

Abstract
Nα-aroyl-N-aryl-phenylalanine amides (AAPs) are RNA polymerase inhibitors with activity against Mycobacterium tuberculosis and non-tuberculous mycobacteria. We observed that AAPs rapidly degrade in microsomal suspensions, suggesting that avoiding hepatic metabolism is critical for their effectiveness in vivo. As both amide bonds are potential metabolic weak points of the molecule, we synthesized 16 AAP analogs in which the amide bonds are shielded by methyl or fluoro substituents in close proximity. Some derivatives show improved microsomal stability, while being plasma-stable and non-cytotoxic. In parallel with the metabolic stability studies, the antimycobacterial activity of the AAPs against Mycobacterium tuberculosis, Mycobacterium abscessus, Mycobacterium avium and Mycobacterium intracellulare was determined. The stability data are discussed in relation to the antimycobacterial activity of the panel of compounds and reveal that the concept of steric shielding of the anilide groups by a fluoro substituent has the potential to improve the stability and bioavailability of AAPs.
Keywords: Phenylalanine amides, AAPs, Mycobacterium abscessus, microsomal stability, RNA polymerase
Graphical Abstract
Nα-aroyl-N-aryl-phenylalanine amides (AAPs) are active against numerous mycobacteria including Mycobacterium tuberculosis and Mycobacterium abscessus. As peptides, they are rapidly degraded in human and murine microsomal suspensions. Adding small substituents to the residues adjacent to the amide bonds, in particular at the anilide bond, results in increased stability.
Introduction
Mycobacterium tuberculosis infections remain a significant global health concern1, posing challenges to healthcare systems worldwide due to their persistence, potential for drug resistance, and the burden they impose on resources for diagnosis, treatment, and prevention2–5. Another growing healthcare concern are non-tuberculous mycobacteria (NTM)6, such as Mycobacterium abscessus (Mabs), that can cause severe infections of various organs, foremost in the respiratory tract7 that often require different treatment approaches in comparison to tuberculosis8–11. Patients with underlying lung conditions, such as chronic obstructive pulmonary disease (COPD), cystic fibrosis (CF)12, or a compromised immune system are particularly susceptible to NTM infections13. As the bacteria are commonly found in the environment, NTM are usually acquired from soil and water sources. Patient-to-patient transmission can occur, and this particularly prominent among CF patients14,15. Especially when Mabs is the causative agent, eradication of NTM infections is difficult owing to its inherent resistance to common antimycobacterial drugs16–18. For this reason, new antimycobacterial agents are urgently needed.
MMV688845 (hereafter MMV) was first described as a hit structure against Mycobacterium tuberculosis (Mtb) and provides a potential chemical scaffold for further modification19. Ebright et al tested MMV anti-NTM properties and displayed in vitro activity against Mycobacterium avium (Mavium)20. Screening the Pathogen Box (Medicines for Malaria Venture, Geneva, Switzerland) against Mabs and Mavium21–23 also identified MMV (Scheme 1A) as a promising hit compound against NTM21–23. The closely related analogs D-AAP1 and D-IX336 were reported to inhibit mycobacterial RNA polymerase (RNAP) by targeting its β and β’ subunits24, thus inhibiting transcription. The in vitro generation and genome sequencing of MMV resistant mutants of Mabs Bamboo suggested that the same holds true for the hit compound25. Rifamycins such as rifampicin, rifabutin and rifapentine are RNAP inhibitors that are currently in clinical use against many mycobacterial infections. Since Mabs exhibits intrinsic resistance mechanisms to rifampicin through C23 ribosylation26–28 and naphthohydroquinone oxidation29, a therapy regimen that includes rifamycins is usually not an option. Consequently, there is a need to develop alternative RNAP inhibitors for anti-Mabs therapy. Cross-resistance of AAPs with rifamycins is unlikely as the target binding site of AAPs has been shown to be different24, which has also been proven by in vitro experiments25. Furthermore, the hit compound MMV was active against a variety of Mabs clinical isolates and exhibits bactericidal activity against Mabs in broth and in a macrophage infection model.10 AAPs are also of interest for the treatment of rifamycin-resistant Mtb, since they retain the effective bactericidal mechanism of RNAP inhibition while being chemically distinct.
Scheme 1.
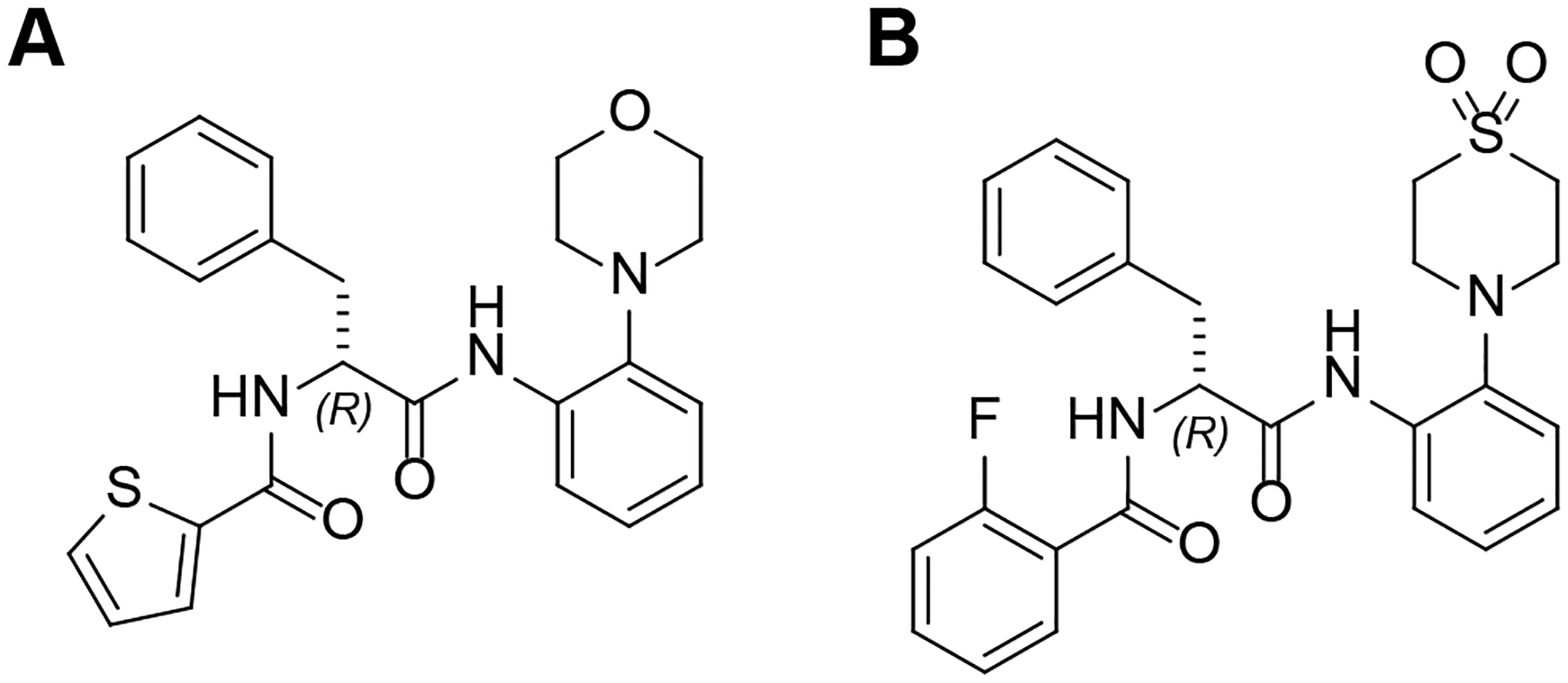
A: Molecular structure of the hit compound MMV. B: Molecular structure of 2.
Recently, we prepared analogs of MMV and obtained compounds with higher antimycobacterial activity, solubility, and plasma stability than the hit compound30. We have established a synthetic pathway that retains the R configuration, which is necessary for the desired antimycobacterial activity, with ee values of 99%30,31. In particular, the introduction of thiomorpholine dioxide instead of morpholine increased the whole cell activity. Compound 2 (Scheme 1B) displays an MIC90 < 1 μM against Mabs and Mtb.
Previous investigations on AAPs revealed that MMV gave insufficient plasma levels after oral administration in male Sprague Dawley rats23,32. We have already presented plasma and microsomal stability data of the hit compound MMV and the highly active compound 24 (in this report called 2) and revealed that MMV degraded quickly in murine plasma, whereas 2 was stable in both murine and human plasma30. Amide moieties of drug candidates are known to be hydrolyzed in blood plasma to form inactive metabolites33. The blood plasma of rodents typically exhibits higher and less specific hydrolase activity than human plasma due to differences in plasma esterases and their expression levels34.
Both MMV and 2 were highly unstable during incubation with human and murine liver microsomes30. AAPs contain two amide bonds which can undergo enzyme-catalyzed hydrolytic cleavage. The anilide is potentially more susceptible, since C-N π-bond overlap of the amide group is weakened by the conjugation of the nitrogen atom with the phenyl group. Serine esterases such as carboxylesterases, which are predominantly found in the liver, are known to catalyze amide cleavage in humans35. Another study also presents evidence for anilide cleavage in liver microsomes resulting in formation of an inactive drug candidate metabolite36.
To address the poor hepatic stability of these compounds, we designed and synthesized a new series of AAP derivatives and performed in vitro characterization of plasma and microsomal stability, microbiological activity assessment against a variety of mycobacteria, cytotoxicity testing as well as solubility screening of the new substances.
Results and Discussion
Derivatization plan for increased metabolic stability
The derivatization strategy is based on analog 2 (Scheme 1B) that we developed, which shows increased in vitro activity against Mabs (MIC90 = 0.78 μM)30 compared to the initial hit MMV (MIC90 = 6.25 μM). To gain structural insight, we subjected 2 to X-ray crystallography (Figure 1). In the crystal, 2 adopts the same conformation with an intramolecular N−H···O hydrogen bond similar to the previously described 2-thiophenoyl analog MMV30. Likewise, the molecules form pseudo centrosymmetric N−H···O hydrogen bond dimers in the solid-state. Owing to the steric demand of the fluorine atom in the ortho position, the 2-fluorobenzene moieties are significantly tilted out of the plane of the attached amide groups. The 1,1-dioxo-1λ6-thiomorpholin-4-yl moiety appears to be primarily responsible for its increased potency and was therefore retained in the new AAPs.
Figure 1.
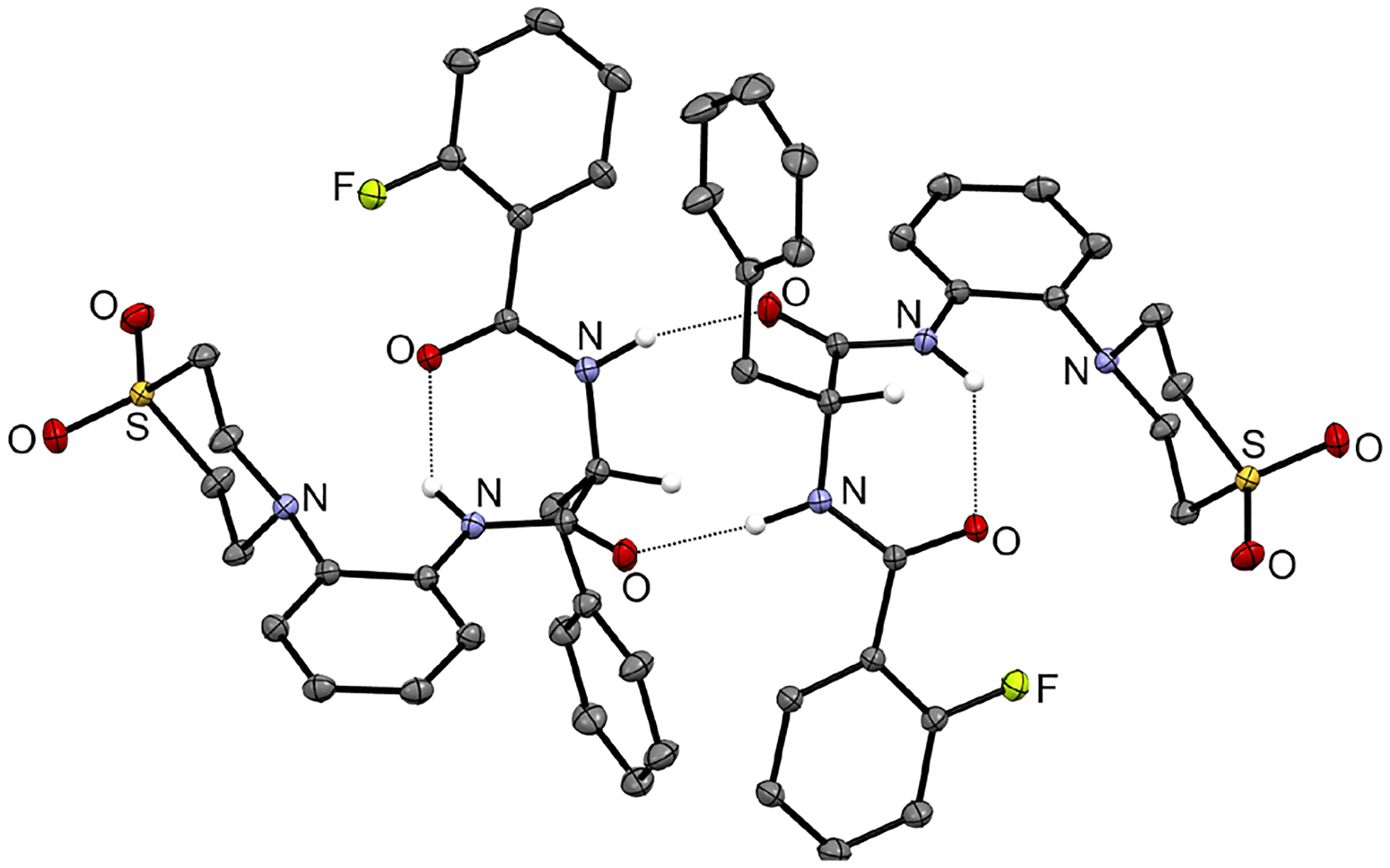
Asymmetric unit of 2. Displacement ellipsoids are drawn at the 50 % probability level. Nitrogen-bound hydrogen atoms and the carbon-bound hydrogen atoms attached to centers of chirality are represented by small spheres of arbitrary radius, otherwise hydrogen atoms are omitted for clarity. Dashed lines represent hydrogen bonds. Colour scheme: C, grey; H, white; N, blue; O, red; F, light green; S, yellow. The crystal structure was refined using NoSpherA2.37,38
We systematically derivatized various positions of the scaffold in the proximity of the amide bonds with the aim of sterically shielding the amide linkage and thereby prevent hydrolysis by amidases and esterases. This involved derivatization of positions 2 and 6, ortho positions to carbonyl, R2 and R3, of aromatic system A as well as the ortho position of aromatic system B (R1) adjacent to the anilide bond (Scheme 2A).
Scheme 2.
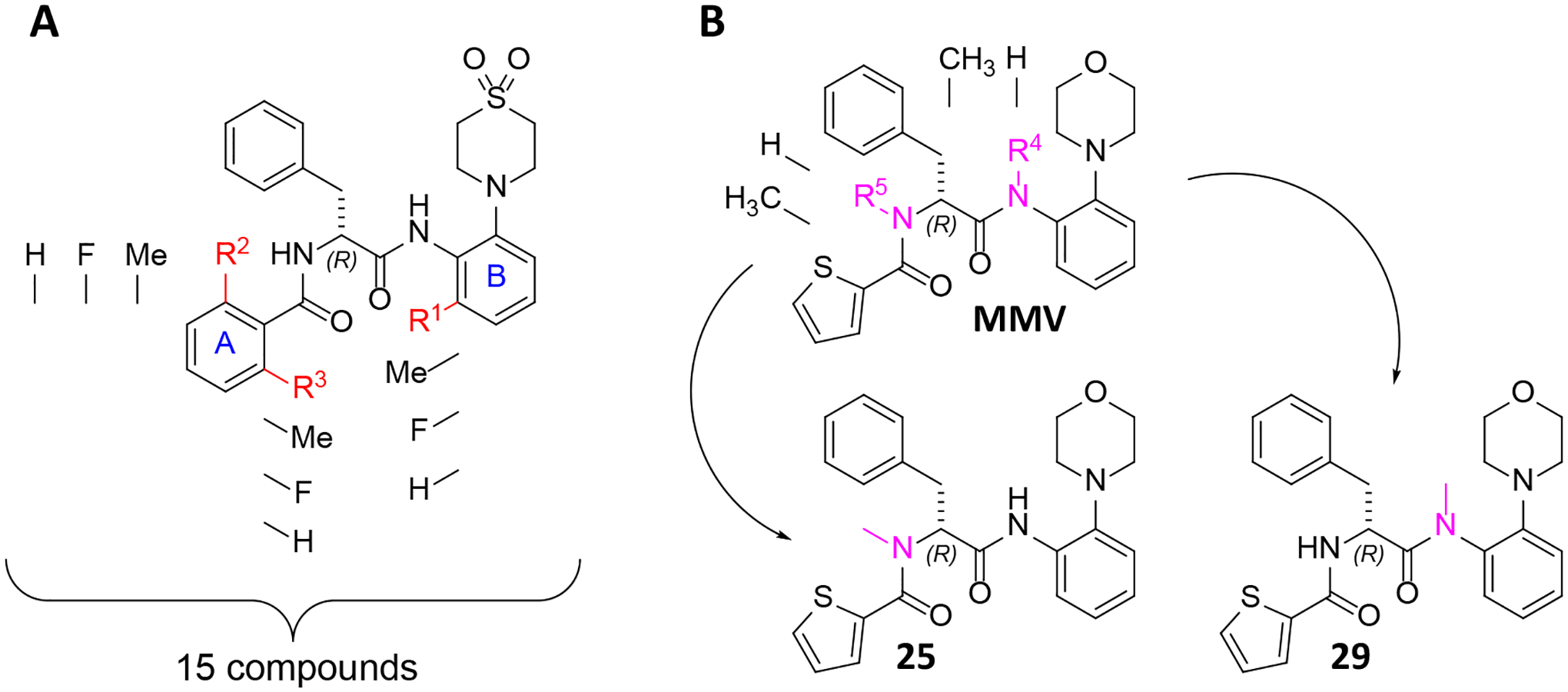
Derivatization strategy of AAPs for increased metabolic stability. A: The scaffold of (2R)-2-[(2-fluorophenyl)formamido]-N-[2-(1,1-dioxo-1λ6-thiomorpholin-4-yl)phenyl]-3-phenylpropanamide is depicted, and the changes at R1, R2, and R3 are indicated. The synthesized derivatives contain either hydrogen, fluoro, or methyl substituents at the respective positions. All conceivable combinations within this range of substituents were synthesized, resulting in a total of 15 compounds. B: Two derivatives of MMV were synthesized by N-methylation at positions R4 and R5. One compound was methylated at R4 (29) and the other one at R5 (25).
This approach was inspired by the development of lidocaine39 in which methyl groups were introduced in order to prevent fast metabolism40–42. Introduction of two methyl groups to procaine amide resulted in a conformational change (a twist of the benzene ring) sufficient to sterically shield the amide bond from hydrolysis by amidases41. Similar to this approach, the introduction of two ortho methyl groups in the aromatic system A could stabilize the amide towards metabolic hydrolysis. The peptides were also N-methylated to increase the stability (Scheme 2B). N-methylation can lead to higher serum stability of peptide like structures43 and a higher stability against peptidases like chymotrypsin, as demonstrated by Haviv et al. in the synthesis of N-methyl leuprolide derivatives44.
Results from Ebright et al. suggest that substituents that are bulkier than hydrogen, fluoro or methyl tend to result in lower antimycobacterial activities (e.g., o-chlorine ↓, 4-indolyl ↓, o-bromine ↓↓, o-ethyl ↓↓↓)20. Thus, we restricted the study to small substituents, preferring fluorine and methyl to chlorine and bromine20 to sterically shield the adjacent amide bond.
To the best of our knowledge, AAPs substituted in the ortho position to the anilide bond at aromatic system B (refer to Scheme 2A) have not been reported so far. In accordance with the approach employed in aromatic system A, we also elected to utilize less voluminous substituents to maximize the likelihood of achieving antimycobacterial activity. The addition of a methyl group is expected to result in a greater shielding effect due to its larger size compared to that of a fluorine atom.11
Synthesis of 4-(2-aminophenyl)- 1λ6-thiomorpholine-1,1-diones
The synthetic sequence commences with the synthesis of an aniline building block substituted with a thiomorpholine dioxide moiety in one ortho-position (Scheme 3). The other ortho-position is substituted with either hydrogen, fluorine atom, or a methyl group (as explained above).
Scheme 3.
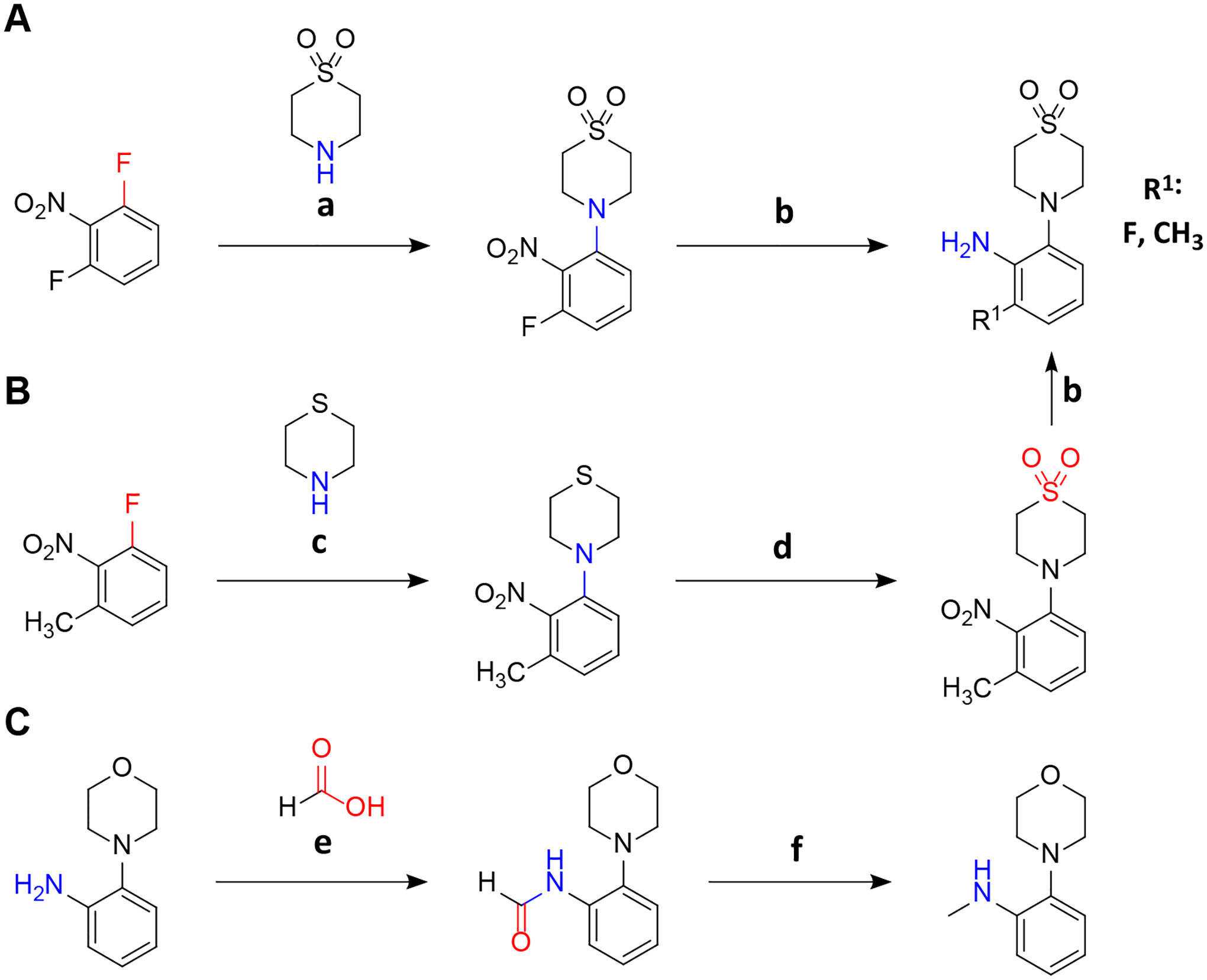
Synthesis of 4-(2-aminophenyl)-1λ6-thiomorpholine-1,1-diones and N-methyl derivatives. A: Synthesis of 3-fluoro derivatives. B: Synthesis of 3-methyl derivatives. C: Synthesis of N-methyl derivatives. a: DIPEA, 3 d, 50 °C; b: EtOH, H2, Pd(OH)2/C 20 %; c: DIPEA, 50 °C, 24 h; d: DCM, −20 °C, mCPBA in DCM added over 30 min; e: HCOOH, HCOONa, room temperature, overnight; f: THF, LiAlH4 1 M in THF dropwise over 30 min, argon, 0 °C to room temperature. For quantities and detailed procedures see Supporting Information.
In the initial step, various halogen-substituted nitrobenzenes were subject to aromatic nucleophilic substitutions. 1,3-Difluoro-2-nitrobenzene reacts smoothly with thiomorpholine dioxide owing to the strong electron deficiency caused by the two fluoro- and nitro substituents (Scheme 3A). The reaction of one equivalent of symmetric 1,3-difluoro-2-nitrobenzene with one equivalent of the nucleophile provides an efficient approach with a 95 % yield of the desired product. The mono-substituted product of the reaction did not undergo a second substitution reaction as it was less electrophilic than 1,3-difluoro-2-nitrobenzene.
Thiomorpholine dioxide is less nucleophilic than the corresponding thioether due to the electron-withdrawing effect of the sulfone group. Consequently, the unsubstituted30 and 3-methyl nitrobenzene which are less electron-deficient than 1,3-difluoro-2-nitrobenzene had to be coupled with nucleophilic thiomorpholine and subsequently oxidized with mCPBA (Scheme 3B). Attempted reactions with thiomorpholine dioxide did not result in the desired product.
The N-methyl aniline moiety was obtained by initial formation of the formic acid amide of 2-morpholinoaniline and subsequent reduction to the secondary amine with LiAlH4 (Scheme 3C)
Amide coupling with N-Boc-(R)-phenylalanine
To couple the previously synthesized anilines with N-Boc-(R)-phenylalanine or N-Boc-N-methyl-(R)-phenylalanine we utilized the coupling agent propane phosphonic acid anhydride (T3P)45. In a former study T3P was found to be highly efficient for the synthesis of the AAP anilide structure while retaining the R configuration of the phenylalanine stereocenter30,46 which is essential for activity31.The reaction produced the desired 6-fluoro and 6-methyl derivatives at the R1 position as well as the N-methylated derivatives (Scheme 4). A notable difference was observed in the yields of the reactions. The unsubstituted derivative yielded 99% product, while the 6-fluoro, 6-methyl and N-methyl derivatives gave yields of 92%, 56% and 33%, respectively. The particularly low yield of the 6-methyl and the N-methyl derivative indicate that the methyl groups sterically hinder the amine from attacking the electrophilic carbon atom.
Scheme 4.
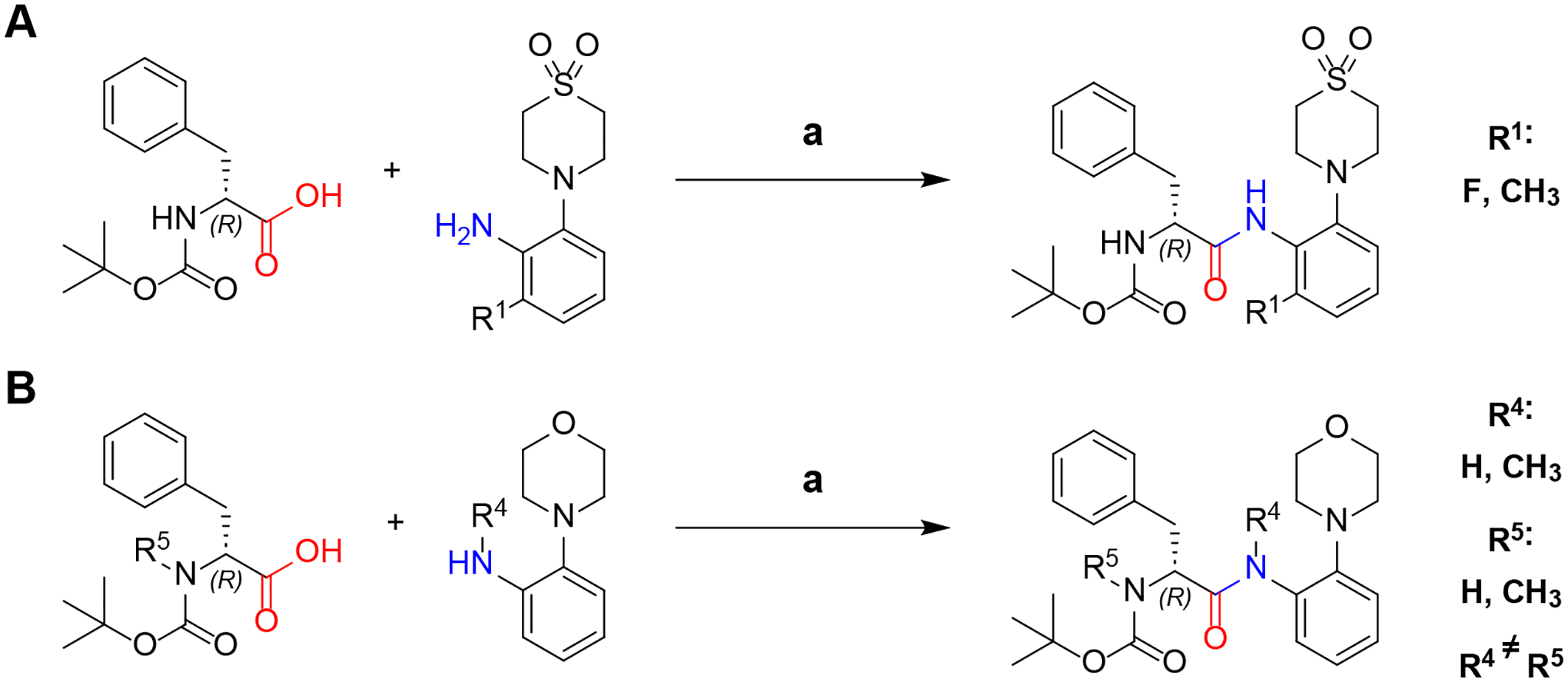
Synthesis of N-Boc-(R)-phenylalanine anilides (A) and N-Boc-N-methyl-(R)-phenylalanine anilides (B). a: EtOAc + pyridine 2:1, T3P 50 % m/v in EtOAc, −20 °C to RT, 20 h. For quantities and detailed procedures see Supporting Information.
Amide coupling with different benzoic acids
The final step of the synthetic sequence involves Boc-deprotection of the N-Boc-(R)-phenylalanine anilides (see Scheme 5) and the N-Boc-N-methyl-(R)-phenylalanine anilides (not shown) and subsequent amide coupling with a benzoic acid derivative. The Boc-deprotected intermediates were used for the amide coupling without further purification. For the formation of amides, 3-(diethoxyphosphoryloxy)-1,2,3-benzotriazin-4(3H)-one (DEPBT) and benzotriazol-1-yloxytripyrrolidinophosphonium hexafluorophosphate (PyBOP) were ≠employed as effective and convenient coupling reagents for the synthesis of AAPs30,31 with retention of stereochemistry. PyBOP was used when conversion with DEPBT resulted in low yields. The conversion of 2,6-dimethylbenzoic acid proved challenging owing to the high degree of steric hindrance. In preliminary experiments, no consumption of the activated 2,6-dimethylbenzoic acid was observed when using DEPBT. Utilizing PyBOP as an alternative coupling reagent9,11 resulted in the formation of the desired products. Nevertheless, the yields remained relatively low at only 38% in two cases. All final products had >95 % purity, as determined by analytical HPLC.
Scheme 5.
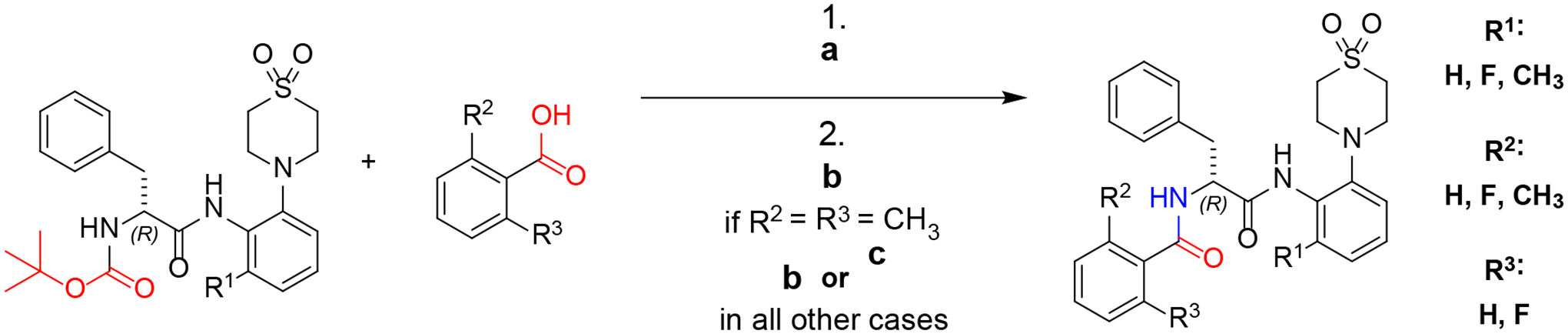
Synthesis of Nα-2-phenoyl-(R)-phenylalanine-2-anilides. a: TFA, DCM, 1 h, RT; b: DMF, PyBOP, overnight, RT; c: Dioxane, DEPBT, overnight, RT. For synthesis of N-methyl compounds methods a and b were utilized (not depicted). For quantities and detailed procedures see Supporting Information.
In vitro plasma stability
All tested substances remained stable in human plasma over the test period of 120 min (as shown in Table 1). MMV exhibits a significant decrease in concentration in murine plasma30. This result is comparable to the murine plasma stability published by Medicines for Malaria Venture (60% remaining substance after 4 h of incubation)32. The only other compound with a comparable fast degradation in murine plasma was 25, which differs from MMV by only one N-methyl group (R5). In contrast, 29 did not show a similar drop in concentration, which is possibly due to the N-methyl group protecting the anilide (R4).
Table 1.
Remaining relative amounts of AAP derivatives after incubation in human and murine plasma for 120 min. Green marked cells indicate over 90 % remaining substance. The values are means of two biological replicates.
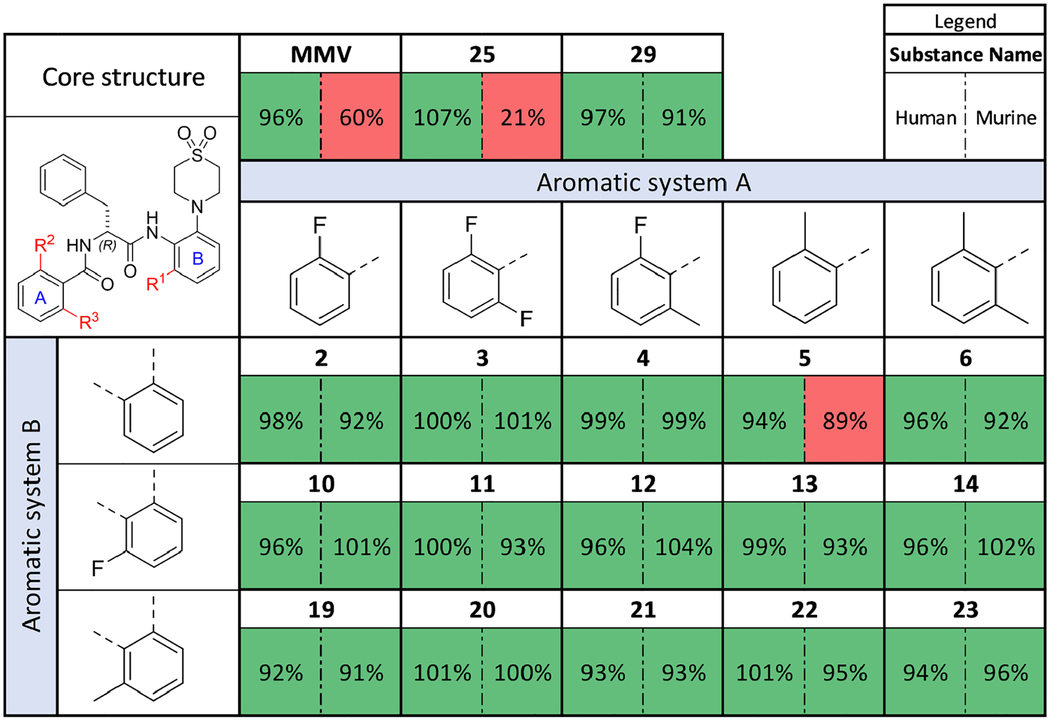
|
Our previous study showed a high human and murine plasma stability of 230. We observed the same for the new derivatives described herein. Altering both the morpholine to thiomorpholine dioxide as well as the Nα-2-thiophenoyl to Nα-2-fluorobenzoyl and Nα-2-methylbenzoyl groups appeared to sufficiently enhance the plasma stability of the compounds.
In vitro microsomal stability
All tested compounds showed a concentration decline in the microsomal suspensions (Table 2) used whereas stability in human microsomal suspensions was higher than in murine microsomal suspensions in every case analyzed (exemplary curves are depicted in Figure 2).
Table 2.
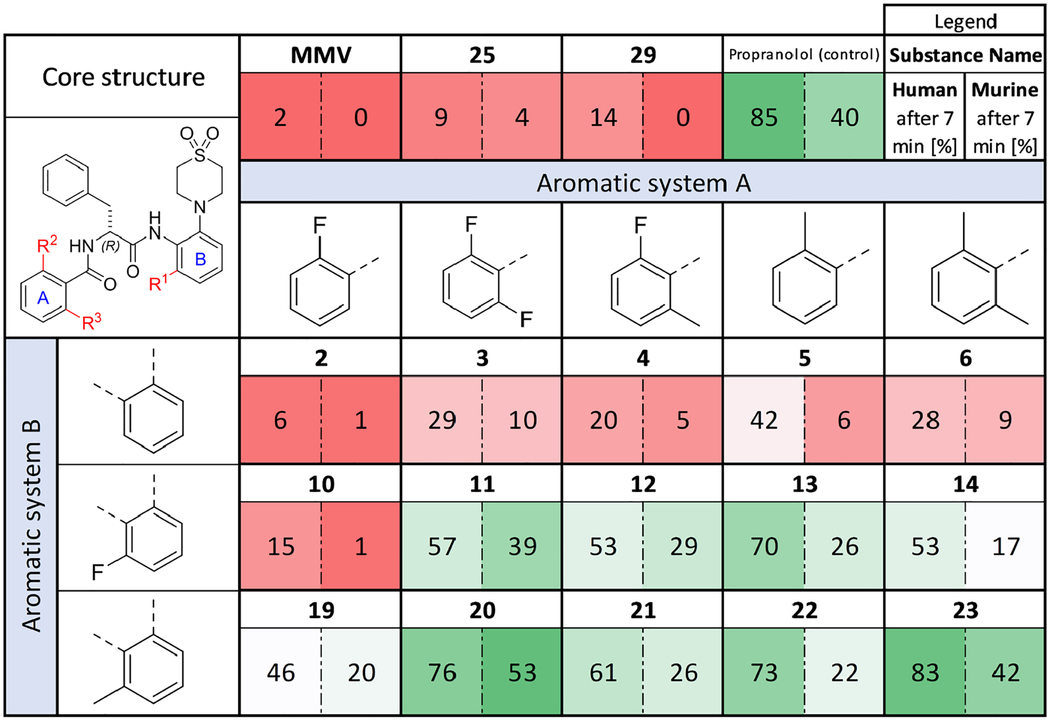
Remaining substance [%] of AAP derivatives in murine and human microsomal suspensions after 7 min of incubation. Color coding compares remaining substance in the human and murine assays. Colors show the difference with respect to the average value. Dark green: highest percentage; light green: over average; white: closest to average; light red: below average, dark red: lowest percentage. The displayed values are means of two biological replicates. Propranolol was included as a reference.
|
Figure 2.
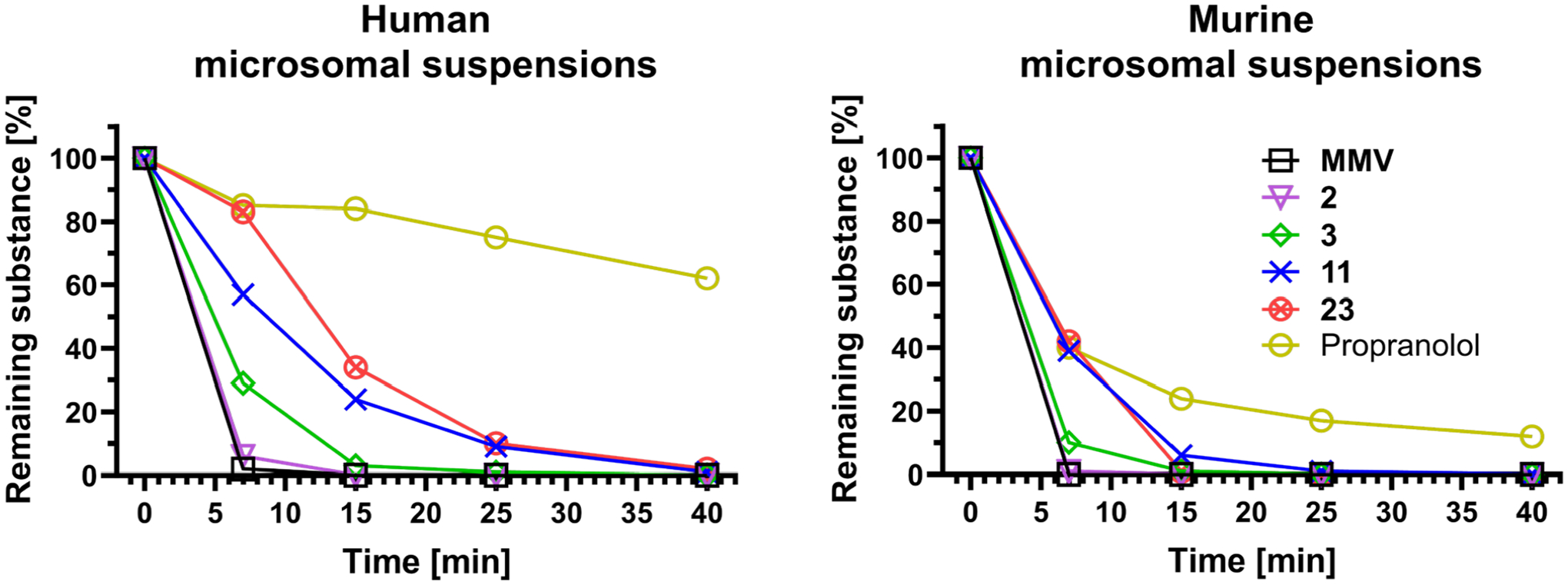
Degradation of a selection of compounds in human and murine microsomal suspensions over 40 min. Compounds 11 and 23 are shielded at aromatic system B with a 6-fluoro or 6-methyl substituent respectively and show higher stability than the unshielded derivatives (3). Displayed values are means of two replicates. Propranolol is shown as reference.
MMV showed particularly low stability in human and murine microsomal suspensions with only respectively 2 % and 0 % remaining after 7 min. These results of our study differ significantly from those published by Medicines for Malaria Ventures for MMV. While they reported half-lives of 129 minutes in human microsomal suspension and 795 minutes in murine microsomal suspension32. Our study found much lower half-lives of 1.2 and 0.9 minutes, respectively. The observed difference may be due to differences in assay conditions. Medicines for Malaria Venture report briefly on the assay conditions used, including a substrate concentration of 0.5 μM, 0.25 mg/mL microsomal proteins, and 50 mM phosphate buffer at pH 7.432. In our assay, we used 2 μM substrate concentration, 0.42 mg/mL microsomal proteins, and 100 mM phosphate buffer at pH 7.4. The authors did not disclose the usage of an NADPH-cofactor system, which is critical for oxidative metabolism catalyzed by microsomal enzymes. If this component is not utilized, it could explain the observed difference in microsomal stability.
The N-methylated compounds 25 and 29 did not exhibit superior performance to MMV. Compound 2, the starting point for the series reported here, showed comparably low stabilities. This result shows that the sole replacement of morpholine by thiomorpholine dioxide and thiophene carboxylic acid amide by 2-fluoro benzoic acid amide does not increase the microsomal stability as was observed for the plasma stability.
Modifying the aromatic system A of 2 to provide further shielding of the adjacent amide bond only showed a slight tendency to increased microsomal stabilities, independent of which substituents were introduced. Increased stabilities were however achieved in combination with fluoro- and methyl-substitutions in aromatic system B. There was only a small increase in stability when 2-fluoro benzoic acid amide (aromatic system A) was used in combination with a fluoro-substituent at aromatic system B (compound 10). Nevertheless, a tendency to higher stabilities could be observed with a methyl group in aromatic system B (compound 19).
For all other substitution patterns in aromatic system A, increased stabilities were observed when combined with either methyl- or fluoro substitutions in aromatic system B. The highest stability increases in human microsomal suspensions (5.5- to 6.5-fold) were observed for 13 and 22 (a combination of Nα-2-methylbenzoyl with 6-fluoro or 6-methyl substitutions, respectively). In contrast, the highest stability increases for murine microsomal suspensions (4- to 5-fold) were obtained with the combination of Nα-2,6-difluorobenzoyl group together with 6-fluoro or 6-methyl substitutions.
Antimycobacterial activity assessment
Growth inhibition testing was performed through microdilution assays for a selection of mycobacteria. The results of the assays are depicted in Table 3.
Table 3.
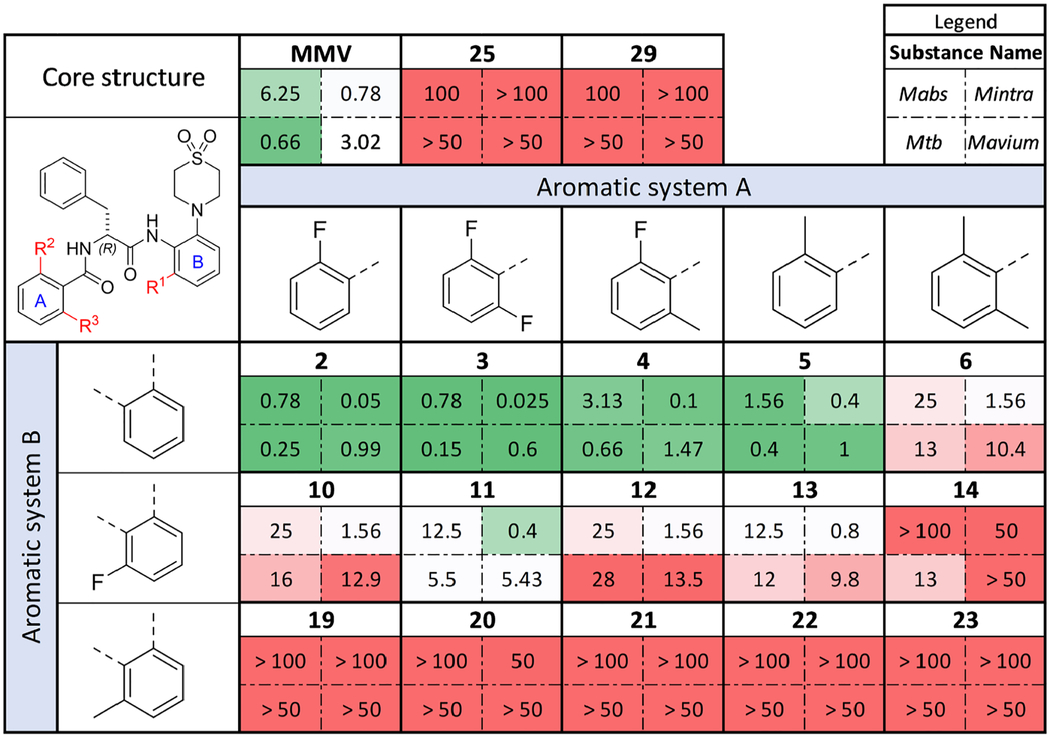
MIC90 values of the new AAP derivatives against different mycobacteria. Mabs = Mycobacterium abscessus ATCC 19977; Mintra = Mycobacterium intracellulare ATCC 35761; Mtb = Mycobacterium tuberculosis H37Rv; Mavium = Mycobacterium avium ssp. hominissuis strain 109 (MAC109). Incubation at 37 °C for three days (Mabs), five days (Mintra) and seven days (Mtb). Experiments were performed in duplicate, results were averaged. Protocols for different strains differ slightly, for detailed information on the methodology see Supporting Information.
|
The activities appear to be largely unaffected when aromatic system A was derivatized. Only the Nα-2,6-dimethylbenzoyl derivative caused an overall decline in activity (see compounds 6 and 14). All other substitution patterns are well accepted, with the tendency that the Nα-2,6-difluorobenzoyl containing compounds showed the highest activities with 3 showing the highest activity (all activities in the nanomolar range). Even in combination with the fluoro-substituted aromatic system B, low micromolar activities against Mavium and Mtb and sub-micromolar activities against Mycobacterium intracellulare (Mintra) were observed (compound 11). Particularly striking were the high activities of this compound class against the Mintra strain that we tested.
Changing the substitution pattern near and at the amide bonds strongly affected activity against the mycobacterial strains tested. The N-methylated compounds 25 and 29 suffered from complete activity loss, independent of the amide bond to which the N-methyl group was added. Likewise, derivatization of aromatic system B caused strong declines in activity. The introduction of a fluoro substituent on the aromatic ring led to increased MIC90 values, whereas the respective methyl substitution resulted in complete loss of activity. Only derivatives that contained a hydrogen atom next to the amide bond at aromatic system B showed improved MIC90 values in comparison to the hit compound.
Cytotoxicity
The compounds synthesized in this study were tested for their single concentration cytotoxicity against an immortalized the human kidney epithelia cell line HEK293. To the best of our knowledge, AAPs have never been tested against this cell line. Cell viability relative to a DMSO-treated control is depicted in Figure 3.
Figure 3.
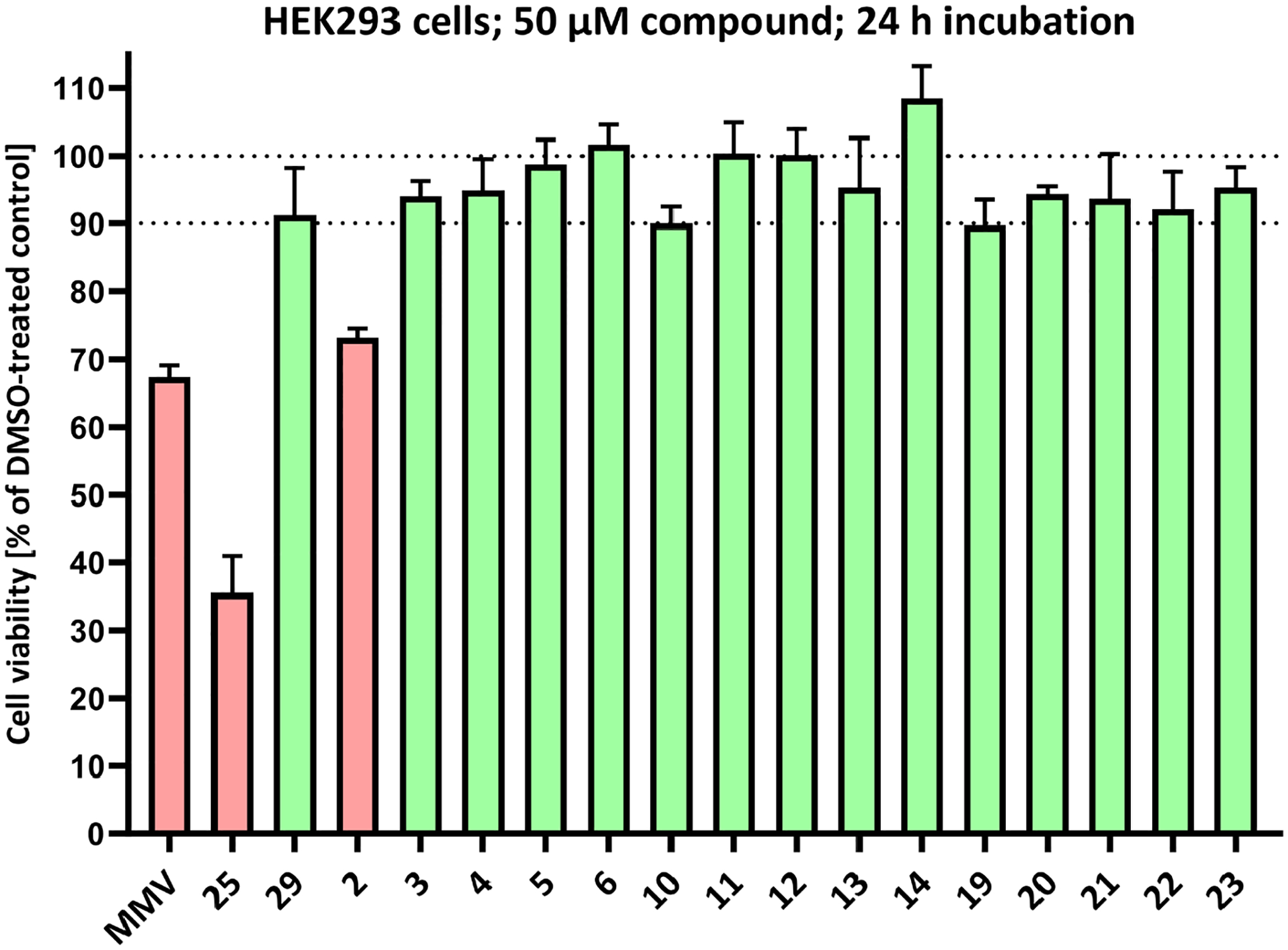
Cytotoxicity against human kidney epithelia cells HEK293. Determination of cell viability was performed at a single compound concentration of 50 μM after 24 h of incubation relative to DMSO-treated cells. The displayed values are means ± SD of triplicates. For detailed information of the protocol, see the Supporting information
Only three compounds caused a relative cell viability lower than 90 %, including the hit compound MMV (67 % viability) and also 2 (73 % viability), which demonstrated high antimycobacterial activities. Both compounds have been tested against a variety of cell lines and displayed no concerning behavior30–32. The N-methyl compound 25 exhibited the highest cytotoxicity with only 35% viability. In contrast, the other N-methyl compound (29) did not demonstrate a high degree of cytotoxicity.
Conclusions
Microsomal stability as a model for hepatic stability was used as an indicator of intrinsic clearance for AAPs. We observed that sterically shielding the amide bonds within the molecular structures of AAPs increases their microsomal stability. In particular, shielding of the anilide bond at the aromatic system B resulted in higher stabilities in microsomal suspensions.
Methyl substituents result in complete loss of whole cell activity, rendering these molecules unsuitable for further efficacy development, although compounds within this group demonstrated improved stabilities against murine (20) and human (22) microsomes. Antimycobacterial activity was observed with fluoro substituents making the respective fluorinated AAPs valuable option for future efficacy development. The compound 11, which carries fluorine atoms at all investigated positions, possess MIC90 values between 0.4 and 12.5 μM depending on the mycobacterial species combined with improved microsomal stabilities.
Modification of the aromatic system A was tolerated well with regard to the potency of the compounds. Nα-2,6-dimethylbenzoyl and Nα-2,6-difluorobenzoyl groups were found to be beneficial to microsomal stability, offering compounds that show higher or equal activities together with higher stabilities in comparison to both the hit compound and the most active compound 2 published so far.
N-methylation of the anilide bond resulted in increased plasma stability, whereas N-methylation of the Nα-2-thiophenoyl groups did not cause the same effect. This suggests that the anilide bond of the hit compound is susceptible to hydrolysis and that it is stabilized by an adjacent N-methyl group.
Based on these results, we conclude that the AAP derivatives investigated have sufficient plasma stability for activity even without N-methylation, which is an improvement compared to the hit compound MMV. Thus, cleavage or metabolization by plasma components probably does not contribute to low in vivo plasma levels to a relevant extent.
These findings support the view that the stability issues of AAPs are probably due to the instability of the amide bonds. Useful additional information is provided by the observation that shielding of the anilide bond in aromatic system B results in increased stability. Future studies on resulting metabolites will aid the design of AAPs for improved stability and activity.
Supplementary Material
Acknowledgements
We would like to thank Nadine Jänckel, Dr. Nadine Taudte and Dr. Jens-Ulrich Rahfeld for providing and maintaining the biosafety level 2 facility, and Dr. Christian Ihling and Antje Herbrich-Peters for measuring the HRMS spectra.
This work was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation)—432291016, the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under award number R01AI132374 and Mukoviszidose Institut gGmbH project number 2202 (Bonn, Germany), the research and development arm of the German Cystic Fibrosis Association Mukoviszidose e. V.
Footnotes
Supporting information for this article is given via a link at the end of the document.
Supporting Information
CCDC 2293688 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/structures.
The authors have cited additional references within the Supporting Information. [18, 30, 31, 35–47]
References
- (1).World Health Organization. Global Tuberculosis Report 2023. https://www.who.int/teams/global-tuberculosis-programme/tb-reports/global-tuberculosis-report-2023 (accessed 2023-12-18).
- (2).Malenfant JH; Brewer TF Rifampicin Mono-Resistant Tuberculosis—A Review of an Uncommon But Growing Challenge for Global Tuberculosis Control. Open Forum Infect Dis 2021, 8 (2). 10.1093/OFID/OFAB018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Shah I; Poojari V; Meshram H Multi-Drug Resistant and Extensively-Drug Resistant Tuberculosis. Indian J Pediatr 2020, 87 (10), 833–839. 10.1007/S12098-020-03230-1/TABLES/2. [DOI] [PubMed] [Google Scholar]
- (4).Seaworth BJ; Griffith DE Therapy of Multidrug-Resistant and Extensively Drug-Resistant Tuberculosis. Microbiol Spectr 2017, 5 (2). 10.1128/MICROBIOLSPEC.TNMI7-0042-2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Conradie F; Diacon AH; Ngubane N; Howell P; Everitt D; Crook AM; Mendel CM; Egizi E; Moreira J; Timm J; McHugh TD; Wills GH; Bateson A; Hunt R; Van Niekerk C; Li M; Olugbosi M; Spigelman M Treatment of Highly Drug-Resistant Pulmonary Tuberculosis. New England Journal of Medicine 2020, 382 (10), 893–902. 10.1056/NEJMOA1901814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Prevots DR; Marshall JE; Wagner D; Morimoto K Global Epidemiology of Nontuberculous Mycobacterial Pulmonary Disease: A Review. Clin Chest Med 2023. 10.1016/J.CCM.2023.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Johansen MD; Herrmann JL; Kremer L Non-Tuberculous Mycobacteria and the Rise of Mycobacterium Abscessus. Nat Rev Microbiol 2020, 18 (7), 392–407. 10.1038/S41579-020-0331-1. [DOI] [PubMed] [Google Scholar]
- (8).Yan M; Brode SK; Marras TK Treatment of the Less Common Nontuberculous Mycobacterial Pulmonary Disease. Clin Chest Med 2023. 10.1016/j.ccm.2023.06.011. [DOI] [PubMed] [Google Scholar]
- (9).Daley CL; Iaccarino JM; Lange C; Cambau E; Wallace RJ; Andrejak C; Böttger EC; Brozek J; Griffith DE; Guglielmetti L; Huitt GA; Knight SL; Leitman P; Marras TK; Olivier KN; Santin M; Stout JE; Tortoli E; Van Ingen J; Wagner D; Winthrop KL Treatment of Nontuberculous Mycobacterial Pulmonary Disease: An Official Ats/Ers/Escmid/Idsa Clinical Practice Guideline. Clinical Infectious Diseases 2020, 71 (4), E1–E36. 10.1093/CID/CIAA241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Nguyen M-VH; Daley CL Treatment of Mycobacterium Avium Complex Pulmonary Disease: When Should I Treat and What Therapy Should I Start? Clin Chest Med 2023. 10.1016/j.ccm.2023.06.009. [DOI] [PubMed] [Google Scholar]
- (11).Holt MR; Baird T Treatment Approaches to Mycobacterium Abscessus Pulmonary Disease. Clin Chest Med 2023. 10.1016/j.ccm.2023.06.010. [DOI] [PubMed] [Google Scholar]
- (12).Baird T; Bell S Cystic Fibrosis-Related Nontuberculous Mycobacterial Pulmonary Disease. Clin Chest Med 2023. 10.1016/j.ccm.2023.06.008. [DOI] [PubMed] [Google Scholar]
- (13).Loebinger MR; Quint JK; van der Laan R; Obradovic M; Chawla R; Kishore A; van Ingen J Risk Factors for Nontuberculous Mycobacterial Pulmonary Disease: A Systematic Literature Review and Meta-Analysis. Chest 2023, 164 (5), 1115–1124. 10.1016/J.CHEST.2023.06.014. [DOI] [PubMed] [Google Scholar]
- (14).Ruis C; Bryant JM; Bell SC; Thomson R; Davidson RM; Hasan NA; van Ingen J; Strong M; Floto RA; Parkhill J Dissemination of Mycobacterium Abscessus via Global Transmission Networks. Nature Microbiology 2021 6:10 2021, 6 (10), 1279–1288. 10.1038/s41564-021-00963-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Aitken ML; Limaye A; Pottinger P; Whimbey E; Goss CH; Tonelli MR; Cangelosi GA; Ashworth Dirac M; Olivier KN; Brown-Elliott BA; McNulty S; Wallace RJ Respiratory Outbreak of Mycobacterium Abscessus Subspecies Massiliense in a Lung Transplant and Cystic Fibrosis Center. Am J Respir Crit Care Med 2012, 185 (2), 231–232. 10.1164/AJRCCM.185.2.231. [DOI] [PubMed] [Google Scholar]
- (16).Johnson MM; Odell JA Nontuberculous Mycobacterial Pulmonary Infections. J Thorac Dis 2014, 6 (3), 210. 10.3978/J.ISSN.2072-1439.2013.12.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Lopeman RC; Harrison J; Desai M; Cox JAG Mycobacterium Abscessus: Environmental Bacterium Turned Clinical Nightmare. Microorganisms 2019, Vol. 7, Page 90 2019, 7 (3), 90. 10.3390/MICROORGANISMS7030090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Nessar R; Cambau E; Reyrat JM; Murray A; Gicquel B Mycobacterium Abscessus: A New Antibiotic Nightmare. Journal of Antimicrobial Chemotherapy 2012, 67 (4), 810–818. 10.1093/JAC/DKR578. [DOI] [PubMed] [Google Scholar]
- (19).Ballell L; Bates RH; Young RJ; Alvarez-Gomez D; Alvarez-Ruiz E; Barroso V; Blanco D; Crespo B; Escribano J; González R; Lozano S; Huss S; Santos-Villarejo A; Martín-Plaza JJ; Mendoza A; Rebollo-Lopez MJ; Remuiñan-Blanco M; Lavandera JL; Pérez-Herran E; Gamo-Benito FJ; García-Bustos JF; Barros D; Castro JP; Cammack N Fueling Open-Source Drug Discovery: 177 Small-Molecule Leads against Tuberculosis. ChemMedChem 2013, 8 (2), 313–321. 10.1002/CMDC.201200428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Ebright Richard H.; Ebright Yon W.; Mandal Soma; Wilde Richard; Li S Preparation of N-Alpha-Aroyl-N-Aryl-Phenylalaninamides as Inhibitors of Bacterial RNA Polymerase and as Antibacterials. WO2015120320 A1 2015-08-13, 2015.
- (21).Richter A; Strauch A; Chao J; Ko M; Av-Gay Y Screening of Preselected Libraries Targeting Mycobacterium Abscessus for Drug Discovery. Antimicrob Agents Chemother 2018, 62 (9). 10.1128/AAC.00828-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Jeong J; Kim G; Moon C; Kim HJ; Kim TH; Jang J Pathogen Box Screening for Hit Identification against Mycobacterium Abscessus. PLoS One 2018, 13 (4), e0195595. 10.1371/JOURNAL.PONE.0195595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Low JL; Wu ML; Aziz DB; Laleu B; Dick T Screening of TB Actives for Activity against Nontuberculous Mycobacteria Delivers High Hit Rates. Front Microbiol 2017, 8 (AUG), 1539. 10.3389/FMICB.2017.01539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Lin W; Mandal S; Degen D; Liu Y; Ebright YW; Li S; Feng Y; Zhang Y; Mandal S; Jiang Y; Liu S; Gigliotti M; Talaue M; Connell N; Das K; Arnold E; Ebright RH Structural Basis of Mycobacterium Tuberculosis Transcription and Transcription Inhibition. Mol Cell 2017, 66 (2), 169–179.e8. 10.1016/j.molcel.2017.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Mann L; Ganapathy US; Abdelaziz R; Lang M; Zimmerman MD; Dartois V; Dick T; Richter A In Vitro Profiling of the Synthetic RNA Polymerase Inhibitor MMV688845 against Mycobacterium Abscessus. Microbiol Spectr 2022, 10 (6). 10.1128/SPECTRUM.02760-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Rominski A; Roditscheff A; Selchow P; Böttger EC; Sander P Intrinsic Rifamycin Resistance of Mycobacterium Abscessus Is Mediated by ADP-Ribosyltransferase MAB_0591. Journal of Antimicrobial Chemotherapy 2017, 72 (2), 376–384. 10.1093/JAC/DKW466. [DOI] [PubMed] [Google Scholar]
- (27).Lan T; Ganapathy US; Sharma S; Ahn Y-M; Zimmerman M; Molodtsov V; Hegde P; Gengenbacher M; Ebright RH; Dartois V; Freundlich JS; Dick T; Aldrich CC Redesign of Rifamycin Antibiotics to Overcome ADP-Ribosylation-Mediated Resistance. Angewandte Chemie 2022, 134 (45), e202211498. 10.1002/ANGE.202211498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Zaw MT; Emran NA; Lin Z Mutations inside Rifampicin-Resistance Determining Region of RpoB Gene Associated with Rifampicin-Resistance in Mycobacterium Tuberculosis. J Infect Public Health 2018, 11 (5), 605–610. 10.1016/J.JIPH.2018.04.005. [DOI] [PubMed] [Google Scholar]
- (29).Ganapathy US; Lan T; Krastel P; Lindman M; Zimmerman MD; Ho HP; Sarathy JP; Evans JC; Dartois V; Aldrich CC; Dick T Blocking Bacterial Naphthohydroquinone Oxidation and Adpribosylation Improves Activity of Rifamycins against Mycobacterium Abscessus. Antimicrob Agents Chemother 2021, 65 (9). https://doi.org/ 10.1128/AAC.00978-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Lang M; Ganapathy US; Mann L; Abdelaziz R; Seidel RW; Goddard R; Sequenzia I; Hoenke S; Schulze P; Aragaw WW; Csuk R; Dick T; Richter A Synthesis and Characterization of Phenylalanine Amides Active against Mycobacterium Abscessus and Other Mycobacteria. J Med Chem 2023. 10.1021/ACS.JMEDCHEM.3C00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Mann L; Lang M; Schulze P; Halz JH; Csuk R; Hoenke S; Seidel RW; Richter A Racemization-Free Synthesis of Nα-2-Thiophenoyl-Phenylalanine-2-Morpholinoanilide Enantiomers and Their Antimycobacterial Activity. Amino Acids 2021, 53 (8), 1187–1196. 10.1007/S00726-021-03044-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Medicines for Malaria Venture. Biological Data and DMPK Data of the Pathogen Box Compounds. https://www.mmv.org/mmv-open/pathogen-box/about-pathogen-box (accessed 2024-01-31).
- (33).Beuchel A; Robaa D; Negatu DA; Madani A; Alvarez N; Zimmerman MD; Richter A; Mann L; Hoenke S; Csuk R; Dick T; Imming P Structure–Activity Relationship of Anti-Mycobacterium Abscessus Piperidine-4-Carboxamides, a New Class of NBTI DNA Gyrase Inhibitors. ACS Med Chem Lett 2022, acsmedchemlett.1c00549. 10.1021/ACSMEDCHEMLETT.1C00549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Bahar FG; Ohura K; Ogihara T; Imai T Species Difference of Esterase Expression and Hydrolase Activity in Plasma. J Pharm Sci 2012, 101 (10), 3979–3988. 10.1002/JPS.23258. [DOI] [PubMed] [Google Scholar]
- (35).Bradshaw PR; Wilson ID; Gill RU; Butler PJ; Dilworth C; Athersuch TJ Metabolic Hydrolysis of Aromatic Amides in Selected Rat, Minipig, and Human In Vitro Systems. Scientific Reports 2018 8:1 2018, 8 (1), 1–8. 10.1038/s41598-018-20464-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Liu L; Halladay JS; Shin Y; Wong S; Coraggio M; La H; Baumgardner M; Le H; Gopaul S; Boggs J; Kuebler P; Davis JC Jr; Charlene Liao X; Lubach JW; Deese A; Gregory Sowell C; Currie KS; Young WB; Cyrus Khojasteh S; C A Hop CE; Wong H Significant Species Difference in Amide Hydrolysis of GDC-0834, a Novel Potent and Selective Bruton’s Tyrosine Kinase Inhibitor. 2011. 10.1124/dmd.111.040840. [DOI] [PubMed] [Google Scholar]
- (37).Midgley L; Bourhis LJ; Dolomanov OV; Grabowsky S; Kleemiss F; Puschmann H; Peyerimhoff N Vanishing of the Atomic Form Factor Derivatives in Non-Spherical Structural Refinement - a Key Approximation Scrutinized in the Case of Hirshfeld Atom Refinement. Acta Crystallogr A Found Adv 2021, 77 (6), 519–533. 10.1107/S2053273321009086. [DOI] [PubMed] [Google Scholar]
- (38).Kleemiss F; Dolomanov OV; Bodensteiner M; Peyerimhoff N; Midgley L; Bourhis LJ; Genoni A; Malaspina LA; Jayatilaka D; Spencer JL; White F; Grundkötter-Stock B; Steinhauer S; Lentz D; Puschmann H; Grabowsky S Accurate Crystal Structures and Chemical Properties from NoSpherA2. Chem Sci 2021, 12 (5), 1675–1692. 10.1039/D0SC05526C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Ruetsch Y; Boni T; Borgeat A From Cocaine to Ropivacaine: The History of Local Anesthetic Drugs. Curr Top Med Chem 2001, 1 (3), 175–182. 10.2174/1568026013395335. [DOI] [PubMed] [Google Scholar]
- (40).Robert A; Schultz JR; Nezamis JE; Lancaster C Gastric Antisecretory and Antiulcer Properties of PGE2, 15-Methyl PGE2, and 16, 16-Dimethyl PGE2. Intravenous, Oral and Intrajejunal Administration. Gastroenterology 1976, 70 (3), 359–370. 10.1016/S0016-5085(76)80147-3. [DOI] [PubMed] [Google Scholar]
- (41).Barreiro EJ; Kümmerle AE; Fraga CAM The Methylation Effect in Medicinal Chemistry. Chem Rev 2011, 111 (9), 5215–5246. 10.1021/CR200060G. [DOI] [PubMed] [Google Scholar]
- (42).Dajani EZ; Driskill DR; Bianchi RG; Collins PW; Pappo R SC-29333: A Potent Inhibitor of Canine Gastric Secretion. Am J Dig Dis 1976, 21 (12), 1049–1057. 10.1007/BF01071862. [DOI] [PubMed] [Google Scholar]
- (43).Van Neer RHP; Dranchak PK; Liu L; Aitha M; Queme B; Kimura H; Katoh T; Battaile KP; Lovell S; Inglese J; Suga H Serum-Stable and Selective Backbone-N-Methylated Cyclic Peptides That Inhibit Prokaryotic Glycolytic Mutases. ACS Chem Biol 2022, 17 (8), 2284–2295. 10.1021/ACSCHEMBIO.2C00403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Haviv F; Fitzpatrick TD; Swenson RE; Nichols CJ; Mort NA; Bush EN; Diaz G; Bammert G; Nguyen A; Rhutasel NS; Nellans HN; Hoffman DJ; Johnson ES; Greer J Effect of N-Methyl Substitution of the Peptide Bonds in Luteinizing Hormone-Releasing Hormone Agonists. J Med Chem 1993, 36 (3), 363–369. 10.1021/JM00055A007. [DOI] [PubMed] [Google Scholar]
- (45).García ALL T3P: A Convenient and Useful Reagent in Organic Synthesis. Synlett 2007, 2007 (08), 1328–1329. 10.1055/S-2007-980339. [DOI] [Google Scholar]
- (46).Dunetz JR; Xiang Y; Baldwin A; Ringling J General and Scalable Amide Bond Formation with Epimerization-Prone Substrates Using T3P and Pyridine. Org Lett 2011, 13 (19), 5048–5051. 10.1021/OL201875Q. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.