

Summary
Transient anoxia causes amnesia and neuronal death. This is attributed to enhanced glutamate release and modeled as anoxia-induced long-term potentiation (aLTP). aLTP is mediated by glutamate receptors and nitric oxide (·NO) and occludes stimulation-induced LTP. We identified a signaling cascade downstream of ·NO leading to glutamate release and a glutamate-·NO loop regeneratively boosting aLTP. aLTP in entothelial ·NO synthase (eNOS)-knockout mice and blocking neuronal NOS (nNOS) activity suggested that both nNOS and eNOS contribute to aLTP. Immunostaining result showed that eNOS is predominantly expressed in vascular endothelia. Transient anoxia induced a long-lasting Ca2+ elevation in astrocytes that mirrored aLTP. Blocking astrocyte metabolism or depletion of the NMDA receptor ligand D-serine abolished eNOS-dependent aLTP, suggesting that astrocytic Ca2+ elevation stimulates D-serine release from endfeet to endothelia, thereby releasing ·NO synthesized by eNOS. Thus, the neuro-glial-endothelial axis is involved in long-term enhancement of glutamate release after transient anoxia.
Subject areas: Molecular biology, Biophysics
Graphical abstract
Highlights
-
•
Glutamate elevated by anoxia boosts evoked glutamate release via ·NO synthesis
-
•
NMDA receptor activation induces ·NO release from neurons and vascular endothelia
-
•
Glutamate activates endothelial ·NO synthesis via astrocytic Ca2+ and D-serine
-
•
Anoxia-induced sustained glutamate release blocks memory-associated stimulation-induced LTP
Molecular biology; Biophysics
Introduction
Following transient brain ischemia caused by a stroke or vascular injury, cell death occurs selectively in hippocampal pyramidal neurons in the CA1 area,1,2 and this is associated with anterograde amnesia reflecting working memory impairment.2 Elevated extracellular glutamate enhances Ca2+ influx through NMDA receptors, thereby activating Ca2+-dependent protease and phospholipase to cause cell death.3 Post-anoxic enhancement of glutamate release from presynaptic terminals is modeled in rodent hippocampal slices, where transient deprivation of oxygen and/or glucose induces long-term potentiation (LTP) of glutamatergic excitatory synaptic transmission at CA1 synapses.4,5 This anoxia-induced LTP (aLTP) is associated with a reduced paired-pulse ratio (PPR; amplitude of the second response relative to the first), suggesting an enhanced exocytic release of glutamate.6,7 In hippocampal slices, aLTP induction can be blocked by the NMDA receptor antagonist 2-amino-5-phosphonovaleric acid (APV)4 or a nitric oxide synthase (NOS) inhibitor,8,9 suggesting the involvement of NMDA receptors and ·NO in aLTP induction.
·NO is a rapidly diffusible volume transmitter with a high diffusion coefficient in brain tissue, which easily permeates the plasma membrane.10 At hippocampal,11 cerebellar,12 and brainstem synapses,13 ·NO released from postsynaptic neurons retrogradely activates presynaptic cyclic guanosine monophosphate (cGMP)-dependent protein kinase (PKG) and its downstream molecules and eventually upregulates phosphatidyl-inositol-4,5-bisphosphate (PIP2), thereby accelerating compensatory endocytosis for recovering vesicles from depletion after massive exocytosis.11,13,14 This ·NO-PKG cascade is initiated by transmitter glutamate activating postsynaptic NMDA receptors, through which Ca2+ enters the cells and binds to calmodulin (CaM). Upon Ca2+ binding, CaM activates NOS to produce ·NO.10,15
Induction of aLTP requires ·NO,8,9 but the detailed mechanism of aLTP production remains unknown. Immediately after anoxic insult, before inflammation starts to activate inducible NOS (iNOS), ·NO can be produced by constitutive NOS isoforms, including nNOS and eNOS. The latter is highly expressed in endothelial cells16,17 but may also be expressed in brain astrocytes.18 Using both extracellular and whole-cell patch-clamp recordings of excitatory synaptic responses from CA1 pyramidal neurons in hippocampal slices, in combination with eNOS knockout (KO) mice and two-photon astrocyte Ca2+ imaging, we addressed the molecular mechanisms underlying induction, expression and maintenance of aLTP, and its impact on the stimulation-induced LTP (sLTP) underlying memory formation.
Results
aLTP induced by transient anoxia is caused by increased transmitter release probability
We first recorded field excitatory postsynaptic potentials (fEPSPs) in the CA1 region of hippocampal slices from C57BL/6 (WT) mice, evoked by a pair of stimulations (50 ms interval) on Schaffer collaterals every 20s (Figure 1A). Transient anoxia was induced in slices by replacing the O2 in artificial cerebrospinal fluid (aCSF) with N2 for 10 min. In line with previsous reports4,6 anoxic insult reduced the fEPSP slope, but, after returning to oxygenated aCSF, it recovered beyond baseline to a stable high level (147%), lasting more than 2.5 h in our experiments (Figure 1B). Consistent with previous reports,6,7 PPR decreased during aLTP, suggesting an increased transmitter release probability.19 Likewise, in whole-cell recordings from pyramidal neurons with patch pipettes containing the ·NO precursor L-arginine (1 mM), EPSCs evoked by Schaffer collateral stimulation showed aLTP (147%) with decreased PPR (Figure 1C), like aLTP of fEPSPs (Figure 1B).
Figure 1.

aLTP induced by transient anoxia is caused by increased transmitter release probability
(A) Schematic illustration of hippocampal slice with recording and stimulation electrodes in CA1 region separated from CA3 by an incision.
(B) Field EPSPs recorded from the CA1 stratum radiatum (SR) in response to Schaffer collateral (SC) stimulations with a pair of pulses (50 ms interval) repeated every 20 s (sample records). Time plots indicate the slope of fEPSPs and bar graphs show mean fEPSC and PPRs (with SEMs, n = 7 slices) at different epochs: (i) before and (ii) at the end of anoxic period (O2 replaced by N2 in aCSF for 10 min, red column), and (iii) 160 min after anoxia. A transient decrease of fEPSP slope during anoxia (18.7 ± 4%) was followed by a long-lasting increase (147.3 ± 12%, recorded up to 180 min after anoxia; n = 7 slices, p < 0.001 in one-way repeated-measures ANOVA). Scale bars: 0.2 mV. 20 ms.
(C) EPSCs in whole-cell recording from individual CA1 pyramidal neurons. After anoxia, EPSC amplitude increased to 147.6 ± 9% (iii, n = 8 cells) accompanied by a decrease in PPR (bar graphs). Asterisks indicate ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 in paired-sample t test. Scale bars; 0.4 nA, 20 ms.
(D) Variance-mean analysis of EPSCs before (sample EPSC traces in black) and 30 min after (red) the onset of anoxia at different Ca2+/Mg2+ concentrations (EPSC amplitudes shown in the upper panel). Variance-mean data plots were fitted to parabola. Bar graphs indicate quantal parameters before and after aLTP expression (from top to bottom; release probability, number of release sites and quantal size, respectively, n = 6 cells, ∗p < 0.05, N.S, not significant difference in paired-sample t test). aCSF contained the low-affinity AMPA receptor antagonist γ-D-glutamylglycine (γ-DGG, 1 mM) throughout to minimize saturation of AMPA receptors. Scale bars; 0.2 mV, 20 ms.
To determine the quantal parameters involved in aLTP, we performed variance-mean analysis20 of EPSCs recorded from CA1 pyramidal neurons (Figure 1D). We induced aLTP by 10 min anoxia in the presence of the low-affinity AMPA receptor antagonist γ-D-glutamylglycine (γ-DGG, 1 mM) to minimize postsynaptic AMPA receptor saturation.21 Variance-mean plots of EPSCs were obtained by changing the extracellular Ca2+/Mg2+ ratio before and after aLTP expression (Figure 1D). The parabolic variance-mean plots indicated that neither the number of release sites with releasable vesicles (N) nor the single vesicular response size (q, calculated from the initial slopes) was changed after aLTP expression, whereas the release probability (Pr, calculated from the mean EPSC amplitude divided by Nq)22 was significantly increased (Figure 1D).
Induction of aLTP is mediated by a presynaptic ·NO signaling cascade
The induction of aLTP reportedly depends upon the activity of NMDA receptors,4,5 ·NO and cGMP-dependent PKG.6,9 Indeed, aLTP induction in CA1 in hippocampal slices was abolished by the NMDA receptor antagonist APV (100 μM, Figure 2A), the ·NO scavenger 2-phenyl-4,4,5,5-tetramethylimidazolineoxyl-1-oxyl-3-oxide (PTIO, 100 μM; Figure 2B), or a PKG inhibitor (KT-5823, 10 μM; Figure 2C).
Figure 2.

Induction of aLTP is mediated by presynaptic ·NO signaling cascade
Block of aLTP induction by (A) APV (100 μM, n = 7 slices), (B) PTIO (100 μM, n = 7), (C) KT-5823 (10 μM, n = 5), (D) Y-27632 (10 μM, n = 7), and (E) PAO (1 μM, n = 7). Sample records of fEPSPs are shown above each time plot of fEPSP slope. Bar graphs indicate normalized fEPSP slopes before (i) and after (ii) anoxic insult.
(F) Rho activator II (1 μg/mL) potentiated fEPSP slope (ii, 139 ± 7%) and occluded aLTP (145.2 ± 8%, n = 5, one-way repeated-measures ANOVA: F(2, 12) = 23.4). Bar graphs indicate PPR (ii, 80.3 ± 5%, iv, 82 ± 6%, n = 5, one-way repeated-measures ANOVA: F(2, 12) = 20.16). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, N.S, no significance. Scale bars; 0.2 mV, 20ms.
(G) ·NO released from the hippocampal CA1 area after 3 min anoxia (OGD) amperometrically monitored using a carbon fiber ·NO sensor probe. An averaged trace from 5 measurements is shown after subtracting the baseline.
(H)·NO signaling cascade in presynaptic terminals causing enhancement of glutamate release probability.
At the calyx of Held in rodent brainstem slices, Ca2+ influx through NMDA receptors activates CaM, which in turn upregulates NOS activity increasing ·NO synthesis.10 The resulting ·NO propagates to the extracellular fluid and enters presynaptic terminals, thereby activating PKG, Rho-kinase (ROCK), and phosphatidylinositol (PtdIns) 5-kinase (PI5K) to upregulate PIP2.13,14 We examined whether this ·NO-PKG-ROCK-PtdIns cascade in presynaptic terminals might likewise be involved in aLTP induction. Both the ROCK inhibitor Y27632 (10 μM; Figure 2D) and the PI4K inhibitor phenylarsine oxide (PAO, 1 μM; Figure 2E) abolished aLTP, whereas the Rho activator II (1 mg/mL) induced LTP and occluded induction of aLTP (Figure 2F). Using an ·NO sensor probe attached to the hippocampal CA1 region in slice,23 we confirmed amperometrically that ·NO is released after a transient anoxic insult (Figure 2G). Thus, the ·NO-PKG-ROCK-PtdIns cascade identified at the calyx of Held13,14 is fully involved in the induction of aLTP at hippocampal CA1 presynaptic terminals (Figure 2H).
A regenerative feedback mechanism maintains aLTP
Previous reports and present results indicate that ·NO is required for aLTP induction, but it remains unknown how aLTP is maintained for hours after expression (Figure 1B). Might there be a presynaptic mechanism by which high release probability is maintained, for example by Ca2+ channel insertion or shortening the Ca2+-secretion coupling distance? More simply, might aLTP be repetitively induced by continuous ·NO production? We tested the latter possibility by applying the aLTP induction blockers approximately 1 h after aLTP expression (Figure 3). During APV application (Figure 3A), established aLTP gradually reversed and within 1h the fEPSP slope returned to the baseline before anoxia. These results suggest that continuous NMDA receptor activation is required for the maintenance of aLTP (Figure 3A). As with APV, inhibition of each step in the presynaptic ·NO signaling cascade using PTIO, KT5823, Y27632, or PAO reversed aLTP (Figures 3B–3E), suggesting that the long-term maintenance of aLTP depends on continuous ·NO signaling.
Figure 3.

Reversal of established aLTP by aLTP induction blockers
Established aLTP was canceled by (A) APV (100 μM, n = 6), (B) PTIO (100 μM, n = 7), (C) KT-5823 (10 μM, n = 5), (D) Y-27632 (10 μM, n = 7), or (E) PAO (1 μM, n = 6). Bar graphs indicate normalized fEPSC slopes and PPRs before (i) and after (iii) aLTP induction and after application of aLTP induction blocker. Scale bars; 0.2 mV, 20 ms.
(F) A positive feedback loop causing regenerative expression of aLTP. Glutamate released by transient anoxia induces ·NO production via activation of NMDA receptors. ·NO then activates presynaptic ·NO signaling cascade to enhance glutamate release. This positive loop can be disrupted by APV, PTIO, KT-5823, Y-27632 or PAO, all of which are aLTP-induction blockers.
Massive release of glutamate from presynaptic terminals into the synaptic cleft causes Ca2+ influx through activated postsynaptic NMDA receptors, thereby stimulating ·NO synthesis. ·NO back-propagates to presynaptic terminals, where it enhances glutamate release from nerve terminals via activation of the ·NO-PKG-ROCK-PtdIns cascade (Figure 2H). Thus, a positive feedback loop is formed between extracellular glutamate and ·NO, by which aLTP is repeatedly induced (Figure 3F), and pharmacological disruption of the positive loop terminates aLTP expression (Figure 3). During transient anoxia, extracellular glutamate concentration ([Glu]o) is also elevated due to glutamate leakage.24 Leaked glutamate may activate NMDA receptors, thereby inducing aLTP via activation of the presynaptic ·NO-PKG-ROCK-PtdIns cascade. In this regard, anoxia-induced glutamate leakage may play a role in triggering the regenerative loop underlying sustained expression of aLTP. Since glutamate leakage is reversed after reoxygenation,24 the enhanced release of glutamate from nerve terminals is more likely driving the regenerative glutamate-·NO loop in the long-term maintenance of aLTP.
Occlusion of sLTP by aLTP was rescued by aLTP induction blockers
Transient anoxia impairs working memory in rodents and in humans.2,25 Working memory requires sLTP, and, in the hippocampal slice model, sLTP induced by high-frequency stimulation and aLTP occlude each other.6 To test whether this occlusion can be rescued by reversing aLTP expression (Figure 3), we induced sLTP at hippocampal CA1 synapses with theta-burst stimulations (TBS) on Schaffer collaterals (Figure 4A). This sLTP was not associated with a change in PPR, consistent with its well-established postsynaptic mechanism.26,27 Tetanic stimulation has been reported to induce ·NO-dependent presynaptic-type sLTP,28 but the ·NO scavenger PTIO had no effect on the TBS-induced postsynaptic sLTP (Figure 4B). As previously reported,6 prior induction of presynaptic aLTP by transient anoxia occluded postsynaptic sLTP (Figure 4C). This occlusion of sLTP by aLTP was rescued by reversing the latter by PTIO (Figure 4D).
Figure 4.

Occlusion of sLTP by aLTP rescued by aLTP induction blockers
(A) TBS-induced LTP of fEPSPs (n = 6) in hippocampal CA1 synapses (stimulation protocol shown in inset). PPR was unchanged during sLTP (lower panel).
(B) PTIO (100 μM) had no effect on the TBS-induced LTP (n = 5).
(C) aLTP (151 ± 9%) occluded TBS-induced LTP (n = 6), with no further potentiation induced by TBS (153 ± 9%, one-way repeated-measures ANOVA).
(D) Block of established aLTP by PTIO rescued TBS-induced LTP (137 ± 8%, n = 6). Bar graphs indicate percentages of fEPSC slope at different epochs relative to controls (i). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, N.S, no significance. Scale bars; 0.2 mV, 20 ms.
·NO released after transient anoxia is derived from both nNOS and eNOS
In response to glutamate, ·NO can be synthesized by nNOS in postsynaptic pyramidal neurons, but ·NO might also be derived from eNOS in endothelial cells or astrocytes.29,30 eNOS is reportedly involved in a presynaptic form of sLTP at hippocampal CA1 synapses.31 We used eNOS-KO mice to determine whether eNOS is involved in aLTP induction and expression. In hippocampal slices from eNOS-KO mice, the magnitude of aLTP (24%, Figure 5A) was half that in wild-type (WT) slices (47%), suggesting that eNOS-derived ·NO contributes to aLTP. Bath application of the nNOS inhibitor NG-propyl-L-arginine (NPA, 1 μM)32 completely abolished aLTP in slices from eNOS-KO mice (Figure 5B). Therefore, both eNOS and nNOS contribute to aLTP induction in WT mice.
Figure 5.

·NO released after transient anoxia derived from nNOS and eNOS
(A) aLTP of fEPSPs induced in hippocampal CA1 synapses from WT mice (147 ± 12%, black, n = 7 slices, duplicated from Figure 1B) and from eNOS-KO mice (125.4 ± 4%, purple, n = 6 slices, superimposed). Bar graphs show the fEPSP slope before (i, black) and 60 min after anoxic insult (iii). Scale bars; 0.4 mV, 20 ms.
(B) The nNOS inhibitor NPA (1 μM) blocked aLTP induced in eNOS-KO mice (n = 5). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, N.S, no significance. Scale bars; 0.4 mV, 20 ms.
(C) Whole-cell washout of intracellular L-arginine in CA1 pyramidal neurons in slices from WT mice attenuated aLTP magnitude to 124 ± 3% (red, n = 8 cells) from control (147 ± 9% black, n = 9). Bath-application of fluoroacetate (FA, 10 mM) abolished the aLTP remaining after L-arginine washout (blue, n = 7). Bar graphs show normalized EPSC amplitude 35 min after anoxia onset (ii) relative to controls before anoxia (i). Scale bars; 0.2nA, 20 ms.
(D) Whole-cell washout of intracellular L-arginine in CA1 pyramidal neurons in slices from eNOS-KO mice abolished aLTP (controls, purple, n = 7; L-Arg-free, red, n = 7). Scale bars; 0.2 nA, 20 ms.
(E) Whole-cell loading of BAPTA (10 mM) in CA1 pyramidal neurons in slices from WT mice attenuated aLTP magnitude to 119 ± 5% (BAPTA, green, n = 7) compared to controls (147 ± 9% black, n = 9). Bath-application of FA (10 mM) abolished the aLTP remaining after BAPTA loading (orange, n = 8). Bar graphs show normalized EPSP amplitude 35 min after anoxic insult (ii) relative to controls before anoxia (i). Scale bar; 0.2 nA, 20 ms.
·NO synthesis from L-arginine by nNOS and eNOS is upregulated by Ca2+-bound CaM. To inhibit ·NO synthesis specifically in neurons, we next washed cytosolic L-arginine out of postsynaptic pyramidal neurons using whole-cell dialysis with L-arginine-free pipette solution before transient anoxia (Figure 5C). Whole-cell L-arginine washout halved the magnitude of aLTP (24% potentiation vs. 47% in controls without L-arginine washout). Likewise, inhibiting neuronal ·NO synthesis by whole-cell loading of the Ca2+ chelator BAPTA (10 mM) reduced the aLTP magnitude (19% potentiation, Figure 5D). Furthermore, in slices from eNOS-KO mice, whole-cell washout of L-arginine from hippocampal CA1 pyramidal neurons completely abolished aLTP (Figure 5E). Thus, these results from whole-cell recording, together with the field recording in the presence of NPA (Figure 5B), consistently suggest that both nNOS and eNOS contribute to aLTP production.
Significant contribution of eNOS to aLTP and partial inhibition of aLTP by blocking ·NO synthesis in neurons together suggest the involvements of non-neuronal cells in aLTP formation. To examine whether astrocytes might be involved in aLTP, we tested the effect of the glia-specific TCA cycle inhibitor fluoroacetate (FA),33,34 which is widely utilized for blocking astrocytic activity.35,36,37 In whole-cell recordings from single pyramidal neurons, in which aLTP was reduced by L-arginine washout (Figure 5C) or BAPTA loading (10 mM, Figure 5D), bath-applied FA (10 mM) abolished the remaining aLTP (Figures 5C and 5D). In field recordings from WT slices, bath application of FA reduced aLTP from 50% to 25% (Figure S1A), whereas, in eNOS-KO mice with 25% aLTP, FA had no additional effect on the aLTP magnitude (27%, Figure S1B). These results together suggest that astrocytic activity is required for the eNOS-dependent component of aLTP.
Absence of eNOS expression in astrocytes
Since the expression of eNOS in hippocampal astrocytes is controversial, we performed immunohistochemical examination of eNOS in CA1 area of the hippocampus. Brain sections from 6-week-old WT and eNOS-KO mice were double-labelled with antibodies against eNOS and the astrocyte marker GFAP. Since the available eNOS antibodies display non-specific reactivity, we identified eNOS-specific staining by comparing labeling in WT and eNOS-KO tissues (Figure 6A). GFAP-positive astrocytes were widely distributed in the stratum radiatum (SR) and stratum lacunosum-moleculare (SLM). In WT tissue sections, the eNOS antibody labeled the vascular endothelia of small blood vessels (BVs) in SR and SLM and larger BVs in SLM. BVs were not labeled in the eNOS-KO sections, suggesting that this signal reflects genuine eNOS expression. At higher magnification, a ring of eNOS-positive vascular endothelial cells was clearly visible in WT sections encircled by GFAP-positive astrocytic endfeet, without an overlap with eNOS labels. The eNOS antibody labeling of pyramidal neurons was not different between WT and eNOS-KO sections, suggesting that this is non-specific labeling. Although the epitope recognized by the eNOS antibody is not highly conserved in nNOS, we cannot rule out cross-reactivity with nNOS. Labeling of GFAP-positive astrocytes with eNOS antibody was very weak and not different between WT and eNOS-KO sections. Densitometric comparison of eNOS immunofluorescence between sections from WT and eNOS-KO mice indicated specific eNOS labeling in vascular endothelia but not in pyramidal neurons or GFAP-positive astrocytes (Figure 6A).
Figure 6.

Absence of eNOS expression in astrocytes and astrocytic Ca2+ transients mirroring aLTP
(A) Immunofluorescent staining of GFAP and eNOS in hippocampal tissue sections. In WT mice (upper panels), eNOS (green) is strongly expressed in blood vessels (BVs) in stratum lacunosum-moleculare (SLM) of CA1, with no overlap with the astrocyte marker GFAP (red), whereas in eNOS-KO mice (lower panels), BVs showed no significant signal besides non-specific background. Rightmost panels show higher magnification views of the boxed regions in merged pictures. GFAP-positive astrocytic endfeet surrounded distinct eNOS signals in vascular endothelia. In eNOS-KO mice, in the absence of eNOS signal, GFAP signals were seen around BVs. The graph in the right panel shows densitometric quantification of fluorescent signals in WT and eNOS-KO mice. Individual fluorescence signal intensities were normalized to that in WT, except for the astrocyte signal, which was normalized to the intensity of WT BVs. The eNOS fluorescence intensity in BVs in WT and eNOS-KO sections was significantly different (p < 0.0001, unpaired t-test), whereas the signal intensity in neurons and astrocytes was not different.
(B) Two-photon Ca2+ imaging of GCaMP6f-expressing astrocytes (outlined in upper panels) from different periods (i-v) before (i), during (ii) and after (iii-v) 10-min anoxia (from time 0). Lower panels show 5 min samples of astrocytic Ca2+ signal intensity (DF/F) at different epochs. After astrocytic Ca2+ signal recovered from anoxia-induced decline (iii), it underwent a significant increase lasting >70 min (iv, Ca2+-aLTP). Bath-application of PTIO (100 μM) at 70 min, after establishment of Ca2+-aLTP, reversed it to the baseline level (v). Bar graphs indicate long-term potentiation of Ca2+ signal after anoxia (iv, 111.26 ± 3.1%) canceled by PTIO (v, 103.51 ± 3.4%; 12 slices, n = 25, one-way repeated-measures ANOVA: F(4, 120) = 14.6).
Astrocytic Ca2+ transients mirror neuronal aLTP
Block by FA of eNOS-dependent aLTP (Figures 5C, 5D, and S1A), but not eNOS-independent aLTP (Figure S1B), suggests that astrocytic activity is required for eNOS-dependent aLTP induction. Since immunohistochemistry indicated that eNOS is predominantly expressed in vascular endothelia (Figure 6A), astrocytes may somehow mediate neuro-vascular coupling to activate eNOS in endothelia. Since transient anoxia reportedly induces astrocytic Ca2+ elevation in hippocampal slices,38,39 we performed two-photon imaging of astrocytic Ca2+ signals during induction and expression of aLTP. Adeno-associated virus expressing the genetically encoded Ca2+ indicator GCaMP6f under control of the astrocyte-specific GfaABC1D promoter40 was injected into SR in hippocampal CA1 in anesthetized mice 2–3 weeks prior to the experiments. Astrocytic Ca2+ signals of stable fluorescence intensity were observed in the SR of GCaMP6f-expressing hippocampal slices at resting state (Figure 6Bi). Transient anoxia (10 min) induced by oxygen-free aCSF significantly reduced the Ca2+ signal (ii, iii) but recovered beyond the baseline after returning to normal aCSF (iii, iv). Like excitatory postsynaptic responses in pyramidal neurons (Figure 1), astrocytic Ca2+ signal underwent an LTP lasting longer than 40 min after transient anoxia (iii, iv). After expression of astrocytic Ca2+-aLTP (iv), reducing ·NO with PTIO (100 μM) reversed it to the baseline (v) like its effect of canceling established aLTP (Figure 3B). Blocking NMDA receptors using APV (100 μM) prevented long-term elevation of astrocytic intracellular Ca2+ in response to transient anoxia (Figure S2) like the effect of APV on aLTP of fEPSPs (Figure 2A). Thus, in response to transient anoxia, intracellular Ca2+ dynamics in astrocytes mirrored aLTP of neuronal EPSPs.
D-serine-mediated astrocyte-endothelium coupling activates ·NO production
Elevated intracellular Ca2+ elevation in astrocytes reportedly triggers the release of the glio-transmitter D-serine,41,42,43 which activates endothelial NMDA receptors,43,44 thereby stimulating ·NO synthesis by eNOS for vasodilation.42,43 We examined whether this glio-endothelial coupling mechanism plays a role in eNOS-dependent aLTP.
We first isolated eNOS-dependent aLTP by abolishing the nNOS-dependent component of aLTP using NPA (1 μM) and confirmed half-size aLTP (Figure 7A). We then pretreated slices with the D-serine catabolic enzyme D-amino acid oxidase (DAAO, 0.1 unit/ml, 20 min) to deplete D-serine43 and found it completely abolished eNOS-dependent aLTP (Figure 7A). In separate whole-cell recordings from pyramidal neurons, we isolated eNOS-dependent aLTP by L-arginine washout (Figure 5C) and tested the effect of DAAO. DAAO treatment completely abolished eNOS-dependent aLTP (Figure 7B). These results from field and whole-cell recordings together suggest that D-serine released from astrocytes in response to intracellular Ca2+ elevation activates endothelial NMDA receptors, thereby triggering ·NO synthesis by eNOS. Thus, these mechanisms coincide with those reported for functional neuro-vascular coupling for vasodilation by ·NO from endothelia.42,43,44
Figure 7.

Astrocyte-endothelium coupling mediated by D-serine
(A) In WT mouse hippocampal CA1 neurons, the nNOS inhibitor NPA attenuated aLTP of fEPSP to 125.5 ± 7% (orange, n = 5). Co-treatment of NPA with DAAO abolished aLTP (105.1 ± 6%, green, n = 6; one-way ANOVA: F(2,15) = 16.21). Bar graph shows the fEPSP slope 60 min after anoxic insult (ii) relative to baseline before anoxia (i). Scale bars; 0.4 mV, 20 ms.
(B) In whole-cell recording with intracellular washout of L-arginine, aLTP was reduced (orange, data reproduced from Figure 5C) but was abolished by pre-treatment with DAAO (104 ± 6%, blue, n = 7; one-way ANOVA: F(2,15) = 15.39). Bar graph shows normalized EPSC amplitude 35 min after anoxic insult relative to baseline before anoxia (i). ∗p < 0.05, ∗∗p < 0.01. Scale bars; 0.2 nA, 20 ms.
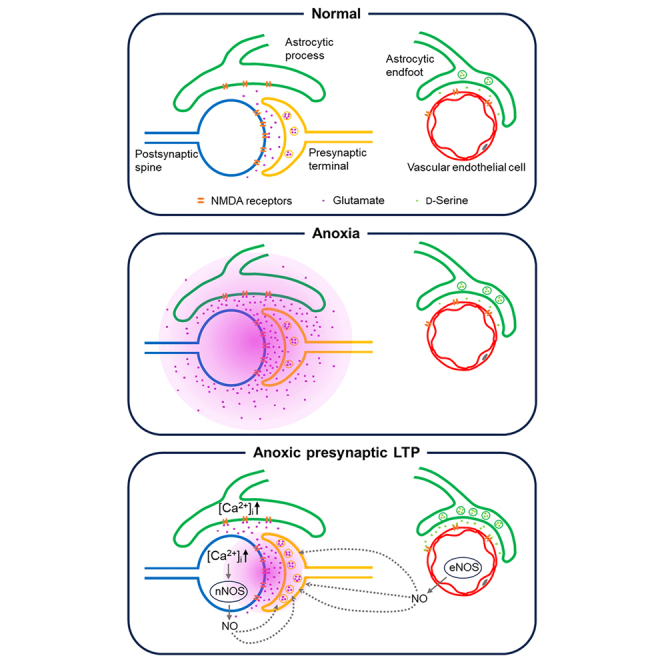
(C) Neuro-glial-endothelial coupling for ·NO production by eNOS. Transient anoxia causes glutamate leakage, triggering the positive feedback loop (B). After anoxia, glutamate leakage ceases and the feedback loop is driven by elevated glutamate release from nerve terminals. High [Glu]o in the synaptic cleft causes Ca2+ elevation throughout astrocytes by propagation from their processes at the synapses to their endfeet at the vascular endothelia and co-release glutamate and D-serine, thereby stimulating eNOS to produce ·NO, which together with that produced by neuronal nNOS contributes to the induction (Figure 2) and expression (Figure 3) of aLTP.
Discussion
In the brain, astrocytes extend their processes to the synaptic cleft to take up neurotransmitter released from presynaptic terminals. Astrocytes also extend processes to BVs, with their endfeet closely apposed to the vascular endothelium, where they constitute the blood-brain barrier (Figure 7C). Using mouse hippocampal slices, we show that this neuro-glial-endothelial axis is essentially involved in the LTP of glutamate release from nerve terminals induced by transient anoxia.
Induction and expression mechanisms of aLTP
At glutamatergic excitatory synapses in CA1 area of rodent hippocampal slices, transient oxygen deprivation induces aLTP lasting longer than 2–3 h (Figure 1B). Variance-mean analysis of EPSCs revealed enhanced transmitter release probability during aLTP (Figure 1F). The induction mechanism of aLTP was pharmacologically dissected to confirm involvements of NMDA receptors, ·NO, and the ·NO-downstream cascade, PKG-ROCK-PtdIns (Figure 2). During transient anoxia, the concentration of extracellular glutamate [Glu]o is greatly increased (e.g., from 1 μM to 100 μM) by leakage owing to reverse operation of glutamate uptake carriers.24,45 Elevated [Glu]o activates neuronal and non-neuronal NMDA receptors, inducing Ca2+ influx and upregulating NOS activity.4,6,8,9 ·NO diffuses into presynaptic terminals, where it activates the ·NO-downstream cascade to elevate PIP2 (Figure 2G),13,14 which can bind to synaptotagmin,46,47 thereby increasing release probability. Thus, aLTP may be induced by [Glu]o elevated by leakage from neuronal and non-neuronal cells during anoxia. After reoxygenation, glutamate leakage is reversed,24 but in and around synaptic cleft massive release of glutamate from nerve terminals activates NMDA receptors thereby producing ·NO. Thus, a positive feedback loop is established that sustains [Glu]o and ·NO, boosting long-term expression of aLTP (Figure 3F). In support of this hypothesis, pharmacological block at each stage of the feedback loop reversed existing aLTP to baseline (Figure 3). Since glutamate leakage is transient during anoxic period,24 it likely plays a role of triggering, but not maintaining, aLTP.
Occlusion of postsynaptic sLTP by presynaptic aLTP
Both aLTP and sLTP depend upon a common molecular cascade, in which activation of postsynaptic NMDA receptors by [Glu]o causes Ca2+ influx and subsequent CaM activation, which can upregulate ·NO synthesis by NOS to induce aLTP,10,15 but it can also activate CaM kinase and induce sLTP.48 When this cascade is preoccupied by full activation of NMDA receptors by massive [Glu]o released from nerve terminals in aLTP (Figure 3F), sLTP may not be inducible using the same cascade (Figure 4C). Disruption of the positive feedback loop by PTIO can reduce [Glu]o elevation and NMDA receptor activation, thereby canceling aLTP expression (Figures 3B and 4D) and rescueing TBS-induced sLTP (Figure 4D). Thus, sLTP “hijacked” by aLTP can be rescued by pharmacological cancellation of aLTP (Figure 3). Since sLTP is a cellular mechanism of memory formation, its occlusion by aLTP may be the underlying cause of post-ischemic impairment of working memory.2 In this regard, pharmacological cancellation of aLTP (Figure 3) could treat amnesia in patients even hours after ischemic insult.
The neuro-glial-endothelial axis is involved in aLTP formation
The magnitude of aLTP in eNOS-KO mice was about half of that in WT mice (Figure 5A). Likewise, the astrocyte metabolic inhibitor FA halved aLTP magnitude in WT mice (Figure S1A). The remaining aLTP was abolished by pharmacological inhibition of nNOS (Figure 5B) or whole-cell washout of the ·NO precursor L-arginine from neuronal cytosol (Figure 5C), suggesting that nNOS and eNOS make comparable contributions to aLTP formation. In eNOS-KO slices, FA had no effect on aLTP (Figure S1B) suggesting that astrocyte activity is required for ·NO synthesis by eNOS in response to anoxia. Since eNOS immunoreactivity was present in vascular endothelia but absent in GFAP-positive astrocytes (Figure 6A), ·NO production in endothelia is likely mediated by astrocytes.
Two-photon Ca2+ imaging of astrocytes in hippocampal slices showed that transient anoxia induced a long-term elevation of astrocytic Ca2+ level with a similar time course and pharmacology to aLTP (Figure 6B). Like aLTP, induction of astrocytic Ca2+ elevation was blocked by APV (Figure S2) and sustained elevation was reversed by PTIO (Figure 6B), suggesting the involvement of NMDA receptors and ·NO in astrocytic Ca2+ responses. NMDA receptors expressed in astrocytes are composed of NR2C49,50 and/or NR2D51,52 subunits and are Ca2+ permeable in their resting state with little Mg2+ block.53 Therefore, it is likely that NMDA receptors in astrocytic processes at the synaptic cleft are activated by glutamate released and/or leaked from nerve terminals, thereby elevating astrocytic Ca2+.
Most astrocytes contact vascular endothelia with their endfeet,54 and astrocytic Ca2+ waves are propagated to their endfeet.55 In functional hyperemia, neurovascular coupling drives vasodilation in response to synaptic activity. This coupling is mediated by astrocytic Ca2+ elevation triggering release of glutamate and D-serine from astrocytic endfeet to activate endothelial ionotropic NMDA receptors,44 thereby stimulating eNOS to synthesize ·NO.42,43 Our results from field and whole-cell recordings suggest that the same neuro-glial-endothlial coupling underlies eNOS-dependent aLTP (Figures 7A and 7B). Since the diffusion coefficient of ·NO is 3300 mm2/s,10 ·NO propagates at 115 mm/s (calculated from the mean square displacement = 4Dt). This is fast enough for ·NO from endothelia to reach CA1 synapses for aLTP induction, which takes tens of minutes. Thus, the neuro-glial-endothelial coupling likely mediates dual pathophysiological functions, vasodilation and aLTP production. The former provides physiological support of brain metabolic demand, whereas the latter operates for impairing synaptic plasticity after ischemic insult.
Limitations of the study
Although we have clarified mechanisms by which glutamate remains elevated long after a transient anoxia in the rodent hippocampal slice model, it remains open whether these mechanisms likewise operate in vivo and in humans exposed to anoxia. In this respect, based upon present results in the slice model, diverse pharmacological trials of rescuing memory impairments in rodents exposed to transient ischemia remain to be tested.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-GFAP | Sigma-Aldrich | Cat# G3893 |
| Rbbit anti-eNOS | Novus Biologicals | Cat# NB300-500 |
| Bacterial and virus strains | ||
| pAAV-GFaABC1D-Flex-LckTag-GCaMP6f | Sirion Biotech | Custom made |
| AAV5.GfaABC1D.PI.Cre.SV40 | Addgene | Plasmid #105603 |
| Chemicals, peptides, and recombinant proteins | ||
| γ- D-Glutamylglycine |
Tocris | Cat# 0112 |
| D-APV | Tocris | Cat# 0106 |
| PTIO | TCI | Cat# A5440 |
| KT-5823 | Tocris | Cat# 1289 |
| Y-27632 dihydrochloride | abcam | Cat# ab120129 |
| Rho activator II | Cytoskeleton | Cat# CN03-B |
| PAO | Sigma-Aldrich | Cat# P3075 |
| NPA hydrochloride | Tocris | Cat# 1200 |
| Fluoroacetate | TCI | Cat# F0030 |
| DAAO | Tokyo Kasei | N/A |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6J | Jackson Laboratory | stock number: 000664 |
| eNOS-KO Mouse: B6.129P2-Nos3tm1Unc/J | Jackson Laboratory | stock number: 002684 |
| Software and algorithms | ||
| Patchmaste | HEKA | https://www.heka.com/ |
| SigmaPlot | Systat software | http://www.systat.de/ |
| IgorPro | WaveMetrics | https://www.wavemetrics.com/ |
| OriginPro | OriginLab | https://www.originlab.com/ |
| MView | Sutter Instrument | https://www.sutter.com/ |
| FIJI (ImageJ) | ImageJ | https://imagej.net/software/ |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Han-Ying Wang (sagaciouswhy@gmail.com)
Materials availability
This study did not generate new unique reagents.
Commercially available materials are denoted in the manuscript and the method details.
Data and code availability
-
•
Data reported in this paper will be shared by the lead contact upon reasonable request.
-
•
No unpublished custom code, software, or algorithm was used in this study.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental model and study participant details
Animals
Animal experiments were conducted in accordance with the guidelines of OIST and were approved by OIST Animal Care and Use Committee. C57BL/6 WT mice or eNOS-KO (B6.129P2-Nos3tm1Unc/J; JAX stock #002684)56 mice of both sexes were raised on a 12h/12h light/dark cycle with ad libitum access to food and water. Mice aged 5–8 weeks at the start of the experiments were used.
Method details
Slice electrophysiology
Hippocampal slices were prepared from 5-8-week-old C57BL/6 mice. Briefly, mice were decapitated under isoflurane anesthesia and their brains were quickly removed. Transverse hippocampal slices (350 μm thick) were cut on a vibroslicer (VT1200S, Leica Microsystem) in ice-cold Ca2+-free artificial cerebrospinal fluid (aCSF) containing (in mM): 200 sucrose, 2.5 KCl, 26 NaHCO3, 1.25 NaH2PO4, 6 MgCl2, 10 glucose, 3 myo-inositol, 2 sodium pyruvate, and 0.5 sodium ascorbate (pH 7.4 when bubbled with 95% O2 and 5% CO2, 310–320 mOsm). An incision was made to separate the CA1 and CA3 regions to suppress epileptiform activity in CA1. Before recording, slices were incubated for 30 min at 36°C–37°C in standard aCSF containing (in mM): 125 NaCl, 2.5 KCl, 26 NaHCO3, 1.25 NaH2PO4, 2 CaCl2, 1 MgCl2, 10 glucose, 3 myo-inositol, 2 sodium pyruvate, and 0.5 sodium ascorbate (pH 7.4 when bubbled with 95% O2 and 5% CO2, 295–305 mOsm), and maintained thereafter for at least 1h at room temperature (24°C–26°C).
Extracellular field recording and whole-cell patch clamp recording were performed in a submerged-type chamber maintained at 31°C–33°C and continuously perfused at 3–4 mL/min with oxygenated aCSF that routinely contained 20 μM bicuculline methiodide (Santa Cruz Biotechnology) to block GABAA receptor response. Paired-pulse stimulation (0.1 ms in length and 50 ms in inter-pulse interval) was given at 0.05 Hz to Schaffer collateral/commissural pathways, with a bipolar tungsten electrode to evoke fEPSPs, which were recorded using a borosilicate glass electrode filled with 1M NaCl (resistance 0.5–1 MΩ). Recordings were made using EPC-10 patch-clamp amplifier controlled by Patchmaster software (HEKA). Data were online low-pass-filtered at 5 kHz and digitized at 50 kHz. Following a stable baseline period of at least 20 min, transient anoxia was introduced by switching the perfusing bath solution to oxygen-free aCSF saturated with 95% N2 and 5% CO2. To evoke activity-dependent LTP, theta-burst stimulation (TBS) comprising 5 bursts, with each burst composed of 5 pulses at 100 Hz, was delivered at 5 Hz for 1s. For whole-cell patch-clamp recording, CA1 pyramidal neurons were visualized using a 40× water immersion objective (Olympus) attached to an upright microscope (BX51WI, Olympus). A tungsten bipolar stimulating electrode was placed in the stratum radiatum ∼200–300 μm away from the recorded neuron. For whole-cell recording of EPSCs, patch pipettes (3–5 MΩ) were filled with an internal solution containing (mM): 120 CsMeSO3, 30 CsCl, 10 HEPES, 0.5 EGTA, 12 disodium phosphocreatine, 1 MgCl2, 5 QX-314, 3 Mg-ATP, 0.3 Na2 -GTP, (pH 7.3–7.4 adjusted with CsOH, 315–320 mOsm) including 1 mM L-arginine unless otherwise specified. Cells were voltage-clamped at −70 mV and series resistance was compensated ∼75% to 9.2 ± 0.5 MΩ (range from 6.8 to 13.7 MΩ) measured from capacitive currents. Data were discarded if the series resistance changed by >20%.
For variance-mean analysis, EPSCs were evoked by a single pulse at 0.05 Hz. To avoid AMPAR saturation, 1mM γ-DGG was added to the aCSF with various extracellular [Ca2+]/[Mg2+]. Fifteen successive EPSCs were collected for constructing a variance-mean plot. In each recording, the release probability Pr, number of functional release sites N, and quantal size q were estimated by fitting a simple parabola equation as follows:
Where Var and Imean represent the variance and mean amplitude of EPSCs respectively. The initial slope of the parabola can be used to estimate q whereas the X intercept of the parabola can be used to indicate Nq. Pr at 2 mM extracellular Ca2+ calculated as I/Nq.
·NO detection
A carbon fiber ·NO sensor probe (tip diameter, 100 mm; ·NO Monitor IMN-111, InterMedical, Japan) was attached to the CA1 area of hippocampal slices. Amperometric ·NO currents released after a transient anoxia with glucose deprivation (OGD, 3 min) were monitored at room temperature.
Distribution of astrocytes and eNOS in hippocampal CA1 area
6-week-old male wild-type and eNOS-KO mice were anesthetized with isoflurane and perfused transcardially with ice-cold phosphate-buffered saline (PBS) and then with ice-cold 4% formaldehyde in PBS, pH 7.4. The brain was removed and incubated overnight in 4% formaldehyde in PBS at 4°C. Fixed brains were washed twice in cold PBS, incubated in 15% sucrose in PBS for 6 h at 4°C, then in 30% sucrose in PBS at 4°C for 48 hours, or until the brain sunk to the bottom of the tube. Brains were then removed from the sucrose solution, rapidly frozen on dry ice, and stored in foil at −80°C before use.
Coronal brain sections (25 μm thick) were cut using a Leica CM3050S cryostat and the sections stored in PBS at 4°C. For staining, sections containing the hippocampus were mounted on polylysine-coated slides and dried at room temperature. Sections were rehydrated in PBS for 10 minutes, blocked for 1 hour in PBS containing 5% normal goat serum and 0.1% Triton X-100, then incubated overnight at 4°C in blocking buffer containing mouse anti-GFAP (1:500; G3893, Sigma) and rabbit anti-eNOS (1:250; NB300-500, Novus Biologicals) primary antibodies. Sections were washed in PBS containing 0.1% Triton X-100 (PBST), then incubated with appropriate Alexa Fluor Plus secondary antibodies (Invitrogen) for 2 hours at room temperature protected from light, then washed again in PBST. To label nuclei, 1 μg/ml DAPI was added to the final wash. Stained sections were mounted and imaged using a Zeiss LSM 780 confocal microscope and Zen 2.1 software (Zeiss). Labelling of eNOS in blood vessels in WT and eNOS KO sections was quantified by densitometry using ImageJ [Rasband, W.S., ImageJ, U. S. National Institutes of Health, Bethesda, Maryland, USA, https://imagej.nih.gov/ij/, 1997–2018.]. The intensity of anti-eNOS fluorescence was measured in cross-sections through the wall of large blood vessels in SLM (4 cross-sections per image; two images per genotype) in images captured using the 63× objective. Non-specific labeling of CA1 pyramidal neurons and GFAP-positive astrocytes was quantified from images taken using the 20× objective (3 regions of equal area were measured per image; 3 images were quantified per genotype).
Targeted expression of a calcium sensor in astrocytes
Adeno-associated virus expressing the genetically-encoded Ca2+ indicator GCaMP6f under control of the astrocyte-specific GfaABC1D promoter40 was injected stereotaxically into the hippocampal CA1 stratum radiatum (SR) of anesthetized 5-8-week-old mice. The Cre/Flex system used in this studies included pAAV-GFaABC1D-Flex-LckTag-GCaMP6f (custom made, Sirion Biotech, 1x1013 genome copies, GC, per ml) and AAV5.GfaABC1D.PI.Cre.SV40 (gift from Baljit Khakh, Addgene plasmid #105603; http://n2t.net/addgene:105603; RRID:Addgene 105603, 4.03 × 1013 GC/ml) which anchors the calcium indicator to the astrocytic plasma membrane and allows to control the density of labeled astrocytes with the Cre/Flex system by injection of the two AAVs at different ratios. For sparse labeling of astrocytes, AAV5.GfaABC1D.PI.Cre.SV40 was diluted 100-fold with saline, and then a 1:1 mixture of the 100-fold diluted AAV5.GfaABC1D.PI.Cre.SV40 and pAAV-GfaABC1D-Flex-LckTag-GCaMP6f was prepared. This AAV mixture (140 nl) was injected into each side of the dorsal-CA1 region of the hippocampus of 5-8-week-old mice (coordinates ML+/− 1.4, AP-1.9, DV-1.65) at a rate of ∼14 nl/5 min with positive air pressure through a beveled glass pipette (15 μm opening). The pipette was left in position for ∼5 min after injection was completed.
Two-photon calcium imaging of sparsely labeled astrocytes in acute brain slices
Two to three weeks after viral injection, acute slices were cut from hippocampal tissues. The slices were transferred to a recording chamber of a custom-built wide-field/two-photon microscope (MOM, Sutter Instruments) and continuously perfused at 3–4 ml/min with oxygenated aCSF at 31°C–33°C. A 25x/N.A.1.05 water immersion objective (Olympus) was used for imaging. To increase the point spread function of excitation, the back aperture of the objective was underfilled (spatial resolution 1 μm × 1 μm x 5 μm).57,58 The GCaMP6f was excited using a Ti:sapphire femtosecond pulsed laser (Vision II, Coherent) at 950 nm with typically 5–8 mW laser power under the objective. Fluorescence was detected by a GaAsP photomultipliers (Hamamatsu) in a spectral window of 490–550 nm (Semrock). The microscope was controlled using MScan software (Sutter Instruments). Movies of 512 x 512 pixels per frame corresponding to 375 × 375 μm2 were recorded at a sampling rate of 30.9 frames/s. Different recording protocols were used depending on the experiment.
For APV experiments the baseline fluorescence was recorded for 10 min in oxygenated aCSF. Subsequently the solution was changed to aCSF containing 100 μM APV and the fluorescence was recorded for 10 min in the presence of APV. Anoxia was then induced by switching aCSF to oxygen-free aCSF saturated with 95% N2 and 5% CO2. After recording the fluorescence signal for 10 min in anoxic condition, the perfusate was returned to oxygenated aCSF and fluorescence was recorded for 60 min thereafter.
For PTIO experiments the baseline fluorescence was recorded for either 5 or 10 min in oxygenated aCSF. Then, anoxia was induced by switching to oxygen-free aCSF. The fluorescence was recorded for 5 or 10 min at different epochs. After 10 min anoxia, the slice was reoxygenated by switching back to oxygenated aCSF. After confirming long-term potentiation of the Ca2+ signal, PTIO (100 μM) was bath-applied and the Ca2+ signal was monitored for >70 min in the presence of PTIO.
The imaging raw data were converted to Tiff format using commercial software (MView, Sutter Instrument). The recordings were preprocessed automatically with a custom macro in ImageJ applied to each recording that corrected movement artifacts (TurboReg plugin [3]), applied a 3D Gaussian filter (sigma = 1 pixel in x, y and t), and temporally binned the data to 10.3Hz. Regions of interest (ROIs) for individual astrocytes were drawn manually in ImageJ and the fluorescence intensity of ROIs was obtained for each time point using MATLAB (Mathworks). Fluorescence values () were normalized to , where the baseline fluorescence () corresponds to the average intensity of each individual ROI over the 10 min baseline recording. No bleaching correction was required.
Data analysis
All values were expressed as mean ± SEM. Responses were normalized to the baseline prior to anoxic LTP induction unless otherwise mentioned. 95% CIs on the difference of the means were considered statistically significant in two-tailed paired-sample t-test, Student’s t test, one-way repeated-measures ANOVA, or one-way ANOVA with a post hoc Bonferroni test (p < 0.05) as appropriate. Details, including sample sizes, can be found in figure legends.
Reagents
All chemicals were prepared from frozen stock solution (stored at −30°C). γ- D-Glutamylglycine (γ-DGG; Tocris); D-(−)-2-Amino-5-phosphonopentanoic acid (APV; Tocris); 2-Phenyl-4,4,5,5-tetramethylimidazoline-3-oxide-1-oxyl (PTIO; TCI); (9S,10R,12R)-2,3,9,10,11,12-Hexahydro-10-methoxy-2,9-dimethyl-1-oxo-9,12-epoxy-1H-diindolo[1,2,3-fg:3′,2′,1′-kl]pyrrolo[3,4-i][1,6]benzodiazocine-10-carboxylic acid, methyl ester (KT-5823; Tocris); Y-27632 dihydrochloride (Y-27632; abcam); Rho activator II (Cytoskeleton); Phenylarsine oxide (PAO; Sigma-Aldrich); Nω-Propyl-L-arginine hydrochloride (NPA; Tocris); Fluoroacetate (FA; TCI); D-amino acid oxidase (DAAO; Tokyo Kasei).
Acknowledgments
This work was supported by the Okinawa Institute of Science and Technology (OIST) to T.T. and B.K. We thank Tetsuya Hori, Kohgaku Eguchi, and Anna Garanzini for technical support. We are also grateful to Toshiya Manabe, Yukiko Goda, and Christian Rosenmund for comments.
Author contributions
H.-Y.W. designed and performed most experiments and data analysis and wrote the paper. H.T. designed and performed experiments including amperometric ·NO measurements. P.N.S. performed immunostaining experiments and wrote the paper. A.E. and B.K. performed astrocyte Ca2+ imaging experiments and data analysis. K.-S.H. instructed and supervised H.-Y.W. for the aLTP experiments. T.T. designed the whole study, organized collaborations, supervised all experiments, and wrote the paper.
Declaration of interests
The authors declare no competing interests.
Published: March 16, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2024.109515.
Contributor Information
Han-Ying Wang, Email: sagaciouswhy@gmail.com.
Tomoyuki Takahashi, Email: ttakahas@oist.jp.
Supplemental information
References
- 1.Kirino T., Tamura A., Sano K. A reversible type of neuronal injury following ischemia in the gerbil hippocampus. Stroke. 1986;17:455–459. doi: 10.1161/01.str.17.3.455. [DOI] [PubMed] [Google Scholar]
- 2.Zola-Morgan S., Squire L.R., Amaral D.G. Human amnesia and the medial temporal region: enduring memory impairment following a bilateral lesion limited to field CA1 of the hippocampus. J. Neurosci. 1986;6:2950–2967. doi: 10.1523/JNEUROSCI.06-10-02950.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Choi D.W. Glutamate neurotoxicity and diseases of the nervous system. Neuron. 1988;1:623–634. doi: 10.1016/0896-6273(88)90162-6. [DOI] [PubMed] [Google Scholar]
- 4.Crépel V., Hammond C., Chinestra P., Diabira D., Ben-Ari Y. A selective LTP of NMDA receptor-mediated currents induced by anoxia in CA1 hippocampal neurons. J. Neurophysiol. 1993;70:2045–2055. doi: 10.1152/jn.1993.70.5.2045. [DOI] [PubMed] [Google Scholar]
- 5.Tekkök S., Krnjević K. Long-term potentiation in hippocampal slices induced by temporary suppression of glycolysis. J. Neurophysiol. 1995;74:2763–2766. doi: 10.1152/jn.1995.74.6.2763. [DOI] [PubMed] [Google Scholar]
- 6.Hsu K.S., Huang C.C. Characterization of the anoxia-induced long-term synaptic potentiation in area CA1 of the rat hippocampus. Br. J. Pharmacol. 1997;122:671–681. doi: 10.1038/sj.bjp.0701409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Takagi H., Kodama K., Saito M., Suzuki H. Presynaptic K+ channel modulation is a crucial ionic basis of neuronal damage induced by ischemia in rat hippocampal CA1 pyramidal neurons. Zool. Sci. (Tokyo) 2003;20:7–11. doi: 10.2108/zsj.20.7. [DOI] [PubMed] [Google Scholar]
- 8.Huang C.C., Hsu K.S. Nitric oxide signalling is required for the generation of anoxia-induced long-term potentiation in the hippocampus. Eur. J. Neurosci. 1997;9:2202–2206. doi: 10.1111/j.1460-9568.1997.tb01387.x. [DOI] [PubMed] [Google Scholar]
- 9.Costa C., Tozzi A., Siliquini S., Galletti F., Cardaioli G., Tantucci M., Pisani F., Calabresi P. A critical role of NO/cGMP/PKG dependent pathway in hippocampal post-ischemic LTP: modulation by zonisamide. Neurobiol. Dis. 2011;44:185–191. doi: 10.1016/j.nbd.2011.06.015. [DOI] [PubMed] [Google Scholar]
- 10.Steinert J.R., Kopp-Scheinpflug C., Baker C., Challiss R.A.J., Mistry R., Haustein M.D., Griffin S.J., Tong H., Graham B.P., Forsythe I.D. Nitric oxide is a volume transmitter regulating postsynaptic excitability at a glutamatergic synapse. Neuron. 2008;60:642–656. doi: 10.1016/j.neuron.2008.08.025. [DOI] [PubMed] [Google Scholar]
- 11.Micheva K.D., Buchanan J., Holz R.W., Smith S.J. Retrograde regulation of synaptic vesicle endocytosis and recycling. Nat. Neurosci. 2003;6:925–932. doi: 10.1038/nn1114. [DOI] [PubMed] [Google Scholar]
- 12.Collado-Alsina A., Ramírez-Franco J., Sánchez-Prieto J., Torres M. The regulation of synaptic vesicle recycling by cGMP-dependent protein kinase type II in cerebellar granule cells under strong and sustained stimulation. J. Neurosci. 2014;34:8788–8799. doi: 10.1523/JNEUROSCI.0103-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eguchi K., Nakanishi S., Takagi H., Taoufiq Z., Takahashi T. Maturation of a PKG-dependent retrograde mechanism for exoendocytic coupling of synaptic vesicles. Neuron. 2012;74:517–529. doi: 10.1016/j.neuron.2012.03.028. [DOI] [PubMed] [Google Scholar]
- 14.Taoufiq Z., Eguchi K., Takahashi T. Rho-kinase accelerates synaptic vesicle endocytosis by linking cyclic GMP-dependent protein kinase activity to phosphatidylinositol-4,5-bisphosphate synthesis. J. Neurosci. 2013;33:12099–12104. doi: 10.1523/JNEUROSCI.0730-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bredt D.S., Snyder S.H. Isolation of nitric oxide synthetase, a calmodulin-requiring enzyme. Proc. Natl. Acad. Sci. USA. 1990;87:682–685. doi: 10.1073/pnas.87.2.682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Blackshaw S., Eliasson M.J.L., Sawa A., Watkins C.C., Krug D., Gupta A., Arai T., Ferrante R.J., Snyder S.H. Species, strain and developmental variations in hippocampal neuronal and endothelial nitric oxide synthase clarify discrepancies in nitric oxide-dependent synaptic plasticity. Neuroscience. 2003;119:979–990. doi: 10.1016/s0306-4522(03)00217-3. [DOI] [PubMed] [Google Scholar]
- 17.Garthwaite G., Bartus K., Malcolm D., Goodwin D., Kollb-Sielecka M., Dooldeniya C., Garthwaite J. Signaling from blood vessels to CNS axons through nitric oxide. J. Neurosci. 2006;26:7730–7740. doi: 10.1523/JNEUROSCI.1528-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gabbott P.L., Bacon S.J. The organisation of dendritic bundles in the prelimbic cortex (area 32) of the rat. Brain Res. 1996;730:75–86. doi: 10.1016/0006-8993(96)00437-4. [DOI] [PubMed] [Google Scholar]
- 19.Katz B., Miledi R. The role of calcium in neuromuscular facilitation. J. Physiol. 1968;195:481–492. doi: 10.1113/jphysiol.1968.sp008469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Clements J.D., Silver R.A. Unveiling synaptic plasticity: a new graphical and analytical approach. Trends Neurosci. 2000;23:105–113. doi: 10.1016/s0166-2236(99)01520-9. [DOI] [PubMed] [Google Scholar]
- 21.Fourcaudot E., Gambino F., Humeau Y., Casassus G., Shaban H., Poulain B., Lüthi A. cAMP/PKA signaling and RIM1alpha mediate presynaptic LTP in the lateral amygdala. Proc. Natl. Acad. Sci. USA. 2008;105:15130–15135. doi: 10.1073/pnas.0806938105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Del Castillo J., Katz B. Quantal components of the end-plate potential. J. Physiol. 1954;124:560–573. doi: 10.1113/jphysiol.1954.sp005129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ledo A., Barbosa R.M., Gerhardt G.A., Cadenas E., Laranjinha J. Concentration dynamics of nitric oxide in rat hippocampal subregions evoked by stimulation of the NMDA glutamate receptor. Proc. Natl. Acad. Sci. USA. 2005;102:17483–17488. doi: 10.1073/pnas.0503624102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Szatkowski M., Attwell D. Triggering and execution of neuronal death in brain ischaemia: two phases of glutamate release by different mechanisms. Trends Neurosci. 1994;17:359–365. doi: 10.1016/0166-2236(94)90040-x. [DOI] [PubMed] [Google Scholar]
- 25.Ohno M., Yamamoto T., Watanabe S. Intrahippocampal administration of the NO synthase inhibitor L-NAME prevents working memory deficits in rats exposed to transient cerebral ischemia. Brain Res. 1994;634:173–177. doi: 10.1016/0006-8993(94)90273-9. [DOI] [PubMed] [Google Scholar]
- 26.Manabe T., Renner P., Nicoll R.A. Postsynaptic contribution to long-term potentiation revealed by the analysis of miniature synaptic currents. Nature. 1992;355:50–55. doi: 10.1038/355050a0. [DOI] [PubMed] [Google Scholar]
- 27.Manabe T., Nicoll R.A. Long-term potentiation: evidence against an increase in transmitter release probability in the CA1 region of the hippocampus. Science. 1994;265:1888–1892. doi: 10.1126/science.7916483. [DOI] [PubMed] [Google Scholar]
- 28.Schuman E.M., Madison D.V. A requirement for the intercellular messenger nitric oxide in long-term potentiation. Science. 1991;254:1503–1506. doi: 10.1126/science.1720572. [DOI] [PubMed] [Google Scholar]
- 29.Barna M., Komatsu T., Reiss C.S. Activation of type III nitric oxide synthase in astrocytes following a neurotropic viral infection. Virology. 1996;223:331–343. doi: 10.1006/viro.1996.0484. [DOI] [PubMed] [Google Scholar]
- 30.Doyle C.A., Slater P. Localization of neuronal and endothelial nitric oxide synthase isoforms in human hippocampus. Neuroscience. 1997;76:387–395. doi: 10.1016/s0306-4522(96)00297-7. [DOI] [PubMed] [Google Scholar]
- 31.Son H., Hawkins R.D., Martin K., Kiebler M., Huang P.L., Fishman M.C., Kandel E.R. Long-term potentiation is reduced in mice that are doubly mutant in endothelial and neuronal nitric oxide synthase. Cell. 1996;87:1015–1023. doi: 10.1016/s0092-8674(00)81796-1. [DOI] [PubMed] [Google Scholar]
- 32.Zhang H.Q., Fast W., Marletta M.A., Martasek P., Silverman R.B. Potent and selective inhibition of neuronal nitric oxide synthase by N omega-propyl-L-arginine. J. Med. Chem. 1997;40:3869–3870. doi: 10.1021/jm970550g. [DOI] [PubMed] [Google Scholar]
- 33.Szerb J.C., Issekutz B. Increase in the stimulation-induced overflow of glutamate by fluoroacetate, a selective inhibitor of the glial tricarboxylic cycle. Brain Res. 1987;410:116–120. doi: 10.1016/s0006-8993(87)80030-6. [DOI] [PubMed] [Google Scholar]
- 34.Swanson R.A., Graham S.H. Fluorocitrate and fluoroacetate effects on astrocyte metabolism in vitro. Brain Res. 1994;664:94–100. doi: 10.1016/0006-8993(94)91958-5. [DOI] [PubMed] [Google Scholar]
- 35.Zhang J.M., Wang H.K., Ye C.Q., Ge W., Chen Y., Jiang Z.L., Wu C.P., Poo M.M., Duan S. ATP released by astrocytes mediates glutamatergic activity-dependent heterosynaptic suppression. Neuron. 2003;40:971–982. doi: 10.1016/s0896-6273(03)00717-7. [DOI] [PubMed] [Google Scholar]
- 36.Henneberger C., Papouin T., Oliet S.H.R., Rusakov D.A. Long-term potentiation depends on release of D-serine from astrocytes. Nature. 2010;463:232–236. doi: 10.1038/nature08673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pougnet J.T., Toulme E., Martinez A., Choquet D., Hosy E., Boué-Grabot E. ATP P2X receptors downregulate AMPA receptor trafficking and postsynaptic efficacy in hippocampal neurons. Neuron. 2014;83:417–430. doi: 10.1016/j.neuron.2014.06.005. [DOI] [PubMed] [Google Scholar]
- 38.Duffy S., MacVicar B.A. In vitro ischemia promotes calcium influx and intracellular calcium release in hippocampal astrocytes. J. Neurosci. 1996;16:71–81. doi: 10.1523/JNEUROSCI.16-01-00071.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dong Q.P., He J.Q., Chai Z. Astrocytic Ca(2+) waves mediate activation of extrasynaptic NMDA receptors in hippocampal neurons to aggravate brain damage during ischemia. Neurobiol. Dis. 2013;58:68–75. doi: 10.1016/j.nbd.2013.05.005. [DOI] [PubMed] [Google Scholar]
- 40.Ortinski P.I., Dong J., Mungenast A., Yue C., Takano H., Watson D.J., Haydon P.G., Coulter D.A. Selective induction of astrocytic gliosis generates deficits in neuronal inhibition. Nat. Neurosci. 2010;13:584–591. doi: 10.1038/nn.2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Papouin T., Henneberger C., Rusakov D.A., Oliet S.H.R. Astroglial versus Neuronal D-Serine: Fact Checking. Trends Neurosci. 2017;40:517–520. doi: 10.1016/j.tins.2017.05.007. [DOI] [PubMed] [Google Scholar]
- 42.Lu L., Hogan-Cann A.D., Globa A.K., Lu P., Nagy J.I., Bamji S.X., Anderson C.M. Astrocytes drive cortical vasodilatory signaling by activating endothelial NMDA receptors. J. Cerebr. Blood Flow Metabol. 2019;39:481–496. doi: 10.1177/0271678X17734100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stobart J.L.L., Lu L., Anderson H.D.I., Mori H., Anderson C.M. Astrocyte-induced cortical vasodilation is mediated by D-serine and endothelial nitric oxide synthase. Proc. Natl. Acad. Sci. USA. 2013;110:3149–3154. doi: 10.1073/pnas.1215929110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim K.S., Jeon M.T., Kim E.S., Lee C.H., Kim D.G. Activation of NMDA receptors in brain endothelial cells increases transcellular permeability. Fluids Barriers CNS. 2022;19:70. doi: 10.1186/s12987-022-00364-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Borisova T. Permanent dynamic transporter-mediated turnover of glutamate across the plasma membrane of presynaptic nerve terminals: arguments in favor and against. Rev. Neurosci. 2016;27:71–81. doi: 10.1515/revneuro-2015-0023. [DOI] [PubMed] [Google Scholar]
- 46.Bai J., Tucker W.C., Chapman E.R. PIP2 increases the speed of response of synaptotagmin and steers its membrane-penetration activity toward the plasma membrane. Nat. Struct. Mol. Biol. 2004;11:36–44. doi: 10.1038/nsmb709. [DOI] [PubMed] [Google Scholar]
- 47.Chen Y., Wang Y.H., Zheng Y., Li M., Wang B., Wang Q.W., Fu C.L., Liu Y.N., Li X., Yao J. Synaptotagmin-1 interacts with PI(4,5)P2 to initiate synaptic vesicle docking in hippocampal neurons. Cell Rep. 2021;34 doi: 10.1016/j.celrep.2021.108842. [DOI] [PubMed] [Google Scholar]
- 48.Tao W., Lee J., Chen X., Díaz-Alonso J., Zhou J., Pleasure S., Nicoll R.A. Synaptic memory requires CaMKII. Elife. 2021;10 doi: 10.7554/eLife.60360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Watanabe M., Inoue Y., Sakimura K., Mishina M. Distinct distributions of five N-methyl-D-aspartate receptor channel subunit mRNAs in the forebrain. J. Comp. Neurol. 1993;338:377–390. doi: 10.1002/cne.903380305. [DOI] [PubMed] [Google Scholar]
- 50.Schipke C.G., Ohlemeyer C., Matyash M., Nolte C., Kettenmann H., Kirchhoff F. Astrocytes of the mouse neocortex express functional N-methyl-D-aspartate receptors. Faseb. J. 2001;15:1270–1272. doi: 10.1096/fj.00-0439fje. [DOI] [PubMed] [Google Scholar]
- 51.Lee M.C., Ting K.K., Adams S., Brew B.J., Chung R., Guillemin G.J. Characterisation of the expression of NMDA receptors in human astrocytes. PLoS One. 2010;5 doi: 10.1371/journal.pone.0014123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Palygin O., Lalo U., Pankratov Y. Distinct pharmacological and functional properties of NMDA receptors in mouse cortical astrocytes. Br. J. Pharmacol. 2011;163:1755–1766. doi: 10.1111/j.1476-5381.2011.01374.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lalo U., Pankratov Y., Kirchhoff F., North R.A., Verkhratsky A. NMDA receptors mediate neuron-to-glia signaling in mouse cortical astrocytes. J. Neurosci. 2006;26:2673–2683. doi: 10.1523/JNEUROSCI.4689-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hösli L., Zuend M., Bredell G., Zanker H.S., Porto de Oliveira C.E., Saab A.S., Weber B. Direct vascular contact is a hallmark of cerebral astrocytes. Cell Rep. 2022;39 doi: 10.1016/j.celrep.2022.110599. [DOI] [PubMed] [Google Scholar]
- 55.Mulligan S.J., MacVicar B.A. Calcium transients in astrocyte endfeet cause cerebrovascular constrictions. Nature. 2004;431:195–199. doi: 10.1038/nature02827. [DOI] [PubMed] [Google Scholar]
- 56.Shesely E.G., Maeda N., Kim H.S., Desai K.M., Krege J.H., Laubach V.E., Sherman P.A., Sessa W.C., Smithies O. Elevated blood pressures in mice lacking endothelial nitric oxide synthase. Proc. Natl. Acad. Sci. USA. 1996;93:13176–13181. doi: 10.1073/pnas.93.23.13176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Roome C.J., Kuhn B. Simultaneous dendritic voltage and calcium imaging and somatic recording from Purkinje neurons in awake mice. Nat. Commun. 2018;9:3388. doi: 10.1038/s41467-018-05900-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Georgiou L., Echeverría A., Georgiou A., Kuhn B. Ca(+) activity maps of astrocytes tagged by axoastrocytic AAV transfer. Sci. Adv. 2022;8 doi: 10.1126/sciadv.abe5371. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Data reported in this paper will be shared by the lead contact upon reasonable request.
-
•
No unpublished custom code, software, or algorithm was used in this study.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.