

Abstract
Incorporation of fluoroalkyl motifs in pharmaceuticals can enhance therapeutic profiles of parent molecules. The hydrofluoroalkylation of alkenes has emerged as a promising route to diverse fluoroalkylated compounds; however, current methods require superstoichiometric oxidants, expensive/oxidative fluoroalkylating reagents, precious metals, and often exhibit limited scope, making a universal protocol that addresses these limitations highly desirable. Here we report the hydrofluoroalkylation of alkenes with cheap, abundant, and available fluoroalkyl carboxylic acids as the sole reagents. Hydrotrifluoro-, difluoro-, monofluoro- and perfluoroalkylation are all demonstrated, with broad scope, mild conditions (redox-neutral) and potential for late-stage modification of bioactive molecules. Critical to success is overcoming the exceedingly high redox-potential of feedstock fluoroalkyl carboxylic acids such as trifluoroacetic acid by leveraging cooperative earth-abundant, inexpensive iron and redox-active thiol catalysis, enabling these to be directly utilized as hydroperfluoroalkylation donors without pre-activation. Preliminary mechanistic studies support the radical nature of this cooperative process.
Main text
The incorporation of fluoroalkyl groups is emerging as a powerful tool in medicinal chemistry, enabling modulation of the biological and physiological activities of lead compounds, which can significantly affect the therapeutic profiles of parent molecules through enhancement of lipophilicity, bioavailability and metabolic stability.1-6 Among diverse fluoroalkyl motifs, the introduction of trifluoromethyl (CF3) and difluoromethyl (CF2H) on aliphatic chains has experienced growing demand in recent decades, serving as competent bioisosteres to methyl and other functional groups in late-stage functionalization (LSF) (Figure 1a).7 Toward installing these functional groups, radical hydrofluoroalkylation of alkenes represents an important class of reactions to access diverse fluorine-containing motifs from cheap and accessible hydrocarbon chemical feedstocks.8-33 Early hydrofluoroalkylation examples utilizing CF3I, CF3SO2Cl and CF3SO2Na have allowed the generation of CF3 radical for olefin coupling upon thermal- or photo-irradiation or with the treatment of redox agents; however, prohibitively harsh conditions and unstable reagents have impeded the general application of these methods,34-36 prompting current efforts to develop bench-stable and easy-to-use electrophilic fluoroalkyl reagents (CF3, CF2H etc.) as potential radical surrogates including Umemoto, Togni and Hu reagents (Figure 1b).37,38 Despite being significantly more operationally-simple compared with previous strategies, poor atom economy, high cost, highly oxidative characteristics, dependence on noble-metal based photoredox catalysts, and requirement of sacrificial base and/or high temperature have limited the applicability of these hydrotrifluoromethylation protocols,9,11,13,15,18 rendering this class of fluoroalkylation reagents less ideal. Recent efforts to overcome some of these challenges have been made by Xie via halogen-atom-transfer (XAT) of fluoroalkyl halides using manganese catalysis; however, superstoichiometric strong and expensive reductant (supersilane) was still required, presenting a barrier to wide adoption of this method.39 Outside of hydrotrifluoromethylation, previous methods of hydrodifluoromethylation have often exhibited limited scope tolerance and required complicated multistep syntheses.19,20 Recent advances by Gouverneur21 and Wu22 have made progress in expanding the scope of hydrodifluoromethylation, though these methods still require stoichiometric use of strong oxidants or sacrificial XAT reagents, rendering these protocols less atom-economic .
Figure 1. Hydrofluoroalkylation of alkenes for accessing valuable fluorinated molecules.

(a) Selected examples of fluorine-containing pharmaceuticals. (b) Fluoroalkyl radicals are challenging to generate directly from fluoroalkyl carboxylic acids due to their high oxidation potential, necessitating the use of complex and expensive precursor reagents. Cooperative photocatalytic hydrofluoroalkylation allows simple, mild, and broadly-applicable syntheses of valuable fluorinated molecules by direct activation of fluoroalkyl carboxylic acids, addressing limitations including the use of noble metals, electrophilic/expensive agents and harsh conditions in previous methods. (c) Postulated mechanism of the photocatalytic hydrofluoroalkylation. Upon light irradiation, homolytic cleavage of fluoroalkyl carboxylate is induced, forming carboxyl radical II and reduced FeII. Radical II undergoes CO2 extrusion and generates fluoroalkyl radical III, which can then engage in radical addition to the alkene and forming an alkyl radical. The thiol co-catalyst can then sequester this transient alkyl radical intermediate via hydrogen atom transfer (HAT) to form hydrofluoroalkylated product and a thiyl radical. Finally, redox interaction between reduced FeII and thiyl radical in the presence of a proton source allows both catalytic cycles to be closed, enabling cooperative photocatalytic hydrofluoroalkylation.
Under this context, fluoroalkyl carboxylic acids (such as trifluoroacetic acid, TFA) are attractive candidates for achieving hydrofluoroalkylation. However, decarboxylation of these reagents to generate perfluoroalkyl fragments has proven challenging, with previous methods requiring extremely forcing conditions such as high temperature (>140 °C with Cu(TFA)2)40 or strong oxidants, providing significant barriers to their use in practical synthesis. Many of these difficulties arise from exceedingly high redox potential of fluoroalkyl carboxylate anions (e.g. CF3CO2Na, E1/2 ox > +2.4V vs SCE).41 Accordingly, direct outer-sphere single electron oxidation of fluoroalkyl carboxylate anions is not viable as these forcing potentials can directly oxidize most organic solvents in preference to the carboxylate substrate. The Pan17 and the Stephenson42 groups have avoided these challenges by using pre-formed or in situ generated redox auxiliaries, allowing for fine-tuning of the reagents’ redox profile to accommodate the electrochemical window for decarboxylation using conventional photoredox catalysts. While enabling, these approaches have several drawbacks, including generation of stoichiometric waste derived from the chloro-oxime auxiliary, expensive noble-metal based photoredox catalysts, and stoichiometric hydrogen-atom-transfer (HAT) reagents for Pan’s hydrotrifluoromethylation. Stephenson’s system also requires the use of noble-metal based photoredox catalysts and uses a pyridinium auxiliary to enable C(sp2)─CF3 construction, with a limited scope of activated alkenes. Together, while this redox-auxiliary, photoredox manifold permits access to perfluoroalkyl radicals, a general, inexpensive, and operationally-simple hydrofluoroalkylation protocol directly using perfluoroalkyl carboxylic acids remains elusive. In an ideal method, diverse fluoroalkyl radicals (CF3, CF2H, CFH2 etc.) could be generated directly from abundant perfluoroalkyl carboxylic acids using earth abundant element catalysts with no additional reagents and carbon dioxide as the sole byproduct. However, we recognized that achieving this aspirational reaction requires a new mechanistic approach.
We hypothesized that we might be able to directly engage perfluoroalkyl carboxylic acids in decarboxylation chemistry through changing the nature of the photochemical step. The primary elementary step of conventional visible light-mediated photoredox catalysis is bimolecular, outer-sphere single electron transfer (OSET) between the photocatalyst and reactants, where matching the redox-potential of the substrate and excited state catalyst is a prerequisite for effective quenching. Interestingly, the direct coordination of a metal and substrate allows for a complementary inner-sphere single electron transfer (ISET) process, where the electron is transferred to -or from- a directly coordinated substrate. Ligand-to-metal charge transfer (LMCT) is one such ISET process, also one of the most studied reaction schemes in visible-light induced homolysis (VLIH), in which matching the redox-potential is not relevant for the light-driven redox process as the mechanism is fundamentally different.43-47 VLIH photoreactivities of 3d transition metals’ (copper,43,46 iron47 etc.) has seen significant development in recent years, providing sustainable alternatives to noble-metal based photocatalysts.44,45 Pioneering work from Wärnmark and coworkers unambiguously showed iron complexes can engage in LMCT, suggesting this mechanism may be accessible in catalysis as well.48 Consistent with this investigation, examples of decarboxylative reactivities via iron-photocatalysis have been demonstrated in some contexts.49-52 Recently, we reported a dual catalytic hydrodecarboxylation protocol which allows for efficient reaction of a variety of carboxylic acids through leveraging the synergistic cooperation of ligand-to-metal charge transfer (LMCT) and hydrogen-atom-transfer (HAT).53 A key differentiator of this reaction from previously-reported acridinium-photocatalyzed decarboxylative protonation (proceeding via an OSET pathway) is the inner-sphere electron transfer process between the iron catalyst and carboxylate substrate bypasses unselective bimolecular oxidation of electron rich functional groups, an unavoidable source of side-reactivity using the acridinium approach. Taking advantage of this pre-association process, we envisioned that a weakly oxidizing iron salt (Fe3+/Fe2+, E1/2 red = +0.53 V vs SCE) can engage in the net photooxidation of challenging substrates (e.g. TFA, E1/2 ox > +2.4 V vs SCE) under visible-light irradiation and overcome the redox-potential mismatching challenge inherent to the photoredox OSET processes, presenting an intriguing opportunity to directly oxidize otherwise inactive fluoroalkyl acids for direct hydrofluoroalkylation of a broad range of alkenes in combination with a suitable HAT cocatalyst.44,45
In our aspirational scheme, the homolytic cleavage of the O─Fe bond of the fluoroalkyl carboxylate I produces a carboxyl radical II and a lower-valent iron species under light irradiation (Figure 1c). While we envision that this process might occur via LMCT, other VLIH processes are also possible.46,47 Radical II can then undergo extrusion of CO2 to access fluoroalkyl radical species III, which would engage in radical addition onto alkene, providing transient carbon-centered radical intermediate. In a parallel manifold, the thiol would function as hydrogen atom transfer (HAT) reagent to furnish the hydrofluoroalkylation product IV and an oxidizing thiyl radical (step B). Finally, the thiyl radical would reoxidize the lower-valent iron intermediate while receiving a proton from another equivalent of acid substrate or H2O, closing both catalytic cycles (step A).
Realizing this design, here we report the first general photocatalytic hydrofluoroalkylation of alkenes via iron-photocatalysis and thiol-catalyzed hydrogen-atom-transfer (HAT). Hydrotri-, di-, mono- and perfluoroalkylation are all demonstrated by leveraging a unified cooperative catalytic system. Aside from the conceptually novel mechanistic pathway, this dual-catalytic design excludes the use of noble metals, stoichiometric oxidants/reductants, non-economical/corrosive electrophilic fluoroalkylating reagents, and allows incorporation of diverse fluoroalkyl moieties, including trifluoromethyl, directly from the parent carboxylic acids (e.g. TFA), an ideal yet exceedingly challenging approach due to the exceptional redox oxidation potential of these precursors. Key to unlocking the direct use of acid precursors is the photoreactivity of iron in light-induced decarboxylation,49-53 addressing the redox-potential mismatching issue in previous methods, while simultaneously allowing redox-neutral operation with an earth abundant thiol cocatalyst. Together, this protocol presents a general approach to accessing diverse fluoroalkylated alkanes under extremely mild conditions, providing powerful tools in direct and modular hydrofunctionalization of alkenes with readily available carboxylic acids.
Results and discussion
Reaction Design and Optimization.
We first set out to explore the possibility of the cooperative photocatalytic hydrotrifluoromethylation by using the pent-4-en-1-yl benzoate as model substrate, trifluoroacetic acid (TFA) as trifluoromethyl (CF3) source, and an earth abundant iron salt and organic thiol in catalytic loading under the irradiation of 390 nm LED light at room temperature (Table 1, for more details, see supplemental information Tables 1-6). Gratifyingly, we found that inexpensive iron diacetate in combination with 2,4,6-triisopropylbenzene thiol (TRIP thiol) or the corresponding disulfide can facilitate this cooperative hydrotrifluoromethylation process in high efficiency, giving 86% or 92% yield respectively, validating our hypothesis that CF3 radical can be generated through iron-photolysis process with cheap feedstock carboxylic acid precursors (e.g. TFA), addressing the redox potential mismatching in previous works engendered by their OSET strategy (Table 1, entry 1).17,42 Interestingly, decarboxylation of aromatic and aliphatic carboxylic acids via copper LMCT has been studied intensively recently,54-56 however, these catalysts are incompetent for decarboxylation of TFA under our conditions, showing the privileged reactivity of our iron/thiol cooperative system (Table 1, entry 2). Next, screening of different iron salts was carried out, indicating Fe(OAc)2 to be the most efficient catalyst (Table 1, entry 3). Interestingly, the loading of TFA could be reduced to 2 equivalents, giving comparable hydrotrifluoromethylated product (Table 1, entry 4). It was found that the loading of base additive is important: no base results in less complete transformation (72% yield), while adding stoichiometric base decreases the yield significantly, possibly due to competitive deprotonation of the thiol, impeding the subsequent HAT process (Table 1, entry 5). Solvent screening indicated weakly- or non-coordinating solvent such as DCM or THF either cannot or sluggishly promote the reaction, while coordinating solvent such as EA or acetone can facilitate the initial iron photolysis, in line with our previous observation (Table 1, entries 6–7).57 To demonstrate the necessary elements of our cooperative hydrofunctionalization protocol, control experiments were next pursued. No conversion was observed in the absence of light or iron salt (Table 1, entries 8–9) and as expected, only trace amount of product could be observed with the omission of thiol catalyst, indicating the synergistic activity between iron and thiol catalysts (Table 1, entry 10). Lastly, low yield was observed by removing water (co-solvent) from the system, indicating water is necessary, likely in regenerating thiol to facilitate hydrogen atom transfer (Table 1, entry 11).
Table 1.
Optimization of alkene hydrotrifluoromethylation.
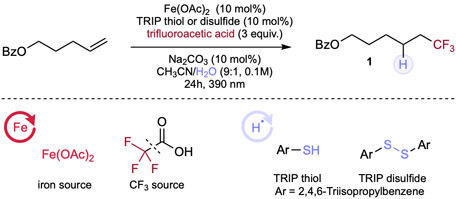
| ||
|---|---|---|
| Entry | Deviation from Standard Conditions | Yield (%)a |
| 1 | none | 86(84)b, 92(89)c |
| 2b | [Cu(MeCN)4]BF4, Cu(OTf)2 | ND |
| 3d | Fe(acac)3, FeCl3·6H2O, Fe(NO3)3·9H2O, Fe(OTf)2, FeCl2 | trace–72 |
| 4 | 2 equiv. of TFA | 74 |
| 5b | no base; 100 mol% of Na2CO3 | 72; 62 |
| 6b | DCM, THF | trace–20 |
| 7b | Acetone, EA | 42–76 |
| 8 | no iron salt | ND |
| 9 | no light | ND |
| 10b | no HAT reagent | trace |
| 11 | no water | 18b, 24c |
Reaction conditions: alkene (0.1mmol, 1.0 equiv.), trifluoroacetic acid (3.0 equiv.), Fe(OAc)2 (10 mol%), TRIP thiol or disulfide (10 mol%), Na2CO3 (10 mol%) and solvent (0.1 M), 24h, RT, 390nm Kessil blue LED. a 1H NMR yield is determined by using CH2Br2 as an internal standard. Isolated yield in the parentheses. b With 10 mol% of TRIP thiol. c With 10 mol% of TRIP disulfide. d With 5 mol% of iron salt and 5 mol% TRIP disulfide.
Hydrotrifluoro- and difluoromethylation of alkenes.
Encouraged by successful optimization of our cooperative hydrotrifluoromethylation, we sought to apply this system onto another attractive fluoroalkyl moiety. The difluoromethyl group (CF2H) has been gathering enormous interest in medicinal and agrochemical science, functioning as important bioisostere to diverse functional groups. Recent efforts by Dolbier,19, Qing,20 Gouverneur,21 and Wu22 have elegantly addressed practicality and scope tolerance issues in previous works; however, the reliance of these methods on multistep syntheses, superstoichiometric strong oxidants, and/or high loading of sacrificial XAT reagents has prevented the development of a mild and sustainable method of hydrodifluoromethylation. Further, it would be ideal to be able to achieve this transformation with a generic catalytic manifold capable of other hydrofluoroalkylation reactions with minimal modification. Thus, we sought to unify hydrodifluoromethylation with our hydrotrifluoromethylation using the same cooperative photocatalytic pathway and were delighted to find our system can achieve both transformations (for optimization of hydrodifluoromethylation protocol, see supplemental information Tables 7-11). We next investigated the scope generality of this hydrofunctionalization system (Table 2). First, aliphatic alkenes were used to demonstrate the systems’ compatibility with simple unactivated alkenes. High yields were obtained for both hydrotrifluoro- (2, 3) and difluoromethylation (28, 29). Protecting groups such as benzoate (30) and base-sensitive tosylate (34) were tolerated; the 2,2,2-trichloroethoxycarbonyl, useful in peptide chemistry and susceptible to reduction, was tolerated in both systems (4, 31). Halo- (5, 6, 32) and other para-substituted arenes (7, 33) exhibited good tolerance to both trifluoro- and difluoromethylation conditions. Notably, most heterocycles behave smoothly in our system; however, competitive direct aryl trifluoromethylation was observed in one case. N-phthalimides (8, 35), dihydrobenzofurans (9, 36), and thiophenes (39) provided corresponding products in moderate to good yields, contrasting the scope limitation in previous protocols when using highly oxidizing fluoroalkylation reagents. In the case of N-methyl-pyrrole (10), we observed competitive radical trifluoromethylation of the heterocyclic moiety (for more details, see supplemental information section 2.12), showing a potential limitation of this protocol.
Table 2.
Hydrotrifluoromethylation and hydrodifluoromethylation of alkenes.
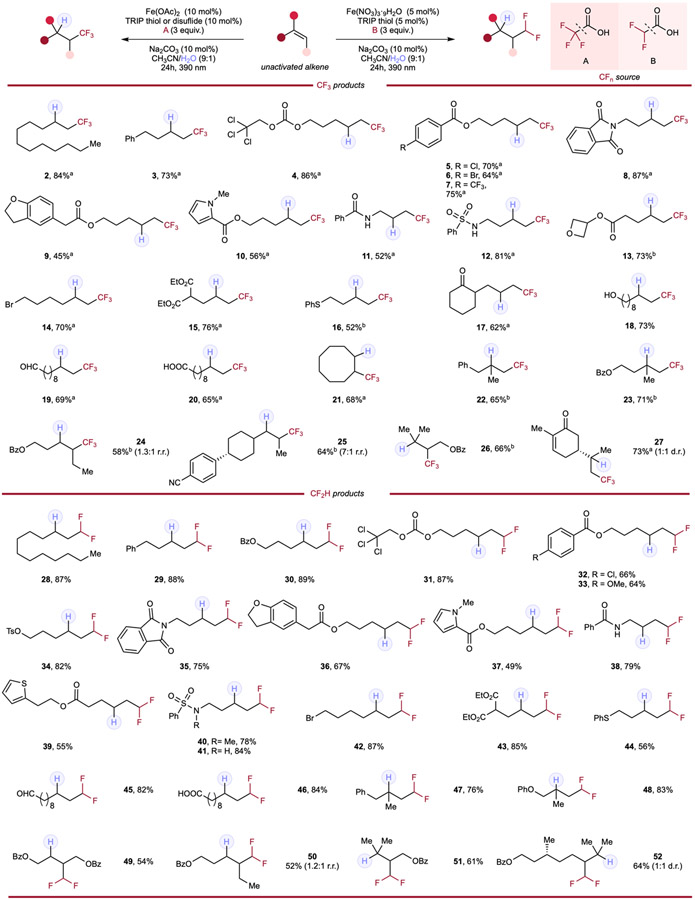
|
Reaction conditions for hydrotrifluoromethylation: alkene (0.1mmol, 1.0 equiv.), trifluoroacetic acid (3.0 equiv.), Fe(OAc)2 (10 mol%), HAT reagent (10 mol%), Na2CO3 (10 mol%) and CH3CN/H2O (9:1, 0.1 M), 24h, RT, 390nm Kessil blue LED. Reaction conditions for hydrodifluoromethylation: alkene (0.1mmol, 1.0 equiv.), difluoroacetic acid (3.0 equiv.), Fe(NO3)3·9H2O (5 mol%), TRIP thiol (5 mol%), Na2CO3 (10 mol%) and CH3CN/H2O (9:1, 0.1 M), 24h, RT, 390nm Kessil blue LED. a With 10 mol% of TRIP thiol. b With 10 mol% of TRIP disulfide.
Having established a large scope of unactivated alkene additions, a series of substrates containing labile functionalities (acid/base-, substitution-, and redox-sensitive groups) were tested. Active proton-containing benzamides (11, 38) and medicinally prevalent sulfonamides with acidic N–H protons (12, 41) as well as N-methylated sulfonamides (40) were also well-preserved in tri- and difluoromethylation. Reactive functionalities including strained-ring system such as oxetane (13) and SN2-labile aliphatic halides (14, 42) remain untouched, giving high yields of corresponding products. Of great importance is that oxidatively-labile functionalities including esters (15, 43), sulfides (16, 44), ketone (17), alcohol (18), aldehydes (19, 45) and non-fluoroalkyl carboxylic acids (20, 46) all produced expected products in moderate to high yields (52–85%), which would be a significant challenge by leveraging photoredox-catalyzed OSET processes with exceptional oxidizing ability as the majority of these functionalities are expected to be decomposed under such extreme redox potentials (especially up to +2.4 V vs SCE). Having observed preservation of a wide range of useful and medicinally important functional handles in both systems, we shifted our efforts to investigate spectrum of alkene substitution tolerance. In the hydrotrifluoromethylation reaction, cyclooctene (21) displayed conversion and reactivity comparable to previous substrates tested. In the difluoromethylation reactions, linear internal alkene (49) displayed similar conversion and yield in non-diastereomeric fashion. While similar to the difluoromethylation counterpart (60) regarding low diastereomeric control (24), disubstituted alkenes show evident preference in hydrotrifluoromethylation when encountering a bulky substituent (25). Not only was good reactivity observed in 1,1-disubstituted alkenes (22, 23, 47, 48) for both hydrotrifluoro- and difluoromethylation, but tri-substituted alkenes (26, 51, 52) also provided hydrotrifluoro- and difluoromethylated products in good yields, offering pathway to a variety of compounds containing fluoroalkyl moieties on relatively sterically-hindered molecules. Finally, (R)-Carvone was utilized as a substrate containing two chemically distinct alkenes: an electron-deficient (activated) and an electron rich alkene (unactivated). The resulting trifluoromethylated compound (27) displayed exclusive conversion at the unactivated site, demonstrating the system’s unique chemoselectivity.
Late-stage hydrotrifluoro- and difluoromethylation.
Understanding the prominent therapeutic profiles engendered by introduction of fluoroalkyl moieties, we next endeavored to investigate an array of alkenes derived from commercially available active pharmaceutical ingredients (APIs) and natural products to evaluate the systems’ viability as a late-stage modification (Table 3). Substrates derived from common nonsteroidal anti-inflammatory (NSAID) drugs such as Ibuprofen (53, 66), Flurbiprofen (55), and Loxoprofen (68) provided moderate to high yields (62–82%) for both fluoromethylation systems. Sulfonamide-containing Probenecid (56) was transformed into its trifluoromethylated derivative in 89% yield; Naproxen (58) demonstrated moderate efficiency in the trifluoromethylation reaction. Alkenes derived from common herbicide 2,4-dichlorophenoxyacetic acid (54) and lipid-lowering Benzafibrate (59) afforded corresponding products in 81% and 78% yield respectively. The sterically congested and bicyclic (−)-Borneol (60, 70) and natural product (L)-Menthol (61, 69) were all efficiently derivatized in both systems, accessing hydrofluoroalkylated products in good yields. The unsaturated fat Oleic acid (63) demonstrated similar conversion and diastereoselectivity as with other internal alkenes tested. A ketal-protected monosaccharide (71) was well-behaved in the difluoromethylation system and heavily delocalized Flavone-type compound (64) underwent efficient trifluoromethylation. Boc-protected Proline (73) also found tolerance in the hydrofluoroalkylation system, giving high yield of difluoromethylated product. Lastly, complex steroids such as the diene derivative of 18β-Glycyrrhetinic acid (65), Estrone (62, 75) and tri-substituted Pregnenolone (72) demonstrated synthetically useful conversion to the respective hydrofluoromethylated products. It is notable that pharmaceuticals, natural products, and herbicides that have innate olefinic functionalities such as Vinclozolin (57, 67), Pregnenolone (72) and Nootkatone (74), can be directly hydrofluoroalkylated, affording corresponding products in moderate to high yields, showcasing ability in direct late-stage functionalization (LSF) of substrate containing olefinic functionalities by our cooperative system. The simplicity and generality of our general strategy in hydrofluoroalkylation, in addition to the low cost, earth-abundance, and low toxicity of iron makes it ideally positioned for medicinal chemistry campaigns, allowing for the efficient and versatile generation of fluoroalkylated drugs/natural products analogues.
Table 3.
Hydrotrifluoromethylation and hydrodifluoromethylation of APIs & natural product alkene derivatives.
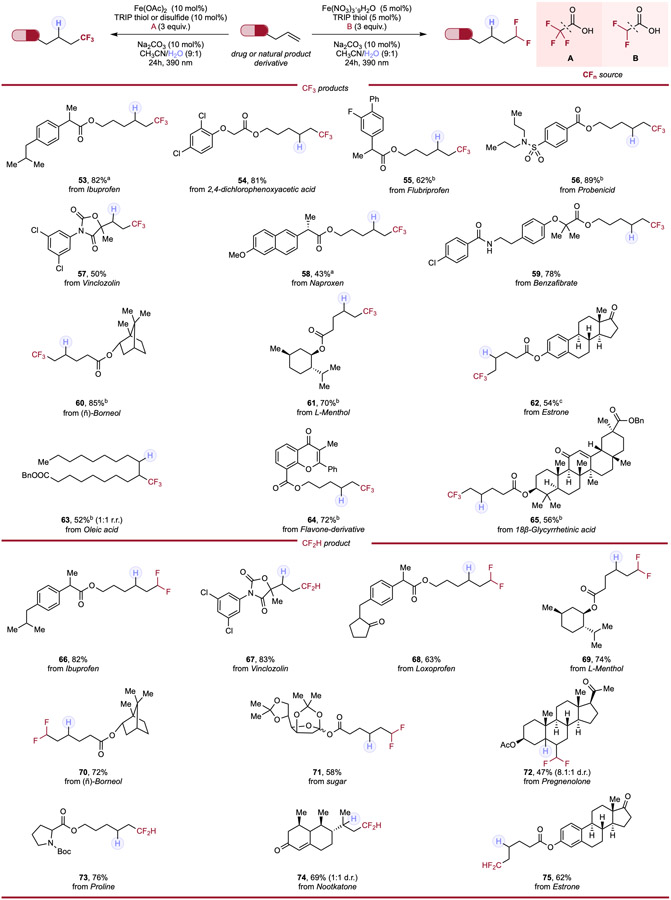
|
Reaction conditions for hydrotrifluoromethylation: alkene (0.1mmol, 1.0 equiv.), trifluoroacetic acid (3.0 equiv.), Fe(OAc)2 (10 mol%), HAT reagent (10 mol%), Na2CO3 (10 mol%) and CH3CN/H2O (9:1, 0.1 M), 24h, RT, 390nm Kessil blue LED. Reaction conditions for hydrodifluoromethylation: alkene (0.1mmol, 1.0 equiv.), difluoroacetic acid (3.0 equiv.), Fe(NO3)3·9H2O (5 mol%), TRIP thiol (5 mol%), Na2CO3 (10 mol%) and CH3CN/H2O (9:1, 0.1 M), 24h, RT, 390nm Kessil blue LED. a With 10 mol% of TRIP thiol. b With 10 mol% of TRIP disulfide.c With Fe(OAc)2 (20 mol%), TRIP thiol (20 mol%) and trifluoroacetic acid (6 equiv.).
Hydromonofluoroalkylation of activated alkenes.
The thorough investigations leveraging tri- and difluoroacetic acid as fluoromethyl radical sources further encouraged us to explore alkene modification using monofluoroacetic acid to achieve monofluoromethylation under an analogous pathway, as monofluoroalkanes are also prevalent in drugs, agrochemicals and functional materials.1-6 Cognizant of the challenges in the preparation of alkyl monofluorides through direct C─F bond formation, we sought to deliver this alternative strategy in accessing diverse monofluoroalkanes with mild conditions and greater modularity. Based on nucleophilic character of the CH2F radical,29 styrene and styrene derivatives were selected as the major pool of substrates to explore the hydromonofluoromethylation (Table 4). Through careful optimization including various solvent conditions, reactant concentrations and other variations, styrene-type activated alkenes (76–88) featuring different functional groups (77, 78) or substitution patterns (80, 81) all provided the monofluoromethylated product smoothly. Interestingly, water as a cosolvent was not beneficial in all these examples, presumably as protonation of the thiol could occur directly from monofluoroacetic acid in these cases. Michael acceptor-type alkenes (82, 83) provided the expected products in moderate yields. Moreover, different monofluoroalkyl derivatives such as 2-phenyl-2-fluoroacetic acid (84) and 2-fluoropropanoic (85) also showed reactivities, giving corresponding products in comparable yields. Styrene-containing drugs/natural products including Gemfibrozil (86), Proline (87) and Palmtic acid (88) were also tested and, to our delight, monofluoromethylated products were all produced in useful yields.
Table 4.
Hydromonofluoroalkylation and hydroperfluoroalkylation of alkenes.
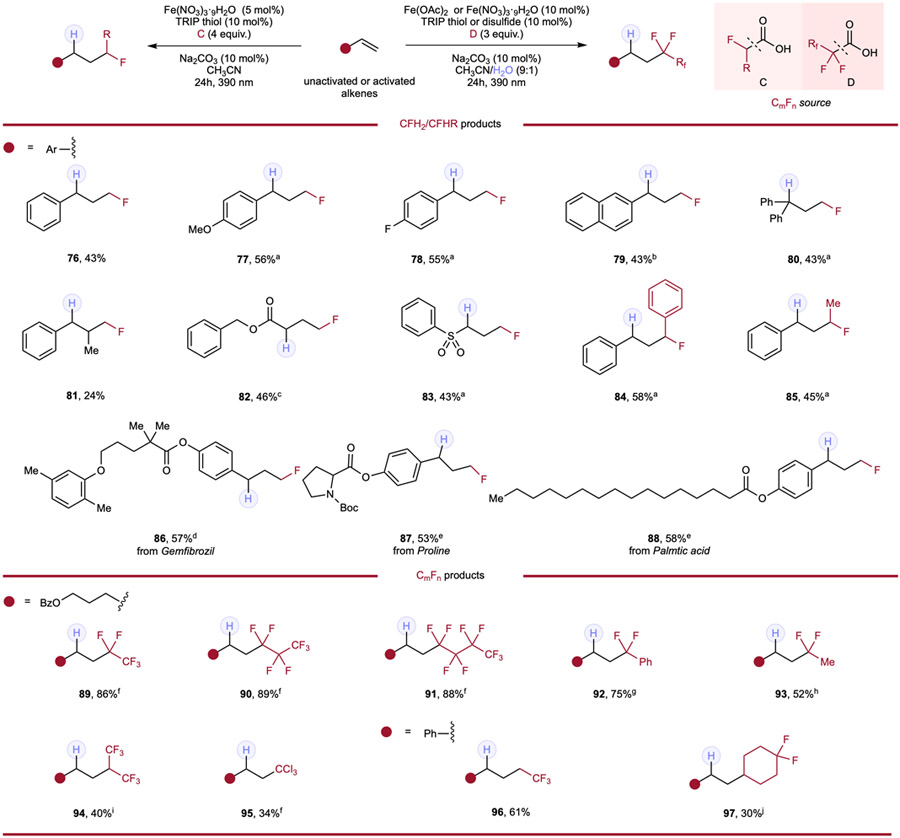
|
Reaction conditions of hydromonofluoromethylation: alkene (0.1mmol, 1.0 equiv.), monofluoroacetic acid (4.0 equiv.), Fe(NO3)3·9H2O (5 mol%), TRIP thiol (10 mol%), Na2CO3 (10 mol%) and CH3CN (0.1 M), 24h, RT, 390nm Kessil blue LED. a 0.2M. b With 10 mol% of H2O in 0.1M solution. c Without thiol. d With 10 mol% of Fe(NO3)3·9H2O in 0.2M solution. e With 10 mol% of Fe(NO3)3·9H2O. Reaction conditions of hydroperfluoroalkylation: alkene (0.1mmol, 1.0 equiv.), perfluoroalkyl carboxylic acid (3.0 equiv.), Fe(OAc)2 (10 mol%), HAT reagent (10 mol%), Na2CO3 (10 mol%) and CH3CN/H2O (9:1, 0.1 M), 24h, RT, 390nm Kessil blue LED. f With 10 mol% of TRIP thiol. g With 10 mol% of Fe(NO3)3·9H2O and 10 mol% of TRIP disulfide. h With 10 mol% of Fe(NO3)3·9H2O and 10 mol% of TRIP thiol. i With 10 mol% of TRIP disulfide. Reaction conditions of hydroalkylation of styrene: styrene (0.1mmol, 1.0 equiv.), carboxylic acid (4.0 equiv.), Fe(NO3)3·9H2O (5 mol%), TRIP thiol (5 mol%), Na2CO3 (10 mol%) and CH3CN (0.1 M), 24h, RT, 390nm Kessil blue LED. j 0.2M.
Scope of hydro(polyfluoro)alkylation of alkenes.
Perfluoroalkyl acids (PFAAs) have become ubiquitous in many commercial and household items, being processed and utilized on industrial scales.58 However, environmental research suggests that PFAAs are highly resistant to biodegradation and toxic to the environment and human health. Based on our findings in using fluoroalkylated acids for alkene hydrofunctionalization, upcycling these industrial pollutants like PFAAs by utilizing them as reagents became an intriguing potential extension of our studies (Table 4). Perfluoropropionic acid (89), perfluorobutanoic acid (90), and perfluoropentanoic acid (91) were all found to be competent fluoroalkyl sources to modify our model substrate in high yields, realizing facile application of these PFAAs. Additionally, difluoroacetic acid derivatives, including phenyldifluoroacetic acid (92) and 2,2-difluoropropanoic acid (93) were also suitable fluoroalkyl sources when applied in similar conditions. Other strong acid derivatives of acetic acid including a fluorine-containing propanoic acid derivative (94), trichloroacetic acid (95), 3,3,3-trifluoropropanoic acid (96), and the fluorocyclohexyl-containing acid (97) all produced their expected products, indicating non-alpha fluorine substituted carboxylic acids could also engage in this cooperative hydrofunctionalization protocol.
Preliminary mechanistic studies.
Bolstered by the broad scope and generality of our cooperative hydrofluoroalkylation protocol, we next sought to perform mechanistic probes to interrogate the elementary steps of this process (Figure 2). We chose to focus on the most challenging hydrotrifluoromethylation as a model for these studies to illustrate our unified strategy in these hydrofluoroalkylation transformations. First, the inclusion of 1 equivalent of radical scavenger 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) in the system, completely suppressed the reaction and alkene starting material was fully recovered, with detection of TEMPO-CF3 adduct on 19F NMR, supporting the presence of radical intermediates (Figure 2, panel A). To further provide support for TFA undergoing VLIH in the presence of Fe(III) species, we pursued a series of control experiments. First, irradiating Fe(NO3)3·9H2O, TFA, and TEMPO in CH3CN/H2O results in homolysis of the acid under visible-light irradiation, indicated by TEMPO-CF3 detection on 19F NMR. Similarly, a mixture of Fe(OAc)2 and TRIP thiol can induce homolysis of TFA under irradiation, again detected by TEMPO trapping of the CF3 radical. We hypothesize this reactivity is possible through initial photochemical formation of disulfide or thiyl radical,59 enabling oxidation of Fe(II) to Fe(III). No TEMPO-CF3 was detected in either case in the absence of light (for more details, see supplemental information section 2.7). With these results in hand, we sought to further support a radical pathway by applying two different radical clock substrates, where N-tosylated-tethered diene furnished 5-exo-trig product 98 in 62% yield and a cyclopropyl-substituted alkene undergoes a facile ring-opening process, offering hydrotrifluoromethylated product 99 in excellent E/Z selectivity. Both entries have provided strong support of a radical process and suggest that the subsequent hydrogen atom transfer step is slower than the rate constant of 2 x 105 s−1 (approx. for 5-exo-trig) in this cooperative system (Figure 2, panel B).
Figure 2. Mechanistic studies and deuterium labeling experiments.

Standard conditions: as described in Table 1, entry 1 with TRIP thiol (a) Radical scavenger. Reduced reactivity and detection of TEMPO-CF3 on 19F NMR both indicated a radical process is likely involved. (b) Radical clock experiments. With two radical clock experiments behaving well, accessing 5-exo-trig or ring-opening products correspondingly, we reasoned that this result correlated with radical reactivity and panel A. (c) Deuterium labeling and KIE experiments. High deuterium incorporation rate provided support that the hydrogen that is delivered in HAT step comes from H2O co-solvent. Primary KIE value indicates that thiol HAT has rate determining character.
Furthermore, deuterium labeling and kinetic isotope effect studies were carried out. As expected from our proposed mechanism, exchanging the H2O cosolvent with D2O results in moderate yield of deuterotrifluoromethylation with good deuterium incorporation efficiency. The lower yield of deuterotrifluoromethylation compared with standard hydrotrifluoromethylation could suggest either slower proton exchange between thiol and D2O or sluggish deuterium atom transfer (DAT) step, leading to less complete conversion of starting material. The parallel reaction of both H2O and D2O reactions provided a KIE value of 2.5,60 suggesting hydrogen atom transfer (HAT) has rate determining character in this transformation (Figure 2, panel C). In line with the kinetic investigation via KIE studies, we sought preliminary kinetic insight into our dual catalytic system using initial rate experiments. These tests reveal zeroth-order dependence on both alkene and trifluoroacetic acid, first-order dependence on iron, and complex dependence on thiol catalyst (for more details, see supplemental information section 2.7). The reaction exhibits positive order in thiol at low concentrations and negative order at high concentrations, suggesting that it has rate determining character at low concentrations (consistent with the observed KIE) and inhibitory effect at higher concentrations (for more details, see supplemental information section 2.7). Additionally, we found the reaction rate to be positively correlated to light intensity. Together, these data are consistent with thiol HAT having rate determining character and thiol concentration being impacted by both iron concentration and light intensity.
Conclusions
We have demonstrated a general catalytic protocol for hydrofluoroalkylation of alkenes with diverse fluoroalkyl carboxylic acids enabled by cooperative iron-photocatalysis and thiol-catalyzed hydrogen atom transfer (HAT). Hydrotrifluoro-, difluoro-, monofluoro- and perfluoroalkylation were all efficiently achieved under a unified pathway. Broad scope, mild, redox-neutral conditions, and the potential for late-stage modification were showcased in these transformations, addressing limitations of previous synthetic methods, including scope tolerance, superstoichiometric use of strong oxidants, or requirement of noble metal catalysts. Importantly, cheap feedstock chemicals such as trifluoroacetic acid (TFA) that have exceedingly high redox potentials can be directly utilized as CF3 source, overcoming challenging redox-potential considerations by applying earth abundant, inexpensive iron catalyst in synergy with a redox-active organic thiol cocatalyst. We expect this cooperative methodology will serve as a powerful tool in modular construction of fluoroalkylated compounds and accelerate the syntheses of fluoroalkylated analogues to commercially available pharmaceuticals and natural products. Future studies of hydrofunctionalization leveraging cooperative iron and thiol catalysis using various nucleophiles are ongoing in our lab.
Methods
General procedures of photocatalytic hydrofluoroalkylation of alkenes.
Fe salt (5 or 10 mol%, 0.05 or 0.1 equiv.), Na2CO3 (10 mol%, 0.1 equiv.) and TRIP disulfide or TRIP thiol (5 or 10 mol%, 0.05 or 0.1 equiv.) (in the case using TRIP thiol, HAT reagent was added via syringe after backfilling with N2) was added in an oven-dried 8-mL test vial containing a Teflon®-coated magnetic stir bar. The vial was evacuated and backfilled with N2 (repeated for 4 times), followed by addition of alkenes (0.1 mmol, 1.0 equiv.) and fluoroalkyl carboxylic acid (0.30 or 0.40 mmol, 3.0 or 4.0 equiv) in MeCN/H2O (9:1, 0.1 M in regard to alkenes) or MeCN (0.1 M in regard to alkenes) via syringe under N2. The reaction mixture was placed under 390nm Kessil® light after sealing the punctured holes of the vial cap with vacuum grease and electric tape/parafilm for better air-tight protection and allowed to react at room temperature for 24 h. Following this, the reaction mixture was filtered through a pad of celite which was subsequently rinsed with DCM. The filtrate was concentrated, and the residue was then purified by flash column chromatography to give the corresponding hydrofluoromethylated products. The details of hydrotrifluoro-, difluoro-, monofluoro and polyfluoroalkylation (the types and equivalent of iron salt, HAT reagents, solvent used in each protocol) are demonstrated in the supplemental information section 1.3.
Supplementary Material
Acknowledgements
J.G.W. acknowledge financial support from CPRIT (RR190025), NIH (R35GM142738), and the Welch Foundation (C-2085). J.G.W. is a CPRIT Scholar in Cancer Research. Dr. Yohannes H. Rezenom (TAMU/LBMS), Dr. Ian M Riddington (UT Austin Mass Spectrometry Facility), and Dr. Christopher L. Pennington (Rice University Mass Spectrometry Facility) are acknowledged for assistance with mass spectrometry analysis.
Footnotes
Competing Interests statement
The authors declare no competing interests.
Data availability
The authors declare that all the data supporting the findings of this research are available within the article and its supplementary information.
References:
- 1.Müller K, Faeh C & Diederich F Fluorine in Pharmaceuticals: Looking Beyond Intuition. Science 317, 1881–1886 (2007). [DOI] [PubMed] [Google Scholar]
- 2.O'Hagan D. Understanding organofluorine chemistry. An introduction to the C─F bond. Chemical Society Reviews 37, 308–319 (2008). [DOI] [PubMed] [Google Scholar]
- 3.Purser S, Moore PR, Swallow S & Gouverneur V Fluorine in medicinal chemistry. Chemical Society Reviews 37, 320–330 (2008). [DOI] [PubMed] [Google Scholar]
- 4.Wang J. et al. Fluorine in Pharmaceutical Industry: Fluorine-Containing Drugs Introduced to the Market in the Last Decade (2001–2011). Chemical Reviews 114, 2432–2506 (2014). [DOI] [PubMed] [Google Scholar]
- 5.Ni C & Hu J The unique fluorine effects in organic reactions: recent facts and insights into fluoroalkylations. Chemical Society Reviews 45, 5441–5454 (2016). [DOI] [PubMed] [Google Scholar]
- 6.Inoue M, Sumii Y & Shibata N Contribution of Organofluorine Compounds to Pharmaceuticals. ACS Omega 5, 10633–10640 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Börgel J & Ritter T Late-Stage Functionalization. Chem 6, 1877–1887 (2020). [Google Scholar]
- 8.Wu X, Chu L & Qing F-L Silver-Catalyzed Hydrotrifluoromethylation of Unactivated Alkenes with CF3SiMe3. Angewandte Chemie International Edition 52, 2198–2202 (2013). [DOI] [PubMed] [Google Scholar]
- 9.Mizuta S. et al. Catalytic Hydrotrifluoromethylation of Unactivated Alkenes. Journal of the American Chemical Society 135, 2505–2508 (2013). [DOI] [PubMed] [Google Scholar]
- 10.Wilger DJ, Gesmundo NJ & Nicewicz DA Catalytic hydrotrifluoromethylation of styrenes and unactivated aliphatic alkenes via an organic photoredox system. Chemical Science 4, 3160–3165 (2013). [Google Scholar]
- 11.Pitre SP, McTiernan CD, Ismaili H & Scaiano JC Metal-Free Photocatalytic Radical Trifluoromethylation Utilizing Methylene Blue and Visible Light Irradiation. ACS Catalysis 4, 2530–2535 (2014). [Google Scholar]
- 12.Choi S. et al. Hydrotrifluoromethylation and iodotrifluoromethylation of alkenes and alkynes using an inorganic electride as a radical generator. Nature Communications 5, 4881 (2014). [DOI] [PubMed] [Google Scholar]
- 13.Egami H, Usui Y, Kawamura S, Nagashima S & Sodeoka M Product Control in Alkene Trifluoromethylation: Hydrotrifluoromethylation, Vinylic Trifluoromethylation, and Iodotrifluoromethylation using Togni Reagent. Chemistry – An Asian Journal 10, 2190–2199 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Straathof NJW, Cramer SE, Hessel V & Noël T Practical Photocatalytic Trifluoromethylation and Hydrotrifluoromethylation of Styrenes in Batch and Flow. Angewandte Chemie International Edition 55, 15549–15553 (2016). [DOI] [PubMed] [Google Scholar]
- 15.Cheng Y & Yu S Hydrotrifluoromethylation of Unactivated Alkenes and Alkynes Enabled by an Electron-Donor–Acceptor Complex of Togni’s Reagent with a Tertiary Amine. Organic Letters 18, 2962–2965 (2016). [DOI] [PubMed] [Google Scholar]
- 16.Cui B. et al. Mn(OAc)3-Mediated Hydrotrifluoromethylation of Unactivated Alkenes Using CF3SO2Na as the Trifluoromethyl Source. The Journal of Organic Chemistry 83, 6015–6024 (2018). [DOI] [PubMed] [Google Scholar]
- 17.Zhang W. et al. Leaving Group Assisted Strategy for Photoinduced Fluoroalkylations Using N-Hydroxybenzimidoyl Chloride Esters. Angewandte Chemie International Edition 58, 624–627 (2019). [DOI] [PubMed] [Google Scholar]
- 18.Jia H, Häring AP, Berger F, Zhang L & Ritter T Trifluoromethyl Thianthrenium Triflate: A Readily Available Trifluoromethylating Reagent with Formal CF3+, CF3•, and CF3− Reactivity. Journal of the American Chemical Society 143, 7623–7628 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tang X-J, Zhang Z & Dolbier WR Jr. Direct Photoredox-Catalyzed Reductive Difluoromethylation of Electron-Deficient Alkenes. Chemistry – A European Journal 21, 18961–18965 (2015). [DOI] [PubMed] [Google Scholar]
- 20.Lin Q-Y, Xu X-H, Zhang K & Qing F-L Visible-Light-Induced Hydrodifluoromethylation of Alkenes with a Bromodifluoromethylphosphonium Bromide. Angewandte Chemie International Edition 55, 1479–1483 (2016). [DOI] [PubMed] [Google Scholar]
- 21.Meyer CF, Hell SM, Misale A, Trabanco AA & Gouverneur V Hydrodifluoromethylation of Alkenes with Difluoroacetic Acid. Angewandte Chemie International Edition 58, 8829–8833 (2019). [DOI] [PubMed] [Google Scholar]
- 22.Zhang Z-Q et al. Difluoromethylation of Unactivated Alkenes Using Freon-22 through Tertiary Amine-Borane-Triggered Halogen Atom Transfer. Journal of the American Chemical Society 144, 14288–14296 (2022). [DOI] [PubMed] [Google Scholar]
- 23.Yu C, Iqbal N, Park S & Cho EJ Selective difluoroalkylation of alkenes by using visible light photoredox catalysis. Chemical Communications 50, 12884–12887 (2014). [DOI] [PubMed] [Google Scholar]
- 24.Sumino S. et al. Photoredox-Catalyzed Hydrodifluoroalkylation of Alkenes Using Difluorohaloalkyl Compounds and a Hantzsch Ester. The Journal of Organic Chemistry 82, 5469–5474 (2017). [DOI] [PubMed] [Google Scholar]
- 25.Supranovich VI, Levin VV, Struchkova MI, Korlyukov AA & Dilman AD Radical Silyldifluoromethylation of Electron-Deficient Alkenes. Organic Letters 19, 3215–3218 (2017). [DOI] [PubMed] [Google Scholar]
- 26.Yu Y-J et al. Sequential C-F bond functionalizations of trifluoroacetamides and acetates via spin-center shifts. Science 371, 1232–1240 (2021). [DOI] [PubMed] [Google Scholar]
- 27.Campbell MW et al. Photochemical C─F Activation Enables Defluorinative Alkylation of Trifluoroacetates and -Acetamides. Journal of the American Chemical Society 143, 19648–19654 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yue W-J, Day CS, Brenes Rucinski AJ & Martin R Catalytic Hydrodifluoroalkylation of Unactivated Olefins. Organic Letters 24, 5109–5114 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hell SM et al. Hydrofluoromethylation of alkenes with fluoroiodomethane and beyond. Chemical Science 12, 12149–12155 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu B-B et al. A General and Efficient Solution to Monofluoroalkylation: Divergent Synthesis of Aliphatic Monofluorides with Modular Synthetic Scaffolds. Angewandte Chemie International Edition 61, e202208938 (2022). [DOI] [PubMed] [Google Scholar]
- 31.Hu C-M & Qiu Y-L Co/Zn bimetal redox system promoted hydroperfluoroalkylation of acrylates with perfluoroalkyl iodides. Tetrahedron Letters 32, 4001–4002 (1991). [Google Scholar]
- 32.Dolbier WR Structure, Reactivity, and Chemistry of Fluoroalkyl Radicals. Chemical Reviews 96, 1557–1584 (1996). [DOI] [PubMed] [Google Scholar]
- 33.Barata-Vallejo S & Postigo A (Me3Si)3SiH-Mediated Intermolecular Radical Perfluoroalkylation Reactions of Olefins in Water. The Journal of Organic Chemistry 75, 6141–6148 (2010). [DOI] [PubMed] [Google Scholar]
- 34.Haszeldine RN 603. The reactions of fluorocarbon radicals. Part I. The reaction of iodotrifluoromethane with ethylene and tetrafluoroethylene. Journal of the Chemical Society (Resumed), 2856–2861 (1949). [Google Scholar]
- 35.Kamigata N, Fukushima T, Terakawa Y, Yoshida M & Sawada H Novel perfluoroalkylation of alkenes with perfluoroalkanesulphonyl chlorides catalysed by a ruthenium(II) complex. Journal of the Chemical Society, Perkin Transactions 1, 627–633 (1991). [Google Scholar]
- 36.Langlois BR, Laurent E & Roidot N Trifluoromethylation of aromatic compounds with sodium trifluoromethanesulfinate under oxidative conditions. Tetrahedron Letters 32, 7525–7528 (1991). [Google Scholar]
- 37.Li M, Wang Y, Xue X-S & Cheng J-P A Systematic Assessment of Trifluoromethyl Radical Donor Abilities of Electrophilic Trifluoromethylating Reagents. Asian Journal of Organic Chemistry 6, 235–240 (2017). [Google Scholar]
- 38.Zhao Y, Huang W, Zhu L & Hu J Difluoromethyl 2-Pyridyl Sulfone: A New gem-Difluoroolefination Reagent for Aldehydes and Ketones. Organic Letters 12, 1444–1447 (2010). [DOI] [PubMed] [Google Scholar]
- 39.Han J. et al. Photoinduced manganese-catalysed hydrofluorocarbofunctionalization of alkenes. Nature Synthesis 1, 475–486 (2022). [Google Scholar]
- 40.Chen M & Buchwald SL Rapid and Efficient Trifluoromethylation of Aromatic and Heteroaromatic Compounds Using Potassium Trifluoroacetate Enabled by a Flow System. Angewandte Chemie International Edition 52, 11628–11631 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Depecker C, Marzouk H, Trevin S. p. & Devynck J Trifluoromethylation of aromatic compounds via Kolbe electrolysis in pure organic solvent. Study on laboratory and pilot scale. New Journal of Chemistry 23, 739–742 (1999). [Google Scholar]
- 42.Beatty JW, Douglas JJ, Cole KP & Stephenson CRJ A scalable and operationally simple radical trifluoromethylation. Nature Communications 6, 7919 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kochi JK Photolyses of Metal Compounds: Cupric Chloride in Organic Media. Journal of the American Chemical Society 84, 2121–2127 (1962). [Google Scholar]
- 44.Abderrazak Y, Bhattacharyya A & Reiser O Visible-Light-Induced Homolysis of Earth-Abundant Metal-Substrate Complexes: A Complementary Activation Strategy in Photoredox Catalysis. Angewandte Chemie International Edition 60, 21100–21115 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Juliá F. Ligand-to-Metal Charge Transfer (LMCT) Photochemistry at 3d-Metal Complexes: An Emerging Tool for Sustainable Organic Synthesis. ChemCatChem 14, e202200916 (2022). [Google Scholar]
- 46.Hossain A, Bhattacharyya A & Reiser O Copper’s rapid ascent in visible-light photoredox catalysis. Science 364, eaav9713 (2019). [DOI] [PubMed] [Google Scholar]
- 47.de Groot LHM, Ilic A, Schwarz J & Wärnmark K Iron Photoredox Catalysis–Past, Present, and Future. Journal of the American Chemical Society 145, 9369–9388 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chábera P. et al. A low-spin Fe(iii) complex with 100-ps ligand-to-metal charge transfer photoluminescence. Nature 543, 695–699 (2017). [DOI] [PubMed] [Google Scholar]
- 49.Li Z, Wang X, Xia S & Jin J Ligand-Accelerated Iron Photocatalysis Enabling Decarboxylative Alkylation of Heteroarenes. Organic Letters 21, 4259–4265 (2019). [DOI] [PubMed] [Google Scholar]
- 50.Feng G, Wang X & Jin J Decarboxylative C─C and C─N Bond Formation by Ligand-Accelerated Iron Photocatalysis. European Journal of Organic Chemistry 2019, 6728–6732 (2019). [Google Scholar]
- 51.Zhang Y, Qian J, Wang M, Huang Y & Hu P Visible-Light-Induced Decarboxylative Fluorination of Aliphatic Carboxylic Acids Catalyzed by Iron. Organic Letters 24, 5972–5976 (2022). [DOI] [PubMed] [Google Scholar]
- 52.Tu J-L et al. Iron-catalyzed ring-opening of cyclic carboxylic acids enabled by photoinduced ligand-to-metal charge transfer. Green Chemistry 24, 5553–5558 (2022). [Google Scholar]
- 53.Lu Y-C & West JG Chemoselective Decarboxylative Protonation Enabled by Cooperative Earth-Abundant Element Catalysis. Angewandte Chemie International Edition 62, e202213055 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xu P, López-Rojas P & Ritter T Radical Decarboxylative Carbometalation of Benzoic Acids: A Solution to Aromatic Decarboxylative Fluorination. Journal of the American Chemical Society 143, 5349–5354 (2021). [DOI] [PubMed] [Google Scholar]
- 55.Chen TQ et al. A Unified Approach to Decarboxylative Halogenation of (Hetero)aryl Carboxylic Acids. Journal of the American Chemical Society 144, 8296–8305 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li QY et al. Decarboxylative cross-nucleophile coupling via ligand-to-metal charge transfer photoexcitation of Cu(ii) carboxylates. Nature Chemistry 14, 94–99 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bian K-J, Kao S-C, Nemoto D, Chen X-W & West JG Photochemical diazidation of alkenes enabled by ligand-to-metal charge transfer and radical ligand transfer. Nature Communications 13, 7881 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Glüge J. et al. An overview of the uses of per- and polyfluoroalkyl substances (PFAS). Environmental Science: Processes & Impacts 22, 2345–2373 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dénès F, Pichowicz M, Povie G & Renaud P Thiyl Radicals in Organic Synthesis. Chemical Reviews 114, 2587–2693 (2014). [DOI] [PubMed] [Google Scholar]
- 60.Simmons EM & Hartwig JF On the Interpretation of Deuterium Kinetic Isotope Effects in C─H Bond Functionalizations by Transition-Metal Complexes. Angewandte Chemie International Edition 51, 3066–3072 (2012). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors declare that all the data supporting the findings of this research are available within the article and its supplementary information.