

Summary
Evolvability is an emergent hallmark of cancer that depends on intra-tumor heterogeneity and genetic variation. Mutations generated by APOBEC3 contribute to genetic variation and tumor evolvability. However, the influence of APOBEC3 on the evolvability of the genome and its differential impact on cancer genes versus non-cancer genes remains unclear. Analyzing over 40,000 human protein-coding transcripts, we identified distinct distribution patterns of APOBEC3A/B TC motifs between cancer and non-cancer genes, suggesting unique associations with cancer. Studying a bat species with numerous APOBEC3 genes, we found distinct motif patterns in orthologs of cancer genes compared to non-cancer genes, as in humans, suggesting APOBEC3 evolution to reduce impacts on the genome rather than the converse. Simulations confirmed that APOBEC3-induced heterogeneity enhances cancer evolution through bimodal patterns of mutations in certain classes of genes. Our results suggest the bimodal distribution of APOBEC-induced mutations can significantly increase cancer heterogeneity.
Subject areas: Evolutionary processes, Bioinformatics, Cancer
Graphical abstract
Highlights
-
•
A biased pattern of potential effects of APOBEC activity found in the human genome
-
•
Bat and human genomes show similar patterns despite the bat having more APOBEC genes
-
•
Cancer genes showed a bimodal pattern, with many genes susceptible to APOBEC activity
-
•
Simulation of the pattern supports APOBEC’s role in the neutral evolution of cancer
Evolutionary processes; Bioinformatics; Cancer
Introduction
Cancer originates when a somatic cell deviates from the cooperative regulation of multicellularity in resident tissue and begins to evolve as an independent entity. During early pre-cancer cell divisions, genetic and epigenetic changes occur and accumulate in the offspring of cancer cells, giving rise to new trait variations and leading to the emergence of subclones. Continuous subclonal diversification, also known as intra-tumor heterogeneity (ITH), allows for somatic evolution by natural selection, particularly in circumstances of competition for limited resources among early cancer cells.1,2,3 Importantly, the presence of genotypic and phenotypic ITH in cancer directly influences evolutionary trajectory and is associated with poor prognosis and treatment failure.4,5 Consequently, determining the fundamental principles governing somatic evolution and the maintenance of ITH in the face of subclonal competition is crucial to understanding cancer initiation and progression, and for developing more effective strategies to prevent cancer or designing therapies.6
The APOBEC (Apolipoprotein B mRNA-editing enzyme catalytic polypeptide-like) genes are a family of enzymes involved in single-stranded DNA (ssDNA) and, to a lesser extent RNA, deamination of C sites.7,8,9,10,11,12,13 Several members of the APOBEC3 subfamilies, particularly APOBEC3A and APOBEC3B, are associated with cancer initiation or progression.14 Many subsequent studies have established APOBEC3A and B as causal for mutations in genomic DNA by targeting ssDNA during the process of replication or transcription.15,16 APOBEC3A and B are the only two APOBEC3s that enter the nucleus,17,18,19 and recent work suggests that APOBEC3A may be more mutagenic despite lower expression levels.20 APOBEC cytidine deamination activity results in C-to-U conversion and, following replication, can generate C-to-T mutations or, more rarely, C-to-A and C-to-G mutations that depend on downstream error-prone DNA repair.21,22 These APOBEC-induced mutations can contribute to the initiation and progression of cancer by disrupting the normal function of genes or increasing the ITH.23,24,25 ITH can provide a basis for the high evolutionary capacity (evolvability) of cancer cells by increasing the probability of having progeny that can survive selection.26,27,28 In a tumor, there are two types of somatic aberrations: founder mutations and progressor mutations. Founder mutations, also called trunk mutations or clonal mutations, are present in all tumor cells belonging to one clone and are caused by an initial carcinogenic event, driving tumor initiation and growth. Progressor mutations, also known as branch or leaf mutations or subclonal mutations, occur at later stages of tumor development, contributing to tumor progression and heterogeneity by introducing additional genetic changes. Distinguishing between these two types of aberrations is vital for understanding the complex genetic landscape behind the evolution of tumors. Therefore, it is crucial to understand how and to what extent different mutational processes can contribute to ITH, influence tumor evolvability and evolutionary trajectory, as well as predict the pattern of evolution (i.e., neutral versus branching) resulting from such mutational processes.29
Analysis of large datasets of genome-level mutations has revealed that diverse mutational processes generate distinct patterns of mutations known as mutational signatures.30,31,32 Alexandrov et al.30,32 conducted a comprehensive study on the patterns of somatic mutations in cancer genomes using whole exome sequencing data. Through their analysis, they identified more than 30 distinct mutational signatures, specifically, single-base substitution (SBS) signatures. These SBS signatures represent characteristic patterns of DNA base substitutions that occur during the development of different cancers. Each signature is defined by the specific types of base substitutions it comprises and the sequence context in which these substitutions tend to occur. These signatures provide insights into the mutational processes that contribute to cancer development and progression. The mutational signatures encompass a range of processes, including those associated with aging, DNA repair deficiencies, and exposure to various mutagens such as tobacco smoke and ultraviolet radiation. While the etiology of many mutational signatures remains unknown, the causes of many mutational signatures are supported by experiments, including two APOBEC signatures. SBS signatures 2 and 13 are associated with APOBEC3-induced mutagenesis and have been observed in various types of cancers, including breast, bladder, and lung cancers.33,34 The association of these signatures with APOBEC activity has been well documented in some cancer studies, particularly in the early stages of breast and ovarian cancer.35,36,37
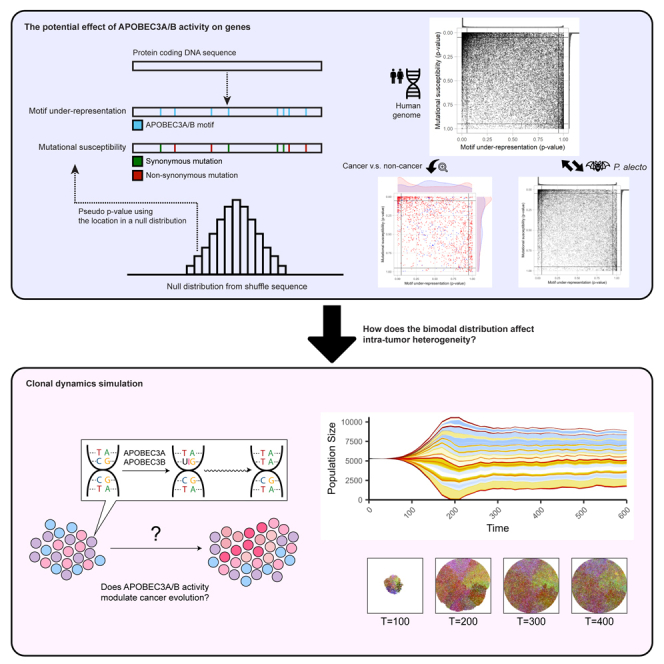
We previously developed a program, Cytidine Deaminase Under-representation Reporter (CDUR), that uses computational statistical methods to evaluate the record of APOBEC evolutionary influence on coding sequences of viral genomes.38,39 Different APOBEC proteins prefer a cytidine target in different motifs such as TC, TCC, or CCC in the case of APOBEC3A/B, 3C, and 3G in single-stranded DNA. The program evaluates whether a coding sequence contains a statistically significant under-representation (scarcity) or over-representation (abundance) of cytidine deaminase mutation motifs. This statistical approach involves creating a null distribution to describe the expected occurrence of mutation motifs within the analyzed sequence (the “subject”) by using codon shuffling methods, for example, rearranging nucleotides at the third codon position, while preserving the amino acid sequence. The subject sequence is then compared to this expected null distribution, and a p value can be calculated for under- or over-representation. In addition to assessing the count of cytidine deaminase mutation motifs, CDUR also computes a related statistic for the number of nonsynonymous mutations occurring at these motifs and the ratio of nonsynonymous mutations to mutation motifs, a measure we describe as mutational susceptibility.
We applied CDUR to study human genes and their relationship to APOBEC motif preferences. In this study, we analyzed motif under-representation and mutational susceptibility of APOBEC3A/B TC hotspots motif of the entire human transcriptome (over 40,000 protein-coding transcripts) and found a distribution that is skewed toward APOBEC3A/B robustness, i.e., tolerance of mutations. Furthermore, we selected cancer genes and non-cancer genes with no point mutations reported in the COSMIC database. We then compared the distributions of APOBEC3A/B motif statistics of cancer and non-cancer groups and found the two distributions were significantly different. The cancer genes had a bimodal distribution containing both under- and over-representation of APOBEC3A/B targeting motifs. We further investigated the APOBEC3A/B motif statistics of orthologs of cancer and non-cancer genes in other species, including bats with the most APOBEC genes across taxa. Interestingly, orthologs with similar protein sequences and likely the same function showed a very broad range of motif under-representation and mutational susceptibility, suggesting genes are labile in terms of these statistics. Our analyses of both human and bat genomes suggested APOBEC targeting preferences predominantly evolved to avoid excessive damage to the genome rather than the genome evolving to accommodate APOBEC mutations. In addition, our simulations suggest that additional APOBEC activity, together with the bimodal distribution of susceptibility to APOBEC, can introduce higher heterogeneity during the clonal evolutionary process in cancer development, providing the best conditions for neutral evolution.
Results
APOBEC3A/B TC hotspots analysis of human genome protein-coding transcripts shows a biased distribution
We hypothesized that APOBEC motif preferences have co-evolved with the human genome. This co-evolution may have caused human genes to evolve so as to avoid APOBEC mutagenesis. At the same time, APOBEC preferences may also have evolved to minimize damage to the human genome. To evaluate the extent to which the human genome will be affected by APOBEC activity, we analyzed how much each gene has significantly fewer mutational motifs (motif under-representation) and depletion of motifs in synonymous positions (defined as the fraction of nonsynonymous mutation motifs, a measure we will refer to as mutational susceptibility) using CDUR.38 Figure 1A shows both measures for APOBEC3A/B TC hotspots for each gene in the human transcriptome (GENCODE v40). Both the motif under-representation and mutational susceptibility measures are heavily skewed, primarily toward the top-left and, to some extent, toward the bottom-right corner of the CDUR plot. One explanation for the high density of transcripts in the top-left corner is that a significant number of transcripts have evolved to avoid APOBEC3A/B mutagenesis such that TC motifs have become under-represented in the sequence and those that remain are inevitably at nonsynonymous sites. At the same time, the APOBEC3A/B TC motif may have evolved to cause less damage to our genome, especially to crucial genes (e.g., DNA repair). We suggest three possible scenarios to explain the high density in the bottom-right corner: (i) the simplest scenario is that these genes are not exposed to APOBEC3A/B so the TC motif abundance of the genes was not subject to selection by APOBEC3A/B activity. (ii) Genes might have followed a distinct strategy to avoid function loss by APOBEC3A/B mutation by increasing the number of TC hotspots and thus making the hotspots less susceptible. (iii) Another scenario is that selection independent of APOBEC3A/B-induced mutagenesis would have led to increased TC hotspots and the underlying cause of decreased mutational susceptibility remains unknown. Several studies have shown that DNA stem-loop structures are hotspots for mutations caused by APOBEC3A/B. We used those as a control to validate our findings. In addition to analyzing TC motifs, we also examined CDUR statistics of VC motifs in hairpin-forming structures, considering substantial evidence indicating that APOBEC3A/B targets these motifs.40 We found the expected distribution in motif under-representation; the reversed trend compared to TC motifs. We also found a similar distribution in mutational susceptibility only in VC motifs in hairpin-forming structures but not in all VC motifs (Figures S1A and S1B). We discovered the distribution of mutational susceptibility of TC motifs in hairpin-forming structures was more uniform, meaning it has lower average mutational susceptibility (Figures S1C and S1D).
Figure 1.

APOBEC3A/B TC hotspots statistics of protein-coding transcripts of the human genome
(A) CDUR analysis on the human genome shows two dense regions at the top-left and right-bottom extremes and relatively sparse dispersion elsewhere.
(B) Genes in the top-left partition of the CDUR plot showed significant enrichment of GO Biological Processes associated with differentiation and development.
(C) Genes in the top-left partition of the CDUR plot showed significant enrichment of KEGG Pathway associated with several cancers, basal cell carcinoma, breast cancer, and gastric cancer.
Genes located in the top-left corner of the CDUR plot display a significant enrichment of KEGG pathways associated with cancer
To understand the two extreme groups of genes discovered in CDUR analyses, we performed a gene enrichment analysis. First, the top-left corner has 6,539 transcripts of 3,869 genes. Gene Ontology (GO) analysis of these genes showed that 5 out of the top 20 ranked by fold enrichment were associated with differentiation and development (cell morphogenesis involved in neuron differentiation, cell morphogenesis involved in differentiation, neuron development, tube development, regulation of multicellular organismal development, epithelium development). These processes are associated with cancer through disruption of differentiation and development-associated genes leading to cancer initiation or at least stimulating cancer progression40,41,42(Figure 1B). Enrichment analysis on the KEGG pathway database showed significant enrichment in several cancers (Figure 1C). To confirm the association between top-left genes and cancer pathways, we randomly selected the same number of genes outside of the top-left corner 20 times and performed KEGG pathway enrichment analysis. We strictly constrained cancer-related pathways as pathways in 6.1 Cancer: overview and 6.2 Cancer: specific types from the KEGG pathway database. With this criterion, the top-left corner has 4 cancer-related KEGG pathways and an average fold enrichment of 2.14. In contrast, KEGG pathway analysis of randomly selected genes showed 1.05 cancer-related KEGG pathways, and 11 out of 20 cases showed no cancer-related pathway. Moreover, the average fold enrichment of cancer-related pathways was 1.46, lower than 1.56, and the minimum cancer-related pathway fold enrichment is in the top left corner. In addition, if we randomly select a square box size of 0.05 in the plot and run the KEGG pathway analysis, it does not show any significant fold enrichment (Figure S2). For GO cellular components, enrichment analysis showed a significant fold enrichment of channel complex and synapse components (Figure S3). Enrichment analysis on the GO Molecular Function term showed a significant fold enrichment of mostly ion channel or transporter activity (Figure S4). This is an intriguing result considering the role of ion channels and transporters in solid tumors and their adaptation to harsh microenvironments such as hypoxia and acidosis.43 Second, the bottom-right corner has 396 transcripts of 230 genes. GO biological processes on these genes did not show any significant enrichment. KEGG pathway enrichment showed herpes simplex virus 1 (HSV-1) infection (Figure S5). HSV-1 genome is a potential substrate of APOBEC3, so abundant TC hotspots with low susceptibility of those genes would be expected. Gene enrichment analysis on the GO cellular component showed mostly collagen-associated terms (Figure S6), and GO molecular functions showed mostly DNA binding activity (Figure S7).
Cancer genes have a distinct bimodal distribution of APOBEC3A/B under-representation
As the KEGG pathway enrichment analysis of the genes in the top-left corner showed a significant fold enrichment of cancer-associated pathways, we further analyzed known cancer genes to understand whether cancer genes indeed share common characteristics under potential APOBEC3A/B mutational activity. For this purpose, the analysis requires a list of cancer genes that thoroughly confirm that mutations in these genes are functionally related to cancers. We used the COSMIC database list of cancer genes, retrieved from the COSMIC Cancer Gene Census dataset.44,45 Genes in the COSMIC Cancer Gene Census are cancer genes systematically curated by professional researchers. For comparative purposes, we generated a list of non-cancer genes collecting protein-coding genes from the NCBI Gene database that do not have any annotated point mutations in the COSMIC Mutation database. While applying the method, we observed significantly fewer non-cancer genes (380) than cancer genes (737). Nevertheless, to maintain a systematic analysis, we retained the non-cancer/cancer criterion. Figure 2A shows the same data as Figure 1A but with cancer genes highlighted in red and non-cancer genes in blue. We found a significant difference in the distribution between cancer and non-cancer genes in the CDUR plot (Figure 2A) (Kolmogorov-Smirnov test for motif-representation axis, p = 2.828 x 10−7; for mutational susceptibility axis, p = 1.175 x 10−4). However, in the motif under-representation axis, the difference of means test between the two groups was not significant (Wilcoxon rank-sum test, p = 0.1047) because of the bimodal shape of the cancer gene distribution compared to the unimodal non-cancer gene distribution. Along the mutational susceptibility axis, cancer genes do have a significantly higher mutational susceptibility than non-cancer genes (p = 4.838 x 10−5). This high mutational susceptibility of cancer genes suggests the human genome did not evolve to avoid APOBEC-induced mutations that potentially cause functional defects. In summary, cancer genes have a bimodal distribution for TC motif under-representation that is distinct from non-cancer genes. Cancer genes also have significantly higher mutational susceptibility (y axis of Figure 2A), consistent with depletion of TC hotspots in synonymous sites. We also compared genes from sex chromosomes and found no significant difference between X and Y chromosomes (Kolmogorov-Smirnov test for motif-representation axis, p = 0.1838; for mutational susceptibility axis, p = 0.4523, Wilcoxon rank-sum test for motif-representation axis, p = 5.515 x 10−2; for mutational susceptibility axis, p = 0.6658). However, the difference between them is inconclusive since there are many more genes in the X (1,676) than in the Y (83) chromosomes (Figure S8).
Figure 2.

Cancer genes have a significantly different distribution in CDUR plots compared to non-cancer genes
(A) Cancer and non-cancer genes show distinct distribution in both motif under-representation and mutational susceptibility (Kolmogorov-Smirnov test for motif-representation axis, P = 2.828 x 10-7; for mutational susceptibility axis, P = 1.175 x 10-4).
(B) Motif under-representation and mutational susceptibility of BRCA1 orthologs show higher variance than sequential mutations of the gene on the CDUR plot.
(C) Standard deviation of motif under-representation comparison between orthologs and sequential mutations shows significantly high variance among orthologs in both cancer genes (196 genes) and non-cancer genes (20 genes).
(D) Standard deviation of mutational susceptibility comparison between orthologs and sequential mutations shows significantly high variance among orthologs in both cancer genes (196 genes) and non-cancer genes (20 genes).
CDUR statistics of orthologous genes from other species are dispersed over a broad range
Given the surprising result of a bimodal distribution for the under-representation of cancer genes, we sought to investigate the extent of natural variation that is possible in these genes. For this, we used the same CDUR measures to consider orthologous genes (orthologs). Orthologs are homologous genes in different species that have highly similar protein sequences. Orthologs are derived from a common ancestor gene and are presumed to have preserved the same function. Orthologs are, therefore, good candidates for evaluating the genotypic heterogeneity of a phenotype. We collected orthologs of 196 cancer genes and 20 non-cancer genes from ten vertebrate species including human (Homo sapiens, Pan troglodytes, Canis lupus familiaris, Gallus gallus, Loxodonta africana, Mus musculus, Physeter catodon, Xenopus tropicalis, Myotis lucifugus, and Petromyzon marinus) and calculated the CDUR statistics as we did for the human genes. As an example, Figure 2B shows ten orthologs dispersed across a wide range in the CDUR plot of breast cancer type 1 susceptibility protein (BRCA1), a well-known tumor suppressor gene that is crucial to DNA damage repair. To investigate the generality of this observation, we compared variation in the CDUR plots of each ortholog set to sequences generated from the corresponding human genes by sequentially mutating TC motifs until we encounter an amino acid change, thus simulating the variation that might be introduced to the human gene by APOBEC3A/B. We found that, regardless of whether the genes are associated with cancer or not, most orthologs showed a much greater variation for both TC motif under-representation and mutational susceptibility when compared to the variation that might be introduced by APOBEC to the human gene (Figures 2C and 2D). Although most of the observed variance in the orthologs is likely unrelated to coevolution with APOBEC (since there are many other evolutionary selection pressures involved), the broad range distribution suggests that genes can evolve a wide range of tolerance to APOBEC3A/B-induced mutagenesis while maintaining their essential functions.
TC hotspot analysis of the bat genome suggests a predominant evolution of APOBEC preferences rather than genomes
To investigate the relationship between the distribution of TC hotspots in the human genome and APOBEC3A/B motif preference further, we performed the same TC hotspot analysis of a bat (Pteropus alecto) genome. Pteropus alecto has undergone an expansion of APOBEC3 genes, leading to 18 APOBEC3 genes, compared to 7 in humans. As in humans, the targeting preference is also predominantly TC.46 Bats are known to be long-lived animals and are expected to have low cancer incidence.47 If APOBEC were a main cancer-initiating factor in bats, we would expect a different TC hotspot distribution compared to the human genome. Instead, the Pteropus alecto genome displayed an overall pattern similar to the human genome, with high density in the top-left and bottom-right corners (Figures 3A and S9). Despite the qualitative similarity in both axes between the human and bat genome distributions, there is a greater under-representation of TC hotspots in the human genome (Wilcoxon rank-sum test for motif under-representation: p < 2.2 x 10−16 and for mutational susceptibility: p = 2.204 x 10−10). We also analyzed the distribution of bat orthologs of both human cancer and non-cancer genes (Figure 3B) and again observed features similar to the human distribution, including the bimodal distribution for TC hotspot under-representation. Furthermore, we analyzed 5 different species, including elephant, chimpanzee, mouse, yeast, and nematode worm (Figure S10). We found no qualitative difference between mammalian species. The yeast genome showed a uniform distribution in both measurements. The C. elegans genome showed that most genes have an over-representation of TC hot spots and low mutational susceptibility. We are interested in bats because of their longevity despite their small size and because the APOBEC gene family has expanded in various species.48 Compared to P. alecto and several other bats, all other species studies have fewer APOBECs, yet results from bats were not qualitatively different than those in other species. These results suggest that the APOBEC TC motif preference may have evolved to avoid harm to its genome, which is also supported by a previous study that the cytotoxicity of APOBEC3A is reduced via interaction with CCT chaperonin complex,51 but the genome itself does not appear to have been greatly shaped by APOBEC activity.
Figure 3.

APOBEC3A/B TC hotspots statistics of protein-coding transcripts of Pteropus alecto genome
(A) CDUR analysis on the bat genome shows two dense regions at the top-left and right-bottom extremes and relatively sparse dispersion elsewhere as in the human genome.
(B) Bat orthologs of human cancer and non-cancer genes show distinct distribution in mutational susceptibility but not in motif under-representation and (Kolmogorov-Smirnov test for motif-representation axis, P = 0.1701; for mutational susceptibility axis, P = 3.169 x 10-5).
Role of APOBEC3A/B in intra-tumor heterogeneity
To better understand the consequences of the bimodal distribution of APOBEC3A/B motif under-representation on tumor evolution and heterogeneity, we performed simulations. We adapted a previous computational model constructed with the Java-based spatial platform HAL49,50 to run simulations comparing clonal dynamics and trajectories over tumor evolution. Briefly, the model starts with cells without any mutations, and at each timestep, cells can divide to produce additional single cells or die. Depending on the average ratio between the division rate and the death rate in a population, the number of cells in a population may increase, decrease, or stay the same. A cell can acquire a single mutation according to the given mutation rate when the cell divides. The mutation can affect the fitness of cells positively, neutrally, and negatively, and the division rate at the next time step will be updated accordingly. First, we compared simulations with only random mutations and with additional APOBEC3A/B mutations where all genes have the same probability to get positive and negative mutations, to evaluate if APOBEC activity alone can increase heterogeneity (Figures 4A and 4B). We found that although APOBEC3A/B activity increases the overall mutation rate, there is no significant difference in intra-tumor heterogeneity (Figures S11A and S11B). Muller plots from each condition show that intra-tumor heterogeneity increases faster when there is APOBEC3A/B, however, the intra-tumor heterogeneity difference at the final time point is negligible (Figures 4C and 4D). Then, we simulated the effect of bimodal distribution of APOBEC3A/B motif under-representation from Figure 1A: the bimodal distribution is more evident in cancer genes compared to non-cancer genes. To investigate the role of the bimodal distribution, we assumed three categories of genes based on their motif under-representation. The genes with “low” and “high” motif under-representation are defined as genes whose motif under-representation is below 0.05 or above 0.95, respectively, and those that fall in between are defined as “mid”. Then, we assigned the probability of genes falling into each category according to the distribution of interest. For example, if we assume motif under-representation of a genome is uniformly distributed, the probability that a gene falls into the “low” or “high” category will be equal to its proportion, 0.05, and 0.9 for the “mid” category. In contrast, if we assume a bimodal distribution based on Figure 1A, the probability for “low,” “mid,” and “high” becomes 0.3, 0.6, and 0.1, respectively. In addition, we set a higher probability of getting a positive mutation than negative mutation for the genes in the “low” and “high” categories to emulate the result of Figure 2A that shows cancer genes mostly fall into “low” and “high” category. During the simulation, we recorded the number of genotypes in the population and computed a heterogeneity metric based on Shannon entropy as in West et al. 2021.50 We found the bimodal distribution leads to higher intra-tumor heterogeneity (Figures 4E and 4F). Furthermore, the difference in means test showed an increasing -log(P) value, indicating that the difference became substantial (Figures S11C and S11D). We found no evidence of linear or branching evolution in our simulation, and we only observed neutral evolution over 600-time steps. Based on our simulations, the effect of APOBEC activity on tumor initiation is comparable to random mutation, however, APOBEC activity with a bimodal distribution of TC hotspots in the genome can increase final tumor heterogeneity.
Figure 4.

Protein-coding transcripts from the human genome display biased distribution in APOBEC3A/B motif representation and mutational susceptibility
The bimodal distribution of APOBEC3A/B motif representation displays higher heterogeneity by spatial clonal dynamics simulation.
(A) The number of genotypes between with and without APOBEC3A/B shows no significant difference.
(B) Heterogeneity between the two simulations with and without APOBEC3A/B shows no significant difference.
(C and D) Muller plots of simulation with and without APOBEC3A/B activity show no significant difference in heterogeneity at the final time point.
(E) The number of genotypes between uniform and bimodal distribution shows an increasing difference over time.
(F) Heterogeneity between uniform and bimodal distribution shows an increasing difference over time.
(G and H) Muller plots of simulation uniform and bimodal distribution show an increasing difference over time.
Discussion
Evolvability is an emerging hallmark of cancer. The presence of genetic and phenotypic ITH is both the main source of evolvability and a significant factor contributing to poor prognosis and treatment failures in cancer patients. Several studies have highlighted the impact of ITH on disease progression and clinical outcomes.4,5 The heterogeneity itself arises through ongoing somatic evolution, driven by genetic mutations and the selection of clones. Different subclones within the tumor can possess distinct genetic alterations represented by a selectable phenotype, leading to variability in their response to their variable tumor microenvironment and increasing tumor fitness in general. Consequently, some subclones may survive and become dominant, while others may be resistant or susceptible to specific selection by tumor microenvironments. Understanding the principles that govern somatic evolution and the maintenance of ITH is crucial to understand tumor initiation, progression, evolution, and evolvability. However, the main unanswered question is how tumor heterogeneity fuels tumor evolution leading to increased cancer cell evolvability.
To answer that question, we designed an analysis pipeline for measuring genome mutation tolerance considering APOBEC as a mutator. Our goal was to discover how random mutation can affect genotypic heterogeneity while maintaining a protein-coding phenotype. We used CDUR to analyze the APOBEC mutation potential of the human genome. We analyzed motif under-representation and mutational susceptibility of the APOBEC3A/B TC hotspot motif for more than 40,000 human protein-coding transcripts. CDUR analyses revealed disproportionate motif under-representation and mutational susceptibility. Transcripts of the human genome were densely concentrated in the top-left or bottom-right corner of the plot, indicating distinct strategies employed by genes to either avoid APOBEC3A/B mutagenesis or increase the number of TC hotspots while reducing mutational susceptibility.39,51 However, APOBEC may have evolved to avoid causing damage to genes instead of genes evolving to avoid APOBEC mutation hotspots. To validate this finding, we extended our research to bats. Despite having many more APOBEC3 genes, hotspot under-representation in Pteropus alecto mirrored that of humans. Such similarity in pattern suggests APOBEC evolution to mitigate impacts on the genome instead of the converse.
Moreover, genes located in the top-left corner of the CDUR plot exhibited significant enrichment in cancer-associated pathways, suggesting a potential link between APOBEC3A/B TC hotspot statistics and cancer formation. However, it is still unclear to what extent human genes have evolved under APOBEC selection and how adaptation to APOBEC is related to their original role in tissue or tumors, or correlated with tumor microenvironment.
Further analysis of cancer genes demonstrated a distinct distribution compared to non-cancer genes. While cancer genes exhibited significantly higher mutational susceptibility, the means of the motif under-representation axis were statistically similar between the two groups. The presence of two peaks at opposite positions in the motif under-representation axis likely contributed to this non-significant comparison. However, a Kolmogorov-Smirnov test comparing frequency distributions (as opposed to just means) confirmed that cancer and non-cancer genes differed significantly in both axes of the CDUR plot.
We propose a hypothesis that addresses why cancer genes exhibit distinct behaviors under APOBEC activity. Because of its involvement in viral defense, APOBEC can induce mutations in its own genome. Under strong selection to minimize genomic mutations induced by APOBEC, there are two scenarios: (1) APOBEC evolves target motifs to reduce activity on genes co-expressed with APOBEC, or (2) genes co-expressed with APOBEC evolve to have fewer targets, preventing mutations caused by APOBEC. We argue that the first scenario is more plausible, and this is supported by experimental evidence by Green et al. 2021.52 First, evolutionary adaptation toward reducing targets for all genes expressed in cells, including APOBEC, seems less likely than a specific mutation occurring in a single gene, in our case, APOBEC. Second, variable gene expression is indispensable for normal cellular activities, therefore selection on a single gene, like APOBEC, is more likely than the evolution of a broad reduction in mutation targets for all co-expressed genes. Given this perspective, APOBEC target preference evolves so as not to harm essential genes in which mutations are fatal, corresponding to the non-cancer genes.
In addition, reflecting on historical viral pandemics such as COVID-19, avian influenza, and the Spanish flu, selection may have favored causing more harm to viruses instead of minimizing APOBEC-induced mutations in the host. Selection to mitigate APOBEC-induced mutations in genes linked to cancer is likely weaker than that favoring inducing mutations detrimental to viruses. This is because individuals must survive viral infections before selection to minimize potential cancer risks associated with APOBEC-induced mutations can take effect. Therefore, it is more likely that APOBEC target preference evolved, not in the context of cancer, but to reduce harm to genes co-expressed during viral infection. We propose the age dependency of antiviral activity (lifelong but critical in youth) vs. cancer explains the high mutational susceptibility of cancer-associated genes in Figure 2A. Why the motif under-representation axis has a bimodal distribution, however, is still unknown.
Additionally, analyses of orthologs from multiple vertebrate species revealed a wide range of variance in motif representation and mutational susceptibility for genes, irrespective of their association with cancer. This observation suggests that genes undergo various evolutionary trajectories that can greatly alter the representation and susceptibility of APOBEC3A/B motifs. The broad distribution of orthologs in the CDUR plot indicates there are multiple strategies for genes to tolerate APOBEC3A/B-induced mutagenesis and maintain their essential functions.
Our simulations showed the effects of APOBEC-driven heterogeneity on cancer cell clonal dynamics and evolutionary trajectories. In cancer, substitution accumulation can occur in two ways: some mutations provide a selective advantage, leading to clonal expansion and dominant subclones, while others arise randomly and have minimal impact on cell fitness. Over time, and with stable or increasing cell population size, these neutral substitutions accumulate, contributing to genetic diversity and intra-tumor heterogeneity. We found APOBEC can significantly influence cancer genes compared to non-cancer genes, leading to a notable increase in neutral intra-tumoral heterogeneity. Neutral evolution refers to the maintenance of genetic changes that arise randomly without conferring a selective advantage or disadvantage to cells. In the context of APOBEC activity, we propose two effects on evolution. First, APOBEC activity increases the occurrence of random mutations within the genome. These substitutions result from the enzymatic activity of APOBEC. Second, APOBEC activity exhibits a biased increase in mutations within a subset of cancer genes compared to non-cancer genes. This bias toward cancer genes further shapes the genetic landscape of the tumor, potentially influencing tumor progression and response to treatment. In summary, APOBEC activity impacts neutral evolution through increased random mutations that generate greater variation, and a preferential increase in mutations within cancer-related genes. Both effects contribute to the genetic diversity and intra-tumor heterogeneity observed in cancer.
Overall, our study provides insights into the APOBEC3A/B TC hotspots in the human genome and highlights the distinct characteristics of cancer genes in the APOBEC3A/B motifs statistics. These findings contribute to our understanding of the interplay between APOBEC3A/B mutagenesis and the development of cancer. We found APOBEC has a significantly stronger influence inducing progressor mutations and mainly on cancer genes compared to non-cancer genes, leading to increased intra-tumoral heterogeneity. Our findings emphasize the critical role of APOBEC as an accelerator of carcinogenesis that amplifies intra-tumoral heterogeneity through-toward neutral evolution, instead of an initiator of the cancer process.
Limitations of the study
This study has several limitations. First, it focuses solely on APOBEC3A/B TC hotspots and does not account for other mutational processes or factors that may contribute to the observed patterns. Second, the reliance on computational analyses and statistical tests introduces inherent assumptions and potential inaccuracies. Furthermore, the study’s conclusions are based on correlations instead of causation, and experimental validation is necessary. The use of CDUR statistics as the primary analysis tool has its own limitations, and alternative or complementary approaches could provide additional insights. Lastly, while the comparative analysis of orthologs across species is informative, species-specific factors may influence mutational dynamics. Overall, while valuable, the findings should be interpreted cautiously, and further research is needed to address these limitations and provide a more comprehensive understanding of the relationship between mutational processes, gene function, and cancer.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited data | ||
| Human protein-coding transcript sequences | GENCODE v40 | https://www.gencodegenes.org/ |
| Mouse protein-coding transcript sequences | GENCODE vm33 | https://www.gencodegenes.org/ |
| Cancer gene census | COSMIC v97 (Nov 2022) | https://cancer.sanger.ac.uk/cosmic |
| COSMIC mutation data | COSMIC v97 (Nov 2022) | https://cancer.sanger.ac.uk/cosmic |
| Human genes list | NCBI Datasets (RefSeq release 218) | https://www.ncbi.nlm.nih.gov/datasets/ |
| Orthologs sequences | NCBI Datasets (RefSeq release 218) | https://www.ncbi.nlm.nih.gov/datasets/ |
| Pteropus alecto gene list | NCBI Datasets (RefSeq release 218) | https://www.ncbi.nlm.nih.gov/datasets/ |
| Software and algorithms | ||
| NCBI Datasets command line tools (CLI v13.x (API v1)) | NCBI Datasets | https://www.ncbi.nlm.nih.gov/datasets/docs/v1/download-and-install/ |
| ShinyGO v. 0.77 | ShinyGO | http://bioinformatics.sdstate.edu/go/ |
| HAL (v.1.1.0) | Hybrid Agent Library: HAL | https://halloworld.org/ |
| CDUR | Shapiro, Meier & MacCarthy, 201838 | https://gitlab.com/maccarthyslab/CDUR |
| R (v. 4.2.3) | R CRAN | https://www.r-project.org/ |
| biomarRt (v. 3.17) | Bioconductor | https://bioconductor.org/packages/release/bioc/html/biomaRt.html |
| Python (v. 3.6; v. 3.9) | Python | https://www.python.org/ |
| BioPython (v. 1.76; v. 1.79) | Biopython | https://biopython.org/ |
| Other | ||
| Python and R analysis scripts for the publication | This paper | https://github.com/sbu-damaghi-ceel/Evolvability_of_cancer-associated_genes_under_APOBEC3AB_selection |
Resource availability
Lead contact
Further information and any related requests should be directed to and will be fulfilled by the lead contact Mehdi Damaghi (Mehdi.Damaghi@stonybrookmedicine.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
-
•
GENCODE transcripts sequences: All human coding sequences used in this work can be acquired from GENCODE (https://www.gencodegenes.org/).
-
•
Cancer mutation data: Cancer genes and all point mutation data in cancer can be acquired from the COSMIC Database (https://cancer.sanger.ac.uk/cosmic).
-
•
Orthologs sequences and annotations: Orthlogs RefSeq ID and sequences can be acquired via NCBI Datasets command line tools (https://www.ncbi.nlm.nih.gov/datasets/docs/v2/download-and-install/).
-
•
Pteropus alecto sequences and annotations: All Pteropus transcripts sequences and annotations can be acquired via NCBI Datasets command line tools (https://www.ncbi.nlm.nih.gov/datasets/docs/v2/download-and-install/).
-
•
Clonal evolution simulations of cancer: Hybrid Automata Library (HAL) is freely available from https://halloworld.org/, and the model Java scripts are available in the following GitHub repository.
-
•
The cytidine deaminase under-representation reporter (CDUR): CDUR is freely available from the GitLab repository (https://gitlab.com/maccarthyslab/CDUR).
-
•
All original scripts to download and process the data are available at https://github.com/sbu-damaghi-ceel/Evolvability_of_cancer-associated_genes_under_APOBEC3AB_selection and are publicly available as of the date of publication.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Method details
Transcripts sequences preprocessing
CDUR requires inputs as FASTA files, with each record being a complete coding sequence starting with the start codon and ending with stop codons. The FASTA file from GENCODE contains protein-coding transcript sequences that have not only coding sequences but also 5’ and 3’ untranslated regions. In addition, several sequences were incomplete coding sequences. To make an analysis only on valid coding sequences, we filtered records in the original GENCODE FASTA file and extracted valid sequences. First, substrings of the sequence were extracted based on annotation in the FASTA file name field. Then, we checked four criteria; 1) whether the length of the coding sequence is multiple of three, 2) whether the sequence starts with ATG, 3) whether the sequence ends with a stop codon, and 4) whether the name contains PAR_Y indicating pseudoautosomal region from Y chromosome. Transcript sequences that passed all criteria were saved in a single FASTA for CDUR input. The aforementioned preprocessing was applied to human and mouse genome analysis.
For other species, including black flying fox (Pteropus alecto), African elephant (Loxodonta africana), chimpanzee (Pan troglodytes), yeast (Saccharomyces cerevisiae), and nematode worm (Caenorhabditis elegans), we used NCBI Datasets command line tools to download all annotated genes information. Then, we used a custom Python script with BioPython Entrez packages to retrieve coding sequences of all valid transcript sequences. The scripts first used previously downloaded gene information to retrieve all protein-coding NCBI gene IDs. In the second step, the script fetches gene ids using the “efetch” command to get the associated RefSeq ID. Third, RefSeq IDs were fetched to get GenBank information, and coding sequences were extracted based on the information. Finally, all valid transcript sequences were saved in a single FASTA file for CDUR input.
APOBEC3A/B motifs statistics analysis with CDUR
We applied the Cytidine Deaminase Under-representation Reporter (CDUR) to evaluate APOBEC3A/B motifs statistics, motif under-representation, and mutational susceptibility, of human transcripts. Briefly, CDUR generates a null model by generating shuffled sequences from a given sequence of interest. Then CDUR computes motif under-representation of the fraction of sequence in the null model, which has fewer motifs than the given sequence. For TC hotspots, we used motif under-representation as the “belowT_C_'' value from the CDUR result. Mutational susceptibility was defined by how probable the sequence is for transition mutations in hotspots to result in amino acid alterations. CDUR results contain a value “repTrFrac_belowT_C_,” which is the ratio of non-synonymous change in TC hotspots, and we used 1-“repTrFrac_belowT_C_” for mutational susceptibility. Preprocessed coding sequences (CDS) of transcripts for humans and bats were passed to CDUR with default settings; the shuffle method is n3, and the number of shuffles is 1,000. We ran CDUR three times on the same sequences, respectively, and the average of the values was used in the analysis.
To analyze VC motifs in hairpin-forming structures, we set strict constraints to minimize search space based on Langenbucher et al. 2021.40 We considered 3 bp NVC loops with a maximum 20 bp stem without bulge, and stem strength greater than or equal to 15. The stem strength was calculated using the algorithm from Langenbucher et al. 2021,40 which sums the pairing strengths: A:T pairs are scored as 1, G:C pairs as 3, and other pairings as 0.
Gene enrichment analysis
Top-left genes were defined as genes with under-representation ≤ 0.05 and mutational susceptibility ≤ 0.05. Bottom-right genes were defined as genes with under-representation ≥ 0.95 and mutational susceptibility ≥ 0.95. top-left and bottom-right genes were extracted using the dplyr R package. Only gene names were saved in separate text files and submitted to ShinyGO 0.7753,54,55 with default parameters; FDR cutoff = 0.05, # pathways to show = 20, Pathway size: Min = 2, and Max = 2,000. We downloaded the enrichment results of the top 20 pathways from GO Biological Process, GO Cellular Component, GO Molecular Function, and KEGG, and bar plots were generated with R package dplyr and ggplot2 using custom R scripts.
Cancer and non-cancer genes
We defined cancer genes as genes in the COSMIC Cancer Census.45 We retrieved RefSeq ID and Ensembl ID with biomaRt to map these genes to GENCODE transcripts. Bat equivalent cancer-associated genes were collected using NCBI Datasets command line tools with Gene ID extracted from COSMIC Cancer Census. Non-cancer genes were defined as genes that rarely get point mutations in the coding region of their transcript. We used custom R and shell scripts for the process to filter out those genes. First, all human reference genes were downloaded from NCBI using NCBI Datasets Command line tools. Then all genes from the NCBI list were checked to see whether genes have been recorded in COSMIC mutation data at least once. Among the genes that did not have any record, we filtered only protein-coding sequences. In addition, cloned genes, such as gene names starting with LOC, were removed, as well as mitochondrial genes. We applied the same procedure to collect bat-equivalent cancer genes to generate bat-equivalent non-cancer genes.
Ten species ortholog genes
Orthologs of human cancer or non-cancer genes were downloaded from NCBI using the NCBI Datasets command lines tool. For each human gene, we retrieved the gene ID and used it to download orthologs. This process downloads all orthologs available. After downloading all orthologs, we selected genes that have orthologs for all ten species. The sequences of the orthologs were preprocessed to pass into CDUR as we did for the Pteropus alecto genome, and CDUR statistics were computed three times per sequence. For variation analysis, we generated mutated sequences from human transcript sequences by simulating sequential C-to-T mutation until the amino acid sequence changes. We performed ten simulations for each transcript, and the standard deviation on both axes was computed.
Heterogeneity comparison simulation
Clonal expansion simulations were performed using a computation model constructed with the Java-based spatial simulation platform HAL.49 The basic scheme of our model was adopted from West et al. 2022.50 We added more parameters and changed the mutation method to capture the effect of human genome distribution by the APOBEC3A/B motif proportion of each gene. The model starts with 100 cells in the center of a circle radius of 100 grid points where each single cell occupies a single grid point. Each cell in our model can have three different types of mutations that have different effects on the fitness (division rate); positive, neutral, and negative mutations. Positive and negative mutations increase or decrease fitness, respectively, and neutral mutations do not have any effect on fitness. During each time step, each cell can divide, producing one additional cell into a grid or die off, introducing a single empty grid point. This process may increase the number of cells in a population if the average division rate is higher than the death rate and decreases in the opposite situation. Probabilities to divide or die were computed using the following equation:
| (Equation 1) |
| (Equation 2) |
where and are the baseline division and death rates, respectively.
The following parameters were used: total number of mutable sites, , positive fitness effect, , negative fitness effect, , random mutation rate, , APOBEC3 mutation rate . APOBEC3 mutation was turned on from time point 0 to 50 to reflect burst expression in the early stage. We used 0.001, 0.01, and 0.1 for the proportion of APOBEC3 for “low,” “mid,” and “high.” To emulate the phenomenon that cancer-associated genes (higher probability to have positive fitness effect) were located in bimodal extreme (“low” and “high”), we set the probability to have mutations that have positive, neutral, and negative fitness to 0.4, 0.5, and 0.1, respectively, and for non-cancer genes (higher probability to have negative fitness effect), we set the probabilities to 0.1, 0.5, and 0.4, respectively.
To count the number of genotypes in the population, the genotype ID of the cell at each time point was tagged using the number of each mutation type by Equation 3,
| (Equation 3) |
where , , and are the number of positive, negative, and neutral mutations, respectively, and and are the maximum number of negative and neutral mutations in the population. In addition, heterogeneity was measured using Shannon entropy as in West et al. 2022, given by:
| (Equation 4) |
where pi is the proportion of cells within the population with genotype ID .
We performed 100 simulations for each condition and used the R base package t-test for the difference of means test for every 50 time points between with and without APOBEC activity, or uniform and bimodal distribution.
Quantification and statistical analysis
Difference of means test and equality test of distribution
We used R base package functions wilcox.test and ks.test with default parameters for the Wilcoxon rank sum test and Kolmogorov-Smirnov test, respectively, to compare motif under-representation and mutational susceptibility distribution between cancer genes and non-cancer genes (Figures 2A, 3B, S8, and S9).
Difference of means test for orthologs and sequential mutations
We used R base package function t.test for paired t-test to compare the mean of the standard deviation of motif under-representation and mutational susceptibility between sequential mutations of genes and orthologs (Figures 2C and 2D).
Heterogeneity difference test for clonal dynamic simulation
For clonal dynamic simulation, we performed a t-test using the R base package function t.test to perform a difference of means test between two conditions. Specifically, for every 50 time points, we compared two distributions consisting of 100 values from 100 simulations of each condition (Figure S11).
Acknowledgments
We gratefully acknowledge funding from the Physical Sciences Oncology Network at the National Cancer Institute (grant U01CA261841) as well as support from the Stony Brook Cancer Center. This work was also supported partly by R01 grant R01CA272601 (NCI). L.M.D was supported in part by NSF-IOS 2032063 and NSF-DEB 1838273.
Author contributions
J-H.S., T.M., and M.D., designed research; J-H.S performed research; J-H.S contributed new reagents/analytic tools; J-H.S. analyzed data; L.M.D. contributed to results interpretation; and J-H.S., T.M., and M.D. wrote the paper. All authors revised the paper.
Declaration of interests
The authors declare no competing interest.
Published: March 6, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2024.109433.
Contributor Information
Thomas MacCarthy, Email: thomas.maccarthy@stonybrook.edu.
Mehdi Damaghi, Email: mehdi.damaghi@stonybrookmedicine.edu.
Supplemental information
References
- 1.Greaves M., Maley C.C. Clonal evolution in cancer. Nature. 2012;481:306–313. doi: 10.1038/nature10762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Korolev K.S., Xavier J.B., Gore J. Turning ecology and evolution against cancer. Nat. Rev. Cancer. 2014;14:371–380. doi: 10.1038/nrc3712. [DOI] [PubMed] [Google Scholar]
- 3.Nowell P.C. The clonal evolution of tumor cell populations. Science. 1976;194:23–28. doi: 10.1126/science.959840. [DOI] [PubMed] [Google Scholar]
- 4.Gerlinger M., Rowan A.J., Horswell S., Math M., Larkin J., Endesfelder D., Gronroos E., Martinez P., Matthews N., Stewart A., et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012;366:883–892. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Swanton C. Intratumor heterogeneity: evolution through space and time. Cancer Res. 2012;72:4875–4882. doi: 10.1158/0008-5472.CAN-12-2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sottoriva A., Kang H., Ma Z., Graham T.A., Salomon M.P., Zhao J., Marjoram P., Siegmund K., Press M.F., Shibata D., Curtis C. A Big Bang model of human colorectal tumor growth. Nat. Genet. 2015;47:209–216. doi: 10.1038/ng.3214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bishop K.N., Holmes R.K., Sheehy A.M., Davidson N.O., Cho S.-J., Malim M.H. Cytidine deamination of retroviral DNA by diverse APOBEC proteins. Curr. Biol. 2004;14:1392–1396. doi: 10.1016/j.cub.2004.06.057. [DOI] [PubMed] [Google Scholar]
- 8.Kohli R.M., Abrams S.R., Gajula K.S., Maul R.W., Gearhart P.J., Stivers J.T. A Portable Hot Spot Recognition Loop Transfers Sequence Preferences from APOBEC Family Members to Activation-induced Cytidine Deaminase. J. Biol. Chem. 2009;284:22898–22904. doi: 10.1074/jbc.M109.025536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Suspène R., Aynaud M.-M., Vartanian J.-P., Wain-Hobson S. Efficient deamination of 5-methylcytidine and 5-substituted cytidine residues in DNA by human APOBEC3A cytidine deaminase. PLoS One. 2013;8 doi: 10.1371/journal.pone.0063461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Adolph M.B., Ara A., Feng Y., Wittkopp C.J., Emerman M., Fraser J.S., Chelico L. Cytidine deaminase efficiency of the lentiviral viral restriction factor APOBEC3C correlates with dimerization. Nucleic Acids Res. 2017;45:3378–3394. doi: 10.1093/nar/gkx066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hultquist J.F., Lengyel J.A., Refsland E.W., LaRue R.S., Lackey L., Brown W.L., Harris R.S. Human and rhesus APOBEC3D, APOBEC3F, APOBEC3G, and APOBEC3H demonstrate a conserved capacity to restrict Vif-deficient HIV-1. J. Virol. 2011;85:11220–11234. doi: 10.1128/JVI.05238-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yu Q., König R., Pillai S., Chiles K., Kearney M., Palmer S., Richman D., Coffin J.M., Landau N.R. Single-strand specificity of APOBEC3G accounts for minus-strand deamination of the HIV genome. Nat. Struct. Mol. Biol. 2004;11:435–442. doi: 10.1038/nsmb758. [DOI] [PubMed] [Google Scholar]
- 13.Holden L.G., Prochnow C., Chang Y.P., Bransteitter R., Chelico L., Sen U., Stevens R.C., Goodman M.F., Chen X.S. Crystal structure of the anti-viral APOBEC3G catalytic domain and functional implications. Nature. 2008;456:121–124. doi: 10.1038/nature07357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rebhandl S., Huemer M., Greil R., Geisberger R. AID/APOBEC deaminases and cancer. Oncoscience. 2015;2:320–333. doi: 10.18632/oncoscience.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brown A.L., Collins C.D., Thompson S., Coxon M., Mertz T.M., Roberts S.A. Single-stranded DNA binding proteins influence APOBEC3A substrate preference. Sci. Rep. 2021;11 doi: 10.1038/s41598-021-00435-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Adolph M.B., Love R.P., Feng Y., Chelico L. Enzyme cycling contributes to efficient induction of genome mutagenesis by the cytidine deaminase APOBEC3B. Nucleic Acids Res. 2017;45:11925–11940. doi: 10.1093/nar/gkx832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mussil B., Suspène R., Aynaud M.-M., Gauvrit A., Vartanian J.-P., Wain-Hobson S. Human APOBEC3A isoforms translocate to the nucleus and induce DNA double strand breaks leading to cell stress and death. PLoS One. 2013;8 doi: 10.1371/journal.pone.0073641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Salamango D.J., McCann J.L., Demir Ö., Brown W.L., Amaro R.E., Harris R.S. APOBEC3B Nuclear Localization Requires Two Distinct N-Terminal Domain Surfaces. J. Mol. Biol. 2018;430:2695–2708. doi: 10.1016/j.jmb.2018.04.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Burns M.B., Lackey L., Carpenter M.A., Rathore A., Land A.M., Leonard B., Refsland E.W., Kotandeniya D., Tretyakova N., Nikas J.B., et al. APOBEC3B is an enzymatic source of mutation in breast cancer. Nature. 2013;494:366–370. doi: 10.1038/nature11881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Petljak M., Green A.M., Maciejowski J., Weitzman M.D. Addressing the benefits of inhibiting APOBEC3-dependent mutagenesis in cancer. Nat. Genet. 2022;54:1599–1608. doi: 10.1038/s41588-022-01196-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Burns M.B., Temiz N.A., Harris R.S. Evidence for APOBEC3B mutagenesis in multiple human cancers. Nat. Genet. 2013;45:977–983. doi: 10.1038/ng.2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mertz T.M., Collins C.D., Dennis M., Coxon M., Roberts S.A. APOBEC-Induced Mutagenesis in Cancer. Annu. Rev. Genet. 2022;56:229–252. doi: 10.1146/annurev-genet-072920-035840. [DOI] [PubMed] [Google Scholar]
- 23.Wu J., Pan T.-H., Xu S., Jia L.-T., Zhu L.-L., Mao J.-S., Zhu Y.-L., Cai J.-T. The virus-induced protein APOBEC3G inhibits anoikis by activation of Akt kinase in pancreatic cancer cells. Sci. Rep. 2015;5 doi: 10.1038/srep12230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rustad E.H., Yellapantula V., Leongamornlert D., Bolli N., Ledergor G., Nadeu F., Angelopoulos N., Dawson K.J., Mitchell T.J., Osborne R.J., et al. Timing the initiation of multiple myeloma. Nat. Commun. 2020;11:1917. doi: 10.1038/s41467-020-15740-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Henderson S., Fenton T. APOBEC3 genes: retroviral restriction factors to cancer drivers. Trends Mol. Med. 2015;21:274–284. doi: 10.1016/j.molmed.2015.02.007. [DOI] [PubMed] [Google Scholar]
- 26.McGranahan N., Swanton C. Biological and therapeutic impact of intratumor heterogeneity in cancer evolution. Cancer Cell. 2015;27:15–26. doi: 10.1016/j.ccell.2014.12.001. [DOI] [PubMed] [Google Scholar]
- 27.Martelotto L.G., Ng C.K.Y., Piscuoglio S., Weigelt B., Reis-Filho J.S. Breast cancer intra-tumor heterogeneity. Breast Cancer Res. 2014;16:210. doi: 10.1186/bcr3658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hu Z., Sun R., Curtis C. A population genetics perspective on the determinants of intra-tumor heterogeneity. Biochim. Biophys. Acta Rev. Canc. 2017;1867:109–126. doi: 10.1016/j.bbcan.2017.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Swanton C., McGranahan N., Starrett G.J., Harris R.S. APOBEC Enzymes: Mutagenic Fuel for Cancer Evolution and Heterogeneity. Cancer Discov. 2015;5:704–712. doi: 10.1158/2159-8290.CD-15-0344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Alexandrov L.B., Nik-Zainal S., Wedge D.C., Aparicio S.A.J.R., Behjati S., Biankin A.V., Bignell G.R., Bolli N., Borg A., Børresen-Dale A.L., et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–421. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nik-Zainal S., Alexandrov L.B., Wedge D.C., Van Loo P., Greenman C.D., Raine K., Jones D., Hinton J., Marshall J., Stebbings L.A., et al. Mutational processes molding the genomes of 21 breast cancers. Cell. 2012;149:979–993. doi: 10.1016/j.cell.2012.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alexandrov L.B., Kim J., Haradhvala N.J., Huang M.N., Tian Ng A.W., Wu Y., Boot A., Covington K.R., Gordenin D.A., Bergstrom E.N., et al. The repertoire of mutational signatures in human cancer. Nature. 2020;578:94–101. doi: 10.1038/s41586-020-1943-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Koutros S., Rao N., Moore L.E., Nickerson M.L., Lee D., Zhu B., Pardo L.A., Baris D., Schwenn M., Johnson A., et al. Targeted Deep Sequencing of Bladder Tumors Reveals Novel Associations between Cancer Gene Mutations and Mutational Signatures with Major Risk Factors. Clin. Cancer Res. 2021;27:3725–3733. doi: 10.1158/1078-0432.CCR-20-4419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chang M.T., Penson A., Desai N.B., Socci N.D., Shen R., Seshan V.E., Kundra R., Abeshouse A., Viale A., Cha E.K., et al. Small-Cell Carcinomas of the Bladder and Lung Are Characterized by a Convergent but Distinct Pathogenesis. Clin. Cancer Res. 2018;24:1965–1973. doi: 10.1158/1078-0432.CCR-17-2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Venkatesan S., Angelova M., Puttick C., Zhai H., Caswell D.R., Lu W.-T., Dietzen M., Galanos P., Evangelou K., Bellelli R., et al. Induction of APOBEC3 Exacerbates DNA Replication Stress and Chromosomal Instability in Early Breast and Lung Cancer Evolution. Cancer Discov. 2021;11:2456–2473. doi: 10.1158/2159-8290.CD-20-0725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Roelofs P.A., Martens J.W.M., Harris R.S., Span P.N. Clinical Implications of APOBEC3-Mediated Mutagenesis in Breast Cancer. Clin. Cancer Res. 2023;29:1658–1669. doi: 10.1158/1078-0432.CCR-22-2861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Leonard B., Hart S.N., Burns M.B., Carpenter M.A., Temiz N.A., Rathore A., Vogel R.I., Nikas J.B., Law E.K., Brown W.L., et al. APOBEC3B upregulation and genomic mutation patterns in serous ovarian carcinoma. Cancer Res. 2013;73:7222–7231. doi: 10.1158/0008-5472.CAN-13-1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shapiro M., Meier S., MacCarthy T. The cytidine deaminase under-representation reporter (CDUR) as a tool to study evolution of sequences under deaminase mutational pressure. BMC Bioinf. 2018;19:163. doi: 10.1186/s12859-018-2161-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shapiro M., Krug L.T., MacCarthy T. Mutational pressure by host APOBEC3s more strongly affects genes expressed early in the lytic phase of herpes simplex virus-1 (HSV-1) and human polyomavirus (HPyV) infection. PLoS Pathog. 2021;17 doi: 10.1371/journal.ppat.1009560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen Y., Verbeek F.J., Wolstencroft K. Establishing a consensus for the hallmarks of cancer based on gene ontology and pathway annotations. BMC Bioinf. 2021;22:178. doi: 10.1186/s12859-021-04105-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Farach A., Ding Y., Lee M., Creighton C., Delk N.A., Ittmann M., Miles B., Rowley D., Farach-Carson M.C., Ayala G.E. Neuronal Trans-Differentiation in Prostate Cancer Cells. Prostate. 2016;76:1312–1325. doi: 10.1002/pros.23221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sillars-Hardebol A.H., Carvalho B., de Wit M., Postma C., Delis-van Diemen P.M., Mongera S., Ylstra B., van de Wiel M.A., Meijer G.A., Fijneman R.J.A. Identification of key genes for carcinogenic pathways associated with colorectal adenoma-to-carcinoma progression. Tumour Biol. 2010;31:89–96. doi: 10.1007/s13277-009-0012-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Damaghi M., West J., Robertson-Tessi M., Xu L., Ferrall-Fairbanks M.C., Stewart P.A., Persi E., Fridley B.L., Altrock P.M., Gatenby R.A., et al. The harsh microenvironment in early breast cancer selects for a Warburg phenotype. Proc. Natl. Acad. Sci. USA. 2021;118 doi: 10.1073/pnas.2011342118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tate J.G., Bamford S., Jubb H.C., Sondka Z., Beare D.M., Bindal N., Boutselakis H., Cole C.G., Creatore C., Dawson E., et al. COSMIC: the Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2019;47:D941–D947. doi: 10.1093/nar/gky1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sondka Z., Bamford S., Cole C.G., Ward S.A., Dunham I., Forbes S.A. The COSMIC Cancer Gene Census: describing genetic dysfunction across all human cancers. Nat. Rev. Cancer. 2018;18:696–705. doi: 10.1038/s41568-018-0060-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hayward J.A., Tachedjian M., Cui J., Cheng A.Z., Johnson A., Baker M.L., Harris R.S., Wang L.-F., Tachedjian G. Differential Evolution of Antiretroviral Restriction Factors in Pteropid Bats as Revealed by APOBEC3 Gene Complexity. Mol. Biol. Evol. 2018;35:1626–1637. doi: 10.1093/molbev/msy048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ricci M., Peona V., Boattini A., Taccioli C. Comparative analysis of bats and rodents’ genomes suggests a relation between non-LTR retrotransposons, cancer incidence, and ageing. Sci. Rep. 2023;13:9039. doi: 10.1038/s41598-023-36006-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jebb D., Huang Z., Pippel M., Hughes G.M., Lavrichenko K., Devanna P., Winkler S., Jermiin L.S., Skirmuntt E.C., Katzourakis A., et al. Six reference-quality genomes reveal evolution of bat adaptations. Nature. 2020;583:578–584. doi: 10.1038/s41586-020-2486-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bravo R.R., Baratchart E., West J., Schenck R.O., Miller A.K., Gallaher J., Gatenbee C.D., Basanta D., Robertson-Tessi M., Anderson A.R.A. Hybrid Automata Library: A flexible platform for hybrid modeling with real-time visualization. PLoS Comput. Biol. 2020;16 doi: 10.1371/journal.pcbi.1007635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.West J., Schenck R.O., Gatenbee C., Robertson-Tessi M., Anderson A.R.A. Normal tissue architecture determines the evolutionary course of cancer. Nat. Commun. 2021;12:2060. doi: 10.1038/s41467-021-22123-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Martinez T., Shapiro M., Bhaduri-McIntosh S., MacCarthy T. Evolutionary effects of the AID/APOBEC family of mutagenic enzymes on human gamma-herpesviruses. Virus Evol. 2019;5:vey040. doi: 10.1093/ve/vey040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Green A.M., DeWeerd R.A., O’Leary D.R., Hansen A.R., Hayer K.E., Kulej K., Dineen A.S., Szeto J.H., Garcia B.A., Weitzman M.D. Interaction with the CCT chaperonin complex limits APOBEC3A cytidine deaminase cytotoxicity. EMBO Rep. 2021;22 doi: 10.15252/embr.202052145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ge S.X., Jung D., Yao R. ShinyGO: a graphical gene-set enrichment tool for animals and plants. Bioinformatics. 2020;36:2628–2629. doi: 10.1093/bioinformatics/btz931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Luo W., Brouwer C. Pathview: an R/Bioconductor package for pathway-based data integration and visualization. Bioinformatics. 2013;29:1830–1831. doi: 10.1093/bioinformatics/btt285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kanehisa M., Furumichi M., Sato Y., Ishiguro-Watanabe M., Tanabe M. KEGG: integrating viruses and cellular organisms. Nucleic Acids Res. 2021;49:D545–D551. doi: 10.1093/nar/gkaa970. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
GENCODE transcripts sequences: All human coding sequences used in this work can be acquired from GENCODE (https://www.gencodegenes.org/).
-
•
Cancer mutation data: Cancer genes and all point mutation data in cancer can be acquired from the COSMIC Database (https://cancer.sanger.ac.uk/cosmic).
-
•
Orthologs sequences and annotations: Orthlogs RefSeq ID and sequences can be acquired via NCBI Datasets command line tools (https://www.ncbi.nlm.nih.gov/datasets/docs/v2/download-and-install/).
-
•
Pteropus alecto sequences and annotations: All Pteropus transcripts sequences and annotations can be acquired via NCBI Datasets command line tools (https://www.ncbi.nlm.nih.gov/datasets/docs/v2/download-and-install/).
-
•
Clonal evolution simulations of cancer: Hybrid Automata Library (HAL) is freely available from https://halloworld.org/, and the model Java scripts are available in the following GitHub repository.
-
•
The cytidine deaminase under-representation reporter (CDUR): CDUR is freely available from the GitLab repository (https://gitlab.com/maccarthyslab/CDUR).
-
•
All original scripts to download and process the data are available at https://github.com/sbu-damaghi-ceel/Evolvability_of_cancer-associated_genes_under_APOBEC3AB_selection and are publicly available as of the date of publication.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.