

Summary
Mitochondria account for essential cellular pathways, from ATP production to nucleotide metabolism, and their deficits lead to neurological disorders and contribute to the onset of age-related diseases. Direct neuronal reprogramming aims at replacing neurons lost in such conditions, but very little is known about the impact of mitochondrial dysfunction on the direct reprogramming of human cells. Here, we explore the effects of mitochondrial dysfunction on the neuronal reprogramming of induced pluripotent stem cell (iPSC)-derived astrocytes carrying mutations in the NDUFS4 gene, important for Complex I and associated with Leigh syndrome. This led to the identification of the unfolded protein response as a major hurdle in the direct neuronal conversion of not only astrocytes and fibroblasts from patients but also control human astrocytes and fibroblasts. Its transient inhibition potently improves reprogramming by influencing the mitochondria-endoplasmic-reticulum-stress-mediated pathways. Taken together, disease modeling using patient cells unraveled novel general hurdles and ways to overcome these in human astrocyte-to-neuron reprogramming.
Keywords: direct neuronal reprogramming, Leigh syndrome, mitochondria, astrocytes, unfolded protein response
Graphical abstract
Highlights
-
•
NDUFS4-mutant patient astrocytes are impaired in reprogramming into neurons
-
•
Unfolded protein response inhibition improves neuronal reprogramming
-
•
Patient iNs are affected in neuronal subtype specification
-
•
Transient unfolded protein inhibition leads to more functional iNs
Sonsalla, Malpartida, et al. investigated the direct neuronal reprogramming of human astrocytes and fibroblasts carrying mutations in NDUFS4. This led to the discovery of the unfolded protein response as a major hurdle whose inhibition improved the conversion rate, leading to more mature neurons.
Introduction
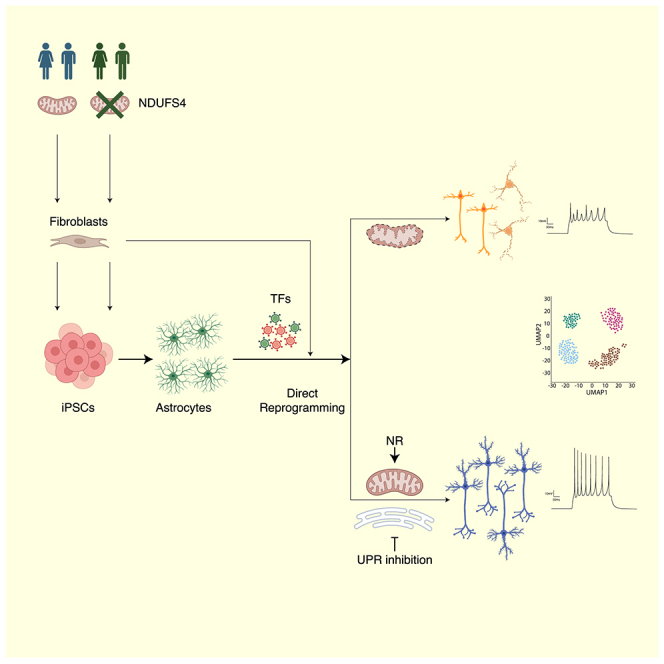
A hallmark of traumatic brain injury and neurodegenerative diseases is the irreversible loss of neurons, leading to cognitive and functional deficits.1 Neurodegeneration is often associated with mitochondrial dysfunction,2,3 causing mitochondrial stress and excessive reactive oxygen species (ROS).2,3 These negative effects for pre-existing neurons also hamper direct neuronal conversion from glial cells.4,5 Astrocytes and neurons differ significantly in their mitochondrial proteome5,6 and the early expression of neuron-enriched mitochondrial proteins improves the induction of neurons.5 However, it is unknown how cells from patients with mitochondrial deficiencies may respond to direct neuronal reprogramming. To model this, we used proliferating astrocytes (pAstros)7 derived from human-induced pluripotent stem cell (iPSC) carrying mutations in nicotinamide adenine dinucleotide (NADH):ubiquinone oxidoreductase subunit S4 (hereafter, NDUFS4), an accessory protein of Complex I (C-I) associated with Leigh syndrome (LS) and mitochondrial C-I deficiency, causing excessive ROS levels.8,9,10,11,12,13 NDUFS4-pAstros from 3 different patients consistently showed deficits in direct neuronal conversion compared with control pAstros and, thus, provided a suitable platform for testing pharmacological treatments aimed at overcoming mitochondria-dependent roadblocks. Interestingly, treatments successful in murine astrocyte-to-neuron reprogramming, e.g., ROS scavengers or Bcl2 co-transduction, were less efficient in human astrocyte-to-neuron conversion. Remarkably, we discovered the unfolded protein response (UPR) and integrated stress response (ISR) as novel, common, and major hurdles in direct neuronal conversion: blocking these pathways allowed high rates of conversion reaching more than 70% success also for patient-derived astrocytes.
Results
Control and NDUFS4-patient iPSCs differentiate equally well into astrocytes
First, we obtained iPSCs from fibroblasts of control donors (hereafter “control”) or from patients with different mutations in NDUFS4 (hereafter “patient”) (Table S1), but all not expressing detectable levels of the protein (Figure S1A). iPSCs were differentiated into fibroblast growth factor (FGF)/epidermal growth factor (EGF)-treated pAstros and leukemia inhibitory factor (LIF)-treated non-pAstros (Figure S1B).7 Immunocytochemistry for the astrocyte proteins S100B and FGFR3 showed no detectable differences between control and patient cells at the differentiation stages analyzed (Figure S1C). Bulk RNA sequencing (RNA-seq) analysis14 of 3 control and 3 patient lines revealed a clear separation between iPSCs and differentiated samples (Figure S1D), with non-pAstro showing lower G1/S and G2 scores compared with iPSCs and pAstros (Figure S1E).15 Remarkably, control and patient samples were intermingled at any given stage (Figures S1D and S1E), also for the unsupervised clustering based on the expression of iPSC16 and astrocyte markers17 (Figure S1F). Pairwise comparison of control and patient cells revealed an overall relatively small subset of transcripts differentially expressed at specific stages (Figures S1G–S1I; Table S2).
Glutamate uptake assay was similar between control and patient pAstros or non-pAstros, both of which took up more glutamate than iPSCs (Figure S1J). Likewise, control and patient pAstros (Figure S1K) and non-pAstros (Figure S1L) had similar oxygen consumption rate (OCR) at resting basal condition, though the basal respiration was higher in pAstros than non-pAstros (Figures S1K and S1L). Applying inhibitors of the electron transport chain (ETC) revealed higher maximal respiration and spare respiratory capacity in control pAstros compared with patient pAstros (Figure S1K), but such difference was not detectable in non-pAstros (Figure S1L), suggesting a different metabolic demand between pAstros and non-pAstros. Together, these data revealed high similarity of control and patient iPSCs during astrocyte differentiation.
NDUFS4 mutations impair direct neuronal reprogramming of pAstros
pAstros were chosen as target cell for reprogramming because NDUFS4-patient pAstros showed functional defects and astrocytes in vivo proliferate following injury18,19 including in human patient brains with hemorrhagic injuries20 (Figure 1A). Control and patient pAstros were transduced with a retrovirus encoding either the red fluorescent protein dsRed alone, as a control, or together with the proneural transcription factors (TFs) Neurogenin-2 (Neurog2/Ngn2), Achaete-scute homolog 1 (Ascl1)4,5,21,22 or their phosphorylation-deficient versions (referred to as pMutNgn2 and pMutAscl1), because of their enhanced neurogenic activity in vivo.23,24,25 Although dsRed-transduced control or patient pAstros did not generate neuron-like cells at 20 days post transduction (20 DPT; Figures 1B and 1D), control pAstros were more efficiently reprogrammed than patient pAstros upon the forced expression of Ngn2 or pMutNgn2 (Figure 1D), consistently across different lines. Remarkably, pMutNgn2 was more efficient than wild-type Ngn2 in control but not in patient cells. The forced expression of Ascl1 or pMutAscl1 led to cell death in both genotypes (Figures 1B and 1D).
Figure 1.

NDUFS4-patient pAstros exhibit impaired neuronal reprogramming
(A) Scheme of direct neuronal reprogramming.
(B and C) Micrographs depicting control (HMGU-1) and patient (#87971) pAstros transduced with the indicated transcription factors (B, single factor; C, two factors) at 20 DPT. Scale bars, 50 μm.
(D and E) Boxplots of reprogramming efficiency of control and patient pAstros with different factors (D, single factor; E, two factors). Efficiency is defined as the ratio of βIII-tubulin+/dsRed+ to dsRed+ cells. Data are shown as median ± IQR. ∗p ≤ 0.05, ∗∗p ≤ 0.01. n = 3–4 independent culture batches per line, 2 lines per control, and 3 lines per patient.
To independently evaluate the impact of mitochondrial dysfunction in direct reprogramming, we transiently treated at 2 DPT dsRed- or Ngn2-transduced control and patient pAstros with OligomycinA, an ATP synthase inhibitor,26 as previously done in mouse astrocyte-to-neuron direct conversion4: almost no induced neuronal cells could be found in both genotypes (Figures S2A and S2B), similar to murine astrocytes,4 thus suggesting that the activity of ATP synthase is essential for the conversion process. Moreover, the transient treatment with the ROS scavenger α-tocopherol,4 also at 2 DPT, neither rescued nor improved the conversion of patient or control pAstros (Figure S2B), suggesting differences in ROS production (either levels or timing) or in scavenging capability between human and murine astrocytes.27 We also tested Bcl2 co-expression that had improved murine astrocyte-to-neuron reprogramming,4 but it had no significant effect in control or patient pAstros compared with Ngn2 or Ascl1 alone (Figures 1C and 1E).
Together, these results suggested that defects in C-I significantly impair the direct neuronal reprogramming of human pAstros and treatments efficient for murine astrocyte-to-neuron reprogramming were not sufficient in the context of human astrocyte direct conversion.
Genetic rescue of C-I function partially rescues the reprogramming deficits of NDUFS4-patient pAstros
To genetically rescue C-I function, we co-expressed atNDB4, an Arabidopsis thaliana NADH dehydrogenase (Figure S2A), in patient pAstros capable of rescuing C-I deficiency in NDUFS4-patient fibroblasts.28 This improved the reprogramming in patient pAstros, either in combination with Ngn2 or pMutNgn2 (Figure S2C) or together with Bcl2 (Figure S2D), but not when co-expressed with Ascl1 (Figures S2C and S2D). However, the rescue of the conversion was only partial, suggesting that other mitochondrial functions are defective during the process. Therefore, we explored other aspects of metabolic stress by targeting different branches of mitochondrial dysfunction.29
Pharmacological treatments toward improving the reprogramming deficits of NDUFS4-patient pAstros
In particular, we transiently treated control and patient pAstros undergoing Ngn2-mediated reprogramming (Figure 2A) with small molecules modulating various aspects of mitochondria function.29 NAD+ precursor nicotinamide riboside (NR) was used to increase NAD+ levels and restore NAD+/NADH balance, disrupted in NDUFS4-patient patients30: remarkably, NR significantly improved reprogramming of both control and patient cells (Figures 2B and 2C), suggesting NAD+ as a limiting factor in pAstros-to-neuron conversion. The treatment with idebenone, a synthetic analog of coenzyme Q1031 acting as antioxidant and improving the transport of electrons in the ETC,32 led to a mild increase in neurons induced from control pAstros (Figures 2B and 2C). Urolithin A (UA), a mitophagy activator used to increase mitochondria turnover,33 had no detectable effect (Figures 2B and 2C).
Figure 2.

Pharmacological treatments improve direct neuronal reprogramming of pAstros
(A) Scheme of direct neuronal reprogramming and the site of action of small molecules. Images adapted from Biorender.com.
(B) Micrographs of control (HMGU-1) and patient (#87971) pAstros transduced with Ngn2 alone (untreated, UT) or in combination with AMG-PERK 44 (AMG), STF-083010 (STF), idebenone, nicotinamide riboside (NR), or urolithin A (UA) treatment at 20 DPT. Scale bars, 50 μm.
(C and D) Boxplots of reprogramming efficiency (C) and neurite length (D) of control and patient pAstros treated with the indicated small molecules. Asterisks indicate statistical significance of the conversion between untreated and small molecules samples within the same genotype; asterisks above bar indicate the significance of the reprogramming between control and patient pAstros within the same treatment. Data are shown as median ± IQR. ∗p ≤ 0.05, ∗∗∗p ≤ 0.001. n = 3 independent culture batches per line; 2 lines per controls (HMGU-1 and HMGU-12) and 3 lines per patient (#79787, #87971, #114107).
As dysfunctional mitochondria have been found to induce stress responses in the endoplasmic reticulum (ER),34,35,36,37 eliciting the UPR and slowing protein translation,38 we investigated the role of ER stress during direct pAstro-to-neuron reprogramming. Specifically, we interfered with the inositol-requiring enzyme 1 (IRE1) branch, which induces the expression of protein folding chaperones by regulating the splicing of X-box binding protein 1 (XBP1) mRNA39,40 (Figure 2A), and the protein kinase R (PKR)-like ER kinase (PERK) branch, which recognizes the accumulation of misfolded proteins and reduces protein synthesis38 by phosphorylating the eukaryotic translation initiation factor 2 alpha (EIF2α) (Figure 2A). Strikingly, inhibiting either pathway with small molecules (STF-083010 [STF] for IRE1 and AMG-PERK 44 [AMG] for PERK) significantly improved the reprogramming from both control and patient pAstros (Figures 2B and 2C). UPR inhibition led to the generation of neuronal cells with longer processes, with AMG having the strongest impact on the proportion of induced neurons (iNs; Figure 2C) and neurite length (Figure 2D). To validate the impact of PERK inhibition, we used GSK2606414 (GSK), a very specific PERK inhibitor41,42: indeed, it improved Ngn2-induced conversion efficiency in both genotypes, although with higher variability in patient pAstros (Figures S2E and S2F). AMG and STF co-treatment did not improve the generation of neuronal cells (Figures S2E and S2F). We further explored if UPR activation is detrimental to direct neuronal reprogramming by treating dsRed or Ngn2-transduced pAstros with tunicamycin (TM), a known UPR inducer,42 for 16 h: even if transiently treated, all Ngn2-transduced cells died within few days (data not shown).
Together, these results revealed ER stress as a major hurdle in the direct neuronal reprogramming of pAstros, as its manipulation greatly increased the conversion into neurons from both control and patient cells.
Induction of UPR during direct neuronal conversion and aggresome formation
Given the above effects of UPR inhibition, we first investigated the responsiveness of pAstros to ER stress and then explored UPR induction during direct reprogramming. Following TM treatment for 16 h (Figure S3A), control and patient pAstros showed an increased expression of downstream targets for the 3 UPR branches (e.g., DDIT3/CHOP, HSPA5/BIP, MANF, and HYOU1; Figures S3B and S3C). Overall, UPR downstream effectors were similarly expressed in homeostatic conditions in control and patient cells (Figure S3D). However, control Ngn2-transduced pAstros showed a more pronounced expression of some UPR targets at 5 DPT (e.g., CHOP, MANF, and HYOU1; Figure 3B) compared with patient Ngn2-transduced cells (Figure 3B). This may be a genuinely higher response in control cells or relate to different timing of this response in patient cells.
Figure 3.

UPR activation and proteostasis during direct neuronal reprogramming of pAstros
(A) Experimental design.
(B) Boxplots depicting the expression of UPR targets in control (left) and patient (right) pAstros at 5 DPT as measured by RT-qPCR. Data are shown as the fold-change relative to dsRed and normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Data are shown as median ± IQR. Each dot represents an independent biological replicate. n = 3–4 independent experiments.
(C) Micrographs of control and patient pAstros transduced with ATF4-YFP sensor and dsRed or Ngn2-dsRed at 3 DPT (left) and 7 DPT (right). Scale bars, 50 μm.
(D) Boxplots depicting mean fluorescent intensity of ATF4-YFP sensor at 3 DPT (left) and 7 DPT (right) in control and patient pAstros. Data are shown as median median ± IQR. n = 3–5 independent batch cultures (2 lines per control and 3 lines per patient). ∗∗p ≤ 0.01.
(E) Micrographs of control and patient pAstros transduced with BFP or Ngn2-BFP with aggresomes labeled with PROTEOSTAT dye at 20 DPT. Scale bars, 50 μm.
(F) Boxplots depicting aggresome detection in control and patient pAstros in different experimental conditions at 20 DPT. Cells are subdivided into Ngn2-A (astrocyte-like) and Ngn2-N (neuronal). Data are shown as median ± IQR. ∗p ≤ 0.05; ∗∗p ≤ 0.01. n = 3 independent biological replicates per line.
To observe the activation of UPR branches during direct neuronal conversion at single-cell level, we employed genetically encoded sensors for ATF4-PERK pathway activation43 or ATF6 pathway.44 Both sensors responded to UPR induction by TM in control and patient pAstros (Figures S3E–S3G): interestingly, ATF6-YFP trended toward a higher intensity in patients compared with control pAstros and its induction following TM treatment was more pronounced than the PERK sensor, though some variability was observed across patient lines (e.g., line #79787) (Figure S3G). When expressed together with dsRed or Ngn2 in control and patient pAstros (3 and 7 DPT, Figure 3A), the PERK sensor showed a trend to higher expression in dsRed-transduced patient pAstros than controls at 3 DPT (Figures 3C and 3D), though this was also highly variable across lines. In Ngn2-transduced control pAstros, PERK sensor was significantly higher compared with dsRed-transduced control cells as well as Ngn2-transduced patient pAstros, which were very similar to their dsRed-positive controls (Figure 3D). At 7 DPT, PERK sensor was similarly low in all conditions tested (Figure 3D), suggesting a dynamic ER-related stress response mostly during early stages of reprogramming. Conversely, no significant difference could be detected using the ATF6 sensor at 3 DPT, both in control and patient pAstros, and not differently induced in dsRed- or Ngn2-expressing pAstros (Figures S3H and S3I), supporting the low induction of ATF6 targets at 5 DPT (Figure 3B). At 7 DPT, however, Ngn2-expressing patient cells showed a trend to higher expression of the ATF6 reporter, suggesting a delayed induction of ATF6-dependent UPR in patient cells (Figure S3I).
As the UPR is induced upon the accumulation of misfolded proteins, we examined misfolded proteins during reprogramming using a dye that intercalates in misfolded proteins turning on red fluorescence45 (Figure 3A). Control and patient pAstros expressing blue fluorescent protein (BFP) alone or with Ngn2, either treated or not with small molecules, were examined at 5 DPT and 20 DPT. Generally, at 5 DPT aggresome accumulation was relatively minor and not significant (Figures S3J and S3K), except for an STF treatment in dsRed-transduced control pAstros and NR treatment, which led to an overall decrease in aggresomes (Figure S3K). At 20 DPT, Ngn2-transduced cells were separated in converted (Ngn2-N [neurons]; yellow dashed line; Figure 3E) and not converted (Ngn2-A [astrocytes]; white dashed line; Figure 3E). Interestingly, aggresome levels were significantly higher in Ngn2-N, compared with BFP and non-converted Ngn2-A cells, irrespective of genotype (Figure 3F). Thus, neuronal conversion indeed increased the aggresome load significantly. However, UPR inhibitors, except STF (Figure 3F), did not lead to a further significant increase in aggresomes in iNs, and NR treatment even reduced aggresome intensity close to the untreated condition (Figure 3F).
Together, the transient treatment with UPR inhibitors potently improves reprogramming with only mild effects, if any, on aggresome formation. However, iNs generally showed an increase in misfolded proteins, which led us to explore induced neurons further at the molecular and physiological level.
Single-cell RNA-seq reveals different dynamics and outcome of reprogramming in control and NDUFS4-patient pAstros
To assess the quality of iNs, we performed single-cell RNA sequencing (scRNA-seq) of fluorescence-activated cell sorting (FACS)-sorted control (HMGU-1) and patient (#87971) pAstros, transduced with either dsRed or Ngn2 in the presence or absence of AMG, as the most effective treatment in improving direct neuronal conversion of pAstros, at 2 time points (5 and 20 DPT) (Figure 4A). Although equal cell numbers per condition were loaded, some conditions were less represented (Figure S4A), probably due to loss during sample preparation. Uniform manifold approximation and projection (UMAP) of the 2 integrated datasets (8,079 cells at 5 DPT and 10,710 cells at 20 DPT), followed by cluster analysis (resolution 0.5), identified 20 clusters (0–19; Figure 4B), with cells well intermingled mainly by the expression profile and not by a priori defined features (genotypes, factors, treatments and time of analysis) (Figures S4C–S4F). Scores for astrocytes, neurons, and vascular-leptomeningeal cells (VLMCs, which appear in iPSC-to-neuron direct differentiation protocol at late stages46; Figure S4B) revealed their specific distribution in distinct UMAP clusters (Figure 4B). Cluster A, characterized by dsRed-transduced cells from both genotypes and DPT (Figures S4G–S4K), had the highest score for astrocyte markers (astrocyte_score, Figure 4C); conversely, cluster D had the highest score for neuronal markers (neuron_score, Figure 4D) and was mainly composed of Ngn2-transduced cells at 20 DPT of both controls and patients (Figures S4C–S4F). Interestingly, clusters B and D were positive for both astrocyte_score and neuron_score (Figures 4C, 4D, S4B, and S4G–S4K), with cluster D more enriched for Ngn2-expressing cells at 20 DPT (Figures S4C–S4F). Thus, cluster A contained dsRed- and non-reprogrammed pAstros, cluster D contained mainly cells with neuronal features, whereas clusters B and C represent 2 intermediate states. Of note, a small subset of cells positive for the proliferation marker KI67 was found within cluster A (Figure 4F), suggesting that most cells do not proliferate at later stages. Quantitative RT-PCR (RT-qPCR) on selected candidates (e.g., HES6, MAP2, and ASS1; Figure S4G) confirmed scRNA-seq results (Figure S4H).
Figure 4.

scRNA-seq analysis of pAstros undergoing Ngn2-mediated direct conversion
(A) Experimental design.
(B) UMAP projection of cells colored by clusters, after combining scRNA-seq of control (HMGU-1) and patient (#87971) cells transduced with dsRed or Ngn2, untreated or treated with AMG, and collected at 5 or 20 DPT. Pie charts show the proportion of control (orange) and patient (blue) within each subcluster.
(C–F) UMAP projection of cells colored by the Astrocyte_score (C), Neuron-score (D), VLMC_score (E), and MKI67 (F).
(G) Venn diagram depicting genes unique to each cluster, common to each cluster comparison, and the top 5 GO terms associated.
(H–K) UMAP projections of cells colored for Glycolysis_score (H), FAO_score (I), ATP5_score (J), and FerroptosisMarker_score (K).
(L and M) UMAP projection of cells within the main cluster D (L) and proportion of cluster composition based on the genotype (M).
(N–Q) GO analysis on biological processes (BP) and molecular function (MF) of genes upregulated (log2FC > 1, padj = 0.01) in cluster 5 compared with cluster 3 (N and O) and vice versa (P and Q).
(R and S) UMAP projection of cells colored by glutamatergic score (R) or GABAergic score (S).
Remarkably, all main clusters contained AMG-treated cells (Figure S4F), but several genes associated to UPR were upregulated following Ngn2 expression (Figures S4I–S4N), supporting RT-qPCR analysis at 5 DPT (Figure 3B). ATF4, a major downstream effector of the PERK pathway,42 was upregulated in Ngn2-expressing cells and further boosted upon AMG treatment, especially at 20 DPT (Figures S4O and S4P). Notably, the concentration of AMG used also induces the activation of the ISR,47 a complex pro-survival signaling pathway induced by extrinsic (e.g., glucose deprivation) and intrinsic stimuli (e.g., unfolded proteins and mitochondrial dysfunction).48,49 Interestingly, EIF2AK4, coding for GCN2 kinase responsible for phosphorylating EIF2α, and known ISR effectors, were more expressed in specific clusters (e.g., ATG5 and ATG7 in clusters C and D; MAP1LC3B in clusters B and D; Figures S4O–S4Q), suggesting different responses to ISR across clusters. Therefore, scRNA-seq analysis indicated that the ISR pathway is also activated during direct neuronal reprogramming.
Metabolic and lipid-peroxidation signature of partially reprogrammed clusters
Next, we evaluated the genes differentially regulated between reprogramming clusters (B, C, and D) over cluster A, containing the starter cells, followed by searching for common and cluster-specific signatures (Figure 4G; Table S3). Such analysis led to identify 34 genes commonly upregulated in clusters B, C, and D (Figure 4G), which are associated to cytoskeleton organization and neuronal morphogenesis (Figure 4G; Table S4); cluster B specifically expressed genes associated to neuronal fate and endothelial development (Figure 4G; Table S5); cluster C was enriched for genes related to extracellular matrix (ECM) organization and development of other structures (e.g., vasculature, Figure 4G; Table S6); and cluster D contained genes relevant for neuronal maturation and functions (Figure 4K; Table S7). This analysis further supports the conclusion that cluster C largely consists of cells failing to reprogram, lacking differential expression of many neurogenic signature genes. Interestingly, cluster B expressed genes of endothelial cells, which was not previously observed in neuronal reprogramming.
Given the metabolic deficits in patient pAstros (Figure S1K) and the fact that the metabolic shift is required for the conversion of mouse astrocytes to neurons,4 we analyzed the expression of genes involved in glycolysis (Figure S4R) and fatty acid oxidation (FAO, Figure S4S). Indeed, glycolysis and FAO scores were enriched in cluster A (dsRed-enriched cluster; Figures 4H and 4I), indicating that cells in all other clusters undergo metabolic conversion to some extent. Conversely, cluster C was enriched for genes associated to oxidative phosphorylation and genes coding for the subunits of the mitochondrial ATP synthase, highly expressed also in cluster D (Figures 4J and S4T; Table S8), suggesting a more complete metabolic shift than cluster B. We then checked ferroptosis in human context, as it is a major hurdle in mouse direct conversion.4 Ferroptosis_score50 was enriched in cluster C, i.e., one of the clusters of cells failing to reprogram (Figures 4K and S4U). This suggests that cluster C is blocked in the conversion process despite the downregulation of glycolysis and FAO related genes and upregulation of ATP-synthesis genes, possibly due to deficits in the induction of the protective machinery. Notably, both clusters B and C contain control and patient cells, although in partially separate subclusters (pie charts in Figure 4B), suggesting that these failed reprogramming states are not genotype specific. Cells in cluster D are clearly the most progressed, as they not only increased the expression of neuronal markers and genes for oxidative phosphorylation but also decreased the expression of astrocyte genes, FAO, and ferroptosis genes, and hence seem to have overcome the metabolic and lipid-peroxidation bottleneck in this process.
Similarities and differences in iNs
We further analyzed cluster D, which contained successfully iNs from both genotypes. Unsupervised clustering identified 3 clusters (Figure 4L) with cluster 3 composed almost exclusively of patient cells and cluster 5 of control cells (Figure 4M). The comparison between these two clusters identified 84 significantly different genes (log2FC > |1|, padj < 0.01); the 45 genes enriched in cluster 5 (Figure S4V; Table S9) contained 6 TFs important for neurogenesis (e.g., BCL11B, ONECUT2, bold genes in Figure S4V)51,52 and genes associated to synapse organization and transmission (biological processes [BPs]; Figure 4N; Table S10) and triphosphate-related functions (molecular functions [MFs]; Figure 4O; Table S11), suggesting the induction of a specific neuronal program. Among the 39 genes more highly expressed in cluster 3, only one TF was present (PBX3; Figure S4W),53 whereas the other genes were associated to translation, rRNA processing, and ECM organization (BP and MF; Figures 4P and 4Q; Tables S12 and S13). These data suggest that patient cells may need to enhance translation to cope with the demands of reprogramming.
All cells in cluster D had a comparable neuronal score and expressed similar levels of mature neuronal markers54 (Figure S4X). However, iNs from cluster 5 had a higher score for glutamatergic neurons (Figures 4R and S4Y), whereas iNs in cluster 3 also expressed GABAergic genes (Figures 4S, S4Z, and S4Z), suggesting a confused neuronal subtype identity. These data thus suggest that mitochondrial dysfunction could affect the neuronal subtype specification, a key result for future therapeutic approaches.
Functional improvement of neuronal activity upon AMG treatment
Given the expression differences between patient and control iNs, we investigated their electrophysiological properties. Ngn2-Bcl2 co-transduced pAstros from different donors (control or patients), either treated with AMG or not, were cultured on a layer of mouse astrocytes for 50–70 DPT (Figures 5A and 5B). AMG-treated cells showed a higher proportion of patient iN firing action potentials over untreated counterparts (Figures 5C–5F). Remarkably, only AMG-treated patient iNs had spontaneous action potentials (Figures 5G and 5H), demonstrating a striking long-term effect of AMG treatment on the functional properties of the patient iNs. Passive (e.g., input resistance; Figure 5I) and active properties (action potential threshold, duration, amplitude, rise-to-fall ratio; Figures 5J–5M) showed no differences between control and patient iNs, suggesting that the above observed gene expression differences do not manifest in electrophysiology readouts, at least under these in vitro conditions. Overall, electrophysiological data revealed that neurons reprogrammed from human control pAstros are indeed functional, whereas those reprogrammed from patient pAstros reached this state only following AMG treatment.
Figure 5.

Electrophysiological analysis of control or NDUFS-4 patient iNs upon AMG treatment
(A) Scheme for electrophysiological analysis of iNs.
(B) Micrographs depicting the morphology of iNs before patch-clamp.
(C and D) Representative traces of action potential upon increasing step depolarization in control (left) or patient (right) reprogrammed neurons following AMG treatment (D) or without (C).
(E and F) Boxplots depicting the proportion of iNs spiking at least 1 (E) or 3 (F) action potentials in the indicated conditions. Each dot represents an independent batch culture. Control Ngn2+Bcl2_noAMG: n = 25 cells; control Ngn2+Bcl2_AMG: n = 25; patient Ngn2+Bcl2_noAMG: n = 15; control Ngn2+Bcl2_AMG: n = 16.
(G) Example of spontaneous synaptic activity recorded from a patient-Ngn2-Bcl2-AMG-treated neuron.
(H) Boxplot depicting the proportion of iNs showing a spontaneous synaptic activity. Each dot represents a biological replicate. Same number of cells as in (E) and (F).
(I–M) Boxplots showing input resistance (I), action potential threshold (J), action potential duration (K), action potential amplitude (L), and rise-to-fall ratio (M) in different conditions. Each dot represents one cell. Same number of cells as in (E) and (F).
Fibroblasts with NDUFS4 mutations are also deficient in direct neuronal reprogramming
To test whether NDUFS4-dependent deficits in direct neuronal conversion are cell-type specific or a general hurdle, we reprogrammed human fibroblasts from 3 control donors and 3 NDUFS4-patients (Table S1; Figure S1A). The overexpression of Ngn2 or pMutNgn2 (Figures S5A–S5C) resulted in lower reprogramming rates55 of both genotypes (Figure S5C) in stark contrast to pAstros conversion (Figure 1). Likewise, Ascl1 poorly reprogrammed human fibroblasts56 (Figures S5B–S5E), whereas pMutAscl1 significantly increased the proportion of iNs from control and patient fibroblasts (Figures S5B–S5E). Remarkably, Bcl2-co-expression improved Ngn2- and pMutAscl1-mediated neuronal conversion from control but not patient fibroblasts (Figures S5D and S5E). Thus, as shown in 3 independent cell lines, NDUFS4 mutations impair direct neuronal conversion irrespective of the starter cells, and this deficit cannot be rescued by Bcl2.
Fibroblast-to-neuron conversion is improved by inhibiting UPR
We then reprogrammed a patient line (# 114107) stably expressing atNDB428: remarkably, this resulted in an improvement of the reprogramming rate to a level close to control (Figures S5F and S5G). As atNDB4 expression could fully rescue neuronal reprogramming in patient fibroblasts but only partially in patient pAstros, these cell types seem to significantly differ in their metabolic needs.
This may suggest that fibroblasts may also differ in their responsiveness to the above treatments. To test this, fibroblasts from one control (NDHFNeo) and one patient (#114107) line were transduced with pMutAscl1 and subsequently transiently treated with the compounds used above (Figure S5A). NR treatment improved the reprogramming into neurons of control but not patient fibroblasts (control untreated median = 44.9%, interquartile range [IQR] = 8.72; control NR median = 48.41%, IQR = 4.94; patient untreated median = 29.41%, IQR = 6.77; patient NR median = 33.52%, IQR = 8.17; Figures S5H and S5I); similarly, UA had a mild impact on the proportion of induced neuronal cells in both control or patient fibroblasts (control UA median = 48.47%, IQR = 7.78; patient UA median = 35.11%, IQR = 6.55; Figures S5H and S5I). Idebenone did not change direct conversion (control idebenone median = 40.92%, IQR = 6.76; patient idebenone median = 22.92%, IQR = 5.37; Figures S5H and S5I), thus indicating coenzyme Q10 is not a limiting factor in the conversion of different human cell types into neurons. However, STF and, most strikingly, AMG treatment significantly improved the reprogramming rate of control (AMG: median = 59.94%, IQR = 6.70; STF median = 51.78%, IQR = 5.44; Figures S5H and S5I) and patient fibroblasts (AMG: median = 47.17, IQR = 3.42; STF median = 37.08%, IQR = 5.32; Figures S5H and S5I). Notably, UA, STF, or AMG-treated iNs had longer neurites (Figure S5J), suggesting a more mature state. Thus, contrary to reprogramming into iPSCs,57 UPR is a limiting factor in the reprogramming into neurons from different starter cells of both control and patients.
Discussion
Mitochondrial dysfunction is common in neurological disorders, either of developmental origin, such as in LS, or related to aging and neurodegenerative disorders.2 Direct neuronal reprogramming of NDUFS4 iPSCs-derived pAstros revealed a severe impairment of the conversion process, independent of the reprogramming factors and the starter cell type. This allowed probing several pharmacological treatments, which revealed the UPR and ISR as major general hurdles in the successful fate conversion. Inhibiting these pathways significantly increased reprogramming not only of patient cells, which turned into more mature and functional neurons, but also control cells, either astrocytes or fibroblasts. Importantly, misfolded proteins did not increase upon UPR inhibition in the emerging neurons but were notably increased in iNs. Thus, this study identified a general major hurdle in neuronal reprogramming for patient and control cells, allowing not only to improve the process considerably but also to highlight for the first time the key importance of proteostasis mechanisms in direct reprogramming.
Astrocyte differentiation and direct neuronal reprogramming of NDUFS4- patient cells
In contrast to neural stem cells (NSCs) and neurons differentiating from SURF1 patients,58 astrocyte differentiation of NDUSF4 iPSCs was rather homogeneous (Figure S1C) and largely comparable to control cells at morphological, marker, and transcriptional levels (Figures S1D–S1I) at any stage analyzed.
Neuronal differentiation is particularly sensitive and vulnerable to mitochondria deficits, as neurogenesis and terminal neuronal differentiation are defective in all LS patient-derived iPSC differentiation paradigms tested so far,58,59,60 mimicking some aspects of the phenotypes of LS patients.61,62 Accordingly, direct neuronal conversion was severely impaired and always lower in NDUFS4-cells compared with control cells, independent on the initial population (pAstros or fibroblasts) and the reprogramming factor employed (Figures 1 and S5), even in presence of Bcl2. Remarkably, the expression of atNDB428 reverted the direct neuronal conversion of patient fibroblasts back to control levels, but achieved only a partial rescue in pAstros (Figure S2), suggesting additional hurdles in astrocytes hampering their neuronal conversion. Of note, our results highlight that pMutNgn2 is most efficient in pAstros, but pMutAscl1 in fibroblasts, thereby further supporting the key role of the starter cell identity in neuronal reprogramming.63,64
Pharmacological treatments improve neuronal conversion of NDUFS4-patient cells
Mitochondrial remodeling is a major hurdle in astrocyte-to-neuron reprogramming due to the slow exchange of mitochondrial proteins5 and excessive ROS production,4 which is further increased in NDUFS4 cells.65,66 However, treatment with a ROS scavenger (α-tocopherol) did not improve the neuronal reprogramming of patient pAstros (Figure S2), consistent with the recently published results on SURF1-NPC (neural precursor cells) differentiation,58 nor in control pAstros, thus highlighting a key difference to murine astrocytes.4,27 Idebenone, acting also as an antioxidant,31 and UA had a mild or even adverse effects on the direct conversion in specific contexts (Figures 2 and S5). One may thus hypothesize that increasing mitochondrial mass58 might be more beneficial than manipulating their turnover.
A trait common to most LS patients is the reduced NAD+ level or NAD+/NADH ratio.58,67,68 As NAD+ is involved in most metabolic pathways, its reduction affects cell viability, differentiation, and aging.69 Consistently, NR treatment increases mitochondrial content in skeletal muscle and brown adipose tissue70 and improves mitochondrial function in a mitochondrial myopathy mouse model.71 Here, NR treatment improved the direct conversion of pAstros and fibroblasts of both genotypes. As enzymes that generate NAD+ (e.g., nicotinamide-nucleotide adenyltransferase, NMNAT) or use it as substrate (e.g., sirtuins) are present in mitochondria, cytosol, and nucleus, treatment with NR could potentially impact each of these compartments. For example, C-I defects in oxidizing NADH to NAD+ lead to a decrease in mitochondrial NAD+.72 NR treatment could contribute to restore mitochondrial homeostasis via regulating SIRT3, a deacetylase important for mitochondrial function under stress,73 as shown for axon degeneration and neurite retraction in vivo.74,75 Similarly, NR could increase the activity of nuclear sirtuins, contributing to regulate histone acetylation and, thus, gene expression.76,77 In addition, NR was the only treatment that significantly reduced the aggresomes seen in patient iNs (Figure 3), and, hence, it could indirectly contribute to improve the direct neuronal reprogramming in both pAstros and fibroblasts.
ER stress, UPR, and ISR as a key limitation in direct neuronal reprogramming
The cross-talk between ER and mitochondria coordinates key cellular functions, such as calcium homeostasis, inflammation signaling, and autophagy, among others.78 Increased mitochondrial activity enhances ROS production, which can lead to ER stress, causing the accumulation of misfolded proteins,38,79,80 increasing the expression of chaperones, reducing translation rate, and mounting an antioxidant response, including Nrf2-mediated transcription.80,81 When persisting, UPR elicits cell death via apoptosis82 and ferroptosis.80,83,84
pAstros from both control and patients responded to ER-stress stimuli (Figure S3): genetic sensors for PERK and ATF6 cascades revealed an overall slightly higher basal level of these pathways in patient cells at an early stage of the Ngn2-mediated conversion (Figures 3D, 3E, S3H, and S3I), with an elevated ATF6 reporter expression at later stages (7 DPT, Figure S3I), suggesting a delayed or prolonged activation of such cascade in patient cells. However, it is worth noting that these levels significantly vary across cell lines, thus indicating that cell-intrinsic features could contribute to their susceptibility to ER-stress stimuli. However, the PERK reporter showed a significant increase in control cells during early stages of reprogramming, highlighting the PERK pathway as a general reaction to the need of so many new proteins. Despite the differences between patient and control cells in UPR reporter activity, the early and short AMG treatment was most beneficial for improving the direct conversion of both control and NDUFS4-patient cells, from pAstros and fibroblasts (Figures 2 and S5): this suggests that the early and transient manipulation is sufficient to improve fate conversion (Figures 3 and S5) and maturation into functional neurons (Figure 5).
Although both IRE1 and PERK transmembrane proteins detect misfolded proteins, their response differs quite substantially: IRE1 stimulates the expression of chaperones to restore proper protein folding,39,40 whereas PERK slows protein translation via EIF2α phosphorylation,38,85,86 and increases ATF4 translation. We employed 2 different small molecules to inhibit PERK: AMG and GSK. Interestingly, both treatments significantly improved the conversion of control pAstros, although AMG had a stronger and more significant effect. At the concentration used here (10 μM), AMG is more active on the ISR cascade: consistently, we detected an increase in ATF447 expression (Figure S4) and some downstream targets (e.g., ATG5 and ATG10; Figures S4O–S4Q). Although UPR can be induced by 3 branches (PERK, IRE1, and ATF6), ISR results from the activation of 4 kinases, among which GCN2 is highly expressed in pAstros (Figures S4O and S4P): as GCN2 is mainly activated by amino acid deprivation,47 it raises the question of whether amino acid depletion plays a role during direct conversion. Therefore, it is reasonable to hypothesize that the successful treatments tested here (NR, AMG, GSK, and STF) act at different levels and might contribute to reduce cellular stress overall, allowing the cells undergoing neuronal conversion to overcome this hurdle.
As both UPR and ISR lead to the reduction in protein translation,54 their inhibition might cause an increase in translation and, subsequently, in aggresomes. Although highly variable, an overall higher signal for aggresomes in patient pAstros compared with control cells could be observed, further increased upon Ngn2 expression at 5 DPT (Figures S3K and S3J). At 20 DPT, iNs had significantly elevated levels of aggresomes, with patient iNs showing the highest levels (Figures 3E and 3F). The transient treatment with UPR inhibitors did not significantly exacerbate aggresome formation (Figure 3F), whereas, remarkably, NR seems to reduce the aggresomes, which might contribute to improve the conversion. Most importantly, these data highlight the crucial importance of proteostasis mechanisms in direct neuronal reprogramming, which could be further optimized with the highest need in cells with mitochondrial dysfunction.
scRNA-seq reveals distinct stages of failed conversion and differences in the maturation of control- and NDUFS4-iNs
scRNA-seq revealed very important and novel aspects of the reprogramming process and the newly generated iNs. First, two intermediate states were observed, both expressing astrocyte and neuronal genes. One such state (cluster B) progressed further in fate conversion as judged by a lower astrocyte_score and higher neuronal_score (Figures 4C and 4D) but retains a higher expression of astrocyte-related metabolic features, such as glycolysis and FAO genes (Figures 4H and 4I), indicative of an incomplete metabolic shift. The other intermediate state (cluster C) progressed further in the metabolic conversion with lower score for glycolysis and FAO and higher for ATP5 genes (Figure 4J and 4L) but also failed to convert possibly by running into issues with ferroptosis, which could eventually lead to cell death.4 Thus, scRNA-seq data revealed the existence of distinct partially reprogrammed states with metabolic or stress-related roadblocks.
Cluster D contained cells that are most advanced in their neuronal identity.54 Remarkably, among the 3 subclusters one (cluster 5) contained mainly control cells and another (cluster 3) mostly patient cells, with the third cluster (cluster 10) containing cells of both genotypes. Cluster 5 was enriched for genes related to a mature neuronal phenotype (Figures 4N and 4O) and expressing mainly glutamatergic-related genes (Figure 4R), whereas iNs in cluster 3 seemed to be less-mature neurons and not fully specified, as they expressed also GABAergic markers (Figure 4S). These results suggest that the patient cells with mitochondrial deficits are impaired in converting to a specific neuronal subtype identity, calling for a careful and comprehensive analysis of the neuronal type emerging from patient cell direct reprogramming. These data also show that further treatment paradigms are needed, as AMG could not rectify this phenotype. Notably, however, patient iNs had normal electrophysiological hallmarks when treated with AMG (Figure 5) but failed to fire action potentials without treatment. These differences were not reflected in the transcriptome data that were, however, acquired earlier and in different culture conditions (no co-culture). Nevertheless, these data further support the loose relationship between the transcriptome and physiological function, as recently suggested by patch-seq analysis.63 Therefore, both aspects need to be carefully monitored for achieving healthy, fully functional neurons of the correct identity.
Taken together, beyond elucidating key regulatory mechanisms of the neuronal reprogramming process, the discovery of the mitochondria-UPR axis in direct neuronal conversion is highly relevant for disease and repair. As protein accumulation and the activated UPR pathways have been tied to neurodegenerative diseases,87 the improvement in reprogramming following transient UPR inhibition in cells carrying NDUFS4 mutations might open a new path for the treatment of neurodevelopmental disorders with mitochondrial deficits and allow neuronal replacement therapy in neurodegenerative disease conditions with mitochondrial deficits. Thus, our work highlights not only the validity of using direct neuronal reprogramming to model mitochondrial disease but also unravels possible novel therapeutic strategies for treating mitochondrial disorders and replacing functional neurons in aging and neurodegeneration conditions where mitochondrial dysfunctions are prevalent.
Limitations of the study
Here, we constitutively expressed the reprogramming factors, as they achieve the best conversion into neurons. However, the constant expression of the proneural reprogramming factors may exacerbate the load on proteostasis mechanisms. Thus, it would be important to test optimized inducible constructs in regard to proteostasis—in particular the formation of misfolded proteins—in direct reprogramming. Notably, tamoxifen-inducible constructs reach high levels of the reprogramming factors fastest,88 but the fusion proteins can also be hypomorphs. Conversely, doxycycline-inducible constructs reach efficient levels of the reprogramming factors later and are often less efficient, but protein is in its native state. Further optimization of such approaches, including RNA delivery, small molecules, or constructs using the degron system, is important to pursue toward reaching the minimum exposure to the reprogramming factors required.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-S100beta | Sigma-Aldrich | Cat# S2532; RRID: AB_477499 |
| Rabbit anti-FGFR3 | Santa Cruz | Cat# sc-123; RRID: AB_631511 |
| Guinea pig anti-β-III-Tubulin | Synaptic systems | Cat# 302-304; RRID:AB_10805138 |
| Mouse anti-MAP2 | Sigma Aldrich | Cat# M4403; RRID:AB_477193 |
| Chicken anti-GFP | Aves Lab | Cat# 1020; RRID:AB_10000240 |
| Rat anti-RFP | Chromotek | Cat# 5F8; RRID: AB_2336064 |
| Rabbit anti-RFP | Rockland | Cat# 600-401-379; RRID: RRID:AB_2209751 |
| Anti-puromycin | Merck | Cat#: MABE343; RRID:AB_2566826 |
| Anti-mouse IgG1, Alexa Fluor 488 | Thermo Fisher Scientific | Cat# A-21121; RRID: AB_2535764) |
| Anti-rabbit, AF546-linked | Thermo Fisher Scientific | Cat# A-11010; RRID: AB_2534077 |
| Anti-chicken, Alexa Fluor 488 | Life Technologies | Cat# A11039; RRID:AB_142924 |
| Anti-guinea pig, Alexa Fluor 647 | Jackson ImmunoResearch | Cat# 705-605-003; RRID:AB_2340436 |
| Anti-rat, Alex Fluor 546 | Life Technologies | Cat# A11081; RRID: AB_2534125 |
| Recombinant DNA | ||
| RV CAG-Neurog2-IRES-DsRedExpress2 | Gascón et al.4 | N/A |
| RV CAG-pMutNeurog2-IRES-DsRedExpress2 | This study | N/A |
| RV CAG-Ascl1-IRES-DsRed | Heinrich et al.22 | N/A |
| RV CAG-pMutAscl1-IRES-DsRedExpress2 | This study | N/A |
| RV pMIG-hBCL2-ires-GFP | Gascón et al.4 | Addgene 3544 |
| RV CAG-DsRedExpress2 | Heinrich et al.22 | N/A |
| RV CAG-GFP | Heinrich et al.22 | N/A |
| RV CAG-Neurog2-IRES-BFP | This study | N/A |
| RV CAG-BFP | This study | N/A |
| RV CAG-ATF4-YFP | This study / Walter et al.43 | N/A |
| RV CAG-ATF6-YFP | This study / Walter et al.44 | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Matrigel® (GFR) Basement Membrane Matrix, LDEV-free | Corning® | Cat# 354230 |
| Geltrex™ LDEV-Free Reduced Growth Factor Basement Membrane Matrix | Thermo Fisher Scientific | Cat# A1413302 |
| mTeSR™1 medium | Stem Cell Technologies | Cat# 85850 |
| Neurobasal Medium | Life Technologies | Cat# 21103049 |
| DMEM/F12 | Gibco | Cat# 11039021 |
| Astrocyte Media | ScienCell | Cat# 1801 |
| Recombinant Human LIF | Alomone Labs | Cat# L-200 |
| Recombinant Human FGF-basic | Peprotech | Cat# AF-100-18B |
| Recombinant Human EGF | Peprotech | Cat# AF-100-15 |
| Recombinant Human Noggin | Peprotech | Cat# 120-10C |
| Recombinant Human PDGF-AA | R&D Systems | Cat# 221-AA |
| Rock inhibitor Y-27632 | Stem Cell Technologies | Cat# 72304 |
| Recombinant Human NT-3 | Peprotech | Cat# 450-03-10 |
| SB431542 | Axon Medchem | Cat# 1661 |
| CHIR9902 | Axon Medchem | Cat# 1386 |
| cAMP | Sigma Aldrich | Cat# D0627 |
| LDN 193189 | Axon Medchem | Cat# 1509 |
| LM 22A4 | R and D sysems | Cat# 4607 |
| Human recombinant GDNF | Novus Biologicals | Cat# NBP2-34888 |
| Poly-L-Ornithine Hydrobromide | Sigma-Aldrich | Cat# P3655 |
| Laminin | Sigma-Aldrich | Cat# L2020 |
| AMG PERK 44 (10mg) | R&D systems | Cat# 5517 |
| STF 083010 (10mg) | R&D systems | Cat# 4509 |
| Idebenone | Sigma – Aldrich | Cat# 67805 |
| Urolithin A | Sigma – Aldrich | Cat# SML1791 |
| Nicotinamide riboside | Biomol | Cat# Cay23132-5 |
| GSK2606414 (10mg) | R&D systems | Cat#5107 |
| Tunicamycin Ready Made Solution 5 mg/mL in DMSO | Sigma - Aldrich | Cat# SML1287-1ML |
| Accutase | Thermo Fisher Scientific | Cat# A1110501 |
| Collagenase Type IV | Stem Cell Technologies | Cat# 07909 |
| Pen-Strep | Lager | Cat# 5000956 |
| MEM Non-Essential Amino Acids Solution (100X) | Thermo Fisher Scientific | Cat# 11140050 |
| N2 supplement | Gibco | Cat# 17502048 |
| B27 supplement | Gibco/Lager | Cat# 5001207 |
| FBS | Life Technologies | Cat# 10272106 |
| EGF | LIFE Technologies | Cat# PHG0311 |
| bFGF | Life Technologies | Cat# 13256029 |
| Oligomycin A | Sigma-Aldrich | Cat# 73351 |
| FCCP | Sigma-Aldrich | Cat# C2920 |
| Rotenone | Sigma-Aldrich | Cat# R8875 |
| Antimycin A | Sigma-Aldrich | Cat# A8674 |
| Triton X-100 | Sigma-Aldrich | Cat# T9284 |
| Bovine Serum Albumine (BSA) | Sigma-Aldrich | Cat# A2153 |
| Trypan Blue | Thermo Fisher Scientific | Cat# 15250061 |
| DMSO | Sigma-Aldrich | Cat# D2438 |
| Critical commercial assays | ||
| Glutamate Assay Kit | Abcam | Cat# ab83389 |
| PicoPure RNA Isolation Kit | Life Technologies | Cat# KIT0204 |
| ExtractME Total RNA Kit | Blirt | Cat# EM31.1-250 |
| Maxima First Strand cDNA synthesis Kit | Thermo Fisher Scientific | Cat# K1641 |
| PowerUp SYBR Green Master Mix | Life Technologies | Cat# A25742 |
| Chromium Next GEM Single Cell 3ʹ Reagent Kits v3.1 (Dual Index) | 10Xgenomics | N/A |
| Cell Multiplexing Oligos (CMOs) | 10Xgenomics | N/A |
| PROTEOSTAT® Aggresome detection kit | Enzo Life Sciences | ENZ-51035-K100 |
| Primers for RT-qPCR | ||
| GAPDH | AGCCACATCGCTCAGACAC | GCCCAATACGACCAAATCC |
| DDIT3 / CHOP | CAGAACCAGCAGAGGTCACA | AGCTGTGCCACTTTCCTTTC |
| PPP1R15A/GADD34 | CCTCTACTTCTGCCTTGTCTCCAG | TTTTCCTCCTTCTTCTCGGACG |
| HMOX1 | ACTGCGTTCCTGCTCAACATC | GCTCTGGTCCTTGGTGTCATG |
| HSPA5/BIP | TGTTCAACCAATTATCAGCAAACTC | TTCTGCTGTATCCTCTTCACCAGT |
| XBP1s | GGAGTTAAGACAGCGCTTGGGGA | TGTTCTGGAGGGGTGACAACTGGG |
| MANF | TCACATTCTCACCAGCCACT | CAGGTCGATCTGC TTGTCATAC |
| BLOC1S1 | CCCAATTTGCCAAGCAGACA | CATCCCCAATTTCCTTGAGTGC |
| PMP22 | CTGGTCTGTGCGTGATGAGTG | TGTAGGCGAAACCGTAGGAG |
| Col6A1 | CCCTCGTGGACAAAGTCAAG | GTTTCGGTCACAGCGGTAGT |
| HERPUD1 | CGTTGTTATGTACCTGCATC | TCAGGAGGAGGACCATCATTT |
| HYOU1 | GCAGACCTGTTGGCACTGAG | TCACGATCACCGGTGTTTTC |
| MAP2 | AGTTCAGGCCCACTCTCCCTCC | GGGAGCCAGAGCTGATTCCC |
| HES6 | AGCCCCTGGTGGAGAAGA | CAGCACTTCGGCGTTCTC |
| GADD45A | TCGGCTGGAGAGCAGAAGAC | CGCTTCGTACACCCCGAC |
| LGALS1 | CTGCCAGATGGATACGAAT | GGCTGATTTCAGTCAAAGG |
| USP18 | ACTCCTTGATTTGCGTTGAC | TTTCCCACGGGTCTTCTT |
| ASS1 | TCCTGGAGAACCCCAAGAAC | CTCAGCAAATTTCAAGCCCA |
| Deposited data | ||
| Bulk-RNAseq | This study | GSE248129 |
| scRNASeq | This Study | GSE248129 |
| Experimental models: Cell lines | ||
| Human Fibroblast NDHFneo | Lonza | Cat# CC-2509 |
| Human Fibroblast #47041 | Micha Drukker | |
| Human Fibroblast #61691 | Micha Drukker | |
| Human Fibroblast Patient (#79787) | Holger Prokisch | |
| Human Fibroblast Patient (#114107) | Holger Prokisch | |
| Human Fibroblast Patient (#114106) | Holger Prokisch | |
| Human IPSC control HMGU#1 | Micha Drukker | |
| Human IPSC control HMGU12 | Micha Drukker | |
| Human IPSC control UKERi82a-R1-002 | ForIPS Consortium | |
| Human IPSC patient (#87791) | Micha Drukker | |
| Human IPSC patient (#79787) | Micha Drukker | |
| Human IPSC patient (#114107) | Micha Drukker | |
| Software and algorithms | ||
| ZEN software | Zeiss | https://www.zeiss.com/microscopy/en/products/software/zeiss-zen-lite.html; RRID:SCR_013672 |
| ThImageJ | ImageJ | https://imagej.net/Downloads; RRID: SCR_003070 |
| GraphPad Prism 8.0 | GraphPad Software |
http://www.graphpad.com; RRID:SCR_002798 |
| Adobe Photoshop | Adobe Photoshop |
https://www.adobe.com; RRID: SCR_014199 |
| Adobe Illustrator | Adobe Illustrator |
https://www.adobe.com; RRID:SCR_010279 |
| Microsoft Excel | Microsoft Excel |
https://www.microsoft.com/en-gb/; RRID:SCR_016137 |
| Affinity Designer | Affinity | RRID:SCR_016952 |
| Affinity Photo | Affinity | RRID:SCR_016951 |
| Seahorse Wave | Agilent Technologies | https://www.agilent.com/en-us/products/cell-analysis-(seahorse)/software-download-for-wave-desktop; RRID:SCR_014526 |
| RStudio | RStudio |
http://www.rstudio.com/; RRID: SCR_000432 |
| TopGO v.2.34.0 | Adrian Alexa and Jorg Rahnenfuhrer.89 topGO: Enrichment Analysis for Gene Ontology. R package version 2.34.0. | https://bioconductor.org/packages/release/bioc/html/topGO.html |
| DESeq2 v. 1.22.2 | Love et al.90 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| Ggplot2 v.3.2.0 | Wickham91 | https://cran.r-project.org/web/packages/ggplot2/index.html |
| fgsea | Korotkevich92 | https://bioconductor.org/packages/release/bioc/html/fgsea.html |
| Cellranger | CellRanger v6.0.0 | 10XGenomics |
| Seurat 4.0 | Hao et al.93 | N/A |
| clusterProfiler | Wu et al.94 | N/A |
| Other | ||
| Aqua Poly/Mount | Polysciences | Cat# 18606 |
| pluriStrainer Mini | pluriselect | Cat# 43-10040-40 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Giacomo Masserdotti (giacomo.masserdotti@helmholtz-munich.de).
Materials availability
Plasmids generated in this study are available upon request.
Data and code availability
-
•
RNASeq data are deposited in GEO with the number GSE248129. Raw data are available upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to re-analyze the data reported in this paper is available from the lead contact upon request.
Experimental model and subject details
Human iPSCs and fibroblasts
The control fibroblast cell line NDHFNeo was obtained from Lonza (Cat. No. CC-2509). The other two control fibroblast cell lines (#47041, #61691) and the three NDUFS4-mutant patient fibroblast cell lines (#79787, #114107, #114106) were obtained from skin biopsies of patients with signed informed consent. The cell line #79787 belongs to a patient with the homozygous frameshift mutation c.462delA (p.Lys154fs) within the NDUFS4 gene. The cell lines #114107 and #114106 belong to patients with a substitution mutation in the NDUFS4 gene, at c.119G>A (p.Trp40) and at c.316C>T (p.Arg106) respectively.
The control iPSC cell line UKERi82a-R1-002 was obtained as part of the ForIPS research consortium. The ethics approval is No. 4120, FAU Erlangen-Nürnberg, Germany. Detailed genetic information can be found in Popp et al..95 The other two control iPSC cell lines (HMGU-1 and HMGU-12) and the three patient iPSC cell lines (#87971, #79787, #114107) were generated by the iPSC core unit (headed by Prof. Dr. Micha Drukker) at the Helmholtz Zentrum München. HMGU-1 and HMGU-12 were derived from the same parental fibroblasts (ATCC CRL-2522), but generated using different mRNA cocktails and at different time (HMGU-1 in 2013 and HMGU-12 in 2017). Control and patient iPSCs were karyotyped and did not show any sign of chromosomal aberrations.
Primary cultures of mouse cortical astrocytes
Mouse astrocytes were isolated and cultured as previously described, with small changes.96,97 First, meninges were removed and grey matter tissue from the cerebral cortex of C57BL/6J mice at postnatal day 5-7 (P5–P7) was dissected and mechanically dissociated. Dissociated cells were centrifuged at 1,400rpm for 5min, re-suspended and plated in a T25 flask for culture. Astrocytes were cultured in mouse astrocyte medium (DMEM/F12 (1:1) plus Glutamax, 10% fetal bovine serum (FBS), penicillin /streptomycin (P/S), 1x B27 serum-free-supplement, 10 ng/ml epidermal growth factor (EGF), and 10 ng/ml basic fibroblast growth factor (bFGF). Typically, cells reached ∼80% confluency after 7 days and were then passaged using trypsin/EDTA and plated on poly-D-lysine coated glass coverslips in a 24-well plate. Fresh mouse astrocyte medium was used to plate the cells at a density of 45,000-55,000 cells per coverslip (12mm diameter). Primary mouse astrocytes were maintained in an incubator at 37°C and with 5% CO2.
Human iPSC culture
iPSCs were cultured on Geltrex™ LDEV-Free Reduced Growth Factor Basement Membrane Matrix coated 6-well plates in mTESR1 medium containing 1× mTESR1 supplement. Media was changed every day. For passaging, cells were incubated with Collagenase Type IV for 5–7 minutes at 37°C. Collagenase was aspirated and fresh mTESR1 (with supplement) medium was added to each well. A cell scraper was used to collect the cells, which were subsequently plated on a fresh 6-well plate at the desired density (e.g., one confluent well split 1: 4 or 1:8).
Generation of human proliferating astrocytes (pAstros) and non-proliferating astrocytes (nonpAstros)
Proliferating astrocytes (pAstros) and lon-gterm cultured non-proliferating astrocytes (non-pAstros) were generated as previously described with minor modifications.7 Briefly, confluent iPSC cultures were dissociated with collagenase, collected with a cell scraper and cultured in suspension to form embryoid bodies. Cells were cultured in mTESR1 with 1× mTESR1 supplement and 10 μM Rock Inhibitor Y-27632 for 24 hours. Then, medium was changed to Astrocyte Medium (AM) supplemented with 20ng/ml Noggin and 10ng/mL PDGFAA for the next two weeks and an additional week with only PDGFAA. Embryoid bodies were then manually dissociated by pipetting and the resulting pAstros were plated on poly-L-ornithine (PLO) and laminin-coated dishes in AM supplemented with 10ng/ml bFGF and 10ng/ml EGF. Upon reaching ∼80% confluency, pAstros were passaged using Accutase. pAstros were maintained in culture until ∼day 45-50 of glial differentiation and subsequently terminally differentiated into non-pAstros in AM supplemented with 10ng/ml LIF. Media was changed every second day.
Human fibroblast culture
Human fibroblasts were cultured in T75 flasks with fibroblast media (DMEM plus Glutamax, P/S (1:100), and FBS 10%). Upon reaching ∼80% confluency, fibroblasts were passaged using Trypsin/EDTA. Media was changed every second day.
Method details
Direct neuronal reprogramming
Two days post-transduction (2 DPT), pAstro media or fibroblast media was replaced with fresh neuronal differentiation media consisting of DMEM/F12 and Neurobasal media (1:1) supplemented with 1x P/S, 1x B27 supplement, 1x N2 supplement and 1x MEM non-essential amino acids (NEAA). Media was supplemented with growth factors and small molecules (adapted from the media used in Drouin-Ouellet et al.98) at the following concentrations: LDN-193189 (0.5 μM), LM-22A4 (2 μM), GDNF (2 ng/ml), CHIR99021 (2 μM), NT3 (10 ng/μl), SB-431542 (10 μM), db-cAMP (0.1 μg/ml), Noggin (50 ng/ml), and valproic acid sodium salt (VPA; 1 mM). Half of the media was replaced with fresh media every second day. From 17 DPT until the end of the experiment, the neuronal media was only supplemented with GDNF, LM-22A4, NT3, and db-cAMP. At 20 DPT cells were fixed with 4% paraformaldehyde (PFA) for 10 min and stored in 1xPBS at 4°C for further analysis. During reprogramming, the cultures were maintained in an incubator at 37°C, 5% O2 and 5% CO2.
Bulk RNA sequencing and analysis
Cells at iPSC, pAstro and nonpAstro stages were collected and RNA was isolated on column using the PicoPure™ RNAextraction kit (Applied Biosystems). RNA quality and concentration were evaluated with an Agilent BioAnalyzer 2100 (Agilent). All samples had a RIN > 9. 10 ng of RNA from each sample was used to generate the RNA-seq libraries using bulk-adapted Prime-seq protocol14,99: cDNA was generated by oligo-dT primers containing well-specific (e.g., sample specific) barcodes and unique molecular identifiers (UMIs). Unincorporated barcode primers were digested using Exonuclease I (Thermo Fisher). cDNA was pre-amplified using KAPA HiFi HotStart polymerase (Roche) and pooled before Nextera libraries were constructed from 0.8 ng of pre-amplified cleaned up cDNA using Nextera XT Kit (Illumina). 3’ ends were enriched with a custom P5 primer (P5NEXTPT5, IDT) and libraries were size selected using 2% E6 Gel Agarose EX Gels (Life Technologies), cut out in the range of 300–800 bp, and extracted using the Monarch DNA Gel Extraction Kit (New England Biolabs) according to manufacturer’s recommendations. Libraries were single end sequenced on an Illumina HiSeq 1500 instrument. On average, we sequenced around 20 million reads/sample. Gene counts were calculated by aligning reads against hg38 genome and Ensembl annotation (release 90, 2017) with STAR aligner (version 2.5.3a100) with GeneCounts option. We analyzed 3 biological replicates per stage per cell line. One sample (#87971, iPSC stage) was analyzed twice. GEO number for this dataset is GSE248120 (GEO for all the datasets is GSE248129).
The analysis was performed using R (3.5.3) and RStudio (version 1.2.1335). See key resources table for packages used. Three main stages were considered (iPSC, pAstros and nonpAstros) belonging to 2 different genotypes (Control and Patient). Control iPSCs and patient iPSCs are listed in Table S1. Per each donor and stage, 3 biological replicates were collected and analyzed. Differential gene expression analysis was performed using DESEQ2 package.90 Venn diagrams in Figure S1 was generated considering all the transcripts with an Ensembl number (total: 58306 transcripts) and with log2FC>1, pvalue<0.01 or Log2FC<-1, pvalue<0.01.
Cell cycle score was generated by summing the normalized gene expression, obtained from DESeq2, of the genes listed in Tirosh15 for G1/S and G2/M. Genes plotted in Figure S1D were selected based on pluripotent stem cell markers16 and known astrocytic markers (e.g., SOX9) and markers identified in Barbar.101
Single cell RNA sample preparation, sequencing and analysis
Control (HMGU-1) and patient (#87971) pAstros, transduced and subjected to the indicated treatments, were collected either at 5 DPT or 20 DPT using Accutase. After 3 steps of washing with 1X PBS, cells were resuspended in DMEM/F12 1X phenol-red free medium and DsRed-positive cells were sorted via Fluorescent Activated Cell Sorting (FACS). Around 80.000 DsRed positive cells per condition were collected; untransduced cells were used to set gates. For cell multiplexing, Cell Multiplexing Oligos (CMOs, 10x Genomics) were used. Briefly, cells were centrifuged for 5 min. at 1.000 rpm and then resuspended in PBS + 0.04% BSA (room temperature (RT)) and spun down at 300 rcf for 5 min at RT. Pellets were resuspended in 100 μl of CMO (thawed to RT), pipette mixed 10-15 times and incubated at RT for 5 min. Cells were washed by adding 1.9 ml PBS + 1% BSA (chilled) and spun down at 300 rcf for 5 min at 4°C. Cell pellet was washed with 2 ml chilled PBS + 1% BSA for a total of two washes. Cells were counted and volume adjusted to contain approximately 600-1500 cells per μl before pooling. Around 43μl was loaded, to target approximately 35.000 cells. Single cell gene expression and CellPlex libraries were generated using the Chromium Single Cell 3′ Reagent Kits v3 in combination with the 3’ CellPlex Kit (10x Genomics). For sequencing, gene expression libraries and CellPlex libraries were pooled at a ratio of 5:1. All libraries were sequenced on an Illumina NovaSeq 6000 with a 100 bp paired end configuration according to manufacturer’s instructions. GEO accession number is GSE248122 for 5 days and GSE248128 for 20 days. GEO accession for all the datasets is GSE248129.
Sequencing data of gene expression and CellPlex libraries was jointly processed running the cellranger multi command (CellRanger v6.0.0) using standard parameters and the human GRCh38 genome from Ensembl.
Single cell analysis was performed in RStudio (4.05) mainly using the package Seurat93 (4.0). Libraries either D5 DPT or D20 DPT were first analyzed separately to filter low quality cells (1000 < nFeature_RNA <9000; percent.mt <15) and remove cells without a clear barcode. The resulting 2 datasets were merged using “SCT” function in Seurat (nfeatures = 3000; k.anchor = 5), and data processed as suggested in Seurat pipeline. Uniform Manifold Approximation and Projection (UMAP) was build using dims=30. Clusters were defined using resolution = 0.5. Cell Cycle score was calculated in Seurat. Differentially expressed genes (DEG) were calculated by specifying the clusters in FindMarkers function (min.pct = 0.25). To generate the Venn diagram in Figure 4, we included all the genes with padj<0.01 and Log2FC>0. To compare cluster 5 and cluster 3 in Figure 4, we included the genes with padj<0.01 and Log2FC>1. Scores were generated by providing a list of genes to the Seurat function “AddModuleScore”. For the ferroptotic_score, we considered the genes marked as “Marker”.50 Gene Ontology (GO) analysis was performed using package ‘‘TopGO’’, considering all differentially expressed genes. As background, a list of all detected genes was used. Top 40 GO terms, ranked by Exact Fisher score (< 0.01), were selected and the top 5 those non-redundant with the highest fold enrichment (over the expected number of genes by TopGO and filtered by number of annotated genes>5) were plotted. For Figures 4O and 4P, we employed the packages “clusterProlifer” and “enrichplot”.
Plasmids and viral production
The plasmids containing Ascl1 or Ngn2 cDNA were previously described.22 Briefly, the coding sequence of the reprogramming factors was cloned downstream of a CAG promoter and followed by an intra-ribosome-entry-site (IRES) together with a DsRed coding sequence. For the aggresome detection analysis, we replaced the DsRed cassette with a cassette coding for the Blue fluorescent protein (BFP). The plasmid containing Bcl2 was previously described.4 The phospho-incompetent forms of Ngn2 and Ascl1 were a kind gift of Prof. Dr. Anna Philpott. cDNA of these phospho-mutants was cloned in the same plasmid used for the wild type forms of Ngn2 and Ascl1. ATF4-YFP and ATF6-YFP43,44 were subcloned from their original backbone to a lentiviral backbone with a CAG promoter. Viral vectors were produced and titered as described.96 Retrovirus and lentivirus were used with a titer of 108-109/ml.
Transduction
pAstros were plated on PLO/laminin-coated glass coverslips and fibroblasts were plated on 0.1% gelatin-coated glass coverslips in 24-well plates at a density of 35,000 cells per well. One day later, cells were transduced with 1μl (108-109 particles/ml) of virus per well and cultured at 37°C, 5% O2 and 5% CO2. Media was changed to neuronal differentiation media 2 DPT, as described above.
Glutamate Uptake Assay
iPSCs, pAstros and nonpAstros were plated on 6-well plates: when confluent, the cells were treated with glutamate to obtain a final concentration of 10μM, 100μM or were left untreated. The cells were incubated for 30 minutes before the supernatant was removed. The glutamate concentration of each supernatant was subsequently analyzed using the Glutamate Assay Kit (ab83389) from Abcam, following manufacturer’s instructions.
Seahorse experiments
pAstros and nonpAstros were plated on XF96 V3-PS 96-well cell culture microplates from Seahorse Bioscience and analyzed the day after plating. The cells were seeded at a density of 20,000 cells per well, and the four corner wells contained only media for background correction. Before the Seahorse analysis, cells were washed once with un-buffered media before incubation with 180μl of bicarbonate-free DMEM in an air incubator without CO2 at 37°C for 30 minutes. The XF96 plate was then placed in the XF96 Extracellular Flux Analyzer (Seahorse Bioscience) and the oxygen consumption rates (OCR) were measured. OCR was measured with no addition as a baseline before adding various compounds to obtain measurements of different respiration states. First, Oligomycin A (1 μM) was added to inhibit ATP synthase, followed by carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP, 0.4 μM) to determine the maximal oxidation capacity, and finally a combination of rotenone (2 μM) and Antimycin A (2.5 μM) to block ETC activity. OCR was measured at three time points for each measurement condition, and ∼16-24 technical replicate wells were plated for each cell line. After the Seahorse Assay finished, the normalization for the number of cells in each well was conducted using the CyQuant Assay.
Three biological replicates from 1 cell line (control = HMGU-1, patient = #87971) were conducted for the Seahorse data in Figure S1. The parameters depicted in Figure S1 are from OCR measurements in different conditions: Basal respiration is calculated from the OCR measurements before the first compound injection; ATP-linked respiration is calculated from the OCR before oligomycin injection minus OCR after oligomycin injection; Maximal respiration is calculated from the OCR after FCCP injection; Spare respiratory capacity is calculated from maximal respiration minus basal respiration.
Pharmacological treatment
The pharmacological treatment of the pAstros and fibroblasts undergoing reprogramming was conducted during the first media change, at 2 DPT. Cells were treated once with each compound at the following final concentrations: α-tocopherol (10 μM), Oligomycin A (1 μg/ml), Urolithin A (2 μg/ml), Nicotinamide riboside (10 μM), Idebenone (1 μg/ml), AMG PERK-44 (10 μM), STF-083010 (10 μM), GSK2606414 (1μM) and Tunicamycin (500ng/ml).
RNA Extraction and Real Time Quantitative PCR (RT-qPCR)
Control and patient pAstros were plated on 6-well plates at ∼500,000 cells per well. The following day, the cells were either transduced or treated with 500ng/ml tunicamycin for 16 hours. Transduced cells were collected at 5 DPT using Accutase. DsRed-positive cells were sorted via Fluorescent Activated Cell Sorting (FACS). Between 20.000 and 100.000 DsRed positive cells were collected per condition; untransduced cells were used to set gates.
RNA was extracted using the EXTRACTME Total RNA Kit (blirt) according to manufacturer’s instructions, including the removal of genomic DNA. 50ng RNA was retro-transcribed using Maxima First Strand cDNA Synthesis Kit (ThermoFisher). Each cDNA sample was diluted 1 to 5 and 1μl was used for each RT-qPCR reaction. RT-qPCR was performed on a QuantStudio™ 6 Flex Real-Time PCR System (Thermo Fisher) using PowerUp SYBR Green Master Mix (Life Technologies) according to manufacturer’s instructions (10μl final volume). The expression of each gene was analyzed in triplicate and data were processed with the ΔΔCt method using GAPDH as a housekeeping gene. Experiment was performed on 2-4 independent biological replicates.
Detection of UPR during Direct neuronal Reprogramming
pAstros were plated on 24-well plates and underwent the neuronal reprogramming paradigm as described above. Briefly, astrocytes were transduced with retroviruses encoding either DsRed or Ngn2-IRES-DsRed as well as ATF4-YFP or ATF6-YFP lentiviruses. 2 DPT, the media was changed to neuronal differentiation media and the cells were treated with Tunicamycin (500ng/ml) as a positive control for 16 hours where necessary. Cells were fixed at 3 DPT and 7 DPT with PFA 4% for 10 minutes at RT. PFA solution was aspirated and cells were washed three times with PBS1X. For immunofluorescent analysis, cells were incubated for 1 hour in blocking solution (3% BSA, 0.5% TritonX-100, PBS1X) and incubated overnight with anti-GFP (to amplify the YFP signal) and anti-RFP antibodies. Cells were then washed 3 times with PBS 1X, stained with secondary antibodies and DAPI for 1 hour at RT, before washing and mounting. The experiment was repeated 3-5 times using pAstros from 2 control lines (HMGU-1 and HMGU-12) and 3 patient lines (#87971, #79787 and #114107). In each biological replicate, a region of interest (ROI) was manually drawn and the mean fluorescent intensity of the YFP signal was analyzed in 30-50 DsRed-positive cells with Fiji. Each dot represents the average for 1 biological replicate.
Aggresome Detection
pAstros were plated on 24-well plates and underwent the neuronal reprogramming paradigm as described above. The main difference is that here we transduced pAstros with retroviruses encoding either BFP or Ngn2-IRES-BFP. At 2 DPT, pAstro media was replaced with neuronal differentiation media and the cells were treated with the indicated small molecules. At 5 DPT and 20 DPT, misfolded proteins were measured using the PROTEOSTAT® Aggresome detection kit (ENZOlifesciences) following the manufacturer’s instructions. Briefly, cells were fixed with PFA 4% for 30 minutes at RT. PFA solution was aspirated, cells were washed once with PBS1X and then permeabilized for 30 minutes with 0.5% Triton X-100, 3 mM EDTA, in 1X Assay Buffer at 4°C. Permeabilization solution was aspirated and PROTEOSTAT® Aggresome Detection Reagent was added in 1X Assay Buffer (1:2000) for 30 minutes at RT. Cells were then washed 3 times with PBS 1X and mounted. Cells were imaged on the same day the assay was performed. The experiment was repeated 3 times using pAstros from the control line HMGU#1 and patient line 79787. In each biological replicate, a region of interest (ROI) was manually drawn and the mean fluorescent intensity of the PROTEOSTAT® signal was analyzed in 30-50 BFP-positive cells with Fiji. For experiments performed on 20 DPT, 30-50 BFP-positive cells were further separated into non-neuronal and neuronal morphology prior to analysis. Each dot represents the average for 1 independent biological experiment.
Morphological analysis of reprogrammed cells
The neurite length of both pAstros- and fibroblast-induced neuronal cells across conditions was analyzed at 20 DPT. The Fiji plugin “Simple Neurite Tracer” was used for the measurement quantification. Neurite length of 10 randomly selected cells were analyzed across 3 biological replicates per each condition.
Immunocytochemistry
Cells were fixed with 4% PFA for 10 min at RT and stored in 1xPBS at 4°C before staining. Cells were incubated in blocking solution (PBS 1x plus 3% Bovine Serum Albumin (BSA) and 0.5% Triton X-100) for 30 minutes and subsequently incubated with primary antibodies (concentrations listed in the key resources table) diluted in blocking solution for 2 hours at RT or overnight at 4°C. After washing twice for 5 minutes with 1xPBS, cells were incubated with the correct species-specific secondary antibodies at a dilution of 1:1000 for 1hr in the dark at RT. DAPI (1:10,000 in blocking solution) was also added during this step to label the nuclei. Cells were washed twice for 5 minutes with 1xPBS and the coverslips were mounted with Aqua Poly/Mount. The cells were imaged at the Axio Observer Z1 epifluorescence microscope (Carl Zeiss) or the LSM710 laser-scanning confocal. Digital images were acquired using the ZEN software (Carl Zeiss) at 20X, 25X or 40X.
Co-culture for electrophysiology
Human pAstros were plated on 6-well plates at ∼210,000 cells per well. The following day, the cells were transduced with 5 μl of virus per well. 2 DPT pAstros were washed twice with PBS 1x and plated on top of mouse astrocytes plated the day before in 24-well plates. The co-cultured cells were maintained in mouse astrocyte conditioned media (media taken from T75 flasks of mouse astrocyte cultures for 3-4 days) and supplemented with the neuronal reprogramming media factors plus 10 μM Rock Inhibitor Y-27632 until 50-70 DPT. Half media was exchanged every second day.
Whole-cell patch-clamp recordings
Whole-cell patch-clamp measurements were performed as previously described.102 Briefly, coverslips with cultured iNeurons were transferred to an organ bath mounted on the stage of an upright microscope (BX-RFA-1-5, Olympus, Japan). A single coverslip was continuously perfused with artificial cerebro-spinal fluid (ASCF) containing (in mM): NaCl (125), KCl (3), NaH2PO4 (1.25), NaHCO3 (25), CaCl2 (2), MgCl2 (2) and D-Glucose (25 mM). The ACSF was saturated with 95% O2 / 5% CO2 to maintain a pH of 7.4. The osmolarity of the ACSF ranged between 305 to 318 mOsmol. The perfusion rate with ACSF was set to 3 mL / min and recordings were performed at 28°C. Cultured cells were visualized with a Dodt contrast tube (DGC, Scientifica, UK) that was attached to the microscope. Successfully transduced neurons were identified by DsRed expression with the help of a fluorescent lamp (pE-300, CooLED, UK) and epifluorescence optics for red fluorescence (filter: ZT635dcrb, Chroma Technology, USA). Images were taken and displayed using a software-operated microscope camera (Evolve 512 Delta, Teledyne Photometrics, USA). The electrodes for whole cell patch-clamp recordings were fabricated from borosilicate glass capillaries (OD: 1.5 mm, ID: 0.86 mm, Hugo Sachs Elektronik-Harvard Apparatus, March-Hugstetten, Germany) and filled with a solution composed of (in mM): K-gluconate (135), KCl (4), NaCl (2), EGTA (0.2), HEPES (10), Mg-ATP (4), Na-GTP (0.5), and phosphocreatine (10). The osmolarity ranged between 288-295 mOsmol, the pH was adjusted to 7.3. The electrodes (resistance: 3 – 5 MΩ) were connected to the headstage of a npi ELC-03XS amplifier (npi, Tamm, Germany). The recorded signals were amplified (x20), filtered at 20 kHz, digitized at a sampling rate of 50 kHz and stored on a computer for off-line analysis. Data acquisition was performed by means of a CED 1401 Power 3 system in conjunction with the Signal6 data acquisition software (Cambridge electronic design, Cambridge, England). The input resistance was determined by injecting at least 10 small hyperpolarizing current steps (500 ms, 1-10 pA) into the cells and by determining the averaged voltage deflection in response to the current injection. The input resistance was then calculated according to Ohm’s law (R= V/I). The properties of single action potentials (AP) were obtained from recordings in which action potentials were elicited by means of just suprathreshold current steps or current ramps (duration: 50 ms). The following AP parameters were analyzed: AP amplitude, AP duration, AP threshold, AP rising slope. The amplitude was determined as the difference between the resting membrane potential and the AP peak voltage. Duration, rising slope and spike threshold were determined according to methods given by Bean103: Single spikes were differentiated, and the spike duration corresponded to the temporal difference between the maximum and the minimum of the differentiated spike. The rising slope was equal to the maximum of the first spike derivative. For the spike threshold analysis, the differentiated spike was plotted as a function of the membrane voltage. In this phase plane, the threshold corresponded to the point where the rising slope of the membrane voltage displayed a sudden increase. To test whether an induced neuron was able to fire repetitive action potentials, at least 20 current steps with increasing amplitudes (duration: 1 s) were injected into the cells. Depending on a cell’s input resistance, these current steps ranged between 1-10 pA. Data analysis was performed using IGOR Pro 6 together with the Neuromatic IGOR plugin.104
Quantification and statistical analysis
The efficiency of neuronal reprogramming for both pAstros and fibroblasts across conditions was analyzed at 20 DPT. The quantification of neuronal-like cells was conducted as previously described4: in short, morphological parameters such as the length of processes and β-III-tubulin immunoreactivity were assessed.
Data were analyzed with Microsoft Excel, GraphPad Prism 8.0 software and linear regression using the “lm” function (R Stats package) in RStudio. Evaluation of the residuals for fitted linear models was performed with the package “DHARMa” (Florian Hartig (2020). DHARMa: Residual Diagnostics for Hierarchical (Multi-Level / Mixed) Regression Models. R package version 0.3.2.0. https://CRAN.R-project.org/package=DHARMa) in RStudio. For the analysis of the intensity of genetic sensors, paired t-test was used when the values of the groups and the differences were normally distributed and the variances were not unequal; otherwise, Wilcoxon signed rank exact test was used.
The number of biological replicates is detailed in the Figure Legends, and the data are plotted as median ± interquartile range (IQR) or mean ± SEM. The significance illustrated in the graphs belong to the corresponding p-values ∗ p≤0.05, ∗∗ p≤0.01, ∗∗∗p≤0.001.
Schemes have been generated or adapted from BioRender.com.
Acknowledgments
We thank Paulina Chlebik and Edina Kepesidis for virus production, Tatiana Simon-Ebert for primary mouse astrocyte cultures, Kalina Draganova for advice on human iPSC culture, Alicia Kemble for advice on human astrocyte differentiation, and Anna Danese and Maria Colomé-Tatché for initial observation of aberrant reprogramming and for demultiplexing bulk RNA-seq. We acknowledge the Core Facility Flow Cytometry and Core Facility Bioinformatics at the Biomedical Center, LMU Munich, the laboratory for functional Genome analysis (Lafuga) at LMU Munich, and the Core Facility Sequencing Core facility of the Helmholtz Institute Munich (HMGU) for providing equipment, services, and expertise. This work was funded by the German Research Foundation TRR274 (no. 408885537, M. Götz), FOR2879/2 (no. 405358801, M. Götz), the SPP 2306 Ferroptosis (project no. 461629173, M. Götz), and SyNergy (EXC2145/Project-ID 390857198, M. Götz). Parts of this work were supported by a New Frontiers in Research Fund Transformation grant to M. Götz, funded through three Canadian federal funding agencies (CIHR, NSERC, and SSHRC), the advanced ERC Neurocentro (no. 885382), the European Union’s Horizon 2020 research and innovation program under grant agreement no. 874758 (NSC Reconstruct to M. Götz), and EN 1093/2-1 (to A.J. and W.E.).
Author contributions
Conceptualization: G.M. and M. Götz; methodology: E.R. (iPSCs generation), G.S. (iPSC-to-pAstro differentiation), and A.B.M. (UPR); investigation: G.S. and A.B.M. (reprogramming experiments), A.B.M. (UPR experiments), G.M. and A.B.M. (FACS for bulk and scRNA-seq); T.R. (whole-cell patch-clamp experiments), M. Gusic (seahorse analysis together with G.S.), G.B. (western blot), S.N. (the first observing an increase in ER stress related signature), A.J. (prime-seq libraries), B.A.H. (de-multiplexed 10× genomics data), and P.S. (demultiplexed bulk RNA-seq data); resources: J.H.M.P., M.D., and W.E.; H.P. (and expertise in mitochondria defects); formal analysis: G.M.; supervision: G.M. and M. Götz; funding acquisition: M. Götz; writing G.S., A.B.M., G.M., M. Götz, and all authors contributed corrections and comments.
Declaration of interests
M. Götz is member of the advisory board of Neuron.
Published: January 23, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.neuron.2023.12.020.
Contributor Information
Magdalena Götz, Email: magdalena.goetz@bmc.med.lmu.de.
Giacomo Masserdotti, Email: giacomo.masserdotti@helmholtz-munich.de.
Supplemental information
References
- 1.Cruz-Haces M., Tang J., Acosta G., Fernandez J., Shi R. Pathological correlations between traumatic brain injury and chronic neurodegenerative diseases. Transl. Neurodegener. 2017;6:20. doi: 10.1186/s40035-017-0088-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stanga S., Caretto A., Boido M., Vercelli A. Mitochondrial Dysfunctions: A Red Thread across Neurodegenerative Diseases. Int. J. Mol. Sci. 2020;21:3719. doi: 10.3390/ijms21103719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Watts M.E., Pocock R., Claudianos C. Brain Energy and Oxygen Metabolism: Emerging Role in Normal Function and Disease. Front. Mol. Neurosci. 2018;11:216. doi: 10.3389/fnmol.2018.00216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gascón S., Murenu E., Masserdotti G., Ortega F., Russo G.L., Petrik D., Deshpande A., Heinrich C., Karow M., Robertson S.P., et al. Identification and Successful Negotiation of a Metabolic Checkpoint in Direct Neuronal Reprogramming. Cell Stem Cell. 2016;18:396–409. doi: 10.1016/j.stem.2015.12.003. [DOI] [PubMed] [Google Scholar]
- 5.Russo G.L., Sonsalla G., Natarajan P., Breunig C.T., Bulli G., Merl-Pham J., Schmitt S., Giehrl-Schwab J., Giesert F., Jastroch M., et al. CRISPR-Mediated Induction of Neuron-Enriched Mitochondrial Proteins Boosts Direct Glia-to-Neuron Conversion. Cell Stem Cell. 2021;28:524–534.e7. doi: 10.1016/j.stem.2020.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fecher C., Trovò L., Müller S.A., Snaidero N., Wettmarshausen J., Heink S., Ortiz O., Wagner I., Kühn R., Hartmann J., et al. Cell-type-specific profiling of brain mitochondria reveals functional and molecular diversity. Nat. Neurosci. 2019;22:1731–1742. doi: 10.1038/s41593-019-0479-z. [DOI] [PubMed] [Google Scholar]
- 7.Santos R., Vadodaria K.C., Jaeger B.N., Mei A., Lefcochilos-Fogelquist S., Mendes A.P.D., Erikson G., Shokhirev M., Randolph-Moore L., Fredlender C., et al. Differentiation of Inflammation-Responsive Astrocytes from Glial Progenitors Generated from Human Induced Pluripotent Stem Cells. Stem Cell Rep. 2017;8:1757–1769. doi: 10.1016/j.stemcr.2017.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bénit P., Beugnot R., Chretien D., Giurgea I., De Lonlay-Debeney P., Issartel J.P., Corral-Debrinski M., Kerscher S., Rustin P., Rötig A., et al. Mutant NDUFV2 subunit of mitochondrial complex I causes early onset hypertrophic cardiomyopathy and encephalopathy. Hum. Mutat. 2003;21:582–586. doi: 10.1002/humu.10225. [DOI] [PubMed] [Google Scholar]
- 9.Budde S.M., van den Heuvel L.P., Janssen A.J., Smeets R.J., Buskens C.A., DeMeirleir L., Van Coster R., Baethmann M., Voit T., Trijbels J.M., et al. Combined enzymatic complex I and III deficiency associated with mutations in the nuclear encoded NDUFS4 gene. Biochem. Biophys. Res. Commun. 2000;275:63–68. doi: 10.1006/bbrc.2000.3257. [DOI] [PubMed] [Google Scholar]
- 10.Budde S.M., van den Heuvel L.P., Smeets R.J., Skladal D., Mayr J.A., Boelen C., Petruzzella V., Papa S., Smeitink J.A. Clinical heterogeneity in patients with mutations in the NDUFS4 gene of mitochondrial complex I. J. Inherit. Metab. Dis. 2003;26:813–815. doi: 10.1023/b:boli.0000010003.14113.af. [DOI] [PubMed] [Google Scholar]
- 11.Budde S.M., van den Heuvel L.P., Smeitink J.A. The human complex I NDUFS4 subunit: from gene structure to function and pathology. Mitochondrion. 2002;2:109–115. doi: 10.1016/s1567-7249(02)00035-1. [DOI] [PubMed] [Google Scholar]
- 12.Petruzzella V., Vergari R., Puzziferri I., Boffoli D., Lamantea E., Zeviani M., Papa S. A nonsense mutation in the NDUFS4 gene encoding the 18 kDa (AQDQ) subunit of complex I abolishes assembly and activity of the complex in a patient with Leigh-like syndrome. Hum. Mol. Genet. 2001;10:529–535. doi: 10.1093/hmg/10.5.529. [DOI] [PubMed] [Google Scholar]
- 13.Ugalde C., Janssen R.J., van den Heuvel L.P., Smeitink J.A., Nijtmans L.G. Differences in assembly or stability of complex I and other mitochondrial OXPHOS complexes in inherited complex I deficiency. Hum. Mol. Genet. 2004;13:659–667. doi: 10.1093/hmg/ddh071. [DOI] [PubMed] [Google Scholar]
- 14.Janjic A., Wange L.E., Bagnoli J.W., Geuder J., Nguyen P., Richter D., Vieth B., Vick B., Jeremias I., Ziegenhain C., et al. Prime-seq, efficient and powerful bulk RNA sequencing. Genome Biol. 2022;23:88. doi: 10.1186/s13059-022-02660-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tirosh I., Izar B., Prakadan S.M., Wadsworth M.H., 2nd, Treacy D., Trombetta J.J., Rotem A., Rodman C., Lian C., Murphy G., et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science. 2016;352:189–196. doi: 10.1126/science.aad0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Giulitti S., Pellegrini M., Zorzan I., Martini P., Gagliano O., Mutarelli M., Ziller M.J., Cacchiarelli D., Romualdi C., Elvassore N., et al. Direct generation of human naive induced pluripotent stem cells from somatic cells in microfluidics. Nat. Cell Biol. 2019;21:275–286. doi: 10.1038/s41556-018-0254-5. [DOI] [PubMed] [Google Scholar]
- 17.Barbar L., Jain T., Zimmer M., Kruglikov I., Sadick J.S., Wang M., Kalpana K., Rose I.V.L., Burstein S.R., Rusielewicz T., et al. CD49f Is a Novel Marker of Functional and Reactive Human iPSC-Derived Astrocytes. Neuron. 2020;107:436–453.e12. doi: 10.1016/j.neuron.2020.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bardehle S., Krüger M., Buggenthin F., Schwausch J., Ninkovic J., Clevers H., Snippert H.J., Theis F.J., Meyer-Luehmann M., Bechmann I., et al. Live imaging of astrocyte responses to acute injury reveals selective juxtavascular proliferation. Nat. Neurosci. 2013;16:580–586. doi: 10.1038/nn.3371. [DOI] [PubMed] [Google Scholar]
- 19.Sirko S., Behrendt G., Johansson P.A., Tripathi P., Costa M., Bek S., Heinrich C., Tiedt S., Colak D., Dichgans M., et al. Reactive glia in the injured brain acquire stem cell properties in response to sonic hedgehog. [corrected] Cell Stem Cell. 2013;12:426–439. doi: 10.1016/j.stem.2013.01.019. [DOI] [PubMed] [Google Scholar]
- 20.Sirko S., Schichor C., Della Vecchia P., Metzger F., Sonsalla G., Simon T., Bürkle M., Kalpazidou S., Ninkovic J., Masserdotti G., et al. Injury-specific factors in the cerebrospinal fluid regulate astrocyte plasticity in the human brain. Nat. Med. 2023;29:3149–3161. doi: 10.1038/s41591-023-02644-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Berninger B., Costa M.R., Koch U., Schroeder T., Sutor B., Grothe B., Götz M. Functional properties of neurons derived from in vitro reprogrammed postnatal astroglia. J. Neurosci. 2007;27:8654–8664. doi: 10.1523/JNEUROSCI.1615-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Heinrich C., Blum R., Gascón S., Masserdotti G., Tripathi P., Sánchez R., Tiedt S., Schroeder T., Götz M., Berninger B. Directing astroglia from the cerebral cortex into subtype specific functional neurons. PLoS Biol. 2010;8 doi: 10.1371/journal.pbio.1000373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ali F., Hindley C., McDowell G., Deibler R., Jones A., Kirschner M., Guillemot F., Philpott A. Cell cycle-regulated multi-site phosphorylation of Neurogenin 2 coordinates cell cycling with differentiation during neurogenesis. Development. 2011;138:4267–4277. doi: 10.1242/dev.067900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ali F.R., Cheng K., Kirwan P., Metcalfe S., Livesey F.J., Barker R.A., Philpott A. The phosphorylation status of Ascl1 is a key determinant of neuronal differentiation and maturation in vivo and in vitro. Development. 2014;141:2216–2224. doi: 10.1242/dev.106377. [DOI] [PubMed] [Google Scholar]
- 25.Woods L.M., Ali F.R., Gomez R., Chernukhin I., Marcos D., Parkinson L.M., Tayoun A.N.A., Carroll J.S., Philpott A. Elevated ASCL1 activity creates de novo regulatory elements associated with neuronal differentiation. BMC Genomics. 2022;23:255. doi: 10.1186/s12864-022-08495-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jastroch M., Divakaruni A.S., Mookerjee S., Treberg J.R., Brand M.D. Mitochondrial proton and electron leaks. Essays Biochem. 2010;47:53–67. doi: 10.1042/bse0470053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gascón S., Masserdotti G., Russo G.L., Götz M. Direct Neuronal Reprogramming: Achievements, Hurdles, and New Roads to Success. Cell Stem Cell. 2017;21:18–34. doi: 10.1016/j.stem.2017.06.011. [DOI] [PubMed] [Google Scholar]
- 28.Catania A., Iuso A., Bouchereau J., Kremer L.S., Paviolo M., Terrile C., Bénit P., Rasmusson A.G., Schwarzmayr T., Tiranti V., et al. Arabidopsis thaliana alternative dehydrogenases: a potential therapy for mitochondrial complex I deficiency? Perspectives and pitfalls. Orphanet J. Rare Dis. 2019;14:236. doi: 10.1186/s13023-019-1185-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Russell O.M., Gorman G.S., Lightowlers R.N., Turnbull D.M. Mitochondrial Diseases: Hope for the Future. Cell. 2020;181:168–188. doi: 10.1016/j.cell.2020.02.051. [DOI] [PubMed] [Google Scholar]
- 30.Lee S., Na J.H., Lee Y.M. Epilepsy in Leigh Syndrome With Mitochondrial DNA Mutations. Front. Neurol. 2019;10:496. doi: 10.3389/fneur.2019.00496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Haefeli R.H., Erb M., Gemperli A.C., Robay D., Courdier Fruh I., Anklin C., Dallmann R., Gueven N. NQO1-dependent redox cycling of idebenone: effects on cellular redox potential and energy levels. PLoS One. 2011;6 doi: 10.1371/journal.pone.0017963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Giorgio V., Schiavone M., Galber C., Carini M., Da Ros T., Petronilli V., Argenton F., Carelli V., Acosta Lopez M.J., Salviati L., et al. The idebenone metabolite QS10 restores electron transfer in complex I and coenzyme Q defects. Biochim. Biophys. Acta Bioenerg. 2018;1859:901–908. doi: 10.1016/j.bbabio.2018.04.006. [DOI] [PubMed] [Google Scholar]
- 33.Ryu D., Mouchiroud L., Andreux P.A., Katsyuba E., Moullan N., Nicolet-Dit-Félix A.A., Williams E.G., Jha P., Lo Sasso G., Huzard D., et al. Urolithin A induces mitophagy and prolongs lifespan in C. elegans and increases muscle function in rodents. Nat. Med. 2016;22:879–888. doi: 10.1038/nm.4132. [DOI] [PubMed] [Google Scholar]
- 34.Moisoi N., Klupsch K., Fedele V., East P., Sharma S., Renton A., Plun-Favreau H., Edwards R.E., Teismann P., Esposti M.D., et al. Mitochondrial dysfunction triggered by loss of HtrA2 results in the activation of a brain-specific transcriptional stress response. Cell Death Differ. 2009;16:449–464. doi: 10.1038/cdd.2008.166. [DOI] [PubMed] [Google Scholar]
- 35.Gӧbel J., Engelhardt E., Pelzer P., Sakthivelu V., Jahn H.M., Jevtic M., Folz-Donahue K., Kukat C., Schauss A., Frese C.K., et al. Mitochondria-Endoplasmic Reticulum Contacts in Reactive Astrocytes Promote Vascular Remodeling. Cell Metab. 2020;31:791–808.e8. doi: 10.1016/j.cmet.2020.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moltedo O., Remondelli P., Amodio G. The Mitochondria-Endoplasmic Reticulum Contacts and Their Critical Role in Aging and Age-Associated Diseases. Front. Cell Dev. Biol. 2019;7:172. doi: 10.3389/fcell.2019.00172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carreras-Sureda A., Kroemer G., Cardenas J.C., Hetz C. Balancing energy and protein homeostasis at ER-mitochondria contact sites. Sci. Signal. 2022;15:eabm7524. doi: 10.1126/scisignal.abm7524. [DOI] [PubMed] [Google Scholar]
- 38.Li A., Song N.J., Riesenberg B.P., Li Z. The Emerging Roles of Endoplasmic Reticulum Stress in Balancing Immunity and Tolerance in Health and Diseases: Mechanisms and Opportunities. Front. Immunol. 2019;10:3154. doi: 10.3389/fimmu.2019.03154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee A.H., Iwakoshi N.N., Glimcher L.H. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol. Cell. Biol. 2003;23:7448–7459. doi: 10.1128/MCB.23.21.7448-7459.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Travers K.J., Patil C.K., Wodicka L., Lockhart D.J., Weissman J.S., Walter P. Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell. 2000;101:249–258. doi: 10.1016/s0092-8674(00)80835-1. [DOI] [PubMed] [Google Scholar]
- 41.Axten J.M., Medina J.R., Feng Y., Shu A., Romeril S.P., Grant S.W., Li W.H., Heerding D.A., Minthorn E., Mencken T., et al. Discovery of 7-methyl-5-(1-{[3-(trifluoromethyl)phenyl]acetyl}-2,3-dihydro-1H-indol-5-yl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (GSK2606414), a potent and selective first-in-class inhibitor of protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK) J. Med. Chem. 2012;55:7193–7207. doi: 10.1021/jm300713s. [DOI] [PubMed] [Google Scholar]
- 42.Smith H.L., Freeman O.J., Butcher A.J., Holmqvist S., Humoud I., Schätzl T., Hughes D.T., Verity N.C., Swinden D.P., Hayes J., et al. Astrocyte Unfolded Protein Response Induces a Specific Reactivity State that Causes Non-Cell-Autonomous Neuronal Degeneration. Neuron. 2020;105:855–866.e5. doi: 10.1016/j.neuron.2019.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Walter F., Schmid J., Düssmann H., Concannon C.G., Prehn J.H. Imaging of single cell responses to ER stress indicates that the relative dynamics of IRE1/XBP1 and PERK/ATF4 signalling rather than a switch between signalling branches determine cell survival. Cell Death Differ. 2015;22:1502–1516. doi: 10.1038/cdd.2014.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Walter F., O'Brien A., Concannon C.G., Düssmann H., Prehn J.H.M. ER stress signaling has an activating transcription factor 6alpha (ATF6)-dependent "off-switch". J. Biol. Chem. 2018;293:18270–18284. doi: 10.1074/jbc.RA118.002121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shimizu T., Taguchi A., Higashijima Y., Kanki Y., Nakaki R., Urade Y., Wada Y. Inhibition of cardiac PERK signaling promotes peripartum cardiac dysfunction. Sci. Rep. 2021;11:18687. doi: 10.1038/s41598-021-98344-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tiklová K., Nolbrant S., Fiorenzano A., Björklund Å.K., Sharma Y., Heuer A., Gillberg L., Hoban D.B., Cardoso T., Adler A.F., et al. Single cell transcriptomics identifies stem cell-derived graft composition in a model of Parkinson's disease. Nat. Commun. 2020;11:2434. doi: 10.1038/s41467-020-16225-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mijit M., Boner M., Cordova R.A., Gampala S., Kpenu E., Klunk A.J., Zhang C., Kelley M.R., Staschke K.A., Fishel M.L. Activation of the integrated stress response (ISR) pathways in response to Ref-1 inhibition in human pancreatic cancer and its tumor microenvironment. Front. Med. (Lausanne) 2023;10:1146115. doi: 10.3389/fmed.2023.1146115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mick E., Titov D.V., Skinner O.S., Sharma R., Jourdain A.A., Mootha V.K. Distinct mitochondrial defects trigger the integrated stress response depending on the metabolic state of the cell. eLife. 2020;9:e49178. doi: 10.7554/eLife.49178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pakos-Zebrucka K., Koryga I., Mnich K., Ljujic M., Samali A., Gorman A.M. The integrated stress response. EMBO Rep. 2016;17:1374–1395. doi: 10.15252/embr.201642195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhou N., Yuan X., Du Q., Zhang Z., Shi X., Bao J., Ning Y., Peng L. FerrDb V2: update of the manually curated database of ferroptosis regulators and ferroptosis-disease associations. Nucleic Acids Res. 2023;51:D571–D582. doi: 10.1093/nar/gkac935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Du H., Wang Z., Guo R., Yang L., Liu G., Zhang Z., Xu Z., Tian Y., Yang Z., Li X., et al. Transcription factors Bcl11a and Bcl11b are required for the production and differentiation of cortical projection neurons. Cereb. Cortex. 2022;32:3611–3632. doi: 10.1093/cercor/bhab437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Goetz J.J., Martin G.M., Chowdhury R., Trimarchi J.M. Onecut1 and Onecut2 play critical roles in the development of the mouse retina. PLoS One. 2014;9 doi: 10.1371/journal.pone.0110194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Moreau M.X., Saillour Y., Cwetsch A.W., Pierani A., Causeret F. Single-cell transcriptomics of the early developing mouse cerebral cortex disentangle the spatial and temporal components of neuronal fate acquisition. Development. 2021;148:dev197962. doi: 10.1242/dev.197962. [DOI] [PubMed] [Google Scholar]
- 54.Bardy C., van den Hurk M., Kakaradov B., Erwin J.A., Jaeger B.N., Hernandez R.V., Eames T., Paucar A.A., Gorris M., Marchand C., et al. Predicting the functional states of human iPSC-derived neurons with single-cell RNA-seq and electrophysiology. Mol. Psychiatry. 2016;21:1573–1588. doi: 10.1038/mp.2016.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu M.L., Zang T., Zou Y., Chang J.C., Gibson J.R., Huber K.M., Zhang C.L. Small molecules enable neurogenin 2 to efficiently convert human fibroblasts into cholinergic neurons. Nat. Commun. 2013;4:2183. doi: 10.1038/ncomms3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pang Z.P., Yang N., Vierbuchen T., Ostermeier A., Fuentes D.R., Yang T.Q., Citri A., Sebastiano V., Marro S., Südhof T.C., et al. Induction of human neuronal cells by defined transcription factors. Nature. 2011;476:220–223. doi: 10.1038/nature10202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Simic M.S., Moehle E.A., Schinzel R.T., Lorbeer F.K., Halloran J.J., Heydari K., Sanchez M., Jullié D., Hockemeyer D., Dillin A. Transient activation of the UPR(ER) is an essential step in the acquisition of pluripotency during reprogramming. Sci. Adv. 2019;5:eaaw0025. doi: 10.1126/sciadv.aaw0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Inak G., Rybak-Wolf A., Lisowski P., Pentimalli T.M., Jüttner R., Glažar P., Uppal K., Bottani E., Brunetti D., Secker C., et al. Defective metabolic programming impairs early neuronal morphogenesis in neural cultures and an organoid model of Leigh syndrome. Nat. Commun. 2021;12:1929. doi: 10.1038/s41467-021-22117-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Galera-Monge T., Zurita-Díaz F., Canals I., Hansen M.G., Rufián-Vázquez L., Ehinger J.K., Elmér E., Martin M.A., Garesse R., Ahlenius H., et al. Mitochondrial Dysfunction and Calcium Dysregulation in Leigh Syndrome Induced Pluripotent Stem Cell Derived Neurons. Int. J. Mol. Sci. 2020;21:3191. doi: 10.3390/ijms21093191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lorenz C., Prigione A. Mitochondrial metabolism in early neural fate and its relevance for neuronal disease modeling. Curr. Opin. Cell Biol. 2017;49:71–76. doi: 10.1016/j.ceb.2017.12.004. [DOI] [PubMed] [Google Scholar]
- 61.Gerards M., Sallevelt S.C., Smeets H.J. Leigh syndrome: Resolving the clinical and genetic heterogeneity paves the way for treatment options. Mol. Genet. Metab. 2016;117:300–312. doi: 10.1016/j.ymgme.2015.12.004. [DOI] [PubMed] [Google Scholar]
- 62.Hong C.M., Na J.H., Park S., Lee Y.M. Clinical Characteristics of Early-Onset and Late-Onset Leigh Syndrome. Front. Neurol. 2020;11:267. doi: 10.3389/fneur.2020.00267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kempf J., Knelles K., Hersbach B.A., Petrik D., Riedemann T., Bednarova V., Janjic A., Simon-Ebert T., Enard W., Smialowski P., et al. Heterogeneity of neurons reprogrammed from spinal cord astrocytes by the proneural factors Ascl1 and Neurogenin2. Cell Rep. 2021;36:109409. doi: 10.1016/j.celrep.2021.109409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bocchi R., Masserdotti G., Götz M. Direct neuronal reprogramming: Fast forward from new concepts toward therapeutic approaches. Neuron. 2022;110:366–393. doi: 10.1016/j.neuron.2021.11.023. [DOI] [PubMed] [Google Scholar]
- 65.Verkaart S., Koopman W.J., van Emst-de Vries S.E., Nijtmans L.G., van den Heuvel L.W., Smeitink J.A., Willems P.H. Superoxide production is inversely related to complex I activity in inherited complex I deficiency. Biochim. Biophys. Acta. 2007;1772:373–381. doi: 10.1016/j.bbadis.2006.12.009. [DOI] [PubMed] [Google Scholar]
- 66.Valsecchi F., Grefte S., Roestenberg P., Joosten-Wagenaars J., Smeitink J.A., Willems P.H., Koopman W.J. Primary fibroblasts of NDUFS4(-/-) mice display increased ROS levels and aberrant mitochondrial morphology. Mitochondrion. 2013;13:436–443. doi: 10.1016/j.mito.2012.12.001. [DOI] [PubMed] [Google Scholar]
- 67.Iannetti E.F., Smeitink J.A.M., Willems P.H.G.M., Beyrath J., Koopman W.J.H. Rescue from galactose-induced death of Leigh Syndrome patient cells by pyruvate and NAD. Cell Death Dis. 2018;9:1135. doi: 10.1038/s41419-018-1179-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Thompson Legault J., Strittmatter L., Tardif J., Sharma R., Tremblay-Vaillancourt V., Aubut C., Boucher G., Clish C.B., Cyr D., Daneault C., et al. A Metabolic Signature of Mitochondrial Dysfunction Revealed through a Monogenic Form of Leigh Syndrome. Cell Rep. 2015;13:981–989. doi: 10.1016/j.celrep.2015.09.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ryall J.G., Dell'orso S., Derfoul A., Juan A., Zare H., Feng X., Clermont D., Koulnis M., Gutierrez-Cruz G., Fulco M., et al. The NAD(+)-dependent SIRT1 deacetylase translates a metabolic switch into regulatory epigenetics in skeletal muscle stem cells. Cell Stem Cell. 2015;16:171–183. doi: 10.1016/j.stem.2014.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cantó C., Houtkooper R.H., Pirinen E., Youn D.Y., Oosterveer M.H., Cen Y., Fernandez-Marcos P.J., Yamamoto H., Andreux P.A., Cettour-Rose P., et al. The NAD(+) precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metab. 2012;15:838–847. doi: 10.1016/j.cmet.2012.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Khan N.A., Auranen M., Paetau I., Pirinen E., Euro L., Forsström S., Pasila L., Velagapudi V., Carroll C.J., Auwerx J., et al. Effective treatment of mitochondrial myopathy by nicotinamide riboside, a vitamin B3. EMBO Mol. Med. 2014;6:721–731. doi: 10.1002/emmm.201403943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lee C.F., Caudal A., Abell L., Nagana Gowda G.A., Tian R. Targeting NAD(+) Metabolism as Interventions for Mitochondrial Disease. Sci. Rep. 2019;9:3073. doi: 10.1038/s41598-019-39419-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Marcus J.M., Andrabi S.A. SIRT3 Regulation Under Cellular Stress: Making Sense of the Ups and Downs. Front. Neurosci. 2018;12:799. doi: 10.3389/fnins.2018.00799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Brown K.D., Maqsood S., Huang J.Y., Pan Y., Harkcom W., Li W., Sauve A., Verdin E., Jaffrey S.R. Activation of SIRT3 by the NAD(+) precursor nicotinamide riboside protects from noise-induced hearing loss. Cell Metab. 2014;20:1059–1068. doi: 10.1016/j.cmet.2014.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sasaki Y., Araki T., Milbrandt J. Stimulation of nicotinamide adenine dinucleotide biosynthetic pathways delays axonal degeneration after axotomy. J. Neurosci. 2006;26:8484–8491. doi: 10.1523/JNEUROSCI.2320-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chakrabarty R.P., Chandel N.S. Mitochondria as Signaling Organelles Control Mammalian Stem Cell Fate. Cell Stem Cell. 2021;28:394–408. doi: 10.1016/j.stem.2021.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Karamanlidis G., Lee C.F., Garcia-Menendez L., Kolwicz S.C., Jr., Suthammarak W., Gong G., Sedensky M.M., Morgan P.G., Wang W., Tian R. Mitochondrial complex I deficiency increases protein acetylation and accelerates heart failure. Cell Metab. 2013;18:239–250. doi: 10.1016/j.cmet.2013.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Missiroli S., Patergnani S., Caroccia N., Pedriali G., Perrone M., Previati M., Wieckowski M.R., Giorgi C. Mitochondria-associated membranes (MAMs) and inflammation. Cell Death Dis. 2018;9:329. doi: 10.1038/s41419-017-0027-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cao S.S., Kaufman R.J. Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease. Antioxid. Redox Signal. 2014;21:396–413. doi: 10.1089/ars.2014.5851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhang Z., Zhang L., Zhou L., Lei Y., Zhang Y., Huang C. Redox signaling and unfolded protein response coordinate cell fate decisions under ER stress. Redox Biol. 2019;25:101047. doi: 10.1016/j.redox.2018.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wang M., Kaufman R.J. Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature. 2016;529:326–335. doi: 10.1038/nature17041. [DOI] [PubMed] [Google Scholar]
- 82.Martucciello S., Masullo M., Cerulli A., Piacente S. Natural Products Targeting ER Stress, and the Functional Link to Mitochondria. Int. J. Mol. Sci. 2020;21:1905. doi: 10.3390/ijms21061905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rutkowski D.T., Arnold S.M., Miller C.N., Wu J., Li J., Gunnison K.M., Mori K., Sadighi Akha A.A., Raden D., Kaufman R.J. Adaptation to ER stress is mediated by differential stabilities of pro-survival and pro-apoptotic mRNAs and proteins. PLoS Biol. 2006;4:e374. doi: 10.1371/journal.pbio.0040374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tabas I., Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell Biol. 2011;13:184–190. doi: 10.1038/ncb0311-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Harding H.P., Zhang Y., Bertolotti A., Zeng H., Ron D. Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol. Cell. 2000;5:897–904. doi: 10.1016/s1097-2765(00)80330-5. [DOI] [PubMed] [Google Scholar]
- 86.Harding H.P., Zhang Y., Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature. 1999;397:271–274. doi: 10.1038/16729. [DOI] [PubMed] [Google Scholar]
- 87.Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012;13:89–102. doi: 10.1038/nrm3270. [DOI] [PubMed] [Google Scholar]
- 88.Masserdotti G., Gillotin S., Sutor B., Drechsel D., Irmler M., Jørgensen H.F., Sass S., Theis F.J., Beckers J., Berninger B., et al. Transcriptional Mechanisms of Proneural Factors and REST in Regulating Neuronal Reprogramming of Astrocytes. Cell Stem Cell. 2015;17:74–88. doi: 10.1016/j.stem.2015.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Alexa A., Rahnenfuhrer J. topGO: Enrichment Analysis for Gene Ontology. 2023. R package version 2.54.0, https://bioconductor.org/packages/topGO.
- 90.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wickham H. ggplot2: Elegant Graphics for Data Analysis. Springer-Verlag New York; 2016. [Google Scholar]
- 92.Korotkevich G., Sukhov V., Sergushichev A. Fast gene set enrichment analysis. Preprint at bioRxiv. http://biorxiv.org/content/early/2016/06/20/060012
- 93.Hao Y., Hao S., Andersen-Nissen E., Mauck W.M., 3rd, Zheng S., Butler A., Lee M.J., Wilk A.J., Darby C., Zager M., et al. Integrated analysis of multimodal single-cell data. Cell. 2021;184:3573–3587.e29. doi: 10.1016/j.cell.2021.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wu T., Hu E., Xu S., Chen M., Guo P., Dai Z., Feng T., Zhou L., Tang W., Zhan L., et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation (Camb) 2021;2:100141. doi: 10.1016/j.xinn.2021.100141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Popp B., Krumbiegel M., Grosch J., Sommer A., Uebe S., Kohl Z., Plotz S., Farrell M., Trautmann U., Kraus C., et al. Need for high-resolution Genetic Analysis in iPSC: Results and Lessons from the ForIPS Consortium. Sci Rep. 2018;8:17201. doi: 10.1038/s41598-018-35506-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Heinrich C., Gascón S., Masserdotti G., Lepier A., Sanchez R., Simon-Ebert T., Schroeder T., Götz M., Berninger B. Generation of subtype-specific neurons from postnatal astroglia of the mouse cerebral cortex. Nat. Protoc. 2011;6:214–228. doi: 10.1038/nprot.2010.188. [DOI] [PubMed] [Google Scholar]
- 97.Heins N., Malatesta P., Cecconi F., Nakafuku M., Tucker K.L., Hack M.A., Chapouton P., Barde Y.A., Götz M. Glial cells generate neurons: the role of the transcription factor Pax6. Nat. Neurosci. 2002;5:308–315. doi: 10.1038/nn828. [DOI] [PubMed] [Google Scholar]
- 98.Drouin-Ouellet J., Lau S., Brattås P.L., Rylander Ottosson D., Pircs K., Grassi D.A., Collins L.M., Vuono R., Andersson Sjöland A., Westergren-Thorsson G., et al. REST suppression mediates neural conversion of adult human fibroblasts via microRNA-dependent and -independent pathways. EMBO Mol. Med. 2017;9:1117–1131. doi: 10.15252/emmm.201607471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Bagnoli J.W., Ziegenhain C., Janjic A., Wange L.E., Vieth B., Parekh S., Geuder J., Hellmann I., Enard W. Sensitive and powerful single-cell RNA sequencing using mcSCRB-seq. Nat. Commun. 2018;9:2937. doi: 10.1038/s41467-018-05347-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Barbar L., Rusielewicz T., Zimmer M., Kalpana K., Fossati V. Isolation of Human CD49f(+) Astrocytes and In Vitro iPSC-Based Neurotoxicity Assays. Star Protoc. 2020;1:100172. doi: 10.1016/j.xpro.2020.100172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Riedemann T., Straub T., Sutor B. Two types of somatostatin-expressing GABAergic interneurons in the superficial layers of the mouse cingulate cortex. PLoS One. 2018;13 doi: 10.1371/journal.pone.0200567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Bean B.P. The action potential in mammalian central neurons. Nat. Rev. Neurosci. 2007;8:451–465. doi: 10.1038/nrn2148. [DOI] [PubMed] [Google Scholar]
- 104.Rothman J.S., Silver R.A. NeuroMatic: An Integrated Open-Source Software Toolkit for Acquisition, Analysis and Simulation of Electrophysiological Data. Front. Neuroinform. 2018;12:14. doi: 10.3389/fninf.2018.00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
RNASeq data are deposited in GEO with the number GSE248129. Raw data are available upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to re-analyze the data reported in this paper is available from the lead contact upon request.