

Abstract
Intervertebral disc degeneration (IVDD) is a prevalent cause of low back pain and a leading contributor to disability. IVDD progression involves pathological shifts marked by low-grade inflammation, extracellular matrix remodeling, and metabolic disruptions characterized by heightened glycolytic pathways, mitochondrial dysfunction, and cellular senescence. Extensive posttranslational modifications of proteins within nucleus pulposus cells and chondrocytes play crucial roles in reshaping the intervertebral disc phenotype and orchestrating metabolism and inflammation in diverse contexts. This review focuses on the pivotal roles of phosphorylation, ubiquitination, acetylation, glycosylation, methylation, and lactylation in IVDD pathogenesis. It integrates the latest insights into various posttranslational modification-mediated metabolic and inflammatory signaling networks, laying the groundwork for targeted proteomics and metabolomics for IVDD treatment. The discussion also highlights unexplored territories, emphasizing the need for future research, particularly in understanding the role of lactylation in intervertebral disc health, an area currently shrouded in mystery.
Introduction
Low back pain (LBP) is a degenerative disease that profoundly impacts the quality of life and places a substantial burden on society [1]. Various factors contribute to LBP, with intervertebral disc degeneration (IVDD) identified as the primary culprit [2]. Aging, prolonged overuse, and back injuries collectively induce degenerative changes in the intervertebral disc (IVD) [3]. These changes encompass the loss of collagen II and proteoglycan content, progressively disrupting normal spinal structure and function and ultimately leading to pain and disability [4]. The cartilage endplate (CEP)—a thin layer of hyaline cartilage situated between the vertebral body and IVD—undergoes structural alterations that impede the delivery of essential nutrients and oxygen to the IVD, emerging as another significant contributor to IVDD [5–6]. This cascade also results in an increase in metabolic intermediates for the biosynthesis of inflammatory and degradative proteins [7–8], activating key factors and inflammatory signaling pathways implicated in catabolic processes. Catabolic and hypoxic microenvironments further contribute to the degradation of the extracellular matrix (ECM) and the loss of homeostasis in the IVD microenvironment [9–10]. Moreover, mounting evidence suggests that IVDD involves low-grade inflammation and metabolic disturbances, prompting a shift in research focus toward metabolic features and changes in disease pathophysiology [11–12].
Nucleus pulposus cells (NPCs) within the degenerative IVD microenvironment and inflammatory conditions undergo pathological transformations marked by metabolic disorders and matrix remodeling [13]. These transformations are characterized by enhanced glycolysis pathways, mitochondrial dysfunction, and cellular senescence [14]. A shift toward glycolysis as the primary energy source results in lactate accumulation and a subsequent decrease in pH within the IVD, leading to tissue acidosis and ECM degradation [15]. Dysfunctional mitochondria generate excessive reactive oxygen species (ROS), which are the byproducts of oxidative phosphorylation [16]. ROS accumulation overwhelms cellular antioxidant defense mechanisms, resulting in oxidative stress and damage to cellular macromolecules, including lipids, proteins, and DNA, which activate inflammatory pathways and contribute to cellular senescence [17]. The accumulation of senescent cells within the disc tissue further exacerbates inflammation and matrix breakdown and impairs tissue repair processes associated with IVDD. In response to an increased nutrient supply, NPCs activate systems to boost synthesis output and promote energy storage and growth [18]. This metabolic response plays a crucial role in maintaining cellular homeostasis and supporting tissue integrity within the IVD [19]. Conversely, starvation triggers metabolic programs that inhibit biosynthetic pathways and activate catabolic energy-generating pathways [20]. These metabolic adaptations under nutrient deprivation conditions may lead to the breakdown of ECM components and compromise disc structural integrity, contributing to IVDD progression. Crucially, the activation of numerous hypoxia-responsive signaling pathways, essential for maintaining mitochondrial homeostasis and acid-base balance, underscores the characterization of IVDD by disturbances in intracellular metabolic regulation [21–22]. However, a detailed understanding of how metabolic alterations are induced during IVDD remains the subject of ongoing investigation.
Metabolism can be regulated through changes in metabolic enzyme expression, substrate concentrations controlling reaction kinetics, or posttranslational modifications (PTMs) of proteins that facilitate these reactions [23–24]. These sequential events collectively govern the initiation, activation, and termination of metabolic reactions, determining the intensity of cell signaling pathways, cellular activities, tissue functions, and overall health and disease [25]. While the first two regulations establish key checkpoints reliant on meticulous regulatory mechanisms at the molecular level, regulation at the posttranslational level serves as a transducer of these metabolic signals and plays a pivotal role [26]. Traditional PTMs, such as phosphorylation and ubiquitination, have been shown to rapidly regulate protein stability and activity, ensuring the appropriate duration and extent of metabolism [27–28]. Unconventional PTMs, such as acetylation, glycosylation, and methylation, are increasingly implicated in the compartmentalization, transport, and physical interactions of key molecules, effectively regulating subcellular structure homeostasis and inflammatory responses [29–31]. This review focuses on the role of PTMs in IVDD (Fig 1). By delving into the mechanisms by which PTMs control metabolism and inflammatory responses in NPCs and chondrocytes, we consolidate the current state of knowledge in this field and provide a foundation for targeting proteomics and metabolomics to prevent IVDD. Furthermore, we identified priority areas for future research, particularly the potential role of lactylation in IVD, an area currently shrouded in mystery.
Fig. 1.

Representative PTMs in IVDD. IVDD is marked by depletion of the ECM, fibrous and dehydrated NP, severe structural alterations of collagen fibers in the AF, extensive cartilage damage, subchondral bone sclerosis, and a significant reduction in IVD height. This intricate process is influenced by various PTMs, including phosphorylation, ubiquitination, acetylation, glycosylation, methylation, and lactylation.
Phosphorylation
Phosphorylation regulates metabolic network
Phosphorylation, arguably the most prevalent and extensively studied PTM, is intricately linked to almost every cellular process [32]. The two primary mechanisms through which phosphorylation governs protein function involve the modulation of protein interactions and changes in protein conformation. This modification occurs through covalent binding of phosphate groups to serine, threonine, and tyrosine residues, catalyzed by protein kinases, and can be reversed by protein phosphatases [33]. Notably, robust communication between key kinases that regulate metabolism is a distinctive feature of the metabolic signaling network. For instance, AMP (adenosine monophosphate)-activated protein kinase (AMPK) and mammalian target of rapamycin (mTOR) jointly control ULK1 [34]. AMPK activation leads to a significant reduction in mTOR phosphorylation, subsequently activating ULK1 and initiating autophagosome assembly [35]. As a result, t-butyl hydroperoxide (TBHP)-induced autophagic influx is restored. Furthermore, AMPK-induced autophagy plays a crucial role in promoting ECM synthesis and inhibiting ECM degradation. This regulation occurs through the control of anabolic genes (e.g., collagen II and aggrecan) and catabolic genes [e.g., matrix metalloproteinase 13 (MMP13) and ADAMTS5], safeguarding against TBHP-induced ECM destruction [36]. Phosphorylated proteomics provides insights into the intricate network of signal processors. For instance, AMPK induces SIRT3 expression in human NPCs by phosphorylating PGC-1α [37]. Recent research has identified acetyl-CoA (coenzyme A) carboxylase (ACC) as a target of AMPK in lipopolysaccharide (LPS)-stimulated rat NPCs [38]. Through ACC phosphorylation, AMPK mitigates the generation of free radicals (ROS and nitric oxide) and production of proinflammatory cytokines (IL-1β and TNF-α). AMPK-mediated ACC activation also preserves aggrecan and collagen II content while inhibiting the expression of major ECM-degrading enzymes [39]. Independent phosphoproteomics studies have revealed that increased proteoglycans in the IVD inhibit the mTOR phosphatidylinositol 3-kinase (PI3K)/AKT signaling pathway, thereby promoting autophagy and reducing apoptosis in rat NPCs. Additionally, these studies have identified elevated BRD4 expression in NPCs of degenerative IVD. Silencing BRD4 activates the AMPK pathway, inhibits mTOR activity, promotes ULK1 phosphorylation, increases the LC3-ii/LC3-i ratio and Beclin-1 levels, and reduces P62 level in NPCs [40]. These findings suggest that autophagy mediated by mTOR activation can mitigate oxidative stress injury and impede IVDD progression. Recently, researchers found overloading-induced activation of ras homolog family member A-protein kinase N, which phosphorylates keratin 8 serine 43, subsequently impeding Golgi apparatus trafficking of the small guanosine triphosphatase RAB33B. This disruption inhibits autophagosome initiation and contributes to IVDD pathogenesis, suggesting a critical role of phosphorylation-regulated autophagy in IVDD [41].
Metabolic stress-dependent phosphorylation
A burgeoning paradigm in cell signaling asserts that beyond merely regulating metabolism, signaling is reciprocally governed by metabolic processes [42]. Intracellular AMP levels increase in response to energy deprivation or metabolic stress stimuli. Elevated AMP binds to the γ-subunit of AMPK and induces a conformational change [43]. This alteration enables the phosphorylation of a threonine residue by a specific kinase on the catalytic α-subunit, ultimately activating AMPK [44]. NPCs dynamically modulate their metabolic rate to establish novel metabolic homeostasis by reconciling adenosine triphosphatase (ATP) demand and supply pathways, potentially regulated by the activation of the mTOR signaling pathway. Intriguingly, NPCs may enhance autophagic flux, eliminate intracellular ROS, and mitigate cellular damage by suppressing mTOR activity through continuous exposure to adverse factors [45]. Crucially, AMPK curtails mTORC1 activity by phosphorylating TSC2 and Raptor, key members of the mTORC1 pathway, thereby restraining protein and lipid synthesis and limiting cell growth [46–47]. In the context of obesity, excess substrate supplied to insulin-sensitive cells induces mitochondrial hyperactivity, leading to ATP overproduction and AMPK inhibition [48]. Concurrently, obesity often coincides with reduced secretion of the endogenous AMPK activator adiponectin, which contributes to IVDD [49]. Furthermore, mTORC1 can sense amino acids via the Rag complex, a regulator of the lysosomal surface [50–52]. AKT activity—a controller of cell growth and proliferation—is directly regulated by metabolism through an ATP-dependent allosteric switch [53]. Investigations have revealed that this ATP-dependent conformational change in AKT governs the pathway of inhibitory phosphatases, modulating kinase activity in response to cellular ATP levels [54–56]. Glycolysis also exerts a profound and direct influence on signaling. For instance, the phosphofructokinase 2 isoform positively regulates glycolytic flux and modulates insulin-like growth factor-1 (IGF-1) [57]. Meanwhile, IGF-1/AKT sustains anabolism and prevents endplate calcification by down-regulating carbonic anhydrase in degenerative CEP (Fig 2). In summary, phosphorylation serves as a docking site for intramolecular and intermolecular protein interactions. It can directly regulate enzyme activity by inducing changes in the protein conformation. Furthermore, phosphorylation governs the subcellular localization and turnover of its targets, influencing signaling in conjunction with other PTMs.
Fig. 2.

Interplay between phosphorylation and metabolism. Growth factors, hypoxia, stress, amino acids, and TNF-α activate mTORC1 phosphorylation, engaging in different signaling pathways that regulate lipid and glucose metabolism, nucleotide metabolism, and autophagy. Activation of mTORC2 is implicated in autophagy, cell survival, osteogenic differentiation, and aging. AMPK promotes apoptosis and senescence, inhibits cell proliferation, and retards cell cycle progression by inhibiting mTOR. Additionally, it inhibits oxidative stress, inflammation, and ECM destruction in IVDD through the activation of ACC. AMPK induces autophagy in IVD by regulating the mTOR-ULK1 pathway, promoting mitophagy, and inhibiting oxidative stress and inflammation in IVDD by activating PGC-1α and SIRT3. Furthermore, AMPK may shield IVDD from intrinsic apoptosis, senescence, and ECM destruction by maintaining mitochondrial homeostasis.
Ubiquitination
Ubiquitin: Inflammation regulator
Ubiquitin—a ubiquitous small regulatory protein in eukaryotes—exerts its influence by covalently attaching to substrates through the coordinated activities of the E1-E2-E3 enzyme ubiquitin thioester cascade [58]. This process involves the participation of E1 ubiquitin-activating enzyme, E2 ubiquitin-conjugating enzymes, and E3 ubiquitin-protein ligases [59–60]. Ubiquitin serves as a crucial regulator of substrate stability and signaling, playing a role in temporal, spatial, and specific contexts with significant implications for health and disease [61–63]. A20—identified as a dual-function ubiquitin regulatory enzyme—possesses ubiquitin E3 ligase and deubiquitinase capabilities and functions as a linear ubiquitin effector [64–65]. Mutations leading to impaired A20 protein function are associated with autoimmune and autoinflammatory phenotypes, given their role as inflammation suppressors. For instance, A20 plays a pivotal role in attenuating NLRP3 inflammasome-mediated apoptosis in NPCs by promoting mitophagy and stabilizing mitochondrial dynamics [66–67]. Targeting NLRP3 ubiquitination has emerged as a potential therapeutic approach for inhibiting NPC apoptosis. Platelet-rich derived exosomes facilitate NLRP3 autophagic degradation by enhancing NLRP3 ubiquitination, thereby reducing IL-1β and caspase-1 production and slowing IVDD progression [68]. Knockdown of ubiquitin-specific peptidase 14 (USP14) to induce NLRP3 ubiquitination inhibits pyroptosis in chondrocytes located in the annulus fibrosus (AF) [69]. X-linked inhibitor of apoptosis protein (XIAP)—an E3 ubiquitin-protein ligase—governs both the cell death pathway and immune signaling events dependent on nuclear factor κB (NF-κB) [70]. It facilitates NOD2 proinflammatory communication by promoting K63-linked RIPK2 polyubiquitination in the NOD2 communication complex, attracting LUBAC, and intensifying the generation of cytokines that are dependent on NF-κB and mitogen-activated protein kinase (MAPK). Reduced XIAP expression is directly correlated with excessive ECM apoptosis and an imbalance in the synthesis of catabolic factors [71]. SIAH1 targets and ubiquitinates XIAP, mediating senescence, apoptosis, increased inflammation, and decreased ECM synthesis in NPCs [72].
Ubiquitination regulates oxidative stress
Maintaining a delicate balance between ROS and antioxidants is crucial for the normal function of IVD [73]. LPS induces ROS production and catabolism in rat endplate chondrocytes, triggering the secretion of inflammatory factors by macrophages and contributing to inflammation-related IVDD [74–75]. This process is dependent on ZNF598-mediated NRF2 ubiquitination [76]. Prussian blue nanoparticles enhance the mitochondrial structure and antioxidant capacity by stabilizing the ubiquitin-proteasome degradation of superoxide dismutase 1 (SOD1). This, in turn, increases mRNA and protein levels related to the oxidoreductase system, ultimately rescuing ROS-induced IVDD [77]. Conditional deletion of the E3 ubiquitin ligase von Hippel-Lindau tumor suppressor (VHL) in the CEP and AF of adult mice results in up-regulation of hypoxia-inducible factor-1α (HIF-1α) expression and age-dependent IVDD [78]. TRIM21 acts as a ubiquitin E3 ligase and drives oxidative stress-induced IVDD by promoting HIF-1α degradation [79]. Furthermore, TBHP induced the up-regulation of TRIM32 expression in rat NPCs, activating the β-catenin signaling pathway through the ubiquitination of Axin1, thereby regulating NPC apoptosis [80]. Lipid peroxidation (LPO) is a consequence of the interaction between ROS and lipids. Ferroptosis—a form of programmed cell death characterized by glutathione depletion and GPX4 inactivation—results in LPO accumulation [81]. Due to similar pathological processes, the interplay between oxidative stress and ferroptosis has been investigated in IVDD [82]. Mass spectrometry revealed that USP11 maintains stability by directly binding and deubiquitinating SIRT3, significantly improving oxidative stress-induced ferroptosis and alleviating IVDD [83]. Additionally, polydopamine (PDA) nanoparticles (NPs) colocalize with GPX4 around mitochondria, inhibiting ubiquitin-mediated degradation and down-regulating malondialdehyde and LPO production [84]. In this process, ROS, LPO, and ferroptosis represent direct responses to oxidative stress, whereas HIF-1α and mitochondrial homeostasis represent indirect responses to oxidative stress (Fig. 3). Ubiquitination regulates the effects of oxidative stress on NPCs and chondrocytes by modulating their metabolic state and cell survival mechanisms. The unique and time-dependent pattern of ubiquitination underscores its significance as a regulatory process that explains alterations in signaling and cellular milieu.
Fig. 3.

Ubiquitination regulation of inflammation and oxidative stress. The process involves successive enzyme interactions: ubiquitin activation and thioester formation by the E1-activating enzyme, ubiquitin trans-thiylation by the E2-conjugating enzyme, and ubiquitin attachment to the substrate assisted by E3 ligase. A20, a bifunctional ubiquitin regulatory enzyme, plays a role in inflammatory factor-induced apoptosis and pyroptosis in NPCs. The pivotal molecular event involves the direct interaction between USP11 and SIRT3, inhibiting oxidative stress-induced ferroptosis, improving IVDD, and alleviating the pain response. PDA NPs, functioning as a biological material, colocalize with GPX4 around the mitochondria to impede ubiquitin-mediated degradation, offering protective effects through the conversion and clearance of phospholipid hydroperoxides.
Acetylation
The multifunction of sirtuins
Acetylation is governed by two classes of enzymes: lysine acetyltransferases, which are responsible for transferring acetyl moieties to lysine residues, and lysine deacetylases, which catalyze acetyl group removal. Among these, sirtuins are a highly conserved class of nicotinamide adenine dinucleotide (NAD+)-dependent histone deacetylases (HDACs) that collectively regulate a diverse array of cellular functions [85–87]. SIRT1, a potent inhibitor of inflammation, deacetylates lysine residues on the p65 subunit of NF-κB, thereby inhibiting its transcriptional activity [88]. SIRT2 plays a crucial role in glucose homeostasis through deacetylation and activation of glucose-6-phosphatase dehydrogenase [89]. SIRT3, a vital mitochondrial deacetylase, plays a pivotal role in promoting fatty acid oxidation, Krebs cycle, and urea cycle [90]. Additionally, SIRT3 provides metabolic benefits and safeguards against metabolic dysregulation and oxidative stress associated with obesity [91]. SIRT4 controls glutamine breakdown [92], fatty acid oxidation [93], and lipid breakdown by deacetylating malonyl-CoA decarboxylase. SIRT5 selectively focuses on modifications other than lysine acetylation, including malonylation [94], succinylation [95], and glutarylation [96]. SIRT6 knockout mice exhibit symptoms of premature aging and succumb to premature death due to severe hypoglycemia [97]. SIRT6 overexpression in male mice has the potential to extend the lifespan by interfering with IGF-1 signaling [98]. By deacetylating the transcription factor GABPβ1, SIRT7 controls the expression of nuclear-encoded mitochondrial genes and provides defense against mitochondrial diseases [99]. Recent studies have highlighted the multifunctional role of sirtuins in IVD metabolism. They coordinate stress responses by collectively regulating substrate clusters rather than by isolating key metabolic enzymes [100]. Moreover, their dependence on NAD+ links their enzymatic activity to the metabolic state of the cells, rendering them stress sensors [101]. Consequently, a deeper exploration of the regulation of IVD metabolism is warranted, especially in maintaining mitochondrial homeostasis.
Acetylation maintains mitochondrial metabolism
Mitochondria play a crucial role in cellular activities by producing ATP through oxidative phosphorylation to ensure normal cellular function [102–103]. However, mitochondrial dysfunction or abnormal electron leakage can lead to oxidative stress, causing damage to cells and triggering cellular senescence [104]. In response to oxidative stress, SIRT2 is up-regulated, leading to deacetylation of FOXO3a and enhanced expression of FOXO3a-targeted genes, ultimately reducing ROS production [105]. As an effector of SIRT2, PGC-1α is indispensable for cell viability and participates in mitochondrial biogenesis and energy regulation [106–107]. SIRT2 protects chondrocytes from apoptosis triggered by oxidative stress by targeting PGC-1α to inhibit mitophagy. By up-regulating antioxidant enzymes such as manganese SOD, SIRT1 deacetylates PGC-1α to suppress oxidative stress [108]. Additionally, SIRT1 removes acetyl groups from FOXO3a, transporting them to the nucleus and enhancing the production of additional antioxidant enzymes and catalase to shield cells against damage induced by oxidative stress [109]. When cellular energy demand increases, the NAD+/NADH ratio also increases, leading to heightened SIRT3 activity [110]. Elevated SIRT3 levels during mitochondrial stress enhance the expression of SOD and catalase, reducing ROS buildup during stressful conditions [111]. By targeting AIFM1, SIRT5 disrupts its interaction with CHCHD4, consequently reducing electron transport chain (ETC) complex subunits and impairing mitochondrial function in NPCs, thereby contributing to IVDD progression under mechanical stress [112]. Notably, mechanical stress refers to the physical loading experienced by NPCs within the IVD. NPCs play a pivotal role in maintaining ECM and metabolic homeostasis within the IVD. Upon exposure to mechanical stress, such as repetitive loading or abnormal mechanical forces, aberrant acetylation may serve as a baroreceptor signal, triggering various metabolic and molecular alterations in NPCs, ultimately culminating in IVDD [113]. In addition to sirtuins, acetylation of other proteins, as revealed by proteomic studies, is involved in cellular metabolism. For instance, KLF5 contributes to cartilage degradation by mediating p65 acetylation in the NF-κB cascade activated by IL-1β [114]. Inhibition of HDAC by GSK3β down-regulates KLF5 and ASK1 and prevents IVDD progression [115]. In response to TBHP stimulation, the increased nuclear movement of HDAC3 strengthened its interaction with peroxisome proliferator-activated receptor γ (PPARγ), and HDAC3 knockdown increased PPARγ acetylation and trans-activation, effectively reversing oxidative stress in NPCs (Fig 4). Notably, considering that cofactors such as acyl-CoA donors are metabolites, the cellular metabolic state also plays a vital role in determining the targets and kinetics of acetylation. However, the relevant content of metabolite-mediated PTMs is beyond the scope of this review [116].
Fig. 4.
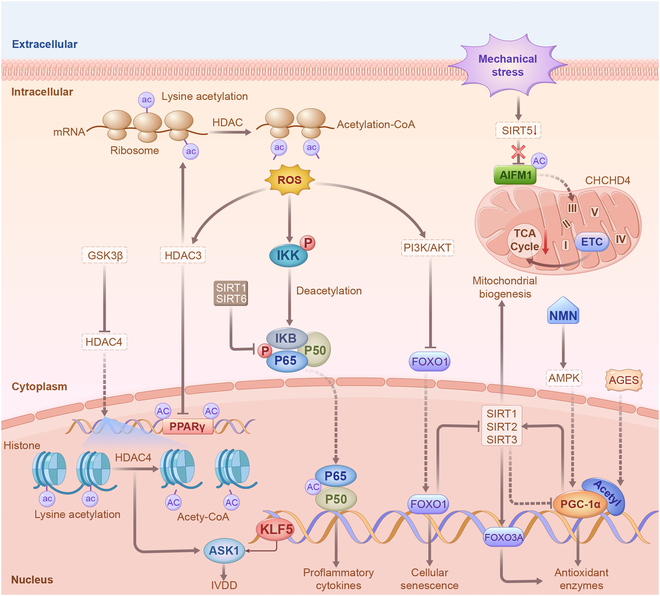
Acetylation regulation of inflammation and maintenance of mitochondrial homeostasis. The deacetylation of lysine in the p65 subunit of NF-κB inhibits its transcriptional activity, mitigating the ensuing inflammatory response. The deacetylation of FOXO3 enhances the expression of FOXO3-dependent antioxidant enzymes and catalase, thereby reducing ROS generation. SIRT1 and SIRT2 elevate the expression of antioxidant enzymes by deacetylating PGC-1α. Mechanical loading increases AIFM1 acetylation in NPCs by suppressing SIRT5 expression, disrupting the interaction between AIFM1 and CHCHD4, and resulting in reduced mitochondrial ETC complex subunits. ROS overproduction induces nuclear translocation of HDAC3, augmenting the association between HDAC3 and PPARγ in NPCs.
Glycosylation
Glycosylation regulates ECM metabolism
Protein glycosylation involves the reaction of sugar chain molecules with protein amino acid side chain active groups catalyzed by glycosyltransferase, forming glycosidic bonds that connect sugar chains to proteins. Based on the type of chemical bonds formed during glycosylation, they can be mainly divided into N-glycosylation and O-glycosylation [117]. N-glycosylation occurs on an asparagine residue in the presence of an asparagine-X-Ser/Thr motif (where X is any amino acid other than proline) and is predominantly found in secreted and membrane-bound glycoproteins. O-glycosylation mainly occurs in the Golgi apparatus on serine or threonine residues via the action of glycosyltransferases [118]. This glycosylation pattern is evident in mucosal secretions and transmembrane glycoproteins found on cell surfaces, particularly where polysaccharides are exposed to the external environment. Within the IVD, structural integrity and biomechanical properties rely on two key constituents of the ECM: aggrecan and collagen II [119]. Aggrecan, a proteoglycan, plays a pivotal role in retaining water molecules within the disc, thereby creating osmotic pressure that helps resist stress [36]. Meanwhile, collagen II provides the disc with tensile strength and structural support, enhancing its overall elasticity and capacity to endure mechanical loads [4]. These components synergistically increase the osmotic pressure of the disc, provide essential structural support, and confer elasticity, collectively enabling the disc to effectively function as a load-bearing structure within the spine. However, in IVDD, the accumulation of glycosylation products chemically modifies the aggregate proteins and collagen II, hindering their ability to repair and renew. This imbalance allows ECM breakdown to outpace the building process [120]. Moreover, the up-regulation of catabolic enzymes in the matrix further promotes ECM degradation and accelerates IVDD progression. Studies have reported that the accumulation of glycosylated alterations in aggrecan and collagen II in the IVD increases with age and is partially influenced by sugars. In individuals with diabetes, increased levels of blood sugar expedite the buildup of glycosylation substances [121]. The buildup of glycosylation in NPCs enhances MMP2 expression through the extracellular signal–regulated kinase (ERK) signaling pathway, leading to ECM degradation. In diabetic IVD, MMP13 is up-regulated by BRD4, transmitting MAPK and NF-κB signals and inducing autophagy [122]. Additionally, BRD4 inhibition prevents ECM degradation in diabetic rats. Interestingly, pyridoxamine, as a glycosylation inhibitor [123], effectively alleviates the up-regulation of ADAMTS5 and MMP13, delaying IVDD progression in diabetic mice.
Glycosylation regulates endoplasmic reticulum stress
The accumulation of glycosylated proteins in IVD tissues affects endoplasmic reticulum (ER) homeostasis, leading to the buildup of unfolded/misfolded proteins, known as ER stress [124]. ER phagocytosis—a form of selective autophagy—involves phagocytosis of specific ER fragments by autophagosomes through specific receptors. These fragments are then transported to lysosomes for degradation, restoring cellular energy levels and ER homeostasis [125]. O-GlcNAc glycosylation, a form of O-glycosylation, and ER-phagy are well-characterized adaptive regulatory mechanisms that help maintain cellular homeostasis and function under various stress conditions. Research has revealed that O-GlcNAc glycosylation and O-GlcNAc transferase expression profiles are significantly increased in degenerated IVD tissues and nutrient-deprived NPCs [126]. Interestingly, increased O-GlcNAc glycosylation abundance can significantly enhance cellular function and promote survival under nutrient-deprived conditions. Additionally, FAM134B-mediated activation of ER phagocytosis is regulated by O-GlcNAc glycosylation, and knockdown of FAM134B, which inhibits ER phagocytosis, significantly counteracts the protective effect of O-GlcNAc glycosylation [127]. As glycosylation products accumulate, ER stress activates the unfolded protein response (UPR) through key transmembrane proteins [128]. After UPR is initiated, the downstream C/EBP homologous protein is transcriptionally activated and controls the expression of apoptosis-related genes, inducing apoptosis under severe ER stress [129–130]. Furthermore, FAM134B overexpression attenuated ROS accumulation, apoptosis, and senescence in NPCs caused by glycosylation products (Fig. 5). However, the current understanding of specific structure–function relationships and the role of specific glycans on target proteins is incomplete. Further global analyses of the enzymatic network coordinating protein glycosylation and assessment of the potential glycosylation energies of specific cell types are required.
Fig. 5.

Glycosylation regulation of ECM metabolism and ER stress. In degenerated IVD, glycosylation products predominantly accumulate in aggrecan and collagen II, hindering their repair and renewal. Consequently, the up-regulation of matrix catabolic enzymes occurs, leading to enhanced ECM degradation and accelerated IVDD. Notably, O-GlcNAc glycosylation and O-GlcNAc transferase expression profiles are significantly increased in degenerated NPCs. Furthermore, FAM134B-mediated activation of ER phagocytosis was modulated by O-GlcNAc glycosylation. Following UPR initiation, the downstream C/EBP homologous protein is transcriptionally activated, controlling the expression of apoptosis-related genes and inducing apoptosis under severe ER stress.
Methylation
Methylation regulates aging-related gene transcription
Protein methylation is a common covalent PTM catalyzed by methyltransferases that add methyl groups from the donor S-adenyl-L-methionine to specific substrates [131]. These enzymes can be divided into lysine methyltransferases (KMTs) and arginine methyltransferases (PRMTs) based on their target residues [132–133]. Abnormal methylation levels often occur in aging cells, and their up-regulation is associated with a poor prognosis [134]. In the context of IVDD, several studies have highlighted the role of protein methylation in regulating cellular processes. For instance, WTAP expression increases in aging NPCs due to a KDM5A-mediated epigenetic increase in promoter H3K4me3, contributing to NORAD posttranscriptional regulation [135]. Another study revealed that the ALKBH5 expression is enhanced during IVDD due to decreased KDM4A-mediated H3K9me3 modification. Functionally, ALKBH5 induces NPC senescence by demethylating DNMT3B transcripts, promoting its expression via reduced YTHDF2 recognition and subsequent degradation due to transcript hypomethylation in vitro and in vivo [136]. DNMT3B, which inhibits COX2 expression by methylating the TRPA1 promoter, is suggested to promote NPC proliferation through the TRPA1/COX2/YAP axis, potentially preventing ECM degradation and alleviating IVDD in rats [137]. KMT2D and H3K4me1 modifications were significantly up-regulated during oxidative stress-induced degeneration of NPCs, and knockdown of KMT2D down-regulated the expression levels of catabolic enzymes, such as MMP3, MMP9, and MMP13 [138], highlighting the crucial role of KMT2D-mediated methylation in the pathological state of IVD [139]. EZH2, which regulates NOX4 expression through H3K27me3 at the promoter, is involved in a feedback loop with NOX4, which regulates NPC senescence [140]. EZH2 overexpression inhibits the expressions of collagen II and aggrecan, thereby promoting the expressions of ADAMTS5 and MMP13. However, SOX9 overexpression can reverse the effect of EZH2 in rat NPCs [141]. Additionally, miR-129-5p expression is reduced, while EZH2 and MAPK1 levels are overexpressed in the lumbar spine tissue of patients with IVDD [142]. Mechanistically, miR-129-5p can down-regulate MAPK1 expression, whereas EZH2 inhibits miR-129-5p by modifying H3K27me3 in the promoter, increasing the release of inflammation and cell senescence factors (Fig 6). Methylation regulation in IVDD primarily focuses on histones, which are closely associated with chromatin remodeling. Histone methylation, particularly at the N-terminal tails of H3 and H4, modifies and remodels chromatin by recruiting specific reading proteins or enzymes, thereby regulating chromatin accessibility and gene transcription. However, an increasing number of studies have reported methylation events in nonhistone proteins, revealing that methylation is involved in various biological processes.
Fig. 6.
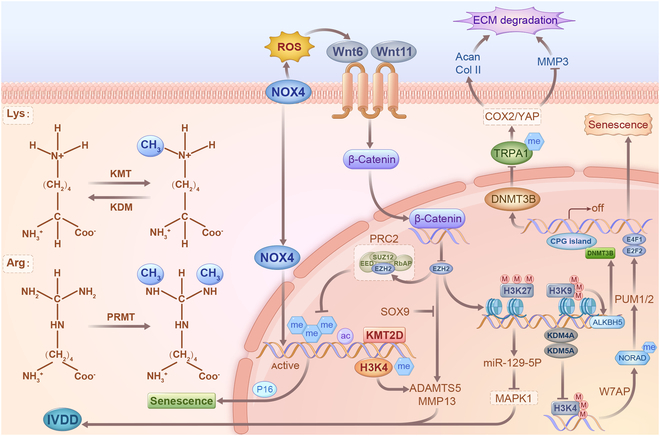
Methylation regulation of age-related metabolism. Methylation undergoes significant alterations in oxidative stress-induced IVDD and is implicated in regulating the expression levels of catabolic enzymes such as MMP3, MMP9, and MMP13. The feedback loop between EZH2 and NOX4 regulates NPC senescence. Additionally, EZH2 overexpression inhibits the expressions of collagen II and aggrecan while promoting the expressions of ADAMTS5 and MMP13. SOX9 overexpression reversed the effects of EZH2 in rat NPCs. Moreover, miR-129-5p overexpression or MAPK1 silencing promotes NPC proliferation and inhibits senescence. Intriguingly, miR-129-5p down-regulates MAPK1 expression, whereas EZH2 inhibits miR-129-5p by modifying H3K27me3 in the promoter, increasing the release of inflammatory and cellular senescence factors.
Fig. 7.

Hypoxia-induced lactate production regulating anti-inflammatory repair. Glucose is taken up by NPCs by the glucose transporter (GluT), and glycolysis converts glucose to pyruvate. Lactic dehydrogenase (LDH) converts pyruvate to lactate, which is exported from NPCs by MCT4. Lactate can be converted to lactate-CoA, which participates in the lactylation of histone and nonhistone proteins. This process regulates the expression of genes encoding anti-inflammatory factors such as NRF2. Conversely, LDH1 converts lactate to pyruvate, which is then converted to acetyl-CoA via pyruvate dehydrogenase (PDH). Pyruvate dehydrogenase kinase (PDK) negatively regulates the entry of acetyl-CoA into the TCA cycle, generating precursors for biosynthesis and/or oxidative phosphorylation for ATP production.
Lactylation
Lactate regulates energy metabolism
Given the avascular nature of IVD, energy metabolism in this tissue primarily relies on glycolysis for survival, leading to a high production of lactate [143]. Lactate is traditionally regarded as a terminal waste product, contributing to lowering pH value within the IVD. This acidic environment inhibits proteoglycan synthesis, increases MMP activation, promotes cellular senescence, and ultimately accelerates IVDD [2,14–15]. Conventionally, in a nutrient-poor and hypoxic setting, heightened cellular demand results in increased accumulation of lactate, which serves as the end product of anaerobic glycolysis. However, recent studies have revealed contrasting responses in healthy IVD. Following exercise, lactate levels exhibited an immediate decrease, slowly and partially normalizing for 1 hour. This intriguing finding suggests that lactate, instead of being regarded solely as a waste product necessitating excretion, may be utilized as a fuel source through alternative pathways [144]. Alterations in this response hold promise as potential noninvasive radiological biomarkers for the early detection of disc degeneration. Moreover, the burgeoning interest in treatments targeting IVD degeneration and regeneration has spurred a deeper exploration of the internal metabolic mechanisms within the disc environment at the molecular level. Elevated lactate levels act as a hypoxia mimetic factor, promoting the biosynthesis of tricarboxylic acid (TCA) cycle intermediates that functionally compete with α-ketoglutarate, which is required for HIF-1α hydroxylation and degradation. During the final steps of glycolysis, HIF-1α controls the flow of lactate and H+ out of NPCs, partially regulating glycolytic flux [145]. HIF-1α increases SLC16A3 expression, which encodes monocarboxylate transporter 4 (MCT4) that couples H+ and lactate. However, rapid suppression of MCT4 results in decreased glycolysis and TCA cycle flow, causing reconfiguration of metabolic processes in NPCs. NPCs exhibit metabolic plasticity, with short-term MCT inhibition leading to up-regulated TCA cycle flux and maintained oxidation–reduction reaction ratios. However, prolonged MCT4 inhibition may impair NPC viability due to cytoplasmic acidification and failure to maintain a high TCA cycle flux. Elimination of MCT4 in mice reproduces the key pathoanatomical characteristics of IVDD in humans, such as the absence of cell phenotypic markers and disruption of ECM integrity [146–147]. Studies on NPCs have revealed that the interplay between metabolic flow and HIF-1α function is regulated by cytosolic lactate levels [148]. Intracellular lactate accumulation enhances HIF-1α function, whereas the activation of MCT4 transcription is controlled by intronic enhancers sensitive to HIF-1α. This suggests a reciprocal relationship between MCT4 activity induced by hypoxia and the stability of HIF-1α in NPCs, with inherent lactate levels controlling this relationship. Therefore, cytosolic lactate levels play a crucial role in modulating the interplay between metabolic flow and HIF-1α function in NPCs.
Lactylation regulates inflammatory microenvironment
Recent scientific discoveries have unveiled how lactate-derived histone lactylation serves as a mediator of gene expression regulation. Lactylation disrupts histone–DNA interactions and activates transcriptional machinery under specific conditions [149]. In NPCs, blocking glycolysis leads to reduced lactate production in the IVD microenvironment, potentially influencing IL-1β production by macrophages. Several studies have demonstrated the significant impact of macrophage-mediated inflammation on NPC function and viability in IVD. For example, macrophage polarization regulates IVDD by modulating cell proliferation, inflammatory mediator secretion, and the ECM [150]. Macrophage infiltration into degenerative IVD is correlated with the severity of disc degeneration and is associated with increased inflammatory cytokine expression and ECM degradation [151]. These studies provide evidence of the indirect effects of macrophages on NPCs through the secretion of proinflammatory mediators and modulation of the disc microenvironment. However, histone lactylation in macrophages, especially in classically activated proinflammatory macrophages, enhances the expression of genes related to wound healing, such as ARG1 [152–153]. Research indicates that proinflammatory macrophages transition to a homeostatic selective activation state during later stages, contributing to inflammation resolution [154]. This aligns with previous findings linking lactate to the polarization of specific macrophages [155]. The absence of direct blood supply in the IVD creates a distinctive microenvironment marked by inadequate oxygen levels and hindered nutrient diffusion [122]. Consequently, cells residing within the IVD, including resident macrophages, predominantly resort to glycolysis as the primary means of energy production under hypoxic conditions, leading to lactate generation as a metabolic byproduct. Lactate accumulation within the compact and oxygen-deprived milieu of IVD has the potential to reshape the epigenetic landscape of resident macrophages. Initially, proinflammatory gene expression relied heavily on glycolysis. However, as time progresses, lactate emerges as a key regulator, with its abundance—stemming from glycolysis—exerting an anti-inflammatory effect through the lactylation of histones at specific target genes [156]. This lactylation-mediated anti-inflammatory and pro-repair response of NPCs warrants further investigation. Moreover, considering the altered TFEB-dependent autophagy observed in IVDD and the regulatory role of lactate in autophagy, it becomes essential to investigate the direct or indirect role of cellular lactate in IVDD [157]. The intricate interplay between lactate, histone lactylation, and inflammation resolution holds potential significance for understanding and addressing IVDD.
Conclusions and Prospects
As previously mentioned, PTMs have been associated with IVDD, outlining their regulatory roles in NPCs and chondrocytes. These PTMs intricately contribute to the regulation of protein function and ensure precise functionality at designated times and locations. Consequently, they introduced novel and diverse mechanisms crucial for sustaining metabolic homeostasis within the IVD. By delving into the mechanisms of PTMs governing metabolism and inflammatory responses in NPCs and chondrocytes, this discussion sheds light on the intricate ways in which PTM-driven alterations in metabolism and inflammation impact IVDD. For instance, by altering the charge state of a protein, phosphorylation influences its interactions with other molecules, thereby regulating key enzyme activities in metabolic pathways. Within the intricate metabolic signaling network, the extensive interplay of key kinases emerges as a prominent characteristic of phosphorylation in regulating IVD metabolism. As an inflammatory regulator, ubiquitin can be conjugated to target proteins to mark them for proteasomal degradation. This process directly influences intracellular protein levels, thereby regulating metabolic status and cell survival mechanisms to counter the impact of oxidative stress and inflammation on the IVD. Abnormal glycosylation levels during IVDD lead to an excessive folding burden on the ER, which is responsible for the correct folding of newly synthesized proteins into functional structures. Sirtuin-mediated acetylation plays a multifunctional regulatory role in metabolism, encompassing energy balance, lipid and glucose metabolism, and resistance to oxidative stress. These molecules have emerged as key regulators of cell metabolism and are essential for maintaining normal metabolism in IVD, particularly mitochondrial homeostasis. The regulatory impact of methylation in IVDD primarily focuses on histones, which induce chromatin to form a condensed structure that limits DNA accessibility, thereby playing a pivotal role in biological processes such as the cell cycle. Lactate, the main product of glycolysis, mediates lactylation in metabolic processes and exhibits anti-inflammatory and repair-promoting effects. Some researchers have highlighted the anti-inflammatory and antioxidative properties of lactate in mitigating disc degeneration and promoting tissue repair in preclinical IVDD models. Strategies aimed at modulating lactate levels, such as the inhibition of lactate dehydrogenase or augmentation of lactate utilization pathways, have demonstrated promise in preclinical studies for attenuating disc degeneration and promoting tissue regeneration. Given the limited number of existing studies, the role of lactylation in IVDD is a key area warranting further research. In summary, these modifications exert regulatory control over IVD metabolism by altering the structure, stability, or interactions of proteins. The specific nature and mechanism of each modification contribute to these differences.
Notably, the interaction of different PTMs leads to joint regulatory effects, contributing to a more intricate and sophisticated regulation of cellular metabolism. The significance of this joint regulation lies in the integration of multiple signaling pathways, ensuring cellular adaptability to complex environmental changes. PTMs exert control over proteins involved in cell signaling either by activating or inhibiting specific signaling pathways. This synergistic effect directly influences the cellular responses to external stimuli, thereby regulating energy metabolism and cell growth. Furthermore, PTMs may occur at specific cellular locations or during particular life cycle stages, achieving spatiotemporal regulation of metabolism. This regulatory approach enhances cellular sensitivity to environmental changes, enabling cells to effectively respond to diverse environmental stressors and physiological requirements. A comprehensive understanding of these PTM interactions not only unravels the complexity of cellular regulatory networks but also promises novel insights for future biomedical research and IVDD treatment. Additionally, metabolites or intermediates generated through glycolysis, the TCA cycle, lipid and amino acid metabolism, and other pathways act as signaling molecules, impacting the PTMs of proteins. Although it is beyond the scope of this review, elucidating these processes holds the potential to deepen our understanding of IVDD’s nature, offering profound considerations for future treatments and management strategies. The knowledge gained from current metabolism-related studies suggests that PTMs may be more prevalent and crucial to cell biology than initially perceived. We posit that given the appropriate substrates and reaction conditions, PTMs occur in all biomolecules, irrespective of their intracellular location. In addition to enzyme availability, substrate availability may dictate the type and extent of PTMs. Consequently, the pursuit of new PTMs should be guided more by substrate availability rather than by enzymes. Future studies should leverage multi-omics mapping sequencing with high spatial resolution, proteomics, and metabolomics for the in-depth phenotypic analysis of IVDD. In this regard, innovative technologies are imperative to capture these PTMs, such as the development of biorthogonal labeling techniques coupled with microscopy imaging or high-resolution mass spectrometry. Establishing frameworks for the detection, treatment, and prevention of challenging human diseases is pivotal in this evolving landscape.
Acknowledgments
We thank the members of their respective research groups and colleagues for valuable feedback and suggestions, and the Home for Researchers (www.home-for-researchers.com).
Funding: This work was supported by the National Natural Science Foundation of China (NSFC) (nos. 82130072 and 82072505), the Fundamental Research Funds for the Central Universities (HUST: YCJJ202201033), and the Natural Science Foundation of Hubei Province, China (2023AFB532).
Author contributions: D.Z. drafted the original manuscript. H.L., Z.D., Q.L., G.L., W.Z., X.Z., and D.W. reviewed and edited the manuscript. Y.S. and C.Y. revised and approved the manuscript. All authors have read and agreed to the published version of the manuscript.
Competing interests: The authors declare that they have no competing interests.
Data availability
Data will be made available on request.
References
- 1.Knezevic NN, Candido KD, Vlaeyen JWS, Zundert JV, Cohen SP. Low back pain. Lancet. 2021;398(10294):78–92. [DOI] [PubMed] [Google Scholar]
- 2.Lee BS, Nault R, Grabowski M, Whiting B, Tanenbaum J, Knusel K, Poturalski M, Emch T, Mroz TE, Steinmetz MP. Utility of repeat magnetic resonance imaging in surgical patients with lumbar stenosis without disc herniation. Spine J. 2019;19(2):191–198. [DOI] [PubMed] [Google Scholar]
- 3.Zheng H, Sun Y, Kong D, Yin M, Chen J, Lin Y, Ma XF, Wang HS, Yuan GJ, Yao M, et al. Deep learning-based high-accuracy quantitation for lumbar intervertebral disc degeneration from MRI. Nat. Commun. 2022;13(1):841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hartvigsen J, Hancock MJ, Kongsted A, Louw Q, Ferreira ML, Genevay S, Hoy D, Karppinen J, Pransky G, Sieper J, et al. What low back pain is and why we need to pay attention. Lancet. 2018;391(10137):2356–2367. [DOI] [PubMed] [Google Scholar]
- 5.Liu L, Huang K, Li W, Qiu R, Fang Y, Huang Y, Zhao S, Lv H, Zhang K, Shan H, et al. Molecular imaging of collagen destruction of the spine. ACS Nano. 2021;15(12):19138–19149. [DOI] [PubMed] [Google Scholar]
- 6.Francisco V, Pino J, González-Gay M, Lago F, Karppinen J, Tervonen O, Mobasheri A, Gualillo O. A new immunometabolic perspective of intervertebral disc degeneration. Nat. Rev. Rheumatol. 2022;18(1):47–60. [DOI] [PubMed] [Google Scholar]
- 7.Dudli S, Sing DC, Hu SS, Berven SH, Burch S, Deviren V, Cheng I, Tay BKB, Alamin TF, Ith MAM, et al. Intervertebral disc/bone marrow cross-talk with Modic changes. Eur. Spine J. 2017;26(5):1362–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thijs G, Geoffrey W, Dahbia MS, Saeed AS, Jill PGU. Nutrient supply and intervertebral disc metabolism. J. Bone Joint Surg. Am. 2006;2:30–35. [DOI] [PubMed] [Google Scholar]
- 9.Francisco V, Ruiz-Fernández C, Pino J, Mera A, González-Gay MA, Gómez R, Lago F, Mobasheri A, Gualillo O. Adipokines: Linking metabolic syndrome, the immune system, and arthritic diseases. Biochem. Pharmacol. 2019;165:196–206. [DOI] [PubMed] [Google Scholar]
- 10.Ruiz-Fernández C, Francisco V, Pino J, Mera A, González-Gay MA, Gómez R, Lago F, Gualillo O. Molecular relationships among obesity, inflammation and intervertebral disc degeneration: Are adipokines the common link? Int. J. Mol. Sci.. 2019;20(8):2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rustenburg CME, Emanuel KS, Peeters M, Lems WF, Vergroesen PA, Smit TH. Osteoarthritis and intervertebral disc degeneration: Quite different, quite similar. JOR Spine. 2018;1(4): Article e1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Choi H, Tessier S, Silagi ES, Kyada R, Yousefi F, Pleshko N, Shapiro IM, Risbud MV. A novel mouse model of intervertebral disc degeneration shows altered cell fate and matrix homeostasis. Matrix Biol. 2018;70:102–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Silagi ES, Shapiro IM, Risbud MV. Glycosaminoglycan synthesis in the nucleus pulposus: Dysregulation and the pathogenesis of disc degeneration. Matrix Biol. 2018;71-72:368–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marei H, Tsai WK, Kee Y, Ruiz K, He J, Cox C, Sun T, Penikalapati S, Dwivedi P, Choi M, et al. Antibody targeting of E3 ubiquitin ligases for receptor degradation. Nature. 2022;610(7930):182–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gorth DJ, Shapiro IM, Risbud MV. Transgenic mice overexpressing human TNF-α experience early onset spontaneous intervertebral disc herniation in the absence of overt degeneration. Cell Death Dis. 2018;10(1):7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Risbud MV, Shapiro IM. Role of cytokines in intervertebral disc degeneration: Pain and disc content. Nat. Rev. Rheumatol. 2014;10(1):44–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mobasheri A, Margaret PR, Oreste G, Sellam J, Peter K, Ursula F. The role of metabolism in the pathogenesis of osteoarthritis. Nat. Rev. Rheumatol. 2017;13(5):302–311. [DOI] [PubMed] [Google Scholar]
- 18.Albert V, Hall MN. mTOR signaling in cellular and organismal energetics. Curr. Opin. Cell Biol. 2015;33:55–66. [DOI] [PubMed] [Google Scholar]
- 19.Steinberg GR, Hardie DG. New insights into activation and function of the AMPK. Nat. Rev. Mol. Cell Biol. 2023;24(4):255–272. [DOI] [PubMed] [Google Scholar]
- 20.Chiarotto A, Ostelo RW, Boers M, Terwee CB. A systematic review highlights the need to investigate the content validity of patient-reported outcome measures for physical functioning in patients with low back pain. J. Clin. Epidemiol. 2018;95:73–93. [DOI] [PubMed] [Google Scholar]
- 21.Das T, Shin SC, Song EJ, Kim EE. Regulation of deubiquitinating enzymes by post-translational modifications. Int. J. Mol. Sci. 2020;21(11):4028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Christian A, Sasa ADM, Daniela T. How pervasive are post-translational and -transcriptional modifications? Trends Cell Biol. 2022;32(6):475–478. [DOI] [PubMed] [Google Scholar]
- 23.Kelsall IR, Zhang J, Knebel A, Arthur JSC, Cohen P. The E3 ligase HOIL-1 catalyses ester bond formation between ubiquitin and components of the Myddosome in mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 2019;116(27):13293–13298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fert-Bober J, Murray CI, Parker SJ, Eyk JEV. Precision profiling of the cardiovascular post-translationally modified proteome: Where there is a will there is a way. Circ. Res. 2018;122(9):1221–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhao M, Yao P, Mao Y, Wu J, Wang W, Geng C, Cheng J, du W, Jiang P. Malic enzyme 2 maintains protein stability of mutant p53 through 2-hydroxyglutarate. Nat. Metab. 2022b;4(2):225–238. [DOI] [PubMed] [Google Scholar]
- 26.Mo J, Meng Z, Kim YC, Park HW, Hansen CG, Kim S, Lim DS, Guan KL. Cellular energy stress induces AMPK-mediated regulation of YAP and the hippo pathway. Nat. Cell Biol. 2015;17(4):500–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mendes KL, Lelis D, Santos SHS. Nuclear sirtuins and inflammatory signaling pathways. Cytokine Growth Factor Rev. 2017;38:98–105. [DOI] [PubMed] [Google Scholar]
- 28.Liu M, Zhang M, Xie M. Molecular mechanisms of anti-inflammatory action of AMPK. Sheng Li Xue Bao. 2018;70(3):329–334. [PubMed] [Google Scholar]
- 29.Dudli S, Fields AJ, Samartzis D, Karppinen J, Lotz JC. Pathobiology of Modic changes. Eur. Spine J. 2016;25(11):3723–3734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Puigdellívol M, Allendorf DH, Brown GC. Sialylation and galectin-3 in microglia-mediated neuroinflammation and neurodegeneration. Front. Cell. Neurosci. 2020;14:162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nalbandian M, Takeda M. Lactate as a signaling molecule that regulates exercise-induced adaptations. Biology. 2016;5(4):38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cohen P. The origins of protein phosphorylation. Nat. Cell Biol. 2022;4(5):E127–E130. [DOI] [PubMed] [Google Scholar]
- 33.Li M, Li S, Zhang L. Phosphorylation promotes the accumulation of PERIOD protein foci. Research. 2023;6:0139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim J, Kundu M, Viollet B, Guan K. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011;13(2):132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kang L, Xiang Q, Zhan S, Song Y, Wang K, Zhao K, Li S, Shao Z, Yang C, Zhang Y. Restoration of autophagic flux rescues oxidative damage and mitochondrial dysfunction to protect against intervertebral disc degeneration. Oxid. Med. Cell. Longev. 2019;2019:7810320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen D, Xia D, Pan Z, Xu D, Zhou Y, Wu Y, Cai N, Tang Q, Wang C, Yan M, et al. Metformin protects against apoptosis and senescence in nucleus pulposus cells and ameliorates disc degeneration in vivo. Cell Death Dis. 2016;7(10): Article e2441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen L, Tu Z, Wang Y, He Y, Wang X, Tao S, Xu YY, Li CR, Wang RL, Yang ZX, et al. ATP5O hypo-crotonylation caused by HDAC2 hyper-phosphorylation is a primary detrimental factor for downregulated phospholipid metabolism under chronic stress. Research. 2022;2022:9834963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang Z, Shen J, Feng E, Jiao Y. AMPK as a potential therapeutic target for intervertebral disc degeneration. Front. Mol. Biosci. 2021;8: Article 789087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu Y, Lin J, Wu X, Guo X, Sun H, Yu B, Shen J, Bai J, Chen Z, Yang H, et al. Aspirin-mediated attenuation of intervertebral disc degeneration by ameliorating reactive oxygen species in vivo and in vitro. Oxid. Med. Cell. Longev. 2019;2019:7189854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen H, Zhou J, Zhang G, Luo Z, Li L, Kang X. Emerging role and therapeutic implication of mTOR signalling in intervertebral disc degeneration. Cell Prolif. 2023;56(1): Article e13338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang D, Shang Q, Mao J, Gao C, Wang J, Wang D, Wang H, Jia H, Peng P, du M, et al. Phosphorylation of KRT8 (keratin 8) by excessive mechanical load-activated PKN (protein kinase N) impairs autophagosome initiation and contributes to disc degeneration. Autophagy. 2023;19(9):2485–2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sean JH, David EJ, Matthias M. Protein phosphorylation: A major switch mechanism for metabolic regulation. Trends Endocrinol. Metab. 2015;26(12):676–687. [DOI] [PubMed] [Google Scholar]
- 43.Yan Y, Mukherjee S, Harikumar KG, Strutzenberg TS, Zhou XE, Suino-Powell K, Xu TH, Sheldon RD, Lamp J, Brunzelle JS, et al. Structure of an AMPK complex in an inactive ATP-bound state. Science. 2021;373(6553):413–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Su Z, Burchfield JG, Yang P, Humphrey SJ, Yang G, Francis D, Yasmin S, Shin SY, Norris DM, Kearney AL, et al. Global redox proteome and phosphoproteome analysis reveals redox switch in AKT. Nat. Commun. 2019;10(1):5486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stefania B, Don B, Matthias W, Timm M, Michael NH. mTOR substrate phosphorylation in growth control. Cell. 2022;185(11):1814–1836. [DOI] [PubMed] [Google Scholar]
- 46.Inoki K, Ouyang H, Zhu T, Lindvall C, Wang Y, Zhang X, Yang Q, Bennett C, Harada Y, Stankunas K, et al. TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell. 2006;126(5):955–968. [DOI] [PubMed] [Google Scholar]
- 47.Inoki K, Zhu T, Guan K. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115(5):577–590. [DOI] [PubMed] [Google Scholar]
- 48.Ye J. Mechanism of insulin resistance in obesity: A role of ATP. Front. Med. 2021;15(3):372–382. [DOI] [PubMed] [Google Scholar]
- 49.Li G, Zhong L, Han L, Wang Y, Li B, Wang D, Zhao Y, Li Y, Zhang Q, Qi L, et al. Genetic variations in adiponectin levels and dietary patterns on metabolic health among children with normal weight versus obesity: The BCAMS study. Int. J. Obes. (Lond). 2022c;46(2):325–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Evavold CL, Hafner-Bratkovič I, Devant P, D’Andrea JM, Ngwa EM, Boršić E, Doench JG, La Fleur MW, Sharpe AH, Thiagarajah JR, et al. Control of gasdermin D oligomerization and pyroptosis by the Ragulator-rag-mTORC1 pathway. Cell. 2021;184(17):4495–4511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sancak Y, Peterson TR, Shaul YD, Lindquist RA, Thoreen CC, Bar-Peled L, Sabatini DM. The rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science. 2008;320(5882):1496–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kim E, Goraksha-Hicks P, Li L, Neufeld TP, Guan K. Regulation of TORC1 by rag GTPases in nutrient response. Nat. Cell Biol. 2008;10(8):935–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Meng Z, Li S, Wang L, Ko H, Lee Y, Jung DY, Okutsu M, Yan Z, Kim JK, Lin JD. Baf60c drives glycolytic metabolism in the muscle and improves systemic glucose homeostasis through Deptor-mediated Akt activation. Nat. Med. 2013;19(5):640–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chan TO, Zhang J, Rodeck U, Pascal JM, Armen RS, Spring M, et al. Resistance of Akt kinases to dephosphorylation through ATP-dependent conformational plasticity. Proc. Natl. Acad. Sci. U.S.A. 2011;108(46):E1120–E1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lin K, Lin J, Wu W, Ballard J, Lee BB, Gloor SL, Vigers GP, Morales TH, Friedman LS, Skelton N, et al. An ATP-site on-off switch that restricts phosphatase accessibility of Akt. Sci. Signal. 2012;5(223):ra37. [DOI] [PubMed] [Google Scholar]
- 56.Lu S, Deng R, Jiang H, Song H, Li S, Shen Q, Huang W, Nussinov R, Yu J, Zhang J. The mechanism of ATP-dependent allosteric protection of Akt kinase phosphorylation. Structure. 2015;23(9):1725–1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Trefely S, Khoo P, Krycer JR, Chaudhuri R, Fazakerley DJ, Parker BL, Sultani G, Lee J, Stephan JP, Torres E, et al. Kinome screen identifies PFKFB3 and glucose metabolism as important regulators of the insulin/insulin-like growth factor (IGF)-1 signaling pathway. J. Biol. Chem. 2015;290(43):25834–25846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zheng Y, Nair SK. YcaO-mediated ATP-dependent peptidase activity in ribosomal peptide biosynthesis. Nat. Chem. Biol. 2023;19(1):111–119. [DOI] [PubMed] [Google Scholar]
- 59.Scheffner M, Nuber U, Huibregtse JM. Protein ubiquitination involving an E1-E2-E3 enzyme ubiquitin thioester cascade. Nature. 1995;373(6509):81–83. [DOI] [PubMed] [Google Scholar]
- 60.David BB, Achim W, Daniel LK, Ivona A. Disorders of ubiquitylation: Unchained inflammation. Nat. Rev. Rheumatol. 2022;18(8):435–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schulman BA, Harper JW. Ubiquitin-like protein activation by E1 enzymes: The apex for downstream signalling pathways. Nat. Rev. Mol. Cell Biol. 2009;10(5):319–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Otten EG, Werner E, Crespillo-Casado A, Boyle KB, Dharamdasan V, Pathe C. Ubiquitylation of lipopolysaccharide by RNF213 during bacterial infection. Nature. 2021;594(7861):111–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pao K, Wood NT, Knebel A, Rafie K, Stanley M, Mabbitt PD, Sundaramoorthy R, Hofmann K, van Aalten DMF, Virdee S. Activity-based E3 ligase profiling uncovers an E3 ligase with esterification activity. Nature. 2018;556(7701):381–385. [DOI] [PubMed] [Google Scholar]
- 64.Wan M, Wang X, Huang C, Xu D, Wang Z, Zhou Y, Zhu Y. A bacterial effector deubiquitinase specifically hydrolyses linear ubiquitin chains to inhibit host inflammatory signalling. Nat. Microbiol. 2019;4(8):1282–1293. [DOI] [PubMed] [Google Scholar]
- 65.Wertz IE, O’Rourke KM, Zhou H, Eby M, Aravind L, Seshagiri S, Wu P, Wiesmann C, Baker R, Boone DL, et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature. 2004;430(7000):694–699. [DOI] [PubMed] [Google Scholar]
- 66.Polykratis A, Martens A, Eren RO, Shirasaki Y, Yamagishi M, Yamaguchi Y, Uemura S, Miura M, Holzmann B, Kollias G, et al. A20 prevents inflammasome-dependent arthritis by inhibiting macrophage necroptosis through its ZnF7 ubiquitin-binding domain. Nat. Cell Biol. 2019;21(6):731–742. [DOI] [PubMed] [Google Scholar]
- 67.Peng X, Zhang C, Zhou Z, Wang K, Gao J, Qian Z, Bao JP, Ji HY, Cabral VLF, Wu XT. A20 attenuates pyroptosis and apoptosis in nucleus pulposus cells via promoting mitophagy and stabilizing mitochondrial dynamics. Inflamm. Res. 2022;71(5–6):695–710. [DOI] [PubMed] [Google Scholar]
- 68.Li X, Zhang P, Yin Z, Xu F, Yang Z, Jin J, Qu J, Liu Z, Qi H, Yao C, et al. Caspase-1 and gasdermin D afford the optimal targets with distinct switching strategies in NLRP1b Inflammasome-induced cell death. Research. 2022;2022:9838341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Peng X, Zhang C, Bao J, Zhu L, Shi R, Xie Z, Wang F, Wang K, Wu XT. A20 of nucleus pulposus cells plays a self-protection role via the nuclear factor-kappa B pathway in the inflammatory microenvironment. Bone Joint Res. 2020;9(5):225–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Qian J, Wang X, Su G, Shu X, Huang Z, Jiang H, Zhu Q. Platelet-rich plasma-derived exosomes attenuate intervertebral disc degeneration by promoting NLRP3 autophagic degradation in macrophages. Int. Immunopharmacol. 2022;110: Article 108962. [DOI] [PubMed] [Google Scholar]
- 71.Hai B, Mao T, Du C, Jia F, Liu Y, Song Q. USP14 promotes pyroptosis of human annulus fibrosus cells derived from patients with intervertebral disc degeneration through deubiquitination of NLRP3. Acta Biochim. Biophys. Sin. 2022;54(11):1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cheng X, Zhang L, Zhang K, Zhang G, Hu Y, Sun X, Zhao C, Li H, Li YM, Zhao J. Circular RNA VMA21 protects against intervertebral disc degeneration through targeting miR-200c and X linked inhibitor-of-apoptosis protein. Ann. Rheum. Dis. 2018;77(5):770–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wang F, Wang L, Gan S, Feng S, Ouyang S, Wang X, Yuan S. SERBP1 promotes stress granule clearance by regulating 26S proteasome activity and G3BP1 ubiquitination and protects male germ cells from thermostimuli damage. Research. 2023;6:0091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhao L, Zhao L, Zhong K, Tong A, Jia D. Targeted protein degradation: Mechanisms, strategies and application. Signal Transduct. Target. Ther. 2022a;7(1):113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Donovan KA, Ferguson FM, Bushman JW, Eleuteri NA, Bhunia D, Ryu S, Tan L, Shi K, Yue H, Liu X, et al. Mapping the degradable kinome provides a resource for expedited degrader development. Cell. 2020;183(6):1714–1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tseng C, Chen B, Han Y, Wang K, Song SH, Chen Z. Advanced glycation end products promote intervertebral disc degeneration by transactivation of matrix metallopeptidase genes. Osteoarthr. Cartil. 2024;32:187–199. [DOI] [PubMed] [Google Scholar]
- 77.Huang B, Wu H, Zheng L, Wei X, Zheng Z, Wu H, Chen J, Shan Z, Liu J, Zhao F. Activation of Nrf2 signaling by 4-octyl itaconate attenuates the cartilaginous endplate degeneration by inhibiting E3 ubiquitin ligase ZNF598. Osteoarthr. Cartil. 2023;31(2):213–227. [DOI] [PubMed] [Google Scholar]
- 78.Zhou M, He S, Liu W, Yang M, Hou Z, Meng Q, Qian ZL. EZH2 upregulates the expression of MAPK1 to promote intervertebral disc degeneration via suppression of miR-129-5p. J. Gene Med. 2022a;24(3): Article e3395. [DOI] [PubMed] [Google Scholar]
- 79.Wang J, Xia D, Lin Y, Xu W, Wu Y, Chen J, Chu J, Shen P, Weng S, Wang X, et al. Oxidative stress-induced circKIF18A downregulation impairs MCM7-mediated anti-senescence in intervertebral disc degeneration. Exp. Mol. Med. 2022;54(3):285–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zheng J, Chang L, Bao X, Zhang X, Li C, Deng L. TRIM21 drives intervertebral disc degeneration induced by oxidative stress via mediating HIF-1α degradation. Biochem. Biophys. Res. Commun. 2021;555:46–53. [DOI] [PubMed] [Google Scholar]
- 81.Boyden LM, Choi M, Choate KA, Nelson-Williams CJ, Farhi A, Toka HR, Tikhonova IR, Bjornson R, Mane SM, Colussi G, et al. Mutations in kelch-like 3 and cullin 3 cause hypertension and electrolyte abnormalities. Nature. 2012;482(7383):98–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Choi J, Lee K, Ingvarsdottir K, Bonasio R, Saraf A, Florens L, Washburn MP, Tadros S, Green MR, Busino L. Loss of KLHL6 promotes diffuse large B-cell lymphoma growth and survival by stabilizing the mRNA decay factor roquin2. Nat. Cell Biol. 2018b;20(5):586–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Metzger MB, Pruneda JN, Klevit RE, Weissman AM. RING-type E3 ligases: Master manipulators of E2 ubiquitin-conjugating enzymes and ubiquitination. Biochim. Biophys. Acta. 2014;1843(1):47–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yang X, Chen Y, Guo J, Li J, Zhang P, Yang H, Rong K, Zhou T, Fu J, Zhao J. Polydopamine nanoparticles targeting ferroptosis mitigate intervertebral disc degeneration via reactive oxygen species depletion, iron ions chelation, and GPX4 ubiquitination suppression. Adv Sci (Weinh). 2023;10(13): Article e2207216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Fang EF, Scheibye-Knudsen M, Brace LE, Kassahun H, Gupta TS, Nilsen H. Defective mitophagy in XPA via PARP-1 hyperactivation and NAD (+)/SIRT1 reduction. Cell. 2014;157(4):882–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Fang EF, Scheibye-Knudsen M, Chua KF, Mattson MP, Croteau DL, Bohr VA. Nuclear DNA damage signalling to mitochondria in ageing. Nat. Rev. Mol. Cell Biol. 2016;17(5):308–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chalkiadaki A, Guarente L. The multifaceted functions of sirtuins in cancer. Nat. Rev. Cancer. 2015;15(10):608–624. [DOI] [PubMed] [Google Scholar]
- 88.Erik V, Marjut L, Hanna F, Saara M, Johanna R, Petri K. Sirtuin1-p53, forkhead box O3a, p38 and post-infarct cardiac remodeling in the spontaneously diabetic Goto-Kakizaki rat. Cardiovasc. Diabetol. 2010;9:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wang Y, Zhou L, Zhao Y, Wang S, Chen L, Liu L, Ling ZQ, Hu FJ, Sun YP, Zhang JY, et al. Regulation of G6PD acetylation by SIRT2 and KAT9 modulates NADPH homeostasis and cell survival during oxidative stress. EMBO J. 2014;33(12):1304–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bell EL, Guarente L. The SirT3 divining rod points to oxidative stress. Mol. Cell. 2011;42(5):561–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hallows WC, Yu W, Smith BC, Devries MK, Ellinger JJ, Someya S. Sirt3 promotes the urea cycle and fatty acid oxidation during dietary restriction. Mol. Cell. 2011;41(2):139–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Haigis MC, Mostoslavsky R, Haigis KM, Fahie K, Christodoulou DC, Murphy AJ, Valenzuela DM, Yancopoulos GD, Karow M, Blander G, et al. SIRT4 inhibits glutamate dehydrogenase and opposes the effects of calorie restriction in pancreatic beta cells. Cell. 2006;126(5):941–954. [DOI] [PubMed] [Google Scholar]
- 93.Laurent G, Boer VCJ, Finley LWS, Sweeney M, Lu H, Schug TT, Cen Y, Jeong SM, Li X, Sauve AA, et al. SIRT4 represses peroxisome proliferator-activated receptor α activity to suppress hepatic fat oxidation. Mol. Cell. Biol.. 2013a;33(22):4552–4561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Peng C, Lu Z, Xie Z, Cheng Z, Chen Y, Tan M, Luo H, Zhang Y, He W, Yang K, et al. The first identification of lysine malonylation substrates and its regulatory enzyme. Mol. Cell. Proteomics. 2011;10(12):M111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Park J, Chen Y, Tishkoff DX, Peng C, Tan M, Dai L, Xie Z, Zhang Y, Zwaans BMM, Skinner ME, et al. SIRT5-mediated lysine desuccinylation impacts diverse metabolic pathways. Mol. Cell. 2013;50(6):919–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Tan M, Peng C, Anderson KA, Chhoy P, Xie Z, Dai L, Park J, Chen Y, Huang H, Zhang Y, et al. Lysine glutarylation is a protein posttranslational modification regulated by SIRT5. Cell Metab. 2014;19(4):605–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Mostoslavsky R, Chua KF, Lombard DB, Pang WW, Fischer MR, Gellon L, Liu P, Mostoslavsky G, Franco S, Murphy MM, et al. Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell. 2006;124(2):315–329. [DOI] [PubMed] [Google Scholar]
- 98.Kanfi Y, Naiman S, Amir G, Peshti V, Zinman G, Nahum L, Bar-Joseph Z, Cohen HY. The sirtuin SIRT6 regulates lifespan in male mice. Nature. 2012;483(7388):218–221. [DOI] [PubMed] [Google Scholar]
- 99.Ryu D, Jo YS, Sasso GL, Stein S, Zhang H, Perino A. A SIRT7-dependent acetylation switch of GABPβ1 controls mitochondrial function. Cell Metab. 2014;20(5):856–869. [DOI] [PubMed] [Google Scholar]
- 100.Laurent G, German NJ, Saha AK, Boer VCJ, Davies M, Koves TR, Dephoure N, Fischer F, Boanca G, Vaitheesvaran B, et al. SIRT4 coordinates the balance between lipid synthesis and catabolism by repressing malonyl CoA decarboxylase. Mol. Cell. 2013b;50(5):686–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lou G, Palikaras K, Lautrup S, Scheibye-Knudsen M, Tavernarakis N, Fang EF. Mitophagy and neuroprotection. Trends Mol. Med. 2020;26(1):8–20. [DOI] [PubMed] [Google Scholar]
- 102.Arenas A, Chen J, Kuang L, Barnett KR, Kasarskis EJ, Gal J, Zhu H. Lysine acetylation regulates the RNA binding, subcellular localization and inclusion formation of FUS. Hum. Mol. Genet. 2020;29(16):2684–2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Cohen TJ, Hwang AW, Restrepo CR, Yuan C, Trojanowski JQ, Lee VMY. An acetylation switch controls TDP-43 function and aggregation propensity. Nat. Commun. 2015;6:5845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zhang G, Deng Y, Xie Q, Ren E, Ma Z, He X, Gao YC, Kang XW. Sirtuins and intervertebral disc degeneration: Roles in inflammation, oxidative stress, and mitochondrial function. Clin. Chim. Acta. 2020;508:33–42. [DOI] [PubMed] [Google Scholar]
- 105.Martínez-Jiménez V, Cortez-Espinosa N, Rodríguez-Varela E, Vega-Cárdenas M, Briones-Espinoza M, Ruíz-Rodríguez VM, López-López N, Briseño-Medina A, Turiján-Espinoza E, Portales-Pérez DP. Altered levels of sirtuin genes (SIRT1, SIRT2, SIRT3 and SIRT6) and their target genes in adipose tissue from individual with obesity. Diabetes Metab. Syndr. 2019;13(1):582–589. [DOI] [PubMed] [Google Scholar]
- 106.Sato Y, Hilbert L, Oda H, Wan Y, Heddleston JM, Chew T, Zaburdaev V, Keller P, Lionnet T, Vastenhouw N, et al. Histone H3K27 acetylation precedes active transcription during zebrafish zygotic genome activation as revealed by live-cell analysis. Development. 2019;146(19):dev179127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Mo F, Zhuang X, Liu X, Yao PY, Qin B, Su Z, Zang J, Wang Z, Zhang J, Dou Z, et al. Acetylation of Aurora B by TIP60 ensures accurate chromosomal segregation. Nat. Chem. Biol. 2016;12(4):226–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ukmar-Godec T, Hutten S, Grieshop MP, Rezaei-Ghaleh N, Cima-Omori M, Biernat J, Mandelkow E, Söding J, Dormann D, Zweckstetter M. Lysine/RNA-interactions drive and regulate biomolecular condensation. Nat. Commun. 2019;10(1):2909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Behera V, Stonestrom AJ, Hamagami N, Hsiung CC, Keller CA, Giardine B, Sidoli S, Yuan ZF, Bhanu NV, Werner MT, et al. Interrogating histone acetylation and BRD4 as mitotic bookmarks of transcription. Cell Rep. 2019;27(2):400–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Reid MA, Dai Z, Locasale JW. The impact of cellular metabolism on chromatin dynamics and epigenetics. Nat. Cell Biol. 2017;19(11):1298–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Wagner GR, Payne RM. Widespread and enzyme-independent Nε-acetylation and Nε-succinylation of proteins in the chemical conditions of the mitochondrial matrix. J. Biol. Chem. 2013;288(40):29036–29045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Cheng A, Yang Y, Zhou Y, Maharana C, Lu D, Peng W, Liu Y, Wan R, Marosi K, Misiak M, et al. Mitochondrial SIRT3 mediates adaptive responses of neurons to exercise and metabolic and excitatory challenges. Cell Metab. 2016;23(1):128–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Mao J, Wang D, Wang D, Wu Q, Shang Q, Gao C, Wang H, Wang H, du M, Peng P, et al. SIRT5-related desuccinylation modification of AIFM1 protects against compression-induced intervertebral disc degeneration by regulating mitochondrial homeostasis. Exp. Mol. Med. 2023;55(1):253–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Xie Z, Jie Z, Wang G, Sun X, Tang P, Chen S, Qin A, Wang J, Fan S. TGF-β synergizes with ML264 to block IL-1β-induced matrix degradation mediated by Krüppel-like factor 5 in the nucleus pulposus. Biochim Biophys Acta Mol Basis Dis. 2018;1864(2):579–589. [DOI] [PubMed] [Google Scholar]
- 115.Xiao L, Gong D, Liang L, Liang A, Liang H, Xu X, Teng H. Inhibition of HDAC4 by GSK3β leads to downregulation of KLF5 and ASK1 and prevents the progression of intravertebral disc degeneration. Clin. Epigenetics. 2021;13(1):53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Li S, Li N, He J, Zhou R, Lu Z, Tao YJ, Guo YR, Wang Y. Molecular basis of KAT2A selecting acyl-CoA cofactors for histone modifications. Research. 2023;6:0109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Tu J, Li W, Yang S, Yang P, Yan Q, Wang S, Lai K, Bai X, Wu C, Ding W, et al. Single-cell transcriptome profiling reveals multicellular ecosystem of nucleus pulposus during degeneration progression. Adv Sci (Weinh). 2022;9(3): Article e2103631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ri A, Agar JN, Amster IJ, Baker MS, Bertozzi CR, Boja ES. How many human proteoforms are there? Nat. Chem. Biol. 2018;14(3):206–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kamatani T, Hagizawa H, Yarimitsu S, Morioka M, Koyamatsu S, Sugimoto M, Kodama J, Yamane J, Ishiguro H, Shichino S, et al. Human iPS cell-derived cartilaginous tissue spatially and functionally replaces nucleus pulposus. Biomaterials. 2022;284: Article 121491. [DOI] [PubMed] [Google Scholar]
- 120.Taylor EL, Collins JA, Gopalakrishnan P, Chubinskaya S, Loeser RF. Age and oxidative stress regulate Nrf2 homeostasis in human articular chondrocytes. Osteoarthr. Cartil. 2023;31(9):1214–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Illien-Junger S, Grosjean F, Laudier DM, Vlassara H, Striker GE, Iatridis JC. Combined anti-inflammatory and anti-AGE drug treatments have a protective effect on intervertebral discs in mice with diabetes. PLOS ONE. 2013;8(5): Article e64302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Tseng C, Han Y, Lv Z, Song Q, Wang K, Shen H, Chen Z. The CRL4DCAF6 E3 ligase ubiquitinates CtBP1/2 to induce apoptotic signalling and promote intervertebral disc degeneration. J. Mol. Med. 2023;101(1–2):171–181. [DOI] [PubMed] [Google Scholar]
- 123.Fields AJ, Berg-Johansen B, Metz LN, Miller S, La B, Liebenberg EC. Alterations in intervertebral disc composition, matrix homeostasis and biomechanical behavior in the UCD-T2DM rat model of type 2 diabetes. J. Orthop. Res. 2015;33(5):738–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Luo R, Li S, Li G, Lu S, Zhang W, Liu H, Lei J, Ma L, Ke W, Liao Z, et al. FAM134B-mediated ER-phagy upregulation attenuates AGEs-induced apoptosis and senescence in human nucleus pulposus cells. Oxid. Med. Cell. Longev. 2021;2021:3843145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Delorme-Axford E, Popelka H, Klionsky DJ. TEX264 is a major receptor for mammalian reticulophagy. Autophagy. 2019;15(10):1677–1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Chan C, Huang D, Huang Y, Hsu S, Kang L, Shen C, Lin WW. Methylglyoxal induces cell death through endoplasmic reticulum stress-associated ROS production and mitochondrial dysfunction. J. Cell. Mol. Med. 2016;20(9):1749–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Luo R, Li G, Zhang W, Liang H, Lu S, Cheung J, Zhang T, Tu J, Liu H, Liao Z, et al. O-GlcNAc transferase regulates intervertebral disc degeneration by targeting FAM134B-mediated ER-phagy. Exp. Mol. Med. 2022;54(9):1472–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Guo R, Liu W, Liu B, Zhang B, Li W, Xu Y. SIRT1 suppresses cardiomyocyte apoptosis in diabetic cardiomyopathy: An insight into endoplasmic reticulum stress response mechanism. Int. J. Cardiol. 2015;191:36–45. [DOI] [PubMed] [Google Scholar]
- 129.Zhang X, Yang Z, Pan T, Long X, Sun Q, Wang P, Li X, Kuang E. SARS-CoV-2 ORF3a induces RETREG1/FAM134B-dependent reticulophagy and triggers sequential ER stress and inflammatory responses during SARS-CoV-2 infection. Autophagy. 2022a;18(11):2576–2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Zhang Z, Wu J, Teng C, Wang J, Yu J, Jin C, Wang L, Wu L, Lin Z, Yu Z, et al. Orientin downregulating oxidative stress-mediated endoplasmic reticulum stress and mitochondrial dysfunction through AMPK/SIRT1 pathway in rat nucleus pulposus cells in vitro and attenuated intervertebral disc degeneration in vivo. Apoptosis. 2022b;27(11–12):1031–1048. [DOI] [PubMed] [Google Scholar]
- 131.Wu Q, Schapira M, Arrowsmith CH, Dalia B. Protein arginine methylation: From enigmatic functions to therapeutic targeting. Nat. Rev. Drug Discov. 2021;20(7):509–530. [DOI] [PubMed] [Google Scholar]
- 132.Cheng X, Roberts RJ. AdoMet-dependent methylation, DNA methyltransferases and base flipping. Nucleic Acids Res. 2001;29(18):3784–3795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.So H, Kim H, Lee J, You C, Yun C, Jeong H, Jin EJ, Jo Y, Ryu D, Bae GU, et al. Protein arginine methyltransferase 1 ablation in motor neurons causes mitochondrial dysfunction leading to age-related motor neuron degeneration with muscle loss. Research. 2023;6:0158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Lucie M, Ha TP, Louisane E, Michael RS, Coralie P, Muriel LR. How protein methylation regulates steroid receptor function. Endocr. Rev. 2022;43(1):160–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Li G, Ma L, He S, Luo R, Wang B, Zhang W, Song Y, Liao Z, Ke W, Xiang Q, et al. WTAP-mediated m6A modification of lncRNA NORAD promotes intervertebral disc degeneration. Nat. Commun. 2022b;13(1):1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Li G, Luo R, Zhang W, He S, Wang B, Liang H, Song Y, Ke W, Shi Y, Feng X, et al. m6A hypomethylation of DNMT3B regulated by ALKBH5 promotes intervertebral disc degeneration via E4F1 deficiency. Clin. Transl. Med. 2022a;12(3): Article e765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Luo Z, Ma Y, Di T, Ma B, Li H, An J. DNMT3B decreases extracellular matrix degradation and alleviates intervertebral disc degeneration through TRPA1 methylation to inhibit the COX2/YAP axis. Aging (Albany NY). 2021b;13(16):20258–20276. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 138.Kamakoti PB, Kaniskan H, Jin J, Gozani O. Epigenetics and beyond: Targeting writers of protein lysine methylation to treat disease. Nat. Rev. Drug Discov. 2021;20(4):265–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Xu W, Zhang X, Liu G, Zhu M, Wu Y, Jie Z, Xie Z, Wang S, Ma Q, Fan S, et al. Oxidative stress abrogates the degradation of KMT2D to promote degeneration in nucleus pulposus. Biochim Biophys Acta Mol basis Dis. 2020;1866(10): Article 165888. [DOI] [PubMed] [Google Scholar]
- 140.Liu C, Liu L, Yang M, Li B, Yi J, Ai X, Zhang Y, Huang B, Li C, Feng C, et al. A positive feedback loop between EZH2 and NOX4 regulates nucleus pulposus cell senescence in age-related intervertebral disc degeneration. Cell Div. 2020;15:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Jiang C, Guo Q, Jin Y, Xu J, Sun Z, Zhu D, Lin JH, Tian NF, Sun LJ, Zhang XL, et al. Inhibition of EZH2 ameliorates cartilage endplate degeneration and attenuates the progression of intervertebral disc degeneration via demethylation of Sox-9. EBioMedicine. 2019;48:619–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Zhou T, Yang X, Chen Z, Yang Y, Wang X, Cao X, Chen C, Han C, Tian H, Qin A, et al. Prussian blue nanoparticles stabilize SOD1 from ubiquitination-proteasome degradation to rescue intervertebral disc degeneration. Adv Sci (Weinh). 2022b;9(10): Article e2105466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Wang D, Robert H, Han C, Zhou C, Brandon C, Malkamaki M. Lactate oxidative phosphorylation by annulus fibrosus cells: Evidence for lactate-dependent metabolic symbiosis in intervertebral discs. Arthritis Res. Ther. 2021;23(1):145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Pushpa BT, Rajasekaran S, Easwaran M, Murugan C, Algeri R, Sri Vijay Anand KS, Mugesh Kanna R, Shetty AP. ISSLS PRIZE in basic science 2023: Lactate in lumbar discs-metabolic waste or energy biofuel? Insights from in vivo MRS and T2r analysis following exercise and nimodipine in healthy volunteers. Eur. Spine J. 2023;32(5):1491–1503. [DOI] [PubMed] [Google Scholar]
- 145.Elizabeth SS, Ernestina S, Irving MS, Makarand VR. Hypoxia and HIFs maintain the metabolic health of cells of the intervertebral disc. Nat. Rev. Rheumatol. 2021;17(7):426–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Vergroesen PA, Kingma I, Emanuel KS, Hoogendoorn RJW, Welting TJ, Royen BJ, van Dieën JH, Smit TH. Mechanics and biology in intervertebral disc degeneration: A vicious circle. Osteoarthr. Cartil.. 2015;23(7):1057–1070. [DOI] [PubMed] [Google Scholar]
- 147.Fine N, Lively S, Séguin CA, Perruccio AV, Kapoor M, Rampersaud R. Intervertebral disc degeneration and osteoarthritis: A common molecular disease spectrum. Nat. Rev. Rheumatol. 2023;19(3):136–152. [DOI] [PubMed] [Google Scholar]
- 148.Silagi ES, Novais EJ, Bisetto S, Telonis AG, Snuggs J, Maitre CLL. Lactate efflux from intervertebral disc cells is required for maintenance of spine health. J. Bone Miner. Res. 2020;35(3):550–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Zhang D, Tang Z, Huang H, Zhou G, Cui C, Weng Y, Liu W, Kim S, Lee S, Perez-Neut M, et al. Metabolic regulation of gene expression by histone lactylation. Nature. 2019a;574(7779):575–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Li X, Luo S, Fan W, Zhou T, Tan D, Tan R. Macrophage polarization regulates intervertebral disc degeneration by modulating cell proliferation, inflammation mediator secretion, and extracellular matrix metabolism. Front. Immunol. 2022;18(13): Article 922173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Teng Y, Huang Y, Yu H, Wu C, Yan Q, Wang Y, Yang M, Xie H, Wu T, Yang H, et al. Nimbolide targeting SIRT1 mitigates intervertebral disc degeneration by reprogramming cholesterol metabolism and inhibiting inflammatory signaling. Acta Pharm Sin B. 2023;13(5):2269–2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Heiden MGV, Cantley LC, Thompson CB. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science. 2009;324(5930):1029–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Zhang Z, Lin J, Nisar M, Chen T, Xu T, Zheng G, Wang C, Jin H, Chen J, Gao W, et al. The Sirt1/P53 axis in diabetic intervertebral disc degeneration pathogenesis and therapeutics. Oxid. Med. Cell. Longev. 2019;2019:7959573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Diskin C, Ryan TAJ, O’Neill LAJ. Modification of proteins by metabolites in immunity. Immunity. 2021;54(1):19–31. [DOI] [PubMed] [Google Scholar]
- 155.Colegio OR, Chu N, Szabo AL, Chu T, Rhebergen AM, Jairam V, Cyrus N, Brokowski CE, Eisenbarth SC, Phillips GM, et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature. 2014;513(7519):559–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Komilova NR, Angelova PR, Berezhnov AV, Stelmashchuk OA, Mirkhodjaev UZ, Houlden H, Gourine AV, Esteras N, Abramov AY. Metabolically induced intracellular pH changes activate mitophagy, autophagy, and cell protection in familial forms of Parkinson’s disease. FEBS J. 2022;289(3):699–711. [DOI] [PubMed] [Google Scholar]
- 157.Liang H, Luo R, Li G, Zhang W, Zhu D, Wu D, Zhou X, Tong B, Wang B, Feng X, et al. Lysine methylation of PPP1CA by the methyltransferase SUV39H2 disrupts TFEB-dependent autophagy and promotes intervertebral disc degeneration. Cell Death Differ. 2023;30(9):2135–2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data will be made available on request.