

Abstract
INTRODUCTION
In 2013, the ALzheimer's and FAmilies (ALFA) project was established to investigate pathophysiological changes in preclinical Alzheimer's disease (AD), and to foster research on early detection and preventive interventions.
METHODS
We conducted a comprehensive genetic characterization of ALFA participants with respect to neurodegenerative/cerebrovascular diseases, AD biomarkers, brain endophenotypes, risk factors and aging biomarkers. We placed particular emphasis on amyloid/tau status and assessed gender differences. Multiple polygenic risk scores were computed to capture different aspects of genetic predisposition. We additionally compared AD risk in ALFA to that across the full disease spectrum from the Alzheimer's Disease Neuroimaging Initiative (ADNI).
RESULTS
Results show that the ALFA project has been successful at establishing a cohort of cognitively unimpaired individuals at high genetic predisposition of AD.
DISCUSSION
It is, therefore, well‐suited to study early pathophysiological changes in the preclinical AD continuum.
Highlights
Prevalence of ε4 carriers in ALzheimer and FAmilies (ALFA) is higher than in the general European population
The ALFA study is highly enriched in Alzheimer's disease (AD) genetic risk factors beyond APOE
AD genetic profiles in ALFA are similar to clinical groups along the continuum
ALFA has succeeded in establishing a cohort of cognitively unimpaired individuals at high genetic AD risk
ALFA is well suited to study pathogenic events/early pathophysiological changes in AD
Keywords: AD continuum , ALFA study, Alzheimer's disease, neurogenetics, neurological diseases, prevention
1. BACKGROUND
In 2013, the ALzheimer's and FAmilies (ALFA) project was launched by the Barcelonaβeta Brain Research Center with the aim of enhancing our understanding of the pathogenesis and pathophysiology of Alzheimer's disease (AD) at its early preclinical stages. 1 The ALFA project consists of the ALFA parent cohort (NCT01835717), and the nested ALFA+ cohort study (NCT02485730). The ALFA parent cohort is composed of 2743 cognitively unimpaired participants aged between 45 and 74 years at the time of recruitment who underwent cognitive testing, clinical history, magnetic resonance imaging (MRI), environmental and lifestyle questionnaires, and blood sampling. A significant feature of ALFA is that it is a genetically enriched cohort of cognitively unimpaired individuals, as ∼50% of them are adult children of patients with AD dementia diagnosed before the age of 75 years. The recruitment strategy of the ALFA parent cohort has resulted in a population that is enriched in genetic risk factors for AD, the most notable of which is the apolipoprotein E (APOE)‐ε4 allele. A subset of the ALFA parent cohort participants were invited to participate in a nested longitudinal long‐term study, which is referred to as ALFA+. This study involved more detailed phenotyping, including fluid (blood and cerebrospinal fluid [CSF]) and positron emission tomography (PET). ALFA+ participants were selected according to their age, sex, and APOE‐ε4 carriership to cover the full range of the AD risk spectrum.
In a pilot study (NCT02198586), ∼575 ALFA parent cohort participants underwent MRI to investigate the effects of APOE variants on different cerebral phenotypes. As a result of this project, we found that ε4‐homozygous displayed an age‐related increase in radial but not axial diffusivity, consistent with a reduced myelin sheath in several white matter regions. 2 , 3 When assessing gray matter volumes in APOE‐ε4 homozygous, we found reductions in brain regions known to undergo atrophy in symptomatic AD stages 4 and dose‐dependent protective effects of the APOE‐ε2 allele. 5 We also showed, as suggested in previous studies, that APOE‐ε4 reversed the association between cognitive performance and brain morphology, similar to aging, suggesting that this risk allele leads to an accelerated biological phenotype of brain aging. 6 Furthermore, ε4‐homozygous displayed reduced connectivity between networks in areas typically susceptible to amyloid deposition in the early AD continuum, and altered effects of amyloid on brain structure. 7 , 8 Additional results indicated that cognitively unimpaired APOE‐ε4 homozygous were at significantly higher risk of having pathological levels of white matter hyperintensities (WHM) than heterozygotes, 9 and observed a protective effect of the ε2 allele on global WMH. 10 We also found a higher prevalence of cerebral microbleeds in APOE‐ε4 carriers, 11 although no association was found between APOE‐ε4 and the enlargement of perivascular spaces. 12
These findings have encouraged us to extend the genetic characterization of the ALFA parent cohort beyond APOE to deepen our understanding of the biological mechanisms associated with genetic predisposition to AD at preclinical stages, which may aid in the implementation of prevention programs. In this article, we present the rationale, methods, and genetic characterization of participants of the ALFA parent cohort. In addition, we assessed AD genetic risk prediction across the spectrum of the disease by including data from the Alzheimer's Disease Neuroimaging Initiative (ADNI) project. 13
2. METHODS
2.1. Design
On the September 17, 2012, a press conference was held where the main aims of the ALFA project were presented along with detailed inclusion and exclusion criteria. 1 Briefly, participants had to be cognitively unimpaired Spanish and/or Catalan‐speaking individuals aged 45–74 who agreed to undergo clinical interviews and questionnaires associated with dementia risk factors, cognitive tests, a blood sample extraction for DNA analysis, and MRI. A total of 2743 individuals were recruited in the ALFA parent cohort, with 86.3% reporting a diagnosis of AD in at least one of their parents. When considering a more strict family history encoding, 47.4% of the ALFA study participants had at least one of their parents that had been diagnosed with AD before the age of 75 years. In total, 2686 participants were genotyped, as 57 individuals were excluded since blood extraction could not be performed or sufficient DNA could not be obtained to perform the genotyping. The genetic data processing procedure is detailed in Figure S1. Summary information on project participants (Figure 1), DNA extraction, genotyping, and data availability is described in Supplementary methods. A subset of ALFA parent cohort (N = 380), referred to as ALFA+, also underwent lumbar puncture to determine biomarkers in CSF, enhanced MRI, more detailed cognitive testing, blood sampling for biomarker determination, 18F‐fluoro‐2‐deoxyglucose PET, amyloid, and tau PET imaging. Participants were classified into A/T groups defined by their biomarker profile according to the A/T framework described in Ref. 14 Briefly, CSF amyloid‐β 42 (Aβ42) levels of ≤1098 pg/ml were designated as A+, and phosphorylated tau (pTau) levels of ≥24 pg/ml were designated as T+. 15
FIGURE 1.
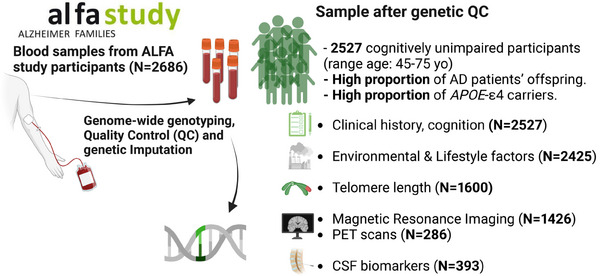
Summarized information of the characteristics of project participants.
RESEARCH IN CONTEXT
Systematic review: The ALFA (ALzheimer and FAmilies) project prospectively follows cognitively unimpaired late‐/middle‐aged participants, most of whom are adult offspring of Alzheimer's disease (AD) patients, with the aim of investigating pathophysiological events that occur during the preclinical stages of AD and to test preventive interventions. We aimed here to genetically characterize ALFA participants and to describe AD risk prediction across the disease spectrum.
Interpretation: The ALFA study population is highly enriched for AD genetic risk factors beyond apolipoprotein E (APOE), defined by individuals in the early stages of AD. ALFA study participants show heterogeneous genetic profiles similar to clinical groups along the AD continuum. Results support that the ALFA parent cohort and, even more, ALFA+ are highly enriched for genetic risk factors for AD. This made them suitable for investigating the early pathophysiological alterations in AD.
Future directions: Integration with other ‐omic data modalities and risk factors, as well as collaborating with global initiatives will be crucial to discover underlying disease mechanisms.
2.2. Genetic quality control and imputation
Quality control (QC) of genetic data was performed with the PLINK software. Samples with a call rate of less than 98%, mismatched genetically determined sex (from X‐chromosome heterogeneity) versus that coming from demographic data, or excess of heterozygosity (four standard deviations from the mean) estimated by an F statistic, were excluded from the analyses. Individuals at high genetic relatedness (at the level of cousins or closer relatives), sharing proportionally more than 18.5% of alleles (IBD > 0.185), were also excluded. After completion of sample level QC, genetic variants with low minor allele frequency (MAF < 1%), a Hardy‐Weinberg equilibrium (p < 10−6), and missingness rates >5% were also excluded. Population stratification was checked by clustering the samples using principal component analysis. After the QC procedure, a total of 2527 cognitively unimpaired individuals from the ALFA parent cohort were genetically characterized.
Imputation of genetic variants was carried out using the Michigan Imputation Server with the Haplotype Reference Consortium Panel (HRC r1.1 2016) 16 , 17 following default parameters and established guidelines. Mismatched and monomorphic genetic variants were removed during pre‐imputation filtering. Genetic variants with invalid alleles were switched and flipped in the genome‐wide association studies (GWAS) sets (Human Reference Panel: GRCh37 hg19 b37 humanG1Kv37). Phasing was performed using the EAGLE software v.2.4, and imputation results were rechecked using standard Michigan imputation server QC parameters (imputation quality >0.2 and MAF >1%).
2.3. Statistical analysis
Differences in demographic characteristics were assessed according to APOE‐ε4 carriership, AT groups and sex, using chi‐square tests for categorical variables and parametric (t‐test, ANOVA) and non‐parametric tests (Wilcox test or Kruskal‐Wallis) for continuous normally and non‐normally distributed variables, as appropriate. Pairwise comparison p‐values were also provided, adjusted for false discovery rate correction.
Polygenic risk scores (PRS) were computed using PRSice version 2 18 to assess polygenicity. PRSice computes PRS by summing all SNP alleles carried by participants, weighting them by the SNP allele effect size estimated in a previous GWAS, and normalizing the score by the total number of SNPs included. PRS were calculated in representative genetic variants per linkage disequilibrium block (LD) (clumped variants), using a cutoff for LD of r2 > 0.1 in a 250‐kb window. For PRS calculation of AD and AD CSF biomarkers, the APOE region was additionally excluded (chr19:45,409,011‐45,412,650; GRCh37/hg19). Results were displayed at a restrictive threshold, 5 × 10−6. A total of 40 PRS were calculated based on recently published GWAS, distributed in 6 main categories of diseases and conditions: AD biomarkers, 19 neurodegenerative diseases, 20 , 21 , 22 cerebrovascular diseases, 23 , 24 brain endophenotypes of AD, 25 , 26 , 27 AD risk factors, 28 , 29 , 30 , 31 , 32 , 33 , 34 , 35 and aging biomarkers. 36 , 37 , 38 , 39 Further details on the PRS can be found in Table S1. In addition, individuals were classified into three groups based on the distribution of PRS for AD, Aβ42, and pTau: low genetic predisposition (PRS percentile 10), intermediate genetic predisposition (percentile 10 < PRS percentile 90), and high genetic predisposition (PRS > percentile 90). Additionally, significant differences in the median values (Wilcoxon test) and variances (Levene test) of the PRS were evaluated between APOE‐ε4 carriers, AT groups in the ALFA+ cohort, and sex. Finally, the AD predisposition in ALFA was compared (pairwise median test) to that across the full disease spectrum in the ADNI database by including controls (CN) (N = 530), amyloid positive individuals with mild cognitive impairment (MCI) (N = 598), and AD dementia patients (N = 205). ALFA+ and ADNI participants were further stratified according to amyloid status (ALFA+: A+ N = 134, A‐ N = 246; ADNI: CN A+ N = 96, MCI A+ N = 264, AD A+ N = 132).
3. RESULTS
3.1. Demographic characteristics and APOE genotype distribution
A characteristic feature of the ALFA study is the high prevalence of APOE‐ε4 carriers compared with the general European population (35.6% vs. 14%; p < 0.001), 40 and the high prevalence of homozygotes in the ALFA+ subsample (Table 1, Figure 2). Significant differences in demographic characteristics were found between APOE‐ε4 non‐carriers (N = 1624; 64.39%) and carriers (N = 898; 35.61%). Non‐carriers were on average older than ε4‐homozygous individuals (p = 0.024). The percentage of women in the non‐carrier group was higher than in the APOE‐ε4 heterozygous one (p = 0.038). We also observed a higher percentage of individuals with a family history of AD in APOE‐ε4 heterozygous compared to non‐carriers (p < 0.001) (Figure 2A, Table S2). In ALFA+, a total of 8% (N = 31) of the individuals were APOE‐ε4 homozygous. Moreover, among APOE‐ε4 carriers, 8.2% of individuals were A+T+ (vs. 7.5% among non‐carriers), 41.3% were A+T‐ (vs. 10.9% among non‐carriers), and 50.5% were A‐T‐ (vs. 81.6% among non‐carriers) (Figure 2B, Table S3, and S4). Notably, there was a higher percentage of A+ individuals among APOE‐ε4 carriers compared with non‐carriers (49.5% vs. 18.4%; p < 0.001). Similar to the overall sample, the proportion of women was higher in the non‐carrier group compared to the APOE‐ε4 heterozygous one (p = 0.029). No significant differences were found in the distribution of individuals according to family history of AD.
TABLE 1.
Demographic characteristics and APOE genotypes distribution in the ALFA parent cohort as well as in the ALFA+ subsample
| ALFA parent cohort (N = 2527) | N | ALFA+ subsample (N = 380) | N | |
|---|---|---|---|---|
| Age (years, median) | 56.00 [51.00;62.00] | 2527 | 57.00 [53.75;61.00] | 380 |
| Sex (n, %): | 2527 | 380 | ||
| Men | 932 (36.88%) | 149 (39.21%) | ||
| Women | 1595 (63.12%) | 231 (60.79%) | ||
| Education (years, median) | 12.00 [10.00;17.00] | 2527 | 12.00 [11.00;17.00] | 380 |
| Mini Mental State Exam (0‐30, median) | 29.00 [28.00;30.00] | 803 | 30.00 [29.00;30.00] | 68 |
| APOE‐ε4 load (n,%) | 2522 | 380 | ||
| Non‐carriers | 1624 (64.39%) | 174 (45.79%) | ||
| One ε4 allele | 810 (32.12%) | 175 (46.05%) | ||
| Two ε4 alleles | 88 (3.49%) | 31 (8.16%) | ||
| APOE‐ε4 status (n, %) | 2522 | 380 | ||
| Non‐carriers | 1624 (64.39%) | 174 (45.79%) | ||
| Carriers | 898 (35.61%) | 206 (54.21%) | ||
| Family history (FH) of AD (n,%) | 2527 | 380 | ||
| No | 294 (11.63%) | 28 (7.37%) | ||
| Yes | 2233 (88.37%) | 352 (92.63%) | ||
| Parental family history of AD (n,%) | 2527 | 380 | ||
| Both | 97 (3.84%) | 19 (5.00%) | ||
| Father | 679 (26.87%) | 107 (28.16%) | ||
| Mother | 1457 (57.66%) | 226 (59.47%) | ||
| No Family History of AD | 294 (11.63%) | 28 (7.37%) | ||
| Family history of AD (n,%) | 2527 | 380 | ||
| age of onset < 75 | 1436 (56.83%) | 232 (61.05%) | ||
| age of onset > = 75 | 866 (34.27%) | 120 (31.58%) | ||
| at least one < 75 | 72 (2.85%) | 16 (4.21%) | ||
| both > = 75 | 45 (1.78%) | 7 (1.84%) | ||
| No FH | 108 (4.27%) | 5 (1.32%) | ||
| Amyloid status (n, %) | 380 | |||
| Aβ42/Aβ40‐ | 246 (64.74%) | |||
| Aβ42/Aβ40+ | 134 (35.26%) | |||
| AT classification (n, %) | 380 | |||
| A‐T‐ | 246 (64.74%) | |||
| A+T‐ | 104 (27.37%) | |||
| A+T+ | 30 (7.89%) | |||
| CSF Aβ142 (pg/ml, median) | 1160.00 [852.80; 1563.50] | 376 | ||
| CSF pTau181 (pg/ml, median) | 14.18 [11.09; 18.59] | 377 | ||
| Centiloids (whole cerebellum) (median) | −1.32 [−6.52; 5.30] | 324 |
Note: A total of 5 individuals presented NA values in APOE‐ε4 characterization. A total of 13 individuals with A‐T+ profile were excluded from the ALFA+ subsample. Available CSF biomarkers data after outliers removal. A total of 56 individuals presented NA values in Centiloids. Amyloid positivity is defined as Aβ42/Aβ40 < 0.071.
Abbreviations: ALFA, ALzheimer's and FAmilies; APOE, apolipoprotein E; CSF, cerebrospinal fluid.
FIGURE 2.
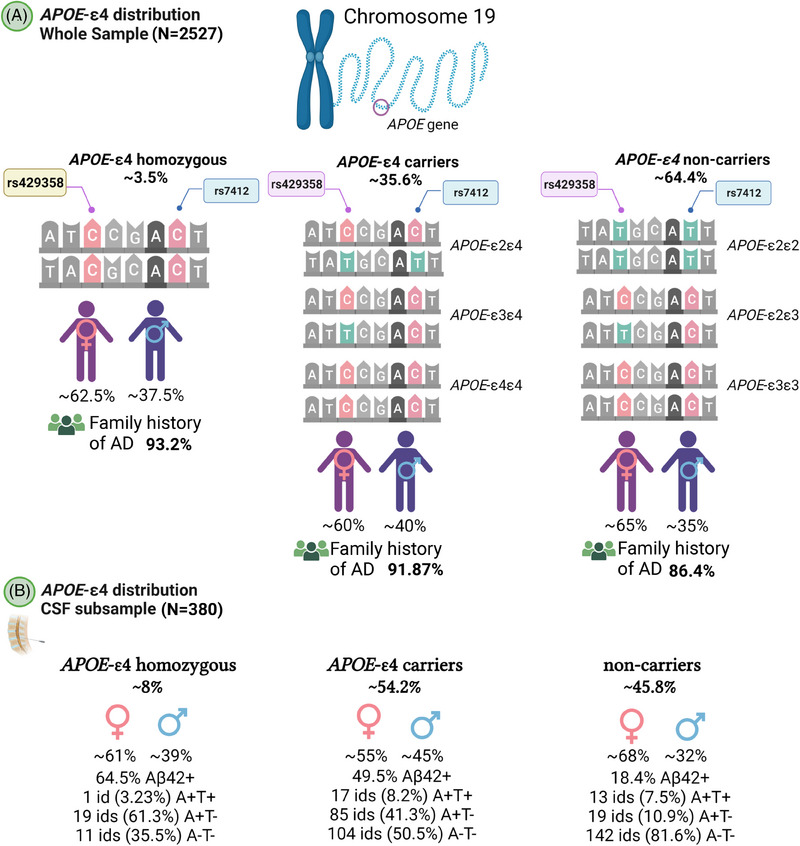
Distribution of APOE‐ε4 carriers in the ALFA and ALFA+ samples. ALFA, ALzheimer's and FAmilies; APOE, apolipoprotein E.
3.2. Polygenic characterization of ALFA participants
PRS of AD (PRS‐AD) showed the highest variability in the sample (IQR = 0.25) as well as frontotemporal dementia (FTD) subtypes (IQR > 0.035), sleep risk factors and biological aging phenotypes (IQR > 0.04), whereas Parkinson's disease (IQR = 3·10−5), cerebral infarction (IQR = 8·10−5), and intracerebral hemorrhage (IQR = 2·10−5) were the main conditions in which subjects had more homogeneous scores (IQR ∼ 0) (Figure 3, Table S5).
FIGURE 3.
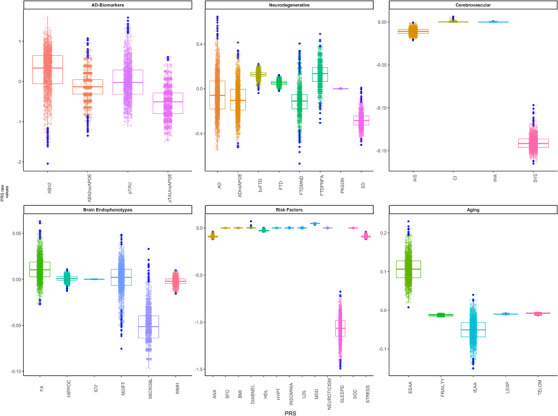
Distribution of genetic scores in the ALFA parent cohort. ALFA, ALzheimer's and FAmilies.
We observed that ε4 homozygous had higher scores for AD and FTD, AD biomarkers, and some brain endophenotypes, risk factors and aging conditions compared with heterozygotes and non‐carriers (Figure S2). Moreover, we observed that A+T‐ individuals had higher scores for PRS‐AD (p = 0.006) as well as lower median PRS of CSF Aβ42 levels than A‐T‐ (p ∼ 0). In addition, A+T‐ showed higher genetic predisposition to WMH than A‐T‐ (p = 0.006). Similarly, A+T‐ showed a higher median for the risk score of microbleeds and longer sleep duration than A‐T‐ (p < 0.018) (Figure S3, Table S6). Finally, only significant differences were found between sex for genetic predisposition to social isolation and sleep duration (Figure S4, Table S7).
3.3. Characteristics of individuals with AD genetic predisposition
We found significant differences in the percentage of ε4 carriers among individuals classified at high (81%), intermediate (33.3%), and low (7.94%) genetic predisposition to AD (p < 0.001) (Table S8). When the APOE region was excluded from the PRS‐AD, the proportion of ε4 carriers and non‐carriers was balanced. When classifying individuals based on both the PRS‐AD and the PRS‐ADno APOE , the proportion of individuals with a positive family history of AD was higher in the group with high genetic predisposition to AD compared to the rest of groups. We did not find any differences in the proportion of women and men across groups. In ALFA+, we also observed a higher percentage of ε4 carriers among individuals at high genetic predisposition to AD (89.39%) compared to individuals at intermediate genetic predisposition (50.53%, p < 0.001) and low (12.90%, p < 0.001) genetic predisposition. In the high genetic group, 21.2% of the individuals were 𝜀4‐homozygous (Table S9). The proportion of A+T‐ and A+T+ was also significantly higher in the group at higher genetic predisposition of AD compared to the intermediate and low groups (p < 0.05). When removing the APOE region from the genetic score computation these significant differences were no longer observed. When we classified individuals according to their genetic predisposition to higher CSF‐Aβ42 levels, we found that the proportion of APOE‐ε4 carriers and, specifically, homozygotes, was significantly higher in the genetically predisposed group to present abnormal levels of CSF‐Aβ42 (low genetic predisposition to higher CSF‐Aβ42 levels) compared to the other groups (p < 0.001). However, when we removed the APOE region from the PRS‐Aβ42, we did not observe significant differences in the percentage of APOE‐ε4 carriers among groups (Table S10). Similarly, we found a higher proportion of APOE‐ε4 carriers, and specifically, homozygotes, in the group of individuals at higher genetic predisposition to display higher CSF‐pTau levels (at risk group) (p < 0.001). However, we did not find these differences when removing the effect of the genetic variants in the APOE region (Table S11). The above results were also assessed for the subgroup of individuals with available CSF biomarkers from the ALFA+ study. Significant differences were also found in the distribution of APOE‐ε4 carriers among genetic groups based on their genetic predisposition to CSF Aβ42 levels (Table S12). As in the whole sample, higher percentages of both APOE‐ε4 heterozygous and homozygous were found in the group at lower genetic predisposition to higher CSF Aβ42 levels compared with the high and intermediate groups (p < 0.001). Moreover, in this group we found a higher percentage of A+ individuals compared with the intermediate (p = 0.008) and high (p = 0.008) genetic groups. In the low genetic group, we found a higher percentage of A+T+ individuals than in the higher group (p = 0.012) but lower than in the intermediate group (p = 0.005). These differences were not observed when removing the APOE region. Significant differences were also observed between genetic groups based on their genetic predisposition to CSF pTau levels (Table S13). As in the whole sample, higher percentages of both APOE‐ε4 carriers and homozygous were found in the group showing high genetic predisposition to higher CSF pTau levels compared with the low and intermediate groups (p < 0.001). Moreover, in the high group, we found a higher percentage of A+ individuals as well as a higher percentage of A+T+ individuals compared with the low and intermediate genetic groups (pairwise comparisons p < 0.05). Non‐significant differences were observed after removing the APOE region.
3.4. AD genetic risk prediction in the whole disease spectrum
The median value of the PRS‐AD increased along the AD continuum (Figure 4). In ADNI (Table S14), significant differences in the median score of PRS‐AD were observed between CN and MCI (p < 0.001) and between CN and AD (p < 0.001). Significant differences were also found between MCI and AD subjects (p < 0.05). When participants from the ALFA parent cohort were included in the spectrum, we found that the median value of the PRS‐AD in ALFA participants was higher than in CN from ADNI (p < 0.001) although was significantly lower than in MCI (p < 0.01) and AD (p < 0.001) (Figure 4A). When ADNI and ALFA+ individuals were stratified by Aβ status, we found that the genetic predisposition to AD was higher in ALFA+ A+ than in ALFA+ A‐ (p < 0.001) (Figure 4B). Moreover, the median PRS‐AD in ALFA+ A+ individuals was higher compared both to CN A‐ (p < 0.001) and CN A+ (p < 0.05). Although there were no significant differences between them, ALFA+ A+ individuals showed a higher median value of the PRS‐AD than MCI A+. Non‐significant differences were found between ALFA+ A+ and AD individuals (Table S15). Comparisons stratifying by other risk factors were assessed (Figure 5). We found significant differences among APOE‐ε4 groups for all disease‐stage groups (Figure 5A). The higher the number of ε4 alleles, the higher the median value of the PRS‐AD. Neither in ALFA A‐ nor in A+ significant differences were found in the median PRS‐AD between ε4 allele heterozygous and homozygous. Nonetheless, in both groups, both ε4 allele heterozygous and homozygous displayed higher values for the median PRS‐AD than non‐carriers (p < 0.001). Differences were also assessed stratifying by A/T profiles (Figure 5B). In the ALFA+ sample, A+T‐ individuals showed higher median PRS‐AD than A‐T‐ (p < 0.001). In ADNI, CN that were A+T+ displayed higher median values than A‐T‐ (p < 0.01). In MCIs, significant differences were found between all pairwise comparisons (p < 0.05), while in AD patients, non‐significant differences were found in the median value of the PRS‐AD among AT groups. Finally, we did not observe significant sex‐differences within ADNI nor in ALFA groups (Figure 5C). Moreover, when removing the APOE region, the median score was significantly lower (p < 0.05) in CN A‐ compared to AD A+ from ADNI (Figure S5A, Table S16). Also, we consistently observed a higher median score in ALFA+ A+, MCI A+, and AD A+ groups, specifically for less restrictive inclusion thresholds (Tables S17–S, 20). An extended characterization of other neurodegenerative and aging‐related scores along the AD continuum can be found in the Supplementary Results documentation.
FIGURE 4.
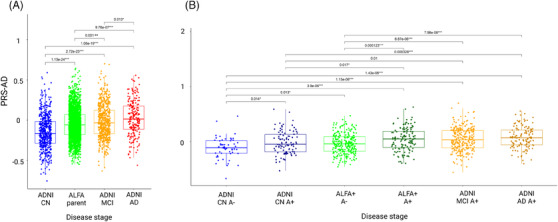
Distribution of genetic scores of AD along the AD continuum. (A) Subgroups from ALFA and ADNI within the AD continuum. (B) Subgroups within the AD continuum stratifying ALFA participants by amyloid status. Pairwise comparisons are assessed to compare the median PRS‐AD (Wilcoxon test) among groups. Significant results at nominal p‐value are displayed (p‐value < 0.05*; p‐value < 0.01**, p‐value < 0.001***). AD, Alzheimer's disease; ADNI, Alzheimer's Disease Neuroimaging Initiative; ALFA, ALzheimer's and FAmilies; PRS‐AD, polygenic risk scores of AD.
FIGURE 5.
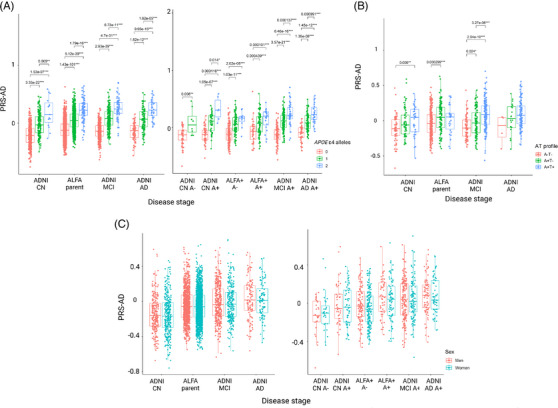
Distribution of genetic scores of AD along the AD continuum stratified by risk factors. (A) Distribution stratified by APOE‐ε4 carriership. (B) Distribution stratified by AT group. (C) Distribution stratified by sex. Pairwise comparisons are assessed to compare the median PRS‐AD (Wilcoxon test) among groups. Significant results at nominal p‐value are displayed (p‐value < 0.05*; p‐value < 0.01**, p‐value < 0.001***). AD, Alzheimer's disease; APOE, apolipoprotein E; PRS‐AD, polygenic risk scores of AD.
4. DISCUSSION
The genetic characterization of ALFA parent cohort participants enabled us to investigate the genetic predisposition to neurodegenerative and other complex diseases in cognitively unimpaired individuals, many of whom were in the preclinical AD continuum. Our results showed a significant enrichment of genetic risk factors for AD in the ALFA parent cohort, further highlighting its suitability for studying early pathophysiological alterations in AD. Specifically, we observed a substantial genetic predisposition to AD in ALFA participants, even in the absence of the APOE‐ε4 allele. These findings suggest that other genetic variants, independent of APOE, may play a role in modulating the risk of AD and potentially compensate for or complement the effect of APOE in genetic predisposition to the disease. Our results align with previous studies, 41 , 42 emphasizing the importance of considering additional APOE‐independent mechanisms when exploring genetic predisposition to AD. This illustrates how the use of PRS can reveal additional pathophysiological pathways with potential for prevention, 43 which is relevant since the pathophysiology of AD has not yet been fully elucidated and the best biological characterization to date focuses on the determination of AD biomarkers. 44 , 45 However, it is important to emphasize the contribution of APOE to the observed genetic enrichment. APOE has been well‐established as a major genetic risk factor for AD, primarily associated with early alterations in Aβ42 metabolism. 46 In our study, individuals at genetic predisposition to AD displayed abnormal CSF Aβ42 levels and were enriched for APOE‐ε4 homozygosity. These findings support the notion that APOE plays a crucial role in the early pathogenesis of AD, while also suggesting that additional genetic variants contribute to the overall genetic risk profile.
Furthermore, comparing the PRS‐AD profiles of the ALFA+ participants, particularly those already on the AD continuum (A+), with clinical groups in ADNI, revealed that the ALFA participants exhibited a higher genetic risk profile than CN in ADNI, and similar to that of the clinical groups (MCI, AD). These results support the notion that the ALFA cohort is well‐suited for detecting early pathogenic events in AD, encompassing both APOE‐associated and independent biological pathways. The study also confirmed that CN individuals with abnormal AD biomarkers had a higher genetic predisposition to AD, as shown by the distribution of PRS‐AD against AT groups. These results suggest that PRS could be used as a proxy for risk stratification of unimpaired individuals. 47 , 48 However, further research is needed to establish the additional information that PRS provide in addition to biomarkers and their utility in research and clinical practice.
Moreover, individuals showing AD pathological changes (A+T‐) showed higher genetic predisposition to AD, WMH, long sleep duration, and microbleed counts compared to subjects with normal levels of AD biomarkers (A‐T‐). We did not detect any differences in PRS variability in T+ individuals, likely due to the low sample size of this group at the moment. These findings suggest different patterns of genetic predisposition in the Alzheimer's continuum and/or increased susceptibility to comorbidities . 49 , 50 For instance, our results imply a possible common pathological mechanism linking genetic predisposition to cerebrovascular disease and sleep measurements to the development of amyloid‐beta pathology at the earliest stages of the AD continuum. Further studies that consider scores regardless of the inclusion of the APOE region will additionally give insights into the biological interpretation of the results.
We observed slight differences in variability of genetic scores between sexes. Analyses using sex‐specific summary statistics will help to better determine the mechanisms associated with such differences and to understand structural and functional differences in the brain, as well as cognitive and age‐related processes. 51 , 52 In addition, previous studies suggest the influence of sex‐specific variants with small effects or more complex mechanisms involving epigenetic changes, gene‐environment interactions, or genetic variants within sex chromosomes that should be incorporated into future studies. 53 , 54
Methodological limitations also should be taken into account, such as the subtle arbitrariness in the selection of the threshold for including genetic variants in the computation of genetic scores, alongside the threshold for categorizing individuals into genetic risk groups.
Our research aims to systematically address these challenges and significantly impact the field by collaborating with global initiatives like the UNITED (Uncovering Neurodegenerative Insights Through Ethnic Diversity) Consortium, 55 the European Prevention of Alzheimer's Dementia (EPAD) cohort, 56 and the Quantitative amyloid PET in Alzheimer's disease (AMYPAD) initiative, 57 and using publicly available genetic data from other resources. 58 This will manifest in increased statistical power and precision in determining genetic variants for score computation, as well as in a more profound understanding of the genetic mechanisms underlying AD worldwide, while also considering diverse populations.
By leveraging the ALFA cohort's substantial genetic risk profile, our research gains a unique advantage in unraveling the initial pathophysiological alterations within the preclinical AD continuum. Through the incorporation of multiple PRSs and focusing on distinct genetic markers, our research not only advances the comprehension of the genetic underpinnings of AD but also reveals promising additional strengths for interventions at an early stage. The complex nature of AD furthermore adds more value to the ALFA cohort, as it propels the utilization of alternative modalities, allowing us to characterize the biological pathways involved in AD processes. This multifaceted approach enhances our understanding of how molecular differences correlate with in vivo neuroimaging phenotypes. 59 Additionally, our incorporation of techniques such as Mendelian Randomization provides a means to establish causal links between modifiable risk factors and AD pathology, including its subsequent consequences, 60 ultimately influencing the potential to establish primary and secondary prevention strategies during the primary and preclinical stages. 61
CONFLICT OF INTEREST STATEMENT
J.L.M. is currently a full‐time employee of Lundbeck and has previously served as a consultant or at advisory boards, or has given lectures in symposia sponsored by the following for‐profit companies: Roche Diagnostics, Genentech, Novartis, Lundbeck, Oryzon, Biogen, Lilly, Janssen, Green Valley, MSD, Eisai, Alector, BioCross, GE Healthcare, ProMIS Neurosciences. J.D.G. has received speaker's or consultant's fees from Philips Nederlands, Roche Diagnostics and Biogen and research support from GE Healthcare, Roche Diagnostics and Hoffmann‐La Roche. M.S.C. has served as a consultant and at advisory boards for Roche Diagnostics International Ltd., has given lectures in symposia sponsored by Roche Diagnostics, S.L.U., Roche Farma, S.A. and Roche Sistemas de Diagnósticos, Sociedade Unipessoal, Lda. and research support from Roche Diagnostics International Ltd. G.S.B. has served as a consultant for Roche Farma, S.A. The remaining authors declare that they have no conflict of interest. Author disclosures are available in the supporting information.
CONSENT STATEMENT
The study was conducted in accordance with the directives of the Spanish Law 14/2007, of July 3 , on Biomedical Research (Ley 14/2007 de Investigación Biomédica). The ALFA study protocol was approved by the Independent Ethics Committee Parc de Salut Mar Barcelona and registered at Clinicaltrials.gov (Identifier: NCT01835717). All participants accepted the study procedures by signing the study's informed consent form that had also been approved by the same Institutional Review Board.
Supporting information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
ACKNOWLEDGMENTS
This publication is part of the ALFA study. The authors express their most sincere gratitude to the ALFA project participants and relatives without whom this research would have not been possible. Collaborators of the ALFA study are: Müge Akinci, Federica Anastasi, Annabella Beteta, Raffaele Cacciaglia, Lidia Canals, Alba Cañas, Carme Deulofeu, Maria Emilio, Irene Cumplido‐Mayoral, Marta del Campo, Carme Deulofeu, Ruth Dominguez, Maria Emilio, Sherezade Fuentes, Marina García, Laura Hernández, Gema Huesa, Jordi Huguet, Laura Iglesias, Esther Jiménez, David López‐Martos, Paula Marne, Tania Menchón, Paula Ortiz‐Romero, Eleni Palpatzis, Wiesje Pelkmans, Albina Polo, Sandra Pradas, Iman Sadeghi, Mahnaz Shekari, Lluís Solsona, Anna Soteras, Laura Stankeviciute, Núria Tort‐Colet, and Marc Vilanova.
We additionally express our sincere gratitude to Prof. Kaj Blennow and Prof. Henrik Zetterberg for their invaluable assistance in acquiring the biomarkers used in the ALFA+ study. We would also like to acknowledge the guidance and expertise of Prof. Roderic Guigó in the acquisition and modelization of the genetic data, and Prof. Anna González‐Neira for her guidance in genetic quality control. Finally, we extend our heartfelt appreciation to Prof. Jordi Camí for his significant contribution to the design of the ALFA parent cohort. The authors thank Roche Diagnostics International Ltd for providing the kits to measure CSF biomarkers, and the laboratory technicians at the Clinical Neurochemistry Lab in Mölndal, Sweden, who performed the analyses. COBAS, COBAS E, and ELECSYS are trademarks of Roche.
Data collection and sharing for this project was funded by the Alzheimer's Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH‐12‐2‐0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie, Alzheimer's Association; Alzheimer's Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol‐Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann‐La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer's Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California.
We thank all the Consortium members involved in the analysis and generation of summary statistics used in this project.
CSF Amyloid Aβ42 (https://www.ebi.ac.uk/gwas/ Accession ID GCST90129599); CSF Tau phosphorylated (https://www.ebi.ac.uk/gwas/ Accession ID GCST90129600); Leukocyte Telomere Length (Requested to corresponding Author); Fractional Anisotropy (http://www.kp4cd.org/dataset_downloads/stroke); Hippocampal volume (http://ftp.ebi.ac.uk/pub/databases/gwas/summary_statistics/GCST90002001‐GCST90003000/GCST90002711/); Mean Diffusivity (http://www.kp4cd.org/dataset_downloads/stroke); Microbleeds (http://www.kp4cd.org/dataset_downloads/stroke); White Matter Hyperintensities (http://www.kp4cd.org/dataset_downloads/stroke); Ischemic Stroke (Requested to corresponding Author); Cerebral infarction (http://geneatlas.roslin.ed.ac.uk); Small Vessel Disease (Requested to corresponding Author); Alzheimer's Disease (https://ctg.cncr.nl/software/summary_statistics); Frontotemporal Dementia and subtypes (https://ifgcsite.wordpress.com/data‐access/); Parkinson's Disease (https://drive.google.com/file/d/1FZ9UL99LAqyWnyNBxxlx6qOUlfAnublN/view?usp=sharing); Semantic Dementia (https://ifgcsite.wordpress.com/data‐access/); Anxiety Disorder (https://ipsych.dk/en/research/downloads/); Body Mass Index (https://portals.broadinstitute.org/collaboration/giant/index.php/GIANT_consortium_data_files); HDL Cholesterol (http://csg.sph.umich.edu/willer/public/lipids2010/); Hyperthyroidism/thyrotoxicosis (http://geneatlas.roslin.ed.ac.uk); Hypothyroidism/myxoedema (http://geneatlas.roslin.ed.ac.uk); Hypertension (http://geneatlas.roslin.ed.ac.uk); Insomnia (https://ctg.cncr.nl/software/summary_statistics); LDL Cholesterol (http://csg.sph.umich.edu/willer/public/lipids2010/); Life Expectancy (http://ftp.ebi.ac.uk/pub/databases/gwas/summary_statistics/GCST003001‐GCST004000/GCST003395/results.UKBiobank_9millionSNPs.parents_lifespan.Pilling_et_al_2016_top_1_percent.txt); Sleep Duration (https://ctg.cncr.nl/software/summary_statistics); Stress Disorder (https://ipsych.dk/en/research/downloads/). Further details can be found in Table S1. Schematic representations were created with Biorender.com. The project leading to these results has received funding from “la Caixa” Foundation (ID 100010434), under agreement LCF/PR/GN17/50300004, the Health Department of the Catalan Government (Health Research and Innovation Strategic Plan (PERIS) 2016‐2020 grant# SLT002/16/00201) and the Alzheimer's Association (Grant AARG‐19‐618265). Additional support has been received from the Universities and Research Secretariat, Ministry of Business and Knowledge of the Catalan Government under grant no. 2021_SGR_00913. All CRG authors acknowledge the support of the Spanish Ministry of Science, Innovation, and Universities to the EMBL partnership, the Centro de Excelencia Severo Ochoa, and the CERCA Programme/Generalitat de Catalunya. NV‐T and OG‐R receive funding from the MCIN/AEI/10.13039/501100011033 and the European Union NextGenerationEU/PRTR, through the Juan de la Cierva Incorporación Programme (IJC2020‐043216‐I and IJC2020‐043417‐I respectively). MS‐C receives the support of a fellowship from “la Caixa” Foundation (ID 100010434) and from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska‐Curie grant agreement No 847648. The fellowship code is LCF/BQ/PR21/11840004.
Vilor‐Tejedor N, Genius P, Rodríguez‐Fernández B, et al. Genetic characterization of the ALFA study: Uncovering genetic profiles in the Alzheimer's continuum . Alzheimer's Dement. 2024;20:1703–1715. 10.1002/alz.13537
Natalia Vilor‐Tejedor and Patricia Genius contributed equally to this study.
The complete list of collaborators of the ALFA study can be found in the acknowledgments section.
Data used in preparation of this article were obtained from the Alzheimer's Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu). As such, the investigators within the ADNI contributed to the design and implementation of ADNI and/or provided data but did not participate in analysis or writing of this report. A complete listing of ADNI investigators can be found at: http://adni.loni.usc.edu/wp‐content/uploads/how_to_apply/ADNI_Acknowledgement_List.pdf
Contributor Information
Natalia Vilor‐Tejedor, Email: nvilor@barcelonabeta.org.
Juan Domingo Gispert, Email: jdgispert@barcelonabeta.org.
REFERENCES
- 1. Molinuevo JL, Gramunt N, Gispert JD, et al. The ALFA project: a research platform to identify early pathophysiological features of Alzheimer's disease. Alzheimer's Dement: Transl Res Clin. 2016;2(2):82‐92. doi: 10.1016/j.trci.2016.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Operto G, Cacciaglia R, Grau‐Rivera O, et al. White matter microstructure is altered in cognitively normal middle‐aged APOE‐ε4 homozygotes. Alzheimers Res Ther. 2018;10(1):48. doi: 10.1186/s13195-018-0375-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Operto G, Molinuevo JL, Cacciaglia R, et al. Interactive effect of age and APOE‐ε4 allele load on white matter myelin content in cognitively normal middle‐aged subjects. Neuroimage Clin. 2019;24:101983. doi: 10.1016/j.nicl.2019.101983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cacciaglia R, Molinuevo JL, Falcón C, et al. Effects of APOE ‐ε4 allele load on brain morphology in a cohort of middle‐aged healthy individuals with enriched genetic risk for Alzheimer's disease. Alzheimers Dement. 2018;14(7):902‐912. doi: 10.1016/j.jalz.2018.01.016 [DOI] [PubMed] [Google Scholar]
- 5. Salvadó G, Ferreira D, Operto G, et al. The protective gene dose effect of the APOE ε2 allele on gray matter volume in cognitively unimpaired individuals. Alzheimers Dement. 2022;18(7):1383‐1395. doi: 10.1002/alz.12487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cacciaglia R, Molinuevo JL, Falcón C, et al. APOE‐ε4 risk variant for Alzheimer's disease modifies the association between cognitive performance and cerebral morphology in healthy middle‐aged individuals. Neuroimage Clin. 2019;23:101818. doi: 10.1016/j.nicl.2019.101818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cacciaglia R, Molinuevo JL, Falcón C, et al. APOE‐ε4 shapes the cerebral organization in cognitively intact individuals as reflected by structural gray matter networks. Cereb Cortex. 2020;30(7):4110‐4120. doi: 10.1093/cercor/bhaa034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cacciaglia R, Salvadó G, Molinuevo JL, et al. Age, sex and APOE‐ε4 modify the balance between soluble and fibrillar β‐amyloid in non‐demented individuals: topographical patterns across two independent cohorts. Mol Psychiatry. 2022;27(4):2010‐2018. doi: 10.1038/s41380-022-01436-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rojas S, Brugulat‐Serrat A, Bargalló N, et al. Higher prevalence of cerebral white matter hyperintensities in homozygous APOE‐ɛ4 allele carriers aged 45‐75: results from the ALFA study. J Cereb Blood Flow Metab. 2018;38(2):250‐261. doi: 10.1177/0271678X17707397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Salvadó G, Brugulat‐Serrat A, Sudre CH, et al. Spatial patterns of white matter hyperintensities associated with Alzheimer's disease risk factors in a cognitively healthy middle‐aged cohort. Alzheimers Res Ther. 2019;11(1):12. doi: 10.1186/s13195-018-0460-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ingala S, Mazzai L, Sudre CH, et al. The relation between APOE genotype and cerebral microbleeds in cognitively unimpaired middle‐ and old‐aged individuals. Neurobiol Aging. 2020;95:104‐114. doi: 10.1016/j.neurobiolaging.2020.06.015 [DOI] [PubMed] [Google Scholar]
- 12. Vilor‐Tejedor N, Ciampa I, Operto G, et al. Perivascular spaces are associated with tau pathophysiology and synaptic dysfunction in early Alzheimer's continuum. Alzheimers Res Ther. 2021;13(1):135. doi: 10.1186/s13195-021-00878-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Weber CJ, Carrillo MC, Jagust W, et al. The Worldwide Alzheimer's Disease Neuroimaging Initiative: ADNI‐3 updates and global perspectives. Alzheimers Dement. 2021;7(1):e12226. doi: 10.1002/trc2.12226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jack CR, Bennett DA, Blennow K, et al. A/T/N: an unbiased descriptive classification scheme for Alzheimer disease biomarkers. Neurology. 2016;87(5):539. doi: 10.1212/WNL.0000000000002923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Milà‐Alomà M, Salvadó G, Gispert JD, et al. ALFA study. Amyloid beta, tau, synaptic, neurodegeneration, and glial biomarkers in the preclinical stage of the Alzheimer's continuum. Alzheimers Dement. 2020;16(10):1358‐1371. doi: 10.1002/alz.12131. Epub 2020 Jun 23. PMID: 32573951; PMCID: PMC7586814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Das S, Forer L, Schönherr S, et al. Next‐generation genotype imputation service and methods. Nat Genet. 2016;48(10):1284‐1287. doi: 10.1038/NG.3656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. McCarthy S, Das S, Kretzschmar W, et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat Genet. 2016;48(10):1279‐1283. doi: 10.1038/NG.3643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Choi SW, O'Reilly PF. PRSice‐2: Polygenic Risk Score software for biobank‐scale data. Gigascience. 2019;8(7). doi: 10.1093/GIGASCIENCE/GIZ082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jansen IE, van der Lee SJ, Gomez‐Fonseca D, et al. Genome‐wide meta‐analysis for Alzheimer's disease cerebrospinal fluid biomarkers. Acta Neuropathol. 2022;144(5):821‐842. doi: 10.1007/s00401-022-02454-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wightman DP, Jansen IE, Savage JE, et al. A genome‐wide association study with 1,126,563 individuals identifies new risk loci for Alzheimer's disease. Nat Genet. 2021;53(9):1276‐1282. doi: 10.1038/s41588-021-00921-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ferrari R, Hernandez DG, Nalls MA, et al. Frontotemporal dementia and its subtypes: a genome‐wide association study. Lancet Neurol. 2014;13(7):686‐699. doi: 10.1016/S1474-4422(14)70065-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nalls MA, Blauwendraat C, Vallerga CL, et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson's disease: a meta‐analysis of genome‐wide association studies. Lancet Neurol. 2019;18(12):1091‐1102. doi: 10.1016/S1474-4422(19)30320-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Canela‐Xandri O, Rawlik K, Tenesa A. An atlas of genetic associations in UK Biobank. Nat Genet. 2018;50(11):1593‐1599. doi: 10.1038/s41588-018-0248-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Malik R, Chauhan G, Traylor M, et al. Multiancestry genome‐wide association study of 520,000 subjects identifies 32 loci associated with stroke and stroke subtypes. Nat Genet. 2018;50(4):524‐537. doi: 10.1038/s41588-018-0058-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Smith SM, Douaud G, Chen W, et al. An expanded set of genome‐wide association studies of brain imaging phenotypes in UK Biobank. Nat Neurosci. 2021;24(5):737‐745. doi: 10.1038/S41593-021-00826-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Persyn E, Hanscombe KB, Howson JMM, Lewis CM, Traylor M, Markus HS. Genome‐wide association study of MRI markers of cerebral small vessel disease in 42,310 participants. Nat Commun. 2020;11(1). doi: 10.1038/S41467-020-15932-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Knol MJ, Lu D, Traylor M, et al. Association of common genetic variants with brain microbleeds: a genome‐wide association study. Neurology. 2020;95(24):e3331‐e3343. doi: 10.1212/WNL.0000000000010852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Meier SM, Trontti K, Purves KL, et al. Genetic variants associated with anxiety and stress‐related disorders: a genome‐wide association study and mouse‐model study. JAMA Psychiatry. 2019;76(9):924‐932. doi: 10.1001/jamapsychiatry.2019.1119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Okbay A, Baselmans BML, De Neve JE, et al. Genetic variants associated with subjective well‐being, depressive symptoms, and neuroticism identified through genome‐wide analyses. Nat Genet. 2016;48(6):624‐633. doi: 10.1038/ng.3552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wray NR, Ripke S, Mattheisen M, et al. Genome‐wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nat Genet. 2018;50(5):668‐681. doi: 10.1038/s41588-018-0090-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Day FR, Ong KK, Perry JRB. Elucidating the genetic basis of social interaction and isolation. Nat Commun. 2018;9(1):2457. doi: 10.1038/s41467-018-04930-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yengo L, Sidorenko J, Kemper KE, et al. Meta‐analysis of genome‐wide association studies for height and body mass index in ∼700000 individuals of European ancestry. Hum Mol Genet. 2018;27(20):3641‐3649. doi: 10.1093/hmg/ddy271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pulit SL, Stoneman C, Morris AP, et al. Meta‐analysis of genome‐wide association studies for body fat distribution in 694 649 individuals of European ancestry. Hum Mol Genet. 2019;28(1):166‐174. doi: 10.1093/hmg/ddy327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Teslovich TM, Musunuru K, Smith AV, et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature. 2010;466(7307):707‐713. doi: 10.1038/nature09270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jansen PR, Watanabe K, Stringer S, et al. Genome‐wide analysis of insomnia in 1,331,010 individuals identifies new risk loci and functional pathways. Nat Genet. 2019;51(3):394‐403. doi: 10.1038/s41588-018-0333-3 [DOI] [PubMed] [Google Scholar]
- 36. Codd V, Wang Q, Allara E, et al. Polygenic basis and biomedical consequences of telomere length variation. Nat Genet. 2021;53(10):1425‐1433. doi: 10.1038/s41588-021-00944-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pilling LC, Atkins JL, Bowman K, et al. Human longevity is influenced by many genetic variants: evidence from 75,000 UK Biobank participants. Aging. 2016;8(3):547‐560. doi: 10.18632/aging.100930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Atkins JL, Jylhävä J, Pedersen NL, et al. A genome‐wide association study of the frailty index highlights brain pathways in ageing. Aging Cell. 2021;20(9):e13459. doi: 10.1111/acel.13459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gibson J, Russ TC, Clarke TK, et al. A meta‐analysis of genome‐wide association studies of epigenetic age acceleration. PLoS Genet. 2019;15(11):e1008104. doi: 10.1371/journal.pgen.1008104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ward A, Crean S, Mercaldi CJ, et al. Prevalence of apolipoprotein E4 genotype and homozygotes (APOE e4/4) among patients diagnosed with Alzheimer's disease: a systematic review and meta‐analysis. Neuroepidemiology. 2012;38(1):1‐17. doi: 10.1159/000334607 [DOI] [PubMed] [Google Scholar]
- 41. Skoog I, Kern S, Najar J, et al. A non‐APOE polygenic risk score for Alzheimer's disease is associated with cerebrospinal fluid neurofilament light in a representative sample of cognitively unimpaired 70‐Year olds. J Gerontol A Biol Sci Med Sci. 2021;76(6):983‐990. doi: 10.1093/gerona/glab030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Luckett ES, Abakkouy Y, Reinartz M, et al. Association of Alzheimer's disease polygenic risk scores with amyloid accumulation in cognitively intact older adults. Alzheimers Res Ther. 2022;14(1):138. doi: 10.1186/s13195-022-01079-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Daunt P, Ballard CG, Creese B, et al. Polygenic risk scoring is an effective approach to predict those individuals most likely to decline cognitively due to Alzheimer's disease. J Prev Alzheimers Dis. 2021;8(1):78‐83. doi: 10.14283/JPAD.2020.64/FIGURES/3 [DOI] [PubMed] [Google Scholar]
- 44. Milà‐Alomà M, Salvadó G, Gispert JD, et al. Amyloid beta, tau, synaptic, neurodegeneration, and glial biomarkers in the preclinical stage of the Alzheimer's continuum. Alzheimers Dement. 2020;16(10):1358‐1371. doi: 10.1002/alz.12131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Van Hulle C, Jonaitis EM, Betthauser TJ, et al. An examination of a novel multipanel of CSF biomarkers in the Alzheimer's disease clinical and pathological continuum. Alzheimers Dement. 2021;17(3):431‐445. doi: 10.1002/alz.12204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Salvadó G, Grothe MJ, Groot C, et al. Differential associations of APOE‐ε2 and APOE‐ε4 alleles with PET‐measured amyloid‐β and tau deposition in older individuals without dementia. Eur J Nucl Med Mol Imaging. 2021;48(7):2212‐2224. doi: 10.1007/s00259-021-05192-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Vlachakis D, Papakonstantinou E, Sagar R, et al. Improving the utility of polygenic risk scores as a biomarker for Alzheimer's disease. Cells. 2021;10(7):1627. doi: 10.3390/cells10071627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Desikan RS, Fan CC, Wang Y, et al. Genetic assessment of age‐associated Alzheimer disease risk: development and validation of a polygenic hazard score. PLoS Med. 2017;14(3):e1002258. doi: 10.1371/journal.pmed.1002258. Erratum in: PLoS Med. 2017 Mar 28;14 (3):e1002289. PMID: 28323831; PMCID: PMC5360219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Maciejewska K, Czarnecka K, Szymański P. A review of the mechanisms underlying selected comorbidities in Alzheimer's disease. Pharmacol Rep. 2021;73(6):1565‐1581. doi: 10.1007/s43440-021-00293-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Grande G, Marengoni A, Vetrano DL, et al. Multimorbidity burden and dementia risk in older adults: the role of inflammation and genetics. Alzheimers Dement. 2021;17(5):768‐776. doi: 10.1002/alz.12237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Deming Y, Dumitrescu L, Barnes LL, et al. Sex‐specific genetic predictors of Alzheimer's disease biomarkers. Acta Neuropathol. 2018;136(6):857‐872. doi: 10.1007/s00401-018-1881-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Dumitrescu L, Mahoney ER, Mukherjee S, et al. Genetic variants and functional pathways associated with resilience to Alzheimer's disease. Brain. 2020;143(8):2561‐2575. doi: 10.1093/brain/awaa209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wang H, Lo MT, Rosenthal SB, et al. Similar genetic architecture of Alzheimer's disease and differential APOE effect between sexes. Front Aging Neurosci. 2021;13:674318. doi: 10.3389/fnagi.2021.674318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Silva T, Zhang W, Young JI, et al. Distinct sex‐specific DNA methylation differences in Alzheimer's disease. Alzheimers Res Ther. 2022;14(1):1‐21. doi: 10.1186/s13195-022-01070-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Adams HHH, Evans TE, Terzikhan N. The Uncovering Neurodegenerative Insights Through Ethnic Diversity consortium. Lancet Neurol. 2019;18(10):915. doi: 10.1016/S1474-4422(19)30324-2 [DOI] [PubMed] [Google Scholar]
- 56. Ritchie CW, Muniz‐Terrera G, Kivipelto M, Solomon A, Tom B, Molinuevo JL. The European Prevention of Alzheimer's Dementia (EPAD) longitudinal cohort study: baseline data release V500.0. J Prev Alzheimers Dis. 2020;7(1):8‐13. doi: 10.14283/jpad.2019.46 [DOI] [PubMed] [Google Scholar]
- 57. Lopes Alves I, Collij LE, Altomare D, et al. Quantitative amyloid PET in Alzheimer's disease: the AMYPAD prognostic and natural history study. Alzheimers Dement. 2020;16(5):750‐758. doi: 10.1002/alz.12069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Bycroft C, Freeman C, Petkova D, et al. The UK Biobank resource with deep phenotyping and genomic data. Nature. 2018;562(7726):203‐209. doi: 10.1038/s41586-018-0579-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Arnatkeviciute A, Fulcher BD, Fornito A. A practical guide to linking brain‐wide gene expression and neuroimaging data. Neuroimage. 2019;189:353‐367. doi: 10.1016/j.neuroimage.2019.01.011 [DOI] [PubMed] [Google Scholar]
- 60. Andrews SJ, Fulton‐Howard B, O'Reilly P, Marcora E, Goate AM, collaborators of the Alzheimer's Disease Genetics Consortium . Causal associations between modifiable risk factors and the Alzheimer's phenome. Ann Neurol. 2021;89(1):54‐65. doi: 10.1002/ana.25918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Rodríguez‐Fernández B, Vilor‐Tejedor N, Arenaza‐Urquijo EM, et al. Genetically predicted telomere length and Alzheimer's disease endophenotypes: a Mendelian randomization study. Alzheimers Res Ther. 2022;14(1):167. doi: 10.1186/s13195-022-01101-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information