

Abstract
Rather than a “short‐term tenant,” the tumor microbiome has been shown to play a vital role as a “permanent resident,” affecting carcinogenesis, cancer development, metastasis, and cancer therapies. As the tumor microbiome has great potential to become a target for the early diagnosis and treatment of cancer, recent research on the relevance of the tumor microbiota has attracted a wide range of attention from various scientific fields, resulting in remarkable progress that benefits from the development of interdisciplinary technologies. However, there are still a great variety of challenges in this emerging area, such as the low biomass of intratumoral bacteria and unculturable character of some microbial species. Due to the complexity of tumor microbiome research (e.g., the heterogeneity of tumor microenvironment), new methods with high spatial and temporal resolution are urgently needed. Among these developing methods, multi‐omics technologies (combinations of genomics, transcriptomics, proteomics, and metabolomics) are powerful approaches that can facilitate the understanding of the tumor microbiome on different levels of the central dogma. Therefore, multi‐omics (especially single‐cell omics) will make enormous impacts on the future studies of the interplay between microbes and tumor microenvironment. In this review, we have systematically summarized the advances in multi‐omics and their existing and potential applications in tumor microbiome research, thus providing an omics toolbox for investigators to reference in the future.
Keywords: cancer biology, host‐microbe interaction, multi‐omics, tumor microbiome, tumor microenvironment
In this review, we have systematically summarized the advances of multi‐omics and their existing and potential applications in tumor microbiome research, thus providing an omics toolbox for investigators to reference in the future.
Highlights
The tumor microbiome plays a significant role in carcinogenesis, cancer development, metastasis, and cancer therapies.
The tumor microbiome has great potential to become a target for early diagnosis and treatment of cancer.
Genomics, transcriptomics, proteomics, and metabolomics are powerful approaches for tumor microbiome studies, by facilitating an understanding of different levels of the central dogma.
Multi‐omics will make enormous impacts on future research of the tumor microbiome.
The development of single‐cell multi‐omics technologies will motivate the progression of the tumor microbiome field.
INTRODUCTION
The earliest known historical records linking cancer and microbes can be traced back to over a century ago [1]. There are currently about 1012 known microbial species on Earth [1, 2], with only 11 having been declared to be “oncomicrobes” (or pathogens known to be carcinogenic to humans) by the International Agency for Research on Cancer (IARC). These tumor microbes cause about 2.2 million cases every year or ~13% of cancer cases worldwide. Furthermore, researchers have found that live microbes exist in many kinds of tumors (Figure 1), including breast cancer [3], colorectal cancer [4, 5], hepatocellular carcinoma [6, 7], pancreatic cancer [8, 9], skin cancer [10], renal cell carcinoma [11], gastric cancer [12], lung cancer [13, 14], prostate cancer [15–17], nasopharyngeal carcinoma [18], and so forth. Notably, microbes in the same type of tumors have striking similarities [19]. Intratumoral microbes have been found to affect the development and treatment of tumors through diverse mechanisms, such as DNA damage, oncogenic pathway activation, antitumor drug catabolism, and immune system modulation (Figure 1) [19–25].
Figure 1.
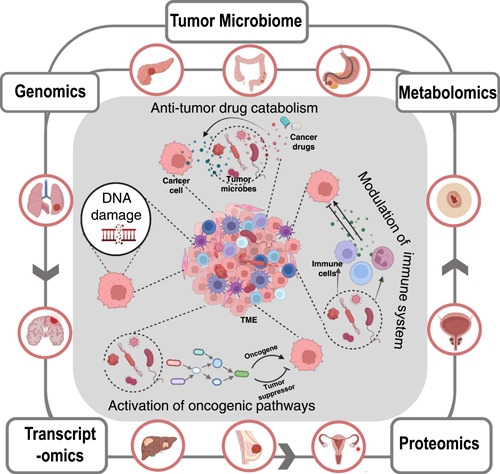
An integrated model showing of the roles of microorganisms in carcinogenesis and tumor development, various types of tumors that the tumor microbiome has been linked to, and multi‐omics research methods of the tumor microbiome. TME, tumor microenvironment.
Research advances have begun to uncover a sliver of the full scope in understanding how the tumor microbiome influences cancer [26]. One well‐established mechanism of these microorganisms is the release of genotoxins that are capable of inducing oncogenic DNA mutations, including colicin (DNA alkylator) from Escherichia coli [27], cytolethal distending toxins (deoxyribonuclease activator) from Gram‐negative bacteria (e.g., Campylobacter jejuni) [28], and toxins that act as reactive oxygen species (ROS) producers from enterotoxigenic Bacteroides fragilis [29]. Furthermore, many regulatory pathways related to cancer pathogenesis can be affected by microbes. For instance, Helicobacter pylori, one of the aforementioned 11 declared oncomicrobes, has been shown to cause carcinogenesis, by inducing inflammation and affecting key intracellular signal pathways that regulate the growth and proliferation of mucosal cells [30]. In addition to influencing tumorigenesis, the microbial community has been implicated in cancer treatment approaches, through the metabolism of anti‐tumor drugs. Gemcitabine, a chemotherapeutic drug commonly used to treat pancreatic ductal adenocarcinoma, can be metabolized to an inactive state by intratumoral bacteria, thereby resulting in cancer drug resistance [31]. Moreover, many microorganisms can affect cancer development and therapies by affecting host immunosurveillance [32]. Therefore, the tumor microbiome is gradually becoming a new focus in the early diagnosis and treatment (especially immuno‐oncology) of cancer [33, 34].
Even though research into the tumor microbiome has made great progress, a number of challenges in this emerging area still remain, such as the low biomass of intratumoral bacteria, unculturable character of some microbial species, and heterogeneity of the tumor microenvironment (TME) [35–37]. Thus, our understanding of the tumor microbiome is still far from sufficient, mainly due to the limits in research technologies. As the tumor microbiome is becoming more and more significant in both basic and translational research, the development of new methods with high spatial and temporal resolution is required. Recently, the integrated application of multi‐omics technologies (combining genomics, transcriptomics, proteomics, and metabolomics) has shown great power in biomedical research and facilitated the understanding of many previously elusive biological processes [38]. To reveal the pathophysiological functions of the tumor microbiome in cancer development and therapies on different levels of the central dogma (Figure 1), multi‐omics (especially single‐cell omics) approaches have begun to be developed and applied [39, 40]. Therefore, in this review, we will systematically summarize the multi‐omics techniques that can be utilized for tumor microbiome research and present our views on the current advantages and limitations of these methodologies.
GENOMICS AND TRANSCRIPTOMICS FOR TUMOR MICROBIOME STUDIES
Genomics and transcriptomics focus on nucleic acids (i.e., DNA and RNA), especially those involved in the regulation of gene expression. At present, a large number of research techniques have been developed to study the genomes and transcriptomes of diverse biological systems. These techniques of note include high throughput sequencing and gene editing (Figure 2). Currently, the most widely employed sequencing techniques include three next‐generation sequencing (NGS) systems (i.e., Illumina, Ion Torrent, BGI) and two third‐generation sequencing techniques (i.e., PacBio and Nanopore) [41]. These sequencing approaches are the basis for the development of a variety of genomic and transcriptomic research methodologies, including DNA sequencing [15], RNA sequencing [42], 16S rRNA sequencing [43], epigenetic techniques (e.g., ChIP‐seq and DNA/RNA methylation sequencing) [44–46], and three‐dimensional genomic technologies [47]. Moreover, recent research advances of gene editing tools have enabled multi‐gene manipulation on a genome‐wide level [48]. As these technologies can easily target nucleic acids, they have become important tools for research into the tumor microbiome and TME, greatly accelerating the identification and traceability of tumor microbes, and helping to reveal how tumor microbes, cancer cells, and the immune system interact [7, 33, 49, 50].
Figure 2.
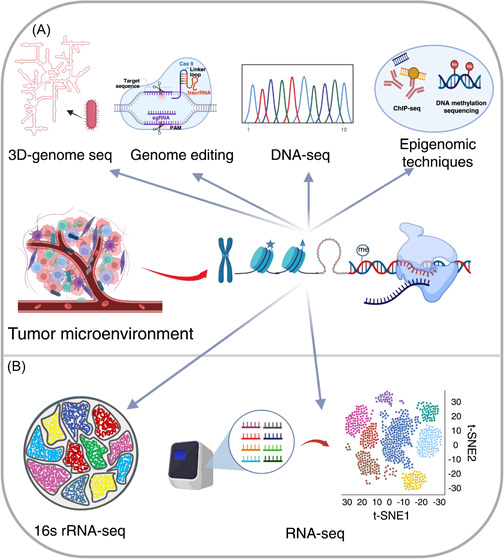
Techniques for genome and transcriptome research of the tumor microenvironment. (A) Techniques for metagenomic sequencing. CHIP‐seq, chromatin immunoprecipitation sequencing; DNA‐seq, DNA sequencing. 16S rRNA‐seq, 16S ribosomal RNA sequencing; RNA‐seq, RNA sequencing.
DNA sequencing (DNA‐seq)
DNA‐seq is one of the most important approaches in microbial genome research, which can be applied to analyze all of the genes of complex samples, such as the human microbiome. DNA‐seq is often used for three types of genomic analyses: whole‐genome sequencing (WGS), whole‐exome sequencing (WES), and targeted sequencing [51, 52]. With the rapid development of DNA technologies, DNA‐seq is becoming a powerful tool to study unculturable microbes, including those in the human microbiome [53, 54]. The sequencing and heterologous expression of genomic libraries significantly facilitates our understanding of metabolic pathways and the physiology of the tumor microbiome [55, 56].
16S rRNA sequencing
16S rRNA sequencing is a sequencing technology that is specifically focused on the 16 S component of the 30 S ribosomal subunit expressed by bacteria and archaea. This sequence contains alternating highly conserved and hypervariable regions [57]. The conserved region can be used to design universal primers for the amplification of target fragments, while analyses of the hypervariable region can be employed to identify specific bacterial species, though some annotations may be below the species level [58]. In the tumor microbiome field, 16 S rRNA sequencing is mainly applied to study the species composition in communities, evolutionary relationships between species, and diversity of microbial communities [19].
RNA sequencing (RNA‐seq)
RNA‐seq is a transcriptome sequencing method, where high‐throughput sequencing methodologies are utilized to study the populations of diverse RNA in complex samples, including messenger RNA (mRNA), transfer RNA (tRNA), noncoding RNA (ncRNA), microRNA (miRNA), etc. Over the past decades, RNA‐seq technologies have developed rapidly and become an indispensable tool to analyze gene expression on the transcriptome level [59]. The application ranges of RNA‐seq have grown with the development of NGS. In the field of RNA biology, RNA‐seq has been widely applied to study single‐cell gene transcription, protein expression, and RNA structures [60, 61]. The advances of RNA‐seq technologies have also facilitated the emergence and development of spatial transcriptomics [62]. Direct long‐read or full‐length RNA‐seq methods, as well as better data analysis methods, have enabled researchers to better understand RNA biology, including the machinery of the transcription process and the impacts of folding and intermolecular interactions on RNA function [63–65]. Overall, RNA‐seq is a powerful tool to investigate the mutual influences of cancer cells and tumor microbiome on each other's pathways by analyzing at the level of gene transcription.
Epigenomic techniques
Epigenetics has been referred to as the investigation of phenotypic changes that do not involve alterations in the DNA sequence. Distinct from genetic regulation, typical epigenetic changes (including DNA/RNA modifications and histone posttranslational modifications) are reversible and do not influence cellular genomic sequences. In eukaryotic cells, nucleosomes are the basic repeating unit of chromatin organization and are comprised of 147 base pairs of DNA spooled 1.5 times around an octamer of core histones (i.e., two copies of H2A, H2B, H3, and H4). The dynamics of covalent modifications on DNA, RNA, and histones are a key mechanism of epigenetic regulation that controls the activation and suppression of specific genes. Epigenetic regulation plays an essential role in regulation of chromatin structure and function during replication, transcription, and repair of DNA damage, which have close relevance to many human diseases, especially cancer [66].
Specifically, DNA methylation alters the genetic transcription without changing the genome sequence, which may result in tumorigenesis when tumor suppressor genes are turned off [67]. A large number of studies have shown that aberrant methylation of DNA is closely related to the occurrence and development of diverse types of tumors [68, 69]. Therefore, changes in DNA methylation levels have been detected as biomarkers for cancer diagnosis [70]. Currently, multiple DNA methylation sequencing techniques have been developed, including whole genome bisulfite sequencing (WGBS) [71], reduced representation bisulfite sequencing (RRBS) [72], oxidative bisulfite sequencing (oxBS‐seq) [73], and cell‐free methylated DNA immunoprecipitation and high‐throughput sequencing (cfMeDIP‐seq) [74].
On the other hand, histone posttranslational modifications (PTMs) include methylation, phosphorylation, acetylation, crotonylation, ubiquitination, glycosylation, glycation, and ADP ribosylation [75]. Various histone PTMs are believed to be involved in the emergence and development of cancers [76]. For example, methylglyoxal synthases (MGSs) derived from microorganisms can biosynthesize excess amount of methylglyoxal in TME [77, 78]. Histone MGO‐glycation has been shown to influence tumor development, by affecting the three‐dimensional architecture of chromatin [79, 80].
Chromatin immunoprecipitation sequencing (ChIP‐seq) is one of the most commonly used methods used to study the effects of histone modifications on gene transcription [81, 82], though it was initially developed to study the interactions between proteins and DNA [83]. Chromatin immunoprecipitation (ChIP), also known as binding site analysis, is a powerful tool to study these interactions in vivo and has been crucial to better understanding epigenetic changes to the genome [84]. ChIP‐seq, which combines ChIP with second‐generation DNA sequencing, can efficiently detect the DNA binding sites of specific histone modifications and transcription factors [85]. The principle of ChIP‐seq is as follows: DNA fragments bound with a protein of interest are enriched and immunoprecipitated by ChIP before being purified, and thereafter, a library is constructed. The enriched DNA fragments are then subjected to high‐throughput sequencing. By applying this method, researchers have accurately located sequences in the genome that interact with specific histones [86] and transcription factors [87]. Importantly for tumor microbiome research, ChIP‐seq is suitable for the (epi)genomic studies of both host and microbe cells.
Three‐dimensional (3D) genomics
The 3D genomics, also known as spatial genomics, is based on the basic information of the 1D genome sequence and studies the 3D structure of genome, as well as the mechanisms of transcriptional regulation mediated by different elements (including transcription factors and DNA/RNA‐binding proteins). In recent years, the technologies developed in this field mainly include Hi‐C (high‐throughput/resolution chromosome conformation capture) [88, 89], Micro‐C (micrococcal nuclease‐based analysis of chromosome folding) [90], Micro‐C XL (micrococcal nuclease‐based analysis of chromosome folding using long x‐linkers) [91], ChIA‐PET (chromatin interaction analysis by paired‐end tag sequencing) [92], and HiChIP (in situ Hi‐C followed by chromatin immunoprecipitation) [93]. As related to tumor microbiome research, while most of these approaches above are focused on eukaryotic genomes of host cells, Hi‐C technology has emerged recently as a powerful tool to investigate bacterial chromosomes, enabling the physical proximity of DNA sequences in a sample to be assayed [94, 95].
Genome editing
Gene editing (also known as genetic modification or genetic engineering) mainly refers to the use of biochemical approaches to modify genomic sequences. This is often done by introducing the target gene fragment to the genome or deleting the specific gene region from the genome, so as to change the genotype of the host cell or strengthen the original genotype [96]. As there is a long history of gene editing, many elegant tools have been developed as “genetic scalpels,” including artificial restriction enzymes [97], zinc finger nucleases (ZFNs) [98], transcription activator‐like effector nucleases (TALENs) [99], clustered regularly interspaced short palindromic repeats (CRISPR) and CRISPR‐associated protein 9 (Cas9) [100–102], CRISPR base editors [103], prime editing systems [104], and programmable addition through site‐specific targeting elements (PASTE) [105]. At present, the CRISPR‐Cas9 technology and the systems derived from it are the mainstream technologies most widely utilized in genome editing [106]. Furthermore, Cas9 endonuclease dead (also known as dead Cas9 or dCas9) is a mutant form of Cas9, that can no longer act as an endonuclease, due to point mutations in its endonuclease domains [107]. dCas9, which serves as a gene location guide, is now widely used in the genome editing of different biological systems through the fusion expression with other functional enzymes (such as cytidine deaminases [108], adenine deaminases [109], and epigenetic writer/eraser enzymes [110, 111]) and other resulting emerging technologies (e.g., epigenome editing) [112, 113].
As for tumor microbiome research, genome editing has been widely applied toward studying both host cells [114] and microbes [115]. For example, it has been reported that the genomic composition of intestinal microorganisms varies widely [116]. Phylogenetic analyses of single nucleotide polymorphisms (SNPs) show significant intraspecific genetic variation in global metagenome samples [117]. There is considerable variation in the genomes of microbial strains, with the same species possibly using similar sequences for different functions [118, 119]. Therefore, to understand the role of genomic variation, editing with CRISPR‐Cas9 is a promising method to modify microbial isolates.
SINGLE‐CELL GENOMICS AND TRANSCRIPTOMICS
Genome and transcriptome methods can achieve higher coverage and lower the risk of artifacts, but their assembly depends on the sequencing depth and community complexity [120]. For example, metagenomic approaches allow researchers to study bacteria in complex communities, but the complexity of these communities means that sequences of low‐abundance bacteria may be suppressed by those of higher abundance organisms, or, in the case of the human microbiome, by mitochondrial DNA [121]. Additionally, genetic information in cells with similar phenotypes may be significantly different and a large amount of low abundance information may be lost in the overall characterization. To make up for these limitations, single cell sequencing technology aims to reveal gene structures and gene expression of a single cell, better reflecting the heterogeneity between cells [122].
Looking at current publications analyzing the tumor microbiota using single cell techniques, there are four main research methods to study the genome and transcriptome: agar plate dilution [123], single‐cell exome sequencing based on fine capillary pipetting separation system coupled with an inverted microscope [124, 125], droplet‐based microfluidics [126], and improved fluorescence in situ hybridization (FISH) [127]. Notably, the first and key step of analyzing single cells is to prepare a single cell suspension from tissue or other sources.
Agar plate dilution
Agar plate dilution refers to the method of obtaining pure culture of a single microorganism by streaking plates and is the most traditional method of obtaining pure cultures of microorganisms. By diluting a mixture of microorganisms or different cell types repeatedly, one can separate diverse colonies into single cells, which will grow into homogenous colonies (Figure 3A). The development of the microfluidic streak plate (MSP) technique builds on traditional plate streaking with the use of microfluidic technology [128]. MSP uses microfluidics to make microdroplets that can be streaked onto dishes to grow single cells. Both traditional agar plate dilution and MSP can be integrated into the high‐throughput screening workflow.
Figure 3.
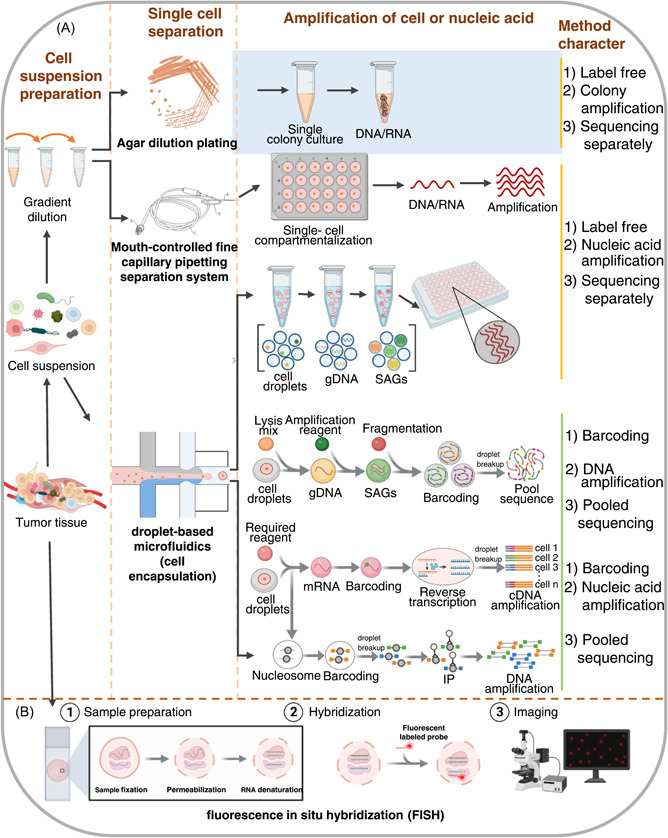
Single‐cell genomic and transcriptomic approaches. (A) Preparation of mixed cell suspension from tumor tissue (left). Workflow of three methods to separate mixture into single cells for genomic and transcriptomic sequencing (middle): agar dilution plating, mouth‐controlled fine capillary pipetting separation system, droplet‐based microfluidics (cell encapsulation). gDNA, guide DNA; SAGs, single‐cell amplified genomes. Comparison of characteristics of each method (right). (B) Workflow of single‐cell fluorescence in situ hybridization (FISH).
Methodology based on inverted microscope and mouth‐controlled fine capillary pipetting system
Serially diluted cell suspensions from tissues can be processed and isolated into single cells using a mouth‐controlled fine capillary system or other single‐cell pipetting systems under an inverted microscope. Isolated single cells are randomly isolated into PCR tubes and recorded as photomicrographs after visual confirmation with a microscope. Single‐cell genomes can then be extracted for whole genome amplification (WGA) [125]. This amplified genome can be used for exome sequencing, as described previously (Figure 3A). This method has been used to study the intra‐tumoral genetic landscape of renal cell carcinoma and adjacent renal tissues, finding that this carcinoma may be more complex than previously thought and promoting the use of more effective cell‐targeted therapies [124]. Notably, this approach may also be applied to study the microbes within individual tumor cells.
Droplet microfluidic approach
The droplet microfluidic approach is mainly used to prepare droplets by using two incompatible liquids, with the generation of droplets being controlled by a microtubule structure and the flow rate ratio between the two liquids [120] (Figure 3A). Driven by a pump with a fixed flow rate, the two liquids enter different microchannels and when they meet, one fluid shears the other, creating micro‐droplets. These droplets are ideal micro‐reaction chambers and can be sized to accommodate a single cell. Individual droplets can be filled, manipulated, split, combined, detected, and classified with these systems, and thousands of individual droplets can be manipulated using microliters of reagents. Droplet microfluidics can separate a large population into individual cells, extracting and segmenting the genome through a series of enzymatic steps. Genetic material is amplified and marked before these single cells are sequenced. This is an integrated technology combining microfluidic technology, DNA barcoding, and sequencing technology.
An encapsulated microorganism can be directly cultured in liquid droplets and allowed to grow to hundreds of cells during the first genome amplification, before being lysed, and the genomic DNA purified. Then, a cell sorter is used to sort beads containing single‐cell amplified DNA into standard PCR plates, followed by reamplification into single‐cell amplified genome (SAG) library. A microfluidic droplet generator has been used to culture mouse gut microbes in agarose gel beads, a typical single‐cell sequencing method based on the SAG‐gel platform [120] (Figure 3A).
The encapsulated single microorganism can also be lysed directly, followed by DNA amplification or RNA reverse transcription and barcoding. For single cell genome sequencing, cells are usually amplified first, then labeled with barcodes [129, 130]. For single cell transcriptome sequencing, cellular RNA is usually labeled first, followed by reverse transcription, and finally complementary DNA (cDNA) amplification [131] (Figure 3A). To study nucleosomes within cells, the nucleosome must first be labeled with a barcode, followed by immunoprecipitation (single‐cell ChIP‐seq) and DNA amplification [132] (Figure 3A). Droplet microfluidic technology can analyze millions of independent reactions. Recently, it has been used for deep sequencing of a single DNA molecule, with the nucleosome being labeled to be analyzed through single‐cell ChIP‐seq and high‐throughput analysis [123, 133, 134].
The ability to sequence cells without having to separately culture microorganisms is a powerful aspect of micro‐droplet microfluidic single cell sequencing. With this method, a metagenome database can be generated to study microbes in tumors and other regions like the intestine. The limitation of this method are that it starts with a single genome copy and the loss of material during enzyme and microfluidics treatment is irretrievable, limiting the maximum available coverage and resulting in significantly lower coverage per cell.
Improved fluorescence in situ hybridization (FISH)
FISH has always been an important tool to study cultured microorganisms, and can be directly applied to identify single bacterial cells, though traditional FISH has been limited by its low resolution. FISH includes the use of single‐stranded oligonucleotide probes to specifically label target RNA (Figure 3B). However, because the degree of fluorescence is directly related to the copy number of target RNA, this technique is most suitable for targeting rRNA. As different regions of 16 S rRNA have different levels of conservation, the probe can be species‐specific or selected according to different classification levels. To overcome low resolution, researchers have developed various methods, including catalyzed reporter deposition‐FISH (CARD‐FISH), also known as tyramide signal amplification (TSA) FISH, which uses horseradish peroxidase (HRP)‐labeled probes to amplify signals exponentially, so that single cells can be fluorescently labeled accurately and specifically [135, 136]. Highly phylogenetic resolution FISH (HiPR‐FISH) is another newly designed method that modifies existing algorithms in single cell automatic image segmentation and performs pixel classification and image optimization filtering [127]. Therefore, these advances have enabled researchers to better study specific microbial populations, visualize and sort small amounts of bacteria and very low copy RNA, and achieve single cell quantification [137]. It is likely that FISH will be used first to identify and locate microorganisms in tumor tissues, before they are sorted and studied using other analysis methods.
PROTEOMICS
Expression and regulation of the genome and transcriptome are directly reflective of tens of thousands of proteins, whose characteristics, including expression levels, PTMs, and protein‐protein interactions, are intricate. Mass spectrometry coupled with liquid chromatography (LC‐MS) is one of the most popular methods used for such analyses [138] and has become a powerful tool for tumor microbiome research [139–141]. There are three different strategies of proteomic analysis: “top‐down,” “middle‐down,” and “bottom‐up.” Specifically, “top‐down” (Figure 4A) [142] directly sends target proteins to mass spectrometry (MS) for analysis, without any pretreatment. “Middle‐down” [143] refers to proteins being partially digested to obtain relatively large peptide fragments, before being sent to MS for further analysis. Finally, “bottom‐up” analysis, also known as shotgun analysis, uses robust methods where the proteins are digested down to 6‐20 amino acids, before being analyzed by MS (Figure 4B) [144]. Among these three strategies, the third is the most widely used by proteome researchers globally, as the bottom‐up analysis strategy is usually more efficient for protein mixtures with high complexity that can contain thousands of mixed proteins of different orders of magnitude [145].
Figure 4.
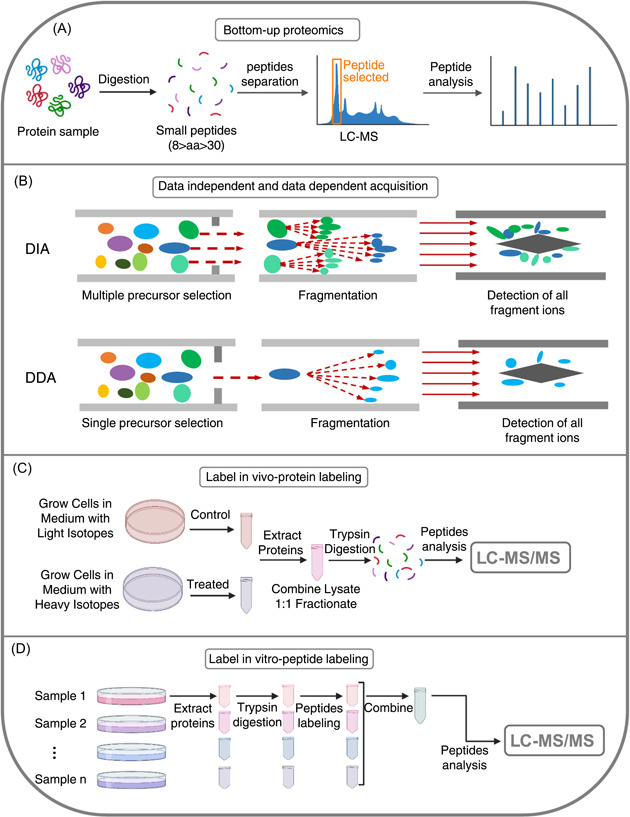
Different techniques of proteomics. (A) Procedure of bottom‐up proteomics. LC‐MS, liquid chromatography‐mass spectroscopy. (B) Data‐independent (DIA) and data‐dependent acquisition (DDA) modes of proteomics. (C) Workflow with SILAC (stable isotope labeling by/with amino acids in cell culture), a method of labeled quantitative proteomics, in vivo. (D) Workflow with TMT (tandem mass tag), a labeling quantitative proteomics method, in vitro.
In proteomics, one of the major aims is to compare samples of interest (such as healthy and diseased tissue) to identify proteins that are differentially expressed and to quantify these differences. There are currently two broad approaches toward generating shotgun MS proteomics data: data‐dependent acquisition (DDA) and data‐independent acquisition (DIA) [146] (Figure 4B). In tandem with MS (MS/MS), the DDA strategy only selects certain peptides of higher signals generated in the first cycle of MS for fragmentation in the second cycle, while with the DIA approach, all peptides generated during the first MS cycle can be fragmented in the second round [147].
DIA has become the most popular choice in research on pathogenic mechanisms and biomarker screening with clinical samples, such as those of gut microbes [148] and tumors [149, 150], as it can obtain an unbiased and deeper look at the proteome of samples, particularly when these samples are from understudied organisms (e.g., tumor microbes) or complex tissues (e.g., tumor tissues). Compared to the DIA approach, DDA is a more widely applied and quantitative method in targeted analyses. Targeted proteomics analysis focuses on a subset of proteins of interest in a sample, mainly through parallel reaction monitoring (PRM) [151] or multiple reaction monitoring (MRM) [152]. MRM, also known as selective reaction monitoring (SRM), is a highly specific and sensitive MS technique that can selectively quantify peptides within complex mixtures. This technique uses a triple quadrupole MS (TQMS, QqQ) that first targets the peptide ion corresponding to the protein of interest, subsequently fragmentating it to produce a range of daughter ions. One (or several) of these fragmented daughter ions can be selected for quantification purposes. PRM is comparable to SRM/MRM, but is more convenient for assay development, due to its absolute quantification of proteins and peptides, based on high‐resolution and high‐precision mass spectrometry [153]. Protein quantification methods generally include absolute quantification and relative quantification. Absolute quantification determines the expression levels of the target peptide in the sample, following the establishment of a standard curve, for which the precise amount of the standard peptide is known [154]. Relative quantification determines fold changes in the expression between two samples by comparing intensities, without acquiring a standard curve.
There are labeled and label‐free methods of the relative quantification approach. Label‐free quantification methods aim to determine the relative amount of protein in two or more biological samples without using a stable isotope label bound to the protein. Commonly used labeled quantification techniques include isobaric tags for relative and absolute quantitation (iTRAQ) [155], tandem mass tag (TMT) [156], and stable isotope labeling by/with amino acids in cell culture (SILAC) [157], and are accomplished by labeling or substituting the corresponding amino acids with isotopic labels. TMT (Figure 4D) and iTRAQ are used for in vitro labeling, while SILAC is an in vivo labeling technology (Figure 4C). The advantage of these labeling methods is that the amount of proteins from different sources can be determined in a single experiment. The SILAC strategy stands out for substituting an isotopically heavy form of an amino acid for the naturally occurring light form. Because labeled and unlabeled samples are combined during the initial steps of sample preparation, SILAC minimizes quantitative error that may result from handling samples in parallel [158]. In addition, the mixing of samples permits a variety of enrichment techniques, including immunoprecipitation. These techniques can improve the detection of changes in abundance for both low‐abundance proteins and PTMs, such as phosphorylation and glycosylation [159–161].
METABOLOMICS
Metabolomics is widely applied to quantitatively analyze all the metabolites in different organisms and to discover the corresponding pathophysiological functions of these metabolites. The application of metabolomics in tumor microbiome research is committed to identifying the metabolic differences of tumor microorganisms and their interactions with the surrounding environment, to enhance the understanding of how tumor microorganisms affect molecular mechanisms related to carcinogenesis. These metabolites mainly include short‐chain fatty acids (SCFAs) [162], amino acid metabolites [163], vitamins [164], bile acids (BAs) [165], lactic acid [166], toxins [167], and formate [168], among others. Many studies have shown that microbial metabolites can significantly affect the occurrence and development of tumors through positive or negative regulation (Figure 5A), by affecting immunity, inflammation, signaling pathways, or by regulating the TME [26].
Figure 5.
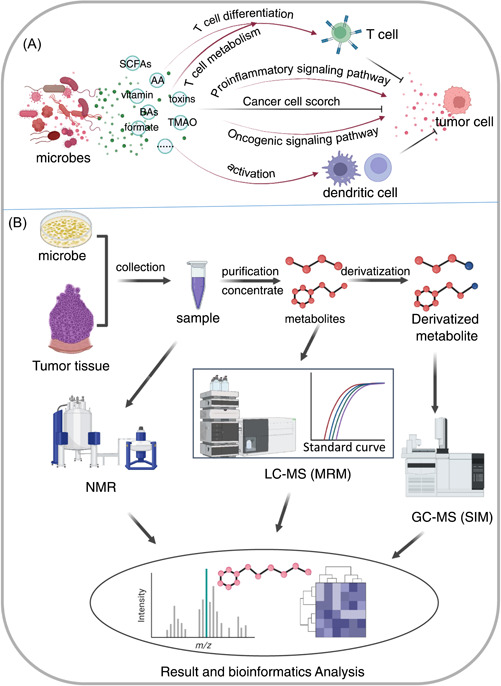
Metabolic methods used in tumor microbiome research. (A) Microbial metabolites affecting tumors through a variety of mechanisms. (B) Three most commonly used approaches of tumor microbe metabolomic research: nuclear magnetic resonance (NMR), liquid chromatography mass spectroscopy (LC‐MS) with multiple reaction monitoring (MRM), and gas chromatography mass spectroscopy (GC‐MS) with selection ion monitoring (SIM).
For example, SCFAs are one of the main metabolites of the intestinal flora. Studies have shown that SCFAs can affect intestinal function and intestinal metabolism positively and negatively. Supplementation of certain SCFA‐producing bacteria has been found to inhibit the development of intestinal tumors [169]. On the other hand, SCFAs are also expected to become a diagnostic and treatment target of CRC [169]. Isovaleric acid (IVA), a SCFA metabolite of intestinal microbes associated with CRC, can promote the expression of proteins that lead to downstream increases in 5‐hydroxytryptamine (5‐HT) [170]. 5‐HT directly acts on cancer stem cells, promoting self‐renewal and increasing intestinal tumor occurrence [171].
The interruption of bacteria producing vitamins B6 and B9 reduces the curative effect of 5‐fluorouracil (5‐FU), one of the most common chemotherapeutic drugs [172]. Additionally, Vitamin E secreted by intestinal bacteria can enter dendritic cells (DCs), enhancing the activation of DCs and promoting T‐cell antitumor immune responses [173].
Other metabolites of microbes can also regulate T‐cell metabolism, such as the secondary BA isoallolithocholic acid, which can enhance oxidative phosphorylation and mitochondrial ROS production, promoting Treg differentiation [174]. Interactions between some refluxed BAs and gut microbes can lead to the production of the secondary BA taurodeoxycholic acid, which promotes the occurrence of gastric cancer [175]. Toxin‐producing bacteria Clostridium difficile can activate signaling pathways in intestinal epithelial progenitor cells, leading to increased production of ROS and tumor‐promoting mucosal immune responses, including the infiltration of activated myeloid cells and an increase in interleukin‐17 (IL‐17) producing lymphocyte subsets [176]. Formate secreted by Fusobacterium nucleatum can activate many signaling pathways, increasing invasiveness of CRC cells [168]. Trimethylamine oxide (TMAO) from intestinal flora enhances CD8+ T‐cell‐mediated antitumor immunity [177]. Gallic acid, another metabolite of intestinal microbes, has been shown to regulate Treg cells and enhance cancer immunotherapy [176], while also causing a malignant phenotype in cells in the presence of p53 mutations [178]. Intestinal bacteria and their metabolites can promote tumors by inducing mutations, enhancing tumor‐promoting inflammatory responses, and recruiting immunosuppressive cells. Intestinal fungi can also induce a tumor‐promoting inflammatory microenvironment and can produce mycotoxins to induce mutations, thus promoting tumors [179].
Analytical and sample preparation methods for tumor microbiome metabolomics
Metabolomics depends on the use of analytical techniques. Two instrument platforms (Figure 5B), nuclear magnetic resonance (NMR) [180] and MS [181], are the first choice at present. NMR is nondestructive, independent of analytical separation techniques, and the sample preparation is relatively fast, easy to operate, and low in cost. Absolute quantification using NMR can also be improved by adding an internal standard compound with a known concentration into the sample [182]. The principle of molecular characterization through NMR is in its strong magnetic field, which interacts with the nucleus of molecules, as they absorb and resonate the electromagnetic spectra of a specific frequency. However, due to the relatively low sensitivity of NMR, complexity of microbial metabolites, and variation in metabolite concentrations, the application of NMR in microbial metabolomics is limited [183].
To overcome these problems, high‐resolution MS has become an indispensable tool in metabolomics research. The technology utilizes various separation techniques, including liquid chromatography (LC), gas chromatography (GC), and capillary electrophoresis (CE), and is widely used in microbial metabolomics [184]. GC‐MS is one of the most commonly used technologies in metabolomics, due to its stable performance, good reproducibility of results, and high peak capacity [185]. This technique is mainly used for volatile and thermally‐stable low molecular weight compounds. However, most microbial metabolites, such as phosphorylated metabolites, are non‐volatile and thermally unstable, and thus, may be degraded when placed at higher temperatures. Therefore, this method is often used to analyze derivatized microbial metabolites, such as those from Propionibacterium fischeri, E. coli, and Bacillus subtilis [186]. Miyamoto et al. has also utilized GC‐TOF‐MS to find biomarkers associated with lung cancer prognosis [187]. CE‐MS is another tool for metabolomics analysis [188]. Compared to GC‐MS and LC‐MS, its advantages include high separation efficiency, small sample volume, and low cost. However, its main disadvantage is that the interface between CE and MS is difficult, so CE‐MS is not widely used. LC‐MS and tandem LC‐MS/MS are important analytical tools in metabolomics. Unlike GC‐MS, LC‐MS can analyze polar and high molecular weight compounds without high temperature or volatility, has a wider coverage of metabolites for analysis, and has more convenient sample preparation [189]. In addition, LC‐MS has a higher sensitivity, even with a small number of samples [190]. The appearance of ultrahigh performance liquid chromatography (UHPLC) has greatly improved the chromatographic resolution of LC‐MS.
Metabolomics research generally falls into two categories: targeted metabolomics and nontargeted metabolomics [191]. Nontargeted refers to the systematic analysis of all metabolites in a sample, while targeted refers to the relative or absolute quantification of metabolic changes, with absolute quantification often using SRM and an established standard curve. TQMS and traditional ion trap mass spectrometers have been widely used in targeted metabolomics research. However, the implementation of nontargeted metabolomics has required more advanced analytical methods, automated spectral data processing, biological data interpretation, and hypothesis generation [192]. High‐resolution tandem mass spectrometers, such as quadrupole time of flight (Q‐TOF) mass spectrometers, Fourier‐transform ion cyclotron resonance (FT‐ICR) mass spectrometers, and ion trap mass analyzer Orbitrap‐based mass spectrometers, are the preferred instruments for nontargeted metabolomics research, as they can clarify structural information and allow for high data acquisition speed [193].
Intracellular metabolism can change rapidly based on time and the external environment [194]. Therefore, methods and conditions of sampling and sample preparation, including time, storage conditions, and other factors, can greatly affect the reproducibility, precision, and accuracy of metabolic detection. Sample preparation occurs in two stages: metabolic quenching and extraction of metabolites. Quenching is the process of rapidly stopping the metabolic activity of cells with limited cell membrane damage [195]. Tissues or cells are collected quickly, immediately frozen in liquid nitrogen, and stored them at −80°C. In addition, other methods like freezing quenching or chemical quenching with acids or cold aqueous methanol solutions are also commonly used.
Extraction is the process of separating metabolites from samples. Extraction can be achieved through physical means, by manual grinding or using a tissue homogenizer. Solvent extraction using organic solvents, such as methanol, chloroform, and acetonitrile, is the more commonly used method, because of its higher extraction efficiency and minimal ionization suppression in LC‐MS analysis [196]. NMR‐based analysis does not need chemical derivatization and the sample preparation is much simpler. Both of these microbiologic metabolic analysis methods have advantages and disadvantages. Researchers should choose the best method or combination of tools to analyze microbial metabolites, according to the characteristics of interested compounds and availability of analysis tools.
SINGLE‐CELL PROTEOMICS AND METABOLOMICS
Single‐cell sequencing is used for the analysis of the cellular genome, transcriptome, and epigenome. Similarly, single‐cell proteome and metabolome studies are also essential for genome and transcriptome research on single cell levels. Unlike single‐cell nucleic acid sequencing where signals can be amplified, single‐cell protein and metabolite analysis signals cannot be. In this type of research, tracing is the biggest challenge, as this is critical to reduce the loss of intracellular analytes during sample processing.
The field of single‐cell proteomics is still in its infancy, and lacks a clear and generally applicable approach [197]. Nonetheless, recent rapid progress in absolute quantification of single‐cell proteins and highly multiplexed protein measurements has relied on the increased sensitivity of MS instruments and the improvements in sample preparation methods to overcome the limitations of traced proteins in single cells.
Orbitrap‐based high‐resolution MS instruments have undergone continuous improvements in ion transmission efficiency, detector sensitivity, duty cycle, and resolution over successive generations of mass spectrometers. Recently, when individual HeLa cells were analyzed on the latest generation of the Orbitrap Eclipse MS, peptide and protein coverage increased by 36% and 20%, respectively [198]. The latest MS instruments have also introduced drift tube‐based ion mobility spectrometry (DT‐IMS) [199, 200], trapped ion mobility spectrometry (TIMS) [201], and high field asymmetric waveform ion mobility spectrometry (FAIMS) [202], which can improve selectivity, separate or filter singly charged ions from multiply charged peptides, and select multiply charged peptides for self‐determination. In terms of identification and quantification with shotgun proteomics, the removal of singly charged species is beneficial for trace sample analysis, since solvent clusters and contaminants that are negligible in batch studies can become very important in low‐input studies. In all cases, LC separation and MS acquisition settings, such as column, flow rate, ion injection time, and automatic gain control (AGC) target settings, in single‐cell analyses tend to be very different from those used for batch studies, ensuring that sufficient reporter ion signal is available for accurate quantification and maximum ion utilization. However, current techniques of single‐cell metabolomics have mainly utilized single‐cell analyses of cultured microbes, and cannot be directly applied to multicellular organisms to study microbe‐cell interactions [203]. Mass spectrometry imaging (MSI) has shown great potential in the field of single‐cell metabolism, by mapping metabolites and other biomolecules in tissues [204].
Matrix‐assisted laser desorption/ionization (MALDI) mass spectrometry, a type of MSI technology, is currently another one of the most commonly used single‐cell metabolic analysis techniques [203, 205–207]. A sample is mixed with a matrix and irradiated with a pulsed laser. The beam protonates or deprotonates the analyte, before sending it into the mass spectrometer. MALDI‐MS measures mass spectra at different points in tissue sections or slides containing plated cells, allowing researchers to create spatial maps of metabolism. MS images using MALDI have been obtained with a spatial resolution below 5 μm [208, 209], which has made studying the molecular distribution of metabolites more viable. MALDI‐MS stands out for requiring less sample preparation and having a higher throughput than other methods, and has been successfully used to reveal the heterogeneity among clonal populations of single‐celled organisms and discover specialized microorganisms and cellular subtypes in tissues [210–212]. The two biggest limitations of MALDI imaging are low metabolite coverage and low ionization efficiency. To overcome low metabolite coverage, Feenstra et al. proposed an MSI approach of using multiple matrixes with different ionizations to target dual polarities, alongside tandem MSI, though the metabolites identified with low ionization energies were still limited [213]. The ionization efficiency can be increased by on‐tissue chemical derivatization [214, 215]. The newer and more sensitive MALDI‐2 was developed to have an enhanced analyte signal through the initiation of an additional ionization process in the gas‐phase laser plume produced via a second laser [216].
These advances suggest that improvements to MSI can increase our understanding of different metabolites at the cellular and subcellular levels. As an evolving technology, MSI still has many challenges to overcome. The data obtained from MSI is complex and can contain signals from known and unknown metabolites [217]. Improvements in software and databases are essential to identify metabolites with a high degree of confidence and to draw accurate biological conclusions. Furthermore, advances in quantification methods will be critical to distinguish results that may be biased by matrix effects or analyte ionization efficiency.
APPLYING MULTI‐OMICS APPROACHES FOR TUMOR MICROBIOME RESEARCH
We are growing more aware that a single field of omics is not comprehensive enough to understand more complicated biological processes, such as those related to the tumor microbiome. Integrated multi‐omics seeks to fill this gap in research and is a potential new direction to explore these topics.
It has been shown that a variety of cytotoxic metabolites produced by microbes can play a vital role in tumorigenesis and tumor development. Using the metabolite methylglyoxal (MGO) as a case study, MGO is known to affect human health in various manners. MGO, a nonenzymatic byproduct of aerobic and anaerobic glycolysis [218, 219] and a microbial metabolite enzymatically synthesized by MGSs [25], can covalently modify proteins without enzymes and regulate cell functions, including metabolism and transcription [79, 220–223]. It has been reported that MGO can modify DNA and RNA by reacting with guanine residues, inducing DNA and RNA damage [218, 220], and directly affect the integrity of the genome by damaging chromosome segregation during mitosis [224]. Additionally, MGO can react with lysine residues of proteins, leading to the formation of advanced glycation end products (AGEs), which in turn, can cause cellular dysfunction and inflammation [224]. MGO is one of the most effective glycating agents in vivo and can glycate the lysine and arginine residues of histones, which can lead to metabolic disturbances, epigenetic changes, and instability of nucleosomes [219–221, 224, 225]. It has been reported that MGO can affect the activity of transcription factors and change gene transcription, as seen through RNA sequencing, and can lead to the accumulation of cell stress factors [221]. The two‐stage model of histone MGO glycosylation suggests that a low amount of MGO is beneficial to the proliferation of cancer cells by promoting complex transcription, while an excessive amount of MGO leads to chromatin cross‐linking, weakening transcription and leading to cell death [221].
Chemical proteomics methods have shown that high concentrations of MGO can induce histone‐histone and histone‐DNA cross‐linking in chromatin, which can impair its kinetic properties [220]. The synthesis and application of chemical probes for the study of MGO‐based glycation can improve the accessibility and feasibility of important tools for tracking, enriching and studying MGO‐based glycation, and advance the understanding of its potential biochemical functions [226]. MGO regulates the occurrence and development of tumors at genomic, transcriptomic, proteomic, and metabolomic levels (Figure 6). Research on MGO has shown that using a single omics method is not enough to comprehensively understand the effects of such proteins in biological research, and presents a case study for other metabolites and proteins that may require a more interdisciplinary multi‐omics approach.
Figure 6.
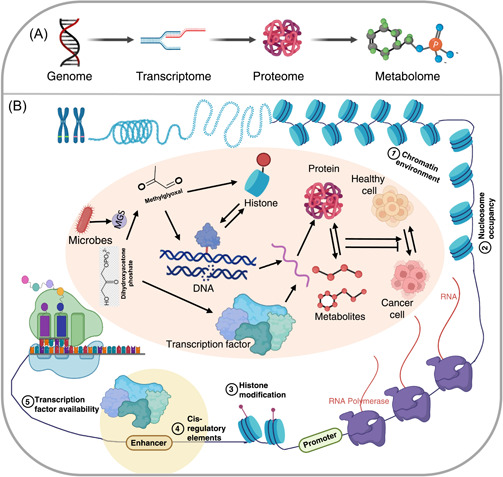
Multi‐omics allows for a better understanding of interrelated disciplines. (A) Flow of omics techniques, which can be combined for multi‐omics, through layers of the central dogma. (B) The metabolite methylglyoxal can affect many levels of the multi‐omics network, including genes, proteins, histones, and chromatin. All of these pathways can contribute to tumor development. MGS, methylglyoxal synthase.
FUTURE PERSPECTIVES
Tumor microbes have been shown to play an essential role in carcinogenesis, development, and treatment [227–229]. The interactions between microbes, tumor cells, and immune cells can greatly affect the TME. Immune cells not only influence the relationship of microbes to anticancer therapies [230], but can also directly modulate the influence of microbes on cancer development [231]. Based on this, combining microbial therapy with immunotherapy has become an effective direction for cancer treatment research [232]. Multi‐omics techniques have been applied to identify cancer biomarkers produced by the tumor cells and microbes in the TME [39, 233, 234], thereby significantly facilitating the development of new diagnostic and treatment strategies. Although many advances have been made in the field through omics approaches, the field is still in its infancy. Many research techniques are immature, such as the application of single‐cell technologies in tumor microbial proteomics and metabolomics research, which is still largely theoretical. Moreover, there is a lack of a unified standard process for multi‐omics studies of the tumor microbiome. All in all, in this review, we have systematically summarized the muti‐omics approaches that can be applied for tumor microbiome research, as well as their advantages and shortcomings. Due to the low biomass of intratumoral microbes and the heterogeneity of TME, we propose that single cell muti‐omics will become the most powerful tool in tumor microbiome research, though there is still a long way to go to develop the corresponding methodologies.
AUTHOR CONTRIBUTIONS
Nan Zhang drafted the manuscript, made the figures, and edited the references. Shruthi Kandalai edited the manuscript and helped to edit the references. Xiaozhuang Zhou helped to make Figure 4 and assisted to edit the references. Farzana Hossain helped to edit the references. Qingfei Zheng proposed the conception, wrote, and edited the manuscript. All authors listed in the paper have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
ACKNOWLEDGMENTS
This study is supported by OSUCCC startup funds for Qingfei Zheng.
Zhang, Nan , Kandalai Shruthi, Zhou Xiaozhuang, Hossain Farzana, and Zheng Qingfei. 2023. “Applying multi‐omics toward tumor microbiome research.” iMeta 2, e73. 10.1002/imt2.73
DATA AVAILABILITY STATEMENT
No new data or script was used in this paper. Supplementary materials (figures, tables, scripts, graphical abstracts, slides, videos, Chinese translated versions and updated materials) may be found in the online DOI or iMeta Science http://www.imeta.science/.
REFERENCES
- 1. Sepich‐Poore, Gregory D. , Zitvogel Laurence, Straussman Ravid, Hasty Jeff, Wargo Jennifer A., and Knight Rob. 2021. “The Microbiome and Human Cancer.” Science 371: eabc4552. 10.1126/science.abc4552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Locey, Kenneth J. , and Lennon Jay T.. 2016. “Scaling Laws Predict Global Microbial Diversity.” Proceedings of the National Academy of Sciences of the United States of America 113: 5970–75. 10.1073/pnas.1521291113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Noguti, Juliana , and Lee Delphine J.. 2019. “Association of Microbes With Breast Cancer.” In Microbiome and Cancer, edited by Erle S. Robertson, 123–49. Humana Press. 10.1007/978-3-030-04155-7 [DOI] [Google Scholar]
- 4. Kwong, Thomas N. Y. , Wang Xiansong, Nakatsu Geicho, Chow Tai Cheong, Tipoe Timothy, Dai Rudin Z. W., Tsoi Kelvin K. K., et al. 2018. “Association Between Bacteremia From Specific Microbes and Subsequent Diagnosis of Colorectal Cancer.” Gastroenterology 155: 383–90. 10.1053/j.gastro.2018.04.028 [DOI] [PubMed] [Google Scholar]
- 5. Wirbel, Jakob , Pyl Paul Theodor, Kartal Ece, Zych Konrad, Kashani Alireza, Milanese Alessio, Fleck Jonas S., et al. 2019. “Meta‐Analysis of Fecal Metagenomes Reveals Global Microbial Signatures That Are Specific for Colorectal Cancer.” Nature Medicine 25: 679–89. 10.1038/s41591-019-0406-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tao, Xuemei , Wang Ning, and Qin Wenxin. 2015. “Gut Microbiota and Hepatocellular Carcinoma.” Gastrointestinal Tumors 2: 33–40. 10.1159/000380895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Huang, Hechen , Ren Zhigang, Gao Xingxing, Hu Xiaoyi, Zhou Yuan, Jiang Jianwen, Lu Haifeng, et al. 2020. “Integrated Analysis of Microbiome and Host Transcriptome Reveals Correlations Between Gut Microbiota and Clinical Outcomes in HBV‐Related Hepatocellular Carcinoma.” Genome Medicine 12: 102. 10.1186/s13073-020-00796-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Leinwand, Joshua C. , and Miller George. 2019. “Microbes as Biomarkers and Targets in Pancreatic Cancer.” Nature Reviews Clinical Oncology 16: 665–66. 10.1038/s41571-019-0276-3 [DOI] [PubMed] [Google Scholar]
- 9. Mima, Kosuke , Nakagawa Shigeki, Sawayama Hiroshi, Ishimoto Takatsugu, Imai Katsunori, Iwatsuki Masaaki, Hashimoto Daisuke, et al. 2017. “The Microbiome and Hepatobiliary‐Pancreatic Cancers.” Cancer Letters 402: 9–15. 10.1016/j.canlet.2017.05.001 [DOI] [PubMed] [Google Scholar]
- 10. Limeta, Angelo , Ji Boyang, Levin Max, Gatto Francesco, and Nielsen Jens. 2020. “Meta‐Analysis of the Gut Microbiota in Predicting Response to Cancer Immunotherapy in Metastatic Melanoma.” JCI Insight 5: 5. 10.1172/jci.insight.140940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Derosa, Lisa , Routy Bertrand, Fidelle Marine, Iebba Valerio, Alla Laurie, Pasolli Edoardo, Segata Nicola, et al. 2020. “Gut Bacteria Composition Drives Primary Resistance to Cancer Immunotherapy in Renal Cell Carcinoma Patients.” European Urology 78: 195–206. 10.1016/j.eururo.2020.04.044 [DOI] [PubMed] [Google Scholar]
- 12. Li, Yonghong , Wang Jia, Wang Mengge, Gao Yongshun, Jin Cheng‐Yun, Shi Xiaojing, Ji Boyang, Wei Yongjun, and Liu Hongmin. 2021. “Microbial Profiling Identifies Potential Key Drivers in Gastric Cancer Patients.” Biotechnology & Biotechnological Equipment 35: 496–503. 10.1080/13102818.2021.1896384 [DOI] [Google Scholar]
- 13. Maddi, Abhiram , Sabharwal Amarpreet, Violante Timothy, Manuballa Sunita, Genco Robert, Patnaik Santosh, and Yendamuri Sai. 2019. “The Microbiome and Lung Cancer.” Journal of Thoracic Disease 11: 280–91. 10.21037/jtd.2018.12.88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mao, Qixing , Jiang Feng, Yin Rong, Wang Jie, Xia Wenjie, Dong Gaochao, Ma Weidong, et al. 2018. “Interplay Between the Lung Microbiome and Lung Cancer.” Cancer Letters 415: 40–48. 10.1016/j.canlet.2017.11.036 [DOI] [PubMed] [Google Scholar]
- 15. Salachan, Paul Vinu , and Sørensen Karina Dalsgaard. 2022. “Dysbiotic Microbes and How To Find Them: A Review of Microbiome Profiling in Prostate Cancer.” Journal of Experimental & Clinical Cancer Research 41: 1–17. 10.1186/s13046-021-02196-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Javier‐DesLoges, Juan , McKay Rana R., Swafford Austin D., Sepich‐Poore Gregory D., Knight Rob, and Parsons J. Kellogg. 2022. “The Microbiome and Prostate Cancer.” Prostate Cancer and Prostatic Diseases 25: 159–64. 10.1038/s41391-021-00413-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ma, Jiayan , Gnanasekar Aditi, Lee Abby, Li Wei Tse, Haas Martin, Wang‐Rodriguez Jessica, Chang Eric Y., Rajasekaran Mahadevan, and Ongkeko Weg M.. 2020. “Influence of Intratumor Microbiome on Clinical Outcome and Immune Processes in Prostate Cancer.” Cancers 12: 2524. 10.3390/cancers12092524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Huang, Tingting , Debelius Justine W., Ploner Alexander, Xiao Xiling, Zhang Tingting, Hu Kai, Zhang Zhe, Wang Rensheng, and Ye Weimin. 2021. “Radiation Therapy–Induced Changes of the Nasopharyngeal Commensal Microbiome in Nasopharyngeal Carcinoma Patients.” International Journal of Radiation Oncology* Biology* Physics 109: 145–50. 10.1016/j.ijrobp.2020.08.054 [DOI] [PubMed] [Google Scholar]
- 19. Nejman, Deborah , Livyatan Ilana, Fuks Garold, Gavert Nancy, Zwang Yaara, Geller Leore T., Rotter‐Maskowitz Aviva, et al. 2020. “The Human Tumor Microbiome is Composed of Tumor Type–Specific Intracellular Bacteria.” Science 368: 973–80. 10.1126/science.aay9189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Erdman, Susan E. , Poutahidis Theofilos, Tomczak Michal, Rogers Arlin B., Cormier Kathleen, Plank Benjamin, Horwitz Bruce H., and Fox James G.. 2003. “CD4+ CD25+ Regulatory T Lymphocytes Inhibit Microbially Induced Colon Cancer in Rag2‐Deficient Mice.” The American Journal of Pathology 162: 691–702. 10.1016/S0002-9440(10)63863-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zeller, Georg , Tap Julien, Voigt Anita Y., Sunagawa Shinichi, Kultima Jens Roat, Costea Paul I., Amiot Aurélien, et al. 2014. “Potential of Fecal Microbiota for Early‐Stage Detection of Colorectal Cancer.” Molecular Systems Biology 10: 766. 10.15252/msb.20145645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rao, Varada P. , Poutahidis Theofilos, Ge Zhongming, Nambiar Prashant R., Boussahmain Chakib, Wang Yan Yan, Horwitz Bruce H., Fox James G., and Erdman Susan E.. 2006. “Innate Immune Inflammatory Response Against Enteric Bacteria Helicobacter Hepaticus Induces Mammary Adenocarcinoma in Mice.” Cancer Research 66: 7395–400. 10.1158/0008-5472.CAN-06-0558 [DOI] [PubMed] [Google Scholar]
- 23. Zackular, Joseph P. , Baxter Nielson T., Iverson Kathryn D., Sadler William D., Petrosino Joseph F., Chen Grace Y., and Schloss Patrick D.. 2013. “The Gut Microbiome Modulates Colon Tumorigenesis.” mBio 4: e00692–00613. 10.1128/mBio.00692-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sivan, Ayelet , Corrales Leticia, and Gajewski Thomas. 2014. “Evidence Implicating the Commensal Microbiota in Shaping Anti‐Tumor Immunity in Melanoma.” Journal for ImmunoTherapy of Cancer 2: 1–1. 10.1186/2051-1426-2-S3-O11 24829758 [DOI] [Google Scholar]
- 25. Cai, Mingwei , Kandalai Shruthi, Tang Xiaoyu, and Zheng Qingfei. 2022. “Contributions of Human‐Associated Archaeal Metabolites to Tumor Microenvironment and Carcinogenesis.” Microbiology Spectrum 10: e0236721. 10.1128/spectrum.02367-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhou, Xiaozhuang , Kandalai Shruthi, Hossain Farzana, and Zheng Qingfei. 2022. “Tumor Microbiome Metabolism: A Game Changer in Cancer Development and Therapy.” Frontiers in Oncology 12: 933407. 10.3389/fonc.2022.933407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Vankemmelbeke, Mireille , Healy Bryan, Moore Geoffrey R., Kleanthous Colin, Penfold Christopher N., and James Richard. 2005. “Rapid Detection of Colicin E9‐induced DNA Damage Using Escherichia coli Cells Carrying SOS Promoter‐Lux Fusions.” Journal of Bacteriology 187: 4900–07. 10.1128/JB.187.14.4900-4907.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Scuron, Monika D. , Boesze‐Battaglia Kathleen, Dlakić Mensur, and Shenker Bruce J.. 2016. “The Cytolethal Distending Toxin Contributes to Microbial Virulence and Disease Pathogenesis by Acting as a Tri‐Perditious Toxin.” Frontiers in Cellular and Infection Microbiology 6: 168. 10.3389/fcimb.2016.00168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kovaleva, Olga V. , Romashin Daniil, Zborovskaya Irina B., Davydov Mikhail M., Shogenov Murat S., and Gratchev Alexei. 2019. “Human Lung Microbiome on the Way to Cancer.” Journal of Immunology Research 2019: 1–6. 10.1155/2019/1394191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bhatt, Aadra P. , Redinbo Matthew R., and Bultman Scott J.. 2017. “The Role of the Microbiome in Cancer Development and Therapy.” CA: A Cancer Journal for Clinicians 67: 326–44. 10.3322/caac.21398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Geller, Leore T. , Barzily‐Rokni Michal, Danino Tal, Jonas Oliver H., Shental Noam, Nejman Deborah, Gavert Nancy, et al. 2017. “Potential Role of Intratumor Bacteria in Mediating Tumor Resistance to the Chemotherapeutic Drug Gemcitabine.” Science 357: 1156–60. 10.1126/science.aah5043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Podack, Eckhard R. , and Munson George P.. 2016. “Killing of Microbes and Cancer by the Immune System With Three Mammalian Pore‐Forming Killer Proteins.” Frontiers in Immunology 7: 464. 10.3389/fimmu.2016.00464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Poore, Gregory D. , Kopylova Evguenia, Zhu Qiyun, Carpenter Carolina, Fraraccio Serena, Wandro Stephen, Kosciolek Tomasz, et al. 2020. “Microbiome Analyses of Blood and Tissues Suggest Cancer Diagnostic Approach.” Nature 579: 567–74. 10.1038/s41586-020-2095-1 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 34. Yu, Zi‐Kun , Xie Rui‐Ling, You Rui, Liu You‐Ping, Chen Xu‐Yin, Chen Ming‐Yuan, and Huang Pei‐Yu. 2021. “The Role of the Bacterial Microbiome in the Treatment of Cancer.” BMC cancer 21: 934. 10.1186/s12885-021-08664-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cornelis, Guy R. , and Rappuoli Rino. 2001. “Host–Microbe Interactions: Bacteria.” Current Opinion in Microbiology 4: 13–15. 10.1016/S1369-5274(00)00157-0 [DOI] [Google Scholar]
- 36. Gao, Weimin , Navarroli Dena, NaimarK Jared, Zhang Weiwen, Chao Shih‐Hui, and Meldrum Deirdre R.. 2013. “Microbe Observation and Cultivation Array (MOCA) for Cultivating and Analyzing Environmental Microbiota.” Microbiome 1: 4. 10.1186/2049-2618-1-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shang, Menglin , Soon Ren Hao, Lim Chwee Teck, Khoo Bee Luan, and Han Jongyoon. 2019. “Microfluidic Modelling of the Tumor Microenvironment for Anti‐Cancer Drug Development.” Lab on a Chip 19: 369–86. 10.1039/c8lc00970h [DOI] [PubMed] [Google Scholar]
- 38. Subramanian, Indhupriya , Verma Srikant, Kumar Shiva, Jere Abhay, and Anamika Krishanpal. 2020. “Multi‐Omics Data Integration, Interpretation, and Its Application.” Bioinformatics and Biology Insights 14: 117793221989905. 10.1177/1177932219899051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Menyhárt, Otília , and Győrffy Balázs. 2021. “Multi‐Omics Approaches in Cancer Research With Applications in Tumor Subtyping, Prognosis, and Diagnosis.” Computational and Structural Biotechnology Journal 19: 949–60. 10.1016/j.csbj.2021.01.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Biswas, Nupur , and Chakrabarti Saikat. 2020. “Artificial Intelligence (AI)‐based Systems Biology Approaches in Multi‐Omics Data Analysis of Cancer.” Frontiers in Oncology 10: 588221. 10.3389/fonc.2020.588221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pervez, Muhammad Tariq , Hasnain Mirza Jawad ul, Abbas Syed Hassan, Moustafa Mahmoud F., Aslam Naeem, and Shah Syed Shah Muhammad. 2022. “A Comprehensive Review of Performance of Next‐Generation Sequencing Platforms.” BioMed Research International 2022: 1–12. 10.1155/2022/3457806 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 42. Liu, Chia‐Hsin , and Peter Di Y.. 2020. “Analysis f RNA Sequencing Data Using CLC Genomics Workbench.” Methods in Molecular Biology 2102: 61–113. 10.1007/978-1-0716-0223-2_4 [DOI] [PubMed] [Google Scholar]
- 43. Zarei, Ali , Javid Hossein, Sanjarian Sara, Senemar Sara, and Zarei Hanieh. 2022. “Metagenomics Studies for the Diagnosis and Treatment of Prostate Cancer.” The Prostate 82: 289–97. 10.1002/pros.24276 [DOI] [PubMed] [Google Scholar]
- 44. Yeh, Wayne W. , Chen Yin‐Quan, Yang Wan‐Shan, Hong Yung‐Chih, Kao Sen, Liu Tze‐Tze, Chen Ting‐Wen, Chang Lung, and Chang Pei‐Ching. 2022. “SUMO Modification of Histone Demethylase KDM4A in Kaposi's Sarcoma‐Associated Herpesvirus‐Induced Primary Effusion Lymphoma.” Journal of Virology 96: e00755–00722. 10.1128/jvi.00755-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Cui, Jianzhou , Sachaphibulkij Karishma, Teo Wen Shiun, Lim Hong Meng, Zou Li, Ong Choon Nam, Alberts Rudi, Chen Jinmiao, and Lim Lina H. K.. 2022. “Annexin‐A1 Deficiency Attenuates Stress‐Induced Tumor Growth Via Fatty Acid Metabolism In Mice: An Integrated Multiple Omics Analysis on the Stress‐Microbiome‐Metabolite‐Epigenetic‐Oncology (SMMEO) Axis.” Theranostics 12: 3794–817. 10.7150/thno.68611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Li, Niannian , Meng Gang, Yang Chunjuan, Li Huiyu, Liu Lin, Wu Yuyun, and Liu Bin. 2022. “Changes In Epigenetic Information During the Occurrence and Development of Gastric Cancer.” The International Journal of Biochemistry & Cell Biology 153: 106315. 10.1016/j.biocel.2022.106315 [DOI] [PubMed] [Google Scholar]
- 47. Horst, Eric N. , Novak Caymen M., Burkhard Kathleen, Snyder Catherine S., Verma Rhea, Crochran Darel E., Geza Izabella A., et al. 2022. “Injectable Three‐Dimensional Tumor Microenvironments To Study Mechanobiology in Ovarian Cancer.” Acta Biomaterialia 146: 222–34. 10.1016/j.actbio.2022.04.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhang, Min , Shi Yibo, Zhang Lihua, Zhu Shiying, Yang Haiquan, Shen Wei, Xia Yuanyuan, and Chen Xianzhong. 2022. “A CRISPR–Cas12a System for Multi‐Gene Editing (CCMGE) and Metabolic Pathway Assembly in Starmerella Bombicola.” Systems Microbiology and Biomanufacturing 2: 665–75. 10.1007/s43393-022-00093-9 [DOI] [Google Scholar]
- 49. Wong‐Rolle, Abigail , Dong Qiang, Zhu Yunhua, Divakar Prajan, Hor Jyh Liang, Kedei Noemi, Wong Madeline, et al. 2022. “Spatial Meta‐Transcriptomics Reveal Associations of Intratumor Bacteria Burden With Lung Cancer Cells Showing a Distinct Oncogenic Signature.” Journal for ImmunoTherapy of Cancer 10: e004698. 10.1136/jitc-2022-004698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Burns, Michael B. , and Blekhman Ran. 2019. “Integrating Tumor Genomics Into Studies of the Microbiome in Colorectal Cancer.” Gut Microbes 10: 547–52. 10.1080/19490976.2018.1549421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Diaz, Luis A. , and Bardelli Alberto. 2014. “Liquid Biopsies: Genotyping Circulating Tumor DNA.” Journal of Clinical Oncology 32: 579–86. 10.1200/JCO.2012.45.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Miller, Eirwen M. , Patterson Nicole E., Zechmeister Jenna Marcus, Bejerano‐Sagie Michal, Delio Maria, Patel Kunjan, Ravi Nivedita, et al. 2017. “Development and Validation of a Targeted Next Generation DNA Sequencing Panel Outperforming Whole Exome Sequencing for the Identification of Clinically Relevant Genetic Variants.” Oncotarget 8: 102033–45. 10.18632/oncotarget.22116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Handelsman, Jo . 2004. “Metagenomics: Application of Genomics to Uncultured Microorganisms.” Microbiology and Molecular Biology Reviews 68: 669–85. 10.1128/MMBR.68.4.669-685.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Schloss, Patrick D. , and Handelsman Jo. 2005. “Metagenomics for Studying Unculturable Microorganisms: Cutting the Gordian Knot.” Genome Biology 6: 229. 10.1186/gb-2005-6-8-229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Borchmann, Sven . 2021. “An Atlas of the Tissue and Blood Metagenome in Cancer Reveals Novel Links Between Bacteria, Viruses and Cancer.” Microbiome 9: 94. 10.1186/s40168-021-01039-4o [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Yachida, Shinichi , Mizutani Sayaka, Shiroma Hirotsugu, Shiba Satoshi, Nakajima Takeshi, Sakamoto Taku, Watanabe Hikaru, et al. 2019. “Metagenomic and Metabolomic Analyses Reveal Distinct Stage‐Specific Phenotypes of the Gut Microbiota in Colorectal Cancer.” Nature Medicine 25: 968–76. 10.1038/s41591-019-0458-7 [DOI] [PubMed] [Google Scholar]
- 57. D'Amore, Rosalinda , Ijaz Umer Zeeshan, Schirmer Melanie, Kenny John G., Gregory Richard, Darby Alistair C., Shakya Migun, et al. 2016. “A Comprehensive Benchmarking Study of Protocols and Sequencing Platforms for 16S rRNA Community Profiling.” BMC Genomics 17: 55. 10.1186/s12864-015-2194-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Johnson, Jethro S. , Spakowicz Daniel J., Hong Bo‐Young, Petersen Lauren M., Demkowicz Patrick, Chen Lei, Leopold Shana R., et al. 2019. “Evaluation of 16S rRNA Gene Sequencing for Species and Strain‐Level Microbiome Analysis.” Nature Communications 10: 5029. 10.1038/s41467-019-13036-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Stark, Rory , Grzelak Marta, and Hadfield James. 2019. “RNA Sequencing: The Teenage Years.” Nature Reviews Genetics 20: 631–56. 10.1038/s41576-019-0150-2 [DOI] [PubMed] [Google Scholar]
- 60. Zheng, Kai , Lin Lingmin, Jiang Wei, Chen Lin, Zhang Xiyue, Zhang Qian, Ren Yi, and Hao Junwei. 2022. “Single‐Cell RNA‐seq Reveals the Transcriptional Landscape in Ischemic Stroke.” Journal of Cerebral Blood Flow & Metabolism 42: 56–73. 10.1177/0271678x211026770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Yip, Shun H. , Sham Pak Chung, and Wang Junwen. 2019. “Evaluation of Tools for Highly Variable Gene Discovery From Single‐Cell RNA‐seq Data.” Briefings in Bioinformatics 20: 1583–89. 10.1093/bib/bby011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Moncada, Reuben , Barkley Dalia, Wagner Florian, Chiodin Marta, Devlin Joseph C., Baron Maayan, Hajdu Cristina H., Simeone Diane M., and Yanai Itai. 2020. “Integrating Microarray‐Based Spatial Transcriptomics and Single‐Cell RNA‐seq Reveals Tissue Architecture in Pancreatic Ductal Adenocarcinomas.” Nature Biotechnology 38: 333–42. 10.1038/s41587-019-0392-8 [DOI] [PubMed] [Google Scholar]
- 63. Parker, Matthew T. , Knop Katarzyna, Sherwood Anna V., Schurch Nicholas J., Mackinnon Katarzyna, Gould Peter D., Hall Anthony Jw, Barton Geoffrey J., and Simpson Gordon G.. 2020. “Nanopore Direct RNA Sequencing Maps the Complexity of Arabidopsis mRNA Processing and m6A Modification.” BioRxiv 9: 706002. 10.7554/eLife.49658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Li, Xiaohan , Yu Kequan, Li Fuyu, Lu Wenxiang, Wang Ying, Zhang Weizhong, and Bai Yunfei. 2022. “Novel Method of Full‐Length RNA‐seq That Expands the Identification of Non‐Polyadenylated RNAs Using Nanopore Sequencing.” Analytical Chemistry 94: 12342–51. 10.1021/acs.analchem.2c01128 [DOI] [PubMed] [Google Scholar]
- 65. Duss, Olivier , Stepanyuk Galina A., Puglisi Joseph D., and Williamson James R.. 2019. “Transient protein‐RNA Interactions Guide Nascent Ribosomal RNA Folding.” Cell 179: 1357–69. 10.1016/j.cell.2019.10.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Zabransky, Daniel J. , Jaffee Elizabeth M., and Weeraratna Ashani T.. 2022. “Shared Genetic and Epigenetic Changes Link Aging and Cancer.” Trends in Cell Biology 32: 338–50. 10.1016/j.tcb.2022.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Li, Nan , Zeng Anqi, Wang Qian, Chen Maohua, Zhu Shaomi, and Song Linjiang. 2022. “Regulatory Function of DNA Methylation Mediated lncRNAs in Gastric Cancer.” Cancer Cell International 22: 227. 10.1186/s12935-022-02648-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Zhang, Linlin , Wang Ran, and Xie Zhengde. 2022. “The Roles of DNA Methylation on the Promotor of the Epstein–Barr Virus (EBV) Gene and the Genome in Patients With EBV‐Associated Diseases.” Applied Microbiology and Biotechnology 106(12): 4413–26. 10.1007/s00253-022-12029-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Zeng, Yunqing , Rong Huimin, Xu Jianwei, Cao Ruyue, Li Shuhua, Gao Yanjing, Cheng Baoquan, and Zhou Tao. 2022. “DNA Methylation: An Important Biomarker and Therapeutic Target for Gastric Cancer.” Frontiers in Genetics 13: 823905. 10.3389/fgene.2022.823905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Roberto, Roberto , Gissi Davide B., Gabusi Andrea, Fabbri Viscardo Paolo, Balbi Tiziana, Tarsitano Achille, and Morandi Luca. 2022. “A 13‐gene DNA Methylation Analysis Using Oral Brushing Specimens as an Indicator of Oral Cancer Risk: A Descriptive Case Report.” Diagnostics 12: 284. 10.3390/diagnostics12020284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Hamaguchi, Yo , Mishima Hiroyuki, Kawai Tomoko, Saitoh Shinji, Hata Kenichiro, Kinoshita Akira, and Yoshiura Koh‐ichiro. 2022. “Identification of Unique DNA Methylation Sites in Kabuki Syndrome Using Whole Genome Bisulfite Sequencing and Targeted Hybridization Capture Followed by Enzymatic Methylation Sequencing.” Journal of Human Genetics 67: 711–20. 10.1038/s10038-022-01083-4 [DOI] [PubMed] [Google Scholar]
- 72. Chapin, Nathaniel , Fernandez Joseph, Poole Jason, and Delatte Benjamin. 2022. “Anchor‐Based Bisulfite Sequencing Determines Genome‐Wide DNA Methylation.” Communications Biology 5: 596. 10.1038/s42003-022-03543-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Zhang, Ze , Lee Min Kyung, Perreard Laurent, Kelsey Karl T., Christensen Brock C., and Salas Lucas A.. 2022. “Navigating the Hydroxymethylome: Experimental Biases and Quality Control Tools for the Tandem Bisulfite and Oxidative Bisulfite Illumina Microarrays.” Epigenomics 14: 139–52. 10.2217/epi-2021-0490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Che, Huiwen , Stanley Kate, Jatsenko Tatjana, Thienpont Bernard, and Vermeesch Joris Robert. 2022. “Expanded Knowledge of Cell‐Free DNA Biology: Potential To Broaden the Clinical Utility.” Extracellular Vesicles and Circulating Nucleic Acids 3: 199–217. 10.20517/evcna.2022.21 [DOI] [Google Scholar]
- 75. Neganova, Margarita E. , Klochkov Sergey G., Aleksandrova Yulia R., and Aliev Gjumrakch. 2022. “Histone Modifications in Epigenetic Regulation of Cancer: Perspectives and Achieved Progress.” Seminars in Cancer Biology 83: 452–71. 10.1016/j.semcancer.2020.07.015 [DOI] [PubMed] [Google Scholar]
- 76. Zhao, Zibo , and Shilatifard Ali. 2019. “Epigenetic Modifications of Histones in Cancer.” Genome Biology 20: 245. 10.1186/s13059-019-1870-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Chakraborty, Sangeeta , Karmakar Kapudeep, and Chakravortty Dipshikha. 2014. “Cells Producing Their Own Nemesis: Understanding Methylglyoxal Metabolism.” IUBMB life 66: 667–78. 10.1002/iub.1324 [DOI] [PubMed] [Google Scholar]
- 78. Zhang, May M. , Ong Cheryl‐lynn Y, Walker Mark J., and McEwan Alastair G.. 2016. “Defence Against Methylglyoxal in Group A Streptococcus: A Role for Glyoxylase I in Bacterial Virulence and Survival in Neutrophils? Pathogens and Disease 74: ftv122. 10.1093/femspd/ftv122 [DOI] [PubMed] [Google Scholar]
- 79. Zheng, Qingfei , Osunsade Adewola, and David Yael. 2020. “Protein Arginine Deiminase 4 Antagonizes Methylglyoxal‐Induced Histone Glycation.” Nature Communications 11: 3241. 10.1038/s41467-020-17066-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Zheng, Qingfei , Omans Nathaniel D., Leicher Rachel, Osunsade Adewola, Agustinus Albert S., Finkin‐Groner Efrat, D'Ambrosio Hannah, et al. 2019. “Reversible Histone Glycation is Associated With Disease‐Related Changes in Chromatin Architecture.” Nature Communications 10: 1289. 10.1038/s41467-019-09192-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Maksimovic, Igor , and David Yael. 2021. “Non‐Enzymatic Covalent Modifications as a New Chapter in the Histone Code.” Trends in Biochemical Sciences 46: 718–30. 10.1016/j.tibs.2021.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Song, Haoyun , Shen Rong, Liu Xiangwen, Yang Xuguang, Xie Kun, Guo Zhao, and Wang Degui. in press. “Histone Post‐Translational Modification and the DNA Damage Response.” Genes & Diseases. 10.1016/j.gendis.2022.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Nakato, Ryuichiro , and Sakata Toyonori. 2021. “Methods for ChIP‐seq Analysis: A Practical Workflow and Advanced Applications.” Methods 187: 44–53. 10.1016/j.ymeth.2020.03.005 [DOI] [PubMed] [Google Scholar]
- 84. Lüdtke, Timo H. , Wojahn Irina, Kleppa Marc‐Jens, Schierstaedt Jasper, Christoffels Vincent M., Künzler Patrick, and Kispert Andreas. 2021. “Combined Genomic and Proteomic Approaches Reveal DNA Binding Sites and Interaction Partners of TBX2 in the Developing Lung.” Respiratory Research 22: 85. 10.1186/s12931-021-01679-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Yu, Miao , Juric Ivan, Abnousi Armen, Hu Ming, and Ren Bing. 2021. “Proximity Ligation‐Assisted ChIP‐seq (PLAC‐Seq).” Methods in Molecular Biology 2351: 181–99. 10.1007/978-1-0716-1597-3_10 [DOI] [PubMed] [Google Scholar]
- 86. O'Geen, Henriette , Echipare Lorigail, and Farnham Peggy J.. 2011. “Using ChIP‐seq Technology to Generate High‐Resolution Profiles of Histone Modifications.” Epigenetics Protocols 791: 265–86. 10.1007/978-1-61779-316-5_20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Mundade, Rasika , Ozer Hatice Gulcin, Wei Han, Prabhu Lakshmi, and Lu Tao. 2014. “Role of ChIP‐seq in the Discovery of Transcription Factor Binding Sites, Differential Gene Regulation Mechanism, Epigenetic Marks and Beyond.” Cell Cycle 13: 2847–52. 10.4161/15384101.2014.949201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Kong, Siyuan , and Zhang Yubo. 2019. “Deciphering Hi‐C: From 3D Genome to Function.” Cell Biology and Toxicology 35: 15–32. 10.1007/s10565-018-09456-2 [DOI] [PubMed] [Google Scholar]
- 89. Lafontaine, Denis L. , Yang Liyan, Dekker Job, and Gibcus Johan H.. 2021. “Hi‐C 3.0: Improved Protocol for Genome‐Wide Chromosome Conformation Capture.” Current Protocols 1: e198. 10.1002/cpz1.198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Akgol Oksuz, Betul , Yang Liyan, Abraham Sameer, Venev Sergey V., Krietenstein Nils, Parsi Krishna Mohan, Ozadam Hakan, et al. 2021. “Systematic Evaluation of Chromosome Conformation Capture Assays.” Nature Methods 18: 1046–55. 10.1038/s41592-021-01248-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Swygert, Sarah G. , Kim Seungsoo, Wu Xiaoying, Fu Tianhong, Hsieh Tsung‐Han, Rando Oliver J., Eisenman Robert N., et al. 2019. “Condensin‐Dependent Chromatin Compaction Represses Transcription Globally During Quiescence.” Molecular Cell 73: 533–46. 10.1016/j.molcel.2018.11.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Li, Guoliang , Sun Tongkai, Chang Huidan, Cai Liuyang, Hong Ping, and Qiangwei, Zhou . 2019. “Chromatin Interaction Analysis With Updated ChIA‐PET Tool (V3).” Genes 10: 554. 10.3390/genes10070554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Di, Giammartino , Campigli Dafne, Polyzos Alexander, and Apostolou Effie. 2022. “Assessing Specific Networks of Chromatin Interactions With Hichip.” Methods in Molecular Biology 2532: 113–41. 10.1007/978-1-0716-2497-5_7 [DOI] [PubMed] [Google Scholar]
- 94. Le, Tung B. K. , Imakaev Maxim V., Mirny Leonid A., and Laub Michael T.. 2013. “High‐Resolution Mapping of the Spatial Organization of a Bacterial Chromosome.” Science 342: 731–34. 10.1126/science.1242059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. DeMaere, Matthew Z. , Liu Michael Y. Z., Lin Enmoore, Djordjevic Steven P., Charles Ian G., Worden Paul, Burke Catherine M., et al. 2020. “Metagenomic Hi‐C of a Healthy Human Fecal Microbiome Transplant Donor.” Microbiology Resource Announcements 9: e01523–19. 10.1128/MRA.01523-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Bak, Rasmus O. , Gomez‐Ospina Natalia, and Porteus Matthew H.. 2018. “Gene Editing on Center Stage.” Trends in Genetics 34: 600–11. 10.1016/j.tig.2018.05.004 [DOI] [PubMed] [Google Scholar]
- 97. Smith, Hamilton O. , and Nathans Daniel. 1973. “A Suggested Nomenclature for Bacterial Host Modification and Restriction Systems and Their Enzymes.” Journal of Molecular Biology 81: 419–23. 10.1016/0022-2836(73)90152-6 [DOI] [PubMed] [Google Scholar]
- 98. Bibikova, Marina , Beumer Kelly, Trautman Jonathan K., and Carroll Dana. 2003. “Enhancing Gene Targeting With Designed Zinc Finger Nucleases.” Science 300: 764. 10.1126/science.1079512 [DOI] [PubMed] [Google Scholar]
- 99. Miller, Jeffrey C. , Tan Siyuan, Qiao Guijuan, Barlow Kyle A., Wang Jianbin, Xia Danny F., Meng Xiangdong, et al. 2011. “A TALE Nuclease Architecture for Efficient Genome Editing.” Nature Biotechnology 29: 143–48. 10.1038/nbt.1755 [DOI] [PubMed] [Google Scholar]
- 100. Jinek, Martin , Chylinski Krzysztof, Fonfara Ines, Hauer Michael, Doudna Jennifer A., and Charpentier Emmanuelle. 2012. “A Programmable dual‐RNA–guided DNA Endonuclease in Adaptive Bacterial Immunity.” Science 337: 816–21. 10.1126/science.1225829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Haurwitz, Rachel E. , Jinek Martin, Wiedenheft Blake, Zhou Kaihong, and Doudna Jennifer A.. 2010. “Sequence‐And Structure‐Specific RNA Processing by a CRISPR Endonuclease.” Science 329: 1355–58. 10.1126/science.1192272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Fonfara, Ines , Le Rhun Anaïs, Chylinski Krzysztof, Makarova Kira S., Lécrivain Anne‐Laure, Bzdrenga Janek, Koonin Eugene V., and Charpentier Emmanuelle. 2014. “Phylogeny of Cas9 Determines Functional Exchangeability of dual‐RNA and Cas9 Among Orthologous Type II CRISPR‐Cas Systems.” Nucleic Acids Research 42: 2577–90. 10.1093/nar/gkt1074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Eid, Ayman , Alshareef Sahar, and Mahfouz Magdy M.. 2018. “CRISPR Base Editors: Genome Editing Without Double‐Stranded Breaks.” Biochemical Journal 475: 1955–64. 10.1042/BCJ20170793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Anzalone, Andrew V. , Randolph Peyton B., Davis Jessie R., Sousa Alexander A., Koblan Luke W., Levy Jonathan M., Chen Peter J., et al. 2019. “Search‐And‐Replace Genome Editing Without Double‐Strand Breaks or Donor DNA.” Nature 576: 149–57. 10.1038/s41586-019-1711-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Yarnall, Matthew T. N. , Ioannidi Eleonora I., Schmitt‐Ulms Cian, Krajeski Rohan N., Lim Justin, Villiger Lukas, Zhou Wenyuan, et al. 2022. “Drag‐And‐Drop Genome Insertion of Large Sequences Without Double‐Strand DNA Cleavage Using CRISPR‐directed Integrases.” Nature Biotechnology: 1–13. 10.1038/s41587-022-01527-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Klompe, Sanne E. , Vo Phuc L. H., Halpin‐Healy Tyler S., and Sternberg Samuel H.. 2019. “Transposon‐Encoded CRISPR‐Cas Systems Direct RNA‐Guided DNA Integration.” Nature 571: 219–25. 10.1038/s41586-019-1323-z [DOI] [PubMed] [Google Scholar]
- 107. Qi, Lei S. , Larson Matthew H., Gilbert Luke A., Doudna Jennifer A., Weissman Jonathan S., Arkin Adam P., and Lim Wendell A.. 2013. “Repurposing CRISPR as an RNA‐guided Platform for Sequence‐Specific Control of Gene Expression.” Cell 152: 1173–83. 10.1016/j.cell.2013.02.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Kim, Hyeran , Kim Sang‐Tae, Ryu Jahee, Kang Beum‐Chang, Kim Jin‐Soo, and Kim Sang‐Gyu. 2017. “CRISPR/Cpf1‐Mediated DNA‐free Plant Genome Editing.” Nature Communications 8: 14406. 10.1038/ncomms14406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Gaudelli, Nicole M. , Komor Alexis C., Rees Holly A., Packer Michael S., Badran Ahmed H., Bryson David I., and Liu David R.. 2017. “Programmable Base Editing of A•T to G•C in Genomic DNA Without DNA Cleavage.” Nature 551: 464–71. 10.1038/nature24644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Hilton, Isaac B. , D'ippolito Anthony M., Vockley Christopher M., Thakore Pratiksha I., Crawford Gregory E., Reddy Timothy E., and Gersbach Charles A.. 2015. “Epigenome Editing by a CRISPR‐Cas9‐based Acetyltransferase Activates Genes From Promoters and Enhancers.” Nature Biotechnology 33: 510–17. 10.1038/nbt.3199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Kearns, Nicola A. , Pham Hannah, Tabak Barbara, Genga Ryan M., Silverstein Noah J., Garber Manuel, and Maehr René. 2015. “Functional Annotation of Native Enhancers With a Cas9–Histone Demethylase Fusion.” Nature Methods 12: 401–3. 10.1038/nmeth.3325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Brocken, Daan J. W. , Tark‐Dame Mariliis, and Dame Remus T.. 2018. “dCas9: A Versatile Tool for Epigenome Editing.” Current Issues in Molecular Biology 26: 15–32. 10.21775/cimb.026.015 [DOI] [PubMed] [Google Scholar]
- 113. Thakore, Pratiksha I. , D'ippolito Anthony M., Song Lingyun, Safi Alexias, Shivakumar Nishkala K., Kabadi Ami M., Reddy Timothy E., Crawford Gregory E., and Gersbach Charles A.. 2015. “Highly Specific Epigenome Editing by CRISPR‐Cas9 Repressors for Silencing of Distal Regulatory Elements.” Nature Methods 12: 1143–49. 10.1038/nmeth.3630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Liu, Ke , Cui Jia‐Jia, Zhan Yan, Ouyang Qian‐Ying, Lu Qi‐Si, Yang Dong‐Hua, Li Xiang‐Ping, and Yin Ji‐Ye. 2022. “Reprogramming the Tumor Microenvironment by Genome Editing for Precision Cancer Therapy.” Molecular Cancer 21: 98. 10.1186/s12943-022-01561-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Jin, Wen‐Bing , Li Ting‐Ting, Huo Da, Qu Sophia, Li Xin V., Arifuzzaman Mohammad, Lima Svetlana F., et al. 2022. “Genetic Manipulation of Gut Microbes Enables Single‐Gene Interrogation in a Complex Microbiome.” Cell 185: 547–62. 10.1016/j.cell.2021.12.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Dohlman, Anders B. , Arguijo Mendoza Diana, Ding Shengli, Gao Michael, Dressman Holly, Iliev Iliyan D., Lipkin Steven M., and Shen Xiling. 2021. “The Cancer Microbiome Atlas: A Pan‐Cancer Comparative Analysis to Distinguish Tissue‐Resident Microbiota From Contaminants.” Cell Host & Microbe 29: 281–98.e285. 10.1016/j.chom.2020.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Shi, Zhou Jason , Dimitrov Boris, Zhao Chunyu, Nayfach Stephen, and Pollard Katherine S.. 2022. “Fast and Accurate Metagenotyping of the Human Gut Microbiome With GT‐Pro.” Nature Biotechnology 40: 507–16. 10.1038/s41587-021-01102-3 [DOI] [PubMed] [Google Scholar]
- 118. Engel, Philipp , Stepanauskas Ramunas, and Moran Nancy A.. 2014. “Hidden Diversity in Honey Bee Gut Symbionts Detected By Single‐Cell Genomics.” PLoS genetics 10: e1004596. 10.1371/journal.pgen.1004596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Medini, Duccio , Donati Claudio, Tettelin Hervé, Masignani Vega, and Rappuoli Rino. 2005. “The Microbial Pan‐Genome.” Current Opinion in Genetics & Development 15: 589–94. 10.1016/j.gde.2005.09.006 [DOI] [PubMed] [Google Scholar]
- 120. Balachandran, M. , Cross K. L., and Podar M.. 2020. “Single‐Cell Genomics and the Oral Microbiome.” Journal of Dental Research 99: 613–20. 10.1177/0022034520907380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Cahn, Jackson K. B. , and Piel Jörn. 2021. “Opening Up the Single‐Cell Toolbox for Microbial Natural Products Research.” Angewandte Chemie International Edition 60: 18412–28. 10.1002/anie.201900532 [DOI] [PubMed] [Google Scholar]
- 122. Wang, Zheng , Chai Chengyan, Wang Rui, Feng Yimei, Huang Lei, Zhang Yiming, Xiao Xia, et al. 2021. “Single‐Cell Transcriptome Atlas of Human Mesenchymal Stem Cells Exploring Cellular Heterogeneity.” Clinical and Translational Medicine 11: e650. 10.1002/ctm2.650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Huang, Xizhi , Li Pengjie, Zhou Mengfan, Li Yiwei, Ou Xiaowen, Chen Peng, Guggenberger Georg, and Liu Bi‐Feng. 2021. “A High‐Throughput Ultrasonic Spraying Inoculation Method Promotes Colony Cultivation of Rare Microbial Species.” Environmental Microbiology 23: 1275–85. 10.1111/1462-2920.15386 [DOI] [PubMed] [Google Scholar]
- 124. Xu, Xun , Hou Yong, Yin Xuyang, Bao Li, Tang Aifa, Song Luting, Li Fuqiang, et al. 2012. “Single‐Cell Exome Sequencing Reveals Single‐Nucleotide Mutation Characteristics of a Kidney Tumor.” Cell 148: 886–95. 10.1016/j.cell.2012.02.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Hou, Yong , Song Luting, Zhu Ping, Zhang Bo, Tao Ye, Xu Xun, Li Fuqiang, et al. 2012. “Single‐Cell Exome Sequencing and Monoclonal Evolution of a JAK2‐Negative Myeloproliferative Neoplasm.” Cell 148: 873–85. 10.1016/j.cell.2012.02.028 [DOI] [PubMed] [Google Scholar]
- 126. Lan, Freeman , Demaree Benjamin, Ahmed Noorsher, and Abate Adam R.. 2017. “Single‐Cell Genome Sequencing at Ultra‐High‐Throughput With Microfluidic Droplet Barcoding.” Nature Biotechnology 35: 640–46. 10.1038/nbt.3880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Shi, Hao , Shi Qiaojuan, Grodner Benjamin, Lenz Joan Sesing, Zipfel Warren R., Brito Ilana Lauren, and De Vlaminck Iwijn. 2020. “Highly Multiplexed Spatial Mapping of Microbial Communities.” Nature 588: 676–81. 10.1038/s41586-020-2983-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Jiang, Cheng‐Ying , Dong Libing, Zhao Jian‐Kang, Hu Xiaofang, Shen Chaohua, Qiao Yuxin, Zhang Xinyue, et al. 2016. “High‐Throughput Single‐Cell Cultivation on Microfluidic Streak Plates.” Applied and Environmental Microbiology 82: 2210–18. 10.1128/AEM.03588-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Lan, Freeman , Haliburton John R., Yuan Aaron, and Abate Adam R.. 2016. “Droplet Barcoding for Massively Parallel Single‐Molecule Deep Sequencing.” Nature Communications 7: 11784. 10.1038/ncomms11784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Macosko, Evan Z. , Basu Anindita, Satija Rahul, Nemesh James, Shekhar Karthik, Goldman Melissa, Tirosh Itay, et al. 2015. “Highly Parallel Genome‐Wide Expression Profiling of Individual Cells Using Nanoliter Droplets.” Cell 161: 1202–14. 10.1016/j.cell.2015.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Tirosh, Itay , Venteicher Andrew S., Hebert Christine, Escalante Leah E., Patel Anoop P., Yizhak Keren, Fisher Jonathan M., et al. 2016. “Single‐Cell RNA‐seq Supports a Developmental Hierarchy in Human Oligodendroglioma.” Nature 539: 309–13. 10.1038/nature20123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Rotem, Assaf , Ram Oren, Shoresh Noam, Sperling Ralph A., Goren Alon, Weitz David A., and Bernstein Bradley E.. 2015. “Single‐Cell ChIP‐seq Reveals Cell Subpopulations Defined By Chromatin State.” Nature Biotechnology 33: 1165–72. 10.1038/nbt.3383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Treutlein, Barbara , Brownfield Doug G., Wu Angela R., Neff Norma F., Mantalas Gary L., Espinoza F Hernan, Desai Tushar J., Krasnow Mark A., and Quake Stephen R.. 2014. “Reconstructing Lineage Hierarchies of the Distal Lung Epithelium Using Single‐Cell RNA‐seq.” Nature 509: 371–75. 10.1038/nature13173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Patel, Anoop P. , Tirosh Itay, Trombetta John J., Shalek Alex K., Gillespie Shawn M., Wakimoto Hiroaki, Cahill Daniel P., et al. 2014. “Single‐Cell RNA‐seq Highlights Intratumoral Heterogeneity in Primary Glioblastoma.” Science 344: 1396–401. 10.1126/science.1254257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Matturro, Bruna , Rossetti Simona, and Leitão Patrícia. 2021. “CAtalyzed Reporter Deposition Fluorescence in Situ Hybridization (CARD‐FISH) for Complex Environmental Samples.” Fluorescence In‐Situ Hybridization (FISH) for Microbial Cells 2246: 129–40. 10.1007/978-1-0716-1115-9_9 [DOI] [PubMed] [Google Scholar]
- 136. Chen, Chun H. , Cho Sung H., Chiang Hsin‐I, Tsai Frank, Zhang Kun, and Lo Yu‐Hwa. 2011. “Specific Sorting of Single Bacterial Cells With Microfabricated Fluorescence‐Activated Cell Sorting and Tyramide Signal Amplification Fluorescence In Situ Hybridization.” Analytical Chemistry 83: 7269–75. 10.1021/ac2013465 [DOI] [PubMed] [Google Scholar]
- 137. Suomalainen, Maarit , and Greber Urs F.. 2021. “Virus Infection Variability by Single‐Cell Profiling.” Viruses 13: 1568. 10.3390/v13081568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Hansen, Steen Honoré , and Reubsaet Leon. 2015. “High‐Performance Liquid Chromatography (HPLC) and High‐Performance Liquid Chromatography–Mass Spectrometry (LC‐MS).” Bioanalysis of Pharmaceuticals: Sample Preparation, Separation Techniques, and Mass Spectrometry: 123–72. 10.1002/9781118716830.ch7 [DOI] [Google Scholar]
- 139. Qian, Xiang , Zhang Hong‐Yan, Li Qing‐Lin, Ma Guan‐Jun, Chen Zhuo, Ji Xu‐Ming, Li Chang‐Yu, and Zhang Ai‐qin. 2022. “Integrated Microbiome, Metabolome, and Proteome Analysis Identifies a Novel Interplay Among Commensal Bacteria, Metabolites and Candidate Targets in Non‐Small Cell Lung Cancer.” Clinical and Translational Medicine 12: e947. 10.1002/ctm2.947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Zhao, Feng , An Rui, Wang Liqian, Shan Jikang, and Wang Xianjun. 2021. “Specific Gut Microbiome and Serum Metabolome Changes In Lung Cancer Patients.” Frontiers in Cellular and Infection Microbiology 11: 11. 10.3389/fcimb.2021.725284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Ma, Ji , Sun Lingqi, Liu Ying, Ren Hui, Shen Yali, Bi Feng, Zhang Tao, and Wang Xin. 2020. “Alter Between Gut Bacteria and Blood Metabolites and the Anti‐Tumor Effects of Faecalibacterium Prausnitzii in Breast Cancer.” BMC Microbiology 20: 82. 10.1186/s12866-020-01739-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Xu, Tian , and Sun Liangliang. 2021. “A Mini Review on Capillary Isoelectric Focusing‐Mass Spectrometry for Top‐Down Proteomics.” Frontiers in Chemistry 9: 651757. 10.3389/fchem.2021.651757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Cassidy, Liam , Helbig Andreas O., Kaulich Philipp T., Weidenbach Kathrin, Schmitz Ruth A., and Tholey Andreas. 2021. “Multidimensional Separation Schemes Enhance the Identification and Molecular Characterization of Low Molecular Weight Proteomes and Short Open Reading Frame‐Encoded Peptides in Top‐Down Proteomics.” Journal of Proteomics 230: 103988. 10.1016/j.jprot.2020.103988 [DOI] [PubMed] [Google Scholar]
- 144. Dupree, Emmalyn J. , Jayathirtha Madhuri, Yorkey Hannah, Mihasan Marius, Petre Brindusa Alina, and Darie Costel C.. 2020. “A Critical Review of Bottom‐Up Proteomics: the Good, the Bad, and the Future of This Field.” Proteomes 8: 14. 10.3390/proteomes8030014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Abidin, Syafiq Asnawi Zainal , Iekhsan, Othman , and Naidu Rakesh. 2021. “Shotgun Proteomics and Mass Spectrometry as a Tool for Protein Identification and Profiling of Bio‐Carrier‐Based Therapeutics on Human Cancer Cells.” Bio‐Carrier Vectors 2211: 233–40. 10.1007/978-1-0716-0943-9_16 [DOI] [PubMed] [Google Scholar]
- 146. Carolina, Fernández‐Costa , Martínez‐Bartolomé Salvador, McClatchy Daniel B., Saviola Anthony J., Yu Nam‐Kyung, and Yates John R.. 2020. "Impact of the Identification Strategy on the Reproducibility of the DDA and DIA Results." Journal of Proteome Research 19: 3153–61. 10.1021/acs.jproteome.0c00153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Chapman, John D. , Goodlett David R., and Masselon Christophe D.. 2014. “Multiplexed and Data‐Independent Tandem Mass Spectrometry for Global Proteome Profiling.” Mass Spectrometry Reviews 33: 452–70. 10.1002/mas.21400 [DOI] [PubMed] [Google Scholar]
- 148. Aakko, Juhani , Pietilä Sami, Suomi Tomi, Mahmoudian Mehrad, Toivonen Raine, Kouvonen Petri, Rokka Anne, Hänninen Arno, and Elo Laura L.. 2019. “Data‐Independent Acquisition Mass Spectrometry in Metaproteomics of Gut Microbiota—Implementation and Computational Analysis.” Journal of Proteome Research 19: 432–36. 10.1021/acs.jproteome.9b00606 [DOI] [PubMed] [Google Scholar]
- 149. Keam, Simon P. , Gulati Twishi, Gamell Cristina, Caramia Franco, Huang Cheng, Schittenhelm Ralf B., Kleifeld Oded, et al. 2018. “Exploring the Oncoproteomic Response of Human Prostate Cancer to Therapeutic Radiation Using Data‐Independent Acquisition (DIA) Mass Spectrometry.” The Prostate 78: 563–75. 10.1002/pros.23500 [DOI] [PubMed] [Google Scholar]
- 150. Song, Yimeng , Zhong Lijun, Zhou Juntuo, Lu Min, Xing Tianying, Ma Lulin, and Shen Jing. 2017. “Data‐Independent Acquisition‐Based Quantitative Proteomic Analysis Reveals Potential Biomarkers of Kidney Cancer.” PROTEOMICS—Clinical Applications 11: 1700066. 10.1002/prca.201700066 [DOI] [PubMed] [Google Scholar]
- 151. Zhu Tiansheng, Zhu Yi, Xuan Yue, Gao Huanhuan, Cai Xue, Piersma Sander R., et al. 2020. “DPHL: A DIA Pan‐Human Protein Mass Spectrometry Library for Robust Biomarker Discovery.” Genomics, Proteomics & Bioinformatics 18: 104–19. 10.1016/j.gpb.2019.11.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Chambers, Andrew G. , Percy Andrew J., Simon Romain, and Borchers Christoph H.. 2014. “MRM for the Verification of Cancer Biomarker Proteins: Recent Applications to Human Plasma and Serum.” Expert review of proteomics 11: 137–48. 10.1586/14789450.2014.877346 [DOI] [PubMed] [Google Scholar]
- 153. Doerr, Allison . 2012. “Targeting With PRM.” Nature Methods 9: 950. 10.1038/nmeth.2193 [DOI] [PubMed] [Google Scholar]
- 154. Kirkpatrick, Donald S. , Gerber Scott A., and Gygi Steven P.. 2005. “The Absolute Quantification Strategy: a General Procedure for the Quantification of Proteins and Post‐Translational Modifications.” Methods 35: 265–73. 10.1016/j.ymeth.2004.08.018 [DOI] [PubMed] [Google Scholar]
- 155. Glen, Adam , Gan Chee S., Hamdy Freddie C., Eaton Colby L., Cross Simon S., Catto James W. F., Wright Phillip C., and Rehman Ishtiaq. 2008. “iTRAQ‐facilitated Proteomic Analysis of Human Prostate Cancer Cells Identifies Proteins Associated With Progression.” Journal of Proteome Research 7: 897–907. 10.1021/pr070378x [DOI] [PubMed] [Google Scholar]
- 156. Yang, Chung S. , Suh Nanjoo, and Kong Ah‐Ng Tony. 2012. “Does Vitamin E Prevent or Promote Cancer? Cancer Prevention Research 5: 701–5. 10.1158/1940-6207.CAPR-12-0045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Everley, Patrick A. , Krijgsveld Jeroen, Zetter Bruce R., and Gygi Steven P.. 2004. “Quantitative Cancer Proteomics: Stable Isotope Labeling With Amino Acids in Cell Culture (SILAC) as a Tool for Prostate Cancer Research.” Molecular & Cellular Proteomics 3: 729–35. 10.1074/mcp.M400021-MCP200 [DOI] [PubMed] [Google Scholar]
- 158. Deng, Jingjing , Erdjument‐Bromage Hediye, and Neubert Thomas A.. 2019. “Quantitative Comparison of Proteomes Using SILAC.” Current Protocols in Protein Science 95: e74. 10.1002/cpps.74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Wang, Xixi , He Yu, Ye Yang, Zhao Xinyu, Deng Shi, He Gu, Zhu Hongxia, Xu Ningzhi, and Liang Shufang. 2018. “SILAC–based Quantitative MS Approach for Real‐Time Recording Protein‐Mediated Cell‐Cell Interactions.” Scientific Reports 8: 8441. 10.1038/s41598-018-26262-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Ibarrola, Nieves , Kalume Dario E., Gronborg Mads, Iwahori Akiko, and Pandey Akhilesh. 2003. “A Proteomic Approach for Quantitation of Phosphorylation Using Stable Isotope Labeling in Cell Culture.” Analytical Chemistry 75: 6043–49. 10.1021/ac034931f [DOI] [PubMed] [Google Scholar]
- 161. Boersema, Paul J. , Geiger Tamar, Wiśniewski Jacek R., and Mann Matthias. 2013. “Quantification of the N‐Glycosylated Secretome By super‐SILAC During Breast Cancer Progression and in Human Blood Samples.” Molecular & Cellular Proteomics 12: 158–71. 10.1074/mcp.M112.023614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Tan, Jian , McKenzie Craig, Potamitis Maria, Thorburn Alison N., Mackay Charles R., and Macia Laurence. 2014. “The Role of Short‐Chain Fatty Acids in Health and Disease.” Advances in Immunology 121: 91–119. 10.1016/B978-0-12-800100-4.00003-9 [DOI] [PubMed] [Google Scholar]
- 163. Heianza, Yoriko , Sun Dianjianyi, Li Xiang, DiDonato Joseph A., Bray George A., Sacks Frank M., and Qi Lu. 2019. “Gut Microbiota Metabolites, Amino Acid Metabolites and Improvements in Insulin Sensitivity and Glucose Metabolism: The POUNDS Lost Trial.” Gut 68: 263–70. 10.1136/gutjnl-2018-316155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Ngo, Bryan , Van Riper Justin M., Cantley Lewis C., and Yun Jihye. 2019. “Targeting Cancer Vulnerabilities With High‐Dose Vitamin C.” Nature Reviews Cancer 19: 271–82. 10.1038/s41568-019-0135-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Reiter, Sinah , Dunkel Andreas, Dawid Corinna, and Hofmann Thomas. 2021. “Targeted LC‐MS/MS Profiling of Bile Acids in Various Animal Tissues.” Journal of Agricultural and Food Chemistry 69: 10572–80. 10.1021/acs.jafc.1c03433 [DOI] [PubMed] [Google Scholar]
- 166. Dietl, Katrin , Renner Kathrin, Dettmer Katja, Timischl Birgit, Eberhart Karin, Dorn Christoph, Hellerbrand Claus, et al. 2010. “Lactic Acid and Acidification Inhibit TNF Secretion and Glycolysis of Human Monocytes.” The Journal of Immunology 184: 1200–1209. 10.4049/jimmunol.0902584 [DOI] [PubMed] [Google Scholar]
- 167. Kumar, Arun , Baruah Aiswarya, Tomioka Masahiro, Iino Yuichi, Kalita Mohan C., and Khan Mojibur. 2020. “Caenorhabditis Elegans: a Model to Understand Host–Microbe Interactions.” Cellular and Molecular Life Sciences 77: 1229–49. 10.1007/s00018-019-03319-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Ternes, Dominik , Tsenkova Mina, Pozdeev Vitaly Igorevich, Meyers Marianne, Koncina Eric, Atatri Sura, Schmitz Martine, et al. 2022. “The Gut Microbial Metabolite Formate Exacerbates Colorectal Cancer Progression.” Nature Metabolism 4: 458–75. 10.1038/s42255-022-00558-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Hou, Huiqin , Chen Danfeng, Zhang Kexin, Zhang Wanru, Liu Tianyu, Wang Sinan, Dai Xin, et al. 2022. “Gut Microbiota‐Derived Short‐Chain Fatty Acids and Colorectal Cancer: Ready for Clinical Translation? Cancer Letters 526: 225–35. 10.1016/j.canlet.2021.11.027 [DOI] [PubMed] [Google Scholar]
- 170. Li, Yuan , Dong Jiali, Xiao Huiwen, Zhang Shuqin, Wang Bin, Cui Ming, and Fan Saijun. 2020. “Gut Commensal Derived‐Valeric Acid Protects Against Radiation Injuries.” Gut Microbes 11: 789–806. 10.1080/19490976.2019.1709387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Tran, Shirley Mei‐Sin , and Mohajeri M Hasan. 2021. “The Role of Gut Bacterial Metabolites in Brain Development, Aging and Disease.” Nutrients 13: 732. 10.3390/nu13030732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Yuan, Lu , Zhang Siruo, Li Huan, Yang Fan, Mushtaq Noosheen, Ullah Shakir, Shi Yi, An Cuihong, and Xu Jiru. 2018. “The Influence of Gut Microbiota Dysbiosis to the Efficacy of 5‐Fluorouracil Treatment on Colorectal Cancer.” Biomedicine & Pharmacotherapy 108: 184–93. 10.1016/j.biopha.2018.08.165 [DOI] [PubMed] [Google Scholar]
- 173. Yuan, Xiangliang , Duan Yimin, Xiao Yi, Sun Kai, Qi Yutao, Zhang Yuan, Ahmed Zamal, et al. 2022. “Vitamin E Enhances Cancer Immunotherapy by Reinvigorating Dendritic Cells Via Targeting Checkpoint SHP1.” Cancer Discovery 12: 1742–59. 10.1158/2159-8290.CD-21-0900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Ivanov, Ivaylo I. , Tuganbaev Timur, Skelly Ashwin N., and Honda Kenya. 2022. “T Cell Responses To the Microbiota.” Annual Review of Immunology 40: 559–87. 10.1146/annurev-immunol-101320-011829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Wang, Shouli , Kuang Junliang, Zhang Hongwei, Chen Wenlian, Zheng Xiaojiao, Wang Jieyi, Huang Fengjie, et al. 2022. “Bile Acid–Microbiome Interaction Promotes Gastric Carcinogenesis.” Advanced Science 9(16): 2200263. 10.1002/advs.202200263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Drewes, Julia L. , Chen Jie, Markham Nicholas O., Knippel Reece J., Domingue Jada C., Tam Ada J., Chan June L., et al. 2022. “Human Colon Cancer‐Derived Clostridioides Difficile Strains Drive Colonic Tumorigenesis in Mice.” Cancer Discovery 12: 1873–85. 10.1158/2159-8290.CD-21-1273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Wang, Hai , Rong Xingyu, Zhao Gan, Zhou Yifan, Xiao Yi, Ma Ding, Jin Xi, et al. 2022. “The Microbial Metabolite Trimethylamine N‐Oxide Promotes Antitumor Immunity in Triple‐Negative Breast Cancer.” Cell Metabolism 34: 581–94. e588. 10.1016/j.cmet.2022.02.010 [DOI] [PubMed] [Google Scholar]
- 178. Kadosh, Eliran , Snir‐Alkalay Irit, Venkatachalam Avanthika, May Shahaf, Lasry Audrey, Elyada Ela, Zinger Adar, et al. 2020. “The Gut Microbiome Switches Mutant p53 From Tumour‐Suppressive to Oncogenic.” Nature 586: 133–38. 10.1038/s41586-020-2541-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Schmitt, Mark , and Greten Florian R.. 2021. “The Inflammatory Pathogenesis of Colorectal Cancer.” Nature Reviews Immunology 21: 653–67. 10.1038/s41577-021-00534-x [DOI] [PubMed] [Google Scholar]
- 180. Emwas, Abdul‐Hamid , Roy Raja, McKay Ryan T., Tenori Leonardo, Saccenti Edoardo, Gowda GA Nagana, Raftery Daniel, et al. 2019. “NMR Spectroscopy for Metabolomics Research.” Metabolites 9: 123. 10.3390/metabo9070123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Liebal, Ulf W. , Phan An N. T., Sudhakar Malvika, Raman Karthik, and Blank Lars M.. 2020. “Machine Learning Applications for Mass Spectrometry‐Based Metabolomics.” Metabolites 10: 243. 10.3390/metabo10060243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Fei, Qiang , Wang Dongfang, Jasbi Paniz, Zhang Ping, Nagana Gowda G. A., Raftery Daniel, and Gu Haiwei. 2019. “Combining NMR and MS With Chemical Derivatization for Absolute Quantification With Reduced Matrix Effects.” Analytical Chemistry 91: 4055–62. 10.1021/acs.analchem.8b05611 [DOI] [PubMed] [Google Scholar]
- 183. Ye, Dongyang , Li Xiaowei, Shen Jianzhong, and Xia Xi. 2022. “Microbial Metabolomics: From Novel Technologies to Diversified Applications.” TrAC Trends in Analytical Chemistry 148: 116540. 10.1016/j.trac.2022.116540 [DOI] [Google Scholar]
- 184. Baidoo, Edward E. K . 2019. “Microbial Metabolomics: a General Overview.” Microbial Metabolomics 1859: 1–8. 10.1007/978-1-4939-8757-3_1 [DOI] [PubMed] [Google Scholar]
- 185. Beale, David J. , Pinu Farhana R., Kouremenos Konstantinos A., Poojary Mahesha M., Narayana Vinod K., Boughton Berin A., Kanojia Komal, et al. 2018. “Review of Recent Developments in GC–MS Approaches to Metabolomics‐Based Research.” Metabolomics 14: 152. 10.1007/s11306-018-1449-2 [DOI] [PubMed] [Google Scholar]
- 186. Koek, Maud M. , Muilwijk Bas, van der Werf Mariët J, and Hankemeier Thomas. 2006. “Microbial Metabolomics With Gas Chromatography/Mass Spectrometry.” Analytical Chemistry 78: 1272–81. 10.1021/ac051683+ [DOI] [PubMed] [Google Scholar]
- 187. Miyamoto, Suzanne , Taylor Sandra, Barupal Dinesh, Taguchi Ayumu, Wohlgemuth Gert, Wikoff William, Yoneda Ken, et al. 2015. “Systemic Metabolomic Changes in Blood Samples of Lung Cancer Patients Identified by Gas Chromatography Time‐Of‐Flight Mass Spectrometry.” Metabolites 5: 192–210. 10.3390/metabo5020192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Çelebier, Mustafa , and Nenni Merve. 2021. “Recent Developments in CE‐MS‐based Metabolomics.” Current Analytical Chemistry 17: 1229–42. 10.2174/1573411016999200709133339 [DOI] [Google Scholar]
- 189. Zeki, Özge Cansın , Eylem Cemil Can, Reçber Tuba, Kır Sedef, and Nemutlu Emirhan. 2020. “Integration of GC–MS and LC–MS for Untargeted Metabolomics Profiling.” Journal of Pharmaceutical and Biomedical Analysis 190: 113509. 10.1016/j.jpba.2020.113509 [DOI] [PubMed] [Google Scholar]
- 190. Krone, Nils , Hughes Beverly A., Lavery Gareth G., Stewart Paul M., Arlt Wiebke, and Shackleton Cedric H. L.. 2010. “Gas Chromatography/Mass Spectrometry (GC/MS) Remains a Pre‐Eminent Discovery Tool in Clinical Steroid Investigations Even in the Era of Fast Liquid Chromatography Tandem Mass Spectrometry (LC/MS/MS).” The Journal of Steroid Biochemistry and Molecular biology 121: 496–504. 10.1016/j.jsbmb.2010.04.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Ribbenstedt, Anton , Ziarrusta Haizea, and Benskin Jonathan P.. 2018. “Development, Characterization and Comparisons of Targeted and Non‐Targeted Metabolomics Methods.” PLoS One 13: e0207082. 10.1371/journal.pone.0207082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Perez De Souza, Leonardo , Alseekh Saleh, Brotman Yariv, and Fernie Alisdair R.. 2020. “Network‐Based Strategies in Metabolomics Data Analysis and Interpretation: From Molecular Networking to Biological Interpretation.” Expert Review of Proteomics 17: 243–55. 10.1080/14789450.2020.1766975 [DOI] [PubMed] [Google Scholar]
- 193. Sun, Shanshan , Zhu Lijun, Hu Yanxi, and Liu Yufeng. 2018. “Studies on the Metabolism of Paeoniflorin in Human Intestinal Microflora by High Performance Liquid Chromatography/Electrospray Ionization/Fourier Transform Ion Cyclotron Resonance Mass Spectrometry and Quadrupole Time‐Of‐Flight Mass Spectrometry.” Journal of Chromatography B 1085: 63–71. 10.1016/j.jchromb.2018.03.042 [DOI] [PubMed] [Google Scholar]
- 194. Alshamleh, Islam , Krause Nina, Richter Christian, Kurrle Nina, Serve Hubert, Günther Ulrich L., and Schwalbe Harald. 2020. “Real‐Time NMR Spectroscopy for Studying Metabolism.” Angewandte Chemie 132: 2324–28. 10.1002/ange.201912919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Yang, Qin , Tao Rui, Yang Bo, Zhang Hao, Chen Yong Q., Chen Haiqin, and Chen Wei. 2018. “Optimization of the Quenching and Extraction Procedures for a Metabolomic Analysis of Lactobacillus plantarum .” Analytical Biochemistry 557: 62–68. 10.1016/j.ab.2017.12.005 [DOI] [PubMed] [Google Scholar]
- 196. Southam, Andrew D. , Pursell Harriet, Frigerio Gianfranco, Jankevics Andris, Weber Ralf J. M., and Dunn Warwick B.. 2021. “Characterization of Monophasic Solvent‐Based Tissue Extractions for the Detection of Polar Metabolites and Lipids Applying Ultrahigh‐Performance Liquid Chromatography‐Mass Spectrometry Clinical Metabolic Phenotyping Assays.” Journal of Proteome Research 20: 831–40. 10.1021/acs.jproteome.0c00660 [DOI] [PubMed] [Google Scholar]
- 197. Vistain, Luke F. , and Tay Savaş. 2021. “Single‐Cell Proteomics.” Trends in Biochemical Sciences 46: 661–72. 10.1016/j.tibs.2021.01.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Cong, Yongzheng , Liang Yiran, Motamedchaboki Khatereh, Huguet Romain, Truong Thy, Zhao Rui, Shen Yufeng, et al. 2020. “Improved Single‐Cell Proteome Coverage Using Narrow‐Bore Packed NanoLC Columns and Ultrasensitive Mass Spectrometry.” Analytical Chemistry 92: 2665–71. 10.1021/acs.analchem.9b04631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Chantipmanee, Nattapong , and Hauser Peter C.. 2021. “Development of Simple Drift Tube Design for Ion Mobility Spectrometry Based on Flexible Printed Circuit Board Material.” Analytica Chimica Acta 1170: 338626. 10.1016/j.aca.2021.338626 [DOI] [PubMed] [Google Scholar]
- 200. Groessl, M. , Graf S., and Knochenmuss R.. 2015. “High Resolution Ion Mobility‐Mass Spectrometry for Separation and Identification of Isomeric Lipids.” Analyst 140: 6904–11. 10.1039/c5an00838g [DOI] [PubMed] [Google Scholar]
- 201. Mast, David H. , Liao Hsiao‐Wei, Romanova Elena V., and Sweedler Jonathan V.. 2021. “Analysis of Peptide Stereochemistry in Single Cells by Capillary Electrophoresis–Trapped Ion Mobility Spectrometry Mass Spectrometry.” Analytical Chemistry 93: 6205–13. 10.1021/acs.analchem.1c00445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Woo, Jongmin , Clair Geremy C., Williams Sarah M., Feng Song, Tsai Chia‐Feng, Moore Ronald J., Chrisler William B., et al. 2022. “Three‐Dimensional Feature Matching Improves Coverage for Single‐Cell Proteomics Based on Ion Mobility Filtering.” Cell systems 13: 426–34.e424. 10.1016/j.cels.2022.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Dueñas, Maria Emilia , and Lee Young Jin. 2021. “Single‐Cell Metabolomics by Mass Spectrometry Imaging.” Cancer Metabolomics 1280: 69–82. 10.1007/978-3-030-51652-9_5 [DOI] [PubMed] [Google Scholar]
- 204. Hansen, Rebecca L. , and Lee Young Jin. 2018. “High‐Spatial Resolution Mass Spectrometry Imaging: Toward Single Cell Metabolomics in Plant Tissues.” The Chemical Record 18: 65–77. 10.1002/tcr.201700027 [DOI] [PubMed] [Google Scholar]
- 205. Dueñas, Maria Emilia , Essner Jeffrey J., and Lee Young Jin. 2017. “3D MALDI Mass Spectrometry Imaging of a Single Cell: Spatial Mapping of Lipids in the Embryonic Development of Zebrafish.” Scientific Reports 7: 14946. 10.1038/s41598-017-14949-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Passarelli, Melissa K. , Newman Carla F., Marshall Peter S., West Andrew, Gilmore Ian S., Bunch Josephine, Alexander Morgan R., and Dollery Colin T.. 2015. “Single‐Cell Analysis: Visualizing Pharmaceutical and Metabolite Uptake in Cells With Label‐Free 3D Mass Spectrometry Imaging.” Analytical Chemistry 87: 6696–702. 10.1021/acs.analchem.5b00842 [DOI] [PubMed] [Google Scholar]
- 207. Taylor, Michael J. , Lukowski Jessica K., and Anderton Christopher R.. 2021. “Spatially Resolved Mass Spectrometry at the Single Cell: Recent Innovations in Proteomics and Metabolomics.” Journal of the American Society for Mass Spectrometry 32: 872–94. 10.1021/jasms.0c00439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Kompauer, Mario , Heiles Sven, and Spengler Bernhard. 2017. “Atmospheric Pressure MALDI Mass Spectrometry Imaging of Tissues and Cells at 1.4‐μm Lateral Resolution.” Nature Methods 14: 90–96. 10.1038/nmeth.4071 [DOI] [PubMed] [Google Scholar]
- 209. Korte, Andrew R. , Yandeau‐Nelson Marna D., Nikolau Basil J., and Lee Young Jin. 2015. “Subcellular‐Level Resolution MALDI‐MS Imaging of Maize Leaf Metabolites by MALDI‐Linear Ion Trap‐Orbitrap Mass Spectrometer.” Analytical and Bioanalytical Chemistry 407: 2301–9. 10.1007/s00216-015-8460-5 [DOI] [PubMed] [Google Scholar]
- 210. Zhang, Linwen , and Vertes Akos. 2018. “Single‐Cell Mass Spectrometry Approaches to Explore Cellular Heterogeneity.” Angewandte Chemie International Edition 57: 4466–77. 10.1002/anie.201709719 [DOI] [PubMed] [Google Scholar]
- 211. Nordhoff, Eckhard , Schürenberg Martin, Thiele Gabriela, Lübbert Christine, Kloeppel Klaus‐Dieter, Theiss Dorothea, Lehrach Hans, and Gobom Johan. 2003. “Sample Preparation Protocols for MALDI‐MS of Peptides and Oligonucleotides Using Prestructured Sample Supports.” International Journal of Mass Spectrometry 226: 163–80. 10.1016/S1387-3806(02)00978-8 [DOI] [Google Scholar]
- 212. Krismer, Jasmin , Sobek Jens, Steinhoff Robert F., Brönnimann Rolf, Pabst Martin, and Zenobi Renato. 2020. “A MALDI‐MS Methodology for Studying Metabolic Heterogeneity of Single Cells in a Population.” Single Cell Metabolism 2064: 113–24. 10.1007/978-1-4939-9831-9_9 [DOI] [PubMed] [Google Scholar]
- 213. Feenstra, Adam D. , Hansen Rebecca L., and Lee Young Jin. 2015. “Multi‐Matrix, Dual Polarity, Tandem Mass Spectrometry Imaging Strategy Applied t a Germinated Maize Seed: Toward Mass Spectrometry Imaging of an Untargeted Metabolome.” Analyst 140: 7293–304. 10.1039/c5an01079a [DOI] [PubMed] [Google Scholar]
- 214. Guo, Shuai , Tang Weiwei, Hu Yu, Chen Yanwen, Gordon Andrew, Li Bin, and Li Ping. 2019. “Enhancement of on‐Tissue Chemical Derivatization by Laser‐Assisted Tissue Transfer for MALDI MS Imaging.” Analytical Chemistry 92: 1431–38. 10.1021/acs.analchem.9b04618 [DOI] [PubMed] [Google Scholar]
- 215. Zang, Qingce , Wang Manjiangcuo, Zhu Ying, Wang Lingzhi, Luo Zhigang, Li Xin, He Jiuming, Zhang Ruiping, and Abliz Zeper. 2021. “Enhanced on‐Tissue Chemical Derivatization With Hydrogel Assistance for Mass Spectrometry Imaging.” Analytical Chemistry 93: 15373–80. 10.1021/acs.analchem.1c03118 [DOI] [PubMed] [Google Scholar]
- 216. Barré, Florian P. Y. , Paine Martin R. L., Flinders Bryn, Trevitt Adam J., Kelly Patrick D., Ait‐Belkacem Rima, Garcia João P., et al. 2019. “Enhanced Sensitivity Using MALDI Imaging Coupled With Laser Postionization (MALDI‐2) for Pharmaceutical Research.” Analytical Chemistry 91: 10840–48. 10.1021/acs.analchem.9b02495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Fiehn, Oliver , Wohlgemuth Gert, Scholz Martin, Kind Tobias, Lee Do Yup, Lu Yun, Moon Stephanie, and Nikolau Basil. 2008. “Quality Control for Plant Metabolomics: Reporting MSI‐compliant Studies.” The Plant Journal 53: 691–704. 10.1111/j.1365-313X.2007.03387.x [DOI] [PubMed] [Google Scholar]
- 218. Sanghvi, Viraj R. , Leibold Josef, Mina Marco, Mohan Prathibha, Berishaj Marjan, Li Zhuoning, Miele Matthew M., et al. 2019. “The Oncogenic Action of nrf2 Depends on De‐Glycation by Fructosamine‐3‐Kinase.” Cell 178: 807–19. 10.1016/j.cell.2019.07.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Galligan, James J. , Wepy James A., Streeter Matthew D., Kingsley Philip J., Mitchener Michelle M., Wauchope Orrette R., Beavers William N., et al. 2018. “Methylglyoxal‐Derived Posttranslational Arginine Modifications Are Abundant Histone Marks.” Proceedings of the National Academy of Sciences of the United States 115: 9228–33. 10.1073/pnas.1802901115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Diao, Xiayao . 2019. “Histone Glycation: Linking Metabolic Perturbation With Epigenetic Misregulation in Cancer.” AIMS Genetics 6: 14–16. 10.3934/genet.2019.2.14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Zheng, Qingfei , Maksimovic Igor, Upad Akhil, and David Yael. 2020. “Non‐Enzymatic Covalent Modifications: a New Link Between Metabolism and Epigenetics.” Protein Cell 11: 401–16. 10.1007/s13238-020-00722-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Ray, Devin M. , Jennings Erin Q., Maksimovic Igor, Chai Xander, Galligan James J., David Yael, and Zheng Qingfei. 2022. “Chemical Labeling and Enrichment of Histone Glyoxal Adducts.” ACS Chemical Biology 17: 756–61. 10.1021/acschembio.1c00864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Zheng, Qingfei , Prescott Nicholas A., Maksimovic Igor, and David Yael. 2019. “(De) Toxifying the Epigenetic Code.” Chemical Research in Toxicology 32: 796–807. 10.1021/acs.chemrestox.9b00013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Donnellan, Leigh , Simpson Bradley, Dhillon Varinderpal S., Costabile Maurizio, Fenech Michael, and Deo Permal. 2021. “Methylglyoxal Induces Chromosomal Instability and Mitotic Dysfunction in Lymphocytes.” Mutagenesis 36: 339–48. 10.1093/mutage/geab028 [DOI] [PubMed] [Google Scholar]
- 225. Galligan, James J. , Mitchener Michelle M., Wauchope Orrette R., Wang Tina, Rose Kristie L., Spiegel David A., and Marnett Lawrence J.. 2015. “Histones Are Major Targets for Modification by Glucose‐Derived Methylglyoxal.” Free Radical Biology & Medicine 87: S24. 10.1096/fasebj.30.1_supplement.586.2 [DOI] [Google Scholar]
- 226. Zheng, Qingfei , Maksimovic Igor, Upad Akhil, Guber David, and David Yael. 2020. “Synthesis of an Alkynyl Methylglyoxal Probe to Investigate Nonenzymatic Histone Glycation.” The Journal of Organic Chemistry 85: 1691–97. 10.1021/acs.joc.9b02504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. Dzutsev, Amiran , Goldszmid Romina S., Viaud Sophie, Zitvogel Laurence, and Trinchieri Giorgio. 2015. "The Role of the Microbiota in Inflammation, Carcinogenesis, and Cancer Therapy." European Journal of Immunology 45: 17–31. 10.1002/eji.201444972 [DOI] [PubMed] [Google Scholar]
- 228. Górska, Agata , Przystupski Dawid, Niemczura Magdalena J., and Kulbacka Julita. 2019. “Probiotic Bacteria: A Promising Tool in Cancer Prevention and Therapy.” Current Microbiology 76: 939–49. 10.1007/s00284-019-01679-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Rahman, Md Mominur , Islam Md Rezaul, Shohag Sheikh, Ahasan Md Tanjimul, Sarkar Nadia, Khan Hosneara, Madalin Hasan Alexandru, Cavalu Simona, and Rauf Abdur. 2022. “Microbiome in Cancer: Role in Carcinogenesis and Impact in Therapeutic Strategies.” Biomedicine & Pharmacotherapy 149: 112898. 10.1016/j.biopha.2022.112898 [DOI] [PubMed] [Google Scholar]
- 230. Reginato, Eleonora . 2014. “Immune Response After Photodynamic Therapy Increases Anti‐Cancer and Anti‐Bacterial Effects.” World Journal of Immunology 4: 1. 10.5411/wji.v4.i1.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Russo, Edda , Taddei Antonio, Ringressi Maria Novella, Ricci Federica, and Amedei Amedeo. 2016. “The Interplay Between the Microbiome and the Adaptive Immune Response in Cancer Development.” Therapeutic Advances in Gastroenterology 9: 594–605. 10.1177/1756283X16635082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232. Huang, Xuehui , Pan Jingmei, Xu Funeng, Shao Binfen, Wang Yi, Guo Xing, and Zhou Shaobing. 2021. “Bacteria‐Based Cancer Immunotherapy.” Advanced Science 8: 2003572. 10.1002/advs.202003572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233. Nicora, Giovanna , Vitali Francesca, Dagliati Arianna, Geifman Nophar, and Bellazzi Riccardo. 2020. “Integrated Multi‐Omics Analyses in Oncology: a Review of Machine Learning Methods and Tools.” Frontiers in Oncology 10: 1030. 10.3389/fonc.2020.01030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234. Bokulich, Nicholas A. , Łaniewski Paweł, Chase Dana M., Caporaso J. Gregory, and Herbst‐Kralovetz Melissa M.. 2022. “Integration of Multi‐Omics Data Improves Prediction of Cervicovaginal Microenvironment in Cervical Cancer.” 18(2): e1009876. 10.1101/2020.08.27.20183426 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No new data or script was used in this paper. Supplementary materials (figures, tables, scripts, graphical abstracts, slides, videos, Chinese translated versions and updated materials) may be found in the online DOI or iMeta Science http://www.imeta.science/.