

Abstract
Biocatalytic hydrogen borrowing represents an environmentally friendly and highly efficient synthetic method. This innovative approach involves converting various substrates into high-value-added products, typically via a one-pot, two/three-step sequence encompassing dehydrogenation (intermediate transformation) and hydrogenation processes employing the hydride shuffling between NAD(P)+ and NAD(P)H. Represented key transformations in hydrogen borrowing include stereoisomer conversion within alcohols, conversion between alcohols and amines, conversion of allylic alcohols to saturated carbonyl counterparts, and α,β-unsaturated aldehydes to saturated carboxylic acids, etc. The direct transformation methodology and environmentally benign characteristics of hydrogen borrowing have contributed to its advancements in fine chemical synthesis or drug developments. Over the past decades, the hydrogen borrowing strategy in biocatalysis has led to the creation of diverse catalytic systems, demonstrating substantial potential for straightforward synthesis as well as asymmetric transformations. This perspective serves as a detailed exposition of the recent advancements in biocatalytic reactions employing the hydrogen borrowing strategy. It provides insights into the potential of this approach for future development, shedding light on its promising prospects in the field of biocatalysis.
Keywords: borrowing hydrogen, biocatalysis, dehydrogenase, reductase, asymmetric catalysis
1. Introduction
Hydrogen borrowing, a redox-neutral reaction, does not require the addition of any additional oxidant or reductant, can be achieved through dehydrogenative oxidation, intermediate transformation, and hydrogenation in one pot to convert simple materials to value-added products (Figure 1A).1 Importantly, this process typically generates water as the sole byproduct, rendering it a highly atom-economic and environmentally friendly reaction. In this system, a catalyst, often a transition metal catalyst or an enzyme, serves as a carrier to extract hydrogen from the raw material during the oxidation step. This extracted hydrogen is temporarily stored and directly reintroduced into the product during the subsequent reduction step, effectively transferring hydrogen from the raw material to the final product. This mechanism is commonly referred to as hydrogen autotransfer.
Figure 1.
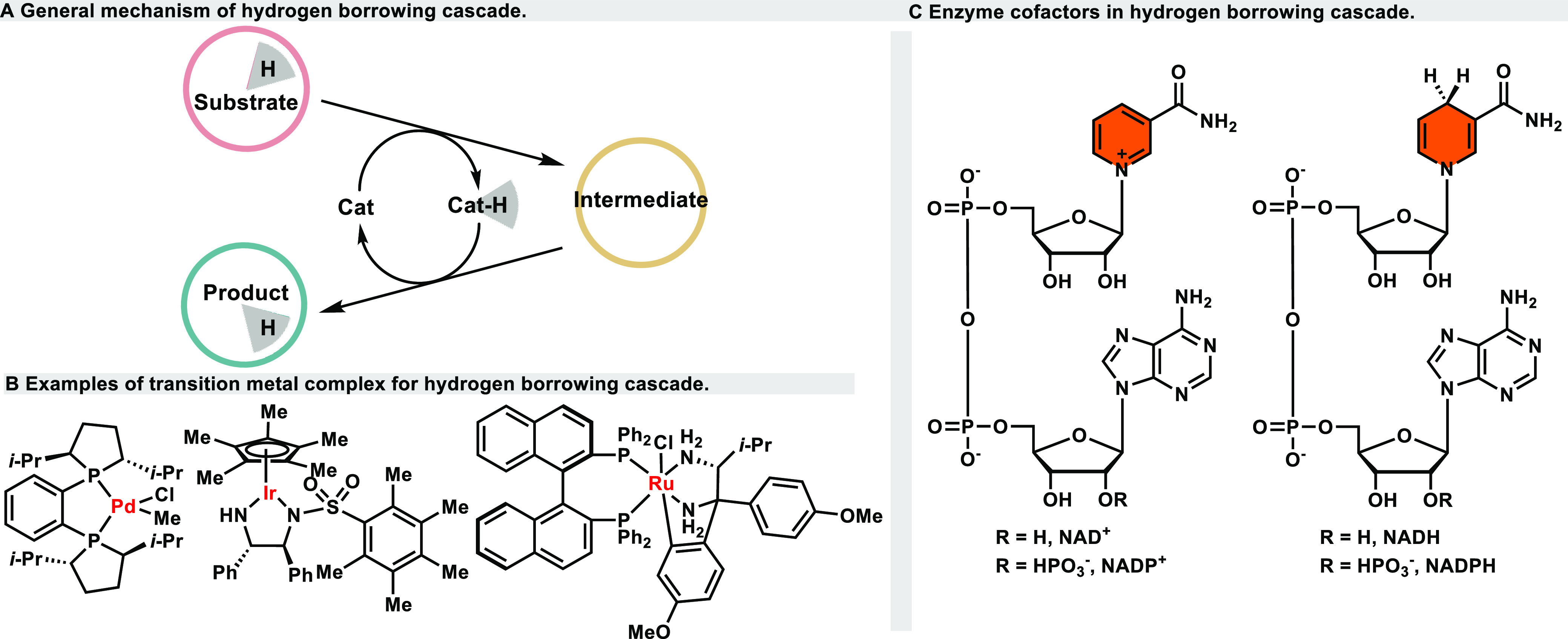
General mechanism and catalysts of hydrogen-borrowing cascade.
With the growing emphasis on the economy and sustainability of chemical synthesis, hydrogen-borrowing reactions catalyzed by transition metals have garnered significant attention. Since the 21st century, there has been extensive research into the application of hydrogen-borrowing reactions for the efficient and selective construction of the C–N bond and C–C bond. This research has made a substantial contribution to the advancement of organic synthesis, yielding a range of highly economical asymmetric synthesis methods and facilitating the creation of novel chiral heterocyclic compounds.2−5 However, this strategy has predominantly relied on the use of expensive transition metals, and has been plagued by challenges related to poor stereoselectivity. Highly selective hydrogen borrowing examples were reported until recently.6
In the meantime, the biocatalytic hydrogen-borrowing strategy has gained substantial popularity due to its outstanding environmental friendliness, high economic efficiency, mild conditions, and precise stereocontrol.7 This process represents a highly innovative artificial biocatalytic linear cascade.8 Currently, biocatalytic hydrogen-borrowing cascades find utility in various applications, including stereoisomer conversion within alcohols, conversion between alcohols and amines, conversion of allylic alcohols to saturated carbonyl counterparts, and conversion of α,β-unsaturated aldehydes to saturated carboxylic acids, etc. In metal-catalyzed reactions, the metal complex plays a dual role as hydride acceptor and donor. Various metals include Pd, Ir, Ru, Ni, and Rh are utilized, and chelated chiral ligands are employed to control stereoselectivity of the transformation (Figure 1B). However, in biocatalytic systems, the hydride shuffling is typically facilitated by coenzymes such as NAD(P)+ and NAD(P)H within the active sites of enzymes. This reliance on coenzymes allows for chemo- and stereoselectivity to be generated within the three-dimensional environment of a specific enzyme pocket (Figure 1C).
This perspective provides a comprehensive overview of the advancements in enzyme-catalyzed hydrogen-borrowing cascade strategies, representing a significant leap toward achieving efficient and remarkably stereoselective transformations. It encompasses a wide range of aqueous, organic, and whole-cell biocatalytic systems and holds the potential for further expansion through the integration of additional enzymes or chemical reactions. The organization of the content follows a chronological order within different reaction types to facilitate a coherent understanding of the developments in this field.
2. Stereoconversion of Alcohol
2.1. Racemization of Alcohols
Racemization is the key step in dynamic kinetic resolution (DKR), especially in the study of alcohols. In addition to utilizing the few racemases that exist in nature, the racemization of alcohol can be accomplished through a hydrogen-borrowing cascade involving a pair of stereocomplementary enzymes or nonstereoselective enzymes.
In 2007, the Kroutil group achieved racemization of a range of aliphatic and aromatic secondary alcohols.9 Two groups of stereocomplementary alcohol dehydrogenases (ADH-″A″/LK-ADH or RE-ADH/LK-ADH) were employed to control the interconversion between chiral alcohols and ketone intermediates (Scheme 1A). The nonstereoselective ADH (PfADH) from Pseudomonas fluorescens was evaluated for its stereoselective error, demonstrating the ability to racemize 2-octanol, albeit at a relatively slow rate.
Scheme 1. Biocatalytic Hydrogen-Borrowing Cascades for the Racemization of Alcohols.

Relative rates were measured from the slope of the decline of ee versus time at the onset of the reaction (conversion <5%), values are expressed as % relative to the natural substrate (S)-2-hydroxy-4-methylpentanoic acid, which was set as standard (100%). Structures that are not racemized are shown in gray.
In 2013, the Musa group achieved the racemization of enantiopure phenyl-ring-containing secondary alcohols using a single secondary alcohol dehydrogenase (W110A TeSADH) from Thermoanaerobacter ethanolicus (Scheme 1B).10 Notably, TeSADH exhibits high tolerance for organic solvents, enabling the racemization of enantiopure alcohols in both monophasic and biphasic media containing high concentrations of organic solvents. For instance, the TeSADH-catalyzed racemization of (R)- and (S)-1-phenyl-2-propyl alcohols occurs in a biphase media containing methyl tert-butyl ether (MTBE) at a concentration of approximately 130 mM. Subsequently, leveraging this property of TeSADH, they employed a sol–gel method to encase W110A TeSADH for the racemization of alcohols.11 When combined with kinetic resolution (KR) catalyzed by lipase B (CALB) from Candida antarctica, a one-pot DKR process for secondary alcohols was achieved using hexane as a solvent resulting in modest conversion (74%) and stereoselectivity (68% ee) (Scheme 1C). It is worth noting that the presence of water in the dry gel was sufficient to maintain the enzyme’s activity. However, the KR process was hampered by the slow diffusion rate of the substrate within the dry gel.
In 2009, the Faber group extended this system to α-hydroxycarboxylic acids employing a pair of stereocomplementary d- and l-α-hydroxy-carboxylate dehydrogenases (HicDH) from L. confusus DSM 20196 and L. paracasei DSM 20008 (Scheme 1D).12 This biocatalytic racemization approach was successfully applied to aliphatic, (aryl)aliphatic, and aromatic α-hydroxyl carboxylic acids, achieving the desired conversions within a 24-h time frame. Importantly, the concentration of α-ketoic acid intermediates could be maintained at a minimum level (<3%) by excluding O2 and adjusting the NADH/NAD+ ratio.
Though the above-mentioned examples were only within alcohols alone, the hydrogen-borrowing cascade racemization strategy could be more diverse. There is potential for investigating the racemization process catalyzed by biocatalytic hydrogen-borrowing cascade with functional groups such as amines, ethers, thiols, or even C–C bonds.
2.2. Stereoinversion of Alcohols
As early as 1949, Caputto et al. made the groundbreaking discovery involving uridine diphosphate (UDP)-galactose 4-epimerase derived from Escherichia coli. This enzyme demonstrated the remarkable ability to facilitate the conversion between UDP-galactose and UDP-glucose (Scheme 2A).13 Subsequent investigations have provided evidence of the enzyme’s mechanism of action, revealing that UDP-galactose 4-epimerase itself is able to catalyze the complete hydrogen-borrowing cycle. The galactose’s hydride is first abstracted by the enzyme-bound NAD+ through an enantioselective manner, followed by the ketopyranose intermediate free rotation and nonenantioselective hydride refund successively.14,15
Scheme 2. Hydrogen-Borrowing Cascades for the Stereo-Inversion of Alcohols.
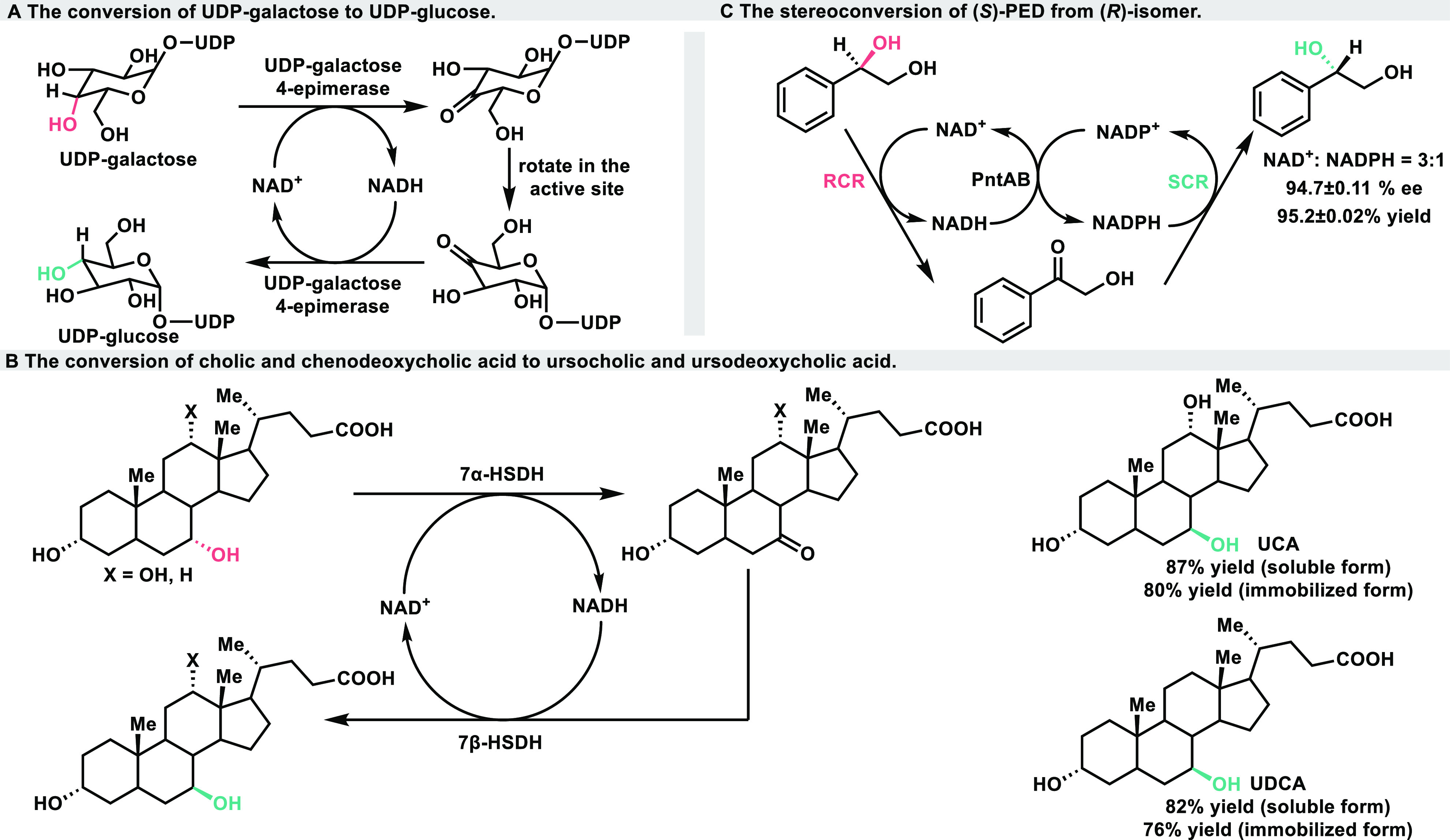
In 2006, the Pedrini group employed a combination of 7α- and 7β-hydroxysteroid dehydrogenases (HSDH) from Xanthomonas maltophilia CBS 897.97 to successfully accomplish the stereoisomerization of cholic and chenodeoxycholic acid.16 This process yielded ursocholic acid (UCA) and ursodeoxycholic acid (UDCA) with impressive yields of 82% and 87% after 20 h, respectively (Scheme 2B). By immobilizing the mixture of 7α- and 7β-HSDH on Sepharose-CL6B and utilizing the enzyme-loaded resin, the desired products were obtained with yields of 80% and 76% over a period of 5 days. Furthermore, after four cycles, the yield of UCA could be increased to 65% after 12 days. Despite a slight drop in productivity, it demonstrated the potential for repeated and sustainable production of these valuable compounds.
In an early study by the Xu group, they discovered that the combination of NAD+ dependent (R)-carbonyl reductase (RCR) and NADPH-dependent (S)-carbonyl reductase (SCR) from Candida parapsilosis CCTCC M203011 facilitated the stereoisomerization of (R)-1-phenyl-1,2-ethanediol.17 The engineered Escherichia coli strain (RS strain) that coexpressed RCR and SCR exhibited a slow and low yield process in the conversion of (R)-PED into its enantiomer. In E. coli, the expression level of the SCR is higher than that of the RCR, and the catalytic efficiency of the SCR significantly exceeds that of the RCR. Consequently, the resulting redox imbalance of intracellular cofactors leads to a low stereoconversion efficiency of the reaction. Furthermore, the system exhibits two distinct cofactor cycles, and the involvement of endogenous cellular metabolism in maintaining the redoxbalance between NAD(P)+ and NAD(P)H suggests that this process does not align with the definition of a borrowing hydrogen cascade. In 2012, an attempt was made to address this challenge. A recombinant E. coli was constructed, incorporating RCR, SCR, PntA, and PntB. The introduction of pyridine nucleotide transhydrogenases (PNTs), which consist of α and β subunits encoded by the PntA and PntB genes (Scheme 2C)18 enabled the simultaneous regeneration of NAD(H) and NADP(H), thereby achieving a balance in intracellular nucleotide levels. Compared to the RS strain, this optimized system significantly improved both the optical purity and yield of the one-step conversion by 51.5% and 80.6%. Additionally, it reduced the reaction time by 7-fold, enhancing the overall efficiency of the process. When both NAD+ and NADPH are added to the reaction mixture, (S)-PED exhibits high optical purity and yield. However, varying NADH/NADP+ ratios result in different bioconversion efficiencies. Optical purity and yield are not ideal when the NAD+/NADPH ratio is lower than 0.5. Conversely, when the ratio is greater than or equal to 3.0, the optical purity and yield of (S)-PED reach their peak values, approximately 97% ee and 95% yield, respectively, of this method.
3. Conversion of Alcohols to Amines
3.1. Conversion of Alcohols to Primary Amines
3.1.1. Three-Enzyme Cascades: Predecessor of Hydrogen-Borrowing Cascades
In 2012, the Kroutil group introduced a pioneering redox-neutral three-enzyme cascades that proved highly successful in achieving the bioamination of primary alcohols.19 The process begins with the conversion of the primary alcohol into ketone intermediates, facilitated by alcohol dehydrogenase (ADH-hT) from Bacillus stearothermophilus. In the subsequent step, ω-transaminase (CV-ωTA) from Chromobacterium violaceum employs l-alanine as the amine donor to convert these ketone intermediates into amine (Scheme 3A). The byproduct of this reaction, pyruvate, is then efficiently regenerated by l-alanine dehydrogenase (AlaDH) from Bacillus subtilis. This regeneration step simultaneously oxidizes NADH to NAD+, effectively linking the oxidation and reduction steps. This system is environmentally friendly, requiring only a catalytic amount of NADH and generating water as the sole byproduct. It shows the capability to perform bioamination on a range of aliphatic primary alcohols and alcohols bearing aromatic moieties, delivering moderate to excellent yields. Furthermore, the successful achievement of the double bioamination of long chain n-alkane-1, ω-diol was accomplished by integrating two redox-neutral cascade systems in series. At a preparative scale, the conversion rate of 1,10-decanediol reached an impressive 94% with isolated yield of 70%, showcasing the potential for practical applications of this method.
Scheme 3. Three-Enzyme Cascades for the Conversion of Alcohols to Amines,

employing crude enzyme.
employing purified enzymes.
In 2013, the Kroutil group made another significant advancement in chiral amine biosynthesis via three-enzyme cascade, successfully accomplishing the bioamination of a range of secondary alcohols with >99% ee (Scheme 3B).20 However, when employing purified enzyme, only 11% amine formation was observed, accompanied by 5% accumulation of ketone intermediates. Conversely, the utilization of crude enzyme preparations containing NADPH oxidase leads to improved amine formation of up to 47%, but ketone accumulation also increased to 25%. However, depending on the amount of NAD+ added, only 2–4% ketones can be formed. Therefore, additional redox equivalents are suspected to existed in the crude lysate, providing additional NADH oxidases to convert NADH to NAD+ before its hydride is consumed in reductive amination, resulting in a large accumulation of ketones that further promote amine production. During the same year, the Skerra group envisioned an extension of this system to enable the double bioamination of dicyclodiol isosorbide (Scheme 3C).21 The intermediate (2S,5S)-5-amino-2-alcohol can be obtained with 7% yield, and the final product of double bioamination was not detected. Further design and optimization of novel ADH variants are necessitated to realize the comprehensive diamination cascade reaction.
In 2014, the Kroutil group combined the strategy with another cofactor self-sufficient biocatalytic cascade module, achieving the conversion of cyclohexanol into the nylon-6 monomer, 6-aminohexanoic acid.22 In module 1, cyclohexanol was converted to ε-caprolactam through the catalysis of alcohol dehydrogenase (sec-ADH) from Lactobacillus brevis and Baeyer–Villiger monooxygenase (BVMO) from Acinetobacter. Utilizing the BVMO C376L M400I mutant, caprolactone was obtained with 75% isolated yield (Scheme 3D). In module 2, the presence of the carboxylic acid moiety in 6-hydroxyhexanoic acid inhibited the oxidation activity of ADHs. To address this challenge, an unprecedented carboxylic acid capping strategy was adopted. Horse liver esterase preparation was used to convert ε-caprolactone into 6-hydroxyhexanoic acid methyl ester preventing the formation of the dead-end intermediate, 6-hydroxyhexanoic acid. Subsequent steps in this process mirrored the aforementioned example,13 involving a hydrogen-borrowing cascade of three enzymes: prim-ADH from Bacillus lipophathermophilus, ω-TA from Paracococcus denitrifying, and AlaDH from Bacillus subtilis. This cascade resulted in the formation of 6-amino ester, which was subsequently hydrolyzed to yield the final product 6-aminocaproic acid, with a total conversion of 24%.
In 2016, the Palacio group published research outlining a similar enzymatic system that facilitated the synthesis of ether amines from alcohols (Scheme 3E).23 This investigation included an analysis of the system’s characteristics through the measurement of reaction balance, followed by optimization. The findings revealed that a high concentration of ammonia enhanced the efficiency of amination, resulting in a conversion rate of up to 60%. However, the enantiomeric excess of the resulting product was not determined.
3.1.2. Dual-Enzyme Hydrogen-Borrowing Cascades
Essentially, the three-enzyme cascade process involves the initial transfer of the hydride to the cosubstrate, followed by its incorporation into the final product. This process does not strictly adhere to the definition of a hydrogen borrowing reaction. However, it serves as a valuable source of inspiration for the development of double-enzyme hydrogen borrowing cascades.
In the three-enzyme amination process, a significant quantity of l- or d-alanine is typically required to serve as a driving force for the reaction. Moreover, there is a clear need for improvements in both the conversion rates and chemoselectivity of secondary alcohols within this system. Consequently, in 2015, in pursuit of efficiently producing amines with exceptional enantiomeric purity while maintaining environmentally friendly conditions, a novel approach, known as the double-enzyme hydrogen-borrowing cascade, was introduced as a more succinct method for synthesizing chiral amines from racemic secondary alcohols, and alanine is no longer required. The Turner group and the Xu group independently reported studies on the conversion of alcohols into enantiopure primary amines using a dual-enzyme hydrogen-borrowing cascade (Scheme 4A). The Turner group employed a pair of stereocomplementary ADHs (Prelog AA-ADH and anti-Prelog LBv-ADH) to oxidize (S)- and (R)-alcohol substrates into corresponding ketone intermediates. In the subsequent reduction step, engineered amine dehydrogenase (Ph-AmDH or Ch1-AmDH) was utilized to transform imine compounds into (R)-amines. This method achieved high conversion rates of aromatic or aliphatic secondary alcohols to chiral amines, reaching up to 99% with 96% ee. Furthermore, primary alcohol conversion could reach 99% (Scheme 4B).24 To achieve maximum or even quantitative conversion in this process, purified enzymes are essential to eliminate the presence of NADH oxidase in the crude enzyme extract. This is crucial to prevent additional NADH production outside the cycle, which can disrupt the cofactor balance within the system, leading to a significant accumulation of ketones, reducing the production of amines. By using purified enzymes, the enzymatic reaction can be tightly controlled, ensuring efficient utilization of cofactors and maximizing the conversion of substrates into desired products. Simultaneously, the Xu group designed a double-enzyme hydrogen-borrowing cascade system by coupling nonstereoselective ADH (ScCR) from Streptomyces coelicolor and an AmDH double mutant (EsLeuDH-DM) from Exiguobacterium sibiricum (Scheme 4C).25 With only one type of ADH, they achieved the conversion of racemic secondary alcohols to chiral amines with excellent conversion rates (94%) and perfect stereoselectivity (>99% ee). However, this approach was limited to simple aliphatic secondary alcohols and phenyl ethanol substrates.
Scheme 4. Dual-Enzyme Hydrogen-Borrowing Cascades for the Conversion of Alcohols to Enantiopure Amines,

Turner group’s double enzyme hydrogen borrowing system.
Xu group’s double enzyme hydrogen borrowing system.
3.1.2.1. System Optimization
Following the initial success of the double-enzyme hydrogen-borrowing cascade, several research groups devoted on optimizing and refining the system for enhanced performance. In 2017, the Turner group employed a semirational design approach to modify NADPH-dependent TeSADH, which is known for its broad substrate specificity and low enantioselectivity. The objective was to enable TeSADH to accept NADH as a cofactor, aligning it with the cofactor dependence of engineered AmDHs.26 This engineering effort resulted in an engineered TeSADH with a remarkable >10 000-fold switch in selectivity from NADPH to NADH. This engineered enzyme was then employed in double enzyme cascade (Scheme 4D), achieving the amination of the substrate alcohol panel from the first generation system with a high conversion rate of up to 90%. In 2020, the Mutti group used the alcohol dehydrogenase activity of l-lysine dehydrogenase (LysEDH) from Geobacillus stearothermophilus to create new enzyme variants.27 By enhancing the ADH catalytic activity of LysEDH, they obtained an enzyme with dual AmDH/ADH activity, and the enzyme’s activity toward the substrate could be modulated by controlling reaction conditions. Under the catalytic action of this enzyme, benzyl alcohol was successfully converted to benzylamine (Scheme 4E), marking the first example of a one-enzyme catalyzed hydrogen-borrowing alcohol amination. However, the ADH activity of the LE-AmDH variant on ketones requires further expansion and optimization.
In 2019, the Mutti group introduced a whole-cell catalytic system that achieved the in vivo amination of alcohols using E. coli strains engineered with ADH and AmDH genes (E. coli/ADH-AmDH strains) (Scheme 4F).28 This approach enabled the conversion of alcohols into chiral amines with yield of up to 80%, and molar yield of up to 15 mM, making it suitable for gram-scale biotransformation. One of the notable advantages of this method is that it enhances enzyme stability and reaction suitability while eliminating the need for enzyme purification and coenzyme supplementation. To maintain the redox balance of cofactors, an appropriate amount of glucose was added to the system. However, it is worth noting that high concentrations of certain amine products can exhibit toxicity and lead to cell death. Therefore, the addition of cosolvents was necessary to extract the amine products efficiently from the reaction mixture. This whole-cell catalytic system represents a promising approach for achieving amination reactions with enhanced efficiency and practicality. It is important to emphasize that in whole-cell systems, the cellular metabolism’s endogenous pathways play a role in maintaining the redox balance between NAD+ and NADH. Consequently, in such systems, the hydrogen utilized in the reduction stage may not exclusively originate from the oxidation stage. Therefore, it is not accurate to categorize this process as a strict hydrogen-borrowing phenomenon.
The overall catalytic efficiency and stability of ADH/AmDH cascade catalytic systems have seen notable improvements through the technique of enzyme coimmobilization (Table 1). In 2018, the Mutti group utilized the affinity adsorption theory to achieve enzyme coimmobilization. When alcohol dehydrogenase (AA-ADH) and chimeric amine dehydrogenase (Ch1-AmDH) were coimmobilized on to controlled porosity glass Fe(III) ion-affinity beads (EziG Fe-Amber),29 the enzyme activity could be retained for up to five cycles. The total turnover times of ADH and AmDH exceeded 4000 and 1000, respectively, which is 2 to 15 times higher than reported data in the literature.23 The amination rate of multiple (S)-configured alcohol substrates reached as high as 95%, with >99% ee. In the same year, the Quin group and the Dannert group also attempted to coimmobilize the biocatalytic cascade enzymes. They employed a self-assembled protein scaffold based on EutM, SpyTag from Streptococcus pyogenes and SpyCatcher, which enabled the fixation of Prelog AA-ADH and Ch1-AmDH on the EutM scaffold through the formation of covalent isopeptidyl bonds between SpyTag and SpyCatcher.30 This approach improved the catalytic efficiency and stability of the enzymes, reducing the time by half to achieve 90% conversion.
Table 1. Co-immobilization of Biocatalytic Cascade Enzymes31.
| entry | 1 | 2 | 3 |
|---|---|---|---|
| carrier | EziG Fe-Amber ion-affinity beads | protein scaffolds | silica nanoparticles (SNPs) |
| theory | affinity adsorption | SpyTag/SpyCatcher | SiBP tag |
| conversiona | 82.4 ± 11% | ∼92.0% | 85.0 ± 0.3% |
| catalyst productivityb | 0.15 ± 0.02 | ∼0.05 | 1.70 ± 0.03 |
| cycles | 5 | nonrecyclable | >8 cycles |
| TONs (ADH)c | 1.9 × 103 ± 2.5 × 102 | ∼3.1 × 103 | 6.3 × 104 ± 9.6 × 102 |
| TONs (AmDH)c | 7.2 × 102 ± 9.6 × 10 | ∼1.2 × 102 | 3.7 × 103 ± 5.6 × 10 |
| ref | 29 | 30 | 31 |
Obtained percentage of (R)-2-aminohexane.
The unit is μmolproduct·h–1·mgenzyme–1.
TON is defined as μmol of converted substrate per μmol of enzyme.
In 2022, the Sun group leveraged the specific binding effect between silica binding peptide (SiBP) and silica to coimmobilize ADH and AmDH on the surface of silica nanoparticles (SNPs).31 This double enzyme coimmobilized system yielded chiral amines at a rate 1.85 times higher than that of the free double enzyme system, enhancing the overall catalytic efficiency. The catalyst productivity was 11–34 times greater than the data reported in the studies mentioned above,29,30 and it exhibited excellent stability and reusability, retaining 87% of the original activity after 8 cycles of use.
3.1.2.2. System Extension
In 2018, based on the successful conversion of cyclohexanols into amines using a double-enzyme hydrogen-borrowing cascade system, the Xu group established a three-step multienzyme cascade system featuring P450 monooxygenase mutant (P450BM3) from Bacillus megaterium and the double-enzyme hydrogen-borrowing cascade (ScCR and EsLeuDH). They adopted a “one-pot, two-step” strategy to achieve C–H amination of the model substrate cyclohexane (Scheme 5A).32 This approach resulted in a cyclohexylamine titer of 14.9 mM, with a product content reaching 92.5%. At the same time, the Xu group constructed a whole cell biocatalyst for the multienzyme system, enabling not only the amination of cyclohexane but also the successful conversion of phenylethylamine into chiral phenylethylamine with >99% ee, although the titer was modest at 2.2 mM. Subsequently, they optimized the system in vitro (Scheme 5B), further demonstrating the feasibility of the multienzyme cascade for catalyzing C–H amination of inert aromatic alkanes.33 This approach yielded a group of substituted phenylethylamine with excellent optical purity (>99% ee) and moderate conversion rate (13–53%). On a preparative scale, (R)-phenylethylamine was obtained with 25% separation yield and >99% ee.
Scheme 5. Borrowing Hydrogen Reaction in Combination with Other Enzyme or Chemocatalysis.

In 2019, the Mutti group combined the double-enzyme hydrogen-borrowing cascade with epoxidation and hydrolysis, utilizing three stereoselectively different NADH-dependent alcohol dehydrogenases, Aa-ADH, Bs-BDHA, and Ls-ADH. They conducted the regioselective oxidation of diols (Scheme 5C), resulting in a continuous multienzyme catalytic conversion of β-methylstyrene to (1R,2R)- and (1S,2R)-phenylpropanolamine with high chemo-, regio-, and stereoselectivity.34 The total yield ranged from 59% to 63%, with diastereomeric ratios (dr) and enantiomeric ratios (er) >99.5%. Subsequently, in 2021, they reported a high regio- and stereoselective synthesis of all phenylpropanolamine stereoisomers from 1-phenylpropane-1,2-diols using a three-enzyme hydrogen-borrowing cascade system (Scheme 5D).35 This cascade system could also synthesize all phenylpropanolamine stereoisomers from β-methylstyrene by combining a styrene monooxygenase with stereocomplementary epoxide hydrolases.
In 2020, the Parmeggiani and Turner groups successfully connected the biocatalytic system to the Buchwald-Hartwig cross-coupling reaction, leading to the conversion of racemic alcohols into chiral anilines (Scheme 5E).36 This cascade proceeds sequentially, meaning that the Buchwald-Hartwig cross-coupling reaction was carried out at the end of the enzymatic cascade. To facilitate this one-pot reaction involving both biocatalysis and chemical synthesis, they introduced TPGS-750-M surfactant into the reaction system, which serves to prevent the inhibition of palladium catalysts from high concentrations of ammonia, salt, or buffers in aqueous media.
3.2. Conversion of Alcohols to Secondary Amines
In 2016, the Ward group developed an NAD(P)H-dependent artificial transfer hydrogenase (ATHase), represented by [Biot-Cp*Ir(L∧L)Cl]⊂Sav K121R (Scheme 6A).37 This artifical transfer hydrogenase utilizes NAD(P)H as hydride source to catalyze the reduction of the intermediate cycloimide, followed by spontaneous cyclization. This innovative approach enabled the conversion of 4-amino-1-phenyl-1-butanol into a cycloamine product with 36.1% yield after 48 h.
Scheme 6. Hydrogen-borrowing cascades for the alkylation of primary amines with alcohols,

Biocatalytic hydrogen-borrowing cascades for the conversion of α-hydroxyl acids to chiral amino acids.
Isolated yield cannot be stated due to impurities in the isolated material.
The Turner group previously discovered a reductive aminase (AspRedAm) from Bacillus megaterium.38 It can directly catalyze the coupling of carbonyl compounds and amines into corresponding imines, and then catalyze the asymmetric reduction of imines, showing good conversion efficiency and enantioselectivity. Based on this discovery, in 2017, they achieved the alkylation of amines using a biocatalytic “hydrogen-borrowing” pathway (Scheme 6B).39 Initially, a single alcohol dehydrogenase with poor enantioselectivity was used to perform oxidative dehydrogenation of primary and racemic secondary alcohols. Subsequently, a variety of primary and secondary amines were synthesized through reductive amination catalyzed by AspRedAm. The conversion rate of aliphatic and aromatic secondary alcohls is up to 99%, and the chiral amines can be directly obtained in up to 97% ee from racemic alcohol precursors. However, this system required a significant excess of amines (20 times more) to drive the reaction, resulting in poor atomic economy. They extended this cascade by designing a biocatalytic pathway for the conversion of cycloalkanes into secondary amines using a “one-pot, two-step” strategy (Scheme 6C).40 The first oxidation step was carried out using cytochrome P450 monooxygenase, followed by amination using ADH and AspRedAm. This approach achieved a titer of 19.6 mM for the amine product, with a temporal-spatial yield of the preparation-grade amination reaction of cyclohexane reaching 2 g·L–1·d–1.
In 2019, the Roiban group obtained an Imine reductases enzyme variant (IRED M3) capable of both reductive amination and chiral resolution of substrate amines through directed evolution. By combining this enzyme with ketoreductase (KRED) from Lactobacillus coleohominis, they successfully prepared the key intermediate of the lysine-specific demethylase-1 (LSD1) inhibitor GSK2879552 using a hydrogen-borrowing cascade system (Scheme 6D).41 At a 5g scale, the product was isolated with a yield of 48.3%, high stereoselectivity (99.5% ee), and purity (97.9%), and the reaction required only 2.2 equiv of amines to drive it.
3.3. Conversion of α-Hydroxyl Acids to Amino Acids
In 2010, the Kroutil group successfully converted racemic mandelic acid into l-phenylglycine through a three-step process involving racemization, enantioselective oxidation and stereoselective reductive amination (Scheme 7A).42 The process began with the selective oxidation of d-mandelic acid catalyzed by d-mandelate dehydrogenase (d-MDH) from Rhodotorula graminis, and subsequent reductive amination was facilitated by l-amino acid dehydrogenase (l-AADH) from Codexis using ammonium salts. During this process, a configuration inversion similar to Mitsunobu stereoinversion occurred, resulting in l-configuration phenylglycine. It is worth noting that d-MDH exhibited high selectivity in this process. Additionally, the use of mandelic acid racemase from Pseudomonas sp. ATCC 12633 enabled the successful transformation of l-mandelic acid into l-phenylglycine. The conversion rate for a 50 mg scale was 94%, with ee exceeding 97%. However, in this multienzyme cascade reaction, d-MDH was sensitive to the product, only a concentration of 5 g/L l-phenylglycine was sufficient to inhibit d-MDH activity and terminate the reaction.
Scheme 7. Simultaneous Enzymatic Synthesis of (S)-3-Fluoroalanine and (R)-3-Fluorolactic Acid.

In 2014, the Xu group identified a new NAD+ dependent d-mandelate dehydrogenase (LbDMDH) from Lactobacillus brevis using genome mining techniques. This enzyme exhibited a catalytic efficiency that was 4.6 times higher than the highest known enzyme (DMDH from Rhodotorula graminis).43,44 Moreover, LbDMDH showed improved stability under high product loading conditions. When the substrate loading exceeded 300 mM, both the substrate and product began to affect the enzyme’s performance. By combining LbDMDH with engineered lactate racemase (PpMRM) from P. putida and leucine dehydrogenase (EsLeuDH) from Exiguobacterium sibiricum DSM 17290, the Xu group successfully achieved the transformation of mandelic acid into four different amino acid derivatives with various substituents (Scheme 7B). At a 1 L scale, they achieved 96.4% conversion of 30.4 g mandelic acid (0.2 M) to l-phenylglycine, with 86.5% isolated yield, 99% ee, and space time yield of 50.4 g·L–1·d–1. Subsequently, they constructed whole-cell biocatalysts coexpressing mandelic acid racemase,45d-mandelic acid dehydrogenase (Lh-DMDH) and l-leucine dehydrogenase (EsLeuDH) enabling the biosynthesis of l-phenylglycine with >99% ee. They used response surface methodology (RSM) to optimize the biosynthesis conditions, achieving a yield of 87.9% and a time-space yield of 79.70 g·L–1·d–1 under high substrate concentrations (300 mM).
In 2018, the Dennig group incorporated the previously mentioned cascades into a multienzyme cascade and successfully achieved the conversion of both canonical and noncanonical α-amino acids from fatty acids through one-pot cascades (Scheme 7C).46 First, cytochrome P450 peroxygenase (P450CLA) from Clostridium acetobutylicum is used to catalyze the regioselective α-hydroxylation of fatty acids using H2O2 as an oxidant. This step yields the corresponding α-hydroxy acids with a conversion rate of up to 95%. A double enzyme hydrogen-borrowing cascade system is then employed. This system involves the conjugation of two stereocomplementary enzymes: α-hydroxyisocaproate dehydrogenase d/l-HIC-DH and l-phenylalanine dehydrogenase (l-Phe-DH) from Rhodococcus sp. These enzymes work together to convert the α-hydroxy acids into the corresponding α-amino acids. The overall process achieves a high conversion rate of up to 99% with >99% ee. It was successfully applied to the conversion of five saturated fatty acids (C6–C10), phenylpropionic acid and phenylbutyric acid. The titer of l-phenylalanine produced in this process reached 2.5 g·L–1.
4. Conversion of Amines to Alcohols
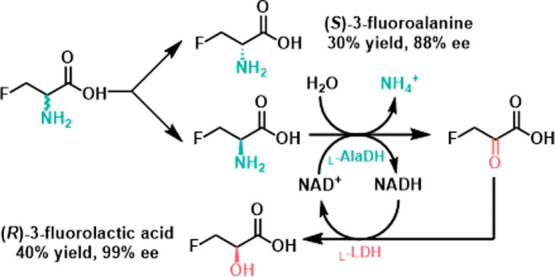
In 2000, the Antunes group utilized enzymes to simultaneously synthesize both (S)-3-fluoroalanine and (R)-3-fluorolactic acid (Scheme 8).47 First, l-alanine dehydrogenase (l-AlaDH) from Bacillus subtilis is used to catalyze the stereoselective oxidative deamination of racemic 3-fluoroalanine, this step oxidizes the (R)-3-fluoroalanine to form the ketoacid intermediate. The reaction is further catalyzed by rabbit muscle l-lactate dehydrogenase (l-LDH), which converts the ketoacid intermediate into final product (R)-3-fluorolactic acid. As a result of this process, (S)-3-fluoroalanine is isolated with 30% yield and 88% ee, while (R)-3-fluorolactic acid is obtained with 40% yield and >99% ee. This demonstrates the simultaneous synthesis of these two valuable compounds with high stereoselectivity. The separation reaction is thermodynamically unfavorable. The energy required for the entire system is provided by the circulatory system, which generates (R)-3-fluorolactic acid.
Scheme 8. Conversion of Allyl Alcohols to Ketones.
5. Conversion of Allyl Alcohols to Ketones
In 2000, the Bruce group achieved the biosynthesis of hydromorphone, a potent analgesic drug, through a hydrogen-borrowing cascade system. This accomplishment involved the use of three enzymes: NADP+-dependent morphine dehydrogenase (MDH), NADH-dependent morphine reductase (MR) from P. putida M10, and soluble pyridine nucleotide transhydrogenase (STH) from Pseudomonas fluorescens (Scheme 9A).48 Morphine is initially oxidized to morphinone by MDH, and this reaction simultaneously reduces NADP+ to NADPH. MR then reduces morphinone to hydromorphone while oxidizing NADH to NAD+. STH plays a critical role in regenerating cofactors (NADP+ and NADH) during the process. As a result of this multienzyme cascade, the yield of hydromorphone reaches 84% when maintaining the MDH:MR:STH ratio at 1:5:0.5. This successful biosynthesis of hydromorphone demonstrates the potential of enzymatic processes for drug synthesis.
Scheme 9. Conversion of Allyl Alcohols to Ketones.

In 2012, the Hollmann group introduced a double-enzyme cascade for the redox isomerization of allyl alcohol (Scheme 9B).49 The isomerization of allyl alcohol into its corresponding ketone was achieved by coupling two enzymes with a preference for cyclic compounds, alcohol dehydrogenase (TADH) from Thermus sp. ATN1 and ene reductase (TsER) from Thermus scotoductus SA-01. Using the redox isomerization of cyclohexanol as the model reaction, the product can be overreduced to cyclohexanol (>20%) with only 5% of the desired product being obtained. By controlling the ratio of TADH to TsER, it is possible to improve the overall yield to 60% with good chemoselectivity (>90%), while this method will possibly be applied into chiral construction of β-substituted ketone substrates.
In 2018, the Deska group conducted a screening of various commercial enzyme preparations, successfully identifying a set of enzymes proficient in enantioselectively converting Achmatowicz-type pyranones into either (S)- or (R)-γ-hydroxy-δ-lactones (Scheme 9C).50 The redox-neutral isomerization process was effectively executed by a single enzyme, utilizing a biocatalytic hydrogen-borrowing cascade strategy. This method demonstrated universal applicability across a spectrum of symmetrically substituted Achmatowicz pyranones. Through a tandem oxidase/peroxidase couples-mediated Achmatowicz rearrangement reaction, it efficiently generated key components of complex polycyclic natural products, such as walterolactone B and osmundalactone. Through the whole-cell transformation within E. coli, the reaction system achieved 71% conversion rate of pyranone substrates. This study provides a streamlined pathway for synthesizing valuable lactones from simple biogenic building blocks, further confirming the multifunctionality of biocatalysis in accessing structurally diverse natural products.
6. Conversion of bis-Aldehydes to Esters
In 2020, the Hall group accomplished the regioselective formation of isochroman-1-one and 4,5-dihydrobenzo[c]oxepin-1(3H)-one through a biocatalytic intramolecular tishchenko-like reaction catalyzed by ADHs with concurrent oxidation and reduction activities (Scheme 10).51 Meanwhile, the regioselectivity of substituted [1,1′-biphenyl]-2,2′-dicarbaldehydes can be controlled by the electronic properties of substituents, and the regioselective synthesis of 2-, 4-, and 8-substituted dibenzo[c,e]oxepin-5(7H)-ones can be achieved.
Scheme 10. Conversion of bis-Aldehydes to Esters.
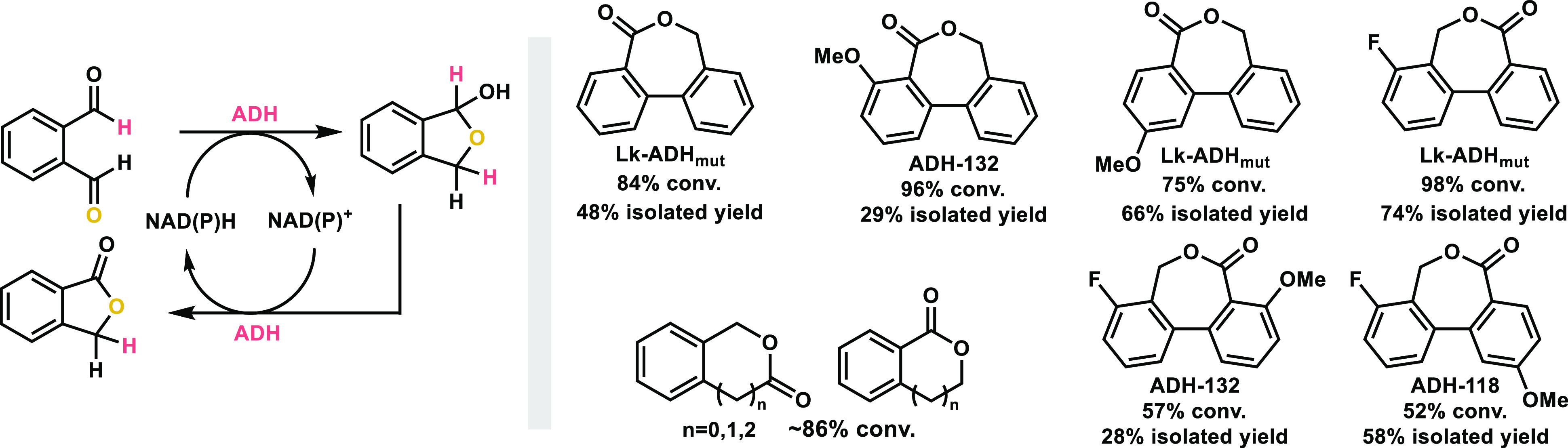
7. Conversion of α,β-Unsaturated Aldehydes to Acids
In 2013, the Hummel group developed a new method to convert α,β-unsaturated aldehydes into enantiomerically pure saturated carboxylic acids.52 This process involves two enzymatic steps. First, the C=C double bond of prochiral α,β-unsaturated aldehydes is reduced to a saturated C–C bond by NADPH-dependent ene reductase (ER) from Gluconobacter oxidans with the formation of the stereogenic center, and then oxidation of the aldehyde group to carboxylic acid is catalyzed by aldehyde dehydrogenase (PP-ALDH) from Pseudomonas putida KT2440, with water as the sole reagent (Scheme 11A). An example of this conversion is the transformation of citral into (S)-citronellic acid, which achieved a high conversion rate of 97% and >99% ee. However, this conversion is carried out using crude preparations (cell extracts) of the enzyme, an effective internal circulation of the nicotinamide cofactor cannot be demonstrated.
Scheme 11. Conversion of α,β-Unsaturated Aldehydes to Acids.

Subsequently, in 2015, the Scrutton group proposed a general strategy for establishing a hydrogen-borrowing biocatalytic cascade, successfully applied to the synthesis of chiral α-substituted carboxylic acids from α-substituted α,β-unsaturated aldehydes (Scheme 11 B).53 They initially screened various enzyme reductases (ER) and aldehyde dehydrogenases (Ald-DH) and identified the necessary enzymes. To establish the hydrogen cascade and optimize reaction conditions, they determined the suitable combination conditions for the two enzymes, including preferred cofactors and optimal pH. Subsequently, they screened enzyme and cofactor concentrations, as well as the enzyme ratio, to maximize conversion rate, chemical selectivity, stereoselectivity, and short reaction time. Finally, they studied the productivity of the cascade reaction with different substrate concentrations. It is noteworthy that this method exhibits broad applicability and can achieve quantitative conversion of various pharmaceutical active ingredients (APIs), including chiral substituted hydrocinnamic acid, fatty acids, heterocyclic rings, and even acetylated amino acids, by adjusting reaction conditions appropriately. Ultimately, (S)-2-methyl-3-phenylpropanoic acid can be prepared on a preparative scale (starting material (E)-2-methyl-3-phenylacrylaldehyde at 100 mg), with quantitative transformation after a 2-h reaction, a chemical selectivity of 96%, and an enantiomeric excess (ee) of >98%, yielding 88% in separation yield.
8. Summary and Perspective
The biocatalytic hydrogen-borrowing cascades offers distinct advantages when compared to traditional synthetic methods, particularly those relying on transition metal catalysis. It has heightened stereoselectivity, exemplary atomic economy, and an environmentally friendly profile. Notably, it effectively overcomes the inherent limitations of transition metal catalysis, including issues related to toxicity, cost, and the imposition of rigorous reaction conditions, thereby demonstrating a marked superiority. Consequently, different types of substrates including alcohols, amines, aldehydes, and ketones could be converted into corresponding products with good efficiency as well as satisfied stereoselectivity. In contrast, in transition metal-catalyzed hydrogen-borrowing systems, the substrates were usually limited to alcohols.
However, it is important to acknowledge that the current landscape of biocatalytic hydrogen-borrowing cascades faces certain challenges and limitations. These primarily encompass the limited variety of enzymes available for hydrogen-borrowing reactions, characterized by their restricted diversity and, in some cases, modest catalytic activity. In comparison to transition metal-catalyzed hydrogen-borrowing cascades, the biocatalytic variant has a narrower range of applicability, with limitations extending to substrate compatibility. It is important to emphasize that the thermodynamic feasibility of the biocatalytic hydrogen-transfer process must be considered. And achieving quantitative conversion would be challenging, this hurdle can be overcome by adjusting reaction conditions such as increasing the amount of coreagents can promote the reaction, or coupling the final product with downstream reactions can drive the consumption of reaction products. Additionally, factors such as enzyme specificity, reaction kinetics, and cofactor regeneration are crucial. Efficient cofactor regeneration systems are particularly necessary to maintain redox balance and sustain catalytic activity over multiple reaction cycles. In the use of crude enzyme preparations or whole-cell catalytic systems, the presence of additional NAD(P)H oxidase enzymes can oxidize NAD(P)H to NAD(P)+ before it is consumed in the reductive amination process. This disrupts the cofactor balance and reduces its effective utilization, resulting in decreased conversion efficiency. This phenomenon presents a challenge to achieving efficient hydrogen-borrowing processes in whole-cell systems, and strictly speaking, the process does not conform to a hydrogen-borrowing cascade. Future research will focus on the discovery of enzymes with new catalytic activities, the adaptation of this strategy to new chemical transformations involving various substrates and reaction conditions, and the enhancement of enzyme catalytic performance and substrate compatibility through techniques such as directed evolution. Furthermore, advancements in the development of biocatalytic hydrogen-borrowing cascade systems and optimization strategies for various reaction parameters will lead to faster and more effective improvements in conversion rate, stereoselectivity, and efficiency. These enhancements increase the applicability of this strategy to a wider range of substrates. These efforts aim to accelerate the widespread adoption of these biocatalytic methods in the industrial sector.
In summary, the future of biocatalytic hydrogen-borrowing cascades holds promise for sustainable and environmentally friendly chemical synthesis. As the repertoire of enzymes and enhanced biocatalytic processes continues to expand, we can anticipate their broader adoption across diverse industrial sectors, marking a shift toward greener and more sustainable chemical production methods.
Acknowledgments
Financial support for this work was provided by the National Natural Science Foundation of China (Grant No. 22371302), the Nonprofit Central Research Institute Fund of Chinese Academy of Medical Sciences (2022-RC350-03), State Key Laboratory of Bioactive Substance and Function of Natural Medicines, and startup funding from State Key Laboratory of Bioactive Substance and Function of Natural Medicines, Institute of Materia Medica, Chinese Academy of Medical Sciences and Peking Union Medical College.
The authors declare no competing financial interest.
Special Issue
Published as part of JACS Auvirtual special issue “Biocatalysis in Asia and Pacific”.
References
- Corma A.; Navas J.; Sabater M. J. Advances in One-Pot Synthesis through Borrowing Hydrogen Catalysis. Chem. Rev. 2018, 118, 1410–1459. 10.1021/acs.chemrev.7b00340. [DOI] [PubMed] [Google Scholar]
- Reed-Berendt B. G.; Latham D. E.; Dambatta M. B.; Morrill L. C. Borrowing Hydrogen for Organic Synthesis. ACS Cent. Sci. 2021, 7, 570–585. 10.1021/acscentsci.1c00125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.; Tao R.; Lin Z. K.; Yang G.; Zhao Y. Redox-enabled direct stereoconvergent heteroarylation of simple alcohols. Nat. Commun. 2021, 12, 5035. 10.1038/s41467-021-25268-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G.; Pan J.; Ke Y. M.; Liu Y.; Zhao Y. Tandem Catalytic Indolization/Enantioconvergent Substitution of Alcohols by Borrowing Hydrogen to Access Tricyclic Indoles. Angew. Chem., Int. Ed. 2021, 60, 20689–20694. 10.1002/anie.202106514. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Diao H.; Hong G.; Edward J.; Zhang T.; Yang G.; Yang B. M.; Zhao Y. Iridium-Catalyzed Enantioconvergent Borrowing Hydrogen Annulation of Racemic 1,4-Diols with Amines. J. Am. Chem. Soc. 2023, 145, 5007–5016. 10.1021/jacs.2c09958. [DOI] [PubMed] [Google Scholar]
- a Gao Y.; Hong G.; Yang B. M.; Zhao Y. Enantioconvergent transformations of secondary alcohols through borrowing hydrogen catalysis. Chem. Soc. Rev. 2023, 52, 5541–5562. 10.1039/D3CS00424D. [DOI] [PubMed] [Google Scholar]; b Zhang X.; Ma W.; Zhang J.; Tang W.; Xue D.; Xiao J.; Sun H.; Wang C. Asymmetric Ruthenium-Catalyzed Hydroalkylation of Racemic Allylic Alcohols for the Synthesis of Chiral Amino Acid Derivatives. Angew. Chem., Int. Ed. 2022, 61, e202203244 10.1002/anie.202203244. [DOI] [PubMed] [Google Scholar]; c Zhang J.; Wang J. Atropoenantioselective Redox-Neutral Amination of Biaryl Compounds through Borrowing Hydrogen and Dynamic Kinetic Resolution. Angew. Chem., Int. Ed. 2018, 57, 465–469. 10.1002/anie.201711126. [DOI] [PubMed] [Google Scholar]; d Yang P.; Zhang C.; Ma Y.; Zhang C.; Li A.; Tang B.; Zhou J. S. Nickel-Catalyzed N-Alkylation of Acylhydrazines and Arylamines Using Alcohols and Enantioselective Examples. Angew. Chem., Int. Ed. 2017, 56, 14702–14706. 10.1002/anie.201708949. [DOI] [PubMed] [Google Scholar]
- a Knaus T.; Mutti F. G. Biocatalytic hydrogen-borrowing cascades. Chim Oggi. 2017, 35 (5), 34–37. [PMC free article] [PubMed] [Google Scholar]; b Tas-sano E.; Hall M. Enzymatic self-sufficient hydride transfer processes. Chem. Soc. Rev. 2019, 48, 5596–5615. 10.1039/C8CS00903A. [DOI] [PubMed] [Google Scholar]
- Schrittwieser J. H.; Velikogne S.; Hall M.; Kroutil W. Artificial Biocatalytic Linear Cascades for Preparation of Organic Molecules. Chem. Rev. 2018, 118, 270–348. 10.1021/acs.chemrev.7b00033. [DOI] [PubMed] [Google Scholar]
- Gruber C. C.; Nestl B. M.; Gross J.; Hildebrandt P.; Bornscheuer U. T.; Faber K.; Kroutil W. Emulation of racemase activity by employing a pair of stereocomplementary biocatalysts. Chem.-Eur. J. 2007, 13 (29), 8271–8276. 10.1002/chem.200700528. [DOI] [PubMed] [Google Scholar]
- Musa M. M.; Phillips R. S.; Laivenieks M.; Vieille C.; Takahashi M.; Hamdan S. M. Racemization of enantiopure secondary alcohols by Thermoanaerobacter ethanolicus secondary alcohol dehydrogenase. Org. Biomol. Chem. 2013, 11, 2911–2915. 10.1039/c3ob27415b. [DOI] [PubMed] [Google Scholar]
- Karume I.; Musa M. M.; Bsharat O.; Takahashi M.; Hamdan S. M.; El Ali B. Dual enzymatic dynamic kinetic resolution by Thermoanaerobacter ethanolicus secondary alcohol dehydrogenase and Candida antarctica lipase B. RSC Advances. 2016, 6, 96616–96622. 10.1039/C6RA18895H. [DOI] [Google Scholar]
- Bodlenner A.; Glueck S. M.; Nestl B. M.; Gruber C. C.; Baudendistel N.; Hauer B.; Kroutil W.; Faber K. Biocatalytic racemization of α-hydroxycarboxylic acids using a stereo-complementary pair of α-hydroxycarboxylic acid dehydrogenases. Tetrahedron 2009, 65, 7752–7755. 10.1016/j.tet.2009.06.051. [DOI] [Google Scholar]
- Caputto R.; Leloir L. F.; Trucco R. E.; Cardini C. E.; Paladini A. C. The Enzymatic Transformation of Galactose into Glucose Derivatives. J. Biol. Chem. 1949, 179, 497–498. 10.1016/S0021-9258(18)56863-0. [DOI] [PubMed] [Google Scholar]
- Thoden J. B.; Hegeman A. D.; Wesenberg G.; Chapeau M. C.; Frey P. A.; Holden H. M. Structural analysis of UDP-sugar binding to UDP-galactose 4-epimerase from. Escherichia coli. Biochemistry 1997, 36, 6294–6304. 10.1021/bi970025j. [DOI] [PubMed] [Google Scholar]
- Thoden J. B.; Holden H. M. Dramatic differences in binding of UDP-galactose and UDP-glucose to UDP-galactose 4-epimerase from Escherichia coli. Biochemistry 1998, 37, 11469–11477. 10.1021/bi9808969. [DOI] [PubMed] [Google Scholar]
- Pedrini P.; Andreotti E.; Guerrini A.; Dean M.; Fantin G.; Giovannini P. P. Xanthomonas maltophilia CBS 897.97 as a source of new 7β- and 7α-hydroxysteroid dehydrogenases and cholylglycine hydrolase: Improved biotransformations of bile acids. Steroids 2006, 71, 189–198. 10.1016/j.steroids.2005.10.002. [DOI] [PubMed] [Google Scholar]
- a Nie Y.; Xu Y.; Mu X. Q.; Wang H. Y.; Yang M.; Xiao R. Purification, characterization, gene cloning, and expression of a novel alcohol dehydrogenase with anti-prelog stereospecificity from Candida parapsilosis. Appli. Environ. Microbiol. 2007, 73, 3759–3764. 10.1128/AEM.02185-06. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Yang M.; Xu Y.; Mu X. Q.; Xiao R. Purification and characterization of a novel carbonyl reductase with high stereo-selectivity. Front. Chem. Eng. China 2007, 1, 404–410. 10.1007/s11705-007-0074-9. [DOI] [Google Scholar]; c Zhang R. Z.; Zhu G. Y.; Zhang W. C.; Cao S.; Ou X. J.; Li X. M.; Bartlam M.; Xu Y.; Zhang X. C.; Rao Z. H. Crystal structure of a carbonyl reductase from Candida parapsilosis with anti-Prelog stereo-specificity. Protein Sci. 2008, 17, 1412–1423. 10.1110/ps.035089.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R.; Xu Y.; Xiao R.; Zhang B.; Wang L. Efficient one-step production of (S)-1-phenyl-1,2-ethanediol from (R)-enantiomer plus NAD(+)-NADPH in-situ regeneration using engineered Escherichia coli. Microb. Cell Factories 2012, 11 (1), 167. 10.1186/1475-2859-11-167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sattler J. H.; Fuchs M.; Tauber K.; Mutti F. G.; Faber K.; Pfeffer J.; Haas T.; Kroutil W. Redox self-sufficient biocatalyst network for the amination of primary alcohols. Angew. Chem., Int. Ed. 2012, 51, 9156–9159. 10.1002/anie.201204683. [DOI] [PubMed] [Google Scholar]
- Tauber K.; Fuchs M.; Sattler J. H.; Pitzer J.; Pressnitz D.; Koszelewski D.; Faber K.; Pfeffer J.; Haas T.; Kroutil W. Artificial multi-enzyme networks for the asymmetric amination of sec-alcohols. Chem.-Eur. J. 2013, 19, 4030–4035. 10.1002/chem.201202666. [DOI] [PubMed] [Google Scholar]
- Lerchner A.; Achatz S.; Rausch C.; Haas T.; Skerra A. Coupled Enzymatic Alcohol-to-Amine Conversion of Isosorbide using Engineered Transaminases and Dehydrogenases. ChemCatChem. 2013, 5, 3374–3383. 10.1002/cctc.201300284. [DOI] [Google Scholar]
- Sattler J. H.; Fuchs M.; Mutti F. G.; Grischek B.; Engel P.; Pfeffer J.; Woodley J. M.; Kroutil W. Introducing an in situ capping strategy in systems biocatalysis to access 6-aminohexanoic acid. Angew. Chem., Int. Ed. 2014, 53, 14153–14157. 10.1002/anie.201409227. [DOI] [PubMed] [Google Scholar]
- Palacio C. M.; Crismaru C. G.; Bartsch S.; Navickas V.; Ditrich K.; Breuer M.; Abu R.; Woodley J. M.; Baldenius K.; Wu B.; Janssen D. B. Enzymatic network for production of ether amines from alcohols. Biotechnol. Bioeng. 2016, 113, 1853–1861. 10.1002/bit.25954. [DOI] [PubMed] [Google Scholar]
- Mutti F. G.; Knaus T.; Scrutton N. S.; Breuer M.; Turner N. J. Conversion of alcohols to enantiopure amines through dual-enzyme hydrogen-borrowing cascades. Science 2015, 349, 1525–1529. 10.1126/science.aac9283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F.-F.; Liu Y.-Y.; Zheng G.-W.; Xu J.-H. Asymmetric Amination of Secondary Alcohols by using a Redox-Neutral Two-Enzyme Cascade. ChemCatChem. 2015, 7, 3838–3841. 10.1002/cctc.201500785. [DOI] [Google Scholar]
- Thompson M. P.; Turner N. J. Two-Enzyme Hydrogen-Borrowing Amination of Alcohols Enabled by a Cofactor-Switched Alcohol Dehydrogenase. ChemCatChem. 2017, 9, 3833–3836. 10.1002/cctc.201701092. [DOI] [Google Scholar]
- Tseliou V.; Schilder D.; Masman M. F.; Knaus T.; Mutti F. G. Generation of Oxidoreductases with Dual Alcohol Dehydrogenase and Amine Dehydrogenase Activity. Chem.-Eur. J. 2021, 27, 3315–3325. 10.1002/chem.202003140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houwman J. A.; Knaus T.; Costa M.; Mutti F. G. Efficient synthesis of enantiopure amines from alcohols using resting E. coli cells and ammonia. Green Chem. 2019, 21, 3846–3857. 10.1039/C9GC01059A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohmer W.; Knaus T.; Mutti F. G. Hydrogen-Borrowing Alcohol Bioamination with Coimmobilized Dehydrogenases. ChemCatChem. 2018, 10, 731–735. 10.1002/cctc.201701366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G.; Quin M. B.; Schmidt-Dannert C. Self-Assembling Protein Scaffold System for Easy in Vitro Coimmobilization of Biocatalytic Cascade Enzymes. ACS Catal. 2018, 8, 5611–5620. 10.1021/acscatal.8b00986. [DOI] [Google Scholar]
- Liu S.; Wang Z.; Chen K.; Yu L.; Shi Q.; Dong X.; Sun Y. Cascade chiral amine synthesis catalyzed by site-specifically co-immobilized alcohol and amine dehydrogenases. Catal. Sci. Technol. 2022, 12, 4486–4497. 10.1039/D2CY00514J. [DOI] [Google Scholar]
- Yu H. L.; Li T.; Chen F. F.; Luo X. J.; Li A.; Yang C.; Zheng G. W.; Xu J. H. Bioamination of alkane with ammonium by an artificially designed multienzyme cascade. Metab. Eng. 2018, 47, 184–189. 10.1016/j.ymben.2018.02.009. [DOI] [PubMed] [Google Scholar]
- Wang H.; Zheng Y. C.; Chen F. F.; Xu J. H.; Yu H. L. Enantioselective Bioamination of Aromatic Alkanes Using Ammonia: A Multienzymatic Cascade Approach. ChemCatChem. 2020, 12, 2077–2082. 10.1002/cctc.201902253. [DOI] [Google Scholar]
- Corrado M. L.; Knaus T.; Mutti F. G. Regio- and stereoselective multi-enzymatic aminohydroxylation of beta-methylstyrene using dioxygen, ammonia and formate. Green Chem. 2019, 21, 6246–6251. 10.1039/C9GC03161H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrado M. L.; Knaus T.; Mutti F. G. High Regio- and Stereoselective Multi-enzymatic Synthesis of All Phenylpropanolamine Stereoisomers from beta-Methylstyrene. Chembiochem 2021, 22, 2345–2350. 10.1002/cbic.202100123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosgrove S. C.; Thompson M. P.; Ahmed S. T.; Parmeggiani F.; Turner N. J. One-Pot Synthesis of Chiral N-Arylamines by Combining Biocatalytic Aminations with Buchwald-Hartwig N-Arylation. Angew. Chem., Int. Ed. 2020, 59, 18156–18160. 10.1002/anie.202006246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto Y.; Kohler V.; Ward T. R. An NAD(P)H-Dependent Artificial Transfer Hydrogenase for Multienzymatic Cascades. J. Am. Chem. Soc. 2016, 138, 5781–5784. 10.1021/jacs.6b02470. [DOI] [PubMed] [Google Scholar]
- Aleku G. A.; France S. P.; Man H.; Mangas-Sanchez J.; Montgomery S. L.; Sharma M.; Leipold F.; Hussain S.; Grogan G.; Turner N. J. A reductive aminase from Aspergillus oryzae. Nat. Chem. 2017, 9, 961–969. 10.1038/nchem.2782. [DOI] [PubMed] [Google Scholar]
- Montgomery S. L.; Mangas-Sanchez J.; Thompson M. P.; Aleku G. A.; Dominguez B.; Turner N. J. Direct Alkylation of Amines with Primary and Secondary Alcohols through Biocatalytic Hydrogen Borrowing. Angew. Chem., Int. Ed. 2017, 56, 10491–10494. 10.1002/anie.201705848. [DOI] [PubMed] [Google Scholar]
- Tavanti M.; Mangas-Sanchez J.; Montgomery S. L.; Thompson M. P.; Turner N. J. A biocatalytic cascade for the amination of unfunctionalised cycloalkanes. Org. Biomol. Chem. 2017, 15, 9790–9793. 10.1039/C7OB02569F. [DOI] [PubMed] [Google Scholar]
- Schober M.; MacDermaid C.; Ollis A. A.; Chang S.; Khan D.; Hosford J.; Latham J.; Ihnken L. A. F.; Brown M. J. B.; Fuerst D.; Sanganee M. J.; Roiban G.-D. Chiral synthesis of LSD1 inhibitor GSK2879552 enabled by directed evolution of an imine reductase. Nat. Catal. 2019, 2, 909–915. 10.1038/s41929-019-0341-4. [DOI] [Google Scholar]
- Resch V.; Fabian W. M. F.; Kroutil W. Deracemisation of Mandelic Acid to Optically Pure Non-Natural L-Phenylglycine via a Redox-Neutral Biocatalytic Cascade. Adv. Synth. Catal. 2010, 352, 993–997. 10.1002/adsc.200900891. [DOI] [Google Scholar]
- Fan C. W.; Xu G. C.; Ma B. D.; Bai Y. P.; Zhang J.; Xu J. H. A novel D-mandelate dehydrogenase used in three-enzyme cascade reaction for highly efficient synthesis of non-natural chiral amino acids. J. Biotechnol. 2015, 195, 67–71. 10.1016/j.jbiotec.2014.10.026. [DOI] [PubMed] [Google Scholar]
- Durham D. R. Initial reactions involved in the dissimilation of mandelate by Rhodotorula graminis. J. Bacteriol. 1984, 160, 778–780. 10.1128/jb.160.2.778-780.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang C. D.; Shi H. L.; Jia Y. Y.; Li X.; Wang L. F.; Xu J. H.; Yao L. G.; Kan Y. C. High level and enantioselective production of L-phenylglycine from racemic mandelic acid by engineered Escherichia coli using response surface methodology. Enzyme Microb. Technol. 2020, 136, 109513. 10.1016/j.enzmictec.2020.109513. [DOI] [PubMed] [Google Scholar]
- Dennig A.; Blaschke F.; Gandomkar S.; Tassano E.; Nidetzky B. Preparative Asymmetric Synthesis of Canonical and Non-canonical α-amino Acids Through Formal Enantioselective Biocatalytic Amination of Carboxylic Acids. Adv. Synth. Catal. 2019, 361, 1348–1358. 10.1002/adsc.201801377. [DOI] [Google Scholar]
- Gonçalves L. P. B.; Antunes O. A. C.; Pinto G. F.; Oestreicher E. G. Simultaneous enzymatic synthesis of (S)-3-fluoroalanine and (R)-3-fluorolactic acid. Tetrahedron: Asymmetry 2000, 11, 1465–1468. 10.1016/S0957-4166(00)00096-3. [DOI] [Google Scholar]
- Boonstra B.; Rathbone D. A.; French C. E.; Walker E. H.; Bruce N. C. Cofactor regeneration by a soluble pyridine nucleotide transhydrogenase for biological production of hydromorphone. Appl. Environ. Microbiol. 2000, 66, 5161–5166. 10.1128/AEM.66.12.5161-5166.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gargiulo S.; Opperman D. J.; Hanefeld U.; Arends I. W.; Hollmann F. A biocatalytic redox isomerisation. Chem. Commun. 2012, 48, 6630–6632. 10.1039/c2cc31947k. [DOI] [PubMed] [Google Scholar]
- Liu Y. C.; Merten C.; Deska J. Enantioconvergent Biocatalytic Redox Isomerization. Angew. Chem., Int. Ed. 2018, 57, 12151–12156. 10.1002/anie.201804911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tassano E.; Merusic K.; Buljubasic I.; Laggner O.; Reiter T.; Vogel A.; Hall M. Regioselective biocatalytic self-sufficient Tishchenko-type reaction via formal intramolecular hydride transfer. Chem. Commun. 2020, 56, 6340–6343. 10.1039/D0CC02509G. [DOI] [PubMed] [Google Scholar]
- Winkler T.; Gröger H.; Hummel W. Enantioselective Rearrangement Coupled with Water Addition: Direct Synthesis of Enantiomerically Pure Saturated Carboxylic Acids from α,β-Unsaturated Aldehydes. ChemCatChem. 2014, 6, 961–964. 10.1002/cctc.201300764. [DOI] [Google Scholar]
- Knaus T.; Mutti F. G.; Humphreys L. D.; Turner N. J.; Scrutton N. S. Systematic methodology for the development of biocatalytic hydrogen-borrowing cascades: application to the synthesis of chiral α-substituted carboxylic acids from α-substituted α, β-unsaturated aldehydes. Org. Biomol. Chem. 2015, 13, 223–233. 10.1039/C4OB02282C. [DOI] [PubMed] [Google Scholar]