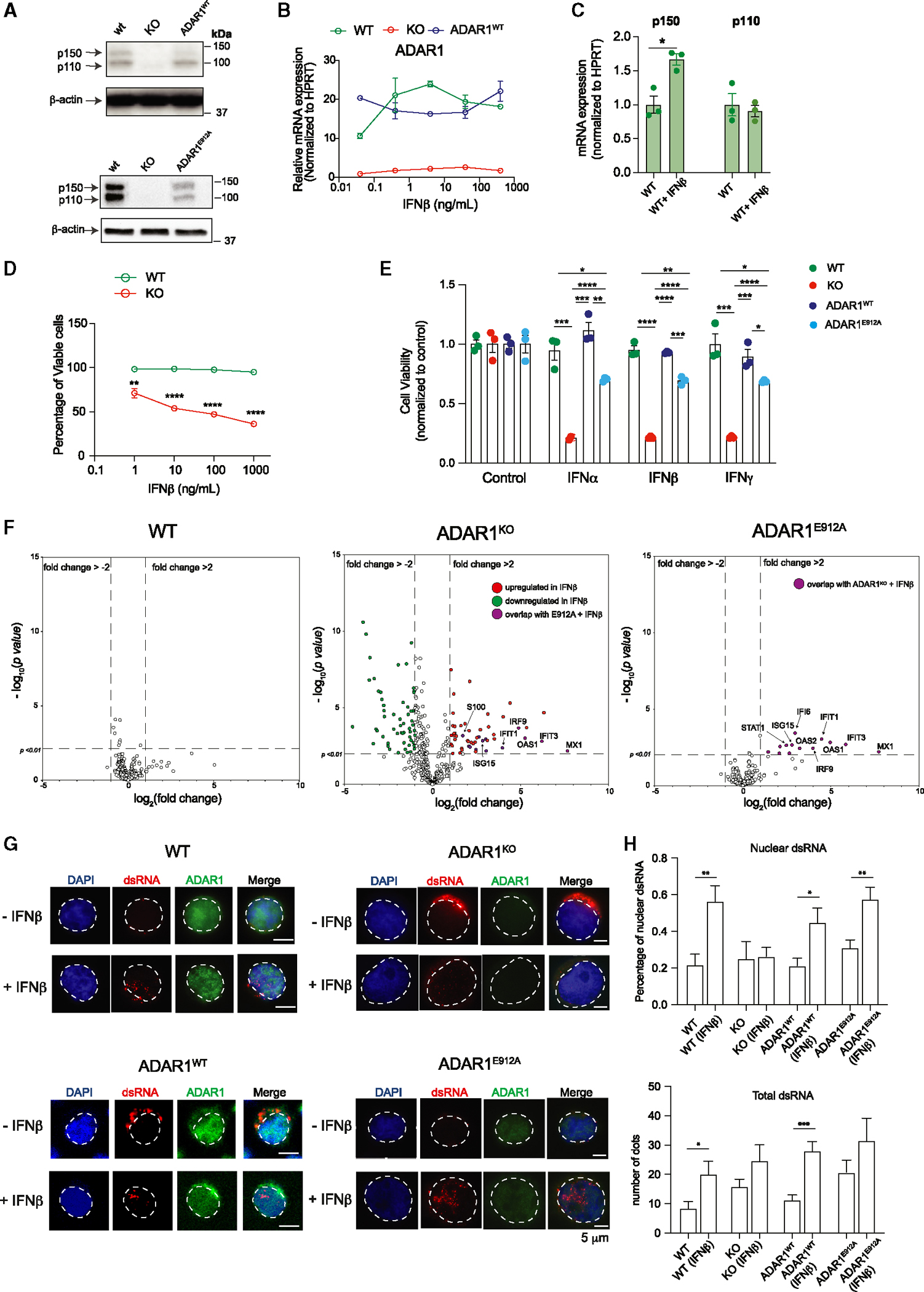
(A) Western blot showing ADAR1 expression in modified Jurkat T-ALL cell lines, including wild-type (WT), ADAR1-KO, re-expressed WT ADAR1 (ADAR1WT), and ADAR1E912A by lentiviral-overexpressing vector. β-Actin was used as loading control.
(B) Jurkat cells were stimulated with various concentrations of IFNβ, and the gene expression of ADAR1 was determined (n = 2 independent experiments).
(C) Quantification of ADAR1 p150 and p110 isoforms upon IFNβ treatment at 10 ng/mL for 24 h (n = 3 independent experiments).
(D) Cell viability was quantified in Jurkat cells upon IFNβ stimulation (n = 3 independent experiments).
(E) Cell viability was quantified in Jurkat cells treated with IFNα (0.1 ng/mL), IFNβ (1 ng/mL), or IFNγ (1 ng/mL) for 48 h (n = 3 independent experiments).
(F) NanoString analysis of gene expression in WT, ADAR1-KO, and ADAR1E912A-overexpressing Jurkat cells stimulated with IFNβ (1 ng/mL, 48 h) (n = 2 independent experiments).
(G) Immunofluorescent staining to detect the localization of dsRNA (J2 antibody) in WT, ADAR1-KO, ADAR1WT, and ADAR1E912A Jurkat cells stimulated with IFNβ (1 ng/mL, 24 h). Scale bars represent 5 μm.
(H) Quantification of total dsRNA dots and percentage of nuclear dsRNA from Jurkat cells treated with IFNβ (1 ng/mL), 24 h, 10 cells/condition.
*p < 0.05, **p < 0.01, and ****p < 0.0001, unpaired Student’s t test. Error bars represent mean with SEM in all graphs.
In addition to MDA5-MAVS signaling, ADAR1 also suppresses cytoplasmic dsRNA sensing through RIG-I and PKR pathways.17,31,66,67 While ADAR1 KO alone does not change MDA5, PKR, and RIG-I levels, IFNβ stimulated the expression of these dsRNA sensors in WT, ADAR1 KO, and ADAR1WT cells (Figures S6B and S6C). Interestingly, knockdown of MDA5 in ADAR1-KO cells abrogated the IFNβ-induced RIG-I activation but increased PKR expression, suggesting that MDA5 may crosstalk with PKR signaling pathways in the combination of ADAR1 loss and IFNβ treatment. When activated, PKR phosphorylates eukaryotic translation initiation factor 2α (eIF2α), which then triggers translation shutdown.17,68,69 Indeed, we reported an elevated level of phosphorylated eIF2α in IFNβ-treated KO cells compared to WT, ADAR1WT, and ADARE912A re-expression conditions (Figure S6D). Further functional validation confirmed that PKR knockdown by shRNA partially rescues cell viability in KO cells but not in WT and ADAR1WT (Figures S6E and S6F). However, no significant rescue effect was seen in ADAR1E912A cells, implying that activation of PKR signaling is independent of ADAR1’s RNA-editing function as previously suggested.17,25
Next, we wanted to examine if the RNA-editing-independent activity of ADAR1 is sufficient in suppressing ISG expression. The expression of a selected panel of 790 ISGs using the NanoString nCounter System was quantified in IFNβ-stimulated WT, ADAR1-KO, and ADAR1E912A cells (Figure 6F). A total of 27 differentially expressed ISGs in ADAR1E912A cells in comparison to 237 ISGs in ADAR1-KO and 30 ISGs in WT cells were reported (Table S5). Surprisingly, ADAR1E912A overexpression was able to rescue all downregulated targets found in the IFNβ-treated KO condition as well as suppress the majority of upregulated ISGs. Approximately 70% (19 out of 27) of ADAR1E912A-regulated targets overlapped with those of ADAR1-KO cells, while only 3.7% (1 out of 27) of targets overlapped with IFN-treated WT cells. These data suggest that ADAR1’s editing-independent function contributes to suppression of ISGs in T-ALL.
The activation of dsRNA-sensing pathways depends on cellular localization and length of the accessible endogenous dsRNA.70 Therefore, we hypothesized that ADAR1 may retain dsRNA via its dsRNA-binding ability in the nucleus to limit the cytosolic dsRNA pool. Immunofluorescent staining using a J2 dsRNA antibody was applied to identify the cellular dsRNA localization prior to and after IFNβ treatment (Figures 6G and 6H). Interestingly, in the absence of IFNβ, ADAR1-KO cells showed an elevated total dsRNA level compared to WT cells without triggering cell death (Figure 6H), suggesting that a certain level of unedited dsRNA is tolerated in the absence of MDA5. WT cells respond to the addition of IFNβ by increasing the total dsRNA level (~2.4-fold) and the percentage of nuclear dsRNA. The elevated nuclear dsRNA level was also observed in ADAR1WT and ADAR1E912A re-expressing cells. In contrast, the level of nuclear dsRNA remained constant prior to and after IFNβ treatment in KO cells, in which the unedited cytosolic dsRNA likely triggers ISG production and the corresponding apoptosis seen in KO cells. To test if this phenomenon applies to other cell types in response to IFN stress, we exposed CUTTL1 (T-ALL) and 293T (embryonic kidney) cell lines to IFNβ for 24 h and analyzed dsRNA location (Figures S7A–S7D). While the total dsRNA level did not increase, the nuclear dsRNA showed a significant increase from 35% to 62% in CUTTL1 and from 32% to 48% in 293T cells. Together, these data reveal an important RNA-editing-independent mechanism of ADAR1 in preventing MDA5-directed dsRNA sensing.
DISCUSSION
T-ALL is an aggressive hematological malignancy that arises from the transformation of lymphoid progenitors with the cooperation of tumor suppressors and oncogenes.71–73 We now understand that RNA modifications such as A-to-I RNA editing are critical in promoting cancer progression and therapeutic resistance.7 We have shown that RNA editing by ADAR1 is an important regulatory mechanism required for HSC maintenance and transformation into myeloid leukemia.15,20,32,41,74 However, the role of ADAR1 in lymphoid neoplasms such as T-ALL has not been well defined. Here, we described a fundamental role of ADAR1 in maintenance of T-ALL LICs. The epitranscriptome landscape revealed elevated A-to-I RNA editing in relapsed patients. Further functional studies demonstrated that loss of ADAR1 leads to impaired survival and self-renewal of LICs, thus improving overall survival in PDX models.
We have previously discovered ADAR1’s contribution to neoplastic transformation of myeloid LICs via several different pathways: (1) regulation of self-renewing microRNA biogenesis, (2) editing of 3′ UTRs of oncogenes to prevent miRNA-directed degradation, (3) editing of coding genes, and (4) induction of oncogenic RNA-splicing events.15,32,33,75 Whether editing of immunogenic dsRNA and suppression of aberrant dsRNA sensing could enhance LIC self-renewal capacities is an important question that has not been extensively addressed. We provide the link between malignant A-to-I RNA editing and suppression of dsRNA sensing as a mechanism promoting LIC self-renewal. Hyper-editing events are commonly observed in ISG genes within intronic regions and 3′ UTRs in T-ALL LICs. Moreover, co-knockdown of MDA5 with ADAR1 rescues LIC self-renewal to various degrees in T-ALL samples. We further reported that the intrinsic expression of MDA5, rather than p150 or p110 isoforms, dictates the level of response to co-knockdown. The ADAR1 p150 isoform specifically prevents innate immune activation by regulating dsRNA-sensing pathways.35,76,77 In contrast, ADAR1 p110 suppresses apoptosis and regulates tissue development by MDA5-independent mechanisms.35,53 This was nicely demonstrated by the ADAR1 p110 isoform KO mouse model where the mice died postnatally but without any upregulation of ISG expression or activation of the MDA5-sensing pathway.63 In this respect, our study suggests that LIC self-renewal may rely entirely on the p150-MDA5 axis and that the p110 isoform is likely dispensable in some patients with T-ALL. In contrast, other T-ALL models with low intrinsic MDA5 levels may utilize both p150 and p110 isoforms in an MDA5-dependent and -independent manner to promote self-renewal. It is possible that LICs possess different levels of dependency on the ADAR1-MDA5 pathway due to the diverse ISG signatures and IFN signaling in patients with T-ALL, as previously reported.60 Future studies are necessary to definitively decouple the isoform-specific function, RNA-editing targets, and pathways regulated in a large cohort of patients with T-ALL and potentially other tumor types.
Lastly, we report an RNA-editing-independent role of ADAR1 in attenuating aberrant dsRNA sensing. To trigger dsRNA sensing by cytosolic MDA5, we surmised that unedited endogenous dsRNA in the absence of ADAR1 must be located in the cytoplasm. Indeed, the deaminase-deficient ADAR1E912A was able to suppress the majority of ISGs in ADAR1-KO cells. It is worth noting that the ADAR1E912A re-expression level is lower than that in WT cells, suggesting that a limited amount of dsRNA binding is sufficient to limit the cytosolic dsRNA pool and suppress IFN signaling. Surprisingly, we noticed that T-ALL cells can tolerate a certain level of unedited cytosolic dsRNA without triggering apoptosis. It is curious how cancer cells set this limit using the complex and diverse regulatory dsRNA-sensing network, which may contribute to the difference in response to cytosolic dsRNA in T-ALL models.
In conclusion, this work highlights the intrinsic difference in how ADAR1 promotes self-renewal among patient samples, in addition to mechanistic details of LIC maintenance. Since ADAR1 is a critical self-renewal factor in normal human HSCs, future studies are necessary to clearly differentiate the molecular mechanisms of ADAR1-regulated self-renewal in normal HSPC and T-ALL LICs. This in turn opens the door for therapeutic targeting of these downstream processes to prevent relapse and therapeutic resistance with the goal to selectively target LICs while sparing HSPCs.
Limitations of the study
This study demonstrated the importance of ADAR1 in T-ALL LIC self-renewal and the hyper-editing landscape in relapsed T-ALL, but our functional studies (in vitro ATO and in vivo PDX) are limited to three patient models due to the inherent rarity of T-ALL. Furthermore, within a population of T-ALL cells, the prevalence of T-ALL LICs is very low, and the variable levels of LICs in each patient have limited the cell yields for downstream functional assays. Thus, future studies should be performed in a large cohort of samples to decipher if ADAR1-MDA5-regulated dsRNA sensing is critical and whether intrinsic IFN signaling dictates the dependency on ADAR1-MDA5.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Qingfei Jiang (q1jiang@health.ucsd.edu).
Materials availability
All custom plasmids generated from this study will be available upon request.
Data and code availability
The RNA-sequencing data used in this study has been deposited (GEO: GSE221112). The TARGET T-ALL datasets used for A-to-I RNA editing analysis can be obtained from dbGaP (phs000464).
This paper does not report original code.
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request
EXPERIMENTAL MODEL AND SUBJECT PARTICIPANT DETAILS
Human subjects
Patient T-ALL samples were obtained from consenting patients at the University of California, San Diego in accordance with an approved human research protections program Institutional Review Board approved protocol (#130794) that meets the requirements as stated in 45 CFR 46.404 and 21 CFR 50.51. De-identified (IRB exempt) human cord blood samples were purchased as purified CD34+ cells from AllCells Inc or StemCell Techologies Inc. Detailed patient information can be found in Table S2.
Animal models and experiments
All mouse studies were conducted under protocols approved by the Institutional Animal Care and Use Committee (IACUC) of the University of California, San Diego and were in compliance with federal regulations regarding the care and use of laboratory animals: Public Law 99–158, the Health Research Extension Act, and Public Law 99–198, the Animal Welfare Act which is regulated by USDA, APHIS, CFR, Title 9, Parts 1, 2, and 3. Immunocompromised Rag2−/−γc−/− mice were bred and maintained in the Sanford Consortium for Regenerative Medicine vivarium according to IACUC approved protocols of the University of California, San Diego. Neonatal mice of both sexes were used in the study. T-ALL CD34+ or CD45+ cells were injected intrahepatically into 2–3 days old neonatal Rag2−/−γc−/− mice. Leukemic engraftment was quantified by FACS analysis-based peripheral blood screening of human CD45+ population starting from week 6 for every 2 weeks until the engraftment exceeded 1%. Mice were then humanely sacrificed, and cells were collected from hematological organs (bone marrow, spleen and thymus) for FACS analysis.
METHOD DETAILS
Patient sample preparation
Peripheral blood mononuclear cells (PBMC) were extracted by Ficoll density centrifugation. Cord blood were purchased as purified CD34+ cells from AllCells Inc or StemCell Techologies Inc. CD45+ and CD34+ cells were purified using magnetic columns (MACS, Miltenyi); CD34+LIN− and CD34−LIN+ FACS sorted with human-specific antibody according to published methods in FACSAria II.15
Cell culture
CUTTL1, SUP-T1 and Jurkat human cell lines were cultured in 37°C in RPMI 1640 supplemented with 10% FBS and 2 mM L-glutamine and maintained. MS5-DLL1 and MS5-DLL4 were maintained in high glucose DMEM with 10% FBS and 1X penicillin-streptomycin according to previous protocol.38,86 293T cells were cultured in 37°C in DMEM supplemented with 10% FBS, 1X penicillin-streptomycin according to ATCC protocol. All cell lines were confirmed to be mycoplasma-free with repeated testing and authenticated by short-tandem repeat (STR) profiling. ADAR1 knockout cell line was generated by Sythego. Wildtype ADAR1 and mutant ADAR1E912A were introduced into the knockout Jurkat cells by transduction of wild-type and mutant ADAR1E912A lentivirus. Stable ADAR1 expression were confirmed by RT-qPCR and western blot every 5 passages.
Lentiviral construct and overexpression
Lentiviral vectors (pLV-shRNA-EGFP or mCherry:T2A:Puro-U6) was purchased (VectorBuilder) and wild-type and mutant ADAR1E912A (pCDH-EF1-T2A-copGFP) were produced according to published protocol.33 All lentivirus was titer by transduction of HEK293T cells and efficiency was assessed by p24 ELISA and RT-qPCR of the 5′ LTR region. Lentiviral transduction of primary T-ALL or cord blood samples was performed at a MOI of 100–200. The cells were cultured for 3–4 days in 96-well plate (2X105-5X105 cells per well) containing StemPro (Life Technologies) media supplemented with human IL-6, stem cell factor (SCF), Thrombopoietin (TPO) and FLT3 (all from R&D Systems). For T-ALL cell lines, the cells were transduced at a MOI of 20–50 in culture media.
ATO 3D organoid culture
ATO organoid experiments were performed as previously described.38 The MS5 mouse stromal cells were engineered to co-express human DLL1 or DLL4 NOTCH ligand and EGFP marker. MS5-DLL1 and -DLL4 were cultured up to 20 passages and the cells will be authenticated every 5 passages by flow of EGFP signal and examining the DLL1 or DLL4 expression by RT-qPCR or flow cytometry. To generate ATOs, 2–5×103 cells of cord blood CD34+ HSPC or T-ALL CD34+ cells were combined with MS5-DLL1/DLL4 cells at 1:20 ratio. The cell mixture is then seeded on a 0.4 μm Millicell transwell insert (EMD Millipore) and placed into 6-well plate with serum-free culture media supplemented by recombinant IL7 (50 ng/mL) and FLT3 (50 ng/mL). ATOs were cultured up to 20 weeks. The cells were harvested by adding staining media (ice-cold PBS with 2% FBS and 2 mM EDTA) to each well and pipetting to dissociate ATOs. Cells were then immunostained with antibodies (Table S1) and analyzed on a BD Aria Fusion and with FlowJo.
Colony formation assay
To measure multi-lineage potentials of T-ALL LICs, CD34+Lin− and CD34−Lin− cells (500 cells per well) were FACS-sorted and cultured in MethoCult media (StemCell Technologies) for 14 days. The differentiated colonies were scored and counted according to previously published protocols.32 To measure self-renewal capacity, colonies were picked and replated in fresh MethoCult media and secondary colonies were counted after an additional 14 days of culture.
Patient derived xenograft transplantation
To establish T-ALL models, freshly ficolled cells were transplanted intrahepatically into neonatal Rag2−/−γc−/− mice (5×105 – 1×106 per pup) according to our preciously published methods.15,33 Bone marrow, spleen and thymus tissues were harvested after 6–20 weeks and stored in liquid nitrogen. For primary and serial transplant assay, CD34+ or CD45+ cells were transduced with lentiviral vectors for 2–3 days. Cells were harvested in staining media, counted, and equal numbers of live cells per condition were transplanted into recipient mice (5×104 – 1×105 per pup). Only live cells are transplanted to account for any effect due to apoptosis. Of note, some pups were lost due to pre-weaning mortality commonly observed in laboratory mouse breeding strains. Transplanted mice were FACS screened for human engraftment in peripheral blood at 6–10 weeks. Once human engraftment was confirmed (>1% human CD45+ cells in peripheral blood), mice were euthanized, and single cell suspensions of hematopoietic tissues (bone marrow, spleen, and thymus) were analyzed by FACS and FlowJo.
Flow cytometry analysis and sorting
Flow staining was performed in staining media for 30 min on ice in the dark. Cells were blocked using FcR block (Biolegend, San Diego, CA) for 15 min before antibody staining with to a final dilution of 1:25. DAPI solution was added before analysis to exclude dead cell debris. Analysis and sorting was performed on BD Aria Fusion, Aria II or Fortessa. Sorted cells were collected into staining media filled FACS tubes or 1.7mL Eppendorf tubes. The LICs are evaluated by the corresponding cell surface markers (Table S1). For intracellular ADAR1 staining, cells were stained with ethidium monoazide (EMA) for 15 min in the dark and then 15 min under light, followed by cell surface staining. After washing in staining buffer, cells were fixed and permeabilizated with an intracellular buffer set (eBioscience, San Diego, CA) and intracellularly stained with an antibody against ADAR1 (Abcam, ab126745) at 1:100 dilution. Secondary antibody of Alexa 488 or Alexa 647 were used to amplify ADAR1 signals.
RNA extraction and RT-qPCR
Total RNA was isolated using RNeasy Micro kit or Mini kit (Qiagen) and the quality was determined by NanoDrop. Complementary DNA was synthesized according to published methods.15 qRT-PCR was performed in duplicate or triplicate on an CFX384 with the use of SYBR GreenER qPCR SuperMix (Invitrogen), 5 ng of template cDNA and 0.2 μM of each forward and reverse primer. Human specific HPRT primers were used as housekeeping control. Quantitative values were obtained from the cycle number (Cq value) using the Bio-Rad Maestro Software. The RT-qPCR primers are shown in Table S6.
Western blots
Cell lysate (10 μg) was mobilized onto a nitrocellulose membrane after electrophoresis on a 10% SDS- acrylamide gel. The membrane was blocked in 5% BSA/20 mM Tris-HCl for 30 min. The blot was incubated with primary antibody in 5% BSA/20 mM Tris-HCl/0.1% Tween 20 overnight at 4°C, followed by secondary HPR-linked Rabbit IgG antibody (Cell Signaling, #70745) for 2 h at room temperature. Membrane was then incubated in SuperSignal West Femto Substrate (ThermoFisher, #34096) for chemiluminescent reading on ChemiDoc System (Bio-Rad).
Interferon stimulation assay
Cells were seeded at a density of 105 in a 12-well plate and treated with a single dose of IFNα, IFNβ, or IFNγ (R&D Systems) at 0.05–500 ng/mL. After 48 h, cells were harvested and analyzed by western blot and RT-qPCR. To measure cell viability, cells were seeded at a density of 2.5×104 in a 96-well plate and treated with IFNβ, followed by MTT assay (Millipore) as described by manufacture’s protocol. 10 mL of MTT labeling reagent was added (final concentration of 5 mg/mL) and incubated in 37°C for 4 h. 100 μL of solubilization solution was added and the cells were incubated overnight. The absorbance was recorded at 570 nm using a microplate reader.
Apoptosis assay
To detect apoptosis activity, Caspase3/7 (CellEvents Caspase 3/7 Green Flow cytometry Assay kit, Invitrogen) was used to quantify active caspase 3 and caspase 7 protein level. Cells was resuspended in 1mL of PBS with 2% FBS and incubated with 1 μL of Caspase3/7 detection reagent for 30 min in the dark at 37°C. 1 μL of 1nM SYTOX AADvanced dead cell staining solution was added prior to flow cytometry acquisition on BD Aria Fusion, Aria II or Fortessa.
Nanostring nCounter
Jurkat cells were collected after 24 h of IFNβ-stimulation and RNA was isolated (RNeasy Plus mini kit, Qiagen). The mRNA levels were directly measured using the Human CAR-T characterization panel kit with additional custom probes (Table S5) from NanoString nCounter gene expression system (NanoString). The differential expression analyses of mRNA were performed using nSolver software (NanoString) and visualized in Prism software.
Immunofluorescence staining
Cells (1–2×103) were harvested in ice-cold PBS and loaded on adhesion slides (Marienfeld Superior) by incubating for 10 min at room temperature. The slides were transferred into a coplin jar containing ice-cold PBS for 5 min and fixed with 4% paraformaldehyde in PBS for 10 min at room temperature. Immunofluorescence was performed by immersing slides in PBST (1x PBS with 0.05% Tween 20). Slides were overlaid with blocking solution (2% fetal bovine serum in PBST) for 1 h at room temperature. After washing, primary antibody was added to the slides and incubated overnight at 4C. Secondary antibody was overlaid to spotted cells for 1 h in the dark. DAPI was added and the slides were sealed with a coverslip. Imaging was performed using a Keyence or Nikon A1R confocal microscope. The intensity and numbers of dsRNA foci were caculated using ImageJ software.
Whole RNA-sequencing
Samples with RNA integrity numbers (RIN) ≥ 7 will be processed using SMART cDNA synthesis and NEBNext paired-end DNA Sample Prep Kit to prepare libraries. RNA-sequencing were performed on NovaSeq 6000 S4 with 150bp paired-end reads. T-ALL RNA sequencing dataset were obtained from data generated by the Therapeutically Applicable Research to Generate Effective Treatments (https://ocg.cancer.gov/programs/target) initiative, phs000464. The data used for this analysis are available at https://portal.gdc.cancer.gov/projects.” The minimal reads per sample was 50M to ensure optimal RNA editing calling.
Transcript and gene quantification and differential expression
Reads were aligned using STAR’s two-pass alignment method, using the GRCh38.84 reference genome and corresponding Ensembl GTF.87,88 STAR was used to output a sorted genome-coordinate based BAM file, as well as a transcriptome-coordinate based BAM file.78 STAR also was used to output the number of reads aligned to each gene for gene expression quantification. STAR settings were based on those used for the ENCODE STAR-RSEM pipeline. The R BioConductor packages EdgeR89 and limma90 were used to implement the limma-voom method for differential expression analysis. Low expressed genes with counts per million (cpm) < 1 in at least 1 of the samples were filtered out and then trimmed mean of M-values (TMM)91 normalization was applied. The infer_experiment.py script from the RSeQC package was used to confirm the strandedness option corresponding to the correct read counts80,79 and to confirm the forward strand probability for input to RSEM. The total reads per million (TPM)92 over the total collapsed exonic regions represent the ‘gene’ expression level. Significant differentially expressed genes (p < 0.05) with log fold value for each comparison was used to perform Gene Set Enrichment Analysis. The R Bioconductor packages DOSE1 and clusterProfile2 was used to implement GSEA and visualize the results based on MSigDb and Disease ontology database. Heatmaps visualize the log2(TPM+1) transformed TPM quantity from RSEM for each feature and were generated using GENE-E with default settings for a row and column clustered heatmap and dendrogram.
RNA editing analysis
Coordinates from the DARNED and RADAR databases were combined and converted to GRCh38 using Crossmap.82–84 The resulting coordinates were used as input to the REDItoolKnown.py script from the REDItools package to determine the number of A, C, G, and T base calls at each coordinate.81 Only coordinates with coverage greater than or equal to 5 in all samples for a given comparison were reported. The percentage of bases called as G at bases with reference A was reported as variant allele frequency (VAF). Coordinates with a percentage G of 0 in all samples for a given sample were not reported. Using percentage G at a coordinate as an input metric, the mean percentage G in each group, the log2 fold change of percentage G of one group versus another, the p values, and minus log10 p values by both the Wilcox and student t-tests were recorded for each coordinate similar to published methods.32 Coordinates were annotated with the name of the closest gene using bedtools closest and bedtools intersect.85
QUANTIFICATION AND STATISTICAL ANALYSIS
All experiments were performed with at least two biological or experimental replicates, with specific number of replicates stated in the figure legends. Unless otherwise stated, the statistical analyses were performed using GraphPad Prism (v7.0) and statistical significance were determined at p value <0.05, with specific statistical test stated in the figure legends.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| β-actin | Abcam | Cat #ab8227; RRID: AB_2305186 |
| ADAR1 | Abcam | Cat #ab126745; RRID: AB_11145661 |
| ADAR1 | Cell Signaling | Cat #14175; RRID: AB_2722520 |
| PARP1 | Abcam | Cat #32138; RRID: AB_777101 |
| PKR | Invitrogen | Cat #PA581499; RRID: AB_2788706 |
| MDA5 | Abcam | Cat #79055; RRID: AB_1640683 |
| RIG-I | Abcam | Cat #180675; RRID: N/A |
| J2 dsRNA antibody | Millipore | Cat #MABE1134; RRID: AB_2819101 |
| eIF1a | Abcam | Cat # ab5369; RRID: AB_304838 |
| Phosphor S51 eIF1a | Abcam | Cat # ab32157; RRID: AB_732117 |
| Goat anti-Rabbit Alexa 488 secondary antibody | ThermoFisher | Cat #A32731; RRID: AB_2633280 |
| Goat anti-Rabbit Alexa 647 secondary antibody | ThermoFisher | Cat #32733; RRID: AB_2633282 |
| CD45-APC | Invitrogen | Cat #MHCD4505; RRID: AB_10372216 |
| CD34-APC | BD | Cat #340441; RRID: AB_400514 |
| CD34-PE | BD | Cat #555822; RRID: AB_396151 |
| CD38-Pecy7 | BD | Cat #335790; RRID: AB_399969 |
| CD8-Pecy 5.5 | BD | Cat #555368; RRID: AB_395771 |
| CD56-Pecy 5.5 | BD | Cat #555517; RRID: AB_395907 |
| CD4-Pecy 5.5 | BD | Cat #555348; RRID: AB_395753 |
| CD3-Pecy 5.5 | BD | Cat #555334; RRID: AB_395741 |
| CD19-Pecy 5.5 | BD | Cat #555414; RRID: AB_395814 |
| CD2-Pecy 5.5 | BD | Cat #555328; RRID: AB_395735 |
| CD14− PeCP-Cy5.5 | BD | Cat #562692; RRID: AB_2737726 |
| Annexin V - PacBlue | Invitrogen | Cat #A35122; RRID: N/A |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| DH5α Competent E. Coli | Invitrogen | Cat #12297016 |
| Stbl2 Competent E. Coli | Invitrogen | Cat #10268019 |
|
| ||
| Biological samples | ||
|
| ||
| T-ALL Patient samples | Obtained through patient consenting at UCSD Rady Children’s Hospital according to the Institutional Review Board-approved protocols. | Table S2 |
|
| ||
| Critical commercial assays | ||
|
| ||
| nCounter CAR-T Kit | NanoString | Cat# 132001; PPL-048 |
| SYBR GreenER qPCR SuperMix | Invitrogen | Cat #11761–500 |
| SuperScript III First-Strand Synthesis SuperMix for qRT-PCR | Invitrogen | Cat #11752–250 |
| Intracellular Fixation and Permeabilization buffer set | eBioscience | Cat #88–8824-00 |
| MTT Cell Proliferation Assay | Sigma Millipore | Cat #11465007001 |
| CellEvent™ Caspase-3/7 Green Flow Cytometry Assay Kit | Invitrogen | Cat #C10427 |
|
| ||
| Deposited data | ||
|
| ||
| NCI TARGET T-ALL | https://portal.gdc.cancer.gov/projects | phs000464 |
| T-ALL LIC shADAR1 | This paper | GSE221112 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| SUP-T1 | ATCC | Cat # CRL-1942; RRID:CVCL_1714 |
| CUTTL1 | Millipore Sigma | Cat # SCC286; RRID:CVCL_4966 |
| Jurkat wildtype | ATCC | Cat # TIB-152; RRID:CVCL_0367 |
| Jurkat ADAR1 knockout | This paper | N/A |
| Jurkat ADAR1 E912A | This paper | N/A |
| 293T | ATCC | Cat #CRL-3216; RRID: CVCL_0063 |
| MS-DLL1 | Crooks laboratory | RRID: CVCL_VR88 |
| MS-DLL4 | Crooks laboratory | N/A |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Rag2−/−γc−/− | Jamieson laboratory | N/A |
|
| ||
| Oligonucleotides | ||
|
| ||
| Primers used in RT-qPCR | This paper | Table S6 |
|
| ||
| Recombinant DNA | ||
|
| ||
| pCDH-EF1-MCS-T2A-copGFP | SBI System Biosciences | Cat #CD521A-1 |
| pCDH-ADAR1 WT | Zipeto et al., 2016 | N/A |
| pCDH-ADAR1E912A | Zipeto et al., 2016 | N/A |
| pLV-shRNA scramble-EGFP:T2A:Puro-U6 | VectorBuilder | Cat #VB191004–1040mdr |
| pLV-shADAR1:T2A:Puro-U6 | VectorBuilder | Cat #VB190813–1162xpr |
| pLV-shADAR1-EGFP:T2A:Puro-U6 | VectorBuilder | Cat #VB191003–1280naj |
| pLV-shMDA5-mCherry:T2A:Puro-U6 | VectorBuilder | Cat#VB231006–1149wvs |
| pLV-shPKR-mCherry:T2A:Puro-U6 | VectorBuilder | Cat #VB220307–1256dsd |
|
| ||
| Software and algorithms | ||
|
| ||
| cutadapt | Martin, 2011 | https://github.com/marcelm/cutadapt |
| samtools | Li et al., 2009 | http://www.htslib.org/ |
| STAR | Dobin et al., 201378 | https://github.com/alexdobin/STAR |
| RSEM | Li & Dewey, 201179 | https://deweylab.github.io/RSEM/ |
| ENCODE long RNA-seq Pipeline | https://github.com/ENCODE-DCC/long-rna-seq-pipeline | |
| RSeQC | Wang et al., 201280 | http://rseqc.sourceforge.net/ |
| REDItools | Picardi & Pesole, 201381 | http://srv00.recas.ba.infn.it/reditools/ |
| CrossMap | Zhao et a., 201482 | http://crossmap.sourceforge.net/ |
| DARNED | Kiran & Baranov, 201083 | http://darned.ucc.ie/ |
| RADAR | Ramaswami &Li, 201484 | http://rnaedit.com/ |
| bedtools | Quinlan & Hall, 201085 | http://bedtools.readthedocs.io/en/latest/ |
| EdgeR | Robinson et al., 2014 | http://bioconductor.org/packages/release/bioc/html/edgeR.html |
| GENE-E | https://software.broadinstitute.org/GENE-E/ | |
| trackViewer | Ou & Zhu, 2019 | https://www.bioconductor.org/packages/release/bioc/html/trackViewer.html |
| Matplotlib | Hunter, 2007 | https://matplotlib.org/ |
| Seaborn | Waskom, 2021 | https://seaborn.pydata.org/index.html |
| gplots | Warnes et al., 2022 | https://github.com/talgalili/gplots |
| DOSE | Yu et al., 2015 | http://bioconductor.org/packages/release/bioc/html/DOSE.html |
| clusterProfiler | Wu et al., 2021 Yu et al., 2021 |
http://bioconductor.org/packages/release/bioc/html/clusterProfiler.html |
Highlights.
Elevated A-to-I RNA modifications are associated with increased risk of relapse
Loss of ADAR1 impairs LIC self-renewal partly via ADAR1-MDA5 axis
Cell-intrinsic level of MDA5 dictates the dependence of LICs on ADAR1-MDA5 axis
RNA-editing-independent activity suppresses ISGs
ACKNOWLEDGMENTS
The authors wish to thank Dr. Robert Signer and Dr. Shizhen Emily Wang for scientific advice and the Scripps Genomics Core, UCSD Institute for Genomic Medicine, and the UCSD Center for Computational Biology & Bioinformatics for library construction, sequencing, and bioinformatics analyses. We are also graceful for Dr. Gay Crooks for sharing the MS5-DLL1 and MS5-DLL4 cells. This work was supported by NIH/NCI K22CA229606 (Q.J.), the Hartwell Foundation (Q.J.), the American Society of Hematology (Q.J.), the Leukemia Research Foundation (Q.J.), UCSD Senate Grants (2021 and 2022, Q.J.), the Altman Clinical and Translational Research Institute (NIH 2UL1TR001442–06 of CTSA, Q.J. and K.M.F.), the JM Foundation (Q.J.), NIH/NCI R01CA205944 (C.J.), The Swedish Childhood Cancer Foundation (Barncancerfonden) (TJ2014–0024 and PR2017–0086, F.H.), and the Märta och Gunnar V. Philipson Foundation (F.H.).
Footnotes
DECLARATION OF INTERESTS
C.J. has served as an advisor for AbbVie, Bristol-Myers Squibb, and Celgene; was a co-founder of Impact Biomedicines and is a co-founder of Aspera Biomedicines; and received royalties from Forty Seven, Inc., and the Jamieson laboratory received unrestricted research grant funding from Janssen, Inc.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2024.113704.
REFERENCES
- 1.Karrman K, and Johansson B (2017). Pediatric T-cell acute lymphoblastic leukemia. Genes Chromosomes Cancer 56, 89–116. [DOI] [PubMed] [Google Scholar]
- 2.Hefazi M, and Litzow MR (2018). Recent Advances in the Biology and Treatment of T Cell Acute Lymphoblastic Leukemia. Curr. Hematol. Malig. Rep. 13, 265–274. [DOI] [PubMed] [Google Scholar]
- 3.McMahon CM, and Luger SM (2019). Relapsed T Cell ALL: Current Approaches and New Directions. Curr. Hematol. Malig. Rep. 14, 83–93. [DOI] [PubMed] [Google Scholar]
- 4.Ma W, Gutierrez A, Goff DJ, Geron I, Sadarangani A, Jamieson CAM, Court AC, Shih AY, Jiang Q, Wu CC, et al. (2012). NOTCH1 signaling promotes human T-cell acute lymphoblastic leukemia initiating cell regeneration in supportive niches. PLoS One 7, e39725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vicente C, and Cools J (2015). The origin of relapse in pediatric T-cell acute lymphoblastic leukemia. Haematologica 100, 1373–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goossens S, and Van Vlierberghe P (2014). Controlling pre-leukemic thymocyte self-renewal. PLoS Genet. 10, e1004881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jiang Q, Crews LA, Holm F, and Jamieson CHM (2017). RNA editing-dependent epitranscriptome diversity in cancer stem cells. Nat. Rev. Cancer 17, 381–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Paul MS, and Bass BL (1998). Inosine exists in mRNA at tissue-specific levels and is most abundant in brain mRNA. EMBO J. 17, 1120–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Levanon EY, Eisenberg E, Yelin R, Nemzer S, Hallegger M, Shemesh R, Fligelman ZY, Shoshan A, Pollock SR, Sztybel D, et al. (2004). Systematic identification of abundant A-to-I editing sites in the human transcriptome. Nat. Biotechnol. 22, 1001–1005. [DOI] [PubMed] [Google Scholar]
- 10.Polson AG, Crain PF, Pomerantz SC, McCloskey JA, and Bass BL (1991). The mechanism of adenosine to inosine conversion by the double-stranded RNA unwinding/modifying activity: a high-performance liquid chromatography-mass spectrometry analysis. Biochemistry 30, 11507–11514. [DOI] [PubMed] [Google Scholar]
- 11.Eisenberg E, and Levanon EY (2018). A-to-I RNA editing - immune protector and transcriptome diversifier. Nat. Rev. Genet. 19, 473–490. [DOI] [PubMed] [Google Scholar]
- 12.Jiang Q, Isquith J, Ladel L, Mark A, Holm F, Mason C, He Y, Mondala P, Oliver I, Pham J, et al. (2021). Inflammation-driven deaminase deregulation fuels human pre-leukemia stem cell evolution. Cell Rep. 34, 108670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Han L, Diao L, Yu S, Xu X, Li J, Zhang R, Yang Y, Werner HMJ, Eterovic AK, Yuan Y, et al. (2015). The Genomic Landscape and Clinical Relevance of A-to-I RNA Editing in Human Cancers. Cancer Cell 28, 515–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nishikura K (2016). A-to-I editing of coding and non-coding RNAs by ADARs. Nat. Rev. Mol. Cell Biol. 17, 83–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jiang Q, Isquith J, Zipeto MA, Diep RH, Pham J, Delos Santos N, Reynoso E, Chau J, Leu H, Lazzari E, et al. (2019). Hyper-Editing of Cell-Cycle Regulatory and Tumor Suppressor RNA Promotes Malignant Progenitor Propagation. Cancer Cell 35, 81–94.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang IX, So E, Devlin JL, Zhao Y, Wu M, and Cheung VG (2013). ADAR regulates RNA editing, transcript stability, and gene expression. Cell Rep. 5, 849–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chung H, Calis JJA, Wu X, Sun T, Yu Y, Sarbanes SL, Dao Thi VL, Shilvock AR, Hoffmann HH, Rosenberg BR, and Rice CM (2018). Human ADAR1 Prevents Endogenous RNA from Triggering Translational Shutdown. Cell 172, 811–824.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rice GI, Kasher PR, Forte GMA, Mannion NM, Greenwood SM, Szynkiewicz M, Dickerson JE, Bhaskar SS, Zampini M, Briggs TA, et al. (2012). Mutations in ADAR1 cause Aicardi-Goutieres syndrome associated with a type I interferon signature. Nat. Genet. 44, 1243–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hartner JC, Walkley CR, Lu J, and Orkin SH (2009). ADAR1 is essential for the maintenance of hematopoiesis and suppression of interferon signaling. Nat. Immunol. 10, 109–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liddicoat BJ, Piskol R, Chalk AM, Ramaswami G, Higuchi M, Hartner JC, Li JB, Seeburg PH, and Walkley CR (2015). RNA editing by ADAR1 prevents MDA5 sensing of endogenous dsRNA as nonself. Science 349, 1115–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mehdipour P, Marhon SA, Ettayebi I, Chakravarthy A, Hosseini A, Wang Y, de Castro FA, Loo Yau H, Ishak C, Abelson S, et al. (2020). Epigenetic therapy induces transcription of inverted SINEs and ADAR1 dependency. Nature 588, 169–173. [DOI] [PubMed] [Google Scholar]
- 22.Dhir A, Dhir S, Borowski LS, Jimenez L, Teitell M, Rötig A, Crow YJ, Rice GI, Duffy D, Tamby C, et al. (2018). Mitochondrial double-stranded RNA triggers antiviral signalling in humans. Nature 560, 238–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kang R, and Tang D (2012). PKR-dependent inflammatory signals. Sci. Signal. 5, pe47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dias Junior AG, Sampaio NG, and Rehwinkel J (2019). A Balancing Act: MDA5 in Antiviral Immunity and Autoinflammation. Trends Microbiol. 27, 75–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hu SB, Heraud-Farlow J, Sun T, Liang Z, Goradia A, Taylor S, Walkley CR, and Li JB (2023). ADAR1p150 prevents MDA5 and PKR activation via distinct mechanisms to avert fatal autoinflammation. Mol. Cell 83, 3869–3884.e7. [DOI] [PubMed] [Google Scholar]
- 26.Bazak L, Haviv A, Barak M, Jacob-Hirsch J, Deng P, Zhang R, Isaacs FJ, Rechavi G, Li JB, Eisenberg E, and Levanon EY (2014). A-to-I RNA editing occurs at over a hundred million genomic sites, located in a majority of human genes. Genome Res. 24, 365–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lamers MM, van den Hoogen BG, and Haagmans BL (2019). ADAR1: “Editor-in-Chief” of Cytoplasmic Innate Immunity. Front. Immunol. 10, 1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brümmer A, Yang Y, Chan TW, and Xiao X (2017). Structure-mediated modulation of mRNA abundance by A-to-I editing. Nat. Commun. 8, 1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mannion NM, Greenwood SM, Young R, Cox S, Brindle J, Read D, Nellåker C, Vesely C, Ponting CP, McLaughlin PJ, et al. (2014). The RNA-editing enzyme ADAR1 controls innate immune responses to RNA. Cell Rep. 9, 1482–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.George CX, Ramaswami G, Li JB, and Samuel CE (2016). Editing of Cellular Self-RNAs by Adenosine Deaminase ADAR1 Suppresses Innate Immune Stress Responses. J. Biol. Chem. 291, 6158–6168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maurano M, Snyder JM, Connelly C, Henao-Mejia J, Sidrauski C, and Stetson DB (2021). Protein kinase R and the integrated stress response drive immunopathology caused by mutations in the RNA deaminase ADAR1. Immunity 54, 1948–1960.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jiang Q, Crews LA, Barrett CL, Chun HJ, Court AC, Isquith JM, Zipeto MA, Goff DJ, Minden M, Sadarangani A, et al. (2013). ADAR1 promotes malignant progenitor reprogramming in chronic myeloid leukemia. Proc. Natl. Acad. Sci. USA 110, 1041–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zipeto MA, Court AC, Sadarangani A, Delos Santos NP, Balaian L, Chun HJ, Pineda G, Morris SR, Mason CN, Geron I, et al. (2016). ADAR1 Activation Drives Leukemia Stem Cell Self-Renewal by Impairing Let-7 Biogenesis. Cell Stem Cell 19, 177–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sun T, Yu Y, Wu X, Acevedo A, Luo JD, Wang J, Schneider WM, Hurwitz B, Rosenberg BR, Chung H, and Rice CM (2021). Decoupling expression and editing preferences of ADAR1 p150 and p110 isoforms. Proc. Natl. Acad. Sci. USA 118, e2021757118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pestal K, Funk CC, Snyder JM, Price ND, Treuting PM, and Stetson DB (2015). Isoforms of RNA-Editing Enzyme ADAR1 Independently Control Nucleic Acid Sensor MDA5-Driven Autoimmunity and Multi-organ Development. Immunity 43, 933–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.de Reuver R, Dierick E, Wiernicki B, Staes K, Seys L, De Meester E, Muyldermans T, Botzki A, Lambrecht BN, Van Nieuwerburgh F, et al. (2021). ADAR1 interaction with Z-RNA promotes editing of endogenous double-stranded RNA and prevents MDA5-dependent immune activation. Cell Rep. 36, 109500. [DOI] [PubMed] [Google Scholar]
- 37.Tang Q, Rigby RE, Young GR, Hvidt AK, Davis T, Tan TK, Bridgeman A, Townsend AR, Kassiotis G, and Rehwinkel J (2021). Adenosine-to-inosine editing of endogenous Z-form RNA by the deaminase ADAR1 prevents spontaneous MAVS-dependent type I interferon responses. Immunity 54, 1961–1975.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Seet CS, He C, Bethune MT, Li S, Chick B, Gschweng EH, Zhu Y, Kim K, Kohn DB, Baltimore D, et al. (2017). Generation of mature T cells from human hematopoietic stem and progenitor cells in artificial thymic organoids. Nat. Methods 14, 521–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xu LD, and Öhman M (2018). ADAR1 Editing and its Role in Cancer. Genes 10, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Song B, Shiromoto Y, Minakuchi M, and Nishikura K (2022). The Role of RNA Editing Enzyme ADAR1 in Human Disease, 13 (Wiley Interdiscip Rev RNA; ), p. e1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tan MH, Li Q, Shanmugam R, Piskol R, Kohler J, Young AN, Liu KI, Zhang R, Ramaswami G, Ariyoshi K, et al. (2017). Dynamic landscape and regulation of RNA editing in mammals. Nature 550, 249–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jain N, Lamb AV, O’Brien S, Ravandi F, Konopleva M, Jabbour E, Zuo Z, Jorgensen J, Lin P, Pierce S, et al. (2016). Early T-cell precursor acute lymphoblastic leukemia/lymphoma (ETP-ALL/LBL) in adolescents and adults: a high-risk subtype. Blood 127, 1863–1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Girardi T, Vicente C, Cools J, and De Keersmaecker K (2017). The genetics and molecular biology of T-ALL. Blood 129, 1113–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu Y, Easton J, Shao Y, Maciaszek J, Wang Z, Wilkinson MR, McCastlain K, Edmonson M, Pounds SB, Shi L, et al. (2017). The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nat. Genet. 49, 1211–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ferrando AA, Neuberg DS, Staunton J, Loh ML, Huard C, Raimondi SC, Behm FG, Pui CH, Downing JR, Gilliland DG, et al. (2002). Gene expression signatures define novel oncogenic pathways in T cell acute lymphoblastic leukemia. Cancer Cell 1, 75–87. [DOI] [PubMed] [Google Scholar]
- 46.Lu WC, Xie H, Yuan C, Li JJ, Li ZY, and Wu AH (2020). Genomic landscape of the immune microenvironments of brain metastases in breast cancer. J. Transl. Med. 18, 327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Passegué E, Jamieson CHM, Ailles LE, and Weissman IL (2003). Normal and leukemic hematopoiesis: are leukemias a stem cell disorder or a reacquisition of stem cell characteristics? Proc. Natl. Acad. Sci. USA 100, 11842–11849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bajaj J, Diaz E, and Reya T (2020). Stem cells in cancer initiation and progression. J. Cell Biol. 219, e201911053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Verovskaya EV, Dellorusso PV, and Passegué E (2019). Losing Sense of Self and Surroundings: Hematopoietic Stem Cell Aging and Leukemic Transformation. Trends Mol. Med. 25, 494–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Armstrong F, Brunet de la Grange P, Gerby B, Rouyez MC, Calvo J, Fontenay M, Boissel N, Dombret H, Baruchel A, Landman-Parker J, et al. (2009). NOTCH is a key regulator of human T-cell acute leukemia initiating cell activity. Blood 113, 1730–1740. [DOI] [PubMed] [Google Scholar]
- 51.Sarma NJ, Takeda A, and Yaseen NR (2010). Colony forming cell (CFC) assay for human hematopoietic cells. J. Vis. Exp. 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jiang Y, Wang Z, Chen X, Wang W, and Wang X (2018). ADAR1 silencing-induced HUVEC apoptosis is mediated by FGFR2 under hypoxia stress. Drug Des. Dev. Ther. 12, 4181–4189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sakurai M, Shiromoto Y, Ota H, Song C, Kossenkov AV, Wickramasinghe J, Showe LC, Skordalakes E, Tang HY, Speicher DW, and Nishikura K (2017). ADAR1 controls apoptosis of stressed cells by inhibiting Staufen1-mediated mRNA decay. Nat. Struct. Mol. Biol. 24, 534–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.de Reuver R, Verdonck S, Dierick E, Nemegeer J, Hessmann E, Ahmad S, Jans M, Blancke G, Van Nieuwerburgh F, Botzki A, et al. (2022). ADAR1 prevents autoinflammation by suppressing spontaneous ZBP1 activation. Nature 607, 784–789. [DOI] [PubMed] [Google Scholar]
- 55.Park SM, Cho H, Thornton AM, Barlowe TS, Chou T, Chhangawala S, Fairchild L, Taggart J, Chow A, Schurer A, et al. (2019). IKZF2 Drives Leukemia Stem Cell Self-Renewal and Inhibits Myeloid Differentiation. Cell Stem Cell 24, 153–165.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.McCormack MP, Shields BJ, Jackson JT, Nasa C, Shi W, Slater NJ, Tremblay CS, Rabbitts TH, and Curtis DJ (2013). Requirement for Lyl1 in a model of Lmo2-driven early T-cell precursor ALL. Blood 122, 2093–2103. [DOI] [PubMed] [Google Scholar]
- 57.Komorowska K, Doyle A, Wahlestedt M, Subramaniam A, Debnath S, Chen J, Soneji S, Van Handel B, Mikkola HKA, Miharada K, et al. (2017). Hepatic Leukemia Factor Maintains Quiescence of Hematopoietic Stem Cells and Protects the Stem Cell Pool during Regeneration. Cell Rep. 21, 3514–3523. [DOI] [PubMed] [Google Scholar]
- 58.Porath HT, Carmi S, and Levanon EY (2014). A genome-wide map of hyper-edited RNA reveals numerous new sites. Nat. Commun. 5, 4726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Carmi S, Borukhov I, and Levanon EY (2011). Identification of widespread ultra-edited human RNAs. PLoS Genet. 7, e1002317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gannon HS, Zou T, Kiessling MK, Gao GF, Cai D, Choi PS, Ivan AP, Buchumenski I, Berger AC, Goldstein JT, et al. (2018). Identification of ADAR1 adenosine deaminase dependency in a subset of cancer cells. Nat. Commun. 9, 5450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ahmad S, Mu X, and Hur S (2021). The Role of RNA Editing in the Immune Response. Methods Mol. Biol. 2181, 287–307. [DOI] [PubMed] [Google Scholar]
- 62.Ishizuka JJ, Manguso RT, Cheruiyot CK, Bi K, Panda A, Iracheta-Vellve A, Miller BC, Du PP, Yates KB, Dubrot J, et al. (2019). Loss of ADAR1 in tumours overcomes resistance to immune checkpoint blockade. Nature 565, 43–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kim JI, Nakahama T, Yamasaki R, Costa Cruz PH, Vongpipatana T, Inoue M, Kanou N, Xing Y, Todo H, Shibuya T, et al. (2021). RNA editing at a limited number of sites is sufficient to prevent MDA5 activation in the mouse brain. PLoS Genet. 17, e1009516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ota H, Sakurai M, Gupta R, Valente L, Wulff BE, Ariyoshi K, Iizasa H, Davuluri RV, and Nishikura K (2013). ADAR1 forms a complex with Dicer to promote microRNA processing and RNA-induced gene silencing. Cell 153, 575–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Orlandi C, Barbon A, and Barlati S (2012). Activity regulation of adenosine deaminases acting on RNA (ADARs). Mol. Neurobiol. 45, 61–75. [DOI] [PubMed] [Google Scholar]
- 66.Yang S, Deng P, Zhu Z, Zhu J, Wang G, Zhang L, Chen AF, Wang T, Sarkar SN, Billiar TR, and Wang Q (2014). Adenosine deaminase acting on RNA 1 limits RIG-I RNA detection and suppresses IFN production responding to viral and endogenous RNAs. J. Immunol. 193, 3436–3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pujantell M, Riveira-Muñoz E, Badia R, Castellví M, Garcia-Vidal E, Sirera G, Puig T, Ramirez C, Clotet B, Esté JA, and Ballana E (2017). RNA editing by ADAR1 regulates innate and antiviral immune functions in primary macrophages. Sci. Rep. 7, 13339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wang Y, and Samuel CE (2009). Adenosine deaminase ADAR1 increases gene expression at the translational level by decreasing protein kinase PKR-dependent eIF-2alpha phosphorylation. J. Mol. Biol. 393, 777–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Costa-Mattioli M, and Walter P (2020). The integrated stress response: From mechanism to disease. Science 368, eaat5314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chen YG, and Hur S (2022). Cellular origins of dsRNA, their recognition and consequences. Nat. Rev. Mol. Cell Biol. 23, 286–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.De Bie J, Demeyer S, Alberti-Servera L, Geerdens E, Segers H, Broux M, De Keersmaecker K, Michaux L, Vandenberghe P, Voet T, et al. (2018). Single-cell sequencing reveals the origin and the order of mutation acquisition in T-cell acute lymphoblastic leukemia. Leukemia 32, 1358–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhang J, Ding L, Holmfeldt L, Wu G, Heatley SL, Payne-Turner D, Easton J, Chen X, Wang J, Rusch M, et al. (2012). The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature 481, 157–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Oshima K, Zhao J, Pérez-Durán P, Brown JA, Patiño-Galindo JA, Chu T, Quinn A, Gunning T, Belver L, Ambesi-Impiombato A, et al. (2020). Mutational and functional genetics mapping of chemotherapy resistance mechanisms in relapsed acute lymphoblastic leukemia. Nat. Can. (Ott.) 1, 1113–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Graveley BR, Brooks AN, Carlson JW, Duff MO, Landolin JM, Yang L, Artieri CG, van Baren MJ, Boley N, Booth BW, et al. (2011). The developmental transcriptome of Drosophila melanogaster. Nature 471, 473–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lazzari E, Mondala PK, Santos ND, Miller AC, Pineda G, Jiang Q, Leu H, Ali SA, Ganesan AP, Wu CN, et al. (2017). Alu-dependent RNA editing of GLI1 promotes malignant regeneration in multiple myeloma. Nat. Commun. 8, 1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ward SV, George CX, Welch MJ, Liou LY, Hahm B, Lewicki H, de la Torre JC, Samuel CE, and Oldstone MB (2011). RNA editing enzyme adenosine deaminase is a restriction factor for controlling measles virus replication that also is required for embryogenesis. Proc. Natl. Acad. Sci. USA 108, 331–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kim S, Bai Y, Fan Z, Diergaarde B, Tseng GC, and Park HJ (2021). The microRNA target site landscape is a novel molecular feature associating alternative polyadenylation with immune evasion activity in breast cancer. Briefings Bioinf. 22, bbaa191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Li B, and Dewey CN (2011). RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinf. 12, 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wang L, Wang S, and Li W (2012). RSeQC: quality control of RNA-seq experiments. Bioinformatics 28, 2184–2185. [DOI] [PubMed] [Google Scholar]
- 81.Picardi E, and Pesole G (2013). REDItools: high-throughput RNA editing detection made easy. Bioinformatics 29, 1813–1814. [DOI] [PubMed] [Google Scholar]
- 82.Zhao H, Sun Z, Wang J, Huang H, Kocher JP, and Wang L (2014). CrossMap: a versatile tool for coordinate conversion between genome assemblies. Bioinformatics 30, 1006–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kiran A, and Baranov PV (2010). DARNED: a DAtabase of RNa EDiting in humans. Bioinformatics 26, 1772–1776. [DOI] [PubMed] [Google Scholar]
- 84.Ramaswami G, and Li JB (2014). RADAR: a rigorously annotated database of A-to-I RNA editing. Nucleic Acids Res. 42, D109–D113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bosticardo M, Pala F, Calzoni E, Delmonte OM, Dobbs K, Gardner CL, Sacchetti N, Kawai T, Garabedian EK, Draper D, et al. (2020). Artificial thymic organoids represent a reliable tool to study T-cell differentiation in patients with severe T-cell lymphopenia. Blood Adv. 4, 2611–2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Guo Y, Dai Y, Yu H, Zhao S, Samuels DC, and Shyr Y (2017). Improvements and impacts of GRCh38 human reference on high throughput sequencing data analysis. Genomics 109, 83–90. [DOI] [PubMed] [Google Scholar]
- 88.Aken BL, Ayling S, Barrell D, Clarke L, Curwen V, Fairley S, Fernandez Banet J, Billis K, García Girón C, Hourlier T, et al. (2016). The Ensembl gene annotation system. Database 2016, baw093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, and Smyth GK (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Robinson MD, and Oshlack A (2010). A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 11, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Mortazavi A, Williams BA, McCue K, Schaeffer L, and Wold B (2008). Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 5, 621–628. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA-sequencing data used in this study has been deposited (GEO: GSE221112). The TARGET T-ALL datasets used for A-to-I RNA editing analysis can be obtained from dbGaP (phs000464).
This paper does not report original code.
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request