

Abstract
Macrocyclic peptides represent promising scaffolds for chemical tools and potential therapeutics. Synthetic methods for peptide macrocyclization are often hampered by C-terminal epimerization and oligomerization, leading to difficult scalability. While chemical strategies to circumvent this issue exist, they often require specific amino acids to be present in the peptide sequence. Herein we report the characterization of Ulm16, a peptide cyclase belonging to the penicillin-binding protein (PBP)-type class of thioesterases that catalyze head-to-tail macrolactamization of nonribosmal peptides. Ulm16 efficiently cyclizes various non-native peptides ranging from 4–6 amino acids with catalytic efficiencies of up to 3 X 106 M−1s−1. Unlike many previously described homologues, Ulm16 tolerates a variety of C- and N-terminal amino acids. The crystal structure of Ulm16, along with modeling of its substrates and site-directed mutagenesis, allows for rationalization of this wide substrate scope. Overall, Ulm16 represents a promising tool for the biocatalytic production of macrocyclic peptides.
INTRODUCTION
Peptides occupy a favorable space between small molecule therapeutics and large biologics. Their lack of immunogenicity, along with their high potency, selectivity, and reasonable manufacturing costs, have led to several therapeutics.1,2 This is well exemplified by clinically relevant drugs such as the antibiotics murepavadin and daptomycin, the immunosuppressant cyclosporine, and the anticancer agent octreotide. However, linear peptides often do not display these selectivities and drug-like properties without additional modifications3. Macrocyclization is of great interest because it often enables greater membrane permeability via elimination of the zwitterionic C and N termini, while also showing increased potency, specificity, and proteolytic stability brought about by defined secondary structure.4 Despite the advantages macrocyclization confers, it remains a synthetically challenging transformation that is often hampered by epimerization at the α-carbon of the C-terminal residue, competing oligomerization, and conformational rigidity precluding remote residue coupling. Together, these factors lead to poor yields and scalability issues.5 These problems are exacerbated as ring size decreases. The ground-state E geometry of the peptide bond prevents the peptides from attaining a conformation favorable to cyclization, making the synthesis and derivatization of pharmaceutically and industrially relevant small cyclic peptides at times impossible.6 This inability to efficiently access small cyclic peptides, such as tetrapeptides, severely limits our ability to further explore bioactive natural product cyclic peptides, which have a variety of interesting bioactivities including tetrapeptide mediated inhibition of the chloroplast F1-ATPase (tentoxin), selective blockage of calcium channels by the cyclic peptide family oncychocins, antagonism of kappa opioid receptors by tetrapeptide CJ-15,208, and inhibition of histone deacetylases (chlamydocin, trapoxin A, apicidin, microsporins, and others).7–10 Additionally, many other antibiotic and anticancer cyclic peptides have been identified but currently have unknown mechanisms of action.6 Further exploration of their mechanisms requires improved access to the natural products and derivatives. More generally, cyclic tetrapeptides are of interest due to their high rigidity and their ability to mimic β-turns in proteins.11 While several strategies have been explored for the chemical synthesis of cyclic tetrapeptides (e.g. pseudo proline protecting groups,12 on-resin cyclization,13 ring contraction,14 and anion assisted cyclization15), they restrict amino acid sequence, leading to the inability to access a larger chemical and conformational space.
The challenges associated with macrocyclization have resulted in an increasing interest in exploring enzymatic cyclization for scalable production of cyclic peptides.16,17 Nature is able to efficiently catalyze the production of linear peptide precursors using multimodular enzyme complexes known as nonribosomal peptide synthetases (NRPSs). These enzymes are frequently implicated in the production of small cyclic peptides including tetra- and pentapeptides18–21. Offloading from the complex and cyclization are typically catalyzed by C-terminal thioesterase (TE) domains or condensation termination (CT) domains.13 While these domains can efficiently catalyze the cyclization of strained cyclic peptides in vivo and have been extensively studied as biocatalysts22, they have failed to reach widespread use due to their low substrate promiscuity,23 an inability to accurately predict cyclization modes (e.g. head-to-tail) via bioinformatics,24 and in the case of CT domains, the strict requirement for a peptidyl carrier protein (PCP) tethered substrate.25,26
Recently, the discovery the penicillin-binding protein (PBP)-type, which share sequence homology to the PBP enzyme family rather than the canonical thioesterase domains, has sparked interest as possible biocatalysts for head-to-tail peptide macrocyclization (Figure 1).27–34 Unlike traditional TE and CT domains, which act in cis to the NRPS, PBP-thioesterases (PBP-TEs) are physically discrete from the assembly line and act in trans.35 Currently, three PBP-TEs have been validated: SurE from Streptomyces albidoflavus,27 which natively catalyzes the offloading and cyclization of two structurally unrelated octa- and decapeptides and in vitro can catalyze the cyclization of peptides ranging from 5–10 amino acids and peptide mimics with the equivalent length of 23 amino acids;20–22,24 PenA from Streptomyces cacaoi, which natively catalyzes the production pentapeptides and has also been shown to take peptides that vary in size and amino acid sequence;32 and WolJ from Streptomyces sp. MST-115088, which natively catalyzes the cyclization of the wollamides, and desotamides in vivo.36 While the substrate scope of WolJ has not been fully explored, it has been shown to catalyze the head-to-tail cyclization of wollamide B (C-Terminal D-Arg) and desotamide B (C-Terminal Gly).34
Fig. 1 |. Cyclic peptide natural products biosynthesized by PBP-type TEs.

A) Selected macrolactams confirmed to be biosynthesized by PBP-type TEs. The residues at the C and N termini of the linear precursors are colored red and blue, respectively. B) Ulleungmycin A, the cyclic peptide natural product produced by Ulm16, the enzyme used in this study. C) PBP-TE containing biosynthetic gene clusters of known natural products. MiBIG or GenBank IDs are indicated.
Note: The chemdraw for the top part of the figure and the AI file for the bottom part of the figure are uploaded separately
Although SurE, PenA, and WolJ have shown great potential as biocatalysts for producing larger cyclic peptides, their ability to access cyclic tetramers has not been demonstrated. Bioinformatics analyses using SurE as a molecular beacon have revealed the presence of SurE homologs in the biosynthetic gene clusters of known and cryptic cyclic peptide natural products.28,35 To explore the potential of other PBP-TEs to cyclize this challenging-to-access core, we generated alpha fold models37 of predicted PBP-TEs from known BGCs such as destoamides (DsaJ), Wollamides (WolJ), mannopeptimycin (MppK), cyclofaulknamycin (FlkO), logicatenamide (Lon18) and the ulleungmycins (Ulm16),35 along with confirmed PBP-TEs PenA, WolJ, and SurE (Extended Data Fig. 1). We also conducted multiple sequence alignments to identify notable differences in regions that were previously hypothesized for substrate selectivity (Supplementary Fig. 1).32 Interestingly, of all the predicted PBP-TEs we screened, Ulm16 displayed three notable features that distinguished it from its homologues: First, it had a remarkably shorter loop region above the active site cleft (Extended Data Fig. 1 and Supplementary Fig. 1, colored red), which was 35 amino acids shorter than the longest (PenA) and 21 shorter than the shortest (MppK). Ulm16 also displays a 16 amino acid loop insertion (Extended Data Fig. 1 and Supplementary Fig. 1, orange, residues 106–122) and a shorter loop region in the lipocalin domain (Extended Data Fig. 1 and Supplementary Fig. 1, yellow), both of which were unique among the known PBP-TEs. Further bioinformatics analyses of the top 500 proteins most like Ulm16 did not reveal any other predicted PBP-TEs missing the loop above the active site cleft, except for those that we hypothesize resulted from horizontal gene transfer and encode a natural product similar or identical to ulleungmycin (Supplementary Figs. 2–3 and Extended Data Fig. 2). Due to the unique sequence of Ulm16, we chose to explore its biocatalytic potential. The ulleungmycins are a group of cyclic peptides that were first discovered in 2017 from Streptomyces sp. KCB13F003.38 However, the mechanism for their offloading from the NRPS has, to this point, not been validated.
In this work, we describe Ulm16, an efficient and promiscuous PBP-TE with a large substrate scope. Ulm16 efficiently catalyzes the production of cyclic hexa-, penta-, and tetrapeptides beyond what has been previously reported for any thioesterase in the literature. Additionally, we present the X-ray crystal structure, docking studies, and mutational analyses to aid in explaining the structural basis behind its substrate promiscuity and increased catalytic activity.
RESULTS
Ulm16 cyclizes a variety of hexapeptides.
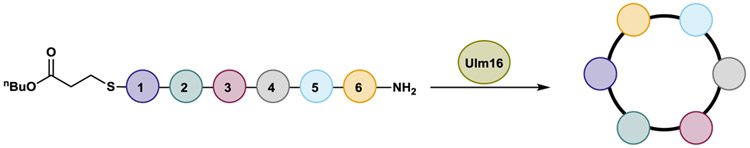
To investigate the substrate scope of Ulm16, a derivative of ulleungymcicin A was synthesized, in which the native, but commercially inaccessible, amino acids D-threo-β-hydroxy asparagine, L-allo-isoleucine, D-homoleucine, and 5-chloro-L-tryptophan were substituted for D-glutamine, L-isoleucine, D-Leucine, and L-tryptophan, respectively (designated Ulm, Extended Data Fig. 3). D-Asparagine, a better substitute for D-threo-β-hydroxy asparagine, was not used due its propensity to rapid asparatamide formation at the C-terminus. While SurE and PenA have previously been examined using peptide thioesters that contained N-Acetylcysteamine (SNAC), a mimic of the phosphopantetheine cofactor employed by the peptidyl carrier protein (PCP) domain of the NRPS,34 recent studies utilizing excised thioesterase domains have shown that other thiols not related to the phosphopantetheine backbone can improve enzyme activity and turnover.39 This led us to explore the Ulm16 peptide with three different C-terminal thiols: N-acetylcysteamine (Ulm-SNAC, 1), methyl 3-mercaptopropionate (Ulm-SMMP, 2), and butyl 3-mercaptopropionate (Ulm-SBMP, 3). Notably, the SBMP-thioester (3) was found to be three times more efficient in processing than the commonly used and expensive SNAC-thioester (1) (Table 1 and Extended Data Fig. 4). For this reason, SBMP-thioesters were used for the rest of the substrate scope, unless otherwise noted. Overall, we discovered that Ulm16 is a highly efficient biosynthetic enzyme,40 exhibiting a catalytic efficiency that is over 100-fold higher than that reported for SurE (kcat/KM = 1.7 X 103 M−1s−1 towards its SNAC linear substrate).31 An alanine scan was performed to determine sites where modifications are tolerated (Table 1, Extended Data Fig. 4). Substitution of internal residues 5 (D-valine) and 3 (L-isoleucine) with D-alanine and L-alanine, respectively, were well tolerated, displaying approximately 80% of the activity as the initial substrate. Surprisingly, D-alanine substitution at position 4 was not as well tolerated (~20% of the activity of the initial substrate). Unfortunately, alanine substitution at residue 2 (L-ornithine) resulted in an insoluble peptide. To investigate modifications at this site, L-ornithine was replaced with the uncharged L-glutamine. While both the SBMP and SMMP thioester for this molecule were too insoluble to obtain full kinetics, the SMMP thioester showed sufficient solubility to determine that the total turnover number (TTN) for 6, which was 9055 with a greater than 20:1 ratio for cyclized over hydrolyzed (Figure 2C and Supplementary Figs. 5-6). While the TTN is lower than that of the initial peptide (TTN for 2 = 22,545), it is noteworthy that an uncharged residue at this site is well tolerated by the enzyme. In addition, Ulm16 tolerates substitution at the C-terminus very well, displaying approximately 60% of the activity observed when the C-terminal D-glutamine is substituted with D-alanine. This contrasts with previously investigated SurE and PenA, which have reduced abilities to cyclize peptides with C-terminal alterations from their native residues.31,32 Modification at the N-terminus is less well tolerated, with an L-alanine substitution displaying only 6% of the catalytic efficiency of the native L-tryptophan. However, its efficiency is still approximately 20-fold better than that of SurE, and little hydrolysis is observed (Supplementary Fig. 13). Similar to other PBP-TEs, Ulm16 cannot tolerate an L-amino acid at the C-terminus. Switching the chirality at the C-terminus (4) results in a molecule that is not processed by the enzyme. Surprisingly, Ulm16 is able to tolerate glycine at the C-terminus (Figure 2C and Supplementary Fig. 14), a property that has only been previously validated with WolJ and a mutant SurEG235L.34 Specifically, both 11 and 12 had appreciable TTNs and showed selectivity for cyclization over hydrolysis, while SurE showed no activity (Supplementary Fig. 17). Taken together, C-terminal D-chirality is not a requirement for Ulm16 activity.
Table 1 |.
Kinetic parameters of Ulm16 against thioester substrates
| |||||||||
|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||
| Substrate | Sequence |
kCat (min−1) (±s.e.m.) | kcat/Km (M−1s−1) (±s.e.m.) | kcat/Km Ratio to (3) | |||||
| 1 | 2 | 3 | 4 | 5 | 6 | ||||
| Ulm-SNAC (1) | dGln | lOrn | lIle | dLeu | dVal | lTrp | 1545 (± 71) | 0.21 (± 0.03) x106 | 0.31 |
| Ulm-SMMP (2) | dGln | lOrn | lIle | dLeu | dVal | lTrp | 2175 (± 109) | 0.43 (± 0.03) x106 | 0.64 |
| Ulm-SBMP (3) | dGln | lOrn | lIle | dLeu | dVal | lTrp | 1921 (± 121) | 0.67 (± 0.18) x106 | 1.00 |
|
| |||||||||
| dQ1LQ (4) | lGln | lOrn | lIle | dLeu | dVal | lTrp | No Reaction | ||
| dQ1DA (5) | dAla | lOrn | lIle | dLeu | dVal | lTrp | 2079 (±163) | 0.39 (± 0.09) x106 | 0.58 |
| lOrn2LQ (6) | dGln | lGln | lIle | dLeu | dVal | lTrp | TTN: 9055 | ||
| lI3LA (7) | dGln | lOrn | lAla | dLeu | dVal | lTrp | 1900 (± 83) | 0.54 (± 0.09) x106 | 0.81 |
| dL4DA (8) | dGln | lOrn | lIle | dAla | dVal | lTrp | 441 (± 31) | 0.13 (± 0.03) x106 | 0.19 |
| dV5DA (9) | dGln | lOrn | lIle | dLeu | dAla | lTrp | 1364 (± 80) | 0.56 (± 0.15) x106 | 0.84 |
| lW6LA (10) | dGln | lOrn | lIle | dLeu | dVal | lAla | 90.1 (± 2.7) | 0.39 (± 0.05) x105 | 0.058 |
|
| |||||||||
| PenA Substrate (13) | dTyr | lArg | lTrp | dVal | lLeu | 3200 (± 390) | 2.0 (± 1.2) x106 | 2.99 | |
| Tetrapeptide (16) | dTyr | dArg | lPhe | lVal | 1160 (± 63) | 3.0 (± 1.2) x106 | 4.48 | ||
Fig 2. Substrate Scope of Ulm16 and SurE.

A) UV traces (214 nm) from UPLC analysis of cyclization of pentapeptide 13 by SurE (240 nM, top, red), Ulm16 (17 nM, middle, green) or no enzyme control (bottom, black) after 4 hours of incubation. Both SurE and Ulm16 are capable of cyclizing 13 but Ulm16 does so more efficiently and with significantly less enzyme. B) UV traces (214 nm) from UPLC analysis of cyclization of tetrapeptide 16 by SurE (240 nM, top, red), Ulm16 (17 nM, middle, green), or no enzyme control (bottom, black) after 4 hours of incubation. Ulm16 completely cyclize while SurE has barely detectable cyclization. C) TTNs (Total Turnover Numbers) of thiopeptide substrates. The top axis represents the TTN values with bars, while the bottom axis represents the ratio of cyclized to hydrolyzed product formed using dots. The values shown in the graph are averages obtained from at least three replicates. TTNs of thiopeptide substrates. The top axis shows the TTN and the bottom axis shows the ratio of cyclized to hydrolyzed product formed. Values are averages of at least three replicates. D) Representation of the Ulm16 tetrapeptide substrate scope showcasing the tolerated amino acid substitutions (>5:1 Cyclic:Hydrolysis) tested at each position.
Ulm16 cyclizes small rings.
Ulm16’s substrate promiscuity with hexapeptides prompted us to explore its activity towards different ring sizes. Ulm16 efficiently cyclized both pentapeptides and tetrapeptides (Table 1 and Figure 2 A-C). Specifically, it was able to cyclize a modified PenA substrate where the C-terminal D-phenylalanine was substituted with a D-tyrosine (13), displaying nearly 3-fold better catalytic efficiency compared to the modified Ulm16 native substrate (3). This modified PenA peptide (13) was also used to directly compare Ulm16 to SurE, since SurE had previously been shown to cyclize the PenA substrate.32 While SurE also had very good catalytic efficiency with this substrate (1.7 × 105 M−1s−1), Ulm16 remains the more efficient enzyme (Extended Data Fig. 5) with approximately 10-fold better activity and approximately 15-fold greater total turnover (Supplementary Figs. 18-19).
Previous work in our lab has focused on investigating the bioactivities of cyclic peptides that were predicted from PBP-TEs.41 However, significant difficulties were encountered in producing cyclic tetrapeptides. To address this issue, we investigated the ability of Ulm16 to cyclize a tetrapeptide consisting of N-terminal L-valine and C-terminal D-serine (14). We found that this peptide was efficiently cyclized by Ulm16, with very little hydrolysis observed (>20:1 cyclic:hydrolyzed) (Figure 2C and Supplementary Fig. 20). These results are particularly noteworthy as no PBP-TEs studied to date have been shown to cyclize tetrapeptides. Validation of this cyclic peptide was carried out by comparison towards a synthetic standard (Supplementary Fig. 21). This led us to investigate the Ulm16 substrate scope with other tetrapeptides by modifying each position and determining the TTN for all substrates. Ulm16 was able to tolerate a wide range of substitutions at the C-terminal residue as long as D-chirality was maintained (peptides 15-18, Figure 2C, Supplementary Table 1, The D-serine substitution for D-threonine (15) resulted in no significant change in cyclized/hydrolyzed ratio or TTN (Supplementary Fig. 22). However, substitution with a D-tyrosine (16) produced a substrate with over 4-fold better catalytic efficiency than the native hexapeptide substrate (Table 1 and Extended Data Fig 6) and a TTN over 10-fold better than the initial tetrapeptide hit (Supplementary Fig. 23). Additionally, this product was once again confirmed as the desired tetrapeptide by comparison with the tetrapeptide standard (Supplementary Fig. 24). Ulm16 also accepted other polar amino acids such as D-glutamine (17), although with an increased susceptibility to hydrolysis (6:1), and a polar charge D-lysine (18) with low variability in TTN (Supplementary Figs. 25-26). However, substitution of D-serine with a negatively charged D-glutamate (19) resulted in a substrate that could not be processed by Ulm16 (Supplementary Fig. 27). As seen with the hexapeptides, modifications of the N-terminal residue were more varied (Supplementary Table 1). Substituting the N-terminal L-valine with L-tryptophan (20) had little effect on TTN and displayed considerable selectivity for cyclized over hydrolyzed products. However, substituting with polar L-glutamine (21) significantly reduced activity (~4-fold reduced TTN compared to L-valine) and completely abolished selectivity for cyclic versus hydrolyzed products (Supplementary Figs. 28-29). A negative charge (L-asparate) at the N-terminus results in hydrolysis, consistent with the observation that a polar amino acid increases hydrolysis. However, blocking the negative charge with a methyl group (22) resulted in a substrate that could be efficiently cyclized by Ulm16 (Supplementary Fig. 30). Interestingly, position 2 (Supplementary Figs. 31-35) showed a preference for polar residues with at least two methylenes between the α-carbon and the polar side chain atom. Substituting D-ornithine with D-glutamine (23), D-glutamic acid (24), or D-2,4-diamino butyric acid (25) resulted in predominantly cyclic products with moderate to high TTNs. However, substitution with D-2,3-diaminopropionic acid (26) or D-histidine (27) resulted in higher levels of linear products. Only one substitution was tested at position 3 (Supplementary Fig. 36), where L-phenylalanine was replaced with polar L-tyrosine (28). Although TTN decreased by approximately 2-fold, the overall ratio of cyclized to hydrolyzed remains unchanged (>20:1). We were unable to explore other polar substitutions at this position, as the linear peptide eluted in the solvent front of the UPLC. Figure 2D illustrates the tetrapeptide substrate scope of Ulm16, indicating that it can accommodate a diverse range of amino acids at positions 1–3 of the tetrapeptide sequence. However, the N-terminal amino acid shows a pronounced preference for non-polar amino acids. Overall, this suggests that Ulm16 has broad specificity for substrate recognition, but also exhibits selectivity for certain amino acid residues.
Stereochemical preorganization into a cyclic conformation can have a significant impact on the ability of a peptide to undergo cyclization. To explore the ability of Ulm16 to cyclize peptides with different stereochemistry, we modified the stereochemistry of tetrapeptide 16, which exhibited the highest total turnover and catalytic efficiency in our study (Figure 2C). We synthesized three other peptides (peptides 29–31), with D-D-D-L, D-L-D-L, and D-D-L-D stereochemistry, respectively. Peptide 31 was synthesized with D-isovaline to prevent epimerization during the final amino acid coupling. Surprisingly, we found that Ulm16 was able to efficiently cyclize the D-D-D-L peptide (peptide 29), with a respectable TTN and selectivity for cyclized over hydrolyzed peptide (~12:1). However, the D-L-D-L stereochemistry (peptide 30) was more prone to hydrolysis, with a cyclization-to-hydrolysis ratio of ~1:1.4. The D-D-L-D stereochemistry (peptide 31) was not efficiently processed by Ulm16, and only hydrolyzed peptide was observed (Supplementary Figs. 37-39). These results are consistent with previous reports that PBP-TEs require a D C-terminal amino acid and L N-terminal amino acid for efficient cyclization activity.31
Ulm16 Crystal Structure and Peptide Modeling
Given the unique ability of Ulm16 to catalyze cyclization of tetrapeptides, we aimed to determine its structure. We have solved the structure of Ulm16 (12–440) to a resolution of 2.07Å in the monoclinic space group P1211 with one molecule in the asymmetric unit (Supplementary Table 3). Architecturally, Ulm16 displays two domains (residues 12–346 and 361–440) connected by a long-unfolded loop (residues 347–360) that forms an indentation in which the catalytic active site is located. The first domain, the PBP domain, contains an α-β-hydrolase fold with a 7-strand all-anti β-sheet flanked by several α-helices, highly reminiscent of β-lactamases and other DD-transpeptidases.42 The second domain contains a lipocalin-like domain with an 8-strand anti-parallel β-barrel fold. Overall, the structure of Ulm16 is highly similar to that of SurE (PDB 6KSU and 6KSV).31 However, there are a few key differences. First, as observed in the sequence alignment (Supplementary Fig. 1), SurE has a 35-residue loop insertion (residues 195–233) bridging the active site cleft, which is significantly shorter in Ulm16. Second, the orientation of the lipocalin-like domain with respect to the PBP domain differs, although it could be, at least in part, due to crystallographic packing differences between the two structures (Figure 3A). Interestingly, this deviation in the lipocalin domain orientation is also predicted in the alpha fold structure for Ulm16, and other PBP-TEs which cyclize shorter sequences (<6 amino acids, Extended Data Fig. 1). Although the 16-amino acid loop insertion in Ulm16 was predicted to form an alpha helix (Extended Data Fig. 1), electron density was not observed for this region. However, given its distance from the active site and lipocalin domain, as well as the number of glutamic acid residues present, we believe this insertion may contribute to the stability of Ulm16.
Fig 3. Crystal structure of Ulm16.

A) Overlay of the crystal structures of SurE (orange) and Ulm16 (blue). The lighter colors indicate the PBP domain and the darker colors indicate the lipocalin domain. Black rectangle indicates the active site. B) Close up of active site of SurE (orange) and Ulm16 (blue) is shown in the top panel. C) hydrogen bonding network with the catalytic serine. D) water molecule that bridges the catalytic serine and the oxyanion hole.
The Ulm16 catalytic tetrad of Ser71, Lys74, Asn179, and His296 overlaps nearly identically to that of SurE (Figure 3B). These enzymes follow a SxxK motif seen in many serine nucleophile transpeptidases/acyltransferases along with a conserved Tyr177 residue seen in type C β-lactamases (Figure 3B-C).42,43 Interestingly, in the active site of Ulm16, the active site Ser71 assumes two rotamers. The distal rotamer with reference to the oxyanion hole engages in a strong hydrogen bonding interaction with Tyr177 at 2.9Å and to a water molecule occupying the oxyanion hole at 3.4Å, as well as a weak interaction with Lys74 at 3.8Å. Additional hydrogen-bonding interactions between Tyr177 and Lys74 (2.9Å), Tyr177 and His296 (3.6Å), along with Asn179 and Lys74 (3.0Å) constitutes a well-organized distal region (Figure 3C). The proximal rotamer to the oxyanion hole engages in a hydrogen bond with a water molecule occupying the oxyanion hole comprised of the amide N-H groups of Ser71, Thr299, and Leu300 (Figure 3D). This arrangement includes an extra hydrogen bond donor compared to that of SurE.
Despite repeated trials, we were unable to obtain a cocrystal structure of Ulm16 bound to a cyclic peptide or a linear peptide substrate. Nonetheless we sought to further understand the ability of Ulm16 to catalyze the cyclization of small peptides. We conducted covalent docking studies on three substrates, the peptide yielding the highest TTN 16 (TTN = 34679) along with its two diastereomers 29 (TTN = 12392) and 30 (TTN = 4323), to gain insights into how the initial stereochemistry of the substrate may affect catalysis. Interestingly, we observed that residues A227, L231, A232, and V309 in PDB:6KSU, which were previously thought to contribute to C-terminal substrate selectivity34, did not align well with Ulm16 due to the significant truncation in its loop sequence. Instead, in substrates 16 and 29, the C-terminal D-tyrosine occupies a hydrophobic pocket comprised of residues Leu275, Trp279, and Phe286 in our docked peptide-bound model (Figure 4A and Supplementary Fig. 41). This pocket aligns well with the pocket where the C-terminal D-leucine binds in the SurE crystal structure (Extended Data Fig. 7). The C-terminal D-tyrosine also engages in hydrogen-bonding interactions with Asp297 and the backbone carbonyl of Leu275. While this result is also observed with peptide 30 (stereochemistry D-L-D-L), variability is observed in the docking results in the initial orientation and binding position of the peptide (Supplementary Fig. 41). This variability could account for the increased hydrolysis rate, contributing to a higher entropic cost associated with cyclization. Additionally, for tetrapeptides 16 and 29, we observed multiple residues, some specifically in the lipocalin domain, within hydrogen bonding distance from the peptide backbone of the substrates (Figure 4B). We hypothesized that these residues (specifically Arg431, Tyr428, and Ser429) help preorganize the substrate into a cyclic conformation. The hydrophobic amino acids phenylalanine at position 3 and valine at the N-termini for all three peptides bind into the hydrophobic region of the lipocalin domain (consisting of amino acids Leu372, Val374, Leu387, Pro388, Tyr428, and Leu433). Covalent docking of peptide 16 with the crystal structure of SurE (6KSV) shows that many of the residues are conserved, such as Tyr443 and Arg446 (Tyr428 and Arg431 of Ulm16). However, the differing position of the lipocalin domain of SurE puts the hydrophobic core of the lipocalin domain out of reach of the tetrapeptide (Extended Data Fig. 7) resulting in a mix of conformational outputs including those which we hypothesized to be unfavorable for cyclization (Extended Data Fig. 8). This agrees with our observations as SurE has shown the ability to cyclize larger peptides but struggles to cyclize tetrapeptides, instead favoring dimerization.32 Additionally, this provides rationalization for why Ulm16 displays a preference towards substrates with a non-polar N-terminal amino acid.
Fig 4. Covelent Docking of Peptide Substrates.

A) Covalent docking of tetrapeptide 16 29 (Pink) with Ulm16. B) Covalent docking of hexapeptide 11 (Tan) with Ulm16. Residues within hydrogen bonding distance are highlighted.
To provide more insight into the substrate scope of Ulm16, two linear hexapeptides were also covalently docked (Ulm, 1 and desotamide A, 11). As expected, 1, which cyclizes without detectable hydrolysis, followed a similar path as the tetrapeptides 16 and 29 and made multiple close contacts throughout the peptide backbone and side chains of the peptide, such as Asp287, which is near the L-ornithine residue of the Ulm peptides, and Thr299 (Ala307 in the SurE crystal structure), which is within hydrogen-bonding distance to a carbonyl of the backbone of peptide 1 (Supplementary Fig. 42). While 1 is two amino acids longer than 16 and 29, the N-terminal tryptophan now occupies the hydrophobic pocket of the lipocalin domain (Leu372, Val374, Leu387, Pro388, Tyr428, and Leu433). Surprisingly, when 11 was docked, it had a completely different orientation (Figure 4B), like that was observed in the conformation we saw with peptide 30 that we hypothesized to be unfavorable and has been observed in previous docking studies with SurE. However, 11 in the docking model is preorganized into a cyclic conformation with the N-terminus being only 3.6 Å away from the C-terminus, while the backbone carbonyls are still within hydrogen-bonding distance of Arg431. Additionally, multiple side chain residues are in position to undergo hydrogen bond and hydrophobic interactions with Asp144/Asp146 and the lipocalin domain residues. The N-terminal L-tryptophan of 11 now resides in the hydrophobic core where the C-terminal D-tyrosine of the tetrapeptides was bound. All this possibly results in its preorganization leading to its favorable cyclization to hydrolysis ratio.
Mutagenesis studies of Ulm16
In the absence of a co-crystal structure, we undertook site-directed mutagenesis of specific residues within alpha-beta hydrolase domain and lipocalin domain of Ulm16 (Supplementary Fig. 43-44). These mutants were subsequently assessed using four tetrapeptides—two with D-D-L-L sequences (14 and 16), along with D-D-D-L (29) and D-L-D-L (30) tetrapeptides all of which had varying TTNs and cyclized to hydrolyzed ratios. Additionally, a hexapeptide (12-MMP) was also tested as the covalent docking predicted it was predisposed to forming a cyclic conformation (Figure 4B). Earlier investigations on PBP-TEs,32 along with our docking studies, suggest that the lipocalin domain is responsible for the ability of the enzyme to induce a cyclic conformation of the peptide. Initially, we removed the lipocalin domain completely and created the Ulm16 1–344 construct. In the absence of this domain, Ulm16 produces very small amounts of cyclic peptide in most cases (Figure 5 and Supplementary Table 2). However, the TTNs are orders of magnitude lower compared to the wild-type enzyme, suggesting that the domain is very important, if not essential, for cyclization. To gain deeper insights into the effect of the lipocalin domain on cyclization, we expressed and purified Ulm16 mutants targeting residues believed to aid substrate preorganization and effective cyclization. These positions (Arg431, Tyr428, and Ser429) are highly conserved throughout PBP cyclases. Notably, docking analyses consistently positioned Arg431 within hydrogen bonding distance of carbonyl groups on the peptide backbone, suggesting a role in preorganizing the peptide into a cyclic conformation (Figure 4B and D). The substitution of arginine with alanine (Arg431Ala) resulted in an enzyme with a greatly reduced cyclized to hydrolyzed ratio (>14-fold change), highlighting the importance of Arg431 in cyclization (see Figure 5 and Supplementary Fig. 45-49). The remaining two mutants (Tyr428Ala and Ser429Ala) yielded variable outcomes. While Ser429Ala exhibited little effect on cyclization—an unexpected result considering its largely conserved nature across other PBP-TEs (Supplementary Fig. 1)— Tyr428Ala led to a minor increase in dimerized peptide formation (Supplementary Table 2) when tested with the tetrapeptide substrates. Notably, the sole peptide greatly affected by this mutation was the hexapeptide 12-SMMP, as Ulm16Y428A lost its ability to efficiently cyclize it (Figure 5 and Supplementary Fig. 45).
Fig. 5. Results from site directed mutagenesis experiments.

A) Close-up of mutated residues (Y428, S429, and R431) located in Domain 2, highlighting their proximity and interactions with the covalently docked peptide 29. B) Heatmap depicting the results of the TTN assay on the left, presenting the overall assay turnover for each peptide juxtaposed with those of the wild-type (WT) protein. On the right, a comparison of cyclization to hydrolysis ratios is shown, juxtaposed with those of the WT protein. The values shown in the graph are averages obtained from at least three replicates. Asterisks reflect the fact that no cyclic peptide was observed and the TTN was calculated based on the amount of hydrolyzed peptide.
For the PBP domain residues, we hypothesized two residues could be playing a role in substrate selectivity: Leu300, which adds increased bulk around the active site, and Asp297, which could be inhibiting the ability of Ulm16 to cyclize peptides with acidic terminal residues. These residues were mutated to glycine and asparagine, respectively, to match MppK, a close homolog of Ulm16 (Supplementary Fig. 2). These mutants were then tested with the same 5 peptides mentioned previously. The Asp297Asn mutant was also tested on peptide 19, a peptide containing a C-terminal glutamic acid. Ulm16L300G overall had lower turnover compared to the wild type protein. However, when tested with peptide 30, an approximately 3-fold increase in cyclized to hydrolyzed ratio was observed (Supplementary Fig. 49). This could result from the decreased bulk in the active site allowing for the peptide to orientate itself into a more favorable conformation. For Asp297Asn, Ulm16 was still unable to perform any catalysis on the peptide containing a C-terminal acidic amino acid, suggesting a more complicated rationale for the substrate scope observed (Supplementary Fig. 50). While further experimental validation through co-crystallization is needed, these computational and mutational studies provide some rationale for the tetrapeptide substrate scope of Ulm16 and its unique ability to constrain linear peptide substrates into preorganized cyclic conformations for catalysis.
DISCUSSION
Cyclic peptide natural products display diverse bioactivities and serve as great therapeutic leads. Unfortunately, accessing cyclic peptides can be chemically challenging, particularly for small cyclic peptides that have high levels of ring strain. Although excised TEs and ribosomally synthesized post-translationally modified peptide cyclases are efficient at cyclizing large peptides44,45 and even proteins46–48, few have demonstrated the ability to efficiently cyclize small rings, such as pentapeptides45, and none have displayed the ability to efficiently cyclize tetrapeptides. Previous studies of PBP-TEs suggest that they are relatively promiscuous in the size of peptides that they can cyclize34. SurE has recently been shown to cyclize peptides as small as five amino acids to rings as large as 23 amino acids using a flexible 2-[2-[2-(amino)ethoxy]ethoxy]acetic acid (AEEA) linker between its canonical N and C-terminal amino acid residues34. Based on our bioinformatics analyses of PBP-TEs, we chose to explore their abilities to cyclize small rings. AlphaFold models37 suggested that Ulm16, the PBP-TE predicted to cyclize the cyclic hexapeptide ulleungymycin, may have a different substrate scope due to the absence of a loop found in other PBP-TEs that cyclize larger peptides. We discovered that Ulm16 efficiently cyclizes a close derivative of the ulleungymycins with much higher efficiency than previously studied PBP-TEs SurE and PenA. It also has a broad substrate range for hexapeptides, with the only major limitations being its requirement for an N-terminal L-amino acid and a C-terminal D-amino acid or glycine. Interestingly, Ulm16 is one of the few PBP-TEs capable of utilizing a C-terminal glycine, with the only other natural example to date being WolJ36. Furthermore, we found that Ulm16 efficiently catalyzes the cyclization of hexa-, penta-, and tetrapeptides, making it the only known PBP-TE to efficiently form a cyclic tetrapeptide.
To rationalize Ulm16’s ability to cyclize peptides with high catalytic efficiency, we solved its crystal structure to a resolution of 2.07 Å and performed covalent docking studies with multiple substrates that displayed varying degrees of cyclization efficiency and cyclization to hydrolysis ratios. Interestingly, the differing position of the lipocalin domain from SurE appears to play a role in organizing the substrate. Specifically, hydrophobic residues appear to form a binding pocket for the N-terminal residues of the docked peptides. Additionally, Arg431 hydrogen bonds with backbone carbonyls of the peptide, possibly aiding in preorganizing the peptide into a conformation that promotes cyclization. The low levels of cyclization observed in Arg431Ala mutants provide strong support for this hypothesis. Taken together, this study provides insight into the structural basis of the unique ability of Ulm16 to efficiently cyclize small peptides, specifically tetrapeptides. The findings presented here contribute to our understanding of the enzymatic mechanisms involved in peptide cyclization and provide a foundation for the development of biocatalytic methods for the cyclization of tetrapeptides with high efficiency.
METHODS
Reagents and materials
Reagents of the purest grade available were purchased from commercial sources and used without further purification: DMF, CH2Cl2, CH3CN (HPLC grade) from Fisher Scientific, N-Methyl-2-pyrrolidone (NMP) from Alfa Aesar. All Amino acids were purchased from Chem-Impex or AAPTECC depending on availability, all other reagents used in this study were purchased from Sigma-Aldrich unless stated otherwise.
AlphaFold2 Modeling
Ulm16 (Accession ATU31793.1), CppA (QQY97180.1), PenA (WP_158102277), SurE (BBZ90014.1), MppK (AAU34204.1), Lon18 (QUJ09165.1), FlkO (AGI87381.1), WolJ (UNO41476.1), and DsaJ (AJW76712.1) were used as inputs for the modeling and carried out utilizing the standard settings in ColabFold v1.5.11
Multiple Sequence Alignment
Ulm16 (Accession ATU31793.1), CppA (QQY97180.1), PenA (WP_158102277), SurE (BBZ90014.1), MppK (AAU34204.1), Lon18 (QUJ09165.1), WolJ (UNO41476.1), and DsaJ (AJW76712.1) were used as inputs for a MUSCLE sequence alignment. The alignment was performed in JalView2 using MUSCLE v3.8.313.
Biosynthetic Gene Cluster Alignments
Biosynthetic gene clusters for ulleungmycin (BGC0001814), desotamide (BGC0001196), surugamide (BGC0001792), and mannopeptimycin (BGC0000388) were downloaded from MiBIG4. The biosynthetic gene clusters for wollamide, longicatenamide, and pentaminomycin were determined by searching for the PBP-TE gene on NCBI and then expanding 20,000 bp on either side then extending until the NRPS was not at the end of the region. The alignments were then generated using Clinker5.
Phylogeny Analysis
Ulm16 (Accession ATU31793.1) was used as the query sequence for a BlastP search performed on April 22, 2023. The top 500 sequences along with CppA (QQY97180.1), PenA (WP_158102277), SurE (BBZ90014.1), MppK (AAU34204.1), Lon18 (QUJ09165.1), WolJ (UNO41476.1), and DsaJ (AJW76712.1) and the outlier AmpC (beta-lactamase from E. coli). MEGA-X6,7 was then used to perform a MUSCLE alignment followed by tree generation using the Maximum Likelihood method (100 bootstraps). Images were generated using MEGA-X.
Ulm16 and SurE cloning for activity assays
The gene for Ulm16 (Supplementary note 1) was amplified via PCR using primer pair Ulm16-Fwd (5′- CAGCAGCCATCATCATCATCATCACatgcacggggactatg-3′) and Ulm16-Rev (5′- CTTTGTTAGCAGCCGGATCTCAGTGtcagtgcgaccgtgcgc-3′) from a pUC57 vector containing ulm16 (synthesized by GeneWiz). The gene for SurE (Supplementary note 2) was amplified via PCR using primer pair SurE-Fwd (5′- CAGCAGCCATCATCATCATCATCACatgggtgccgagggg-3′) and SurE-Rev (5′- CTTTGTTAGCAGCCGGATCTCAGTGtcagagccggtgcatgg-3′) from Streptomyces albus J1074 genomic DNA. The uppercase letters represent sequence homology to the plasmid pET28B, and the lowercase letters represent sequence homology to the gene to be amplified. PCR amplification was performed with Q5 polymerase. Each reaction contained the following: 5 μl of 5× Q5 buffer, 200 μM dNTPs, 500 nM of forward and reverse primer, 1 ng pUC57-Ulm16 plasmid, or 100 ng S. albus genomic DNA, 5 μl of Q5 High GC Enhancer, 0.25 μl Q5 polymerase and nuclease free water to a final volume of 25 μl. Thermocyclers were operated under the following program: (1) initial denaturation at 98 °C for 30 s; (2) 98 °C for 10 s; (3) 72 °C for 20 s; (4) 72 °C for 40 s ; (5) repeat steps 2–4 for a total of 34 cycles with a −0.3 °C in annealing temperature (step 4) per cycle; (6) 72 °C for 2 min and (7) hold at 4 °C. The products were analyzed by electrophoresis through a 1.5% agarose gel with the Gene Ruler 1kb Plus Ladder (Thermo Fisher Scientific) used as a control and showed only a single band at 1.5 kb. The PCR product was cleaned utilizing the GeneJET PCR Purification Kit (Thermo Fisher Scientific) using the standard protocol. The plasmid pET28b was PCR amplified using primers pET28-Fwd (5′-GTGATGATGATGATGATGGCTGCTGCCCATGG −3′) and pET28B-Rev (5′-CACTGAGATCCGGCTGCTAACAAAGCCCG-3′). PCR amplification was performed with Q5 polymerase. Each reaction contained the following: 5 μl of 5× Q5 buffer, 200 μM dNTPs, 500 nM of forward and reverse primer, 50 ng pET28b, 5 μl of Q5 High GC Enhancer, 0.5 μl Q5 polymerase and nuclease free water to a final volume of 25 μl. Thermocyclers were operated under the following program: (1) initial denaturation at 98 °C for 30 s; (2) 98 °C for 10 s; (3) 72 °C for 20 s; (4) 72 °C for 2 min 45 s ; (5) repeat steps 2–4 for a total of 34 cycles; (6) 72 °C for 2 min and (7) hold at 4 °C. The products were analyzed by electrophoresis through a 1.5% agarose gel with the Gene Ruler 1kb Plus Ladder used as a control and showed only a single band at 5 kb. The PCR product was cleaned utilizing the GeneJET PCR Purification Kit using the standard protocol. The gene fragment was then cloned into pET28b using NEB Gibson Assembly master mix to yield pET28b-Ulm16. Specifically, 70 ng of Ulm16 and 40 ng of linearized pET28b were combined (5:1 insert:vector) with an equal volume of master mix and incubated at 50 °C for 60 minutes. NEB High Efficiency cells were then transformed with 2 μl of the mix before plating on LB plates supplemented with kanamycin (50 μg mL−1). Colonies were then grown overnight in 5 mL of LB supplemented with kanamycin at 37 °C on an orbital shaker. Plasmids were isolated using the Thermo Scientific GeneJET Plasmid Miniprep Kit and verified by Sanger sequencing (GeneWiz).
Ulm16 and SurE overexpression and purification
pET28b-Ulm16 or pET28b-SurE was introduced into BL21(DE3) competent cells by electroporation. Transformed cells were selected on LB agar plates supplemented with kanamycin (50 μg mL−1). Single colonies were picked from a fresh plate and inoculated in 5 mL LB with 50 μg mL−1 kanamycin, and grown for 16 hours at 37 °C on an orbital shaker. The overnight culture was used to inoculate 1L of LB with 50 μg mL−1 kanamycin, and the culture was grown at 37 °C with shaking (250 r.p.m.) until OD600 = 0.6. The culture was then cooled on ice before addition of 0.2 mM isopropyl-1-thio-β-D-galactopyranoside (IPTG) and grown at 18 °C with shaking (250 r.p.m.) for 18 hours. Cells were collected via centrifugation (5,000 x g for 10 minutes at 4 °C) and frozen overnight at −80 °C. Cell pellets were thawed on ice for 30 minutes before addition of 25 mL of lysis buffer (50 mM Tris-HCl, 300 mM NaCl, 10 mM imidazole, 10% glycerol (v/v), pH 8.0). The cell suspension was mixed with 1 mg mL−1 lysozyme, 85 μg RNase A, 300 units of DNaseI, and 100 μM phenylmethylsulfonyl fluoride for 30 minutes at 4 °C. The cell suspension was lysed via sonication for 1 minute (10 second pulses, 30 second rest) at 4 °C. The lysate was cleared by centrifugation at 17,000 x g for 30 minutes at 4°C, and the supernatant was transferred and mixed with 1 mL of HisPur Ni-NTA resin (Thermo Scientific) at 4 °C on an orbital shaker. The resin was loaded onto a 10 mL Pierce centrifuge column and washed with 20 mL of wash buffer (50 mM Tris-HCl, 300 mM NaCl, 20 mM imidazole, and 10% glycerol (v/v), pH 8.0). Protein was eluted with 20 mL of elution buffer (50 mM Tris-HCl, 300 mM NaCl, 300 mM imidazole, and 10% glycerol (v/v), pH 8.0), and 1.0 mL fractions were collected. Eluted fractions were assessed by SDS-PAGE using a 4–15% TGX Mini-PROTEAN gel (Bio-Rad) and stained with Coomassie stain (see Supplementary Figure 40). Fractions containing pure protein were pooled and concentrated using an Amicon Ultra-15 centrifugal filter with 30 kDa cutoff (EMD Millipore). Samples were then exchanged into storage buffer (25 mM Tris-HCl, 50 mM NaCl, 5% glycerol (v/v), 1 mM DTT, pH 8.0). The protein concentration was determined using the Pierce™ BCA Protein Assay Kit (Thermo Scientific) following the standard protocol.
Ulm16 and SurE Kinetic Assays
To determine the kinetic parameters of Ulm16, a 50 μL reaction mixture containing 20 mM Tris-HCl (pH 8.0) and 5% DMSO, 24 nM Ulm16, and 12–800 μM substrate was incubated for 2 minutes at 30 °C. For reaction mixtures containing dL3dA (7) and lW1lA (10), 40 nM enzyme was incubated with the substrate at 30°C for 2 and 3 minutes, respectively. The reaction mixture was quenched with 50 μL acetonitrile and centrifuged at 21,000 x g for 10 minutes. Then, 5 μL was loaded onto a CORTECS T3 Column, 120Å, 1.6 μm, 2.1 mm x 50 mm (Waters) and samples were separated by UHPLC with monitoring at 214 nm. The mobile phases A and B were H2O+0.1% formic acid (FA) and acetonitrile+0.1% FA, respectively, and samples were eluted by gradient mode: 0% to 40% for mobile phase B in 11 minutes with a flow rate of 0.5 ml min−1. Waters_connect (Unifi) software version 3.0.0.15 was used in the analyses of the spectra.The peptide concentrations were estimated based on the extinction coefficient ε (214nm) of a synthetic standard. The ε (214nm) values for all SBMP variants were assumed to be equal except for W1A (10), PenA Substrate (13), and dTyrTP (16), for which their initial velocity was calculated assuming that ε (214 nm) values are equal for SBMP substrate and corresponding enzymatic product. All reactions were carried out in triplicate. Kinetic parameters were estimated using the Michaelis-Menten equation and the curve-fitting program PRISM 9 (see Extended Data Figs. 4-6). All enzymatic products shown in Supplementary Fig. 4 were characterized by ESI-MS/MS analysis (Supplementary Figs. 51-76).
Ulm16 Hexapeptide Cyclization Assays
A 50 μL reaction mixture containing 20 mM Tris-HCl (pH 8.0), 5% DMSO, 50 nM Ulm16, and 280 μM substrate was incubated at 30°C for 4 hours. The reaction mixtures were quenched with 50 μL of acetonitrile and centrifuged at 21,000 x g for 10 minutes. Five microliters of the resulting solution were loaded onto a CORTECS T3 Column, 120Å, 1.6 μm, 2.1 mm X 50 mm (Waters) and separated by UPLC with monitoring at 214 nm. The mobile phases used were H2O+0.1% FA and acetonitrile+0.1% FA, and the samples were eluted by gradient mode: 0% to 40% for mobile phase B in 11 minutes with a flow rate of 0.5 ml min−1. All reactions were carried out in triplicate.
Ulm16 Total Turnover Number Assays
A 100 μL reaction mixture containing 20 mM Tris-HCl (pH 8.0), 5% DMSO, and a concentration of Ulm16 and substrate dependent on the peptide (see Supplementary Table 1 for wild type and Supplementary Table 2 for mutant concentrations) was incubated at 30 °C for 4 hours. The reaction mixtures were quenched with 100 μL of acetonitrile and centrifuged at 21,000 x g for 10 minutes. Five microliters of the resulting solution were loaded onto a CORTECS T3 Column, 120 Å, 1.6 μm, 2.1 mm × 50 mm (Waters) and separated by UPLC with monitoring at 214 nm. The mobile phases used were H2O + 0.1% FA and acetonitrile + 0.1% FA. Tetra- and pentapeptides were eluted by gradient mode: 0% to 40% mobile phase B in 11 minutes at a flow rate of 0.5 mL min−1. Unless stated otherwise, all reactions were carried out in triplicate. And TTN was calculated using equation 1 below8.
| (1) |
Ulm16 Cloning, Purification for Crystallization and Mutants
A Ulm16FL and Ulm1612−440 construct was PCR amplified from the pET-28b plasmid harboring the full-length ulm16 using a PCR premix kit (Bioneer) and subcloned into pGEX-6p-1 plasmid (GE Healthcare) using BamHI/XhoI restriction digest and ligation (New England Biolabs). All mutants were created in the Ulm16FL construct (Supplementary Table 4). Plasmid propagation was mediated using chemical competent E. coli DH5α cells. The resulting expression plasmid was sequence verified by Sanger Sequencing (Genewiz) and transformed into chemically competent E. coli BL21 (DE3) (Novagen) to express glutathione S-transferase (GST) fusion protein harboring an HRV-3C PreScission protease site. An overnight culture from a single clone or glycerol stock was grown for 16 h at 37°C and then diluted 1:100 in the expression LB cultures containing 100 μg/mL ampicillin. Cells were grown in LB media at 37°C until an OD600nm of 0.5–0.6 and induced with 0.5 mM IPTG. Expression was carried out for 18 h at 18°C and then harvested by centrifugation at 7000 rpm. The cells were then resuspended in lysis buffer (50 mM Tris pH 8.0, 400 mM NaCl) and subject to high pressure disruption by French press. The lysed resuspension was clarified by ultracentrifugation at 100,000 x g at 4 °C and applied to a gravity chromatography column (Bio-Rad) containing glutathione-agarose resin (Thermo-Fisher Scientific). The resin was washed with 20 column volumes of lysis buffer and protein was eluted with 5 column volumes of GST elution buffer (50 mM Tris pH 7.8, 400 mM NaCl, and 30 mM reduced glutathione). The eluted fusion protein was dialyzed subsequently against two rounds of 4L dialysis buffer (50 mM Tris pH 8.0, 500 mM NaCl, 1 mM DTT) at 4°C while being incubated with GST-tagged PreScission protease as recommended by the manufacturer (GE Healthcare) overnight. The dialyzed sample was concentrated and further purified by Superdex75 size exclusion chromatography using FPLC buffer (50 mM Tris pH 7.8, 100 mM NaCl, 1 mM DTT). Fractions of interest were pooled and applied to pre-equilibrated glutathione resin to remove excess tag and protease contamination repeatedly until homogeneity was achieved, as assessed by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). Purified Ulm1612−440 protein was concentrated to 25 mg/mL for further experimentation.
Crystallization and Data Processing
Crystals were grown by sitting drop vapor diffusion method at 21°C in 0.2 M sodium acetate trihydrate, 0.1 M Tris-hydrochloride pH 8.5, and 30% polyethylene glycol 4000 as the crystallization solution. This solution was initially recorded from the commercial screen Crystal Screen 1 (Hampton Research). These conditions led to crystal forms that needed no further optimization and diffracted to 2.07 Å at the Stanford Synchrotron Radiation Lightsource (SSRL) at the Stanford Linear Accelerator Center (SLAC) on the 12–2 (λ=0.98) beam line. Data was processed and scaled using HKL3000 in the monoclinic space group P12119.Further information can be found in Supplementary Table 3.
Structure Determination and Refinement
The Ulm1612−440 structure was determined by maximum-likelihood molecular replacement10 using the program PHASER in the Phenix suite11,12. A homology model of Ulm1612−440 was developed using SWISS-MODEL13 with the primary sequence of Ulm1612−440 and the tertiary structure of SurE (PDB 6KSU). Molecular replacement was carried out using an input of two components representing the PBP domain (residues 25–345) and the lipocalin domain (residues 362–440). Model building and refinement of the solution was carried out using COOT14 and Phenix suite’s phenix refine where the current crystallographic R and Rfree are 0.180 and 0.222 respectively. Analysis of the Ramachandran plot indicated that 97.8% of residues fall in the most favored regions, 2.2% of residues fell in the allowed regions, and none were observed in the disallowed region. Electron density is well resolved for the entire molecule with the exception of residues 105–125 and 422–423 where no electron density observed for those regions. A total of 234 water molecules were observed in the asymmetric unit. An average B factor of 40.0Å2 was observed for all atoms in the asymmetric unit. The final structure of Ulm1612−440 was validated using MolProbity15 and deposited in the Protein Data Bank (PDB code for the coordinate and reflection files correspond to the code 8FEK). Residues where poor electron density was observed were either not modelled or modelled as alanines.
Docking Methods.
For the covalent docking simulation, the structure of Ulm16 and SurE (PDB 6KSV) was refined using the Protein Preparation Wizard in Maestro (Schrödinger Suite 2022–4, Schrödinger LLC), using PROPKA in the hydrogen bond assignment step with the pH set at 8.0. For SurE the SNAC-D-Leu was removed from the active site prior to docking. Peptides docked in this study were made as C-Terminal Acyl fluorides and suitable 3D coordinates and states were generated using LigPrep. Peptides were covalently docked using Glide through precision mode and nucleophilic C-F substitution as the reaction type. The center of the grid box for the simulation was set around the active site serine of Ulm16 and post-docking minimization was conducted for 200 poses. The top 5 poses (by MMGBSA) for each peptide were chosen an analyzed using Pymol.
Peptide synthesis using a sulfonamide safety-catch resin
Attachment of the first amino acid:
0.4 g (0.37 mmol) of 4-sulfamylbutyryl AM resin (Novabiochem) and an Fmoc-protected amino acid (1.47 mmol) were used to initiate the reaction. The resin was placed in a 5 mL fritted polypropylene syringe (Torviq), washed with CHCl3, and cooled to −20 °C. The resin was then swelled for 15 minutes. After that, the Fmoc-protected amino acid was dissolved in CHCl3 (3 mL), and i-Pr2EtN (1.84 mmol) was added to the resin. The mixture was allowed to mix for 10 minutes at −20 °C. Once cooled, PyOxim (1.47 mmol) was added to the reaction mixture, and the mixture was left to stand at −20 °C for 8 hours with occasional shaking. The resin was then washed with CH2Cl2 (3 × 5 mL) and MeOH (3 × 5 mL) and dried under vacuum for 1 hour. To determine the resin loading, an aliquot (1–3 mg) of the dried resin was treated with piperidine-DMF (1:4). The UV absorbance of the piperidine-dibenzofulvene adduct was observed at 301 nm (ε = 7800 M−1 cm−1).
Solid-phase peptide synthesis (SPPS):
All manual SPPS and cleavage steps were carried out using 5 mL fritted polypropylene syringes (Torviq) as reaction vessels. Pre-loaded 4-sulfamylbutyryl AM resin (0.05 mmol) was swelled in DMF for 15 minutes, drained, and treated with piperidine-DMF (1:4, 3 mL, 1 × 15 minutes). The resin was then filtered and washed with DMF (2 × 3 mL) and CH2Cl2 (2 × 3 mL). In a separate flask, DIC (0.25 mmol) was added to a solution of Fmoc-AA-OH (0.25 mmol) and Oxyma Pure (0.25 mmol) in DMF (1 mL). After a 5-minute preactivation period, the resulting solution was added to the resin, and the mixture was agitated for 1 hour. The resin was then filtered and washed with DMF (3 × 2 mL) and CH2Cl2 (3 × 2 mL). The Kaiser ninhydrin test was performed to determine reaction completion. Deprotection and coupling cycles were repeated until the desired peptide sequence was complete. The last coupled amino acid was N-Boc-protected to block the N-terminus until final peptide cleavage. For amino acids following an arginine, PyOxim (0.25 mmol) and i-Pr2EtN (0.3 mmol) were used in place of oxyma/DIC.
Activation and thiolysis of acyl-sulfonamide bond:
After the last amino acid was attached, the resin (0.05 mmol) was washed with NMP (4 × 5 mL) and allowed to swell in NMP for 15 minutes. The resin was then filtered and a solution of NMP, i-Pr2EtN (0.75 mmol), and iodoacetonitrile (1.5 mmol), previously filtered through an alumina basic plug, was added to the resin. The mixture was allowed to shake for 24 hours. The resin was then washed with NMP (4 × 5 mL) and DMF (5 × 3 mL). The resulting activated N-acylsulfonamide resin was swollen in DMF for 15 minutes, filtered, and then treated with a 50:50 thiol (2.5 mmol) DMF solution containing catalytic sodium thiophenolate (0.025 mmol) for 24 hours. The resin was then filtered and washed with DMF (3 × 1 mL). The combined filtrate and washes were collected in a 20 mL scintillation vial, and the solvent was removed by rotary evaporation followed by lyophilization.
Global deprotection of peptide thioesters:
the crude peptide was subjected to global deprotection by treating it with a mixture of trifluoroacetic acid (TFA), water, and triisopropylsilane (TIPS) in a ratio of 90:5:5 for 1.5 hours. The TFA solution was then removed by a stream of air, and the peptide was precipitated by adding diethyl ether at −20 °C. The resulting precipitate was collected by centrifugation, and the supernatant was discarded. The peptide was then lyophilized to obtain a dry powder. The crude product was characterized by ultra-performance liquid chromatography-mass spectrometry (UPLC-MS) or high-performance liquid chromatography (HPLC). If the purity of the crude product was >90% as determined by monitoring the absorbance at 214 nm, it was used in the cyclization assay without further purification.
Peptide synthesis using a NDbz safety-catch resin
Attachment of Fmoc-MeDbz-OH:
A 5 mL fritted polypropylene syringe containing 0.05 mmol of Fmoc-Gly Rink amide resin (Chem-Impex) was washed with DMF (3 × 5 mL) and allowed to swell for 15 minutes. A solution of Fmoc-MeDbz-OH (synthesized according to a previously published route)16(0.25 mmol), Oxyma (0.25 mmol), and DIC (0.25 mmol) in 3 mL of DMF was then added to the resin and allowed to shake for 1 hour. The resin was filtered and washed with CH2Cl2 (3 × 5 mL) and DMF (3 × 5 mL). Loading efficiency was assumed to be 100% based on a negative ninhydrin test, and the resin was subsequently used in solid-phase peptide synthesis.
Solid-phase peptide synthesis:
Manual SPPS was carried out as previously described for the sulfonamide safety-catch resin.
Activation and thiolysis:
The resin was washed with CH2Cl2 (3 × 5 mL) and allowed to swell for 15 minutes. The resin was then treated with 1 mL of 0.5 M 4-nitrophenyl chloroformate (TCI America) in CH2Cl2 and shaken for 2 hours. The resin was subsequently washed with CH2Cl2 (3 × 5 mL) and DMF (3 × 5 mL). A solution of 3 mL of 0.5 M i-Pr2EtN in DMF was added to the resin and allowed to react for 15 minutes. The resin was then filtered, washed with DMF (1 × 5 mL), and exposed to fresh 0.5 M i-Pr2EtN in DMF solution, repeating this process until the solution no longer turned yellow. The resin was then swollen in DMF for 15 minutes, filtered, and treated with a 50:50 solution of thiol (2.5 mmol) in DMF, shaken for 24 hours. The resin was then filtered and washed with DMF (3 × 1 mL). The combined filtrate and washes were collected in a 20 mL scintillation vial and subjected to rotary evaporation and lyophilization.
Global deprotection of peptide thioesters:
deprotection and UPLC-MS or HPLC analysis was carried out as previously described above.
Extended Data
Extended Data Fig. 1. Alpha fold models of predicted and validated PBP-TEs.

compared to the SurE Crystal structure (RMSD). The alpha fold model of SurE was used as a control and was compared to its known crystal structure. Highlighted in red is the loop region, which is shorter in the Ulm16 sequence, in yellow is the lipocalin loop previously hypothesized to play a role in substrate selectivity, and in orange is the sequence insertion only found in Ulm16 (residues 106–122).
Extended Data Fig. 2. Biosynthetic gene clusters of PBP-TEs that lack the loop region.

These biosynthetic gene clusters all share high homology to the ulleungmycin biosynthetic gene cluster and are predicted to produce natural products identical or very similar to the ulleungmycins.
Extended Data Fig. 3. Ulm peptide structure.

The Ulleungmycin A sequence was modified to incorporate only commercially available amino acids, resulting in a new sequence referred to as ‘Ulm’. The changed amino acids are highlighted in grey, while the C-Terminal amino acid is highlighted in red and the N-Terminal amino acid is highlighted in blue.
Extended Data Fig. 4. Michaelis-Menten plots of Ulm16 kinetics for three peptide thioesters and alanine scans of the ‘Ulm’ peptide.

The names of the substrates used are indicated above each plot, and the enzyme concentrations employed are provided in the methods section. The data is summarized in of the main text. The plots represent the mean of triplicate experiments, and the error bars indicate the standard error of the mean (S.E.M).
Extended Data Fig. 5. Michaelis-Menten plots of Ulm16 and SurE kinetics for a common substrate (13).

The names of the substrates used are indicated above each plot, and the enzyme concentrations employed are provided in the methods section. The plots represent the mean of triplicate experiments, and the error bars indicate the standard error of the mean (S.E.M).
Extended Data Fig. 6. Michaelis-Menten plot of Ulm16 incubated with tetrapeptide 16.

The names of the substrates used are indicated above each plot, and the enzyme concentrations employed are provided in the methods section. The data is summarized in Table 1 of the main text. The plots represent the mean of triplicate experiments, and the error bars indicate the standard error of the mean (S.E.M).
Extended Data Fig. 7. Covalent docking of tetrapeptide 16 with SurE and comparison with Ulm16.

(a, b) Side-by-side of lowest MMGBSA score poses for SurE-16 (orange, A) and Ulm16–16 (blue, B). Peptide 16 is noticeably farther away from the hydrophobic pocket of the lipocalin domain in SurE due to differing angle of the lipocalin domain. (c) Overlay of Ulm16–16 (Sky blue-Light blue) and SurE-16 (Orange-Light Orange) highlights residues that we believe are key for binding and cyclization. Residues L284/L300 and D306/D297 are in the alpha beta hydrolase domain, and we hypothesize are key in binding the C-terminal D-amino acid. Residues R446/R431 and Y443/Y428 are in the lipocalin domain. We hypothesize that they help in cyclization of the peptide. The alpha beta hydrolase domain has been hidden for clarity. (d) Overlay of the C-terminal SNAC-D-Leu (orange) from the SurE substrate bound crystal structure (6SKV) and C-terminal covalently docked D-Tyr from peptide 16 (light orange). (e) Overlay of the C-Terminal SNAC-D-Leu (orange) from the SurE substrate bound crystal structure (6SKV) and C-terminal covalently docked D-Tyr from peptide 16 with Ulm16 (blue) showing they are occupying the same pocket.
Extended Data Fig. 8. Side-by-side comparison of top 5 lowest MMGBSA scoring peptides from covalent docking of 16 with SurE (light orange, A) and Ulm16 (light blue, B).

Peptide 16 exhibits limited access to the hydrophobic pocket within the lipocalin domain of SurE, resulting in a notable diversity of poses generated. Conversely, in the case of Ulm16, distinct conformations of the lipocalin domain facilitate a more proximal interaction with the hydrophobic pocket, leading to a predominant binding orientation consistently observed across outputs. Residues L286/L300 and D306/D297 are in the alpha-beta-hydrolase domain while Y443/Y428 is in the lipocalin domain.
Supplementary Material
ACKNOWLEDGMENTS
We thank A. Alwali and L. Wilbanks for their helpful discussions and aid with UPLC-MS and protein expression, respectively, C. Martinez-Brokaw and M. Hostetler for their advice on chemical synthesis, and G. Buechel for her initial work on peptide synthesis. This work was funded by the National Institutes of Health (1R35GM138002 to E.I.P. and 1F31CA275390 to R.S.P.). Zachary Logan Budimir acknowledges the National Science Foundation for support under the Graduate Research Fellowship Program (GRFP) under grant number DGE-1842166. The authors acknowledge the support from the Purdue Center for Cancer Research, NIH grant P30 CA023168.
Footnotes
Author Contributions Statement
Z.L.B., C.N.E., and J.J.A. synthesized all the peptides used in this study. A.E. and C.N.E. performed NMR analyses of the peptides. Z.L.B., R.S.P., and H.M.R.C. expressed the proteins. Z.L.B. performed the protein assays. R.S.P. performed the crystallography studies. Z.L.B and R.S.P. performed the docking studies. Z.L.B., R.S.P., C.D., and E.I.P. conceived of the ideas and wrote the paper, with input from all authors. Project management and funding, C.D. and E.I.P.
Competing Interest Statement
The authors declare no competing interests.
DATA AVAILABILITY
Ulm16 12–440 coordinates and processed diffraction data has been deposited into the Protein Data Bank under the accession code 8FEK. Other structures used in this manuscript are available at the PDB (6KSU and 6KSV). Protein sequences used in this study are from the NCBI database: Ulm16 (Accession ATU31793.1), CppA (QQY97180.1), PenA (WP_158102277), SurE (BBZ90014.1), MppK (AAU34204.1), Lon18 (QUJ09165.1), FlkO (AGI87381.1), WolJ (UNO41476.1), and DsaJ (AJW76712.1). PDB files of Alphafold models generated this for this study are publicly available and can be accessed utilizing the following DOI. The data that support the findings of this study is available within the main text and its Supplementary Information files. Data is also available from the corresponding author upon request.
REFERENCES
- 1.Driggers EM, Hale SP, Lee J. & Terrett NK The exploration of macrocycles for drug discovery — an underexploited structural class. Nat. Rev. Drug Discov 7, 608–624 (2008). [DOI] [PubMed] [Google Scholar]
- 2.Huang Y, Wiedmann MM & Suga H. RNA Display Methods for the Discovery of Bioactive Macrocycles. Chem. Rev 119, 10360–10391 (2019). [DOI] [PubMed] [Google Scholar]
- 3.Tsomaia N. Peptide therapeutics: Targeting the undruggable space. Eur. J. Med. Chem 94, 459–470 (2015). [DOI] [PubMed] [Google Scholar]
- 4.Malde AK, Hill TA, Iyer A. & Fairlie DP Crystal Structures of Protein-Bound Cyclic Peptides. Chem. Rev 119, 9861–9914 (2019). [DOI] [PubMed] [Google Scholar]
- 5.White CJ & Yudin AK Contemporary strategies for peptide macrocyclization. Nat. Chem 3, 509–524 (2011). [DOI] [PubMed] [Google Scholar]
- 6.Sarojini V, Cameron AJ, Varnava KG, Denny WA & Sanjayan G. Cyclic Tetrapeptides from Nature and Design: A Review of Synthetic Methodologies, Structure, and Function. Chem. Rev 119, 10318–10359 (2019). [DOI] [PubMed] [Google Scholar]
- 7.Du L, Risinger AL, King JB, Powell DR & Cichewicz RH A Potent HDAC Inhibitor, 1-Alaninechlamydocin, from a Tolypocladium sp. Induces G2/M Cell Cycle Arrest and Apoptosis in MIA PaCa-2 Cells. J. Nat. Prod 77, 1753–1757 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Isolation and Structural Elucidation of Cyclic Tetrapeptides from Onychocola sclerotica. https://pubs.acs.org/doi/epdf/10.1021/np3000987 doi: 10.1021/np3000987. [DOI] [PubMed]
- 9.Chloroplast coupling factor 1: A species-specific receptor for tentoxin. https://www.pnas.org/doi/10.1073/pnas.73.7.2245 doi: 10.1073/pnas.73.7.2245. [DOI] [PMC free article] [PubMed]
- 10.CJ-15, 208, a Novel Kappa Opioid Receptor Antagonist from a Fungus, Ctenomyces serratus ATCC15502. https://www.jstage.jst.go.jp/article/antibiotics1968/55/10/55_10_847/_article. [DOI] [PubMed]
- 11.Chung BKW, White CJ, Scully CCG & Yudin AK The reactivity and conformational control of cyclic tetrapeptides derived from aziridine-containing amino acids. Chem. Sci 7, 6662–6668 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Skropeta D, Jolliffe KA & Turner P. Pseudoprolines as Removable Turn Inducers: Tools for the Cyclization of Small Peptides. J. Org. Chem 69, 8804–8809 (2004). [DOI] [PubMed] [Google Scholar]
- 13.Alcaro MC et al. On-resin head-to-tail cyclization of cyclotetrapeptides: optimization of crucial parameters. J. Pept. Sci 10, 218–228 (2004). [DOI] [PubMed] [Google Scholar]
- 14.Meutermans WDF et al. Difficult Macrocyclizations: New Strategies for Synthesizing Highly Strained Cyclic Tetrapeptides. Org. Lett 5, 2711–2714 (2003). [DOI] [PubMed] [Google Scholar]
- 15.Vidović N. et al. Chloride-Assisted Peptide Macrocyclization. Org. Lett 22, 2129–2134 (2020). [DOI] [PubMed] [Google Scholar]
- 16.Jing X. & Jin K. A gold mine for drug discovery: Strategies to develop cyclic peptides into therapies. Med. Res. Rev 40, 753–810 (2020). [DOI] [PubMed] [Google Scholar]
- 17.Abdalla MA Medicinal significance of naturally occurring cyclotetrapeptides. J. Nat. Med 70, 708–720 (2016). [DOI] [PubMed] [Google Scholar]
- 18.Li Y-H et al. Putative Nonribosomal Peptide Synthetase and Cytochrome P450 Genes Responsible for Tentoxin Biosynthesis in Alternaria alternata ZJ33. Toxins 8, 234 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Witte TE, Villeneuve N, Boddy CN & Overy DP Accessory Chromosome-Acquired Secondary Metabolism in Plant Pathogenic Fungi: The Evolution of Biotrophs Into Host-Specific Pathogens. Front. Microbiol 12, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xu H-M et al. Tataricins A and B, two novel cyclotetrapeptides from Aster tataricus, and their absolute configuration assignment. Tetrahedron Lett. 54, 1380–1383 (2013). [Google Scholar]
- 21.Ma G-L et al. Biosynthesis of Tasikamides via Pathway Coupling and Diazonium-Mediated Hydrazone Formation. J. Am. Chem. Soc 144, 1622–1633 (2022). [DOI] [PubMed] [Google Scholar]
- 22.Kohli RM, Walsh CT & Burkart MD Biomimetic synthesis and optimization of cyclic peptide antibiotics. Nature 418, 658–661 (2002). [DOI] [PubMed] [Google Scholar]
- 23.Trauger JW, Kohli RM, Mootz HD, Marahiel MA & Walsh CT Peptide cyclization catalysed by the thioesterase domain of tyrocidine synthetase. Nature 407, 215–218 (2000). [DOI] [PubMed] [Google Scholar]
- 24.Horsman ME, Hari TPA & Boddy CN Polyketide synthase and non-ribosomal peptide synthetase thioesterase selectivity: logic gate or a victim of fate? Nat. Prod. Rep 33, 183–202 (2016). [DOI] [PubMed] [Google Scholar]
- 25.Gao X. et al. Cyclization of fungal nonribosomal peptides by a terminal condensation-like domain. Nat. Chem. Biol 8, 823–830 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hoyer KM, Mahlert C. & Marahiel MA The Iterative Gramicidin S Thioesterase Catalyzes Peptide Ligation and Cyclization. Chem. Biol 14, 13–22 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kuranaga T. et al. Total Synthesis of the Nonribosomal Peptide Surugamide B and Identification of a New Offloading Cyclase Family. Angew. Chem 130, 9591–9595 (2018). [DOI] [PubMed] [Google Scholar]
- 28.Zhou Y. et al. Investigation of penicillin binding protein (pbp)-like peptide cyclase and hydrolase in surugamide non-ribosomal peptide biosynthesis. Cell Chemical Biology 26, (2019). [DOI] [PubMed] [Google Scholar]
- 29.Thankachan D. et al. A trans -Acting Cyclase Offloading Strategy for Nonribosomal Peptide Synthetases. ACS Chem. Biol 14, 845–849 (2019). [DOI] [PubMed] [Google Scholar]
- 30.Matsuda K. et al. SurE is a trans -acting thioesterase cyclizing two distinct non-ribosomal peptides. Org. Biomol. Chem 17, 1058–1061 (2019). [DOI] [PubMed] [Google Scholar]
- 31.Matsuda K. et al. Heterochiral coupling in non-ribosomal peptide macrolactamization. Nat. Catal 3, 507–515 (2020). [Google Scholar]
- 32.Matsuda K, Fujita K. & Wakimoto T. PenA, a penicillin-binding protein-type thioesterase specialized for small peptide cyclization. J. Ind. Microbiol. Biotechnol 48, kuab023 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fazal A, Wheeler J, Webb ME & Seipke RF The N-terminal substrate specificity of the SurE peptide cyclase. Org. Biomol. Chem 20, 7232–7235 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kobayashi M, Fujita K, Matsuda K. & Wakimoto T. Streamlined Chemoenzymatic Synthesis of Cyclic Peptides by Non-ribosomal Peptide Cyclases. J. Am. Chem. Soc 145, 3270–3275 (2023). [DOI] [PubMed] [Google Scholar]
- 35.Fazal A, Webb ME & Seipke RF The Desotamide Family of Antibiotics. Antibiotics 9, 452 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Booth TJ et al. Bifurcation drives the evolution of assembly-line biosynthesis. Nat. Commun 13, 3498 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mirdita M. et al. ColabFold: making protein folding accessible to all. Nat. Methods 19, 679–682 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Son S. et al. Genomics-Driven Discovery of Chlorinated Cyclic Hexapeptides Ulleungmycins A and B from a Streptomyces Species. J. Nat. Prod 80, 3025–3031 (2017). [DOI] [PubMed] [Google Scholar]
- 39.Gao D-W et al. A Polyketide Cyclase That Forms Medium-Ring Lactones. J. Am. Chem. Soc 143, 80–84 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bar-Even A. et al. The Moderately Efficient Enzyme: Evolutionary and Physicochemical Trends Shaping Enzyme Parameters. Biochemistry 50, 4402–4410 (2011). [DOI] [PubMed] [Google Scholar]
- 41.Hostetler MA et al. Synthetic Natural Product Inspired Cyclic Peptides. ACS Chem. Biol 16, 2604–2611 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Walsh C. & Wencewicz TA Antibiotics: Challenges, mechanisms, opportunities. (ASM Press, 2016). [Google Scholar]
- 43.Dubus A, Normark S, Kania M. & Page MGP The Role of Tyrosine 150 in Catalysis of .beta.-Lactam Hydrolysis by AmpC .beta.-Lactamase from Escherichia coli Investigated by Site-Directed Mutagenesis. Biochemistry 33, 8577–8586 (1994). [DOI] [PubMed] [Google Scholar]
- 44.McIntosh JA et al. Circular Logic: Nonribosomal Peptide-like Macrocyclization with a Ribosomal Peptide Catalyst. J. Am. Chem. Soc 132, 15499–15501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ludewig H. et al. Characterization of the Fast and Promiscuous Macrocyclase from Plant PCY1 Enables the Use of Simple Substrates. ACS Chem. Biol 13, 801–811 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nguyen GKT et al. Butelase-mediated cyclization and ligation of peptides and proteins. Nat. Protoc 11, 1977–1988 (2016). [DOI] [PubMed] [Google Scholar]
- 47.Hemu X, Qiu Y, Nguyen GKT & Tam JP Total Synthesis of Circular Bacteriocins by Butelase 1. J. Am. Chem. Soc 138, 6968–6971 (2016). [DOI] [PubMed] [Google Scholar]
- 48.Nguyen GKT et al. Butelase 1: A Versatile Ligase for Peptide and Protein Macrocyclization. J. Am. Chem. Soc 137, 15398–15401 (2015). [DOI] [PubMed] [Google Scholar]
Methods-Only References
- 49.Mirdita M. et al. ColabFold: making protein folding accessible to all. Nat. Methods 19, 679–682 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Waterhouse AM, Procter JB, Martin DMA, Clamp M. & Barton GJ Jalview Version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics 25, 1189–1191 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Edgar RC MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Terlouw BR et al. MIBiG 3.0: a community-driven effort to annotate experimentally validated biosynthetic gene clusters. Nucleic Acids Res. 51, D603–D610 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gilchrist CLM & Chooi Y-H clinker & clustermap.js: automatic generation of gene cluster comparison figures. Bioinformatics 37, 2473–2475 (2021). [DOI] [PubMed] [Google Scholar]
- 54.Jones DT, Taylor WR & Thornton JM The rapid generation of mutation data matrices from protein sequences. Bioinformatics 8, 275–282 (1992). [DOI] [PubMed] [Google Scholar]
- 55.Kumar S, Stecher G, Li M, Knyaz C. & Tamura K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol 35, 1547–1549 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schmidt JJ et al. A Versatile Chemoenzymatic Synthesis for the Discovery of Potent Cryptophycin Analogs. ACS Chem. Biol 15, 524–532 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Minor W, Cymborowski M, Otwinowski Z. & Chruszcz M. HKL-3000: the integration of data reduction and structure solution – from diffraction images to an initial model in minutes. Acta Crystallogr. D Biol. Crystallogr 62, 859–866 (2006). [DOI] [PubMed] [Google Scholar]
- 58.Rossmann MG The molecular replacement method. Acta Crystallogr. A 46, 73–82 (1990). [DOI] [PubMed] [Google Scholar]
- 59.Adams PD et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr 66, 213–221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liebschner D. et al. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr. Sect. Struct. Biol 75, 861–877 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Waterhouse A. et al. Swiss-model: Homology modelling of protein structures and complexes. Nucleic Acids Research 46, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Emsley P, Lohkamp B, Scott WG & Cowtan K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr 66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chen VB et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr 66, 12–21 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Blanco-Canosa JB, Nardone B, Albericio F. & Dawson PE Chemical protein synthesis using a second-generation n-acylurea linker for the preparation of peptide-thioester precursors. Journal of the American Chemical Society 137, 7197–7209 (2015). [DOI] [PubMed] [Google Scholar]
- 65.Matsuda K. et al. Heterochiral coupling in non-ribosomal peptide macrolactamization. Nat. Catal 3, 507–515 (2020). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Ulm16 12–440 coordinates and processed diffraction data has been deposited into the Protein Data Bank under the accession code 8FEK. Other structures used in this manuscript are available at the PDB (6KSU and 6KSV). Protein sequences used in this study are from the NCBI database: Ulm16 (Accession ATU31793.1), CppA (QQY97180.1), PenA (WP_158102277), SurE (BBZ90014.1), MppK (AAU34204.1), Lon18 (QUJ09165.1), FlkO (AGI87381.1), WolJ (UNO41476.1), and DsaJ (AJW76712.1). PDB files of Alphafold models generated this for this study are publicly available and can be accessed utilizing the following DOI. The data that support the findings of this study is available within the main text and its Supplementary Information files. Data is also available from the corresponding author upon request.