

Abstract
Eighteen novel RNA viruses were found in Crassostrea hongkongensis. Phylogenic analysis shows evidence of recombination between major genes of viruses. Picobirnaviruses are ubiquitous and abundant in oysters.
INTRODUCTION
Oysters (phylum Mollusca, class Bivalvia, order Pterioida) are globally distributed shellfish and are an important marine biological resource that is available to humans. Oysters have high nutritional value and are the most farmed shellfish in the world. As the largest oyster producer, China produced 82,593,752 tons of oysters in 2019, accounting for 85.3% of the world's total output. Being filter feeders, oysters can filter up to 5 L of seawater through their gills every hour and enrich suspended microorganisms and particles by factors of a thousand to a hundred thousand times their seawater concentrations, making it easy for viruses to accumulate in oysters. Oysters have a clustered and sessile lifestyle and bring stable and lasting improvements to nearshore marine environments by, for example, reducing water turbidity and purifying water [1, 2]. However, oysters have evidently no acquired immune system [3], which may further increase the probability of virus transmission among oysters.
In 1972, Farley et al. found herpes virus infection in invertebrates in the United States, and showed that oyster deaths caused by the herpes virus were more common in high‐temperature conditions; the virus was named Ostreid herpesvirus‐1 (OsHV‐1) [4]. The mortality rate of OsHV‐1‐infected shellfish seedlings and young shellfish is >90%, which is very harmful to the oyster industry. In addition to OsHV‐1, other oyster‐associated viruses have been reported, including a Papovaviridae virus that causes oyster “Oocystitis,” which leads to egg and gamete cell hypertrophy, and gill necrosis virus, an Iridoviridae virus that may have been the main cause of mass death of the bivalve Crassostrea angulata population in the late 1960s [5, 6]. Moreover, Togaviridae, Reoviridae, and Picornaviridae viruses have also been reported in shellfish hosts [6]. Most of the studies on these viruses were confined to pathological and electron microscopic observations, and no in‐depth reports have been published so far. Norovirus, hepatitis A virus, and astrovirus have been found in farmed oysters, but these viruses are not pathogens of oysters [7]. Research progress on viruses that are pathogenic to oysters is still very slow; therefore, the identification of oyster pathogens is a top priority for oyster disease prevention and control.
With the development of high‐throughput sequencing technologies, methods such as viromics and meta‐transcriptomics have overcome the dependence of traditional virology studies on host cell culture and greatly improved the efficiency of the discovery and identification of new viruses in invertebrates [8]. For example, 1445 RNA viruses with complete genomes were found by transcriptome analysis of more than 220 invertebrate species from nine animal phyla [9], which greatly expanded the understanding of the virus community. Seven complete RNA virus genomes were obtained from Crassostrea gigas and Mytilus galloprovincialis host transcriptome data and classified as Picornavirales; six of them were new viruses [10]. Intracellular RNA libraries of California sea hare (Aplysia californica) and frog (Microhyla fissipes) were sequenced and the complete genomes of two novel viruses of Nidovirales were found [11]. A comparative study on healthy and infected starfish identified a suspected pathogen of Parvoviridae and confirmed that it was also widely present in plankton and marine sediments [12]. Genome fragments of 117 RNA viruses that contained RdRp genes distributed in nine viral families or orders were identified in 58 invertebrate species across three seas [13].
The Data set of Oyster Virome (DOV) was reported by Jiang et al. [14]. DOV, which contains 728,784 contigs (≥800 bp) of nonredundant virus operation taxa (vOTU) and 3473 high‐quality viral genomes, provided the first comprehensive description of oyster viral community structure. Among them, 4958 RNA virus‐related vOTUs were found to be particularly noteworthy [14]. This study used bioinformatics tools to analyze the genomes of 18 oyster‐associated RNA viruses among the RNA virus‐related vOTUs in DOV. The results provide an important reference for the expansion of the DOV and the identification of oyster viral pathogens.
RESULTS
Eighteen novel RNA viruses were found in oysters
We used 18 RNA virus sequences from the DOV obtained previously [14] for deep analysis. Because the 18 RNA virus sequences were very different they could not be reliably aligned, and therefore could not be used to construct a unified and reliable phylogenetic tree. Therefore, we constructed a clustering network based on the similarity of the encoded RdRp and capsid protein sequences. The RdRp protein sequences of the 18 oyster‐associated RNA viruses and related viruses in the nr database clustered roughly into five groups (Figure 1A), which means they belonged to five families or orders (Sobelivirales, Picornavirales, Leviviridae, Durnavirales, and Yanvirus) (Supporting Information: Table S1). Only 10 of the 18 genomes were annotated with capsid proteins, which were clustered into three groups (Figure 1B, Supporting Information: Table S1) (Sobelivirales‐Weivirus, Picornavirales, and Leviviridae).
Figure 1.
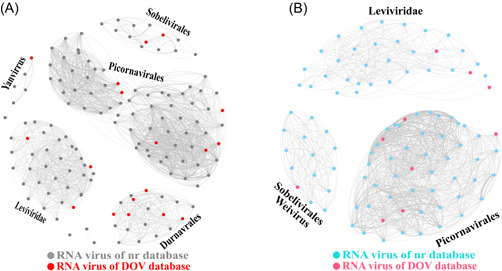
The variety of novel RNA viruses found in oysters. (A) Clustering network of 147 RdRp protein sequences. (B) Clustering network of 99 capsid protein sequences. The networks were visualized using the Fruchterman‐Reingold algorithm in Gephi (version 0.9.2). Dots represent different sequences. Edges indicate that the DIAMOND BLASTP scores between the connected dots were ≥57 (A) and ≥43.5 (B).
Evidence of gene exchange among RNA viruses
Sobelivirales are RNA viruses that are found in plants or invertebrates and have a sense, non‐segmented genome of 4‐4.6 kb [13, 15]. We found two oyster‐associated Sobelivirales viruses (Figures 1 and 2A). Huangsha sobemo‐like virus HSd1‐611299 was most closely related to Beihai sobemo‐like virus 6 (YP_00933713), which was found in a mixed sample of superphylum Lophotrochozoa; the AAI of their RdRp sequences was 93.11% and their capsid proteins were also on the same branch (Figure 2B), but the AAI of the capsid protein sequences was slightly lower at 89.23%. Therefore, we think that these two viruses are different strains of the same virus. Tanwei sobemo‐like virus TWr1‐33874 clustered with Beihai sobemo‐like virus 7, which was found in phylum Arthropoda, but the AAI of the RdRp sequences was <30%. Like arthropods that feed on plants, bivalves such as oysters can also feed on aquatic plants or algae. Sobemoviruses were once considered to be plant‐specific viruses, but they have now been found in both arthropods and mollusks, providing a basis for the transformation of the virus in different trophic hosts [13].
Figure 2.
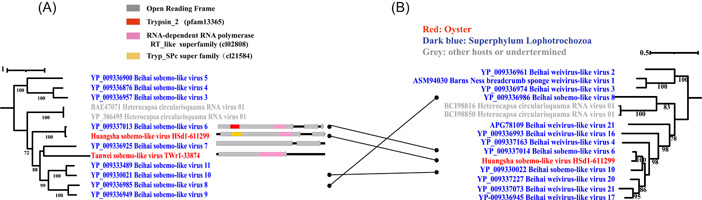
Phylogenic analysis shows evidence of recombination between major viral proteins. Phylogenetic trees of RdRp (A) and capsid proteins (B) of oyster‐associated Sobelivirales. The maximum likelihood phylogenetic tree was constructed using IQtree (version 2.1.4) with the sequences. ModelFinder was set as MFP and 1000 ultrafast bootstrap replicates were used. Bootstrap values >70 are shown. The domains in the genome structure were annotated using the NCBI Conserved Domain Database.
Weiviruses are RNA viruses that were identified from invertebrates [9]. However, in the phylogenetic tree constructed with annotated ten capsid protein sequences and corresponding results of NCBI BLASTP, we found that Huangsha sobemo‐like virus HSd1‐611299, Beihai sobemo‐like virus 6, Beijing sobemo‐like virus 8, and Beihai sobemo‐like virus 10 clustered with the capsid proteins of Weiviruses (Figure 2B), whereas the phylogenetic tree constructed with the RdRp sequences did not contain any Weiviruses (Figure 2A). This finding implies that the capsid protein genes of sobemo‐like viruses and Weiviruses may have a common origin. The recombination between the capsid protein gene and RDRP provided clear evidence.
Picornavirales were found to be the most abundant RNA viruses in coastal water [16, 17]. We also found six oyster‐associated Picornavirales viruses in this study (Figure 3). Oyster picorna‐like virus T8S1‐348502 was closely related to RNA virus (NP_944776) from Heterosigma akashiwo (Rhaphidophyceae), and oyster picorna‐like virus ZHr1‐40939 and oyster picorna‐like virus Vis1‐51363 were closely related to Wenzhou picorna‐like virus 5 (YP_009337362) and Beihai picorna‐like virus 31 (APG78919), respectively, which were found in mixed samples of superphylum Lophotrochozoa. The genetic relationship between oyster picorna‐like virus SZr1‐211549 and known viruses was distant. Oyster picorna‐like virus Vis1‐91049 was most closely related to Beihai picorna‐like virus 29 (YP_009337362) from the chelate subphylum Arthropoda, and oyster picorna‐like virus TWr1‐22141 was most closely related to Wenzhou picorna‐like virus 10 (APG785830) from Arthropoda (subphylum Crustacea). However, the AAI of the RdRp sequences among the six Picornavirales viruses and the unclassified Picornavirales viruses was <90; therefore, we think they are all new viruses.
Figure 3.
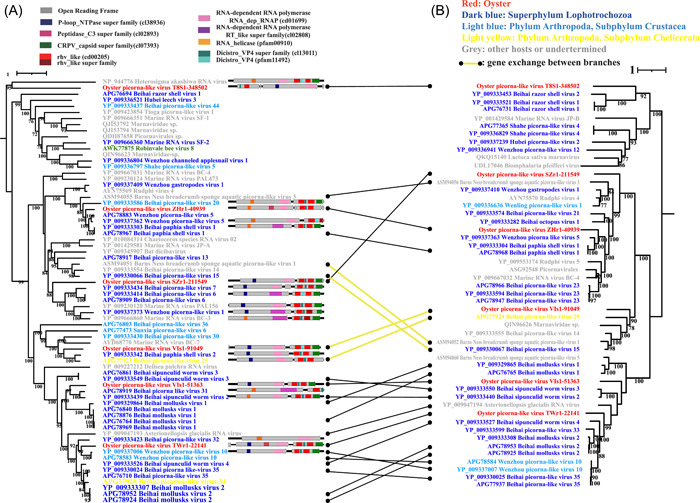
Comparison of the topological structure between the RdRp and capsid phylogenic trees of picornaviruses found in oysters. The trees were constructed using Iqtree (version 2.1.4) based on the RdRp (A) and capsid protein (B) sequences. ModelFinder was set to MFP and 1000 ultrafast bootstrap replicates were used. Bootstrap values >70 are shown.
When comparing the topological structure between the RdRp and capsid phylogenic trees of picornaviruses (Figure 3), although there is no evidence of recombination among the major clades, we still observed the gene exchange among some small branches (Figure 3, yellow lines). It is different from Picornavirales that the recombination was not found even among small branches on the phylogenetic tree of Leviviridae (Supporting Information: Figure S1).
Leviviridae is a kind of single‐stranded RNA virus that can infect a variety of Gram‐negative bacteria. Leviviridae shares the same core genome, which spans 3.4–4.3 kb and encodes a subunit of RdRp, mature protein, and coat protein [18]. We found three strains of Leviviridae viruses in this study, and all of their genomes encoded these three proteins (Supporting Information: Figure S1A). Among them, Taishan Levi‐like virus T4S1‐79710 and Huangsha Levi‐like virus HSd1‐59787, which were found in Guangdong, were most closely related to Beihai Levi‐like virus 28 (APG7701), which was found in Beihai, Guangxi, and Hubei Levi‐like virus 4 (APG77248), which was found in Hubei, respectively. However, the AAIs of their RdRp sequences were only 61.49% and 44.07%, respectively. We believe that the two strains belong to the newly discovered Leviviridae. Taishan Levi‐like virus T4S1‐672536, which was found in Crassostrea hongkongensis in Taishan, Guangdong, was closely related to Beihai Levi‐like virus 17 (APG77031), which was found in the crustacean subphylum of Beihai in Guangxi; the AAI of their RdRp sequences was 96.04% (Supporting Information: Figure S1A) and the AAI of their capsid protein sequences was 97.61% (Supporting Information: Figure S1B). Because the AAIs of these proteins were >95%, we think that these two viruses are different strains of the same virus.
Ubiquitous and abundant oyster‐associated picobirnaviruses
Durnavirales are double‐stranded RNA viruses that can infect both vertebrates and invertebrates. In this study, we found six oyster‐associated Durnavirales viruses that clustered in a branch with unclassified Picobirnaviridae viruses (Supporting Information: Figure S2). Their genomes all contained a conserved RT_like superfamily domain (cl02808), but the number of ORFs was different (1 ≤ ORFs ≥ 4) (Supporting Information: Table S1). The AAI of the RdRp sequences of oyster‐associated RNA virus ZHd1‐112402, oyster picobirna‐like virus SZr1‐72709, and oyster picobirna‐like virus Yjd1‐298692 with the closest viruses was <60%. For oyster picobirna‐like virus ZHr1‐41827, oyster picobirna‐like virus Yjr1‐11446, and oyster picobirna‐like virus Yjr1‐2332 no closely related sequences were found in the NCBI nr database, and the AAI of the RdRp sequences between oyster picobirna‐like virus Yjr1‐11446 and oyster picobirna‐like virus Yjr1‐2332 was <90% (Supporting Information: Figure S3). Therefore, we think that the six viruses of picobirnaviridae found in this study are all new.
We calculated the abundance of these viruses in 54 oyster virus libraries from a variety of sources (Supporting Information: Table S2). Among them, oyster‐associated RNA virus ZHd1‐112402 was found in 24 libraries. The highest FPKM values were 36276.74 in library ChQZ1511Rb and 11132.95 in library ChQZ1511Ra. Oyster picobirna‐like virus YJd1‐298692 was found in 13 libraries, and the highest FPKM value was 3124.75 in library ChTW1511Ra (Supporting Information: Table S2). These two viruses are the most widely distributed and abundant of the 18 newly discovered RNA viruses, showing that picobirnavirus is an important member of the oyster.
Yanviruses are positive‐stranded or double‐stranded RNA viruses [9]. In addition to the virus found above, the oyster yanvirus‐like virus SZr1‐117762 was also found in this study. Although it was closely related to Wenzhou yanvirus‐like virus 2 (Supporting Information: Figure S4A), the average amino acid identity (AAI) of their RdRp sequences was only 68.57%. Therefore, we infer that the oyster yanvirus‐like virus SZr1‐117762 is new. Wenzhou yanvirus‐like virus 2 was derived from mixed samples of superphylum Lophotrochozoa, including Bivalvia, Gastropoda, Cephalopoda, Polychaeta, Oligochaeta, Hirudinea, and Sipuncula Phascolosoma esculenta, and Sipunculus nudus, which was composed of seven groups, and which was similar to the oyster sample from oyster yanvirus‐like virus SZr1‐117762. For oyster yanvirus‐like virus SZr1‐117762, although the RdRp domain was not detected by CDD, the RdRp sequence alignment results showed high AAI in the conserved RdRp domain (Supporting Information: Figure S4B), indicating that the RdRp of this virus had an atypical RdRp domain.
DISCUSSION
Viruses are the most abundant biomasses in oceans, and mollusks, which are types of shellfish, are the largest group of animals in oceans. However, the intersecting field of shellfish and viruses is poorly understood. Virome sequencing has been widely used to analyze many biological and environmental samples, highlighting the potential of high‐throughput sequencing technologies for detecting new viruses [19]. In this study, we used virome technology to identify new RNA viruses in C. hongkongensis and found 17 new RNA viruses that showed only 30%‐70% similarity to their closest viruses, highlighting the genetic diversity of marine RNA viruses (Supporting Information: Table S1). However, two key technical issues remain to be solved in the classification and identification of new virus genomes. On the one hand, because the identification of viruses depends mainly on similarity searches in public databases, the ability to find and identify different or unknown viruses is highly restricted. On the other hand, the classification of RNA viruses is usually based on the highly conserved RdRp protein sequences. However, we found an asynchronous pattern between RdRp genes and capsid protein genes (Figure 1), and that recombination between the capsid protein gene and the RdRp gene may occur in RNA viruses (Figures 2 and 3). Therefore, using a single gene, such as RdRp, to infer the history of RNA viruses has major limitations.
Viruses from the same family or host species can infect species of different phyla or even different kingdoms at the same time. Such events are called host sharing and host switching. Studying host sharing and host switching events can help in the discovery of potential zoonotic viruses and prevent the occurrence of new epidemic diseases; for example, ranaviruses (family Iridoviridae) [20] isolated from reptiles, amphibians, and fish, and the cross‐species transmission of the novel coronavirus (SARS‐CoV‐2) [21, 22]. Our phylogeny results indicate that some of the viruses identified in this study may have host‐sharing characteristics; for example, the sobemo‐like virus was found in arthropods and mollusks, as well as in plants, and oyster picorna‐like virus T8S1‐348502 was found in Picornavirales clustered with Heterosigma akashiwo RNA virus (NP_944776). This may be due to the host transformation of Heterosigma akashiwo by oyster filtering of microalgae in water as food [23].
Viruses from oyster samples have been identified previously. For example, 26 new RNA virus genomes were assembled from the public transcriptome data of C. gigas and Crassostrea corteziensis. They included mainly Dicistroviridae, Picornavirales, herpes‐like viral family viruses, and the algae‐infecting viruses Heterosigma akashiwo and Chaetoceros socialis f. radians RNA virus 1 [10, 24]. Four RNA virus genomic fragments from oyster (C. gigas) samples have also been reported [13], and 33 novel RNA viruses were identified from mixed bivalve samples, including two oyster species C. hongkongensis and C. ariakensis [9]. The 33 viruses were distributed in Narnaviridae (nara‐like), Yanvirus (yanvirus‐like), Weivirus (weivirus‐like), Totiviridae (toti‐like), Tombusviridae (tombus‐like), Picornavirales (picorna‐like), and Nodaviridae (noda‐like). In addition, Birnaviridae RNA viruses were found in shellfish. A virus from Japanese pearl oysters (Pinctada fucata) presenting mass mortality was isolated, named “Marine birnavirus” (MABV) [25]. And aquabirnaviruses were reported from Geoduck clams (Panope abrupta), and littleneck clams (Protothaca staminea) collected in Alaska [26]. However, only one of the RNA viruses identified in this study had RdRp and capsid protein sequences that shared high AAIs with the RdRp and capsid protein sequences of these viruses (AAI > 90%); the other 17 viruses are quite different. Furthermore, we found two virus types, Sobelivirales (sobemo‐like) and Leviviridae (levi‐like), that had not been identified previously in oysters. In our previous mining of DOV data, we found that there were a large number of unclassified circoviruses in oysters [14].
RNA viruses found in oysters also exist in seawater and other marine animals. For example, the Picorna‐like viruses were found to be the most abundant RNA viruses in coastal water [16, 17] and were also found in marine fish [27] and shrimp [28]. Zhang et al. found Duranvirales and Sobemo‐like viruses in gastropods and crustaceans, respectively [13]. The white spot syndrome virus, the viral nervous necrosis virus, the marine birnavirus, and the viral hemorrhagic septicemia virus can be detected in both shellfish (including oysters) and seawater by nested PCR [29]. Although many studies have shown that the microbiota in oysters is mainly disturbed and influenced by the external environment [30, 31], it is significantly different from the environment. It indicates that the internal environment of oysters has a selective effect on their inner microbial community [14, 32]. All these data indicate that oysters have rich, diverse, and unique viral groups that are very different from the viruses found in marine invertebrates so far. Oysters can be regarded as repositories and vectors of marine viruses because of their filter‐feeding methods, low levels of immune defense mechanisms, and high‐density sessile lifestyles. Further studies on the community structure and function of bivalve viruses will greatly help in understanding their role in coastal microflora regulation, disease transmission, and the protection and restoration of coastal ecosystems.
CONCLUSION
The characteristics of 18 RNA virus genomes found in oysters are summarized in this study. Seventeen of them are new virus species, which effectively expands the diversity of the oyster RNA viruses described so far. The common host transformation or host sharing of viruses in invertebrates, and the discovery that the capsid protein genes of sobemo‐like viruses and Weiviruses may have undergone recombination and exchange or have a common origin, have added to the understanding of oyster‐associated viruses.
METHODS
Sequence assembling and virus discovery
We constructed 54 oyster virus libraries from a variety of sources, including nine‐time points, seven sites (Qinzhou, Guangxi, Yangjiang, Zhuhai, Tanwei areas of Huidong, Lianjiang, Shenzhen), and two tissue types [14]. By virome sequencing of oysters (Crassostrea hongkongensis) cultured in many coastal areas of South China, we obtained approximately 2.5 billion reads [33]. Fastp (version 0.20.0) [34] was used to remove low‐quality sequences and adapters for quality control, and the reads were assembled into contigs using MEGAHIT (version 1.2.9) [35, 36]. DIAMOND (version 0.9.24.125) [37] was used to align and annotate the contigs with the National Center for Biotechnology Information (NCBI) nonredundant protein (nr) database as the reference. We classified the annotated sequences using MEGAN6 [38]. Finally, 18 virus genome sequences were identified as suspected RNA viruses and were screened for deep analysis.
Open reading frame (ORF) prediction and annotation
ORFs were predicted in the eighteen virus genomes using Cenote‐Taker2 [39]. NCBI BLASTP [40, 41] was used to align the ORF sequences to the nr database with e‐value cutoff set as 10−5. The protein sequences with the highest consistency were inversely aligned with the virus genome sequences using NCBI tBLASTN [40, 41] to verify the integrity of the ORF predictions. We also carried out domain‐based searching using the NCBI Conserved Domain Database (CDD) [42, 43] with an expected value threshold of 0.001. SnapGene (version 4.3.6) was used to visualize the structure of the genomes.
Similarity clustering analysis
We took the top 10 RdRp sequences and top 10 capsid protein sequences from the BLASTP results based on their total scores and used DIAMOND [37] to align them. Then, we used Gephi (version 0.9.2) [44] to construct clustering networks based on the scores.
Phylogenetic tree construction based on RdRp and capsid protein sequences
We used MAFFT [45] for multiple sequence alignment, TrimAL [46] to remove ambiguous areas, and IQtree (version 2.1.4) [47] to build maximum likelihood phylogenetic trees based on the RdRp and capsid protein sequences. ModelFinder [48] was set to MFP (for ModelFinder Plus) and 1000 ultrafast bootstrap replicates were used. iTOL (version 6.5.2) (https://itol.embl.de) [49] was used for visualization.
Analysis of the abundance of viruses
To calculate the relative abundance of each virus, we combined the 18 virus genome sequences and used the Salmon (version 0.13.1) [50] index command to generate a reference genome data set. Then, we used the Salmon quant command to map the clean reads of all the oyster virome libraries (PRJCA007058) one by one to the reference genome. Finally, we counted the number of mapped reads for each library. The relative abundance of each virus was calculated according to the adjusted FPKM (Fragments Per Kilobase of exon model per Million mapped fragments) as follows:
where genome reads is the number of mapped reads; total reads is the total number of reads obtained by sequencing, in millions; and genome length is the length of the genome, in Kb. Total FPKM is the sum of the FPKM of each library.
AUTHOR CONTRIBUTIONS
Peng Zhu: Validation, Formal analysis, Investigation, Resources, Data Curation Visualization, Writing—Original Draft, Writing—Review & Editing. Guang‐Feng Liu and Chang Liu: Conceptualization, Methodology, Data Curation. Li‐Ling Yang, Min Liu, and Ke‐Ming Xie: Formal analysis, Investigation, Visualization. Shao‐Kun Shi: Investigation, Resources. Mang Shi: Conceptualization, Methodology, Validation, Writing—Review & Editing, Funding acquisition. Jing‐Zhe Jiang: Conceptualization, Methodology, Visualization, Resources, Data Curation, Visualization, Writing—Original Draft, Writing—Review & Editing, Supervision, Project administration, Funding acquisition. All authors have read the final manuscript and approved it for publication.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Supporting information
Supporting Information.
Supporting information.
ACKNOWLEDGMENTS
We thank Margaret Biswas, PhD, from Liwen Bianji (Edanz) (www.liwenbianji.cn/) for editing the English text of a draft of this manuscript. This project was supported by the Key‐Area Research and Development Program of Guangdong Province (no. 2022B1111030001); the Natural Science Foundation of China (31972847) to Jiang J.‐Z.; the Central Public‐Interest Scientific Institution Basal Research Fund, CAFS (nos. 2020TD42 and 2021SD05) to Jiang J.‐Z.; the Guangdong Provincial Special Fund for Modern Agriculture Industry Technology Innovation Teams (no. 2019KJ141) to Jiang J.‐Z.; the Earmarked Fund (no. CARS‐49) to Ye L.‐T.; Shenzhen Science and Technology Program (no. KQTD20200820145822023) to Shi M.; Guangdong Provience “Pearl River Talent Plan” Innovation and Entrepreneurship Team Project (no. 2019ZT08Y464) to Shi M. The funders had no role in the study design, data collection, analysis, decision to publish, or manuscript preparation.
Contributor Information
Mang Shi, Email: shim23@mail.sysu.edu.cn.
Jing‐Zhe Jiang, Email: jingzhejiang@gmail.com.
DATA AVAILABILITY STATEMENT
The data set supporting the results of this article has been deposited in the Genome Sequence Archive (GSA) under BioProject accession code PRJCA007058 [https://ngdc.cncb.ac.cn/gsub/submit/bioproject/subPRO010366/overview] and all RNA virus genetic sequences have been deposited in Genome Warehouse in National Genomics Data Center (NGDC) (Members and Partners 2021) under accession GWHBJCN01000000, GWHBJCM01000000, GWHBJCK01000000, GWHBJCJ01000000, GWHBJCI01000000, GWHBJCH01000000, GWHBJCG01000000, GWHBJCF01000000, GWHBJCE01000000, GWHBJCD01000000, GWHBJCC01000000, GWHBJCB01000000, GWHBJCA01000000, GWHBJBZ01000000, GWHBJBX01000000, GWHBJBW01000000, GWHBJBV01000000, GWHBJBT01000000, that are publicly accessible at https://bigd.big.ac.cn/gwh.
REFERENCES
- 1. Powell, Daniel , Subramanian Sankar, Suwansa‐ard Saowaros, Zhao Min, O'Connor Wayne, Raftos David, and Elizur Abigail. 2018. “The Genome of the Oyster Saccostrea Offers Insight Into the Environmental Resilience Of Bivalves.” DNA Research 25: 655–65. 10.1093/dnares/dsy032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Olalemi, A. , Baker‐Austin C., Ebdon J., and Taylor H.. 2016. “Bioaccumulation and Persistence of Faecal Bacterial and Viral Indicators in Mytilus edulis and Crassostrea gigas .” International Journal of Hygiene and Environmental Health 219: 592–98. 10.1016/j.ijheh.2016.06.002 [DOI] [PubMed] [Google Scholar]
- 3. Li, Jun , Zhang Yang, Zhang Yuehuan, Mao Fan, Xiang Zhiming, Xiao Shu, Ma Haitao, Yu Ziniu. 2017. “The First Invertebrate NFIL3 Transcription Factor With Role in Immune Defense Identified From the Hong Kong Oyster, Crassostrea Hongkongensis.” Developmental & Comparative Immunology 76: 1–8. 10.1016/j.dci.2017.05.011 [DOI] [PubMed] [Google Scholar]
- 4. Farley, C. Austin , Banfield William G., Kasnic George, and Foster Walter S.. 1972. “Oyster Herpes‐Type Virus.” Science 178: 759–60. 10.1126/science.178.4062.759 [DOI] [PubMed] [Google Scholar]
- 5. Elston, R. A. , and Wilkinson M. T.. 1985. “Pathology, Management and Diagnosis of Oyster Velar Virus Disease (OVVD.” Aquaculture 48: 189–210. 10.1016/0044-8486(85)90124-3 [DOI] [Google Scholar]
- 6. Renault, Tristan , and Novoa Beatriz. 2004. “Viruses Infecting Bivalve Molluscs.” Aquatic Living Resources 17: 397–409. 10.1051/alr:2004049 [DOI] [Google Scholar]
- 7. Delmotte, Jean , Chaparro Cristian, Galinier Richard, de Lorgeril Julien, Petton Bruno, Stenger Pierre‐louis, Vidal‐Dupiol Jeremie, et al. 2020. “Contribution of Viral Genomic Diversity to Oyster Susceptibility in the Pacific Oyster Mortality Syndrome.” Frontiers in Microbiology 11: 1579. 10.3389/fmicb.2020.01579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhang, Yong‐Zhen , Chen Yanmei, Wang Wen, Qin Xin‐Chen, and Holmes Edward C.. 2019. “Expanding the RNA Virosphere by Unbiased Metagenomics.” Annual Review of Virology 6: 119–39. 10.1146/annurev-virology-092818-015851 [DOI] [PubMed] [Google Scholar]
- 9. Shi, Mang , Lin Xian‐Dan, Tian Jun‐Hua, Chen Liang‐Jun, Chen Xiao, Li Ci‐Xiu, Qin Xin‐Cheng, et al. 2016. “Redefining the Invertebrate RNA Virosphere.” Nature 540: 539–43. 10.1038/nature20167 [DOI] [PubMed] [Google Scholar]
- 10. Rosani, Umberto , and Gerdol Marco. 2017. “A Bioinformatics Approach Reveals Seven Nearly‐Complete RNA‐Virus Genomes in Bivalve RNA‐seq Data.” Virus Research 239: 33–42. 10.1016/j.virusres.2016.10.009 [DOI] [PubMed] [Google Scholar]
- 11. Bukhari, Khulud , Mulley Geraldine, Gulyaeva Anastasia A., Zhao Lanying, Shu Guocheng, Jiang Jianping, and Neuman Benjamin W.. 2018. “Description and Initial Characterization Of Metatranscriptomic Nidovirus‐Like Genomes From the Proposed New Family Abyssoviridae, and From a Sister Group to the Coronavirinae, the Proposed Genus Alphaletovirus.” Virology 524: 160–71. 10.1016/j.virol.2018.08.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jackson, Elliot W. , Wilhelm Roland C., Johnson Mitchell R., Lutz Holly L., Danforth Isabelle, Gaydos Joseph K., Hart Michael W., and Hewson Ian. 2020. “Diversity of Sea Star‐Associated Densoviruses and Transcribed Endogenous Viral Elements of Densovirus Origin.” Journal of virology 95: 95. 10.1128/JVI.01594-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhang, Yu‐Yi , Chen Yicong, Wei Xiaoman, and Cui Jie. 2022. “Viromes in Marine Ecosystems Reveal Remarkable Invertebrate RNA Virus Diversity.” Science China Life Sciences 65: 426–37. 10.1007/s11427-020-1936-2 [DOI] [PubMed] [Google Scholar]
- 14. Jiang, Jing‐Zhe , Fang Yi‐Fei, Wei Hong‐Ying, Zhu Peng, Liu Min, Yang Li‐Ling, Guo Ying‐Xiang, et al. 2022. “Remarkable Virus Diversity and Organized Community in Filter‐Feeding Oysters Varied From Those of the Ocean Virome.” 10.21203/rs.3.rs-1344035/v1 [preprint]. [DOI]
- 15. Sõmera, Merike , Fargette Denis, Hébrard Eugénie, and Sarmiento Cecilia. 2021. “ICTV Virus Taxonomy Profile: Solemoviridae 2021.” Journal of General Virology 102. 10.1099/jgv.0.001707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Culley, Alexander I. , Lang Andrew S., and Suttle Curtis A.. 2006. “Metagenomic Analysis of Coastal RNA Virus Communities.” Science 312: 1795–98. 10.1126/science.1127404 [DOI] [PubMed] [Google Scholar]
- 17. Vlok, Marli , Lang Andrew S., and Suttle Curtis A.. 2019. “Marine RNA Virus Quasispecies Are Distributed Throughout the Oceans.” mSphere 4: e00157–00119. 10.1128/mSphereDirect.00157-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gorzelnik, Karl V. , Cui Zhicheng, Reed Catrina A., Jakana Joanita, Young Ry, and Zhang Junjie. 2016. “Asymmetric Cryo‐EM Structure of the Canonical Allolevivirus Qβ Reveals a Single Maturation Protein and the Genomic ssRNA In Situ.” Proceedings of the National Academy of Sciences of the United States of America 113: 11519–24. 10.1073/pnas.1609482113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Filipa‐Silva, Andreia , Parreira Ricardo, Martínez‐Puchol Sandra, Bofill‐Mas Sílvia, Barreto Crespo Maria Teresa, and Nunes Mónica. 2020. “The Unexplored Virome of Two Atlantic Coast Fish: Contribution of Next‐Generation Sequencing to Fish Virology.” Foods 9: 1634. 10.3390/foods9111634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jancovich, James K. , Bremont Michel, Touchman Jeffrey W., and Jacobs Bertram L.. 2010. “Evidence for Multiple Recent Host Species Shifts Among the Ranaviruses (Family Iridoviridae).” Journal of Virology 84: 2636–47. 10.1128/JVI.01991-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhou, Peng , Yang Xing‐Lou, Wang Xian‐Guang, Hu Ben, Zhang Lei, Zhang Wei, Si Hao‐Rui, et al. 2020. “A Pneumonia Outbreak Associated With a New Coronavirus Of Probable Bat Origin.” Nature 579: 270–73. 10.1038/s41586-020-2012-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Liu, Ping , Jiang Jing‐Zhe, Wan Xiu‐Feng, Hua Yan, Li Linmiao, Zhou Jiabin, Wang Xiaohu, et al. 2020. “Are Pangolins the Intermediate Host of the 2019 Novel Coronavirus (SARS‐CoV‐2).” PLOS Pathogens 16: e1008421. 10.1371/journal.ppat.1008421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Le, Tuan Son , Southgate Paul C., O'Connor Wayne, Abramov Tomer, Shelley Daniel, Sang V. Vu, and Kurtböke D. İpek. 2020. “Use of Bacteriophages to Control Vibrio Contamination of Microalgae Used as a Food Source for Oyster Larvae During Hatchery Culture.” Current Microbiology 77: 1811–20. 10.1007/s00284-020-01981-w [DOI] [PubMed] [Google Scholar]
- 24. Rosani, Umberto , Shapiro Maxwell, Venier Paola, and Allam Bassem. 2019. “A Needle in A Haystack: Tracing Bivalve‐Associated Viruses in High‐Throughput Transcriptomic Data.” Viruses 11: 205. 10.3390/v11030205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Suzuki, Satoru , Kamakura Maki, and Kusuda Riichi. 1998. “Isolation of Birnavirus From Japanese Pearl Oyster Pinctada fucata .” Fisheries Science 64: 342–43. 10.2331/fishsci.64.342 [DOI] [Google Scholar]
- 26. Meyers, T. R. , Burton T., Evans W., and Starkey N.. 2009. “Detection of Viruses and Virus‐Like Particles in Four Species of Wild and Farmed Bivalve Molluscs in Alaska, USA, From 1987 to 2009.” Diseases of Aquatic Organisms 88: 1–12. 10.3354/dao02154 [DOI] [PubMed] [Google Scholar]
- 27. Geoghegan, Jemma L. , Di Giallonardo Francesca, Wille Michelle, Ortiz‐Baez Ayda Susana, Costa Vincenzo A., Ghaly Timothy, Mifsud Jonathon C. O., et al. 2021. “Virome Composition in Marine Fish Revealed by Meta‐Transcriptomics.” Virus Evolution 7: 7. 10.1093/ve/veab005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Liu, Shuang , Xu Tingting, Wang Chong, Jia Tianchang, and Zhang Qingli. 2021. “A Novel Picornavirus Discovered in White Leg Shrimp Penaeus Vannamei.” Viruses 13: 2381. 10.3390/v13122381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kim, Kwang Il , Kwon Woo Ju, Kim Young Chul, Kim Myoung‐Sug, Hong Suhee, and Do Jeong Hyun. 2016. “Surveillance of Aquatic Animal Viruses in Seawater and Shellfish in Korea.” Aquaculture 461: 17–24. 10.1016/j.aquaculture.2016.03.053 [DOI] [Google Scholar]
- 30. Lokmer, Ana , and Mathias Wegner Karl. 2014. “Hemolymph Microbiome of Pacific Oysters in Response to Temperature, Temperature Stress and Infection.” The ISME Journal 9: 670–82. 10.1038/ismej.2014.160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nguyen, Viet Khue , King William L., Siboni Nachshon, Mahbub Khandaker Rayhan, Dove Michael, O'Connor Wayne, Seymour Justin R., and Labbate Maurizio. 2020. “The Sydney Rock Oyster Microbiota is Influenced By Location, Season and Genetics.” Aquaculture 527: 735472. 10.1016/j.aquaculture.2020.735472 [DOI] [Google Scholar]
- 32. Stevick, Rebecca J. , Post Anton F., and Gómez‐Chiarri Marta. 2021. “Functional Plasticity in Oyster Gut Microbiomes Along a Eutrophication Gradient in an Urbanized Estuary.” Animal Microbiome 3: 5. 10.1186/s42523-020-00066-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Liling, Yang , Yingxiang Guo, Hongying Wei, Meng Wang, Yifei Fang, Peng Zhu, and Jingzhe Jiang. 2022. “Identification of a Novel Oyster‐Related Circovirus Genome Compa‐Rative Genome Analysis of Oyster‐Related Circoviruses.” South China Fisheries Science 18: 65. 10.12131/20210260 [DOI] [Google Scholar]
- 34. Chen, Shifu , Zhou Yanqing, Chen Yaru, and Gu Jia. 2018. “Fastp: an Ultra‐Fast All‐In‐One FASTQ Preprocessor.” Bioinformatics 34: i884–i890. 10.1093/bioinformatics/bty560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Li, Dinghua , Liu Chi‐Man, Luo Ruibang, Sadakane Kunihiko, and Lam Tak‐Wah. 2015. “MEGAHIT: an Ultra‐Fast Single‐Node Solution for Large and Complex Metagenomics Assembly Via Succinct De Bruijn Graph.” Bioinformatics 31: 1674–76. 10.1093/bioinformatics/btv033 [DOI] [PubMed] [Google Scholar]
- 36. Li, Dinghua , Luo Ruibang, Liu Chi‐Man, Leung Chi‐Ming, Ting Hing‐Fung, Sadakane Kunihiko, Yamashita Hiroshi, and Lam Tak‐Wah. 2016. “MEGAHIT v1.0: A Fast and Scalable Metagenome Assembler Driven By Advanced Methodologies and Community Practices.” Methods 102(3–11): 3–11. 10.1016/j.ymeth.2016.02.020 [DOI] [PubMed] [Google Scholar]
- 37. Buchfink, Benjamin , Xie Chao, and Huson Daniel H.. 2014. “Fast and Sensitive Protein Alignment Using DIAMOND.” Nature Methods 12: 59–60. 10.1038/nmeth.3176 [DOI] [PubMed] [Google Scholar]
- 38. Huson, Daniel H. , Beier Sina, Flade Isabell, Górska Anna, El‐Hadidi Mohamed, Mitra Suparna, Ruscheweyh Hans‐Joachim, and Tappu Rewati. 2016. “MEGAN Community Edition—Interactive Exploration and Analysis of Large‐Scale Microbiome Sequencing Data.” PLoS Computational Biology 12: e1004957. 10.1371/journal.pcbi.1004957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tisza, Michael J. , Belford Anna K., Domínguez Huerta Guillermo, Bolduc Benjamin, and Buck Christopher B.. 2020. “Cenote‐Taker 2 Democratizes Virus Discovery and Sequence Annotation.” Virus Evolution 7: 7. 10.1093/ve/veaa100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Altschul, Stephen F. , Gish Warren, Miller Webb, Myers Eugene W., and Lipman David J.. 1990. “Basic Local Alignment Search Tool.” Journal of Molecular Biology 215: 403–10. 10.1016/S0022-2836(05)80360-2 [DOI] [PubMed] [Google Scholar]
- 41. Altschul, Stephen F. , Madden Thomas L., Schäffer Alejandro A., Zhang Jinghui, Zhang Zheng, Miller Webb, and Lipman David J.. 1997. “Gapped BLAST and PSI‐BLAST: A New Generation of Protein Database Search Programs.” Nucleic Acids Research 25: 3389–402. 10.1093/nar/25.17.3389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lu, Shennan , Wang Jiyao, Chitsaz Farideh, Derbyshire Myra K., Geer Renata C., Gonzales Noreen R., Gwadz Marc, et al. 2019. “CDD/SPARCLE: The Conserved Domain Database In 2020.” Nucleic Acids Research 48: D265–D268. 10.1093/nar/gkz991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Marchler‐Bauer, A. , and Bryant S. H.. 2004. “CD‐Search: Protein Domain Annotations On the Fly.” Nucleic Acids Research 32: W327–31. 10.1093/nar/gkh454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bastian, Mathieu , Heymann Sebastien, and Jacomy Mathieu. 2009. “Gephi: An Open Source Software for Exploring and Manipulating Networks.” Proceedings of the International AAAI Conference on Web and Social Media 3: 361–62. 10.13140/2.1.1341.1520 [DOI] [Google Scholar]
- 45. Katoh, K. , and Standley D. M.. 2013. “MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability.” Molecular Biology and Evolution 30: 772–80. 10.1093/molbev/mst010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Capella‐Gutierrez, S. , Silla‐Martinez J. M., and Gabaldon T.. 2009. “TrimAl: A Tool for Automated Alignment Trimming in Large‐Scale Phylogenetic Analyses.” Bioinformatics 25: 1972–73. 10.1093/bioinformatics/btp348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Minh, Bui Quang , Schmidt Heiko A., Chernomor Olga, Schrempf Dominik, Woodhams Michael D., von Haeseler Arndt, and Lanfear Robert. 2020. “IQ‐TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era.” Molecular Biology and Evolution 37: 1530–34. 10.1093/molbev/msaa015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kalyaanamoorthy, Subha , Minh Bui Quang, Wong Thomas K. F., von Haeseler Arndt, and Jermiin Lars S.. 2017. “ModelFinder: Fast Model Selection for Accurate Phylogenetic Estimates.” Nature Methods 14: 587–89. 10.1038/nmeth.4285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Letunic, Ivica , and Bork Peer. 2016. “Interactive Tree Of Life (ITOL) v3: An Online Tool for the Display and Annotation of Phylogenetic and Other Trees.” Nucleic Acids Research 44: W242–W245. 10.1093/nar/gkw290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Patro, Rob , Duggal Geet, Love Michael I., Irizarry Rafael A., and Kingsford Carl. 2017. “Salmon Provides Fast and Bias‐Aware Quantification of Transcript Expression.” Nature Methods 14: 417–19. 10.1038/nmeth.4197 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information.
Supporting information.
Data Availability Statement
The data set supporting the results of this article has been deposited in the Genome Sequence Archive (GSA) under BioProject accession code PRJCA007058 [https://ngdc.cncb.ac.cn/gsub/submit/bioproject/subPRO010366/overview] and all RNA virus genetic sequences have been deposited in Genome Warehouse in National Genomics Data Center (NGDC) (Members and Partners 2021) under accession GWHBJCN01000000, GWHBJCM01000000, GWHBJCK01000000, GWHBJCJ01000000, GWHBJCI01000000, GWHBJCH01000000, GWHBJCG01000000, GWHBJCF01000000, GWHBJCE01000000, GWHBJCD01000000, GWHBJCC01000000, GWHBJCB01000000, GWHBJCA01000000, GWHBJBZ01000000, GWHBJBX01000000, GWHBJBW01000000, GWHBJBV01000000, GWHBJBT01000000, that are publicly accessible at https://bigd.big.ac.cn/gwh.