

Abstract
Background
Checkpoint kinase 1 (CHK1) has dual roles in both the DNA damage response and in the innate immune response to genotoxic stress. The combination of CHK1 inhibition and immune checkpoint blockade has the potential to enhance anti-tumoral T-cell activation.
Methods
This was an open-label phase 1 study evaluating the CHK1 inhibitor prexasertib and the anti-PD-L1 antibody LY3300054. After a lead-in of LY3300054 (Arm A), prexasertib (Arm B) or the combination (Arm C), both agents were administered intravenously at their respective recommended phase 2 doses (RP2Ds) on days 1 and 15 of a 28-day cycle. Flow cytometry of peripheral blood was performed before and during treatment to analyze effects on immune cell populations, with a focus on T cell subsets and activation. Plasma cytokines and chemokines were analyzed using the Luminex platform.
Results
Among seventeen patients enrolled, the combination was tolerable at the monotherapy RP2Ds, 105 mg/m2 prexasertib and 700 mg LY3300054. Dose-limiting toxicities included one episode each of febrile neutropenia (Arm C) and grade 4 neutropenia lasting > 5 days (Arm B). One patient had immune-related AST/ALT elevation after 12 cycles. Three patients with CCNE1-amplified, high-grade serous ovarian cancer (HGSOC) achieved partial response (PR), 2 lasting > 12 months; a fourth such patient maintained stable disease > 12 months. Analysis of peripheral blood demonstrated evidence of CD8 + T-cell activation in response to treatment.
Conclusions
Prexasertib in combination with PD-L1 blockade was tolerable and demonstrated preliminary activity in CCNE1-amplified HGSOC with evidence of cytotoxic T-cell activation in patient blood samples.
Trial registration
ClinicalTrials.gov identifier: NCT03495323. Registered April 12, 2018.
Supplementary Information
The online version contains supplementary material available at 10.1007/s00262-021-02910-x.
Keywords: Ovarian cancer, Immune checkpoint, Checkpoint kinase 1, Cyclin E1, PD-L1
Introduction
The DNA damage response and immune response pathways are evolutionarily conserved and interrelated. The immune system plays a critical role in eliminating and controlling early tumor growth. Upregulation of PD-L1 in tumor cells has been implicated in immune tolerance and immune escape [1]. PD-L1 expression has been shown to be differentially regulated at several crucial timepoints during the DNA damage response. Accumulating evidence shows that DNA damage induces cell surface expression of PD-L1 and that PD-L1 upregulation is mediated by activation of the cell cycle checkpoint kinases ataxia telangiectasia mutated (ATM), ataxia telangiectasia and RAD3-related (ATR), and checkpoint kinase 1 (CHK1) [2, 3]. CHK1, in particular, has been shown to be a central relay point for switch from a DNA damage response to an immune response, promoting the upregulation of PD-L1 expression in response to DNA damage through STAT1/3 and interferon receptor factor 1 (IRF1)-mediated transcription of PD-L1 mRNA [3].
Disruption of the DNA damage response can also lead to upregulation of PD-L1 expression. Collapse of replication forks occurring during the S-phase checkpoint in response to CHK1 inhibition results in accumulation of DNA fragments and generation of cytosolic DNA, which activates a type I interferon response, involving activation of the cGAS-STING pathway, nuclear factor-kappa B mediated release of inflammatory cytokines, and upregulation of PD-L1 expression [4–7]. Additionally, dying cancer cells and the production of neoantigens trigger the release of damage-associated molecular pattern pathways and activation of a type II interferon response, further reinforcing constitutive upregulation of PD-L1 expression [8].
Based on these emerging data, we conducted a phase 1 trial combining the CHK1 inhibitor prexasertib and the PD-L1 antibody LY3300054 in patients with advanced solid tumors, incorporating profiling of peripheral blood to explore the immune response to this DNA damaging agent alone and in combination with immune checkpoint blockade.
Patients and methods
Study population
Patients with advanced solid tumors without approved curative therapy or effective palliative therapy, ≥ 18 years of age, with an Eastern Cooperative Oncology Group (ECOG) performance status 0–1 and measurable disease per Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1 [9] were eligible for the study. All patients were required to have adequate organ function defined by: absolute neutrophil count ≥ 1.5 × 109/L, platelet count ≥ 100 × 109/L, total bilirubin ≤ 1.5 × institutional upper limit of reference range (ULRR), aspartate aminotransferase (AST) and alanine aminotransferase (ALT) ≤ 2.5 × ULRR, creatinine ≤ 1.5 × institutional upper limit of reference range (ULRR) or creatinine clearance ≥ 60 mL/min by Crockcroft-Gault formula, free thyroxine within institutional normal limits, and QTc ≤ 470 ms on screening ECG. Patients were required to have completed previous cancer therapy at least 3 weeks prior to study entry (6 weeks for nitrosureas or mitomycin C). Prior treatment with PD-L1 antibody was permitted if not the most recent therapy prior to enrollment. Subjects who had received radiation to > 25% of the marrow or more than 4 lines of cytotoxic chemotherapy were excluded. Additional exclusion criteria included any prior Grade 3 immune-related adverse events, immune-related neurologic or ocular toxicities, or immune-related toxicities of any grade which required permanent discontinuation of prior immune therapy. The presence of untreated brain metastases or carcinomatous meningitis, pregnancy, or active infection (including Hepatitis B, C, or HIV) was excluded.
Study design and treatment administration
The primary objective of this study was to determine the safe and tolerable dose of the combination of prexasertib and LY3300054. Secondary objectives included characterization of changes in immune cell subsets and cytokine profiles in peripheral blood and tumor biopsies in response to treatment, and assessment of differences in immune phenotype between responders and non-responders. Starting dose levels for both prexasertib and LY3300054 were at the RP2D for the individual agents. Based on expected neutropenia with prexasertib administration, secondary prophylactic growth factor support was permitted beginning with cycle 1. Patients were randomized to one of three administration schedules: lead-in (cycle 0) of LY3300054 alone (Cohort A), prexasertib alone (Cohort B), or the combination (Cohort C), designed to accommodate pharmacokinetic (PK) and pharmacodynamic (PD) assessment of single-agent activity versus the combination. Thereafter, both agents were administered each as a 1 h intravenous infusion on days 1 and 15 of a 28-day cycle. Up to 6 patients could be enrolled to the respective arm with observance of a dose-limiting toxicity (DLT). An expansion cohort with paired tumor biopsies was planned but enrollment was not completed due to internal re-prioritization by the drug manufacturer to discontinue further development of prexasertib necessitating early closure of the study due to limited remaining drug availability.
Dose-limiting toxicity definitions and study assessments
Safety was assessed via monitoring of toxicities during the lead-in cycle 0 + cycle 1 according to National Cancer Institute Common Terminology Criteria for Adverse Events (NCI-CTCAE) v.4.03. Dose-limiting toxicities (DLT) were defined as grade 4 neutropenia > 5 days despite growth factor support or febrile neutropenia, grade 4 thrombocytopenia, and any grade 3–4 non-hematologic toxicities related to study drug and occurring during the lead-in and/or cycle 1. Grade 3 ≥ nausea, vomiting, diarrhea, electrolyte derangements, or rise in creatinine were considered dose-limiting if refractory to management and not improved to grade ≤ 2 within 48 h. Any grade 3 or 4 colitis or non-infectious pneumonitis, grade 4 immune-related toxicities, and any onset of Stevens-Johnson Syndrome, toxic epidermal necrolysis, skin necrosis, or bullous or hemorrhagic skin lesions were considered dose-limiting. Any non-laboratory grade 3 immune-related adverse event not downgraded to Grade 2 within 3 days despite optimal medical management was considered dose-limiting. Increases in AST or ALT to > 3 times institutional ULN and concurrent increase in total bilirubin to > 2 × institutional ULN were considered dose-limiting. Additionally, inability to tolerate 100% of scheduled prexasertib and LY3300054 during the lead-in and cycle 1 was considered dose-limiting. After review of tolerability for each cohort dose schedule by the Safety Review Committee in conjunction with the sponsor, a single dose administration schedule was selected as the RP2D for further planned enrollment. An expansion cohort of up to 10 patients was planned at the RP2D to further evaluate safety, tolerability, and PD endpoints.
A physical examination, assessment of vital signs, pertinent tumor biomarker assessments, hematology, and chemistry assessments were performed at screening, on day 1 of the lead-in cycle, and on days 1 and 15 of every subsequent cycle. Additional safety labs including CBC with differential were collected from patients on days 8 and 22 of the first two cycles due to anticipated myelosuppression with treatment. Thyroid function tests were performed at screening and at the start of each cycle. Standard 12-lead ECGs were obtained at screening, at the start of the first two cycles, and as clinically indicated thereafter.
Pharmacokinetic assessments
Abbreviated plasma samples were collected in 4 mL plastic Vacutainer tubes with spray dried sodium heparin (prexasertib samples) and K3EDTA tubes (LY3300054 samples) on C1D1 pre-dose, at 1 h at the completion of the prexasertib infusion, and 1 h after completion of the LY3300054 infusion; C3D1 pre-dose and at 1 h at the completion of the prexasertib infusion; and pre-dose every three cycles starting with C6D1 for all three arms. For Arm C, additional plasma samples were collected during the lead-in cycle pre-dose, at 1 h at the completion of the prexasertib infusion, 1 h and 2 h after completion of the LY3300054 infusion. Blood collection tubes were centrifuged to harvest the plasma which was stored in cryovials at −80 °C. Prexasertib plasma concentrations were quantified using a validated high-pressure liquid chromatography-mass spectrometry/mass spectrometry method. LY3300054 plasma concentrations were quantified using a validated electrochemiluminescent method. Pharmacokinetic data were analyzed using population pharmacokinetic analysis.
Pharmacodynamic assessments
Whole blood samples were collected during the lead-in period prior to initiation of therapy on C0D1, on C0D2 after the first lead-in administration of drug, on C0D8, C1D8, C1D1, and C2D1, and at the point of progression. C0D1, C0D2, and C2D1 samples were chosen for analysis based on published kinetics of T-cell activation [10]. Peripheral blood mononuclear cells (PBMC) samples were thawed and surface stained with fluorochrome-conjugated monoclonal antibodies against lineage markers (CD3, CD4, CD8, CD19, CD25, CD56, CD127), memory markers (CD45RA, CCR7) and functional markers (CD69, CD71). Fluorescence minus one (FMO) controls were used for compensation, as previously described [11, 12]. Two flow cytometry antibody panels were developed. These are described along with gating strategies in Supplemental Figure 1. Samples were fixed, then acquired within 24 h of staining on a four laser BD Fortessa X20. FCS files were analyzed by manual gating using FlowJo v10.0.8 for Mac (BD, Franklin Lakes, NJ). Data are presented as cellular population frequency based on percentage of viable cells.
Patient plasma samples were thawed and prepared for soluble analyte assay according to previously published methods [13, 14]. Analytes were measured on a Luminex FLEXMAP3D (Luminex, Austin, TX) per the manufacturer’s protocol. Soluble IL6, IL3, IL7, IL10, VEGFα, IL1β, IL4, TNFβ, FGFβ, IL5, IL12p70, TGFα, TNFα, ENA78, GCSF, GMCSF, IFNγ, IL1α, IL1β, IL1RA, IL2, IL8, IL17, MCP1, MIP1α, and MIP1β were tested. Out of all the soluble markers measured, 15 markers were within detectable range and could be quantified by extrapolation of MFIs to the respective standard curve between lower limit of quantitation and upper limit of quantitation. Analyte concentration for each patient was calculated using standard curves. Fold changes were calculated as a ratio relative to the patient’s baseline (C0D1) [15–17].
Statistical analyses
Summaries of patient demographics, disease, and prior treatment characteristics are presented descriptively. Changes in immune subsets by flow analysis and cytokine expression levels are expressed using fold changes relative to C0D1. As a surrogate for response, we associated patients’ time on study with changes in immune cell subsets. Patients were classified as deriving clinical benefit (DCB) if they had best response of stable disease or partial response and remained on study for at least 100 days (N = 8). Those patients who did not derive clinical benefit (Non-DCB) remained on study for less than 100 days due to disease progression (N = 7). The immunomodulatory effects of prexasertib alone versus in combination were investigated using Arms B and C. All comparative, correlative statistical analyses of two groups were based on two-sided Wilcoxon rank-sum tests with significance level of 0.05. There were no adjustments for multiple comparisons. Heatmaps were based on hierarchical clustering using R package “heatmap.2” (R version 3.5.3).
Results
Patient disposition and characteristics
A total of 17 patients were enrolled between June 2018 through January 2020 (Table 1); 15 patients were enrolled to the dose optimization phase of the study, and two were enrolled to the expansion phase of the study prior to early closure by the sponsor. All patients were evaluable for both response and toxicity. Three patients received at least 12 cycles of treatment. The majority of patients had high-grade serous ovarian carcinoma (HGSOC) (14/17); six of these patients had tumors harboring CCNE1 amplification on targeted next-generation sequencing. Three patients with CCNE1-amplified HGSOC achieved a partial response (PR), remaining on study for 7 months, 13 months, and 20 months, respectively; a fourth patient with CCNE1-amplified HGSOC maintained stable disease (SD) lasting > 12 months. RECIST v1.1 response for the subset of ovarian cancer patients is shown for each patient (Fig. 1).
Table 1.
Patient demographics
| Patient demographics | |
|---|---|
| Patients enrolled/treated | 17 |
| Median age in years (range) | 57 (25–76) |
|
Median number of prior therapies (range) Prior immunotherapy |
3 (0–4)* 2 |
| Diagnoses | |
| Ovarian (incl. high-grade serous ovarian cancer—10; low-grade serous ovarian cancer—2; clear cell ovarian cancer—2) | 14 |
| Soft tissue sarcoma (incl. uterine leiomyosarcoma, malignant solitary fibrous tumor) | 2 |
| Colorectal cancer | 1 |
*One patient enrolled with 5 prior systemic therapies, one line was on an experimental trial
Fig. 1.

Waterfall Plot of Clinical Response in Ovarian Cancer Patients (n = 14) on Study. Best Response by RECIST v1.1 is shown. Histologies included: *clear cell (n = 2), ǂlow-grade serous (n = 2), and †high-grade serous ovarian cancer (n = 10); pertinent mutations of interest, where known, are indicated. Days on study and best response are shown on the x-axis
Adverse events
The most common adverse events (AEs) attributed to treatment and occurring in ≥ 10% of patients are summarized in (Table 2), reported as highest grade observed. A total of 382 adverse events of any grade and any causality were captured for the trial. Of these adverse events, 170 or 83% were Grade 1/2. Most common adverse events occurring in greater than 50% of patients include anemia (82%), neutropenia (88%), decreased white blood cell count (82%), decreased platelet count (53%), fatigue (53%), and abdominal pain (53%) consistent with previous reports for prexasertib monotherapy [18, 19]. Decreases in lymphocyte count were also observed but the majority were grade 1 or 2. There were 2 DLTs, including one event of febrile neutropenia on Arm C and one event of prolonged grade 4 neutropenia lasting > 5 days on Arm B. A third patient also developed fever in the setting of viral illness and neutropenia at C4D15, outside of the DLT window. One patient developed immune-related grade 3 elevation in AST and ALT after 12 cycles, requiring mycophenolate for management, necessitating discontinuation of therapy.
Table 2.
Summary of adverse events attributed to study treatment
| Dose cohort | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Adverse events* | Cohort A (n = 3) |
Cohort B (n = 8) |
Cohort C (n = 6) |
Total (%) | |||||||||
| Gr 1 | Gr 2 | Gr 3 | Gr 4 | Gr 1 | Gr 2 | Gr 3 | Gr 4 | Gr 1 | Gr 2 | Gr 3 | Gr 4 | ||
| Blood and lymphatic system disorders | |||||||||||||
| Anemia | 1 | – | 1 | – | 2 | 2 | 2 | – | – | 5 | 1 | – | 14 (82%) |
| Febrile neutropenia | – | – | – | – | – | – | – | – | – | – | 2 | – | 2 (12%) |
| Endocrine disorders | |||||||||||||
| Hypothyroidism | – | – | – | – | 1 | – | – | – | – | 1 | – | – | 2 (12%) |
| Gastrointestinal disorders | |||||||||||||
| Abdominal pain | 1 | 1 | – | – | 2 | 1 | – | – | 4 | – | – | – | 9 (53%) |
| Ascites | – | 2 | – | – | – | – | – | – | – | – | – | – | 2 (12%) |
| Bloating | 1 | – | – | – | 1 | – | – | – | 1 | – | – | – | 3 (18%) |
| Constipation | – | – | – | – | 1 | 2 | – | – | 1 | – | – | – | 4 (24%) |
| Diarrhea | 1 | – | – | – | 3 | – | – | – | 4 | – | – | – | 8 (47%) |
| Dry mouth | 1 | – | – | – | 1 | – | – | – | – | – | – | – | 2 (12%) |
| Dyspepsia | 1 | – | – | – | – | – | – | – | – | – | – | – | 1 (6%) |
| Flatulence | 1 | – | – | – | 1 | – | – | – | 1 | – | – | – | 3 (18%) |
| Gastroesophageal reflux | – | 2 | – | – | – | – | – | – | – | – | – | – | 2 (12%) |
| Gastroparesis | 1 | – | – | – | – | – | – | – | – | – | – | – | 1 (6%) |
| Oral mucositis | 1 | – | – | – | 1 | 1 | – | – | 2 | – | – | – | 5 (29%) |
| Oral dysesthesia | 1 | – | – | – | 1 | – | – | – | – | – | – | – | 2 (12%) |
| Nausea | 1 | – | – | – | 1 | 1 | – | – | 4 | – | – | – | 7 (41%) |
| Vomiting | 1 | – | – | – | – | – | – | – | 2 | – | – | – | 3 (18%) |
| Immune system disorders | |||||||||||||
| Allergic reaction | – | – | – | – | – | – | – | – | 1 | 1 | – | – | 2 (12%) |
| Infections/Infestations | |||||||||||||
| Sepsis | – | – | – | – | – | – | – | – | – | – | – | 1 | 1 (6%) |
| Metabolism and nutrition | |||||||||||||
| Anorexia | 3 | – | – | – | 1 | – | – | – | 2 | – | – | – | 6 (35%) |
| Hypokalemia | – | – | – | – | 1 | 1 | – | – | 1 | – | – | – | 3 (18%) |
| Investigations | |||||||||||||
| Alkaline phosphatase ↑ | 1 | – | – | – | 1 | 1 | – | – | 3 | – | – | – | 7 (41%) |
| AST ↑ | – | – | – | – | 3 | – | – | – | – | – | 1 | – | 4 (24%) |
| ALT ↑ | – | – | – | – | 2 | – | – | – | – | – | 1 | – | 3 (18%) |
| Bilirubin ↑ | – | – | – | – | – | – | – | – | 1 | 1 | – | – | 2 (12%) |
| Creatinine ↑ | 1 | – | – | – | 1 | – | – | – | – | – | – | – | 2 (12%) |
| Platelet count ↓ | – | 1 | – | – | – | – | 3 | – | 3 | 1 | – | 1 | 9 (53%) |
| Lymphocyte count ↓ | 2 | 1 | – | – | 1 | 1 | 2 | – | – | 2 | 2 | – | 11 (65%) |
| White blood cell count ↓ | – | – | 1 | 1 | – | 1 | 2 | 3 | – | – | 2 | 4 | 14 (82%) |
| Neutrophil count ↓ | 1 | – | – | 2 | – | – | – | 6 | – | – | – | 6 | 15 (88%) |
| General/Administration site | |||||||||||||
| Infusion related reaction | – | – | – | – | – | 1 | – | – | 1 | 1 | – | – | 3 (18%) |
| Fatigue | 2 | – | – | – | 2 | 1 | – | – | 3 | 1 | – | – | 9 (53%) |
| Flu like symptoms | – | – | – | – | – | 1 | – | – | 1 | – | – | – | 2 (12%) |
| Chills | 1 | – | – | – | 1 | – | – | – | 2 | – | – | – | 4 (24%) |
| Fever | 1 | – | – | – | 1 | – | – | – | 2 | – | – | – | 4 (24%) |
| Malaise | – | – | – | – | 1 | 1 | – | – | – | – | – | – | 2 (12%) |
| Localized edema | – | – | – | – | 1 | – | – | – | 1 | – | – | – | 2 (12%) |
| Non-cardiac chest pain | – | – | – | – | 1 | – | – | – | – | – | – | – | 1 (6%) |
| Nervous System | |||||||||||||
| Dizziness | 1 | – | – | – | 1 | – | – | – | 1 | – | – | – | 3 (18%) |
| Headache | – | – | – | – | 1 | 2 | – | – | – | – | – | – | 3 (18%) |
| Respiratory/Thoracic/Mediastinal | |||||||||||||
| Dyspnea | – | – | – | – | – | – | – | – | – | 1 | – | – | 1 (6%) |
| Cough | – | – | – | – | 1 | – | – | – | 1 | – | – | – | 2 (12%) |
| Musculoskeletal/Connective tissue | |||||||||||||
| Arthralgia | – | – | – | – | – | 1 | – | – | – | – | – | – | 1 (6%) |
| Back pain | – | – | – | – | – | – | – | – | 1 | – | – | – | 1 (6%) |
| Myalgia | 1 | – | – | – | 2 | – | – | – | 2 | – | – | – | 5 (29%) |
| Skin/Subcutaneous | |||||||||||||
| Pruritus | 1 | – | – | – | 1 | – | – | – | 1 | – | – | – | 3 (18%) |
| Rash maculo-papular | – | 1 | – | – | – | 2 | – | – | 1 | – | – | – | 4 (24%) |
*All adverse events represent the number of patients experiencing the adverse event felt to be related to study treatment, reported as worst grade for each patient
Pharmacokinetic analyses
Prexasertib exposures ranged from 320 to 1230 ng/mL at 60 min after dosing in the majority of patients, within range of previously published data [18]. LY3300054 exposures ranged from 125 to 472 µg/mL at 120 min after dosing and 48–133 µg/mL pre-dose with subsequent cycles (Supplementary Figure 2).
Prexasertib treatment causes changes in T-cell proliferation patterns and cytokine profile
Immune cell subset analysis and functional status using soluble analyte and flow cytometry assays from PBMC samples of the 15 patients enrolled to the dose optimization are shown for C0D2 (Fig. 2a) and C2D1 (Fig. 2b). Cohort A with a lead-in of LY3300054 alone was used as a comparative control to exclude changes due to immune checkpoint blockade. Decrease in circulating CD4 + Tregs at C0D2 compared to baseline were statistically different between cohort B (5/6) versus cohort C (1/6; P = 0.04). Changes in frequency of activated CD8 + T cells positive for transferrin receptor (CD71), Natural Killer T (NKT) cells, and CD8 + NKT cells were also seen at C2D1. CD71 + CD8 + T cells were increased in 12/15 patients indicating expansion of an activated CD8 T cell subset in response to combination treatment. By comparison, the frequency of T cell memory subsets at C2D1 including CD8 + T central memory (TCM), CD8 + T effector memory (TEM), CD8 + T effector memory CD45RA + (TEMRA), CD4 + TEM cells, and CD4 + TCM were decreased in a majority of patients at C2D1, as expected post-antigen exposure.
Fig. 2.

Flow Cytometry of Patient-derived Peripheral Blood Samples. Panel a: shows C0D2 compared to baseline after a single lead-in dose of LY3300054 alone (Cohort A), prexasertib alone (Cohort B), or the combination of LY3300054 and prexasertib (Cohort C). Panel b: shows data for the corresponding samples from C2D1. Data are normalized to baseline pre-treatment samples and expressed as fold changes
Cytokine analysis revealed similar patterns of immune activation with increase in IL-2 and IL-7 (Fig. 3) consistent with previously published results [20] and supporting the patterns of T cell subset proliferation seen in this study. IFNγ levels were below the limits of detection and could not be interpreted. Profiles were compared between patients who derived clinical benefit and remained on study for at least 100 days (shown in red), and patients who did not derive clinical benefit remaining on study for less than 100 days due to disease progression (shown in blue). Levels of IL-6, IL-7, and IL-8 tended to be overall higher in patients who achieved clinical benefit compared to those who did not, although levels did not reach statistical significance.
Fig. 3.

Cytokine analysis of Patient-derived Plasma Samples. Shown are Luminex soluble analyte fold change of C0D2 and C2D1 samples normalized to each patient baseline pre-treatment sample. Clinical benefit (red) denotes patients who remained on study ≥ 100 days, and no clinical benefit (blue) denotes patients who remained on study < 100 days
Peripheral blood samples could not be compared to tumor biopsies in the two patients enrolled to the dose expansion phase of the study. For one patient, biopsy was considered a high-risk, and the procedure was aborted; for the second patient, the on-treatment biopsy contained no tumor due to robust response two weeks into treatment and accordingly could not be compared to the pre-treatment biopsy.
Antitumor activity in CCNE1-amplified high-grade serous ovarian cancers
Of the 17 patients enrolled, 14 had recurrent ovarian cancer. Histologies included clear cell (n = 2), low-grade serous (n = 2), and high-grade serous (n = 10). Eight patients remained on study for at least 4 cycles (≥ 100 days), 3 patients remained on study for at least 12 months. Notable responses occurred in CCNE1-amplified high-grade serous ovarian cancers, including the three patients who achieved a PR (Fig. 1). Immune subset analysis for these patients showed significant interpatient variability, though patterns of T-cell proliferation including increases in CD71 + CD8 + T cells and NK cells were seen in the majority of patients (Fig. 4). Notable T-cell activation response was seen in Patient #10 who achieved a durable PR on study lasting past 12 cycles. Lymphocyte lineage analysis shows expansion of naïve CD4 + and CD8 + memory cells at C2D1 (Fig. 5a and b). Additionally, this patient had significant elevation of IL-2 on cytokine profiling (Fig. 5c). Sample CT images of ongoing PR after 12 cycles is shown in Fig. 5d.
Fig. 4.
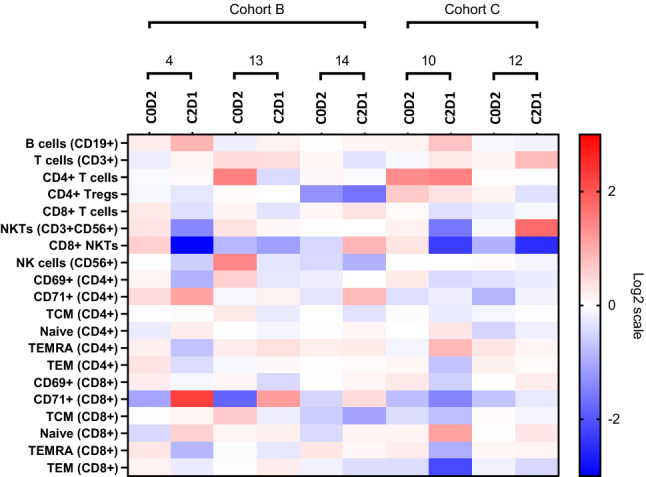
Immune Subset Analysis in CCNE1 Ovarian Cancer Patients. Comparison of immune subsets for the five CCNE1 ovarian cancer patients enrolled to the dose optimization phase of the study. Four of 5 patients derived clinical benefit and remained on study for > 100 days; patient #10 and 13 achieved a PR. One additional CCNE1 ovarian cancer patient was enrolled to the dose expansion phase of the study also achieved a PR, data not shown. Notable signatures include decrease in CD4 + Tregs and CD8 + NKT cells and increase in CD71 + CD8 + cells at C2D1 compared to C0D2 in 4/5 patients
Fig. 5.

Clinical Response and Corresponding Immune Subset and Cytokine Analysis in Patient #10: CCNE1-amplified ovarian cancer. Panel a: Lymphocyte lineage population for Patient #10 based on percentage of parent population (viable cells). Panel b: Shown are subset of CD8 + and CD4 + T memory cells as a function of CD3 + CD4 + and CD3 + CD8 + parent population. Panel c: Heatmap of C2D1 cytokine analytes normalized to baseline samples for this patient compared to patient 13 who also achieved a PR and remained on study for 20 cycles. Panel d: Shown are sample CT images demonstrating durable PR at 12 cycles
Discussion
In this study, we report the immune modulating effect of CHK1 inhibition with prexasertib in combination with anti-PDL1 antibody LY3300054. We additionally show prexasertib and LY3300054 can be given safely in combination at the RP2D of the individual agents and report preliminary antitumor activity in CCNE1-amplified HGSOC.
The design of this study, incorporating a lead-in dose, allowed for pharmacodynamic assessment of peripheral T-cell activation and cytokine signatures after a single dose of prexasertib (Cohort B) compared to the combination (Cohort C; Fig. 2). After a single lead-in dose of prexasertib, circulating CD4 + T regulatory cells (Tregs) and CD8 + NKT cells decreased compared to baseline in 5/6 patients in Cohort B. Although leukopenia has previously been reported with prexasertib [18], decrease in counts typically occur 5–8 days after drug exposure. As samples were obtained on C0D2, within 24 h after dosing, decrease in Tregs and NKT cells are thought to reflect direct immune modulating activity of CHK1 inhibition on T cells rather than an artifact of lymphodepletion as previously reported [21]. Additionally, as both Tregs and NKT cell have been shown to suppress immune surveillance [22], a decrease in both of these cell lines after a single dose of prexasertib in the absence of immune checkpoint blocking antibody further supports a regulatory role for CHK1 in immunity. NK cells were also increased in 3/6 patients in Cohort B, supporting early activation of the innate immune response in response to CHK1 inhibition. At C2D1, after the combination of prexasertib and LY3300054 had been administered, an increase in frequency of CD71 + CD8 + T cells was seen in 4/6 patients in cohort B and in 3/6 patients in cohort C, indicating that the majority of patients had expansion of an activated CD8 + T cell subset in response to treatment.
The significance of decreases in the frequency of T cell memory subsets at C2D1 remains unclear as changes neither correlated with response nor non-response. It is conceivable that this earlier timepoint may fall between post-antigen exposure and pre-proliferation and is in line with published kinetics of T-cell activation [10]. Cytokine analysis confirmed the changes seen in T-cell activation signatures. Soluble IL7 increased during treatment in 9/15 patients at C2D1 (Fig. 3). This cytokine has been shown to induce B and CD8 + T cell proliferation [23]. Levels of IL-17 also increased compared to baseline, though this is difficult to interpret in the setting of G-CSF administration which was required to support neutropenia in 14/17 patients and did not correlate with response. Subset analyses of the two patients who achieved a PR lasting > 12 cycles (Patient #10 and #13) compared to two patients who experienced rapid clinical progression after 1 cycle (Patient #3 and #6) showed no consistent differences in T-cell activation signatures but did trend towards higher expression of CD69 + and CD71 + in the two patients who achieved a PR. Additionally, IL-2 was found to be significantly elevated in P#10, and is consistent with T-cell activation signatures in this patient (Fig. 5).
Interestingly, Patient #10 was the only patient to experience immune-related toxicities on study. This patient experienced grade 2 hypothyroidism after 10 cycles followed by grade 3 immune-related elevation in AST and ALT after 12 cycles which initially responded and improved to grade 1 with high-dose steroid administration but required initiation of mycophenolate for complete resolution. IL-2 has been implicated in the development of severe irAEs [24] and may be more a predictive biomarker of irAE rather than response in this setting.
In this study, clinical benefit was seen in 8/14 recurrent ovarian cancer patients. Three CCNE1-amplified HGSOC patients achieved a PR, a fourth CCNE1-amplified HGSOC maintained SD for more than 12 months. These responses are not thought to be attributed to LY3300054 alone as previous response rates of immune checkpoint blockade monotherapy in recurrent ovarian cancers have been modest, averaging 8–10% [25, 26]. Responses are also not attributed to prexasertib alone. In clinical studies of prexasertib monotherapy in recurrent high-grade serous ovarian cancers (HGSOC), approximately one-third of patients achieved a partial response and median duration of response was only 7.4 months [27]. Both the depth and duration of response seen in this study are attributed to additive activity of the combination of prexasertib and LY3300054.
Our findings of T-cell activation and cytokine signatures in response to treatment with prexasertib and LY3300054 support an immunomodulatory role for CHK1 inhibition. In contrast to previous reports where an increase in immunosuppressive Tregs were seen at C1D15 [21], we saw an early decrease in CD4 + Tregs and CD8 + NKT cells after a single dose of prexasertib at C0D2 which would suggest a more proximal immunoregulatory response with CHK1 inhibition. The discrepancy in Treg response may suggest a dynamic response to CHK1 inhibition that varies with time from exposure and may require the addition of immune checkpoint blockade to further reinforce T-cell activation signatures. Further correlation with patient-derived tumor biopsies is needed to confirm changes in tumor immunity in response to treatment with prexasertib with and without reinforcement with anti-PD-L1 antibody. The decision to halt further agent development and early study closure precluded our ability to collect additional tumor biopsies in a planned expansion cohort. Treatment of recurrent ovarian cancers presents a therapeutic challenge and combination immune-based therapies remain an unmet need. The encouraging responses seen in this study warrant further investigation for immune-based therapies in CCNE1 amplified ovarian cancers.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
We thank the participating patients and their families for their invaluable contributions.
Author contributions
All authors contributed to the study conception and design. Material preparation, data collection, and analysis were performed by KTD, CM, ET, AG-H, and MS. The first draft of the manuscript was written by KTD and CM, and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Funding
Funding for this study was partially supported by institutional grant from Eli Lilly.
Availability of data and material
All data generated or analyzed during this study are included in this article and supplementary information files.
Declarations
Conflict of interest
KTD has served as a consultant/advisory board member Seattle Genetics and QED Therapeutics; consulting fees from Jackson Laboratories; and has received commercial research grants (to institution) from Eli Lilly. CM is an employee of Moderna Therapeutics at the time of this submission. ET is an employee of Fluidigm at the time of this submission. GIS has received research funding from Eli Lilly, Merck KGaA/EMD-Serono, Merck, and Sierra Oncology. He has served on advisory boards for Pfizer, Eli Lilly, G1 Therapeutics, Roche, Merck KGaA/EMD-Serono, Sierra Oncology, Bicycle Therapeutics, Fusion Pharmaceuticals, Cybrexa Therapeutics, Astex, Almac, Ipsen, Bayer, Angiex, Daiichi Sankyo, Seattle Genetics, Boehringer Ingelheim, ImmunoMet, Asana, Artios, Atrin, and Concarlo Holdings. In addition, he holds a patent entitled, “Dosage regimen for sapacitabine and seliciclib,” also issued to Cyclacel Pharmaceuticals, and a pending patent, entitled, “Compositions and Methods for Predicting Response and Resistance to CDK4/6 Inhibition,” together with Liam Cornell. SK, AP, AA, JH, AG-H, and MS declare no relevant financial or non-financial interests to disclose.
Ethical approval
This study was conducted in accordance with the International Conference on Harmonization, Good Clinical Practice guidelines, and the ethical principles outlined in the Declaration of Helsinki 2008. The study protocol, any amendments, informed consent, and other information that required pre-approval were reviewed and approved by the Institutional Review Board, in accordance with the International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use Good Clinical Practice and applicable country-specific requirements, including US 21 Code of Federal Regulations 312.3(b) for constitution of independent ethics committees.
Consent for publication
All participants provided written informed consent prior to study entry.
Footnotes
The results of this study were presented at the Society for Immunotherapy of Cancer (SITC) 34th Annual Conference on November 6th-10th, 2019 at the Walter E. Washington Convention Center in Washington, D.C.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Khanh T. Do and Claire Manuszak contributed equally to this manuscript.
References
- 1.Dunn GP, et al. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002;3(11):991–998. doi: 10.1038/ni1102-991. [DOI] [PubMed] [Google Scholar]
- 2.Permata TBM, et al. Base excision repair regulates PD-L1 expression in cancer cells. Oncogene. 2019;38(23):4452–4466. doi: 10.1038/s41388-019-0733-6. [DOI] [PubMed] [Google Scholar]
- 3.Sato H, et al. DNA double-strand break repair pathway regulates PD-L1 expression in cancer cells. Nat Commun. 2017;8(1):1751. doi: 10.1038/s41467-017-01883-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sen T, et al. Targeting DNA damage response promotes antitumor immunity through STING-Mediated T-cell activation in small cell lung cancer. CancerDiscov. 2019;9(5):646–661. doi: 10.1158/2159-8290.CD-18-1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Parkes EE, et al. Activation of STING-dependent innate immune signaling by S-phase-specific DNA damage in breast cancer. J Natl Cancer Inst. 2017;109(1):199–208. doi: 10.1093/jnci/djw199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Harding SM, et al. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature. 2017;548(7668):466–470. doi: 10.1038/nature23470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mackenzie KJ, et al. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature. 2017;548(7668):461–465. doi: 10.1038/nature23449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu J, et al. Plasma cells from multiple myeloma patients express B7–H1 (PD-L1) and increase expression after stimulation with IFN-{gamma} and TLR ligands via a MyD88-, TRAF6-, and MEK-dependent pathway. Blood. 2007;110(1):296–304. doi: 10.1182/blood-2006-10-051482. [DOI] [PubMed] [Google Scholar]
- 9.Eisenhauer EA, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1) Eur J Cancer. 2009;45(2):228–247. doi: 10.1016/j.ejca.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 10.Ho LP, et al. The road to memory: an early rest for the long journey. J Immunol. 2013;191(11):5603–5614. doi: 10.4049/jimmunol.1301175. [DOI] [PubMed] [Google Scholar]
- 11.Manuszak C, et al. Standardized 11-color flow cytometry panel for the functional phenotyping of human T regulatory cells. J Biol Methods. 2020;7(2):e131. doi: 10.14440/jbm.2020.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Patel T, et al. Development of an 8-color antibody panel for functional phenotyping of human CD8+ cytotoxic T cells from peripheral blood mononuclear cells. Cytotechnology. 2018;70(1):1–11. doi: 10.1007/s10616-017-0106-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cunningham RA, et al. Detection of clinically relevant immune checkpoint markers by multicolor flow cytometry. J Biol Methods. 2019;6(2):e114. doi: 10.14440/jbm.2019.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Holland M, et al. Separation, banking, and quality control of peripheral blood mononuclear cells from whole blood of melanoma patients. Cell Tissue Bank. 2018;19(4):783–790. doi: 10.1007/s10561-018-9734-x. [DOI] [PubMed] [Google Scholar]
- 15.Sridharan V, et al. Effects of definitive chemoradiation on circulating immunologic angiogenic cytokines in head and neck cancer patients. J Immunother Cancer. 2016;4:32. doi: 10.1186/s40425-016-0138-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schoenfeld JD, et al. Pneumonitis resulting from radiation and immune checkpoint blockade illustrates characteristic clinical, radiologic and circulating biomarker features. J Immunother Cancer. 2019;7(1):112. doi: 10.1186/s40425-019-0583-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Davids MS, et al. Ipilimumab for patients with relapse after allogeneic transplantation. N Engl J Med. 2016;375(2):143–153. doi: 10.1056/NEJMoa1601202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hong D, et al. Phase I study of LY2606368, a checkpoint kinase 1 inhibitor, in patients with advanced cancer. J ClinOncol. 2016;34(15):1764–1771. doi: 10.1200/JCO.2015.64.5788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee JM, et al. Prexasertib, a cell cycle checkpoint kinase 1 and 2 inhibitor, in BRCA wild-type recurrent high-grade serous ovarian cancer: a first-in-class proof-of-concept phase 2 study. Lancet Oncol. 2018;19(2):207–215. doi: 10.1016/S1470-2045(18)30009-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li Y, et al. Discovery and preclinical characterization of the antagonist anti-PD-L1 monoclonal antibody LY3300054. J Immunother Cancer. 2018;6(1):31. doi: 10.1186/s40425-018-0329-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lampert EJ, et al. Clinical outcomes of prexasertib monotherapy in recurrent BRCA wild-type high-grade serous ovarian cancer involve innate and adaptive immune responses. J Immunother Cancer. 2020;8(2):e000516. doi: 10.1136/jitc-2019-000516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Terabe M, Berzofsky JA. Tissue-specific roles of NKT cells in tumor immunity. Front Immunol. 2018;9:1838. doi: 10.3389/fimmu.2018.01838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rosenberg SA, et al. IL-7 administration to humans leads to expansion of CD8+ and CD4+ cells but a relative decrease of CD4+ T-regulatory cells. J Immunother. 2006;29(3):313–319. doi: 10.1097/01.cji.0000210386.55951.c2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lim SY, et al. Circulating cytokines predict immune-related toxicity in melanoma patients receiving anti-PD-1-based immunotherapy. Clin Cancer Res. 2019;25(5):1557–1563. doi: 10.1158/1078-0432.CCR-18-2795. [DOI] [PubMed] [Google Scholar]
- 25.Disis ML, et al. Avelumab (MSB0010718C), an anti-PD-L1 antibody, in patients with previously treated, recurrent or refractory ovarian cancer: a phase Ib, open-label expansion trial. J ClinOncol. 2015;33:5509. doi: 10.1200/jco.2015.33.15_suppl.5509. [DOI] [Google Scholar]
- 26.Matulonis UA, et al. Antitumor activity and safety of pembrolizumab in patients with advanced recurrent ovarian cancer: results from the phase II KEYNOTE-100 study. Ann Oncol. 2019;30(7):1080–1087. doi: 10.1093/annonc/mdz135. [DOI] [PubMed] [Google Scholar]
- 27.Lee JM, et al. A phase II study of the cell cycle checkpoint kinases 1 and 2 inhibitor (LY2606368; prexasertibmonomesylate monohydrate) in sporadic high-grade serous ovarian cancer (HGSOC) and germline BRCA mutation-associated ovarian cancer (gBRCAm+ OvCa) Ann Oncol. 2016;27(Suppl 6):296. doi: 10.1093/annonc/mdw374.02. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed during this study are included in this article and supplementary information files.