

Abstract
Blood-based biomarkers hold great promise to revolutionize the diagnostic and prognostic work-up of Alzheimer’s disease (AD) in clinical practice. This is very timely, considering the recent development of anti-amyloid-β (Aβ) immunotherapies. Several assays for measuring phosphorylated tau (p-tau) in plasma exhibit high diagnostic accuracy in distinguishing AD from all other neurodegenerative diseases in patients with cognitive impairment. Prognostic models based on plasma p-tau levels can also predict future development of AD dementia in patients with mild cognitive complaints. The use of such high-performing plasma p-tau assays in the clinical practice of specialist memory clinics would reduce the need for more costly investigations involving cerebrospinal fluid samples or positron emission tomography. Indeed, blood-based biomarkers already facilitate identification of individuals with pre-symptomatic AD in the context of clinical trials. Longitudinal measurements of such biomarkers will also improve the detection of relevant disease-modifying effects of new drugs or lifestyle interventions.
A neuropathological diagnosis of AD is based on the presence of widespread cortical plaques containing Aβ fibrils in combination with neuronal neurofibrillary tangles and neuropil threads containing hyperphosphorylated tau1. Tau-containing tangles restricted to the medial temporal lobe are found in most people older than 60 years. In AD, Aβ plaques start to accumulate 10–30 years before dementia onset, and these changes are thought to facilitate the spread of pathological tau species from the medial temporal lobe throughout the neocortex2. The mechanism by which Aβ aggregates drive tau spread and accumulation is not yet known but could involve increased tau phosphorylation and secretion of soluble tau forms3. Even though tau pathology affects different cortical regions in a rather stereotypic order2, there is evidence that spreading of tau might occur along four main trajectories, resulting in four main tau patterns that are associated with somewhat different clinical syndromes and prognoses4.
A relatively large number of drugs have been developed against Aβ and tau. Immunotherapies targeting aggregated Aβ have recently been shown to be very effective at removing Aβ fibrils from the brains of patients with AD, which has been associated with beneficial clinical effects5–7. For example, lecanemab was recently approved for clinical use in the USA to slow down the clinical deterioration of symptomatic patients with AD7. With the introduction of effective disease-modifying therapies in clinical practice, we urgently need scalable and cost-effective methods for accurate diagnosis of patients with early AD. Unfortunately, the diagnostic work-up of AD is rather mediocre when biomarkers are not used to support the clinical diagnosis. In specialized memory clinics, the misdiagnosis of AD is around 25–30% when not using AD-specific biomarkers2. However, the vast majority of individuals with AD are managed in primary care where >50% of AD cases are not routinely recognized or correctly diagnosed, resulting in suboptimal treatment and care, which is especially problematic in light of emerging disease-modifying treatments for AD2. However, the recent development of blood-based biomarkers (BBMs) for AD holds promise to revolutionize the diagnostic work-up of AD in clinical practice globally but also to improve the design of clinical trials for the earliest stages of AD. In this Review, we will briefly discuss current cerebrospinal fluid (CSF) and imaging biomarkers for AD, which are already used in certain specialist memory clinics. Next, we will discuss recently developed BBMs for AD and how they can be used in both specialist memory clinics and primary care as well as in clinical trials. An overview of current fluid and imaging biomarkers is given in Table 1.
Table 1 |.
Current overview of candidate biomarkers with relevance to Alzheimer’s disease
| Fluid biomarkers | Imaging biomarkers | |
|---|---|---|
| Aβ | Clinical: Aβ42, Aβ42/Aβ40, Aβ42/p-tau Experimental: oligomers of Aβ |
Clinical: Aβ PET imaging (for example, 11C[PiB]) |
| AD-like tau | Clinical: p-tau217, p-tau181 Experimental: p-tau231, p-tau212, p-tau205, MTBR-tau and others |
Clinical: tau PET imaging (for example, 18F[flortaucipir]) |
| Neurodegeneration | Clinical: NfL Experimental: neurogranin, NPTX2, SNAP-25, GAP-43, β-synuclein, 14-3-3 and others |
Clinical: vMRI, FDG PET Experimental: dMRI, ASL |
| Astrocytic response | Clinical: GFAP Experimental: YKL-40 |
Experimental: deprenyl PET and others |
| Microglial response | Experimental: sTREM2, TAM receptors and others | Experimental: TSPO PET and others |
The table divides biomarkers into those that can be measured in fluids (cerebrospinal fluid (CSF) and/or blood) and by brain imaging (positron emission tomography (PET) or magnetic resonance imaging (MRI)). It is important to note that not all fluid biomarkers are relevant to Alzheimer’s disease (AD) when measured in blood but only when measured in CSF (such as neurogranin, soluble triggering receptor expressed on myeloid cells 2 (sTREM2) and YKL-40 (also known as chitinase-3-like protein 1 (CHI3L1)), because they are expressed to a high degree outside the brain as well. The table also indicates biomarkers that might be used in clinical practice and those that are still more experimental. Aβ, amyloid-β; ASL, arterial spin labeling; dMRI, diffusion MRI; FDG PET, fluorodeoxyglucose PET; GAP-43, growth-associated protein, 43 kDa; GFAP, glial fibrillary acidic protein; MTBR, microtubule-binding region; NfL, neurofilament light; NPTX2, neuronal pentraxin 2; PiB, Pittsburgh compound B; SNAP-25, synaptosomal-associated protein, 25 kDa; TSPO, translocator protein, 18 kDa; vMRI, volumetric MRI.
Current imaging- and CSF-based biomarkers for AD
Imaging-based biomarkers
There are several positron emission tomography (PET) tracers that can detect the load of Aβ fibrils in the brain. Three Aβ PET tracers (flutemetamol, florbetapir and florbetaben) are approved for clinical use, and several large-scale studies have shown high concordance between the in vivo uptake of these PET tracers and the density of Aβ plaques as determined post-mortem2. A normal Aβ PET scan result rules out AD as the underlying etiology in most patients with cognitive symptoms; an abnormal Aβ PET scan is indicative of AD in a younger patient with cognitive symptoms, but, in an older patient, such a result should be interpreted with caution, considering that about 40% of individuals aged 90 years have Aβ plaques in the brain8.
Several PET tracers can detect the load of insoluble tau aggregates in the brain2. One tau PET tracer (flortaucipir) is approved for clinical use in the USA. This tracer has been validated against neuropathology, and it can reliably detect the density of both neurofibrillary tangles and neuropil threads9,10, although it lacks the sensitivity to reliably detect the earliest tau stages (restricted to the medial temporal lobe)10. Tau PET has shown excellent diagnostic accuracy for distinguishing AD dementia from most other neurodegenerative diseases11, and it has been suggested that this method can be used to rule in AD in patients with cognitive impairment even at older ages, considering the high specificity of neocortical tau PET retention for patients with AD12. In a recent study, cognitively unimpaired individuals with both positive Aβ PET and positive tau PET had 20× and 40× increased probabilities of developing mild cognitive impairment (MCI) and dementia, respectively, compared to those with normal PET scans13. Cognitively normal individuals with positive Aβ PET but negative tau PET had a very minor risk of developing cognitive impairment13. Together, these results support the National Institute on Aging-Alzheimer’s Association (NIA-AA) research framework for AD, which states that individuals with both Aβ and tau pathology should be labeled as AD independent of cognitive status (that is, including cognitively unimpaired individuals)14.
CSF-based biomarkers
Aβ and tau can also be measured in CSF2. CSF Aβ42 levels and especially the ratios of Aβ42/Aβ40 or Aβ42/p-tau correlate strongly with Aβ PET status15,16 and AD neuropathology17. Several CSF Aβ and p-tau assays on high-performing, fully automated platforms are currently used in clinical practice16,18. Given the high degree of agreement between Aβ PET and CSF Aβ, there is usually no need to perform both investigations on the same patient19.
Tau can be phosphorylated at more than 40 different positions. Tau phosphorylation at threonine 181 (p-tau181) is increased in CSF in AD but not in other neurodegenerative diseases, including other tauopathies20. Other p-tau isoforms have also been investigated extensively in CSF, and there is converging evidence that p-tau217 levels exhibit stronger associations with both tau tangle and Aβ plaque load than levels of p-tau181 and p-tau231 (refs. 21,22), although some results indicate that the assay setup may be more important than the phosphorylation site as such23,24. Furthermore, CSF p-tau217 levels might distinguish AD dementia from other dementias with even higher accuracy than other p-tau isoforms, and this has improved prognostic utility21,22,25,26.
Comparing PET- and CSF-based Aβ and tau measures
According to the NIA-AA research framework for AD, Aβ pathology (A) can be determined using either Aβ PET or CSF Aβ in an interchangeable fashion14. This is likely to be correct in most situations27, but there are subtle differences between these two measures. First, levels of CSF Aβ42, and potentially also Aβ42/Aβ40, change somewhat earlier than Aβ PET signals; this is also the case for Aβ42/Aβ40 levels in blood samples28–30. Furthermore, the Aβ PET signal increases with disease progression as it measures insoluble Aβ-laden plaques, whereas, in CSF and blood, the Aβ42/Aβ40 ratio decreases with development of pathology.
However, using tau PET and CSF p-tau interchangeably for tau pathology (T) seems to be more complex; for example, in cognitively unimpaired populations, more individuals are identified as T-positive when using CSF p-tau versus tau PET27. This is because p-tau levels in CSF and plasma start to increase much earlier than the tau PET signal reaches the threshold for detection during the preclinical stages of AD31,32. In fact, Aβ-positive individuals who are positive for CSF p-tau but still negative for tau PET might represent a population with early AD who are just about to start accumulating tau aggregates in the neocortex33. It has therefore been suggested that the NIA-AA research framework be updated to include p-tau and tau PET as separate biomarker entities, that is, using ‘APT’ instead of ‘AT’, where P stands for p-tau (measuring the levels of soluble hyperphosphorylated tau) and T stands for tau PET (measuring the density of insoluble tau fibrils)33.
Markers of neurodegeneration
Finally, according to the NIA-AA research framework, markers of neurodegeneration (N) provide additional information about disease status14. Hippocampal volume and/or cortical thickness of temporoparietal regions can be determined using structural magnetic resonance imaging (MRI) and reflect the disease stage of AD. Furthermore, several fluid biomarkers of neurodegeneration have emerged. For example, CSF levels of total tau (t-tau) reflect axonal degeneration and injury. Disorders with rapid neurodegeneration, such as Creutzfeldt–Jakob disease and autoimmune encephalitis, are characterized by normal CSF p-tau but a more pronounced increase in t-tau34,35 than that found in AD (which has a slower clinical course). Similarly, in acute neuronal injury such as stroke and acute brain trauma, CSF t-tau shows a temporary increase associated with severity of the neuronal damage and long-term clinical outcome, while p-tau remains relatively normal36,37. Another promising neurodegeneration biomarker is neurofilament light (NfL), which reflects axonal degeneration and injury of the longer myelinated axons of the brain and spinal cord structures, irrespective of cause. NfL levels in CSF are especially increased in amyotrophic lateral sclerosis, frontotemporal dementia and atypical parkinsonian disorders but also in AD38. Importantly, in most neurodegenerative disorders, higher levels of NfL are associated with faster disease progression and higher brain atrophy rates38,39. NfL can therefore be regarded as a measure of the intensity of ongoing neurodegeneration. Even though a substantial number of CSF markers for neurodegeneration and neuroinflammation have been developed over the past decade (Table 1), only Aβ, tau and NfL seem to provide clinically relevant prognostic information in the context of AD40.
BBMs for AD and related disorders
As in CSF, plasma levels of Aβ42/Aβ40 are associated with the presence of Aβ plaques in the brain as determined by neuropathology41. In many studies across several platforms, including different immunoassays and mass spectrometry-based assays, the plasma Aβ42/Aβ40 ratio is lower in Aβ-positive groups than in Aβ-negative groups, regardless of cognitive status of the cohort42–47. However, the performance of different plasma Aβ42/Aβ40 assays varies substantially, and a recent head-to-head comparison showed that certain mass spectrometry-based assays could detect Aβ pathology with areas under the receiver operating characteristic curve (AUC) of 0.84–0.87, whereas many commonly used immunoassays performed much worse (AUC, 0.64–0.69)48. Adding APOE genotype to plasma Aβ42/Aβ40-based prediction models increases the AUC by about 10% (refs. 45,47,48). The assays with better diagnostic performance are characterized by superior control of measurement error. Still, these relatively high-performing Aβ42/Aβ40 assays exhibit only modest correlations between the levels in plasma and CSF (rs of 0.56–0.65)48, probably because much of the Aβ in plasma is derived from peripheral sources49.
Several high-sensitivity assays have recently been developed that can reliably detect different p-tau isoforms in plasma, including p-tau181 (refs. 50–53), p-tau217 (refs. 54,55) and p-tau231 (ref. 56). These assays performed well in detecting AD as defined using neuropathology50–54,56. A few head-to-head comparisons of these assays using plasma from patients with cognitive complaints showed that assays quantifying plasma p-tau217 are somewhat better at detecting AD pathology and predicting future development of AD dementia57–60. The best-performing p-tau217 assay showed a high correlation between plasma and CSF levels, with a correlation coefficient of 0.89 (ref. 57). Plasma p-tau231, on the other hand, seems to start increasing at very low Aβ plaque levels56,61,62. These results are congruent with recent studies showing that plasma p-tau231 is associated with Aβ plaque load but not tau tangle load41,63. By contrast, p-tau181 and p-tau217 were associated with both Aβ plaques and tau tangles, with p-tau217 showing stronger correlations with both pathologies41,64. Our current understanding is that Aβ plaques might induce hyperphosphorylation and secretion of tau, which in turn might promote tau aggregation and formation of tau tangles3,64–66. However, there is currently no tangle-specific tau plasma marker, but recent developments in CSF markers hold great promise, especially those measuring the microtubule-binding region (MTBR) of tau67.
Similar to CSF NfL, plasma NfL is a measure of active neurodegeneration in several neurodegenerative disorders68. Plasma NfL levels generally correlate well with the levels in CSF69. NfL levels are associated with neurodegeneration in AD, but the effect size is smaller for plasma than for CSF, as is the case in other neurodegenerative diseases, for example, Huntington’s disease70.
Glial fibrillary acidic protein (GFAP), which probably reflects reactive astrocytes, can be reliably measured in both blood and CSF. Plasma levels of GFAP are increased in individuals with early Aβ pathology71–73 and can predict subsequent cognitive decline and conversion to AD dementia in cognitively unimpaired individuals74 and in patients with MCI75. Plasma GFAP levels are also increased in other neurodegenerative diseases, including frontotemporal dementia associated with progranulin mutations76. It is currently unclear whether plasma GFAP levels correlate with the number of reactive astrocytes as determined post-mortem using immunohistochemistry or antemortem using PET.
BBMs for diagnosis and prognosis of cognitively impaired patients in specialist memory clinics
BBMs as diagnostic biomarkers in clinical practice
Once anti-Aβ therapies (for example, lecanemab) can be used in patients with MCI or mild dementia, it will be crucial that a highly accurate yet time- and cost-effective diagnostic workflow for AD is in place. BBMs hold great promise in this respect (Fig. 1). In clinics without access to Aβ PET or CSF AD biomarkers, implementation of accurate AD BBMs will improve the diagnostic work-up quite substantially compared to the care as usual of today. In specialist clinics with access to CSF and/or PET, BBMs will speed up the diagnostic process and substantially reduce costs. BBMs will probably be sufficient to support or reject an AD diagnosis in most patients with MCI or dementia; only those patients with uncertain BBM outcomes are likely to need confirmatory testing with Aβ PET or CSF AD biomarkers (Fig. 1). Indeed, a recent study showed that a diagnostic algorithm based on plasma p-tau217 resulted in an accurate AD diagnosis in about 80% of patients with MCI, whereas around 20% had uncertain blood biomarker results and needed further confirmatory testing with CSF AD biomarkers77. A newly developed, highly accurate mass spectrometry assay for p-tau217 might result in fewer patients with uncertain biomarker outcomes, reducing the need for CSF and PET even further57.
Fig. 1 |. Suggested blood-based biomarker-based workflow for Alzheimer’s disease diagnostics.
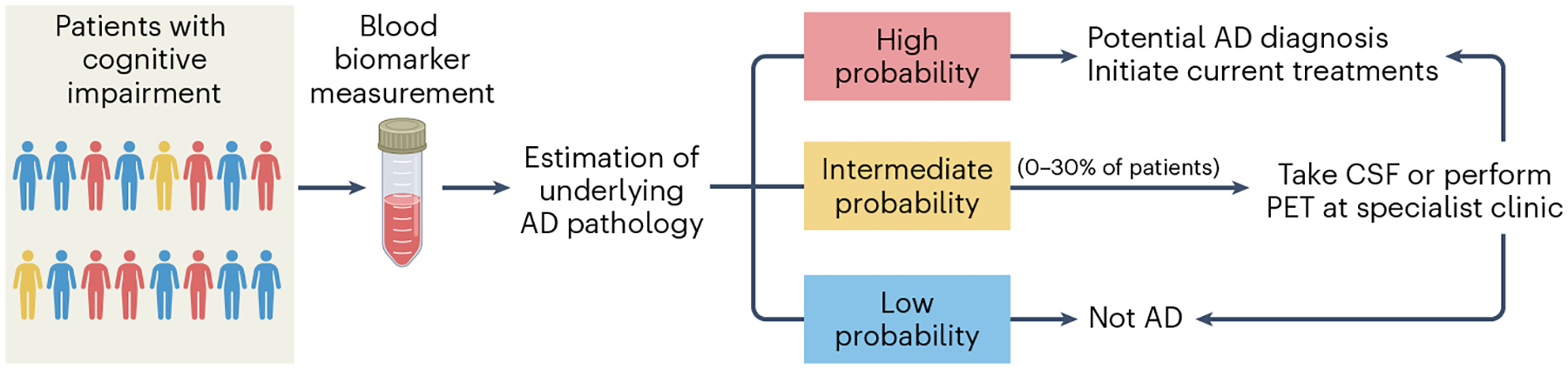
Patients with cognitive complaints undergo blood sampling as part of the standard diagnostic work-up. High-performing blood Alzheimer’s disease (AD) biomarkers (for example, p-tau217) are used to determine the individual-level probability of having AD. For patients deemed to have a very low probability based on blood-based biomarkers (BBMs), another cause of the symptomatology should be sought. For patients deemed to have a very high probability based on BBMs, appropriate treatments might be initiated. Patients with an intermediate probability, whose BBM results lie in an uncertain ‘gray zone’, might be referred for confirmatory testing with either cerebrospinal fluid (CSF) or positron emission tomography (PET) AD biomarkers. The percentage of individuals in such a ‘gray zone’ will depend on the accuracy of the blood-based diagnostic algorithm (very-high-performing BBM assays will have few results ending up in the ‘gray zone’).
An important question is which plasma biomarkers for AD should be implemented in the assessment of patients with MCI and dementia. Although plasma GFAP and NfL levels are increased in patients with MCI or dementia due to AD, they are unlikely to contribute substantially to accurate detection of AD pathology when combined with high-performing plasma p-tau and Aβ42/Aβ40 assays78,79. By contrast, several different p-tau variants, including p-tau181, p-tau217 and p-tau231, are clearly increased in the plasma of patients with MCI or dementia due to AD, and these can be used to distinguish AD from other neurodegenerative diseases with high diagnostic accuracy, often on par with PET and CSF AD biomarkers (for reviews, see, for example, refs. 2,80–82). Plasma p-tau217 is the tau variant that shows the largest fold increase in individuals with symptomatic AD, with increases of about 300–700% compared to both healthy individuals and patients with other neurodegenerative diseases54. Therefore, the clinical performance of this biomarker is less susceptible to test–retest variability when compared to many other plasma biomarkers83, and the effects of comorbidities (for example, kidney dysfunction) on plasma p-tau217 levels are minor84 (see below). The latter is especially true when the p-tau217/t-tau217 ratio is used as quantified using mass spectrometry85. Together, these characteristics of plasma p-tau217 result in robust clinical performance of this biomarker for detection of AD in patients with MCI or dementia in clinical practice (Fig. 2). However, plasma levels of p-tau217 are very low in healthy individuals, and it might therefore be challenging to establish this biomarker on many of the fully automated platforms used in clinical practice today, as has been the case for the Roche Elecsys platform79. Although plasma p-tau217 is currently the best-performing diagnostic biomarker for symptomatic AD, there are also high-performing assays for plasma p-tau181 (refs. 57,58), and plasma levels of p-tau181 are generally higher than p-tau217 and therefore easier to measure on fully automated platforms79.
Fig. 2 |. An overview of key blood-based biomarkers used in the diagnostic or prognostic work-up of Alzheimer’s disease.
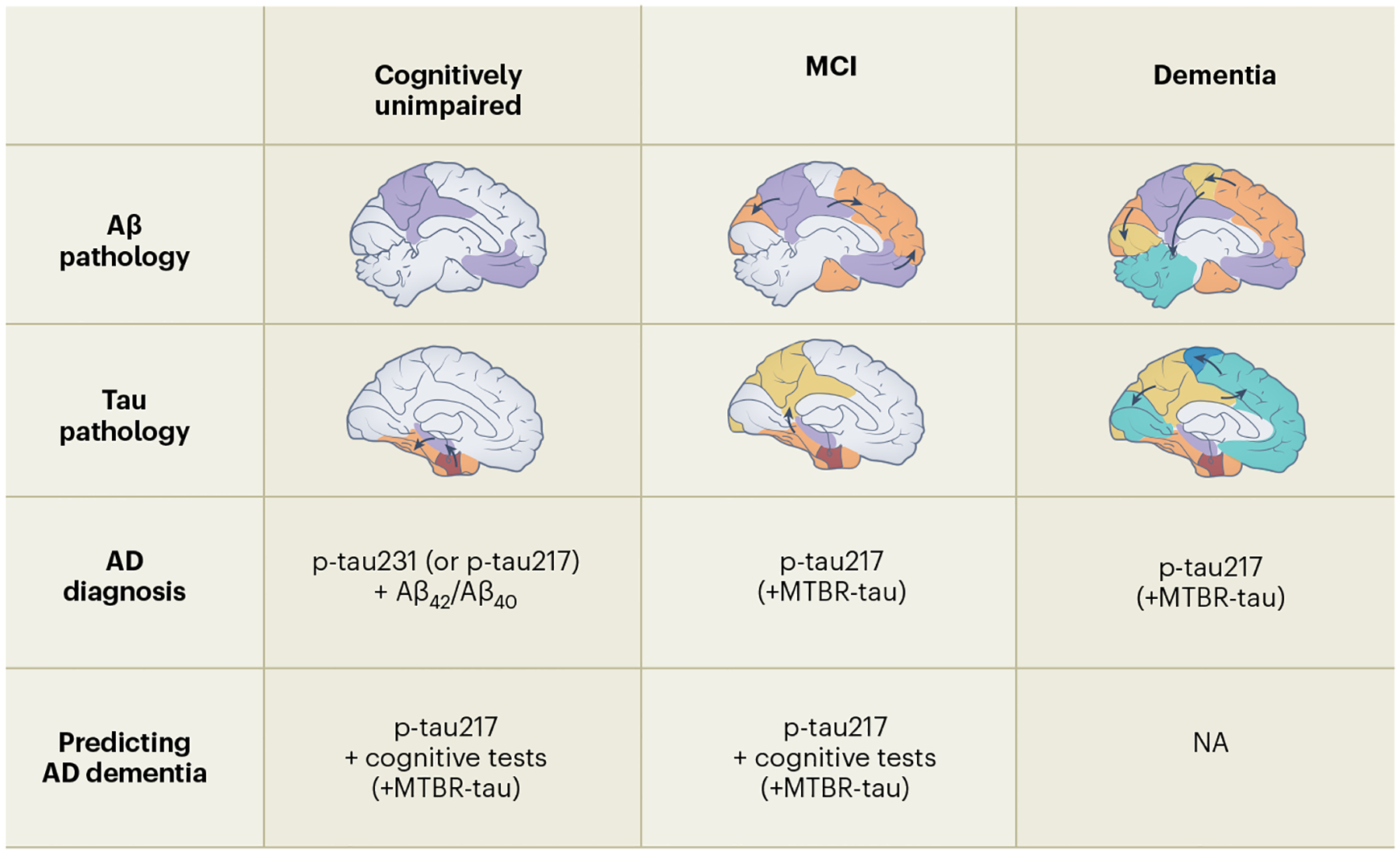
The top two rows depict the evolution of amyloid-β (Aβ) and tau pathological brain changes during the different disease stages of Alzheimer’s disease (AD)2. The third row shows that high-performing p-tau217 assays will probably be sufficient for detection of AD brain pathological changes in patients with cognitive impairment (mild cognitive impairment (MCI) or dementia)54. However, during the preclinical stages of the disease (when individuals are still cognitively unimpaired), p-tau231 and Aβ42/Aβ40 are especially important to detect AD brain changes61. The bottom row shows that plasma p-tau217 is very important for high-performing prognostic algorithms predicting subsequent development of AD dementia in cognitively unimpaired individuals and patients with MCI90,98. Furthermore, in such prognostic algorithms, brief cognitive tests also contribute to the predictive accuracy. Future studies are needed to determine whether new fluid tau markers such as microtubule-binding region (MTBR)-tau will add diagnostic and prognostic information when combined with the already established markers. NA, not applicable.
When it comes to plasma Aβ42/Aβ40 levels, the very modest drop of 8–15% in symptomatic AD2 means that this biomarker has low performance and robustness in routine clinical settings, even if analytical variability and systematic bias are kept at a minimum86, and few current Aβ42/Aβ40 assays fulfill this requirement, resulting in large variability in the clinical performance of different plasma Aβ assays48. Nevertheless, high-performing plasma Aβ42/Aβ40 assays can contribute to plasma p-tau-based diagnostic algorithms that are designed to detect AD pathology in patients with MCI78.
A recent consensus paper proposed that high-performing BBMs can already be used in specialist clinics to facilitate detection of AD pathology in patients with MCI or dementia87. Importantly, BBMs should be combined with a thorough clinical assessment, including psychiatric and neurological examinations, cognitive testing and structural brain imaging. BBMs should never replace such investigations, and they should only be used in patients with cognitive impairment for whom AD is a possible diagnosis and where such a diagnosis will probably change the management of the patient87. These recommendations are primarily based on the risk that false-positive results could lead to anxiety, depression or rash behavior; even a 5% false-positive rate would mean thousands of people would be inappropriately diagnosed with AD if the tests were used in broad screening ahead of identification of objective cognitive impairment.
BBMs as prognostic biomarkers in clinical practice
Information about individual-level prognosis is of key interest for patients with mild cognitive complaints as well as for their care partners and responsible physicians88. Higher baseline plasma p-tau217 and p-tau181 levels in patients with mild cognitive complaints are associated with subsequent progression to AD dementia51,89–91. Combining continuous values of plasma p-tau217 (or p-tau181) levels with performance on a few brief cognitive tests outperforms predictions made by dementia experts and performs similar to CSF-based prognostic models when predicting development of AD dementia within 2–6 years in patients with mild cognitive complaints90 (Fig. 2). An easy-to-use online tool based on plasma p-tau217 and three brief cognitive tests can be used to determine the prognosis of individual patients, and similar tools are likely to be used in clinical practice in the near future90. Neither plasma NfL nor Aβ42/Aβ40 contributed much when predicting the development of AD dementia90,91. However, plasma NfL might have a value when predicting future decline of global cognition in patients with MCI or dementia: prognostic models based on plasma p-tau and NfL can predict changes in global cognition (Mini-Mental State Examination (MMSE) and Clinical Dementia Rating Scale Sum of Boxes (CDR-SB)) in patients with MCI, with performances similar to those of models based on CSF biomarkers91. However, a recent study showed that tau PET imaging may have an even greater value for predicting global cognitive decline in patients with MCI or dementia and that plasma NfL is the only plasma biomarker that provides any additional prognostic information92. However, tau PET is costly and currently not widely available in clinical practice.
BBMs as prescreening biomarkers in clinical trials
Plasma AD biomarkers will also facilitate recruitment of patients with MCI or dementia due to AD for clinical trials. About 40–60% of patients with MCI and 20–30% of those with clinically diagnosed AD dementia do not have brain Aβ pathology2. Thus, when recruiting patients with prodromal AD or mild AD dementia for clinical trials, prescreening individuals for, for example, plasma p-tau217, would reduce the need for confirmatory investigations involving Aβ PET or CSF AD biomarkers (Fig. 3). Such prescreening with high-performing BBMs is likely to be more cost effective in patients with MCI than in patients with dementia, considering the lower prevalence of Aβ positivity in MCI. In certain interventional AD trials, such as trials evaluating lifestyle interventions, a high-performing plasma biomarker might be enough to confirm AD pathology, removing the need for CSF and PET altogether, which would substantially reduce the costs and increase the scalability of such trials.
Fig. 3 |. Suggested workflow for inclusion of study participants into preclinical Alzheimer’s disease trials.
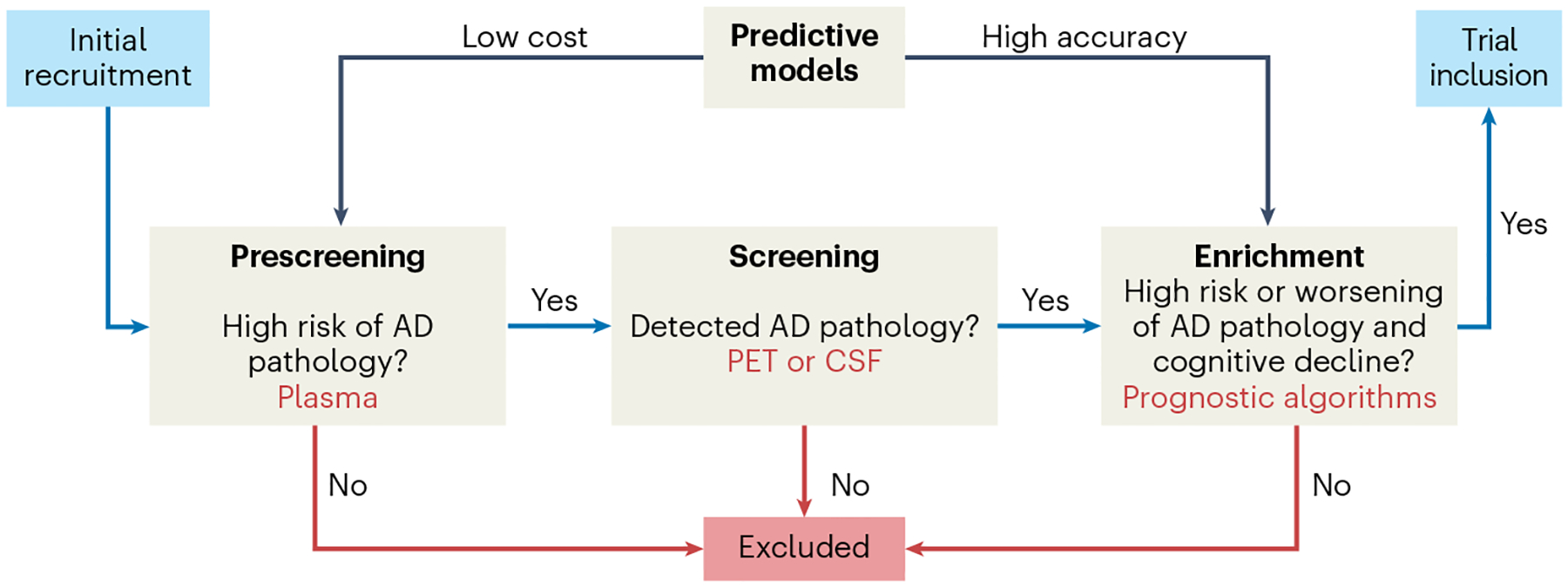
In the ‘prescreening’ step, a diagnostic algorithm based on blood-based biomarkers (BBMs) for Alzheimer’s disease (AD) identifies cognitively normal individuals as being at low risk or high risk of having pre-symptomatic (preclinical) AD. In the ‘screening’ step, individuals deemed high risk will undergo further tests, involving amyloid-β (Aβ) positron emission tomography (PET) or cerebrospinal fluid (CSF) AD biomarkers, to confirm or rule out the presence of AD pathology. The prescreening step with BBMs will result in substantial time and cost savings, as far fewer CSF or PET tests will be needed to identify a certain number of individuals with preclinical AD. In the ‘enrichment’ step, a prognostic algorithm can be used to identify individuals who are likely to subsequently exhibit more severe spread of tau pathology and cognitive decline, so that the population to be included in the trial is enriched for such individuals. This latter enrichment step enables preclinical AD trials with shorter durations and/or fewer study participants.
BBMs for diagnosis and prognosis of cognitively unimpaired individuals in specialist memory clinics
BBMs as diagnostic biomarkers in clinical practice
High-performing assays for plasma p-tau181, p-tau217, p-tau231, Aβ42/Aβ40 and GFAP but not NfL can detect relatively well AD-related pathological changes in cognitively normal individuals and in patients with subjective cognitive decline (for reviews, see, for example, refs. 2,81,82). A recent study analyzed all these plasma biomarkers in preclinical AD and showed that plasma p-tau231 and Aβ42/Aβ40 could be used to detect the earliest AD brain changes61. Indeed, a combination of plasma p-tau and Aβ42/Aβ40 was found to be the best biomarker combination for detection of amyloid pathology in cognitively unimpaired individuals, and high-performing Aβ42/Aβ40 assays might contribute more to the diagnostic work-up in this very early disease stage than to later disease stages78. Although there is currently no obvious clinical need to detect AD in cognitively normal individuals, this might change when phase 3 trials evaluating anti-amyloid therapies, such as lecanemab (NCT04468659) and donanemab (NCT05026866), will read out in 2027–2028. That said, the use of BBMs might be considered in certain patients with subjective cognitive decline, for whom cognitive test results are still normal but the patient history indicates a gradual cognitive deterioration. Such patients could be investigated with BBMs in clinical practice similar to patients with MCI (see above).
BBMs as prescreening biomarkers in clinical trials
Even if AD BBMs will not be widely used for cognitively normal individuals in clinical practice in the foreseeable future, they will be a gamechanger for clinical trials conducted in patients with preclinical AD. As only 10–30% of individuals aged 60–80 years are positive for amyloid PET or CSF Aβ8, a large number of PET (or CSF) examinations is currently needed to identify a sufficient number of individuals for phase 3 trials focusing on preclinical AD. For the A4 (Anti-Amyloid Treatment in Asymptomatic Alzheimer’s) trial (the first phase 3 trial for preclinical AD), it took 3.5 years and more than 4,000 amyloid PET scans to identify 1,169 participants eligible for the study. As shown in Fig. 3, a prescreening step with high-performing BBMs could greatly reduce the number of PET (or CSF) investigations. Using high-performing plasma Aβ42/Aβ40 assays in this way indeed resulted in substantial cost and time savings46,93,94. This was particularly evident if the plasma test was incorporated early in the enrollment process, even before the screening visit93. As mentioned above, combining plasma Aβ42/Aβ40 with p-tau231 (ref. 61) (or p-tau217 (ref. 78)) levels might result in even more efficient detection of preclinical AD. Several large-scale phase 3 anti-Aβ trials already use plasma Aβ42/Aβ40 (NCT05026866) or p-tau217 (NCT04468659) to identify individuals with a high probability of having preclinical AD.
As shown in Fig. 3, efficient clinical trials also need to enrich the preclinical AD population for those that will probably worsen in the primary outcome over a reasonable time period (3–5 years). This is because many individuals with preclinical AD do not deteriorate over 5–10 years or even during their lifespan95–97, and, without enrichment for vulnerable individuals, very large and extended trials would be needed. Power calculations indicate that, if only amyloid positivity is included as a requirement, about 2,000 participants are needed per group to detect a treatment effect of 25% over 4 years using a cognitive composite measure optimized for preclinical AD as a primary endpoint95. In two independent cohorts, plasma p-tau217 levels could accurately predict future cognitive decline in preclinical AD; in this setting, plasma p-tau217 performed better than other plasma and CSF biomarkers (p-tau231, p-tau181, GFAP and NfL) or amyloid PET98. Importantly, power calculations revealed that using plasma p-tau217 levels to enrich for cognitively normal individuals likely to show cognitive decline resulted in large reductions in required sample sizes.
Tau pathology has consistently been shown to be more strongly associated with clinical deterioration than with Aβ pathology, even in cognitively unimpaired individuals13,99. Therefore, future phase 2 trials might use accumulation of tau pathology over time (as measured with longitudinal tau PET) as a more precise primary outcome than cognitive measures, which exhibit high intra-individual variation. Of note, the increase in tau PET signal over time in amyloid-positive AD populations is modest. However, plasma p-tau217 was recently shown to accurately predict future accumulation of tau aggregates in the brain, and a combination of p-tau217 and tau PET at baseline could be used to substantially reduce the needed sample sizes by >40% when using longitudinal tau PET as the primary outcome in preclinical AD trials100.
Potential use of BBMs in primary care settings
Most patients with cognitive symptoms are managed in primary care rather than in specialist clinics. Although few studies in primary care settings have systematically evaluated the accuracy of AD diagnoses against a valid reference standard (for example, dementia expert diagnoses supported by CSF or PET), it seems that about 50–70% of patients with cognitive impairment are currently not recognized or correctly diagnosed in primary care, due to lack of easily accessible, time- and cost-effective, and accurate diagnostic tools101. The problem is even worse in early stages of the disease, that is, in patients with subjective cognitive decline or MCI, because there are no accurate methods for personalized prognosis of AD in primary care. This leads to patients not receiving appropriate diagnostic and prognostic information and also results in suboptimal treatment strategies and care. Misdiagnosis can also lead to unnecessary care seeking and costly investigations due to diagnostic uncertainty. Considering that CSF and PET cannot be used in primary care, AD BBMs have the potential to finally provide primary care physicians with adequate tools to provide their patients with an accurate diagnostic and prognostic work-up.
Several prospective studies are currently evaluating AD BBMs in primary care. For example, a study in Sweden that includes 800 patients with cognitive symptoms at primary care centers evaluates whether AD BBMs can be analyzed prospectively in primary care using pre-defined cutoffs in a diverse population in which many patients have several comorbidities, whether diagnosis and treatment of patients improve by adding AD BBMs to the ‘care as usual’ and whether BBMs can be used to predict future development of AD dementia in non-demented individuals with cognitive complaints in primary care. Regulatory authorities in many countries will probably require such studies before AD BBMs can be widely implemented in primary care settings, which is why the Alzheimer’s Association appropriate-use recommendations do not yet endorse the use of AD BBMs in primary care87. Once BBMs for AD have been validated in primary care, education packages regarding when to use the biomarkers, what they represent, how to interpret the results and what to do with the results must be developed in close collaboration between primary care physicians, dementia experts and patient representatives87.
BBMs for monitoring disease progression
Fluid biomarkers and brain-imaging methods are increasingly being used as outcome measures in clinical trials evaluating disease-modifying therapies for AD and other neurodegenerative disorders. The use of such surrogate endpoints will be especially important in preclinical AD trials, for which very large and long-term studies are needed when using a clinical outcome such as cognitive function95. Biomarker outcomes predicting clinical beneficial effects could shorten the duration and/or reduce the size of future preclinical AD trials. Aβ PET but not yet any AD-related fluid biomarker is deemed by the US Food and Drug Administration (FDA) to be a ‘reasonably likely surrogate endpoint’, which means that it is ‘supported by strong mechanistic and/or epidemiologic rationale, but the amount of clinical data available is not sufficient to show that they are a validated surrogate endpoint’ (ref. 102). Such a biomarker can be used to support the FDA’s Accelerated Approval Program. However, only validated surrogate endpoints can be used as a primary endpoint in pivotal trials used for full FDA approval, and no AD biomarker currently meets this definition.
Many of the fluid tau and neurodegeneration biomarkers discussed above are more or less directly related to disease progression. The best-established biomarker for general neurodegeneration is NfL68,103. The magnitude of NfL increases in CSF and/or plasma reflects the intensity of the neurodegenerative process and predicts imaging and clinical evidence of disease progression104,105. In AD, high NfL levels are associated with longitudinal neurodegeneration as determined by MRI; however, this is only obvious at more advanced dementia stages105. Such associations are clearer in other neurodegenerative diseases such as multiple sclerosis106, amyotrophic lateral sclerosis107 and frontotemporal dementia108, in which NfL levels are generally much higher than in AD68. Interestingly, disease-modifying treatment in, for example, multiple sclerosis and spinal muscular atrophy reduces NfL levels, and the reductions correlate with the clinical efficacy of the intervention109,110. In anti-Aβ antibody trials for AD, attenuated increases of CSF NfL have been reported111,112, but no such results have been obtained thus far for plasma NfL113. NfL may be a better surrogate marker for neurodegenerative disease other than AD, considering the modest increases in plasma NfL in AD and considering the fact that many older individuals have other brain pathologies (for example, TDP-43) that are more related to increased NfL levels than AD.
Early studies showed that people with clearly increased CSF tau levels had faster AD progression, suggesting that this marker, similar to NfL, reflects the intensity of the neurodegenerative process in AD114,115 but in an AD-specific rather than in a general neurodegeneration-reflecting manner. Similarly, studies with new blood tests for p-tau forms2,80–82 showed that longitudinal changes in plasma p-tau levels are associated with both brain atrophy and cognitive decline in AD populations116–118. Importantly, promising anti-Aβ antibody trials have shown treatment-induced reductions in plasma p-tau markers associated with less clinical deterioration, supporting disease modification and slowing of the neurodegenerative process5,113. In clinical practice, it is possible that certain plasma p-tau forms will be used to assess the effect of anti-Aβ antibody treatments for both treatment evaluation and disease-monitoring purposes. One could even envision yearly plasma p-tau testing to detect reoccurrence of disease activity, if and when treatment with anti-Aβ antibodies for 1–2 years eventually becomes a reality.
In addition to p-tau and NfL, other markers of disease intensity that predict AD progression and have shown promising results in clinical trials include plasma GFAP. Plasma GFAP levels increase over time in AD75, and clear reductions are observed after efficient removal of Aβ plaques by anti-Aβ immunotherapy113. Furthermore, CSF and plasma Aβ42/Aβ40 ratios have been suggested to detect drug target engagement of anti-Aβ antibodies. However, therapeutic antibodies may change the half-life of the biomarkers, making data interpretation difficult119, as has been reported for biofluid-based tau biomarkers in anti-tau antibody trials120.
Few longitudinal studies have performed head-to-head comparisons of different plasma AD biomarkers. Recently, we reported that plasma p-tau217 increases more clearly over 4–6 years in preclinical and prodromal AD than Aβ42/Aβ40, p-tau181, p-tau231, GFAP and NfL; p-tau217 also had the strongest associations with brain atrophy and cognitive decline in two independent cohorts61. If replicated in other studies, this might indicate that plasma p-tau217 could be a key biomarker for detecting disease-modifying effects in drug trials and other interventional studies (for example, involving physical activity) targeting preclinical and/or prodromal AD stages.
Standardization, robustness and clinical cutoffs of BBMs
Standardization
Before biomarker-based diagnostic tests can be introduced into routine clinical practice, biomarker standardization and the development of certified reference materials and guidelines are essential to assure high quality of laboratory test results (and thereby patient care and safety), specifically the accuracy of diagnostic classifications.
For the core AD CSF biomarkers, a working group under the International Federation of Clinical Chemistry and Laboratory Medicine has led standardization efforts121. These have resulted in mass spectrometry methods for CSF Aβ42 that have been approved by the Joint Committee for Traceability in Laboratory Medicine as reference measurement procedures. They have also resulted in three certified reference materials (low, medium and high Aβ42 levels) intended to be used to calibrate the commercially available immunoassay, thereby harmonizing levels across assays122. Similar standardization efforts have been initiated for AD BBMs. A first round-robin study (which aims to verify a new method and compare results across methods and laboratories) on Aβ methods showed disappointingly poor correlations across plasma Aβ42 assays (r = 0.41–0.54), including mass spectrometry methods, whereas correlations for Aβ40 assays were better (r = 0.59–0.79)123. Using the Aβ42/Aβ40 ratio did not improve correlations123, and another study obtained similar results48. When the same immunoassays are applied for CSF samples, correlations are generally very high (r = 0.94–0.99)124. In contrast to plasma Aβ42, correlations between different high-performing plasma p-tau assays are tight57,60.
A more widespread launch and implementation of the AD blood biomarkers for clinical use will require not only analytical standardization but also ensuring that blood biomarkers can be measured on the type of laboratory analyzers available in non-specialized, smaller hospital laboratories. Methods for the measurement of these AD BBMs on high-precision, fully automated instruments have been published79, and other assay formats have been released as laboratory-developed tests for potential clinical implementation.
Standardization of sample-collection procedures is also crucial for clinical implementation. Pre-analytical sample-handling procedures have been examined extensively for CSF biomarkers, as such factors may affect biomarker values125. For blood biomarkers, the same type of sampling tubes (EDTA plasma) should be used for all biomarkers; all the blood biomarkers can withstand up to three freeze–thaw cycles126,127. In contrast to CSF Aβ, plasma Aβ is not sensitive to collection tubes made of glass, and tubes with a gel separator can be used. Importantly, both Aβ42 and Aβ40 are unstable in whole blood, with levels decreasing already after 2 h; samples should therefore be centrifuged early (optimally within 1 h) and plasma should be separated, after which it can be stored at +4 °C for up to 6 h before freezing126,128.
Robustness
Robustness describes a biomarker’s ability to classify patients with high consistency and high clinical accuracy80,83,86. For a biomarker to be suitable for clinical use, its levels should be clearly higher (or lower) in AD samples than in all relevant differential diagnostic groups, resulting in high diagnostic sensitivity and specificity (Box 1). The effect size, meaning the difference in mean biomarker levels between patients with and without AD or the pathology (for example, brain amyloidosis) divided by the pooled standard deviation, needs to be much larger than the total measurement variability of the BBM. The total variability depends on biological variability, variability induced by variations in pre-analytical handling of blood samples and the analytical variability inherent to any measurement technique and to drifts or changes over time (Box 1). In other words, a robust biomarker can withstand the variability and bias across measurements that occur in clinical routine. As examples, the pregnancy test for urine human chorionic gonadotropin (HCG) has very high robustness, because levels in pregnant compared with non-pregnant women are more than 1,000-fold different. Also the Aβ42/p-tau and Aβ42/Aβ40 ratios are very robust when measured in CSF129. By contrast, the Aβ42/Aβ40 ratio has very low robustness when measured in plasma, as the ratio is only 0.9-fold lower in patients with brain amyloidosis than in amyloid PET-negative cognitively normal older people48.
Box 1. Factors governing the clinical robustness of a biomarker.
| Total measurement variability | Clinical biomarker performance | |||
|---|---|---|---|---|
| Factors that may lead to variability in measured concentrations include | The clinical diagnostic performance of a biomarker depends on | |||
| Biological factors | Patient-centered factors | Differences across patients may affect biomarker levels. Examples: age, sex, race/ethnicity, genetics (for example, APOE genotype), obesity, comorbidities (for example, kidney dysfunction), medication (for example, enzyme inhibitors). | Biomarker effect size | The effect size, either an increase or a decrease, of the biomarker is assessed using, for example Cohen’s d, defined as the difference in mean between patients with and without (unaffected control individuals or patients with other diseases) the disease or pathology (for example, brain amyloidosis), divided by the pooled standard deviation. |
| Within-individual factors | Temporary influences on biomarker levels in an individual may include hydration status, diurnal variability, stress and concurrent minor infections. | The effect size is linked to other clinical biomarker performance characteristics including Sensitivity (the probability that the test is positive in those who have the disease or pathology) Specificity (the probability that the test is negative in those without the disease or pathology) Positive predictive value (the probability that a patient has the disease or pathology when the test is positive) Negative predictive value (the probability that a patient does not have the disease or pathology when the test is negative) |
||
| Technical and analytical factors | Pre-analytical factors | Fluid-collection and -processing methods may affect the measured level of the biomarker. Examples include type of collection tube, time to centrifugation, shipment time and temperature, and possible freeze-thaw. Pre-analytical conditions may vary between samples, between centers and over the season. | The effect size for a biomarker may be smaller in patients with preclinical or early stages of the disease (with limited amounts of pathology) than in patients with advanced disease (and extensive pathology). | |
| Analytical performance of biomarker assay | Analytical variability means the inevitable differences in concentrations measured present for any measurement technique, both within a run and between runs. Analytical variability is measured through fit-for-purpose validation experiments and includes, for example, within-run precision, between-run repeatability and accuracy (which can be assessed when reference standards exist). |
|||
| Bias | Differences or drifts in measured concentrations between laboratories, batches of reagents or changes in analytical equipment (for example, liquid chromatography columns or instruments) | |||
| Biomarker performance and robustness | ||||
| Prospective biomarker studies | Clinical biomarker performance characteristics depend on the conditions under which they were assessed. Biomarker performance will be higher in selected research cohorts (with a high proportion of typical patients with AD and healthy controls) than in unselected primary care or memory clinic cohorts and also depending on how samples were analyzed (batch analysis with identification of the optimal cutoff with the biomarker data in hand versus clinical routine-like multiple analyses with a pre-set cutoff). Thus, clinical biomarker performance characteristics need to be assessed in prospective clinical trials, in settings resembling the conditions in future clinical routine, meaning a clinical cohort representative of the intended use population and with analyses performed continuously and patients classified using a pre-set cutoff into those with the disease or pathology and those without. | |||
| Clinically robust biomarker | A clinically robust biomarker has a total measurement error that is substantially lower than the biomarker effect size. This biomarker gives an accurate and consistent classification of patients into those who have the disease (or pathology, for example, brain amyloidosis) and those who do not. |
|||
Factors that may affect biomarker measurements are shown in Box 1. Factors that contribute to biological variation can influence classification accuracy and may need consideration when establishing cutoffs (see below). In addition, both pre-analytical (for example, time to centrifugation) and analytical (assay imprecision) factors and drifts or bias in values across rounds of measurements will also add to the total measurement variability (Box 1).
A blood biomarker such as the plasma Aβ42/Aβ40 ratio, which exhibits a modest change of 8–15% in amyloid-positive cases, may be problematic even if the total measurement variability is lower than 5–10% (ref. 86). This small effect size, combined with the total error in plasma Aβ42 assays, means that this biomarker has low robustness. This may induce difficulties if the plasma Aβ42/Aβ40 ratio is introduced as a clinical routine test. By contrast, plasma p-tau217 levels are increased 300–700% in symptomatic AD54.
Biomarker robustness can be tested through simulations that test the influence of increasing the analytical total error of blood biomarker measurements on clinical classifications. Such simulations have shown that even minor increases in total error strongly affect the performance of the plasma Aβ42/Aβ40 ratio as a biomarker to identify brain Aβ pathology but not that of other blood biomarkers (NfL, GFAP and p-tau181)130. A second study found that introducing a 10% bias had a large effect on performance of the plasma Aβ42/Aβ40 ratio but not the CSF p-tau/Aβ42 ratio when they were used as biomarkers of amyloid positivity86. A third study showed that, even though plasma Aβ42/Aβ40 has lower test–retest variability than plasma p-tau217, NfL and GFAP, plasma p-tau217 was least affected by this test–retest variability with a change in diagnostic accuracy of <1% (ref. 83). The better robustness is due to p-tau217 having a substantially higher effect size than the other BBMs83. Consequently, plasma p-tau217 and p-tau181 seem to be robust AD BBMs83,130. Of note, the robustness might depend on disease stage: the effect size increases with severity of pathology, because there is a gradual increase in fold change from preclinical AD to prodromal AD, with the highest levels in AD dementia50,51,53,54. Thus, even if these biomarkers are very robust in symptomatic AD, they might be less robust in detecting preclinical AD, which may have implications for prescreening in preclinical AD trials (see above).
Clinical cutoffs
Regarding the clinical diagnostic performance of the biomarkers, it should be noted that, in principle, all data published thus far come from retrospective studies, in which all samples were analyzed in batch, after which the optimal cutoff was identified and descriptive data on the performance were calculated (AUC, sensitivity and specificity). To generate data on the ‘real-life’ diagnostic performance, prospective studies are needed, with fixed biomarker cutoffs set before the start of the study and biomarkers analyzed on a routine (daily or weekly) basis, allowing the obtained biomarker results to be influenced by all the components constituting the true total measurement variability (Box 1).
For use in clinical practice, biomarkers need well-defined and widely accepted clinical cutoffs. Ideally, each biomarker should have a cutoff value established based on the discrimination between clinical groups (or established proxies for neuropathology) or, alternatively (and commonly used in laboratory medicine), based on the 95th percentile of values in a well-characterized control group131.
Baseline physiological levels of brain proteins in blood depend on various non-disease-associated factors. For example, blood NfL levels are strongly age dependent132. Studies assessing sex differences in blood biomarkers have shown inconsistent results. Although it is common to have age- or sex-specific normative ranges for laboratory tests used in clinical practice, comorbidities are typically left as risks or contraindications to the test or simply need to be considered by the patient’s physician during interpretation of the test result. Indeed, several comorbidities (for example, chronic kidney disease and obesity) are associated with increases in plasma p-tau84 and plasma Aβ40, Aβ42, NfL and GFAP133, even though Aβ42/Aβ40 (ref. 134) and p-tau217/t-tau217 (ref. 85) ratios seem to be unaffected. Of note, although associations between blood biomarkers and comorbidities may be statistically significant in large clinical or population-based cohorts, it is important to describe the magnitude of such effects, especially the effect size135, and whether it is of clinical relevance. Indeed, in two large clinical cohorts, plasma NfL and GFAP and, to a lesser degree, p-tau were associated with kidney dysfunction and body mass index, but these potential confounders had no clinically meaningful effects on either prediction of brain pathophysiology or future cognitive change134. In line with these results, chronic kidney disease, obesity and other comorbidities affect the reference ranges for the AD blood biomarkers only slightly84,134. As mentioned above, biomarker cutoffs in laboratory medicine are not routinely adjusted for comorbid disorders, but it is useful to understand their influence on biomarker results, as they might confound interpretation at an individual patient level (for example, in a patient with severe kidney disease and obesity).
Although common laboratory tests (for example, hemoglobin, platelet count and γ-glutamyl transferase) show differences across racial or ethnic groups136, reference intervals for normality are usually developed predominantly with white populations and not separately for different subpopulations. Possible differences in blood biomarker levels across racial or ethnicity groups have also been discussed, but recent large studies on the BBMs Aβ42, Aβ40, t-tau, p-tau and NfL found that levels were similar across white, Black and Spanish-speaking Americans137,138. These results suggest that the same cutoff for AD BMMs can be used across racial or ethnicity groups. However, further studies are needed to assess possible physiological differences in blood biomarker levels across ethnic groups, also adjusting for socioeconomic status and comorbidities.
When BBM levels are close to the established cutoffs, the interpretation is more uncertain. Patients with such uncertain results could be referred for confirmatory CSF or PET testing (Fig. 1). Indeed, categorization of individuals into low-probability (‘non-AD’), high-probability (‘AD’) and intermediate-probability (‘gray zone’) groups has been suggested for the most common AD biomarkers, and a combined model using several markers resulted in fewer patients in the intermediate-probability (‘gray zone’) group83 (Fig. 1). A similar classification system is used for a test available for clinical use in the US, a probability score based on combining APOE genotype, age and plasma Aβ42/Aβ40 ratio139. As mentioned above, a p-tau217-based diagnostic algorithm could classify about 80% of patients with MCI correctly as having or not having AD, with 20% ending up in the intermediate-probability (‘gray zone’) group77.
Future directions
AD is a common disease for which promising drugs are now emerging that may slow or even stop Aβ-triggered breakdown of neuronal networks. Disease-modifying drugs with different targets (for example, anti-tau therapeutics and synapse stabilizers) are also underway. The emerging availability of this broader range of potentially disease-modifying drug candidates directed against distinct pathogenic mechanisms in the AD process resembles recent developments in, for example, rheumatology, for which effective targeted treatments started to become available 20 years ago and have now been implemented in clinical practice in close collaboration between primary healthcare physicians and specialists using biomarker-supported personalized medicine approaches. We envision similar developments in AD in the next few years, and the recently developed BBMs will play a very important role in this process.
We envision that individuals presenting to primary care physicians with cognitive concerns will be first examined according to standard clinical procedures, starting with an evaluation of the patient’s medical history, present comorbidities and duration of cognitive symptoms, a basic neurological examination and brief cognitive testing. The clinician can subsequently make a request for BBM testing after having discussed its potential implications with the patient and his or her relatives. Elevated levels of plasma p-tau would suggest that AD pathology is responsible for the observed cognitive impairment, whereas normal plasma p-tau levels would indicate non-AD causes. If p-tau is normal, increased blood NfL concentration could suggest the presence of non-AD neurodegeneration. We must stress, however, that BBMs might help the clinician in decision making but should in no case substitute a proper neurological assessment. Indeed, confirmatory diagnosis in specialist care settings will continue to be important for some time for many patient populations, but, in the future, it will probably be possible to accurately diagnose and treat many of these patients in primary care only.
The ability of plasma p-tau measurements to identify AD pathophysiology in individuals with symptomatic disease demonstrates the potential of this marker for identifying and recruiting Aβ-positive symptomatic participants for clinical trials. In addition, we expect that blood p-tau will be important for the recruitment of pre-symptomatic Aβ-positive cohorts, which will result in reduced rates of negative PET scans and substantial cost and time savings. Plasma p-tau biomarkers will also be useful to evaluate effects of therapeutic intervention: significant decreases in plasma p-tau concentration or a reduction in the rate of increase over time could indicate beneficial effects of anti-Aβ treatments.
The discussions above point to a revolution in the next 2–4 years, in which widespread and routine analyses of blood p-tau become routine practice in clinical assessments and research studies, probably combined with (1) high-performing assays of plasma Aβ42/Aβ40 ratio for preclinical AD (Fig. 2), (2) brief digital cognitive testing for prognosis (Fig. 2) and (3) plasma NfL when suspecting non-AD neurodegenerative diseases. However, several outstanding challenges must be addressed. We need to obtain analytical standardization and quality control to provide a framework with which biotechnical companies and clinical laboratories can ascertain that they produce valid biomarker results. We need to demonstrate biomarker validity in diverse cohorts. Finally, we need to perform studies to prospectively generate real-world clinical data on the performance of blood-based AD biomarkers, especially in primary care settings. We do not yet know how observations from such cohorts will translate to the setting of routine memory clinics, which see patients with greater heterogeneity in demographics, disease presentation and comorbidities. Therefore, whether blood p-tau can be used as a single marker or replace CSF biomarkers that have been tested in larger varieties of disease conditions remains unclear. Realistically, we might need to exercise caution in projecting immediate diagnostic use of blood p-tau levels as a CSF substitute until large-scale clinical characterization studies have been performed. Finally, we want to stress that we need to develop blood biomarkers for non-AD brain pathologies, especially for pathological changes in TDP-43, 3R tau, 4R tau, α-synuclein and cerebrovascular changes as well as synaptic dysfunction.
Acknowledgements
O.H. was supported by the Swedish Research Council (202200775), ERA PerMed (ERAPERMED2021-184), the Knut and Alice Wallenberg Foundation (2017-0383), the Strategic Research Area MultiPark (Multidisciplinary Research in Parkinson’s disease) at Lund University, the Swedish Alzheimer Foundation (AF-980907), the Swedish Brain Foundation (FO2021-0293), the Parkinson Foundation of Sweden (1412/22), the Cure Alzheimer’s Fund, the Konung Gustaf V:s och Drottning Victorias Frimurarestiftelse, the Skåne University Hospital Foundation (2020-O000028), Regionalt Forskningsstöd (2022-1259) and the Swedish federal government under the ALF agreement (2022-Projekt0080). K.B. is supported by the Swedish Research Council (2017-00915 and 2022-00732; 1 January 2023 to 31 December 2026), the Alzheimer Drug Discovery Foundation, USA (RDAPB-201809-2016615), the Swedish Alzheimer Foundation (AF-930351, AF-939721 and AF-968270), Hjärnfonden, Sweden (FO20170243 and ALZ2022-0006), the Swedish state under the agreement between the Swedish government and the county councils, the ALF agreement (ALFGBG-715986 and ALFGBG-965240), the European Union Joint Program for Neurodegenerative Disorders (JPND2019-466-236), the National Institutes of Health, USA (grant 1R01AG068398-01), the Alzheimer’s Association 2021 Zenith Award (ZEN-21-848495) and the Alzheimer’s Association 2022-2025 grant (SG-23-1038904 QC). H.Z. is a Wallenberg Scholar supported by grants from the Swedish Research Council (2018-02532), the European Union’s Horizon Europe research and innovation program under grant agreement 101053962, Swedish State Support for Clinical Research (ALFGBG-71320), the Alzheimer Drug Discovery Foundation, USA (201809-2016862), the AD Strategic Fund and the Alzheimer’s Association (ADSF-21-831376-C, ADSF-21-831381-C and ADSF-21-831377-C), the Bluefield Project, the Olav Thon Foundation, the Erling-Persson Family Foundation, Stiftelsen för Gamla Tjänarinnor, Hjärnfonden, Sweden (FO2022-0270), the European Union’s Horizon 2020 research and innovation program under the Marie Skłodowska-Curie grant agreement 860197 (MIRIADE), the European Union Joint Programme, Neurodegenerative Disease Research (JPND2021-00694) and the UK Dementia Research Institute at UCL (UKDRI-1003).
Competing interests
O.H. has acquired research support (for the institution) from ADx, Avid Radiopharmaceuticals, Biogen, Eli Lilly, Eisai, Fujirebio, GE Healthcare, Pfizer and Roche. In the past 2 years, he has received consultancy and/or speaker fees from AC Immune, Amylyx, ALZpath, BioArctic, Biogen, Cerveau, Eisai, Fujirebio, Genentech, Novartis, Roche and Siemens. K.B. has served as a consultant on advisory boards or on data-monitoring committees for Abcam, Axon, BioArctic, Biogen, JOMDD–Shimadzu, Julius Clinical, Lilly, MagQu, Novartis, Ono Pharma, Pharmatrophix, Prothena, Roche Diagnostics and Siemens Healthineers and is a cofounder of Brain Biomarker Solutions in Gothenburg (BBS), which is a part of the GU Ventures Incubator Program, outside the work presented in this paper. H.Z. has served on scientific advisory boards and/or as a consultant for AbbVie, Acumen, Alector, Alzinova, ALZpath, Annexon, Apellis, Artery Therapeutics, AZTherapies, CogRx, Denali, Eisai, NervGen, Novo Nordisk, Passage Bio, Pinteon Therapeutics, Prothena, Red Abbey Labs, reMYND, Roche, Samumed, Siemens Healthineers, Triplet Therapeutics and Wave; has given lectures in symposia sponsored by Cellectricon, Fujirebio, AlzeCure, Biogen and Roche; and is a cofounder of Brain Biomarker Solutions in Gothenburg (BBS), which is a part of the GU Ventures Incubator Program (outside the work presented in this paper). J.D. is an inventor on patents or patent applications of Eli Lilly relating to the assays, methods, reagents and/or compositions of matter related to measurement of p-tau217. J.D. has served as a consultant for AbbVie, Genotix Biotechnologies, Gates Ventures, Karuna Therapeutics, ALZpath and Cognito Therapeutics and received research support from ADx NeuroSciences, Fujirebio, ALZpath, Roche Diagnostics and Eli Lilly in the past 2 years. J.D. has received speaker fees from Eli Lilly.
References
- 1.DeTure MA & Dickson DW The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener 14, 32 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hansson O Biomarkers for neurodegenerative diseases. Nat. Med 27, 954–963 (2021). [DOI] [PubMed] [Google Scholar]
- 3.Pichet Binette A et al. Amyloid-associated increases in soluble tau relate to tau aggregation rates and cognitive decline in early Alzheimer’s disease. Nat. Commun 13, 6635 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vogel JW et al. Four distinct trajectories of tau deposition identified in Alzheimer’s disease. Nat. Med 27, 871–881 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Budd Haeberlein S et al. Two randomized phase 3 studies of aducanumab in early Alzheimer’s disease. J. Prev. Alzheimers Dis 9, 197–210 (2022). [DOI] [PubMed] [Google Scholar]
- 6.Mintun MA et al. Donanemab in early Alzheimer’s disease. N. Engl. J. Med 384, 1691–1704 (2021). [DOI] [PubMed] [Google Scholar]
- 7.van Dyck CH et al. Lecanemab in early Alzheimer’s disease. N. Engl. J. Med 388, 9–21 (2023). [DOI] [PubMed] [Google Scholar]
- 8.Jansen WJ et al. Prevalence estimates of amyloid abnormality across the Alzheimer disease clinical spectrum. JAMA Neurol. 79, 228–243 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Smith R, Wibom M, Pawlik D, Englund E & Hansson O Correlation of in vivo [18F]flortaucipir with postmortem Alzheimer disease tau pathology. JAMA Neurol. 76, 310–317 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fleisher AS et al. Positron emission tomography imaging with [18F]flortaucipir and postmortem assessment of Alzheimer disease neuropathologic changes. JAMA Neurol. 77, 829–839 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ossenkoppele R et al. Discriminative accuracy of [18F]flortaucipir positron emission tomography for Alzheimer disease vs other neurodegenerative disorders. JAMA 320, 1151–1162 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ossenkoppele R & Hansson O Towards clinical application of tau PET tracers for diagnosing dementia due to Alzheimer’s disease. Alzheimers Dement. 17, 1998–2008 (2021). [DOI] [PubMed] [Google Scholar]
- 13.Ossenkoppele R et al. Amyloid and tau PET-positive cognitively unimpaired individuals are at high risk for future cognitive decline. Nat. Med 28, 2381–2387 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jack CR Jr. et al. NIA-AA Research Framework: toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 14, 535–562 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Janelidze S et al. CSF Aβ42/Aβ40 and Aβ42/Aβ38 ratios: better diagnostic markers of Alzheimer disease. Ann. Clin. Transl. Neurol 3, 154–165 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hansson O et al. CSF biomarkers of Alzheimer’s disease concord with amyloid-β PET and predict clinical progression: a study of fully automated immunoassays in BioFINDER and ADNI cohorts. Alzheimers Dement. 14, 1470–1481 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mattsson-Carlgren N et al. Cerebrospinal fluid biomarkers in autopsy-confirmed Alzheimer disease and frontotemporal lobar degeneration. Neurology 98, e1137–e1150 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gobom J et al. Validation of the LUMIPULSE automated immunoassay for the measurement of core AD biomarkers in cerebrospinal fluid. Clin. Chem. Lab. Med 60, 207–219 (2022). [DOI] [PubMed] [Google Scholar]
- 19.Palmqvist S et al. Detailed comparison of amyloid PET and CSF biomarkers for identifying early Alzheimer disease. Neurology 85, 1240–1249 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Blennow K & Zetterberg H Biomarkers for Alzheimer’s disease: current status and prospects for the future. J. Intern. Med 284, 643–663 (2018). [DOI] [PubMed] [Google Scholar]
- 21.Janelidze S et al. Cerebrospinal fluid p-tau217 performs better than p-tau181 as a biomarker of Alzheimer’s disease. Nat. Commun 11, 1683 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leuzy A et al. Comparing the clinical utility and diagnostic performance of CSF p-Tau181, p-Tau217, and p-Tau231 assays. Neurology 97, e1681–e1694 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Karikari TK et al. Head-to-head comparison of clinical performance of CSF phospho-tau T181 and T217 biomarkers for Alzheimer’s disease diagnosis. Alzheimers Dement. 17, 755–767 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Suarez-Calvet M et al. Novel tau biomarkers phosphorylated at T181, T217 or T231 rise in the initial stages of the preclinical Alzheimer’s continuum when only subtle changes in Aβ pathology are detected. EMBO Mol. Med 12, e12921 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hanes J et al. Evaluation of a novel immunoassay to detect p-tau Thr217 in the CSF to distinguish Alzheimer disease from other dementias. Neurology 95, e3026–e3035 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mielke MM et al. Comparison of CSF phosphorylated tau 181 and 217 for cognitive decline. Alzheimers Dement. 18, 602–611 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mattsson-Carlgren N et al. The implications of different approaches to define AT(N) in Alzheimer disease. Neurology 94, e2233–e2244 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schindler SE et al. High-precision plasma β-amyloid 42/40 predicts current and future brain amyloidosis. Neurology 93, e1647–e1659 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Palmqvist S, Mattsson N, Hansson O & Alzheimer’s Disease Neuroimaging Initiative. Cerebrospinal fluid analysis detects cerebral amyloid-β accumulation earlier than positron emission tomography. Brain 139, 1226–1236 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Palmqvist S et al. Cerebrospinal fluid and plasma biomarker trajectories with increasing amyloid deposition in Alzheimer’s disease. EMBO Mol. Med 11, e11170 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mattsson-Carlgren N et al. Aβ deposition is associated with increases in soluble and phosphorylated tau that precede a positive tau PET in Alzheimer’s disease. Sci. Adv 6, eaaz2387 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Janelidze S et al. Associations of plasma phospho-tau217 levels with tau positron emission tomography in early Alzheimer disease. JAMA Neurol. 78, 149–156 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Groot C et al. Phospho-tau with subthreshold tau-PET predicts increased tau accumulation rates in amyloid-positive individuals. Brain 10.1093/brain/awac329 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bastiaansen AEM et al. Autoimmune encephalitis resembling dementia syndromes. Neurol. Neuroimmunol. Neuroinflamm 8, e1039 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Riemenschneider M et al. Phospho-tau/total tau ratio in cerebrospinal fluid discriminates Creutzfeldt–Jakob disease from other dementias. Mol. Psychiatry 8, 343–347 (2003). [DOI] [PubMed] [Google Scholar]
- 36.Hesse C et al. Transient increase in total tau but not phospho-tau in human cerebrospinal fluid after acute stroke. Neurosci. Lett 297, 187–190 (2001). [DOI] [PubMed] [Google Scholar]
- 37.Ost M et al. Initial CSF total tau correlates with 1-year outcome in patients with traumatic brain injury. Neurology 67, 1600–1604 (2006). [DOI] [PubMed] [Google Scholar]
- 38.Khalil M et al. Neurofilaments as biomarkers in neurological disorders. Nat. Rev. Neurol 14, 577–589 (2018). [DOI] [PubMed] [Google Scholar]
- 39.Preische O et al. Serum neurofilament dynamics predicts neurodegeneration and clinical progression in presymptomatic Alzheimer’s disease. Nat. Med 25, 277–283 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Salvado G et al. Optimal combinations of CSF biomarkers for predicting cognitive decline and clinical conversion in cognitively unimpaired participants and mild cognitive impairment patients: a multi-cohort study. Alzheimers Dement. 10.1002/alz.12907 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Salvadó G et al. Specific associations between plasma biomarkers and postmortem amyloid plaque and tau tangle loads. EMBO Mol. Med 10.15252/emmm.202217123 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nakamura A et al. High performance plasma amyloid-β biomarkers for Alzheimer’s disease. Nature 554, 249–254 (2018). [DOI] [PubMed] [Google Scholar]
- 43.Ovod V et al. Amyloid β concentrations and stable isotope labeling kinetics of human plasma specific to central nervous system amyloidosis. Alzheimers Dement. 13, 841–849 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Janelidze S et al. Plasma β-amyloid in Alzheimer’s disease and vascular disease. Sci. Rep 6, 26801 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li Y et al. Validation of plasma amyloid-β 42/40 for detecting Alzheimer disease amyloid plaques. Neurology 98, e688–e699 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Palmqvist S et al. Performance of fully automated plasma assays as screening tests for Alzheimer disease-related β-amyloid status. JAMA Neurol. 76, 1060–1069 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.West T et al. A blood-based diagnostic test incorporating plasma Aβ42/40 ratio, ApoE proteotype, and age accurately identifies brain amyloid status: findings from a multi cohort validity analysis. Mol. Neurodegener 16, 30 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Janelidze S et al. Head-to-head comparison of 8 plasma amyloid-β 42/40 assays in Alzheimer disease. JAMA Neurol. 78, 1375–1382 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Roher AE et al. Amyloid β peptides in human plasma and tissues and their significance for Alzheimer’s disease. Alzheimers Dement. 5, 18–29 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mielke MM et al. Plasma phospho-tau181 increases with Alzheimer’s disease clinical severity and is associated with tau- and amyloid-positron emission tomography. Alzheimers Dement. 14, 989–997 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Janelidze S et al. Plasma p-tau181 in Alzheimer’s disease: relationship to other biomarkers, differential diagnosis, neuropathology and longitudinal progression to Alzheimer’s dementia. Nat. Med 26, 379–386 (2020). [DOI] [PubMed] [Google Scholar]
- 52.Karikari TK et al. Blood phosphorylated tau 181 as a biomarker for Alzheimer’s disease: a diagnostic performance and prediction modelling study using data from four prospective cohorts. Lancet Neurol. 19, 422–433 (2020). [DOI] [PubMed] [Google Scholar]
- 53.Thijssen EH et al. Diagnostic value of plasma phosphorylated tau181 in Alzheimer’s disease and frontotemporal lobar degeneration. Nat. Med 26, 387–397 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Palmqvist S et al. Discriminative accuracy of plasma phospho-tau217 for Alzheimer disease vs other neurodegenerative disorders. JAMA 324, 772–781 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Barthelemy NR, Horie K, Sato C & Bateman RJ Blood plasma phosphorylated-tau isoforms track CNS change in Alzheimer’s disease. J. Exp. Med 217, e20200861 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ashton NJ et al. Plasma p-tau231: a new biomarker for incipient Alzheimer’s disease pathology. Acta Neuropathol. 141, 709–724 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Janelidze S et al. Head-to-head comparison of 10 plasma phospho-tau assays in prodromal Alzheimer’s disease. Brain 10.1093/brain/awac333 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ashton NJ et al. Plasma and CSF biomarkers in a memory clinic: head-to-head comparison of phosphorylated tau immunoassays. Alzheimers Dement. 10.1002/alz.12841 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mielke MM et al. Comparison of plasma phosphorylated tau species with amyloid and tau positron emission tomography, neurodegeneration, vascular pathology, and cognitive outcomes. JAMA Neurol. 78, 1108–1117 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bayoumy S et al. Clinical and analytical comparison of six Simoa assays for plasma p-tau isoforms p-tau181, p-tau217, and p-tau231. Alzheimers Res. Ther 13, 198 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ashton NJ et al. Differential roles of Aβ42/40, p-tau231 and p-tau217 for Alzheimer’s trial selection and disease monitoring. Nat. Med 28, 2555–2562 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mila-Aloma M et al. Plasma p-tau231 and p-tau217 as state markers of amyloid-β pathology in preclinical Alzheimer’s disease. Nat. Med 28, 1797–1801 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Therriault J et al. Association of phosphorylated tau biomarkers with amyloid positron emission tomography vs tau positron emission tomography. JAMA Neurol. 80, 188–199 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mattsson-Carlgren N et al. Soluble p-tau217 reflects amyloid and tau pathology and mediates the association of amyloid with tau. EMBO Mol. Med 13, e14022 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sato C et al. Tau kinetics in neurons and the human central nervous system. Neuron 98, 861–864 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kaeser SA et al. CSF p-tau increase in response to Aβ-type and Danish-type cerebral amyloidosis and in the absence of neurofibrillary tangles. Acta Neuropathol. 143, 287–290 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Horie K, Barthelemy NR, Sato C & Bateman RJ CSF tau microtubule binding region identifies tau tangle and clinical stages of Alzheimer’s disease. Brain 144, 515–527 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ashton NJ et al. A multicentre validation study of the diagnostic value of plasma neurofilament light. Nat. Commun 12, 3400 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gisslen M et al. Plasma concentration of the neurofilament light protein (NfL) is a biomarker of CNS injury in HIV infection: a cross-sectional study. EBioMedicine 3, 135–140 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rodrigues FB et al. Mutant huntingtin and neurofilament light have distinct longitudinal dynamics in Huntington’s disease. Sci. Transl. Med 12, eabc2888 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Verberk IMW et al. Combination of plasma amyloid β(1–42/1–40) and glial fibrillary acidic protein strongly associates with cerebral amyloid pathology. Alzheimers Res. Ther 12, 118 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pereira JB et al. Plasma GFAP is an early marker of amyloid-β but not tau pathology in Alzheimer’s disease. Brain 144, 3505–3516 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Benedet AL et al. Differences between plasma and cerebrospinal fluid glial fibrillary acidic protein levels across the Alzheimer disease continuum. JAMA Neurol. 78, 1471–1483 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Verberk IMW et al. Serum markers glial fibrillary acidic protein and neurofilament light for prognosis and monitoring in cognitively normal older people: a prospective memory clinic-based cohort study. Lancet Healthy Longev. 2, e87–e95 (2021). [DOI] [PubMed] [Google Scholar]
- 75.Cicognola C et al. Plasma glial fibrillary acidic protein detects Alzheimer pathology and predicts future conversion to Alzheimer dementia in patients with mild cognitive impairment. Alzheimers Res. Ther 13, 68 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Heller C et al. Plasma glial fibrillary acidic protein is raised in progranulin-associated frontotemporal dementia. J. Neurol. Neurosurg. Psychiatry 91, 263–270 (2020). [DOI] [PubMed] [Google Scholar]
- 77.Brum WS et al. A two-step workflow based on plasma p-tau217 to screen for Aβ pathology with further confirmatory testing only in uncertain cases. Nat. Aging (in the press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Janelidze S et al. Detecting amyloid positivity in early Alzheimer’s disease using combinations of plasma Aβ42/Aβ40 and p-tau. Alzheimers Dement. 18, 283–293 (2022). [DOI] [PubMed] [Google Scholar]
- 79.Palmqvist S et al. An accurate fully automated panel of plasma biomarkers for Alzheimer’s disease. Alzheimers Dement. 10.1002/alz.12751 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Karikari TK et al. Blood phospho-tau in Alzheimer disease: analysis, interpretation, and clinical utility. Nat. Rev. Neurol 18, 400–418 (2022). [DOI] [PubMed] [Google Scholar]
- 81.Teunissen CE et al. Blood-based biomarkers for Alzheimer’s disease: towards clinical implementation. Lancet Neurol. 21, 66–77 (2022). [DOI] [PubMed] [Google Scholar]
- 82.Leuzy A et al. Blood-based biomarkers for Alzheimer’s disease. EMBO Mol. Med 14, e14408 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Cullen NC et al. Test–retest variability of plasma biomarkers in Alzheimer’s disease and its effects on clinical prediction models. Alzheimers Dement. 19, 797–806 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mielke MM et al. Performance of plasma phosphorylated tau 181 and 217 in the community. Nat. Med 28, 1398–1405 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Janelidze S, Barthélemy NR, He Y, Bateman RJ & Hansson O Mitigating the associations of kidney dysfunction with blood biomarkers of Alzheimer disease by using a phosphorylated tau to total tau ratio. JAMA Neurol. 10.1001/jamaneurol.2023.0199 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Rabe C et al. Clinical performance and robustness evaluation of plasma amyloid-β42/40 prescreening. Alzheimers Dement. 10.1002/alz.12801 (2022). [DOI] [PubMed] [Google Scholar]
- 87.Hansson O et al. The Alzheimer’s Association appropriate use recommendations for blood biomarkers in Alzheimer’s disease. Alzheimers Dement. 18, 2669–2686 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mank A et al. Identifying relevant outcomes in the progression of Alzheimer’s disease; what do patients and care partners want to know about prognosis? Alzheimers Dement. 7, e12189 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Karikari TK et al. Diagnostic performance and prediction of clinical progression of plasma phospho-tau181 in the Alzheimer’s Disease Neuroimaging Initiative. Mol. Psychiatry 26, 429–442 (2021). [DOI] [PubMed] [Google Scholar]
- 90.Palmqvist S et al. Prediction of future Alzheimer’s disease dementia using plasma phospho-tau combined with other accessible measures. Nat. Med 27, 1034–1042 (2021). [DOI] [PubMed] [Google Scholar]
- 91.Cullen N et al. Individualized prognosis of cognitive decline and dementia in mild cognitive impairment based on plasma biomarker combinations. Nat. Aging 1, 114–123 (2021). [DOI] [PubMed] [Google Scholar]
- 92.Smith R et al. Tau-PET is superior to phospho-tau when predicting cognitive decline in symptomatic AD patients. Alzheimers Dement. 10.1002/alz.12875 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Schindler SE et al. Using Alzheimer’s disease blood tests to accelerate clinical trial enrollment. Alzheimers Dement. 10.1002/alz.12754 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Cullen N et al. Plasma amyloid-β42/40 and apolipoprotein E for amyloid PET pre-screening in secondary prevention trials of Alzheimer’s disease. Brain Commun. 5, fcad015 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Insel PS et al. Determining clinically meaningful decline in preclinical Alzheimer disease. Neurology 93, e322–e333 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.van der Kall LM et al. Association of β-amyloid level, clinical progression, and longitudinal cognitive change in normal older individuals. Neurology 96, e662–e670 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Donohue MC et al. Association between elevated brain amyloid and subsequent cognitive decline among cognitively normal persons. JAMA 317, 2305–2316 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Mattsson-Carlgren N et al. Prediction of longitudinal cognitive decline in preclinical Alzheimer disease using plasma biomarkers. JAMA Neurol. 10.1001/jamaneurol.2022.5272 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ossenkoppele R et al. Accuracy of tau positron emission tomography as a prognostic marker in preclinical and prodromal Alzheimer disease: a head-to-head comparison against amyloid positron emission tomography and magnetic resonance imaging. JAMA Neurol. 78, 961–971 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Leuzy A et al. Biomarker-based prediction of longitudinal tau positron emission tomography in Alzheimer disease. JAMA Neurol. 79, 149–158 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bradford A, Kunik ME, Schulz P, Williams SP & Singh H Missed and delayed diagnosis of dementia in primary care: prevalence and contributing factors. Alzheimer Dis. Assoc. Disord 23, 306–314 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.US Food and Drug Administration. Surrogate Endpoint Resources for Drug and Biologic Development https://www.fda.gov/drugs/development-resources/surrogate-endpoint-resources-drug-and-biologic-development (2018).
- 103.Ning L & Wang B Neurofilament light chain in blood as a diagnostic and predictive biomarker for multiple sclerosis: a systematic review and meta-analysis. PLoS ONE 17, e0274565 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zetterberg H et al. Association of cerebrospinal fluid neurofilament light concentration with Alzheimer disease progression. JAMA Neurol. 73, 60–67 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Mattsson N, Andreasson U, Zetterberg H, Blennow K & Alzheimer’s Disease Neuroimaging I Association of plasma neurofilament light with neurodegeneration in patients with Alzheimer disease. JAMA Neurol. 74, 557–566 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Disanto G et al. Serum neurofilament light: a biomarker of neuronal damage in multiple sclerosis. Ann. Neurol 81, 857–870 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Benatar M et al. Validation of serum neurofilaments as prognostic and potential pharmacodynamic biomarkers for ALS. Neurology 95, e59–e69 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Staffaroni AM et al. Temporal order of clinical and biomarker changes in familial frontotemporal dementia. Nat. Med 28, 2194–2206 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Delcoigne B et al. Blood neurofilament light levels segregate treatment effects in multiple sclerosis. Neurology 94, e1201–e1212 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Olsson B et al. NfL is a marker of treatment response in children with SMA treated with nusinersen. J. Neurol 266, 2129–2136 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Salloway S et al. A trial of gantenerumab or solanezumab in dominantly inherited Alzheimer’s disease. Nat. Med 27, 1187–1196 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Swanson CJ et al. A randomized, double-blind, phase 2b proof-of-concept clinical trial in early Alzheimer’s disease with lecanemab, an anti-Aβ protofibril antibody. Alzheimers Res. Ther 13, 80 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Pontecorvo MJ et al. Association of donanemab treatment with exploratory plasma biomarkers in early symptomatic Alzheimer disease: a secondary analysis of the TRAILBLAZER-ALZ randomized clinical trial. JAMA Neurol. 79, 1250–1259 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Buchhave P et al. Cerebrospinal fluid levels of β-amyloid 1–42, but not of tau, are fully changed already 5 to 10 years before the onset of Alzheimer dementia. Arch. Gen. Psychiatry 69, 98–106 (2012). [DOI] [PubMed] [Google Scholar]
- 115.Samgard K et al. Cerebrospinal fluid total tau as a marker of Alzheimer’s disease intensity. Int. J. Geriatr. Psychiatry 25, 403–410 (2010). [DOI] [PubMed] [Google Scholar]
- 116.Mattsson-Carlgren N et al. Longitudinal plasma p-tau217 is increased in early stages of Alzheimer’s disease. Brain 143, 3234–3241 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Hansson O et al. Plasma phosphorylated tau181 and neurodegeneration in Alzheimer’s disease. Ann. Clin. Transl. Neurol 8, 259–265 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Moscoso A et al. Time course of phosphorylated-tau181 in blood across the Alzheimer’s disease spectrum. Brain 144, 325–339 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Portelius E et al. Ex vivo 18O-labeling mass spectrometry identifies a peripheral amyloid β clearance pathway. Mol. Neurodegener 12, 18 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Yanamandra K et al. Anti-tau antibody administration increases plasma tau in transgenic mice and patients with tauopathy. Sci. Transl. Med 9 eaal2029 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kuhlmann J et al. CSF Aβ1–42—an excellent but complicated Alzheimer’s biomarker—a route to standardisation. Clin. Chim. Acta 467, 27–33 (2017). [DOI] [PubMed] [Google Scholar]
- 122.Boulo S et al. First amyloid β1–42 certified reference material for re-calibrating commercial immunoassays. Alzheimers Dement. 16, 1493–1503 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Pannee J et al. The global Alzheimer’s Association round robin study on plasma amyloid β methods. Alzheimers Dement. 13, e12242 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Andreasson U et al. Commutability of the certified reference materials for the standardization of β-amyloid 1–42 assay in human cerebrospinal fluid: lessons for tau and β-amyloid 1–40 measurements. Clin. Chem. Lab. Med 56, 2058–2066 (2018). [DOI] [PubMed] [Google Scholar]
- 125.Hansson O et al. The Alzheimer’s Association international guidelines for handling of cerebrospinal fluid for routine clinical measurements of amyloid β and tau. Alzheimers Dement. 17, 1575–1582 (2021). [DOI] [PubMed] [Google Scholar]
- 126.Rozga M, Bittner T, Batrla R & Karl J Preanalytical sample handling recommendations for Alzheimer’s disease plasma biomarkers. Alzheimers Dement. 11, 291–300 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Verberk IMW et al. Characterization of pre-analytical sample handling effects on a panel of Alzheimer’s disease-related blood-based biomarkers: results from the Standardization of Alzheimer’s Blood Biomarkers (SABB) working group. Alzheimers Dement. 18, 1484–1497 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Winston CN et al. Evaluation of blood-based plasma biomarkers as potential markers of amyloid burden in preclinical Alzheimer’s disease. J. Alzheimers Dis 92, 95–107 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Leuzy A et al. Robustness of CSF Aβ42/40 and Aβ42/p-tau181 measured using fully automated immunoassays to detect AD-related outcomes. Alzheimers Dement. 10.1002/alz.12897 (2023). [DOI] [PubMed] [Google Scholar]
- 130.Benedet AL et al. The accuracy and robustness of plasma biomarker models for amyloid PET positivity. Alzheimers Res. Ther 14, 26 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Hampel H et al. Developing the ATX(N) classification for use across the Alzheimer disease continuum. Nat. Rev. Neurol 17, 580–589 (2021). [DOI] [PubMed] [Google Scholar]
- 132.Simren J et al. Establishment of reference values for plasma neurofilament light based on healthy individuals aged 5–90 years. Brain Commun. 4, fcac174 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Syrjanen JA et al. Associations of amyloid and neurodegeneration plasma biomarkers with comorbidities. Alzheimers Dement. 18, 1128–1140 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Pichet Binette A et al. Confounding factors of Alzheimer’s disease plasma biomarkers and their impact on clinical performance. Alzheimers Dement. 10.1002/alz.12787 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Sullivan GM & Feinn R Using effect size—or why the P value is not enough. J. Grad. Med. Educ 4, 279–282 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Lim E, Miyamura J & Chen JJ Racial/ethnic-specific reference intervals for common laboratory tests: a comparison among Asians, Blacks, Hispanics, and white. Hawaii J. Med. Public Health 74, 302–310 (2015). [PMC free article] [PubMed] [Google Scholar]
- 137.Brickman AM et al. Plasma p-tau181, p-tau217, and other blood-based Alzheimer’s disease biomarkers in a multi-ethnic, community study. Alzheimers Dement. 17, 1353–1364 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Windon C et al. Comparison of plasma and CSF biomarkers across ethnoracial groups in the ADNI. Alzheimers Dement. 14, e12315 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Hu Y et al. Assessment of a plasma amyloid probability score to estimate amyloid positron emission tomography findings among adults with cognitive impairment. JAMA Netw. Open 5, e228392 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]