

Conspectus
RNA modifications found in most RNAs, particularly in tRNAs and rRNAs, reveal an abundance of chemical alterations of nucleotides. Over 150 distinct RNA modifications are known, emphasizing a remarkable diversity of chemical moieties in RNA molecules. These modifications play pivotal roles in RNA maturation, structural integrity, and the fidelity and efficiency of translation processes. The catalysts responsible for these modifications are RNA-modifying enzymes that use a striking array of chemistries to directly influence the chemical landscape of RNA. This diversity is further underscored by instances where the same modification is introduced by distinct enzymes that use unique catalytic mechanisms and cofactors across different domains of life. This phenomenon of convergent evolution highlights the biological importance of RNA modification and the vast potential within the chemical repertoire for nucleotide alteration. While shared RNA modifications can hint at conserved enzymatic pathways, a major bottleneck is to identify alternative routes within species that possess a modified RNA but are devoid of known RNA-modifying enzymes. To address this challenge, a combination of bioinformatic and experimental strategies proves invaluable in pinpointing new genes responsible for RNA modifications. This integrative approach not only unveils new chemical insights but also serves as a wellspring of inspiration for biocatalytic applications and drug design. In this account we present how comparative genomics and genome mining, combined with biomimetic synthetic chemistry, biochemistry, and anaerobic crystallography can be judiciously implemented to address unprecedented and alternative chemical mechanisms in the world of RNA modification. We illustrate these integrative methodologies through the study of tRNA and rRNA modifications, namely dihydrouridine, 5-methyluridine, queuosine, 8-methyladenosine, 5-carboxymethylamino-methyluridine or 5-taurinomethyluridine, each dependent on a diverse array of redox chemistries, often involving organic compounds, organometallic complexes, and metal coenzymes. We explore how vast genome and tRNA databases empower comparative genomic analyses and enable the identification of novel genes which govern RNA modification. Subsequently, we describe how the isolation of a stable reaction intermediate can guide the synthesis of a biomimetic to unveil new enzymatic pathways. We then discuss the usefulness of a biochemical ‘shunt’ strategy to study catalytic mechanisms and to directly visualize reactive intermediates bound within active sites. While we primarily focus on various RNA-modifying enzymes studied in our laboratory, with a particular emphasis on the discovery of a SAM-independent methylation mechanism, the strategies and rationale presented herein are broadly applicable for the identification of new enzymes and the elucidation of their intricate chemistries. This account offers a comprehensive glimpse into the evolving landscape of RNA modification research and highlights the pivotal role of integrated approaches to identify novel enzymatic pathways.
Graphical Abstract
1. Introduction
RNAs ensure the decoding of the genetic information stored in DNA into proteins and are involved in many crucial biological pathways including the control of gene expression5,6. After their biogenesis during transcription, the newly transcribed RNAs undergo several processing steps that functionalize them into mature RNAs to fulfill their diverse biological roles7,8. One of these steps is the incorporation of several distinct chemical groups at the base and/or ribose of specific nucleotides, thus converting a monotonous polymer into a highly decorated molecule9,10. This biological process is termed posttranscriptional RNA modification. To understand these RNA modification processes, also known as the epitranscriptome or RNA epigenetics, a broad range of techniques are required to identify and quantify these chemical marks transcriptome wide, to establish the chemistry of their biosynthesis, and to understand their biological roles11.
RNA modification is currently the subject of intense efforts not only given their functional relevance in RNA structure formation12–14 and their implication as molecular determinants for numerous cellular fates and interactions, but also given their role in the adaptation of gene expression to changing metabolic regimes and stress15–20. To date, more than 150 RNA modifications are known and it is likely that this number will increase with new breakthroughs in technologies dedicated to the analysis of modifications on the whole transcriptome, such as chemical labeling methods, mass spectrometry (MS) analysis and high-throughput sequencing21–23(HTS) including nanopore24,25. This great diversity in chemical groups inevitably implies new chemistries as well as diverse molecular mechanisms of protein/RNA interactions that are yet to be unraveled. Understanding the chemical mechanisms that drive these modifications is fundamental if we hope to target them for future therapeutic approaches26,27. Indeed, a dysfunction in RNA modifications is recognized as a cause of aging28 and a driver for a large number of severe disorders29. Most commonly, mutations in the genes coding the enzymes that catalyze the RNA modifications will disrupt the enzymatic activity leading to a hypomodified RNA and a disease state classified as an RNA modopathy30.
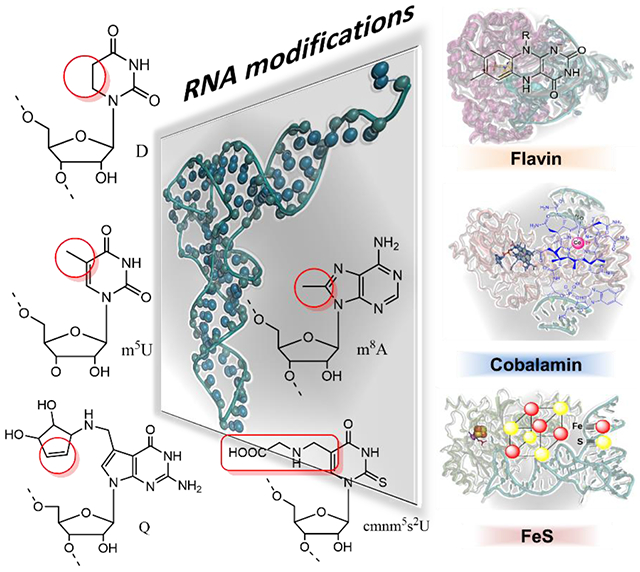
RNA modifications are catalyzed by highly selective enzymes able to specifically recognize their substrate among the millions of RNAs in the cell and to modify a defined single atom contained within the substrate. Throughout evolution, these outstanding catalysts have developed unique and often complex strategies to catalyze RNA modification chemistry. RNA modifying enzymes often rely on organic cofactors such as flavin31 or inorganic cofactors such as iron-sulfur clusters, mostly 4Fe-4S32, or even organometallic cofactors such as cobalamin33 (Figure 1), that synergize with the polypeptide to promote unexpected reactivities.
Figure 1. RNA modifications dependent on redox coenzymes.
Left side : biosynthetic RNA modifications for: (i) the reduction of uridine to dihydrouridine (D) catalyzed by the dihydrouridine synthases (Dus), (ii) the oQ reduction to Q by QueG and QueH, (iii) the reductive methylation of uridine into m5U by TrmFO, (iv) the carboxymethylaminomethylation of uridine into cmnm5U catalyzed by MnmE/mnmG complex (v) the methylation of adenine to m8A by RlmN. (vi) The chemical groups of each modification are boxed in red and the respective enzyme(s) that catalyze each reaction are indicated. Right side: Biochemical coenzymes used for RNA modifications: flavins (FMN and FAD) in orange, cobalamin (CbI) in blue and its metal in magenta, while the cubic 4Fe-4S cluster is represented with Fe atoms in red and sulfur in yellow.
In this account, we will discuss how combining comparative genomics, phylogeny, X-ray crystallography under anaerobiosis, and synthetic chemistry can foster the discovery of new RNA modification enzymes associated with unprecedented chemistries. We will describe how these strategies have led us to decipher different biosynthetic mechanisms for various nucleotide modifications that rely on redox reactions such as queuosine, ribothymidine or dihydrouridine, and highlight the tools that nature has at its disposal to catalyze identical modifications with alternative chemistries, notably through convergent evolutionary events. We will also emphasize how our studies can inform the mechanisms of other complex RNA modifications.
2. Comparative genomics, a powerful approach in the search for alternative and novel mechanisms of RNA modifications
Comparative genomic approaches are powerful tools in the search for new enzymes34. These methods are ideal to identify the functions of genes and to discover new enzymes in Bacteria and Archaea35,36. Since tRNA modification genes are not only widespread but also conserved among specific kingdoms or phyla, in silico comparative approaches that combine different types of genomic and post-genomic evidence, such as physical clustering, phyletic searches, phenotype/expression/localization data or 3D-structures (Figure 2), have successfully identified over 30 missing genes in the last 20 years. The availability of over 200,000 genomes with complete sequences has increased the depth of comparative approaches but a major limiting factor in the field of RNA modification is the limited availability of precise mapping and identification of modifications in all tRNAs from a given organism. Indeed, the atlas of tRNA modifications is currently available for just a handful of species in MODOMICS10. Nevertheless, we are optimistic that the recent progress in MS and HTS-sequencing analytical tools will increase this number in the near future 21.
Figure 2. Comparative genomic pipeline to identify missing tRNA modification genes.
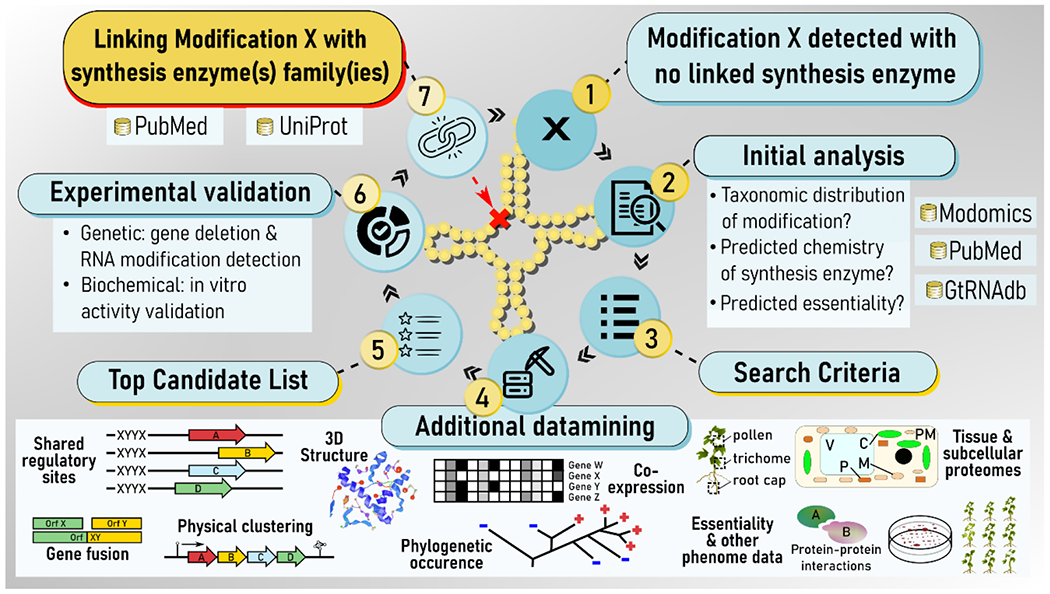
The search starts by mapping tRNA modifications to known enzymes to identify globally or locally missing enzymes (1). After an initial datamining step (2) to define search criteria (3), a combination of in silico tools that integrate different types of in silico and omics data (4) can be used to generate a candidate list (5) that will be experimentally validated (6) before deposition in databases as a novel tRNA modification genes (7). Modomics is a database of RNA modification. GtRNAdb is a Genomic tRNA database.
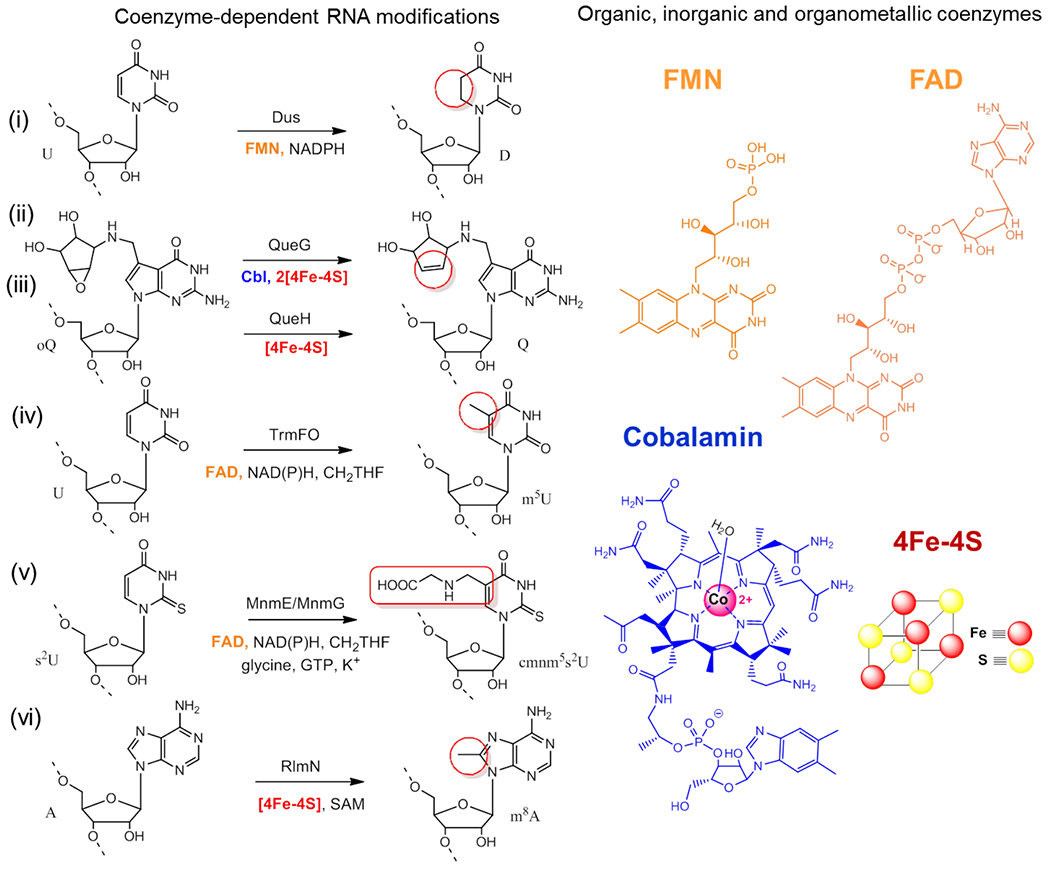
Among the tRNA-modifying enzyme genes identified by comparative genomics, we uncovered the dihydrouridine synthases (Dus) of E. coli responsible for dihydrouridine (D) incorporation in tRNAs37. The guiding strategy for this identification was the analysis of 3307 clusters of orthologous groups available at that time with the comparative search criteria that the gene encoding the D biosynthetic enzyme must be absent in Pyrococcus abyssi, which does not have D in its tRNAs, but present in E. coli, S. cerevisiae and B. subtilis. This led to the identification of a family of flavin mononucleotide (FMN)-dependent flavoenzymes composed of three subfamilies, termed DusA, DusB and DusC. These enzymes use NAD(P)H as a reductant to catalyze the hydrogenation of the C5=C6 double bond of certain uridines in tRNAs, through a hydride transfer from the flavin to the substrate RNA38,39 (Figure 3). Each Dus is in charge of the synthesis of at least one D residue and the complete site specificity of this enzyme family was recently resolved in our laboratory40. Unexpectedly, phylogenetic analysis shows a complex distribution of Dus in bacteria and we demonstrated that gram-positive bacteria rely on a single Dus homolog of type B to insert all D residues into their tRNAs, highlighting the multisite specificity of this enzyme subfamily41.
Figure 3. Illustration of different redox-dependent chemistries involved in RNA modifications.
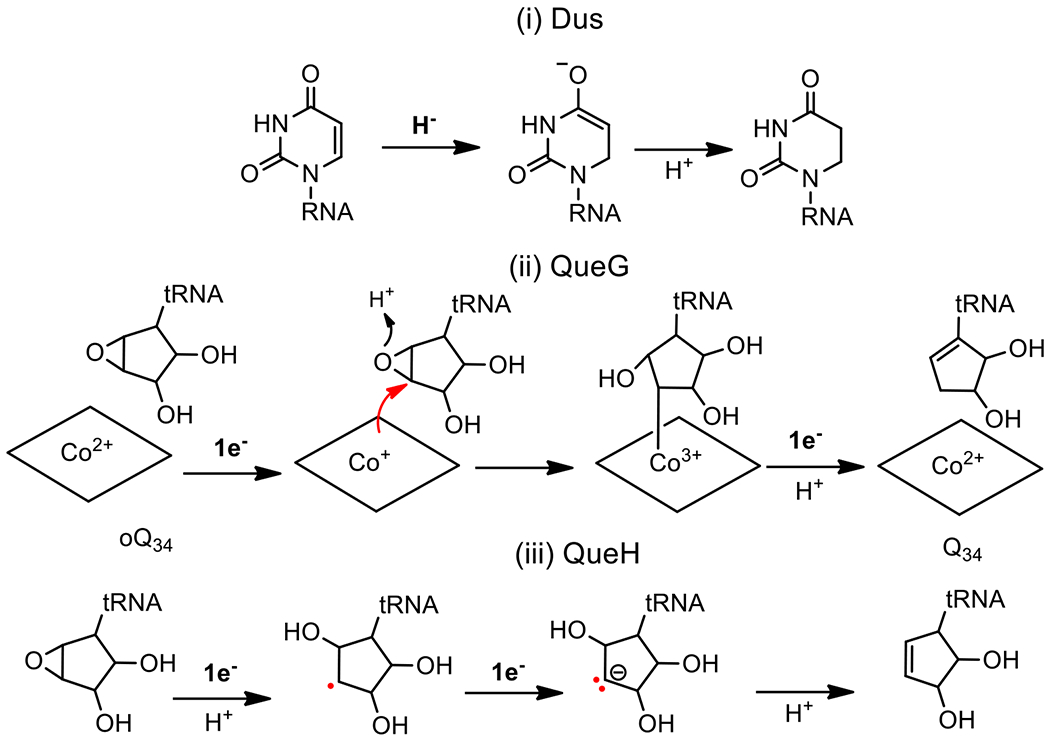
(i) Hydride reduction: Dus catalyzes the reduction of the ethylenic uridine bond of tRNAs and mRNAs by hydride transfer derived from the N5-FMNH− to the sp2 C6 carbon of uracil and by protonating the C5 using the thiol of a conserved site cysteine that acts as an acid. (ii) Covalently catalyzed reduction: epoxyqueuosine reductase, QueG, is the predominant oQ34 enzyme in nature that catalyzes the reduction of the epoxy-cyclopentanediol moiety of oQ34 to the cyclopentenediol of Q34 by covalent cobalamin-dependent catalysis. The reduction requires two x 1e- from the 4Fe-4S clusters of the protein while an aspartic acid from the active site functions as an acid. (iii) Reduction by electron transfer: QueH is the alternative enzyme to QueG but proceeds via a different mechanism based on sequential electron transfer from the single 4Fe-4S center of the protein.
Comparative genomics can also reveal alternative biosynthetic pathways in certain tRNA modifications such as the recent discovery of a new class of FeS enzymes involved in the last step of the queuosine formation at position 34 of tRNAs (Q34)3. The Q base is a complex modification of guanosine present at the wobble position of tRNA anticodons His, Tyr, Asn and Ala. This modification involves three consecutive steps and is found in both eubacteria and eukaryotes. In most organisms the last step of Q synthesis consists of the reduction of epoxyqueuosine (oQ34) to Q34 catalyzed by the cobalamin- and two FeS cluster-dependent enzyme QueG. The proposed enzymatic mechanism of QueG entails a redox-dependent covalent chemistry orchestrated by the cobalamin wherein two electrons and a proton are exchanged in a sequential order to ensure catalytic turnover33,42,43 (Figure 3), although an alternative mechanism was recently proposed44. Analysis of the distribution of QueG in 1792 genomes of various eubacteria revealed that around half lacked the queG gene but still retained Q34 in their tRNAs. Furthermore, all of these bacteria carried the tgt and queA genes further hinting at the existence of a new gene family with an oQ34 reductase function. In bacteria, genes for enzymes involved in successive biosynthetic routes are often physically clustered and it was this assumption that was tested to search for genes near tgt and queA in organisms that did not possess queG. This approach successfully identified a new class of enzyme called QueH that is cobalamin independent and functions with a single 4Fe4S cluster and an additional undefined metal to catalyze the conversion of oQ34 to Q3445. In the case of QueH, the postulated mechanism differs from that of QueG and involves a novel protonation-assisted radical mechanism with the intervention of a transient carbanion (Figure 3), a challenging chemistry that requires more extensive experimental validation.
The comparative genomics approach has also facilitated the discovery of an alternative RNA methylation pathway to the prevalent S-Adenosyl-L-methionine (SAM)-dependent catalysis that relies on the classical SN2-type mechanism. The gene encoding this SAM-independent methyltransferase was later identified by comparative genomics using as search criteria the fact that this alternative methylation pathway exists in some Gram-positive bacteria species, whereas a SAM-dependent enzyme is used in Eukarya, in Gamma-proteobacteria and in a few Archaea46. This led to the identification of an alternative class of folate and flavin-dependent methyltransferases exemplified by TrmFO for the methylation of U54 to m5U54 in Gram-positive tRNAs and then RlmFO involved in the methylation of U1939 to m5U1939 in rRNAs of mollicutes47,48. We will see in the following sections of this account how this work paved the way for the discovery of a new chemistry for RNA modification.
3. The use of a synthetic mimic and reconstitution strategy to uncover an atypical RNA methylation mechanism
The study of an enzymatic mechanism often requires various chemical labeling strategies of the substrate and to monitor the incorporation of the labeled atoms into the final product. One complementary approach is the use of synthetic biomimic intermediates to activate an enzyme of interest. This approach is especially useful for enzymes that rely on a coenzyme for catalysis such as flavoproteins. Indeed, one can produce the apoprotein and attempt to reconstitute the holoenzyme with various synthetic analogues of the natural coenzyme, either to decipher a chemical mechanism or to develop new and original catalysts for biotechnological purposes49. In this section, we will revisit key steps that allowed us to apply this strategy to TrmFO to demonstrate its complex methylation mechanism, likely shared with its homologous proteins RlmFO, MnmG and MTO1.
The discovery of reductive RNA methylation required for the formation of m5U54 in Gram-positive bacterial tRNAs and m5U1939 in Mollicutes rRNAs began with the intriguing observation that the freshly purified Bacillus subtilis recombinant enzyme TrmFO (BsTrmFO) was capable of methylating tRNA transcripts in vitro in the absence of exogenous carbon donor and reductant50. This led us to hypothesize that the enzyme was co-purified with the tRNA methylating agent. To identify the active methylating species, we used diverse spectroscopies as well as LC-MS to observe a covalent flavin/protein adduct formed of an exocyclic methylene sandwiched between the N5 of the reduced FAD and the sulfur of a strictly conserved cysteine (found in all TrmFO, C51 in TtTrmFO and C53 in BsTrmFO, RlmFO, MnmG and MTO1). We will refer to this flavin/protein adduct as FAD-CH2-S-TrmFO (Figure 4)4. Mutation of this cysteine yielded a TrmFO purified with an oxidized flavin and this mutant was inactive even when folate and NAD(P)H or sodium dithionite (used as an artificial flavin reducing agent) were added in the reaction medium. This clearly demonstrated that the cysteine is not only essential for the stabilization of the FAD-CH2-S-TrmFO covalent adduct but also an indispensable component in the RNA methylation reaction. This reductive methylation occurs in two steps and differs from the SAM-dependent reductive methylation where a methyl group is transferred directly through an SN2 mechanism. First, the methylene is released from the adduct and transferred to the RNA and second, this methylene is reduced to a methyl group by the reduced flavin likely via a hydride transfer. This implies that the thioester bond is broken to presumably generate a flavin iminium or carbinolamine that is the genuine methylating agent (FAD=CH2 or FAD-CH2OH, see below for further details). Nevertheless, the FAD-CH2-S-TrmFO adduct is stable when stored at −80°C but decays to FAD under air exposure with a kinetic constant of 0.007 min−1 at pH 7 and 23°C and this decomposition is strongly accelerated by the acidity of the solvent51.
Figure 4. Strategy for the activation of an apoprotein version of a folate and flavin-dependent methyltransferase by a synthetic biomimetic for reductive methylation of tRNA.
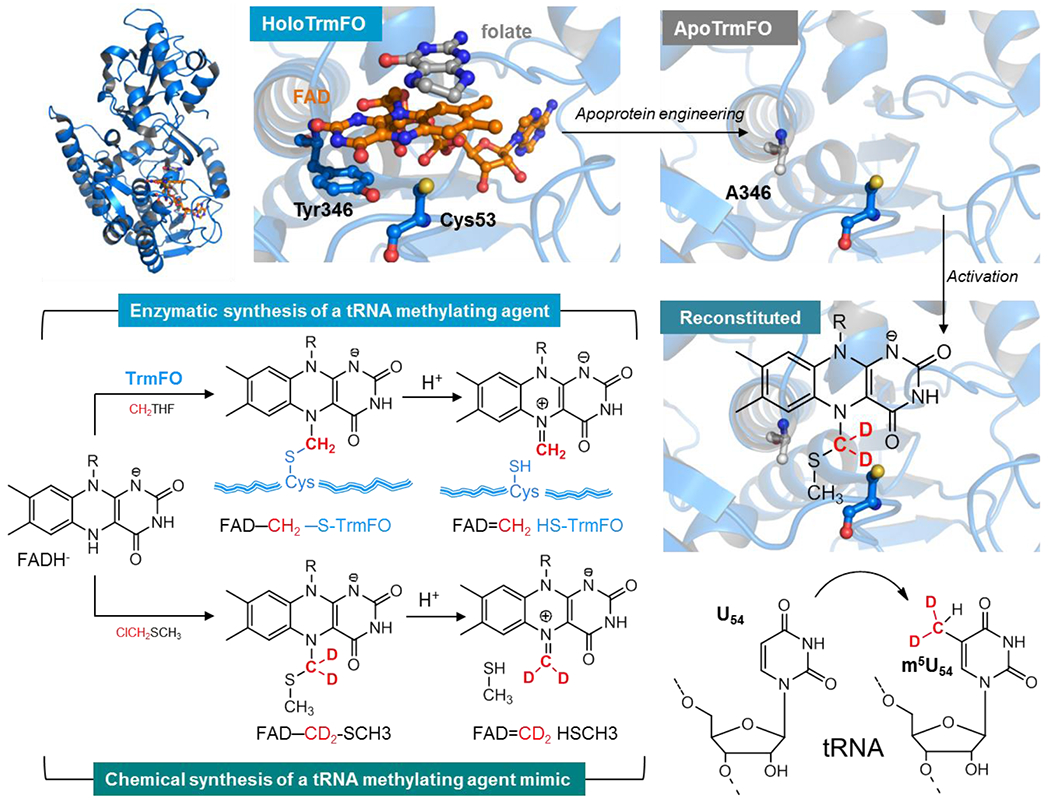
Top right shows the crystal structure of T. thermophilus TrmFO in complex with tetrahydrofolate (gray) (PDB: 3G5R). The zoom on a section of the active site shows the FAD coenzyme in orange, the folate derivative (in grey) whose pteridine stacks with the si-face of the FAD isoalloxazine while on the re-side lies the conserved tyrosine Y346 (numbering based on the sequence of BsTrmFO) engaged in π−π interaction with the flavin. The active site cysteine that stabilizes the flavin methylating intermediate FAD-CH2-S-CH3 (copurified with the Bs enzyme) faces the N5-FAD. This intermediate decomposes under protonation of the sulfur atom to give the FAD=CH2 species the bona fide C5-U methylating agent. Top left is a model of the flavin site of TtTrmFO showing the Y346A mutation artificially generated in PyMOL to illustrate the apoprotein. This apoprotein is reconstituted with the synthetic biomimetic flavin that acts as methylating agent and with the methylene deuterated to track its transfer on the substrate. Once reconstituted, the apoenzyme is activated for specific reductive methylation of the C5-U54 tRNA. This is shown by the incorporation of CD2 in red into the tRNA and its conversion to a methyl group via hydride transfer from the flavin to CHD2.
To prove that the observed FAD-CH2-S-R species is a tRNA methylating agent, we relied on the synthesis of a mimetic compound in the form of FAD-CH2-SCH3 to reconstitute TrmFO’s activity. This mimic of the covalent adduct was produced by reacting under anaerobic conditions FADH−, derived from the reduction of free FAD with dithionite, with chloromethylmethyl sulfide (ClCH2SCH3)2. The deuterated form of this mimic, FAD-CD2-SCH3 was also synthesized to ascertain that it is indeed the electrophilic CH2 held between two heteroatoms (originated from ClCH2SCH3) that is the source of carbon in RNA methylation (Figure 4). Mass spectrometry analyses revealed that the synthesized mimic breaks down under acidic conditions to FAD=CH2, the flavin iminium species that can be further reduced by NaBH4 to FAD-CH32. This is in agreement with computational calculations that support the formation of the flavin iminium by protonation of the sulfur of the FAD-CH2-S-CH3 adduct, an energetically favorable process (−79 kJ/mol)51. This is a barrierless and exothermic process (ΔE ~ −80 kJ/mol) that favors the rupture of the C-S bond, to yield the flavin iminium with the formation of CH3-SH and H2O as byproducts.
Having synthesized FAD-CH2-SCH3 to promote FAD=CH2 formation, it was essential to produce the vector that carries this mimic, namely the apoprotein version of TrmFO. Classical techniques for the production of apoflavoproteins rely on prolonged dialysis of holoenzymes under acidic conditions. However, these methods were unsuccessful for TrmFO not only due to the presence of various covalent adducts but also because of the high affinity of the enzyme for oxidized FAD. Indeed, even the C53A mutation that promotes an adduct-free enzyme did not produce sufficient yields of the apoprotein form for reconstitution experiments. To circumvent this difficulty, we opted for another strategy, namely to weaken the coenzyme/protein bond by mutagenesis while maintaining sufficient affinity of the resulting apoTrmFO for FAD to reconstitute it with a synthetic mimic. Through extensive analysis of the crystal structure of the Thermus thermophilus enzyme as well as a homology model of BsTrmFO, we noticed the presence of a conserved tyrosine (Y346 for BsTrmFO and Y343 for TtTrmFO) that engages in a π−π interaction with the si-face of the isoalloxazine52; making this tyrosine an attractive candidate for mutagenesis (Figure 4). Conclusively, the Y346A and Y346F mutants yielded apoproteins that could still bind to FAD with KD of 0.3 ± 0.06 and 0.6 ± 0.1 μM, respectively53. Additional spectroscopic studies showed that this specific tyrosine strongly quenches the FAD fluorescence. We and others explained this quenching by showing that the excited FAD abstracts very rapidly (0.43 ps) an electron from Y346, yielding an unprecedented FAD˙−/Y346OH˙+ radical pair54,55. The radical pair then decays by charge recombination, mostly in 3-4 ps, without any deprotonation of the Y346OH˙+ radical. Presumably, the H-bond between Y346 and the amide group of C53 increases the pKa of Y346OH˙+ and slows down its deprotonation53,55.
Beyond these spectroscopic considerations, we showed that these mutants can be reconstituted with the synthetic FAD-CH2-S-CH3 (Figure 4) although the Y346A mutant was more stable than its Y346F counterpart (see references detailing this stability difference53,56,57). Once reconstituted, the excess of mimic was removed by size exclusion chromatography, and we performed methylation assays by directly incubating the reconstituted enzyme with a tRNA transcript under anoxia in a glove box. To detect the methylation, we digested the tRNA with RNAse A and analyzed the mixture of nucleotide fragments by MALDI-MS. This approach clearly showed that only the mixture of apoprotein reconstituted with FAD-CD2-S-CH3 and in the presence of tRNA yielded labeled (CD2H)5U54 (Figure 4). In contrast, the mimic or the enzyme alone or the apoenzyme reconstituted with FAD-CH3 did not have this methylase activity. Thus, this original approach allowed us to demonstrate that the apoTrmFO enzyme was activated by the synthetic mimic FAD-CH2-SCH3 and that the redox flavin coenzyme directly mediates the carbon transfer in the reductive methylation of tRNA.
4. The formaldehyde shunt strategy and X-ray crystallography to visualize a flavin-based methylating agent
To obtain molecular details on how flavin mediates carbon transfer, we attempted to crystallize BsTrmFO with the synthetic mimic but without success. As an alternative strategy, we used ThyX from Thermus thermophilus as a model system for C5-uracil flavin- and folate-dependent methylations. Indeed, ThyX is a thymidylate synthase that uses the same substrates as TrmFO to catalyze a reductive methylation of uracil C5 in the monomer dUMP (instead of tRNA for TrmFO) to produce dTMP. Although ThyX is a potential target for future antibiotics due to its structural and mechanistic divergence from the human thymidylate synthase58–62, ThyX’s chemical mechanism remains debated with several models proposed. In parallel to our work on TrmFO, the Kohen’s group revisited the mechanism of ThyX63. They used quench-flow to trap a reaction intermediate identified as FAD-CH2-dUMP adduct, which reinforces the notion that flavin can be a nucleotide methylating agent. To study this methylating species, we developed a novel approach based on a formaldehyde shunt, analogous to the peroxide shunt employed in mechanistic studies of cytochromes P450 to bypass the heme reduction and molecular oxygen fixation steps64. We reasoned that the degree of oxidation of methylene in CH2THF is the same as that of formaldehyde (CH2O) and thus the latter can replace, in theory, the complex folate molecule to activate the methylation reaction65. As hypothesized, our steady-state activities and presteady-state kinetics monitored by stopped-flow under anaerobiosis showed that ThyX can use CH2O directly as a source of methylene for its methylation reaction, instead of CH2THF1. This was also unambiguously demonstrated with 13CH2O labeling reactions coupled to NMR and mass spectrometry analyses of the reaction products that confirmed the incorporation of 13CH2 at the C5 uracil of dUMP (Figure 5). The mechanism of CH2O activation was ascertained by UV/Visible spectroscopy and kinetics suggesting that reduced flavin activates formaldehyde to form a carbinolamine-like adduct (Figure 5). The observed rate constant for the formation of this intermediate increases linearly with CH2O concentration, consistent with a reversible bimolecular reaction (kon = 1.1 M−1.s−1, koff = 0. 022 s−1, KD of ~ 20 mM), while rate constants for flavin oxidation was kox = 0.03 s−1.
Figure 5. Formaldehyde shunt to activate an apoprotein of a folate and flavin-dependent methyltransferase.
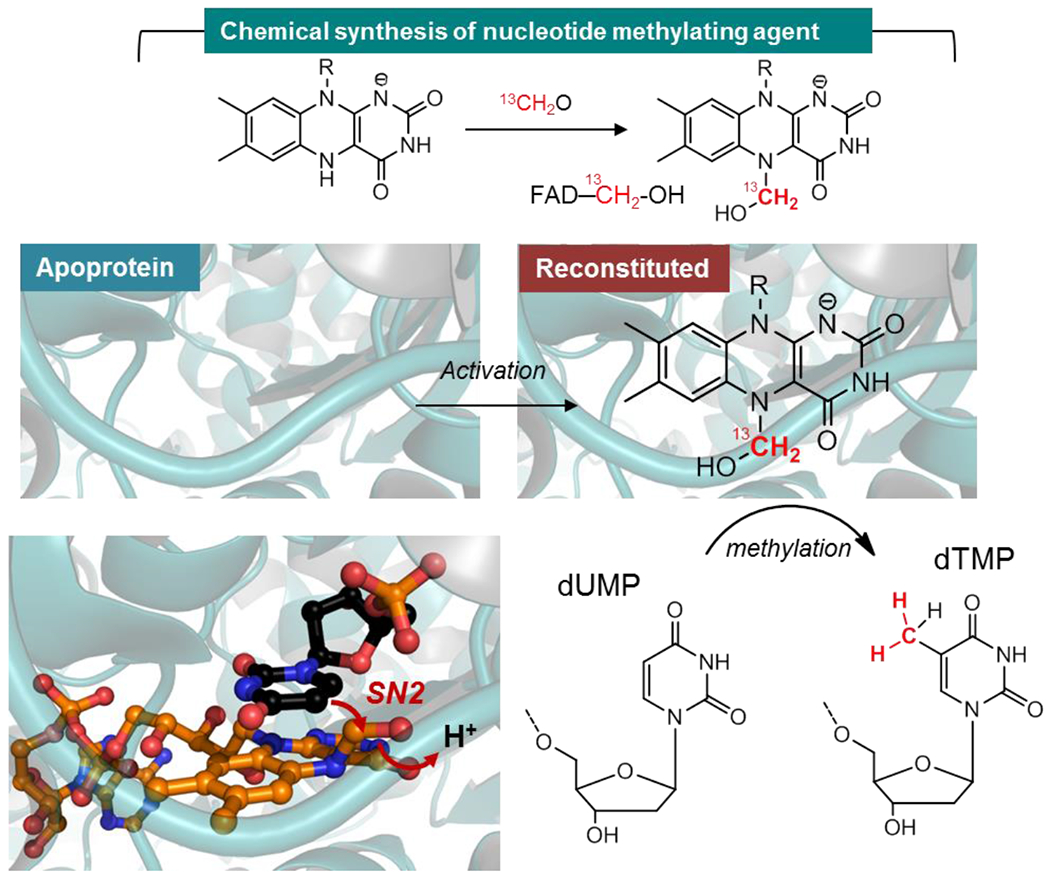
FADH− (reduced flavin), obtained by the reduction of oxidized FAD with dithionite, activates 13C-labeled formaldehyde via the N5 of the isoalloxazine to give an air-labile flavin carbinolamine. The latter can be reconstituted in an apoprotein version of Thermus thermophilus thymidylate synthase (ThyX) that uses flavin and folate to catalyze the reductive methylation of the uracil C5 of dUMP to dTMP. This reaction is similar to that of TrmFO or RlmFO. Once reconstituted with the synthetic carbinolamine under anaerobic conditions, the methyltransferase is active and can readily methylate dUMP. The crystallographic structure of this carbinolamine intermediate (orange) is shown in complex with dUMP (black) by structural alignment. Once activated, the C5 uracil of the substrate attacks the electrophilic methylene of the carbinolamine allowing its transfer from the flavin to dUMP.
To obtain additional evidence that a flavin carbinolamine is the methylating agent, we attempted to crystallize ThyX with this adduct. Two approaches were chosen: the first one consisted in soaking directly crystals of the reduced enzyme with CH2O while the second approach was to crystallize a version of ThyX reconstituted with a synthetic flavin carbinolamine1. Since the methylating agent is sensitive to oxidation, our crystallization efforts were done under strict anaerobic conditions. Remarkably, both approaches yielded crystals that diffracted to 2.8 and 2 Å resolutions for the structures of apo-ThyX reconstituted with the synthetic flavin carbinolamine and the reduced ThyX incubated with CH2O, respectively. The presence of the flavin adduct in both crystals was further verified by measuring UV/visible spectra directly on the crystals. An additional electron density on the FAD was observed in crystallo and attributed to a CH2OH group attached to the N5 atom of the isoalloxazine ring, which adopts a butterfly conformation significantly bent along its C10a–C4a axis with a dihedral angle of ~12° indicative of a reduced flavin (Figure 5). The N5 atom of FAD is pyramidal, consistent with an sp3 hybridized nitrogen. The visualized carbinolamine group adopts a similar axial position, oriented toward the si-face of FAD and interacts with a water molecule. Our structures are superimposable with that of the ThyX/dUMP/folate ternary complex previously reported66, allowing us to provide a model for a catalytically relevant species with the methylene of the flavin carbinolamine located only at 2.4 Å from the C5-uracil receiving atom (Figure 5).
Collectively, this led us to propose that the observed flavin-carbinolamine species is likely the methylene donor in the flavin-dependent nucleotide methylation via an acid-catalyzed SN2 process that releases water to form a methylene-uracil moiety (Figure 4). As this carbinolamine is theoretically in equilibrium with the corresponding flavin-iminium species, the latter could, as previously proposed67, be the actual electrophile. However, this alternative scenario is unlikely since the flavin-iminium is highly unstable and was shown to react with water rapidly to form the corresponding flavin carbinolamine68. Moreover, efficient attack of nucleophiles on the π-system of carbonyls or imines occurs along the so-called Bürgi–Dunitz trajectory, with the nucleophile attacking the unsaturated carbon with an obtuse angle of ~107° with respect to the C–X bond (X being the leaving group)69. In contrast, the sp3 hybridized carbinolamine presents a favorable distance and geometry for the in-line attack of the C5 carbon of dUMP for C–C bond formation and water displacement. Therefore, these stereoelectronic considerations lead us to favor the carbinolamine as the actual carbon transfer agent in the nucleotide’s methylation.
5. Flavin mediated methylene transfer beyond nucleotide methylation
Our recent findings on ThyX inform us not only on the methylation of dUMP but also on other flavin- and folate CH2THF-dependent RNA modifying enzymes that converge on modifying the C5 uridine (Figure 6). While the intervention of carbinolamine (or iminium) flavin as a methylating agent in TrmFO is now taken for granted, it is likely that this same species is also used by homologous systems such as RlmFO for the biosynthesis of m5U1939 in rRNA, MnmE/MnmG for cmnm5s2U34 in bacterial tRNA as well as GTPBP3/MTO1 for τm5U34 in mitochondrial tRNA (Figure 6). Indeed, the Bandarian’s group recently proposed that for MnmE/MnmG of E. coli uses a carbinolamine wherein FAD=CH2 is believed to act as alkylating agent70. In their recent study, Bommisetti et al, successfully reconstituted the activity of the MnmE/MnmG complex in vitro and showed by 13CH2THF labeling experiments that the methylene appended directly to the C5 of U34 in cmnm5s2U34 originates from CH2THF. Similar observations were previously made in vivo for both MnmE/MnmG in E. coli71 and GTPBP3/MTO172 in human cells. Given the commonalities with TrmFO and ThyX, the flavin carbinolamine could be the mediating species in the methylene transfer for MnmE/MnmG and GTBP3/MTO1 but this remains to be proven experimentally.
Figure 6. Potential involvement of flavin as an alkylating agent in other RNA modification reactions.
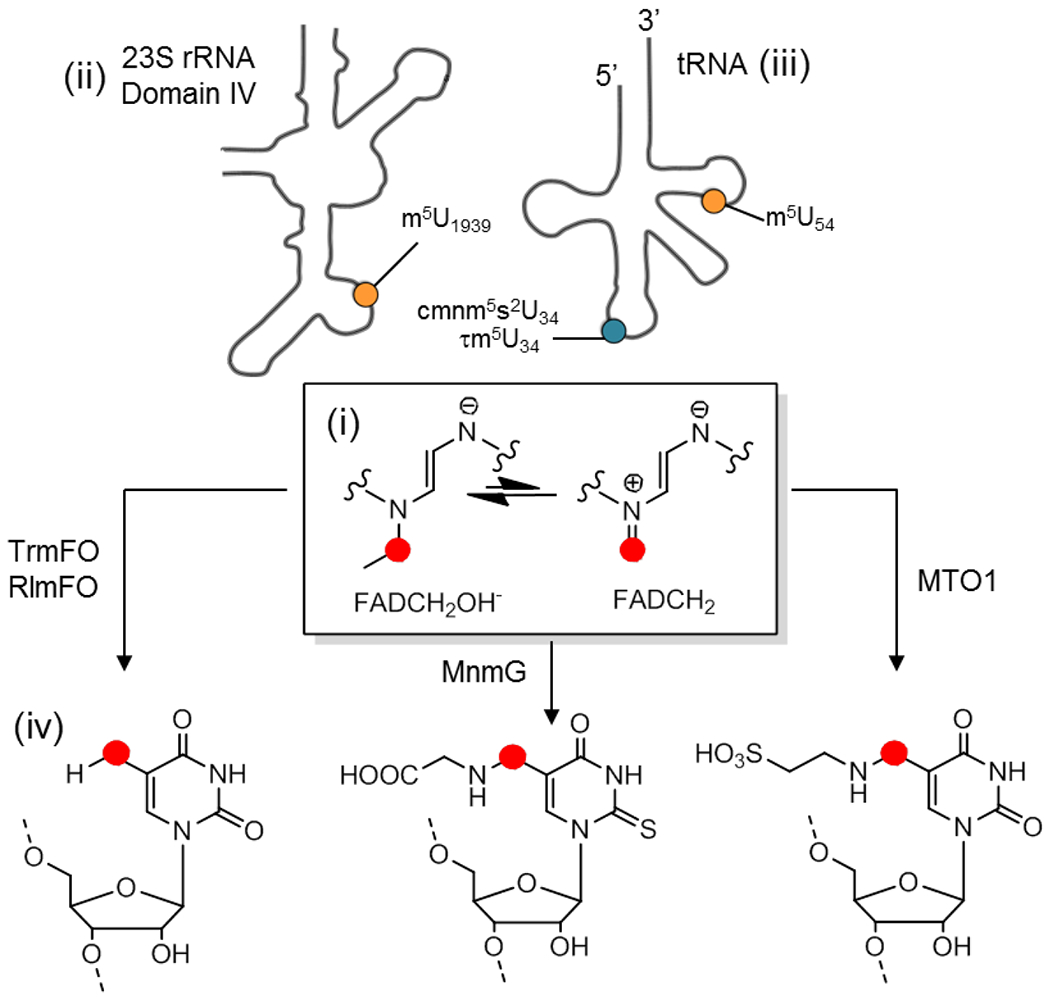
(i) In the center is shown the enediamine function of the reduced flavin carbinolamine in equilibrium with its iminium counterpart. (ii) Top left side is represented the 23S domain of mollicutes rRNA showing the localization of m5U1939 (in yellow) catalyzed by RlmFO. (iii) Top right side is a schematic of tRNA showing the localization of m5U54 (in yellow) at the level of the TpsiC loop catalyzed by TrmFO, cmnm5U34 (in blue) in bacteria catalyzed by the MnmE/MnmG complex, and τm5U34 (in blue) catalyzed by the mitochondrial GTPBP3/MTO1 complex at the anticodon loop. MnmG and MTO1 are the flavoenzyme components of the respective MnmEG and MTO1/GTPBP3 complexes and both flavoenzymes are homologues of TrmFO. (iv) Bottom is the chemical structure of the various modifications that may involve flavin as a methylene (red circle) transfer agent.
Unlike TrmFO or RlmFO where the methylene is converted to methyl by hydride transfer from the reduced flavin (FADH−), in the case of cmnm5s2U or τm5U34 the modification is coupled to the C5-uracil by a methylene (Figure 6). If we assume a mechanism where the flavin mediates the methylene transfer, then for cmnm5s2U or τm5U34 the flavin would catalyze a non-redox reaction. From that perspective, once the methylene transfer is performed, the resulting FADH− can be recharged with methylene from the CH2THF and re-engage in a new catalytic cycle without the consumption of an additional reducing equivalent. Indeed, the N5 atom in sp3 of FADH− acts as a nucleophile (not the case for the N5 in sp2 of oxidized FAD) and a reducing equivalent is theoretically required only once to generate the initial FADH−, the latter acting as a virtuous catalyst in the absence of oxidation.
6. Conclusion and outlooks
The study of RNA modifications paved the discovery of new enzymatic mechanisms highlighting the chemical richness that nature has evolved to functionalize RNA macromolecules. This illustrates the diversity in enzymatic pathways that stemmed from evolutionary convergence to synthesize the same modification in distinct kingdoms of life. Comparative genomics and genome mining are powerful methodologies at the disposal of biochemists to uncover alternative enzymatic mechanisms in the biosynthesis of RNA modifications such as Q34 and m5U54 to name a few. The isolation of stable reaction intermediates (often by luck) can also guide and act as clues for mechanistic study such as the discussed methylations of inert sp2 carbon in the biosynthesis of m5U or m8A in tRNA and rRNA. From this perspective, it is beneficial to purify different orthologous proteins since some species are more prone to stabilize reaction intermediates especially during recombinant protein expression. For instance, TrmFO from Thermus Thermophilus purified with oxidized FAD but the Bacillus subtilis protein co-purified with the flavin-methylene adduct. Even in the absence of an isolated enzymatic intermediate, the synthesis of a postulated reactive species coupled to the reconstitution of the apoprotein with the synthetic biomimetic is an ideal strategy to validate a proposed mechanism. This combined approach of synthetic chemistry and biochemistry supplemented with structural approaches (such as X-ray crystallography or cryogenic electron microscopy) can help simplify and untangle enzymatic reactions that depend on multiple cofactors and substrates. As an example, these approaches allowed us to uncover a novel flavin species, the flavin carbinolamine, as a mediator of methylene transfer in the flavin- and folate-dependent methylation reactions of tRNA U54 and dTMP.
The identification of a flavin carbinolamine for nucleotide methylation revealed unsuspected facets of the old flavin coenzyme known to harbor significant chemical versatility and further evidenced by recent discoveries73. The strategy we have outlined here may prove useful and is readily applicable to unravel other complex mechanisms of tRNA modifications such as cmnm5U34 and tm5U34, both of which are essential for translational fidelity. Moreover, since flavin was one of the first organic cofactors, its reactivity can inform us about prebiotic chemistry74. It’s possible that flavin was among the first cofactors used by RNAs to perform complex chemistries, following the RNA world hypothesis. This is hinted by the recent discovery of an RNA aptamer that modulates flavin’s reduction potential75. Since ribozymes with SAM-dependent methylation activity76,77 were recently discovered, it is tempting to speculate that ancient ribozymes might have used a flavin carbinolamine (similar to TrmFO and ThyX) for methylation during the early phases of life on Earth.
Lastly, all the modifications we have addressed in this account rely on various redox chemistries. Despite the importance of the reducing agent in redox reactions, the origin of reducing equivalents remains enigmatic for several RNA modifications. This is also the case for reactions that employ electrons to activate their substrate, such as many radical SAM systems. For instance, it has always been difficult to reduce TrmFO with NAD(P)H in vitro. In contrast, MnmG is readily reduced by NADH while this has not been studied yet for RlmFO or MTO1. This raises the more general question of the physiological electron donor required for the biosynthesis of many RNA modifications, opening a broad and exciting avenue to explore the connection between the epitranscriptome and the redox metabolism.
Acknowledgments
The authors thank all past and present members of our laboratories who participated in all of our projects. We would like to thank Susana Roque-Malo for Figure 2 graphic design. This research is funded by ANR/DFG grant DERASE (#20-CE92-0030) to DH and by grants GM070641 and GM132254 to V.d.C.L.
Biographies
Biographies
Charles Bou-Nader:
Charles Bou-Nader was born in 1990 Saint-Claude, France. He obtained a B.S. in chemistry in 2011 from the Saint-Joseph University in Beirut, Lebanon. He then moved back to France and received two Master’s degrees, one from the University Lille 1 in Chemistry in 2013 and one from the University Pierre and Marie Curie in Chemistry at the Interface of Biology in 2014. He earned his Ph.D. in Biochemistry and Structural Biology in 2017 from the University Pierre and Marie Curie under the supervision of Dr. Djemel Hamdane and Dr. Marc Fontecave at the Collège de France in Paris, France. He then pursued his postdoctoral work at the National Institute of Diabetes and Digestive and Kidney Disease at the NIH in Bethesda, USA under the supervision of Dr. Jinwei Zhang. His research interests include mechanistic aspects of RNA structure and RNA recognition by proteins. Starting as an assistant professor in fall 2023 at Emory University, his laboratory will focus on understanding the functions of R-loops in genome integrity and gene regulation.
Ludovic Pecqueur:
Ludovic Pecqueur received his Ph.D. from University Joseph Fourier (Grenoble, France) in 2005. Following postdoctoral training in the laboratory of Biological NMR at University of Southampton (U.K.) and at the Laboratory of Enzymology and Structural Biochemistry (France), he is currently employed as a scientific officer in the Laboratory of Chemistry of Biological Processes at the Collège de France.
Valérie de Crécy-Lagard:
After a bachelor’s degree at Ecole Polytechnique in 1987 and a Ph.D. in microbial genetics at the Pasteur Institute (Paris) in 1991, Valérie de Crécy-Lagard worked in diverse academic and industrial settings using the power of bacterial genetics to study primary and secondary metabolism as well as mechanisms of regulation by proteolysis. In the past 25 years, her work has focused on combining comparative genomic analysis with experimental methods to discover the function of the many ‘unknowns’ found in sequenced genomes, first at the Scripps Research Institute and then, since 2004, in the Microbiology and Cell Science Department at the University of Florida where she is now Distinguished Professor. This led to solving many long-standing mysteries, particularly in the fields of coenzyme metabolism and transfer RNA (tRNA) modification. In parallel, she collaborates with biotech groups on using long-term cultures to evolve microorganisms with specific traits.
Djemel Hamdane:
Djemel Hamdane was born in 1978 in Bondy, France. He received his Master degree from University of Paris XIII in 2003 and earned his Ph.D. from the university of Paris-Sud Orsay in 2005 under the supervision of Dr Michael Marden. He pursued postdoctoral studies at University of Michigan, Ann Arbor under the supervision of Prof. Lucy Waskell. In 2008, he began his independent research career as CNRS researcher at the laboratory of Enzymology and Structural biochemistry at Gif-sur-Yvette and then at the College De France in Paris. He is now research Director of the CNRS and his research interests include the chemical, biochemical and structural studies of post-transcriptional RNA modifications with an emphasis on redox-dependent enzyme catalysis.
Footnotes
The authors declare no competing financial interest.
References
- (1).Bou-Nader C; Stull FW; Pecqueur L; Simon P; Guerineau V; Royant A; Fontecave M; Lombard M; Palfey BA; Hamdane D: An enzymatic activation of formaldehyde for nucleotide methylation. Nature communications 2021, 12, 4542–4550. [DOI] [PMC free article] [PubMed] [Google Scholar]; This article describes the first crystallographic structure of the nucleotide methylating agent of a flavin-folate methyltransferase.
- (2).Bou-Nader C; Cornu D; Guerineau V; Fogeron T; Fontecave M; Hamdane D: Enzyme Activation with a Synthetic Catalytic Co-enzyme Intermediate: Nucleotide Methylation by Flavoenzymes. Angew Chem Int Ed Engl 2017, 56, 12523–12527. [DOI] [PubMed] [Google Scholar]; Synthesis and activation of a tRNA methyltransferase by a biomimetic of the flavin coenzyme involved in uridine methylation.
- (3).Zallot R; Ross R; Chen WH; Bruner SD; Limbach PA; de Crecy-Lagard V: Identification of a Novel Epoxyqueuosine Reductase Family by Comparative Genomics. ACS chemical biology 2017, 12, 844–851. [DOI] [PMC free article] [PubMed] [Google Scholar]; Discovery of a new alternative FeS cluster-dependent epoxyqueuosine reduction pathway.
- (4).Hamdane D; Argentini M; Cornu D; Golinelli-Pimpaneau B; Fontecave M: FAD/folate-dependent tRNA methyltransferase: flavin as a new methyl-transfer agent. Journal of the American Chemical Society 2012, 134, 19739–19745. [DOI] [PubMed] [Google Scholar]; Identification of flavin as an RNA methylation cofactor.
- (5).Mattick JS: RNA regulation: a new genetics? Nat Rev Genet 2004, 5, 316–323. [DOI] [PubMed] [Google Scholar]
- (6).Yao RW; Wang Y; Chen LL: Cellular functions of long noncoding RNAs. Nature cell biology 2019, 21, 542–551. [DOI] [PubMed] [Google Scholar]
- (7).Schimmel P: The emerging complexity of the tRNA world: mammalian tRNAs beyond protein synthesis. Nature reviews. Molecular cell biology 2018, 19, 45–58. [DOI] [PubMed] [Google Scholar]
- (8).Hopper AK: Transfer RNA post-transcriptional processing, turnover, and subcellular dynamics in the yeast Saccharomyces cerevisiae. Genetics 2013, 194, 43–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).McCown PJ; Ruszkowska A; Kunkler CN; Breger K; Hulewicz JP; Wang MC; Springer NA; Brown JA: Naturally occurring modified ribonucleosides. Wiley interdisciplinary reviews. RNA 2020, 11, e1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Boccaletto P; Stefaniak F; Ray A; Cappannini A; Mukherjee S; Purta E; Kurkowska M; Shirvanizadeh N; Destefanis E; Groza P; Avsar G; Romitelli A; Pir P; Dassi E; Conticello SG; Aguilo F; Bujnicki JM: MODOMICS: a database of RNA modification pathways. 2021 update. Nucleic acids research 2022, 50, D231–D235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Moshitch-Moshkovitz S; Dominissini D; Rechavi G: The epitranscriptome toolbox. Cell 2022, 185, 764–776. [DOI] [PubMed] [Google Scholar]
- (12).Lewis CJ; Pan T; Kalsotra A: RNA modifications and structures cooperate to guide RNA-protein interactions. Nature reviews. Molecular cell biology 2017, 18, 202–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Harcourt EM; Kietrys AM; Kool ET: Chemical and structural effects of base modifications in messenger RNA. Nature 2017, 541, 339–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Lorenz C; Lunse CE; Morl M: tRNA Modifications: Impact on Structure and Thermal Adaptation. Biomolecules 2017, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Yi CQ; Pan T: Cellular Dynamics of RNA Modification. Accounts Chem Res 2011, 44, 1380–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).El Yacoubi B; Bailly M; de Crécy-Lagard V: Biosynthesis and Function of Posttranscriptional Modifications of Transfer RNAs. Annu Rev Genet 2012, 46, 69–95. [DOI] [PubMed] [Google Scholar]
- (17).Roundtree IA; Evans ME; Pan T; He C: Dynamic RNA Modifications in Gene Expression Regulation. Cell 2017, 169, 1187–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Zhao BXS; Roundtree IA; He C: Post-transcriptional gene regulation by mRNA modifications. Nat Rev Mol Cell Bio 2017, 18, 31–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Helm M; Alfonzo JD: Posttranscriptional RNA Modifications: Playing Metabolic Games in a Cell’s Chemical Legoland. Chemistry & biology 2014, 21, 174–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).de Crécy-Lagard V; Jaroch M: Functions of Bacterial tRNA Modifications: From Ubiquity to Diversity. Trends Microbiol 2021, 29, 41–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Helm M; Motorin Y: Detecting RNA modifications in the epitranscriptome: predict and validate. Nat Rev Genet 2017, 18, 275–291. [DOI] [PubMed] [Google Scholar]
- (22).Kimura S; Dedon PC; Waldor MK: Comparative tRNA sequencing and RNA mass spectrometry for surveying tRNA modifications. Nature chemical biology 2020, 16, 964–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Kimura S; Srisuknimit V; Waldor MK: Probing the diversity and regulation of tRNA modifications. Current opinion in microbiology 2020, 57, 41–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Pratanwanich PN; Yao F; Chen Y; Koh CWQ; Wan YK; Hendra C; Poon P; Goh YT; Yap PML; Chooi JY; Chng WJ; Ng SB; Thiery A; Goh WSS; Goke J: Identification of differential RNA modifications from nanopore direct RNA sequencing with xPore. Nature biotechnology 2021, 39, 1394–1402. [DOI] [PubMed] [Google Scholar]
- (25).Leger A; Amaral PP; Pandolfini L; Capitanchik C; Capraro F; Miano V; Migliori V; Toolan-Kerr P; Sideri T; Enright AJ; Tzelepis K; van Werven FJ; Luscombe NM; Barbieri I; Ule J; Fitzgerald T; Birney E; Leonardi T; Kouzarides T: RNA modifications detection by comparative Nanopore direct RNA sequencing. Nature communications 2021, 12, 7198–7215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Boriack-Sjodin PA; Ribich S; Copeland RA: RNA-modifying proteins as anticancer drug targets. Nat Rev Drug Discov 2018, 17, 435–453. [DOI] [PubMed] [Google Scholar]
- (27).Cayir A: RNA modifications as emerging therapeutic targets. Wires Rna 2022, 13, e1702. [DOI] [PubMed] [Google Scholar]
- (28).McMahon M; Forester C; Buffenstein R: Aging through an epitranscriptomic lens. Nature Aging 2021, 1, 335–346. [DOI] [PubMed] [Google Scholar]
- (29).Torres AG; Batlle E; de Pouplana LR: Role of tRNA modifications in human diseases. Trends Mol Med 2014, 20, 306–314. [DOI] [PubMed] [Google Scholar]
- (30).Suzuki T: The expanding world of tRNA modifications and their disease relevance. Nat Rev Mol Cell Bio 2021, 22, 375–392. [DOI] [PubMed] [Google Scholar]
- (31).Lombard M; Hamdane D: Flavin-dependent epitranscriptomic world. Archives of biochemistry and biophysics 2017, 632, 28–40. [DOI] [PubMed] [Google Scholar]
- (32).Kimura S; Suzuki T: Iron-sulfur proteins responsible for RNA modifications. Bba-Mol Cell Res 2015, 1853, 1272–1283. [DOI] [PubMed] [Google Scholar]
- (33).Dowling DP; Miles ZD; Kohrer C; Maiocco SJ; Elliott SJ; Bandarian V; Drennan CL: Molecular basis of cobalamin-dependent RNA modification. Nucleic acids research 2016, 44, 9965–9976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Hanson AD; Pribat A; Waller JC; de Crécy-Lagard V: ‘Unknown’ proteins and ‘orphan’ enzymes: the missing half of the engineering parts list--and how to find it. The Biochemical journal 2009, 425, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Koonin EV; Galperin MY: In Sequence - Evolution - Function: Computational Approaches in Comparative Genomics: Boston, 2003. [PubMed] [Google Scholar]
- (36).Osterman A; Overbeek R: Missing genes in metabolic pathways: a comparative genomics approach. Current opinion in chemical biology 2003, 7, 238–251. [DOI] [PubMed] [Google Scholar]
- (37).Bishop AC; Xu J; Johnson RC; Schimmel P; de Crecy-Lagard V: Identification of the tRNA-dihydrouridine synthase family. The Journal of biological chemistry 2002, 277, 25090–25095. [DOI] [PubMed] [Google Scholar]
- (38).Rider LW; Ottosen MB; Gattis SG; Palfey BA: Mechanism of dihydrouridine synthase 2 from yeast and the importance of modifications for efficient tRNA reduction. The Journal of biological chemistry 2009, 284, 10324–10333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Bregeon D; Pecqueur L; Toubdji S; Sudol C; Lombard M; Fontecave M; de Crecy-Lagard V; Motorin Y; Helm M; Hamdane D: Dihydrouridine in the Transcriptome: New Life for This Ancient RNA Chemical Modification. ACS chemical biology 2022, 17, 1638–1657. [DOI] [PubMed] [Google Scholar]
- (40).Bou-Nader C; Montemont H; Guerineau V; Jean-Jean O; Bregeon D; Hamdane D: Unveiling structural and functional divergences of bacterial tRNA dihydrouridine synthases: perspectives on the evolution scenario. Nucleic acids research 2018, 46, 1386–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Faivre B; Lombard M; Fakroun S; Vo CD; Goyenvalle C; Guerineau V; Pecqueur L; Fontecave M; De Crecy-Lagard V; Bregeon D; Hamdane D: Dihydrouridine synthesis in tRNAs is under reductive evolution in Mollicutes. RNA biology 2021, 18, 2278–2289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Miles ZD; McCarty RM; Molnar G; Bandarian V: Discovery of epoxyqueuosine (oQ) reductase reveals parallels between halorespiration and tRNA modification. Proceedings of the National Academy of Sciences of the United States of America 2011, 108, 7368–7372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Miles ZD; Myers WK; Kincannon WM; Britt RD; Bandarian V: Biochemical and Spectroscopic Studies of Epoxyqueuosine Reductase: A Novel Iron-Sulfur Cluster- and Cobalamin-Containing Protein Involved in the Biosynthesis of Queuosine. Biochemistry 2015, 54, 4927–4935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Greenhalgh ED; Kincannon W; Bandarian V; Brunold TC: Spectroscopic and Computational Investigation of the Epoxyqueuosine Reductase QueG Reveals Intriguing Similarities with the Reductive Dehalogenase PceA. Biochemistry 2022, 61, 195–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Li Q; Zallot R; MacTavish BS; Montoya A; Payan DJ; Hu Y; Gerlt JA; Angerhofer A; de Crecy-Lagard V; Bruner SD: Epoxyqueuosine Reductase QueH in the Biosynthetic Pathway to tRNA Queuosine Is a Unique Metalloenzyme. Biochemistry 2021, 60, 3152–3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Urbonavicius J; Skouloubris S; Myllykallio H; Grosjean H: Identification of a novel gene encoding a flavin-dependent tRNA:m5U methyltransferase in bacteria--evolutionary implications. Nucleic acids research 2005, 33, 3955–3964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Lartigue C; Lebaudy A; Blanchard A; El Yacoubi B; Rose S; Grosjean H; Douthwaite S: The flavoprotein Mcap0476 (RlmFO) catalyzes m5U1939 modification in Mycoplasma capricolum 23S rRNA. Nucleic acids research 2014, 42, 8073–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Sirand-Pugnet P; Bregeon D; Beven L; Goyenvalle C; Blanchard A; Rose S; Grosjean H; Douthwaite S; Hamdane D; Crecy-Lagard V: Reductive Evolution and Diversification of C5-Uracil Methylation in the Nucleic Acids of Mollicutes. Biomolecules 2020, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Fruk L; Kuo CH; Torres E; Niemeyer CM: Apoenzyme Reconstitution as a Chemical Tool for Structural Enzymology and Biotechnology. Angew Chem Int Edit 2009, 48, 1550–1574. [DOI] [PubMed] [Google Scholar]
- (50).Hamdane D; Guerineau V; Un S; Golinelli-Pimpaneau B: A Catalytic Intermediate and Several Flavin Redox States Stabilized by Folate-Dependent tRNA Methyltransferase from Bacillus subtilis. Biochemistry 2011, 50, 5208–5219. [DOI] [PubMed] [Google Scholar]
- (51).Hamdane D; Bruch E; Un S; Field M; Fontecave M: Activation of a unique flavin-dependent tRNA-methylating agent. Biochemistry 2013, 52, 8949–8956. [DOI] [PubMed] [Google Scholar]
- (52).Nishimasu H; Ishitani R; Yamashita K; Iwashita C; Hirata A; Hori H; Nureki O: Atomic structure of a folate/FAD-dependent tRNA T54 methyltransferase. Proceedings of the National Academy of Sciences of the United States of America 2009, 106, 8180–8185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Hamdane D; Bou-Nader C; Cornu D; Hui-Bon-Hoa G; Fontecave M: Flavin-Protein Complexes: Aromatic Stacking Assisted by a Hydrogen Bond. Biochemistry 2015, 54, 4354–4364. [DOI] [PubMed] [Google Scholar]
- (54).Nag L; Sournia P; Myllykallio H; Liebl U; Vos MH: Identification of the TyrOH(center dot+) Radical Cation in the Flavoenzyme TrmFO. Journal of the American Chemical Society 2017, 139, 11500–11505. [DOI] [PubMed] [Google Scholar]
- (55).Dozova N; Lacombat F; Bou-Nader C; Hamdane D; Plaza P: Ultrafast photoinduced flavin dynamics in the unusual active site of the tRNA methyltransferase TrmFO. Physical Chemistry Chemical Physics 2019, 21, 8743–8756. [DOI] [PubMed] [Google Scholar]
- (56).Hamdane D; Velours C; Cornu D; Nicaise M; Lombard M; Fontecave M: A chemical chaperone induces inhomogeneous conformational changes in flexible proteins. Physical Chemistry Chemical Physics 2016, 18, 20410–20421. [DOI] [PubMed] [Google Scholar]
- (57).Bou-Nader C; Pecqueur L; Cornu D; Lombard M; Dezi M; Nicaise M; Velours C; Fontecave M; Hamdane D: Power of protein/tRNA functional assembly against aberrant aggregation. Physical Chemistry Chemical Physics 2017, 19, 28014–28027. [DOI] [PubMed] [Google Scholar]
- (58).Koehn EM; Fleischmann T; Conrad JA; Palfey BA; Lesley SA; Mathews II; Kohen A: An unusual mechanism of thymidylate biosynthesis in organisms containing the thyX gene. Nature 2009, 458, 919–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Mishanina TV; Koehn EM; Conrad JA; Palfey BA; Lesley SA; Kohen A: Trapping of an intermediate in the reaction catalyzed by flavin-dependent thymidylate synthase. Journal of the American Chemical Society 2012, 134, 4442–4448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Conrad JA; Ortiz-Maldonado M; Hoppe SW; Palfey BA: Detection of intermediates in the oxidative half-reaction of the FAD-dependent thymidylate synthase from Thermotoga maritima: carbon transfer without covalent pyrimidine activation. Biochemistry 2014, 53, 5199–5207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Mishanina TV; Corcoran JM; Kohen A: Substrate activation in flavin-dependent thymidylate synthase. Journal of the American Chemical Society 2014, 136, 10597–10600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Stull FW; Bernard SM; Sapra A; Smith JL; Zuiderweg ER; Palfey BA: Deprotonations in the Reaction of Flavin-Dependent Thymidylate Synthase. Biochemistry 2016, 55, 3261–3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Mishanina TV; Yu L; Karunaratne K; Mondal D; Corcoran JM; Choi MA; Kohen A: An unprecedented mechanism of nucleotide methylation in organisms containing thyX. Science 2016, 351, 507–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Munro AW; McLean KJ; Grant JL; Makris TM: Structure and function of the cytochrome P450 peroxygenase enzymes. Biochemical Society transactions 2018, 46, 183–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Kallen RG; Jencks WP: The mechanism of the condensation of formaldehyde with tetrahydrofolic acid. The Journal of biological chemistry 1966, 241, 5851–5863. [PubMed] [Google Scholar]
- (66).Koehn EM; Perissinotti LL; Moghram S; Prabhakar A; Lesley SA; Mathews II; Kohen A: Folate binding site of flavin-dependent thymidylate synthase. Proceedings of the National Academy of Sciences of the United States of America 2012, 109, 15722–15727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Matthews RG a. D., J. T.: Providing One-Carbon Units for Biological Methylations: Mechanistic Studies on Serine Hydroxymethyltransferase, Methylenetetrahydrofolate Reductase, and Methyltetrahydrofolate-Homocysteine Methyltransferase. Chemical Reviews 1990, 90, 1275–1290. [Google Scholar]
- (68).Kemal C; Bruice TC: The chemistry of an N5-methyl-1,5-dihydroflavin and its aminium cation radical. Journal of the American Chemical Society 1976, 98, 3955–3964. [DOI] [PubMed] [Google Scholar]
- (69).Burgi HB; Dunitz JD; Shefter E: Geometrical Reaction Coordinates .2. Nucleophilic Addition to a Carbonyl Group. Journal of the American Chemical Society 1973, 95, 5065–5067. [Google Scholar]
- (70).Bommisetti P; Young A; Bandarian V: Elucidation of the substrate of tRNA-modifying enzymes MnmEG leads to in vitro reconstitution of an evolutionarily conserved uridine hypermodification. The Journal of biological chemistry 2022, 298, 102548–102562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Moukadiri I; Prado S; Piera J; Velazquez-Campoy A; Bjork GR; Armengod ME: Evolutionarily conserved proteins MnmE and GidA catalyze the formation of two methyluridine derivatives at tRNA wobble positions. Nucleic acids research 2009, 37, 7177–7193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Morscher RJ; Ducker GS; Li SHJ; Mayer JA; Gitai Z; Sperl W; Rabinowitz JD: Mitochondrial translation requires folate-dependent tRNA methylation. Nature 2018, 554, 128–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Piano V; Palfey BA; Mattevi A: Flavins as Covalent Catalysts: New Mechanisms Emerge. Trends in biochemical sciences 2017, 42, 457–469. [DOI] [PubMed] [Google Scholar]
- (74).Kirschning A: Coenzymes and Their Role in the Evolution of Life. Angew Chem Int Ed Engl 2021, 60, 6242–6269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Samuelian JS; Gremminger TJ; Song Z; Poudyal RR; Li J; Zhou Y; Staller SA; Carballo JA; Roychowdhury-Saha M; Chen SJ; Burke DH; Heng X; Baum DA: An RNA aptamer that shifts the reduction potential of metabolic cofactors. Nature chemical biology 2022, 18, 1263–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Scheitl CPM; Ghaem Maghami M; Lenz AK; Hobartner C: Site-specific RNA methylation by a methyltransferase ribozyme. Nature 2020, 587, 663–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Jiang HY; Gao YQ; Zhang L; Chen DR; Gan JH; Murchie AIH: The identification and characterization of a selected SAM-dependent methyltransferase ribozyme that is present in natural sequences. Nat Catal 2021, 4, 872–881. [Google Scholar]