

Abstract
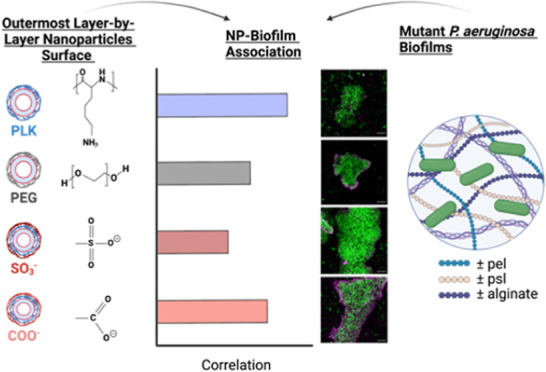
Pseudomonas aeruginosa biofilms comprise three main polysaccharides: alginate, psl, and pel, which all imbue tolerance against exogenous antimicrobials. Nanoparticles (NPs) are an exciting new strategy to overcome the biofilm matrix for therapeutic delivery applications; however, zero existing FDA approvals for biofilm-specific NP formulations can be attributed to the complex interplay of physiochemical forces at the biofilm-NP interface. Here, we leverage a set of inducible, polysaccharide-specific, expressing isogenic P. aeruginosa mutants coupled with an assembled layer-by-layer NP (LbL NP) panel to characterize biofilm-NP interactions. When investigating these interactions using confocal microscopy, alginate-layered NPs associated more than dextran-sulfate-layered NPs with biofilms that had increased alginate production, including biofilms produced by mucoid P. aeruginosa isolates from people with cystic fibrosis. These differences were further confirmed in LbL NPs layered with polysaccharide- or hydrocarbon-based polymers with pendent carboxylate or sulfate functional groups. These data suggest carboxylated NP surfaces have enhanced interactions specifically with mucoid biofilms as compared to sulfated surfaces and lay the foundation for their inclusion as a design element for increasing biofilm-NP interactions and efficacious drug delivery.
Keywords: cystic fibrosis, bionano, surface interaction, polysaccharide, layer-by-layer
Introduction
Bacterial infectious diseases represent an increasingly urgent public threat. Bacterial infections affect 17 million people, cause approximately 550,000 deaths, and cost the United States healthcare system billions of dollars each year.1 Furthermore, estimates from the Center for Disease Control and the National Institute of Health indicate that 65–80% of bacterial infections are biofilm-associated.2 Biofilms are sessile, extracellular matrix-enclosed microbial masses that adhere to biological or nonbiological surfaces.3 Microbes produce a matrix, termed the extracellular polymeric substance (EPS), which may account for up to 90% of the dry weight of a biofilm.4 The EPS permits microbes to evade host defenses, tolerate external stresses, and withstand 100- to 1000-fold higher concentrations of antibiotics, resulting in recurrent and chronic infections.5−7 Targeting and overcoming the EPS is a critical aspect of new therapeutic solutions to combat antibiotic recalcitrance in biofilm-based infections.
Biofilm-based infections have been largely studied using the model Gram-negative opportunistic pathogen model microbe Pseudomonas aeruginosa because of its ubiquitous role in chronic infections throughout the body. The dense EPS matrix produced by P. aeruginosa acts as a barrier and is composed primarily of proteins, lipids, extracellular DNA (eDNA), and polysaccharides in the form of a water-swollen network. These components attenuate antibiotic penetration due to their mechanical and physicochemical properties, including intermolecular forces of attraction such as electrostatics, hydrogen bonding, and van der Waals hydrophobic forces.8−10 The three main EPS polysaccharides produced by P. aeruginosa include alginate, psl, and pel. Alginate overproduction by mucoid P. aeruginosa has been well-characterized,11 particularly in the airways of people with cystic fibrosis (CF),12 and increasing evidence points to pivotal and distinct roles that both psl13,14 and pel15,16 play in the biofilm structure and protection against antimicrobials.17 Clinical samples of sputum from people colonized with P. aeruginosa have varied polysaccharide compositions,18 indicating the potential for more directed therapeutic approaches depending on biofilm matrix composition. Ultimately, strategies aimed at eradicating biofilms via enhanced antibiotic efficacy must be designed with these three essential polysaccharides in mind.
Nanoparticles (NPs) have recently been established as an exciting new approach to treating biofilm infections.19 NPs combine controllable sizes, high loading capabilities, and nonspecific accumulation in vivo to deliver increased local concentrations of therapeutics throughout biofilms.20 Indeed, numerous studies have reported successful encapsulation of various classes of antibiotics and their subsequent enhanced activity,21−28 and there are FDA-approved NP-encapsulating antimicrobial formulations such as amphotericin B liposomes for injection29 and amikacin liposome inhalation suspension.30 However, these therapies are simply liposome-encapsulated drugs, which primarily reduce off-target toxicity when delivered systemically, as opposed to increasing cargo efficacy through targeted delivery mechanisms. Consequently, there is no therapy currently on the market to combat P. aeruginosa-based biofilm infections due to the limited benefits compared to traditional treatments. For that reason, next-generation antimicrobial NP vehicles must include additional functionality, such as surface modifications, to increase the level of biofilm-NP association.
Screening polysaccharide-NP interactions remains an underdeveloped avenue with immense potential. However, synthesizing a panel of NPs with unique, high-density surface functionalities remains difficult through traditional chemical strategies. The platform layer-by-layer (LbL) electrostatic assembly technique overcomes this limitation and affords a modular, robust approach to constructing chemically diverse NPs.31−33 Charged colloidal templates are sequentially incubated with oppositely charged polyions, facilitating the formation of a uniform particle with a distinct outermost surface. Testing families of LbL NPs can illuminate previously unknown interactions imbued by distinct charged functional groups.34 Here, we screened NP interactions against biofilms composed of each of the three canonical polysaccharides using confocal microscopy and quantified biofilm-NP co-occurrence as a metric of interaction. Using these data, we established a general strategy for designing polysaccharide-specific associating NPs with the goal of enhancing targeted drug delivery and efficacy.
Materials and Methods
Materials and Reagents
All materials were purchased from Millipore-Sigma (Burlington, MA) unless otherwise specified. 18 kDa molecular weight (MW) poly-l-lysine (PLK) hydrochloric acid was purchased from Alamanda Polymers (Huntsville, AL). Very low-viscosity sodium alginate, with a MW < 75 kDa, was purchased from NovaMatrix (Sandvika, Norway). 6–15 kDa MW dextran sulfate (DXS) sodium salt was purchased from Millipore-Sigma. 10–20 kDa MW ∼ 20% COOH carboxymethyl-dextran sodium salt was purchased from Biosynth (San Diego, CA). 2–5 kDa MW poly(vinyl sulfonic acid, sodium salt) and 8 kDa MW polyacrylic acid (PAA) were both purchased from Millipore-Sigma.
Lipids and plant-based cholesterol were purchased from Avanti Polar Lipids, Inc. (Birmingham, AL). The chloroform used to resuspend was purchased from Macron Fine Chemicals (Radnor, PA). Cholesterol was resuspended in a 50 mg/mL chloroform solution. 1,2-distearoyl-sn-glycero-3-phosphocholine (18:0 DSPC), 1,2-distearoyl-sn-glycero-3-phospho-(1′-rac-glycerol) (18:0 DPSG), 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (ammonium salt) (18:0 PEG2000 PE), and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-(Cyanine 5) (18:0 Cy5 PE), were resuspended in a 25 mg/mL chloroform solution, 25 mg/mL solution of 65:35 vol/vol chloroform/methanol solution, 10 mg/mL chloroform solution, or a 1 mg/mL chloroform solution, respectively. Membranes used for liposome extrusion were purchased from GE Healthcare (Chicago, IL), and tangential flow filtration (TFF) hollow modules were purchased from Repligen (Waltham, MA). Teflon-coated tubing, sizes 13 and 16, used for filtration, was purchased from Saint-Gobain (Waltham, MA).
Glycerol was purchased from VWR Chemicals BDH. The nutrient agar used for making Luria–Bertani (LB) agar plates was purchased from BD Difco. 24-well glass bottom SensoPlates were purchased from Thomas Scientific (Swedesboro, NJ), and SYTO9 dye for staining was purchased from Invitrogen (Waltham, MA).
Nanoparticle Formulation
DSPC, DSPG, and cholesterol were added into a 50 mL round-bottom flask at a 1:1:1 mol/mol/mol ratio. Dried thin films were created by heating the round-bottom at 50 °C, spinning, and pumping the pressure down to <25 mTorr for at least 1 h using a rotary evaporator system (Buchi). For NPs used in confocal microscopy, 0.4 mol % DSPE-Cy5 lipid was added to replace DSPC, resulting in a 32.9:33.3:33.4:0.4 DSPC/DSPG/Chol/DSPE-Cy5 molar ratio.
After the evaporation of all organic solvents, the round-bottomed flask was partially submerged in a sonicator bath (Branson 1800 Ultrasonic Bath) filled with 65 °C water, and the thin film was resuspended at 1 mg/mL using fresh 65 °C milli-Q water while sonicating. The resulting colloidal suspension was extruded at 65 °C by using an Averin LipsosFast LF-50 liposome extruder. Liposomes were first passed three times through a 200 nm filter, then three times through a 100 nm filter, and finally at least once through a 50 nm filter.
Nanoparticle Layering and Purification
NPs were layered by pipetting, while sonicating, equal volumes of a colloidal template (e.g., liposome or layered liposome) into a solution of oppositely charged polymer in an Eppendorf tube for 3–5 s at a predetermined ratio. This ratio was based on titrations of increasing polymer concentrations to determine which causes a minimal size difference, defined as a PDI <0.3, and a zeta potential >|30 mV|, all measured using dynamic light scattering (Malvern). All polymers were dissolved at 10 mg/mL in milli-Q water and then further diluted using 50 mM HEPES and 40 mM NaCl, as this optimal layering solution was taken from previous literature.35 The PLK polycation optimal ratio was found to be 0.25–0.33:1 wt/wt polymer/NP, and the remaining polyanion ratios were anywhere from 0.25: to 1:1 wt/wt polymer/NP.
NPs were immediately purified via the TFF method using the KrosFlo II (Spectrum Laboratories) system, as previously described.36 Porous 100 kDa membranes were washed with milli-Q water prior to use. Crude NP solutions were passed through either microKros filters (if the solution volume was less than 8 mL total) at 13 mL/min or midiKros filters at 70 mL/min. NPs were washed with at least 5 times the volume of NP solution, as measured by the permeate mass. For layering PLK, membranes were coated with a closed-loop wash of 0.1 mg/mL PLK for 5 min prior to purification, since the anionic, modified poly(ether sulfone) membrane could disrupt the positive PLK layering. After purification, membranes were stored in 70% ethanol to maintain integrity and sterility.
Bacteria Culture
PA14 and clinical isolates of mucoid P. aeruginosa were kindly donated by the Yonker Lab (Mass General Brigham IBC: 2011B000789). All other bacterial strains used, as described in Table S1, were kindly donated by the Lory Lab. Mueller–Hinton broth supplemented with 25 μg/mL calcium chloride and 12.5 μg/mL magnesium chloride as cations (CAMHB) was used as the primary medium, except for the clinical isolates, which were all grown in standard LB broth.37 20% frozen glycerol stocks were made for all bacteria, streaked out onto LB agar plates, and single colonies were picked and incubated in CAMHB at 37 °C while shaking at 225 rpm. Subcultures were taken after overnight incubation, and all assays used normalized bacteria that were growing during the mid-log phase (4–6 h of growth) unless otherwise stated. For any bacterial washing, suspensions were pelleted at 6000 rcf for 5 min.
Crystal Violet Staining
The procedure was adapted from O’Toole with slight modifications.38 Bacteria were normalized to 0.01 OD600, and 100 μL were plated in each well of a 96-well plate. For determining the role inducer concentration plays in biomass production, 10 μL of 10-fold concentrated inducer solution, either arabinose or isopropyl β-d-1-thiogalactopyranoside, was spiked into the media before growth in the 96-well plate. The plate was incubated at 37 °C in a humid environment for 24 h. The plate was vigorously washed three times with water and patted dry, and then 125 μL of a 0.1% crystal violet solution was added to each well. The plate was stained for 15 min at room temperature in the dark and then washed 5 more times with water. The plate was blotted dry and then air-dried for at least 24 h at room temperature in the dark. To solubilize, 125 μL of 30% glacial acetic acid was added to each well and incubated for 15 min in the dark at room temperature, and 100 μL of the 125 μL was transferred to a fresh polypropylene plate. Biomass was recorded as the absorbance measured at 570 nm.
Confocal Microscopy Imaging
Biofilms were grown by plating 1 mL of OD600 0.02 bacteria in CAMHB with or without arabinose in each well of a 24-well glass bottom plate. Wells were statically incubated for 48 h in a humid chamber. Biofilms were washed three times by adding 1 mL of medium and then removing 1 mL of medium to ensure that the biofilm remained hydrated. At the final washing step, the medium was removed so that 0.9 mL of medium remained and topped with 100 μL of 50 μg/mL normalized NPs. NPs were incubated statically at 37 °C for 4 h in a humid chamber. Wells were again washed three times, leaving 0.9 mL of medium in the final wash to top up with 100 μL of 20 μM SYTO9 dye. Dye was incubated statically at room temperature for 30 min in the dark, washed three more times, and ended with 1 mL of medium at the final step before imaging.
Image acquisition was performed by using a confocal laser scanning microscope (800, Zeiss) with a 63× oil immersion magnification lens and a step size of 0.5 μm. SYTO9 was read at an excitation of 488 nm and emission of 510 nm, whereas the Cy5 dye was read at an excitation of 633 nm and emission of 670 nm. Image LUTs were adjusted linearly in order to improve the image contrast using FIJI or ZEN software.
Pearson’s coefficient was calculated using the BiofilmQ software,39 where cells were segmented in both fluorescence channels of each image. Pearson’s correlation coefficient was measured in fluorescence channel 1 with fluorescence channel 2. All calculations were global, with a range of 20. Intensity plot profiles were calculated using FIJI software, and plot profile analysis was performed on five random rectangles of equal height through the length of the representative biofilm image in Figure 4. Image LUTs were adjusted linearly in order to improve image contrast using FIJI or ZEN software.
Figure 4.

Confocal microscopy uncovers differences in the association of ALG, DXS, PEG, and PLK NP with mucA22 biofilms. Biofilms produced by mucA22 (green) are incubated with NPs (magenta) for 4 h, washed, and z-stack images are taken using a confocal microscope at 63× magnification. Images show the maximum intensity throughout the entire z-stack of the biofilm, edited with ZEN software. Scale bar = 20 μm.
Biofilm CFU Enumeration and MBEC Determination
Biofilms were grown on the minimum biofilm eradication concentration (MBEC) assay kit (previously known as the Calgary Biofilm Device),40 as described by the manufacturer protocol. Briefly, 150 μL of log-phase bacteria in CAMHB normalized to OD600 0.02 were inoculated in each well and incubated for 48 h in a humid environment while shaking at 110 rpm. After 48 h, the biofilms were equilibrated for 30 min in a new 96-well plate with 200 μL of phosphate-buffered saline (PBS) in each well, and then transferred to a sonicator bath and sonicated on high for 30 min. Serial dilutions from the removed biofilms were plated onto LB agar plates and counted.
For MBEC determination, biofilms were similarly grown on the MBEC assay kit, but for 24 h instead of 48 h. After growth, biofilms were washed by placing the lid in a new 96-well plate with PBS for 10 s, and then the lid containing the biofilms was introduced to a challenge plate in which each well had varied concentrations of NP, or free PLK, in CAMHB. After a 16 h incubation with NPs, biofilms were equilibrated for 30 min in a new 96-well plate with 200 μL of CAMHB and sonicated for another 30 min. The peg lid was replaced with a traditional plastic 96-well plate lid, and bacteria were allowed to grow in a static, humid 37 °C container for 16–18 h. OD600 measurements were taken, and the MBEC was determined via the concentration at which bacteria did not have measurable turbidity.
Statistics
All statistics used are described in each figure caption and analyzed using GraphPad Prism v8, using either a student’s t-test or a one-way analysis of variance (ANOVA) with Tukey’s correction for multiple comparisons. Outliers are calculated by finding the interquartile range (IQR) of a given dataset and checking if a data point lies outside 1.5 * IQR + third quartile or 1.5 * IQR – first quartile.
Results
Inducible Polysaccharide-Expressing Mutants Produce Varied Biofilm Biomass
To investigate the role of each of the three canonical polysaccharides (alginate, pel, and psl) that plays in NP-biofilm interactions, we leveraged six isogenic PAO1 strains (Table S1). These strains include one that lacks production of all three polysaccharides (ΔΔΔ), a strain that overproduces alginate and is characterized as mucoid (mucA22),41,42 one that nonspecifically overproduces all three polysaccharides (ΔwspF),43 and strains that inducibly produce a single polysaccharide (ΔΔpsl, ΔΔpel, or ΔΔalg). The ΔΔpsl, ΔΔpel, and ΔΔalg strains were assayed to determine the proper concentration of inducer that maximized biofilm biomass production, and this concentration remained constant throughout subsequent experiments (Figure S1). Relative biofilm biomass production after 24 h was measured for each mutant isogenic strain (Figure 1). Note that while subsequent experiments use 48 h old biofilms instead of 24 h old biofilms, previous seminal studies using similar mutants grown in flow cells have shown analogous results for biofilms grown for up to 5 days.44−46
Figure 1.

Crystal violet staining of biofilms grown for 24 h. N = 3 biological replicates with at least 8 technical replicates in each, where bars and error bars are averages and standard deviations, respectively.
As expected, the ΔΔΔ mutant had low biomass production, as this strain cannot make any of the three main polysaccharides and does not attach to the plastic surface.44 The converse was also true in that ΔwspF had the highest biomass production. Furthermore, in agreement with previous literature,44 the ΔΔpsl strain had higher relative biomass as compared to ΔΔpel due to a deficiency in ΔΔpel attachment. Finally, there was an increase in mucA22 biomass production as compared to the wild-type PAO1. While originally hypothesized that the mucA22 mutant would have a statistically larger relative biomass as compared to PAO1, previous studies have shown that alginate does not have a significant role in the biofilm matrix at early stages (i.e., 24 h) of development.45,46
Assembling a Family of Distinct Nanoparticle Surface Chemistries
In order to probe NP interactions with the group of isogenic PAO1 mutants, a panel of 4 distinct outer surface chemistry NPs was synthesized and assembled via the electrostatic LbL assembly method (Figure 2A). The panel included NPs with outermost surfaces with different chemical compositions composed of homopolypeptides, naturally occurring polysaccharides, or poly(ethylene) glycol (PEG). Depending on their outermost surface, NPs were either: (1) cationic (PLK); (2) anionic [sodium alginate (ALG), DXS]; or (3) neutral (liposomes assembled with 5 mol % lipid functionalized PEG). All of the layered NPs had comparable hydrodynamic diameters ranging from 141 to 206 nm and distinct zeta potentials, which are used as a proxy for NP surface charge (Figure 2B). The PEG-functionalized liposome had a smaller hydrodynamic diameter (80 nm) than the layered formulations, but the relative size difference from ∼100 to ∼200 nm in PEGylated and carboxylated NPs was not previously shown to effect diffusivity, and therefore we believe in association capabilities, through biofilms produced by two different Pseudomonas species.47,48 All of the assembled NPs also had polydispersity indices less than 0.3 (Figure S2), which indicates their relative monodispersity. Note that the larger hydrodynamic diameter of the alginate-layered NP is most likely due to the increased swelling of this highly hydrated biopolymer.49
Figure 2.
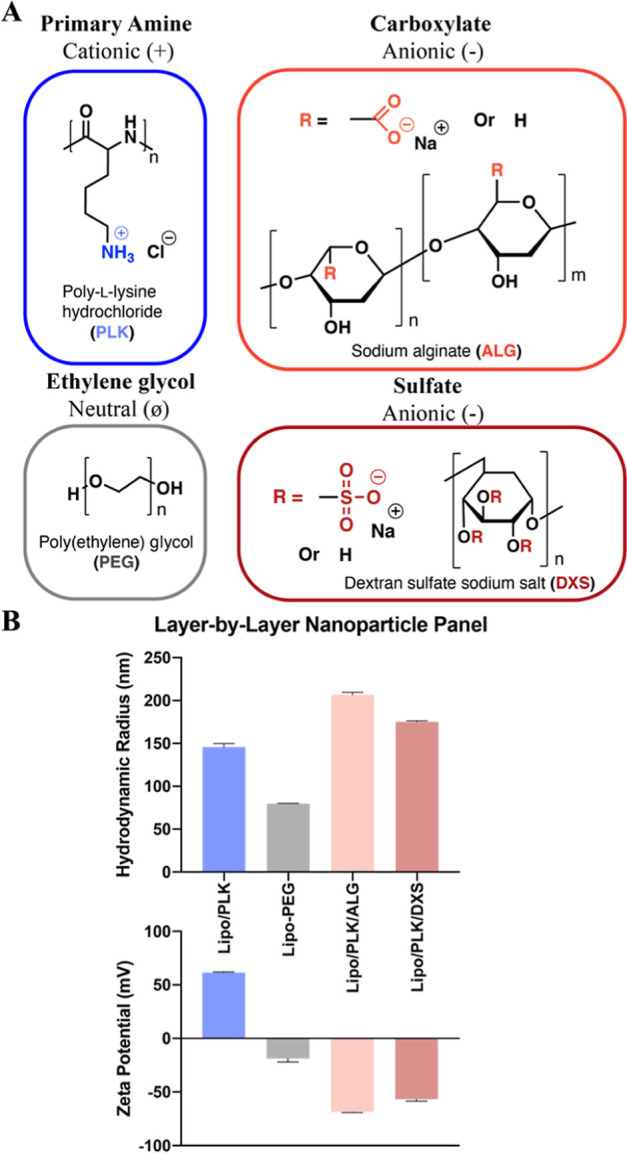
Assembled panel of LbL NPs spans a diverse outer layer chemical range. (A) Chemical structures of each polymer used. (B) Hydrodynamic diameter and zeta potential measurements of 4 distinct surface chemistry NPs. N = 3 technical replicates for NP physiochemical characterization, where bars and error bars are averages and standard deviations, respectively.
We use PLK NPs as a positive control for interactions, as the positively charged surface has already been reported to interact with high affinity to the biofilm matrix due to electrostatic interactions with anionic biofilm components (alginate, eDNA, microbial membranes, etc.).50,51 However, positively charged NPs are toxic52 and rapidly sequestered from the bloodstream53 when systemically delivered in vivo, thereby disadvantaging their use in the clinic, which is paramount when developing new therapeutic strategies.
Microscopy Reveals Enhanced Biofilm-NP Association of PLK and ALG NPs
To more accurately discern differences in local biofilm-NP interactions, we used confocal microscopy to illuminate the discrete NP associations with each biofilm. We excluded imaging biofilm-NP interactions with both the ΔΔΔ and ΔwspF mutants because ΔΔΔ does not attach to the glass bottom in sufficient quantities to provide reliable measurements. ΔwspF does not differentially produce any singular component (i.e., alginate, psl, or pel), so conclusions drawn from microscopy data would be primarily driven by biomass differences and not polysaccharide composition. Bacteria were grown for 48 h and all strains had similar CFU/mL when grown as 48 h old biofilms on the MBEC assay kit (Figure S3). After this 48 h growth, NPs were incubated for 4 h, and the biofilm-NP association was quantified using Pearson’s coefficient. Pearson’s coefficient measures the fractional colocalization of pixels and calculates the relative overlap of two fluorophores54 (i.e., biofilm and NP). It should be noted that the viability dye used, SYTO9, stains bacterial nuclei and DNA and not the polysaccharide components of the biofilm. Therefore, any pixel overlap is most likely between the microbes embedded within the biofilm and the Cyanine5(Cy5)-tagged liposomal core; however, for all Pearson’s coefficient calculations, the association of Cy5 and SYTO9 will be referred to as the association of biofilms and NPs.
As seen in Figure 3, most bacterial strains follow a general trend in which the positively charged PLK outer surface has the highest association with the biofilm, followed by ALG, PEG, and then DXS with the lowest association. The highest association of PLK with all strains imparted credence for the use of Pearson’s coefficient as a means of quantifying biofilm-NP interactions, though we are not suggesting the use of PLK as a delivery vehicle due to its aforementioned toxicity. Importantly, the MBEC of the Lipo/PLK formulation for all five strains was >50 μg/mL, higher than any concentration used throughout this work, and therefore should not have an impact on biofilm viability (Figure S4). We note that the addition of PA14 for these data was chosen because PA14 is the other well-characterized P. aeruginosa wild-type strain55 known to be more virulent56 and used for modeling airway infections. PA14 relies exclusively on pel for biofilm production15 as compared to PAO1, which tends to rely more on psl,57 providing a natural contrast between the two different biofilm architectures, though with confounding genetic differences.
Figure 3.

Biofilm-NP colocalization using Pearson’s coefficient reveals a strain-specific increase in ALG NP biofilm association as compared to DXS. Statistics are calculated within each bacterial strain. One-way ANOVAs with Tukey’s correction for multiple comparisons were used, where * means p < 0.05, ** means p < 0.01, and **** means P < 0.0001. N = 3 independent biofilms were tested, and N = 5 images were taken per biofilm, though co-occurrence data of 1.0 were considered artifacts of the image analysis software and were excluded. Note that the open circles in mucA22 are shown as representative images in Figure 4.
ALG interacted significantly more with biofilms produced by PA14 and the mucoid mucA22 compared to DXS (p = 0.0073 and 0.0074, respectively). Counterintuitively, liposomes assembled with PEGylated lipids did not show the lowest interactions, an initial hypothesized outcome of these studies because PEG is known to be antifouling.58−60 This can potentially be explained by the fact that while effective water coordination gives PEG some steric protection from protein adsorption, the polymer itself can also exhibit interactions with other molecules such as the polysaccharides in the matrix via hydrogen bonding, van der Waals, and other secondary interactions.61,62 When plotting the pixel intensity profiles as a function of the distance across the biofilm, both ALG and PEG NPs tended to accumulate at the biofilm edges, indicating potential NP association with surface matrix biopolymers, whereas DXS had a more uniform distribution throughout the film (Figure S5). Finally, PLK NPs showed the highest pixel intensity at the center of the biofilm.
Alginate and Carboxylated, LbL NPs Have Enhanced Mucoid Biofilm Interactions as Compared to DXS and Sulfated NPs
In order to further validate the increased association of biofilms with our alginate-layered NPs, biofilms were grown from clinically characterized mucoid P. aeruginosa isolates from the airways of people with CF, and biofilm biomass was quantified for each strain (Figure S6). Similar to previous experiments, biofilms were grown for 48 h, and either ALG or DXS NPs were incubated for 4 h. This further subset of NPs was chosen for their in vivo biocompatibility and natural comparison of negatively charged functional groups. Importantly, in four of the five ex vivo clinical strains, there was a statistically significant enhancement of ALG NP colocalization with the biofilms as compared to DXS NPs (Figure 5). For clinical strain 4277, which had a similar Pearson’s coefficient for both NP formulations, there was also a relatively low association for both NPs, indicating low association overall.
Figure 5.

Alginate NPs associated with clinically isolated CF mucoid P. aeruginosa-produced biofilms. (A) Pearson’s coefficient was calculated for ALG and DXS NPs interacting with biofilms produced from isolated mucoid P. aeruginosa strains. N = 3 independent biofilms were tested, and at least N = 5 images were taken per biofilm, though co-occurrence data of 1.0 were considered artifacts of the image analysis software and were excluded. Colored asterisks below the significance lines denote outlier data points. Statistics are calculated using an unpaired student’s t-test, where * means p < 0.05, ** means p < 0.01, and **** means P < 0.0001. Note that the open circles are images shown in the next panel. (B) Confocal imaging of biofilms formed by strain 4305, portrayed as the maximum intensity throughout the entire z-stack of the biofilm, edited with ZEN software. Scale bar = 20 μm.
Identical Polymer Backbone Chemistry Confirms Carboxylate-Enhanced Biofilm Interactions
Intrigued by the surface chemistry differences between ALG and DXS, we sought to find a more similar carboxylated and sulfated polymer comparison since ALG and DXS have distinct backbone structures, incurring confounding variables. Therefore, we turned to two sets of polymers that have similar backbones: carboxymethyl dextran (CMD) and DXS, and PAA and poly(vinyl) sulfate (PVS) (Figure 6A). Strikingly, when looking at Pearson’s coefficients of LbL NPs assembled with identical polymer backbones but different functional groups, the same trends of increased biofilm association with carboxylated surfaces are observed (Figure 6B). While there is no statistical significance when comparing CMD and DXS, there is a strong trend toward CMD having higher colocalization. These two polymers have the same dextran backbone, but they vary in their functional group density as determined by their manufacturers, making direct comparisons difficult because of dissimilar intermolecular forces of attraction, such as hydrogen bonding. Therefore, by comparing MW-matched, simple hydrocarbon backbone PAA and PVS polymers, we can isolate NP association differences uniquely due to the functional group. Indeed, when comparing NPs terminated with either PAA or PVS, significant differences exist between the associations of biofilms and carboxylate outer surfaces.
Figure 6.

Carboxylated NPs have an increased biofilm association compared to sulfated NPs. (A) Chemical structure of either the dextran or vinyl hydrocarbon backbone, showing functional group differences between CMD and DXS, as well as PAA and PVS. (B) Pearson’s coefficient with biofilms produced by mucA22 or PAO1 of CMD and DXS or PAA and PVS. Student’s t-test was used for each pair of polymer backbones, where ** means p < 0.01. N = 4 independent biofilms were tested with N = 4–5 images per biofilm, though colocalization data of 1.0 were considered artifacts of the image analysis software and were excluded.
Discussion
The delivery of high local concentrations of therapeutics throughout the biofilm matrix is vital in combating chronic biofilm-based infections. Positively charged antibiotics, such as tobramycin and other aminoglycosides, have been shown to be sequestered by negatively charged polysaccharides (i.e., alginate) and eDNA in biofilms.63 Furthermore, increased alginate production leads to a higher MBEC of aminoglycosides as they become attenuated by the negative matrix.8,64 NPs can provide both a shield for encapsulated drugs, thereby potentially protecting them from sequestration, and an active targeting mechanism to target and retain therapeutics at the site of infection. Coupling these two modalities signifies a shift in treatment options from traditional antibiotics. Therefore, overcoming this matrix for enhanced cargo delivery requires knowledge about the interactions at the interface of the biofilm and NP that govern the ultimate fate of the carrier.
This is the first work that we are aware of that uses isogenic P. aeruginosa mutants to focus on precisely investigating what roles alginate, psl, and pel play in interacting with a diverse range of NP outer surface chemistries. By leveraging the electrostatic LbL assembly method, we simultaneously produced a library of distinct NP outer surface chemistries, generating uniform NPs that vary only in their outermost surface identity while retaining the same colloidal template. The NP library throughout this work spanned a diverse range of charges (positive, negative, and neutral), backbones (ethylene glycol, hydrocarbon, and polysaccharide), and functional group identities (carboxylates, sulfates, and amines). Therefore, we could draw distinct conclusions based on matching specific polysaccharide and NP surface chemistry interactions with fewer confounding variables as compared to traditional screens.
Pearson’s coefficient measurements of biofilm-NP co-occurrence illuminated key differences in how distinct NP surfaces interact with the matrix. For example, the highest biofilm association occurred when using a positively charged outermost surface, PLK, which acted as a control to ensure the validity of these measurements. However, PLK causes mammalian cell toxicity53 and cannot be used as systemic delivery vehicles in the clinic, requiring a new vehicle design to maximize biofilm interactions and actively retain the nanocarrier at the site of infection. Further analysis from the Pearson’s coefficient data revealed that the difference in colocalization magnitude of ALG and DXS (Figure 3) trended with increased alginate production. Calculated differences of ALG and DXS Pearson’s coefficient in strains that do not produce alginate (ΔΔpel and ΔΔpsl, −0.01 ± 0.21 and 0.03 ± 0.16, respectively), to normal alginate production (PAO1, 0.07 ± 0.21), to the overproducing alginate mucoid mutant (mucA22, 0.13 ± 0.10) uncover this relationship. Significant differences in the association of ALG and DXS with biofilms produced by four out of five of the clinically characterized mucoid isolate strains strengthen these results (Figure 5).
The conclusion that carboxylated NPs associate with mucoid biofilms at early time points more than sulfated NPs is strengthened as NPs coated with polymers containing a simple hydrocarbon backbone that varies only in side-chain functionality (i.e., carboxylate or sulfate) showed statistical differences in association (Figure 6B). This experiment removed backbone molecular interactions present in the dextran backbone as a potential variable influencing biofilm-NP interactions since NPs were physiochemically similar (Figure S7), highlighting only the role of the functional group. One explanation is that the outermost alginate layer of the ALG NPs can more easily be incorporated into biofilms and shed from the NP surface, exposing a now cationic PLK surface, which would have an increased co-occurrence. However, if this were the case, ALG should have enhanced associations across all strains tested, as alginate can be incorporated into all these biofilms, but this is not the case for ΔΔpel and ΔΔpsl. Also, the deconstruction of the LbL NPs occurs via shedding polyplexes of cationic and anionic polymers, not discrete layers from the NP surface.65 We hypothesize that the increased hydrogen bonding capabilities of the ALG NPs drive the molecular interactions responsible for the increased interactions, but further studies are required to reach definitive conclusions.
Conclusions
These results are significant in designing NP carriers that can associate precisely with mucoid biofilms, which are correlated to poorer clinical outcomes,66−68 and potentially deliver high local concentrations of therapeutics. The FDA gave sodium alginate a generally regarded as safe designation, and ALG was statistically insignificant as compared to PLK when comparing colocalization with biofilms produced by four of the five laboratory strains tested, including the mucoid mucA22 biofilm. These results suggest that the rational design of NP surface functionalization impacts accumulation within the EPS matrix62,69 and potentially greater retention. More work quantifying biofilm viability using NPs encapsulating antibiotics that are sequestered by the matrix, such as aminoglycosides, is required to further validate the feasibility of these carriers. Ultimately, these results lay the foundation for designing carboxylate-containing polyelectrolyte NP-based delivery therapeutics with enhanced biofilm interactions to eradicate mucoid biofilm infections.
Acknowledgments
We would like to thank Simone Douglas-Green for her thoughtful review and comments on this manuscript. E.D.-Y. acknowledges the National Research Foundation, Prime Minister’s Office, Singapore, for support under its Campus for Research Excellence and Technological Enterprise (CREATE) programme. E.D.-Y. also acknowledges the Cystic Fibrosis Foundation (004527H222) for funding part of this work.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsami.3c18656.
Inducer-related biomass production, NP polydispersity, biofilm CFU/mL enumeration, MBEC assay of Lipo/PLK, pixel profile plots of NPs within biofilms, crystal violet biomass staining of clinical isolate strains, physiochemical characterization of similar backbone polymer LbL NPs, and relative polysaccharide production of the mutants (PDF)
The authors declare the following competing financial interest(s): P.T.H. is a co-founder and member of the board of LayerBio, a member of the Scientific Advisory Board of Moderna, and a member of the Board of Alector, Advanced Chemotherapy Technologies, and Burroughs-Wellcome Fund. All other authors report no competing interest.
Supplementary Material
References
- Wolcott R. D.; Rhoads D. D.; Bennett M. E.; Wolcott B. M.; Gogokhia L.; Costerton J. W.; Dowd S. E. Chronic Wounds and the Medical Biofilm Paradigm. J. Wound Care 2010, 19 (2), 45–53. 10.12968/jowc.2010.19.2.46966. [DOI] [PubMed] [Google Scholar]
- Wolcott R.; Dowd S. The Role of Biofilms: Are we Hitting the Right Target?. Plast. Reconstr. Surg. 2011, 127, 28S–35S. 10.1097/PRS.0b013e3181fca244. [DOI] [PubMed] [Google Scholar]
- Sauer K.; Stoodley P.; Goeres D. M.; Hall-Stoodley L.; Burmølle M.; Stewart P. S.; Bjarnsholt T. The Biofilm Life Cycle: Expanding the Conceptual Model of Biofilm Formation. Nat. Rev. Microbiol. 2022, 20 (10), 608–620. 10.1038/s41579-022-00767-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flemming H.-C.; Neu T. R.; Wozniak D. J. The EPS matrix: the “House of Biofilm Cells. J. Bacteriol. 2007, 189 (22), 7945–7947. 10.1128/JB.00858-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flemming H.-C.; van Hullebusch E. D.; Neu T. R.; Nielsen P. H.; Seviour T.; Stoodley P.; Wingender J.; Wuertz S. The Biofilm Matrix: Multitasking in a Shared Space. Nat. Rev. Microbiol. 2023, 21 (2), 70–86. 10.1038/s41579-022-00791-0. [DOI] [PubMed] [Google Scholar]
- Koo H.; Allan R. N.; Howlin R. P.; Stoodley P.; Hall-Stoodley L. Targeting Microbial Biofilms: Current and Prospective Therapeutic Strategies. Nat. Rev. Microbiol. 2017, 15 (12), 740–755. 10.1038/nrmicro.2017.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arciola C. R.; Campoccia D.; Montanaro L. Implant Infections: Adhesion, Biofilm Formation and Immune Evasion. Nat. Rev. Microbiol. 2018, 16 (7), 397–409. 10.1038/s41579-018-0019-y. [DOI] [PubMed] [Google Scholar]
- Tseng B. S.; Zhang W.; Harrison J. J.; Quach T. P.; Song J. L.; Penterman J.; Singh P. K.; Chopp D. L.; Packman A. I.; Parsek M. R. The Extracellular Matrix Protects Pseudomonas aeruginosa Biofilms by Limiting the Penetration of Tobramycin. Environ. Microbiol. 2013, 15 (10), 2865–2878. 10.1111/1462-2920.12155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma D.; Misba L.; Khan A. U. Antibiotics versus Biofilm: An Emerging Battleground in Microbial Communities. Antimicrob. Resist. Infect. Control 2019, 8 (1), 76. 10.1186/s13756-019-0533-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uruén C.; Chopo-Escuin G.; Tommassen J.; Mainar-Jaime R. C.; Arenas J. Biofilms as Promoters of Bacterial Antibiotic Resistance and Tolerance. Antibiotics 2020, 10 (1), 3. 10.3390/antibiotics10010003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryder C.; Byrd M.; Wozniak D. J. Role of Polysaccharides in Pseudomonas aeruginosa Biofilm Development. Curr. Opin. Microbiol. 2007, 10 (6), 644–648. 10.1016/j.mib.2007.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doggett R. G. Incidence of Mucoid Pseudomonas aeruginosa from Clinical Sources. Appl. Microbiol. 1969, 18 (5), 936–937. 10.1128/am.18.5.936-937.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billings N.; Ramirez Millan M.; Caldara M.; Rusconi R.; Tarasova Y.; Stocker R.; Ribbeck K. The Extracellular Matrix Component psl Provides Fast-Acting Antibiotic Defense in Pseudomonas aeruginosa Biofilms. PLoS Pathog. 2013, 9 (8), e1003526 10.1371/journal.ppat.1003526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S.; Liu X.; Liu H.; Zhang L.; Guo Y.; Yu S.; Wozniak D. J.; Ma L. Z. The Exopolysaccharide Psl-eDNA Interaction Enables the Formation of a Biofilm Skeleton in Pseudomonas aeruginosa. Environ. Microbiol. Rep. 2015, 7 (2), 330–340. 10.1111/1758-2229.12252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colvin K. M.; Gordon V. D.; Murakami K.; Borlee B. R.; Wozniak D. J.; Wong G. C.; Parsek M. R. The pel Polysaccharide can Serve a Structural and Protective Role in the Biofilm Matrix of Pseudomonas aeruginosa. PLoS Pathog. 2011, 7 (1), e1001264 10.1371/journal.ppat.1001264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jennings L. K.; Storek K. M.; Ledvina H. E.; Coulon C.; Marmont L. S.; Sadovskaya I.; Secor P. R.; Tseng B. S.; Scian M.; Filloux A.; Wozniak D. J.; Howell P. L.; Parsek M. R. Pel is a Cationic Exopolysaccharide that Cross-Links Extracellular DNA in the Pseudomonas aeruginosa Biofilm Matrix. Proc. Natl. Acad. Sci. U.S.A. 2015, 112 (36), 11353–11358. 10.1073/pnas.1503058112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulaz S.; Vitale S.; Quinn L.; Casey E. Nanoparticle-Biofilm Interactions: the Role of the EPS Matrix. Trends Microbiol. 2019, 27 (11), 915–926. 10.1016/j.tim.2019.07.004. [DOI] [PubMed] [Google Scholar]
- Jennings L. K.; Dreifus J. E.; Reichhardt C.; Storek K. M.; Secor P. R.; Wozniak D. J.; Hisert K. B.; Parsek M. R. Pseudomonas aeruginosa Aggregates in Cystic Fibrosis Sputum Produce Exopolysaccharides that Likely Impede Current Therapies. Cell Rep. 2021, 34 (8), 108782. 10.1016/j.celrep.2021.108782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makabenta J. M. V.; Nabawy A.; Li C.-H.; Schmidt-Malan S.; Patel R.; Rotello V. M. Nanomaterial-Based Therapeutics for Antibiotic-Resistant Bacterial Infections. Nat. Rev. Microbiol. 2021, 19 (1), 23–36. 10.1038/s41579-020-0420-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez Gomez A.; Hosseinidoust Z. Liposomes for Antibiotic Encapsulation and Delivery. ACS Infect. Dis. 2020, 6 (5), 896–908. 10.1021/acsinfecdis.9b00357. [DOI] [PubMed] [Google Scholar]
- Deiss-Yehiely E.; Cárcamo-Oyarce G.; Berger A. G.; Ribbeck K.; Hammond P. T. pH-Responsive, Charge-Reversing Layer-by-Layer Nanoparticle Surfaces Enhance Biofilm Penetration and Eradication. ACS Biomater. Sci. Eng. 2023, 9 (8), 4794–4804. 10.1021/acsbiomaterials.3c00481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meers P.; Neville M.; Malinin V.; Scotto A.; Sardaryan G.; Kurumunda R.; Mackinson C.; James G.; Fisher S.; Perkins W. Biofilm Penetration, Triggered Release and in vivo Activity of Inhaled Liposomal Amikacin in Chronic Pseudomonas aeruginosa Lung Infections. J. Antimicrob. Chemother. 2008, 61 (4), 859–868. 10.1093/jac/dkn059. [DOI] [PubMed] [Google Scholar]
- Pushparaj Selvadoss P.; Nellore J.; Balaraman Ravindrran M.; Sekar U.; Tippabathani J. Enhancement of Antimicrobial Activity by Liposomal Oleic Acid-Loaded Antibiotics for the Treatment of Multidrug-Resistant Pseudomonas aeruginosa. Artif. Cells Nanomed. Biotechnol. 2018, 46 (2), 268–273. 10.1080/21691401.2017.1307209. [DOI] [PubMed] [Google Scholar]
- Webb M. S.; Boman N. L.; Wiseman D. J.; Saxon D.; Sutton K.; Wong K. F.; Logan P.; Hope M. J. Antibacterial Efficacy Against an in vivo Salmonella typhimurium Infection Model and Pharmacokinetics of a Liposomal Ciprofloxacin Formulation. Antimicrob. Agents Chemother. 1998, 42 (1), 45–52. 10.1128/aac.42.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil J. S.; Devi V. K.; Devi K.; Sarasija S. A Novel Approach for Lung Delivery of Rifampicin-Loaded Liposomes in Dry Powder Form for the Treatment of Tuberculosis. Lung India 2015, 32 (4), 331. 10.4103/0970-2113.159559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alipour M.; Halwani M.; Omri A.; Suntres Z. E. Antimicrobial Effectiveness of Liposomal Polymyxin B Against Resistant Gram-Negative Bacterial Strains. Int. J. Pharm. 2008, 355 (1–2), 293–298. 10.1016/j.ijpharm.2007.11.035. [DOI] [PubMed] [Google Scholar]
- Yang Z.; Liu J.; Gao J.; Chen S.; Huang G. Chitosan Coated Vancomycin Hydrochloride Liposomes: Characterizations and Evaluation. Int. J. Pharm. 2015, 495 (1), 508–515. 10.1016/j.ijpharm.2015.08.085. [DOI] [PubMed] [Google Scholar]
- Alhajlan M.; Alhariri M.; Omri A. Efficacy and Safety of Liposomal Clarithromycin and its Effect on Pseudomonas aeruginosa Virulence Factors. Antimicrob. Agents Chemother. 2013, 57 (6), 2694–2704. 10.1128/AAC.00235-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hann I. M.; Prentice H. G. Lipid-Based Amphotericin B: a Review of the Last 10 Years of Use. Int. J. Antimicrob. Agents 2001, 17 (3), 161–169. 10.1016/S0924-8579(00)00341-1. [DOI] [PubMed] [Google Scholar]
- Shirley M. Amikacin Liposome Inhalation Suspension: A review in Mycobacterium avium complex Lung Disease. Drugs 2019, 79 (5), 555–562. 10.1007/s40265-019-01095-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correa S.; Dreaden E. C.; Gu L.; Hammond P. T. Engineering Nanolayered Particles for Modular Drug Delivery. J. Controlled Release 2016, 240, 364–386. 10.1016/j.jconrel.2016.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkekhia D.; Hammond P. T.; Shukla A. Layer-by-Layer Biomaterials for Drug Delivery. Annu. Rev. Biomed. Eng. 2020, 22 (1), 1–24. 10.1146/annurev-bioeng-060418-052350. [DOI] [PubMed] [Google Scholar]
- Decher G. Fuzzy Nanoassemblies: Toward Layered Polymeric Multicomposites. Science 1997, 277 (5330), 1232–1237. 10.1126/science.277.5330.1232. [DOI] [Google Scholar]
- Correa S.; Boehnke N.; Barberio A. E.; Deiss-Yehiely E.; Shi A.; Oberlton B.; Smith S. G.; Zervantonakis I.; Dreaden E. C.; Hammond P. T. Tuning Nanoparticle Interactions with Ovarian Cancer Through Layer-by-Layer Modification of Surface Chemistry. ACS Nano 2020, 14 (2), 2224–2237. 10.1021/acsnano.9b09213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correa S.; Boehnke N.; Deiss-Yehiely E.; Hammond P. T. Solution Conditions Tune and Optimize Loading of Therapeutic Polyelectrolytes into Layer-by-Layer Functionalized Liposomes. ACS Nano 2019, 13 (5), 5623–5634. 10.1021/acsnano.9b00792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correa S.; Choi K. Y.; Dreaden E. C.; Renggli K.; Shi A.; Gu L.; Shopsowitz K. E.; Quadir M. A.; Ben-Akiva E.; Hammond P. T. Highly Scalable, Closed-Loop Synthesis of Drug-Loaded, Layer-by-Layer Nanoparticles. Adv. Funct. Mater. 2016, 26, 991–1003. 10.1002/adfm.201504385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornforth D. M.; Diggle F. L.; Melvin J. A.; Bomberger J. M.; Whiteley M. Quantitative Framework for Model Evaluation in Microbiology Research Using Pseudomonas aeruginosa and Cystic Fibrosis Infection as a Test Case. mBio 2020, 11 (1), e03042-19 10.1128/mBio.03042-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Toole G. A. Microtiter Dish Biofilm Formation Assay. J. Visualized Exp. 2011, (47), 2437. 10.3791/2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann R.; Jeckel H.; Jelli E.; Singh P. K.; Vaidya S.; Bayer M.; Rode D. K. H.; Vidakovic L.; Díaz-Pascual F.; Fong J. C. N.; Dragoš A.; Lamprecht O.; Thöming J. G.; Netter N.; Häussler S.; Nadell C. D.; Sourjik V.; Kovács Á. T.; Yildiz F. H.; Drescher K. Quantitative Image Analysis of Microbial Communities with BiofilmQ. Nat. Microbiol. 2021, 6 (2), 151–156. 10.1038/s41564-020-00817-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceri H.; Olson M. E.; Stremick C.; Read R. R.; Morck D.; Buret A. The Calgary Biofilm Device: New Technology for Rapid Determination of Antibiotic Susceptibilities of Bacterial Biofilms. J. Clin. Microbiol. 1999, 37 (6), 1771–1776. 10.1128/JCM.37.6.1771-1776.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathee K.; Ciofu O.; Sternberg C.; Lindum P. W.; Campbell J. I. A.; Jensen P.; Johnsen A. H.; Givskov M.; Ohman D. E.; Søren M.; Høiby N.; Kharazmi A. Mucoid Conversion of Pseudomonas aeruginos by Hydrogen Peroxide: a Mechanism for Virulence Activation in the Cystic Fibrosis Lung. Microbiology 1999, 145 (6), 1349–1357. 10.1099/13500872-145-6-1349. [DOI] [PubMed] [Google Scholar]
- Pulcrano G.; Iula D. V.; Raia V.; Rossano F.; Catania M. R. Different Mutations in mucA Gene of Pseudomonas aeruginosa Mucoid Strains in Cystic Fibrosis Patients and Their Effect on algU Gene Expression. New Microbiol. 2012, 35 (3), 295–305. [PubMed] [Google Scholar]
- Hickman J. W.; Tifrea D. F.; Harwood C. S. A Chemosensory System that Regulates Biofilm Formation Through Modulation of Cyclic Diguanylate Levels. Proc. Natl. Acad. Sci. U.S.A. 2005, 102 (40), 14422–14427. 10.1073/pnas.0507170102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colvin K. M.; Irie Y.; Tart C. S.; Urbano R.; Whitney J. C.; Ryder C.; Howell P. L.; Wozniak D. J.; Parsek M. R. The pel and psl Polysaccharides Provide Pseudomonas aeruginosa Structural Redundancy Within the Biofilm Matrix. Environ. Microbiol. 2012, 14 (8), 1913–1928. 10.1111/j.1462-2920.2011.02657.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wozniak D. J.; Wyckoff T. J. O.; Starkey M.; Keyser R.; Azadi P.; O’Toole G. A.; Parsek M. R. Alginate is Not a Significant Component of the Extracellular Polysaccharide Matrix of PA14 and PAO1 Pseudomonas aeruginosa Biofilms. Proc. Natl. Acad. Sci. U.S.A. 2003, 100 (13), 7907–7912. 10.1073/pnas.1231792100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hentzer M.; Teitzel G. M.; Balzer G. J.; Heydorn A.; Molin S.; Givskov M.; Parsek M. R. Alginate Overproduction Affects Pseudomonas aeruginosa Biofilm Structure and Function. J. Bacteriol. 2001, 183 (18), 5395–5401. 10.1128/JB.183.18.5395-5401.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peulen T.-O.; Wilkinson K. J. Diffusion of Nanoparticles in a Biofilm. Environ. Sci. Technol. 2011, 45 (8), 3367–3373. 10.1021/es103450g. [DOI] [PubMed] [Google Scholar]
- Forier K.; Messiaen A.-S.; Raemdonck K.; Deschout H.; Rejman J.; De Baets F.; Nelis H.; De Smedt S. C.; Demeester J.; Coenye T.; Braeckmans K. Transport of Nanoparticles in Cystic Fibrosis Sputum and Bacterial Biofilms by Single-Particle Tracking Microscopy. Nanomedicine 2013, 8 (6), 935–949. 10.2217/nnm.12.129. [DOI] [PubMed] [Google Scholar]
- Lee K. Y.; Mooney D. J. Alginate: Properties and Biomedical Applications. Prog. Polym. Sci. 2012, 37 (1), 106–126. 10.1016/j.progpolymsci.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X.; Yeh Y.-C.; Giri K.; Mout R.; Landis R. F.; Prakash Y. S.; Rotello V. M. Control of Nanoparticle Penetration into Biofilms Through Surface Design. Chem. Commun. 2015, 51 (2), 282–285. 10.1039/C4CC07737G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benoit D. S. W.; Sims K. R.; Fraser D. Nanoparticles for Oral Biofilm Treatments. ACS Nano 2019, 13 (5), 4869–4875. 10.1021/acsnano.9b02816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudsen K. B.; Northeved H.; Kumar Ek P.; Permin A.; Gjetting T.; Andresen T. L.; Larsen S.; Wegener K. M.; Lykkesfeldt J.; Jantzen K.; Loft S.; Møller P.; Roursgaard M. In vivo Toxicity of Cationic Micelles and Liposomes. Nanomed. Nanotechnol. Biol. Med. 2015, 11 (2), 467–477. 10.1016/j.nano.2014.08.004. [DOI] [PubMed] [Google Scholar]
- Salvador-Morales C.; Zhang L.; Langer R.; Farokhzad O. C. Immunocompatibility Properties of Lipid-Polymer Hybrid Nanoparticles with Heterogeneous Surface Functional Groups. Biomaterials 2009, 30 (12), 2231–2240. 10.1016/j.biomaterials.2009.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adler J.; Parmryd I. Quantifying colocalization by correlation: The Pearson Correlation Coefficient is Superior to the Mander’s Overlap Coefficient. Cytometry Part A 2010, 77A (8), 733–742. 10.1002/cyto.a.20896. [DOI] [PubMed] [Google Scholar]
- Grace A.; Sahu R.; Owen D. R.; Dennis V. A. Pseudomonas aeruginosa Reference Strains PAO1 and PA14: A Genomic, Phenotypic, and Therapeutic review. Front. Microbiol. 2022, 13, 1023523. 10.3389/fmicb.2022.1023523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikkelsen H.; McMullan R.; Filloux A. The Pseudomonas aeruginosa Reference Strain PA14 Displays Increased Virulence Due to a Mutation in ladS. PLoS One 2011, 6 (12), e29113 10.1371/journal.pone.0029113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L.; Hu Y.; Liu Y.; Zhang J.; Ulstrup J.; Molin S. Distinct Roles of Extracellular Polymeric Substances in Pseudomonas aeruginosa Biofilm Development. Environ. Microbiol. 2011, 13 (7), 1705–1717. 10.1111/j.1462-2920.2011.02503.x. [DOI] [PubMed] [Google Scholar]
- Klibanov A. L.; Maruyama K.; Torchilin V. P.; Huang L. Amphipathic Polyethylene glycols Effectively Prolong the Circulation Time of Liposomes. FEBS Lett. 1990, 268 (1), 235–237. 10.1016/0014-5793(90)81016-H. [DOI] [PubMed] [Google Scholar]
- Gref R.; Domb A.; Quellec P.; Blunk T.; Müller R. H.; Verbavatz J. M.; Langer R. The Controlled Intravenous Delivery of Drugs using PEG-coated Sterically Stabilized Nanospheres. Adv. Drug Delivery Rev. 1995, 16 (2–3), 215–233. 10.1016/0169-409X(95)00026-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Cano C.; Carril M. Recent Developments in the Design of Non-Biofouling Coatings for Nanoparticles and Surfaces. Int. J. Mol. Sci. 2020, 21 (3), 1007. 10.3390/ijms21031007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Israelachvili J. The Different Faces of Poly(ethylene glycol). Proc. Natl. Acad. Sci. U.S.A. 1997, 94 (16), 8378–8379. 10.1073/pnas.94.16.8378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitale S.; Rampazzo E.; Hiebner D.; Devlin H.; Quinn L.; Prodi L.; Casey E. Interaction between Engineered Pluronic Silica Nanoparticles and Bacterial Biofilms: Elucidating the Role of Nanoparticle Surface Chemistry and EPS Matrix. ACS Appl. Mater. Interfaces 2022, 14 (30), 34502–34512. 10.1021/acsami.2c10347. [DOI] [PubMed] [Google Scholar]
- Nichols W. W.; Dorrington S. M.; Slack M. P.; Walmsley H. L. Inhibition of Tobramycin Diffusion by Binding to Alginate. Antimicrob. Agents Chemother. 1988, 32 (4), 518–523. 10.1128/AAC.32.4.518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco-Cabra N.; Movellan J.; Marradi M.; Gracia R.; Salvador C.; Dupin D.; Loinaz I.; Torrents E. Neutralization of Ionic Interactions by Dextran-Based Single-Chain Nanoparticles Improves Tobramycin Diffusion into a Mature Biofilm. npj Biofilms Microbiomes 2022, 8 (1), 52. 10.1038/s41522-022-00317-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barberio A. E.; Smith S. G.; Correa S.; Nguyen C.; Nhan B.; Melo M.; Tokatlian T.; Suh H.; Irvine D. J.; Hammond P. T. Cancer Cell Coating Nanoparticles for Optimal Tumor-Specific Cytokine Delivery. ACS Nano 2020, 14 (9), 11238–11253. 10.1021/acsnano.0c03109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langan K. M.; Kotsimbos T.; Peleg A. Y. Managing Pseudomonas aeruginosa Respiratory Infections in Cystic Fibrosis. Curr. Opin. Infect. Dis. 2015, 28 (6), 547–556. 10.1097/QCO.0000000000000217. [DOI] [PubMed] [Google Scholar]
- Li Z.; Kosorok M. R.; Farrell P. M.; Laxova A.; West S. E.; Green C. G.; Collins J.; Rock M. J.; Splaingard M. L. Longitudinal Development of Mucoid Pseudomonas aeruginosa Infection and Lung Disease Progression in Children with Cystic Fibrosis. JAMA 2005, 293 (5), 581–588. 10.1001/jama.293.5.581. [DOI] [PubMed] [Google Scholar]
- Henry R. L.; Mellis C. M.; Petrovic L. Mucoid Pseudomonas aeruginosa is a Marker of Poor Survival in Cystic Fibrosis. Pediatr. Pulmonology 1992, 12 (3), 158–161. 10.1002/ppul.1950120306. [DOI] [PubMed] [Google Scholar]
- Kondo A.; Higashitani K. Adsorption of Model Proteins with Wide Variation in Molecular Properties on Colloidal Particles. J. Colloid Interface Sci. 1992, 150 (2), 344–351. 10.1016/0021-9797(92)90204-Y. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.